Note: Descriptions are shown in the official language in which they were submitted.
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
FORMULATIONS OF L-ORNITHINE PHENYLACETATE
INCORPORATION BY REFERENCE TO PRIORITY APPLICATIONS
[0001] The present application claims the benefit of priority to U.S.
Provisional
Patent Application No. 62/255,300, filed November 13, 2015; U.S. Provisional
Patent
Application No. 62/276,754, filed January 8, 2016; and U.S. Application No.
15/133,087, filed
April 19, 2016; all which are hereby expressly incorporated by reference in
their entireties.
BACKGROUND
Field
[0002] The present application relates to pharmaceutical compositions
comprising
oral formulations of L-ornithine phenylacetate and methods of administration
and the use for
treating hyperammonemia in patients having various acute and chronic liver
diseases and
disorders, for example, acute liver failure, liver cirrhosis, liver
decompensation, portal
hypertension, hepatic encephalopathy, or patients with urea cycle disorders.
Description
[0003] Chronic liver disease is characterized by the gradual destruction
of liver tissue
over time, whereby healthy and regenerating liver tissue is slowly replaced
with scar and
necrotic tissue. This is known as liver cirrhosis. Normal liver function is
impaired and the scar
tissue progressively diminishes blood flow through the liver. As normal
regenerating liver tissue
is lost, nutrients, hormones, drugs and toxins are no longer effectively
processed. This can result
in symptoms including abnormal clearance of proteins absorbed through the
intestinal tract,
leading to accumulation of ammonia; abnormal excretion, leading to an
accumulation of
bilirubin in the blood, producing jaundice; increased sinusoidal pressure,
leading to fluid
accumulation in the abdomen (ascites); and portal hypertension (and
portosystemic shunting)
wherein scarred liver tissue acts as a barrier to blood flow, leading to
increased portal blood
pressure and oesophageal varices.
[0004] Patients with chronic liver disease can be in a fairly stable
clinical state and
exhibit few or no symptoms. However, such patients are at risk of an abrupt
deterioration in
their condition which can lead to acute-on-chronic liver failure. This
transition from a
"compensated" state, where the liver is able to function, albeit at a reduced
level, to a
"decompensated" state, where liver function fails, involves the effect of
precipitating events.
-1-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
Precipitating events associated with chronic liver disease include
gastrointestinal bleeding,
infection (sepsis), portal vein thrombosis and dehydration.
[0005] Hepatic encephalopathy (HE) is a common complication of
decompensated
cirrhosis; it has a significant negative effect on survival even after liver
transplantation and is
associated with irreversible impairment in cognitive function. An estimated 60-
70% of cirrhotic
subjects have at least subtle signs of neurocognitive impairment, and HE is
the principal
diagnosis in hospitalized subjects. Overt HE has a prevalence of approximately
30% in the
cirrhotic population, and accounts for about 150,000 hospitalizations annually
in the United
States.
[0006] Hepatic encephalopathy (HE) is a complex neuropsychiatric
disorder that
occurs in diverse clinical situations such as acute or chronic liver disease
and spontaneous
portosystemic venous shunting. In the early stages of hepatic encephalopathy
subtle mental
changes occur such as poor concentration, confusion and disorientation. In
severe cases, hepatic
encephalopathy can lead to stupor, coma, brain swelling (cerebral edema) and
death. In the case
of patients who develop HE as a result of chronic liver disease, the onset of
HE is often the
result of a clinically precipitating event such as gastrointestinal bleeding,
sepsis (infection),
portal vein thrombosis or dehydration.
[0007] Gastrointestinal bleeding and portosystemic shunting allows toxic
substances,
which are usually metabolized by the liver, to bypass the liver, enter the
systemic circulation and
cross the blood-brain barrier to exert direct or indirect neurotoxic effects
on the central nervous
system. Ammonia accumulation is thought to play an important role in the
progression of
hepatic encephalopathy and multiorgan failure (respiratory failure,
cardiovascular failure, kidney
failure). In addition to ammonia, septicaemia (or bacterial peritonitis) which
develops soon after
a gastrointestinal bleed is also likely to be a contributing factor to hepatic
encephalopathy.
[0008] Liver decompensation can then lead to multi-organ failure and
hepatic
encephalopathy. In the early stages of hepatic encephalopathy subtle mental
changes such as
poor concentration or the inability to construct simple objects occurs. In
severe cases, hepatic
encephalopathy can lead to stupor, coma, brain swelling and death.
[0009] Urea cycle disorder or urea cycle defect is a genetic disorder
caused by a
deficiency of one of the enzymes in the urea cycle which is responsible for
removing ammonia
from the blood stream. Normally, the urea is transferred into the urine and
removed from the
body. In urea cycle disorders, the nitrogen accumulates in the form of
ammonia, a toxic
substance, and is not removed from the body. It has been reported that sodium
phenylbutyrate
may be used in the management of this condition. See, for example, Batshaw, M.
L. et al.,
-2-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
"Alternative pathway therapy for urea cycle disorders: twenty years later," J.
Pediatr. (2001) 138
(1 Suppl): S46-S55.
[0010] A common therapy for patients with hepatic encephalopathy
involves
strategies to reduce the concentration of ammonia. These include restriction
of dietary protein
intake; administration of lactulose, neomycin, L-ornithine L-aspartate (LOLA),
or sodium
benzoate; and cleansing enemas. There are currently marketed products that
contain
phenylacetic acid (e.g., AMMONULO) or prodrugs of phenylacetic acid, e.g.,
phenylbutyrate
(BUPHENYLO) or glycerol phenylbutyrate (RAVICTIO) as the ammonia scavenger
(binding
agent) for the treatment of hyperammonemia due to urea cycle disorder (UCDs).
RAVICTIO
has also been evaluated in clinical trials and shown preliminary efficacy for
the treatment of
hepatic encephalopathy. See, for example, Rockey D. et al., "Randomized,
Double-Blind,
Controlled Study of Glycerol Phenylbutyrate in Hepatic Encephalopathy,"
Hepatology, 2014,
59(3):1073-1083. In addition, L-ornithine phenylacetate has been reported to
be an effective
treatment of hyperammonemia and hepatic encephalopathy. Jalan et al., reported
a clinical
study where the data showed that L-ornithine phenylacetate is useful in
ammonia lowering. See
Jalan et al., "L-Ornithine phenylacetate (OP): a novel treatment for
hyperammonemia and
hepatic encephalopathy," Med Hypotheses 2007; 69(5):1064-69. See also, U.S.
Publication
Nos. 2008/0119554, 2010/0280119, and 2013/0211135, each of such is hereby
incorporated by
reference in its entirety.
[0011] L-ornithine phenylacetate has been granted orphan drug status by
the United
States Food and Drug Administration and was granted fast track designation for
the treatment of
hyperammonemia and resultant hepatic encephalopathy. Currently, L-ornithine
phenylacetate is
under clinical investigation for the treatment of overt HE in patients with
decompensated liver
cirrhosis. Patients receive continuous intravenous infusion of L-ornithine
phenylacetate at doses
of 10, 15 or 20 g per day for 5 days depending on the baseline severity of the
liver impairment.
[0012] Typically, L-ornithine phenylacetate has excellent solubility in
water or
aqueous solution. In all the known clinical studies of L-ornithine
phenylacetate for treating
acute or chronic liver diseases, L-ornithine phenylacetate is administered by
intravenous
infusion over a period of time, for example, from 1 day or up to five days in
human studies.
There exists a need to develop alternative administration routes to improve
patient convenience.
SUMMARY
[0013] Some embodiments of the present disclosure relate to oral
pharmaceutical
formulations, comprising L-ornithine phenylacetate in an oral dosage of about
0.1 g to about 10
g, and one or more pharmaceutically acceptable excipients or carriers. In some
embodiments,
-3-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
the formulation provides an immediate release profile of L-omithine
phenylacetate upon oral
administration. In some embodiment, the oral dosage of L-omithine
phenylacetate is from about
2 g to about 8 g. In one embodiment, the oral dosage of L-omithine
phenylacetate is about 5 g.
In another embodiment, the oral dosage of L-omithine phenylacetate is about
2.5 g. In some
other embodiment, the oral pharmaceutical formulation provides controlled
release of L-
ornithine phenylacetate.
[0014] Some
embodiments of the present disclosure relate to methods of treating or
ameliorating hyperammonemia comprising administering to a subject in need
thereof an oral
pharmaceutical formulation comprising L-omithine phenylacetate as described
herein. In some
embodiments, the subject has acute liver failure or chronic liver diseases.
In some
embodiments, the subject has liver cirrhosis or liver decompensation. In some
embodiments, the
subject has hepatic encephalopathy. In still some embodiments, the subject has
portal
hypertension. In some further embodiments, the chronic liver disease or liver
cirrhosis is
classified as Child-Pugh A, B or C.
[0015] Some
embodiments of the present disclosure relate to methods of treating
hyperammonemia comprising administering to a subject in need thereof an oral
pharmaceutical
formulation comprising L-omithine phenylacetate, where the pharmaceutical
formulation
provides a plasma Cmax of phenylacetic acid ranging from about 10 ug/mL to
about 150 ug/mL.
In some embodiments, the pharmaceutical formulation also provides a plasma
Cmax of
phenylacetylglutamine ranging from about 5 ug/mL to about 100 ug/mL. In some
embodiments,
the oral pharmaceutical formulation of L-omithine phenylacetate provides a
controlled release of
L-omithine phenylacetate after administration. In some other embodiments, the
oral
pharmaceutical formulation of L-omithine phenylacetate provides an immediate
release of L-
ornithine phenylacetate after administration. In some embodiments, the
pharmaceutical
formulation comprise L-omithine phenylacetate in an oral dosage of about 0.1 g
to about 10 g.
BRIEF DESCRIPTION OF THE DRAWINGS
[0016] FIG.
1 is a line chart depicting the in vivo plasma pharmacokinetic profiles of
phenylacetic acid (PAA) in humans after administration of controlled-release
Formulations A,
B, and C, RAVICTIO, and an immediate-release oral formulation of L-omithine
phenylacetate.
[0017] FIG.
2 is a line chart depicting the in vivo surrogate plasma
pharmacodynamic profiles of phenylacetylglutamine (PAGN) in humans after
administration of
controlled-release Formulations A, B, and C, RAVICTIO, and an immediate-
release oral
formulation of L-omithine phenylacetate.
-4-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
[0018] FIG. 3 is a line chart depicting the in vivo plasma
pharmacokinetic profiles of
phenylacetic acid (PAA) in subjects with chronic liver disease classification
Child-Pugh class A
after administration of a single dose of 5g L-omithine phenylacetate under
four different
treatments.
[0019] FIG. 4 is a line chart depicting the in vivo surrogate plasma
pharmacodynamic profiles of phenylacetylglutamine (PAGN) in subjects with
chronic liver
disease classification Child-Pugh class A after administration of a single
dose of 5g L-omithine
phenylacetate under four different treatments.
DETAILED DESCRIPTION OF THE EMBODIMENTS
[0020] Some embodiments of the present disclosure are directed to oral
formulations
of L-omithine phenylacetate. Some embodiments of the formulations provide a
low dose
formulation, using much lower doses of equivalent phenylacetate as compared to
RAVICTIO.
Some such embodiments are an immediate release formulation. Other embodiments
of the
formulations provide a controlled release or extended release system.
Definitions
[0021] The section headings used herein are for organizational purposes
only and are
not to be construed as limiting the subject matter described.
[0022] Unless defined otherwise, all technical and scientific terms used
herein have
the same meaning as is commonly understood by one of ordinary skill in the
art. The use of the
term "including" as well as other forms, such as "include", "includes," and
"included," is not
limiting. The use of the term "having" as well as other forms, such as "have",
"has," and "had,"
is not limiting. As used in this specification, whether in a transitional
phrase or in the body of
the claim, the terms "comprise(s)" and "comprising" are to be interpreted as
having an open-
ended meaning. That is, the above terms are to be interpreted synonymously
with the phrases
"having at least" or "including at least." For example, when used in the
context of a process, the
term "comprising" means that the process includes at least the recited steps,
but may include
additional steps. When used in the context of a compound, composition,
formulation, or device,
the term "comprising" means that the compound, composition, formulation, or
device includes
at least the recited features or components, but may also include additional
features or
components.
[0023] As used herein, common organic abbreviations are defined as
follows:
AUC Area under the curve
-5-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
AUCo-t Area under the concentration vs. time curve from time =
0 (zero)
to time of last quantifiable concentration
AUC0-inf Area under the plasma concentration with time curve
extrapolated
to infinite time
CL Total plasma clearance
C12 Drug concentration at 12 hr after drug administration
Cmax Maximum plasma concentration
Absolute bioavailability value (%)
hr Hour(s)
IR Immediate release
ORN Ornithine
PAA Phenylacetic acid (or the conjugate base phenylacetate)
PAGN Phenylacetylglutamine
PD Pharmacodynamic
PK Pharmacokinetic
[0024] The term "immediate release" as used herein, has its ordinary
meaning as
understood by those skilled in the art and thus includes, by way of non-
limiting example, release
of a drug from a dosage form in a relatively brief period of time after
administration.
[0025] The term "controlled release" and the term "extended release" as
used herein,
each has its ordinary meaning as understood by those skilled in the art and
thus includes, by way
of non-limiting example, controlled release of a drug from a dosage form over
an extended
period of time. For example, in some embodiments, controlled release or
extended release
formulations are those that have a release rate that is substantially longer
than that of a
comparable immediate release form. The two terms can be used interchangeably.
[0026] The term "about" as used herein, refers to a quantity, value,
number,
percentage, amount, or weight that varies from the reference quantity, value,
number,
percentage, amount, or weight by a variance considered acceptable by one of
ordinary skill in
the art for that type of quantity, value, number, percentage, amount, or
weight. In various
embodiments, the term "about" refers to a variance of 20, 15, 10, 9, 8, 7, 6,
5, 4, 3, 2 or 1%
relative to the reference quantity, value, number, percentage, amount, or
weight.
[0027] The term "oral dosage form" as used herein, has its ordinary
meaning as
understood by those skilled in the art and thus includes, by way of non-
limiting examples, a
formulation of a drug or drugs in a form orally administrable to a human,
including pills, tablets,
cores, capsules, caplets, loose powder, liquid solution or suspension.
-6-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
[0028] The term "phenylacetic acid" as used herein, is also known as
benzeneacetic
OH
acid or 2-phenylacetic acid). It has the following chemical structure: 40
[0029] The term "phenylacetate" as used herein, refers to the anionic
form of
0
1.1
phenylacetic acid with the following chemical structure: 0
[0030] The term "L-omithine phenylacetate" as used herein, refer to a
compound
consisting of L-omithine cation and phenylacetate anion. It has the following
chemical
0
0
H' (OH
1101 0
structure: NH2
[0031] The term "phenylacetylglutamine" as used herein, refers to the
product
formed by the conjugation of phenylacetic acid and glutamine. It is a common
metabolite that
ONH
1,
-%-s-1
HO, ...-- =.,.
N
can be found in human urine. It has the following chemical structure: 0
[0032] As used herein, the term "percent conversion of phenylacetate to
phenylacetylglutamine over 24 hours" refers to the mass percent of
phenylacetate administered
to a patient that is converted to phenylacetylglutamine collected over 24
hours in the urine.
[0033] The term "pharmaceutically acceptable carrier" or
"pharmaceutically
acceptable excipient" includes any and all solvents, dispersion media,
coatings, antibacterial and
antifungal agents, isotonic and absorption delaying agents and the like. The
use of such media
and agents for pharmaceutically active substances is well known in the art.
Except insofar as
any conventional media or agent is incompatible with the active ingredient,
its use in the
therapeutic compositions or formulations is contemplated. Supplementary active
ingredients can
also be incorporated into the compositions or formulations. In addition,
various adjuvants such
as are commonly used in the art may be included. These and other such
compounds are
described in the literature, e.g., in the Merck Index, Merck & Company,
Rahway, NJ.
Considerations for the inclusion of various components in pharmaceutical
compositions are
described, e.g., in Gilman et al. (Eds.) (1990); Goodman and Gilman's: The
Pharmacological
Basis of Therapeutics, 8th Ed., Pergamon Press.
[0034] The term "pharmaceutically acceptable salt" refers to salts that
retain the
biological effectiveness and properties of the compounds of the preferred
embodiments and,
-7-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
which are not biologically or otherwise undesirable. In many cases, the
compounds of the
preferred embodiments are capable of forming acid and/or base salts by virtue
of the presence of
amino and/or carboxyl groups or groups similar thereto. Pharmaceutically
acceptable acid
addition salts can be formed with inorganic acids and organic acids. Inorganic
acids from which
salts can be derived include, for example, hydrochloric acid, hydrobromic
acid, sulfuric acid,
nitric acid, phosphoric acid, and the like. Organic acids from which salts can
be derived include,
for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic
acid, maleic acid,
malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic
acid, cinnamic acid,
mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic
acid, salicylic acid,
and the like. Pharmaceutically acceptable base addition salts can be formed
with inorganic and
organic bases. Inorganic bases from which salts can be derived include, for
example, sodium,
potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper,
manganese, aluminum,
and the like; particularly preferred are the ammonium, potassium, sodium,
calcium and
magnesium salts. Organic bases from which salts can be derived include, for
example, primary,
secondary, and tertiary amines, substituted amines including naturally
occurring substituted
amines, cyclic amines, basic ion exchange resins, and the like, specifically
such as
isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine,
and ethanolamine.
Many such salts are known in the art, as described in WO 87/05297, Johnston et
al., published
September 11, 1987 (incorporated by reference herein in its entirety).
[0035] "Subject" as used herein, means a human or a non-human mammal,
e.g., a
dog, a cat, a mouse, a rat, a cow, a sheep, a pig, a goat, a non-human primate
or a bird, e.g., a
chicken, as well as any other vertebrate or invertebrate.
[0036] "Treat," "treatment," or "treating," as used herein refers to
administering a
pharmaceutical composition/formulation for prophylactic and/or therapeutic
purposes. The term
"prophylactic treatment" refers to treating a patient who is not yet suffering
from a disease, but
who is susceptible to, or otherwise at risk of, a particular liver disease,
whereby the treatment
reduces the likelihood that the patient will develop a liver disease. The term
"therapeutic
treatment" refers to administering treatment to a patient already suffering
from a liver disease.
[0037] The compositions or formulations described herein are preferably
provided in
unit dosage form. As used herein, a "unit dosage form" is a
composition/formulation containing
an amount of a compound that is suitable for administration to an animal,
preferably mammal
subject, in a single administration, according to good medical practice. The
preparation of a
single or unit dosage form however, does not imply that the dosage form is
administered once
per day or once per course of therapy, or that the unit dosage form contains
all of the dose to be
administered at a single time. Such dosage forms are contemplated to be
administered once,
-8-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
twice, thrice or more per day, and may be given more than once during a course
of therapy,
though a single administration is not specifically excluded. In addition,
multiple unit dosage
forms may be administered at substantially the same time to achieve the full
dose intended (e.g.,
two or more tablets may be swallowed by the patient to achieve a complete
dose). The skilled
artisan will recognize that the formulation does not specifically contemplate
the entire course of
therapy and such decisions are left for those skilled in the art of treatment
rather than
formulation.
[0038] As used herein, the act of "providing" includes supplying,
acquiring, or
administering (including self-administering) a composition described herein.
[0039] As used herein, the term "administering" a drug includes an
individual
obtaining and taking a drug on their own. For example, in some embodiments, an
individual
obtains a drug from a pharmacy and self-administers the drug in accordance
with the methods
provided herein.
[0040] In any of the embodiments described herein, methods of treatment
can
alternatively entail use claims, such as Swiss-type use claims. For example, a
method of treating
fibrosis with a composition can alternatively entail the use of a composition
in the manufacture
of a medicament for the treatment of fibrosis, or the use of a composition for
the treatment of
fibrosis.
[0041] Those skilled in the art will understand that pharmacokinetic
parameters may
be determined by comparison to a reference standard using clinical trial
methods known and
accepted by those skilled in the art, e.g., as described in the examples set
forth herein. Since the
pharmacokinetics of a drug can vary from patient to patient, such clinical
trials generally involve
multiple patients and appropriate statistical analyses of the resulting data
(e.g., ANOVA at 90%
confidence). Comparisons of pharmacokinetic parameters can be on a dose-
adjusted basis, as
understood by those skilled in the art.
Low Dose Formulations
[0042] Some embodiments of the present disclosure relate to ORAL
pharmaceutical
formulations, comprising L-ornithine phenylacetate in an dosage of about 0.1 g
to about 10 g,
and one or more pharmaceutically acceptable excipients or carriers. In some
embodiments, the
formulation provides an immediate release profile of L-ornithine phenylacetate
upon
administration (for example, an immediate-release oral formulation in the form
of a liquid
solution or suspension). Other embodiments provide a controlled-release or
extended release
profile. In preferred embodiments, the pharmaceutical formulation is an oral
pharmaceutical
formulation. In some embodiments, the L-ornithine phenylacetate is in a dosage
of about 0.5 g,
-9-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
about 1 g, about 1.5 g, about 2 g, about 2.5 g, about 3 g, about 3.5 g, about
4 g, about 4.5 g,
about 5 g, about 5.5 g, about 6 g, about 6.5 g, about 7 g, about 7.5 g, about
8 g, about 8.5 g,
about 9 g, about 9.5 g, or about 10 g, or in a dosage range defined by any of
the two preceding
values (for example, about 1 g to about 9 g, about 2 g to about 8 g, about 3 g
to about 7g, about
4 g to about 6 g, about 1 g to about 6 g, about 1 g to about 5 g, about 1 g to
about 4 g, about 1 g
to about 3 g, about 2 g to about 6 g, about 2 g to about 5 g, or about 2 g to
about 4 g). In one
embodiment, the oral dosage is about 2.5 g. In another embodiment, the oral
dosage is about 5
g.
[0043] In some embodiments, the pharmaceutical formulation is in a
single unit
dosage form. In some other embodiments, the pharmaceutical formulation is in
two or more unit
dosage forms (i.e., a divided dose). For example, where an oral dosage is
about 5 g, it may be
provided in the form of four or five tablets, each containing about 1.25 g or
1 g of L-ornithine
phenylacetate. In some embodiments, the unit dosage form is a tablet, a
capsule, a pill, pellets,
free-flowing powder, or liquid. In one embodiment, the unit dosage form is a
liquid solution
compring 5 g of L-ornithine phenylacetate.
[0044] In some embodiments, the pharmaceutical formulation provides
conversion of
phenylacetate to phenylacetylglutamine over 24 hours of greater than about
30%, greater than
about 40%, greater than about 50%, greater than about 60%, greater than about
70%, greater
than about 80%, or greater than about 90%. In some further embodiments, the
formulation
provides conversion of phenylacetate to phenylacetylglutamine over 24 hours of
greater than
about 80%. In some embodiments, the conversion efficiency is determined based
on excreted
urinary phenylacetylglutamine.
[0045] In some embodiments, the pharmaceutical formulation provides
conversion of
phenylacetate to phenylacetylglutamine over 12 hours of greater than about
30%, greater than
about 40%, greater than about 50%, greater than about 60%, greater than about
70%, greater
than about 80%, or greater than about 90%. In some further embodiments, the
formulation
provides conversion of phenylacetate to phenylacetylglutamine over 12 hours of
greater than
about 60%. In some embodiments, the conversion efficiency is determined based
on excreted
urinary phenylacetylglutamine.
[0046] The low dose pharmaceutical formulations described herein may be
administered by any suitable route, for example, it may be administered by
oral, intravenous,
intragastric, intraperitoneal or intravasular routes. In a preferred
embodiment, the
pharmaceutical formulation of L-ornithine is an oral dosage form, for example,
a oral solution.
In another embodiment, the pharmaceutical formulation is an intravenous dosage
form.
-10-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
Methods of Treatment
[0047] Some
embodiments of the present disclosure relate to methods of treating or
ameliorating hyperammonemia comprising orally administering to a subject in
need thereof a
pharmaceutical formulation comprising an effective amount of L-ornithine
phenylacetate, in
particular the oral pharmaceutical formulation as described herein. In some
embodiments, the
subject has acute liver failure or chronic liver diseases. In some
embodiments, the subject has
liver cirrhosis or liver decompensation. In some such embodiment, the chronic
liver disease or
liver cirrhosis has a classification of Child-Pugh class A, B or C. Some
embodiments comprise
identifying a subject as having a liver disease with a classification of Child-
Pugh A and then
administering a composition as described herein. Some embodiments comprise
identifying a
subject as having a liver disease with a classification of Child-Pugh B and
then administering a
composition as described herein. In some embodiments, the subject has hepatic
encephalopathy.
Some embodiments comprise identifying a subject as having a liver disease with
a classification
of Child-Pugh C and then administering a composition as described herein. In
still some
embodiments, the subject has portal hypertension. In some embodiments, the
subject has a urea
cycle disorder. In some other embodiment, the subject has recently
discontinued treatment of
lactulose, for example, the subject has discontinued treatment of lactulose
for 1 day, 2 days, 3
days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, 4 weeks, or longer.
[0048] In
some embodiments of the methods described herein, the methods described
herein provide a plasma Cmax of phenylacetic acid ranging from about 10 pg/mL
to about 150
pg/mL. In some such embodiments, the plasma Cmax of phenylacetic acid is from
about 20
pg/mL to about 140 pg/mL. In some such embodiments, the plasma Cmax of
phenylacetic acid
is from about 30 pg/mL to about 130 pg/mL. In some such embodiments, the
plasma Cmax of
phenylacetic acid is from about 40 pg/mL to about 120 pg/mL. In some further
embodiments,
the plasma Cmax of phenylacetic acid is from about 50 pg/mL to about 110
pg/mL.
[0049] In
some embodiments of the methods described herein, the plasma Cmax of
metabolite phenylacetylglutamine ranges from about 5 pg/mL to about 100 pg/mL.
In some such
embodiments, the plasma Cmax of metabolite phenylacetylglutamine is from about
10 pg/mL to
about 80 pg/mL. In
some such embodiments, the plasma Cmax of metabolite
phenylacetylglutamine is from about 20 pg/mL to about 60 pg/mL. In some such
embodiments,
the plasma Cmax of metabolite phenylacetylglutamine is from about 25 pg/mL to
about 50
pg/mL. In some further embodiments, the plasma Cmax of metabolite
phenylacetylglutamine is
from about 30 pg/mL to about 45 pg/mL.
[0050] Some
embodiments of the present disclosure relate to methods of treating
hyperammonemia comprising administering to a subject in need thereof an oral
pharmaceutical
-11-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
formulation comprising L-omithine phenylacetate, where the pharmaceutical
formulation
provides a plasma Cmax of phenylacetic acid ranging from about 10 ng/mL about
150 ng/mL.
In particular, the oral pharmaceutical composition comprising L-omithine
phenylacetate
provides a plasma Cmax of phenylacetic acid of about 10 ng/mL, about 15 ng/mL,
about 20
ng/mL, about 25 ng/mL, about 30 ng/mL, about 35 ng/mL, about 40 ng/mL, about
45 ng/mL,
about 50 ng/mL, about 55 ng/mL, about 60 ng/mL, about 65 ng/mL, about 70
ng/mL, about 75
ng/mL, about 80 ng/mL, about 85 ng/mL, about 90 ng/mL, about 95 ng/mL, about
100 ng/mL,
about 105 ng/mL, about 110 ng/mL, about 115 ng/mL, about 120 ng/mL, about 125
ng/mL,
about 130 ng/mL, about 135 ng/mL, about 140 ng/mL, about 145 ng/mL, or about
150 ng/mL,
or a range as defined by any of the two preceding values. In one embodiment,
the plasma Cmax
level of phenylacetic acid is from about 20 ng/mL to about 140 ng/mL. In
another embodiment,
the plasma Cmax level of phenylacetic acid is from about 30 ng/mL to about 130
ng/mL. In still
another embodiment, the plasma Cmax level of phenylacetic acid is from about
40 ng/mL to
about 120 ng/mL. In a further embodiment, the plasma Cmax level of
phenylacetic acid is from
about 50 ng/mL to about 110 ng/mL. In some embodiments, the plasma AUC04 or
AUCof of
phenylacetic acid is from about 100 to about 1000 hr*ng/mL, from about 150
hr*ng/mL to
about 900 hr*ng/mL, from about 200 hr*ng/mL to about 800 hr*ng/mL, from about
250
hr*ng/mL to about 700 hr*ng/mL, from about 300 hr*ng/mL to about 650 hr*ng/mL,
from
about 350 hr*ng/mL to about 600 hr*ng/mL, or from about 400 hr*ng/mL to about
550
hr*ng/mL. In some embodiments, the pharmaceutical formulation also provides a
plasma Cmax
of phenylacetylglutamine ranging from about 5 ng/mL to about 100 ng/mL. In
particular, the
oral pharmaceutical composition comprising L-omithine phenylacetate provides a
plasma Cmax
of phenylacetylglutamine of about 5 ng/mL, about 10 ng/mL, about 15 ng/mL,
about 20 ng/mL,
about 25 ng/mL, about 30 ng/mL, about 35 ng/mL, about 40 ng/mL, about 45
ng/mL, about 50
ng/mL, about 55 ng/mL, about 60 ng/mL, about 65 ng/mL, about 70 ng/mL, about
75 ng/mL,
about 80 ng/mL, about 85 ng/mL, about 90 ng/mL, about 95 ng/mL, or about 100
ng/mL, or a
range as defined by any of the two preceding values. In one embodiment, the
plasma Cmax of
phenylacetylglutamine is from about 10 ng/mL to about 80 ng/mL. In another
embodiment, the
plasma Cmax of phenylacetylglutamine is from about 20 ng/mL to about 60 ng/mL.
In yet
another embodiment, the plasma Cmax of phenylacetylglutamine is from about 25
ng/mL to
about 50 ng/mL. In some embodiments, the plasma AUCo_t of
phenylacetylglutamine is from
about 25 hr*ng/mL to about 500 hr*ng/mL, from about 50 hr*ng/mL to about 300
hr*ng/mL,
from about 100 hr*ng/mL to about 200 hr*ng/mL, or from about 120 hr*ng/mL to
about 180
hr*ng/mL. In some embodiments, the plasma AUCo_inf of phenylacetylglutamine is
from about
25 hr*m/mL to about 500 hr*m/mL, or from about 50 hr*m/mL to about 400
hr*m/mL, from
-12-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
about 75 hr*pg/mL to about 300 hr*pg/mL, from about 100 hr*pg/mL to about 250
hr*pg/mL,
or from about 150 hr*pg/mL to about 200 hr*pg/mL.
[0051] In
some embodiments of the methods described herein, the oral
pharmaceutical composition is administered under fasting condition. In
some other
embodiments, the oral pharmaceutical composition is administered under fed
condition, for
example, concurrently or within 60 minutes after a meal.
[0052] In
some embodiments of the methods described herein, the oral
pharmaceutical formulation of L-omithine phenylacetate provides a controlled
release of L-
ornithine phenylacetate after administration. In some other embodiments, the
oral
pharmaceutical formulation of L-omithine phenylacetate provides an immediate
release of L-
ornithine phenylacetate after administration.
[0053] In
some embodiments of the methods described herein, L-omithine
phenylacetate is administered in an amount from about 0.1 g to about 50 g per
day, from about
0.5 g to about 45 g per day, from about 1 g to about 40 g per day, from about
1.5 g to about 35 g
per day, from about 2 g to about 30 g per day, from about 2.5 g to about 25 g
per day, from
about 3 g to about 20 g per day, or from about 5 g to about 15 g per day. In
some embodiments,
the pharmaceutical formulation is for administration at least once a day. In
some further
embodiments, the pharmaceutical formulation is for administration two or more
times per day.
In one embodiment, the pharmaceutical formulation is for thrice daily oral
administration.
[0054] In
some embodiments of the methods described herein, L-omithine
phenylacetate is administered as a single dose in an amount from about 1.0 g
to about 10.0 g. In
some further embodiments, L-omithine phenylacetate is administered as a single
dose in an
amount from about 2 g to about 8 g. In various other embodiments, L-omithine
phenylacetate is
administered as a single dose in a range of about 1 g to about 9 g, about 2 g
to about 8 g, about 3
g to about 7g, about 4 g to about 6 g, about 1 g to about 6 g, about 1 g to
about 5 g, about 1 g to
about 4 g, about 1 g to about 3 g, about 2 g to about 6 g, about 2 g to about
5 g, or about 2 g to
about 4 g. In one embodiment, L-omithine phenylacetate is administered as a
single dose in an
amount about 2.5 g. In another embodiment, L-omithine phenylacetate is
administered as a
single dose in an amount about 5 g. In some such embodiment, the
pharmaceutical formulation
containing such amount of L-omithine phenylacetate is in a single oral dosage
form. In some
other such embodiments, the pharmaceutical formulation containing such amount
of L-omithine
phenylacetate is in two or more unit dosage forms. For example, some
embodiments comprise
administering 1 to 5 unit dosage forms each comprising from about 0.1 g to
about 2 g of L-
ornithine phenylacetate, or about 2 to 4 unit dosage forms each comprising
from about 0.5 g to
about 1.25 g of L-omithine phenylacetate. Some embodiments comprise
administering 4 unit
-13-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
dosage forms each comprising about 1.25 g of L-ornithine phenylacetate. Some
embodiments
comprise administering 5 unit dosage forms each comprising about 1 g of L-
ornithine
phenylacetate. Some other embodiments comprise administering 1 unit dosage
form comprising
about 5 g of L-ornithine phenylacetate. In one embodiment, the pharmaceutical
formulation is
administered three times a day. For example, where multiple unit dosage forms
are
administered at a time, the multiple unit dosage administration is repeated
three time a day. In
another embodiment, the pharmaceutical formulation is administered once a day.
[0055] In some embodiments of the methods described herein, the
pharmaceutical
formulation provides conversion of phenylacetate to phenylacetylglutamine over
24 hours of
greater than about 30%, greater than about 40%, greater than about 50%,
greater than about
60%, greater than about 70%, greater than about 80%, or greater than about
90%. In some
further embodiments, the pharmaceutical formulation provides conversion of
phenylacetate to
phenylacetylglutamine over 24 hours of greater than about 80%. In some
embodiments, the
conversion efficiency is determined based on excreted urinary
phenylacetylglutamine.
[0056] In some embodiments of the methods described herein, the
pharmaceutical
formulation provides conversion of phenylacetate to phenylacetylglutamine over
12 hours of
greater than about 30%, greater than about 40%, greater than about 50%,
greater than about
60%, greater than about 70%, greater than about 80%, or greater than about
90%. In some
further embodiments, the formulation provides conversion of phenylacetate to
phenylacetylglutamine over 12 hours of greater than about 60%. In some
embodiments, the
conversion efficiency is determined based on excreted urinary
phenylacetylglutamine.
[0057] In any embodiment of the plasma Cmax or AUC value described
herein, such
value may be selected from a mean or median plasma Cmax or AUC value. In some
embodiments, the plasma Cmax and AUC described herein is achieved after a
single dose
administration of an oral pharmaceutical formulation of L-ornithine
phenylacetate. In some
other embodiments, the plasma Cmax and AUC described herein is a stead-state
plasma Cmax
and AUC achieved after mutiple doses administration of an oral pharmaceutical
formulation of
L-ornithine phenylacetate. In some embodiments, the plasma Cmax and AUC
described herein
are measured at fasting state. In some other embodiments, these PK parameters
are measured at
fed state.
[0058] Some examples of substances that can serve as pharmaceutically-
acceptable
carriers or excipients thereof, are sugars, such as lactose, glucose and
sucrose; starches, such as
corn starch and potato starch; cellulose and its derivatives, such as sodium
carboxymethyl
cellulose, ethyl cellulose, and methyl cellulose; powdered tragacanth; malt;
gelatin; talc; solid
lubricants, such as stearic acid and magnesium stearate; calcium sulfate;
vegetable oils, such as
-14-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
peanut oil, cottonseed oil, sesame oil, olive oil, corn oil and oil of
theobroma; polyols such as
propylene glycol, glycerine, sorbitol, mannitol, and polyethylene glycol;
alginic acid;
emulsifiers, such as the TWEENS; wetting agents, such sodium lauryl sulfate;
coloring agents;
flavoring agents; tableting agents, stabilizers; antioxidants; preservatives;
pyrogen-free water;
isotonic saline; and phosphate buffer solutions.
[0059] In some embodiments, the oral dosage form of L-ornithine
phenylacetate may
be in the form of a liquid, in particular a liquid solution. The oral dosage
formulation may also
comprise conventional pharmaceutical compatible adjuvants, excipients or
carriers, including
those commonly used in the oral solution formulation as discussed herein.
[0060] In some embodiments, the oral formulation described herein
provides for
lower doses than previously expected. For example, RAVICTI (glycerol
phenylbutyrate, a pre-
prodrug of phenylacetate) was found in clinical studies at a dose of 6 mL
(delivering about 1.02
g/mL of phenylbutyrate) twice daily to lower the incidence of hepatic
encephalopathy events.
Both the immediate release and the controlled release oral pharmaceutical
formulations of L-
ornithine phenylacetate described herein provide similar percentage of PAGN
urinary excretion,
permitting use of substantially lower API doses, compared to RAVICTI or other
phenylacetate
formulations.
EXAMPLES
[0061] The following examples, including experiments and results
achieved, are
provided for illustrative purposes only and are not to be construed as
limiting the present
application.
Example 1 ¨ Phase I Pharmacokinetic Studies in Healthy Humans
[0062] An open-label, five-treatment, five-period single-dose crossover
Phase 1
human clinical study was conducted to evaluate the pharmacokinetics of
phenylacetic acid and
phenylacetylglutamine after a single dose of three extended-release oral
dosage forms of L-
ornithine phenylacetate in comparison to RAVICTI (glycerol phenylbutyrate), a
prodrug of
phenylacetic acid. The study also compared the pharmacokinetics and safety of
single doses of
the three extended-release oral dosage forms of L-ornithine phenylacetate in
comparison to a
single dose of an immediate release oral solution of L-ornithine
phenylacetate.
[0063] The five treatments are listed as follows: each of Treatment A,
B, and C
refers to a single oral dose of 10 g Formulations A, B and C (each is an
equivalent of about 5 g
PAA) and the components of these formulations are summarized in Table 1 below;
Treatment D
refers to a single oral dose of 6 mL RAVICTI (equivalent of about 5 g PAA);
Treatment E
-15-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
refers to a single oral dose of an immediate release formulation of 5 g L-
ornithine phenylacetate
(equivalent of about 2.5 g PAA).
Table 1.
Component Quantity (g/dose)
Formulation Formulation Formulation
A
Drug Layered Core Pellets (g)
L-ornithine phenylacetate 10.00 10.00 10.00
Sugar Spheres (500-600 pm) 17.57 17.57 17.57
Talc 0.50 0.50 0.50
Hy droxy propy lmethylcel lulose 0.50 0.50 0.50
Total (Dry weight) Core Pellets 28.57 28.57 28.57
Extended Release Coating (g)
Ethyl cellulose 4.54 11.02 NA
Dibutyl Sebacate 0.50 1.22 NA
Eudragit NM 30D NA NA 11.42
Talc NA NA 8.57
Total Dry Weight per Unit Dose 33.6 40.8 48.6
[0064] The primary objective is to assess the plasma profiles and
pharmacokinetics
of phenylacetic acid (a potent ammonia scavenger), ornithine and
phenylacetylglutamine (the
end-product responsible for clearing ammonia) following a single oral dose of
three extended-
release formulations of L-ornithine phenylacetate in comparison to an oral
solution of L-
ornithine phenylacetate and a prodrug of phenylacetic acid (glycerol
phenylbutyrate,
RAVICTIO) in healthy human subjects. The secondary objective is to determine
the safety,
tolerability, and palatability of three extended-release formulations in
healthy subjects.
[0065] Eligible male or female adult healthy subjects were enrolled to
first receive
four treatments (Treatments A-D) over 4 dosing periods in a crossover fashion
using a balanced
4x4 Latin Square design with an at least 7-day washout interval between
treatments followed by
receiving Treatment E for all subjects in the fifth (last) dose period after a
minimum 7-day
washout interval. Following dosing in each dose period, subjects underwent
serial blood and
urine sampling up to 24 hrs post dose for PK assessment.
PK Assessments
[0066] In the dose periods where Treatment A, B, C, or D was
administered (the ER
formulations of L-ornithine phenylacetate or RAVICTIO), venous blood samples
(5 mL each)
were collected at the following time points: immediately (within 15 mins)
prior to dosing, and
then at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, 20, and 24 hrs
post dose. In the dose period
-16-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
where the immediate release formulation of L-omithine phenylacetate was
administered (Period
5), venous blood samples (5 mL each) were collected at the following time
points: immediately
(within 15 mins) prior to dosing, and then at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5,
3, 3.5, 4, 4.5, 5, 6, 7,
8, 10, and 12 hrs post-dose.
[0067] In addition, urine samples were collected at the following time
intervals:
within 1 hour prior to dosing (spot sample), and then cumulatively over the
intervals of 0-4, 4-8,
8-12, and 12-24 hrs post dose. Plasma samples were separated by centrifugation
within 1 hour of
blood collection and stored at approximately ¨80 C until analyzed. The total
urine volume for
each collection interval were measured and recorded and aliquots of urine were
stored at
approximately ¨80 C until analyzed.
Bioanalytical Methods
[0068] Plasma samples were analyzed for concentrations of phenylacetic
acid
(PAA), phenylacetylglutamine (PAGN) and omithine (ORN) using a validated LC-
MS/MS
method. All urine samples were analyzed for concentrations of PAGN using a
validated LC-
MS/MS method.
Endpoints
[0069] Pharmacokinetics: Plasma concentration vs. time profiles of
phenylacetate,
omithine, and phenylacetylglutamine following a single oral dose of each of
the study drug were
analyzed by noncompartmental PK methods. Pharmacokinetic parameters that were
determined
include Cmax, tmax, AUCO-t, AUCO-.o, C12 and ti/2. The amount of PAGN excreted
in urine and the
percent of PAA dose excreted in urine as PAGN over each collection interval
and the entire 24
hr interval were also determined.
[0070] FIG. 1 and FIG. 2 illustrate the mean plasma profiles of PAA and
PAGN,
respectively, from this Phase 1 study. FIG. 1 demonstrates the Mean Plasma PAA
concentration vs. time curves following administration of a single oral dose
of the controlled-
release formulations in comparison to the immediate-release solution and
glycerol
phenylbutyrate. FIG. 2 demonstrates the Mean Plasma PAGN concentration vs.
time curves
following administration of a single oral dose of the controlled-release
formulations in
comparison to the immediate-release solution and glycerol phenylbutyrate.
[0071] The mean maximum concentration (Cmax) of plasma PAA from three
extended-release formulations ranged from approximately 50 to 90 pg/mL
occurring at various
time points over 4 to 9 hours after dosing. For comparison, RAVICTI produced
a mean plasma
PAA Cmax of approximately 10 pg/mL at 4 to 6 hours after dosing. The plasma
PAA data after
a single oral dose of 6mL RAVICTI are consistent with published data in
healthy subjects. In
-17-
CA 03004331 2018-05-03
WO 2017/083758
PCT/US2016/061678
addition, PAA exposure with the extended-release formulations of L-ornithine
phenylacetate
showed lower inter-subject variability than RAVICTI .
[0072] Plasma profiles of PAGN, the end-product of ammonia scavenging,
also
demonstrated a similar pattern as the PAA profiles. The mean Cmax of plasma
PAGN from the
three extended-release formulations of L-ornithine phenylacetate ranged from
approximately 30
to 45 pg/mL occurring at various time points over 4 to 10 hours after dosing.
For comparison,
RAVICTI produced a mean plasma PAGN Cmax of about 20 to 25 pg/mL at
approximately 5
hours. These data are also consistent with the published data in healthy
subjects.
[0073] The total urinary excretion data of PAGN over 24 hours is
summarized in
Table 2 below. The mean PAGN excretion amount was comparable for Treatment A
through C,
each with about 80% PAA converted to PAGN excretion over 24 hours. In
contrast, Treatment
D with RAVICTI only showed about 40% conversion efficiency compared to
Treatment A
through C at approximately the same molar PAA dose (in the case of RAVICTI ,
PAA is
provided from the glycerol phenylbutyrate prodrug). It was surprisingly
observed that the
immediate release formulation Treatment E also exhibited about 80% conversion
efficiency,
which provided a similar mean PAGN excretion amount at approximately half the
molar dose of
PAA administered in the RAVICTI arm.
Table 2.
Treatment Statistics Total PAGN PAA Equivalents Percint PAA
Amount Excreted Excreted over 24 Dose
Excreted
over 24 hours (G) hours (G) over
24 hours (%)
A N 12 12 12
Mean 8.26
83,8
Median 8.47 4,37 86,0
%CV 14.7 14.7 14.7
12 12 12
Mean 717 4.00
Methan 7.77 4...00
%CV 10,7 101 10,8
12 12 12
Mean 9j.i3 4.39 Set
Me6an 8.39 4.33 85.3
WI L 11.3 11.3 11.3
12 12
Mean 4.11 2.12 42..7
Medial 4.06209 42,1
----------- %CV 24,9 24,9 -------------------- 24..9
12 12 12
Mean 4.õ01 2.7 81.5
Methan 4.14 2.13 84.0
%CV , 152 15.1 15.1 :
-18-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
[0074] Conclusion: The controlled-release and immediate release
formulations were
well tolerated throughout the study, with no toxicities or serious adverse
events observed. The
results showed a robust, extended-release pattern for all three extended-
release formulations
with mean plasma PAA concentrations exceeding those achieved with RAVICTI
(glycerol
phenylbutyrate) at all time points for at least 24 hours post dose. In
addition, mean plasma
PAGN concentrations and urinary PAGN excretion were greater for all three
extended-release
dosage forms than for RAVICTI at approximately the same molar PAA dose. It
also
demonstrated that urinary PAGN excretion for the immediate release formulation
of L-ornithine
phenylacetate was approximately twice as efficient as for RAVICTI .
Example 2 ¨ Phase I Pharmacokinetic Studies in Child-Pugh Class A Subjects
[0075] In this example, a single-dose, partially randomized clinical
study to evaluate
g L-ornithine phenylacetate oral solution administered under fed conditions,
fasting
conditions, or under fasting conditions following discontinuation of lactulose
in 5 subjects with
cirrhosis (Child-Pugh class A). The purpose is to determine the
pharmacokinetics of PAA and
PAGN following a single 5 g dose of L-ornithine phenylacetate oral solution
administered under
fed conditions, fasting conditions, or under fasting conditions following
discontinuation of
lactulose as compared to a single 5 g intravenous dose of L-ornithine
phenylacetate under
fasting conditions in subjects with cirrhosis (Child-Pugh class A).
[0076] The treatments are summarized as follows: Treatment A is a single
5 g oral
dose of L-ornithine phenylacetate oral solution administered under fasting
conditions; Treatment
B is a single 5 g oral dose of L-ornithine phenylacetate oral solution
administered under fed
conditions; Treatment C is a single 5 g intravenous dose of L-ornithine
phenylacetate solution
infused over 1 hour under fasting conditions; and Treatment D is a single 5 g
oral dose of L-
ornithine phenylacetate oral solution administered under fasting condition
following
discontinuation of lactulose.
[0077] Eligible subjects received a single dose of study drug on Day 1.
Subjects were
confined at the Phase 1 unit from admission on Day -1 until the final blood
sample for
pharmacokinetic assessment is obtained. In Dosing Period 1, all subjects
received intravenous L-
ornithine phenylacetate (Treatment C), and in Dosing Period 4, all subjects
received a single
dose of L-ornithine phenylacetate oral solution following discontinuation of
lactulose
(Treatment D). Treatments A and B were administered during Dosing Periods 2
and 3 in a
randomized fashion. At the end of Dosing Periods 1, 2, and 3, subjects
returned to the clinic for
the next dosing period. At the end of Dosing Period 3, all subjects
discontinued lactulose. There
was a minimum 4-day washout interval between consecutive dosing periods.
-19-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
Pharmacokinetic Assessments
[0078]
Following each oral dose (Treatments A, B, and D), venous blood samples (5
mL each) was collected at the following time points: immediately (within 15
minutes) prior to
dosing, and then at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, 8,
10, and 12 hours post-dose.
For Treatment D (Dosing Period 4), an additional blood sample was obtained at
24 hours post-
dose. Following the intravenous dose (Treatment C), venous blood samples (5 mL
each) were
collected at the following time points: immediately (within 15 minutes) prior
to the start of
infusion, and then at 0.5 hours after the start of infusion, and immediately
prior to the end of
infusion, subsequently at 10, 20, 30, 45 and 60 minutes after the end of
infusion, and then at 1.5,
2, 2.5, 3, 4, 6, 8, 10, 12, and 24 hours after the end of infusion.
[0079] In
addition, urine samples were collected for each treatment at the following
time intervals: within 1 hour prior to dosing (spot sample), and then
cumulatively over the
intervals of 0-4, 4-8, and 8-12 hours post-dose. For Treatments C and D
(Dosing Periods 1 and
4), urine were also collected over the 12-24 hour post-dose interval.
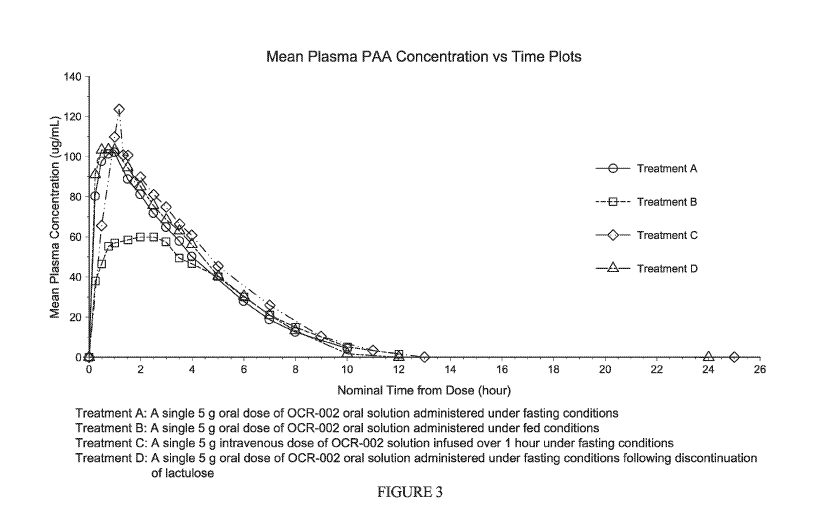
[0080] FIG. 3
and FIG. 4 illustrate the mean plasma profiles of PAA and PAGN,
respectively, from this Phase 1 study. FIG. 3 demonstrates the Mean Plasma PAA
concentration vs. time curves following administration of four treatments
(Treatments A, B, C
and D) described above. FIG. 4 demonstrates the Mean Plasma PAGN concentration
vs. time
curves following administration of the four treatments (Treatments A, B, C and
D) described
above. The pharmacokinetic parameters of PAA and PAGN are summarized in Table
3 and
Table 4.
Table 3. Summary of Pharmacokinetic Parameter Estimates of PAA
----- T, --------------------------------
I _________________________________________________________________________ 1
CL
Treatntot Stati 'sets T c¨
(pafriti...) ll'Aq 0111 {IlettVraL) (ikepoiln):4 Mr 4.43r)
4
,N 4 4 .4 .4 4 4 4
C mem VI 3 1.16 /.99 473.4 6/4:9 893
5..97
Median 109 U18 2;66 495,0 550.9 6,79 4.75
CV% :30,3 221 23,9 43,5 --- 42A.
46..5 Sts.1.7
_
N 5 5 5 5 6 5 4
A Mean 105 5:92 1.38 439.8 453.2: 3.43 88.0
Median /16 em I.2,41 4314 445õ6 3..39 96.2
CV% 276 25,4 14.9 476 46. 39.6 -- 3..94 1
N 6 5 5 6 5 6 4
B klea41 66.1 t55 1,36 M0,1 3n.7 4..15
ao.4
Mediw :952 1.60 1.31 347,7 363.3 4.26 61.8
CV % 216 71.7 122 434 + 42.1 42,5 7.4.
õ.......................+....
N 5 5 5 5 6 Ei. 4
D mem 113 D.66 /..53 457.9 483.0 4.9f)
103.0 1
, kialim 113 a.77 1 Af) 515.0 557:5 4.59 00. 2
I
1 CV% ... 23.4 5.9.9 '166 : 36.7 .. 364 .. 36.7 1:3.9
1
, ........................................................................ ,
-20-
CA 03004331 2018-05-03
WO 2017/083758
PCT/US2016/061678
Table 4. Summary of Pharmacokinetic Parameter Estimates of PAGN
PAGMP AA
Tmattnt,rxt 1*-
Suliw.c% 0.4MIL.1 110) MO (hr#44634) ihrf$90rtU
N .4 '4 4 4 4 4. 4 ... ,
C Mew 24$ 3.05 2.58
0,224
filtd.ian 24:3 3.6/ 2.43 1587 1854 14.92 0207 1
CV%
=+ .6.0 303 . 14..3 28.7 241
3311 36.3
.. ;
N .?-_ 5 .5 5 S 6 5 1
A merarl 26,0 4.02 2.35 161:1 199..5 16.7g
6.256 1
iklectian 244 4.02 2.8:1 168.4 208. 5 1 0.3I .0235
CV% 19.7 25.2 40.9 2es x9 ------- 41.4 96.f1
1
N .5 5 .5. 5 5 . 6 5 ;
B Mem 23:9 4:42 .2.78 '102,0 203.8
0:304 ;
Median 23,7 403 3.01 162.0 2104 2311. 0.208 1
4. CV% 16:5 '16:1 321 32,4 34,0 42.2 36:8 ;
;
N
Wan 1 27:4 313 1 2.6g 177.1 205.9 1.4.A Ø237
iMedi43 1 26, I 403 i 2.70 172.0 214,3 13. 0247
CV% 24.6. 10.0 .zu 32.0 31 .4 . 31,6 36,7
[0081] The plasma exposure data and PK parameter estimates of PAA in
Child-Pugh
A subjects after a single oral dose of 5 g of L-ornithine phenylacetate were
as expected based on
projections from previous data in healthy subjects.
[0082] Based on a small number of subjects, the study showed that after
a single oral
dose of 5 g of L-ornithine phenylacetate to Child-Pugh A subjects, mean peak
plasma PAA
concentration (Cmax) was slight (20%) lower and overall plasma exposure to PAA
(AUCo)
was about 30% higher than that in healthy subjects. The slightly higher AUC
value in Child-
Pugh A subjects is most likely due to slower metabolism of PAA in Child-Pugh A
subjects and
longer elimination half life of PAA which increased from about 0.9 hr in
healthy subjects to 1.4
hr in Child-Pugh A subjects. There was large intersubject variability in
plasma PAA exposure
after either intravenous or oral administration of L-ornithine phenylacetate.
[0083] After an oral dose of L-ornithine phenylacetate, PAA was almost
completely
bioavailable, as shown by the absolute bioavailability value (F) of 96% for
PAA, determined in
Child-Pugh A subjects after a single 5 g oral dose of L-ornithine
phenylacetate in comparison to
an intravenous dose.
[0084] Subjects on lactulose appeared to have similar PAA and PAGN
plasma
profiles and pharmacokinetics from administration of a single oral dose of L-
ornithine
phenylacetate before and after lactulose has been washed out.
[0085] Plasma exposure and pharmacokinetic profiles of PAGN were
comparable
between the four treatments, i.e., after a single oral dose of 5 g L-ornithine
phenylacetate
intravenously or orally with or without food or without lactulose in Child-
Pugh A subjects.
Mean AUCo_inf of PAGN was slightly (-10%) lower in Child-Pugh A subjects than
in healthy
-21-
CA 03004331 2018-05-03
WO 2017/083758 PCT/US2016/061678
subjects. Mean plasma half life of PAGN in Child-Pugh subjects, 2.6 hrs, was
longer than that in
healthy subjects, 1.4 hours.
Table 5. Summary of Urinary Excretion Data of PAGN
Duration of ' Total PAGhl Total PAA
Percent PAA
Urine
Tremmorst ' Amount Equivalents Oose
Excreted
I Statistics Collection
Excreted (G) Excreted
interval
N 4 4 4
C I Me m 24 hour 3.86 1.99 783
1 Medim 3.91 2,02 79.4
1 CV% 22.9 _______________________________________ 23,1 ________ 23,1
N 5 5 c
...I
,
,
A I Mean 12 hour 3.31 1.70 67,1
1 Median 3.16 1.63 642
i CV% 22,3 222 22.2
N 5 5 5
.
a I Mean 12 hour 3,45 1.
1.73 MO
1 Medhal 3.36 68.2
i CV% 10.0 _______________________________________ 19,0 _________ 19,0
. _
N 5 5 5
D 1 Mem 24 hour 4.17 2.15 847
1 Median 42 2.07 81 6
[0086]
Urinary excretion data showed that the mean percent PAA dose recovered in
urine as PAGN was 78.3% after a single IV dose of L-ornithine phenylacetate,
and 84.7% after a
single oral dose of L-ornithine phenylacetate (Treatment D). The lower % PAA
dose recovered
in urine as PAGN in Treatments A and B could be due to the shorter urine
collection interval,
i.e., 12 hours.
-22-