Note: Descriptions are shown in the official language in which they were submitted.
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
LYOPHILIZED PHARMACEUTICAL COMPOSITIONS
CROSS REFERENCE
[0001] This Application claims the benefit of United States Provisional
Application No.
62/188,025, filed July 2, 2015, which is incorporated herein by reference in
its entirety.
BACKGROUND
[0002] DNA methylation is a post replicative chemical modification of DNA.
Different cancers
can be stratified by their abnormal DNA methylation profiles (degree of global
or specific DNA
methylation) and the hypermethylation of specific genes can be associated with
the prognosis for
gastric, lung, esophageal, pancreatic, and colon cancer. DNA methylation
patterns can also be
used to predict response or resistance to therapy in glioma and melanoma.
Azacitidine and
decitabine are two FDA approved hypomethylating agents (HMAs) that exert their
therapeutic
effect by inhibiting DNA methylation levels.
[0003] Dinucleotide compounds derived from decitabine for the development of
therapies for
similar indications have been described in U.S. Patent No. 7,700,567 and its
equivalent
W02007041071. Drug formulations containing dinucleotide compounds of the type
described in
W02007041071 are disclosed in W02013033176. The disclosure in each of US
7,700,567,
W02007041071 and W02013033176 is incorporated by reference in its entirety.
[0004] Lyophilization, often referred to as freeze drying, is a method of
dehydration in which a
solvent-containing substrate is frozen and then subjected to a vacuum so that
the solvent is
removed by sublimation, i.e. direct conversion from the the solid frozen state
into the gaseous
state.
INCORPORATION BY REFERENCE
[0005] Each patent, publication, and non-patent literature cited in the
application is hereby
incorporated by reference in its entirety as if each was incorporated by
reference individually.
SUMMARY OF THE INVENTION
[0006] In some embodiments, the invention provides a method of preparing a
lyophilized
pharmaceutical composition, the method comprising dissolving a compound of
formula (1):
1
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
NH2
N N
L
N 0
HO ________________________
i) 0
1 N...........õ/".\.
< I NH
0 =P-0 H
)) N--------NNH 2
\
cØ..
OH (1),
or a pharmaceutically-acceptable salt thereof, in a solvent comprising
dimethylsulfoxide
(DMSO) to form a solution, wherein the solvent is then removed by a freeze-
drying process to
give a lyophilized product, wherein the freeze-drying process comprises: (i) a
first freezing stage
in which the solution is frozen by reducing the temperature thereof to a
temperature of no
greater than about -20 C; (ii) a first warming stage in which the temperature
of the frozen
solution is raised to a temperature in the range from about -15 C to about 5
C, wherein the
temperature in the range from about -15 C to about 5 C keeps the solution
frozen; (iii) a second
freezing stage in which the temperature of the solution is lowered to a
temperature of no greater
than about -20 C; (iv) a primary drying stage, wherein the primary drying
stage comprises a
sublimation step in which the DMSO is removed by sublimation from the solution
in its frozen
state under reduced pressure to give a partially dried product; and (v) a
secondary drying stage
in which the DMSO is removed by evaporation from the partially dried product
in a non-frozen
state under reduced pressure to give the lyophilized product.
[0007] In some embodiments, the invention provides a pharmaceutical
composition prepared by
a process comprising the steps of: dissolving a compound of formula (1):
2
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
NH2
N N
L
NO
HO __
i) 0
1 N...........õ/"\..
< I NH
0 =P-0 H
oI N--------NNH 2
\
cØ..
OH (1), or a pharmaceutically-acceptable salt
thereof, in a
solvent comprising dimethylsulfoxide (DMSO) to form a solution, wherein the
solvent is then
removed by a freeze-drying process to give a lyophilized product, wherein the
freeze-drying
process comprises: (i) a first freezing stage in which the solution is frozen
by reducing the
temperature thereof to a temperature of no greater than about -20 C; (ii) a
first warming stage in
which the temperature of the frozen solution is raised to a temperature in the
range from about -
15 C to about 5 C, wherein the temperature in the range from about -15 C to
about 5 C keeps
the solution frozen; (iii) a second freezing stage in which the temperature of
the solution is
lowered to a temperature of no greater than about -20 C; (iv) a primary
drying stage, wherein
the primary drying stage comprises a sublimation step in which the DMSO is
removed by
sublimation from the solution in its frozen state under reduced pressure to
give a partially dried
product; and (v) a secondary drying stage in which the DMSO is removed by
evaporation from
the partially dried product in a non-frozen state under reduced pressure to
give the lyophilized
product.
BRIEF DESCRIPTION OF THE FIGURES
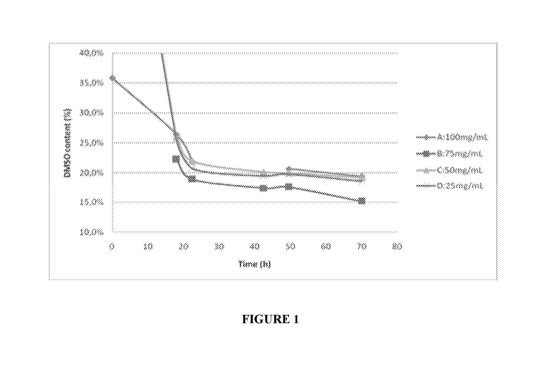
[0008] FIGURE 1 is a plot of DMSO removal with time as the lyophilization
process of the
invention progresses. DMSO removal profiles for four formulations A, B, C and
D of different
concentrations are shown in FIGURE 1.
DETAILED DESCRIPTION
[0009] This application relates to lyophilized pharmaceutical compositions
containing a
dinucleotide derived from decitabine and to methods for the preparation and
use of decitabine-
derived dinucleotide compositions.
3
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[0010] The present invention relates to improved lyophilized compositions
containing a
compound of formula (1) or a pharmaceutically acceptable salt thereof, and to
a method of
preparing the improved lyophilized pharmaceutical compositions using a freeze
drying process.
The invention also provides the use of the lyophilized pharmaceutical
compositions in medicine
and in particular their use in the treatment of cancers.
[0011] The present disclosure provides improved methods for lyophilization of
a substrate
comprising a non-aqueous solvent, for example, DMSO and a compound of formula
(1), or a
pharmaceutically-acceptable salt thereof. Generally, the methods involve two
freezing stages
with an intermediate warming stage (annealing stage) between the two freezing
stages. The
methods can be used for removal of the non-aqueous solvent from the substrate.
In some
particular embodiments, the compound within the substrate is a compound of
formula (1):
NH2
N N
L
NO
HO _______________________
i) 0
1 N............õ/".
< I NH
0 =P-0 H
oI N NH2
\
cØ..
OH
or a pharmaceutically acceptable salt thereof. The present disclosure also
provides lyophilized
compositions comprising a compound of formula (1) or a pharmaceutically
acceptable salt
thereof. In addition, the present disclosure provides uses of the lyophilized
pharmaceutical
compositions in medicine, particularly in the treatment of cancers.
[0012] It has been found that by using two freezing stages and an intermediate
warming stage
(annealing stage) between the two freezing stages, DMSO can be removed much
more quickly
during the subsequent primary drying stage and that, consequently, the length
of the secondary
drying stage can be significantly reduced. Without wishing to be bound by any
theory, it is
believed that the intermediate warming stage can provide increased porosity,
thereby enabling
the DMSO to sublime more readily. Thus, much more of the DMSO is removed
during the
primary drying stage.
[0013] Freeze Drying Microscopy (FDM) studies on the formulations have shown
that, even at
4
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
temperatures below -30 C, on occasion, there can be some residual non-frozen
solvent or co-
solvent present. The term "frozen" as used herein therefore includes a state
in which there is
present a solid structure formed from solvent and/or co-solvent molecules but
there can also be
present some solvent and/or co-solvent in non-frozen, or liquid, form.
Method for preparing lypophilized pharmaceutical composition.
[0014] The methods provided herein include a method of preparing a lyophilized
pharmaceutical
composition comprising a compound, for example, a compound of formula (1)) or
a
pharmaceutically-acceptable salt thereof, which method comprises dissolving
the compound of
formula (1) or the pharmaceutically acceptable salt thereof in a non-aqueous
solvent comprising
dimethylsulfoxide and optionally one or more co-solvents to form a solution,
and then removing
the solvent and any co-solvents by a freeze-drying process to give a
lyophilized product;
wherein the freeze-drying process comprises one or more of the following
stages: (i) a first
freezing stage in which the solution is frozen by reducing the temperature
thereof to a
temperature of no greater than -20 C; (ii) a first warming stage in which the
temperature of the
frozen solution is raised to a temperature in the range from -15 C to 5 C at
which the solution
remains in a frozen state; (iii) a second freezing stage, which occurs after
the first warming stage
and in which the temperature of the solution in its frozen state is lowered to
a temperature of no
greater than -20 C; (iv) a primary drying stage comprising a sublimation step
in which
dimethylsulfoxide and one or more co-solvents when present are removed by
sublimation from
the solution in its frozen state under reduced pressure to give a partially
dried product; and (v) a
secondary drying stage in which dimethylsulfoxide and one or more co-solvents
when present
are removed by evaporation from the partially dried product in a non-frozen
state under reduced
pressure to give the lyophilized product.
[0015] The sequence of freezing and intermediate warming stages (i), (ii), and
(iii) can be
repeated one or more times before proceeding to the primary drying stage (iv).
For example, a
first sequence of stages (i), (ii), and (iii) can be followed by a second
sequence of stages (i), (ii),
and (iii), and optionally by third and fourth sequences of stages (i), (ii),
and (iii) before
proceeding to the primary drying stage (iv).
[0016] The method of the invention can, for example, reduce the overall time
for the freeze-
drying process by at least a day and, in some embodiments of the invention, by
up to two days.
The method of the invention can further allow reconstitution of the solution
more readily than
compositions prepared using methods that omit the intermediate warming stage.
For example, in
some embodiments of the invention as defined herein, the reconstitution time
of the
compositions can be reduced from a time in excess of 30 minutes to a time of
less than 20
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
minutes and, in some embodiments, a time of less than 10 minutes.
[0017] The freeze-drying procedure can be carried out in a lyophilization
apparatus. The
lyophilization apparatus can have a chamber in which lyophilization containers
(e.g.
lyophilization vials) containing solution can be placed for freeze-drying. The
chamber can be
connected to a vacuum source (e.g. a vacuum pump) to enable the pressure
within the chamber
to be reduced. The apparatus can also have components for freezing or heating
the contents of
the chamber. Prior to freeze-drying, a bulk solution of the compound of
formula (1) in DMSO
and optionally one or more co-solvents can be prepared and filtered through a
filter (e.g. a
sterilising filter) before aliquots are filled into lyophilization containers
(e.g. lyophilization vials)
and transferred to the lyophilization apparatus. Prior to transfer to the
lyophilization apparatus,
the containers can be partially stoppered to prevent contamination but still
permit escape of the
solvent during the freeze-drying process.
[0018] In the following paragraphs, parameters of the freeze-drying process
are set out in more
detail with reference to particular embodiments, sets, subsets, ranges and
individual values for
each parameter. For the avoidance of doubt, each embodiment, set, subset,
range and individual
value defined in relation to one parameter of the freeze-drying process can be
combined with
each embodiment, set, subset, range and individual value defined in relation
to any other
parameter of the freeze-drying process. This application therefore discloses
all combinations of
the embodiments, sets, subsets, ranges and individual values for each
parameter of the freeze-
drying process.
[0019] The temperatures referred to above and elsewhere herein in relation to
the parameters of
the lyophilization process are the temperatures of the shelves in the
lyophilization apparatus.The
shelves are typically cooled by cooling fluids, the temperatures of which are
monitored and
provide a method of determining the shelf temperatures. The temperature
measuments obtained
from the cooling fluids can be cross-checked against temperatures obtained
directly from the
product in the lyophilization containers by inserting temperature probes into
selected
lyophilization containers.
[0020] In the first freezing stage (i), the solution can be frozen by reducing
the temperature
thereof to a temperature of no greater than about -20 C, for example, the
temperature can be
reduced to a value of no greater than about -30 C (or no greater than about -
35 C, or no greater
than about -40 C, or no greater than about -41 C, or no greater than about -
42 C, or no greater
than about -43 C, or no greater than about -44 C). For example, the solution
can be frozen by
reducing the temperature to a value in the range from about -40 C to about -
50 C, or about -42
C to about -48 C, or about -43 C to about -47 C, or about -44 C to about -
46 C, e.g. at
about -45 C.
6
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[0021] The first freezing stage can involve a temperature ramping step wherein
the temperature
is reduced from an initial (e.g. ambient) temperature to a target temperature
over a first time
period, for example over a period of up to about 2 hours or up to about 1.5
hours or up to 1.25
hours, e.g. about 1 hour.
[0022] Once the target temperature has been reached, the frozen solution can
be held at the
target temperature for a second time period, for example up to about 3 hours,
or up to about 2.5
hours or up to about 2 hours, e.g. about 1.5 hours.
[0023] Following the first freezing stage, the solution can be subjected to a
first warming stage
in which the temperature of the frozen solution is raised to a temperature in
the range -15 C and
4 C at which the solution remains in a frozen state. For example, the frozen
solution can be
warmed to a temperature in the range from about -5 C to about 5 C, or from
about -3 C to
about 3 C, or from about -2 C to about 2 C, or from about -1 C to about 1
C, for example at
about 0 C.
[0024] The first warming stage can involve a first time period over which the
frozen solution is
warmed to a target temperature and a second time period over which the frozen
solute is held at
the target temperature. For example, the first time period over which the
frozen solution is
warmed to a target temperature can be up to about 2 hours, or up to about 1.75
hours, or up to
about 1.5 hours, for example, about 1.3 hours.
[0025] Following the first warming stage, the still-frozen solution can be
subjected to a second
freezing stage in which the temperature of the solution in its frozen state is
lowered to a
temperature of no greater than about -20 C. The temperature can be reduced to
a value of no
greater than about -30 C (or no greater than about -35 C, or no greater than
about -40 C, or no
greater than about -41 C, or no greater than about -42 C, or no greater than
about -43 C, or no
greater than about 44 C). For example, the temperature of the frozen solution
can be reduced to
a value in the range from about -40 C to about -50 C, or about -42 C to
about -48 C, or about
-43 C to about -47 C, or about -44 C to about -46 C, for example, at about
-45 C.
[0026] After the second freezing stage, the frozen solution can be subjected
to a primary drying
stage comprising a sublimation step in which dimethylsulfoxide and one or more
co-solvents
when present are removed by sublimation from the solution in its frozen state
under reduced
pressure to give a partially dried product. In the primary drying stage, the
frozen solution can be
warmed to facilitate faster sublimation of the DMSO, whilst maintaining the
solution in a frozen
state. For example, the frozen solution can be warmed to a temperature in the
range from -25 C
to 0 C, or from -22 C to -2 C, e.g. from about -20 C to about -5 C.
[0027] In the primary drying stage, the frozen solution can be warmed in
steps. For example, in
a first warming step, the temperature can be raised from a temperature of no
greater than about -
7
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
30 C to a temperature in the range from about -25 C to about -19 C (e.g.
about -20 C), and
then held at that temperature for a defined holding period. At this
temperature, residual unfrozen
solvent and/or co-solvent can be removed by evaporation.
[0028] In a second warming step the temperature can be raised from a
temperature in the range
from about -25 C to about -19 C (e.g. about -20 C), to a temperature in the
range from about
-10 C to about 0 C (e.g. about -5 C) and then held at that temperature for
further defined
holding period. It will be appreciated that further intermediate warming
stages and holding
periods can be added to the first and second warming stages. As an alternative
to warming the
frozen solution in stages, warming can be carried out in a continuous manner
until a required
target temperature is attained.
[0029] At the beginning of the primary drying period, the pressure in the
vessel containing the
frozen solution can be reduced (typically from atmospheric pressure) to a
pressure at which
removal of the DMSO and optionally other co-solvents can take place. The
pressure can be
reduced to a pressure of lower than 1 mBar, for example, below 500 Bar, or
less than 100
Bar, or less than 50 Bar. For example, the pressure can be reduced to a
pressure of less than
20 Bar, or less than 10 Bar, or from 1 to 10 Bar, or from 4 to 8 Bar, e.g.
about 6 Bar.
[0030] The primary drying stage can involve an initial pressure-reducing stage
in which the
temperature is held constant and the pressure is reduced to a target value,
followed by warming
of the frozen solution as defined above. Alternatively, the reduction in
pressure and the warming
of the frozen solution can be carried out simultaneously.
[0031] The primary drying stage can take from about 20 to about 60 hours, for
example, from
about 30 to about 50 hours.
[0032] The progress of the primary drying stage can be monitored by one or
more sensors or
gauges present in a lyophilization chamber of the lyophilization apparatus.
The sensors or
gauges (such as a Pirani gauge) can be used to measure one or more parameters
within the
chamber, whereby defined changes in the one or more parameters can indicate
the progress of
the primary drying and provide a means of determining when sublimation of DMSO
and
optionally any co-solvents has been completed. For example, a sensor or gauge
can measure
pressure within the chamber or the conductivity of gas in the chamber.
[0033] During the sublimation process, the temperature must be below the
critical temperature
and pressure of the product so that the product remains frozen. Sublimation is
a direct solid-to-
gas DMSO phase change. If the conditions are above the critical temperature
and pressure, the
product is not frozen and, instead, is a liquid and the DMSO can change from a
liquid-to-gas
(boils). It is disadvantageous for the DMSO to boil instead of sublime.
[0034] The primary drying stage can be performed under pressures of from about
5 Bar to
8
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
about 40 Bar. The freezing temperature of the product at these pressures is
about -2 C to about
-4 C. The primary drying stage can be performed at temperatures from about -3
C to about -9
C. At this temperature range, the vapor pressure is adequate for a quick
sublimation, which
leads to a better product. In some embodiments, the pressure is about 20 Bar.
In some
embodiments, the temperature is about -6 C.
[0035] Once sublimation of the DMSO has ceased, or has fallen below a certain
level, the
secondary drying stage is initiated. In the secondary drying stage,
dimethylsulfoxide and one or
more co-solvents when present are removed by evaporation from the partially
dried product in a
non-frozen state under reduced pressure to give a lyophilized product. Thus,
in the secondary
drying stage, a reduced pressure environment is maintained and the partially
dried product is
heated to a temperature at which it is no longer frozen. As the boiling point
of DMSO is about
189 C, the partially dried product can be heated to a temperature of at least
about 40 C, more
usually at least about 45 C, for example at least about 50 C, or at least
about 55 C. In some
embodiments, the partially dried product is heated to a temperature in the
range from about 55
C to about 70 C, for example, about 65 C.
[0036] The secondary drying stage can involve one or more temperature ramping
steps in which
the partially dried product is heated to a target temperature, each
temperature ramping step being
followed by a temperature holding step. In one embodiment, there is a single
temperature
ramping step followed by a single temperature holding step.
[0037] During the secondary drying stage, unfrozen solvent molecules are
removed to give a
lyophilized product containing only low levels of residual DMSO.
[0038] It is advantageous for the secondary drying stage to be performed at a
temperature of
about 30 C to about 65 C, for example, about 40 C.
[0039] At the end of the secondary drying stage, an inert gas such as nitrogen
is admitted into
the lyophilization chamber and the containers (e.g. vials) containing the
lyophilized product are
fully sealed (e.g. by means of stopper and optionally also a cap) under inert
gas.
[0040] The freeze-drying procedure can be carried out on a solution of a
compound of the
formula (1) or a pharmaceutically acceptable salt thereof in a non-aqueous
solvent comprising
dimethylsulfoxide and optionally one or more co-solvents.
[0041] In some embodiments, water contamination is avoided at any stage.
Without being bound
by theory, it is believed that hydrate formation particularly disrupts the
product's structure that
becomes not conducive to easy reconstitution.
[0042] In some embodiments of the invention, substantially no co-solvents are
present; i.e. the
solvent consists essentially of DMSO.
[0043] In other embodiments of the invention, one or more of the other non-
aqueous co-solvents
9
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
can be present. Where a co-solvent is present, the total volume of co-solvent
can typically
constitute no more than about 25% (v/v) of the total solvent. More usually,
the total volume of
co-solvent, when present, constitutes no more than about 20%, or no more than
about 15%, or
no more than about 10%, or no more than about 5% by volume of the total volume
of solvent.
For example, the total volume of co-solvent, can constitute from about 0%
(v/v) to about 5%
(v/v) of the total volume of solvent.
[0044] The solution to be lyophilized can contain an amount of the compound of
formula (1) or
the pharmaceutically acceptable salt thereof in the range from about 5 mg/ml
to about 200
mg/ml, for example, in the range from about 10 mg/ml to about 150 mg/ml. For
example, the
solution can contain from about 20 mg/ml to about 120 mg/ ml, or from about 22
mg/ml to about
110 mg/ml, or from about 25 mg/ml to about 105 mg/ml, or from about 25 mg/ml
to about 100
mg/ml of the the compound of formula (1) or the pharmaceutically acceptable
salt thereof.
[0045] In some embodiments, the solution contains from about 40 mg/ml to about
110 mg/ml, or
from about 50 mg/ml to about 105 mg/ml of the compound of formula (1) or the
pharmaceutically acceptable salt thereof.
[0046] In a particular embodiment of the invention, the solution contains
either 75 mg/ml; or
100 mg/ml of a sodium salt of the compound of formula (1).
[0047] Non-limiting examples of pressures that can be used during a method of
the invention
include about 1 Bar, about 2 Bar, about 3 Bar, about 4 Bar, about 5 Bar,
about 6 Bar,
about 7 Bar, about 8 Bar, about 9 Bar, about 10 Bar, about 15 Bar, about
20 Bar, about
25 Bar, about 30 Bar, about 35 Bar, about 40 Bar, about 45 Bar, about 50
Bar, about 55
Bar, about 60 Bar, about 65 Bar, about 70 Bar, about 80 Bar, about 90
Bar, and about
100 Bar.
Lyophilized pharmaceutical compositions.
[0048] The invention provides a lyophilized pharmaceutical composition, which
is preparable by
(or prepared by) a freeze-drying process as described herein.
[0049] The lyophilized pharmaceutical compositions of the invention are
characterized by
enhanced solubility relative to known lyophilized formulations of compounds of
the formula (1)
and their salts. Accordingly, in another embodiment, the invention provides a
lyophilized
pharmaceutical composition comprising a compound of formula (1) or a
pharmaceutically
acceptable salt thereof, which is obtainable by a freeze-drying process as
defined herein and
which has a dissolution time, at ambient temperature, and without the aid of
mechanised stirring,
in a non-aqueous solvent containing 65% (v/v) propylene glycol; 25% (v/v)
glycerine; and 10%
(v/v) ethanol, of no greater than 20 minutes.
[0050] In some embodiments, the lyophilized pharmaceutical composition has a
dissolution time
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
in the non-aqueous solvent of no greater than 15 minutes, or no greater than
12 minutes.
[0051] In particular embodiments, the lyophilized pharmaceutical composition
has a dissolution
time in the non-aqueous solvent of no greater than 10 minutes.
[0052] The lyophilized pharmaceutical compositions of the invention are also
characterised by
reduced levels of residual DMSO solvent. Accordingly, in another embodiment,
the invention
provides a lyophilized pharmaceutical composition comprising a compound of
formula (1) or a
pharmaceutically acceptable salt thereof, which is obtainable by a freeze-
drying process as
defined herein and wherein, in an amount of lyophilized composition obtained
from 1 gram of
solution, there is a residual DMSO content of no greater than 20 mg, or no
greater than 19 mg. It
will be appreciated that the reference to "solution" means the solution of the
pharmaceutically
acceptable salt thereof in a solvent comprising dimethylsulfoxide and
optionally one or more co-
solvents. The solvent can be non-aqueous, anhydrous or substantially-
anhydrous.
[0053] In another embodiment, there is provided a lyophilized pharmaceutical
composition
comprising a compound of formula (1) or a pharmaceutically acceptable salt
thereof, which is
obtainable by a freeze-drying process as defined herein and wherein any
residual DMSO is
present in the composition in an amount corresponding to no more than 35 mg
per 100 mg
equivalent of the free base of the compound of formula (1).
[0054] The term "100 mg equivalent of the free base" refers to the amount by
weight of free
base that can be present or, when the compound of formula (1) is in the form
of a salt, to the
amount by weight of the free base contained within the salt. For example, the
amount of residual
DMSO per 100 mg equivalent of the free base is no more than about 32 mg, or no
more than
about 31 mg, for example in the range from about 15 mg to about 35 mg, or from
about 20 mg to
about 32 mg, or from about 25 mg to about 30 mg.
[0055] In another embodiment, there is provided a lyophilized pharmaceutical
composition
comprising a compound of formula (1) or a pharmaceutically acceptable salt
thereof, which is
obtainable by a freeze-drying process as defined herein and which: (a) has a
dissolution time, at
ambient temperature, and without the aid of mechanised stirring, in a solvent
containing 65%
(v/v) propylene glycol; 25% (v/v) glycerine; and 10% (v/v) ethanol, of no
greater than 20
minutes (or no greater than 15, or 12 or 10 minutes); and (b) has a residual
DMSO content such
that, in an amount of lyophilized composition obtained from 1 gram of
solution, the residual
DMSO content is no greater than 20 mg, or no greater than 19 mg. The solvent
can be non-
aqueous, anhydrous or substantially-anhydrous.
[0056] The lyophilized pharmaceutical compositions of the invention, i.e. the
compositions
obtainable by the freeze-drying process as defined herein, can also be
characterised with regard
to their enhanced porosity, and increased specific surface area compared to
known
11
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
compositions. The specific surface area can be measured using known techniques
such as the
Brunauer¨Emmett¨Teller (BET) adsorption method.
[0057] The lyophilized pharmaceutical compositions of the invention can be
provided in sealed
containers such as vials (e.g. glass vials), optionally containing a
protective atmosphere of an
inert gas such as nitrogen or argon. The sealed containers can be opened when
required and the
contents reconstituted by dissolving in a reconstitution solvent, such as a
non-aqueous,
anhydrous or substantially-anhydrous solvent, prior to administration to a
patient. Examples of
solvents in which the lyophilized pharmaceutical compositions of the invention
can be
reconstituted are disclosed in W02013033176.
[0058] In a further aspect therefore, the invention provides a sealed
pharmaceutical container
containing a lyophilized pharmaceutical composition as defined herein. The
sealed
pharmaceutical container can be, for example, a vial fitted with a stopper and
optionally
additional components (such as a collar) for holding the stopper in place. The
sealed container
can optionally contain a protective atmosphere of an inert gas such as
nitrogen or argon.
[0059] In a particular embodiment, the invention provides a sealed
pharmaceutical container
containing a lyophilized pharmaceutical composition as defined herein wherein
the composition
contains the compound of formula (1) or a pharmaceutically acceptable salt
thereof in an amount
corresponding to approximately 100 mg equivalent of the free base of the
compound of formula
(1), and wherein no more than 35 mg of residual DMSO is present in the
composition.
Reconstituted formulations prepared from the lyophilized pharmaceutical
compositions.
[0060] The lyophilized pharmaceutical compositions of the invention can be
reconstituted in
solvents, such as non-aqueous, anhydrous or substantially-anhydrous solvents,
to give injectable
liquid compositions for administration to a subject. The liquid compositions
can be for
administration by subcutaneous injection. Accordingly, in a further aspect,
the invention
provides a method for preparing an injectable liquid composition, which method
comprises
dissolving a lyophilized pharmaceutical composition as defined herein in a
solvent, particularly
a non-aqueous solvent.
[0061] Non-limiting examples of suitable solvents include propylene glycol,
glycerin, ethanol,
and any combination of the foregoing. The formulations can be prepared as non-
aqueous
formulations. The formulations can be anhydrous or substantially anhydrous.
[0062] A mixture of solvents can contain a percentage of propylene glycol on
either a mass or a
volume basis. In some embodiments, the percentage of propylene glycol can be
at least about
10%, at least about 20%, at least about 30%, at least about 40%, at least
about 50%, at least
about 10%, at least about 20%, at least about 30%, at least about 40%, or at
least about 50%. In
12
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
some embodiments, the percentage of propylene glycol can be at most 90%, at
most 80%, at
most 70%, at most 60%, at most about 90%, at most about 80%, at most about
70%, or at most
about 60%. In some embodiments, the percentage of propylene glycol can be
about 30% to
about 90%, about 45% to about 85%, about 55% to about 75%, about 60% to about
70%, about
30% to about 90%, about 45% to about 85%, about 55% to about 75%, or about 60%
to about
70%. In some embodiments, the percentage of propylene glycol can be 30%, 35%,
40%, 45%,
50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, about 30%, about 35%, about 40%,
about
45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75%, about
80%, about
85%, or about 90%.
[0063] A mixture of solvents can contain a percentage of glycerin on either a
mass or a volume
basis. In some embodiments, the percentage of glycerin can be at least 5%, at
least 10%, at least
15%, at least 25%, at least 30%, at least about 5%, at least about 10%, at
least about 15%, at
least about 25%, or at least about 30%. In some embodiments, the percentage of
glycerin can be
at most 70%, at most 60%, at most 50%, at most 40%, at most 30%, at most about
70%, at most
about 60%, at most about 50%, at most about 40%, or at most about 30%. In some
embodiments, the percentage of glycerin can be 0% to 50%, 5% to 45%, 15% to
35%, 20% to
30%, 0% to about 50%, about 5% to about 45%, about 15% to about 35%, or about
20% to
about 30%. In some embodiments, the percentage of glycerin can be 0%, 5%, 10%,
15%, 20%,
25%, 30%, 35%, 40%, 45%, 50%, about 5%, about 10%, about 15%, about 20%, about
25%,
about 30%, about 35%, about 40%, about 45%, or about 50%.
[0064] A mixture of solvents can contain a percentage of ethanol on either a
mass or a volume
basis. In some embodiments, the percentage of ethanol can be at least 1%, at
least 3%, at least
5%, at least 10%, at least 15%, at least about 1%, at least about 3%, at least
about 5%, at least
about 10%, or at least about 15%. In some embodiments, the percentage of
ethanol can be at
most 30%, at most 25%, at most 20%, at most 15%, at most 10%, at most about
30%, at most
about 25%, at most about 20%, at most about 15%, or at most about 10%. In some
embodiments, the percentage of ethanol can be 0% to 30%, 0% to 25%, 0% to 20%,
5% to 15%,
0% to about 30%, 0% to about 25%, 0% to about 20%, or about 5% to about 15%.
In some
embodiments, the percentage of ethanol can be 0%, 1%, 2%, 3%, 4%, 5%, 6%, 7%,
8%, 9%,
10%, 11%, 12%, 13%, 14%, 15%, about 1%, about 2%, about 3%, about 4%, about
5%, about
6%, about 7%, about 8%, about 9%, about 10%, about 11%, about 12%, about 13%,
about 14%,
or about 15%.
[0065] In some embodiments, a solvent or a mixture of solvents comprises 45%
to 85%
propylene glycol, 5% to 45% glycerin, and 0% to 30% ethanol. In some
embodiments, a solvent
or a mixture of solvents comprises about 45% to about 85% propylene glycol,
about 5% to about
13
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
45% glycerin, and 0% to about 30% ethanol. In some embodiments, a solvent or a
mixture of
solvents consists essentially of 45% to 85% propylene glycol, 5% to 45%
glycerin, and 0% to
30% ethanol. In some embodiments, a solvent or a mixture of solvents consists
essentially of
about 45% to about 85% propylene glycol, about 5% to about 45% glycerin, and
0% to about
30% ethanol. In some embodiments, a solvent or a mixture of solvents is 45% to
85% propylene
glycol, 5% to 45% glycerin, and 0% to 30% ethanol. In some embodiments, a
solvent or a
mixture of solvents is about 45% to about 85% propylene glycol, about 5% to
about 45%
glycerin, and 0% to about 30% ethanol.
[0066] In some embodiments, a solvent or a mixture of solvents comprises 55%
to 75%
propylene glycol, 15% to 35% glycerin, and 0% to 20% ethanol. In some
embodiments, a
solvent or a mixture of solvents comprises about 55% to about 75% propylene
glycol, about
15% to about 35% glycerin, and 0% to about 20% ethanol. In some embodiments, a
solvent or a
mixture of solvents consists essentially of 55% to 75% propylene glycol, 15%
to 35% glycerin,
and 0% to 20% ethanol. In some embodiments, a solvent or a mixture of solvents
consists
essentially of about 55% to about 75% propylene glycol, about 15% to about 35%
glycerin, and
0% to about 20% ethanol. In some embodiments, a solvent or a mixture of
solvents is 55% to
75% propylene glycol, 15% to 35% glycerin, and 0% to 20% ethanol. In some
embodiments, a
solvent or a mixture of solvents is about 55% to about 75% propylene glycol,
about 15% to
about 35% glycerin, and 0% to about 20% ethanol.
[0067] In some embodiments, a solvent or a mixture of solvents comprises 60%
to 70%
propylene glycol; 20% to 30% glycerin; and 5% to 15% ethanol. In some
embodiments, a
solvent or a mixture of solvents comprises about 60% to about 70% propylene
glycol; about
20% to about 30% glycerin; and about 5% to about 15% ethanol. In some
embodiments, a
solvent or a mixture of solvents consists essentially of 60% to 70% propylene
glycol; 20% to
30% glycerin; and 5% to 15% ethanol. In some embodiments, a solvent or a
mixture of solvents
consists essentially of about 60% to about 70% propylene glycol; about 20% to
about 30%
glycerin; and about 5% to about 15% ethanol. In some embodiments, a solvent or
a mixture of
solvents is 60% to 70% propylene glycol; 20% to 30% glycerin; and 5% to 15%
ethanol. In
some embodiments, a solvent or a mixture of solvents is about 60% to about 70%
propylene
glycol; about 20% to about 30% glycerin; and about 5% to about 15% ethanol.
[0068] In some embodiments, a solvent or a mixture of solvents comprises 65%
propylene
glycol; 25% glycerin; and 10% ethanol. In some embodiments, a solvent or a
mixture of
solvents comprises about 65% propylene glycol; about 25% glycerin; and about
10% ethanol. In
some embodiments, a solvent or a mixture of solvents consists essentially of
65% propylene
glycol; 25% glycerin; and 10% ethanol. In some embodiments, a solvent or a
mixture of
14
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
solvents consists essentially of about 65% propylene glycol; about 25%
glycerin; and about 10%
ethanol. In some embodiments, a solvent or a mixture of solvents is 65%
propylene glycol; 25%
glycerin; and 10% ethanol. In some embodiments, a solvent or a mixture of
solvents is about
65% propylene glycol; about 25% glycerin; and about 10% ethanol.
Excipients.
[0069] A pharmaceutical composition of the invention can be a combination of
any
pharmaceutical compounds described herein with other chemical components, such
as carriers,
stabilizers, diluents, dispersing agents, suspending agents, thickening
agents, and/or excipients.
The pharmaceutical composition facilitates administration of the compound to
an organism.
Pharmaceutical compositions can be administered in therapeutically-effective
amounts as
pharmaceutical compositions by various forms and routes including, for
example, intravenous,
subcutaneous, intramuscular, oral, rectal, aerosol, parenteral, ophthalmic,
pulmonary,
transdermal, vaginal, otic, nasal, and topical administration.
[0070] A pharmaceutical composition can be administered in a local or systemic
manner, for
example, via injection of the compound directly into an organ, optionally in a
depot or sustained
release formulation. Pharmaceutical compositions can be provided in the form
of a rapid release
formulation, in the form of an extended release formulation, or in the form of
an intermediate
release formulation. A rapid release form can provide an immediate release. An
extended
release formulation can provide a controlled release or a sustained delayed
release.
[0071] For oral administration, pharmaceutical compositions can be formulated
readily by
combining the active compounds with pharmaceutically-acceptable carriers or
excipients. Such
carriers can be used to formulate tablets, powders, pills, dragees, capsules,
liquids, gels, syrups,
elixirs, slurries, and suspensions, for oral ingestion by a subject.
[0072] Pharmaceutical preparations for oral use can be obtained by mixing one
or more solid
excipient with one or more of the compounds described herein, optionally
grinding the resulting
mixture, and processing the mixture of granules, after adding suitable
auxiliaries, if desired, to
obtain tablets or dragee cores. Cores can be provided with suitable coatings.
For this purpose,
concentrated sugar solutions can be used, which can contain an excipient such
as gum arabic,
talc, polyvinylpyrrolidone, carbopol gel, polyethylene glycol, and/or titanium
dioxide, lacquer
solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or
pigments can be added
to the tablets or dragee coatings, for example, for identification or to
characterize different
combinations of active compound doses.
[0073] Pharmaceutical preparations which can be used orally include push-fit
capsules made of
gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer,
such as glycerol or
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
sorbitol. In some embodiments, the capsule comprises a hard gelatin capsule
comprising one or
more of pharmaceutical, bovine, and plant gelatins. A gelatin can be alkaline-
processed. The
push-fit capsules can contain the active ingredients in admixture with filler
such as lactose,
binders such as starches, and/or lubricants such as talc or magnesium stearate
and, stabilizers. In
soft capsules, the active compounds can be dissolved or suspended in suitable
liquids, such as
fatty oils, liquid paraffin, or liquid polyethylene glycols. Stabilizers can
be added. All
formulations for oral administration are provided in dosages suitable for such
administration.
[0074] For buccal or sublingual administration, the compositions can be
tablets, lozenges, or
gels.
[0075] Parenteral injections can be formulated for bolus injection or
continuous infusion. The
pharmaceutical compositions can be in a form suitable for parenteral injection
as a sterile
suspension, solution or emulsion in oily or aqueous vehicles, and can contain
formulatory agents
such as suspending, stabilizing and/or dispersing agents. Pharmaceutical
formulations for
parenteral administration include aqueous solutions of the active compounds in
water-soluble
form. Suspensions of the active compounds can be prepared as oily injection
suspensions.
Suitable lipophilic solvents or vehicles include fatty oils such as sesame
oil, or synthetic fatty
acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous
injection suspensions
can contain substances which increase the viscosity of the suspension, such as
sodium
carboxymethyl cellulose, sorbitol, or dextran. The suspension can also contain
suitable
stabilizers or agents which increase the solubility of the compounds to allow
for the preparation
of highly concentrated solutions. Alternatively, the active ingredient can be
in powder form for
constitution with a suitable vehicle, for example, sterile pyrogen-free water,
0.9% saline, or 5%
dextrose in water, before use.
[0076] The active compounds can be administered topically and can be
formulated into a variety
of topically administrable compositions, such as solutions, suspensions,
lotions, gels, pastes,
medicated sticks, balms, creams, and ointments. Such pharmaceutical
compositions can contain
solubilizers, stabilizers, tonicity enhancing agents, buffers and
preservatives.
[0077] Formulations suitable for transdermal administration of the active
compounds can
employ transdermal delivery devices and transdermal delivery patches, and can
be lipophilic
emulsions or buffered aqueous solutions, dissolved and/or dispersed in a
polymer or an
adhesive. Such patches can be constructed for continuous, pulsatile, or on
demand delivery of
pharmaceutical compounds. Transdermal delivery can be accomplished by means of
iontophoretic patches. Additionally, transdermal patches can provide
controlled delivery. The
rate of absorption can be slowed by using rate-controlling membranes or by
trapping the
compound within a polymer matrix or gel. Conversely, absorption enhancers can
be used to
16
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
increase absorption. An absorption enhancer or carrier can include absorbable
pharmaceutically
acceptable solvents to assist passage through the skin. For example,
transdermal devices can be
in the form of a bandage comprising a backing member, a reservoir containing
compounds and
carriers, a rate controlling barrier to deliver the compounds to the skin of
the subject at a
controlled and predetermined rate over a prolonged period of time, and
adhesives to secure the
device to the skin or the eye.
[0078] For administration by inhalation, the active compounds can be in a form
as an aerosol, a
mist, or a powder. Pharmaceutical compositions are conveniently delivered in
the form of an
aerosol spray presentation from pressurized packs or a nebuliser, with the use
of a suitable
propellant, for example, dichlorodifluoromethane, trichlorofluoromethane,
dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case
of a pressurized
aerosol, the dosage unit can be determined by providing a valve to deliver a
metered amount.
Capsules and cartridges of, for example, gelatin for use in an inhaler or
insufflator can be
formulated containing a powder mix of the compounds and a suitable powder base
such as
lactose or starch.
[0079] The compounds can also be formulated in rectal compositions such as
enemas, rectal
gels, rectal foams, rectal aerosols, suppositories, jelly suppositories, or
retention enemas,
containing conventional suppository bases such as cocoa butter or other
glycerides, as well as
synthetic polymers such as polyvinylpyrrolidone and PEG. In suppository forms
of the
compositions, a low-melting wax such as a mixture of fatty acid glycerides or
cocoa butter can
be used.
[0080] In practicing the methods of treatment or use provided herein,
therapeutically-effective
amounts of the compounds described herein are administered in pharmaceutical
compositions to
a subject having a disease or condition to be treated. In some embodiments,
the subject is a
mammal such as a human. A therapeutically-effective amount can vary widely
depending on
the severity of the disease, the age and relative health of the subject, the
potency of the
compounds used, and other factors. The compounds can be used singly or in
combination with
one or more therapeutic agents as components of mixtures.
[0081] Pharmaceutical compositions can be formulated using one or more
physiologically-
acceptable carriers comprising excipients and auxiliaries, which facilitate
processing of the
active compounds into preparations that can be used pharmaceutically.
Formulation can be
modified depending upon the route of administration chosen. Pharmaceutical
compositions
comprising a compounds described herein can be manufactured, for example, by
mixing,
dissolving, granulating, dragee-making, levigating, emulsifying,
encapsulating, entrapping, or
compression processes.
17
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[0082] The pharmaceutical compositions can include at least one
pharmaceutically acceptable
carrier, diluent, or excipient and compounds described herein as free-base or
pharmaceutically-
acceptable salt form. The methods and pharmaceutical compositions described
herein include
the use of crystalline forms (also known as polymorphs), and active
metabolites of these
compounds having the same type of activity.
[0083] Methods for the preparation of compositions comprising the compounds
described herein
include formulating the compounds with one or more inert, pharmaceutically-
acceptable
excipients or carriers to form a solid, semi-solid, or liquid composition.
Solid compositions
include, for example, powders, tablets, dispersible granules, capsules,
cachets, and suppositories.
Liquid compositions include, for example, solutions in which a compound is
dissolved,
emulsions comprising a compound, or a solution containing liposomes, micelles,
or
nanoparticles comprising a compound as disclosed herein. Semi-solid
compositions include, for
example, gels, suspensions and creams. The compositions can be in liquid
solutions or
suspensions, solid forms suitable for solution or suspension in a liquid prior
to use, or as
emulsions. These compositions can also contain minor amounts of nontoxic,
auxiliary
substances, such as wetting or emulsifying agents, pH buffering agents, and
other
pharmaceutically-acceptable additives.
[0084] Non-limiting examples of dosage forms suitable for use in the invention
include feed,
food, pellet, lozenge, liquid, elixir, aerosol, inhalant, spray, powder,
tablet, pill, capsule, gel,
geltab, nanosuspension, nanoparticle, microgel, suppository troches, aqueous
or oily
suspensions, ointment, patch, lotion, dentifrice, emulsion, creams, drops,
dispersible powders or
granules, emulsion in hard or soft gel capsules, syrups, phytoceuticals,
nutraceuticals, and any
combination thereof.
[0085] Non-limiting examples of pharmaceutically-acceptable excipients
suitable for use in the
invention include granulating agents, binding agents, lubricating agents,
disintegrating agents,
sweetening agents, glidants, anti-adherents, anti-static agents, surfactants,
anti-oxidants, gums,
coating agents, coloring agents, flavouring agents, coating agents,
plasticizers, preservatives,
suspending agents, emulsifying agents, anti-microbial agents, plant cellulosic
material and
spheronization agents, and any combination thereof.
[0086] A composition of the invention can be, for example, an immediate
release form or a
controlled release formulation. An immediate release formulation can be
formulated to allow the
compounds to act rapidly. Non-limiting examples of immediate release
formulations include
readily dissolvable formulations. A controlled release formulation can be a
pharmaceutical
formulation that has been adapted such that drug release rates and drug
release profiles can be
matched to physiological and chronotherapeutic requirements or, alternatively,
has been
18
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
formulated to effect release of a drug at a programmed rate. Non-limiting
examples of controlled
release formulations include granules, delayed release granules, hydrogels
(e.g., of synthetic or
natural origin), other gelling agents (e.g., gel-forming dietary fibers),
matrix-based formulations
(e.g., formulations comprising a polymeric material having at least one active
ingredient
dispersed through), granules within a matrix, polymeric mixtures, and granular
masses.
[0087] The disclosed compositions can optionally comprise from about 0.001% to
about 0.005%
weight by volume pharmaceutically acceptable preservatives. One non-limiting
example of a
suitable preservative is benzyl alcohol.
[0088] In some, a controlled release formulation is a delayed release form. A
delayed release
form can be formulated to delay a compound's action for an extended period of
time. A delayed
release form can be formulated to delay the release of an effective dose of
one or more
compounds, for example, for about 4, about 8, about 12, about 16, or about 24
hours.
[0089] A controlled release formulation can be a sustained release form. A
sustained release
form can be formulated to sustain, for example, the compound's action over an
extended period
of time. A sustained release form can be formulated to provide an effective
dose of any
compound described herein (e.g., provide a physiologically-effective blood
profile) over about
4, about 8, about 12, about 16 or about 24 hours.
[0090] Non-limiting examples of pharmaceutically-acceptable excipients can be
found, for
example, in Remington: The Science and Practice of Pharmacy, Nineteenth Ed
(Easton, Pa.:
Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical
Sciences,
Mack Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. and Lachman,
L., Eds.,
Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and
Pharmaceutical
Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams &
Wilkins1999),
each of which is incorporated by reference in its entirety.
[0091] The disclosed methods include administration of a decitabine derivative
dinucleotide, or
a pharmaceutically acceptable salt thereof, in combination with a
pharmaceutically acceptable
carrier. The carrier can be selected to minimize any degradation of the active
ingredient and to
minimize any adverse side effects in the subject.
[0092] The compound of formula (I) or a pharmaceutically acceptable salt
thereof herein can be
conveniently formulated into pharmaceutical compositions composed of one or
more
pharmaceutically acceptable carriers. See e.g., Remington 's Pharmaceutical
Sciences, latest
edition, by E.W. Martin Mack Pub. Co., Easton, PA, which discloses typical
carriers and
conventional methods of preparing pharmaceutical compositions that can be used
in conjunction
with the preparation of formulations of the compound described herein and
which is
incorporated by reference herein. Such pharmaceuticals can be standard
carriers for
19
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
administration of compositions to humans and non-humans, including solutions
such as, saline
and buffered solutions at physiological pH. Other compositions can be
administered according
to standard procedures. For example, pharmaceutical compositions can also
include one or more
additional active ingredients such as antimicrobial agents, anti-inflammatory
agents, and
anesthetics.
[0093] Non-limiting examples of pharmaceutically-acceptable carriers include,
but are not
limited to, saline, Ringer's solution and dextrose solution. The pH of the
solution can be from
about 5 to about 8, and can be from about 7 to about 7.5. Further carriers
include sustained
release preparations such as semipermeable matrices of solid hydrophobic
polymers containing
the compound of formula (I) or a pharmaceutically-acceptable salt thereof,
where the matrices
are in the form of shaped articles, e.g., films, liposomes, microparticles, or
microcapsules.
[0094] The disclosed methods relate to administering the compound of formula
(I) or a
pharmaceutically acceptable salt thereof as part of a pharmaceutical
composition. In various
embodiments, compositions of the invention can comprise a liquid comprising an
active agent in
solution, in suspension, or both. Liquid compositions can include gels. In one
embodiment, the
liquid composition is aqueous. Alternatively, the composition can take form of
an ointment. In
another embodiment, the composition is an in situ gellable aqueous
composition. In some
embodiments, the composition is an in situ gellable aqueous solution.
[0095] Pharmaceutical formulations can include additional carriers, as well as
thickeners,
diluents, buffers, preservatives, and surface active agents in addition to the
compounds disclosed
herein. Pharmaceutical formulations can also include one or more additional
active ingredients
such as antimicrobial agents, anti-inflammatory agents, and anesthetics.
[0096] An excipient can fill a role as simple and direct as being an inert
filler, or an excipient as
used herein can be part of a pH stabilizing system or coating to insure
delivery of the ingredients
safely to the stomach.
[0097] The compound of formula (I) or a pharmaceutically-acceptable salt
thereof can also be
present in liquids, emulsions, or suspensions for delivery of active
therapeutic agents in aerosol
form to cavities of the body such as the nose, throat, or bronchial passages.
The ratio of the
compound of formula (I) or a pharmaceutically-acceptable salt thereof to the
other compounding
agents in these preparations can vary as the dosage form requires.
[0098] Depending on the intended mode of administration, the pharmaceutical
compositions
administered as part of the disclosed methods can be in the form of solid,
semi-solid or liquid
dosage forms, such as, for example, tablets, suppositories, pills, capsules,
powders, liquids,
suspensions, lotions, creams, gels, or the like, for example, in unit dosage
form suitable for
single administration of a precise dosage. The compositions can contain, as
noted above, an
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
effective amount of the compound of formula (I) or a pharmaceutically-
acceptable salt thereof in
combination with a pharmaceutically-acceptable carrier and, in addition, can
include other
medicinal agents, pharmaceutical agents, carriers, adjuvants, diluents, etc.
[0099] For solid compositions, nontoxic solid carriers include, for example,
pharmaceutical
grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin,
talc, cellulose,
glucose, sucrose, and magnesium carbonate.
Pharmaceutically Acceptable Salts.
[00100] The compound of formula (1) and the pharmaceutically acceptable salts
thereof can be
prepared by the methods described in W02013033176 and as described below in
the Examples.
[00101] In each of the foregoing aspects and embodiments of the invention, the
compound of
formula (1) can be used in the form of a salt or a non-salt.
[00102] Pharmaceutically-acceptable salts include, for example, acid-addition
salts and base-
addition salts. The acid that is added to a compound to form an acid-addition
salt can be an
organic acid or an inorganic acid. A base that is added to a compound to form
a base-addition
salt can be an organic base or an inorganic base. In some embodiments, a
pharmaceutically-
acceptable salt is a metal salt. In some embodiments, a pharmaceutically-
acceptable salt is an
ammonium salt.
[00103] Acid addition salts can arise from the addition of an acid to a
compound described
herein. In some embodiments, the acid is organic. In some embodiments, the
acid is inorganic.
Non-limiting examples of suitable acids include hydrochloric acid, hydrobromic
acid,
hydroiodic acid, nitric acid, nitrous acid, sulfuric acid, sulfurous acid, a
phosphoric acid,
nicotinic acid, isonicotinic acid, lactic acid, salicylic acid, 4-
aminosalicylic acid, tartaric acid,
ascorbic acid, gentisinic acid, gluconic acid, glucaronic acid, saccaric acid,
formic acid, benzoic
acid, glutamic acid, pantothenic acid, acetic acid, propionic acid, butyric
acid, fumaric acid,
succinic acid, citric acid, oxalic acid, maleic acid, hydroxymaleic acid,
methylmaleic acid,
glycolic acid, malic acid, cinnamic acid, mandelic acid, 2-phenoxybenzoic
acid, 2-
acetoxybenzoic acid, embonic acid, phenylacetic acid, N-cyclohexylsulfamic
acid,
methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-
toluenesulfonic acid, 2-
hydroxyethanesulfonic acid, ethane-1,2-disulfonic acid, 4-
methylbenzenesulfonic acid,
naphthalene-2-sulfonic acid, naphthalene-1,5-disulfonic acid, 2-
phosphoglyceric acid, 3-
phosphoglyceric acid, glucose-6-phosphoric acid, and an amino acid.
[00104] Non-limiting examples of suitable acid addition salts include a
hydrochloride salt, a
hydrobromide salt, a hydroiodide salt, a nitrate salt, a nitrite salt, a
sulfate salt, a sulfite salt, a
phosphate salt, a hydrogen phosphate salt, a dihydrogen phosphate salt, a
carbonate salt, a
21
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
bicarbonate salt, a nicotinate salt, an isonicotinate salt, a lactate salt, a
salicylate salt, a 4-
aminosalicylate salt, a tartrate salt, an ascorbate salt, a gentisinate salt,
a gluconate salt, a
glucaronate salt, a saccarate salt, a formate salt, a benzoate salt, a
glutamate salt, a pantothenate
salt, an acetate salt, a propionate salt, a butyrate salt, a fumarate salt, a
succinate salt, a citrate
salt, an oxalate salt, a maleate salt, a hydroxymaleate salt, a methylmaleate
salt, a glycolate salt,
a malate salt, a cinnamate salt, a mandelate salt, a 2-phenoxybenzoate salt, a
2-acetoxybenzoate
salt, an embonate salt, a phenylacetate salt, an N-cyclohexylsulfamate salt, a
methanesulfonate
salt, an ethanesulfonate salt, a benzenesulfonate salt, a p-toluenesulfonate
salt, a 2-
hydroxyethanesulfonate salt, an ethane-1,2-disulfonate salt, a 4-
methylbenzenesulfonate salt, a
naphthalene-2-sulfonate salt, a naphthalene-1,5-disulfonate salt, a 2-
phosphoglycerate salt, a 3-
phosphoglycerate salt, a glucose-6-phosphate salt, and an amino acid salt.
[00105] Metal salts can arise from the addition of an inorganic base to a
compound described
herein. The inorganic base consists of a metal cation paired with a basic
counterion, such as, for
example, hydroxide, carbonate, bicarbonate, or phosphate. The metal can be an
alkali metal,
alkaline earth metal, transition metal, or main group metal. Non-limiting
examples of suitable
metals include lithium, sodium, potassium, cesium, cerium, magnesium,
manganese, iron,
calcium, strontium, cobalt, titanium, aluminum, copper, cadmium, and zinc.
[00106] Non-limiting examples of suitable metal salts include a lithium salt,
a sodium salt, a
potassium salt, a cesium salt, a cerium salt, a magnesium salt, a manganese
salt, an iron salt, a
calcium salt, a strontium salt, a cobalt salt, a titanium salt, an aluminum
salt, a copper salt, a
cadmium salt, and a zinc salt.
[00107] Ammonium salts can arise from the addition of ammonia or an organic
amine to a
compound described herein. Non-limiting examples of suitable organic amines
include triethyl
amine, diisopropyl amine, ethanol amine, diethanol amine, triethanol amine,
morpholine, N-
methylmorpholine, piperidine, N-methylpiperidine, N-ethylpiperidine, dibenzyl
amine,
piperazine, pyridine, pyrrazole, pipyrrazole, imidazole, pyrazine, pipyrazine,
ethylenediamine,
N,N'-dibenzylethylene diamine, procaine, chloroprocaine, choline, dicyclohexyl
amine, and N-
methylglucamine.
[00108] Non-limiting examples of suitable ammonium salts include is a triethyl
amine salt, a
diisopropyl amine salt, an ethanol amine salt, a diethanol amine salt, a
triethanol amine salt, a
morpholine salt, an N-methylmorpholine salt, a piperidine salt, an N-
methylpiperidine salt, an
N-ethylpiperidine salt, a dibenzyl amine salt, a piperazine salt, a pyridine
salt, a pyrrazole salt, a
pipyrrazole salt, an imidazole salt, a pyrazine salt, a pipyrazine salt, an
ethylene diamine salt, an
N,N'-dibenzylethylene diamine salt, a procaine salt, a chloroprocaine salt, a
choline salt, a
dicyclohexyl amine salt, and a N-methylglucamine salt.
22
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00109] One particular example of a salt of the compound of formula (1) is a
sodium salt.
Therapeutic Uses.
[00110] The lyophilized pharmaceutical compositions according to the present
invention can be
used to treat a wide variety of diseases that are sensitive to the treatment
with decitabine,
including those described herein.
[00111] Accordingly, in other aspects, the invention provides: (i) a
lyophilized pharmaceutical
composition as defined herein for use in medicine; (ii) a lyophilized
pharmaceutical composition
as defined herein for use in the treatment of a disease as defined herein;
(iii) a method of treating
a disease as defined herein, which method comprises mixing a lyophilized
pharmaceutical
composition as defined herein with a pharmaceutically acceptable solvent and
administering an
effective amount of the mixture to a subject in need thereof; (iv) the use of
a lyophilized
pharmaceutical composition as defined herein for the manufacture of a
medicament for the
treatment of a disease as defined herein; (v) a method of treating cancer in a
patient in need
thereof, which method comprises reconstituting the lyophilized pharmaceutical
composition as
defined herein in a pharmaceutically acceptable solvent to give a liquid
formulation containing a
compound of formula (1) or a pharmaceutically acceptable salt thereof, and
administering a
therapeutically effective amount of the liquid formulation to the patient.
[00112] Examples of diseases that can be treated using the lyophilized
pharmaceutical
compositions of the present invention include those involving undesirable or
uncontrolled cell
proliferation. Such indications include benign tumors, various types of
cancers such as primary
tumors and tumor metastasis, restenosis (e.g. coronary, carotid, and cerebral
lesions),
hematological disorders, abnormal stimulation of endothelial cells
(atherosclerosis), insults to
body tissue due to surgery, abnormal wound healing, abnormal angiogenesis,
diseases that
produce fibrosis of tissue, repetitive motion disorders, disorders of tissues
that are not highly
vascularized, and proliferative responses associated with organ transplants.
[00113] Generally, cells in a benign tumor retain their differentiated
features and do not divide
in a completely uncontrolled manner. A benign tumor is usually localized and
nonmetastatic.
Specific types benign tumors that can be treated using the present invention
include
hemangiomas, hepatocellular adenoma, cavernous haemangioma, focal nodular
hyperplasia,
acoustic neuromas, neurofibroma, bile duct adenoma, bile duct cystanoma,
fibroma, lipomas,
leiomyomas, mesotheliomas, teratomas, myxomas, nodular regenerative
hyperplasia, trachomas
and pyogenic granulomas.
[00114] In a malignant tumor cells become undifferentiated, do not respond to
the body's
growth control signals, and multiply in an uncontrolled manner. The malignant
tumor is
23
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
invasive and capable of spreading to distant sites (metastasizing). Malignant
tumors are
generally divided into two categories: primary and secondary. Primary tumors
arise directly
from the tissue in which they are found. A secondary tumor, or metastasis, is
a tumor which is
originated elsewhere in the body but has now spread to a distant organ. The
common routes for
metastasis are direct growth into adjacent structures, spread through the
vascular or lymphatic
systems, and tracking along tissue planes and body spaces (peritoneal fluid,
cerebrospinal fluid,
etc.)
[00115] Examples of cancers are carcinomas, for example carcinomas of the
bladder, breast,
colon, kidney, epidermis, liver, lung, oesophagus, gall bladder, ovary,
pancreas, stomach, cervix,
thyroid, prostate, gastrointestinal system, or skin, hematopoieitic tumours
such as leukaemia, B-
cell lymphoma, T-cell lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma,
hairy cell
lymphoma, or Burkett's lymphoma; hematopoieitic tumours of myeloid lineage,
for example
acute and chronic myelogenous leukaemias, myelodysplastic syndrome, or
promyelocytic
leukaemia; thyroid follicular cancer; tumours of mesenchymal origin, for
example fibrosarcoma
or habdomyo sarcoma; tumours of the central or peripheral nervous system, for
example
astrocytoma, neuroblastoma, glioma or schwannoma; melanoma; seminoma;
teratocarcinoma;
osteosarcoma; xeroderma pigmentosum; keratoctanthoma; thyroid follicular
cancer; or Kaposi's
sarcoma.
[00116] Specific types of cancers or malignant tumors, either primary or
secondary, that can be
treated using this invention include bladder cancer, breast cancer, ovarian
cancer, skin cancer,
bone cancer, prostate cancer, liver cancer, lung cancer, brain cancer, cancer
of the larynx, gall
bladder, pancreas, rectum, parathyroid, thyroid, adrenal, neural tissue, head
and neck, colon,
stomach, bronchi, kidneys, basal cell carcinoma, squamous cell carcinoma of
both ulcerating
and papillary type, metastatic skin carcinoma, osteo sarcoma, Ewing's sarcoma,
veticulum cell
sarcoma, myeloma, giant cell tumor, small-cell lung tumor, gallstones, islet
cell tumor, primary
brain tumor, acute and chronic lymphocytic and granulocytic tumors, hairy-cell
tumor, adenoma,
hyperplasia, medullary carcinoma, pheochromocytoma, mucosal neuronms,
intestinal
ganglloneuromas, hyperplastic corneal nerve tumor, marfanoid habitus tumor,
Wilm's tumor,
seminoma, ovarian tumor, leiomyomater tumor, cervical dysplasia and in situ
carcinoma,
neuroblastoma, retinoblastoma, soft tissue sarcoma, malignant carcinoid,
topical skin lesion,
mycosis fungoide, rhabdomyosarcoma, Kaposi's sarcoma, osteogenic and other
sarcoma,
malignant hypercalcemia, renal cell tumor, polycythermia vera, adenocarcinoma,
glioblastoma
multiforma, leukemias, lymphomas, malignant melanomas, epidermoid carcinomas,
and other
carcinomas and sarcomas.
[00117] In one embodiment, the cancer is selected from myelodysplastic
syndrome, acute
24
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
myelogenous leukaemia, ovarian cancer, liver cancer, and colorectal cancer.
[00118] Hematologic disorders include abnormal growth of blood cells which can
lead to
dysplastic changes in blood cells and hematologic malignancies such as various
leukemias.
Examples of hematologic disorders include but are not limited to acute myeloid
leukemia, acute
promyelocytic leukemia, acute lymphoblastic leukemia, chronic myelogenous
leukemia, the
myelodysplastic syndromes, and sickle cell anemia.
[00119] Treatment of abnormal cell proliferation due to insults to body tissue
during surgery can
be possible for a variety of surgical procedures, including joint surgery,
bowel surgery, and
cheloid scarring. Diseases that produce fibrotic tissue include emphysema.
[00120] Repetitive motion disorders that can be treated using the present
invention include
carpal tunnel syndrome. An example of cell proliferative disorders that can be
treated using the
invention is a bone tumor.
[00121] The proliferative responses associated with organ transplantation that
can be treated
using this invention include those proliferative responses contributing to
potential organ
rejections or associated complications. Specifically, these proliferative
responses can occur
during transplantation of the heart, lung, liver, kidney, and other body
organs or organ systems.
[00122] Abnormal angiogenesis that can be treated using this invention include
those abnormal
angiogenesis accompanying rheumatoid arthritis, ischemic-reperfusion related
brain edema and
injury, cortical ischemia, ovarian hyperplasia and hypervascularity,
(polycystic ovary
syndrome), endometriosis, psoriasis, diabetic retinopaphy, and other ocular
angiogenic diseases
such as retinopathy of prematurity (retrolental fibroplastic), muscular
degeneration, corneal graft
rejection, neuroscular glaucoma and Oster Webber syndrome.
[00123] Diseases associated with abnormal angiogenesis require or induce
vascular growth. For
example, corneal angiogenesis involves three phases: a pre-vascular latent
period, active
neovascularization, and vascular maturation and regression. The identity and
mechanism of
various angiogenic factors, including elements of the inflammatory response,
such as
leukocytes, platelets, cytokines, and eicosanoids, or unidentified plasma
constituents have yet to
be revealed.
[00124] In some embodiments, the lyophilized pharmaceutical compositions of
the present
invention can be used for treating diseases associated with undesired or
abnormal angiogenesis.
The method comprises administering to a patient suffering from undesired or
abnormal
angiogenesis the pharmaceutical formulations of the present invention alone,
or in combination
with anti-neoplastic agent whose activity as an anti-neoplastic agent in vivo
is adversely affected
by high levels of DNA methylation. The particular dosage of these agents
required to inhibit
angiogenesis and/or angiogenic diseases can depend on the severity of the
condition, the route of
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
administration, and related factors that can be decided by the attending
physician. Generally,
accepted and effective daily doses are the amount sufficient to effectively
inhibit angiogenesis
and/or angiogenic diseases.
[00125] The lyophilized pharmaceutical compositions of the present invention
can be used to
treat a variety of diseases associated with undesirable angiogenesis such as
retinal/choroidal
neuvascularization and corneal neovascularization. Examples of
retinal/choroidal
neuvascularization include, but are not limited to, Bests diseases, myopia,
optic pits, Stargarts
diseases, Pagets disease, vein occlusion, artery occlusion, sickle cell
anemia, sarcoid, syphilis,
pseudoxanthoma elasticum carotid abostructive diseases, chronic
uveitis/vitritis, mycobacterial
infections, Lyme's disease, systemic lupus erythematosis, retinopathy of
prematurity, Eales
disease, diabetic retinopathy, macular degeneration, Bechets diseases,
infections causing a
retinitis or chroiditis, presumed ocular histoplasmosis, pars planitis,
chronic retinal detachment,
hyperviscosity syndromes, toxoplasmosis, trauma and post-laser complications,
diseases
associated with rubesis (neovascularization of the angle) and diseases caused
by the abnormal
proliferation of fibrovascular or fibrous tissue including all forms of
proliferative
vitreoretinopathy. Examples of corneal neuvascularization include, but are not
limited to,
epidemic keratoconjunctivitis, Vitamin A deficiency, contact lens overwear,
atopic keratitis,
superior limbic keratitis, pterygium keratitis sicca, sjogrens, acne rosacea,
phylectenulosis,
diabetic retinopathy, retinopathy of prematurity, corneal graft rejection,
Mooren ulcer, Terrien's
marginal degeneration, marginal keratolysis, polyarteritis, Wegener
sarcoidosis, Scleritis,
periphigoid radial keratotomy, neovascular glaucoma and retrolental
fibroplasia, syphilis,
Mycobacteria infections, lipid degeneration, chemical burns, bacterial ulcers,
fungal ulcers,
Herpes simplex infections, Herpes zoster infections, protozoan infections and
Kaposi sarcoma.
[00126] In some embodiments, the lyophilized pharmaceutical compositions of
the present
invention can be used for treating chronic inflammatory diseases associated
with abnormal
angiogenesis. The method comprises administering to a patient suffering from a
chronic
inflammatory disease associated with abnormal angiogenesis the pharmaceutical
formulations of
the present invention alone, or in combination with an anti-neoplastic agent
whose activity as an
anti-neoplastic agent in vivo is adversely affected by high levels of DNA
methylation. The
chronic inflammation depends on continuous formation of capillary sprouts to
maintain an
influx of inflammatory cells. The influx and presence of the inflammatory
cells produce
granulomas and thus, maintains the chronic inflammatory state. Inhibition of
angiogenesis using
the pharmaceutical formulations of the present invention can prevent the
formation of the
granulomas, thereby alleviating the disease. Examples of chronic inflammatory
disease include,
but are not limited to, inflammatory bowel diseases such as Crohn's disease
and ulcerative
26
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
colitis, psoriasis, sarcoidois, and rheumatoid arthritis.
[00127] Inflammatory bowel diseases such as Crohn's disease and ulcerative
colitis are
characterized by chronic inflammation and angiogenesis at various sites in the
gastrointestinal
tract. For example, Crohn's disease occurs as a chronic transmural
inflammatory disease that
most commonly affects the distal ileum and colon but can also occur in any
part of the
gastrointestinal tract from the mouth to the anus and perianal area. Patients
with Crohn's
disease generally have chronic diarrhea associated with abdominal pain, fever,
anorexia, weight
loss and abdominal swelling. Ulcerative colitis is also a chronic,
nonspecific, inflammatory and
ulcerative disease arising in the colonic mucosa and is characterized by the
presence of bloody
diarrhea. These inflammatory bowel diseases are generally caused by chronic
granulomatous
inflammation throughout the gastrointestinal tract, involving new capillary
sprouts surrounded
by a cylinder of inflammatory cells. Inhibition of angiogenesis by the
pharmaceutical
formulations of the present invention should inhibit the formation of the
sprouts and prevent the
formation of granulomas. The inflammatory bowel diseases also exhibit extra
intestinal
manifectations, such as skin lesions. Such lesions are characterized by
inflammation and
angiogenesis and can occur at many sites other the gastrointestinal tract.
Inhibition of
angiogenesis by the lyophilized pharmaceutical compositions of the present
invention should
reduce the influx of inflammatory cells and prevent the lesion formation.
[00128] Sarcoidois, another chronic inflammatory disease, is characterized as
a multi-system
granulomatous disorder. The granulomas of this disease can form anywhere in
the body and,
thus, the symptoms depend on the site of the granulomas and whether the
disease is active. The
granulomas are created by the angiogenic capillary sprouts providing a
constant supply of
inflammatory cells. By using the lyophilized pharmaceutical compositions of
the present
invention to inhibit angionesis, such granulomas formation can be inhibited.
Psoriasis, also a
chronic and recurrent inflammatory disease, is characterized by papules and
plaques of various
sizes. Treatment using the pharmaceutical formulations of the present
invention can reduce the
likelihood of the formation of new blood vessels necessary to maintain the
characteristic lesions
and provide the patient relief from the symptoms.
[00129] Rheumatoid arthritis (RA) is also a chronic inflammatory disease
characterized by non-
specific inflammation of the peripheral joints. It is believed that the blood
vessels in the
synovial lining of the joints undergo angiogenesis. In addition to forming new
vascular
networks, the endothelial cells release factors and reactive oxygen species
that lead to pannus
growth and cartilage destruction. The factors involved in angiogenesis can
actively contribute
to, and help maintain, the chronically inflamed state of rheumatoid arthritis.
Treatment using the
pharmaceutical formulations of the present invention alone or in conjunction
with other anti-RA
27
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
agents can reduce the likelihood of the formation of new blood vessels
necessary to maintain the
chronic inflammation and provide the RA patient relief from the symptoms.
[00130] In some embodiments, the lyophilized pharmaceutical compositions of
the present
invention can be used for treating diseases associated with abnormal
hemoglobin synthesis. The
method comprises administering the pharmaceutical formulations of the present
invention to a
patient suffering from disease associated with abnormal hemoglobin synthesis.
Decitabine-
containing formulations stimulate fetal hemoglobin synthesis because the
mechanism of
incorporation into DNA is associated with DNA hypomethylation. Examples of
diseases
associated with abnormal hemoglobin synthesis include, but are not limited to,
sickle cell
anemia and P-thalassemia.
[00131] In some embodiments, the lyophilized pharmaceutical compositions of
the present
invention can be used to control intracellular gene expression. The method
comprises
administering the pharmaceutical formulations of the present invention to a
patient suffering
from disease associated with abnormal levels of gene expression. DNA
methylation is associated
with the control of gene expression. Specifically, methylation in or near
promoters inhibit
transcription while demethylation restores expression. Examples of the
possible applications of
the described mechanisms include, but are not limited to, therapeutically
modulated growth
inhibition, induction of apoptosis, and cell differentiation.
[00132] In some embodiments, the lyophilized pharmaceutical compositions of
the invention
can be used in the treatment of patients with genetic mutations associated
with tumor
hypermethylation such as patients with tumor types which contain the succinate
dehydrogenase
(SDH) mutation or deficiency which includes patients with non-KIT mutated
gastrointestinal
stromal tumors (GIST).
[00133] Gene activation facilitated by the lyophilized pharmaceutical
compositions of the
present invention can induce differentiation of cells for therapeutic
purposes. Cellular
differentiation is induced through the mechanism of hypomethylation. Examples
of
morphological and functional differentiation include, but are not limited to
differentiation
towards formation of muscle cells, myotubes, cells of erythroid and lymphoid
lineages.
[00134] Myelodysplastic syndromes (MDS) are heterogeneous clonal hematopoietic
stem cell
disorders associated with the presence of dysplastic changes in one or more of
the hematopoietic
lineages, including dysplastic changes in the myeloid, erythroid, and
megakaryocytic series.
These changes result in cytopenias in one or more of the three lineages.
Subjects afflicted with
MDS typically develop complications related to anemia, neutropenia
(infections), or
thrombocytopenia (bleeding). Generally, from about 10% to about 70% of
subjects with MDS
develop acute leukemia. Representative myelodysplastic syndromes include acute
myeloid
28
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
leukemia, acute promyelocytic leukemia, acute lymphoblastic leukemia, and
chronic
myelogenous leukemia.
[00135] Acute myeloid leukemia (AML) is the most common type of acute leukemia
in adults.
Several inherited genetic disorders and immunodeficiency states are associated
with an
increased risk of AML. These include disorders with defects in DNA stability
leading to random
chromosomal breakage, such as Bloom's syndrome, Fanconi's anemia, Li-Fraumeni
kindreds,
ataxia-telangiectasia, and X-linked agammaglobulinemia.
[00136] Acute promyelocytic leukemia (APML) represents a distinct subgroup of
AML. This
subtype is characterized by promyelocytic blasts containing the 15; 17
chromosomal
translocation. This translocation leads to the generation of a fusion
transcript comprising a
retinoic acid receptor sequence and a promyelocytic leukemia sequence.
[00137] Acute lymphoblastic leukemia (ALL) is a heterogenerous disease with
distinct clinical
features displayed by various subtypes. Reoccurring cytogenetic abnormalities
have been
demonstrated in ALL. The most common associated cytogenetic abnormality is the
9; 22
translocation leading to development of the Philadelphia chromosome.
[00138] In a particular embodiment, the the lyophilized pharmaceutical
compositions of the
present invention can be used to treat an MDS, for example an MDS selected
from AML,
APML and ALL.
[00139] It will be appreciated that in each of the foregoing therapeutic uses,
the lyophilized
pharmaceutical compositions of the invention will typically be reconstituted
in a suitable solvent
as defined herein before administration to a subject, e.g. a mammalian subject
such as a human
patient.
Dosing and Administration.
[00140] Doses of lyophilized pharmaceutical compositions of the invention,
reconstituted or
mixed as necessary with a pharmaceutically acceptable solvent or solvent
mixture as defined
herein can be administered to a subject by methods known in the art. Non-
limiting examples of
methods of administration include subcutaneous injection, intravenous
injection, and infusion.
[00141] A dose of a formulation contains an amount that is therapeutically-
effective for treating
a disease. A therapeutically-effective amount of a compound of the invention
can be expressed
as mg of the compound per kg of subject body mass. In some embodiments, a
therapeutically-
effective amount is 1-1,000 mg/kg, 1-500 mg/kg, 1-250 mg/kg, 1-100 mg/kg, 1-50
mg/kg, 1-25
mg/kg, or 1-10 mg/kg. In some embodiments, a therapeutically-effective amount
is 5 mg/kg, 10
mg/kg, 25 mg/kg, 50 mg/kg, 75 mg/kg, 100 mg/kg, 150 mg/kg, 200 mg/kg, 250
mg/kg, 300
mg/kg, 400 mg/kg, 500 mg/kg, 600 mg/kg, 700 mg/kg, 800 mg/kg, 900 mg/kg, 1,000
mg/kg,
29
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
about 5 mg/kg, about 10 mg/kg, about 25 mg/kg, about 50 mg/kg, about 75 mg/kg,
about 100
mg/kg, about 150 mg/kg, about 200 mg/kg, about 250 mg/kg, about 300 mg/kg,
about 400
mg/kg, about 500 mg/kg, about 600 mg/kg, about 700 mg/kg, about 800 mg/kg,
about 900
mg/kg, or about 1,000 mg/kg.
[00142] A compound described herein can be present in a composition in a range
of from about
1 mg to about 5 mg, from about 5 mg to about 10 mg, from about 10 mg to about
15 mg, from
about 15 mg to about 20 mg, from about 20 mg to about 25 mg, from about 25 mg
to about 30
mg, from about 30 mg to about 35 mg, from about 35 mg to about 40 mg, from
about 40 mg to
about 45 mg, from about 45 mg to about 50 mg, from about 50 mg to about 55 mg,
from about
55 mg to about 60 mg, from about 60 mg to about 65 mg, from about 65 mg to
about 70 mg,
from about 70 mg to about 75 mg, from about 75 mg to about 80 mg, from about
80 mg to about
85 mg, from about 85 mg to about 90 mg, from about 90 mg to about 95 mg, from
about 95 mg
to about 100 mg, from about 100 mg to about 125 mg, from about 125 mg to about
150 mg,
from about 150 mg to about 175 mg, from about 175 mg to about 200 mg, from
about 200 mg to
about 225 mg, from about 225 mg to about 250 mg, or from about 250 mg to about
300 mg.
[00143] A compound described herein can be present in a composition in an
amount of about 1
mg, about 5 mg, about 10 mg, about 15 mg, about 20 mg, about 25 mg, about 30
mg, about 35
mg, about 40 mg, about 45 mg, about 50 mg, about 55 mg, about 60 mg, about 65
mg, about 70
mg, about 75 mg, about 80 mg, about 85 mg, about 90 mg, about 95 mg, about 100
mg, about
125 mg, about 150 mg, about 175 mg, about 200 mg, about 225 mg, about 250 mg,
or about 300
mg.
[00144] In some embodiments, a therapeutically-effective amount can be
administered 1-35
times per week, 1-14 times per week, or 1-7 times per week. In some
embodiments, a
therapeutically-effective amount can be administered 1-10 times per day, 1-5
times per day, 1
time, 2 times, or 3 times per day.
[00145] It is envisaged that the lyophilized pharmaceutical compositions of
the invention will be
useful either alone or in combination therapy with other chemotherapeutic
agents or radiation
therapy in the prophylaxis or treatment of a range of proliferative disease
states or conditions.
Examples of such disease states and conditions are set out above.
[00146] The lyophilized pharmaceutical compositions of the invention, whether
administered
alone, or in combination with anti-cancer agents and therapies such as
radiotherapy, are
generally administered to a subject in need of such administration, for
example a human or
animal patient, preferably a human.
[00147] Examples of chemotherapeutic agents that can be co-administered with
the lyophilized
pharmaceutical compositions of the invention as defined herein include but are
not limited to
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
topoisomerase I inhibitors; other antimetabolites; tubulin targeting agents;
DNA binder and
topoisomerase II inhibitors; alkylating agents; monoclonal antibodies; anti-
hormones; signal
transduction inhibitors; proteasome inhibitors; DNA methyl transferase
inhibitors; cytokines;
interferons; interleukins; retinoids; chromatin targeted therapies, e.g. HDAC
or HAT
modulators; T-cell activating agents, including immunomodulating antibodies;
cancer vaccines;
hormonal agents; plant-derived agents; biologic agents; immunomodulating
agents;
radiotherapy; and other therapeutic or prophylactic agents; for example agents
that reduce or
alleviate some of the side effects associated with chemotherapy; for example
anti-emetic agents
and agents that prevent or decrease the duration of chemotherapy-associated
neutropenia and
prevent complications that arise from reduced levels of red blood cells or
white blood cells, such
as erythropoietin (EPO), granulocyte macrophage-colony stimulating factor (GM-
CSF), and
granulocyte-colony stimulating factor (G-CSF).
[00148] In one embodiment, the lyophilized pharmaceutical compositions of the
invention are
used in combination with (or further comprise) inhibitors of histone
deacetylase (HDAC) to
further modulate transcription of genes, e.g., to reestablish transcription of
genes silenced by
hypermethylation and acetylation of histones, in a synergistic manner.
[00149] Inhibitors of HDACs include, but are not limited to, the following
structural classes: 1)
hydroxamic acids, 2) cyclic peptides, 3) benzamides, and 4) short-chain fatty
acids. Examples of
hydroxamic acids and hydroxamic acid derivatives, include trichostatin A
(TSA),
suberoylanilide hydroxamic acid (SAHA), oxamflatin, suberic bishydroxamic acid
(SBHA), m-
carboxy-cinnamic acid bishydroxamic acid (CBHA), and pyroxamide. TSA was
isolated as an
antifungi antibiotic (Tsuji et al (1976) J. Antibiot (Tokyo) 29:1-6) and found
to be a potent
inhibitor of mammalian HDAC (Yoshida et al. (1990) J. Biol. Chem. 265:17174-
17179). The
finding that TSA-resistant cell lines have an altered HDAC evidences that this
enzyme is an
important target for TSA. Other hydroxamic acid-based HDAC inhibitors, SAHA,
SBHA, and
CBHA are synthetic compounds that are able to inhibit HDAC at micromolar
concentration or
lower in vitro or in vivo. Glick et al. (1999) Cancer Res. 59:4392-4399. These
hydroxamic acid-
based HDAC inhibitors all possess an essential structural feature: a polar
hydroxamic terminal
linked through a hydrophobic methylene spacer (e.g. 6 carbon at length) to
another polar site
which is attached to a terminal hydrophobic moiety (e.g., benzene ring).
[00150] Cyclic peptides used as HDAC inhibitors are mainly cyclic
tetrapeptides. Examples of
cyclic peptides include, but are not limited to, trapoxin A, apicidin and
FR901228. Trapoxin A is
a cyclic tetrapeptide that contains a 2-amino-8-oxo-9,10-epoxy-decanoyl (AOE)
moiety. Kijima
et al. (1993) J. Biol. Chem. 268:22429-22435. Apicidin is a fungal metabolite
that exhibits
potent, broad-spectrum antiprotozoal activitity and inhibits HDAC activity at
nanomolar
31
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
concentrations. Darkin-Rattray et al. (1996) Proc. Natl. Acad. Sci. USA.
93;13143-13147.
FR901228 is a depsipeptide that is isolated from Chromobacterium violaceum,
and has been
shown to inhibit HDAC activity at micromolar concentrations.
[00151] Examples of benzamides include but are not limited to MS-27-275. Saito
et al. (1990)
Proc. Natl. Acad. Sci. USA. 96:4592-4597. Examples of short-chain fatty acids
include but are
not limited to butyrates (e.g., butyric acid, arginine butyrate and
phenylbutyrate (PB)). Newmark
et al. (1994) Cancer Lett. 78:1-5; and Carducci et al. (1997) Anticancer Res.
17:3972-3973. In
addition, depudecin which has been shown to inhibit HDAC at micromolar
concentrations
(Kwon et al. (1998) Proc. Natl. Acad. Sci. USA. 95:3356-3361) also falls
within the scope of
histone deacetylase inhibitor of the present invention.
[00152] In one embodiment, an alkylating agent is used in combination with the
lyophilized
pharmaceutical compositions of the invention. Examples of alkylating agents
include
bischloroethylamines (nitrogen mustards, e.g. chlorambucil, cyclophosphamide,
ifosfamide,
mechlorethamine, melphalan, uracil mustard), aziridines (e.g. thiotepa), alkyl
alkone sulfonates
(e.g. busulfan), nitro soureas (e.g. carmustine, lomustine, streptozocin),
nonclas sic alkylating
agents (altretamine, dacarbazine, and procarbazine), platinum compounds
(carboplastin and
cisplatin).
[00153] In another embodiment the lyophilized pharmaceutical composition of
the invention is
used in combination with a platinum compound such as cisplatin or carboplatin.
[00154] In another embodiment, the lyophilized pharmaceutical composition of
the invention is
used in combination with a member of the retinoids superfamily such as all-
trans-retinol, all-
trans-retinoic acid (tretinoin), 13-cis retinoic acid (isotretinoin) and 9-cis-
retinoic acid.
[00155] In a further embodiment, the lyophilized pharmaceutical composition of
the invention is
used in combination with a hormonal agent such as a synthetic oestrogen (e.g.
diethylstibestrol),
antiestrogen (e.g. tamoxifen, toremifene, fluoxymesterol and raloxifene),
antiandrogen
(bicalutamide, nilutamide, flutamide), aromatase inhibitor (e.g.,
aminoglutethimide, anastrozole
and tetrazole), ketoconazole, goserelin acetate, leuprolide, megestrol acetate
and mifepristone.
[00156] In yet another embodiment, the lyophilized pharmaceutical composition
of the
invention is used in combination with a plant-derived agent such as a vinca
alkaloid (e.g.,
vincristine, vinblastine, vindesine, vinzolidine and vinorelbine),
camptothecin (20(S)-
camptothecin, 9-nitro-20(S)-camptothecin, and 9-amino-20(S)-camptothecin), a
podophyllotoxin (e.g., etoposide (VP-16) and teniposide (VM-26)), and taxane
(e.g., paclitaxel
and docetaxel).
[00157] In a particular embodiment, the lyophilized pharmaceutical composition
of the
invention is used in combination with a taxane such as paclitaxel and
docetaxel.
32
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00158] In another embodiment, lyophilized pharmaceutical compositions of the
invention can
be used in combination with an anthracycline, such as daunorubicin or
idarubicin.
[00159] In a further embodiment, the lyophilized pharmaceutical composition of
the invention is
used in combination with a biological agent such as an immuno-modulating
protein (e.g. a
cytokine), a monoclonal antibody against a tumour antigen, a tumour suppressor
gene or a
cancer vaccine.
[00160] Examples of interleukins that can be used in combination with the
lyophilized
pharmaceutical composition of the invention include, but are not limited to,
interleukin 2 (IL-2),
and interleukin 4 (IL-4), interleukin 12 (IL-12). Examples of interferons that
can be used in
conjunction with the lyophilized pharmaceutical composition of the invention
include, but are
not limited to, interferon [alpha], interferon [beta] ibroblast interferon)
and interferon [gamma]
(fibroblast interferon). Examples of such cytokines include, but are not
limited to erythropoietin
(epoietin), granulocyte-CSF (filgrastim), and granulocyte, macrophage-CSF
(sargramostim).
Immuno-modulating agents other than cytokines include, but are not limited to
bacillus
Calmette-Guerin, levamisole, and octreotide.
[00161] Examples of monoclonal antibodies against tumour antigens that can be
used in
conjunction with the the lyophilized pharmaceutical composition of the
invention include, but
are not limited to, HERCEPTIN(R) (Trastruzumab), RITUXAN(R) (Rituximab),
MYLOTARG(R) (anti-CD33), and CAMPATH(R) (anti-CD52).
[00162] In a further embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with a cancer vaccine, for example a cancer vaccine
selected from a
CTA cancer vaccine, such as a vaccine based on a CTA antigen selected from: NY-
ESO-1,
LAGE-1, MAGE-Al, -A2, -A3, -A4, -A6, -A10, -Al2, CT7, CT10, GAGE1-6, GAGE 1-2,
BAGE, SSX1-5, SSX 2, HAGE, PRAME, RAGE-1, XAGE-1, MUC2, MUC5B and HMW-
MAA. Non-limiting examples of CTA vaccines include those based on MAGE-A3 (for
example
recMAGE-A3), NY-ESO-1 and PRAME.
[00163] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with a T-cell activating agent, for example a T-
cell activating agent
which is an antibody (optionally a mAb), for example selected from: (a) a
CD137 agonist; (b) a
CD40 agonist; (c) an 0X40 agonist; (d) a PD-1 mAb; (e) a PD-L1 mAb; (f) a CTLA-
4 mAb;
and (g) combinations of (a)-(f). In some embodiments, the ancillary
therapeutic component is
Tremelimumab or Ipilimumab.
[00164] In another embodiment, the the lyophilized pharmaceutical composition
of the
invention can be used in combination with carboplatin for the treatment of
platinum-resistant
recurrent ovarian cancer.
33
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00165] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in the treatment of hepatocellular carcinoma (e.g. post sorafenib
failures).
[00166] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with irinotecan for the treatment of metastatic
colon cancer.
[00167] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with 5-fluorouracil (5-FU), leuocovorin,
oxaliplatin for the treatment
of metastatic colon cancer.
[00168] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with cytarabine and fludarabine for the treatment
of pediatric
relapsed/refractory AML.
[00169] In another embodiment, the lyophilized pharmaceutical composition of
the invention
can be used in combination with a JAK2 inhibitor for the treatment of
myoproliferative
neoplasms.
[00170] The lyophilized pharmaceutical composition of the invention and any
other therapeutic
agents can be presented separately or presented together in a pharmaceutical
package, kit or
patient pack.
[00171] The lyophilized pharmaceutical composition of the invention and
combinations with
other therapeutic agents or radiation therapies as described above can be
administered over a
prolonged term to maintain beneficial therapeutic effects or can be
administered for a short
period only. Alternatively, they can be administered in a pulsatile or
continuous manner.
[00172] The lyophilized pharmaceutical composition of the invention can be
administered in an
effective amount, i.e. an amount that is effective to bring about the desired
therapeutic effect
either alone (in monotherapy) or in combination with one or more
chemotherapeutic agents or
radiation therapy. For example, the "effective amount" can be a quantity of
compound which,
when administered to a subject suffering from cancer, slows tumour growth,
ameliorates the
symptoms of the disease and/or increases longevity.
[00173] The amount of the lyophilized pharmaceutical composition of the
invention
administered to the subject can depend on the type and severity of the disease
or condition and
on the characteristics of the subject, such as general health, age, sex, body
weight and tolerance
to drugs. The skilled person is able to determine appropriate dosages
depending on these and
other factors. Effective dosages for commonly used anti-cancer drugs and
radiation therapy are
well known to the skilled person.
Purity of Compounds of the Invention.
34
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00174] Any compound herein can be purified. A compound herein can be least 1%
pure, at
least 2% pure, at least 3% pure, at least 4% pure, at least 5% pure, at least
6% pure, at least 7%
pure, at least 8% pure, at least 9% pure, at least 10% pure, at least 11%
pure, at least 12% pure,
at least 13% pure, at least 14% pure, at least 15% pure, at least 16% pure, at
least 17% pure, at
least 18% pure, at least 19% pure, at least 20% pure, at least 21% pure, at
least 22% pure, at
least 23% pure, at least 24% pure, at least 25% pure, at least 26% pure, at
least 27% pure, at
least 28% pure, at least 29% pure, at least 30% pure, at least 31% pure, at
least 32% pure, at
least 33% pure, at least 34% pure, at least 35% pure, at least 36% pure, at
least 37% pure, at
least 38% pure, at least 39% pure, at least 40% pure, at least 41% pure, at
least 42% pure, at
least 43% pure, at least 44% pure, at least 45% pure, at least 46% pure, at
least 47% pure, at
least 48% pure, at least 49% pure, at least 50% pure, at least 51% pure, at
least 52% pure, at
least 53% pure, at least 54% pure, at least 55% pure, at least 56% pure, at
least 57% pure, at
least 58% pure, at least 59% pure, at least 60% pure, at least 61% pure, at
least 62% pure, at
least 63% pure, at least 64% pure, at least 65% pure, at least 66% pure, at
least 67% pure, at
least 68% pure, at least 69% pure, at least 70% pure, at least 71% pure, at
least 72% pure, at
least 73% pure, at least 74% pure, at least 75% pure, at least 76% pure, at
least 77% pure, at
least 78% pure, at least 79% pure, at least 80% pure, at least 81% pure, at
least 82% pure, at
least 83% pure, at least 84% pure, at least 85% pure, at least 86% pure, at
least 87% pure, at
least 88% pure, at least 89% pure, at least 90% pure, at least 91% pure, at
least 92% pure, at
least 93% pure, at least 94% pure, at least 95% pure, at least 96% pure, at
least 97% pure, at
least 98% pure, at least 99% pure, at least 99.1% pure, at least 99.2% pure,
at least 99.3% pure,
at least 99.4% pure, at least 99.5% pure, at least 99.6% pure, at least 99.7%
pure, at least 99.8%
pure, or at least 99.9% pure.
EXAMPLES
EXAMPLE 1. Preparation of a lyophilized formulation of a sodium salt of the
compound of
formula (1).
[00175] The sodium salt of the compound of formula (1) was dissolved in DMSO
at a defined
concentration using an overhead mixer in an appropriately sized stainless
steel (SS) vessel.
Upon complete solubilization of the drug in DMSO, samples of the bulk solution
were tested
using a UV or HPLC in-process method to determine that the amount of the
sodium salt of the
compound of formula 1 was within 95-105% of the target concentration. The bulk
solution was
filtered through a series of two pre-sterilized 0.2 micron sterilizing filters
that were DMSO-
compatible, and collected into a 2L SS surge vessel. The filtration rate was
continuously
adjusted by visual monitoring of quantity available for filling in the surge
vessel. One gram
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
aliquots of the filtered bulk solution were then filled into 5 cc
depyrogenated, clear glass vials.
Each vial was automatically and partially stoppered on the fill line with a
fluoropolymer coated,
chlorobutyl rubber lyo stopper that was pre-sterilized. The product vials were
transferred to a
lyophilizer under aseptic transfer conditions for initiation of a
lyophilization cycle. The
lyophilizer used was a pilot scale lyophilizer, Lyobeta 35, IMA-Telstar, which
has 1.02 m2 of
chamber space, an ice capacity of 35 kg, 22kg/24hr for condenser capacity.
[00176] Vials containing the solution were lyophilized using the cycle
parameters set out below
in TABLE 1.
TABLE 1 ¨ Lyophilization cycle operating parameters
Stage Event Temperature/Pressure/Time
T ( C) P Time (h)
Load 5 Atm 0.0
First freezing stage Ramp temperature -45 Atm 1.0
First freezing stage Hold temperature -45 Atm 1.5
First warming stage Ramp temperature 0 Atm 1.3
First warming stage Hold temperature 0 Atm 2.0
Second freezing stage Ramp temperature -45 Atm 2.0
Second freezing stage Hold temperature -45 Atm 2.0
Primary drying stage Decrease and hold
-45 6 bar 4.0
pressure
Primary drying stage Ramp temperature -20 6 bar 3.0
Primary drying stage Hold temperature -20 6 bar 12.0
Primary drying stage Ramp temperature -5 6 bar 3.0
Primary drying stage Hold temperature -5 6 bar 24.0
Secondary drying stage Ramp temperature 65 6 bar 6.0
Secondary drying stage Hold temperature 65 6 bar 15.0
[00177] Upon completion of the lyophilization cycle, the lyophilizer was back-
filled with
nitrogen, and the vials were completely and automatically stoppered. Vials
were aseptically
transferred to an isolator where each of the vials was automatically capped
with a blue
aluminum flip-off cap. Vials were visually inspected before proceeding with
sampling for
release testing, and the labeling and packaging operation. Vials were kept at
2-8 C until ready.
Each vial was labeled for its content.
36
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
EXAMPLE 2. Comparative Tests.
[00178] I. Lyophilized formulations made by the process of the invention:
Bulk solutions were made containing the sodium salt of the compound of formula
(1) at four
different concentrations in DSMO and the resulting solutions (designated A to
D) were filled
into lyophilization vials and subjected to lyophilization using the protocol
described above in
Example 1. Pirani and Baratron gauges were used to determine the end of the
primary drying
(sublimation) stage. FIGURE 1 shows the progressive reduction in DMSO content
over time
during the primary and secondary drying stages.
[00179] Following lyophilization, the lyophilized samples were analysed for
purity (% purity by
HPLC), DMSO residual content, and residual moisture. The samples were
reconstituted by
dissolving them in the non-aqueous solvent system described in TABLE 2 below
and the
reconstitution time and appearance of the reconstituted formulations were
analysed.
TABLE 2 - Solvent for reconstitution
% of each Grade Function
ingredient
Propylene glycol 65 NF, PhEur Solvent
Glycerin 25 NF, PhEur Solvent
Alcohol/Ethanol 10 USP, PhEur Thinning agent
The results of the analyses are set out in TABLE 3 below.
Results for four different concentrations, n=1
TABLE 3
Sample ID ¨> A
(100 (75 (50 (25
Analysis mg/mL) mg/mL) mg/mL) mg/mL)
% Purity by HPLC (API purity
93.6%) 93.2 93.1 93.2 93.2
DMSO residual solvent % 19.4 15.1 19.2 20.8
Residual Moisture <LOQ <LOQ <LOQ <LOQ
Reconstitution time (manual) 17 min 40s 12 min 51s 12 min 49s 18 min 51s
Appearance of the reconstituted
Clear solution, slightly yellow
solution
LOQ = limit of quantitation
[00180] II. Comparative formulations:
Bulk solutions of the sodium salt of the compound of formula (1) at a
concentration of 100
37
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
mg/mL were subjected to lyophilization using the apparatus described in
Example 1 above but a
different temperature profile which did not include the first warming stage
during the freezing of
the solution but included freezing the formulation at different freezing
rates.
The characteristics of the Comparative formulations prepared in this way are
shown in TABLE
4 below.
Table 4
Identification N ¨> FP1 FP2 FP3
Analysis Specification Result
Firm cake with
Appearance of Compact cake Compact cake
cracks adhering to
the cake (all Description detached from the detached from the
the bottom of the
vials) walls walls
vial
Appearance of Clear solution with Clear solution with Clear
solution with
the particles sticking to particles sticking to
particles sticking to
reconstituted Clear solution the walls the walls the
wall
solution and free of particles >30 min for > 30 min for >
30 min for
time for complete complete complete
reconstitution dissolution dissolution dissolution
Water content Below 0.02% 0.005% .001%
(tentati1 % 0
ve)
Residual Report result for
19.1% (FP1-9) 19.4% (FP2-9) 19.4% (FP3-9)
Solvent DMSO information
[00181] III. Comparison of results obtained from the formulations described in
I and II
The results shown in step I above demonstrate that when an intermediate
warming stage ("first
warming stage") is included during the freezing of the solution prior to
primary drying in
accordance with the invention, the result is a lyophilized dry formulation
which can be
reconstituted in under 20 minutes and under 15 minutes in some cases.
[00182] By comparison, the Comparative formulations FP1, FP2 and FP3 described
in II above,
made by a process that omitted the intermediate warming stage, took longer to
reconstitute (over
30 minutes). Without wishing to be bound by any theory, it is believed that
the intermediate
warming stage has the effect of increasing the porosity of the lyophilized
product and increasing
the surface area available for contact with solvent molecules, thereby
increasing the solubility of
the formulations.
[00183] IV. Comparison of drying times with Example 4 in W02013/033176
Example 4 in W02013/033176 describes the lyophilization of a solution of the
sodium salt of
the compound of formula (1) using the cycle parameters shown in TABLE 5 below.
38
CA 02991167 2017-12-29
WO 2017/004538
PCT/US2016/040730
Table 5
Stage Event Temperature/Pressure/Time
T ( C) P Time
(minutes)
Freezing stage Ramp temperature -40 Atm 133
Freezing stage Hold temperature -40 Atm 360
Primary drying stage Ramp temperature and
-5 100
mTorr 117
pressure
Primary drying stage Hold temperature and
-5 100
mTorr 1440
pressure
Primary drying stage Ramp temperature 10 100 mTorr 50
Primary drying stage Hold temperature 10 100 mTorr 1440
Secondary drying stage Ramp temperature and
30 50 mTorr 67
pressure
Secondary drying stage Hold temperature and
30 50 mTorr 1440
pressure
Secondary drying stage Ramp temperature 60 50 mTorr 100
Secondary drying stage Hold temperature 60 50 mTorr 1440
Total lyophilization
6587 minutes = 109 hours and 47 minutes
time
[00184] The total lyophilization time for the formulation described in Example
4 of
W02013/033176 is 109 hours and 47 minutes. By comparison, the total
lyophilization time for
the formulation of the invention as described in I above was 76.8 hours, i.e.
over 30 hours
shorter than the total lyophilization time for Example 4 in W02013/033176.
Most of the
difference is accounted for by the significantly reduced secondary drying
stage of the process of
the present invention compared to the process described in Example 4 of
W02013/033176 (21
hours vs 50.78 hours). In the process of the present invention, an
intermediate (first) warming
stage is interposed between two freezing stages when the solution is initially
frozen, and this is
believed to result in a much more porous structure from which DMSO can more
readily sublime
during the primary drying stage. Thus, a greater proportion of the DMSO is
removed during the
primary drying stage with the result that much shorter secondary drying stage
can be employed.
[00185] Therefore, in summary, the process of the present invention can reduce
the time
necessary to produce a lyophilized product that has greatly enhanced
dissolution characteristics.
EXAMPLE 3. Larger scale studies on the 75 mg/ml and 100 mg/ml formulations A
and B.
39
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00186] The results obtained in the experiments described in Example 2 showed
that the lowest
residual DMSO levels were obtained with formulation B in which a bulk solution
containing a
concentration of 75 mg/mL of active compound was lyophilized. Confirmatory
studies were
therefore carried out on 75 mg/ml and 100 mg/ml solutions of the sodium salt
of the compound
of formula (1) in DMSO. The lyophilization was carried out at a 100 vial
scale, and analysis was
carried out on multiple samples. The protocol used was as described in Example
1. The
properties of the resulting lyophilized products were as shown in TABLE 6
below.
TABLE 6
Sample ID¨*
Analysis 100 mg/mL 75 mg/mL
Residual DMSO % w/w, n=3 17.4 (24.2 mg/vial) 18.7% (25.4 mg/vial)
Reconstitution time (min), n=3 8 min* 8 min*
Appearance, n=3 Clear and colourless**
Water Content, n=2 <LOQ <LOQ
Assay %w/w, n=2 107.8 105
* The reconstitution time does not include dissipation of bubbles which might
take about an
additional 10 minutes. However, the reconstitution was carried out manually
and did not require
mechanised mixing apparatus.
** Although not seen in this instance, there can be occasions when the
solutions are slightly
hazy and/or slightly off-white to yellow in color.
[00187] The results in TABLE 6 demonstrate that the process of the invention
can be used to
prepare lyophilized formulations that have a reconstitution time of less than
ten minutes
(excluding the time taken for bubbles to clear) and that reconstitution can be
carried out
manually without the need for mechanized mixers.
EXAMPLE 4. Preparation of the sodium salt of the compound of formula (1).
[00188] The sodium salt of the compound of formula (1) was prepared as
described in US
7700567 (the content of which is hereby incorporated by reference) by coupling
ls (where R1 =
carbamate protective group) with phosphoramidite building block ld:
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
0
N H
< I
1;1 .....õ--õ,........ ......c.,....õ,.... _.õ-R,
N 1st'
HO
.c..Øm.
0
Contro9erl-pore glass
R, -NH
N N
0
NC 1,,o
I
--......,..õ,...õ.õõNõ,....õ.õ.õ--
ld
[00189] A protected 2'-deoxyguanosine-linked CPG solid support ls (where R1 =
tert-butyl
phenoxyacetyl) was coupled with 2-2.5 equivalents of phenoxyacetyl decitabine
phosphoramidite (1d, where R1 = phenoxyacetyl) in the presence of 60% of 0.3 M
benzylthiotetrazole activator (in acetonitrile) for 10 minutes. The CPG solid
support containing
protected DpG dinucleotide was treated with 20 mL of 50 mM K2CO3 in methanol
for 1 hour
and 20 minutes. The coupled product was oxidized, the protective group was
removed, and the
resultant compound was washed, filtered, and purified by the AKTA Explorer 100
HPLC with a
Gemini C18 preparative column (Phenomenex), 250x21.2 mm, 101.tm with guard
column
(Phenomenex), 50x21.2mm, 101.tm, with 50 mM triethylammonium acetate (pH 7) in
MilliQ
water (Mobile Phase A) and 80% acetonitrile in MilliQ water (Mobile Phase B),
with 2% to
20/25% Mobile Phase B in column volumes.
[00190] The ESI-MS (-ve) of DpG dinucleotide 2b:
41
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
to-r2
N
N
HO
0
0 N
e47/ tiJH
NH2
0 ________________________________ 0
OH
2b
where X+ = triethylammonium (calculated exact mass for the neutral compound
C181-124N9010P
is 557.14), exhibited m/z 556.1 [M-HT and 1113.1 for [2M-H] - (see mass
spectrum in Figure 31
of US 7700567).
[00191] The sodium salt of the compound of formula (1), i.e. DpG dinucleotide
2b, where X+ =
sodium, was obtained by re-dissolving the triethylammonium salt in 4 mL water,
0.2 mL 2M
NaC104 solution. When 36 mL acetone was added, the dinucleotide precipitated.
The solution
was kept at -20 C for several hours and centrifugated at 4000 rpm for 20
minutes. The
supernatant was discarded and the solid was washed with 30 mL acetone followed
by an
additional centrifugation at 4000 rpm for 20 minutes. The precipitate, which
was dissolved in
water and freeze dried, exhibited m/z 556.0 [M-HT (see mass spectrum in Figure
36 of US
7700567).
EMBODIMENTS
[00192] The following non-limiting embodiments provide illustrative examples
of the invention,
but do not limit the scope of the invention.
[00193] Embodiment 1. A method of preparing a lyophilized pharmaceutical
composition, the
method comprising dissolving a compound of formula (1):
42
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
NH2
N N
L
N 0
HO ________________________
i) 0
1 N...........õ/".\.
< I NH
0 =P-0 H
oI N--------NNH 2
\
cØ..
OH (1),
or a pharmaceutically-acceptable salt thereof, in a solvent comprising
dimethylsulfoxide
(DMSO) to form a solution, wherein the solvent is then removed by a freeze-
drying process to
give a lyophilized product, wherein the freeze-drying process comprises: (i) a
first freezing stage
in which the solution is frozen by reducing the temperature thereof to a
temperature of no
greater than about -20 C; (ii) a first warming stage in which the temperature
of the frozen
solution is raised to a temperature in the range from about -15 C to about 5
C, wherein the
temperature in the range from about -15 C to about 5 C keeps the solution
frozen; (iii) a second
freezing stage in which the temperature of the solution is lowered to a
temperature of no greater
than about -20 C; (iv) a primary drying stage, wherein the primary drying
stage comprises a
sublimation step in which the DMSO is removed by sublimation from the solution
in its frozen
state under reduced pressure to give a partially dried product; and (v) a
secondary drying stage
in which the DMSO is removed by evaporation from the partially dried product
in a non-frozen
state under reduced pressure to give the lyophilized product.
[00194] Embodiment 2. The method of embodiment 1, wherein the compound of
formula (1) is
in the form of a sodium salt.
[00195] Embodiment 3. The method of any one of embodiments 1-2, wherein the
solvent is non-
aqueous.
[00196] Embodiment 4. The method of any one of embodiments 1-3, wherein the
lyophilized
pharmaceutical composition has a dissolution time, at ambient temperature, and
without the aid
of mechanised stirring, in a non-aqueous solvent containing 65% (v/v)
propylene glycol; 25%
(v/v) glycerine; and 10% (v/v) ethanol, of no greater than about 20 minutes.
[00197] Embodiment 5. The method of any one of embodiments 1-4, wherein in an
amount of
43
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
the lyophilized pharmaceutical composition obtained from 1 gram of the
solution, there is a
residual DMSO content of no greater than about 20 mg.
[00198] Embodiment 6. The method of any one of embodiments 1-5, wherein any
residual
DMSO present in the lyophilized pharmaceutical composition is in an amount
corresponding to
no more than 35 mg per 100 mg equivalent of a free base of the compound of
formula (1).
[00199] Embodiment 7. The method of any one of embodiments 1-6, further
comprising
packing the lyophilized pharmaceutical in a sealed pharmaceutical container.
[00200] Embodiment 8. The method of any one of embodiments 1-7, further
comprising
dissolving the lyophilized pharmaceutical composition in a solvent to form an
injectable liquid
composition.
[00201] Embodiment 9. The method of embodiment 8, wherein the solvent is a non-
aqueous
solvent.
[00202] Embodiment 10. The method of any one of embodiments 1-9, wherein the
solution
further comprises a co-solvent.
[00203] Embodiment 11. The method of embodiment 1, further comprising
reconstituting the
lyophilized pharmaceutical composition in a pharmaceutically acceptable
solvent to give a liquid
formulation containing a compound of formula (1) or the pharmaceutically
acceptable salt
thereof.
[00204] Embodiment 12. The method of any one of embodiments 1-11, wherein the
reduced
pressure in the primary drying stage is from about 5 Bar to about 40 Bar.
[00205] Embodiment 13. The method of any one of embodiments 1-12, wherein the
temperature
in the primary drying stage is from about -3 C to about -9 C.
[00206] Embodiment 14. The method of any one of embodiments 1-13, wherein the
temperature
in the secondary drying stage is from about 30 C to about 65 C.
[00207] Embodiment 15. A pharmaceutical composition prepared by a process
comprising the
steps of: dissolving a compound of formula (1):
44
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
NH2
N N
L
N 0
HO ________________________
i) 0
1 N...........õ/"\..
< I NH
0 =P-0 H
oI N--------NNH 2
\
cØ..
OH (1),
or a pharmaceutically-acceptable salt thereof, in a solvent comprising
dimethylsulfoxide
(DMSO) to form a solution, wherein the solvent is then removed by a freeze-
drying process to
give a lyophilized product, wherein the freeze-drying process comprises: (i) a
first freezing stage
in which the solution is frozen by reducing the temperature thereof to a
temperature of no
greater than about -20 C; (ii) a first warming stage in which the temperature
of the frozen
solution is raised to a temperature in the range from about -15 C to about 5
C, wherein the
temperature in the range from about -15 C to about 5 C keeps the solution
frozen; (iii) a second
freezing stage in which the temperature of the solution is lowered to a
temperature of no greater
than about -20 C; (iv) a primary drying stage, wherein the primary drying
stage comprises a
sublimation step in which the DMSO is removed by sublimation from the solution
in its frozen
state under reduced pressure to give a partially dried product; and (v) a
secondary drying stage
in which the DMSO is removed by evaporation from the partially dried product
in a non-frozen
state under reduced pressure to give the lyophilized product.
[00208] Embodiment 16. The pharmaceutical composition of embodiment 15,
wherein the
compound of formula (1) is in the form of a sodium salt.
[00209] Embodiment 17. The pharmaceutical composition of any one of
embodiments 15-16,
wherein the solvent is non-aqueous.
[00210] Embodiment 18. The pharmaceutical composition of any one of
embodiments 15-17,
wherein the lyophilized pharmaceutical composition has a dissolution time, at
ambient
temperature, and without the aid of mechanised stirring, in a non-aqueous
solvent containing
65% (v/v) propylene glycol; 25% (v/v) glycerine; and 10% (v/v) ethanol, of no
greater than
about 20 minutes.
CA 02991167 2017-12-29
WO 2017/004538 PCT/US2016/040730
[00211] Embodiment 19. The pharmaceutical composition of any one of
embodiments 15-18,
wherein in an amount of the lyophilized pharmaceutical composition obtained
from 1 gram of
the solution, there is a residual DMSO content of no greater than about 20 mg.
[00212] Embodiment 20. The pharmaceutical composition of any one of
embodiments 15-19,
wherein any residual DMSO present in the lyophilized pharmaceutical
composition is in an
amount corresponding to no more than 35 mg per 100 mg equivalent of a free
base of the
compound of formula (1).
[00213] Embodiment 21. The pharmaceutical composition of any one of
embodiments 15-20,
the process further comprising packing the lyophilized pharmaceutical in a
sealed
pharmaceutical container.
[00214] Embodiment 22. The pharmaceutical composition of any one of
embodiments 15-21,
the process further comprising dissolving the lyophilized pharmaceutical
composition in a
solvent to form an injectable liquid composition.
[00215] Embodiment 23. The pharmaceutical composition of embodiment 22,
wherein the
solvent is a non-aqueous solvent.
[00216] Embodiment 24. The pharmaceutical composition of any one of
embodiments 15-23,
wherein the solution further comprises a co-solvent.
[00217] Embodiment 25. The pharmaceutical composition of embodiment 15, the
process
further comprising reconstituting the lyophilized pharmaceutical composition
in a
pharmaceutically acceptable solvent to give a liquid formulation containing a
compound of
formula (1) or the pharmaceutically acceptable salt thereof.
[00218] Embodiment 26. The pharmaceutical composition of any one of
embodiments 15-25,
wherein the reduced pressure in the primary drying stage is from about 5 Bar
to about 40 Bar.
[00219] Embodiment 27. The pharmaceutical composition of any one of
embodiments 15-26,
wherein the temperature in the primary drying stage is from about -3 C to
about -9 C.
[00220] Embodiment 28. The pharmaceutical composition of any one of
embodiments 15-27,
wherein the temperature in the secondary drying stage is from about 30 C to
about 65 C.
46