Note: Descriptions are shown in the official language in which they were submitted.
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
TITLE OF THE INVENTION
IN VITRO ARTIFICIAL LYMPH NODE FOR SENSITIZATION AND EXPANSION OF T
CELLS FOR THERAPY AND EPITOPE MAPPING
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims priority to U.S. Provisional Application Serial No.
62/138,684, filed March 26, 2015, and to U.S. Provisional Application Serial
No. 62/138,969,
filed March 26, 2015, the contents of each of which are incorporated by
reference herein in
their entireties.
BACKGROUND OF THE INVENTION
HER2+ invasive breast cancer (IBC) patients with residual disease following
neoadjuvant chemotherapy have an anti-HER2 Type 1 T helper (Thl) cell immune
deficit
and a significant risk of recurrent disease. It has been shown that anti-HER2
CD4+ T-cell
responses incrementally decrease along the breast cancer continuum ¨ a robust
response in
healthy donors and patients with benign disease, a depressed response in
patients with
HER2+ ductal carcinoma in situ, and a nearly absent response in patients with
HER2+ IBC.
The lifetime risk of breast cancer development is nearly one in eight. The erb-
B2 oncogene (HER-2/neu) is a molecular driver that is Overexpressed in a
significant number
of breast, ovarian, gastric esophageal, lung, pancreatic, prostate and other
solid tumors. HER2
overexpression ("HER2ws"), a molecular oncodriver in several tumor types
including
approximately 20-25% of breast cancers (Meric, F., et al., J. Am Coll. Surg.
194:488-501
(2002)), is associated with an aggressive clinical course, resistance to
chemotherapy, and a
poor overall prognosis in BC. See, Henson, E.S., Clin. Can. Res. 12:845-53
(2006) ("Henson,
et al.") and Wang, G.S., Mol. Med. Rep. 6:779-82 (2012). In incipient BC, HER2
overexpression is associated with enhanced invasiveness (Roses, R.E., et al.,
Cancer
Epidemiol. Biomarkers & Prey. 18(5):1386-9 (2009)), tumor cell migration (Wolf-
Yadlin, A.,
et al., Molecular Systems Biology 2:54 (2006)), and the expression of
proangiogenic factors
(Wen, X.F., et al., Oncogene 25:6986-96 (2006)), suggesting a critical role
for HER2 in
promoting a tumorigenic environment. In a retrospective analysis of ductal
carcinoma in situ
("DCIS") patients, DCIS lesions overexpressing HER2 were over six times as
likely to be
associated with invasive breast cancer than were DCIS lesions without HER2
overexpression.
Although molecular targeting therapies targeting HER2, (i.e.,
Herceptin /trastuzumab), in combination with chemotherapy, have significantly
improved
1
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
survival in HER2Pc's BC patients (Piccart-Gebhart., M.J., et al., N Eng. .I.
Med. 353:1659-72
(2005)), a substantial proportion of patients become resistant to such
therapies (Pohlmann,
P.R., et al., Clin. Can. Res. 15:7479-91 (2009) ("Pohlman, et al.")).
Strategies to identify
patient subgroups at high risk of treatment failure, as well as novel
approaches to improve
response rates to HER2-targeted therapies, are needed. Although molecular
targeting
therapies targeting HER2, i.e., trastuzumab, has resulted in tremendous
positive clinical effect
in this type of breast cancer, the almost universal resistance to the existing
HER2 therapies in
advanced disease states, plus disease relapse in a sizeable proportion of
women who receive
the targeted therapy prove the need for additional strategies targeting HER2.
The promise of
vaccines that activate the immune system against HER2 which seek to mitigate
tumor
progression and preventing recurrence while encouraging, is yet to be fully
realized. There
remains a need for additional tests and therapies to diagnose and treat HER2
breast cancer.
The role of systemic anti-HER2 CD4+Th1 responses in HER2-driven breast
tumorigenesis, have been recently elucidated. There has been identified a
progressive loss of
anti-HER2 CD4+ Thl response across a tumorigenic continuum in HER2Ps -breast
cancer,
which appears to be HER2-specific and regulatory T-cell (Treg)-independent.
Specifically,
there is an inverse correlation of anti-HER2 CD4I Thl responses with HER2
expression and
disease progression. Thl reactivity profiles show a significant stepwise
decline in anti-HER2
Thl immunity across a continuum (HD (healthy donors)aBD (benign breast
biopsy)AHER2neg-DCIS (ductal carcinoma in situ)aHER2neg-IBC (invasive breast
cancer)A1IER2wsDCISAHER2P"-IBC (invasive breast cancer) in HER2P0' breast
tumorigenesis. See, Datta, J., et al., OncoImmunology 2015 (in press) and U.S.
Ser. No.
14/658,095 filed March 13, 2015 (collectively hereinafter, "Datta, et al.").
The depressed
anti-HER2 Thl responses in HER2Ps-invasive breast cancer were differentially
restored after
HER2-pulsed type-1 polarized dendritic cell ("DC1") vaccinations, but the
depressed
responses were not restored following HER2-targeted therapy with trastuzumab
and
chemotherapy ("T/C") or by other standard therapies such as surgical resection
or radiation.
Id. The restored anti-HER2 Thl responses also appear to be durable for at
least about six
months or longer.
The expansion of T lymphocyte subsets (CD4 or CD81) is an essential step to
gain enough T cells to perform adoptive therapy, or to identify epitopes on
target antigens for
peptide-based vaccines. Expansion of T cells, in principle is a simple
process. However, in
practice, many technical problems exist including poor levels of expansion,
premature
activation-induced cell death (apoptosis), or loss of antigen specificity
and/or function.
2
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
Part of the problem lies in the inability to replicate, in vitro, the
environment
inside the body where antigen-specific T cell expansion occurs, which is the
lymph node.
These are specialized tissues that contain a number of different cell types
apart from T
lymphocytes including antigen-presenting dendritic cells and stromal cells
such as epithelial
cells. Each of these cell types plays a different role (both currently defined
and as yet
incompletely characterized) by providing contact-dependent signals (surface
receptors) and
soluble signals (cytokines) important for T cell growth and maintenance of
cell function.
There remains a need for new methods of treating cancer. Accordingly, there
is a need in the art to have additional immunotherapeutic approaches for
treating or
preventing breast cancer and other malignancies. The present invention
fulfills this need.
SUMMARY OF THE INVENTION
In one embodiment, the present invention provides a method of expanding a T
cell. In one embodiment, the method comprises contacting the T cell with one
or more of a
dendritic cell, at least two cytokines, and a T cell growth factor.
In one embodiment, the T cell is contacted with an antigen thereby generating
an antigen specific T cell.
In one embodiment, the dendritic is type I dendritic cell.
In one embodiment, the at least two cytokines is interleukin-7 (IL-7) and
interleukin-15 (IL-15).
In one embodiment, the T cell growth factor is interleukin-2 (1L-2).
In one embodiment, the method comprises: a) contacting the T cell with
autologous type I dendritic cells DCs in combination with an antigen in vitro
thereby
generating an antigen specific T cell; b) contacting the antigen specific T
cells with IL-7 and
IL-5 to generate a stimulated antigen specific T cell; c) contacting the
stimulated antigen
specific T cell with IL-2 thereby generating an expanded antigen specific T
cell population
that maintains antigen specificity and cellular function.
In one embodiment, the invention provides a T cell generated by a method of
the invention.
In one embodiment, the invention provides a population of cultured expanded
antigen specific T cell exhibiting antitumor activity generated by a method
according to
claims 1-6, wherein the cells are expanded to a number sufficient for
effective therapy in a
mammal.
3
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
In one embodiment, the invention provides a population of cultured expanded
antigen specific T cell exhibiting antitumor activity generated by a method of
the invention,
wherein the cells are expanded to a number sufficient for effective epitope
mapping.
In one embodiment, the invention provides an isolated polynucleotide
encoding a T cell receptor (TCR) that is derived from an antigen specific T
cell, wherein the
antigen specific T cell is generated by a method of the invention.
In one embodiment, the invention provides a method of immunotherapy
comprising administering a T cell to a subject in need thereof, wherein the T
cell is generated
by a method of the invention.
In one embodiment, the invention provides a method of expanding a T cell
population which comprises at least one T cell obtained from a blood sample
from a subject
who has been vaccinated against an antigen, comprising the step of: contacting
the T cell with
one or more of a dendritic cell or a precursor thereof, at least two
cytokines, and a T cell
growth factor.
In one embodiment, the blood sample contains at least one T cell of the
population specific for the vaccine antigen and at least one DC precursor.
In one embodiment, the DC precursor is pulsed with the antigen and activated
to an antigen-specific type I dendritic cell (DC1) and then co-cultured with
the T cell to
generate an antigen-specific DC1.
In one embodiment, the at least two cytokines is interleukin-7 (IL-7) and
interleukin-15 (IL-15).
In one embodiment, the T cell growth factor is interleukin-2 (IL-2).
In one embodiment, the method comprises: a) co-culturing the T cell from the
patient sample with the antigen-specific T cell autologous type I dendritic
cell (DC1) in vitro;
b) contacting the cells from step a) with IL-7 and IL-5 to generate a
stimulated antigen-
specific T cell; c) subsequently contacting the stimulated antigen specific T
cell with IL-2,
thereby generating an expanded antigen specific T cell population that
maintains antigen
specificity and cellular function.
ln one embodiment, the method further comprises repeating step a) through c)
from one to at least three additional times to generate further expanded
antigen-specific T cell
populations.
In one embodiment, T cell is CD4+.
In one embodiment, the antigen is HER2.
4
CA 02981033 2017-09-26
WO 2016/154508
PCT/11S2016/024146
In one embodiment, the invention provides a method of expanding a CD4+ T
cell population which comprises at least one CD4+ T cell obtained from a blood
sample from
a breast cancer patient who has been vaccinated against HER2, comprising the
step of:
contacting the CD4+ T cell with one or more of a dendritic cell or a precursor
thereof, at least
two cytokines, and a T cell growth factor.
In one embodiment, at least one DC precursor in the sample is pulsed with
MHC class H HER2 peptide and is contacted with the CD4+ T cell.
In one embodiment, the method comprises: a) co-culturing the T cell from
claiml 1 with the HER2-pulsed DC1; b) contacting the cells from step a) with
IL-7 and 1L-5
to generate a stimulated antigen-specific T cell; c) subsequently contacting
the stimulated
antigen specific T cell with IL-2, thereby generating an expanded antigen
specific T cell
population that maintains antigen specificity and cellular function.
In one embodiment, the method further comprises repeating step a) through c)
from one to at least three additional times to generate further expanded
antigen-specific T cell
populations.
BRIEF DESCRIPTION OF THE DRAWINGS
The following detailed description of preferred embodiments of the invention
will be better understood when read in conjunction with the appended drawings.
For the
purpose of illustrating the invention, there are shown in the drawings
embodiments which are
presently preferred. It should be understood, however, that the invention is
not limited to the
precise arrangements and instrumentalities of the embodiments shown in the
drawings.
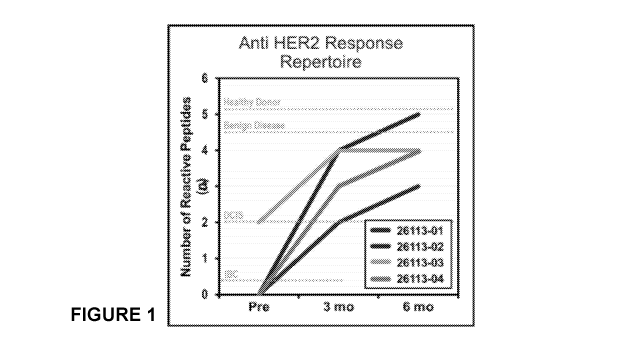
Figure 1 shows anti-HER2 Thl response repertoire of four HER2 IBC
patients with residual disease following neoadjuvant therapy who received
adjuvant HER2-
pulsed DC1 vaccines. Each patient is depicted in a different color and shows
number of
reactive peptides (n) (also referred to as "response repertoire") pre-vaccine,
3-months-post
vaccine, and 6-months post vaccine. Mean response repertoire increased from
0.5 1 peptides
pre-vaccination to 3.25 0.96 peptides at 3 months post-vaccination (p=0.01)
and 410.8
peptides at 6 months post-vaccination (p=0.01).
Figure 2 shows anti HER2 Thl cumulative response of four HER2' IBC
patients with residual disease following neoadjuvant therapy who received
adjuvant HER2-
pulsed DC1 vaccines. Each patient is depicted in a different color and shows
cumulative
response (SEC/106 cells) pre-vaccine, 3-months-post vaccine, and 6-months post
vaccine.
Patient mean cumulative response improved from 36.5138.3 SFC/106pre-
vaccination to
5
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
151.0 60.0 SFC/106 at 3 months post vaccination (p=0.04) and 198.4 39.7
SFC/106 at 6
months post vaccination (1)=0.02).
Figure 3 and Figure 4 show a direct comparison between CD4+T cells co-
cultured with HER2-specific DC1's from patients vaccinated with HER2
peptidepulsed DC1
vaccines stimulated with IL-2 versus those stimulated with IL-2/7/15 for two
different
patients, respectively. Immature DC's ("iDC's") from the respective patients
were pulsed
with the following MHC class II peptides: peptide 42-56 (SEQ ID NO: 1),
peptide 98-114
(SEQ ID NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide 776-790 (SEQ ID
NO: 4)
and matured to DC1's. The resulting HER2-pulsed DC1's were then co-cultured
with CD4' T
cells and stimulated with IL-2 alone or with IL-2/7/15 as indicated. The red
outline boxes
indicate the specific peptide and stimulation protocol for which specificity
is shown (greater
than 2:1 ratio of specific antigen:control antigen IFN-y production). For each
set of
peptide/stimulation protocol: "Control antigen" shows non-specific iDC's co-
cultured with
control antigen; "Specific antigen" represents anti-HER2 CD4+ T cells co-
cultured with
iDC's that were pulsed with HER2 antigen/peptide; and ¨Ice11" represents anti-
HER2 CD4+
T cells in culture medium. Graphs showing fold expansion (defined as number of
T cells post
expansion/number of T cells pre expansion) are shown at right, respectively.
Specificity was
measured by antigen-specific IFN-y production by ELISA.
Figure 5 and Figure 6 show specific responses followed by nonspecific
immune responses: Figure 5 shows a specific response following a first
stimulation/expansion with HER2-specific DC1's and Figure 6 shows the
subsequent loss of
that specific response after the second stimulation/expansion with non-
specific anti
CD3/CD28. The first stimulation of CD4+T cells with HER2-specific DC1s
resulted in
multiple specific immune responses as shown by red outline boxes in Figure 5.
Figure 6
shows the second stimulation of the HER2-specific CD4+ T cells with a non-
specific anti-
CD3/CD28 stimulus resulted in a four-fold expansion (side graph), but with a
loss of
specificity in three fourths of the peptide groups. iDC's from patients were
pulsed with the
following MHC class II peptides: peptide 42-56 (SEQ ID NO: 1), peptide 98-114
(SEQ ID
NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide 776-790 (SEQ ID NO: 4) and
matured
to DC1's. The resulting HER2-pulsed DC1's were then co-cultured with CD4' T
cells and
stimulated with IL-2 alone or with IL-2/7/15 as indicated.
Figure 7 and Figure 8 show non-specific immune response followed by
specific immune responses: Figure 7 shows non-specific expansion of CD4+T
cells. Figure 8
shows failure to obtain specificity following subsequent stimulation with HER2-
specific
6
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
DC1's. The first stimulation of CD4' T cells with non-specific anti CD3/CD28
resulted in a
3.8 fold expansion (Figure 7). The second stimulation of the non-specific
CD4'T cells with
HER2-specific DC1's failed to result in a specific immune response (Figure 8).
iDC's from
patients were pulsed with the following MHC class If peptides: peptide 42-56
(SEQ ID NO:
1), peptide 98-114 (SEQ ID NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide
776-790
(SEQ ID NO: 4) and matured to DC1's. The resulting HER2-pulsed DC1's were then
co-
cultured with CD4+ T cells and stimulated with IL-2 alone or with IL-2/7/15 as
indicated.
Figures 9A and 9B show in vitro primary/first expansion of HER2specific Thl
cells comparing CD4' T cells co-cultured with HER2-specific DC1's expanded
with 1L-2
versus those expanded with IL-2/7/15. Immature DC's ("iDC's") were pulsed with
the
following MHC class II peptides: peptide 42-56 (SEQ ID NO: 1), peptide 98-114
(SEQ ID
NO: 2), peptide 776-790 (SEQ ID NO: 4), and peptide 927-941 (SEQ ID NO: 5),
and
matured to DC1's. The resulting HER2-pulsed DC1's were then co-cultured with
CD4+T
cells and stimulated with IL-2 alone or with IL-2/7/15 as indicated. The red
outline boxes
(Figure 9B) indicate the specific peptide and stimulation protocol for which
specificity is
shown (greater than 2:1 ratio of specific antigen:control antigen IFN-y
production). For each
set of peptide/stimulation protocol: "Control Antigen" shows non-specific
iDC's cocultured
with control antigen; "Specific Antigen" represents anti-HER2 CD4+ T cells co-
cultured with
iDC's that were pulsed with HER2 antigen/peptide; and "T cells" represents
anti-HER2
CD4+ T cells in culture medium. Figure 9A shows mean fold expansion (defined
as number
of T cells post expansion/number of T cells pre expansion) of Thl cells was
significantly
better when stimulated with IL-2, IL-7, and IL-15 than with IL-2 alone (2.6
0.75 vs
1.0 0.12; p=0.001). Figure 9B shows specificity for the various
peptide/expansion protocols
as measured by antigen-specific IFN-y production by ELISA. Both stimulation
with IL-2, IL-
7, and IL-15 and with IL-2 alone resulted in a specific Thl response in the
same HER2
peptide 776-790.
Figures 10A and 10B show in vitro secondary/second expansion of HER2-
pulsed DC1's versus anti-CD3/CD28. Re-stimulation of Thl cells with HER2-
peptide pulsed
DC1s and anti-CD3/CD28 each resulted in a similar fold expansion (3.9 1.0
vs.4.3+2.0
p=0.7) (Figure 10A). However, Figure 10B shows stimulation of the 'Thl cells
with HER2-
specific DC's enhanced the specific Thl response; whereas non-specific
stimulation with
anti-CD3/CD28 resulted in an overall loss of HER2-peptide specificity. The
following MHC
class II peptides: peptide 42-56 (SEQ ID NO: 1), peptide 98-114 (SEQ ID NO:
2), peptide
776-790 (SEQ ID NO: 4), and peptide 927-941 (SEQ ID NO: 5) were used. The red
outline
7
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
boxes (Figure 10B) indicate the specific peptide and stimulation protocol for
which
specificity is shown (greater than 2:1 ratio of specific antigen:control
antigen IFN-y
production) (i.e., DC1 restimulation of peptide 42-56-and peptide 776-790
¨specific Thl
cells. For each set of peptide/stimulation protocol: "Control Antigen" shows
non-specific
iDC's co-cultured with control antigen; "Specific Antigen" represents anti-
HER2 CD4+ T
cells co-cultured with iDC's that were pulsed with HER2 antigen/peptide; and
"T cells"
represents anti-HER2 CD4+ T cells in culture medium.
Figures 11A and 11B show tertiary/third expansion of the Thl cells with
HER2-pulsed DC1's ( peptide 42-56 (SEQ ID NO: 1), peptide 98-114 (SEQ ID NO:
2),
peptide 776-790 (SEQ ID NO: 4), and peptide 927-941 (SEQ ID NO: 5) were used).
Following a third stimulation with indicated HER2-specific DC1s, both mean
fold expansion
(4.32 0.5, 43.7-fold cumulative expansion ( Figure 11A) and antigen
specificity (Figure 11B)
increased again, specifically all four peptides show specificity and increased
IFN-y
production.
Figures 12 through15 show sequential results of repeated in vitro stimulation
(4 times) of HER2-specifc CD4+Th1 cells with IL-2/7/15. For all Figures 12-15
the
respective left panels show peptide specificity by IFN-y production ("Tel" is
a tetanus patient
control); respective right panels show fold expansion for the specific
HER2peptides used. In
Figure 12 two additional MHC-class II peptides were used to pulse iDC's:
peptide 927-941
(SEQ ID NO: 5); and peptide 1 166-1 180 (SEQ ID NO: 6) in addition to the
other four used in
above figures. However, as seen in the fold expansion results (Figure 12,
right panel),
peptide 328-345-specific and peptide 1166-1180-specific Thl cells did not
produce enough
cells for further expansion, thus only HER2 Thl cells specific to the
remaining four peptides
were so used. Sequentially, Figure 12 for the first stimulation shows
specificity only for
peptide 776-790-specific Thl cells; Figure 13 for the second stimulation shows
an increase,
specificity for peptide 42-56-and peptide 776-790-specific Thl cells; Figure
14 for the third
expansion shows specificity for all four peptides, and Figure 15 for the
fourth expansion
shows loss of specificity for one of the peptides (peptide 927-941) leaving
three remaining
HER2-specific peptides.
Figure 16 shows cumulative fold expansion of the four expansions shown in
Figures 12-15 for all the HER2-specific Thl cells, with the last bar of each
group (dots)
showing cumulative fold expansion.
DETAILED DESCRIPTION
8
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
This invention relates to a method of creating a microenvironment in vitro for
culture expansion of antigen-specific helper-T cells. The expanded antigen-
specific T cells
can be used for a variety of therapeutic and research purposes, for example
adoptive T cell
therapy for cancer or other conditions and or for the identification of epi
topes on target
antigens to foster the production of peptide-based vaccines.
In one embodiment, the invention comprises the use of autologous type I
dendritic cells (DCs) in combination with a protein or peptide antigen to
stimulate T cells in
vitro. After stimulation, at least two soluble factors (e.g., cytokines) are
added to the T cells.
In some instances, the at least two soluble factors are Interleukin-7 (IL-7)
and Interleukin-15
(IL-15). Following the addition of the soluble factors to the T cells, a T
cell growth factor is
added. In some instances, the T cell growth factor is Interleukin-2 (IL-2).
The soluble factors,
in addition to those naturally produced by the Type I DCs, support the
proliferation and
acquisition/maintenance of T cell function. This process of stimulation can be
repeated in
weekly cycles until T cells are of sufficient numbers for therapy or epitope
scanning/mapping. In one embodiment, the T cells are expanded to a level
necessary for
adoptive therapy and epitope mapping studies while maintaining antigen
specificity and
cellular function.
Definitions
Unless defined otherwise, all technical and scientific terms used herein have
the same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Although any methods and materials similar or equivalent to
those
described herein can be used in the practice or testing of the present
invention, the preferred
methods and materials are described.
Generally, the nomenclature used herein and the laboratory procedures in cell
culture, molecular genetics, organic chemistry, and nucleic acid chemistry and
hybridization
are those well-known and commonly employed in the art.
Standard techniques are used for nucleic acid and peptide synthesis. The
techniques and procedures are generally performed according to conventional
methods in the
art and various general references (e.g., Sambrook and Russell, 2012,
Molecular Cloning, A
Laboratory Approach, Cold Spring Harbor Press, Cold Spring Harbor, NY, and
Ausubel et
al., 2012, Current Protocols in Molecular Biology, John Wiley & Sons, NY),
which are
provided throughout this document.
9
CA 02981033 2017-09-26
WO 2016/154508 PCT/1.182016/024146
The nomenclature used herein and the laboratory procedures used in analytical
chemistry and organic syntheses described below are those well-known and
commonly
employed in the art. Standard techniques or modifications thereof are used for
chemical
syntheses and chemical analyses.
As used herein, each of the following terms has the meaning associated with it
in this section.
The articles "a" and "an" are used herein to refer to one or to more than one
(i.e., to at least one) of the grammatical object of the article. By way of
example, "an
element" means one element or more than one element.
"About" as used herein when referring to a measurable value such as an
amount, a temporal duration, and the like, is meant to encompass variations of
+20%, or
1C0/0, or 5%, or 1%, or 0.1% from the specified value, as such variations
are appropriate
to perform the disclosed methods.
The term "abnormal" when used in the context of organisms, tissues, cells or
components thereof, refers to those organisms, tissues, cells or components
thereof that differ
in at least one observable or detectable characteristic (e.g., age, treatment,
time of day, etc.)
from those organisms, tissues, cells or components thereof that display the
"normal"
(expected) respective characteristic. Characteristics which are normal or
expected for one cell
or tissue type, might be abnormal for a different cell or tissue type.
The term "activation", as used herein, refers to the state of a cell following
sufficient cell surface moiety ligation to induce a noticeable biochemical or
morphological
change. Within the context of T cells, such activation refers to the state of
a T cell that has
been sufficiently stimulated to induce cellular proliferation. Activation of a
T cell may also
induce cytokine production and performance of regulatory or cytolytic effector
functions.
Within the context of other cells, this term infers either up or down
regulation of a particular
physico-chemical process. The term "activated T cells" indicates T cells that
are currently
undergoing cell division, cytokine production, performance of regulatory or
cytolytic effector
functions, and/or has recently undergone the process of "activation."
"Adjuvant therapy" for breast cancer as used herein refers to any treatment
given after primary therapy (i.e., surgery) to increase the chance of long-
term survival.
"Neoadjuvant therapy" is treatment given before primary therapy.
The term "antigen" or "ag" as used herein is defined as a molecule that
provokes an immune response. This immune response may involve either antibody
production, or the activation of specific immunologically-competent cells, or
both. The
CA 02981033 2017-09-26
WO 2016/154508 PCT/IJS2016/024146
skilled artisan will understand that any macromolecule, including virtually
all proteins or
peptides, can serve as an antigen. Furthermore, antigens can be derived from
recombinant or
genomic DNA. A skilled artisan will understand that any DNA, which comprises a
nucleotide
sequences or a partial nucleotide sequence encoding a protein that elicits an
immune response
therefore encodes an "antigen" as that term is used herein. Furthermore, one
skilled in the art
will understand that an antigen need not be encoded solely by a full length
nucleotide
sequence of a gene. It is readily apparent that the present invention
includes, but is not limited
to, the use of partial nucleotide sequences of more than one gene and that
these nucleotide
sequences are arranged in various combinations to elicit the desired immune
response.
Moreover, a skilled artisan will understand that an antigen need not be
encoded by a "gene"
at all. It is readily apparent that an antigen can be generated synthesized or
can be derived
from a biological sample. Such a biological sample can include, but is not
limited to a tissue
sample, a tumor sample, a cell or a biological fluid.
The term "agent," "ligand," or "agent that binds a cell surface moiety," as
used
herein, refers to a molecule that binds to a defined population of cells. The
agent may bind
any cell surface moiety, such as a receptor, an antigenic determinant, or
other binding site
present on the target cell population. The agent may be a protein, peptide,
antibody and
antibody fragments thereof, fusion proteins, synthetic molecule, an organic
molecule (e.g., a
small molecule), a carbohydrate, or the like. Within the specification and in
the context of T
cell stimulation, antibodies and natural ligands are used as prototypical
examples of such
agents.
The terms "agent that binds a cell surface moiety" and "cell surface moiety",
as used herein, are used in the context of a ligand/anti-ligand pair.
Accordingly, these
molecules should be viewed as a complementary/anti-complementary set of
molecules that
demonstrate specific binding, generally of relatively high affinity.
"An antigen presenting cell" (APC) is a cell that are capable of activating T
cells, and includes, but is not limited to, monocytes/macrophages, B cells and
dendritic cells
(DCs).
"Antigen-loaded APC" or an "antigen-pulsed APC" includes an APC, which
has been exposed to an antigen and activated by the antigen. For example, an
APC may
become Ag-loaded in vitro, e.g., during culture in the presence of an antigen.
The APC may
also be loaded in vivo by exposure to an antigen. An "antigen-loaded APC" is
traditionally
prepared in one of two ways: (1) small peptide fragments, known as antigenic
peptides, are
"pulsed" directly onto the outside of the APCs; or (2) the APC is incubated
with whole
11
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
proteins or protein particles which are then ingested by the APC. These
proteins are digested
into small peptide fragments by the APC and are eventually transported to and
presented on
the APC surface. In addition, the antigen-loaded APC can also be generated by
introducing a
polynucleotide encoding an antigen into the cell.
"Anti-HER2 response" is the immune response specifically against HER2
protein.
The term "anti-tumor effect" as used herein, refers to a biological effect
which
can be manifested by a decrease in tumor volume, a decrease in the number of
tumor cells, a
decrease in the number of metastases, an increase in life expectancy, or
amelioration of
various physiological symptoms associated with the cancerous condition. An
"anti-tumor
effect" can also be manifested by the ability of the peptides,
polynucleotides, cells and
antibodies of the invention in prevention of the occurrence of tumor in the
first place.
The term "autoimmune disease" as used herein is defined as a disorder that
results from an autoimmune response. An autoimmune disease is the result of an
inappropriate and excessive response to a self-antigen. Examples of autoimmune
diseases
include but are not limited to, Addision's disease, alopecia areata,
ankylosing spondylitis,
autoimmune hepatitis, autoimmune parotitis, Crohn's disease, diabetes (Type
I), dystrophic
epidermolysis bullosa, epididymitis, glomerulonephritis, Graves' disease,
Guillain-Barr
syndrome, Hashimoto's disease, hemolytic anemia, systemic lupus erythematosus,
multiple
sclerosis, myasthenia gravis, pemphigus vulgaris, psoriasis, rheumatic fever,
rheumatoid
arthritis, sarcoidosis, scleroderma, Sjogren's syndrome,
spondyloarthropathies, thyroiditis,
vasculitis, vitiligo, myxedema, pernicious anemia, ulcerative colitis, among
others.
As used herein, the term "autologous" is meant to refer to any material
derived
from the same individual to which it is later to be re-introduced into the
individual.
"Allogeneic" refers to a graft derived from a different animal of the same
species.
The term "B cell" as used herein is defined as a cell derived from the bone
marrow and/or spleen. B cells can develop into plasma cells which produce
antibodies.
"CD4+ Thl cells," "Th1 cells," "CIA+ T-helper type lcells," "CD4+T cells,"
and the like are defined as a subtype of T-helper cells that express the
surface protein CD4
and produce high levels of the cytokine IFN-y. See also, "T-helper cells."
The term "cancer" as used herein is defined as a hyperproliferation of cells
whose unique trait¨loss of normal control--results in unregulated growth, lack
of
differentiation, local tissue invasion, and/or metastasis. Examples include
but are not limited
12
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
to, breast cancer, prostate cancer, ovarian cancer, cervical cancer, skin
cancer, pancreatic
cancer, colorectal cancer, renal cancer, liver cancer, brain cancer, lymphoma,
leukemia, lung
cancer, germ-cell tumors, and the like.
A "co-stimulatory signal", as used herein, refers to a signal, which in
combination with a primary signal, such as TCR/CD3 ligation, leads to T cell
proliferation
and/or upregulation or downregulation of key molecules.
A "co-stimulatory signal", as used herein, refers to a signal, which in
combination with a primary signal, such as TCR/CD3 ligation, leads to T cell
proliferation
and/or upregulation or downregulation of key molecules.
A "co-stimulatory molecule" refers to the cognate binding partner on a T cell
that specifically binds with a co-stimulatory ligand, thereby mediating a co-
stimulatory
response by the T cell, such as, but not limited to, proliferation.
"Co-stimulatory ligand," as the term is used herein, includes a molecule on an
antigen presenting cell (e.g., a dendritic cell, B cell, and the like) that
specifically binds a
cognate co-stimulatory molecule on a T cell, thereby providing a signal which,
in addition to
the primary signal provided by, for instance, binding of a TCR/CD3 complex
with an MHC
molecule loaded with peptide, mediates a T cell response, including, but not
limited to,
proliferation, activation, differentiation, and the like. A co-stimulatory
ligand can include, but
is not limited to, CD7, B7-1 (CD80), B7-2 (CD86), PD-L1, PD-L2, 4-1BBL, OX4OL,
inducible costimulatory ligand (ICOS-L), intercellular adhesion molecule
(ICAM), CD3OL,
CD40, CD70, CD83, HLA-G, MICA, MICB, HVEM, lymphotoxin beta receptor, 3/TR6,
ILT3, ILT4, HVEM, an agonist or antibody that binds Toll ligand receptor and a
ligand that
specifically binds with B7-H3.
"Cumulative response" means the combined immune response of a patient
group expressed as the total sum of reactive spots (spot-forming cells "SFC"
per 106 cells
from IFN-y ELISPOT analysis) from all 6 MHC class II binding peptides from a
given
patient group.
The term "dendritic cell" (DC) is an antigen presenting cell existing in vivo,
in
vitro, ex vivo, or in a host or subject, or which can be derived from a
hematopoietic stem cell
or a monocyte. Dendritic cells and their precursors can be isolated from a
variety of lymphoid
organs, e.g., spleen, lymph nodes, as well as from bone marrow and peripheral
blood. The
DC has a characteristic morphology with thin sheets (lamellipodia) extending
in multiple
directions away from the dendritic cell body. Typically, dendritic cells
express high levels of
MHC and costimulatory (e.g., B7-1 and B7-2) molecules. Dendritic cells can
induce antigen
13
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
specific differentiation of T cells in vitro, and are able to initiate primary
T cell responses in
vitro and in vivo.
"DC vaccination," "DC immunization," "DC1 immunization," and the like
refer to a strategy using autologous dendritic cells to hamess the immune
system to recognize
specific molecules and mount specific responses against them.
"DC-1 polarized dendritic cells," "DC 1s" and "type-1 polarized DCs" refer to
mature DCs that secrete Thl-driving cytokines, such as IL-12, IL-18, and IL-
23. DC's are
fully capable of promoting cell-mediated immunity. DC1s are pulsed with HER2
MI-IC class
II-binding peptides in preferred embodiments herein.
As used herein, an "activated DC" is a DC that has been exposed to a Toll-like
receptor agonist. The activated DC may or may not be loaded with an antigen.
The term "mature DC" as used herein, is defined as a dendritic cell that
expresses molecules, including high levels of MHC class II, CD80 (B7.1) and
CD86 (B7.2),
In contrast, immature dendritic cells express low levels of MHC class II, CD80
(B7.1) and
CD86 (B7.2) molecules, yet can still take up an antigen. "Mature DC" also
refers to an
antigen presenting cell existing in vivo, in vitro, ex vivo, or in a host or
subject that is DC1-
polarized (i.e., fully capable of promoting cell-mediated immunity).
A "disease" is a state of health of an animal wherein the animal cannot
maintain homeostasis, and wherein if the disease is not ameliorated then the
animal's health
continues to deteriorate.
A "disorder" in an animal is a state of health in which the animal is able to
maintain homeostasis, but in which the animal's state of health is less
favorable than it would
be in the absence of the disorder. Left untreated, a disorder does not
necessarily cause a
further decrease in the animal's state of health.
A disease or disorder is "alleviated" if the severity or frequency of at least
one
sign or symptom of the disease or disorder experienced by a patient is
reduced.
"Effective amount" or "therapeutically effective amount" are used
interchangeably herein, and refer to an amount of a compound, formulation,
material, or
composition, as described herein effective to achieve a particular biological
result. Such
results may include, but are not limited to, the inhibition of virus infection
as determined by
any means suitable in the art.
As used herein "endogenous" refers to any material from or produced inside
an organism, cell, tissue or system.
14
CA 02981033 2017-09-26
WO 2016/154508
PCT/11S2016/024146
As used herein, the term "exogenous" refers to any material introduced from
or produced outside an organism, cell, tissue or system.
The term "epitope" as used herein is defined as a small chemical molecule on
an antigen that can elicit an immune response, inducing B and/or T cell
responses. An antigen
can have one or more epitopes. Most antigens have many epitopes; i.e., they
are multivalent.
In general, an epitope is roughly five amino acids and/or sugars in size. One
skilled in the art
understands that generally the overall three-dimensional structure, rather
than the specific
linear sequence of the molecule, is the main criterion of antigenic
specificity and therefore
distinguishes one epitope from another.
"HER2" is a member of the human epidermal growth factor receptor
("EGFR") family. HER2 is overexpressed in approximately 20-25% of human breast
cancer
and is expressed in many other cancers.
"HER2 binding peptides," "HER2 MHC class II binding peptides," "binding
peptides," "peptide antigens," "HER2 peptides," "immunogenic MHC class II
binding
peptides," "antigen binding peptides," "HER2 epitopes," "reactive peptides,"
and the like as
used herein refer to MHC Class II peptides derived from or based on the
sequence of the
HER2/neu protein, a target found on approximately 2025% of all human breast
cancers and
their equivalents. HER2 extracellular domain "ECD" refers to a domain of HER2
that is
outside of a cell, either anchored to a cell membrane, or in circulation,
including fragments
thereof. HER2 intracellular domain "ICD" refers to a domain of the HER2/neu
protein within
the cytoplasm of a cell. According to a preferred embodiment HER2 epitopes or
otherwise
binding peptides comprise 6 HER2 binding peptides which include 3 HER2 ECD
peptides
and 3 HER2 ICD peptides. Preferred HER2 ECD peptides comprise: Peptide 42-56:
HLDMLRHLYQGCQVV (SEQ ID NO: 1); Peptide 98-114: RLRIVRGTQLFEDNYAL
(SEQ ID NO: 2); and Peptide 328-345: TQRCEKCSKPCARVCYGL (SEQ ID NO: 3);
Preferred HER2 ICD peptides comprise: Peptide 776-790: GVGSPYVSRLLGICL (SEQ ID
NO: 4); Peptide 927-941: PAREIPDLLEKGERL (SEQ ID NO: 5); and Peptide 1166-
1180:
TLERPKTLSPGKNGV (SEQ ID NO: 6).
"HER2P S" is the classification or molecular subtype of a type of breast
cancer
as well as numerous other types of cancer. HER2 positivity is currently
defined by gene
amplification by FISH (fluorescent in situ hybridization) assay and 2+ or 3+
on intensity of
pathological staining.
"HER2neg" is defined by the lack of gene amplification by FISH, and can
encompass a range of pathologic staining from 0 to 2+ in most cases.
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
"Homologous" as used herein, refers to the subunit sequence similarity
between two polymeric molecules, e.g., between two nucleic acid molecules,
e.g., two DNA
molecules or two RNA molecules, or between two polypeptide molecules. When a
subunit
position in both of the two molecules is occupied by the same monomeric
subunit, e.g., if a
position in each of two DNA molecules is occupied by adenine, then they are
completely or
100% homologous at that position. The percent homology between two sequences
is a direct
function of the number of matching or homologous positions, e.g., if half
(e.g., five positions
in a polymer ten subunits in length) of the positions in two compound
sequences are
homologous then the two sequences are 50% identical, if 90% of the positions,
e.g., 9 of 10,
are matched or homologous, the two sequences share 90% homology. By way of
example,
the DNA sequences 51ATTGCC3' and 5'TATGGC3' share 50% homology.
In addition, when the terms "homology" or "identity" are used herein to refer
to the nucleic acids and proteins, it should be construed to be applied to
homology or identity
at both the nucleic acid and the amino acid sequence levels.
The term "inhibit," as used herein, means to suppress or block an activity or
function, for example, about ten percent relative to a control value.
Preferably, the activity is
suppressed or blocked by 50% compared to a control value, more preferably by
75%, and
even more preferably by 95%. "Inhibit," as used herein, also means to reduce a
molecule, a
reaction, an interaction, a gene, an mRNA, and/or a protein's expression,
stability, function or
activity by a measurable amount or to prevent entirely. Inhibitors are
compounds that, e.g.,
bind to, partially or totally block stimulation, decrease, prevent, delay
activation, inactivate,
desensitize, or down regulate a protein, a gene, and an mRNA stability,
expression, function
and activity, e.g., antagonists.
As used herein, an "instructional material" includes a publication, a
recording,
a diagram, or any other medium of expression which can be used to communicate
the
usefulness of the compositions and methods of the invention. The instructional
material of the
kit of the invention may, for example, be affixed to a container which
contains the nucleic
acid, peptide, and/or composition of the invention or be shipped together with
a container
which contains the nucleic acid, peptide, and/or composition. Alternatively,
the instructional
material may be shipped separately from the container with the intention that
the instructional
material and the compound be used cooperatively by the recipient.
Interleukin 2 ("IL-2" or "IL2") is an interleukin, a type of cytokine
signaling
molecule in the immune system. IL2 is the principal T cell growth and
proliferation factor.
16
CA 02981033 2017-09-26
WO 2016/154508 PCT/111S2016/024146
Interleukin 7 ("IL-7" or "IL2") is a hematapoietic growth factor produced by
stromal epithelial cells in lymph nodes. IL-7 is essential for lymphocyte
proliferation and
survival.
Interleukin 15 ("IL-15" or "IL2") is a T cell growth activation and survival
factor. IL-15 is produced by fibroblasts, dendritic cells and macrophages.
"Isolated" means altered or removed from the natural state. For example, a
nucleic acid or a peptide naturally present in a living animal is not
"isolated," but the same
nucleic acid or peptide partially or completely separated from the coexisting
materials of its
natural state is "isolated." An isolated nucleic acid or protein can exist in
substantially
purified form, or can exist in a non-native environment such as, for example,
a host cell.
"Loaded" with a peptide, as used herein, refers to presentation of an antigen
in
the context of an MHC molecule.
The term "major histocompatibility complex" or "MHC" as used herein is
defined as a specific cluster of genes, many of which encode evolutionary
related surface
proteins involved in antigen presentation, which are among the most important
determinants
of histocompatibility. Class I MHC, or MHC class I, function mainly in antigen
presentation
to CD8 T lymphocytes. Class II MHC, or MHC class II, function mainly in
antigen
presentation to CIA+ T lymphocytes (T-helper cells).
"Mature DC" as used herein means a dendritic cell that expresses molecules,
including high levels of MHC class II, CD80 (B7.1) and CD86 (B7.2) molecules.
In contrast,
immature DCs ("iDCs") express low levels of MHC class II, CD80 (B7.1) and CD86
(B7.2)
molecules, yet can still take up an antigen. "Mature DC" also refers to an
antigen presenting
cell existing in vivo, in vitro, ex vivo,or ina host or subject that may also
be DC1 -polarized
(i.e., fully capable of promoting cell-mediated immunity.)
"Metrics" of CDeThl responses (or Thl responses) are defined for each
subject group analyzed for anti-HER2 CIA+ Thl immune response: (a) overall
anti-HER2
responsivity (expressed as percent of subjects responding to reactive
peptide); (b)
response repertoire (expressed as mean number of reactive peptides (n)
recognized by each
subject group); and (c) cumulative response (expressed as total sum of
reactive spots (spot-
forming cells "SFC" per 106cells from IFN-y ELISPOT analysis) from 6 MHC Class
II
binding peptides from each subject group.
By the term "modulating," as used herein, is meant mediating a detectable
increase or decrease in the level of a response in a subject compared with the
level of a
response in the subject in the absence of a treatment or compound, and/or
compared with the
17
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
level of a response in an otherwise identical but untreated subject. The term
encompasses
perturbing and/or affecting a native signal or response thereby mediating a
beneficial
therapeutic response in a subject, preferably, a human.
"Non-equivocal HER2neg is defined as non-gene amplified and 0 or 1+ on
pathologic staining. "Equivocal HER2neg" is defined as non-gene amplified but
2+ on
pathologic staining.
As used herein, a "population" includes reference to an isolated culture
comprising a homogenous, a substantially homogenous, or a heterogeneous
culture of cells.
Generally, a "population" may also be regarded as an "isolated" culture of
cells.
As used herein, a "recombinant cell" is a host cell that comprises a
recombinant polynucleotide.
"Responsivity" or "anti-1-1ER2 responsivity" are used interchangeably herein
to mean the percentage of subjects responding to at least 1 of 6 binding
peptides.
"Response repertoire" is defined as the mean number ("n") of reactive
peptides recognized by each subject group.
"Sample" or "biological sample" as used herein means a biological material
from a subject, including but is not limited to organ, tissue, exosome, blood,
plasma, saliva,
urine and other body fluid. A sample can be any source of material obtained
from a subject.
By the term "specifically binds," as used herein, is meant a molecule, such as
an antibody, which recognizes and binds to another molecule or feature, but
does not
substantially recognize or bind other molecules or features in a sample.
By the term "stimulation," is meant a primary response induced by binding of
a stimulatory molecule with its cognate ligand thereby mediating a signal
transduction event,
such as, but not limited to, signal transduction via the TCR/CD3 complex.
Stimulation can
mediate altered expression of certain molecules, such as downregulation of TGF-
13, and/or
reorganization of cytoskeletal structures, and the like.
A "stimulatory ligand," as used herein, means a ligand that when present on an
antigen presenting cell (e.g., a dendritic cell, a B-cell, and the like) can
specifically bind with
a cognate binding partner (referred to herein as a "stimulatory molecule") on
a T cell, thereby
mediating a primary response by the T cell, including, but not limited to,
activation, initiation
of an immune response, proliferation, and the like.
A "stimulatory molecule," as the term is used herein, means a molecule on a T
cell that specifically binds with a cognate stimulatory ligand present on an
antigen presenting
cell.
18
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
The terms "subject," "patient," "individual," and the like are used
interchangeably herein, and refer to any animal, or cells thereof whether in
vitro or in situ,
amenable to the methods described herein. In certain non-limiting embodiments,
the patient,
subject or individual is a human.
As used herein, a "substantially purified" cell is a cell that is essentially
free of
other cell types. A substantially purified cell also refers to a cell which
has been separated
from other cell types with which it is normally associated in its naturally
occurring state. In
some instances, a population of substantially purified cells refers to a
homogenous population
of cells. In other instances, this term refers simply to cell that have been
separated from the
cells with which they are naturally associated in their natural state. In some
embodiments, the
cells are cultured in vitro. In other embodiments, the cells are not cultured
in vitro.
The term "targeted therapies" as used herein refers to cancer treatments that
use drugs or other substances that interfere with specific target molecules
involved in cancer
cell growth usually while doing little damage to normal cells to achieve an
anti-tumor effect.
Traditional cytotoxic chemotherapy drugs, by contrast, act against all
actively dividing cells.
In breast cancer treatment monoclonal antibodies, specifically
trastuzumab/Herceptin ,
targets the HER2/neu receptor.
"T/C" is defined as trastuzumab and chemotherapy. This refers to patients that
receive both trastuzumab and chemotherapy before/after surgery for breast
cancer.
The term "T cell" as used herein is defined as a thymus-derived cell that
participates in a variety of cell-mediated immune reactions.
The terms "T-helper cells," "helper T cells," "Th cells," and the like are
used
herein with reference to cells indicates a sub-group of lymphocytes (a type of
white blood
cell or leukocyte) including different cell types identifiable by a skilled
person in the art. In
particular, T-helper cells are effector T-cells whose primary function is to
promote the
activation and functions of other B and T lymphocytes and/or macrophages.
Helper T cells
differentiate into two major subtypes of cells known as "Thl" or "Type 1" and
"Th2" or
"Type 2" phenotypes. These Th cells secrete cytokines, proteins, or peptides
that stimulate or
interact with other leukocytes. "Thl cell," "CD4+Th1 cell," "CD4+T-he1per
typel cell,"
"CD4' T cell" and thelike as used herein refer to a mature T-cell that has
expressed the
surface glycoprotein CD4. CD4+ T-helper cells become activated when they are
presented
with peptide antigens by MHC class 11 molecules which are expressed on the
surface of
antigen-presenting peptides ("APCs") such as dendritic cells. Upon activation
of a CD4+T
helper cell by the MHC-antigen complex, it secretes high levels of cytokines
such as
19
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
interferon-y ("IFN-y"). Such cells are thought to be highly effective against
certain disease-
causing microbes that live inside host cells, and are critical in antitumor
response in human
cancer.
The term "qtotoxic T cell" or "CD8}T cell or "killer T cell" is a T
lymphocyte that kills target cells such as cancer cells, cells that are
infected, or cells that are
damaged in other ways.
"Treg" "Treg" and "regulatory T-cells" are used herein to refer to cells which
are the policemen of the immune system, and which act to regulate the
anticancer activities of
the immune system. They are increased in some cancers, and are mediators in
resistance to
immunotherapy in these cancer types.
The term "therapeutic" as used herein means a treatment and/or prophylaxis.
A therapeutic effect is obtained by suppression, remission, or eradication of
a disease state.
"Therapeutically effective amount" is an amount of a compound of the
invention, that when administered to a patient, ameliorates a symptom of the
disease. The
amount of a compound of the invention which constitutes a "therapeutically
effective
amount" will vary depending on the compound, the disease state and its
severity, the age of
the patient to be treated, and the like. The therapeutically effective amount
can be determined
routinely by one of ordinary skill in the art having regard to his own
knowledge and to this
disclosure.
The terms "treat," "treating," and "treatment," refer to therapeutic or
preventative measures described herein. The methods of "treatment" employ
administration
to a subject, in need of such treatment, a composition of the present
invention, for example, a
subject afflicted a disease or disorder, or a subject who ultimately may
acquire such a disease
or disorder, in order to prevent, cure, delay, reduce the severity of, or
ameliorate one or more
symptoms of the disorder or recurring disorder, or in order to prolong the
survival of a
subject beyond that expected in the absence of such treatment.
The term "vaccine" as used herein is defined as a material used to provoke an
immune response after administration of the material to an animal, preferably
a mammal, and
more preferably a human. Upon introduction into a subject, the vaccine is able
to provoke an
immune response including, but not limited to, the production of antibodies,
cytokines and/or
other cellular responses.
Ranges: throughout this disclosure, various aspects of the invention can be
presented in a range format. It should be understood that the description in
range format is
merely for convenience and brevity and should not be construed as an
inflexible limitation on
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
the scope of the invention. Accordingly, the description of a range should be
considered to
have specifically disclosed all the possible subranges as well as individual
numerical values
within that range. For example, description of a range such as from 1 to 6
should be
considered to have specifically disclosed subranges such as from 1 to 3, from
1 to 4, from 1
to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual
numbers within that
range, for example, 1, 2, 2.7, 3, 4, 5, 5.3, and 6. This applies regardless of
the breadth of the
range.
Description
The invention relates to replicating the environment of the lymph node for
generating a therapeutic amount of T cells for adoptive therapy. The T cells
can also be used
for the identification of epitopes on target antigens to foster the production
of peptide-based
vaccines.
In one embodiment, the invention provides an in vitro environment that
replicates the environment of the lymph node. In one embodiment, replication
of the lymph
node comprises supplying one or more of the following elements to the culture
conditions:
type 1 dendritic cells, 1L-15, IL-7, and IL-2.
Type 1 dendritic cells process and present peptide antigens to T cells and
supply so-called "costimulatory molecules" including surface-expressed CD80
and CD86
(which bind to CD28 counter-receptor on T cells), as well as CD40 (which
interacts with
CD4OL on T cells). In addition the DCs produce soluble factors such as
Interleukin-12 (IL-
12) which supports long life (anti-apoptotic factor) as well as Interferon-
gamma production
(T cell function). DCs produce a number of other factors critical to T cell
development. DCs
are normally found in lymph nodes and are of known importance to T cell
activation and
expansion.
IL-15 is a T cell growth activation and survival factor. IL-15 is produced by
fibroblasts, dendritic cells and macrophages.
IL-7 is a factor produced by stromal epithelial cells in lymph nodes. IL-7 is
essential for lymphocyte proliferation and survival.
IL2 is the principal T cell growth and proliferation factor.
Accordingly, the invention provides compositions and methods for combining
the cytokines and type of dendritic cells to generate desirable T cells. In
one embodiment, the
T cells are expanded to a level necessary for adoptive therapy and epitope
mapping studies
while maintaining antigen specificity and cellular function.
21
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
In one aspect, the invention relates to the discovery that 1-IER2 invasive
breast
cancer ("IBC") patients with residual disease following neoadjuvant
chemotherapy have an
anti-HER2 Type 1 T helper (Thl) cell immune deficit and a significant risk of
recurrent
disease. It was shown in Datta, et al. that anti-HER2 CD4"T-ce11 responses
incrementally
decrease along the breast cancer continuum ¨ a robust response in healthy
donors and patients
with benign disease, a depressed response in patients with HER2+ ductal
carcinoma in situ,
and a nearly absent response in patients with HER2+ IBC. Herein is explored
the role of (A)
adjuvant type 1-polarized dendritic cell ("DC1") vaccination and (B) methods
of expanding
antigen-specific t-cells for adoptive T-cell transfer in restoring anti-HER2
Thl immunity.
The present embodiments also relate to a method of creating a
microenvironment in vitro for culture expansion of antigen-specific CD4+ or
CD8' T cells.
The expanded antigen-specific T cells can be used for a variety of therapeutic
and research
purposes, for example adoptive T cell therapy for cancer or infectious disease
such as chronic
viral infections other conditions and/or for the identification of epitopes on
target antigens to
foster the production of peptide-based vaccines.
In one embodiment, the invention comprises the use of autologous type I
dendritic cells ("DC1s") in combination with a protein or peptide antigen to
stimulate T cells
in vitro. After stimulation, at least two soluble factors (e.g., cytokines)
are added to the T
cells. In some instances, the at least two soluble factors are Interleukin-7
("IL-7") and
Interleukin-15 ("IL-15"). Following the addition of the soluble factors to the
T cells, a T cell
growth factor is added. In some instances, the T cell growth factor is
Interleukin-2 ("IL-2").
The soluble factors, in addition to those naturally produced by the DC1s,
support the
proliferation and acquisition/maintenance of T cell function. This process of
stimulation can
be repeated in weekly cycles until T cells are of sufficient numbers for
therapy or epitope
scanning/mapping. In certain embodiments, the T cells are expanded to a level
necessary for
adoptive therapy and epitope mapping studies while maintaining antigen
specificity and
cellular function.
In Vivo Thl Response to HER-2-Pulsed DC1 Vaccination: Restoration of Anti-
HER2CD4+
Thl Response in IBC Patients
In addition to the identification of a progressive loss of anti-HER2 CD4+ Thl
response across a tumorigenic continuum in HERD' -breast cancer, as taught by
Datta, et al.,
the depressed anti-HER2 Thl responses in HER2P s-invasive breast cancer were
differentially
restored after HER2-pulsed type-1 polarized dendritic cell ("DC1")
vaccinations. The
22
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
depressed responses were not restored following HER2targeted therapy with
trastuzumab and
chemotherapy ("T/C") or by other standard therapies such as surgical resection
or radiation.
The restored anti-HER2 Thl responses appear to be durable for at least about
six months or
considerably longer.
HER2+ IBC patients with residual disease following neoadjuvant therapy
received adjuvant HER2-pulsed DC1 vaccines. Immune responses were generated
from
PBMCs pulsed with HER2 Class II peptides by measuring IFN-y production via
ELISPOT.
Responses were evaluated on the three metrics of CD4+ Thl response: (1) the
overall anti-
HER2 responsivity (responding to ?._1 peptide), (2) the number of reactive
peptides (response
repertoire), and (3) the cumulative response across the 6 HER2 peptides. Pre-
vaccination Thl
responses were compared with 3-month and 6month post-vaccination responses.
Datta, et al., describes the methods of making DC1 vaccines. . See, also,
Koski, G. K., et al., J Immonother. 35(1): 54 (2012) ("Koski, et al.");
Sharma, A., et al.,
Cancer 118(17):4354 (2012) ("Sharma, et al."); Fracol, M., et al., Ann. Surg.
Oncol.
20(10):3233 (2013); Lee, M. K. 4th, et al., Expert Rev. 8(11):e74698 (2013);
Czemiecki, B.J.,
et al., Cancer Res. 67(4):1842 (2007); Czemiecki, B. J., et al., Cancer Res.
67(14):6531
(2007); and U.S. Published Application US 2013/0183343 A1. Briefly, patients'
monocytes
are first separated from other white blood cells by leukapheresis and
elutriation. These
monocytes are then cultured in serum-free medium ("SFM") with granulocyte-
macrophage
colony-stimulating factor ("GM-CSF") and interleukin ("IL")-4 to become
immature
dendritic cells ("iDCs"). These cells are then preferably pulsed with six HER2
MHC class II
binding peptides, and in the present case, binding peptides identified by SEQ
ID NOS: 1-6,
and then interferon ("IFN")-y and lipopolysaccharide ("LPS") are added to
complete the
maturing and activation process to achieve full DC activation to DC1s before
injecting back
into the patient. See, Fracol, M., et al., Ann. Surg. Oncol. 20(10):3233
(2013). In the case of
HLA-A2N6 patients, half of the cells are pulsed with a MHC class I binding
peptide and the
other half with a different MHC class 1 binding peptide.
Datta, et al. also describe blood tests/assays which generate a circulating
anti-
cancer CD4+ Thl response (i.e., IFN-y-secreting) and the resulting IFN-y
production is
detected and measured. Such blood tests were performed on patients pre-DC1
vaccination,
and 3-months and 6 months post-vaccination. In preferred embodiments, subject
blood
samples containing CD4+Th1 cells and antigen-presenting cells or precursors
thereof are
pulsed with MHC class II immunogenic peptides based on the type of cancer the
subject is
afflicted with and which are capable of inducing an immune response in said
subject.
23
=
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
Preferably the antigen-presenting cells or precursors thereof are mature or
immature dendritic
cells or monocyte precursors thereof. In particularly preferred embodiments,
the cancer is
preferably HER2-expressing and the mammalian subject is preferably a human,
and more
preferably the cancer is HER2Ns breast cancer and the human subject is a
female.
A preferred embodiment is provided for generating a circulating antiHER2
CD4+Thl response in a mammalian subject by isolating unexpanded peripheral
blood
mononuclear cells ("PBMCs") from a subject and pulsing the PBMCs with a
composition
comprising HER2-derived MHC class II antigenic binding peptides capable of
generating an
immune response in the subject. Without wishing to be bound by any particular
theory, when
the binding peptides are presented to CD4+Thl cells that are present in the
PBMC sample
they activate the CD4+Th1 cells and the activated CD4' Thl cells produce
interferon-y
("IFN-y"). DC1s (type-1 polarized dendritic cells) derived from precursor
pluripotent
monocytes contained in the subject's PBMC sample are antigen-presenting cells
("APCs")
which upon exposure to the binding peptides become antigen-loaded APCs which
present the
MHC class II antigen binding peptides to the subject's CD4+Th teens in the
sample thereby
activating the CD4+Th1 cells to produce/secrete 1FN-y. The IFN-y thereby
produced is
subsequently measured for analysis.
In the present case, according to this preferred embodiment each patient's
PBMC's were pulsed with 6 HER2-specific MHC class II peptides, in particular,
those
having sequences identified by SEQ ID NOs: 1-6. IFN-y produced by anti-HER2
CD4' Thl
cells was detected and measured via IFN-y enzyme-linked immunospot ("ELISPOT")
assay.
In particularly preferred embodiments for HER2P'5 cancers, DCs, immature or
type-1 polarized DC1s, are pulsed with a composition comprising 6 MHC class II
binding
peptides derived from or based on HER2 that are capable of generating an
immune response
in a patient. HER2 MHC class II binding peptides or epitopes include:
Peptide 42-56: HLDMLRHLYQGCQVV (SEQ ID NO: 1);
Peptide 98-114: RLRIVRGTQLFEDNYAL (SEQ ID NO: 2);
Peptide 328-345: TQRCEKCSKPCARVCYGL (SEQ ID NO: 3);
Peptide 776-790: GVGSPYVSRLLGICL (SEQ ID NO: 4);
Peptide 927-941: PAREIPDLLEKGERL (SEQ ID NO: 5); and
Peptide 1166-1180: TLERPKTLSPGKNGV (SEQ ID NO: 6).
In embodiments where donors have A2.1 blood type HER2 MHC class I
peptides or epitopes include:
Peptide 369-377: KIFGSLAFL (SEQ ID NO: 7); and
24
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
Peptide 689-697: RLLQETELV (SEQ ID NO: 8).
Datta, et al. also describe an alternate preferred embodiment, wherein a
circulating anti HER2 CD4+ Thl response is generated in a mammalian subject by
co-
culturing previously unstimulated purified CD4+T-ce11s from a subject blood
sample with
autologous immature or mature dendritic cells ("iDCs" or mature "DCs") pulsed
with a
composition comprising HER2-derived MHC class II antigenic binding peptides
capable of
generating an immune response in the subject. Without wishing to be bound by
any particular
theory, when the binding peptides are presented to CD4+Thl cells present in
the T-cell
sample they activate the CD4TTh1 cells and the activated CD4 Thl cells
produce/secrete
IFN-y. The immature DCs are matured to DC1's, which present the MHC class II
binding
peptides to the subject's CD4+Th1 cells that are present in the sample thereby
activating the
CD4+Thl cells to produce IFN-y, which is subsequently measured for analysis.
In both alternate preferred embodiments for generating anti-HER2 immune
response in a subject, IFN-y produced by anti-HER2 CD4+ Thl cells is detected
and
measured via IFN-y enzyme-linked immunospot ("ELISPOT") assay, although it
should be
understood by one skilled in the art that other detection methods may be used.
For example,
flow cytometry, enzyme-linked immunosorbent assay ("ELISA"), and
immunofluorescence
("IF") can be used for monitoring immune response. Alternatively, in instances
of immune
monitoring of patients, it can be advantageous to measure the ratio of IFN-y
to 1L-10 as
opposed to, or in addition to, a straight IFN-y test such as ELISPOT which
shows total CD4'
cell spots. Such testing would be particularly advantageous for patients at
risk. Further, the
use of immunofluorescence provides other ways to measure and visualize immune
response
via use of ELISPOT readers that read results by fluorescence. In such
instances the results
can be arranged to show 2, 3, or more cytokines/other secreted immune
molecules, each
showed in a different color, in the same patient sample. In the present case
IFN-y ELISPOT
was used.
Although a presently preferred embodiment features six HER2 MHC class Il
binding peptides/epitopes, other possible MHC class II HER2 peptides can be
used in the
present embodiments in that any components of the entire HER2 molecule can be
used as a
source for other binding peptides so long as they are sufficiently
immunologically active in
patients.
Responsivity: Pre-vaccination, only one IBC patient produced an immune
response, defined as >20 SFC/106 cells in an experimental well after
subtracting unstimulated
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
background. Compared with pre-vaccination results, all vaccinated IBC patients
produced an
immune response, defined as >2-fold increase in anti-HER2 IFN-ywsThl
responses.
Response Repertoire: Figure 1 shows mean repertoire increased from 0.5 1
peptides pre-vaccination to 3.25+0.96 peptides at 3 months (p=0.01) and 4 0.8
peptides at 6
months (p=0.01) in the IBC patients.
Cumulative response: Figure 2 shows mean cumulative response in the
patients improved from 36.5+38.3 SFC/106pre-vaccination to 151.0+60.0 SFC/106
at 3
months (p=0.04) and 198.4+39.7 SFC/106at 6 months (p=0.02).
There are many other HER2P ' solid cancers in addition to breast cancer, such
as, for example, brain, bladder, esophagus, lung, pancreas, liver, prostate,
ovarian, colorectal,
and gastric, and others, for which the materials and methods of the
embodiments described
herein can be used for diagnosis and treatment. Therefore the six anti-HER2
binding peptides
described above may be used in accordance with the herein embodiments to
generate immune
responses capable of detection and useful for diagnostics for these and other
HER2-
expressing cancers.
Vaccines can be developed to target HER2-expressing tumors using the same
anti-HER2 binding peptides described above or may employ any composition of
HER2 that
is immunogenic such as, for example, DNA, RNA, peptides, or proteins or
components
thereof such as the ICD and ECD domains. For example, subjects can be
vaccinated against
the whole HER2 protein and the six above-referenced binding peptides can be
used to
monitor the patient's immune response. Similarly vaccines can be developed for
other types
of cancer such as other members of the HER2 family which includes HER1, HER3,
and c-
MET.
Although the present preferred embodiments are directed to treating and
diagnosing HER2P ' breast cancer in women it should be readily appreciated by
the skilled
artisan that the present embodiments are not limited to female humans. The
presently
preferred embodiments includes male humans, for example, HER2expressing
prostate cancer,
as well as other mammalian subjects
The identified anti-I-IER2 CD4+Th1 response decrement allows the detected
immune response generated in such blood tests to be used as a cancer
diagnostic/response
predictor alone or, as in the example here, in tandem with the use of
specialized vaccines to
restore a patient's immune response. The preferred embodiments described
herein thus shift
the focus of cancer diagnosis and therapy to patient immunity and use of blood
tests to
determine and/or predict the immune response against a cancer, including
patients at risk for
26
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
recurrence, as opposed to diagnosis and treatment methods that rely on
identification of
tumor cells.
In vitro expansion of HER2-specific TH1 cells
In vitro, HER2-specific Thl cells were generated by co-culture with HER2-
peptide pulsed DC1s and expanded using IL-2 alone or IL-2, IL-7, and IL-15.
Thl cells were
subsequently expanded either by repeat HER2-peptide pulsed DC1 co-culture or
via anti-
CD3/CD28 stimulation. Fold expansion was defined as: (#Tcells post expansion /
#T-cells
pre expansion); specificity was measured by antigen specific IFN-y production
by ELISA.
The present embodiments related to T cell expansion are in no way limited to
CD4+T cells. Thus the present embodiments provide methods for growing chimeric
antigen
receptor T cells ("CART cells"), cytotoxic T lymphocytes (CD8's) as well as
all other kinds
of T cells. See, for example, Dana, J., et al., "CD4+ T-helper Type 1
Cytokines and
Trastuzumab Facilitate CD8T T-cell Targeting of HER-2/neuexpressing Cancers"
Cancer
Immunol. Res. (2015).
Embodiments relate to replicating the environment of the lymph node for
generating a therapeutic amount of antigen-specific T cells, either helper
(CD41) or cytotoxic
(CD8+), for adoptive therapy for cancer or other conditions. The expanded
antigen-specific T
lymphocytes can also be used for the identification of epitopes on target
antigens to foster the
production of peptide-based vaccines.
A present embodiment provides an in vitro environment that replicates the
environment of the lymph node. In that embodiment, replication of the lymph
node comprises
supplying one or more of the following elements to the culture conditions:
type 1 dendritic
cells, IL-15, IL-7, and IL-2.
Type 1 dendritic cells process and present peptide antigens to T cells and
supply so-called "costimulatory molecules" including surface-expressed CD80
and CD86
(which bind to CD28 counter-receptor on T cells), as well as CD40 (which
interacts with
CD4OL on T cells). In addition, the DCs produce soluble factors such as
Interleukin-12 ("IL-
12") which supports long life (anti-apoptotic factor) as well as IFN-y
production (T cell
function). DCs produce a number of other factors critical to T cell
development. DCs are
normally found in lymph nodes and are of known importance to T cell activation
and
expansion.
IL-15 is a T cell growth activation and survival factor. IL-15 is produced by
fibroblasts, dendritic cells and macrophages.
27
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
IL-7 is a factor produced by stromal epithelial cells in lymph nodes. IL7 is
essential for lymphocyte proliferation and survival.
IL2 is the principal T cell growth and proliferation factor.
Accordingly, the embodiments provide compositions and methods for
combining the particular cytokines and type of dendritic cells while also
using particular
timing and sequence of lymphocyte addition to generate desirable T cells. In
preferred
embodiments, T cells are expanded to a level necessary for adoptive therapy
and epitope
mapping studies while maintaining antigen specificity and cellular function.
Sources of T Cells
Prior to expansion, a source of T cells is obtained from a subject. Examples
of
subjects include humans, dogs, cats, mice, rats, and transgenic species
thereof. Preferably, the
subject is a human. T cells can be obtained from a number of sources,
including peripheral
blood mononuclear cells, bone marrow, lymph node tissue, spleen tissue, and
tumors. In
certain embodiments of the present invention, any number of T cell lines
available in the art,
may be used. In certain embodiments of the present invention, T cells can be
obtained from a
unit of blood collected from a subject using any number of techniques known to
the skilled
artisan, such as ficoll separation. In one preferred embodiment, cells from
the circulating
blood of an individual are obtained by apheresis or leukapheresis. The
apheresis product
typically contains lymphocytes, including T cells, monocytes, granulocytes, B
cells, other
nucleated white blood cells, red blood cells, and platelets. In one
embodiment, the cells
collected by apheresis may be washed to remove the plasma fraction and to
place the cells in
an appropriate buffer or media for subsequent processing steps. In one
embodiment of the
invention, the cells are washed with phosphate buffered saline (PBS). In an
alternative
embodiment, the wash solution lacks calcium and may lack magnesium or may lack
many if
not all divalent cations. After washing, the cells may be resuspended in a
variety of
biocompatible buffers, such as, for example, Ca-free, Mg-free PBS.
Alternatively, the
undesirable components of the apheresis sample may be removed and the cells
directly
resuspended in culture media.
In another embodiment, T cells are isolated from peripheral blood by lysing
the red blood cells and depleting the monocytes, for example, by
centrifugation through a
PERCOLLTM gradient. Alternatively, T cells can be isolated from umbilical
cord. In any
event, a specific subpopulation of T cells can be further isolated by positive
or negative
selection techniques.
28
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
Enrichment of a T cell population by negative selection can be accomplished
using a combination of antibodies directed to surface markers unique to the
negatively
selected cells. A preferred method is cell sorting and/or selection via
negative magnetic
immunoadherence or flow cytometry that uses a cocktail of monoclonal
antibodies directed to
cell surface markers present on the cells negatively selected. For example, to
enrich for CD4+
cells by negative selection, a monoclonal antibody cocktail typically includes
antibodies to
CD14, CD20, CD1 lb, CD16, HLA-DR, and CD8.
For isolation of a desired population of cells by positive or negative
selection,
the concentration of cells and surface (e.g., particles such as beads) can be
varied. In certain
embodiments, it may be desirable to significantly decrease the volume in which
beads and
cells are mixed together (i.e., increase the concentration of cells), to
ensure maximum contact
of cells and beads. For example, in one embodiment, a concentration of 2
billion cells/ml is
used. In one embodiment, a concentration of 1 billion cells/m1 is used. In a
further
embodiment, greater than 100 million cells/ml is used. In a further
embodiment, a
concentration of cells of 10, 15, 20, 25, 30, 35, 40, 45, or 50 million
cells/ml is used. In yet
another embodiment, a concentration of cells from 75, 80, 85, 90, 95, or 100
million cells/ml
is used. In further embodiments, concentrations of 125 or 150 million cells/ml
can be used.
Using high concentrations can result in increased cell yield, cell activation,
and cell
expansion.
T cells for stimulation can also be frozen after the washing step, which does
not require the monocyte-removal step. While not wishing to be bound by
theory, the freeze
and subsequent thaw step provides a more uniform product by removing
granulocytes and to
some extent monocytes in the cell population. After the washing step that
removes plasma
and platelets, the cells may be suspended in a freezing solution. While many
freezing
solutions and parameters are known in the art and will be useful in this
context, in a non-
limiting example, one method involves using PBS containing 20% DMSO and 8%
human
serum albumin, or other suitable cell freezing media. The cells are then
frozen to -80 C at a
rate of 1 per minute and stored in the vapor phase of a liquid nitrogen
storage tank. Other
methods of controlled freezing may be used as well as uncontrolled freezing
immediately at -
20 C or in liquid nitrogen.
Activation and Expansion of T Cells
29
CA 02981033 2017-09-26
WO 2016/154508
PCT/1JS2016/024146
Generally, T cells of the invention are expanded under conditions that
replicate the lymph node. In one embodiment, replication of the lymph node
comprises
supplying one or more of the following elements to the culture conditions:
type 1 dendritic
cells, IL-15, IL-7, and IL-2. In one embodiment, antigen specific T cells can
be expanded in
the presence of one or more of type 1 dendritic cells, IL-15, IL-7, and IL-2.
In one embodiment, the T cells may be stimulated as described herein, such as
by contacting with a DC. The DC is able to provide supply a costimulatory
molecule to the T
cell. After the T cells are contacted with DCs, the T cells are cultured in
the presence of IL-
15, 1L-7, and IL-2.
In some instances, the T cells are cultured with a mixture comprising one or
more of DCs, IL-15, IL-7, and IL-2. In one embodiment of the present
invention, the mixture
may be cultured for several hours (about 3 hours) to about 14 days or any
hourly integer
value in between. In another embodiment, the mixture may be cultured for 21
days. In one
embodiment of the invention the T cells are cultured for about eight days. In
another
embodiment, the T cells are cultured together for 2-3 days. Several cycles of
stimulation may
also be desired such that culture time of T cells can be 60 days or more.
Conditions
appropriate for T cell culture include an appropriate media (e.g., Minimal
Essential Media or
RPMI Media 1640 or, X-vivo 15, (Lonza)) that may contain factors necessary for
proliferation and viability, including serum (e.g., fetal bovine or human
serum), interleukin-2
(IL-2), insulin, IFN-y, IL-4, IL-7, GM-CSF, IL-10, IL-12, IL-15, TGFf3, and
TNF-a or any
other additives for the growth of cells known to the skilled artisan. Other
additives for the
growth of cells include, but are not limited to, surfactant, plasmanate, and
reducing agents
such as N-acetyl-cysteine and 2-mercaptoethanol.
Media can include RPMI 1640, AIM-V, DMEM, MEM, a-MEM, F-12, X-
Vivo 15, and X-Vivo 20, Optimizer, with added amino acids, sodium pyruvate,
and vitamins,
either serum-free or supplemented with an appropriate amount of serum (or
plasma) or a
defined set of hormones, and/or an amount of cytokine(s) sufficient for the
growth and
expansion of T cells. Antibiotics, e.g., penicillin and streptomycin, are
included only in
experimental cultures, not in cultures of cells that are to be infused into a
subject. The target
cells are maintained under conditions necessary to support growth, for
example, an
appropriate temperature (e.g., 37 C) and atmosphere (e.g., air plus 5% CO2).
In one embodiment, the T cells are expanded to a level necessary for adoptive
therapy and epitope mapping studies while maintaining antigen specificity and
cellular
function. Accordingly, any cell number is within the context of the present
invention. Cells
CA 02981033 2017-09-26
WO 2016/154508 PCT/1182016/024146
stimulated by the methods of the present invention are activated as shown by
the induction of
signal transduction, expression of cell surface markers and/or proliferation.
One such marker
appropriate for T cells is IFNy production which is an important
immunomodulating
molecule. The production of IFNy is extremely beneficial in amplifying the
immune
response.
With respect to T cells, the T cell populations resulting from the various
expansion methodologies described herein may have a variety of specific
phenotypic
properties, depending on the conditions employed. Such phenotypic properties
include
enhanced expression of CD25, CD154, IFN-y and GM-CSF, as well as altered
expression of
CD137, CD134, CD62L, and CD49d. The ability to differentially control the
expression of
these moieties may be very important. For example, higher levels of surface
expression of
CD154 on "tailored T cells," through contact with CD40 molecules expressed on
antigen-
presenting cells (such as dendritic cells, monocytes, and even leukemic B
cells or
lymphomas), will enhance antigen presentation and immune function. Such
strategies are
currently being employed by various companies to ligate CD40 via antibodies or
recombinant
CD4OL. The approach described herein permits this same signal to be delivered
in a more
physiological manner, e.g., by the T cell. The ability to increase IFN-y
secretion by tailoring
the T cell activation process could help promote the generation of Thl-type
immune
responses, important for anti-tumor and anti-viral responses. Like CD154,
increased
expression of GM-CSF can serve to enhance APC function, particularly through
its effect on
promoting the maturation of APC progenitors into more functionally competent
APC, such as
dendritic cells. Altering the expression of CD137 and CD134 can affect a T
cell's ability to
resist or be susceptible to apoptotic signals. Controlling the expression of
adhesion/homing
receptors, such as CD62L and/or CD49d and/or CCR7 may determine the ability of
infused T
cells to home to lymphoid organs, sites of infection, or tumor sites.
The phenotypic properties of T cell populations of the present invention can
be
monitored by a variety of methods including standard flow cytometry methods
and ELISA
methods known by those skilled in the art.
Those of ordinary skill in the art will readily appreciate that the cell
stimulation methodologies described herein may be carried out in a variety of
environments
(i.e., containers). For example, such containers may be culture flasks,
culture bags, or any
container capable of holding cells, preferably in a sterile environment. In
one embodiment of
the present invention a bioreactor is also useful. For example, several
manufacturers
31
CA 02981033 2017-09-26
WO 2016/154508
PCT/11S2016/024146
currently make devices that can be used to grow cells and be used in
combination with the
methods of the present invention. See for example, Celdyne Corp., Houston, TX;
Unisyn
Technologies, Hopkinton, MA; Synthecon, Inc., Houston, TX; Aastrom
Biosciences, Inc.,
Ann Arbor, MI; Wave Biotech LLC, Bedminster, NJ. Further, patents covering
such
bioreactors include U.S. Patent Nos: 6,096,532; 5,985,653; 5,888,807;
5,190,878, which are
incorporated herein by reference.
In one embodiment, a bioreactor with a base rocker platform is used, for
example such as "The Wave" (Wave Biotech LLC, Bedminster, NJ), that allows for
varying
rates of rocking and at a variety of different rocking angles. The skilled
artisan will
recognize that any platform that allows for the appropriate motion for optimal
expansion of
the cells is within the context of the present invention. In certain
embodiments, the methods
of stimulation and expansion of the present invention provide for rocking the
culture
container during the process of culturing at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16,
17, 18, 19, or 20 rocks per minute. In certain embodiments, the methods of
stimulation and
expansion of the present invention provide for the angle of the rocking
platform to be set at
1.5., 2., 2.5., 3., 3.5., 4., 4.5., 5., 5.5., 6., 6.5., 7., 7.5., "o,
6 8.5 , or 9.0 .
In certain embodiments, the capacity of the bioreactor container ranges from
about 0.1 liter to about 200 liters of medium. The skilled artisan will
readily appreciate that
the volume used for culture will vary depending on the number of starting
cells and on the
final number of cells desired. In particular embodiments, the cells of the
present invention,
such as T cells are seeded at an initial concentration of about 0.2 X 106
cells/rill to about 5 X
106 cells/ml, and any concentration therebetween. In one particular
embodiment, the cells
may be cultured initially in a static environment and transferred to a
bioreactor on a rocking
platform after 1, 2, 3, 4, 5, 6, 7, 8, or more days of culture. In a related
embodiment, the
entire process of stimulation, activation, and expansion takes place in a
bioreactor comprising
a rocking platform and an integrated magnet, as described above. Illustrative
bioreactors
include, but are not limited to, "The Wave".
In one particular embodiment, the cell stimulation methods of the present
invention are carried out in a closed system, such as a bioreactor, that
allows for perfusion of
medium at varying rates, such as from about 0.1 ml/minute to about 10
ml/minute.
Accordingly, in certain embodiments, the container of such a closed system
comprises an
outlet filter, an inlet filter, and a sampling port for sterile transfer to
and from the closed
system. In other embodiments, the container of such a closed system comprises
a syringe
pump and control for sterile transfer to and from the closed system. Further
embodiments
32
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
provide for a mechanism, such as a load cell, for controlling media in-put and
out-put by
continuous monitoring of the weight of the bioreactor container. In one
embodiment the
system comprises a gas manifold. In another embodiment, the bioreactor of the
present
invention comprises a CO2 gas mix rack that supplies a mixture of ambient air
and CO2 to the
bioreactor container and maintains the container at positive pressure. In
another embodiment,
the bioreactor of the present invention comprises a variable heating element.
In one embodiment, media is allowed to enter the container starting on day 2,
3, 4, 5, or 6 at about 0.5 to 5.0 liters per day until the desired final
volume is achieved. In one
preferred embodiment, media enters the container at 2 liters per day starting
at day 4, until the
volume reaches 10 liters. Once desired volume is achieved, perfusion of media
can be
initiated. In certain embodiments, perfusion of media through the system is
initiated on about
day 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 of culture. In one embodiment,
perfusion is initiated
when the volume is at about 0.1 liter to about 200 liters of media. In one
particular
embodiment, perfusion is initiated when the final volume is at 4, 5, 6, 7, 8,
9, 10, or 20 liters
or higher volume. The rate of perfusion can be from about .5 ml/minute to
about 10
ml/minute. In certain embodiments, the perfusion rate is about 1, 2, 2.5, 3,
3.5, 4, 4.5, 5, 5.5,
6, 6.5, 7, 7.5, or 8.0 mls/minute.
In a further embodiment of the present invention, the cells, such as T cells,
are
cultured for up to 5 days in a closed, static system and then transferred to a
closed system that
comprises a rocking element to allow rocking of the culture container at
varying speeds.
In certain aspects, the methodologies of the present invention provide for the
expansion of cells, such as T cells, to a concentration of about between 6 X
106 cell/ml and
about 90 X 106 cells/ml in less than about two weeks. In particular the
methodologies herein
provide for the expansion of T cells to a concentration of about 10, 15, 20,
25, 30, 35, 40, 45,
50, 55, 60, 65, 70, 75, 80, or 85 X 106 cells/ml and all concentrations
therein. In certain
embodiments, the cells reach a desired concentration, such as any of those
listed above, by
about day 5, 6, 7, 8, 9, 10, 11, or 12 of culture. In one embodiment, the T
cells expand by at
least about 1.5 fold in about 24 hours from about day 4 to about day 12 of
culture. In one
embodiment, the cells, such as T cells, expand from a starting number of cells
of about 100 X
106 to a total of about 500 X 109 cells in less than about two weeks. In
further embodiments,
the T cells expand from a starting number of cells of about 500 X 106 to a
total of about 500
X 109 cells in less than about two weeks. In related embodiments, the cells
expand from a
starting number of about 100 - 500 X 106 to a total of about 200, 300, or 400
X 1 09 cells in
less than about two weeks.
33
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
Therapy
In certain embodiments, a population of T cells is first contacted with
antigen,
and then subjected to a mixture of the invention comprising one or more of
DCs, 1L-15, 1L-7,
and IL-2. In one particular embodiment, the antigen-specific T cells are
induced by
vaccination of a patient with a particular antigen, either alone or in
conjunction with an
adjuvant or pulsed on dendritic cells. Antigen-specific cells for use in
expansion using the
stimulation method of the invention may also be generated in vitro.
Another aspect of the present invention provides a method for expanding
antigen specific T cells, comprising contacting a population of T cells with
an antigen for a
time sufficient to induce activation of T cells specific to said antigen;
contacting said
population of antigen-specific T cells ex vivo with a mixture comprising one
or more of DCs,
1L-15, IL-7, and IL-2 under conditions and for time sufficient to induce
proliferation of T
cells specific to said antigen, thereby expanding antigen-specific T cells. In
one embodiment,
the antigen is a tumor antigen. In another embodiment, the antigen is pulsed
on or expressed
by an antigen-presenting cell. In another embodiment, the population of T
cells is contacted
with said antigen ex vivo. In another embodiment, the method comprises at
least one round of
peptide-MHC tetramer sorting of said antigen-specific T cells. In certain
embodiments, the
method of the present invention further comprises at least one round of
peptide-MHC
tetramer magnetic selection of said antigen-specific T cells.
Another aspect of the present invention provides a method for the treatment of
cancer comprising administering to a cancer patient antigen-specific T cells
expanded
according to the methods provided herein.
The cells generated according to the present invention can also be used to
treat
autoimmune diseases. Examples of autoimmune disease include but are not
limited to,
Acquired Immunodeficiency Syndrome (AIDS, which is a viral disease with an
autoimmune
component), alopecia areata, ankylosing spondylitis, antiphospholipid
syndrome,
autoimmune Addison's disease, autoimmune hemolytic anemia, autoimmune
hepatitis,
autoimmune inner ear disease (AIED), autoimmune lymphoproliferative syndrome
(ALPS),
autoimmune thrombocytopenic purpura (ATP), Behcet's disease, cardiomyopathy,
celiac
sprue-dermatitis hepetiformis; chronic fatigue immune dysfunction syndrome
(CFIDS),
chronic inflammatory demyelinating polyneuropathy (CIPD), cicatricial
pemphigold, cold
agglutinin disease, crest syndrome, Crohn's disease, Degos' disease,
dermatomyositis-
juvenile, discoid lupus, essential mixed cryoglobulinemia, fibromyalgia-
fibromyositis,
34
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
Graves' disease, Guillain-Barre syndrome, Hashimoto's thyroiditis, idiopathic
pulmonary
fibrosis, idiopathic thrombocytopenia purpura (ITP), IgA nephropathy, insulin-
dependent
diabetes mellitus, juvenile chronic arthritis (Still's disease), juvenile
rheumatoid arthritis,
Meniere's disease, mixed connective tissue disease, multiple sclerosis,
myasthenia gravis,
pernacious anemia, polyarteritis nodosa, polychondritis, polyglandular
syndromes,
polymyalgia rheumatica, polymyositis and dermatomyositis, primary
agammaglobulinemia,
primary biliary cirrhosis, psoriasis, psoriatic arthritis, Raynaud's
phenomena, Reiter's
syndrome, rheumatic fever, rheumatoid arthritis, sarcoidosis, scleroderma
(progressive
systemic sclerosis (PSS), also known as systemic sclerosis (SS)), Sjogren's
syndrome, stiff-
man syndrome, systemic lupus erythematosus, Takayasu arteritis, temporal
arteritis/giant cell
arteritis, ulcerative colitis, uveitis, vitiligo and Wegener's granulomatosis.
The cells generated according to the present invention can also be used to
treat
inflammatory disorders. Examples of inflammatory disorders include but are not
limited to,
chronic and acute inflammatory disorders. Examples of inflammatory disorders
include
Alzheimer's disease, asthma, atopic allergy, allergy, atherosclerosis,
bronchial asthma,
eczema, glomerulonephritis, graft vs. host disease, hemolytic anemias,
osteoarthritis, sepsis,
stroke, transplantation of tissue and organs, vasculitis, diabetic retinopathy
and ventilator
induced lung injury.
The present invention also provides methods for preventing, inhibiting, or
reducing the presence of a cancer or malignant cells in an animal, which
comprise
administering to an animal an anti-cancer effective amount of the anti-tumor
cells of the
invention.
The cancers contemplated by the present invention, against which the immune
response is induced, or which is to be prevented, inhibited, or reduced in
presence, may
include but are not'limited to melanoma, non-Hodgkin's lymphoma, Hodgkin's
disease,
leukemia, plasmocytoma, sarcoma, glioma, thymoma, breast cancer, prostate
cancer, colo-
rectal cancer, kidney cancer, renal cell carcinoma, pancreatic cancer,
esophageal cancer, brain
cancer, lung cancer, ovarian cancer, cervical cancer, multiple myeloma,
hepatocellular
carcinoma, nasopharyngeal carcinoma, ALL, AML, CML, CLL, and other neoplasms
known
in the art.
Alternatively, compositions as described herein can be used to induce or
enhance responsiveness to pathogenic organisms, such as viruses, (e.g., single
stranded RNA
viruses, single stranded DNA viruses, double-stranded DNA viruses, HIV,
hepatitis A, B, and
C virus, HSV, CMV, EBV, HPV), parasites (e.g., protozoan and metazoan
pathogens such as
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
Plasmodia species, Leishmania species, Schistosoma species, Trypanosoma
species), bacteria
(e.g., Mycobacteria, Salmonella, Streptococci, E. coli, Staphylococci), fungi
(e.g., Candida
species, Aspergillus species) and Pneumocystis carinii.
The immune response induced in the animal by administering the subject
compositions of the present invention may include cellular immune responses
mediated by
CD8+ T cells, capable of killing tumor and infected cells, and CD4+ T cell
responses.
Humoral immune responses, mediated primarily by B cells that produce
antibodies following
activation by CD4+ T cells, may also be induced. A variety of techniques may
be used for
analyzing the type of immune responses induced by the compositions of the
present
invention, which are well described in the art; e.g., Coligan et al., Current
Protocols in
Immunology, John Wiley & Sons Inc., 1994.
When "an immunologically effective amount," "an anti-tumor effective
amount," "a tumor-inhibiting effective amount," or "therapeutic amount" is
indicated, the
precise amount of the compositions of the present invention to be administered
can be
determined by a physician with consideration of individual differences in age,
weight, tumor
size, extent of infection or metastasis, and condition of the patient. It can
generally be stated
that a pharmaceutical composition comprising the subject cells of the
invention, may be
administered at a dosage to be determined during appropriate clinical trials.
Cells of the
invention may also be administered multiple times at these dosages. The
optimal dosage and
treatment regime for a particular patient can readily be determined by one
skilled in the art of
medicine by monitoring the patient for signs of disease and adjusting the
treatment
accordingly.
Cells of the invention can be administered in dosages and routes and at times
to be determined in appropriate clinical trials. Cell compositions may be
administered
multiple times at dosages within these ranges. The cells of the invention may
be combined
with other methods. The cells of the invention for administration may be
autologous,
allogeniec or xenogenic to the patient undergoing therapy. If desired, the
treatment may also
include administration of mitogens (e.g., PHA) or lymphokines, cytokines,
and/or
chemokines (e.g., GM-CSF, IL-4, IL-13, F113-L, RANTES, MIP1-a, etc.) as
described herein
to enhance induction of the immune response.
The administration of the cells of the invention may be carried out in any
convenient manner. The cells of the present invention may be administered to a
patient
subcutaneously, intradermally, intramuscularly, by intravenous (i.v.)
injection, or
36
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
intraperitoneally. In some instances, the cells of the invention are
administered to a patient by
intradermal or subcutaneous injection. In other instances, the cells of the
invention are
administered by i.v. injection. In other instances, the cells of the invention
are injected
directly into a tumor or lymph node.
The cells of the invention can also be administered using any number of
matrices. The present invention utilizes such matrices within the novel
context of acting as an
artificial lymphoid organ to support, maintain, or modulate the immune system,
typically
through modulation of T cells. Accordingly, the present invention can utilize
those matrix
compositions and formulations which have demonstrated utility in tissue
engineering.
Accordingly, the type of matrix that may be used in the compositions, devices
and methods
of the invention is virtually limitless and may include both biological and
synthetic matrices.
In one particular example, the compositions and devices set forth by U.S. Pat.
Nos.
5,980,889; 5,913,998; 5,902,745; 5,843,069; 5,787,900; or 5,626,561 are
utilized, as such
these patents are incorporated herein by reference in their entirety. Matrices
comprise
features commonly associated with being biocompatible when administered to a
mammalian
host. Matrices may be formed from natural and/or synthetic materials. The
matrices may be
non-biodegradable in instances where it is desirable to leave permanent
structures or
removable structures in the body of an animal, such as an implant; or
biodegradable. The
matrices may take the form of sponges, implants, tubes, telfa pads, fibers,
hollow fibers,
lyophilized components, gels, powders, porous compositions, or nanoparticles.
In addition,
matrices can be designed to allow for sustained release of seeded cells or
produced cytokine
or other active agent. In certain embodiments, the matrix of the present
invention is flexible
and elastic, and may be described as a semisolid scaffold that is permeable to
substances such
as inorganic salts, aqueous fluids and dissolved gaseous agents including
oxygen.
A matrix is used herein as an example of a biocompatible substance. However,
the current invention is not limited to matrices and thus, wherever the term
matrix or matrices
appears these terms should be read to include devices and other substances
which allow for
cellular retention or cellular traversal, are biocompatible, and are capable
of allowing
traversal of macromolecules either directly through the substance such that
the substance
itself is a semi-permeable membrane or used in conjunction with a particular
semi-permeable
substance.
In one aspect of the present invention, the cells of the invention can be used
in
vivo as an adjuvant as described in U.S. Pat. No. 6,464,973. In a further
embodiment, the
cells of the invention can be used as a vaccine to induce an immune response
in vivo against
37
CA 02981033 2017-09-26
WO 2016/154508
PCT/1JS2016/024146
an antigen of interest such as those described herein (e.g., tumor antigens,
viral antigens,
autoantigens, etc). In one embodiment the cells of the invention can be used
to generate an
immune response in vivo, either administered alone or in combination with
other immune
regulators and in combination with other known therapies.
Identification of Epitopes
In one embodiment, the invention provides for compositions and methods to
expand antigen-specific T-cells. The antigen-specific T-cells can be expanded
according to
the invention which comprises contacting the T cell with one or more of DCs,
IL-15, IL-7,
and IL-2. The expanded T cells can be used to identify antigen-specific T-cell
receptors
(TCRs) and epitopes derived therefrom. For example, TCRs from the expanded T
cells can be
cloned. The cloned TCRs present a promising tool for the development of
specific adoptive
T-cell therapies to treat a desired disease or disorder. For example, the
cloned TCRs can be
used to generate peptides/antigens useful for vaccines.
In addition to their role in combating infections, T cells have also been
implicated in the destruction of cancerous cells. Autoimmune disorders have
also been linked
to antigen-specific T cell attack against various parts of the body. One of
the major problems
hampering the understanding of and intervention on the mechanisms involved in
these
disorders is the difficulty in identifying T cells specific for the antigen to
be studied.
TCRs are closely related to antibody molecules in structure, and they are
involved in antigen binding although, unlike antibodies, they do not recognize
free antigen;
instead, they bind antigen fragments which are bound and presented by antigen-
presenting
molecules. An important group of antigen-presenting molecules are the MHC
class I and
class II molecules that present antigenic peptides and protein fragments to T
cells.
Variability in the antigen binding site of a TCR is created in a fashion
similar
to the antigen binding site of antibodies, and also provides specificity for a
vast number of
different antigens. Diversity occurs in the complementarity determining
regions (CDRs) in
the N-terminal domains of the disulfide-linked alpha (a) and beta (f)), or
gamma (y) and delta
(A), polypeptides of the TCR. The CDR loops are clustered together to form an
MHC-
antigen-binding site analogous to the antigen-binding site of antibodies,
although in TCRs,
the various chains each contain two additional hypervariable loops as compared
to antibodies.
TCR diversity for specific antigens is also directly related to the MHC
molecule on the APC's
surface to which the antigen is bound and presented to the TCR.
38
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
In the embodiments described elsewhere herein, a peptide of the invention can
be located within the MHC molecule of a dendritic cell in order to generate
suitable T-cells.
In some embodiments, the MHC molecule is loaded with the peptide
extracellularly by
incubating cells at 37 C, 5% CO2 for 4 hours with varying concentrations of
peptide, then
washed once in serum-free RPMI. However, in alternative embodiments, the
antigen
presenting cells are transfected with a polynucleotide encoding a fusion
protein comprising
the peptide connected to at least an MHC Class I molecule alpha chain by a
flexible linker
peptide. Thus, when expressed, the fusion protein results in the peptide
occupying the MHC
Class I binding groove. Suitable MHC Class I molecules and costimulatory
molecules are
available from public databases. Further details of the synthesis of such a
fusion molecule
may be found in Mottez et al, J Exp Med. 1995 Feb. 1; 181(2):493-502, which is
incorporated
herein by reference. The advantage of expressing a fusion protein of the
peptide and the
MHC molecule is that a much higher concentration of peptide is displayed on
the surface of
the antigen presenting cells.
In some situations in the preparation of T-cells, it is preferred that there
is an
HLA match between the antigen presenting cells and the T-cells. That is to say
the antigen-
presenting cells display an MHC molecule of an allele for which the donor of
the T-cells is
HLA positive. In some embodiments, this is achieved by obtaining the antigen
presenting
cells from a first individual and the T-cells from a second individual wherein
the first and
second individuals have an HLA match.
However, in alternative embodiments, the antigen presenting cells and the T-
cells are obtained from the same individual but the antigen presenting cells
are transfected
with polynucleotides encoding the MHC molecule of a similar HLA allele. In
some
embodiments, the polynucleotide encodes a protein which encodes the MHC
molecule
connected to the peptide via a linker. There are numerous HLA Class I alleles
in humans and
the MHC molecule displayed by the antigen presenting cells, may, in principle,
be of any of
these alleles. However, since the HLA-A*0201 allele is particularly prevalent,
it is preferred
that the MHC molecule be of this allele. However, any HLA-A2 allele is usable
or other
alleles such as HLA-Al, HLA-A3, HLA-A 11 and HLA-A24 may be used instead.
In further embodiments of the present invention there is provided a method for
preparing T-cells suitable for delivery to a patient suffering from cancer.
The method
comprises providing dendritic cells expressing an HLA molecule of a first HLA
allele and
locating a peptide in the binding groove of the HLA molecule. The peptide may
or may not
39
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
be a peptide of the invention. T-cells are then primed with the dendritic
cells, the T-cells
being obtained or obtainable from an individual who is HLA matched for a first
HLA allele.
As described elsewhere herein, the dendritic cells may either be obtained from
a first donor
individual and the T-cells from a second donor individual wherein the first
and second donor
individuals are HLA matched. The advantage of using a dendritic cell, rather
than a non-
professional antigen presenting cell is that it results in a much higher
stimulus of the T-cells
In these embodiments, it is preferred that the peptide is a cell type specific
peptide, that is to say a peptide that is obtained from a protein which is
only expressed, or is
expressed at a much higher level (e.g. at least 10X higher concentration) in
specific cells than
in other cell types.
The T-cells prepared in accordance with the invention are administered to
patients in order to treat cancer in the patients. In principle, the T-cells
of the invention are
capable of being used for the treatment of many different types of cancer
including leukemia,
lymphomas such as non-Hodgkin lymphoma, multiple myeloma and the like.
Thus, in some embodiments of the present invention, pharmaceutical
preparations are provided comprising a T-cell of the invention and a
pharmaceutically
acceptable carrier, diluent or excipient, further details of which may be
found in
Remmington's Pharmaceutical Sciences in US Pharmacopeia, 1984 Mack Publishing
Company, Easton, Pa., USA.
As discussed elsewhere herein, the HLA allele of the MHC molecule used to
present the peptide to the T-cells is an HLA allele also expressed by the
patient and therefore
when the T-cells are administered to the patient, they recognize the peptide
displayed on
MHC molecules of that HLA allele.
In some altemative embodiments, multiple sets (e.g. 2 or 3 sets) of T-cells
are
provided, each T-cell being specific for a different peptide. In each case,
the T-cells are
allogeneic, as described elsewhere herein, that is to say the HLA allele of
the MHC molecule
on which the peptide is displayed during preparation of the T-cells is an 1-
ILA allele which is
not expressed in the donor individual from whom the T-cells are obtained. The
peptides may
all be from the same cell specific protein or may be from different proteins
but specific for
the same cell type. The peptide may or may not be a peptide of the invention.
In some
embodiments, the multiple sets of T-cells are administered simultaneously but
in other
embodiments they are administered sequentially.
Reference has been made to the preparation and provision of T-cells.
However, it is to be appreciated that the important feature of the T-cells is
the T-cell receptor
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
(TCR) which is displayed on the T-cells and, more specifically, the
specificity of the T-cell
receptor for the complex of the peptide and the MHC molecule. Therefore, in
some
alternative embodiments of the invention, following the preparation of T-cells
as described
elsewhere herein, the T-cell receptors of T-cells specific for a certain
peptide when
complexed with an MHC molecule of a particular allele are harvested and
sequenced. A
cDNA sequence encoding the T-cell receptor is then generated and which can be
used to
express the T-cell receptor recombinantly in a T-cell (e.g. the patient's own
T-cells or T-cells
from a donor). For example, the cDNA may be incorporated into a vector such as
a viral
vector (e.g. a retroviral vector), lentiviral vector, adenoviral vector or a
vaccinia vector.
Alternatively, a non-viral approach may be followed such as using naked DNA or
lipoplexes
and polyplexes or mRNA in order to transfect a T-cell.
Thus a T-cell which is "obtainable" from a donor individual includes a T-cell
which is obtained recombinantly in the manner described elsewhere herein
because the
recombinantly expressed TCR is naturally produced.
Since transfected T-cells also display their endogenous TCRs, it is preferred
that the T-cells are pre-selected, prior to transfection, to eliminate T-cells
that would give rise
to graft-versus-host disease. In some embodiments, the T-cells are pre-
selected such that the
specificity of their endogenous TCRs is known. For instance, T-cells are
selected which are
reactive with glypican-3. In other embodiments, the T-cells are obtained from
the patient and
thus are naturally tolerized for the patient. This approach can only be
adopted where the T-
cells of the patient are healthy.
In some alternative embodiments, the T-cell receptor, as a whole, is not
recombinantly expressed but rather the regions of the T-cell receptor which
are responsible
for its binding specificity are incorporated into a structure which maintains
the confirmation
of these regions. More specifically, complementarity determining regions
(CDRs) 1 to 3 of
the T-cell receptor are sequenced and these sequences are maintained in the
same
conformation in the recombinant protein.
In one embodiment, the expanded T cells provide a source for cloning TCRs
and epitopes/antigens associated therewith. The epitopes/antigens identified
can be used to
generate a vaccine. In one embodiment the vaccination antigens can be
constructed by
modifying a polypeptide (e.g. the target antigen) at specific amino acid
positions identified by
epitope mapping. Thus the method of the invention includes identifying
relevant positions for
modification in the target antigen by epitope mapping, modifying the target
antigen at
41
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
relevant positions to produce variants, and including the variants in separate
candidate
preparations.
Vaccination antigen polypeptides may be epitope mapped by a number of
methods, including those disclosed in detail in W000/26230 and W001/83559. In
brief,
these methods comprises a database of epitope pattems (determined from an
input of peptide
sequences, known to bind specifically to anti-protein antibodies) and an
algorithm to analyze
3-D structure of a given protein against the epitope pattern database. This
will determine the
possible epitopes on that protein, and the preference of each amino acid in
the protein
sequence to be part of epitopes.
In another aspect, methods are provided for identifying candidate MHC class
II epitopes. In certain embodiments, candidate epitopes can be identified
using a computer-
implemented algorithm for candidate epitope identification. Such computer
programs
include, for example, the TEPITOPE program (see, e.g., Hammer et al., Adv.
Immunol
66:67-100 (1997); Sturniolo et al., Nat. Biotechnol. 17:555-61 (1999); Manici
et al., J Exp.
Med. 189:871-76 (1999); de Lalla et al., J. Immunol. 163:1725-29 (1999);
Cochlovius et al.,
J. lmmunol. 165:4731-41 (2000); the disclosures of which are incorporated by
reference
herein), as well as other computer implemented algorithms.
The computer-implemented algorithm for candidate epitope identification can
identify candidate epitopes in, for example, a single protein, in a very large
protein, in a
group of related proteins (e.g., homologs, orthologs, or polymorphic
variants), in a mixtures
of unrelated proteins, in proteins of a tissue or organ, or in a proteome of
an organism. Using
this approach, it can be possible to interrogate complex tissues or organisms
based on
sequence information for expressed proteins (e.g., from deduced open reading
frame or a
cDNA library), in addition to analysis of known candidate molecular targets,
as an efficient,
sensitive and specific approach to identification of T cell epitopes.
Following identification of candidate epitopes, peptides or pools of peptides
can be formed that correspond to the candidate epitope(s). For example, once a
candidate
epitope is identified, overlapping peptides can be prepared that span the
candidate epitope, or
portions thereof, to confirm binding of the epitope by the MHC class II
molecule, and, as
necessary, to refine the identification of that epitope. Altematively, pools
of peptides can be
prepared including a plurality of candidate epitopes identified using a
computer-implemented
algorithm for candidate epitope identification.
T cell epitope peptide
42
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
A T cell epitope of the invention is a short peptide that can be derived from
a
protein antigen. Antigen presenting cells can directly bind antigen via
surface MHC
molecules and/or internalize antigen and process it into short fragments which
are capable of
binding MHC molecules. The specificity of peptide binding to the MHC depends
on specific
interactions between the peptide and the peptide-binding groove of the
particular MHC
molecule.
Peptides which bind to MHC class I molecules are usually between 6 and 30,
more usually between 7 and 20 amino or between 8 and 15 amino acids in length.
The amino-
terminal amine group of the peptide makes contact with an invariant site at
one end of the
peptide groove, and the carboxylate group at the carboxy terminus binds to an
invariant site at
the other end of the groove. Thus, typically, such peptides have a hydrophobic
or basic
carboxy terminus and an absence of proline in the extreme amino terminus. The
peptide is in
an extended confirmation along the groove with further contacts between main-
chain atoms
and conserved amino acid side chains that line the groove. Variations in
peptide length are
accommodated by a kinking in the peptide backbone, often at proline or glycine
residues.
Peptides which bind to MHC class II molecules are usually at least 10 amino
acids, for example about 13-18 amino acids in length, and can be much longer.
These
peptides lie in an extended confirmation along the MHC II peptide-binding
groove which is
open at both ends. The peptide is held in place mainly by main-chain atom
contacts with
conserved residues that line the peptide-binding groove.
The peptide of the present invention may be made using chemical methods.
For example, peptides can be synthesized by solid phase techniques (Roberge J
Y et al (1995)
Science 269: 202-204), cleaved from the resin, and purified by preparative
high performance
liquid chromatography. Automated synthesis may be achieved, for example, using
the ABI
431 A Peptide Synthesizer (Perkin Elmer) in accordance with the instructions
provided by the
manufacturer.
The peptide may alternatively be made by recombinant means or by cleavage
from a longer polypeptide. For example, the peptide may be obtained by
cleavage from full-
length glypican-3 protein. The composition of a peptide may be confirmed by
amino acid
analysis or sequencing.
The peptides of the invention can be tested in an antigen presentation system
which comprises antigen presenting cells and T cells. For example, the antigen
presentation
system may be a murine splenocyte preparation, a preparation of human cells
from tonsil or
43
CA 02981033 2017-09-26
WO 2016/154508
PCT/1JS2016/024146
PBMC. Alternatively, the antigen presentation system may comprise a particular
T cell
line/clone and/or a particular antigen presenting cell type.
T cell activation may be measured via T cell proliferation (for example using
3H-thymidine incorporation) or cytokine production. Activation of TH1-type
CD4+ T cells
can, for example be detected via IFN7 production which may be detected by
standard
techniques, such as an ELISPOT assay.
Measurement of antigen-specific T cells during an immune response are
important parameters in vaccine development, autologous cancer therapy,
transplantation,
infectious diseases, inflammation, autoimmunity, toxicity studies, and the
like. Peptide
libraries are crucial reagents in monitoring of antigen-specific T cells. The
present invention
provides improved methods for the use of a peptide library in analysis of T
cells in samples
including diagnostic, prognostic and immune monitoring methods. Furthermore
the use of a
peptide library in anti-tumor therapy are described elsewhere herein,
including isolation of
antigen-specific T cells capable of inactivation or elimination of undesirable
target cells or
isolation of specific T cells capable of regulation of other immune cells. The
present
invention also relates to MEC multimers comprising one or more tumor derived
peptides.
The identification of particular antigenic peptides provides new opportunities
for the development of diagnostic and therapeutic strategies against cancer.
Advantageously,
identification of novel T cell epitopes enable the production of MHC class I
and class II
multimers, tetramers and pentamers, useful as analytical tools delivering both
increased
sensitivity of immuno-monitoring. In addition, the detection of antigen
specific CTL in the
periphery of individuals at risk of disease recurrence is a useful diagnostic
tool.
Accordingly, in a further aspect, the invention provides an MEC multimer,
tetramer or a pentamer comprising at least one of the MHC class I or II
glypican-3 peptide
epitopes as described herein.
The invention also provides compositions and methods for identifying
peptides useful for cancer therapy. Peptide sequences from a candidate protein
predicted to
bind to HLA-A*0201 can be identified by a computer algorithm. Peptides are
selected for
synthesis according to predicted affinity with a cut-off value of 500 nM or
less, but also
higher values may be chosen. Peptides are synthesized and binding to HLA-
A*0201 can be
confirmed using biochemical assays. Peptide binding is compared with the
binding achieved
with a pass/fail control peptide, designated 100%, and with a positive control
peptide.
Corresponding HLA-A*0201-peptide multimers are also synthesized for peptides
with a
44
CA 02981033 2017-09-26
WO 2016/154508
PCT/1.JS2016/024146
binding affinity above the pass/fail control peptide. These peptides are
tested for the ability to
generate a T cell line specifically reacting with the specific peptide-HLA-
A*0201 complex.
The cell line can be referred to as multimer-, tetramer-, or pentamer-positive
T cells.
Multimer positive cells indicate a high immunogenicity for the corresponding
peptide.
Additional responses can be measured to assess production of the cytokine
interferon-y,
degranulation and killing of target cells.
In some embodiments, the peptides of the invention can be administered
directly to a patient as a vaccine. Thus the peptides of the invention are
immunogenic
epitopes of specific proteins and are used in order to elicit a T-cell
response to their
respective proteins. In some embodiments, the polypeptide of the invention is
administered
directly to a patient as a vaccine. For example, in a patient that has
leukemia, a polypeptide
comprising a peptide from a hematopoietic cell specific protein is
administered to the patient
in order to elicit a T-cell response to the protein. The T-cell response leads
to death of
hematopoietic cells, including the cancerous cells, but is specific to these
cells and does not
result in an immune response to other cell types.
It is also to be noted that, in many patients, directly administering such a
peptide will not elicit a T-cell response because the cell specific protein is
a "self-protein"
and any T-cells that are capable of binding the polypeptide when presented on
an MHC
molecule of the HLA alleles of the patient are tolerized. That is to say T-
cells that would be
reactive are either destroyed in the thymus of the patient during the
selection process or are
inactivated through central or peripheral tolerance mechanisms. Therefore, it
is preferred that
the peptides of the invention are used to generate T-cells obtained, or
obtainable, from an
allogeneic donor individual. This individual should preferably be HLA negative
for an HLA
allele of which the patient is HLA positive. For example, if the patient is
HLA positive for
the HLA allele HLA-A*0201 then T-cells are obtained from an individual who is
negative for
HLA-A*0201. It is generally preferred that the donor individual is otherwise
HLA-identical
to the patient. Antigen presenting cells (APCs) are then provided which
display MHC
molecules of the HLA-A*0201 allele and which are loaded with the peptide. The
T-cells of
the donor individual are then primed with the APCs and the resulting cells are
allowed to
proliferate.
The proliferated T-cells which are capable of binding the peptide of the
invention when in complex with the HLA-A*0201 antigen are then enriched using
artificial
structures which comprise a plurality of peptide-MHC molecules (e.g. pentamers
or
CA 02981033 2017-09-26
WO 2016/154508 PCT/1182016/024146
tetramers). The T cells specific for the particular peptide-HLA-A*0201 complex
within the
mixture of T cells have the capacity to bind to these structures when mixed
with them. The T
cells are subsequently mixed with magnetic beads with the capacity to bind the
artificial
structures. The artificial structures and the T cells bound to them are then
removed from the
remainder of the mixture by magnetic attraction of the beads.
Therapy
The antigen specific T cell can be administered to an animal as frequently as
several times daily or it may be administered less frequently, such as once a
day, once a
week, once every two weeks, once a month, or even less frequently, such as
once every
several months or even once a year or less. The frequency of the dose will be
readily apparent
to the skilled artisan and will depend upon any number of factors, such as,
but not limited to,
the type and severity of the disease being treated, the type and age of the
animal, etc.
An antigen specific T cell may be co-administered with the various other
compounds (cytokines, chemotherapeutic and/or antiviral drugs, among many
others).
Alternatively, the compound(s) may be administered an hour, a day, a week, a
month, or even
more, in advance of an antigen specific T cell, or any permutation thereof.
Further, the
compound(s) may be administered an hour, a day, a week, or even more, after
administration
of an antigen specific T cell, or any permutation thereof The frequency and
administration
regimen will be readily apparent to the skilled artisan and will depend upon
any number of
factors such as, but not limited to, the type and severity of the disease
being treated, the age
and health status of the animal, the identity of the compound or compounds
being
administered, the route of administration of the various compounds and the
antigen specific T
cell, and the like.
In the method of treatment, the administration of the composition of the
invention may be for either "prophylactic" or "therapeutic" purpose. When
provided
prophylactically, the composition of the present invention is provided in
advance of any
symptom, although in particular embodiments the vaccine is provided following
the onset of
one or more symptoms to prevent further symptoms from developing or to prevent
present
symptoms from becoming worse. The prophylactic administration of composition
serves to
prevent or ameliorate any subsequent infection or disease. When provided
therapeutically, the
pharmaceutical composition is provided at or after the onset of a symptom of
infection or
disease. Thus, the present invention may be provided either prior to the
anticipated exposure
to a disease-causing agent or disease state or after the initiation of the
infection or disease.
46
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
An effective amount of the composition would be the amount that achieves
this selected result of enhancing the immune response, and such an amount
could be
determined as a matter of routine by a person skilled in the art. For example,
an effective
amount of for treating an immune system deficiency against cancer or pathogen
could be that
amount necessary to cause activation of the immune system, resulting in the
development of
an antigen specific immune response upon exposure to antigen. The term is also
synonymous
with "sufficient amount."
The effective amount for any particular application can vary depending on
such factors as the disease or condition being treated, the particular
composition being
administered, the size of the subject, and/or the severity of the disease or
condition. One of
ordinary skill in the art can empirically determine the effective amount of a
particular
composition of the present invention without necessitating undue
experimentation.
EXPERIMENTAL EXAMPLES
The invention is further described in detail by reference to the following
experimental examples. These examples are provided for purposes of
illustration only, and
are not intended to be limiting unless otherwise specified. Thus, the
invention should in no
way be construed as being limited to the following examples, but rather,
should be construed
to encompass any and all variations which become evident as a result of the
teaching
provided herein.
Without further description, it is believed that one of ordinary skill in the
art
can, using the preceding description and the following illustrative examples,
make and utilize
the present invention and practice the claimed methods. The following working
examples
therefore, specifically point out the preferred embodiments of the present
invention, and are
not to be construed as limiting in any way the remainder of the disclosure.
Example 1: Restoration Of Anti-HER2 CD4+ Thl Responses In DC1 Vaccinated
Breast
Cancer Patients
The following studies were designed to explore the role of adjuvant type 1-
polarized dendritic cell ("DC1") vaccination.
DC1Vaccination of HER2+ IBC Patients with Residual Disease Following
Neoadjuvant
Therapy
47
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
Four HER2 IBC patients with residual disease following neoadjuvant therapy
received adjuvant HER2-pulsed DC1 vaccines. Thl immune responses of each
patient were
determined pre-DC1 vaccination, 3-months post-DC vaccination, and 6-months
post-
vaccination and were generated from patient PBMCs pulsed with six HER2 Class
II peptides
(SEQ ID NOS:1-6) by measuring IFNy production via ELISPOT as described above.
Autologous DC1 vaccines were prepared as described above. Responses were
evaluated on:
(1) the overall anti-HER2 responsivity (responding to ?,1 peptide), (2) the
number of reactive
peptides (response repertoire), and (3) the cumulative response across the six
HER2 peptides.
Results:
Responsivity: Pre-vaccination, only one IBC patient produced an immune
response, defined as >20 SFC/106 cells in an experimental well after
subtracting unstimulated
background. Compared with pre-vaccination results, all vaccinated IBC patients
produced an
immune response, defined as >2-fold increase in anti-HER2 IFNyPos Thl
responses.
Response Repertoire: Mean repertoire increased from 0.511 peptides pre-
vaccination to 3.2510.96 peptides at 3 months (p=0.01) and 410.8 peptides at 6
months
(p=0.01) (Figure 1).
Cumulative response: Mean cumulative response improved from 36.5138.3
SFC/106pre-vaccination to 151.0160.0 SFC/106 at 3 months (p=0.04) and
198.4139.7
SFC/106 at 6 months (p=0.02) (Figure 2).
Conclusions:
HER2-pulsed DC1 vaccination of HER2+ IBC patients with residual disease
following treatment with neoadjuvant chemotherapy boosts anti-HER2 Thl immune
responses.
The anti-HER2 Thl immune responses increase in both breadth (response
repertoire) and depth (cumulative response).
Example 2: In Vitro Expansion of HER2-Specific Thl Cells
The following studies were designed to explore the role of adoptive T-cell
transfer in restoring anti-HER2 Thl immunity. T cells are expanded to a level
necessary for
adoptive therapy and epitope mapping studies while maintaining antigen
specificity and
cellular function.
48
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
Briefly, in vitro, HER2-specific Thl cells were generated by coculture with
HER2-peptide pulsed DC's and expanded using IL-2 alone or IL-2, IL7, and IL-
15. Thl cells
were subsequently expanded either by repeat HER2-peptide pulsed DC1 co-culture
or via
anti-CD3/CD28 stimulation. Fold expansion was defined as: (#T-cells post
expansion / #T-
cells pre expansion); specificity was measured by antigen specific IFNy
production by
ELISA.
As will be shown herein, repeated co-culture of CD4+T cells with HER2
peptide pulsed DC1s stimulated with IL-2, IL-7, and IL-15 results in a
significant expansion
of highly specific anti-HER2 Thl cells, providing a potential population of
cells for adoptive
transfer. Co-culture with specific peptide specific DC1s and IL-2, IL-7, and
IL-15 stimulation
may mimic the lymph node environment and may be used to significantly expand
any
population of antigen specific Thl cells.
Without wishing to be bound by any particular theory, it is believed that when
a subject is vaccinated against a protein antigen (for example, a tumor target
antigen), blood
can be removed from the subject after vaccination and collected. The collected
blood contains
dendritic cell precursors as well as low levels of T cells specific for the
tumor target antigen.
DC precursors and T cells are separated from each other. DC precursors can be
loaded/pulsed
with tumor target protein/antigen and then activated to DC1 status. The
antigen-specific
DC1s can then be co-cultured with the T cells, and cytokines (IL-15, IL-7 and
IL-2) are
added to the co-culture in appropriate sequence. This cycle can be repeated
weekly until T
cells grow to sufficient numbers (e.g. 1X109). The T cells can then be
supplied to the original
subject, infusing them with a large quantity of T cells that their body could
not produce
naturally. This large army of antigen-specific T cells can have strong
antitumor activity.
Method for Expanding T Cells
HER2 Specific DC1 Preparation:
DC precursors were obtained from HER2 breast cancer patients (DCIS) who
were vaccinated with HER2 peptide-pulsed DC1 vaccines, as described
previously. DC
precursors were obtained via tandem leukapheresis/countercurrent centrifugal
elutriation.
DCs were incubated at 3x106 cells in lml Macrophage Serum-free Medium (SFM-
Gibco Life
Technologies, Carlsbad, CA) with GM-CSF 50 ng/ml (Berlex, Richmond, CA) at 37
C. DCs
were pulsed with a single HER2 peptide antigen (42-56, 98-114, 328-345, 776-
790, 927-
941, 1166-1180 (SEQ ID NOS 1-6)); 20m/m1) 48-72hrs after the cells were
initially plated.
For maturation, DCs were further activated 6 hours later with IFN-y (1000
U/ml) and the
49
CA 02981033 2017-09-26
WO 2016/154508 PCMJS2016/024146
following day with lipopolysaccharide ("LPS") (lOng/m1). HER2 specific DC1s
were
harvested 6 hours after LPS administration at the point of maximum IL-12
production.
CD4+ T Cell Preparation:
Lymphocytes were also obtained from previously vaccinated (HER2pulsed
DC1 vaccination) breast cancer patients via tandem
leukapheresis/countercurrent centrifugal
elutriation. CD4+T-ce11s were purified by negative selection using Human CD4+
T Cell
Enrichment Kit (Stemcell Technologies; Vancouver BC, Canada). CD4+ T-cells
were
resuspended at 2x106cells/m1 in culture medium (ISOCOVE's Medium, 1% L-
Glutamine,
1% Pen/Strep, 1% Sodium Pyruvate, 1% non-essential amino acids, Mediatech;
Manassas,
VA and 5% heat inactivated human AB serum)
DC 1-CD4+ Co-Culture:
DC1s were plated with CD4+ T-cells at a 1:10 ratio (2x105DC1s/m1 with
2x106CD4+T-ce11s/m1) in 24-well plates and incubated at 37 C. Recombinant
Human IL-7
(lOng/m1) and IL-15 (lOng/m1) (BioLegend; San Diego, CA) were added 48-72hrs
after co-
culture. Twenty-four hours after adding IL-7 and IL-15, Recombinant Human IL-2
(5U/m1)
was added.
HER2 Specific iDCs for Testing and HER2 Specific DC1s for Restimulation:
Two additional groups of DCs were prepared as described above. In one
group, each well was pulsed with a single peptide antigen (20ug/m1), and was
considered as
immature DCs ("iDCs"). In the second group, each well was pulsed with a single
peptide
antigen (20 g/m1) and matured to DC 1s as described above. Seven to nine days
following the
previous DC1 co-culture, the HER2 specific CD4+ T-cells were harvested. The T-
cells were
co-cultured with iDCs for ELISA testing. Interferon gamma production was
measured by
ELISA assay according to manufacturer's recommendations and protocols. The T-
cells were
also co-cultured with DC1s and stimulated with IL-7/15 and IL-2 as described
above. The
cycle was repeated with co-culture of CD4+T-cells with HER2 specific DC1s a
total of 4
times.
Expansion method outline:
Develop HER2-specific DC1s as previously described
Day 1: Culture monocytes (1 well of each peptide)
CA 02981033 2017-09-26
WO 2016/154508
PCT/1JS2016/024146
Day 4: Mature HER2-specific DC1 s
AM: pulse with antigen (20 g/m1).
PM (6hrs later): add IFN-y
Day 5: Purify CD4+ T-cells & DC1 co-culture.
AM: LPS
6 hrs later: Co-Culture: 2x106CD4+ T cells with 2x105DC1s
*48-72 hrs after co-culture: add IL-7 (lOng/m1) and IL-15 (lOng/m1);
24 hrs after adding IL-7/15: add IL-2 (5U/m1);
(In the IL-2 alone condition, IL2 was added 72-96 hrs after co-culture (at the
same time as
IL-2 was added to the IL2/7/15 group).
Develop BOTH HER2-specific iDCs for ELISA testing as well as HER2-specific DC
1s for
restimulation
Day 9-11: Culture monocytes (2 wells of each peptide)
48 hrs after culturing monocytes: process HER2-specific immature DCs and
mature
DC1 s
iDCs: Pulse with antigen (20 g,/m1) (given at the same time to future iDCs
and DC's)
DC 1s: AM: pulse with antigen (20p.g/m1).
PM (6 hrs later): add IFN-g
The next day: iDC co-culture & DC1 co-culture (i.e.: 7-9 days following DC1 co-
culture)
DC Is: AM: LPS
Harvest HER2 specific CD4+Tcells
Harvest HER2 specific iDCs
4 Co-Culture: 2x106CD4+Tce11s with 2x105 iDCs
Harvest HER2 specific DC1 s
Co-Culture: 2x106CD4+ Tcells with 2x105DC1s
The next day: Run ELISA on iDC co-culture
Repeat process above; resume at IL7/15 stimulation (*)
Results:
51
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
The above-referenced methods were used in the experiments and studies for
which the results are shown in Figures 3-16:
Figure 3 and Figure 4 show a direct comparison between CD4+T cells co-
cultured with HER2-specific DC1's stimulated with IL-2 versus those stimulated
with IL-
2/7/15 for two different patients who had received HER2-pulsed DC1
vaccination,
respectively.
Briefly, immature DC's ("iDC's") from the respective patients were pulsed
with the following MHC class II peptides: peptide 42-56 (SEQ ID NO: 1),
peptide 98-114
(SEQ ID NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide 776790 (SEQ ID NO:
4) and
matured to DC l's. The resulting HER2-pulsed DC l's were then co-cultured with
CD4+T
cells and stimulated with IL-2 alone or with IL-2/7/15 as indicated. The red
outline boxes
indicate the specific peptide and stimulation protocol for which specificity
is shown (greater
than 2:1 ratio of specific antigen:control antigen IFN-y production). Thus in
Figure 3
specificity was shown for peptide 42-56-IL2/7/15 protocol; peptide 98-114 ¨IL-
2 protocol
and IL-2/7/15 protocol, peptide 328-345 both protocols, and peptide 776-790,
both protocols.
In Figure 4 specificity was shown for peptide 776-790, both protocols, only.
Graphs showing
fold expansion (defined as number of T cells post expansion/number of T cells
pre
expansion) are shown at right in each figure. In general, fold expansion was
greater for the
IL-2/7/15 stimulation than for IL-2 stimulation alone as shown by the
respective fold
expansion data. Specificity was measured by antigen-specific IFN-y production
by ELISA.
Figures 5 and Figure 6 show specific to non-specific immune responses:
Figure 5 shows a specific response following a first stimulation/expansion
with HER2-
specific DC l's and Figure 6 shows the subsequent loss of that specific
response after the
second stimulation/expansion with non-specific anti CD3/CD28.
The first stimulation of CD4+ T cells with HER2-specific DC1s resulted in
multiple specific immune responses as shown by red outline boxes in Figure 5:
peptide 42-
56, IL2/7/15 protocol; peptide 98-114, both protocols, peptide 328-345, both
protocols, and
peptide 776-790, both protocols. Figure 6 shows the second stimulation of the
HER2-specific
CD4+T cells with a non-specific anti CD3/CD28 stimulus resulted in a four-fold
expansion
(side graph), but with a loss of specificity in three fourths of the peptide
groups (only peptide
328-345 showed specificity after CD3/28 expansion). iDC's from patients were
pulsed with
the following MHC class II peptides: peptide 42-56 (SEQ ID NO: 1), peptide 98-
114 (SEQ
ID NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide 776-790 (SEQ ID NO: 4)
and
52
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
matured to DC1's as described above. The resulting HER2-pulsed DC1's were then
co-
cultured with CD4+ T cells and stimulated with IL-2 alone or with IL2/7/15 as
indicated.
Figure 7 and Figure 8 show non-specific immune response followed by
specific immune responses: Figure 7 shows non-specific expansion of CD4+T
cells. Figure 8
shows failure to obtain specificity following subsequent stimulation with HER2-
specific
DC1's. The first stimulation of CD4+ T cells with non-specific anti CD3/CD28
resulted in a
3.8 fold expansion (Figure 7). The second stimulation of the non-specific
CD4+T cells with
HER2-specific DC l's failed to result in a specific immune response (Figure
8). iDC's from
patients were pulsed with the following MHC class II peptides: peptide 42-56
(SEQ ID NO:
1), peptide 98-114 (SEQ ID NO: 2), peptide 328-345 (SEQ ID NO: 3), and peptide
776-790
(SEQ ID NO: 4) and matured to DC l's as described above. The resulting
HER2pulsed DC l's
were then co-cultured with CD4+ T cells and stimulated with IL-2 alone or with
IL-2/7/15 as
indicated.
Figures 9A and 9B show in vitro primary/first expansion of HER2-specific
Thl cells comparing CD4+ T cells co-cultured with HER2-specific DC1's expanded
with IL-2
versus those expanded with 1L-2/7/15. iDC's were pulsed with the following MHC
class 11
peptides: peptide 42-56 (SEQ ID NO: 1), peptide 98-114 (SEQ ID NO: 2), peptide
776-790
(SEQ ID NO: 4) and peptide 927-941 (SEQ ID NO: 5), and) and matured to DC I
's. The
resulting HER2-pulsed DC1's were then co-cultured with CD4+ T cells and
stimulated with
IL-2 alone or with IL2/7/15 as indicated. The red outline boxes (Figure 9B)
indicate the
specific peptide and stimulation protocol for which specificity is shown
(greater than 2:1 ratio
of specific antigen:control antigen IFN-y production). Figure 9A shows mean
fold expansion
(defined as number of T cells post expansion/number of T cells pre expansion)
of Thl cells
was significantly better when stimulated with IL-2, IL-7, and IL-15 than with
IL-2 alone
(2.6 0.75 vs 1.0 0.12; p=0.001). Figure 9B shows specificity for the various
peptide/expansion protocols as measured by antigen-specific IFN-y production
by ELISA.
Both stimulation with IL-2, IL-7, and IL-15 and with IL-2 alone resulted in a
specific Thl
response in the same HER2 peptide 776-790.
Primary expansion summary: IL-2 vs. IL-2/7/15. Mean fold expansion of Thl
cells was significantly better when stimulated with IL-2, IL-7, and IL-15 than
with IL-2 alone
(2.6 0.75 vs 1.0 0.12; p=0.001) (Figure 9A). Both stimulation with IL-2, IL-7,
and IL-15
and with IL-2 alone resulted in a specific Thl response in the same HER2
peptide (peptide
776-790) (Figure 9B).
53
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
Figures 10A and 10B show in vitro secondary/second expansion of HER2-
pulsed DC l's versus anti-CD3/CD28. Re-stimulation of Thl cells with HER2-
peptide pulsed
DC1s and anti-CD3/CD28 each resulted in a similar fold expansion (3.911.0
vs.4.3 2.0
p=0.7) (Figure 10A). However, Figure 10B shows stimulation of the Thl cells
with HER2-
specific DC1s enhanced the specific Thl response; whereas non-specific
stimulation with
anti-CD3/CD28 resulted in an overall loss of HER2-peptide specificity. The
following MHC
class II peptides were used: peptide 42-56 (SEQ ID NO: 1), peptide 98-114 (SEQ
ID NO: 2),
peptide 776790 (SEQ ID NO: 4) and peptide 927-941 (SEQ ID NO: 5). The red
outline
boxes (Figure 10B) indicate the specific peptide and stimulation protocol for
which
specificity is shown (greater than 2:1 ratio of specific antigen:control
antigen IFN-y
production) (i.e., DC1 restimulation of peptide 42-56-specific Th1Cells and
peptide 776-790
¨specific Thl cells.
Secondary expansion summary: HER2-peptide pulsed DC1 vs. anti-
CD3/CD28. Re-stimulation of Thl cells with HER2-peptide pulsed DC1s and
antiCD3/CD28
each resulted in a similar fold expansion (3.911.0 vs. 4.312.0, p=0.7) (Figure
10A). However,
stimulation of the Thl cells with HER2-specific DC1s enhanced the specific Thl
response,
whereas non-specific stimulation with antiCD3/CD28 resulted in an overall loss
of HER2-
peptide specificity (Figure 10B).
Tertiary expansion of the Thl cells with HER2-pulsed DC1's Following a
third stimulation with indicated HER2-specific DC1s, both mean fold expansion
(4.3210.5,
43.7-fold cumulative expansion (Figure 11A) and antigen specificity (Figure
11B) increased
again, specifically all four peptides ( peptide 42-56 (SEQ ID NO: 1), peptide
98-114 (SEQ
ID NO: 2), peptide 776-790 (SEQ ID NO: 4) and peptide 927-941 (SEQ ID NO: 5))
show
specificity and increased IFN-y production.
Tertiary expansion summary: HER2-peptide pulsed DC1. Following a third
stimulation with HER2-specific DC1s, both mean fold expansion (4.32 0.5, 43.7-
fold
cumulative expansion) (Figure 11A) and specificity-both the number of specific
peptides and
IFN-y production (Figure 11B) increased again.
Overall it is seen In Figures 9B, 10B, and 11B, which have the same numbers
of T cells, the IFN-y production goes up by logs from expansion to expansion.
It was also
seen that in the second stimulation, non-specific CD3/CD28 (Figure 10B) there
was an
overall loss of specificity. The cells were expanded as Thl phenotype with 50-
200-fold
expansion (Figures 9A, 10A, and 11A) that became more specific and stronger
with each
stimulation.
54
CA 02981033 2017-09-26
WO 2016/154508 PCT/US2016/024146
Figures 1 2-1 5 show sequential results of repeated in vitro stimulation (4
times)
of HER2-specifc CD4T Thl cells with IL-2/7/15. For all Figures 12-15 the
respective left
panels show peptide specificity by 1FN-7 production ("Tet" is a tetanus
patient control);
respective right panels show fold expansion for the specific HER2-peptides
used. In Figure
12 an additional MEC-class II peptide was used to pulse iDC's: peptide 1166-
1180 (SEQ ID
NO: 6) in addition to the other five used in the above studies. However, as
seen in the fold
expansion results (Figure 12, right panel), peptide 328-345-specificThl cells
and peptide
1166-1180-specific Thl cells did not produce enough cells for further
expansion thus only
HER2 Thl cells specific to the remaining four peptides were used.
Sequentially, Figure 12 for the first stimulation shows specificity only for
peptide 776-790-specific Thl cells; Figure 13 for the second stimulation shows
an increase,
specificity for peptide 42-56 in addition to peptide 776-790specific Thl
cells; Figure 14 for
the third expansion shows specificity for all four peptide-specific Thl cells
(peptide 42-56,
peptide 98-114, peptide 776-790, and peptide 927-941), and Figure 15 for the
fourth
expansion shows loss of specificity for one of the peptides (peptide 927-941)
leaving three
remaining HER2-specific peptides (peptide 42-56, peptide 98-114, and peptide
776-790).
Figure 16 shows cumulative fold expansion of the four expansions shown in
Figures 12-15 for all the HER2-specific Thl cells, with the last bar of each
group (dots)
showing cumulative fold expansion. Average cumulative fold expansion was over
100-fold.
Conclusions:
Repeated co-culture of CD4+T cells with HER2-peptide pulsed DC1s
stimulated with IL-2, IL-7, and IL-15 results in a significant expansion of
highly specific anti
HER2 Thl cells, providing a potential population of cells for adoptive
transfer.
Each stimulation out of the total four stimulations resulted in both increased
fold expansion and increased antigen specificity, without reaching a limit of
either. Indeed
there was shown a 100-400-fold expansion.
Co-culture with peptide specific DC1s and IL-2, IL-7, and IL-15 stimulation
may mimic the lymph node environment and be used to significantly expand any
population
of antigen specific 'Thl cells. Those skilled in the art will readily
recognize that the present
embodiments related to T cell expansion are in no way limited to CD4+t cells.
Thus the
present embodiments provide methods for growing CART cells, cytotoxic T
lymphocytes
(CD8+'s) as well as all other kinds of T cells.
CA 02981033 2017-09-26
WO 2016/154508 PCT/1JS2016/024146
The results presented herein demonstrate that HER2-pulsed DC1 vaccination
of HER2+ IBC patients with residual disease following treatment with
neoadjuvant
chemotherapy boosts anti-HER2 Thl immune responses. The antiHER2 Thl immune
response increases in both breadth (response repertoire) and depth (cumulative
response).
Adoptive transfer of HER2-specific Thl cells may serve a role in resurrecting
the CD4+ Thl
immune response. Repeated co-culture with HER2-peptide pulsed DC1s stimulated
with IL-
2, IL-7, and IL-15 can result in significant expansion of highly specific anti-
HER2 Thl cells.
Example 3: Anti-HER2 CD4+ Thl Responses Can Be Restored in DC 1 Vaccinated
Breast
Cancer Patients
The expansion of T cell subsets is an essential step to gain enough T cells to
perform adoptive therapy, or to identify epitopes on target antigens for
peptide-based
vaccines. Expansion of T cells, in principle is a simple process. However, in
practice, many
technical problems exist including poor levels of expansion, premature
activation-induced
cell death (apoptosis) or loss of antigen specificity and/or function.
Part of the problem lies in the inability to replicate, in vitro, the
environment
inside the body where antigen-specific T cell expansion occurs, which is the
lymph node.
These are specialized tissues that contain a number of different cell types
apart from T cells
including antigen-presenting dendritic cells, stromal cell such as epithelial
cells. Each of
these cell types play a different role by providing contact-dependent signals
(surface
receptors) and soluble signals (cytokines) important for T cell growth and
maintenance of cell
function.
Experiments were designed to explore the role of (I) adjuvant type 1-polarized
dendritic cell (DC1) vaccination and (2) adoptive T-cell transfer in restoring
anti-HER2 Thl
immunity. T cells are expanded to a level necessary for adoptive therapy and
epitope
mapping studies while maintaining antigen specificity and cellular function.
The materials and methods employed in these experiments are now described.
Preparation of fully activated DC
Freshly elutriated myeloid monocytes were cultured in 6 well microplates
(12x106 cells/well). Culture medium consisted of Serum Free Medium (SFM
Invitrogen
Carlsbad CA). The final concentration of added GMCSF was 5Ong/m1 and of IL4 is
1000
U/ml. Cells were cultured overnight at 37 C in 5% CO2. In some batches, the
cells were
56
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
pulsed with the adequate peptides after 16-20 hr and cultured for additional 6-
8 hr, after
which 1000U/ml IFN-y was added. Dendritic cells were matured with TLR agonist
LPS
(TLR 4, lOng/m1) or R848 (TLR8, 11.1g/m1).
To induce the production of the Thl-polarizing cytokine IL-12, the DCs are
activated with combinations of the cytokine IFN-y, or the TLR agonists
bacterial LPS and/or
R848. This should induced T cells that produce IFN-y. Alternatively, the DCs
can be
activated with combinations of ATP, bacterial LTA, LPS and prostaglandin E2
(PGE2). This
can cause IL-23, IL-6 and IL-1I3 to be amplified, leading an immune response
dominated by
IL-17 and IL-22-secreting Th17 cells.
Expansion method
HER2-specific DC1s as described elsewhere herein
Day 1: Culture monocytes (1 well of each peptide)
Day 4: Mature HER2-specific DC's
AM: pulse with antigen (20ug/m1)
PM (6hrs later): add IFN-y
Day 5: Purify CD4+ T-cells & DC1 co-culture.
AM: LPS
6hrs later: Co-Culture: 2x10A6 CD4+ T cells with 2x10^5 DC1s
*48-72hrs after co-culture: add IL-7 (lOng/rn1) and IL-15 (lOng/m1)
24hrs after adding IL-7/15: add IL-2 (5U/m1)
(In the IL-2 alone condition, IL2 was added 72-96hrs after co-culture (at the
same time as IL-
2 was added to the IL2/7/15 group)
Vaccination
HER2+ IBC patients with residual disease following neoadjuvant therapy
received adjuvant HER2-pulsed DC1 vaccines. Immune responses were generated
from
PBMCs pulsed with HERZ Class II peptides by measuring IFNy production via
ELISPOT.
Responses were evaluated on: (1) the overall anti-HER2 responsivity
(responding to >1
peptide), (2) the number of reactive peptides (response repertoire), and (3)
the cumulative
response across the six HER2 peptides. Pre-vaccination Thl responses were
compared with
3-month and 6-month responses.
57
CA 02981033 2017-09-26
WO 2016/154508
PCT/US2016/024146
Production of HER2-specific Thl cells
In vitro, HER2-specific Thl cells were generated by co-culture with HER2-
peptide pulsed DC1s and expanded using IL-2 alone or 1L-2, IL-7, and IL-15.
Thl cells were
subsequently expanded either by repeat HER2-peptide pulsed DC1 co-culture or
via anti-
CD3/CD28 stimulation. Fold expansion was defined as: (4T-cells post expansion
/ 4T-cells
pre expansion); specificity was measured by antigen specific IFNy production
by ELISA.
The results of the experiments disclosed herein are now described.
In vivo - Thl response to HER2-pulsed DC1 vaccination:
Responsivity: Pre-vaccination, only one IBC patient produced an immune
response, defined as >20 SFC/2*105 cells in an experimental well after
subtracting
unstimulated background. Compared with pre-vaccination results, all vaccinated
IBC patients
produced an immune response, defined as >2-fold increase in anti-HER2
IFINlyposThl
responses.
Response Repertoire: Mean repertoire increased from 0.5 1 peptides pre-
vaccination to 3.25 0.96 peptides at 3 months (p=0.01) and 4 0.8 peptides at 6
months
(p=0.01) (Figure 1).
Cumulative response: Mean cumulative response improved from 36.5 38.3
SFC/2*105 pre-vaccination to 151.0 60.0 SFC/2*105 at 3 months (p=0.04) and
198.4 39.7
SFC/2*105 at 6 months (p=0.02) (Figure 2).
In vitro - expansion of HER2-specific Thl cells:
Primary expansion: IL-2 vs. IL-2/7/15. Mean fold expansion of Thl cells was
significantly better when stimulated with IL-2, IL-7, and IL-15 than with IL-2
alone
(2.610.75 vs 1.0 0.12; p=0.001) (Figure 9A). Both stimulation with IL-2, IL-7,
and IL-15
and with IL-2 alone resulted in a specific Thl response in the same HER2
peptide (Figure
9B).
Secondary expansion: HER2-peptide pulsed DC1 vs. anti-CD3/CD28. Re-
stimulation of Thl cells with HER2-peptide pulsed DC1s resulted in a 3.9-fold
expansion,
10.1-fold cumulative expansion; similarly, subsequent stimulation with anti-
CD3/CD28
resulted in a 4.4-fold expansion, 11.5-fold cumulative expansion (p=0.5,
Figure 10A). Re-
stimulation of the Thl cells with HER2-specific DC1s enhanced the specific Thl
response;
58
CA 02981033 2017-09-26
WO 2016/154508
PCT/1JS2016/024146
whereas non-specific stimulation with anti-CD3/CD28 resulted in an overall
loss of HER2-
peptide specificity (Figure 10B).
Tertiary expansion: HER2-peptide pulsed DC1. Following a third stimulation
with HER2-specific DC1s, both mean fold expansion (4.32 0.5; Figure 11A) and
specificity
(Figure 11B) increased again.
Fourth expansion: The T cells can be expanded for a fourth round of
stimulation. It was observed that the T cells continued to expand and maintain
T cell function
(Figure 12).
. The results presented herein demonstrate that HER2-pulsed DC1 vaccination
of HER2+ IBC patients with residual disease following treatment with
neoadjuvant
chemotherapy boosts anti-HER2 Thl immune responses. The anti-HER2 Thl immune
responses increase in both breadth (response repertoire) and depth (cumulative
response).
Adoptive transfer of HER2-specific Thl cells may serve a role in resurrecting
the CD4+ Thl
immune response. Repeated co-culture with HER2-peptide pulsed DC1s stimulated
with IL-
2, 1L-7, and IL-15 can result in significant expansion of highly specific anti-
HER2 Thl cells.
Without wishing to be bound by any particular theory, it is believed that when
a subject is vaccinated against a protein antigen (for example, a tumor target
antigen), blood
can be removed from the subject after vaccination. The blood containing
dendritic cell
precursors as well as low levels of T cells specific for the tumor target
antigen can be
collected. DC precursors and T cells is separated from each other. DC
precursors can be
loaded with tumor target protein and then activated. They can then be co-
cultured with the T
cells, and cytokines (IL-15, IL-7 and IL-2) added to the co-culture in
appropriate sequence.
This cycle can be repeated weekly until T cells grow to sufficient numbers
(e.g. 1X109). The
T cells can then be supplied to the original subject, infusing them with a
large quantity of T
cells that their body could not produce naturally. This large army of antigen-
specific T cells
can have strong anti-tumor activity.
The disclosures of each and every patent, patent application, and publication
cited herein are hereby incorporated herein by reference in their entirety.
While this invention
has been disclosed with reference to specific embodiments, it is apparent that
other
embodiments and variations of this invention may be devised by others skilled
in the art
without departing from the true spirit and scope of the invention. The
appended claims are
intended to be construed to include all such embodiments and equivalent
variations.
59