Note: Descriptions are shown in the official language in which they were submitted.
METHODS FOR GENERATING HIGH TITER HELPER-FREE
PREPARATIONS OF RECOMBINANT AAV VECTORS
This application is a divisional application of Canadian Patent Application
Number
2,302,992 entitled "Methods for Generating High Titer Helper-Free Preparations
of
Recombinant AAV Vectors", which is the national phase of PCT International
Application
Number PCT/US98/18600 filed September 4, 1998 and entered into the Canadian
National Phase March 3, 2000.
FIELD OF THE INVENTION
The present invention relates generally to the field of recombinant adeno-
associated virus (AAV) vectors and preparations thereof that can be used for
gene transfer.
More specifically, it relates to methods for generating high titer
preparations of
recombinant AAV vectors that are substantially free of helper virus (e.g.
adenovirus) as
well as cellular proteins.
BACKGROUND
Adeno-associated viruses (AAV) have unique features that make them attractive
as
vectors for gene therapy. Adeno-associated viruses infect a wide range of cell
types.
However, they are non-transforming, and are not implicated in the etiology of
any human
disease. Introduction of DNA to recipient host cells generally leads to long-
term
persistence and expression of the DNA without disturbing the normal metabolism
of the
cell.
There are at least three desirable features of a recombinant AAV vector
preparation
for use in gene transfer, especially in human gene therapy. First, it is
preferred that the
vector should be generated at titers sufficiently high to transducer an
effective proportion
of cells in the target issue. Gene therapy in vivo typically requires a high
number of vector
particles. For example, some treatments may require in excess of 108
particles, and
treatment of cystic fibrosis by direct delivery to the airway may require in
excess of 1010
particles. Second, it is preferred that the vector preparations should be
essentially free of
replication-competent AAV (i.e. phenotypically wild-type AAV which can be
replicated
in the presence of helper virus or helper virus functions). Third, it is
preferred that the
rAAV vector preparation as a whole be essentially free of other viruses (such
as a helper
1
CA 2995542 2018-02-15
virus used in AAV production) as vell as helper virus and cellular proteins,
and other
components such as lipids and carbohydrates, so as to minimize or eliminate
any risk of
generating an immune response in the context of gene therapy. This latter
point is
especially significant in the context of AAV because AAV is a "helper-
dependent" virus
la
CA 2995542 2018-02-15
that requires co-infection with a helper virus (typically adenovirus) or other
provision of
helper virus functions in order to be effectively replicated and packaged
during the process
of AAV production; and, moreover, adenovirus has been observed to generate a
host
immune response in the context of gene therapy applications (see, e.g., Byrnes
et al.,
Neuroscience 66:1015, 1995; McCoy et al., Human Gene Therapy 6:1553, 1995; and
Barr
et at., Gene Therapy 2:151, 1995). The methods of the present invention
address these and
other desirable features of rAAV vector preparations, as described and
illustrated in detail
below.
General reviews of AAV virology and genetics are available elsewhere. The
reader
may refer inter alia to Carter, "Handbook of Parvoviruses", Vol. I, pp. 169-
228 (1989),
and Berns, "Virology", pp. 1743-1764, Raven Press, (1990). What follows is a
brief
synopsis for the convenience of the reader. AAV is a replication-defective
virus, which
means that it relies on a helper virus in order to complete its replication
and packaging
cycle in a host cell. The AAV genome generally comprises the packaging genes
rep and
cap, with other necessary functions being provided in trans from the helper
virus and the
host cell.
AAV particles are comprised of a proteinaceous capsid having three capsid
proteins, VP1, VP2 and VP3, which enclose a ¨4.6 kb linear single-stranded DNA
genome. Individual particles package only one DNA molecule strand, but this
may be
either the plus or minus strand. Particles containing either strand are
infectious, and
replication occurs by conversion of the parental infecting single strand to a
duplex form,
and subsequent amplification, from which progeny single strands are displaced
and
packaged into capsids. Duplex or single-strand copies of AAV genomes
(sometimes
referred to as "proviral DNA" or "provirus") can be inserted into bacterial
plasmids or
phagemids, and transfected into adenovirus-infected cells.
By way of illustration, the linear genome of serotype AAV2 is terminated at
either
end by an inverted terminal repeat (ITR) sequence. Between the ITRs are three
transcription promoters p5, p19, and p40 that are used to express the rep and
cap genes
(Laughlin et al., 1979, Proc. Natl. Acad. Sci. USA, 76:5567-5571). ITR
sequences are
required in cis and are sufficient to provide a functional origin of
replication, integration
into the cell genome, and efficient excision and rescue from host cell
chromosomes or
2
CA 2995542 2018-02-15
recombinant plasmids. The rep and cap gene products provide functions for
replication
and encapsidation of viral genome, respectively, and it is sufficient for them
to be present
in trans.
The rep gene is expressed from two promoters, p5 and pI9, and produces four
proteins designated Rep78, Rep68, Rep52 and Rep40. Only Rep78 and Rep68 are
required for AAV duplex DNA replication, but Rep52 and Rep40 appear to be
needed for
progeny, single-strand DNA accumulation (Chejanovsky et at., Virology 173:120,
1989).
Rep68 and Rep78 bind specifically to the hairpin conformation of the AAV ITR
and
possess several enzyme activities required for resolving replication at the
AAV termini.
Rep78 and Rep68, also exhibit pleiotropic regulatory activities including
positive and
negative regulation of AAV genes and expression from some heterologous
promoters, as
well as inhibitory effects on cell growth. The cap gene encodes capsid
proteins VP1, VP2,
and VP3. These proteins share a common overlapping sequence, but VP I and VP2
contain
additional amino terminal sequences transcribed from the p40 promoter by use
of alternate
initiation codons. All three proteins are required for effective capsid
production.
AAV genomes have been introduced into bacterial plasmids by procedures such as
GC tailing (Samulski et al., 1982, Proc. Natl. Acad. Sci. USA, 79:2077-2081),
addition of
synthetic linkers containing restriction endonuclease cleavage sites (Laughlin
et al., 1983,
Gene, 23:65-73) or by direct, blunt-end ligation (Senapathy & Carter, 1984, J.
Biol.
Chem., 259:4661-4666). Transfection of such AAV recombinant plasmids into
mammalian cells with an appropriate helper virus results in rescue and
excision of the
AAV genome free of any plasmid sequence, replication of the rescued genome and
generation of progeny infectious AAV particles.
Recombinant AAV vectors comprising a heterologous polynucleotide of
therapeutic interest may be constructed by substituting portions of the AAV
coding
sequence in bacterial plasmids with the heterologous polynucleotide. General
principles of
rAAV vector construction are also reviewed elsewhere. See, e.g., Carter, 1992,
Current
Opinions in Biotechnology, 3:533-539; and Muzyczka, 1992, Curr. Topics in
Microbiol.
and Immunol., 158:97-129). The AAV ITRs are generally retained, since
packaging of the
vector requires that they be present in cis. However, other elements of the
AAV genome,
in particular, one or more of the packaging genes, may be omitted. The vector
plasmid can
3
CA 2995542 2018-02-15
be packaged into an AAV particle by supplying the omitted packaging genes in
trans 'via
an alternative source.
In one approach, the sequence flanked by AAV ITRs (the rAAV vector sequence),
and the AAV packaging genes to be provided in trans, are introduced into the
host cell in
separate bacterial plasmids. Examples of this approach are described in
Ratschin et al.,
Mol. Cell. Biol. 4:2072 (1984); Hermonat et al., Proc. Natl. Acad, Sci. USA,
81:6466
(1984); Tratschin et al., Mol. Cell. Biol. 5:3251 (1985); McLaughlin et at.,
J. Virol,,
62:1963 (1988); and Lebkowski et al., 1988 Mol. Cell. Biol., 7:349 (1988).
Samulski et al.
(1989, J. Virol., 63:3822-3828) have described a packaging plasmid called
pAAV/Ad,
which consists of Rep and Cap encoding regions enclosed by ITRs from
adenovirus.
Human airway epithelial cells from a cystic fibrosis patient have been
transduced with an
AAV vector prepared using the pAAV/Ad packaging plasmid and a plasmid
comprising
the selective marker gene neo expressed via the AAV p5 promoter (Flotte et
al., Am. J.
Respir. Cell. Mol, Biol. 7:349, 1992).
A second approach is to provide either the vector sequence, or the AAV
packaging
genes, in the form of an episomal plasmid in a mammalian cell used for AAV
replication.
For example, U.S. Patent 5,173,414 describes a cell line in which the vector
sequence is
present as a high-copy episomal plasmid, The cell lines can be transduced with
the trans-
complementing AAV functions rep and cap to generate preparations of AAV
vector. This
approach is not ideal, because the copy number per cell cannot be rigorously
controlled
and episomal DNA is much more likely to undergo rearrangement, leading to
production
of vector byproducts.
A third approach is to provide either the vector sequence, or the AAV
packaging
genes, or both, stably integrated into the genome of the mammalian cell used
for
replication.
One exemplary technique is outlined in international patent application WO
95/13365 (Targeted Genetics Corporation and Johns Hopkins University) and
corresponding U.S. Patent No. 5,658,776 (by Flotte et al.). This example uses
a
mammalian cell with at least one intact copy of a stably integrated rAAV
vector, wherein
the vector comprises an AAV ITR and a transcription promoter operably linked
to a target
polynucleotide, but wherein the expression of rep is limiting, In a preferred
embodiment,
4
CA 2995542 2018-02-15
an AAV packaging plasmid comprising the rep gene operably linked to a
heterologous
AAV is introduced into the cell, and then the cell is incubated under
conditions that allow
replication and packaging of the AAV vector sequence into particles.
A second exemplary technique is outlined in patent application WO 95/13392
(Trempe et al.). This example uses a stable mammalian cell line with an AAV
rep gene
operably linked to a heterologous promoter so as to be capable of expressing
functional
Rep protein. In various preferred embodiments, the AAV cap gene can be
provided stably
as well or can be introduced transiently (e.g. on a plasmid). A recombinant
AAV vector
can also be introduced stably or transiently.
Another exemplary technique is outlined in patent application WO 96/17947 (by
Targeted Genetics Corporation, J. Allen). This example uses a mammalian cell
which
comprises a stably integrated AAV cap gene. and a stably integrated AAV rep
gene
operably linked to a heterologous promoter and inducible by helper virus. In
various
preferred embodiments, a plasmid comprising the vector sequence is also
introduced into
the cells (either stably or transiently). The rescue of AAV vector particles
is then initiated
by introduction of the helper virus.
These various examples address the issue of providing AAV at sufficiently high
titer, minimizing recombination between vector and packaging components, and
reducing
or avoiding the potential difficulties associated with the expression of the
AAV rep gene in
mammalian cell line (since the Rep proteins can not only limit their own
expression but
can also affect cellular metabolism). However, packaging of an AAV vector into
viral
particles still relies on the presence of a suitable helper virus for AAV or
the provision of
helper virus functions. Helper viruses capable of supporting AAV replication
are
exemplified by adenovirus, but include other viruses such as herpes and pox
viruses. The
presence of significant quantities of infectious helper virus in a preparation
of AAV
vectors is problematic in that the preparation is intended for use in human
administration.
Even the presence of non-replicative helper virus components can cause an
unacceptable
immunological reaction in the treated subject.
The potential problems elicited by helper virus antigen have been illustrated
in
several recent studies. Byrnes et al. (Neuroscience 66:1015, 1995) injected an
El-region
deleted, non-replicating human adenovirus type 5 into the brains of inbred
rats. An
CA 2995542 2018-02-15
inflammation response was observed that was attributed to the particles
administered rather
than to expression of new viral proteins due to viral infection of the cells.
Presence of the
virus was associated with increase in MHC Class I expression and a heavy
infiltration of
macrophages and T cells. McCoy et al. (Human Gene Therapy 6:1553, 1995)
instilled the
lungs of mice with intact adenovirus, adenovirus with incomplete genomes, or
adenovirus
inactivated with ultraviolet light. All induced pulmonary inflammation, and
the number of
inflammatory cells in the lung tissue was quantitatively similar for all three
forms of the
virus. Comparative experiments using adenovirus constructs in normal and
immune-
deficient mice performed by Barr et al. (Gene Therapy 2:151, 1995) indicate
that the anti-
adenovirus immune response is primarily T-cell mediated and gives rise to a
memory
response that affects subsequent doses.
Accordingly, in the development of recombinant AAV vectors such as those for
use
in gene therapy, there is a need for strategies that minimize the amount of
helper virus, as
well as helper virus proteins and cellular proteins, present in the final
preparation, while at
the same time still achieving a high titer of AAV so that the methods can be
effectively
employed on a scale that is suitable for the practical application of gene
therapy
techniques.
Since high titers of rAAV vector preparations are particularly useful, but the
production of high titers of rAAV, particularly in large-scale procedures, can
lead to the
generation of significant quantities of contaminating helper virus (e.g.
adenovirus or
"Ad"). helper virus proteins (e.g. Ad proteins), and/or cellular proteins, it
became
especially important to design scalable methods for the production of rAAV
that can be
used for the generation of high-titer preparations that are substantially free
of
contaminating virus and/or viral or cellular proteins. The present disclosure
provides
methods for achieving these competing goals and demonstrates that such
techniques can be
employed for the large-scale production of recombinant AAV vector
preparations.
SUMMARY OF THE INVENTION
This invention provides methods and materials for generating high titer
preparations of adeno-associated virus (AAV) that are substantially free of
helper virus,
helper virus proteins, and cellular proteins and other components.
6
CA 2995542 2018-02-15
Embodiments of the invention include but are not limited to the following:
A method of generating a population of recombinant adeno-associated virus
(rAAV) particles, comprising the steps of: a) providing an AAV producer cell
that
comprises: (i) one or more AAV packaging genes, wherein each said AAV
packaging
gene encodes an AAV replication or encapsidation protein; (ii) a recombinant
AAV
(rAAV) pro-vector that comprises a heterologous non-AAV polynucleotide flanked
by at
least one AAV inverted terminal repeat (ITR); and (iii) a helper virus for
AAV; b)
incubating the producer cell provided in step a) under conditions that are
permissive for
replication of AAV; c) 1ysing the producer cell after the incubation of step
b) to produce
an AAV producer cell lysate; and d) chromatographing the AAV producer cell
lysate of
step c) on a plurality of ion-exchange resins comprising at least one
positively-charged
anion exchange resin and at least one negatively-charged cationic exchange
resin to
generate a purified population of rAAV vector particles, or chromatographing
the AAV
producer cell lysate of step c) on an anion exchange resin followed by
tangential flow
filtration (TFF).
According to one aspect of the present invention there is provided a method of
generating a purified population of recombinant adeno-associated virus (rAAV)
particles,
comprising the steps of: a) providing an AAV producer cell that comprises: (i)
one or more
AAV packaging genes, wherein each said AAV packaging gene encodes an AAV
replication
or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-vector that
comprises a
heterologous non-AAV polynucleotide flanked by at least one AAV inverted
terminal repeat
(ITR); and (iii) a helper virus for AAV or a polynucleotide sequence of said
helper virus that
encodes at least one helper virus function; b) incubating the producer cell
provided in step a)
under conditions that are permissive for replication of AAV; c) lysing the
producer cell after
the incubation of step b) to produce an AAV producer cell lysate; d)
chromatographing the
AAV producer cell lysate of step c) on a positively-charged anion exchange
media; or
chromatographing the AAV producer cell lysate of step c) on a negatively-
charged cation
exchange media, and collecting a fraction containing rAAV particles; and e)
chromatographing the fraction of step d) on an exchange media opposite in
charge to that used
iin step d) and collecting a fraction containing rAAV particles; and whereby
said purified
population of rAAV particles is generated.
7
CA 2995542 2018-02-15
According to a further aspect of the present invention there is provided a
method of
generating a purified population of recombinant adeno-associated virus (rAAV)
particles,
comprising the steps of: a) providing an AAV producer cell that comprises: (i)
one or more
AAV packaging genes, wherein each said AAV packaging gene encodes an AAV
replication
or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-vector that
comprises a
heterologous non-AAV polynucleotide flanked by at least one AAV inverted
terminal repeat
(ITR); and (iii) a helper virus for AAV or a polynucleotide sequence of said
helper virus that
encodes at least one helper virus function; b) incubating the producer cell
provided in step a)
under conditions that are permissive for replication of AAV; c) lysing the
producer cell after
the incubation of step b) to produce an AAV producer cell lysate; d)
chromatographing the
AAV producer cell lysate of step c) on at least one positively-charged anion
exchange media;
and e) purifying the chromatographic fractions containing rAAV particles of
step d) by
tangential flow filtration to generate said purified population of rAAV vector
particles.
A method of generating a population of rAAV particles !as described above,
wherein
said helper virus is an adenovirus or a temperature-sensitive helper virus,
and said step of
incubating the producer cell is conducted at a temperature that is permissive
for replication
of AAV but non-permissive for replication of the temperature-sensitive helper
virus.
A method of generating a population of rAAV particles, wherein incubating the
producer cell is conducted in a vessel selected from the group consisting of a
tissue culture
flask, a roller bottle, a spinner flask, a tank reactor, a fermentor, and a
bioreactor, optionally
using a microcarrier, and preferably using a suspension-adapted mammalian cell
line.
A method of generating a population of recombinant adeno-associated virus
(rAAV)
particles, comprising the steps of: a) providing an AAV producer cell that
comprises: (i)
one or more AAV packaging genes, wherein each said AAV packaging gene encodes
an
AAV replication or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-
vector
that comprises a heterologous non-AAV polynucleotide flanked by at least one
AAV
inverted terminal repeat (ITR); and (iii) a helper virus for AAV or a
polynucleotide
sequence of said helper virus that encodes at least one helper virus function;
b) subjecting
the producer cell provided in step a) to a sub-lethal stress; and c)
incubating the stressed
7a
CA 2995542 2018-02-15
producer cell of step b) under conditions that are permissive for replication
of AAV.
Possible forms of sub-lethal stress may be selected but are not limited to
those in the group
consisting of a nutritional stress, an osmotic stress, a pH stress, a
temperature stress, an
aerobic stress, a mechanical stress, a radiational stress and a toxic stress.
A non-limiting
example by which nutritional stress is imposed is by culturing the producer
cells in a medium
that is deficient in one or more amino acids. Additional illustrations are
provided below.
A method of generating a population of rAAV particles, wherein said purified
population of rAAV vector particles is substantially free of replication-
competent AAV and
of helper virus and cellular proteins.
A method of generating a population of recombinant adeno-associated virus
(rAAV)
particles, comprising the steps of: a) providing an AAV producer cell that
comprises: (i)
one or more AAV packaging genes, wherein each said AAV packaging gene encodes
an
AAV replication or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-
vector
that comprises a heterologous non-AAV polynucleotide flanked by at least one
AAV
inverted terminal repeat (ITR); and (iii) a helper virus for AAV; b)
incubating the
producer cell provided in step a) under conditions that are permissive for
replication of
AAV and which comprise inducing a sub-lethal stress in the AAV producer cell;
c) lysing
the producer cell after the incubation of step b) to produce an AAV producer
cell lysate; and
d) purifying the AAV producer cell lysate to generate a population of
recombinant adeno-
associated virus (rAAV) particles. Suitable purification methods include those
described
elsewhere in this disclosure. An exemplary purification procedure comprises
chromatographing the AAV producer cell lysate of step c) on at least one
chromatographic
resin selected from the group consisting of a positively-charged anion
exchange resin and a
negatively-charged cationic exchange resin to generate a purified population
of rAAV vector
particles (preferred methods include anion exchange followed by cation
exchange or
tangential flow filtration (TFF)). Illustrative chromatographic procedures,
including ion
exchange chromatography, and chromatographic purification on heparin sulfate
are provided
below by way of example.
A host cell for producing recombinant adeno-associated virus (rAAV) particles
at
high efficiency, comprising: a) one or more AAV packaging genes, wherein each
said AAV
packaging gene encodes an AAV replication or encapsidation protein; b) a
heterologous
8
CA 2995542 2018-02-15
polynucleotide introduced into said host cell using an rAAV pro-vector,
wherein the
rAAVpro-vector comprises the heterologous polynucleotide flanked by at least
one AAV
inverted terminal repeat (ITR) and is deficient in said AAV packaging gene(s);
c) a helper
virus such as a temperature-sensitive helper virus (tsHV) for AAV, wherein
said tsHV is
temperature-sensitive for self-replication.
A population of rAAV particles, produced according to any of the production
methods of this invention. Preferably, the population of particles contains no
more than
about one infectious adenovirus particles per thousand infectious rAAV
particles,
preferably less than one per 106 rAAV, still more preferably less than about
one in 109.
Also provided are high-throughput assay techniques which can be used, for
example, in the titering of virus preparations as well as in the screening of
agents that
affect viral replication.
These and other embodiments of the invention are outlined in the description
that
follows.
In one aspect, there is provided a method of generating a population of
recombinant adeno-associated virus (rAAV) particles, comprising the steps of:
(a)
providing an AAV producer cell that is a mammalian cell and comprises: (i) one
or more
AAV packaging genes, wherein each said AAV packaging gene encodes an AAV
replication or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-vector
that
comprises a heterologous non-AAV polynucleotide flanked by at least one AAV
inverted
terminal repeat (ITR); and (iii) a helper virus for AAV or a polynucleotide
sequence of
said helper virus that encodes at least one helper virus function; and (b)
incubating the
producer cell of step (a) under conditions that are permissive for replication
of AAV and
which comprise inducing a sub-lethal stress in the producer cell so as to
enhance AAV
production level; wherein the sub-lethal stress comprises one or more
conditions selected
from: (I) a nutritional stress imposed by culturing the producer cell in a
medium that is
deficient for serum; (II) a temperature stress imposed by culturing the
producer cell for 3-6
days at: (i) a temperature lower than the optimum growth temperature of the
producer cell;
or (ii) a temperature higher than the optimum growth temperature of the
producer cell;
(III) an osmotic stress imposed by: (i) culturing the producer cell in a
hypoosmotic
medium; or (ii) culturing the producer cell in a hyperosmotic medium; (IV) a
pH stress,
wherein the pH stress comprises subjecting the producer cell to a pH of above
pH7.2
continuously during culture; and (V) a toxic stress, wherein the toxic stress
comprises
9
CA 2995542 2018-02-15
exposing the producer cell to a genotoxic agent selected from a chemical
carcinogen,
radiation, UV, a metabolic inhibitor of DNA synthesis and a drug that affects
topoisomerases.
In another aspect, there is provided a method of generating a population of
recombinant adeno-associated virus (rAAV) particles, comprising the steps of:
(a)
providing an AAV producer cell that is a mammalian cell and comprises: (i) one
or more
AAV packaging genes, wherein each said AAV packaging gene encodes an AAV
replication or encapsidation protein; (ii) a recombinant AAV (rAAV) pro-vector
that
comprises a heterologous non-AAV polynucleotide flanked by at least one AAV
inverted
terminal repeat (ITR); and (iii) a helper virus for AAV or a polynucleotide
sequence of
said helper virus that encodes at least one helper virus function; (b)
incubating the
producer cell provided in step (a) under conditions that are permissive for
replication of
AAV and which comprise inducing a sub-lethal stress in the AAV producer cell;
(c) lysing
the producer cell after the incubation of step (b) to produce an AAV producer
cell lysate;
and (d) purifying the AAV producer cell lysate to generate a population of
recombinant
adeno-associated virus (rAAV) particles; wherein the sub-lethal stress
comprises one or
more conditions selected from: (I) a nutritional stress imposed by culturing
the producer
cell in a medium that is deficient for serum; (II) a temperature stress
imposed by culturing
the producer cell for 3-6 days at: (i) a temperature lower than the optimum
growth
temperature of the producer cell; or (ii)a temperature higher than the optimum
growth
temperature of the producer cell; (III) an osmotic stress imposed by: (i)
culturing the
producer cell in a hypoosmotic medium; or (ii) culturing the producer cell in
a
hyperosmotic medium; (IV) a pH stress, wherein the pH stress comprises
subjecting the
producer cell to a pH of above pH7.2 continuously during culture; and (V) a
toxic stress,
wherein the toxic stress comprises exposing the producer cell to a genotoxic
agent selected
from a chemical carcinogen, radiation, UV, a metabolic inhibitor of DNA
synthesis and a
drug that affects topoisomerases.
In another aspect, there is provided a method of generating a population of
rAAV
particles comprising the steps of: (a) providing an AAV producer cell that is
a mammalian
cell and comprises: (i)one or more AAV packaging genes, wherein each said AAV
packaging gene encodes an AAV replication or encapsidation protein; (ii) a
recombinant
AAV (rAAV) pro-vector that comprises a heterologous non-AAV polynucleotide
flanked
by at least one AAV inverted terminal repeat (ITR); and (iii) a helper virus
for AAV or a
9a
CA 2995542 2018-02-15
polynucleotide sequence of said helper virus that encodes at least one helper
\jilts function:
and (b) incubating the producer cell of step (a) under conditions that are
permissive for
replication ofAAV and which comprise inducing a nutritional stress in the
producer cell so as
to enhance AAV production level; wherein said nutritional stress is imposed by
culturing the
producer cells in a medium that is deficient in one or more amino acids.
In another aspect, there is provided a method of generating a population of
recombinant adeno-associated virus (rAAV) particles, comprising the steps of:
(a) providing
an AAV producer cell that is a mammalian cell and comprises: (i) one or more
AAV
packaging genes, wherein each said AAV packaging gene encodes an AAV
replication or
encapsidation protein; (ii) a recombinant AAV (rAAV) pro-vector that comprises
a
heterologous non-AAV polynucleotide flanked by at least one AAV inverted
terminal repeat
(ITR); and (iii) a helper virus for AAV or a polynucleotide sequence of said
helper virus that
encodes at least one helper virus function; (b) incubating the producer cell
provided in step
(a) under conditions that are permissive for replication of AAV and which
comprise inducing
a nutritional stress in the AAV producer cell; (c) lysing the producer cell
after the incubation
of step (b) to produce an AAV producer cell lysate; and (d) purifying the AAV
producer cell
lysate to generate a population of recombinant adeno-associated virus (rAAV)
particles;
wherein said nutritional stress is imposed by culturing the producer cells in
a medium that is
deficient in one or more amino acids.
In another aspect, there is provided a method of generating a population of
recombinant adeno-associated virus (rAAV) particles, comprising the steps of:
(a) providing an AAV producer cell that is a mammalian cell and
comprises:
(i) one or more AAV packaging genes, wherein each said AAV packaging
gene encodes an AAV replication or encapsidation protein;
(ii) a recombinant AAV (rAAV) pro-vector that comprises a heterologous
non-AAV polynucleotide flanked by at least one AAV inverted terminal
repeat (ITR); and
(iii) a helper virus for AAV or a polynucleotide sequence of said helper
virus that encodes at least one helper virus function; and
(b) incubating the producer cell of step (a) under conditions that are
permissive for
replication of AAV and which comprise inducing a sub-lethal stress in the
producer
cell so as to enhance AAV production level;
9b
CA 2995542 2018-02-15
wherein the sub-lethal stress comprises one or more conditions selected from:
(I) a nutritional stress imposed by culturing the producer cell in a medium
that is deficient
for serum;
(II) a temperature stress imposed by culturing the producer cell for 3-6 days
at:
(i) a temperature lower than the optimum growth temperature of the producer
cell; or
(ii) a temperature higher than the optimum growth temperature of the
producer
cell; and
(III) a pH stress, wherein the pH stress comprises subjecting the producer
cell to a pH of
above pH7.2 continuously during culture.
In another aspect, there is provided a method of generating a population of
recombinant adeno-associated virus (rAAV) particles, comprising the steps of:
(a) providing an AAV producer cell that is a mammalian cell and comprises:
(i) one or more AAV packaging genes, wherein each said AAV packaging
gene encodes an AAV replication or encapsidation protein;
(ii) a recombinant AAV (rAAV) pro-vector that comprises a heterologous
non-AAV polynucleotide flanked by at least one AAV inverted terminal
repeat (ITR); and
(iii) a helper virus for AAV or a polynucleotide sequence of said helper
virus that encodes at least one helper virus function;
(b) incubating the producer cell provided in step (a) under conditions that
are
peimissive for replication of AAV and which comprise inducing a sub-lethal
stress in
the AAV producer cell;
(c) lysing the producer cell after the incubation of step (b) to produce an
AAV
producer cell lysate; and
(d) purifying the AAV producer cell lysate to generate a population of
recombinant adeno-associated virus (rAAV) particles;
wherein the sub-lethal stress comprises one or more conditions selected from:
(I) a nutritional stress imposed by culturing the producer cell in a medium
that is deficient for
serum;
(II) a temperature stress imposed by culturing the producer cell for 3-6 days
at:
9c
CA 2995542 2018-02-15
(i) a temperature lower than the optimum growth temperature of the producer
cell; or
(ii) a temperature higher than the optimum growth temperature of the
producer
cell: and
(III) a pll stress, wherein the pH stress comprises subjecting the producer
cell to a p1! of
above pI17.2 continuously during culture.
In another aspect, there is provided a high-throughput assay for determining
the
infectious titer of a preparation containing a virus that can replicate in a
mammalian cell,
comprising the steps of:
a) providing an array of culture \ -ells each comprising an aliquot of
mammalian cells and an
aliquot of the virus preparation to be titered;
b) incubating the cells and virus of step a) to allow replication of said
virus:
c) lysing said cells to produce a multiplicity of lysates containing viral
polynucleotides;
d) transferring the multiplicity of lysates from step c) to a membrane that
binds nucleic ac ids
to produce a membrane-bound array of nucleic acids; and
e) hybridizing the membrane-bound array of nucleic acids of step d) with a
viral-specific
probe and then determining the relative amount of viral nucleic acid
replicated in each of said
culture wells.
In another aspect, there is provided a high-throughput method of screening for
agents that
affect replication of a virus in a mammalian cell, comprising the steps of
a) providing an array of culture µvells each comprising an aliquot of
mammalian cells, an
aliquot of the virus and an optionally an aliqout of the agent;
b) incubating the cells, virus, and optional agent of step b) to allow
replication of said
virus:
c) ly-sing said cells to produce a multiplicity of lysates containing viral
polynucleotides;
d) transferring the multiplicity of lysates from step c) to a membrane that
binds nucleic
acids to produce a membrane-bound array of nucleic acids;
e) hybridizing the membrane-bound array of nucleic acids of step d) with a
viral- specific probe
and then determining the relative amount of viral nucleic acid replicated in
each of said culture wells.
9d
CA 2995542 2018-02-15
BRIEF DESCRIPTION OF THE FIGURES
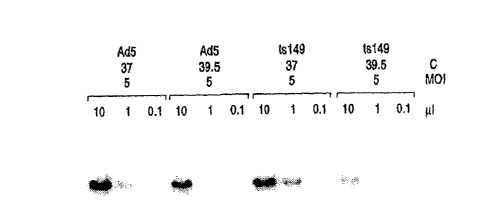
Figure 1 is a half-tone reproduction of a Southern analysis for rAAV Vector
production. using a probe for a model CF therapeutic gene contained in the
vector. The
prominent band at 1.4 kb indicates presence of rAAV in the preparation. Helper
function was
supplied by adenovirus subtype 5 (Ad5) or by the adenovirus temperature-
sensitive strain
ts149.
Figure 2 is a half-tone reproduction of a slot-blot analysis for rAAV vector
production, to quantitate the level ofrAAV present in each preparation. When
helper function
is supplied by ts149, the amount ofrAAV produced under standard culture
conditions is
several logs below that produced in the presence of AdS.
Figure 3 is a half-tone reproduction of a Southern analysis for rAAV,
indicating that
increasing the level of ts149 does not improve the level of rAAV production.
Figure 4 is a bar graph indicating a dramatic increase in the amount of rAAV
produced in the presence of ts149 (hatched bars) if culture periods are
extended beyond 5
days. This is in marked contrast to the substantial decrease in rAAV that
occurs beyond
9e
CA 2995542 2018-02-15
day 5 when non-temperature sensitive adenovirus is used to supply helper
function (solid
bars)-.
Figure 5 is a line graph showing the viable cell density (VCD) of HeLa S3
cells
grown in suspension culture at 37 C (circles) or 32 C (squares).
Figure 6 is a line graph showing the effect of tangential flow filtration at
two
different rates on HeLa S3 cells grown in suspension culture.
Figure 7 is a bar graph showing the production of tsl 49 detected in infected
HeLa
S3 cells cultured for 3-7 days in suspension at the permissive temperature of
32 C,
compared with the level detected at day 7 after microfluidization (MF).
Figure 8 is a combination graph showing the purification of ts149 by anion
exchange chromatography on PI matrix, eluted with a linear 900-1300 meq NaCI
gradient
at pH 8Ø
Figure 9 is a combination graph showing the purification of Adenovirus on PI
anion-exchange matrix, eluted with a 800-1300 meq gradient of NaCI at pH8Ø
Bars:
Viral activity measured in an infectivity assay; Solid line: A280 (a measure
of total protein);
Dotted line: buffer conductivity (ms).
Figure 10 is a combination graph showing the separation of Adenovirus and
recombinant AAV. The upper panel shows separation on PI anion-exchange matrix,
cluted
with a 0-1000 meq gradient of NaCI at pH 8Ø The lower panel shows subsequent
separation of Adenovirus from contaminants on HS cation-exchange matrix.
eluted with a
0-500 meq gradient of NaCI at pH 8Ø
Figure 11 is two bar graphs, showing the effect of fetal bovine serum levels
(FBS)
in the culture medium on rAAV production. Serum deficiency in the culture
medium is
one of a number of stress factors that the producer cells can be subjected to
in order to
enhance the production of viral particles.
Figure 12 is a half-tone reproduction of a SDS-polyacrylamide gel analysis for
AAV proteins during purification steps. The AAV preparation was subjected to
tangential
flow filtration after chromatography on an anion exchange column (POROS 50
PI). The
silver stained gel shows the highly purified AAV capsid proteins, VP I , VP2,
and VP3 in
the final bulk material.
* Trade-Mark
CA 2995542 2018-02-15
Figure 13 is a chromatogram showing concentration of AAV on a heparin sulfate
column. The sharp peak in absorbance at 280 rim (left-hand axis) at about 18
minutes
elution time represents the AAV fraction (after anion exchange and tangential
flow
filtration) as eluted from heparin sulfate with a linear gradient of 0 to 1M
NaC1
(conductivity in ms shown on right-hand axis).
DETAILED DESCRIPTION
It is an object of this invention to provide methods and materials for
generating
high titer preparations of adeno-associated virus (AAV) that are substantially
free of helper
virus, helper virus proteins, and cellular proteins and other components.
Various methods for the generation and processing of AAV particles in
mammalian
cells are described in detail below, and illustrations of the use of such
techniques are
provided in the Examples following.
By way of introduction, it is typical to employ a host or "producer" cell for
rAAV
vector replication and packaging. Such a producer cell (usually a mammalian
host cell)
generally comprises or is modified to comprise several different types of
components for
rAAV production. The first component is a recombinant adeno-associated viral
(rAAV)
vector genome (or "rAAV pro-vector") that can be replicated and packaged into
vector
particles by the host packaging cell. The rAAV pro-vector will normally
comprise a
heterologous polynucleotide (or "transgene"), with which it is desired to
genetically alter
another cell in the context of gene therapy (since the packaging of such a
transgene into
rAAV vector particles can be effectively used to deliver the transgene to a
variety of
mammalian cells). The transgene is generally flanked by two AAV inverted
terminal
repeats (ITRs) which comprise sequences that are recognized during excision,
replication
and packaging of the AAV vector, as well as during integration of the vector
into a host
cell genome. A second component is a helper virus that can provide helper
functions for
AAV replication. Although adenovirus is commonly employed, other helper
viruses can
also be used as is known in the art. Alternatively, the requisite helper virus
functions can
be isolated genetically from a helper virus and the encoding genes can be used
to provide
helper virus functions in trans. The AAV vector elements and the helper virus
(or helper
virus functions) can be introduced into the host cell either simultaneously or
sequentially
11
CA 2995542 2018-02-15
in any order. The final components for AAV production to be provided in the
producer
cell are "AAV packaging genes" such as AAV rep and cap genes that provide
replication
and encapsidation proteins, respectively. Several different versions of AAV
packaging
genes can be provided (including wild-type rep-cap cassettes as well as
modified rep
and/or cap cassettes in which the rep and/or cap genes can be left under the
control of the
native promoters or operably linked to heterologous promoters. Such AAV
packaging
genes can be introduced either transiently or stably into the host packaging
cell, as is
known in the art and described in more detail below.
After culturing the host cells under conditions that permit AAV replication
and
encapsidation, the cells and sub-cellular fractions can be processed to
generate high titer
preparations of adeno-associated virus (AAV) that are substantially free of
helper virus,
helper virus proteins, and cellular proteins. Detailed descriptions of
processing techniques
and illustrative protocols employing such techniques are provided below.
Definitions
A "vector" as used herein refers to a macromolecule or association of
macromolecules that comprises or associates with a polynucleotide and which
can be used
to mediate delivery of the polynucleotide to a cell. Illustrative vectors
include, for
example, plasmids, viral vectors, Liposomes and other gene delivery vehicles.
"AAV" is an abbreviation for adeno-associated virus, and may be used to refer
to
the virus itself or derivatives thereof. The term covers all subtypes and both
naturally
occurring and recombinant forms, except where required otherwise. The
abbreviation
"rAAV" refers to recombinant adeno-associated virus, also referred to as a
recombinant
AAV vector (or "rAAV vector").
An "rAAV vector" as used herein refers to an AAV vector comprising a
polynucleotide sequence not of AAV origin (i.e., a polynucleotide heterologous
to AAV),
typically a sequence of interest for the genetic transformation of a cell. In
preferred vector
constructs of this invention, the heterologous polynucleotide is flanked by at
least one,
preferably two AAV inverted terminal repeat sequences (ITRs). The term rAAV
vector
encompasses both rAAV vector particles and rAAV vector plasmids.
12
CA 2995542 2018-02-15
An "AAV virus" or "AAV viral particle" refers to a viral particle composed of
at
least one AAV capsid protein (preferably by all of the capsid proteins of a
wild-type AAV)
and an encapsidated polynucleotide. If the particle comprises a heterologons
polynucleotide (i.e. a polynucleotide other than a wild-type AAV genome such
as a
transgene to be delivered to a mammalian cell), it is typically referred to as
an "rAAV
vector particle" or simply an "rAAV vector".
"Packaging" refers to a series of intracellular events that result in the
assembly and
encapsidation of an AAV particle.
AAV "rep" and "cap" genes refer to polynucleotide sequences encoding
replication
and encapsidation proteins of adeno-associated virus. They have been found in
all AAV
serotypes examined, and are described below and in the art. AAV rep and cap
are referred
to herein as AAV "packaging genes-.
A "helper virus" for AAV refers to a virus that allows AAV (e.g. wild-type
AAV)
to be replicated and packaged by a mammalian cell. A variety of such helper
viruses for
AAV are known in the art. including adenoviruses, herpesviruses and poxviruses
such as
vaccinia. The adenoviruses encompass a number of different subgroups, although
Adenovirus type 5 of subgroup C is most commonly used. Numerous adenoviruses
of
human, non-human mammalian and avian origin are known and available from
depositories such as the ATCC. Viruses of the herpes family include, for
example. herpes
simplex viruses (HSV) and Epstein-Barr viruses (EBV), as well as
cytomegaloviruses
(CMV) and pseudorabies viruses (PRV). which are also available from
depositories such
as ATCC.
The term "tsHV" refers to a temperature-sensitive helper virus, which can
provide
helper functions for AAV replication and packaging but is temperature-
sensitive with
respect to its own replication (i.e. it can replicate at a "permissive"
temperature but
replicates at lower efficiency, or preferably not at all, at a "non-
permissive" temperature).
The ability of the tsHV to provide help for AAV replication may also be
temperature
sensitive, but preferred tsHV for use with this invention efficiently support
AAV
replication at temperatures at which AAV can replicate but which are non-
permissive for
replication of the tsHV. Examples of such tsHV are described below.
13
CA 2995542 2018-02-15
An "infectious" virus or viral particle is one that comprises a polynucleotide
component which it is capable of delivering into a cell for which the viral
species is
trophic. The term does not necessarily imply any replication capacity of the
virus. Assays
for counting infectious viral particles are described elsewhere in this
disclosure and in the
art.
A "replication-competent" virus (e.g. a replication-competent AAV, sometimes
abbreviated as "RCA") refers to a phenotypically wild-type virus that is
infectious, and is
also capable of being replicated in an infected cell (i.e. in the presence of
a helper virus or
helper virus functions). In the case of AAV, replication competence generally
requires the
presence of functional AAV packaging genes. Preferred rAAV vectors as
described herein
are replication-incompetent in mammalian cells (especially in human cells) by
virtue of the
lack of one or more AAV packaging genes. Preferably, such rAAV vectors lack
any AAV
packaging gene sequences in order to minimize the possibility that RCA are
generated by
recombination between AAV packaging genes and an incoming rAAV vector.
Preferred
rAAV vector preparations as described herein are those which contain few if
any RCA
(preferably less than about 1 RCA per 102 rAAV particles, more preferably less
than about
1 RCA per 104 rAAV particles, still more preferably less than about 1 RCA per
10B rAAV
particles, even more preferably less than about 1 RCA per 10 rAAV particles,
most
preferably no RCA).
The term "polynucleotide" refers to a polymeric form of nucleotides of any
length,
including deoxyribonucleotides or ribonucleotides, or analogs thereof. A
polynucleotide
may comprise modified nucleotides, such as methylated nucleotides and
nucleotide
analogs, and may be interrupted by non-nucleotide components. If present,
modifications
to the nucleotide structure may be imparted before or after assembly of the
polymer. The
term polynucleotide, as used herein, refers interchangeably to double- and
single-stranded
molecules. Unless otherwise specified or required, any embodiment of the
invention
described herein that is a polynucleotide encompasses both the double-stranded
form and
each of two complementary single-stranded forms known or predicted to make up
the
double-stranded form,
A "gene" refers to a polynucleotide containing at least one open reading frame
that
is capable of encoding a particular protein after being transcribed and
translated.
14
CA 2995542 2018-02-15
"Recombinant", as applied to a polynucleotide means that the polynucleotide is
the
product of various combinations of cloning, restriction or ligation steps, and
other
procedures that result in a construct that is distinct from a polynucleotide
found in nature.
A recombinant virus is a viral particle comprising a recombinant
polynucleotide. The
terms respectively include replicates of the original polynucleotide construct
and progeny
of the original virus construct.
A "control element" or "control sequence" is a nucleotide sequence involved in
an
interaction of molecules that contributes to the functional regulation of a
polynucleotide,
including replication, duplication, transcription, splicing, translation, or
degradation of the
polynucleotide. The regulation may affect the frequency, speed, or specificity
of the
process, and may be enhancing or inhibitory in nature. Control elements known
in the art
include, for example, transcriptional regulatory sequences such as promoters
and
enhancers. A promoter is a DNA region capable under certain conditions of
binding RNA
polymerase and initiating transcription of a coding region usually located
downstream (in
the 3' direction) from the promoter.
"Operatively linked" or "operably linked" refers to a juxtaposition of genetic
elements, wherein the elements are in a relationship permitting them to
operate in the
expected manner. For instance, a promoter is operatively linked to a coding
region if the
promoter helps initiate transcription of the coding sequence. There may be
intervening
residues between the promoter and coding region so long as this functional
relationship is
maintained.
An "expression vector" is a vector comprising a region which encodes a
polypeptide of interest, and is used for effecting the expression of the
protein in an
intended target cell. An expression vector also comprises control elements
operatively
linked to the encoding region to facilitate expression of the protein in the
target. The
combination of control elements and a gene or genes to which they are operably
linked for
expression is sometimes referred to as an "expression cassette," a large
number of which
are known and available in the art or can be readily constructed from
components that are
available in the art.
"Heterologous" means derived from a genotypically distinct entity from that of
the
rest of the entity to which it is being compared. For example, a
polynucleotide introduced
CA 2995542 2018-02-15
by genetic engineering techniques into a plasmid or vector derived from a
different species
is a heterologous polynucleotide. A promoter removed from its native coding
sequence
and operatively linked to a coding sequence with which it is not naturally
found linked is a
heterologous promoter.
"Genetic alteration" refers to a process wherein a genetic element is
introduced into
a cell other than by mitosis or meiosis. The element may be heterologous to
the cell, or it
may be an additional copy or improved version of an element already present in
the cell.
Genetic alteration may be effected, for example, by transfecting a cell with a
recombinant
plasmid or other polynucleotide through any process known in the art, such as
electroporation, calcium phosphate precipitation, or contacting with a
polynucleotide-
liposome complex. Genetic alteration may also be effected, for example, by
transduction
or infection with a DNA or RNA virus or viral vector. Preferably, the genetic
element is
introduced into a chromosome or mini-chromosome in the cell; but any
alteration that
changes the phenotype and/or genotype of the cell and its progeny is included
in this term.
A cell is said to be "stably" altered, transduced, or transformed with a
genetic
sequence if the sequence is available to perform its function during extended
culture of the
cell in vitro. In preferred examples, such a cell is "inheritably" altered in
that a genetic
alteration is introduced which is also inheritable by progeny of the altered
cell.
The terms "polypeptide", "peptide" and "protein" are used interchangeably
herein
to refer to polymers of amino acids of any length. The terms also encompass an
amino
acid polymer that has been modified: for example, disulfide bond formation,
glycosylation.
lipidation, or conjugation with a labeling component.
Polypeptides such as "CFTR", "p53", "E I A" and the like, when discussed in
the
context of gene therapy and compositions therefor, refer to the respective
intact
polypeptide, or any fragment or genetically engineered derivative thereof,
that retains the
desired biochemical function of the intact protein. Similarly, references to
CFTR, p53,
El A genes, and other such genes for use in gene therapy (typically referred
to as
"transgenes" to be delivered to a recipient cell), include polynucleotides
encoding the
intact polypeptide or any fragment or genetically engineered derivative
possessing the
desired biochemical function.
16
CA 2995542 2018-02-15
An "isolated" plasmid, virus, or other substance refers to a preparation of
the
substance devoid of at least some of the other components that may also be
present where
the substance or a similar substance naturally occurs or is initially prepared
from. Thus,
for example, an isolated substance may be prepared by using a purification
technique to
enrich it from a source mixture. Enrichment can be measured on an absolute
basis, such as
weight per volume of solution, or it can be measured in relation to a second,
potentially
interfering substance present in the source mixture. Increasing enrichments of
the
embodiments of this invention are increasingly more preferred. Thus, for
example, a
2-fold enrichment is preferred, 10-fold enrichment is more preferred, 100-fold
enrichment
is more preferred, 1000-fold enrichment is even more preferred.
A preparation of AAV is said to be "substantially free" of helper virus if the
ratio of
infectious AAV particles to infectious helper virus particles is at least
about 102:1;
preferably at least about 104:1. more preferably at least about I0':1; still
more preferably at
least about 108:1. Preparations are also preferably free of equivalent amounts
of helper
virus proteins (i.e, proteins as would be present as a result of such a level
of helper virus if
the helper virus particle impurities noted above were present in disrupted
form). Viral
and/or cellular protein contamination can generally be observed as the
presence of
Coomassie staining bands on SDS gels (e.g. the appearance of bands other than
those
corresponding to the AAV capsid proteins VPI, VP2 and VP3).
"Efficiency" when used in describing viral production, replication or
packaging
refers to useful properties of the method: in particular, the growth rate and
the number of
virus particles produced per cell. "High efficiency" production indicates
production of at
least 100 viral particles per cell: preferably at least about 10,000 and more
preferably at
least about 100,000 particles per cell, over the course of the culture period
specified.
An "individual" or "subject" treated in accordance with this invention refers
to
vertebrates, particularly members of a mammalian species, and includes but is
not limited
to domestic animals, sports animals, and primates, including humans.
"Treatment" of an individual or a cell is any type of intervention in an
attempt to
alter the natural course of the individual or cell at the time the treatment
is initiated. For
example, treatment of an individual may be undertaken to decrease or limit the
pathology
caused by any pathological condition, including (but not limited to) an
inherited or induced
17
CA 2995542 2018-02-15
genetic deficiency, infection by a viral, bacterial, or parasitic organism, a
neoplastic or
aplastic condition, or an immune system dysfunction such as autoimmunity or
immunosuppression. Treatment includes (but is not limited to) administration
of a
composition, such as a pharmaceutical composition, and administration of
compatible cells
that have been treated with a composition. Treatment may be performed either
prophylactically or therapeutically; that is, either prior or subsequent to
the initiation of a
pathologic event or contact with an etiologic agent.
General techniques
The practice of the present invention will employ, unless otherwise indicated,
conventional techniques of molecular biology, virology, animal cell culture
and
biochemistry which are within the skill of the art Such techniques are
explained fully in
the literature. See, for example, "Molecular Cloning: A Laboratory Manual",
Second
Edition (Sambrook, Fritsch & Maniatis. 1989); "Animal Cell Culture" (R.I.
Freshney, ed.,
1987); "Gene Transfer Vectors for Mammalian Cells" (J.M. Miller & M.P. Gatos,
eds.,
1987); "Current Protocols in Molecular Biology" (RM. Ausubel et al., eds.,
1987);
"Current Protocols in Protein Science" (John E Coligan, et al. eds. Wiley and
Sons, 1995);
and "Protein Purification: Principles and Practice" (Robert K. Scopes,
Springer-Verlag,
1994).
Selection and Preparation of AAV Vector and AAV Packaging Genes
A recombinant AAV vector of this invention comprises a heterologous (i.e. non-
AAV) polynucleotide of interest in place of the AAV rep and/or cap genes that
normally
make up the bulk of the AAV genome. As in the wild-type AAV genome, however,
the
rAAV pro-vector is preferably flanked by two AAV inverted terminal repeats
(ITRs) as
noted above. Variations in which an rAAV construct is flanked by a only a
single
(typically modified) ITR have also been described in the art and can be
employed in
connection with the present invention.
18
CA 2995542 2018-02-15
Adeno-associated viruses of any serotype are suitable, since the various
serotypes
are functionally and structurally related, even at the genetic level (see,
e.g., Blacklow, pp.
165-174 of "Parvoviruses and Human Disease" J.R. Pattison, ed. (1988); and
Rose,
Comprehensive Virology 3:1, 1974). All AAV serotypes apparently exhibit
similar
replication properties mediated by homologous rep genes; and all generally
bear three
related capsid proteins such as those expressed in AAV2. The degree of
relatedness is
further suggested by heteroduplex analysis which reveals extensive cross-
hybridization
between serotypes along the length of the genome; and the presence of
analogous self-
annealing segments at the termini that correspond to ITRs. The similar
infectivity patterns
also suggest that the replication functions in each serotype are under similar
regulatory
control. Among the various AAV serotypes, AAV2 is most commonly employed.
An AAV vector of this invention will typically comprise a polynucleotide that
is
heterologous to the AAV. The polynucleotide is typically of interest because
of a capacity to
provide a function to a target cell in the context of gene therapy, such as up-
or down-
regulation of the expression of a certain phenotype. Such a heterologous
polynucleotide or
"transgene", will generally be of sufficient length to provide the desired
function or encoding
sequence. For encapisdation within AAV2 particles, the transgene will
preferably be less
than about 5kb although other serotypes and/or modifications may be employed
to allow
larger sequences to packaged into the AAV viral particles.
Where transcription of the heterologous polynucleotide is desired in the
intended
target cell, it can be operably linked to its own or to a heterologous
promoter, depending for
example on the desired level and/or specificity of transcription within the
target cell, as is
known in the art. Various types of promoters and enhancers are suitable for
use in this
context. Constitutive promoters provide an ongoing level of gene
transcription, and are
preferred when it is desired that the therapeutic polynucleotide be expressed
on an ongoing
basis. Inducible promoters generally exhibit low activity in the absence of
the inducer, and
are up-regulated in the presence of the inducer. They may be preferred when
expression is
desired only at certain times or at certain locations, or when it is desirable
to titrate the level
of expression using an inducing agent. Promoters and enhancers may also be
tissue-specific:
that is, they exhibit their activity only in certain cell types, presumably
due to gene regulatory
elements found uniquely in those cells.
19
CA 2995542 2018-02-15
Illustrative examples of promoters are the SV40 late promoter from simian
virus 40,
the Baculovirus polyhedron enhancer/promoter element, Herpes Simplex Virus
thymidine
kinase (HSV tk), the immediate early promoter from cytomegalovirus (CMV) and
various
retroviral promoters including LTR elements. Inducible promoters include heavy
metal ion
inducible promoters (such as the mouse mammary tumor virus (mMTV) promoter or
various
growth hormone promoters), and the promoters from T7 phage which are active in
the
presence of T7 RNA polymerase. By way of illustration, examples of tissue-
specific
promoters include various surfactin promoters (for expression in the lung),
myosin promoters
(for expression in muscle), and albumin promoters (for expression in the
liver). A large
variety of other promoters are known and generally available in the art, and
the sequences for
many such promoters are available in sequence databases such as the GenBank.
database.
Where translation is also desired in the intended target cell, the
heterologous
polynucleotide will preferably also comprise control elements that facilitate
translation (such
as a ribosome binding site or "RBS" and a polyadenylation signal).
Accordingly, the
heterologous polynucleotide will generally comprise at least one coding region
operatively
linked to a suitable promoter, and may also comprise, for example, an
operatively linked
enhancer, ribosome binding site and poly-A signal. The heterologous
polynucleotide may
comprise one encoding region, or more than one encoding regions under the
control of the
same or different promoters. The entire unit, containing a combination of
control elements
and encoding region, is often referred to as an expression cassette.
The heterologous polynucleotide is integrated by recombinant techniques into
or
preferably in place of the AAV genomic coding region (i.e. in place of the AAV
rep and
cap genes), but is generally flanked on either side by AAV inverted terminal
repeat (ITR)
regions. This means that an ITR appears both upstream and downstream from the
coding
sequence, either in direct juxtaposition, preferably (although not
necessarily) without any
intervening sequence of AAV origin in order to reduce the likelihood of
recombination that
might regenerate a replication-competent AAV genome. Recent evidence suggests
that a
single ITR can be sufficient to carry out the functions normally associated
with
configurations comprising two ITRs (WO 94/13788), and vector constructs with
only one
ITR can thus be employed in conjunction with the packaging and production
methods of
the present invention.
CA 2995542 2018-02-15
The native promoters for rep are self-regulating, and can limit the amount of
AAV
particles produced. The rep gene can also be operably linked to a heterologous
promoter,
whether rep is provided as part of the vector construct, or separately. Any
heterologous
promoter that is not strongly down-regulated by rep gene expression is
suitable; but
inducible promoters are preferred because constitutive expression of the rep
gene can have
a negative impact on the host cell. A large variety of inducible promoters are
known in the
art; including, by way of illustration, heavy metal ion inducible promoters
(such as
metallothionein promoters); steroid hormone inducible promoters (such as the
MMTV
promoter or growth hormone promoters); and promoters such as those from T7
phage
which are active in the presence of T7 RNA polymerase. An especially preferred
sub-class
of inducible promoters are those that are induced by the helper virus that is
used to
complement the replication and packaging of the rAAV vector, A number of
helper-virus-
inducible promoters have also been described, including the adenovirus early
gene
promoter which is inducible by adenovirus El A protein; the adenovirus major
late
promoter; the herpesvirus promoter which is inducible by herpesvirus proteins
such as
VP16 or 1CP4; as well as vaccinia or poxvirus inducible promoters.
Methods for identifying and testing helper-virus-inducible promoters have been
described in a commonly-owned copending application published as W096/17947 by
Targeted Genetics Corporation (Allen et al.). Thus. methods are known in the
art to
determine whether or not candidate promoters are helper-virus-inducible, and
whether or
not they will be useful in the generation of high efficiency packaging cells.
Briefly, one
such method involves replacing the p5 promoter of the AAV rep gene with the
putative
helper-virus-inducible promoter (either known in the art or identified using
well-known
techniques such as linkage to promoter-less "reporter" genes). The AAV rep-cap
genes
(with p5 replaced), preferably linked to a positive selectable marker such as
an antibiotic
resistance gene, are then stably integrated into a suitable host cell (such as
the I-IeLa or
A549 cells exemplified below). Cells that are able to grow relatively well
under selection
conditions (e.g. in the presence of the antibiotic) are then tested for their
ability to express
the rep and cap genes upon addition of a helper virus. As an initial test for
rep and/or cap
expression, cells can be readily screened using immunofluorescence to detect
Rep and/or
Cap proteins. Confirmation of packaging capabilities and efficiencies can then
be
21
CA 2995542 2018-02-15
determined by functional tests for replication and packaging of incoming rAAV
vectors.
Using this methodology, a helper-virus-inducible promoter derived from the
mouse
metallothionein gene has been identified as a suitable replacement for the p5
promoter, and
used for producing high titers of rAAV particles (as described in WO 96/17947,
Targeted
Genetics Corporation).
Given the relative encapsidation size limits of various AAV genomes, insertion
of a
large heterologous polynucleotide into the genome necessitates removal of a
portion of the
AAV sequence. Removal of one or more AAV genes is in any case desirable, to
reduce
the likelihood of generating replication-competent AAV ("RCA"). Accordingly,
encoding
or promoter sequences for rep, cap, or both, are preferably removed, since the
functions
provided by these genes can be provided in trans.
The resultant vector is referred to as being "'defective" in these functions.
In order
to replicate and package the vector, the missing functions are complemented
with a
packaging gene, or a plurality thereof, which together encode the necessary
functions for
the various missing rep and/or cap gene products. The packaging genes or gene
cassettes
are preferably not flanked by AAV ITRs and preferably do not share any
substantial
homology with the rAAV genome. Thus, in order to minimize homologous
recombination
during replication between the vector sequence and separately provided
packaging genes, it
is desirable to avoid overlap of the two polynucleotide sequences. The level
of homology
and corresponding frequency of recombination increase with increasing length
of the
homologous sequences and with their level of shared identity. The level of
homology that
will pose a concern in a given system can be determined theoretically and
confirmed
experimentally, as is known in the art. Typically, however, recombination can
be
substantially reduced or eliminated if the overlapping sequence is less than
about a 25
nucleotide sequence if it is at least 80% identical over its entire length, or
less than about a
50 nucleotide sequence if it is at least 70% identical over its entire length.
Of course, even
lower levels of homology are preferable since they will further reduce the
likelihood of
recombination. It appears that, even without any overlapping homology, there
is some
residual frequency of generating RCA. Even further reductions in the frequency
of
generating RCA (e.g. by nonhomologous recombination) can be obtained by
"splitting" the
replication and encapsidation functions of AAV, as described by Allen et al.
in U.S. patent
22
CA 2995542 2018-02-15
application 08/769,728, filed 18 Dec. 1996, published internationally as
W098/27204 on
25 June 1998 (Targeted Genetics Corporation)).
The rAAV vector construct, and the complementary packaging gene constructs can
be implemented in this invention in a number of different forms. Viral
particles, plasmids,
and stably transformed host cells can all be used to introduce such constructs
into the
packaging cell, either transiently or stably.
In certain embodiments of this invention, the AAV vector and complementary
packaging gene(s), if any, are provided in the form of bacterial plasmids, AAV
particles, or
any combination thereof. In other embodiments, either the AAV vector sequence.
the
packaging gene(s), or both, are provided in the form of genetically altered
(preferably
inheritably altered) eukaryotic cells. The development of host cells
inheritably altered to
express the AAV vector sequence, AAV packaging genes, or both, provides an
established
source of the material that is expressed at a reliable level.
A variety of different genetically altered cells can thus be used in the
context of this
invention. By way of illustration, a mammalian host cell may be used with at
least one
intact copy of a stably integrated rAAV vector. An AAV packaging plasmid
comprising at
least an AAV rep gene operably linked to a promoter can be used to supply
replication
functions (as described in a co-owned application by Flotte et al., now U.S.
Patent
5,658,776). Alternatively, a stable mammalian cell line with an AAV rep gene
operably
linked to a promoter can be used to supply replication functions (see, e.g.,
Trempe et al.,
(USSN 08(362,608, 9 Jan. 1995, W095/13392, 18 May 1995); Burstein et al.,
(USSN
08/770,122, filed 18 Dec. 1996, W098/23018, 25 June 1998); and Johnson et al.,
(USSN
08/254,358, filed 6 June 1994, issued as U.S. No. 5,656,785, 19 Aug. 1997)).
The AAV
cap gene, providing the encapsidation proteins as described above, can be
provided
together with an AAV rep gene or separately (see, e.g., the above-referenced
applications
and patents as well as Allen et al., USSN 08/769,728, filed 18 Dec. 1996,
W098/27204 on
25 June 1998 (Targeted Genetics Corporation)). Other combinations are possible
and
included within the scope of this invention.
23
CA 2995542 2018-02-15
Introduction of Genetic Material Into Cells
As is described in the art, and illustrated both herein and in the references
cited
above, genetic material can be introduced into cells (such as mammalian
"producer" cells
for the production of AAV) using any of a variety of means to transform or
transduce such
cells. By way of illustration, such techniques include for example
transfection with
bacterial plasmids, infection with viral vectors, electroporation, calcium
phosphate
precipitation, and introduction using any of a variety of lipid-based
compositions (a
process often referred to as "lipofection"). Methods and compositions for
performing
these techniques have been described in the art and are widely available.
Selection of suitably altered cells may be conducted by any technique in the
art.
For example, the polynucleotide sequences used to alter the cell may be
introduced
simultaneously with or operably linked to one or more detectable or selectable
markers as
is known in the art. By way of illustration, one can employ a drug resistance
gene as a
selectable marker. Drug resistant cells can then be picked and grown, and then
tested for
expression of the desired sequence - i.e., a packaging gene product, or a
product of the
heterologous polynucleotide, as appropriate. Testing for acquisition,
localization and/or
maintenance of an introduced polynucleotide can be performed using DNA
hybridization-
based techniques (such as Southern blotting and other procedures as known in
the art).
Testing for expression can be readily performed by Northern analysis of RNA
extracted
from the genetically altered cells, or by indirect immunofluorescence for the
corresponding
gene product. Testing and confirmation of packaging capabilities and
efficiencies can be
obtained by introducing to the cell the remaining functional components of AAV
and a
helper virus, to test for production of AAV particles. Where a cell is
inheritably altered
with a plurality of polynucleotide constructs, it is generally more convenient
(though not
essential) to introduce them to the cell separately, and validate each step
seriatim.
References describing such techniques include those cited herein.
Selection and Preparation of Helper Virus
As discussed above, AAV is a parvovirus that is defective for self-
replication, and
must generally rely on a helper virus to supply certain replicative functions.
A number of
such helper viruses have been identified, including adenoviruses, herpes
viruses (including
24
CA 2995542 2018-02-15
but not limited to HSV I , cytomegalovirus and HHV-6), and pox viruses
(particularly
vaccinia). Any such virus may be used with this invention.
Frequently, the helper virus will be an adenovirus of a type and subgroup that
can
infect the intended host cell, Human adenovirus of subgroup C, particularly
serotypes 1, 2,
4, 6, and 7, are commonly used. Serotype 5 is generally preferred.
The features and growth patterns of adenovirus are known in the art. The
reader
may refer, for example, to Horowitz. "Adenoviridae and their replication", pp
771-816 in
"Fundamental Virology", Fields et al., eds. The packaged adenovirus genome is
a linear
DNA molecule, linked through adenovirus ITRs at the left- and right-hand
termini through
a terminal protein complex to form a circle. Control and encoding regions for
early,
intermediate, and late components overlap within the genome. Early region
genes are
implicated in replication of the adenovirus genome, and are grouped depending
on their
location into the El, E2, E3, and E4 regions.
Although not essential, in principle it is desirable that the helper virus
strain be
defective for replication in the subject ultimately to receive the genetic
therapy. Thus, any
residual helper virus present in an rAAV preparation will be replication-
incompetent.
Adenoviruses from which the ElA or both the El A and the E3 region have been
removed
are not infectious for most human cells. They can be replicated in a
permissive cell line
(e.g. the human 293 cell line) which is capable of complementing the missing
activity.
Regions of adenovirus that appear to be associated with helper function, as
well as regions
that do not, have been identified and described in the art (see, e.g., P.
Colosi et al.,
W097/17458;
Use of a Conditionally-Sensitive Helper Virus
As described herein, a "conditionally-sensitive" helper virus can also be
employed
to provide helper:virus activity. Such a helper virus strain must minimally
have the
property of being able to support AAV replication in a host cell under at
least one set of
conditions where it itself does not undergo efficient genomic replication.
Where helper
virus activity is supplied as intact virus particles, it is also generally
necessary that the
virus be capable of replication in a host cell under a second set of
conditions. The first set
of conditions will differ from the second set of conditions by a readily
controllable feature,
CA 2995542 2018-02-15
such as the presence or absence of a required cofactor (such as a cation), the
presence or
absence of an inhibitory drug, or a shift in an environmental condition such
as temperature.
Most conveniently, the difference between the two conditions is temperature,
and such a
conditionally-sensitive virus is thus referred to as a temperature-sensitive
helper virus
(tsHV).
For the purposes of this disclosure, a "temperature-sensitive" or "ts" helper
virus is
one which is capable of replicating its genetic material in a eukaryotic cell
at a certain
temperature range (the "permissive" temperature range), typically about 15 -35
C and
preferably about 20-32 C. However, at the "non-permissive" temperature, even
when
other conditions are kept the same, the rate of replication of genetic
material is
substantially lower, at least 10-fold lower; usually at least about 100-fold
lower; and
preferably at least about 1000-fold lower. This temperature is typically about
35 -50 C,
generally about 42 C. In a typical example of such a ts helper virus, the
virus is capable of
efficient replication at relatively low temperatures such as temperatures of
about 20-32 C,
but is incapable of efficient replication at relatively high temperatures such
as temperatures
of about 37-42 C. It is understood that the virus-infected cell may
nonetheless exhibit
some metabolic processes attributable to the virus at the non-permissive
temperature,
including but not limited to helper function for AAV production.
A temperature-sensitive helper virus can be produced in bulk quantities by
culturing infected cells at a permissive temperature. AAV vector can then be
produced by
culturing cells comprising vector elements and the temperature-sensitive
helper virus at a
non-permissive temperature. The vector preparation will be substantially free
of helper
virus components.
A large number of temperature-sensitive adenovirus variants have been
described
in the art; see, e.g., the variants described by Ensinger et al. (J. Virol.
10:328, 1972);
Williams et al. (J. Gen Viral. 11:95, 1971); Ishibashi (Proc. Natl. Acad. Sci.
USA 65:304,
1970); Lundholm et al. (Virology 45:827, 1971); and Shiroki et al., (Virology
61:474,
1974); amongst others. Complementation analysis indicates that such variants
fall into a
plurality of different complementation groups (Ginsberg et al., Cold Spring
Harbor Symp.
Quant. Biol. 34:419, 1974). This suggests that a number of steps in the
adenovirus
replicative cycle may be rendered temperature-sensitive.
26
CA 2995542 2018-02-15
Since helper function for AAV replication requires that only part of the
adenovirus
cycle be intact, testing for helper function of various mutants at the non-
permissive
temperature provides a means for mapping the helper function. For example,
Ishibashi et
al. (Virology 45:317, 1971) reported that temperature-sensitive avian
adenovirus variants
support replication of AAV I and AAV2. Ito et al. reported that temperature-
sensitive
mutant ts13 of human adenovirus 7 (Ad7ts13) helps AAV replication at the non-
permissive temperature as efficiently as the wild strain. Drake et al.
(Virology 60: 230.
1974) reported complementation of AAV4 antigen synthesis by 3 groups of
temperature-
sensitive mutants of herpes simplex virus type 1 (HSV I). Handa et al. (J.
Gen. Viro.
29:239, 1975) reported helper activity for AAV1 virus production by human
adenovirus
mutants Ad5ts36, Ad5ts125, Ad5ts149, Adl2tsA275, Adl 2tsB221, and Adl2tsC295.
strove et al. (Virology 104:502, 1980) reported that temperature sensitive
mutants
Ad5ts125, Ad5ts135, Ad5ts157, Ad5ts116, and Ad5ts142, and the host range
mutants hr6
but not hr3 support AAV replication. Mayor et al. (J. Gen Virol. 35:545, 1977)
reported
that Ad3 1 ts13 but not Ad3lts94 supported AAV1 production at the non-
permissive
temperature.
Straus et al. (Proc. Natl. Acad. Sci. USA 73:742, 1976) reported that Ad5ts125
supported AAV2 replication under conditions where the adenovirus did not
itself replicate.
They used this property to study DNA intermediates formed during AAV
replication.
Myers et al. (J. Virol. 35:65, 1980) performed a quantitative study on helper
function, and
showed that Ad5ts149 supported the production of 20,000 infectious AAV
particles per
cell at the non-permissive temperature, whereas Ad5ts107 produced only ¨100
particles
per cell. Since Ad5ts107 has a mutation in the 72 kDa DNA binding protein
encoding
region, they concluded that this protein played a role in the AAV RNA
expression. More
recently, Carter et al. (Virology 191:473, 1992) proposed that a fully
functional 72 kDa
protein is required for quantitative post-transcriptional expression of the
AAV rep and cap
genes.
As outlined in the background section, the existence of temperature-sensitive
adenovirus has been known for quite some time. However, there has been no
effective
teaching or suggestion regarding the actual use of conditional helper viruses
in the
generation of recombinant AAV vectors, such as those that might be used for
gene therapy.
27
CA 2995542 2018-02-15
Part of the explanation may be the difficulty in obtaining workable titers of
AAV
when using recombinant vectors. Among other things, AAV Rep proteins
apparently
down-regulate their own expression through the p5 promoter (Tratschin et al.,
Mol. Cell
Biol. 6:2884, 1986). In addition, it has been observed that the expression of
the rep gene
in packaging cell lines such as those that might be used for the production of
recombinant
AAV vector, tends to inhibit the growth and/or metabolism of the cell (see,
e.g., Targeted
Genetics Corporation, W096/17947, by Allen et al.).
The differences between the generation of wild-type AAV and recombinant AAV
vectors tend to be quite dramatic when considered in terms of production. In
particular, it
has been observed that production of recombinant AAV vectors tends to be
substantially
lower that production of wild-type AAV particles, and that the presence or
generation of
even small amounts of contaminating wild-type AAV tends to result in a
preferential
production of wild-type virus that can eventually outnumber the recombinant
AAV
vectors.
These phenomena are further illustrated by the results described in Examples 1
and
2 of this disclosure, and in Figure 1. The adenovirus temperature-sensitive
mutant ts149 is
reported elsewhere to support AAV particle replication (Myers et al., J.
Virol. 35:65,
1980). However, Example 2 shows that when this mutant is used to support the
production
of an AAV vector with a heterologous promoter under standard conditions, the
level of
production is several orders of magnitude lower than is supported by wild-type
adeno virus.
This disclosure shows that temperature-sensitive helper virus can indeed be
used to
prepare recombinant AAV vectors at workable titers, overcoming the apparent
production
obstacles. The descriptions that follow illustrate how to select a temperature-
sensitive
helper virus and optimize conditions to provide sufficient AAV for the
purposes of gene
therapy.
In particular, it is shown that extending the replication period for AAV when
using
tsAd as helper dramatically increases the amount of AAV vector that is
produced
(Example 3). This is counter-intuitive, because extending the replication
period when
using wild-type Ad in the same way decreases the amount of AAV vector by at
least an
order of magnitude. A practitioner of skill in the art seeking to optimize
conditions for
28
CA 2995542 2018-02-15
AAV production would logically go to shorter culture times and higher
concentrations of
helper virus; both of which are shown herein to be ineffective.
This invention further provides improved culture and separation methods for
preparing quantitative amounts of temperature-sensitive adenovirus. While not
strictly
required for the practice of certain embodiments of this invention,
preparations of
temperature-sensitive adenovirus obtained by these methods are particularly
suited for
production of AAV, inter alia, for the purposes of gene therapy.
Condition-sensitive variants of the selected strain of helper virus may be
generated
by an appropriate mutagenization and selection strategy. For example, virus
may be
mutagenized with nitrosoguanidine, nitrous acid, hydroxylamine, or 5-bromo-2-
deoxyuridine. Candidates are selected that can multiply in a suitable
eukaryotic cell under
the desired permissive conditions. but not under the desired non-permissive
conditions. As
an illustration, adenovirus temperature-sensitive mutants can be obtained that
multiply,
e.g., at 32 C, but not at 39.5 C. Plaguing efficiency ratios at 39.5 C versus
32 C are
preferably less than 104 and more preferably less than 10. Further
illustration of suitable
selection processes for temperature-sensitive adenovirus can be found, for
example, in
Ensinger et al., J. Virol. 10:328, 1972; and Williams et al., J. Gen Virol.
11:95, 1971.
Description of adenovirus variants which are not temperature-sensitive, but
host-range
sensitive, can be found in Harrison et al., Virology 77:319, 1977. Temperature-
sensitive
mutants effective for use in this invention can be prepared, for example, from
alternative
helper viruses like herpes simplex 1 (HSV1), or herpes simplex 2 (HSV2). See,
e.g.,
Schaffer et al., Virology 52:57, 1973 for HSV1; Esparza et al., Virology
57:554, 1974 for
HSV2. As indicated in the background section, a large number of condition-
sensitive
helper viruses have been described, and can be obtained from the scientists
who developed
or described them or from a public depository.
Not all condition-sensitive variants of the aforelisted viruses will work with
the
present invention. In particular, the strain must be rendered condition-
sensitive at a stage
in its replicative cycle such that the function that is blocked under non-
permissive
conditions is not one that is required for high-efficiency replication of AAV.
The choice of
which helper virus strain to use can be made by reference to both the known
biology of the
helper virus and the replicative requirements of AAV.
29
CA 2995542 2018-02-15
An exemplary helper virus for use with this invention is the temperature-
sensitive
adenovirus ts149 of the Ad5 serotype (Ad5ts149). As shown in the example
section, under
optimized conditions, this strain can be used to produce rAAV at levels that
match or
exceed those supported by wild-type Ad5. The ts149 has a single transition of
C-G to A-T
at position 7563 (Roovers etal., Virus Genes 4:53, 1990). This results in a
change of
amino acid leucine at residue 411 of the DNA polymerase to phenylalanine. The
DNA
polymerase is contained within the E2 transcription unit of adenovirus.
However, other ts
mutants mapping to this region are less suitable. In particular, the E2
transcription unit
also comprises the encoding region for the 72 kDa DNA binding protein (DBP). A
strain
that produces no detectable DBP (Add/802) supports AAV replication, but at a
level that is
reduced by an order of magnitude (Carter et al., Virology 191:473, 1992).
Adts125, which
also comprises a mutation mapping to the DBP encoding region, support AAV
replication
(Straus et al., J. Virol. 17:140, 1976), although the levels are generally
much lower than
with wild-type Ad5 (Myers et al., J. Virol. 35:65, 1980). Accordingly,
suitable
temperature-sensitive adenovirus vectors for use in this invention include
those for which
the sensitivity maps to the E2A region of the genome, preferably to the DNA
polymerase
encoding region.
The artisan can readily determine which viral strains are suitable for use as
helper
virus by conducting an rAAV replication assay using a panel of candidate
helper virus
strains in a candidate cell under conditions that are non-permissive for self-
replication of
the helper. For temperature-sensitive variants, screening is done at the non-
permissive
temperature according to the known properties of the strain. Non-permissive
temperatures
are generally higher than permissive temperatures, typically about 35 -50 C,
preferably
38 -45 C, more preferably about 39.5 C. Variants supporting AAV replication at
a level
that is within one order of magnitude of that supported by the corresponding
wild-type
virus is preferred. In conducting the screening, the artisan should
incorporate the other
teachings of this disclosure. In particular, screening by culturing for times
that give peak
AAV replication with wild-type virus is insufficient. A kinetic matrix should
be set up in
which the candidate helper viruses are used for longer periods, and then
compared with the
wild-type virus at peak harvest time. A more detailed illustration of this
analysis is
provided in Example 3 of this disclosure.
CA 2995542 2018-02-15
Once a suitable helper virus strain has been selected, it may be implemented
in this
invention in a number of different forms. Viral particles, viral plasmids, and
stably
transformed host cells can all be used.
In one embodiment, the genome of the helper virus (or minimally, the regions
of
the helper virus genome encoding helper function) is introduced into the host
cell to be
used for replication of the rAAV vector in the form of a DNA plasmid, or a
plurality of
plasmids that provide complementary functions. Procedures for experimental
manipulation
of adenovirus are known in the art. The reader is referred to Graham et al.,
"Manipulation of
adenovirus vectors". In: Murray EJ, ed Methods in molecular biology: Gene
transfer and
expression protocols, vol7. Clifton, NJ: The Human Press, 1991:109-128, which
provides
detailed protocols for propagation, titration, and purification of adenovirus,
cotransfection
and in vivo recombination. Adenovirus plasmids are available commercially from
Microbix
Biosystems Inc., Toronto, Canada.
In another embodiment, the host cell is stably transfected with adenovirus
genes, or
genetically altered to provide the requisite functions for rAAV replication.
Alternatively,
the host cell may be genetically altered with only a portion of the adenovirus
genome, and
is subsequently infected or transfected with an adenovirus particle or
plasmid. Patent
applications WO 95/27071 and WO 95/34671 describe host cells inheritably
altered to
provide adenovirus function, which complements the replicative property of
various
defective adenovirus constructs.
In yet another embodiment, the host cell used for AAV replication is infected
with
a helper virus which is capable of self-replication, but not under non-
permissive
conditions. Any preparation of the requisite strain providing a sufficient MOI
may be
used. In keeping with GMP and other regulatory requirements, and to facilitate
scale-up
for commercial purposes, preparations of helper virus preferably comprise a
high density
of infectious particles and are substantially free of cellular debris and
other contaminants.
Desirable properties include the following:
= A density of at least 106, preferably at least about 10g, more preferably
at least
about 10 IU/ml, as determined in a TCID50 assay.
31
CA 2995542 2018-02-15
a A ratio of adenovirus DNA to total protein or adenovirus hexon
that indicates that
at least 10%, preferably at least about 50%, more preferably at least about
80% of the viral
particles contain adenovirus DNA.
a Less than 20%, preferably less than about 10%, more preferably
less than about
1% contamination by non-adenovirus material at the protein or DNA level, as
detected by
SDS gels stained for protein, or agarose gels of restriction nuclease digests
stained with
ethidium bromide.
0 A total of at least 109, preferably at least about 10' ', more
preferably at least about
10" IU per production batch.
Helper virus may be prepared in any cell that is permissive for viral
replication.
For adenovirus, preferred cells include 293 cells and HeLa cells.
Traditionally, when these
cells have been used for replication of adenovirus, they have been used in
plate cultures.
However, as shown in Example 4, these methods generally support replication of
temperature-sensitive adenovirus at levels that are one or two logs lower than
for wild-type
adenovirus.
Accordingly, it is preferable to employ culture techniques that permit an
increase in
seeding density. 293 cells and HeLa cell variants are available that have been
adapted to
suspension culture. HeLa is preferable for reasons of cell growth, viability,
and
morphology in suspension. As shown in Example 5, these cells can be grown at
sufficient
density (2 x 106 per ml) to make up for the lower replication rate of the
temperature-
sensitive adenovirus strain. Once established, cells are infected with the
virus and cultured
at the permissive temperature for a sufficient period; generally 3-7 days and
typically about
days.
Tangential flow filtration is a technique used in the art for processing large
volumes
of mammalian cells for the purpose of perfusing, concentrating, and harvesting
them. See,
e.g., Dorin et al., Biotechnol. Prog. 6:494, 1990; Maiorella et al.,
Biotechnol. Bioeng.
37:121, 1991. It is recommended that this technique be used with suspension
cultures for
the preparation of helper virus for use in this invention. Example 5
demonstrates that
HeLa S3 cells withstand shear forces of 750-1500 sec-1, permitting
concentration of the
cells and diafiltration of spent media.
32
CA 2995542 2018-02-15
Virus is harvested from the culture either from the spent media or by
microfluidization of the cells. The level of helper virus produced in the
culture is typically
at least 10 IU/ml, and preferably at least about 3 x 107 IU/ml.
Helper virus prepared according to the foregoing description may be used
directly
for infecting host cells used for rAAV replication. More usually, the virus is
isolated and
concentrated before use. Current methods for purifying and concentrating
helper virus
typically involve isopynic CsC1 gradients. This method is time and labor
intensive,
requires numerous open processing steps, and is difficult to scale up.
Instead, purification
by chromatography is recommended. The reader is referred generally to Prior et
al.,
Pharmaceut. Technol. 19:30, 1995; and Huyghe etal., Human Gene Therapy 6:1403,
1995.
Particularly preferred for isolation of temperature-sensitive strains of
adenovirus is anion
exchange chromatography, especially on a resin of polyethyleneimine using a
continuous
NaC1 gradient at pH 7.4. A detailed illustration of the polyethyleneimine
separation
method is provided in Example 6.
Providing a Host Cell Comprising Helper Virus and AAV
Several criteria influence selection of cells for use in producing rAAV
particles as
described herein. As an initial matter, the cell must be permissive for
replication and
packaging of the rAAV vector when using the selected helper virus. However,
since most
mammalian cells can be productively infected by AAV, and many can also be
infected by
helper viruses such as adenovirus, it is clear that a large variety of
mammalian cells and
cell lines effectively satisfy these criteria. Among these, the more preferred
cells and cell
lines are those that can be easily grown in culture so as to facilitate large-
scale production
of recombinant AAV vector preparations. Again, however, many such cells
effectively
satisfy this criterion. Where large-scale production is desired, the choice of
production
method will also influence the selection of the host cell, For example, as
described in
more detail below and in the art, some production techniques and culture
vessels or
chambers are designed for growth of adherent or attached cells, whereas others
are
designed for growth of cells in suspension. In the latter case, the host cell
would thus
preferably be adapted or adaptable to growth in suspension. However, even in
the case of
cells and cell lines that are regarded as adherent or anchorage-dependent, it
is possible (as
33
CA 2995542 2018-02-15
described below) to derive suspension-adapted variants of an anchorage-
dependent
parental line by serially selecting for cells capable of growth in suspension.
Where a temperature-sensitive helper virus is used, the cell must be able to
effectively replicate the rAAV vector under conditions that are non-permissive
for
replication of the helper virus. By way of illustration, when adenovirus ts149
is used as a
ts helper virus (as described and ilustrated below), the cell must be capable
of supporting
rAAV replication and packaging at temperatures well above 32 C, preferably
about
39.5 C. Human 293 cells are an example of a cell line fulfilling these
criteria but
numerous other cells and cell lines are capable of replicating rAAV at this
relatively
elevated temperature.
Ultimately, the helper virus, the rAAV vector sequence, and all AAV sequences
needed for replication and packaging must be present in the same cell. Where
one or more
AAV packaging genes are provided separately from the vector, a host cell is
provided that
comprises: (i) one or more AAV packaging genes, wherein each said AAV
packaging gene
encodes an AAV replication or encapsidation protein; (ii) a heterologous
polynucleotide
introduced into said host cell using an rAAV vector or pro-vector, wherein
said rAAV
vector or pro-vector comprises said heterologous polynucleotide flanked by at
least one
AAV ITR and is deficient in said AAV packaging gene(s); and (iii) a helper
virus or
sequences encoding the requisite helper virus functions. It should be noted,
however, that
one or more of these elements may be combined on a single replicon. By way of
illustration, a helper virus can also comprise an rAAV pro-vector or an AAV
packaging
gene.
The helper virus is preferably introduced into the cell culture at a level
sufficient to
infect most of the cells in culture, but can otherwise be kept to a minimum in
order to limit
the amount of helper virus present in the resulting preparation. A
multiplicity of infection
or "MOI" of 1-100 may be used, but an MOI of 5-10 is typically adequate.
Similarly, if the AAV vector and/or packaging genes are transiently introduced
into
the packaging cell (as opposed to being stably introduced), they are
preferably introduced
at a level sufficient to genetically alter most of the cells in culture.
Amounts generally
required are of the order of 10 g per 106 cells, if supplied as a bacterial
plasmid; or 108
34
CA 2995542 2018-02-15
particles per 105 cells, if supplied as an AAV particle. Determination of an
optimal amount
is an exercise of routine titration that is within the ordinary skill of the
artisan.
These elements can be introduced into the cell, either simultaneously, or
sequentially in any order. Where the cell is inheritably altered by any of the
elements, the
cell can be selected and allowed to proliferate before introducing the next
element.
In one preferred embodiment, the helper virus is introduced last into the cell
to rescue
and package a resident rAAV vector. The cell will generally already be
supplemented to the
extent necessary with AAV packaging genes. Preferably, either the rAAV vector
or the
packaging genes, and more preferably both are stably integrated into the cell.
It is readily
appreciated that other combinations are possible. Such combinations are
included within
the scope of the invention.
Once the host cell is provided with the requisite elements, the cell is
cultured under
conditions that are permissive for the replication AAV, to allow replication
and packaging
of the rAAV vector. Culture time is preferably adjusted to correspond to peak
production
levels, and is typically 3-6 days. Preferably, at least 100 viral particles
are produced per cell;
more preferably at least about 1000 per cell, still more preferably at least
about 10,000 per
cell. Preferably, at least 0.5 x 106, more preferably at least about 1 x 106,
even more
preferably at least about 2 x 106 RU/ml AAV vectors are produced per 2 x 105
cells during
the culture period. Optionally, large-scale production methods such as
suspension culture
and tangential flow filtration may be used. AAV particles are then collected,
and isolated
from the cells used to prepare them.
Preparations of rAAV particles of the present invention preferably comprise a
high
density of infectious AAV particles and are substantially free of helper
virus, helper virus
proteins and cellular debris and other contaminants. Desirable properties
include the
following:
= A concentration of at least 10', preferably at least about 108, more
preferably at
least about 109 RU/ml, as determined in a replication assay or quantitative
hybridization
comparison with a known standard.
= No more than 103, preferably no more than about 102, more preferably no
more
than about 10' infectious particles of helper virus per 108 RU of rAAV
particles.
CA 2995542 2018-02-15
= Less than 5%, preferably less than about 1%, more preferably less than
about
0.01%, even more preferably less than about 0.001% contamination by helper
virus on a
protein basis (wt/wt), detected either by densitornetric analysis of SDS gels,
or by
immunoassay for helper virus specific protein (such as hexon or penton-fiber
of adenovirus).
to Less than 5%, preferably less than about 1%, more preferably
less than about
0.01%, even more preferably less than about 0.001% contamination by helper
virus or
cellular protein (wt/wt), detected either by densitometric analysis of SDS
gels, or by
immunoassay for helper virus or cellular specific proteins.
= Preferably, the preparation is also substantially free of other potential
cellular
components such as cellular lipids, carbohydrates and/or nucleic acids.
The methods outlined in this disclosure are suitable for preparing small
experimental
batches, or preparative batches of 10-100 liters or more. For large scale
batch preparations,
the following property is also desirable:
* A total of at least 1010, preferably 1012, and more preferably 10" RU of
AAV
vector particles in the preparation.
Optionally, rAAV vectors may be further processed to enrich for rAAV
particles,
deplete helper virus particles, or otherwise render them suitable for
administration to a
subject. Purification techniques may include isopynic gradient centrifugation,
and
chromatographic techniques. Reduction of infectious helper virus activity may
include
inactivation by heat treatment or by pH treatment as is known in the art.
Other processes
may include concentration, filtration, diafiltration, or mixing with a
suitable buffer or
pharmaceutical excipient. Preparations may be divided into unit dose and multi
dose
aliquots for distribution, which will retain the essential characteristics of
the batch, such as
the homogeneity of antigenic and genetic content, and the relative proportion
of
contaminating helper virus.
Exemplary techniques for generating preparations of helper virus and AAV
exhibiting various desirable properties as described above are provided in the
following
sections and in the subsequent examples.
Various methods for the determination of the infectious titer of a viral
preparation are
known in the art. However, a preferred method for titer determination is a
high-throughput
titering assay as provided herein. In an exemplary high-throughput titering
assay, an array of
36
CA 2995542 2018-02-15
culture wells each comprising an aliquot of mammalian cells and an aliquot of
virus
preparation (as well as control wells comprising e.g., cells alone, virus
alone and null) is
established. The array of culture wells may, for example, be in the form of a
microtiter
vessel. Typically, aliquots (e.g., serially diluted aliquots) of the virus
preparation to be titered
are added to the cells, and then the cells and virus are incubated under
conditions that allow
for replication of the virus (typically growth conditions suitable for the
mammalian host cell).
Following replication of the virus, viral nucleic acid is generally released
by lysis of the
mammalian cells (using conditions or agents that promote lysis as necessary).
In preferred
embodiments, nucleic acid (including viral nucleic acid) in the multiplicity
of lysates is
transferred and fixed to a membrane under conditions that bind nucleic acid
(washing as
appropriate to remove proteins and other contaminants). The membrane
preferably is a
replicate or mirror image of the culture array in which the individual wells
of the original
array are subsequently represented by "pools" of nucleic acid (from the lysate
of each culture
well) that are bound at corresponding positions on the membrane. Hybridizing
the
membrane with a labeled virus-specific (or viral-insert-specific) probe can
then be used to
identify and quantify the relative amount of viral-specific nucleic acid in
each of the points
on the array, and by correspondence, in each of the original culture wells.
Conditions and
materials for nucleic acid transfer, binding, washing and hybridizing can be
adapted from
routine molecular biological techniques such as "dot blot" hybridization (as
described in the
art, see. e.g. the molecular biological techniques in Sambrook et al., supra,
and Ausubel et al.,
supra). Illustrative applications of these techniques are presented below.
These methods thus provide a high-throughput infectivity assay which can be
use.,d
in the determination of the infectious titer of a virus preparation. As shown
in Exairtple 4,
virus titers determined by this rapid and quantitative method closely
correspond 'to the
titers determined by more classical techniques. In addition, however, this
higt-throughput
method allows for the concurrent processing and analysis of many viral
rep'tication
reactions and thus has many others uses, including for example the screening
of cell lines
permissive or non-permissive for viral replication and infectivity, as with as
the screening
of agents that affect viral replication, as discussed further below.
37
CA 2995542 2018-02-15
Preferred Helper Virus Production and Purification Techniques for use in the
Present Invention
In various preferred aspects of the present invention, production and
purification
methods are employed for the generation of helper virus suitable for use in
the production
of rAAV vectors as described herein. A commonly used helper virus for the
production of
AAV is adenovirus, typically Ad5, although other helper viruses can also be
employed as
discussed herein and in the art.
For purposes of illustration, it is convenient to divide the discussion of
virus
production and purification into "upstream" and "downstream" phases. The
"upstream"
process generally refers to the production of the virus in suitable host cells
and release or
removal of the virus from the cells to produce a "crude" virus preparation
such as a lysate.
"Downstream" processing can be employed to purify the crude virus preparation
(e.g. to
isolate it away from cellular proteins and/or other contaminants).
A variety of techniques are known for the production and processing of helper
viruses, including adenovirus (e.g., CsC1 centrifugation, as well as other
techniques such as
those described in WO 96/27677). Helper virus produced using such techniques
can then
be employed in the production of rAAV vectors as described herein.
The following sections describe, for purposes of illustration, techniques that
can be
employed for the production of adenovirus although other techniques are known
in the art
and can be employed herein.
(i) Helper Virus Upstream
Helper virus, such as Ad5, can be readily produced by infecting mammalian
cells
(e.g. human cells). In illustrative examples described below, cells are grown
in media and
culture vessels suitable for growth of the host cell, concentrated prior to
infection, and then
infected with helper virus (e.g. at an MOI of 1-5) with gentle stirring.
Following infection,
cells can be resuspended in fresh medium and incubated for an additional
period of time
(typically about 2 days) in order to allow for replication and packaging of
the helper virus.
Following incubation, cells can be harvested and lysed to release the helper
virus.
Following lysis, the cell lysate is preferably treated with a nuclease to
degrade free nucleic
38
CA 2995542 2018-02-15
acid (e.g. cellular nucleic acid) without degrading nucleic acid that is
encapsidated in viral
particles. The lysate can be clarified (e.g. by filtration and/or
centifugation), and can also
be subjected to further purification techniques in order to purify and
concentrate the helper
virus in the preparation, as described and illustrated below.
As an illustrative example of such a process, cells can be grown in media at a
density of about 1 x 10 cell/ml in a vessel such as a spinner flask. After
incubation, cells
can then be concentrated to about 10 cells/ml, and infected with Ad5 at 1-2
infectious
units/cell with gentle stirring. Cells can then be resuspended in medium at
about 106
cells/ml, and allowed to produce virus over an incubation period of about 2
days. Cells
can then be harvested, resuspended in medium or buffer (e.g., at about 5 x 106
cells/ml),
and then disrupted, e.g. by mechanical lysis such as by passaging through a
mierofluidizer
at 8000 psi or equivalent technique (e.g. freeze-thaw or sonication). The
lysate can be
treated with a nuclease (e.g., Benzonase) for one hour at 37 C. The lysate can
be clarified
through a filter, such as a 1.0u filter, or by centrifugation. Analogous
techniques and
modifications thereof are further described below.
(ii) Helper Virus Downstream
Preferred techniques for the downstream processing of helper virus, such as
adenovirus, employ ion-exchange chromatographic procedures for the
purification of the
helper virus.
By way of illustration, the adenovirus filtrate as described above can be
loaded on
an anion-exchange resin, such as an N-charged amino or imino resin (e.g. POROS
50 PI,
or any DEAE, TMAE, tertiary or quaternary amine, or PEI-based resin) in a
chromatography column equilibrated with buffer (such as TMEG, also referred to
herein as
Chromatography Buffer A: 50 mM Iris (pH 8.0), 5 mM MgCl2, 1mM EDTA, 5%
glycerol).
The column can then be washed with multiple column volumes of TMEG (e.g. 5-6
volumes), followed by multiple volumes of a saline wash (e.g. 5-6 volumes of
TMEG with
800mM NaC1 (Chromatography Buffer "B": 60 % TMEG and 40% TMEG with 2M
NaC1). The Adenovirus can be eluted with TMEG with 1300 triM NaCI. (35%
Chromatography Buffer A, 65% Chromatography Buffer B).
39
CA 2995542 2018-02-15
The peak of adenovirus can be identified in the fractions by an infectivity
assay or
by a nucleic acid hybridization or immunoassay, as have been described in the
art. The
peak can be sterile filtered through a 0.2u sterile filter. Optionally, the
peak can be
concentrated by tangential-flow filtration, for example in a Filtron
Ultrasette-or Millipore
Pellicon unit. The peak or concentrate may be diafiltered in this system into
a suitable
buffer, such as PBS + 5% Sucrose. Alternatively, the adenovirus can be left in
elution
buffer. The final adenovirus product can be sterile filtered through a 0.2
filter and stored
for use. As described and illustrated herein, a temperature-sensitive helper
virus (such as a
temperature-sensitive adenovirus) can also be employed.
Examples describing the preparation and use of such helper viruses are
provided
below for purposes of further illustration.
Preferred AAV Production and Purification Techniques for use in the Present
Invention
As with helper virus, it is convenient for purposes of illustration to divide
the
discussion of AAV production and purification into "upstream" and "downstream"
process
phases; with the "upstream" process generally refering to the production of
AAV in
suitable host cells and release or removal of the virus from the cells to
produce a "crude"
AAV preparation. "Downstream" processing can be employed to purify the AAV
preparation (e.g. to isolate AAV away from cellular proteins and/or other
contaminants).
In preferred aspects of the present invention, upstream and downstream
processing
of AAV are conducted in a manner designed to substantially reduce and/or
eliminate
contaminating cellular proteins, as well as any contaminating helper virus
(e.g. Ad) or
helper virus proteins, any of which might contribute to elicitation of an
immune response if
present at substantial levels in the final rAAV vector preparation to be used
for gene
transfer.
The following sections describe, for purposes of illustration, techniques that
can be
employed for the production of AAV.
* Trade-Mark
CA 2995542 2018-02-15
(i) AAV Upstream Processing
AAV vector can be produced from a mammalian cell line that contains the
necessary AAV packaging genes (e.g. an AAV rep and cap gene); a recombinant
AAV
(rAAV) pro-vector that comprises a heterologous non-AAV polynucleotide flanked
by at
least one AAV inverted terminal repeat (1TR); and a helper virus for AAV (e.g.
an
adenovirus). These components can be intoduced into the cell in a variety of
configurations, as described above and illustrated below. Since AAV can be
replicated and
packaged in any of a variety of mammalian cells, there are a large number of
cell lines that
can be modified and employed for the production of AAV.
By way of illustration. AAV vector can be produced from ace!! line, such as
"C12"
(as described by K.R. Clark et al., Gene Therapy, 3: 1124-1132, 1996) or the
"C137.5" line
(described in a commonly-owned copending application by Targeted Genetics
Corporation, J. Allen et al.. WO 96/17947), that has been engineered to
contain a rep
and/or a cap construct, as well as a vector construct. Optionally, a cell line
such as C12 or
c137 that contains a rep and/or a cap construct can be transfected with a
plasmid that
contains a vector construct. such as ptgAAV-CF. Or a cell can be transfected
with a
plasmid that contains rep and cap. such as pRS5, as well as a plasmid that
contains a vector
construct. The cell can be infected with Adenovirus, or transfected with DNA
that
contains adenovirus genes.
A variety of such AAV "producer- cells can be generated, as described in the
references cited herein and in the art.
The AAV producer cells can be grown under conditions (including media,
temperature and the like) that are generally suitable for growth of the
mammalian cells,
which are generally also permissive for the replication of AAV. For example,
DMEM/F12
suspension medium is preferred for growth of the cells and DMEM medium alone
is
preferred for AAV vector production. As is known in the art, some cell types
and cell lines
tend to be attachment-dependent, whereas others are capable of growth in
suspension; and
many attachment-dependent cells can also be "adapted" to growth in suspension
by cycling
of the cells under suspension conditions as a means of enriching for and
ultimately
selecting for variants that are capable of suspension growth. Accordingly,
growth of cells
for AAV production can be conducted in any of a variety of vessels, depending
in part on
41
CA 2995542 2018-02-15
whether the selected producer cell line is relatively attachment dependent or
is suspension
adapted. Such vessels for the growth of attachment-dependent cells include,
for example,
tissue culture flasks, roller bottles, microcarriers and bioreactors (such as
hollow-fiber,
packed-bed or fluidized-bed bioreactors). Vessels for suspension-adapted
mammalian cell
lines include, for example, spinner flasks, tank reactors and air lift
fermentors.
AAV replication proceeds for a period of time as well as to a point in the
growth
cycle where viral production is optimal, preferable mid- to late-logarithmic
growth
(typically one to three days), after which time the cells can be harvested and
lysed to
release progeny virus. For example, cells can be resuspended in growth media
to about 1-
x106 cells/ml, and allowed to produce for 48 hours. Cells can then be
harvested (e.g. by
centrifugation), and resuspended in buffer (e.g., TMEG (or "Chromatography
Buffer A"):
50 mM Tris, pH 8.0, 5 mM MgCl, 1mM EDTA, 5% Glycerol) at about 1-10 x 106
cells/ml.
AAV can replicate to high copy number (e.g. 105 - 106 genomes/cell) in
transduced
cells if the necessary AAV Rep proteins and helper virus functions are
provided relatively
simultaneously. If Cap proteins are also provided, AAV particles are assembled
in the
nucleus of the infected cells where they tend to be assembled in crystalline
arrays. The
first step in product recovery is therefore cellular disruption. Although
freeze-thawing
and/or sonication can be used to disrupt the cells (as with adenovirus), such
techniques are
not very suitable to large-scale preparation. Mechanical lysis, using
techniques such as
microfluidization are thus preferable in those regards. Detergents and other
chemical
agents can also be employed to mediate or facilitate lysis. Treatment of
lysates with
nucleases (such as Benzonase) has been found to be helpful for reducing
viscosity and
improving filterability. Clarification, e.g. by microfiltration to separate
vector from at least
some portion of the cellular debris, is also helpful for promoting recovery
and purification.
By way of illustration, cells can be mechanically lysed after the incubation
period
by sequential passaging through a microfluidizer (typically at about 8000 psi,
using two
passages). Other commonly-employed techniques include freeze-thaw cycling and
sonication, as is known in the art. The lysate can also be treated with a
nuclease to degrade
nucleic acid (such as cellular or viral nucleic acid) that is not effectively
"protected" by
virtue of being packaged into a viral particle. We typically employ Benzonase
digestion
42
CA 2995542 2018-02-15
for about one hour at 37 C. The lysate can also be clarified. Methods for
clarification
include passage through a filter, such as a 1Øt filter, and centrifugation.
Tangential flow filtration (TFF) can be beneficially employed for processing
and
harvesting large volumes of cells. TFF can be used to perfuse, concentrate and
harvest
animal cells. For example, TFF can be used to process cells under laminar flow
conditions
at average wall shear rates of up to 3000 per second (see, e.g., Maiorella,
B., et al.,
Biotechnology and Bioengineering, 37: 121-126, 1991). Large-scale
concentration of
viruses using TFF ultrafiltration has been described by R. Paul et al. Human
Gene
Therapy, 4:609-615, 1993.
Illustrative production runs employing such techniques are described below.
(ii) AAV Downstream Processing
As described above, it would be particularly advantageous to obtain
preparations of
AAV that are substantially free of helper virus particles (such as Ad
particles). In addition,
AAV vector preparations will preferably also be substantially free of helper
virus and
cellular proteins (which can also be immunogenic). However, there is a further
set of
constraints that influence the suitability of techniques for AAV production.
Namely, in
order to be particularly useful for the production of AAV for gene therapy, it
is most
desirable for the techniques to be "scalable", i.e. applicable in conjunction
with large-scale
manufacturing devices and procedures. This latter set of constraints
effectively reduces or
eliminates the utility of available standard techniques such as cesium
chloride separation
(which is not well-suited to large-scale preparation procedures).
We have discovered a combination of procedures that are both scalable and
remarkably effective for the generation of AAV preparations that are
substantially free of
helper virus particles, as well as helper virus and cellular proteins and
other such
contaminants. Our preferred combination of procedures employs ion exchange
chromatographic procedures which contrast with various procedures mentioned in
the art
for the potential purification of, e.g., AAV or Ad. In particular, such
procedures as
described in the art typically employ a single type of ionic separation,
sometimes in
combination with other sorts of chromatographic procedures (see, e.g., K.
Tamayose et al.,
Human Gene Therapy 7: 507-513 (1996), and W096/27677, Sept. 12, 1996).
However, in
43
CA 2995542 2018-02-15
the case of AAV production, we have found that a combination of sequential
opposing ion
exchange chromatography is particularly effective for the generation of AAV
preparations
that are substantially free of helper virus particles and proteins, as well as
cellular proteins.
In view of these discoveries, it appears that AAV is not only "adapted" to
both
anion exchange and cation exchange chromatography, but that such a combination
of both
opposing ionic exchanges is particularly effective for eliminating all of the
various particle
and protein contaminants that typically occur in the generation or AAV vector
preparations. Any of a variety of cation and anion exchange resins can be
employed in
conjuction with these procedures, the fundamental properties of which are the
availability
of negatively- and positively-charged groups, respectively, to which AAV can
bind at least
to some degree (most preferably to a degree that differs substantially from
the relative
binding affinity of one or more of the contaminants referred to above, i.e. Ad
particles and
proteins, as well as mammalian cellular proteins). Without wishing to be bound
by theory,
it is believed that the anionic exchange step is particularly effective for
separating AAV
from Adenovirus; whereas both steps (but especially the cationic exchange
step) are
believed to be particularly effective for separating AAV from cellular
proteins. We have
also employed anion exchange followed by tangential flow filtration, as
described and
illustrated below. As further described below, we have found AAV preparations
can be
highly concentrated by chromatography on heparin sulfate.
By way of illustration, a clarified AAV lysate as described above can be
loaded on
an positively charged anion-exchange column. such as an N-charged amino or
imino resin
(e.g. POROS 50 PI, or any DEAR. TMAE, tertiary or quaternary amine, or PEI-
based
resin) or a negatively charged cation-exchange column (such as HS, SP, CM or
any sulfo-,
phospho- or carboxy-based cationic resin). The column can be washed with a
buffer (such
as chromatography buffer A/TMEG). The column can be eluted with a gradient of
increasing NaCI concentration and fractions can be collected and assayed for
the presence
of AAV and/or contaminants.
Other procedures can be used in place of or, preferably, in addition to the
above-
described anion and cation exchange procedures, based on inter-molecular
associations
mediated by features other than charge as is known in the art. Such other
procedures
include intermolecular associations based on ligand-receptor pairs (such as
antibody-
44
CA 2995542 2018-02-15
antigen or lectin-carbohydrate interactions), as well as separations based on
other attributes
of the molecules, such as molecular sieving chromatography based on size
and/or shape.
To take just a single example, the filtrate or partially purified AAV
preparation may be
loaded on a column that contains an AAV-specific antibody. This column can
bind AAV.
The column can be rinsed with buffer to remove contaminating proteins, and
then eluted
with a gradient or step of increasing NaCI concentration and fractions can be
collected.
Alternatively, such a column can be eluted with a buffer of different pH than
that of the
loading buffer.
The peaks of AAV and adenovirus can be identified in the fractions by
infectivity
assays or by a nucleic acid hybridization or immunoassays. The peaks can be
pooled, and
the pool can be diluted or dialyzed or diafi ltered with a buffer (e.g. TMEG
or equivalent)
to reduce the salt concentration.
This pool can be injected on a positively charged anion-exchange column and/or
a
negatively charged cation-exchange column (such as those referred to above).
The column
can be washed with a buffer (such as chromatography buffer A/TMEG). The column
can
be eluted with a gradient of increasing NaCI concentration and fractions can
be collected.
The peaks of AAV and adenovirus can be identified in the fractions by an
infectivity assay
or by a nucleic acid hybridization or immunoassay. The peaks can be pooled
based on the
results of any of these assays.
The pool of AAV-containing fractions eluted from an anion exchange column as
described above can be concentrated and purified by tangential flow filtration
(TFF), for
example in a Filtron Ultrasette or Millipore Pellicon unit. A membrane of
suitable
molecular weight cut-off (such as a 100,00 or 300,000 cut-off), is typically
composed of a
polymer such as regenerated cellulose or polyethersulfone. The preparation is
filtered
through the membrane, and the product is retained. The retained material can
be
diatiltered using the membrane with successive washes of a suitable buffer
such as
Ringer's Balanced Salt Solution + 5% glycerol. The final sample is highly
enriched for the
product and can be sterile filtered through a 0.211 filter and stored for use.
In the purification and concentration of AAV with tangential flow filtration
from
post-anionic exchange column material, the 300,000 molecular weight cut-off
membrane
CA 2995542 2018-02-15
has resulted in higher yields of replicative units than the 100,000 molecular
weight cut-off
membrane.
An additional step that can be employed for removal of adenovirus, if desired,
involves treating the eluant pool with a heat inactivation step (as described
herein) and
then filtration (e.g. prior to subjecting the preparation to TFF). However, we
have found
that the "anion exchange-to-TFF" procedure described above resulted in an AAV
preparation that was free of detectable adenovirus, and resulted in better
yields of purified
AAV.
Illustrative production runs employing such techniques are described below.
Altering the Growth Conditions of the AAV Producer Cells to Enhance Production
During the course of our production tests with AAV in various media and
culture
vessels, we typically monitored the cultures with respect various growth
and/or metabolic
parameters such as cell density, availability of glucose and amino acids, and
the production
of metabolic by-products such as ammonia and lactic acid. Such components can
be
readily monitored using standard techniques such as HPLC and enzymatic assays,
as
described in the art.
As described in the Examples below, we discovered that certain amino acids,
particularly aspartate and glutamate, were rapidly depleted in both batch and
perfusion
cultures. Indeed, in various batch and perfusion experiments, we have observed
that from
90 to 99% of the available asp and glu is substantially eliminated after 24 to
48 hours in
such cultures. Since the levels of asp and glu appeared to be sub-optimal in
such media,
we therefore provided additional amounts of either or both amino acid. Culture
maintenance and optimization techniques such as these have been routinely
applied in the
context of large-scale bioproduction (see, e.g., Glacken, M.W., et alõ
Biotechnology and
Bioengineering, 28: 1376-1389, 1986; Glacken, M.W., Bio/Technology 6: 1041-
1050,
1988; Bibila, T.A., etal., Biotechnol. Prog., 10:87-96, 1994; and Borys, M.C.,
etal.,
Biotechnology and Bioengineering, 43: 505-514, 1994).
To our surprise, replacement of these depleted amino acids resulted in a sharp
drop
in AAV production. For example, in experiments described below, supplementing
the
standard medium (DMEM ) with additional asp and glu drove production
efficiency down
46
CA 2995542 2018-02-15
by more than an order of magnitude (from about 1800 DNase-resistant particles
(DRP) per
cell to about 140 DRP per cell), although viability was slightly enhanced.
Another common component of media for the growth of mammalian producer cells
is a component of serum, such as fetal bovine serum (FBS), which is typically
included in
media at a level of about 10%. As described below, when the serum level for
AAV
production was increased (to 20%), AAV vector production dropped by more than
2-fold.
In contrast, when the cells were subjected to increasingly lower levels of
serum, AAV
vector production increased dramatically. For example, when serum levels were
reduced
to one-tenth of the normal starting levels (i.e. to 1%), vector production
increased by more
than 4-fold.
Without wishing to be bound by theory, it now appears that stressing the
producer
cells, either metabolically or by other means as described below, can
dramatically enhance
the production of AAV vector.
Stress can be effectively characterized, and tested, on the basis of the
negative
effect of the stress condition or stress agent on cellular growth and/or
metabolism. In
effect, stress can be achieved by the introduction of any condition or agent
that inhibits
cellular growth and/or metabolism, or by altering the level of a pre-exisiting
condition or
agent such that it becomes sub-optimal with respect to cellular growth and/or
metabolism.
A large variety of such conditions are known and/or apparent, including
nutritional stress
(one or more nutrients present at sub-optimal levels for growth and/or
metabolism),
temperature stress (sub-optimal temperature, which may include growing the
cells at lower
or higher temperatures, or subjecting the cells to temporary temperature
shocks such as
cold shock or heat shock), osmotic stress (sub-optimal osmotic level, which
may be
hypoosmotie or hyperosmotic), pH stress (sub-optimal pH which may be acidic or
alkaline), aeration stress (e.g., sub-optimal levels of oxygen or gas
exchange), mechanical
stress (e.g., shear stress as occurs in culture mixing), radiation stress, and
toxic stress
(presence of one or more chemicals or other agents that inhibits growth and/or
metabolism). With most if not all of such agents and conditions, it is
possible to subject
the cells to the stress continuously, or temporarily. By way of illustration,
in the case of
temperature stress, the cells can be grown at temperatures that are above or
below the
optimum (typically the optimum is approximately the normal body temperature of
the
47
CA 2995542 2018-02-15
animal from which the cells are derived), or the cells can be subjected to a
temporary
temperature shock, such as a cold shock or a heat shock. Presently preferred
examples of
such stress conditions include; nutritional stresses, such as amino acid or
serum limitation,
the alteration of aeration levels and agitation, the alteration of osmotic
levels (e.g. using
non-metabolizable carbohydrates such as sorbitol), and inclusion of chemical
agents, such
as saturated aliphatic carboxylic acids (e.g., propionic, butyric, isobutryic,
valeric and
caproic acids and their salts with organic or inorganic bases), N,N'-
diacylated diamines
(such as pentamethylenebisacetamide, hexamethylenebisacetamide and
heptamethylenebisacetamide), organic sulfur compounds (such as
dimethylsulfoxide), and
glucocorticoids (such as hydrocortisone dexamethasone, prednisolone,
aldosterone,
triamcinolone and cortexolone). Other such agents include genotoxic agents
such as
chemical carcinogens, UV, heat shock, metabolic inhibitors of DNA synthesis
(e.g.,
hydroxyurea, methotrexate, aphidicolin, drugs that affect topoisomerases
(e.g., amsacrine,
campthecin, etoposide and novobiocin).
As noted above, the producer cells can also be subjected to sub-lethal stress
by
altering pH. As exemplified below, we found that pH stress induced by
elevating medium
pH not only increased AAV, but it also caused a dramatic shift in the relative
proportions
of AAV that were released into the culture medium. As further described below,
this
technique can thus be used to facilitate AAV purification as well as enhance
production.
Illustrative procedures for optimizing the production of AAV by employing
various
stress conditions are provided below: as are results demonstrating that the
application of a
variety of different stress conditions can be used to effectively enhance AAV
production
levels.
Use of rAAV for gene therayy
Embodied in this invention are vector compositions comprising polynucleotides
with a therapeutically relevant genetic sequence. AAV viral vectors of this
invention can
be used' for administration to an individual for purposes of gene therapy.
Suitable diseases
for gene therapy include but are not limited to those induced by viral,
bacterial, or parasitic
infections, various malignancies and hyperproliferative conditions, autoimmune
conditions, and congenital deficiencies.
48
CA 2995542 2018-02-15
Gene therapy can be conducted to enhance the level of expression of a
particular
protein either within or secreted by the cell. Vectors of this invention may
be used to
genetically alter cells either for gene marking, replacement of a missing or
defective gene,
or insertion of a therapeutic gene. Alternatively, a polynucleotide may be
provided to the
cell that decreases the level of expression. This may be used for the
suppression of an
undesirable phenotype, such as the product of a gene amplified or
overexpressed during the
course of a malignancy, or a gene introduced or overexpressed during the
course of a
microbial infection. Expression levels may be decreased by supplying a
therapeutic
polynucleotide comprising a sequence capable, for example, of forming a stable
hybrid with
either the target gene or RNA transcript (antisense therapy), capable of
acting as a ribozyme
to cleave the relevant mRNA. or capable of acting as a decoy for a product of
the target gene.
Of particular interest is the correction of the genetic defect of cystic
fibrosis, by
supplying a properly functioning cystic fibrosis transmembrane conductance
regulator
(CFTR) to the airway epithelium. Alione et al. (J. Virol. 70:3235. 1996) and
Conrad et. al.
(Gene Therapy: in press. 1996) have shown stable in vivo CFTR gene transfer to
the
primate lung using single-dose AAV vectors. There are a variety of CFTR
polypeptides
that are capable of reconstructing CFTR functional deficiencies in cells
derived from cystic
fibrosis patients. Rich et at.. Science. 253: 205 (1991) described a CFTR
derivative
missing amino acid residues 708-835, that was capable of transporting chloride
and
capable of correcting a naturally occurring CFTR defect. Egan et al.. Nature,
358:581
(1992) described another CFTR derivative (comprising about 25 amino acids from
an
unrelated protein followed by the sequence of native CFTR beginning at residue
119) that
was also capable of restoring electrophysiological characteristics of normal
CFTR. Arispe
et al., Proc. Natl. Acad. Sci. USA 89: 1539 (1992) showed that a CFTR fragment
comprising residues 433-586 was sufficient to reconstitute a correct chloride
channel in
lipid bilayers. Sheppard et al., Cell 76: 1091 (1994) showed that a CFTR
polypeptide
truncated at residue 836 to about half its length was still capable of
building a regulated
chloride channel. Thus, AAV vectors with encoding sequences for native CFTR
protein,
and mutants and fragments thereof, are all preferred embodiments of this
invention.
49
CA 2995542 2018-02-15
Also of particular interest is the correction of the p53 tumor suppressor
gene,
locally defective in certain tumor types, by supplying a properly functioning
p53 gene to
the tumor site (Huyghe et al., Human Gene Therapy 6:1403, 1995).
Compositions of this invention may be used in vivo as well as ex vivo. In vivo
gene
therapy comprises administering the vectors of this invention directly to a
subject.
Pharmaceutical compositions can be supplied as liquid solutions or
suspensions, as
emulsions, or as solid forms suitable for dissolution or suspension in liquid
prior to use. For
administration into the respiratory tract, a preferred mode of administration
is by aerosol,
using a composition that provides either a solid or liquid aerosol when used
with an
appropriate aerosolizer device. Another preferred mode of administration into
the respiratory
tract is using a flexible fiberoptic bronchoscope to instill the vectors.
Typically, the viral
vectors are in a pharmaceutically suitable pyrogen-free buffer such as
Ringer's balanced salt
solution (pH 7.4). Although not required, pharmaceutical compositions may
optionally be
supplied in unit dosage form suitable for administration of a precise amount.
An effective amount of virus is administered, depending on the objectives of
treatment. An effective amount may be given in single or divided doses. Where
a low
percentage of transduction can cure a genetic deficiency, then the objective
of treatment is
generally to meet or exceed this level of transduction. In some instances,
this level of
transduction can be achieved by transduction of only about 1 to 5% of the
target cells, but is
more typically 20% of the cells of the desired tissue type, usually at least
about 50%,
preferably at least about 80%, more preferably at least about 95%, and even
more preferably
at least about 99% of the cells of the desired tissue type. As a guide, the
number of vector
particles present in a single dose given by bronchoscopy will generally be at
least about
1 x 10', and is more typically 5 x 10% 1 x 10', and on some occasions 1 x 10"
particles,
including both DNAse resistant and DNAse susceptible particles. In terms of
DNAse
resistant particles, the dose will generally be between 1 x 106 and I x 10'4
particles, more
generally between about 1 x 1 Os and 1 x 1012 particles. The treatment can be
repeated as
often as every two or three weeks, as required, although treatment once in 180
days may be
sufficient.
The effectiveness of the genetic alteration can be monitored by several
criteria.
Samples removed by biopsy or surgical excision may be analyzed by in situ
hybridization,
CA 2995542 2018-02-15
PCR amplification using vector-specific probes, RNAse protection,
inununohistology, or
immunofluorescent cell counting. When the vector is administered by
bronchoscopy, lung
function tests may be performed, and bronchial lavage may be assessed for the
presence of
inflammatory cytokines. The treated subject may also be monitored for clinical
features, and
to determine whether the cells express the function intended to be conveyed by
the
therapeutic polynucleotide.
The decision of whether to use in vivo or ex vivo therapy, and the selection
of a
particular composition, dose, and route of administration will depend on a
number of
different factors, including but not limited to features of the condition and
the subject being
treated. The assessment of such features and the design of an appropriate
therapeutic
regimen is ultimately the responsibility of the prescribing physician.
The foregoing description provides, inter alia, methods for generating high
titer
preparations of recombinant AAV vectors that are substantially free of helper
virus (e.g.
adenovirus) and cellular proteins. It is understood that variations may be
applied to these
methods by those of skill in this art without departing from the spirit of
this invention.
The examples presented below are provided as a further guide to a practitioner
of
ordinary skill in the art, and are not meant to be limiting in any way.
EXAMPLES
EXAMPLE 1
ILLUSTRATIVE PRODUCTION OF RECOMBINANT AAV VECTOR USING A WILD-TYPE
HELPER VIRUS (AD5) AND A TEMPERATURE-SENSITIVE HELPER VIRUS (AD TS149)
This example illustrates the use of a wild-type helper virus (Ad5) and a
temperature-sensitive helper virus (Ad ts149) to provide helper functions for
the
replication of a recombinant AAV vector particle comprising a model
therapeutic gene.
The ptgAAVCF plasmid consists of the left hand AAV2 ITR; a full length cystic
fibrosis transmembrane regulator cDNA; a synthetic polyadenylation sequence
based on
the mouse 13-globin polyadenylation sequence; AAV2 sequences downstream of the
cap
coding sequences; and the right-hand AAV2 ITR in a pBR322 plasmid backbone
(Afione
et al., 1996). The pGEM-RS5 packaging plasmid was derived from the pHIVrep
plasmid
51
CA 2995542 2018-02-15
(Antoni et al., 1991) and consists of the U3 and R regions from the HIV-1 LTR;
the rep
and cap regions from AAV2 including the p19 and p40 promoters; pBR322 and pGEM
plasmid sequences for bacterial replication and selection; and a small region
of human Ain
repetititve cellular DNA upstream of the HIV LTR.
Adenovirus type .5 was grown from a stock obtained from the American Type
Culture Collection (Rockville, Md). Ad5ts149 (Ensinger et at., J. Virol.
10:328, 1972)
was obtained from Harold S. Ginsberg.
Working stocks of Ad5 and Ad5ts149 (ts149) were produced at 37 C and 32 C;
respectively, by infecting 293-1 cells at a multiplicity of infection (MOD of
5 and 1;
respectively. After 4 hours the cultures were refed with fresh medium and
incubated at
37 C in a humidified 10% CO1 incubator. After seventy-two hours, cells were
removed,
pelleted at 1000 g at 15 C and resuspended in PBS containing 0.1 g/L of MgCl2
and 0.1
g/L CaC12. The cell suspension was then frozen and thawed three times. sheared
three
times through an 18 gauge needle and clarified by centrifugation at 1000 g at
15 C. The
clarified lysate was then treated with IDNase I at a final concentration of 2
mg/ml for 30
minutes at 37 C. The treated lysate was layered on a discontinuous step
gradient of CsC1
in water comprising 4.0 ml of CsCI (1.25 g/cm') layered over 2.0 ml of CsC1
(1.40 g/cin3)
in water and centrifuged at 35,000 RPM for I to 2 hours in a Beckman SW41
rotor. The
adenovirus band from each tube was removed, pooled and diluted in 1.35 g/cm3
CsC1 in
water and centrifuged overnight at 35.000 RPM in a Beckman SW55 rotor. The
adenovirus band was pooled. adjusted to 10% glycerol and dialyzed extensively
against 10
mM Tris pH 7.5 buffer supplemented with 1.0 mM MgCl2 and 10% glycerol.
293-1 cells (ATCC CRL 1573) were maintained in 1-flasks in a humidified 10%
CO, incubator in DMEM high glucose medium (JRH) supplemented with 10% fetal
bovine
serum (FBS, Hyclone). For this example, the 293-1 cells were inoculated at 4.4
x 104
cells/cm2 in tissue culture flasks with DMEM supplemented with 10% FBS and 2.0
mM L-
glutamine ,and incubated for twenty-four hours at 37 C in a humidified 10% CO2
incubator.
The cells (about 10 cells per flask) were then infected with working stocks of
either Ad5 or ts149 for 1 hour at a MOI of 5, followed by transient
transfection of vector
and packaging plasmids. Transient co-transfection of ptgAAVCF vector plasmid
and
52
CA 2995542 2018-02-15
pGEM-RS5 helper plasmid was performed using LIPOFECTAMINETm (Gibco). In that
process, 37.5 lag of each plasmid along with 150 1.1.1 LIPOFECTAMINETm were
mixed and
diluted in 4.75 ml of serum-free MEM. The adenovirus inoculum was removed and
the
plasmid- LIPOFECTAMINETm mixture was added to the cells and incubated for four
hours in a 5% CO2 incubator at the appropriate temperature. The plasmid-
LIPOFECTAMINETm mixture was removed from the culture after four hours and
replaced
with fresh medium.
Cells infected with wild-type virus were cultured at 37 C and cells infected
with
Adts149 were incubated at 39.5 C. After 72 hours, the cells were harvested,
pelleted and
resuspended in 10 mM Tris pH 7.5. The suspension was then lysed by sonication
in a ice-
water bath using a Branson cup-horn sonicator utilizing four 15 second pulses
and assayed
for rAAVCF and adenovirus production.
EXAMPLE 2
QUANTITATION OF RAAV AND ADENOVIRUS TITERS IN VECTOR PREPARATIONS
Cell lysates from the preceding example were assayed for production of rAAVCF
vector by C37 replication assay and analyzed for adenovirus production by slot-
blot
hybridization.
HeLa C37 was constructed to allow inducible expression of AAV Rep proteins for
rAAV vector replication. Briefly, an AAV Rep/Cap expression cassette
consisting of the
mouse metallothionein I promoter, AAV2 rep and cap genes and AAV transcription
termination site was constructed. Also included in the plasmid was a neomycin
resistance
gene under the control of the SV40 early promoter, SV40 small T intron and the
SV40
polyadenylation signal. HeLa cells were transfected with the plasmid and
clones were
selected in G4I8. A panel of clones was screened by a rAAV vector
amplification assay.
One clone, C37, demonstrated consistent and dose dependent amplification of
rAAV
vector following transduction and adenovirus infection.
Detection of replicating vector is accomplished by DNA isolation followed by
hybridization to a CFTR probe. In detail, HeLa C37 cells were inoculated at
4.4 x 104
cells/cm2 in tissue culture flasks with DMEM supplemented with 10% FBS and 2.0
mM L-
53
CA 2995542 2018-02-15
glutamine and incubated for twenty-four hours at 37 C in a humidified
incubator at 5 %
CO2. The cells were then inoculated with adenovirus (MOI = 5) and dilutions of
rAAVCI7
sample for 72 hours. Cells were harvested by scraping and prepared for
Southern blot
analysis. Total cellular DNA was prepared, digested with EcoRI,
electrophoresed on a 1%
agarose gel, transferred to a nylon 66 membrane followed by hybridization with
a 32P-
labeled human CFTR cDNA restriction fragment. This probe detects an
approximately 1.5
kb fragment from the AAVCF vector (corresponding to the predicted I .488 kb
EcoRI
fragment). Vector replication was quantitated relative to an endogenous
genomic CFTR
band and is expressed as replication units. One replication unit (RU) is
defined as a signal
intensity equivalent to that of the endogenous genomic CFTR band which is
approximately
1.8 kb. In some experiments, linear regression of serially diluted known
vector standards
was used to extrapolate and calculate vector concentration in samples.
The adenovirus DNA slot blot assay was conducted as follows. Aliquots of
samples were denatured in 0.4M NaOH, 10 mM EDTA with 1.0 Rim] salmon sperm
DNA at 65 C. Samples and adenovirus standards were diluted and filtered onto
nylon
membranes using a slot blot manifold and washed with 0.4M NaOH. The filter was
hybridized with a 32P-labeled probe corresponding to the adenovirus El A gene
sequence.
The entire Ad5 genome is available on Genbank at accession number X02996. We
used a
1 kb Ssp1-XbaI fragment (corresponding to nucleotides 339-1339) and analyzed
the blots
on a phosphorimager (Molecular Dynamics). One genome equivalent was considered
to
be equivalent to one adenovirus particle.
Figure 1 shows the results of the replication assay for rAAVCF vector in
lysates
prepared with Ad5 or ts149 at permissive (37 C) and non-permissive
temperatures
(39.5 C). Production of recombinant vector was supported by ts149 at 39.5 C
but
productivity was approximately 2 to 3 fold less than Ad5.
Figure 2 shows the results of the slot blot assay to determine the quantity of
adenovirus. Production of adenovirus genomes was reduced 3-4 logs by use of
the
temperature sensitive mutant as compared to wild-type.
54
CA 2995542 2018-02-15
EXAMPLE 3
OPTIMIZATION OF HELPER FUNCTION TO IMPROVE RAAV PRODUCTION
This example illustrates various attempts to improve the level of rAAV
obtained
when using temperature-sensitive helper virus. Increasing infection levels of
the helper
virus was unhelpful, but adjusting the kinetics was surprisingly effective.
The effects of increasing multiplicity of infection on vector production was
evaluated first. 293-1 cells were infected with either Ad5 at a MOI of 5 or
ts149 at various
MOI, followed by transient co-transfection with vector and packaging plasmids.
After 72
hours, the cells were lysed and assayed for production of rAAVCF vector by C37
vector
replication assay and analyzed for adenovirus production by slot-blot
hybridization. An
additional 96 hour time point was collected for cells infected with ts149 at a
MO! of 5.
Figure 3 shows the results of the rAAVCF replication assay conducted on cell
lysates prepared with ts149 at various MOI. Increasing the MO1 of ts149 did
not restore
vector productivity to levels observed with Ad5 (as shown by the intensity of
the 1.4 kb
hybridization band). However, a higher level of vector production was observed
at the 96
hour time point. The concentration of ts149 in the lysate detected by slot
blot analysis
increased with increasing MO!, but were still 3 to 4 logs lower compared with
Ad5.
Following the observation of increased vector productivity with ts149 at 96
hours
in the previous experiment, a time course and production kinetic study was
performed.
293-1 cells were infected with either Ad5 or ts149 at a MOI of 5 followed by
transient co-
transfection with vector and packaging plasmids. Cells infected with Ad5 and
ts149 were
cultured at 37 C and 39.5 C; respectively, for six days. Lysates from days
3, 4, 5 and 6
were assayed for vector production by vector replication assay and analyzed
for adenovirus
by slot-blot hybridization.
Figure 4 illustrates the kinetics of vector production. Solid bars represent
lysates
produced using wild-type Ad5 as helper; hatched bars represent lysates
produced using
ts149 as helper. Maximal vector production when using Ad5 was ¨2.0 x 106RU/ml,
peaking at day 4. At this time point, the vector production obtained using
ts149 was less
than ¨0.3 x I 06RU/ml. On day 5, however, there was a dramatic alteration in
the relative
efficacy of the two helper viruses. Vector production supported by Ad5 fell to
below
CA 2995542 2018-02-15
0.3 x 106 RU/ml. In contrast, vector production supported by ts149 jumped to
over 2 x 106
RU/ml. Adenovirus genome levels observed when using ts149 were significantly
lower
than with Ad5.
EXAMPLE 4
ASSAYING VIRAL TITERS AND HIGH-THROUGHPUT ASSAY TECHNIQUES
The temperature-sensitive and wild-type adenovirus stocks used in the
preceding
examples were produced in 293-1 cells in tissue culture flasks. In this
example, the levels
of adenovirus being produced by 293-1 cells was quantified by TCID50 endpoint
assay or
infectivity assay.
The TCID50 assay was conducted as follows: 1.0 x 103 293-I cells were plated
into
96-well microtiter plates and infected with serial dilutions of adenovirus
stock and allowed
to incubate at 37 C in a humidified 5% CO2 incubator. Eight replicates of 100
id of each
dilution were inoculated onto the cells. Three days after infection the cells
were methanol
fixed, washed with PBS and stained with FITC-conjugated anti-hexon antibody
(Biodesign) followed by propidium iodide staining to visualize cell nuclei,
After rinsing
with PBS, the plate was examined under a fluorescent microscope and scored for
the
presence of hexon containing cells. Titer at endpoint was calculated using a
Poisson
distribution. A dilution of virus that yields 50% of replicate samples hexon
positive has
0.5 IU/100 MI inoculum. Infectious titer is the product of the reciprocal of
this dilution, 0.5
IU/100 id and 10 (conversion factor to ml) to give the final infectious titer
per mi.
A high-throughput microtiter infectivity assay to measure infectious virus was
conducted as follows. Aliquots (10 1.11) of serially diluted cell-free
supernatants were
inoculated onto HeLa cells grown in 96-well microtiter plates. After three
days, infected
cells were treated and lysed with a denaturation solution (addition of I/10th
volume of 4.0
M NaOH, 10 pg/m1 salmon sperm DNA and 100 mM EDTA). Lysate was transferred to
a
Silent Monitor BiodyneB*Plate (Pall) and vacuum filtered onto the nylon
membrane. The
membrane was washed, denatured, hybridized with 32P-labeled adenovirus ETA
cDNA
restriction fragment and analyzed on a phosphorimager (Molecular Dynamics),
Linear
* Trade-Mark 56
CA 2995542 2018-02-15
regression analysis of serially diluted adenovirus standards was used to
calculate infectious
adenovirus titers in samples, using adenovirus standards titered by the TCID50
assay.
Specific virus productivity was calculated by normalizing infectious virus
titers in
the lysate to cell numbers at the time of infection. Results are shown in
Table 1:
TABLE 1: Adenovirus Production
Adenovirus Cell line Specific
productivity Assay
(1U/cell)
Ad5 293-1 125 TC1D5c,
HeLa S3 400 ICID50
Ad5ts149 293-1 10 TC1D50
293-1 16 microtiter
infectivity
293-1 15 microtiter
infectivity
293-1 10 microtiter
infectivity
These results show that specific production of Adts149 in 293-1 cells was one
to
two logs lower than Ad5.
An Ad5 virus preparation of known titer showed a linear range extending from
12.5
to 500 IU/well based on linear regression in the microtiter infectivity assay.
Combining a viral infectivity assay with a microtiter array format as
described
above resulted in a technique which is both rapid and quantitative, and which
is highly
suitable to automation.
The high-throughput infectivity assay as described above can also be applied
to
assaying other viruses (e.g., rAAV and wtAAV). The assay can be performed
essentially
as described above using appropriate mammalian cells (e.g., HeLa C37 cells for
rAAV or
293 cells for wtAAV) and under conditions permissive for the replication of
the virus to be
assayed (e.g., in the presence of helper virus for rAAV and wtAAV); and then
lysates can
be prepared and nucleic acids in said lysates can be transferred to a membrane
as described
above. Hybridization of the membrane containing the array of bound nucleic
acid pools
(each pool being released from the cells of the corresponding culture well) is
typically
performed with a suitable virus-specific probe (e.g., a probe specific for AAV
rep and/or
57
CA 2995542 2018-02-15
cap might be used to detect IMAM!, or a probe specific for an inserted
transgene might be
used in the case of a recombinant AAV vector).
The above-described high-throughput infectivity assay exhibited a linear
response
in the determination of rAAV titers over a relatively broad range of
concentrations. For
example, when a viral preparation of known titer (as determined by a modified
infectious
center assay) was serially diluted 1:2, starting from 2400 infectious units or
"IU"/well, and
used as a standard for the titer determination of two purified tgAAVCF
preparations of
unknown titer each of which was serially diluted 1:5, the microtiter assay
showed a linear
range extending from 75 to 600 IU/well based on linear regression. The
determination of
the titer of wtAAV preferably employed a limiting dilution format (for
example, when
eight serial limiting dilutions of a wtAAV preparation of known titer were
assayed, the
titer determined by the microtiter assay was essentially the same as that
determined by the
standard TCID50 assay, 3 x 109111/m1).
Either with limiting dilution or by comparison to a known standard, an
infectious
virus titer can be determined which corresponds to the titers determined by
more classical
techniques (e.g., the infectious center assay or the TCID50 50% end-point
analysis).
Besides its use in the determination of viral titers, this high-throughput
infectivity assay has
many others uses, including, but not limited to, the screening of cell lines
permissive or
non-permissive for viral replication and infectivity (e.g. by including
various mammalian
cells or variants thereof in different wells of a microtiter array); as well
as the screening of
agents that affect viral replication (e.g. by including various agents in
different wells of a
microtiter array as described above and determining the effect of the agents
on the
resulting infectious titer of virus). Among other things, the ability to
rapidly screen for
agents or conditions that enhance viral infectivity and/or replication is
particularly useful
in the context of developing or optimizing the production of viral vectors.
Conversely, the
ability to rapidly screen for agents or conditions that repress viral
infectivity/replication is
quite useful in the context of identifying anti-viral therapeutics.
58
CA 2995542 2018-02-15
EXAMPLE 5
DEVELOPNIENT OF SUSPENSION CULTURES FOR PRODUCING HELPER VIRUS
The preceding example shows that the levels of temperature-sensitive
adenovirus
produced by conventional culture techniques is low. This limits the ability to
use
temperature-sensitive adenovirus as helpers in production of AAV vectors. The
present
example provides an improved method that allows for the production of
temperature-
sensitive adenovirus in much higher amounts. Central to the improvement is the
use of
host cells grown in suspension culture.
293 N3S and HeLa S3 are suspension variants of the 293-1 human embryonic
kidney and HeLa human epitheloid carcinoma cell lines; respectively.
Suspension
cultivation was performed in 500 ml spinner flasks (Bellco) with working
volumes of 250
to 300 ml. HeLa S3 (ATCC 2.2-CCL) cells were maintained in DMEM/F-12 with 15
mM
HEPES supplemented with 7.5% FBS and 2.0 mM L-glutamine. 293-1 N3S (Microbix
Biosystems Inc.) were passaged in Joklik MEM supplemented with 7.5% FBS and
2.0 mM
L-glutamine. Spinner-flasks were agitated at 50-65 RPM.
Growth performance was assessed in the following experiment. 293 N3S and
HeLa S3 were serially passaged in suspension in replicate 500 ml spinner
flasks and cell
growth and viability was monitored. Flasks were inoculated at cell densities
of 2 to 5 x105
cells/m1 and then cultured for 2 to 3 days. To control for seeding density
differences,
population doubling levels (PDLs) were compared for replicate cultures. The
average PDL
was 2.0 0.49 (mean SD.) and 1.1 0.62 for HeLa S3 and 293 N3S;
respectively (n =
14). Higher cell doublings were consistently observed with the HeLa S3 cells.
Cell
morphology in suspension was dramatically different for the two lines. HeLa
cells grew as
single cells or small aggregates. In contrast, 293 N3S cells formed large
aggregates of 50
to 100 cells each. Significant numbers of non-viable cells were observed in
the center of
the large clumps. Stocks were subcultivated by centrifugation followed by
gentle
disruption with a pipette releasing the non viable cells from the aggregates.
Initial culture
viabilities of 293 N3S were consistently lower compared to HeLa S3.
Based on cell growth, viability and morphology in suspension, the FieLa S3
cell
line was selected for further process development. Growth and viability at
permissive
59
CA 2995542 2018-02-15
temperatures were evaluated. HeLa S3 cells were seeded into 500 ml spinner
flasks at 5 x
105 cells/ml, and monitored daily for seven days.
Figure 5 shows the viable cell density (VCD) of HeLa S3 cells, grown at 32 C
(squares) and 37 C (circles). Bars about the 32 C time points indicate the
range of values
observed in replicate 500 ml spinner flasks. Cells grown at 37 C peaked at 2.5
x 106
cells/m1 on day 5, whereas cells grown at 32 C peaked at 2 x 106 cells/m1 on
day 6.
Viability (determined by trypan blue exclusion) was at least about 90%
throughout.
Tangential flow or cross flow filtration is a versatile technique for a wide
variety of
large scale biopharmaceutical applications including concentration or removal
of cells,
concentration of macromolecules and media/buffer exchange. Tangential flow
processing
is required for concentrating cells for infection and for harvesting infected
cells at large
scale.
The effect of laminar shear on cell viability in tangential flow filtration
was
evaluated by concentrating and diafiltering the HeLa S3 cells. HeLa S3 cells
were
inoculated at a density of 4 x105 cells/ml in three liter Applikon bioreactors
and cultured to
2 x 106 cells/ml in DMEM/F-12 with 15 mM HEPES (JRH) supplemented with 7.5%
FBS,
2.0 mM glutamine, 1 X MEM amino acids. 1 X MEM non-essential amino acids, 0.1%
Pluronic polyol F-68 and 2 g/L glucose. Bioreactor working volume was two
liters.
Dissolved oxygen, pH, temperature and agitation were controlled at 60%
(relative to air
saturation), 7.2, 37 C and 100 rpm ; respectively, using the FERMCONTm (Scius
Corporation) controller system.
Tangential flow filtration experiments were performed with mixed cellulose
ester
hollow fiber membranes (Microgon). Pore size and surface area was 0.211 and
725 cm2;
respectively. A 0.21.1 filter was selected to retain cells while allowing
passage of spent
media. Cells were pumped (Cole Palmer) through the inside diameter of the
hollow fibers.
Recirculation rates were adjusted to provide average wall shear rates of 750
and 1500
sec . Once the crossfiow was established, permeate flow control of 30
and 90 ml/min;
respectively, was achieved by a pump (Cole Palmer) located on the permeate
line. During
cell concentration, permeate withdrawal continued until the desired fold
concentration was
achieved. During diafiltration, media feed entering the bioreactor was
activated until the
CA 2995542 2018-02-15
desired fold medium exchange was achieved. Viable cells were counted before
and after
each treatment.
Figure 6 shows the growth curves of HeLa S3 cells before and after tangential
flow
processing in an exemplary experiment. Two liters of cells were cultured in 3
liter
bioreactors. On day 3 (arrow), cells were concentrated seven fold from the 2-
liter working
volume, diafiltered against six volumes of growth medium and brought up to the
original
working volume. The results show that the cells were not damaged by wall shear
of 750
sec"' (squares) and 1500 sec" (circles), and continued to grow to high cell
densities.
Suspension cultures of HeLa S3 cells were then tested as host cells for ts149
production. or their ability in 300 ml suspension culture was investigated.
HeLa S3 cells
from 300 ml suspension culture (1x10" cells/m1) were centrifuged, concentrated
and
infected with ts149 (M01 = 3), After 1 hour. the culture was transferred to a
spinner-flask,
resuspended in media and cultured for seven days at 32 C. The HeLa S3 cells
continued to
grow from about 1 x 10' cells/m1 at the time of infection to about 2 x
105cells/m1 by day 5.
Viability decreased to ¨60 % on day 7.
Figure 7 shows the production of ts149 by HeLa S3 cell cultures. The culture
was
sampled daily, and lysates were prepared by freeze-thaw for analysis of virus
production
by the adenovirus infectivity assay. Virus production reached ¨4.5 x 107 IU/m1
of culture
by about day 3-5. On day 7. the cells were collected by centrifugation,
resuspended in
TMEG buffer and lysed by microfluidization (MF). The infectious titer of the
microfluidized lysate was comparable to those of the freeze-thaw lysate sample
indicating
recovery by microfluidization was comparable to freeze-thaw methods.
EXAMPLE 6
IMPROVED PURIFICATION METHOD FOR THE PRODUCTION OF A TEMPERATURE-
SENSITIVE HELPER VIRUS (AD TS149)
Purification using CsC1 gradients is burdensome for large scale production.
This
example illustrates the purification of ts149 by ion exchange chromatography.
Chromatography was performed on a Perseptive Biosystem BIOCADTM
chromatography workstation. The resin used was a polyethyleneimine (PI) weak
anion
61
CA 2995542 2018-02-15
exchanger (POROSTm 50 PI). The column was equilibrated with TMEG (50 rtiM
Tris, pH
8.0, 5 mM MgC12, I mM EDTA, 5 % glycerol). Chromatography was monitored on-
line
for pH, conductivity and optical density at 280 run.
Suspension HeLa S3 infected with ts149 at a MOI of 2 was harvested and
centrifuged. The pellet was resuspended in TMEG and lysed by cavitation at
3000 PSI
using a microfluidizer (Microfluidics). Lysate was clarified by filtration
through a 51.1
syringe filter (Millex SV) followed by a 0.45 tt syringe filter (Acrodisc).
Clarified lysate
was loaded onto a 1.6 ml POROSTM 50 PI anion exchange column run at 1 ml/min.
The
column was washed with 10 column volumes of TMEG with 900 mM NaC1, and the
ts149
was eluted with a linear gradient from 900 to 1300 mM NaCI. Fractions of 0.5
ml were
collected and assayed by infectivity assay and slot blot for the presence of
adenovirus.
Figure 8 shows the results of the infectivity assay conducted on consecutive
column fractions. The majority of the infectious adenovirus was found in
fractions 26 to
28, coincident to the peak of absorbance eluting at about 100 ins at
approximately 25
minutes. The ts149 eluted just prior to the large peak at higher salt
concentration.
Infectivity and slot blot assays conducted in parallel confirmed particles and
infectious
virus were in the same peak fractions.
Lysate and PI peak fractions were also assayed for total protein by the
Bradford
method. Protein concentration was 1.8 mg/ml in the lysate and less than 30.0
n/m1 in the
PI pool. The virions were separated from the majority of cellular protein in a
single step
and eluted as a single peak. The virions showed very high affinity for the PI
matrix, as
evidenced by the relatively high salt concentration required to elute them
from the column.
Large-scale production method for temperature-sensitive helper virus can
incorporate all the improvements described in these examples. In one
illustration, virus
production would comprise the following steps:
= Cell culture in suspension bioreactor
= Concentration/Medium exchange
= Infection with helper virus
= Virus production
= Harvest: Concentration/Diafiltration
= Lysis by microfluidization
62
CA 2995542 2018-02-15
e PI ion-exchange chromatography
= Concentration/Diafiltration
e Sterile filtration
This type of approach is inherently scalable and amenable to current Good
Manufacturing Practices.
Additional exemplary illustrations of such techniques are provided below.
EXAMPLE 7
COMPARISON OF FIRST AND SECOND GENERATION PROCESSES FOR HELPER VIRUS
PRODUCTION
A. Illustrative First Generation Helper Virus Production and Processing
In an exemplary "first generation" process for helper virus production,
mammalian
cells were grown in 40 T225 flasks, and then infected with Ad5 at an MOI of
about I.
After incubating, the cells were harvested by centrifugation, and lysed by
freeze-thawing
and passage through a needle. The lysate was subjected to treatment with DNase
I and
then run on a step CsC1 gradient and isopycnic gradient. Purified material was
dialyzed
and sterile filtered.
Using this first generation process, we obtained approximately lx10'2
particles (or
approx. lx le infectious units) from 4x108 cells.
B. Illustrative Second Generation Helper Virus Production and Processing
In an exemplary "second generation" process for helper virus production,
mammalian cells (HeLa S3) were grown in 10 liter bioreactors, and then
infected with Ad5
(from ATCC, subsequently plaque-purified on 293 cells, serially expanded on
HeLa S3
cells and double purified by CsC1 gradient centrifugation) at an MOI of about
I. After
incubating, the cells were concentrated and harvested by diafiltration, and
lysed by
microfluidization. The lysate was subjected to treatment with Benzonase
(nuclease) and
then filtered. The filtrate was then run on an anion exchange column
(PI)concentrated and
diafiltered, and finally sterile filtered,
63
CA 2995542 2018-02-15
Using this second generation process, we obtained approximately I x10"
particles
(or approx. 5x10'2 infectious units) from lx10'' cells.
Figure 9 illustrates the results of the downstream processing of helper virus
using
anion exchange chromatography as described above. Bars: Viral activity
measured in an
infectivity assay; Solid line: Am; Dotted line: buffer conductivity (ms).
As is apparent from comparing the fractionation of viral activity versus A280
absorbance, these processing procedures resulted in a substantial separation
of the helper
virus from the bulk of contaminating materials which would be expected to
contain cellular
proteins and nucleic acids.
EXAMPLE 8
COMPARISON OF FIRST AND SECOND GENERATION PROCESSES FOR PRODUCTION OF
RECOMBINANT AAV VECTORS
A. Illustrative First Generation rAAV Production and Processing
In an exemplary "first generation" process for rAAV vector production,
mammalian cells were grown in 40 T225 flasks, and then infected with Ad5 at an
MOI of
about 5. After incubating, the cells were harvested by centrifugation. and
lysed by
sonication. The lysate was subjected to treatment with DNase I and then run on
a series of
two Csa gradients. Purified material was dialyzed and sterile filtered. Using
this first
generation process, we obtained approximately 5x10`) replicative units RUs
from 4x108
cells.
B. Illustrative Second Generation rAAV Production and Processing
In an exemplary "second generation" process for rAAV vector production,
mammalian cells were grown in 10 liter bioreactors, and then infected with Ad5
at an MOI
of about 5. After incubating, the cells were concentrated and harvested by
diafiltration,
and lysed by microfluidization. The lysate was subjected to treatment with
Benzonase
(nuclease) and then filtered. The filtrate was then run on anion exchange
column, followed
by a cation exchange column. Eluant fractions containing AAV were pooled,
concentrated
64
CA 2995542 2018-02-15
and diafiltered, and finally sterile filtered. This second generation process
is expected to
yield greater than lx10" replicative units RUs from lx101 cells.
Figure 10 show the results of sequential fractionation on ion exchange
columns:
first, on an anion exchange matrix (upper panel), and then on a cation
exchange matrix
(lower panel). Bars: Viral activity measured in an infectivity assay for
either Adenovirus
or AAV; Solid line: A280 (a measure of total protein); Dotted line: buffer
conductivity
(ins). As is apparent from the analyzed fractions, it is possible to obtain
extremely high
levels of separation between AAV and Adenovirus, as well as between AAV and
A280-
absorbing material (largely proteins) using the techniques of the present
invention. In
particular, the results revealed that AAV vectors can be retained on both
anionic and
cationic exchange columns. and that the differential elution of AAV using both
anionic and
cationic exchange resulted in dramatically enhanced ability to separate AAV
from all of
the major contaminants of interest (including Adenovirus as well as cellular
proteins).
In another exemplary second generation process for rAAV vector production, the
filtrate was prepared as described above and was then run on an anion exchange
column,
followed by pooling of eluant fractions containing AAV, and then subjecting
the pooled
anion exchange eluants to tangential flow filtration (TFF). As described
below, this anion
exchange to TFF procedure was found to result in a highly conentrated and
purified
preparation of AAV.
Detailed analysis of AAV obtained using such second generation technology,
using
techniques as described above and in the art (including infectivity assays,
slot blot analyses
and SDS gel electrophoresis) provided further confirmation that the material
was of high
quality and substantially free of contaminating adenovirus particles (and
adenovirus
protein and DNA), and also substantially free of contaminating cellular
proteins and DNA.
SDS gels revealed the presence of bands corresponding to VP1, VP2 and VP3
(i.e. the
AAV capsid proteins). No other bands were visible after Coomassie staining.
These data
are consistent with the results of the column fractionation analyses as
depicted in Figures
10-11.
As an illustrative anion exchange to TFF procedure, the following is an
examplary
purification and concentration process starting with one liter of pooled
fractions from
anion exchange chromatography, If desired (as noted above), this pool can be
subjected to
CA 2995542 2018-02-15
heat inactivation followed by a filtration step (e.g., using a 0.22 j.trn
filter). For tangential
flow filtration (TFF), we employed a sanitized Pellicon XL system equipped
with a
300,000 molecular weight cut-off membrane which was operated at 40/0 for inlet
and
outlet pressures. One liter of pooled material was loaded into the system at a
500 ml
volume and then concentrated to 250 ml. Diafiltration was performed with 5
diavolumes
(1250 ml) of Modified Ringer's Solution + 5% glycerol. Following
diafiltration, the
retentate was concentrated to a final volume of 14 ml. Total process time was
approximately 3.25 hours (not including sanitization time). Silver-stained SDS
gels, slot
blots, and infectivity assays confirmed that the AAV preparation (which
contained
approximately 10' replicative units) was substantially free of contaminating
adenovirus as
well as adenoviral and cellular proteins.
The following are results from such a procedure showing infectious and total
virus
titer as RU (replicative units) and DRP (DNAse-resistant particles)
respectively:
300K TFF Volume Total RU Total DRP P/I %RU %DRP
Input Pool 1000 ml 8.9x 101 3.1 x 1014 3483 100
100
Purified Bulk 12.5 ml 7.3 x 1010 2.3 x 3103 82 74
The data presented in Figure 12 illustrates the results of an AAV production
run
using tangential flow filtration after an anion exchange column. Material
purified on the
POROS 50 PI column was concentrated using a 300,000 molecular weight cut-off
membrane (Millipore Pellicon XL). The concentrated material was diafiltered
with five
successive volumes of Ringer's Balanced Salt Solution + 5% glycerol. The
material was
then concentrated on the membrane 10-fold. Figure 12, a half-tone reproduction
of an SDS
polyacrylamide gel stained with a silver stain, shows the highly-purified AAV
capsid
proteins, VP1 (85 kD), VP2 (72 kD), and VP3 (62 kD), in the final purified
bulk material.
As is apparent from the data presented herein, these second generation
techniques
for the preparation and purification of AAV result in substantially improved
methods as
compared with those described previously.
Exemplary media for growing the Adenovirus helper and for preparing rAAV are
detailed in the following Table:
66
CA 2995542 2018-02-15
TABLE 2:
Ad medium rAAV medium
'INORGANIC SALTS
CaCL 116.61 _
CuSO4*5H20 0.00125
0,00125
Fe(NO3)3*9H20 - 0.05 0.05
FeSO4=7H20
0.4170.417
KCL 311.8 311.8
,MgC12 28.61
,MgSO4 48.84
. _____________________________________________________________________ '
NaCl ,;
iNaHCO3 2200 2200
NaH2PO4.1120 62.5 62.5
_ ______________________________________________________________________
Na2HPO4 71.02
71.02k
_ ______________________________________________________________________
'Zn2SO4 = =7H20 0.4315
0.4315
,OTHER COMPONENTS
'Glucose 4500 4500
'HEPES 3575 3575
-Hypozanthine Na 2.39 2.39t
,
LInoleic acid 0.042 0.042
,
Lipoic acid 0.105 0.105
Phenol Red, Na Salt
'Putrescine.2HCL= 0.081 0.081
Sodium Pyruvate 55 55
,Pluronic Polyol F-68 100 100
AMINO ACIDS
L- Alanine 4,455 4,455
_
L-Arginine=HCL 273.9 273.9
,L-Asparagine+120 22.5 22.5
L-Aspartic 19.95 19.95
L-Cysteine=HCL.H20 17.56 17.55
L-Cystinel2HCL 52.29 52.29
L-Glutamic acid 22.05 22.05
L-Glutamine, 657 657
Glycine , 26.25 26.25
L-HiStidine=HCL!H20 73.48 73.48
Usoleucine 106.97
106.97
,
L-Leucine 111.45
111,451
H
L-Lysine=HCL 163.75
163.75õ
L-Methionine : 32.34 32.34
L-Phenytalanine , , 68.48
68.48:
.L-PrOline= 17.25 17,25
67
CA 2995542 2018-02-15
_____________________________________________________ Ad medium rAAV medium
L-Serine 36.75 36,75
.
L-Threonine 101.05
101,05
L-Tiyptophan19.22 19.22 =,
. _
L-Tyrosine 91.79 91.79
L-Valine 99.65 99.65
VITAMINS
d-Blotin0.00365 0.00365
. - -
D-Ca Pantothenate 2.24 1.00
Choline Chloride 8.98 8,98
Folic Acid 2.65 2.65
myo-inositoi
1 12.6 12.6
Niacinamide 2.0185
2.0185
Pyridoxal=FICL 2 2
Pyridoxine=HCL 0.031 0.031
,
Riboflavin 0.219 0.219
' Thiamine-HCL 2.17 2.17
Thymidine 0.365 0.365
Vitamine B12 0.68 0.68
* add appropriate amount of NaCI for osmolatity of 300 mOsM (+ or - 20
elOsM)
68
CA 2995542 2018-02-15
C. Purification of AAV Vector Using Heparin Sulfate Chromatography
As discussed above, chromatographic techniques can be employed to further
purify
and concentrate AAV preparations in acccordance with the present invention. By
way of
illustration, a preparation of AAV which is in crude form (e.g. lysate), or
which has been
eluted from an anion-exchange or cation-exchange column ancUor concentrated by
tangential flow filtration can be purified by binding to a column comprising
heparin
sulfate. The AAV can then be eluted from such a column using a buffer
containing a salt
(e.g. a linear gradient of NaCI).
As illustrative of the use of heparin sulfate chromatography, AAV obtained
from a
"PI" pool (as described below in Example 9) was first concentrated four-fold
and
diafiltered into TMEG + 100mM NaCI using a 300K tangential flow filtration
membrane.
The concentrate was then injected on a 1 ml heparin sulfate column (Phannacia
"Hi-Trai;k
Heparin" column), and eluted using a linear gradient of NaCI,
Figure 13 is a chromatogram showing the resulting concentration of AAV on the
heparin sulfate column. The sharp peak in absorbance at 280 nm (left-hand
axis) at about
18 minutes elution time represents the AAV fraction as eluted from heparin
sulfate with a
linear gradient of 0 to 1M NaCI (conductivity in ms shown on right-hand axis).
EXAMPLE 9
RECOMBINANT AAV VECTOR PRODUCTION AND TESTING
In another set of production runs. we used 3-4x109 cells grown in a Cell
Factory,
using DMEM + 10% FBS as the growth medium. Cells were infected with Ad at an
MOI
of about 20, and harvested at 72 hours post-infection. Harvested cells were
suspended in
TMEG + NaCI at a concentration of about 5x10 cells/ml. After mechanical lysis
(microfluidization, 2 passes at 8000 psi), lysates were treated with Benzonase
(25 units/ml,
37 degrees C, one hour), and then filtered through a 5 micron filter (Pall
Profile II).
As art exemplary anion exchange column, we employed the POROS 50 PI column
(available from Perseptive Biosystems). Briefly, the filtrate was loaded onto
the column in
about 100 ml and eluted with a gradient of NaC1 to 500mM. Fractions determined
(by
* Trade-Mark
69
CA 2995542 2018-02-15
infectivity assay) to contain the majority of the AAV were collected and
pooled (referred
to as the "PI pool").
The PI pool was then diluted about 1:7 in TMEG and loaded on a 50 ml Toso Haas
SP650C column, and eluted with a gradient to 500 mM NaCI. Fractions determined
(by
infectivity assay) to contain the majority of the AAV were collected and
pooled (referred
to as the "SP pool"). The SP pool was concentrated using a Centriprepq OK
filter, and then
was sterilized by passage through a 0.2 micron filter.
The results revealed that the recombinant AAV was essentially free of
detectable
infectious adenovirus (as determined by limit of detection analysis with
serial
amplification on 293 cells and TCID50 assay). The preparation was also
essentially free of
adenoviral DNA (as determined by slot blot analysis), essentially free of
cellular proteins
(as determined by SDS-PAGE gel analysis), of cellular DNA (determined by PCR
analysis), and was also essentially free of phenotypically wild-type AAV (as
determined
by serial amplification and Southern analysis).
EXAMPLE 10
THE ENHANCEMENT OF AAV PRODUCTION BY NUTRITIONAL STRESS
As discussed above, it is believed that AAV production can be enhanced using
any
of a variety of agents and/or conditions that effectively stress (or de-
optimize) growth or
metabolism of the AAV producer cells. In this example, it is shown that the
depletion of
certain amino acids as occurs during culture is associated with a relative
enhancement in
AAV production; and, conversely, that media supplements to remove the
nutritional stress
actually result in a dramatic reduction in vector yield.
(a) Nutritional stress during batch and perfusion culture
JL14 cells were inoculated at about 4x105 cells/ml in 2 liter bioreactors and
grown
in the rAAV medium shown in Table 2 in either batch mode or by perfusion
(using
tangential flow filtration, day 1 at 0.4 volumes/day, days 2-3 at 1.2 vol/day,
day 4 at 2
vol./day and day 5 at 4 vol./day). Cultures were monitored for cell density,
glucose, lactate
and amino acids using standard techniques.
* Trade-Mark
CA 2995542 2018-02-15
The analyses revealed that cell density peaked in batch culture at lx1011
cells/ml on
day 2, and in perfusion culture at 8x10 cells/ml on day 6. Glucose was not
limiting in
either case (>1 g/1) and lacate was not inhibitory.
However, amino acid analysis revealed that both glutamate and aspartate were
rapidly depleted in both batch and perfusion cultures, as shown in the
following Tables:
TABLE 3:
Amino acid analysis of BATCH culture medium
(time course ¨ pima)
Nivi ¨ day 0 day 1 day 2 day 3 day 4
Aspartic Acid 133 ' 96 10 4 9 7
,
-
Threonine 119 687 644 606 552 533
_
Serine 105 - 271 230 157 117 98
-
Asparagine 132 - 130 113 96 68 69
Glutamic Acid 147 - 90 2 1 1 I
Glutamine 146 - 3424 2987 2450 ' 1989
1843
Proline 115 135 143 162 164 185
Glycine 75 288 241 194 151 130
,
,
Alanine 89 189 306 438 644 681
Valine 117 692 631 518 417 342
Cystine 121 143 133 120 ' 107
99
Methionine 149 160 132 100 74 58
_
Isoleucine 131 617 531 383 264 182
Leucine 131 645 538 374 248 '
161 '
Tryosine 181 407 379 ' 355 329
315 '
_
- Phenylalanine 165 323 289 259 231 214
,
Tryptophan 204 ' 47 ' 41 33 28 26
_
Ammonia 17 ' 760 816 941 1021 -
1033
Omthinine 71 89 110 128 144
-
Lysine Ha 572 521 463 415 384
'
_
Histidine 155 ' 276 257 243 229 211
..
Arginine 174 1020 943 870 791 747
71
CA 2995542 2018-02-15
TABLE 4:
Amino acid analysis of PERFUSION culture medium
(Time course ¨ ,urnol/L)
MW day 0 day 1 day 2 day 3 - day 4
days day 6
AsPartic Acid - 133 - 95 12 5 10 10 10 10
,
Threonine 119 - 709 691 560 596 651 641
657
Serine 105 . 281 264 ' 147 156 180 199
185
- ¨ _
Asparagine 132 130 124 75 78 109 119
119
Glutamic Acid 147 88 - 1 0 ' 1 1 1 0 -
¨ _
Glutamine 146 * 3525 3299 2517 2640 2906
2986 3082
Proline ' 115 145 165 163 174 177 157
171 -
Glycine 75 304 -- 267 189 205 217 227
230
Alanine 89 190 340 341 384 _ 423 333
330
Valin _ e 117 678 635 485 500 532 551
561
Cystine 121 ' 141 136 112 118 123 119
- 118
Meth ionine 149 157 133 91 99 107 108
107
Isoleucine 131 616 543 369 401 430 432
442
_
-Leucine 131 649 554 364 400 430 438
444
Tryosine 181 413 398 328 355 379 373
386
¨
Phenylalanine 165 336 316 1 244 268 291 287
292
I _
Tryptophan 204 ' 58 ' 47 36 41 48 44 47
-,
Ammonia 17 831 1182 956 1202 1219 931
990
Omthinine 42 97 74 115 112 56 44
Lysine HO 718 651 528 594 643 617
628
Histicline 155 - 284 . 310 223 243 262 266
265
Arginine 174 1058 948 826 901 978 974
1016
72
CA 2995542 2018-02-15
(b) Nutritional Stress Associated With Enhanced AAV Production
Follow-up studies were performed to confirm the importance of the relative
paucity
of glutamate and aspartate in the culture media. JLI4 cells were taken from a
spinner flask
and divided into two sets. Each set was innoculated with 3 x 109 infectious
units of 170-37
Ad 5. One set of cells was resuspended at 10 cells/mL in rAAV medium (Table 2)
containing 10% FBS and 1% L-glutamine (300 rnL), The other was resuspended in
rAAV
medium containing 10% FBS, 1% L-glutamine, 10 mgiL aspartatic acid, and 110
mg/L
glutamic acid.
Each set was incubated at 37 degrees for 72 hours in a spinner flask. The
cells
were harvested, microflidized twice at 8000 psi, Benzonased, plated into an
infectivity
assay, harvested and probed.
Results showed that the control spinner flask produced 6.2 RUs per cell. The
spinner flask supplemented with aspartic and glutamic acid produced 0.94 RUs
per cell.
This indicates that when depletion of aspartic acid and glutamic acid is
prevented
by providing these amino acids in excess. rAAV production is compromised due
to the
failure to subject the cells to nutritional stress.
Further tests were performed using a HeLa-derived cell line D6 which has an
integrated rAAV vector (1TR-(CMV promoter)-(3-gal reporter gene)-1TR), as well
as
copies of the wild-type AAV rep and cap genes.
The cells were seeded at 5 x 10" cells per T-225 flask in 30 mL complete DMEM
(10% FBS, 2 mM L-Glutamine), and incubated at 37 degrees in 10% CO2 for 2
days,
whereupon the cells reached a density of 2 x 107 cells per flasks Cells in two
duplicate
flasks were infected with Ad 5 at an MO! of 10. One flask contained complete
DMEM,
the other contained complete DMEM supplemented with 5 x aspartic acid and
glutamic
acid. Cells were harvested and counted after 72 hours of culture.
The complete DMEM yielded 2,6 x 10' cells with fig% viability. The
aspartate/glutamate supplemented medium yielded 3.8 x 107 cells with 91%
viability.
Cells were resuspended, sonicated, treated with Benzonase (25 U(ML),
clarified, and
assayed by slot blot analysis.
73
CA 2995542 2018-02-15
Results were as follows: D6 virus was produced in complete (unsupplemented)
DMEM at 1.8 x 10' DRP/ml, (1800 DRP per cell). D6 virus was produced in
aspartate/glutatrnate supplemented DMEM at 1,4 x 109 DRP/ml, (140 DRP/cell).
EXAMPLE 11
RECOMBINANT AAV VECTOR PRODUCTION UNDER SERUM STRESS
As an example of rAAV production under stress conditions, we have used reduced-
serum stress in conjunction with techniques as described above. Briefly, JL14
cells were
grown in spinner flasks in modified DMEM +10% FBS in continuous serial culture
mode,
and were split every 3-4 days. Cells from suspension culture were placed into
16 Nunc
Cell Factories, 10-stack, at 3x10s cells/factory on a three- to four-day
rotation. The
medium used for growth had a ten-fold reduction in serum (i.e. DMEM + 1% FBS)
thereby
placing the cells under serum stress.
At 24 hours after seeding, the medium in the factories was removed and fresh
medium containing 3x109 Ad units/ml was added. After 72 hours of culture at 37
degrees,
the cells were dislodged from the factories by gentle tapping, medium
containing cells was
collected and the cells were pelleted and resuspended in TMEG + 100 mM NaCI,
and then
lysed by passage through a microfluidizer at 8000 psi. The lysate was
clarified through a 5
micron filter and the clarified lysate was loaded on a 500 ml PI anion exhange
column.
The column was eluted with a gradient of increasing NaCI (up to 500 mM) in
TMEG
buffer. Fractions were collected and assayed using a Clone 37 assay as
described by Allen
et al. (W096/17947, supra). The fractions containing most of the AAV vector
were then
pooled and concentrated 10-fold using a Centriprep centrifugal concentrator at
1000 x g for
30 minutes. The concentrated material was dialyzed against Ringer's Balanced
Salt
Solution with 5% glycerol, and stored at -70 degrees C. The AAV was assayed by
the
Clone 37 assay, as well as by slot blot and SDS-PAGE. The material may also be
assayed
for the presence of adenovirus, adenoviral proteins, and cellular DNA, as well
as other
potential contaminants.
Figure 11 shows the results obtained using GAK-0003 producer cells set up in T-
225 flasks at 10 cells per flask, and innoculated on Day 2 with DAB-003
adenovirus at an
MOI of 10. Different flasks were cultured for 72 hours at 37 degrees in fresh
DMEM
74
CA 2995542 2018-02-15
containing a different percentage of FBS, as shown in the figure. On Day 5,
each flask was
harvested, the cells were counted, resuspended, sonicated, Benzonased, and
plated to
measure vector production as before.
Optimal vector producton was observed at a FBS percentage of 1%. Accordingly,
medium that is deficient in FBS (less than 2.5%, preferably less than 2% but
more than
0%) is preferred as a condition for subjecting the producer cells to serum
stress.
EXAMPLE 12
RECOMBINANT AAV VECTOR PRODUCTION UNDER pH STRESS
As a further example of rAAV production under stress conditions, we have used
pH
stress in conjunction with techniques as described above. Briefly, AAV
producer cells
were grown in bioreactors as described above. Cells were then infected with
Ad5 at
MOI=10 and inoculated into low-serum media (as in Example 11) in suspension in
1.5 liter
bioreactors. Cultures were maintained at various elevated pH levels (from 7.2
to 8.0).
Cultures were then monitored daily for cell number, viability, glucose
consumption, lactate
production, pH, osmolarity and AAV production. As shown below, there was an
increase
in AAV production when the pH was elevated to 7.4; coupled with an even more
dramatic
increase in the number of AAV particles released into the supematant (which
increased as
pH was elevated):
Culture pH Cell- Supernatant Total % Cell- % in
associated Particles Particles associated Supernatant
Particles
7.2 4.70E+ 12 1.90E + 09 4.70E+ 12 100% 0%
7.4 6.50E+ 12 1.30E+ 13 1.95E + 13 33% 67%
7.6 3.40E+ 12 1.50E+ 13 1.84E + 13 18% 82%
8.0 1.30E+ 12 1.50E+ 13 1.63E+ 13 8% 92%
In sum, as pH was raised, we observed a sharp increase in the number of AAV
particles released into the supernatant, and a shift in the percentage of
supernatant:cell-
associated particles (from nearly all cell-associated at pH 7.2 to mostly
supernatant (92%)
at pH 8.0). The ability to recover AAV particles directly from the supernatant
without the
need for lysing the producer cells represents a powerful advantage in terms of
AAV
production and purification. AAV isolated from the supernatant using pH stress
can be
CA 2995542 2018-02-15
readily concentrated and purified using techniques as described herein (e.g,
ion-exchange
chromatography and/or tangential-flow filtration).
76
CA 2995542 2018-02-15