Note: Descriptions are shown in the official language in which they were submitted.
1
Tesofensine for reduction of body weight in Prader-Willi patients
Technical field
The present invention relates to the use of Tesofensine for the reduction of
body weight
or the reduction of hyperphagia in patients suffering from Prader-Willi
syndrome
(PWS).
Background
Prader-Willi syndrome or PWS is a complex genetic condition that affects many
parts
of the body. In infancy, this condition is characterized by weak muscle tone
(hypotonia),
feeding difficulties, poor growth, and delayed development. Beginning in
childhood,
affected individuals develop an insatiable appetite, which leads to chronic
overeating
(hyperphagia) and obesity. Therefore, weight reduction is critical for PWS
patients
suffering from obesity. Thus, there is impetus for finding new and alternative
ways of
treating and managing obesity resulting from PWS.
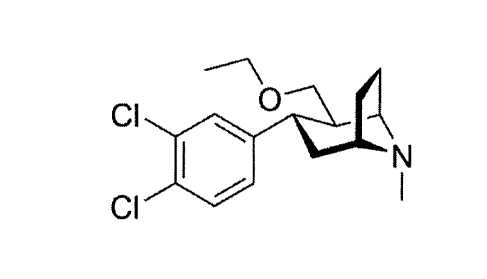
Tesofensine, i.e. [(1R,2R,3S,5S)-3-(3,4-dichloropheny1)-2-(ethoxymethyl)- 8-
methyl-8-
azabicyclo[3.2.1]octane], first described in WO 97/30997, is a triple
monoamine
reuptake inhibitor in development for the treatment of obesity.
Tesofensine effectively produces a weight loss in obese individuals of about
twice of
that seen with currently marketed anti-obesity drugs. Results from clinical
studies with
Tesofensine also showed that the compound has a good safety profile and is
well
tolerated. However, though no clinically relevant cardiovascular adverse
events or
changes in either blood pressure or pulse were seen, some cardiovascular
effects were
measured with slight increases in heart rate and trends in blood pressure.
Although
such small effects have no immediate risk to the patient, some medical and
regulatory
concerns have been raised based on observational studies, that even small
changes in
cardiovascular parameters may have long term implications on patients
benefit/risk
evaluation.
Preclinical and clinical data suggest that appetite suppression is an
important
mechanism by which Tesofensine exerts its robust weight reducing effect.
Notably, the
strong hypophagic response (i.e. less appetite, decreased feeding) to
Tesofensine
36782-064
CA 3029052 2019-01-07
2
treatment is demonstrated to be linked to central stimulation of noradrenergic
and
dopaminergic neurotransmission. However, the sympathomimetic mode of action of
Tesofensine may also associate with the elevated heart rate and blood pressure
observed in clinical settings.
Beta blockers, (8-blockers, beta-adrenergic blocking agents, beta antagonists,
beta-
adrenergic antagonists, beta-adrenoreceptor antagonists, or beta adrenergic
receptor
antagonists) are a class of drugs that are typically used for the management
of cardiac
arrhythmias, protecting the heart from a second heart attack (myocardial
infarction)
after a first heart attack (secondary prevention), and, in certain cases,
hypertension.
Beta blockers are also well known for their reductive effect on heart rate.
Metoprolol, i.e. 1 -(lsopropylamino)-344-(2-methoxyethyl)-phenoxy]- propan-2-
ol,
branded under various trade names, is a selective p1 (adrenergic) receptor
blocker
normally used in the treatment of various disorders of the cardiovascular
system, and
in particular hypertension.
Carvedilol (( )-[3-(9H-carbazol-4-yloxy)-2-hydroxypropyl][2-(2-
methoxyphenoxy)ethyl]amine) is a mixed, i.e. nonselective alpha and beta
blocker. It is
marketed under various trade names and is traditionally used in the treatment
of mild to
severe congestive heart failure (CHF) and high blood pressure.
WO 2013/120935 describes treatment of obesity by co-administration of
tesofensine
and metoprolol in order to ameliorate drug-induced elevation of blood pressure
or
increase in heart rate.
The serum half-life of tesofensine is nine days (Bara-Jimenez W, Dimitrova T,
Sherzai
A, Favit A, Mouradian MM, Chase TN (2004). "Effect of monoamine reuptake
inhibitor
NS 2330 in advanced Parkinson's disease". Mov Disord 19 (10): 1183-6.). In
comparison, the half-life of beta blockers is quite short with metoprolol in
the order of 3-
4 hours and carvedilol about 7 to 10 hours. Therefore simultaneous daily
administration
of these two drugs is likely to induce high fluctuations in the serum levels
of the beta
blocker and potentially recurrent temporary absence of therapeutic efficacy of
the beta
blocker.
36782-064
CA 3029052 2019-01-07
3
Summary
The present inventors discovered an encouraging efficacy signal, particularly
the effect
on hyperphagia in a study on nine adult patients with PWS. Several patients
discontinued prematurely due to adverse events. It is assumed that these
adverse
events were driven by unexpectedly high plasma concentrations of tesofensine
Because of these observations the current inventors contemplate treatment of
Prader-
Willi patients at a daily dose of 0.250 mg or less.
Thus, the present invention relates to the use of tesofensine for reduction of
body
weight in patients with PWS.
In a first aspect, the present disclosure relates to a method of reduction of
body weight
or reduction of hyperphagia in Prader-Willi patients comprising administering
Tesofensine or a pharmaceutically acceptable salt thereof in a daily dosage of
0.250
mg or less.
In a second aspect, the present disclosure relates to a pharmaceutical
composition
comprising no more than 0.25 mg Tesofensine; and 5 to 100 mg of ER beta
blocker,
such as Metoprolol; and 1 to 25 mg of IR beta blocker, such as Metoprolol.
Description of Drawings
Figure 1: An overview of the number of patients from the two countries Hungary
(HU)
and Czech Republic (CZ), including treatment allocation, sex, age, weight at
baseline
and patients who completed the study can be seen. The term "IMP" stands for
Investigation Medicinal Product and corresponds to the co-administration of
tesofensine (0.50 mg)/metoprolol (50 mg). Figure la displays the change in
body
weight over time in kilograms, whereas Figure lb displays the change in body
weight in
/), calculated as ((body weight at visit 2, 5, 9 or 14) - (body weight at
visitl )) / (body
weight at visit 1) x 100. Visits were performed on days 1, 7, 28, 56 and 91
(visits 1, 2,
5, 9 and 14). The bars indicate standard deviations.
Figure 2: The individual hyperphagia scores over time for individual patients
receiving
either the IMP (A) or the placebo (B).
36782-064
CA 3029052 2019-01-07
4
Figure 3: Change in waist circumference calculated at visit 2 (day 7), visit 5
(day 28),
visit 9 (day 56) and visit 14 (day 91) by the following formula: change in
waist
circumference = (waist circumference at day 7, 28, 56, or 91 after start of
treatment) ¨
(waist circumference at baseline).
Figure 4: The average tesofensine trough plasma concentrations from the TIPO-1
can
be seen. With a dose of 0.5 mg Tesofensine, a trough plasma concentration of
10
ng/ml was achieved.
Definitions
Extended release ¨ ER - also known as sustained-release [SR], extended-release
[ER,
XR, XL], and controlled-release [CR], is a mechanism used in pill tablets or
capsules to
dissolve a drug over time in order to be released slower and steadier into the
bloodstream.
Immediate release ¨ IR. The drug is released (dissolved) immediately after
ingestion.
Detailed description
Disclosed herein is a method of reduction of body weight or reduction of
hyperphagia in
Prader-Willi patients comprising administering Tesofensine or a
pharmaceutically
acceptable salt thereof in a daily dosage of less than 0.250 mg. In one
embodiment of
the invention, Tesofensine is administered in combination with a beta blocker.
The composition as described herein is useful as a medicament, e.g. for the
reduction
of body weight or reduction of hyperphagia in Prader-Willi patients.
Prader-Willi syndrome (PWS) is a genetic condition that affects many parts of
the body.
Infants with PWS have severe hypotonia (low muscle tone), feeding
difficulties, and
slow growth. In later infancy or early childhood, affected children typically
begin to eat
excessively and become obese. Other signs and symptoms often include short
stature, hypogonadism, developmental delays, cognitive impairment, and
distinctive
behavioral characteristics such as temper tantrums, stubbornness, and
obsessive-
compulsive tendencies. PWS is caused by missing or non-
36782-064
CA 3029052 2019-01-07
5
working genes on chromosome 15. Most cases are not inherited and occur
randomly.
Rarely, a genetic change responsible for PWS can be inherited.
Prader-Willi syndrome is characterized by severe infantile hypotonia with poor
suck
and failure to thrive; hypogonadism causing genital hypoplasia and pubertal
insufficiency; characteristic facial features; early-childhood onset obesity
and
hyperphagia; developmental delay/mild intellectual disability; short stature;
and a
distinctive behavioral phenotype. Sleep abnormalities and scoliosis are
common.
Growth hormone insufficiency is frequent, and replacement therapy provides
improvement in growth, body composition, and physical attributes. Management
is
otherwise largely supportive.
Consensus clinical Prader-Willi diagnostic criteria exist, but diagnosis
should be
confirmed through genetic testing. Prader-Willi syndrome is due to absence of
paternally expressed imprinted genes at 15q11.2-q13 through paternal deletion
of this
region (65-75% of individuals), maternal uniparental disomy 15 (20-30%), or an
imprinting defect (1-3%). Parent-specific DNA methylation analysis will detect
>99% of
individuals. However, additional genetic studies are necessary to identify the
molecular
class. There are multiple imprinted genes in this region, the loss of which
contribute to
the complete phenotype of Prader-Willi syndrome. However, absence of a small
nucleolar organizing RNA gene, SNORD116, seems to reproduce many of the
clinical
features. Sibling recurrence risk is typically <1%, but higher risks may
pertain in certain
cases. Prenatal diagnosis is available.
The term "hyperphagia" is used to express excessive hunger or desire for food
often
resulting in "overeating". It can be caused by disorders such as diabetes,
Kleine¨Levin
syndrome (a malfunction in the hypothalamus) and the genetic disorder
Prader¨Willi
syndrome. There are several approaches frequently used to describe
hyperphagia:
= By quantifying "overeating" as energy intake relative to a control group;
eating
beyond amount predicted for body size and body composition; and evaluating
food intake pre- vs. post-treatment (e.g., before and after people with leptin
deficiency are given recombinant leptin);
= Relative to a control group, by evaluating "hunger" (e.g., with visual
analog
scales in patients with PWS and controls); time to reach satiation relative to
a
control group; and duration of satiety;
36782-064
CA 3029052 2019-01-07
6
= Measuring preoccupation with food or "hyperphagic drive"; food seeking
behaviors (e.g., night eating, etc.); and
= Evaluating psychological symptoms such as distress and functional
impairment.
Clinical assessment of hyperphagia is done using a "Hyperphagia Questionnaire
for
Clinical Trials (HQ-CT)" as described by Fehnel et al. in "Development of the
Hyperphagia Questionnaire for Use in Prader-Willi Syndrome Clinical Trials" at
the
ISPOR 20th Annual International Meeting.
Due to the particular combination of extended and immediate release forms of a
beta
blocker in combination with tesofensine as described herein the composition of
the
present disclosure effectively alleviates cardiovascular side-effects of
tesofensine while
maintaining the therapeutic efficacy of tesofensine.
In one embodiment the method of the present disclosure is useful for the
reduction of
body weight or hyperphagia in Prader-Willi patients.
In one embodiment, the method of the present disclosure reduces body weight of
the
Prader-Willi patient by at least 3% after two months of treatment, such as
between 5%
and 10% or between 3% and 6%.
In one embodiment, the method of the present disclosure results in a reduction
of the
waist circumference of the Prader-Willi patient by at least 4 cm after 56 days
of
treatment, such as between 4 and 6 cm or between 6 and 10 cm.
In one embodiment, the method according to the present disclosure is used for
reduction of body weight or hyperphagia in Prader-Willi patients suffering
from
hypothalamic obesity (HO). HO typically in occurs in patients with tumors and
lesions in
the medial hypothalamic region. Hypothalamic dysfunction can lead to
hyperinsulinemia and leptin resistance. These patients have often suffered
damage to
the hypothalamus. Damage to the hypothalamus has long been known to promote
excessive eating (hyperphagia) and weight gain, termed "hypothalamic obesity."
This
form of weight gain is often not responsive to diet and exercise.
36782-064
CA 3029052 2019-01-07
7
In one embodiment the composition of the present disclosure is for use in a
method of
decreasing liver fat and/or visceral adiposity. Reduction of liver fat and/or
visceral
adiposity has been shown to be effective in the treatment of fatty liver
disorders.
Tesofensine significantly decreases waist circumference and sagittal diameter
(Astrup
et al., 2008, Lancet 372:1906-13); hence tesofensine is capable of reducing
visceral
adiposity.
The method of the present disclosure is preferably administered to a subject
in need
thereof once a day. However, in certain embodiments, the composition may be
administered more than once a day, such as twice a day or alternatively less
than once
a day, such as once every second or third day depending on the specific
formulation
and concentration of the individual components of the composition. The subject
treated
is preferably a human, such as an adult human aged 18 or older.
In one embodiment the present disclosure relates to use of the composition as
disclosed herein in the manufacture of a medicament for the reduction of body
weight
or hyperphagia in Prader-Willi patients.
In one embodiment, the method of the present disclosure results in a steady
state
Tesofensine serum concentration of from 5 to 15 ng/mL.
In an aspect of the invention, the method of reducing body weight or reducing
hyperphagia in Prader Willi patients comprises:
a. a first composition comprising an extended release (ER) composition of
an active pharmaceutical ingredient (API) selected from the beta-blocker
or a pharmaceutically acceptable salt thereof,
b. a second composition comprising an active pharmaceutical ingredient
(API) selected from Tesofensine or a pharmaceutically acceptable salt
thereof, and
c. a third composition comprising an immediate release (IR) composition of
an active pharmaceutical ingredient (API) selected from a beta blocker or
a pharmaceutically acceptable salt thereof.
In one aspect, the invention concerns a pharmaceutical composition comprising
said
first composition, second composition and third composition. In one
embodiment, said
36782-064
CA 3029052 2019-01-07
8
pharmaceutical composition comprises no more than 0.250 mg of Tesofensine, or
a
pharmaceutically acceptable salt thereof; and 5 to 100 mg of ER beta blocker,
such as
Metoprolol; and 1 to 25 mg of IR beta blocker, such as Metoprolol.
The beta blocker may for example be metoprolol or carvedilol or
pharmaceutically
acceptable salts thereof. These include the phosphate, succinate, maleate,
sulfate,
glutarate, lactate, benzoate, and mandelate salts.
The in vitro bio-dissolution profile (as determined by USP Type II apparatus,
rotating
paddle, with 500 ml of Phosphate buffer at pH 7.4, 37 C set at rotating speed
of 50
rpm) of the beta blocker is preferably as follows:
Dissolution time Range
1 hour 10-35%
4 hours 25-45%
8 hours 45-65%
hours >80%
15 For example, the combined in vitro bio-dissolution profile of metoprolol
preferably has a
dissolution profile lying within one or more of the following release ranges
for different
metoprolol IR:ER ratios at various time points (as determined by USP Type II
apparatus, rotating paddle, with 900 ml of Phosphate buffer at pH 7.4, 37 C
set at
rotating speed of 75 rpm).
Dissolution Calculated Dissolution Calculated Dissolution Overall
time dissolution ranges dissolution ranges range
10 mg (10:100) 25mg (25:100)
IR+100mg I R+100mg
ER ER
1 hour 13% 10-20% 23% 20-30% 10-30%
4 hours 29% 20-40% 38% 30-50% 20-50%
8 hours 53% 40-65% 58% 50-70% 40-70%
24 hours 88% >80% 90% >80% >80%
Dissolution Calculated Dissolution Calculated Dissolution Overall
time dissolution ranges dissolution ranges range
10 mg (10:100) 25mg (25:100)
IR+100mg IR+100mg
36782-064
CA 3029052 2019-01-07
9
ER ER
1 hour 13% 10-20% 23% 20-30% 10-30%
4 hours 29% 20-40% 38% 25-50% 20-50%
8 hours 53% 40-65% 58% 40-70% 40-70%
20 hours 88% >80% 90% >80% >80%
In general the tesofensine of the composition is dissolved within Y2-1 hour.
The in vitro
dissolution profile with tesofensine under the conditions above is at least
80% of the
API within 45 minutes.
Many physiological factors influence both the gastrointestinal transit time
and the
release of a drug from a controlled release dosage form, and thus influence
the uptake
of the drug into the systemic circulation. A sustained-release dosage form
should
release the beta blocker at a controlled rate such that the amount of active
ingredient
available in the body to treat the condition is maintained at a relatively
constant level
over an extended period of time. The release of an active ingredient from a
controlled
release dosage form is generally controlled by diffusion through a coating.
It is likewise important that part of the beta blocker is released rapidly so
that a
therapeutically effective level of the beta blocker is reached rapidly.
Tesofensine
The pharmaceutical composition described herein comprises an active
pharmaceutical
ingredient (API) selected from tesofensine or a pharmaceutically acceptable
salt
thereof.
Tesofensine R1R,2R,3S,5S)-3-(3,4-dichloropheny1)-2-(ethoxymethyl)-8-methyl-8-
azabicyclo[3.2.1]octane] is a centrally acting triple monoamine re-uptake
inhibitor (MRI)
with intrinsic inhibitory activity on noradrenaline, serotonin and dopamine
transporter
function. When corrected for placebo and diet effects, long-term Tesofensine
treatment
produces a weight loss of about 10% in obese patients, which is twice as much
as that
achieved by currently marketed anti-obesity drugs.
The chemical structure of Tesofensine is
36782-064
CA 3029052 2019-01-07
10
0
CI
CI
Preclinical and clinical data suggest that appetite suppression is an
important
mechanism by which Tesofensine exerts its robust weight-reducing effect. In
addition,
Tesofensine has also been demonstrated to increase nocturnal energy
expenditure in
human subjects. These findings have recently been corroborated and extended in
preclinical settings, demonstrating that Tesofensine induces a robust and
sustained
weight loss in a rat model of diet-induced obesity (D10) of which the long-
lasting
reduction in body weight is caused by appetite suppression with a gradual
increase in
energy expenditure. Notably, the hypophagic effect of Tesofensine in DIO rats
is
critically dependent on stimulated al adrenoceptor activity, and to a less
extend
dopamine D1 receptor function, indicating that enhancement of central
noradrenergic
and dopaminergic neurotransmission constitute important mechanisms underlying
the
robust appetite-suppressing effect of Tesofensine.
Overall, chronic Tesofensine treatment is associated with minor adverse
events, and
with minimal cardiovascular effects, suggesting that Tesofensine may generally
be a
well-tolerated long-term treatment for obesity. However, dose-dependent
elevations in
heart rate and significant increases in blood pressure have been reported in
obese
individuals. The long-term implications of such Tesofensine-induced
cardiovascular
effects are not known and can potentially play a role in the benefit/risk
evaluation of
patients treated with Tesofensine.
Beta blockers
The present invention involves the use of beta blockers in certain
embodiments. The
beta blocker may be any conventional beta blocker known in the art.
Preferably, the
beta blocking drug is selected from the following groups of compounds, which
groups
of compounds are known in the art and may be commercially available under
different
brand names, or may be obtained as described in the literature.
36782-064
CA 3029052 2019-01-07
11
Non-selective beta blockers
In one embodiment, the beta blocker is a non-selective beta blocker. Examples
of non-
selective beta blockers include alprenolol, amosulalol, bucindolol, carteolol,
levobunolol, mepindolol, metipranolol, nadolol, oxprenolol, penbutolol,
pindolol,
propranolol, sotalol and timolol.
In one embodiment, the beta blocker is selected from the group consisting of
alprenolol, amosulalol, bucindolol, carteolol, levobunolol, mepindolol,
metipranolol,
nadolol, oxprenolol, penbutolol, pindolol, propranolol, sotalol, timolol and
pharmaceutically acceptable salts thereof.
Beta 1-selective beta blockers
In another embodiment, the beta blocker is a beta 1-selective beta blocker.
Examples of beta 1-selective beta blockers include acebutolol, atenolol,
betaxolol,
bisoprolol, esmolol, landiolol, metoprolol and nebivolol.
In one embodiment, the beta blocker is selected from the group consisting of
acebutolol, atenolol, betaxolol, bisoprolol, esmolol, landiolol, metoprolol,
nebivolol and
pharmaceutically acceptable salts thereof.
In a particular embodiment, the beta blocker is metoprolol or a
pharmaceutically
acceptable salt thereof.
Mixed alpha and beta blockers
In a still further embodiment, the beta blocker is a mixed alpha and beta
blocker.
Examples of mixed alpha and beta blockers include carvedilol, celiprolol and
labetalol.
In one embodiment, the beta blocker is selected from the group consisting of
carvedilol,
celiprolol, labetalol and pharmaceutically acceptable salts thereof.
In a particular embodiment, the beta blocker is carvedilol or a
pharmaceutically
acceptable salt thereof.
36782-064
CA 3029052 2019-01-07
12
Beta 2-selective beta blockers
In a still further embodiment, the beta blocker is a beta 2-selective beta
blocker.
One example of a beta 2-selective beta blocker is butaxamine.
In one embodiment, the beta blocker is butaxamine or a pharmaceutically
acceptable
salt thereof.
Pharmaceutically acceptable salts
Examples of pharmaceutically acceptable salts include, without limitation, the
non-toxic
inorganic and organic acid addition salts such as the hydrochloride, the
hydrobromide,
the nitrate, the perchlorate, the phosphate, the sulphate, the formate, the
acetate, the
aconate, the ascorbate, the benzene- sulphonate, the benzoate, the cinnamate,
the
citrate, the embonate, the enantate, the fumarate, the glutamate, the
glycolate, the
lactate, the maleate, the malonate, the mandelate, the methanesulphonate, the
naphthalene-2-sulphonate, the phthalate, the salicylate, the sorbate, the
stearate, the
succinate, the tartrate, the toluene-p-sulphonate, and the like. Such salts
may be
formed by procedures well known and described in the art.
Examples of pharmaceutically acceptable cationic salts of an API include,
without
limitation, the sodium, the potassium, the calcium, the magnesium, the zinc,
the
aluminium, the lithium, the choline, the lysinium, and the ammonium salt, and
the like,
of an API containing an anionic group. Such cationic salts may be formed by
procedures well known and described in the art.
In the context of this disclosure the "onium salts" of N-containing compounds
are also
contemplated as pharmaceutically acceptable salts. Preferred "onium salts"
include the
alkyl-onium salts, the cycloalkyl-onium salts, and the cycloalkylalkyl-onium
salts.
In one embodiment of the present disclosure, Tesofensine is selected from the
free
base, the citrate salt and the tartrate salt.
Suitable pharmaceutically acceptable salts of metoprolol include any of the
salts
mentioned herein and preferably include the tartrate, succinate, fumarate or
benzoate
36782-064
CA 3029052 2019-01-07
13
salts and especially the succinate salt. The S-enantiomer of metoprolol or a
salt
thereof, particularly the benzoate salt or the sorbate salt, may also be used.
Similarity factors
Similarity factor (f2) is a recognized method for the determination of the
similarity
between the dissolution profiles of a reference and a test compound.
Similarity factor
(f2) is a logarithmic transformation of the sum of squared error. The
similarity factor (f2)
is 100 when the test and reference profiles are identical and approaches zero
as the
dissimilarity increases. The similarity factor has also been adapted to apply
to the
determination of the similarity between the dissolution profiles of a
reference and test
compound as they relate to modified release formulations, such as those
exemplified
herein.
The f2 similarity factor has been adopted in the SUP AC guidelines and by the
FDA
guidance on dissolution testing of immediate release dosage forms (FDA
Guidance for
Industry, Dissolution Testing of Immediate Release Solid Oral Dosage Forms,
FDA,
(CDER), August 1997 (Dissolution Tech. 4, 15-22, 1997)).
Preferably the pharmaceutical composition has a beta blocker in vitro
dissolution profile
generated using the USP Type ll apparatus, rotating paddle method as described
herein with a similarity factor (f2) between 50 and 100 when calculated using
one of the
examples from Figure 1 or Figure 3 as the reference profile.
API amounts and ratios
The ratio of extended release beta blocker, such as metoprolol, to immediate
release
beta blocker may be 75-95:25-5. Suitably, the beta blocker, such as
metoprolol, in a
dosage form is approximately an 80:20 ratio of extended release to immediate
release
amounts. In another embodiment, the beta blocker, such as metoprolol, is in an
approximate 90:10 or 100:10 ratio of extended to immediate release amounts. In
still
another embodiment the ratio is approximately 80:20 or 75:25. Explained
differently, for
unit dosage form, such as a tablet, containing 40 mg beta blocker, such as
metoprolol,
the beta blocker may be present in an amount of about 30 mg in the extended
release
phase and about 10 mg in the immediate release phase. For a unit dosage form
36782-064
CA 3029052 2019-01-07
14
comprising 22 mg beta blocker, such as metoprolol, the beta blocker ER may be
present in an amount of 20 mg and the beta blocker IR may be present in an
amount of
2 mg. For example, in one embodiment, the ratios of extended release to
immediate
release phase represent the proportional amount of each layer in a bi-layer
dosage
form. In another embodiment, the ratios represent the amount of metoprolol in
the
extended release intragranular component versus the immediate release
extragranular
component of a single layer dosage form. The ratios and amounts mentioned in
the
current paragraph apply well to metoprolol as the beta-blocker.
Preferably one dosage form comprises an amount of beta blocker, such as
metoprolol,
ER of 5-200 mg, such as 25-200 mg, such as 5-100 mg API, such as 15-100 mg of
API, preferably 15-50 mg, such as 15-40 mg, such as 5-50 mg, such as 5-20 mg,
for
example about 8 mg, about 20 mg or about 40 mg. In one embodiment, one dosage
form comprises an amount of beta blocker, such as metoprolol, ER of no more
than
200 mg API, such as no more than 150 mg, such as no more than 100 mg, such as
no
more than 50 mg, such as no more than 20 mg.
Other beta-blockers may require lower dosages. In this case one dosage form
may
comprise an amount of beta blocker, such as carvedilol, ER of 5-40 mg of API,
such as
10-20 mg of API, preferably 12-20, for example about 15 mg.
The amount of tesofensine per dosage form (in the second composition) is
generally
between 0.010-0.250 mg API, between 0.01-0.250 mg, for example 0.025-0.200 mg,
such as 0.040-0.125, such as 0.040-0.120 mg for example about 0.050 mg, 0.075
mg,0.100 mg, 0.125 mg or 0.250 mg. In one embodiment, the amount of
tesofensine
per dosage form is no more than 0.25 mg API, such as no more than 0.250 mg,
such
as no more than 0.200 mg, such as no more than 0.150 mg, such as no more than
0.125 mg. The dose of Tesofensine is based on the amount of the free base, but
pharmaceutically relevant salts of Tesofensine may also be used in amounts
equivalent
to the doses of the free base disclosed herein.
The amount of beta blocker, such as metoprolol, IR per dosage form may be from
1-25
mg API, such as 1-15 mg, for example 3-15 mg, such as 4-10 mg, such as 5-10
mg,
such as 1-10 mg, such as 1-5 mg, such as 2-5 mg, for example about 2 mg, about
5
36782-064
CA 3029052 2019-01-07
15
mg, about 10 mg, about 6 mg, or about 8 mg. In one embodiment, the amount of
beta
blocker, such as metoprolol, IR per dosage form is no more than 25 mg API,
such as
no more than 20 mg, such as no more than 15 mg, such as no more than 10 mg,
such
as no more than 5 mg. The amount of beta blocker is based on metoprolol
tartrate, but
other pharmaceutically relevant salts of metoprolol may also be used in
amounts
equivalent to the doses of metoprolol tartrate disclosed herein.
Thus one dosage form may comprise 5-100 mg ER beta blocker, such as
metoprolol,
2-25 mg IR beta blocker, such as metoprolol, and 0.010-0.250 mg tesofensine;
for
example 5-50 mg ER beta blocker, such as metoprolol, 2-15 mg IR beta blocker,
such
as metoprolol, and 0.025-0.250 mg tesofensine; for example 5-40 mg ER beta
blocker,
such as metoprolol, 2-10 mg IR beta blocker, such as metoprolol, and 0.025-
0.075
tesofensine; for example 5-40 mg ER beta blocker, such as metoprolol, 2-10 mg
IR
beta blocker, such as metoprolol, and 0.125-0.250 mg tesofensine; for example
20-40
mg ER beta blocker, such as metoprolol, 5-10 mg IR beta blocker, such as
metoprolol,
and 0.050-0.250 tesofensine; for example 30-60 mg ER beta blocker, such as
metoprolol, 8-15 mg IR beta blocker, such as metoprolol, and 0.050-0.250
tesofensine;
for example 5-20 mg ER beta blocker, such as metoprolol, 2-5 mg IR beta
blocker,
such as metoprolol, and 0.125-0.250 tesofensine; for example 5-20 mg ER beta
blocker, such as metoprolol, 2-15 mg IR beta blocker, such as metoprolol, and
0.025-
0.250 tesofensine; for example 5-20 mg ER beta blocker, such as metoprolol, 2-
5 mg
IR beta blocker, such as metoprolol, and 0.025-0.150 tesofensine.
In one embodiment, the dosage form comprises 25-200 mg ER metoprolol, 5-50 mg
IR
metoprolol, and 0.250 mg Tesofensine or less, for example 50-125 mg ER
metoprolol,
10-25 mg IR metoprolol, and 0.025-0.100 mg tesofensine, for example 75-80 mg
ER
metoprolol, 10-15 mg IR metoprolol, and 0.025-0.175 tesofensine.
In one embodiment, the dosage form comprises 5-200 mg ER metoprolol, 1-50 mg
IR
metoprolol, and 0.250 mg Tesofensine or less; for example 5-125 mg ER
metoprolol, 1-
25 mg IR metoprolol, and 0.025-0.125 mg tesofensine, for example 5-20 mg ER
metoprolol, 1-10 mg IR metoprolol, and 0.025-0.175 tesofensine.
In one embodiment, one dosage form comprises 8-20 mg ER beta blocker, such as
metoprolol, and 2-5 mg IR beta blocker, such as metoprolol and tesofensine. In
one
36782-064
CA 3029052 2019-01-07
16
embodiment, one dosage form comprises 8-20 mg ER beta blocker, such as
metoprolol, and 2-5 mg IR beta blocker, such as metoprolol and no more than
0.25 mg
tesofensine.
In one embodiment, the dosage form comprises 8 mg of ER metoprolol; 2 mg of IR
metoprolol; and 0.125 mg tesofensine. In one embodiment, the dosage form
comprises
20 mg of ER metoprolol; 5 mg of IR metoprolol; and 0.250 mg tesofensine.
In one embodiment the beta blocker is metoprolol and the amount of the two
APIs in
the three phases of the current dosage form are present in the following
absolute
amounts.
Metoprolol ER Metoprolol IR Tesofensine IR
20-150 mg 5-50 mg 0.010-0.250 mg
20-100 mg 5-25 mg 0.010-0.150 mg
30-80 mg 5-20 mg 0.025-0.080 mg
5-50 mg 1-10 mg 0.100-0.250 mg
5-50 mg 1-10 mg 0.075-0.150 mg
80 mg 20 mg 0.025-0.250 mg
40 mg 10 mg 0.250 mg
mg 5 mg 0.050 mg
15 Multi-Layer Dosage Form
The extended release phase may be part of a multiple layer tablet, such as a
bi or tri-
layer dosage form.
20 In one embodiment, the dosage form comprises a tri-layer dosage unit
having an
extended release (ER) phase layer with a beta blocker, such as metoprolol or
carvedilol, and one immediate release phase layer with a beta blocker, such as
metoprolol or carvedilol and another immediate release layer with tesofensine.
The ER
phase contains a therapeutically effective amount of the beta blocker, such as
metoprolol or carvedilol, suitably in granulate form.
36782-064
CA 3029052 2019-01-07
17
In other embodiments, the dosage form is a bi-layer tablet having an ER phase
layer
with a beta blocker, such as metoprolol or carvedilol and one immediate
release layer
with both the betablocker (such as metoprolol or carvedilol) and tesofensine.
Extended release phase
Extended release compositions of beta blockers, such as metoprolol or
pharmaceutically acceptable salts of metoprolol are known the art. Non-
limiting
examples of disclosures of such compositions are found in: WO 2015/004617, WO
2013/084089, WO 2013/ 030725, WO 2012/052834, WO 2011/143420, WO
2007/09770, WO 2004/069234, WO 2007/110753, WO 2007/029070, WO
2008/012346, and WO 2007/048233. Such extended release compositions typically
involve coating the API with an extended release layer that provides an
approximated
zero-order rate of dissolution of the API.
In one embodiment, the extended release beta blocker, such as metoprolol, is
formulated as pellets with pharmaceutically acceptable excipients such as for
example
binders, film coating polymers, plasticizers, starch, glidants, and
disintegrants.
An extended release formulation of carvedilol is also known from US 8,101,209
(Flame!
Technologies).
Inert core
In some embodiments, the pellets comprise an initial core (inert core) coated
with a
layer of a beta blocker, such as metoprolol or a metoprolol salt, and further
coated with
an extended release layer.
As used herein the term initial core refers to a pharmaceutically acceptable
core for use
in pharmaceutical formulations which core is inert.
In one embodiment there is provided a pharmaceutical composition for extended
release comprising pellets coated with a beta blocker, such as metoprolol or a
metoprolol salt, wherein each coated pellet comprises a) an inert core
comprising at
least 50% (w/w) of soluble substance; b) a drug layer comprising the beta
blocker, such
36782-064
CA 3029052 2019-01-07
18
as metoprolol, which layer covers the inert core; and c) a controlled release
layer
thereon.
In another embodiment there is provided a pharmaceutical composition wherein
the
release rate of drug from the pellets part of the pharmaceutical composition
comprising
a tabletted or encapsulated composition of a multitude of pellets is
controlled by the
amount or the percentage of the initial core/spheres of the pellets.
Preferably, the
amount of initial core is from about 15% to about 35% by weight of the
controlled
release coated pellets before tableting or capsule filling, such as from 20-
30%.
In another embodiment the inert core is strengthened by applying a sub-coat on
the
initial core/sphere. In pharmaceutical compositions wherein pellets comprising
the drug
are compressed into tablets, the drug pellets are mixed with powder excipients
to form
a tableting blend. However, the size of the drug coated pellets, often larger
than the
particle size of the powder excipients, can cause a lack of uniformity of the
tableting
blend. The preferred uniformity of the tableting blend is such that the
average assay of
samples of the tableting blend each weighing the equivalent of one tablet lies
within the
range of 90 to 110 percent of the label dose and the relative standard
deviation of the
individual assays is less than or equal to 5 percent. The size of the drug
pellets is
therefore preferably small. When layering a large amount of drug on a small
initial core
a high degree of stress is exerted on the initial core. This stress may cause
attrition
particularly when the inert core comprises sugar spheres. To provide a higher
degree
of physical strength of the inert core without changing the dissolution rate
of drug
coated pellets, a sub-coat may be applied on an initial core/sphere.
Preferably, the
amount of the sub-coat is from about 10% to about 40% of the total weight of
the sub-
coated inert core, more preferably the amount of sub-coat is from about 15% to
about
30% of the total weight of the sub- coated inert core, most preferably the
amount of
sub-coat is about 16% to about 20% of the total weight of the sub-coated inert
core.
The inert core of each of the pellets in the pharmaceutical composition may
comprise
from about 50% to about 100% (per weight) of soluble substance. Preferably the
inert
core comprises from about 70% to about 90% (per weight) of soluble substances.
A
preferred initial core comprises a sugar sphere. Sugar spheres have been used
in the
pharmaceutical industry as excipients. Such sugar spheres used in
pharmaceutical
compositions generally contain not more than 92% of sucrose, calculated on the
dried
36782-064
CA 3029052 2019-01-07
19
basis, the remainder consisting of maize starch. Commonly sugar spheres with a
core
size larger than 500 pm are used. The core size of the inert cores, preferably
a sugar
sphere, is between about 50 pm and about 500 pm, preferably between about 100
pm
and about 400 pm, more preferably from about 250 pm to about 350 pm.
The inert core may comprise an initial core/sphere that is sub-coated with a
layer of a
plasticized film coating polymer. This sub-coating of an initial core/sphere
provides
physical strength to the inert core. The film coating polymer may be a
hydrophobic or a
hydrophilic polymer, or a combination of the two. Suitable film coating
polymers can be
cellulose derivative polymers or polymethacrylate polymers. Further,
hydrophobic
polymers or hydrophilic plasticizers, or a combination of several plasticizers
can be
used to plasticize the film coating polymers. These compounds of the polymeric
sub-
coat are mixed with solvents prior to their application onto the initial
core/sphere.
Suitable solvents for use in mixing the polymeric sub-coating compounds are
selected
from ethanol, isopropyl alcohol, acetone and purified water. For example a
mixture of
ethanol, acetone and water is preferred for use in mixing a mixture of the
preferred
sub-coating compounds EthylCellulose (as a film coating polymer), and
plasticizers
Dibyutyl Sebacate and Polyethylene Glycol (EC, DBS and PEG).
Preferably, the initial core/sphere is a sugar sphere which is sub coated with
a mixture
of polymers such as cellulose derivatives e.g. ethylcellulose and triethyl
citrate,
polyethylene glycol, dibutyl sebacate, and dibutyl phthalate, and wherein the
sub-
coating layer on the initial core/sphere does not alter the release rate of
the drug for the
pharmaceutical composition. A preferred sub-coat on the sugar spheres
comprises
ethyl cellulose as a hydrophobic film coating polymer and a combination of two
or more
plasticizers, at least one hydrophilic and at least one hydrophobic
plasticizer. Suitable
plasticizers may include for example polyethylene glycols, citrate esters,
dibutyl
sebacate, diethyl phthalate, and triacetin. Preferred plasticizers are
polyethylene glycol
and dibutyl sebacate as the hydrophilic and hydrophobic plasticizers
respectively.
Preferably, the sub-coat comprises about 75% to about 85% ethyl cellulose,
about 10%
to about 20% polyethylene glycol and about 3% to about 7% dibutyl sebacate by
weight of the sub-coat. More preferably, the sub-coat comprises 80% ethyl
cellulose,
15% polyethylene glycol and 5% dibutyl sebacate by weight of the sub-coat.
36782-064
CA 3029052 2019-01-07
20
Alternatively the core may be an insoluble core onto which the active
ingredient has
been deposited for example by spraying. It may be made from silicon dioxide,
glass or
plastic resin particles. Suitable types of plastic material are
pharmaceutically
acceptable plastics such as polypropylene or polyethylene preferably
polypropylene.
Such insoluble cores may have a diameter in the range of 0.01-2 mm, preferably
in the
range of 0.05-1.0 mm and more preferably in the range of 0.1-0.7 mm.
Beta blockers for Extended Release
In one embodiment, a beta blocker, such as Metoprolol or its acceptable
pharmaceutical salt, may be applied on the inert core. No use of "Class 2"
solvents (as
defined by the FDA) is required to apply the active pharmaceutical ingredient
(API),
drug, onto the inert core forming a drug coated pellet. The FDA defines "Class
2"
solvents as having inherent toxicity. The active ingredient is dispersed in
water,
preferably together with an acceptable binder excipient such as, but not
limited to,
polyvinyl pyrrolidone, cellulose derivatives polymers, or starch.
The beta blocker, such as metoprolol may be applied as a dispersion rather
than a
solution. Therefore it is preferred that the drug substance has physical
properties that
will allow a high yield in preparing drug coated pellets. Therefore, the drug
substance
preferably has a particle size distribution such that the d(0.9) value is less
than about
80 pm. Preferably, the d(0.9) value for the particle size distribution of the
drug
substance is less than about 50 urn, more preferably less than about 30 m. As
a
result, a concentrated dispersion for application can be produced which may
shorten
the production time.
The drug coated pellets may comprise from about 40% to about 90% (per weight)
of
the drug layer, preferably from about 50% to about 80% (per weight), more
preferably
from about 55% to about 75% (per weight).
Other beta blockers, such as Carvedilol or salts thereof, may be applied in a
similar as
indicated for Metoprolol.
36782-064
CA 3029052 2019-01-07
21
Controlled release layer
The last layer applied on the pellets is a layer which controls the release of
the active
pharmaceutical ingredient. Pellets that have been coated with a controlled
release
layer may have a size between about 200 pm and about 800 pm. Preferably, the
controlled release layer coated pellets have a size ranging from about 300 pm
to about
700 pm, more preferably from about 400 pm to about 600 pm. In addition, the
controlled release layer may comprise water soluble and insoluble components.
Such
components may be film forming polymers and plasticizers. For example, a film
comprising a polymeric layer may be applied onto the drug coated pellets.
In the following three different types of extended release coatings are
described.
First extended release coating.
In one embodiment the extended release film coat comprises i) an acrylic
polymer ii) a
surfactant and iii) sodium stearyl fumarate, wherein the film coat has been
deposited
from a water containing liquid.
Typically a film coating composition comprises
a) 25 to 35% by weight of an acrylic polymer dispersion
b) 0.1 to 4% by weight of a surfactant
c) 0.1 to 4% sodium stearyl fumarate and
d) a water-containing liquid to 100%.
In one embodiment there is provided film coatings which are suitable for
giving
extended release. Suitably the acrylic polymer used in this case comprises
homogeneous particles wherein the polymer or copolymer has Tg<room temperature
in
aqueous dispersion but has Tg>room temperature in the dry state. Suitable
polymers
comprise acrylic acid and esters thereof particularly the methyl, ethyl,
propyl and butyl
esters; and methacrylic acid and esters thereof particularly the methyl,
ethyl, propyl and
butyl esters. Particularly preferred polymers are those provided under the
tradenames
Eudragit L30DO (Rohm Pharma) or Eudragit FS3ODO (Rohm Pharma). Optionally
further anti-tacking agents may be required.
36782-064
CA 3029052 2019-01-07
22
Suitably the amount of the acrylic polymer in the film coating composition is
in the
range of 15 to 50% by weight. Preferably the amount of the acrylic polymer in
the film
coating composition is in the range of 20 to 40% by weight. More preferably
the amount
of the acrylic polymer in the film coating composition is in the range of 25
to 35% by
weight.
Suitably the surfactant is one of the following: a nonionic surfactant, like
sorbitan esters
(Span series); polysorbates (Tween series); polyoxyethylated glycol monoethers
(like
the Brij series); polyoxyethylated alkyl phenols (like the Triton series or
the lgepal
series); alkyl glucosides (e g dodecylmaltoside); sugar fatty acid esters (e g
sucrose
laurate); saponins; etc: or mixtures thereof; ampholytic surfactants, like
betaines;
anionic surfactants, like sulphated fatty alcohols eg sodium dodecylsulphate
SDS;
sulphated polyoxyethylated alcohols; others like dioctyl sulphosuccinate; bile
salts (e g
dihydroxy bile salts like sodium deoxycholate, trihydroxy bile salts like
sodium
glycocholate, etc); fusidates (e g sodium dihydrofusidate); etc cationic
surfactants, like
ammonium compounds; soaps, fatty acids, and lipids and their salts, like
alkanoic
acids; (e g octanoic acid, oleic acid); monoglycerides (eg monolein),
phospholipids
which are neutral or positively or negatively charged (eg dialkyl
phosphatidylcholine,
dialkyl phosphatidylserine, etc); etc; more preferably the surfactant is a
nonionic
surfactant. Most preferably the surfactant is nonoxynol 100.
Suitably the amount of the surfactant in the film coating composition is in
the range of
0.05 to 8% by weight. Preferably the amount of the surfactant in the film
coating
composition is in the range of 0.1 to 6% by weight. More preferably the amount
of the
surfactant in the film coating composition is in the range of 0.5 to 4% by
weight.
In a most preferred embodiment the acrylic polymer and the surfactant are
provided by
Eudragit NE3OD in compositions, a film coats or formulations defined
previously.
Suitably the amount of the sodium stearyl fumarate in the film coating
composition is in
the range of 0.05 to 8% by weight. Preferably the amount of sodium stearyl
fumarate in
the film coating composition is in the range of 0.1 to 6% by weight. More
preferably the
amount of sodium stearyl fumarate in the film coating composition is in the
range of 0.5
to 4% by weight.
36782-064
CA 3029052 2019-01-07
23
Suitably the water-containing liquid comprises water and a water miscible
organic liquid
for example lower alkanols e.g. ethanol, propanol or isopropanol. From a
safety point of
view is preferred that the proportion of the organic is kept to a minimum but
small
amounts are tolerable for example in the range of 0 to 20% by volume.
Preferably the
liquid is water.
The film-coating composition is particularly suitable for use as an aqueous
film-coating
composition wherein the film-coat is applied using water as the liquid. When
the liquid
is water the latex is preferably a poly(ethylacrylate-co-methylmethacrylate)
copolymer,
for example Eudragit NE30D (Rohm Pharma). This process is particularly
advantageous as it negates the need to use environmentally unacceptable
organic
solvents, some of which also present processing problems due to their
inflammablility,
while also eliminating many of the problems experienced with aqueous coatings
described above.
Second extended release coating
Alternatively, the film may comprise at least one film coating polymer and can
be
plasticized with one or more plasticizers. These plasticizers may differ from
each other
in their degree of solubility (hydrophobicity/hydrophilicity). By changing the
ratio
between the plasticizers and the film coating polymer, or the ratio between
the different
plasticizers (if more than one is used), one can control the rate of the
release of the
drug from the pellets. The controlled release layer of the beta blocker ER may
comprise a hydrophobic film coating polymer such as for example ethylcellulose
and a
combination of at least two plasticizers, at least one hydrophilic and one
hydrophobic
plasticizer, for example polyethylene glycol and dibutyl sebacate. Preferably,
the ratio
of hydrophobic to hydrophilic plasticizer in the controlled release layer of
the
pharmaceutical composition is from 3:1 to 1:3, more preferably the ratio is
1:1.
Furthermore, the controlled release layer may comprise at least about 70%
water
insoluble compounds (per weight of the controlled release layer). Preferably,
the
controlled release layer comprises at least about 80% and more preferably at
least
about 90% water insoluble compounds (per weight of the controlled release
layer).
Suitable water insoluble compounds are for example cellulose derived polymers.
These
controlled release layer compounds are mixed with solvents prior to their
application
36782-064
CA 3029052 2019-01-07
24
onto the drug coated pellets. Suitable solvents for use in mixing the
controlled release
layer compounds are selected from ethanol, isopropyl alcohol, acetone and
purified
water. A mixture of ethanol, acetone and water is preferred for use in mixing
the
controlled release layer compounds especially where the controlled release
layer
compounds are a mixture of ethylcellulose, dibutyl sebacate and polyethylene
glycol.
The method of preparing the beta blocker ER component may comprise sub-coating
an
initial core/sphere forming an inert core. Sub-coating an initial core/sphere
comprises
mixing a film coating polymer with one or more plasticizers in a solvent
forming a
coating mixture. Such mixture may be a solution, suspension or slurry for
applying a
coating layer on a surface. The coating mixture is applied to the initial
core/sphere
forming a sub-coated initial core/sphere which is used as an inert core. The
film coating
polymer may be a hydrophobic or a hydrophilic polymer, or a combination of the
two.
Suitable film coating polymers can be cellulose derivative polymers or
polymethacrylate
polymers, preferably ethylcellulose. The amount of ethylcellulose is
preferably from
about 75% to about 85% more preferably about 80% of the total amount of the
weight
of the sub-coat. Further, hydrophobic polymers or hydrophilic plasticizers, or
a
combination of several plasticizers can be used to plasticize the film coating
polymers.
These compounds of the polymeric sub-coat are mixed with solvents prior to
their
application onto the initial core/sphere. Suitable solvents for use in mixing
the
polymeric sub-coating compounds are selected from ethanol, isopropyl alcohol,
acetone and purified water. A mixture of ethanol, acetone and water is
preferred for
use in mixing the polymeric sub-coating compounds.
Suitable plasticizers for use in sub-coating an initial core/sphere are
selected from
polyethylene glycol, dibutyl sebacate, and dibutyl phthalate. Preferred
plasticizers are
polyethylene glycol and dibutyl sebacate as the hydrophilic and hydrophobic
plasticizers respectively. Preferred amounts of plasticizers used in the
method are
about 10% to about 20% polyethylene glycol and 3% to about 7% dibutyl sebacate
by
weight of the sub-coat. More preferably, about 15% polyethylene glycol and 5%
dibutyl
sebacate as plasticizer.
For the extended release coat, the amount of ethylcellulose is preferably from
about
75% to about 85% more preferably about 80% of the total amount of the weight
of the
coat. Suitable plasticizers for use in the ER-coating are selected from
polyethylene
36782-064
CA 3029052 2019-01-07
25
glycol, dibutyl sebacate, and dibutyl phthalate. Preferred plasticizers are
polyethylene
glycol and dibutyl sebacate as the hydrophilic and hydrophobic plasticizers
respectively. Preferred amounts of plasticizers used in the method are about
5% to
about 20% polyethylene glycol and dibutyl sebacate by weight of the ER-coat.
More
preferably, about 10% polyethylene glycol and 10% dibutyl sebacate as
plasticizer.
In one embodiment, a metoprolol ER tablet comprises:
Material Weight Percent total
pellet weight
Sub-coated pellets
Sugar spheres (250-355 pm) 598.00 22.3
Ethyl cellulose 7 cps 92.00 3.4
Polyethylene glycol 400 17.25 0.6
Dibutyl sebacate 5.75 0.2
Drug layer
Metoprolol succinate 1092.50 40.9
Polyvinyl pyrrolidone povidone (PVP K- 276 10.3
30)
Controlled release film layer
Ethyl cellulose 100 cps 473.8 17.7
Polyethylene glycol 400 59.23 2.2
Dibutyl sebacate 59.23 2.2
In a preferred method of preparing the beta blocker ER part of the
composition, the
method comprises the following steps; a) providing sugar spheres as initial
cores; b)
coating the sugar spheres with a sub-coat comprising mixing a film of a
hydrophobic
polymer, a soluble (hydrophilic) plasticizer, and an insoluble (hydrophobic)
plasticizer
with a solvent mixture of e.g. acetone, ethanol 95%, and water and spraying
the
mixture onto the sugar spheres to create a sub-coat on the sugar spheres
resulting in
an inert core; c) coating the sub-coated sugar spheres (inert cores) with a
drug layer
comprising mixing the drug, such as metoprolol succinate, and a binder,
preferably
povidone (PVP K-30) with preferably water, forming an aqueous dispersion and
applying the dispersion onto the sub-coated pellets (inert cores) forming drug
coated
pellets; d) applying a third layer on the drug coated pellets comprising
dissolving a
36782-064
CA 3029052 2019-01-07
26
hydrophobic film coating polymer, an hydrophilic plasticizer and an
hydrophobic
plasticizer in a solvent mixture of e.g. acetone, ethanol 95%, and water
forming a
mixture and spraying the mixture onto the drug coated pellets to create
controlled
release drug coated pellets; e) mixing the controlled release drug coated
pellets with a
powder mixture of one or more excipients forming a final blend; f) compressing
the final
blend into tablets or filling the final blend into capsules; and g) optionally
film coating
the tablets for cosmetic purposes.
In this method the hydrophobic polymer is preferably ethyl cellulose (EC), the
soluble/hydrophilic plasticizer is preferably polyethylene glycol (PEG), and
the
insoluble/hydrophobic plasticizer is preferably dibutyl sebacate (DBS).
Further, in
preparing a mixture for coating the sugar spheres with a sub-coat, and the
drug coated
pellets with a controlled release layer, ethyl cellulose is preferably first
dissolved in
acetone and ethanol 95%, then PEG and DBS are added, followed by adding water
and mixing the solution till it is homogenized. Preferably, the spraying of a
solution or
dispersion onto sugar spheres or drug coated pellets in the method uses a
fluidized
bed coater with a Wurster insertion. Furthermore, the binder, used in coating
the sub-
coated sugar spheres with a drug layer, facilitates binding of the drug to the
inert core
of sub-coated sugar spheres. Moreover, in this method the ratio of powder
mixture to
controlled release drug coated pellets in the final tableting blend is
preferably from
about 20% to about 60% (by weight), more preferably from about 30% to about
50%
(by weight), most preferably from about 35% to about 45% (by weight). As a
result a
uniform final tableting blend and tablets are produced.
Third extended release coating
An extended release phase may comprise at least one high viscosity
hypromellose
(HPMC) ingredient. HPMC is a water soluble matrix- forming polymer used to
provide
an extended release effect of metoprolol. The viscosity of the HPMC used in
the ER
phase may be up to 100.000 centipoise such as in the range of about 3500-6000
cps.
An extended release layer with a therapeutically effective amount of a beta
blocker,
such as metoprolol or carvediol, can be made with high viscosity hypromellose
alone.
36782-064
CA 3029052 2019-01-07
27
In other embodiments, the extended release layer comprises a therapeutically
effective
amount of a beta blocker, such as metoprolol or carvediol, at least one high
viscosity
hypromellose, at least one binding agent, a low viscosity hypromellose, at
least one
modified starch, and optionally one or more other pharmaceutically acceptable
intragranular components including but not limited to a second
pharmaceutically
acceptable active ingredient, other pharmaceutically acceptable excipients
and/or
adjuvants. In one embodiment, the ratio of high- viscosity hypromellose to low
viscosity
hypromellose is about 3.3 to about 0.85. In another embodiment the ratio of
high to low
is about 3:1.
Suitably, the viscosity of the low viscosity hypromellose is in the range of
about 10-30
centipoises. In another embodiment the low viscosity is about 15 centipoises.
The amount of at least one binding agent in the extended release phase of a
bilayer
tablet may be from about 0.5% to about 3% w/w. In one embodiment there are at
least
two binding agents present in the ER phase. Suitably the amount of at least
one
modified starch in the extended release phase of the bilayer tablet is from
about 0.5%
to about 3% w/w. In one embodiment, the amount of modified starch is about 1%
w/w
of the ER phase. In one embodiment there are at least two modified starches
present
in the ER phase. Suitably, the modified starch is pre-gelatinized.
Suitably, the amount of the high viscosity hypromellose present in the
extended release
phase is from about 3%> to about 7%> of the extended release phase formulation
weight. In another embodiment, the amount of high viscosity hypromellose is
from
about 4% to about 6%. In still other embodiments, an amount of >20%
hypromellose is
used in the extended release phase.
In yet another embodiment the amount of high viscosity HPMC is present in an
amount
of about 5% w/w extended release phase formulation weight.
Suitably, the amount of the low viscosity hypromellose present in the extended
release
phase is from about 0.5% to about 3% of the extended release phase formulation
weight. In another embodiment, the amount of low viscosity hypromellose is
from about
1% to about 2% of the extended release phase formulation weight.
36782-064
CA 3029052 2019-01-07
28
Alternatively, the total amount of cellulosic derivatives of HPMC present in
the ER
granulate range from about 3% to about 10% by weight of the total amount of
extended
release components. This encompasses both the high and the low viscosity
HPMC's.
In one embodiment the ER phase comprises metoprolol, povidone, pre-gelatinized
corn
starch, and a high and low viscosity HPMC.
In one embodiment the ER phase comprises carvedilol, povidone, pre-gelatinized
corn
starch, and a high and low viscosity HPMC.
Tablets and capsules
The film coated beads or spheres may be provided in sachets or formulated as a
capsule, for example a hard gelatin capsule, or compressed to form tablets
using
known methods with the optional addition of other pharmaceutically acceptable
additives and with the addition of the beta blocker IR and tesofensine
components
herein described. Coated beads to be compressed into a tablet are obtained by
conventional techniques known to those skilled in the art.
Also, during this process suitable other agents can be added. For example,
during the
tabletting step suitable fillers, eg microcrystalline cellulose, lactose
monohydrate, talc.
sodium stearyl fumarate etc can be utilised to give acceptable compression
characteristics of the formulation, e g hardness of the tablet.
These additives can be granulated in one of the conventional granulation
methods.
However, preferably there is provided a set of additives, for example a powder
mixture
that can be directly compressed into tablets. Such powder mixture serves as a
filler,
cushioning, disintegrant, glidant, and lubricant mixture. Furthermore, the
ratio of
controlled release drug coated pellets to additives in the final (e.g.
tableting) blend of
the present pharmaceutical composition is of particular importance to prepare
a
uniform product e.g. tablets.
To prepare a uniform product, preferably at least 50% (by weight) of the
powder
mixture may have particle sizes between about 30 pm to about 800 pm,
preferably
from about 80 pm to about 600 pm, more preferably from about 100 pm to about
300
36782-064
CA 3029052 2019-01-07
29
pm. More preferably, at least 65% (by weight) of the powder mixture has
particle sizes
between about 30 pm to about 800 pm, preferably from about 80 pm to about 600
pm,
more preferably from about 100 pm to about 300 pm. Most preferably, at least
80% (by
weight) of the powder mixture has particle sizes between about 30 pm to about
800
pm, preferably from about 80 pm to about 600 pm, most preferably from about
100 pm
to about 300 pm.
Furthermore, the amount of controlled release drug coated pellets in the final
tableting
blend is preferably from about 20% to about 60% (by weight) in order to
prepare such
uniform product. More preferably, the amount of controlled release drug coated
pellet in
the final tableting blend is from about 30% to about 50% (by weight), most
preferably
from about 35% to about 45% (by weight).
Suitable powder mixtures comprise, but are not limited to, mixtures of two or
more of
the following compounds; Starlac(R) (a spray-dried compound consisting of 85%
alpha-
lactose monohydrate and 15% maize starch dry matter available from Meggle),
Cellactose(R) (a spray-dried compound consisting of 75% alpha-lactose
monohydrate
and 25% cellulose powder dry matter available from Meggle), Parteck(R) (A
Directly
Compressible Sorbitol available from Merck KGaA), Crospovidone, Silicon
Dioxide,
Magnesium Stearate, Talc, Zinc Stearate, Polyoxyethylene Stearate, Stearic
Acid,
sodium stearyl fumarate Cellulose derivatives, microcrystalline cellulose and
lactose
monohydrate.
If the dosage form is a bi- or tri-layer tablet, the immediate release
layer(s) may be
compressed directly on a previously partly compressed extended release layer,
or
alternatively, the extended release layer may be compressed onto previously
partly
compressed immediate release layer(s).
The compositions can be formulated by conventional methods of admixture such
as
granulating, blending, filling and compressing. For example, tablets can be
produced
by a wet granulation process, where the immediate release phase and extended
release phase are separately prepared. Suitably, for either the immediate
release or
extended release phase, the active drug substance and excipients are screened
and
mixed in a high shear mixer granulator or fluid bed dryer. The blend is
granulated by
the addition of a granulating solution (typically purified water,
disintegration agent
36782-064
CA 3029052 2019-01-07
30
dissolved/dispersed in purified water, or drug dissolved/dispersed in purified
water or a
suitable solvent) sprayed into the high shear mixer granulator or fluid bed
dryer. If
desired wetting agents e.g., surfactants can be added. The resulting granules
(optionally pelletized) are dried usually with residual moisture of 1-5% by
tray, fluid bed
or microwave drying techniques. The dried granules are milled to produce a
uniform
particle size, the granules are blended with extragranular excipients as
necessary,
typically a lubricant and glidant (e.g., magnesium stearate, silicon dioxide).
The
separately prepared immediate release and extended release granules can then
be
compressed together using a rotary tablet press (such as a bilayer tablet
press) if
desired. If the dosage form is a single layer tablet, then the extended
release granules
are admixed with the immediate release extragranular components and compressed
together using a rotary tablet press, etc. These resulting tablets can all be
coated in a
pan coater typically with a 1-5% aqueous film coat, followed by a wax
polishing.
Alternatively tablets can be produced by a direct compression process.
Suitably the
active drug substance and excipients for the immediate release and extended
release
phases are separately screened and mixed in a suitable blender e.g., a cone,
cube or
V- blender. Other excipients are added as necessary, and further blended. The
separately prepared immediate release and extended release phases can be
combined
and compressed together using a rotary tablet press as hereinbefore described.
The
resulting tablets can be coated in a pan coater.
Tablets can also be prepared by using both methods of wet granulation and
direct
compression. For example the extended release phase can be prepared by wet
granulation as described herein, while the immediate release phase can be
prepared
by blending the excipients for direct compression. The two phases can then be
combined and compressed together as hereinbefore described.
Immediate release phase(s)
The immediate release phase(s) may be prepared by combining a directly
compressible commercially available grade of the beta blocker, such as
metoprolol,
and tesofensine with a lubricant, and one or more disintegrating agents if
necessary or
desired. Binders and other excipients and/or adjuvants may be included in the
immediate release layer(s), also if necessary or desired. The beta blocker and
36782-064
CA 3029052 2019-01-07
31
tesofensine in the immediate release layer may be combined with a modified
starch
such as a pre-gelatinized starch, e.g., corn starch, polyethylene glycol, and
a
disintegrant, or super disintegrant such as croscarmellose sodium or Explotab
, a
binder such as methylcellulose or hypromellose polymer, plasticizer, pigment
and a
lubricant.
The immediate release phases may comprise two different layers of the beta
blocker
and tesofensine, respectively. Alternatively, the immediate release phases may
be
combined into one and the same layer. The immediate release phases may also be
formulated into an extragranular phase of a tablet or be granulated into one
or two
different immediate release granules. For tesofensine, the preferred
formulation is a
granulation of tesofensine compared to direct compression of tesofensine as
the dose
is relatively low.
Monolith Dosage Form
In one embodiment, there is only a single layer tablet having an extended
release intra-
granular phase and two immediate release extra-granular phases. The extended
release phase will be comprised of an intra-granular component of the beta
blocker and
excipients as described above. These components form the ER granulate. The ER
blend could be made into pellets and compressed accordingly with the extra-
granular
immediate release blend.
A suitable extra-granular component or phase, i.e., the immediate release
phases, may
be prepared by combining a directly compressible commercially available grade
of a
beta blocker, such as metoprolol, and tesofensine citrate with a lubricant,
and one or
more disintegrating agents if necessary or desired. As mentioned above for
tesofensine
the preferred process is to prepare a granulate of tesofensine before
compression.
Binders and other excipients and/or adjuvants may be included in the extra-
granular
phase if necessary or desired. Alternatively, an extra-granular component can
be
prepared by combining the beta blocker, such as metoprolol, and tesofensine
with a
modified starch, such as a pre-gelatinized starch, e.g., corn starch, a
disintegrant or
super disintegrant, such as croscarmellose sodium, a binder and a lubricant.
36782-064
CA 3029052 2019-01-07
32
Excipients
The present compositions may include components that functions as a binder or
binding agent. Suitably, the binding agent may comprise a first binding agent
and a
second binding agent. Suitable binding agents for use herein include
conventional
binding agents used in the art such as gelatin, starches, povidone, polymers
and
cellulose derivatives or combinations thereof.
Suitably, the starch, is of vegetable origin, such as corn (or maize) starch,
modified
corn starch, wheat starch, modified wheat starch, potato starch, or pre-
gelatinized
starch e.g., available commercially as Starch 1500 G or Prejel; or a
combination of two
or more thereof.
If the binding agent includes a cellulosic derivative such as hydroxypropyl
cellulose
(HPC) (of low to medium viscosity) e.g., as may be available commercially
under the
brand name Klucel from the AquaIon division of Hercules Inc., Dow Chemical
Company e.g., Klucel GF, Klucel JF, Klucel LF and Klucel EF; microcrystalline
cellulose (MCC), carboxymethylcellulose (MC), sodium carboxymethylethyl
cellulose;
or a combination of two or more thereof. Combinations of a cellulosic
derivative with
other binding agents noted above are also envisaged. Generally the total
amount of
cellulosic derivatives present in the granulate are in an amount ranging from
about 3%
to about 10% by weight of the extended release components. It is recognized in
the art
that certain cellulosic derivatives, such as hypromellose, will have varying
roles in a
formulation, depending upon the amount used. For example hypromellose (low or
medium viscosity) may function as a binding agent, a coating agent, or as a
matrix
forming agent.
While a binding agent is present as an intra-granular component, it is
recognized that a
modest amount of binding agent e.g., up to about an additional 3.0%>- 10.0% by
weight of the intra-granular binding agent content of the composition, may
also be
present extra-granularly.
In one embodiment, suitably the starch is pre-gelatinized starch. Pre-
gelatinized starch
is a starch that has been chemically and/or mechanically processed. Typically
pre-
gelatinized starch contains 5% of free amylase, 15% of free amylopectin, and
80%
36782-064
CA 3029052 2019-01-07
33
unmodified starch. Pre-gelatinized starch may be obtained from corn (or
maize), potato
or rice starch.
The granulate provides an intimate admixture of a combination of ingredients
and may
then be mixed with one or more pharmaceutically acceptable extra-granular
components of the composition i.e., with any pharmaceutically acceptable
ingredient
e.g., a diluent, flavor, sweetening agent, binder, disintegrant, glidant,
lubricant, anti-
adherent, anti-static agent, anti-oxidant, desiccant, or a second
pharmaceutically
acceptable active agent. It is recognized that these same ingredients may be
present
both as an intra-granular and as an extra-granular ingredient.
As noted above there are other inactive ingredients that may optionally be
employed in
relatively small quantities, which include lubricants, flow agents, and
binders that
facilitate compression.
Suitable disintegrating agents include a non-super disintegrant, a super
disintegrant or
a combination of both. Suitable non- super disintegrants include conventional
disintegrants such as starch (corn or maize), pre-gelatinized starch e.g.,
Starch 1500
G, clays (e.g. VEEGUM (Vanderbilt Minerals, LLC) or Bentonite (an absorbent
aluminium phyllosilicate clay consisting mostly of montmorillonite)),
microcrystalline
cellulose, cellulose or powdered cellulose. It is recognized in the art, that
some
excipients may perform more than one role in a given pharmaceutical
formulation. For
example certain excipients, e.g., starches including pre- gelatinized starch,
and
microcrystalline cellulose (hereinbefore identified as binding agents)
function as both
binders and disintegrants.
A "super disintegrant" represents a class of disintegrating agent which may
generally
be used in lower amounts in pharmaceutical preparations, as compared to
conventional disintegrants. Examples of super disintegrants include sodium
starch
glycolate, the sodium salt of carboxymethyl starch, modified cellulose and
cross-linked
polyvinyl pyrrolidone. Sodium starch glycolate is available commercially under
the trade
names Explotab (Edward Mendell Co. JRS Pharma), Primojel (Generichem Corp;
DFE Pharma) and Tablo (Blanver, Brazil). An example of modified cellulose
includes
croscarmellose sodium, the sodium salt of carboxymethyl cellulose.
Croscarmellose
sodium is available commercially under the trade names AcDiSol (FMC Corp.),
36782-064
CA 3029052 2019-01-07
34
Nymcel ZSX (Nyma, Netherlands), PrimeHose (Avebe, Netherlands), Solutab
(Blanver, Brazil). An example of a cross-linked polyvinyl pyrrolidone includes
crospovidone, and is commercially available under the trade names Kollidon CL
or
Kollidon CL-M (Basf Corp.), and Polyplasdone XL (ISP Corp; Ashland). A
suitable
super disintegrants includes croscarmellose sodium or sodium starch glycolate
(e.g.
Explotab (JRS Pharma)) or a combination thereof. A super disintegrant may be
used
extragranularly, in an amount ranging from about 0.5% to about 5.0% by weight
of the
composition. Suitable preservative or antimicrobial agents for use include
potassium
sorbate or a paraben, i.e., one or more hydroxy benzoic acid esters e.g.,
methyl, ethyl,
propyl or butyl, suitably singularly or as mixtures. Parabens are commercially
available
under the Nipa brand name, e.g., Nipasept sodium (Aako BV).
Suitable lubricants include magnesium, calcium or sodium stearate, stearic
acid or talc
that may be added in suitable amounts. In one embodiment the lubricant is
magnesium
stearate.
Suitable flow agents include silicon dioxide (e.g. Cab-O-Sile (Cabot
Corporation),
SyloidTM (W.R. Grace & Co.)) and colloidal silicon dioxide (Aerosil (Evonik
Resource
Efficiency GmbH)), that may be added in an amount from about 0.5% to about 1%
by
weight.
The compressed tablet may further comprise a film coat e.g., hypromellose or
polyvinyl
alcohol-part.hydrolised (PVA). Suitably the film coat is a transparent film
coat e.g., a
dye, although an opaque film coat e.g., as obtained when using a film coat in
combination with an opacifier or a pigment such as titanium dioxide or a lake
may also
be used. For example one commercially available film coat is an Opadry
coating
system from Colorcon.
Examples
Example 1. Phase 2a trial entitled "A double-blind, randomized, placebo-
controlled,
multiple-dose, multi-centre safety and efficacy study of co-administration of
tesofensine/metoprolol in subjects with Prader-Willi syndrome (PWS) step 1"
36782-064
CA 3029052 2019-01-07
35
This exploratory Phase 2a trial comprised in of a total of nine adult patients
with
Prader-Willi syndrome (PWS) of which six patients received co-administration
of
tesofensine (0.50 mg)/metoprolol (50 mg) (A, active treatment - Tesomet) and
three
patients received placebo (P). The initial plan was to complete 10-20 adult
subjects
with PWS. However, ultimately nine patients were randomized as more subjects
were
not available at the two participating sites.
The main objective of this study was to examine the effect of co-
administration of
tesofensine/metoprolol on body weight in subjects with PWS. Further objectives
were:
= To establish a pharmacokinetic profile of tesofensine and metoprolol in
subjects
with PWS.
= To examine the effect of co-administration of tesofensine/metoprolol on
glycaemic control and lipid profile in subjects with PWS
= To examine the effect of co-administration of tesofensine/metoprolol on HR
and
BP in subjects with PWS
= To examine the effects of co-administration of tesofensine/metoprolol on
body
composition in subjects with PWS
= To evaluate overall safety and tolerability of co-administration of the
tesofensine/metoprolol in subjects with PWS
Subiects
Minimum of 10 and maximum of 15 randomized adult subjects and minimum of 10
and
maximum of 15 randomized pediatric subjects with PWS, in total minimum of 20
and
maximum of 30 randomized subjects.
= Number of planned: 10 -15 adults +10 -15 children with PWS
= Number of completed: 10 - 15 adult +10 - 15 children with PWS
Site Treatment Visit 1 Visit 2 Visit 3 Visit
4 Visit 5 Visit 6 Visit 7 Visit
(day 0) (day 7) (day 14) (day 21) (day 28) (day (day 8
35) 42) (day
49)
Site Placebo 1 1 1 0 0 0 0 0
01 CZ IMP 3 3 3 3 3 2 2 2
Site Placebo 2 2 2 2 2 1 3 2
02 IMP 3 3 3 3 3 3 3 3
36782-064
CA 3029052 2019-01-07
36
HU
Total Placebo 3 3 3 2 2 1 3 2
IMP 6 6 6 6 6 5 5 5
Site Treatment Visit 9 Visit 10 Visit 11 Visit 12 Visit 13 Visit
14 Visit 15
(day 56) (day 63) (day 70) (day 77) (day 84) (day 91) (day
105)
Site 01 Placebo 0 0 0 0 0 0 0
CZ IMP 2 1 1 1 1 1 1
Site 02 Placebo 2 2 2 2 2 2 2
HU IMP 3 3 1 1 1 1 1
Total Placebo 2 2 2 2 2 2 2
IMP 5 4 2 2 2 2 2
Table 1: Number of patients per visit.
Method low
Two-centre, double-blind, placebo-controlled, randomized, and multiple-dose
clinical
study. Study medication will be administered for 91 days:
= 10-15 adult subjects with PWS will be treated.
= Data Safety Monitoring Board (DSMB) review - following the completion of the
treatment of the adult subjects, unblinded efficacy, safety, PK data as well
as all
data from the study in subjects with type 2 diabetes (TM001) will be reviewed
by an independent DSMB. Following the DSMB's approval the study will
proceed to:
= Step 2 - 10-15 children with PWS, see Example 2.
Arm 1) tesofensine 0.50 mg + metoprolol 50 mg administered once a day, in the
morning with a meal
or
Arm 2) Placebo tablets matching tesofensine + metoprolol administered once a
day, in
the morning with a meal
Each tablet will be formulated separately; a currently available commercial
formulation
of extended-release metoprolol will be used.
In the study a mean trough plasma concentration of - 20 ng/ml was achieved
with a
dosing of 0.5 mg tesofensine which can also be seen in the below Table 2.
36782-064
CA 3029052 2019-01-07
37
TM002 study V5(n=6) V9 (n=5) V14(n=2)
Tesofensine [ng/mL] 18 9 21 8 23 1
Table 2: Mean trough plasma concentration of tesofensine
Two patients had concentrations of 34.1, 34.2 ng/mL, respectively.
Unexpectedly, the patients treated with Tesomet in the Step 1 of the study
were
exposed to on average two to four times higher concentrations than anticipated
from
previous clinical trials.
The observed very high plasma concentration in this study may be explained in
part by
a lower metabolic rate and clearance of tesofensine in patients with PWS and
also
potentially by the extremely high body fat percentage in these patients.
Efficacy:
The clinical study achieved a positive outcome on the primary endpoint with a
clinically
meaningful reduction in weight for patients treated with Tesomet compared to
placebo,
see figure lb. After 8 weeks the mean change in body weight was 5.00% (n=5)
for
patients receiving Tesomet compared to 0.46% (n=2) for patients receiving
placebo.
After 12 weeks the change in body weight was 6.76% (n=2) for patients
receiving
Tesomet compared to 0.75 % (n=2) for patients receiving placebo. The average
weight
reduction was equal to 4.78 kg after 8 weeks and 7.95 kg after 12 weeks for
patients
receiving Tesomet, see figure la. There was a significant variance in weight
loss
between patients where two patients experienced a weight loss of up to 7 and
14 kg
and one patient achieved a weight loss of 2 kg. There was a substantial
reduction in
waist circumference of 7.3 cm after 8 weeks and 10 cm after 12 weeks for
patients
treated with Tesomet. The variance was higher on this parameter and there was
also a
reduction in the placebo group with 4 cm and 6.5 cm for the two time points,
respectively.
A significant reduction in the craving for food in patients treated with
Tesomet was
seen. The total score fell from 10 (n=6) at baseline to 1 (n=5) after 8 weeks
and to 0
(n=2) after 14 weeks where 0 is equivalent to no observations of hyperphagia.
After
just one week (V2) of treatment the score fell from 10 at baseline to 5.67
(n=6)
equivalent to a reduction of 43%. The observed hyperphagia score in the
placebo
36782-064
CA 3029052 2019-01-07
38
group varied over time due to the low number of subjects (n=2), but it did not
change
substantially from baseline. The individual measurements over time can be seen
in
figure 2.
Example 2. Phase 2a trial entitled "A double-blind, randomized, placebo-
controlled,
multiple-dose, multi-centre safety and efficacy study of co-administration of
tesofensine/metoprolol in subjects with Prader-Willi syndrome (PWS) step 2"
Arm 1) tesofensine 0.125 mg + metoprolol 25 mg administered once a day, in the
morning with a meal ¨ first, an initial titration dose of tesofensine 0.0625
mg +
metoprolol 25 mg will be given for the first 4 weeks, in the morning with a
meal.
Following a favorable review of all safety data for each subject by the
investigator,
tesofensine 0.125 mg + metoprolol 25 mg will be given for the final 9 weeks
(the above
proposed dosing plan can be adjusted by the DSMB, if needed)
or
Arm 2) Placebo tablets matching tesofensine + metoprolol administered once a
day, in
the morning with a meal.
If, following the increase to 0.125/25 mg, the IMP is not tolerated; the
investigator can
reduce the dose back to 0.0625/25 mg, or stop the IMP completely.
Each tablet will be formulated separately; a currently available commercial
formulation
of extended-release metoprolol will be used.
36782-064
CA 3029052 2019-01-07