Note: Descriptions are shown in the official language in which they were submitted.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
1
TOLEROGENIC DNA VACCINE
TECHNICAL FIELD
The present invention relates to tolerogeneic DNA immuno-therapy vaccines for
reducing antigen-specific T cell reactivity.
BACKGROUND
According to traditional vaccine approaches, purified protein/antigen is
injected in a
person/patient/animal in order to stimulate immune responses specifically to
that
protein/antigen. This vaccine approach tends to impact primarily antibody
production, while
the T cells tend not to be significantly affected, other than to generate T
cell memory of the
antigen. Traditional vaccine approaches are thus not considered suitable in
connection with
treatment and/or prevention of T cell driven diseases such as e.g. Type 1
diabetes (Ti D), as
activation of T cells, especially CD8+ T cells, are considered the causative
agent of this
disease. Experimental approaches with tolerogenic, protein-based vaccines have
targeted
primarily antibody producing B cells rather than disease relevant T cells.
DNA based vaccines, in contrast to protein-based vaccines, are usually
plasmids
encoding particular antigens ¨ these plasmids are taken up by cells in the
host's body
("transfected"). These transfected host cells then produce the antigen and
process the
antigen into small fragments (T-cell epitopes) for presentation to the immune
system, in
particular to circulating T cells. As T cells can only detect these small
antigen fragments and
not whole proteins, this approach preferentially leads to a modification of T
cell responses,
especially for CD8+ T cells (or cytotoxic T cells), the key drivers of e.g.
T1D pathology. Thus,
DNA vaccines, rather than protein vaccines, are suitable for inducing T cell
responses. While
no DNA vaccines are currently available for human use, there are three
stimulatory plasmid
DNA vaccines licensed for veterinary use, inducing immunity to Equine
Infectious Anemia
Virus, West Nile Virus, and certain canine cancers.
In contrast to stimulatory DNA vaccines, tolerogenic DNA immuno-therapy
vaccines
are intended to suppress immune reactivity towards an antigen, rather than
activating
immune responses against it. These vaccines do not stimulate immunity against
the encoded
antigen, or change the type of stimulation (as e.g. antigenic desensitization
vaccination
approaches for allergies does), but instead cause depletion, and/or lack of
function, and/or
death of self-reactive T cells. In order to do so, the antigen must be
presented to the immune
system without co-stimulation or inflammatory effects, which would otherwise
prime
stimulatory immune responses. This approach of presenting an antigen to be
ignored by the
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
2
immune system, or tolerized against, could be of value in treating autoimmune
diseases, as
the specific mechanism of the disease would thus be targeted rather than
systemically
suppressing the entire immune response. A tolerogenic DNA immuno-therapy
vaccine is thus
a mild method of modulating undesired immune responses.
The end goal of a T1D-specific tolerogenic DNA immuno-therapy vaccine is to
preserve beta cell function and endogenous insulin production. This may occur
through
prevention or delay of disease (especially valuable in pediatric and young
adult cohorts
where monitoring is difficult and "normalcy" of life is a major patient
driver) or extension of the
"honey moon phase" of minimal monitoring and insulin usage that often occurs
for the first six
months after T1D diagnosis.
While DNA based vaccines are known to be safe, none of the (stimulatory or
tolerogenic) DNA vaccines that have been tested in clinical studies have
sufficient potency
as a stand-alone approach for treatment of e.g. Ti D. To!erogenic DNA vaccines
known in
the art showed little efficacy and typically required highly artificial
systems to induce the
desired effects. There is thus a need in the art for tolerogenic DNA immuno-
therapy vaccines
with significantly increased potency, without compromising the safety profile
and preferably
also without requiring an inconvenient administration regimen.
SUMMARY
The present invention relates to a multi-cistronic vector/plasmid which co-
.. expresses/encodes a cellularly retained antigen, such as insulin, as well
as secreted immune
modifiers such as TGF-[3, IL-10, and optionally IL-2. The present invention
furthermore
relates to DNA immuno-therapy vaccines comprising such plasmids as well as
such
pharmaceutical formulations and kits thereof. The present invention finally
relates to the
medicinal use of such products as well as methods for producing such plasmids.
The plasmids/DNA immuno-therapy vaccines herein have therapeutic potential in
treatment of autoimmune diseases that are mainly T cell driven, such as e.g.
type 1 diabetes
(Ti D).
In one aspect the present invention provides plasmid which encodes:
i. an insulin antigen;
ii. TGF-[3; and
iii. IL-10.
BRIEF DESCRIPTION OF DRAWINGS
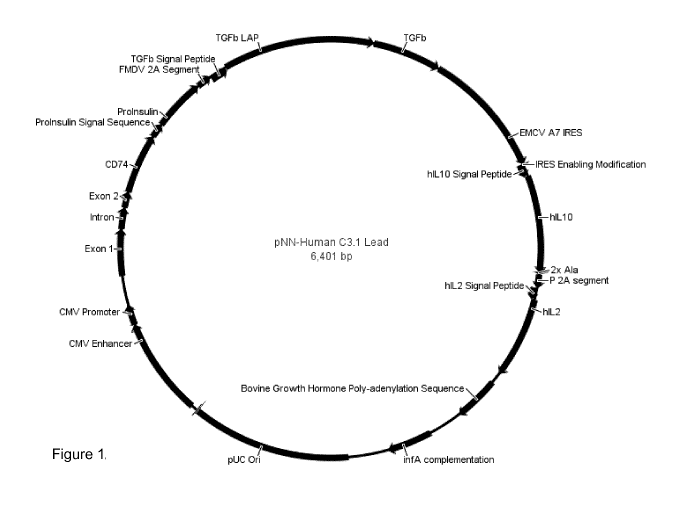
Figure 1. Circular plasmid map.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
3
Figure 2. mRNA and translated protein map for the vector products of the
plasmid from Fig 1.
Figure 3. Plasmid shear stability on three injection passages via a G30
needle.
Figure 4. Confirmation of plasmid retention phenotype by growth at 30 C
(passages 1-50
using 17 hours incubation and passages 51-100 using 22 hours incubation).
DESCRIPTION
The inventor of the present invention has herein provided a single vector
which
drives expression of multiple secreted cytokines, as well as an cellularly
retained antigen,
from a single promoter/multi-cistronic mRNA.
DNA immuno-therapy vaccination with a single vector encoding all components of
the therapy in a single cell is highly preferred over immuno-therapy
vaccination with a
mixture of separate vectors/plasmids each driving expression of single
components, as
random transfection of cells with different vectors does not guarantee
expression of all
components, or even any specific ratio of components, from a given, specific
transfected cell.
Transfection of a single multi-cistronic plasmid/vector results in a
specifically
engineered local environment/micro-environment around the transfected cell. In
this way,
combinations of immuno-modulators can be added to the antigen such that they
potentiate
the desired immunologic effect of single T cells without the requirement of
high systemic
immuno-modulator doses that could otherwise cause adverse events and broad
immunosuppression.
This local restriction of immuno-modulator production of host cells
transfected with
the DNA immuno-therapy vaccine allows for the safe use of highly potent
cytokine hormones,
which are synergistic for modification of T cell responses, but cannot be
dosed either
frequently enough for effect, and/or titrated to give the desired response,
without
unacceptable adverse events.
For example, Interleukin-10 (IL-10) and Transforming Growth Factor-beta1 (TGF-
131)
are both known to be able to induce regulatory T cells (Tregs) from naïve CD4+
T cells.
However, the IL-10/TGF-131 combination provides a synergistic effect (15 to 20
fold more
efficacious) in inducing Tregs than either of the two cytokines alone
(US6083919 A) and this
combination furthermore results in immune tolerance in a broader population of
target cells
than either cytokine alone (Zeller JC, Panoskaltsis-Mortari A, Murphy WJ, et.
al. 1999 J
lmmunol. 163(7):3684-91).
Additionally, Interleukin-2 (IL-2) is known to both expand and stabilize Tregs
but
may on the other hand also contribute to inflammatory responses. The
combination of IL-2
and IL-10, however, results in suppressive Tregs rather than inflammatory
stimulation. As
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
4
circulating T cells encounter cells that are transfected with the DNA immuno-
therapy vaccine
herein, they are temporarily exposed to sub-optimal concentrations of IL-10
and IL-2. The
circulating T cells are slightly biased toward tolerance, and if they are also
reactive toward
the co-expressed antigen (e.g. insulin) they will bind to the transfected cell
and thus receive a
longer duration of immuno-modulator exposure and in addition they will also
receive another
signal that programs/re-educates them for suppressive effects. In this way,
those T cells
which are responsive to the encoded antigen are selectively re-educated to a
suppressive
phenotype when they encounter the transfected cell.
The plasmids/vectors/DNA immuno-therapy vaccines herein are thus designed for
induction of antigen specific Tregs accumulating at sites of autoimmunity to
dampen disease
(e.g. the pancreas in Ti D) rather than to directly impact disease through the
expressed
cytokine hormones.
In addition to an antigen (insulin in the example of Ti D), the
vector/operon/plasmid
herein encodes at least two cytokines (e.g. TGF-[31 and IL-10) which together
synergistically
suppress antigen presenting cells, as well as T cell function, and drive
induction of Tregs.
This effect is enhanced if it also occurs in combination with effective
exposure to antigen.
In one embodiment, TGF-[31 is in a constitutively active form that does not
require
processing or an inflammatory environment for function. While Tregs can be
produced from
naïve T cells via exposure to antigen and TGF-[31, Tregs are, however,
"plastic" meaning that
they can de-differentiate and convert into Th17 effector cells and then cause
more, not less,
autoimmune destruction. The combination of IL-10 with TGF-[31, in addition to
being a more
potent immuno-modulator, suppresses the environment that would produce
pathogenic Th17
cells rather than Tregs.
In one embodiment, the multi-cistronic vector herein also encodes IL-2 in
addition to
antigen, TGF-131, IL-10. IL-2 expands Treg numbers and stabilizes their
phenotype (prevents
Treg cells from de-differentiation into effector T cells) and thus increases
their functional
lifespan in inflamed target tissues.
These three cytokines (TGF-[31, IL-10, and IL-2), in combination with antigen,
thus
have well-known synergistic effects for inducing tolerance by the following
mechanisms: (i)
significantly enhanced generation of antigen-specific suppressive Tregs, (ii)
longer Treg
lifespan, and (iii) greater efficacy per individual Treg cell in suppressing
inflammation/auto-
reactivity. However, the required concentrations of systemically infused
purified cytokine
would have a number of serious, or maybe even lethal, side effects, such as:
(i) lethal fibrosis
from excess TGF-131, (ii) flu-like symptoms, (iii) capillary leak syndrome
from excess IL-2, (iv)
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
broad immunosuppression leading to chronic infections, (v) enhanced tumor
development as
well as (vi) anemia from excess IL-10.
By co-expressing these cytokines from the same vector/plasmid, and therefore
by
the same cell presenting the antigen to the immune system, the vector achieves
the desired
5 local environment for tolerance induction without systemic action and
corresponding side-
effects that would otherwise result from high-dose purified cytokine
administration.
Injection of "naked"/"bare" plasmid/vector DNA (vector and buffer alone) has a
very
low uptake and transfection rate - fewer than one in about 100,000 plasmid
molecules
transfects a cell, while the rest are degraded and thus without any biological
effect. This
extremely low inefficiency of transfection provides a safety mechanism for
distributing and
limiting the transfected cells.
Administration of systemically active quantities of any of these cytokines,
either by
administration of mature proteins or by high-efficiency viral vector
transduction, would be
difficult, if not impossible, to titrate for a safe and effective dose.
Limiting the total exposure to
a very small systemic dose distributed in a few high expressing micro-
environments leads to
a highly advantageous safety and efficacy profile.
The combination of antigen and these three cytokines herein produces an
efficient
protection from T1D development and even appears to be able to stably reverse
disease
progression. Due to the low transfection efficiency of the bare DNA
plasmid/vector injection,
very few cells produce these recombinant proteins and there is thus no
detectable change in
serum cytokine levels from plasmid/vector encoded cytokines ¨ and therefore no
detectable
immune stimulation or immuno-suppression toward any other antigens than the
antigen
encoded by the plasmid/vector (pre-proinsulin). This results in a desirable
safety profile.
Normally, DNA vaccines perform poorly in connection with subcutaneous (s.c.)
injection and are therefore typically administered using intramuscular
injection (often with
electroporation) or alternatively using intradermal jet injection requiring a
cumbersome device
as well as significant maintenance and calibration. As most side effect issues
with
intramuscular injection are adjuvant-related (injection site irritation) they
are therefore not a
concern for the bare DNA immuno-therapy vaccine format herein. Additionally,
the volumes
injected are usually relatively small and therefore do not cause significant
muscle distension
and pain. In one embodiment, the volumes injected are 1 ml or less. In another
embodiment,
the volumes injected are approximately 0.6 or 0.5 ml. Regardless, the multi
cytokine
plasmid/vector provided herein unexpectedly appears to provide protection from
T1D even
when administered through the s.c. route, thereby allowing multiple potential
dosing formats
.. for patients.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
6
In addition to providing local synergy, by encoding all three or four of the
translated
products by a single plasmid/vector and a single promoter, the regulatory
burden and drug
substance release criteria are furthermore simplified with the provision of
the multi-cistronic
plasmid herein.
In contrast, if each of the protein products is produced from a separate
plasmid, then
the synergistic value of co-expression from the same transfected cell would
then potentially
be lost or reduced as each plasmid/vector transfection would be an independent
event, likely
targeting different cells. If the three to four recombinant proteins are
produced from two,
three, or four individual plasmids/vectors, any synergistic effects in the
local environment of
the transfected cell are potentially lost; in addition, several individual
clinical trials would thus
be necessary (one for each plasmid and each combination). Producing all
proteins from a
single plasmid/vector and single mRNA relieves the requirements to test
multiple individual
molecules and determining ideal co-packaging ratios inherent to a multiple
plasmid/vector
format.
Any vector formats suitable for the present invention can be used herein, such
as
plasmids (replicating or passive), mini-circles, linear vectors (MiLVs), viral
vectors (both
integrating [e.g. lentiviral] and non-integrating [e.g. adenoviral]), cosmids,
bacterial artificial
chromosomes (BACs), human artificial chromosomes (HACs), etc.
Furthermore, any permissible transfection enhancement method can be used
herein: e.g. electoporation, sonoporation (ultrasound enhancement, with or
without
microbubble contrast enhancement), lipid/polymer aggregates, hydrodynamics
(pressure via
high injection volume), bio-ballistics / gene-gun (deposition through skin via
compressed
gas), etc.
In one embodiment, non-replicating episomal plasmid DNA is used herein due to:
i)
multiple copies of mRNA derived from a single plasmid transfection, and ii)
extended stability
and function of plasmid nucleic acids over mRNA and other DNA vector formats.
Thus, while
both mRNA and DNA-based expression systems can provide intracellular delivery
and co-
localization, plasmid based systems provide greater control and persistence of
dosing.
In one embodiment, plasmids/vectors encode four proteins:
i) an antigen,
ii) TGF beta 1 (TGF-131),
iii) Interleukin-10, and
iv) Interleukin-2.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
7
In one embodiment, the antigen is an endosomally-targeted T1D relevant
antigen,
such as insulin or GAD65. Endosomal targeting can be done via e.g. a li/CD74
fusion, a
LIMPII/SCARB fusion, or a transferrin receptor fusion.
In one embodiment, TGF-[31 is in an activated form.
Expression of four proteins from one plasmid/vector is possible e.g. if the
desired
sequences are separated either with A) separate promoters, B) an IRES
(Internal Ribosome
Entry Site) sequences which recruit a new ribosome to translate each segment,
or C) viral 2A
sequences (e.g. FMDV 2A or TaV 2A sequences) which are translated and induce a
ribosomal pause/skip which results in production of separate polypeptides from
a single open
reading frame. However, in practice, each of these strategies is complex and
difficult to
enable.
Expression of four independent proteins from a single plasmid/vector is most
easily
achieved by having a separate promoter for each gene. However, this format has
significant
disadvantages in that it A) results in a very large, unstable, and hard to
produce plasmid due
to the excess length of multiple promoters, B) results in unpredictable
behaviour of the
translated proteins relative to each other (they are no longer produced in
fixed ratios to each
other), C) each promoter may be independently silenced, leading to selective
expression of
some genes but not others required for full efficacy, and D) a lack of
regulatory simplicity. In
contrast, IRES elements and 2A sequences operate on the mRNA and translation
levels and
reproducibly co-express fixed ratios of each protein from a single promoter.
Each of the four classes of IRES elements has different co-factor requirements
for
function as well as different sequence requirements for the downstream gene to
be
translated. For instance, the EMCV (EndoMyoCarditis Virus) IRES is a 630 base
pair type 1
IRES which utilizes all eukaryotic translation initiation factors while the
CrPv (Cricket
Paralysis virus) IRES is a 200 base pair type 4 IRES that has no required
cofactors but
utilizes a non-standard initiation codon.
When IRES elements from different classes are utilized, they interfere with
each
other such that each type of IRES element can only be used once in each
plasmid, and when
used together, different types of IRES elements attenuate each other (decrease
in efficacy)
in ways that are difficult to predict.
Furthermore, shuffling the gene / IRES combinations result in unpredictable
ratios of
translated products as the interactions of the genes with the IRES elements
are not static but
context dependant on the flanking nucleotide sequences. In addition, IRES
elements impose
restrictions on the first few amino acid positions at or immediately following
initiation. For
instance, the CrPv IRES requires that the first amino acid be an alanine
rather than the
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
8
standard methionine and the EMCV IRES cannot tolerate P, W, C, R, or K amino
acids within
the first three codons. In one embodiment, to accommodate the N-terminal amino
acid
restrictions imposed by the EMCV IRES, the DNA vaccine contains a three
Alanine
extension to the N-terminal of the IL-10 gene.
In addition, each IRES element comprises a substantial number of base pairs,
ranging from 230 bp to over 700 bp; the inclusion of multiple IRES elements
thus increases
the size and complexity of plasmids/vectors to the extent that many become
unstable and
difficult to be industrially produced due to spontaneous deletions and
recombinations.
Further, due to the high degree of secondary structure that IRES elements
impart on the
transcribed mRNAs that contain them, they increase the probability of
activating pathogen
recognition receptors (Dabo S, Meurs EF. 2012 Viruses 4(11):2598-635.) in the
transfected
cell and producing stimulatory effects counter to the tolerance induction that
is intended.
2A sequences, unlike IRES elements, do not interact with each other and
therefore
provide stable and consistent performance. However, they are translated
themselves and
therefore affect the folding, function, and stability of the final translated
protein products. All
2A sequences result in a significant C-terminal fusion (19-22 aa) onto the
Send of the
sequences to be separated and also begin the 3' sequence with a proline. Some
proteins are
permissive of these modifications and some are not, leading to practical
restrictions to the
use of 2A sequences. For instance, the Interleukin-10 product is permissive of
the 2A tail but
both Interleukin-2 and TGF-[31 mis-fold and lose function if expressed
upstream of a 2A tag.
Therefore, while it is possible to express several independent proteins
separated by 2A
sequences, two of the four proteins herein cannot terminate in 2A tags and
therefore other
strategies must be utilized.
As each type of 2A amino acid sequence modifies ribosomal function during
protein
translation, it will have different efficiencies in the two core properties of
the 2A family namely
(i) separation of the juxtaposed gene products and (ii) processivity (re-
initiation) into the
second gene product. Different 2A sequences have different efficiencies at
generating the
ribosomal pause that breaks the peptide backbone (resulting in the two
separate proteins) as
well as different efficiencies at re-initiating the peptide synthesis of the
second gene product.
The ability of the 2A sequences to separate protein products and re-initiate
protein
translation are dependent on the 2A amino acid sequence (Donnelly ML, Hughes
LE, Luke
G, et. al. 2001 J Gen Virol. 82(Pt 5):1027-41). Small variations in 2A amino
acid sequences
result in significantly different mixes of separated and fused flanking gene
products, ranging
from under 5% (>95% fused) to completely separated (0% fused or 100%
separated).
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
9
Furthermore, the inventor has herein discovered that adjacent amino acid
sequences encoding the two flanking protein products also affect efficiency of
re-initiation
and separation of the 2A sequences, leading to significant deviations from
reported results.
Re-initiation efficiency thus varies depending on the type of 2A amino acid
sequence used as
well as the environment provided by the adjacent amino acid sequences, and
thus the ratio
of the pre-2A gene product and separation of the proteins will be determined
by both the 2A
amino acid sequence used and its context.
In one embodiment, "FM DV 2A" is inserted between the antigen encoding
sequence and the TGF-[31 encoding sequence herein; resulting in 100%
separation, as well
as a 1:1 ratio, of the protein products.
In another embodiment, "TaV 2A" may be inserted between the IL-10 encoding
sequence and the IL-2 encoding sequence herein, resulting in about 50%
separate products
as well as a 10 to 6 ratio of the protein products. Each transfected cell thus
delivers a
relatively low dose of Interleukin-2, that is incapable of stimulating
effector T cells, and a
higher dose of Interleukin-10 to bias the T cells toward the Treg phenotype.
Since the
production of fused IL-10/1L-2 is disadvantageous, attempts to engineer
increased cleavage
efficiency of the TaV 2A segment were made. An attempt to precede the 2A
segment with an
"insulator segment", which is an element that extends the translated region
upstream of the
TaV 2A to reduce upstream sequence impact on the 2A element, did not improve
separation.
In a different attempt to solve the fusion problem, an upstream uncoupler
segment with a
translated protein sequence of GSG was added; however, this approach resulted
only in an
incremental improvement of cleavage efficiency.
As such, cytokine fusions, resulting from separation of the IL-10 and IL-2
encoding
genes by a TAV 2A, are likely to be immunogenic,
In a further embodiment, the vector/plasmid herein has a "P 2A" segment.
Separation of the IL-10 and IL-2 encoding genes by a P2A results in complete
or near-
complete separation of the protein products as well as a ratio of at least
twice as much (or
maybe even up to four or five times as much) IL-10 compared to IL-2.
In order to address the shortcomings of the IRES-only and 2A-only systems
described above, the four cDNA sequences herein (antigen, TGF-[31, IL-10, IL-
2) are
arranged in pairs before and after a single IRES. Each pair is further
separated by a 2A
sequence, which induces ribosomal skipping and production of independent
proteins from
each sequence in the polyprotein pair. As TGF-[31 and IL-2 may not be on the N-
terminal
side of the fusion, one of them must terminate at the central IRES site and
the other one
must end the translated portion of the mRNA sequence.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
The chronology/sequence of expressed proteins and IRES/2A elements herein may
therefore be as follows: (i) Antigen, (ii) FMDV 2A, (iii) TGF beta 1, (iv)
IRES, (v) IL-10, (vi) P
2A, and (vii) IL-2. As a consequence, all four proteins can be independently
expressed from
a single operon/gene segment in a stable and predictable fashion. As each of
these proteins
5 is expressed from a single mRNA, the ratios of each product are fixed ¨
it is not possible to
generate an excess of IL-2 over IL-10 for instance.
Besides using a combination of IRES and 2A elements for separation of encoded
genes, an alternative solution herein could be use of a bidirectional promoter
to generate 2
mRNAs ¨ these mRNAs would each encode a pair of proteins rather than all four
in one
10 mRNA molecule. Equivalent arrangements may therefore be constructed
utilizing pairs of
expression cassettes appropriately arranged around a bidirectional mammalian
promoter and
utilizing separating 2A sequences and/or IRES elements. This approach is,
however,
associated with disadvantages, primarily due to the large size of bi-
directional promoters but
also a potential increased regulatory burden having separate mRNA elements
included in
one medicinal product. Preferred embodiments herein therefore utilize a single
promoter and
a combination of IRES and 2A elements rather than a bidirectional promoter.
In theory, some 2A sequences could be replaced with intracellular endogenous
protease sensitive sequences. However, the inventor has herein discovered that
such
proteases are associated with significant disadvantages (e.g. lack of reported
function
resulting in secretion of fused protein products).
In order for the antigen to be processed and presented to the immune system
within
the local environment of the plasmid encoded cytokine hormones, the antigen
must be
retained within the transfected cell. In the case of type 1 diabetes,
production of active insulin
would potentially lead to undesirable lowering of blood glucose if it were to
be secreted or
otherwise released from the transfected cell.
In order to avoid antigen secretion, any secretion signals can be removed from
the
antigen encoding sequence, e.g. remove secretion signal coding sequence from
the nucleic
acid sequence encoding pre-proinsulin, thus proinsulin rather than pre-
proinsulin would be
generated, thus allowing the antigen to accumulate inside the transfected
cell. While this
translated antigen product (e.g. insulin) would not be actively secreted, it
could be released
during lysis due to necrosis resulting from attack by CD8+ T cells.
Additionally, the signal
sequence of insulin is a region known to contain disease-relevant epitopes
(potentially
inducing auto-immunity) and the inclusion of the signal sequence therefore
ensures broader
tolerance induction and a higher probability of reducing disease.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
11
Additionally, cytoplasmic retention of antigen only allows for processing via
the
proteasome and presentation via the MHC class I pathway, which detects
intracellular
pathogens via CD8+ T cells. As CD4+ T cells are significant contributors to
pro-inflammatory
cytokines and most, if not all, autoimmunity suppressing Tregs are CD4+,
broadening the
presentation of antigen to include MHC class II, which is recognized by CD4+ T
cells, may be
advantageous.
MHC class II processing and CD4+ T cell stimulation normally do not include
intracellular antigen, as access to this pathway is via endocytosis of extra
cellular antigen.
Normally, protein products produced within a transfected cell are only
presented via the
default intracellular! proteasomal processing pathway and MHC class I,
resulting in CD8+ T
cell effects but not CD4+ T cell effects. In order to target both CD4+ and
CD8+ T cells for
immunomodulation the preferred embodiment also includes factors leading to MHC
class II
presentation.
In principle, to induce MHC class II presentation, the antigen can be fused to
any
.. partner that directs the fusion to an endosomal compartment, but there are
functional
differences in activity and exposure. Transferrin receptor, also known as iron
transporting
protein receptor, fusions cycle from the plasma membrane/extracellular space
to the
endosome and therefore may also expose other immune cells to whole antigen,
such as B
cells, macrophages, etc.. LimplUSCARB fusions target directly to the endosome,
but
preferentially to the early endosome and sometimes result in over processing
and total
destruction of the antigen. Ii (CD74) fusions, utilizing the same chaperone
signal that MHC
class II uses for late endosome localization, deliver the antigen and MHC
class II to the same
vesicles at the same developmental stage and maximize the likelihood of
effectively
presenting antigen in the context of MHC class II. Additionally, even with
endosomal sorting
.. from Ii fusions, the preproinsulin secretion sequence must be rendered
inactive or the
antigen would also be secreted and lost prior to processing.
Blockade of insulin antigen secretion has alternatively been accomplished
herein by
mutating two amino acids required for secretion tag removal by the SRP (Signal
Recognition
Particle) on the Rough Endoplasmic Reticulum. Ala (A) to Glu (E) mutations
completely
abolish pre-proinsulin maturation and secretion, while maintaining the
required epitope
structure of the antigen for best tolerance induction.
In one embodiment, the plasmid DNA vaccine is used herein. The plasmid is
grown/replicated for example in E. coil, and isolated/purified from the media,
and
subsequently formulated in liquid formulations e.g. water, saline, PBS liquid
formulations, or
.. as a lyophilized powder for intrademal jet injection, intranasal
administration, or inhalation. In
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
12
one embodiment, the plasmid herein is formulated in an aqueous pharmaceutical
formulation
optionally comprising stabilisers. Any suitable microbial system may be
utilized for plasmid
production.
Stabilizers in the formulation include, but are not limited to, chelating
agents, such
as EDTA, EGTA, or DPTA for scavenging Mg and Fe' which may otherwise be
involved
in degradation of DNA, and/or citrate, which protects the plasmid from non-
specific
degradation effects. In one embodiment, the plasmid herein may be formulated
in isotonic
PBS or alternatively TRIS + citrate + EDTA. Such plasmids have the advantages
of being
stable, easy to produce and being safe and convenient in use.
In another embodiment, delivery agents, such as virus, lipids, liposomes, co-
packaging etc., could be added in connection with the present invention.
However, the use of
delivery agents herein may have potential problems with immunity, viral
integration, etc.
Definitions
Antigen: the DNA immuno-therapy vaccine herein encodes an antigen. The antigen
herein can be any type of immunogenic disease-associated protein or fragment
thereof that
can be recognized by the T cell component of the immune system. For example,
in the case
of type 1 diabetes treatment or prevention, an insulin antigen may be used. In
one example,
the insulin antigen is the InsB 9-23 immunodominant peptide. For multiple
sclerosis DNA
immuno-therapy vaccines herein, a myelin basic protein (MBP), myelin
oligodendrocyte
protein (MOG), and/or proteolipid protein (PLP) antigen may be used as
antigen. Similar
protein antigen encoding sequences for representative antigens from alopecia,
polymyositis/dermatomyositis, celiac sprue, and protein allergens (e.g. peanut
protein ara h
2) are also examples of antigens suitable for use in the DNA immuno-therapy
vaccines
herein.
Antigen targeting: In one embodiment, antigen herein is endosomally targeted.
Antigens herein include whole protein, secretion-deficient pre-proteins, or a
functional or
immuno-dominant peptide fragment thereof.
For example, insulin antigen herein is an antigen for use in immune modulatory
therapy and not a glucose lowering agent. It should therefore not be fully
processed/matured
or secreted in order to make sure that it is presented on MHC molecules to
circulatory T
cells. The DNA immuno-therapy vaccine herein does therefore not result in
increased insulin
levels in the blood but rather results in an increased presentation of
antigens to the immune
system, in particular the T cells.
Therefore, insulin antigen herein can be small immuno-dominant peptide
encoding
fragments (e.g. insulin B chain 9-23 peptide, including shifted register
peptides displaying
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
13
equivalent T cell epitopes), whole proinsulin, which lacks the required
secretion sequence
but otherwise intact, or pre-proinsulin muteins that contain the secretion
sequence but are
modified to prevent secretory function.
Examples of Insulin antigens herein include:
Mouse proinsulin (SEQ ID NO 1):
FVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEG
SLQKRGIVEQCCTSICSLYQLENYCN
Human proinsulin (SEQ ID NO 2):
FVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQ
PLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Modified mouse pre-proinsulin that is not secreted (substitutions in relation
to wt pre-
proinsulin shown with bold and underline (SEQ ID NO 3)):
MALWMRLLPLLALLALWGPDPEQEFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDL
QVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Modified Human pre-proinsulin that is not secreted (substitutions in relation
to wt
pre-proinsulin shown with bold and underline (SEQ ID NO 4)):
MALWMRLLPLLALLALWGPDPEQEFVNQHLCGSHLVEALYLVCGERGFFYTPKTR
REAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Mouse wt pre-proinsulin (SEQ ID NO 5):
ALWMRLLPLLALLALWGPDPAQAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQ
VGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Human wt pre-proinsulin (SEQ ID NO 6):
MALWMRLLPLLALLALWGPDPAQAFVNQHLCGSHLVEALYLVCGERGFFYTPKTR
REAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Insulin peptide "InsB 9-23" identical between mouse and human:
SHLVEALYLVCGERG (SEQ ID NO 7)
Modified InsB 9-23 (substitutions in relation to wt InsB 9-23 shown with bold
and
underline (SEQ ID NO 8) and (SEQ ID NO 27)):
SHLVEALYLVCGEEG and SHLVEALYLVCGGEG
Insulin antigens herein may thus accumulate in the cytosol of the transfected
host
cell and can thus be presented via MHC class I, or be released upon cytolysis.
Endosomal targeting resulting in MHC class II presentation may be accomplished
herein via fusion of the antigen sequence with leader sequences which form
transmembrane
segments with cytoplasmic "YXX0" sequences, in which Y is tyrosine, X is any
amino acid,
and 0 is a bulky hydrophobic amino acid such as tryptophan or isoleucine,
"[DE]XXXL[LI]"
CA 03042321 2019-04-30
WO 2018/083111
PCT/EP2017/077949
14
where D and E are aspartic or glutamic acid respectively, while L and I are
leucine and
isoleucine respectively, or "DXXLL" endosomal/lysosomal sorting signals, which
are
underlined in the following exemplary sequences. Protein domains that include
these signals
therefore target or cycle to the endosome/lysosome include: transferrin
receptor, Limpll, or
CD74, also known as Invariant chain, MHC II chaperone, or Ii, or any similar
domain.
Examples of endosomal targeting domains herein include, but are not limited
to:
Mouse CD74 / Invariant chain (Ii) endosomal targeting domain (SEQ ID NO 9):
MDDQRDLISNHEQLPILGNRPREPERCSRGALYTGVSVLVALLLAGQATTAYFLYQ
QQGRLDKLTITSQNLQLESLRMKLP
Human CD74 / Invariant chain (Ii) endosomal targeting domain (SEQ ID NO 10):
MHRRRSRSCREDQKPVMDDQRDLISNNEQLPMLGRRPGAPESKCSRGALYTGFS
ILVTLLLAGQATTAYFLYQQQGRLDKLTITSQNLQLESLRMKLP
Type 1 diabetes: Type 1 diabetes (Ti D) is considered to be a chronic
autoimmune
disease, where auto-aggressive T cells infiltrate the islets of Langerhans in
the pancreas and
play an important role by specifically destroying the insulin-producing beta-
cell population.
Once a significant number of islet cells are destroyed, reduced amounts of
insulin, or no
insulin at all, will result in insulin deficiency and hyper-glycemia in the
patient. T1D patients
are thus unable to produce enough insulin and need regular injections of the
hormone are
needed throughout life. Some Type 1 Diabetes patients are diagnosed with "type
1.5
Diabetes", "latent autoimmune diabetes"/LADA, "double diabetes" etc., which
are diabetes
diseases carrying symptoms of both Type 1 Diabetes and Type 2 Diabetes ¨ all
diabetes
diseases carrying trains of both Type 1 and Type 2 Diabetes are thus also
contained in the
term "Type 1 Diabetes" herein.
To!erogenic DNA vaccine: DNA-based immuno-therapy vaccines/vectors/plasmids
herein are designed to switch off or down-regulate the part of the immune
system
responsible for destroying normal healthy "self" cells and thus prevent or
ameliorate T cell-
based autoimmunity.
The term "DNA immuno-therapy vaccine" as used herein is intended to mean a
compound or composition comprising a DNA molecule and which is administered to
a
subject in order to reduce the risk of said subject developing one or more
diseases.
In some embodiments, DNA based immuno-therapy vaccines herein are
plasmids/vectors encoding particular antigens. Following vaccination, these
plasmids are
taken up by, in other words, transfected into antigen presenting cells in the
host's body. The
"transfected" host cells then produce the antigen and present small fragments
of the antigen
to the immune system, in particular the T cells. This approach leads to a
modification of
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
specific T cell responses to the encoded antigen as well as minimal
modification to immune
responses to other (non-encoded or "irrelevant") antigens. Only a very few
host cells are
typically transformed with the DNA vaccine plasmid/vector herein, meaning that
likely fewer
than one out of hundred thousand, one out of five hundred thousand, or even
fewer than one
5 out of a million plasmid/vector molecules eventually enter a host cell.
DNA vaccines herein
thus represent a very mild and specific approach for modulating immune
responses to
antigens such as insulin in T1D patients or patients at risk of developing Ti
D.
Plasmid: A plasmid is a small DNA molecule that is most commonly found in
bacteria as small, circular, double-stranded DNA molecules. Artificial
plasmids are widely
10 used as vectors in molecular cloning, serving to drive the replication
of recombinant DNA
sequences within host organisms. Plasmids can be engineered to be suitable for
use as
immuno-therapy DNA vaccines. Plasmids are considered replicons, a unit of DNA
capable of
replicating autonomously within a suitable host. Plasmids can be transmitted
from one
bacterium to another bacterium, which could be of the same or different
bacterial species via
15 three main mechanisms: transformation, transduction, and conjugation.
DNA vaccine
plasmids can be taken up by a host cell by passive transformation ¨ usually at
a relatively
low rate. The plasmids herein replicate efficiently - but do not drive protein
expression - in
bacteria. The plasmids herein furthermore drive protein expression - but not
replication of
plasmid - in humans and other mammals, e.g. mice. In one embodiment, a pVAX1
vector
(Invitrogen/LifeTechnologies) is used as a scaffold herein for inserting the
elements that are
part of the present invention. Other suitable vector scaffolds herein include
any vector
backbone containing a eukaryotic promoter element, a prokaryotic high copy
origin of
replication, and a selection system for plasmid maintenance.
Selection gene and selection system: In one aspect, DNA immuno-therapy
vaccines
herein comprise a selection gene/selection marker for manufacturing purposes.
The
selectable marker herein is e.g. a gene that confers resistance to a cell
toxin - e.g. an
antibiotic such as ampicillin, kanamycin, chloramphenicol, streptomycin, etc.
Other types of suitable selection systems herein include e.g. conditional
lethal
silencing systems (e.g. CcdA/CcdB or ParD/ParE Hok/Sok type systems), or
sequences that
complements a genomic defect in the production cell strain and thus permits
growth of an
otherwise inviable host (e.g. dapD- or pyrF- auxotrophic complementation, infA-
translation
initiation complementation, etc.)
Production cells harbouring the plasmid/DNA vaccine, which includes the
selection
marker, will survive when exposed to the toxin/antibiotic/condition, while
those that have
failed to take up plasmid sequences will die. As such, in one embodiment, DNA
vaccines
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
16
herein comprises the nucleic acid sequence encoding a selection marker in
order to provide
for higher yield/purity and more efficient production/replication in
production cells, such as
E.coli.
While antibiotic selection is a common laboratory strategy there may be
advantages
associated with antibiotic-free selection systems ¨ e.g. in relation to more
efficient regulatory
processes. While vectors which do not contain a selection mechanism such as
minicircles,
synthetic linear vectors, etc., can also be used herein, these implementations
are associated
with certain drawbacks in production, in particular due to increased
production and quality
control costs.
Examples of complementation ("rescue") strategies are known in the prior art,
however these strategies suffer from various disadvantages.
Metabolic complementation systems such as dapD [lysine biosynthesis] or pyrF
[uridine biosynthesis] systems, often result in "cross-feeding" during high
density E.coli
production, where a plasmid-containing bacterium will produce and secrete an
excess of the
required compound and thereby "relaxing" the selection pressure for
neighbouring bacteria
without the plasmid.
Another example of a suitable selection system herein are plasmids encoding
essential proteins, such as infA, encoding IF1 / Initiation Factor 1 which is
required for protein
synthesis. In this selection system, cross-feeding does not occur because the
infA protein is
not secreted. However, it is not possible to further modify the plasmid or
expand plasmid-
deficient cells as there is no way to exogenously complement the required
protein/infA (J
Bacteriol. 1994 Jan;176(1):198-205 and J Biotechnol. 2004 Jul 1;111(1):17-30).
In order to circumvent the disadvantages associated with the infA selection
system,
an alternative selection system has been provided herein with a temperature-
sensitive
translation switch (or "thermosensor") from the invasion protein gene prfA of
L.
monocytogenes (Cell. 2002 Sep 6;110(5):551-61). By placing the hairpin forming
portion of
an RNA "thermosensor" sequence upstream of the E.coli genomic copies of infA
via standard
recombination technology, expression thereof becomes regulated via control of
the
fermentation temperature, enabling slow growth of plasmid free cells at 37 C,
and rapid cell
death at temperatures <30 C. Transformation of the engineered thermo sensitive
E.coli
production strain with plasmids expressing wt infA thus allow full normal
growth rates at all
temperatures, allowing for plasmid-free expansion at 37 C as well as stringent
selection for
plasmid at 30 C. Additionally, this system generates no selective pressure for
wt E.coli to
retain the plasmid and it is thus lost within 8 hours in culture ¨ ensuring no
environmental
persistence of the therapeutic plasmid.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
17
wt E.coli infA nucleotide sequence (SEQ ID NO 11):
ATGGCCAAAGAAGACAATATTGAAATGCAAGGTACCGTTCTTGAAACGTTGCCT
AATACCATGTTCCGCGTAGAGTTAGAAAACGGTCACGTGGTTACTGCACACATCTCCGG
TAAAATGCGCAAAAACTACATCCGCATCCTGACGGGCGACAAAGTGACTGTTGAACTGA
CCCCGTACGACCTGAGCAAAGGCCGCATTGTCTTCCGTAGTCGCTGA
wt E.coli I F1 protein sequence resulting from translation of the infA gene
(initial
methionine/M not included in prfA fusion - (SEQ ID NO 12)):
MAKEDNIEMQGTVLETLPNTMFRVELENGHVVTAHISGKMRKNYIRILTGDKVTVEL
TPYDLSKGRIVFRSR
E. colt production cell lines used herein for production of DNA immuno-therapy
vaccine plasmids may thus harbour the following thermo sensitive prfA
nucleotide sequence:
wt L.monocytogenes prfA ("thermo sensor hairpin") nucleotide sequence (Shine
Dalgarno underlined, ATG start bolded - (SEQ ID NO 13)):
TGTAAAAAACATCATTTAGCGTGACTTTCTTTCAACAGCTAACAATTGTTGTTAC
TGCCTAATGTTTTTAGGGTATTTTAAAAAAGGGCGATAAAAAACGATTGGGGGATGAGAA
ATGAACGCTCAA
wt L.monocytogenes prfA protein sequence (fused upstream of E.coli IF1 ¨
resulting
from translation of SEQ ID NO 13):
MNAQ
Origin of replication ("On"): The origin of replication, also called the
replication origin,
is a particular sequence in a genome at which replication of the DNA strand is
initiated. In
one embodiment, origin of replication sites herein includes the "pUC On" which
allows
replication in the bacterial E. colt production cell line - but not in the
mammalian host cells,
i.e., cells from the body of the vaccinated subject/person/patient. Other
suitable bacterial
replication origins herein include but are not limited to: R6K, pBR322, ColE1,
pMB1, 15A,
pSC101, etc. In one aspect, the origin of replication herein is a high copy
version which
yields a high plasmid/biomass ratio for more efficient production. Vectors
which do not
contain an origin of replication, such as minicircles, synthetic linear
vectors, etc., can also be
used herein.
Promoter: A promoter is a region of DNA that initiates transcription of a
particular
gene. Promoters are located near the transcription start sites of genes, on
the same strand
and upstream on the DNA, which towards the 5' region of the sense strand. For
the
transcription to take place, the RNA polymerase must attach to the DNA near a
gene.
Promoters contain specific DNA sequences, such as response elements, that
provide a
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
18
secure initial binding site for RNA polymerase and for transcription factors
that recruit RNA
polymerase. Transcription factors have specific activator or repressor
sequences that attach
to specific promoters and regulate gene expression. Promoters thus represent
critical
elements that can work in concert with other regulatory regions, such as
enhancers,
silencers, boundary elements/insulators, to direct the level of transcription
of a given gene. A
classical promoter drives the production of a single messenger RNA (mRNA),
whereas
bidirectional promoters herein drive the production of two mRNAs immediately
adjacent to
the promoter, both upstream and downstream of the promoter.
In one embodiment, eukaryotic promoters are used herein. Eukaryotic promoters
do
not necessarily obey the one gene/one promoter rule, such as several viral
promoters as well
as promoters that exhibit broad expression (i.e. do not have narrow cell type
specificities
such as neuron-only expression). Examples of promoters herein that are capable
of driving
broad transcription of large multi-gene mRNA molecules include: the viral CMV
immediate-
early (1E) and SV40 promoters; endogenous EFla, PGK1, Ubc, and beta actin
promoters;
and synthetic promoters such as the CAG hybrid promoter. Many other suitable
mammalian
promoters exist and more are being designed via synthetic biology efforts. Any
promoter that
results in the desired expression characteristics in human cells may be used
in the DNA
immu no-therapy vaccine plasmids herein.
Enhancers: Enhancers are DNA elements that increase the efficiency of
promoters
in producing mRNA transcripts. The enhancers herein may be matched (e.g. SV40
enhancer/CMV promoter) or unmatched. Any suitable enhancer/promoter
combination for
eukaryotic function can be used herein.
Eukaryotic translation start: The eukaryotic translation start sequence is
usually
referred to as the "Kozak" consensus sequence. The Kozak sequence on an mRNA
molecule is recognized by the ribosome as the translational start site, from
which a protein is
encoded. The eukaryotic ribosome requires this sequence, or a variation
thereof, to initiate
protein translation. Kozak sequences are degenerate or variable and rarely
match consensus
sequences. In fact, consensus Kozak sequences are typically less efficient
than wild type
variants isolated from mammalian mRNAs. While weak Kozak sequences are
regularly
isolated from native mRNAs and likely play a role in translational control of
low abundance
proteins, DNA immuno-therapy vaccines herein preferably encode a medium or
high
efficiency Kozak sequence. Examples of useful Kozak sequences herein comprise
the
following nucleotide sequence: gccRccATGG (SEQ ID NO 14), where lower case
bases are
the most common nucleotides but may vary while upper case nucleotides are
fixed (R is the
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
19
I U PAC uncertainty code for A or G bases), and the ATG indicates the
translational start site
of Methionine codon at position +1.
Endosome sorting signal: An endosome is a membrane-bounded compartment
inside eukaryotic cells. Some proteins can be transported to endosomes and
therein be
degraded into peptide fragments. The peptide fragments can bind to MHC
molecules present
in the endosome to form MHC/peptide complexes, which can subsequently be
transported to
the cell surface in order to be presented to circulating T cells, particularly
CD4+ T
cells.Sorting of proteins to endosomes is mediated by signals present within
the cytosolic
domains of the proteins. The endosomal signals are usually short linear amino
acid
sequences. Antigens herein are preferably targeted to the endosomes using an
endosome
sorting signal, such as e.g. YXXO, [DE]XXXL[L1], or DXXLL endosomal/lysosomal
sorting
signals. Endosome sorting signals include various naturally occurring or
synthetic endosomal
sorting signals. Examples herein include the endosome sorting signals present
on
Cd74/invariant chain/li, LimplUSCARB, or transferrin receptor. Any endosomal
targeting
domain which is pharmaceutically acceptable and provides the desired function
may be
utilized. Fusion of such endosomal targeting domains to the antigens directs
them to the
endosomal compartment upon translation for increased efficacy. Endosomal
sorting of
antigens confers processing and presentation to the immune system in MHC class
II
complexes, in addition to constitutive presentation in MHC class I complexes,
for more
complete and robust induction of tolerance and possible expansion of Tregs
(which cannot
be accomplished via MHC class I / antigen complexes). In one embodiment,
tolerogenic DNA
vaccines herein encode a fusion of the antigen with the CD74/invariant
chain/li to drive
endosomal targeting and presentation of the antigen via MHC class II.
lntrons: lntrons are non-coding sequences within an mRNA. It is known that
some
introns significantly increase translation and function of mRNA. Accordingly,
the inclusion of
intron sequences may also be used herein. Standard introns, such as beta-
globin, or any
intron obeying mammalian splicing conventions, such as MCM7, may be utilized.
In one
embodiment, DNA immuno-therapy vaccine vectors herein comprise sequences
encoding
one or more introns. In another embodiment, DNA immunotherapy vaccine vectors
herein do
not possess sequences encoding introns.
Ribosomal pause tag: In connection with the present invention, it may be an
advantage to include one or more ribosomal pause tag sequence(-s) between the
protein
coding sequences in the DNA immuno-therapy vaccine vector/plasmid herein in
order to
separate protein products.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
An example is the viral "FMDV 2A tag" (Foot-and-mouth disease virus 2A tag).
The
translated amino acid sequence of FMDV 2A is APVKQTLNFDLLKLAGDVESNPGP - (SEQ
ID NO 15). FMDV 2A tag is capable of pausing and reinitiating the ribosome.
The ratio of
translated product before and after the FMDV 2A tag is close to 1:1 and the
resulting protein
5 products are normally completely separated. These types of ribosome tags
have previously
been used in connection with co-expression of two different domains, e.g.
heavy chain and
light chain in recombinant antibody production. However, the inventor of the
present
invention has made the surprising discovery that they are useful in connection
with multi-
cistronic DNA vaccines both for separation of flanking products and for
control of the ratios of
10 expressed proteins due to inherent efficiencies of ribosomal re-
initiation. Sequence tags
which favour a 1:1 ratio of translated products are herein preferably inserted
between two
protein encoding sequences that should preferably be produced in (or close to)
a 1:1 ratio
such as e.g. an insulin antigen and a potent cytokine such as e.g. TGF-[3.
Another example of a ribosomal pause tag sequence herein is the viral sequence
15 tag "TaV 2A" (Thosea asigna virus 2A - translated amino acid sequence of
TaV 2A:
RAEGRGSLLTCGDVEENPGP (SEQ ID NO 16). The ratio of translated product
before/upstream and after/downstream of this tag is reported to be 50:1 (or
close to). The
inventor of the present invention has made the surprising discovery that while
this type of tag
can be used to control expression levels in cases where it is vital that one
translated product
20 absolutely dominates another, the separation of flanking cytokine
products is less than 50%
relative to the sequences disclosed in literature and the expression ratio is
thus about 10:6.
In connection with the present invention, a 2A type of ribosomal pause tag
sequence should
preferably result in different expression levels of two proteins encoded by
the same
vector/plasmid. Expression of small amounts of a pleiotropic cytokine (such as
IL-2) relative
to an anti-inflammatory cytokine, such as IL-10, is desirable herein and fused
products are
not desirable.
A further example of a ribosomal pause tag amino acid sequence herein is the
viral
sequence "P 2A" (Porcine teschovirus-1 2A, ATNFSLLKQAGDVEENPGP - (SEQ ID NO
17)). P 2A sequences function appropriately when inserted between IL-10 and IL-
2 herein,
resulting in near complete separation with an expression ratio of >5:1 between
IL-10 and IL-
2.
Alternatively, proteinase sensitive sequences, allowing for endogenous
cleavage
between plasmid expressed poly proteins, may be used herein. A furin sensitive
sequence
(recognizing RAKR motifs) or carboxypeptidase sensitive sequence (recognizing
RRRR,
RKRR, or RRKR motifs) may be used herein for separating protein products.
However, the
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
21
inventor of the present invention has made the surprising discovery that
neither furin nor
carboxypeptidase cleavage sequences result in separated products herein ¨ thus
leading to
secretion of undesired 1L-10/1L-2 fusion proteins.
TGF-14/81 (Transforming growth factor beta/81): TGF-8 is a secreted protein
that
controls proliferation, cellular differentiation, and other functions in most
cells. TGF-8 is a
very potent cytokine with significant effects on cell fate and phenotype in a
context-
dependent manner, e.g. depending upon the other cytokine signals received
contemporaneously. Endogenous TGF-8 is produced in a latent form associated
with the
outer membrane surface of the producing cell and requires activation (e.g. by
inflammatory
macrophages expressing CD36 and plasmin proteinase) for maturation and release
of the
active form. In one embodiment, TGF-8 herein is a modified form that is
constitutively active.
This is achieved by replacing the cysteines at positions 223 and 225 with
amino acids
incapable of forming disulfide bridges. For example, serine or valine are used
to replace
cysteines at positions 223 and 225. This results in an active pro-protein
structure that is
released into the local microenvironment.
Human endogenous TGF-81sequence - SEQ ID NO 18:
MPPSGLRLLLLLLPLLWLLVLTPGRPAAGLSTCKTIDMELVKRKRIEAIRGQILSKLRLASPPS
QGEVPPGPLPEAVLALYNSTRDRVAGESAEPEPEPEADYYAKEVTRVLMVETHNEIYDKFK
QSTHSIYMFFNTSELREAVPEPVLLSRAELRLLRLKLKVEQHVELYQKYSNNSWRYLSNRLL
APSDSPEWLSFDVTGVVRQWLSRGGEIEGFRLSAHCSCDSRDNTLQVDINGFTTGRRGDL
ATIHGMNRPFLLLMATPLERAQHLQSSRHRRALDTNYCFSSTEKNCCVRQLYIDFRKDLGW
KWIHEPKGYHANFCLGPCPYIWSLDTQYSKVLALYNQHNPGASAAPCCVPQALEPLPIVYYV
GRKPKVEQLSNMIVRSCKCS.
Modified human TGF-81sequence that is constitutively active and secreted
(substitutions in relation to wt TGF-81 shown with bold and underline) - SEQ
ID NO 19:
MPPSGLRLLLLLLPLLWLLVLTPGRPAAGLSTCKTIDMELVKRKRIEAIRGQILSKLRLASPPS
QGEVPPGPLPEAVLALYNSTRDRVAGESAEPEPEPEADYYAKEVTRVLMVETHNEIYDKFK
QSTHSIYMFFNTSELREAVPEPVLLSRAELRLLRLKLKVEQHVELYQKYSNNSWRYLSNRLL
APSDSPEWLSFDVTGVVRQWLSRGGEIEGFRLSAHVSVDSRDNTLQVDINGFTTGRRGDL
ATIHGMNRPFLLLMATPLERAQHLQSSRHRRALDTNYCFSSTEKNCCVRQLYIDFRKDLGW
KWIHEPKGYHANFCLGPCPYIWSLDTQYSKVLALYNQHNPGASAAPCCVPQALEPLPIVYYV
GRKPKVEQLSNMIVRSCKCS.
Another modified human TGF-81 sequence that may be used is SEQ ID NO 25:
MPPSGLRLLLLLLPLLWLLVLTPGRPAAGLSTCKTIDMELVKRKRIEAIRGQILSKLRLASPPS
QGEVPPGPLPEAVLALYNSTRDRVAGESAEPEPEPEADYYAKEVTRVLMVETHNEIYDKFK
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
22
QSTHSIYMFFNTSELREAVPEPVLLSRAELRLLRLKLKVEQHVELYQKYSNNSWRYLSNRLL
APSDSPEWLSFDVTGVVRQWLSRGGEIEGFRLSAHSSSDSRDNTLQVDINGFTTGRRGDL
ATIHGMNRPFLLLMATPLERAQHLQSSRHRRALDTNYCFSSTEKNCCVRQLYIDFRKDLGW
KWIHEPKGYHANFCLGPCPYIWSLDTQYSKVLALYNQHNPGASAAPCCVPQALEPLPIVYYV
GRKPKVEQLSNMIVRSCKCS.
Terminator sequence: a transcription terminator is a section of a nucleic acid
sequence that marks the end of a gene during transcription. Release of the
transcriptional
complex frees RNA polymerase and related transcriptional machinery to begin
transcription
of new mRNAs. Additionally, the same cellular factors add a non-templated
"poly-A tail"
which significantly enhances the lifetime and functionality of the mRNA. An
example of a
suitable transcription terminator herein includes the "bGH_PA" terminator,
CGACTGTGCCTTCTAGTTGCCAGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTG
ACCCTGGAAGGTGCCACTCCCACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCA
TTGTCTGAGTAGGTGTCATTCTATTCTGGGGGGTGGGGTGGGGCAGGACAGCAAGGGG
GAGGATTGGGAAGACAATAGCAGGCATGCTGGGGATGCGGTGGGCTCTATGG (SEQ ID
NO 20).
Any acceptable terminator sequence may be utilized herein. Variations include
use
of two different flanking terminator sequences in the instance of
bidirectional promoters
producing two oppositely-oriented mRNAs.
In one embodiment the plasmid of the invention has the sequence as set out in
SEQ
ID NO 24.
In a second embodiment the plasmid of the invention has the sequence SEQ ID NO
26: full (non-annotated) plasmid sequence
GACTCTTCGCGATGTACGGGCCAGATATACGCGTTGACATTGATTATTGACTAG
TTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGT
TACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCATTG
ACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAA
TGGGTGGACTATTTACGGTAAACTGCCCACTTGGCAGTACATCAAGTGTATCATATGCC
AAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCGCCTGGCATTATGCCCAGT
ACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATTAGTCATCGCTATTA
CCATGGTGATGCGGTTTTGGCAGTACATCAATGGGCGTGGATAGCGGTTTGACTCACG
GGGATTTCCAAGTCTCCACCCCATTGACGTCAATGGGAGTTTGTTTTGGCACCAAAATC
AACGGGACTTTCCAAAATGTCGTAACAACTCCGCCCCATTGACGCAAATGGGCGGTAG
GCGTGTACGGTGGGAGGTCTATATAAGCAGAGCTCTCTGGCTAACTAGAGAACCCACT
GCTTACTGGCTTATCGAAATTAATACGACTCACTATAGGGAGACCCAAGCTGGCTAGCG
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
23
TTTAAACTTAAGCTTGGTACCGAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTCTGC
ACTGCAGCTCGCATCTCTCCTTCACGCGCCCGCCGCCCTACCTGAGGCCGCCATCCAC
GCCGGTTGAGTCGCGTTCTGCCGCCTCCCGCCTGTGGTGCCTCCTGAACTGCGTCCGC
CGTCTAGGTAAGTTTAAAGCTCAGGTCGAGACCGGGCCTTTGTCCGGCGCTCCCTTGG
AGCCTACCTAGACTCAGCCGGCTCTCCACGCTTTGCCTGACCCTGCTTGCTCAACTCTA
GGTAAGTTAATGAGACAGATAGAAACTGGTCTTGTAGAAACAGAGTAGTCGCCTGCTTT
TCTGCCAGGTGCTGACTTCTCTCCCCTGGGCTTTTTTCTTTTTCTCAGGTTGAAAAGAAG
AAGACGAAGAAGACGAAGAAGACAAACCGTCGTCGACTGCCATGCGCCGCTGATTAAC
GCCGCCACCATGGCCCACCGACGCAGATCCAGAAGCTGCCGTGAGGACCAGAAGCCC
GTGATGGATGATCAGAGGGACCTTATCTCTAACAATGAACAACTGCCAATGCTCGGCAG
ACGGCCTGGGGCCCCGGAGAGCAAGTGCAGCAGAGGAGCCTTGTACACGGGGTTCTC
CATTTTAGTGACTCTCCTTCTCGCCGGCCAAGCTACCACCGCCTACTTTCTGTACCAACA
GCAAGGCAGACTAGACAAACTGACAATCACAAGCCAGAACCTTCAGCTGGAGTCTCTGC
GGATGAAGCTGCCCGCTTTGTGGATGAGATTGCTTCCTCTACTTGCTCTCCTGGCGCTC
TGGGGACCTGACCCCGAGCAAGAGTTTGTTAATCAGCACCTGTGTGGGAGTCATCTGG
TGGAGGCACTCTATTTAGTGTGCGGAGAGAGGGGCTTCTTCTACACTCCAAAGACCAGA
CGGGAGGCCGAAGACCTTCAAGTGGGGCAAGTAGAACTGGGTGGCGGACCCGGTGCC
GGGAGCCTTCAGCCGCTCGCCCTGGAGGGCTCTCTTCAGAAACGCGGCATCGTGGAG
CAGTGTTGCACATCCATTTGCTCACTCTACCAGCTGGAGAACTACTGCAACGGAAGCGG
AGTGAAGCAGACGTTGAATTTTGATTTGTTGAAGTTGGCGGGGGATGTGGAGAGCAATC
CGGGGCCGATGCCCCCTAGTGGCCTCAGACTTTTGTTATTGTTATTACCGCTTTTATGG
CTCTTGGTGCTGACACCGGGCCGTCCGGCTGCTGGCTTGTCGACTTGTAAGACAATTG
ATATGGAATTGGTGAAACGAAAACGGATTGAGGCCATCCGAGGACAGATTTTGAGCAAG
CTGCGGCTTGCCTCGCCACCCTCGCAAGGGGAAGTCCCACCCGGACCTCTACCAGAAG
CAGTCCTAGCGCTGTACAACAGTACAAGAGATAGAGTGGCCGGGGAATCCGCAGAACC
AGAGCCTGAGCCTGAAGCCGATTATTATGCAAAGGAAGTGACTAGGGTCCTGATGGTC
GAGACCCATAACGAAATCTACGACAAATTCAAACAAAGTACCCACTCTATCTACATGTTC
TTCAACACCAGTGAGCTAAGAGAAGCCGTGCCCGAACCTGTGCTTCTTTCCCGCGCAG
AACTCCGCCTCTTGAGACTCAAATTGAAAGTTGAACAACACGTAGAGCTTTACCAGAAAT
ACTCTAATAATTCATGGCGATATCTTTCTAATCGTCTCCTCGCCCCATCTGACAGCCCTG
AATGGCTCTCCTTCGACGTTACGGGAGTTGTGCGCCAGTGGCTCAGCAGAGGCGGAGA
GATAGAGGGCTTTCGGCTGAGCGCACATAGCTCTAGCGACTCAAGGGACAACACATTG
CAAGTGGATATTAACGGTTTTACAACTGGACGGAGAGGGGACCTGGCGACCATCCACG
GCATGAATAGACCTTTCCTGCTGCTGATGGCTACTCCCCTGGAGAGGGCACAGCACTTA
CAGTCTTCCAGACACCGGCGCGCCCTGGATACAAACTACTGCTTCAGCTCCACCGAAAA
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
24
GAACTGTTGCGTGCGGCAGCTGTACATTGACTTCAGAAAGGATCTGGGCTGGAAGTGG
ATTCATGAGCCCAAGGGGTATCATGCCAACTTCTGTCTTGGGCCATGCCCATACATCTG
GTCACTGGATACCCAGTACTCCAAAGTTCTGGCCTTGTACAATCAACACAACCCTGGAG
CTTCCGCCGCTCCTTGCTGTGTGCCCCAAGCCCTAGAGCCCCTGCCCATCGTTTATTAT
GTCGGACGCAAGCCCAAAGTAGAACAGCTATCAAATATGATCGTGAGAAGCTGCAAGTG
TAGCTGATAAACGCGTCGAGCATGCATCTAGGGCGGCCAATTCCGCCCCTCTCCCCCC
CACCCCTCTCCCTCCCCCCCCCCTAACGTTACTGGCCGAAGCCGCTTGGAATAAGGCC
GGTGTGCGTTTGTCTATATGTTATTTTCCACCATATTGCCGTCTTTTGGCAATGTGAGGG
CCCGGAAACCTGGCCCTGTCTTCTTGACGAGCATTCCTAGGGGTCTTTCCCCTCTCGCC
AAAGGAATGCAAGGTCTGTTGAATGTCGTGAAGGAAGCAGTTCCTCTGGAAGCTTCTTG
AAGACAAACAACGTCTGTAGCGACCCTTTGTAGACAGCGGAACCCCCCACCTGGCGAT
AGATGCCTCTGCGGCCAAAAGCCACGTGTATAAGATACACCTGCAAAGGCGGCACAAC
CCCAGTGCCACGTTGTGAGTTGGATAGTTGTGGAAAGAGTCAAATGGCTCTCCTCAAGC
GTATTCAACAAGGGGCTGAAGGATGCCCAGAAGGTACCCCATTGTATGGGATCTGATCT
GGGGCCTCGGTGCACATGCTTTACATGTGTTTAGTCGAGGTTAAAAAACGTCTAGGCCC
CCCGAACCACGGGGACGTGGTTTTCCTTTGAAAAACACGATGATAATATGATGCACAGC
TCAGCACTGCTCTGTTGCCTGGTCCTCCTGACTGGGGTGAGGGCCAGCCCAGGCCAG
GGCACCCAGTCTGAGAACAGCTGCACCCACTTCCCAGGCAACCTGCCTAACATGCTTC
GAGATCTCCGAGATGCCTTCAGCAGAGTGAAGACTTTCTTTCAAATGAAGGATCAGCTG
GACAACTTGTTGTTAAAGGAGTCCTTGCTGGAGGACTTTAAGGGTTACCTGGGTTGCCA
AGCCTTGTCTGAGATGATCCAGTTTTACCTGGAGGAGGTGATGCCCCAAGCTGAGAACC
AAGACCCAGACATCAAGGCGCATGTGAACTCCCTGGGGGAGAACCTGAAGACCCTCAG
GCTGAGGCTACGGCGCTGTCATCGATTTCTTCCCTGTGAAAACAAGAGCAAGGCCGTG
GAGCAGGTGAAGAATGCCTTTAATAAGCTCCAAGAGAAAGGCATCTACAAAGCCATGAG
TGAGTTTGACATCTTCATCAACTACATAGAAGCCTACATGACAATGAAGATACGAAACGG
GAGCGGCGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCT
GGACCTATGTACAGAATGCAGCTGCTGAGCTGCATCGCCCTGAGCCTGGCCCTGGTGA
CCAACAGCGCACCCACGTCCTCTAGCACCAAGAAGACCCAGTTACAGTTGGAGCATCTA
CTTTTAGACCTGCAAATGATTTTGAACGGCATCAACAACTACAAGAATCCTAAACTTACT
CGCATGCTTACCTTCAAATTTTACATGCCCAAGAAGGCCACCGAACTGAAGCACTTGCA
ATGTCTGGAGGAAGAACTCAAGCCGCTGGAGGAAGTTCTCAACCTCGCGCAGTCCAAG
AATTTCCACCTCCGGCCAAGAGACCTGATCAGTAACATTAATGTGATAGTGCTGGAGCT
GAAGGGAAGCGAGACTACATTTATGTGCGAGTACGCCGATGAAACCGCTACAATCGTC
GAGTTCCTGAATAGATGGATCACATTTTGCCAGTCAATTATCTCTACTCTGACATGATAA
CTCGAGGTCTAGAGGGCCCGTTTAAACCCGCTGATCAGCCTCGACTGTGCCTTCTAGTT
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
GCCAGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGGAAGGTGCCACT
CCCACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGAGTAGGTGTCAT
TCTATTCTGGGGGGTGGGGTGGGGCAGGACAGCAAGGGGGAGGATTGGGAAGACAAT
AGCAGGCATGCTGGGGATGCGGTGGGCTCTATGGCTTCTACTGGGCGGTTTTATGGAC
5 AGCAAGCGAACCGGAATTGCCAGCTGGGGCGCCCTCTGGTAAGGTTGGGAAGCCCTG
CAAAGTAAACTGGATGGCTTTCTCGCCGCCAAGGATCTGATGGCGCAGGGGATCAAGC
TCTGATCAAGAGACAGGATGAGGATCGTTTCGCATGGCCAAAGAAGACAATATTGAAAT
GCAAGGTACCGTTCTTGAAACGTTGCCTAATACCATGTTCCGCGTAGAGTTAGAAAACG
GTCACGTGGTTACTGCACACATCTCCGGTAAAATGCGCAAAAACTACATCCGCATCCTG
10 ACGGGCGACAAAGTGACTGTTGAACTGACCCCGTACGACCTGAGCAAAGGCCGCATTG
TCTTCCGTAGTCGCTGATAAATTATTAACGCTTACAATTTCCTGATGCGGTATTTTCTCCT
TACGCATCTGTGCGGTATTTCACACCGCATACAGGTGGCACTTTTCGGGGAAATGTGCG
CGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAA
TAACCCTGATAAATGCTTCAATAATAGCACGTGCTAAAACTTCATTTTTAATTTAAAAGGA
15 TCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTT
CCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCT
GCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGC
CGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATA
CCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCA
20 CCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAA
GTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCG
GGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAA
CTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGG
CGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTC
25 CAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAG
CGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACG
CGGCCTTTTTACGGTTCCTGGGCTTTTGCTGGCCTTTTGCTCACATGTTCTT.
In a third embodiment the plasmid of the invention has the sequence SEQ ID NO
28: full (non-annotated) plasmid sequence
GACTCTTCGCGATGTACGGGCCAGATATACGCGTTGACATTGATTATTGACTAG
TTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGT
TACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCATTG
ACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAA
TGGGTGGACTATTTACGGTAAACTGCCCACTTGGCAGTACATCAAGTGTATCATATGCC
AAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCGCCTGGCATTATGCCCAGT
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
26
ACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATTAGTCATCGCTATTA
CCATGGTGATGCGGTTTTGGCAGTACATCAATGGGCGTGGATAGCGGTTTGACTCACG
GGGATTTCCAAGTCTCCACCCCATTGACGTCAATGGGAGTTTGTTTTGGCACCAAAATC
AACGGGACTTTCCAAAATGTCGTAACAACTCCGCCCCATTGACGCAAATGGGCGGTAG
GCGTGTACGGTGGGAGGTCTATATAAGCAGAGCTCTCTGGCTAACTAGAGAACCCACT
GCTTACTGGCTTATCGAAATTAATACGACTCACTATAGGGAGACCCAAGCTGGCTAGCG
TTTAAACTTAAGCTTGGTACCGAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTCTGC
AGCTCGCATCTCTCCTTCACGCGCCCGCCGCCCTACCTGAGGCCGCCATCCACGCCGG
TTGAGTCGCGTTCTGCCGCCTCCCGCCTGTGGTGCCTCCTGAACTGCGTCCGCCGTCT
AGGTAAGTTTAAAGCTCAGGTCGAGACCGGGCCTTTGTCCGGCGCTCCCTTGGAGCCT
ACCTAGACTCAGCCGGCTCTCCACGCTTTGCCTGACCCTGCTTGCTCAACTCTAGGTAA
GTTAATGAGACAGATAGAAACTGGTCTTGTAGAAACAGAGTAGTCGCCTGCTTTTCTGC
CAGGTGCTGACTTCTCTCCCCTGGGCTTTTTTCTTTTTCTCAGGTTGAAAAGAAGAAGAC
GAAGAAGACGAAGAAGACAAACCGTCGTCGACTGCCATGCGCCGCTGATTAACGCCGC
CACCATGGCCCACCGACGCAGATCCAGAAGCTGCCGTGAGGACCAGAAGCCCGTGAT
GGATGATCAGAGGGACCTTATCTCTAACAATGAACAACTGCCAATGCTCGGCAGACGGC
CTGGGGCCCCGGAGAGCAAGTGCAGCAGAGGAGCCTTGTACACGGGGTTCTCCATTTT
AGTGACTCTCCTTCTCGCCGGCCAAGCTACCACCGCCTACTTTCTGTACCAACAGCAAG
GCAGACTAGACAAACTGACAATCACAAGCCAGAACCTTCAGCTGGAGTCTCTGCGGATG
AAGCTGCCCGCTTTGTGGATGAGATTGCTTCCTCTACTTGCTCTCCTGGCGCTCTGGGG
ACCTGACCCCGAGCAAGAGTTTGTTAATCAGCACCTGTGTGGGAGTCATCTGGTGGAG
GCACTCTATTTAGTGTGCGGAGAGAGGGGCTTCTTCTACACTCCAAAGACCAGACGGG
AGGCCGAAGACCTTCAAGTGGGGCAAGTAGAACTGGGTGGCGGACCCGGTGCCGGGA
GCCTTCAGCCGCTCGCCCTGGAGGGCTCTCTTCAGAAACGCGGCATCGTGGAGCAGTG
TTGCACATCCATTTGCTCACTCTACCAGCTGGAGAACTACTGCAACGGAAGCGGAGTGA
AGCAGACGTTGAATTTTGATTTGTTGAAGTTGGCGGGGGATGTGGAGAGCAATCCGGG
GCCGATGCCCCCTAGTGGCCTCAGACTTTTGTTATTGTTATTACCGCTTTTATGGCTCTT
GGTGCTGACACCGGGCCGTCCGGCTGCTGGCTTGTCGACTTGTAAGACAATTGATATG
GAATTGGTGAAACGAAAACGGATTGAGGCCATCCGAGGACAGATTTTGAGCAAGCTGC
GGCTTGCCTCGCCACCCTCGCAAGGGGAAGTCCCACCCGGACCTCTACCAGAAGCAGT
CCTAGCGCTGTACAACAGTACAAGAGATAGAGTGGCCGGGGAATCCGCAGAACCAGAG
CCTGAGCCTGAAGCCGATTATTATGCAAAGGAAGTGACTAGGGTCCTGATGGTCGAGA
CCCATAACGAAATCTACGACAAATTCAAACAAAGTACCCACTCTATCTACATGTTCTTCA
ACACCAGTGAGCTAAGAGAAGCCGTGCCCGAACCTGTGCTTCTTTCCCGCGCAGAACT
CCGCCTCTTGAGACTCAAATTGAAAGTTGAACAACACGTAGAGCTTTACCAGAAATACTC
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
27
TAATAATTCATGGCGATATCTTTCTAATCGTCTCCTCGCCCCATCTGACAGCCCTGAATG
GCTCTCCTTCGACGTTACGGGAGTTGTGCGCCAGTGGCTCAGCAGAGGCGGAGAGATA
GAGGGCTTTCGGCTGAGCGCACATAGCTCTAGCGACTCAAGGGACAACACATTGCAAG
TGGATATTAACGGTTTTACAACTGGACGGAGAGGGGACCTGGCGACCATCCACGGCAT
GAATAGACCTTTCCTGCTGCTGATGGCTACTCCCCTGGAGAGGGCACAGCACTTACAGT
CTTCCAGACACCGGCGCGCCCTGGATACAAACTACTGCTTCAGCTCCACCGAAAAGAA
CTGTTGCGTGCGGCAGCTGTACATTGACTTCAGAAAGGATCTGGGCTGGAAGTGGATT
CATGAGCCCAAGGGGTATCATGCCAACTTCTGTCTTGGGCCATGCCCATACATCTGGTC
ACTGGATACCCAGTACTCCAAAGTTCTGGCCTTGTACAATCAACACAACCCTGGAGCTT
CCGCCGCTCCTTGCTGTGTGCCCCAAGCCCTAGAGCCCCTGCCCATCGTTTATTATGTC
GGACGCAAGCCCAAAGTAGAACAGCTATCAAATATGATCGTGAGAAGCTGCAAGTGTAG
CTGATAAACGCGTCGAGCATGCATCTAGGGCGGCCAATTCCGCCCCTCTCCCCCCCAC
CCCTCTCCCTCCCCCCCCCCTAACGTTACTGGCCGAAGCCGCTTGGAATAAGGCCGGT
GTGCGTTTGTCTATATGTTATTTTCCACCATATTGCCGTCTTTTGGCAATGTGAGGGCCC
GGAAACCTGGCCCTGTCTTCTTGACGAGCATTCCTAGGGGTCTTTCCCCTCTCGCCAAA
GGAATGCAAGGTCTGTTGAATGTCGTGAAGGAAGCAGTTCCTCTGGAAGCTTCTTGAAG
ACAAACAACGTCTGTAGCGACCCTTTGTAGACAGCGGAACCCCCCACCTGGCGATAGA
TGCCTCTGCGGCCAAAAGCCACGTGTATAAGATACACCTGCAAAGGCGGCACAACCCC
AGTGCCACGTTGTGAGTTGGATAGTTGTGGAAAGAGTCAAATGGCTCTCCTCAAGCGTA
TTCAACAAGGGGCTGAAGGATGCCCAGAAGGTACCCCATTGTATGGGATCTGATCTGG
GGCCTCGGTGCACATGCTTTACATGTGTTTAGTCGAGGTTAAAAAACGTCTAGGCCCCC
CGAACCACGGGGACGTGGTTTTCCTTTGAAAAACACGATGATAATATGATGCACAGCTC
AGCACTGCTCTGTTGCCTGGTCCTCCTGACTGGGGTGAGGGCCAGCCCAGGCCAGGG
CACCCAGTCTGAGAACAGCTGCACCCACTTCCCAGGCAACCTGCCTAACATGCTTCGA
GATCTCCGAGATGCCTTCAGCAGAGTGAAGACTTTCTTTCAAATGAAGGATCAGCTGGA
CAACTTGTTGTTAAAGGAGTCCTTGCTGGAGGACTTTAAGGGTTACCTGGGTTGCCAAG
CCTTGTCTGAGATGATCCAGTTTTACCTGGAGGAGGTGATGCCCCAAGCTGAGAACCAA
GACCCAGACATCAAGGCGCATGTGAACTCCCTGGGGGAGAACCTGAAGACCCTCAGGC
TGAGGCTACGGCGCTGTCATCGATTTCTTCCCTGTGAAAACAAGAGCAAGGCCGTGGA
GCAGGTGAAGAATGCCTTTAATAAGCTCCAAGAGAAAGGCATCTACAAAGCCATGAGTG
AGTTTGACATCTTCATCAACTACATAGAAGCCTACATGACAATGAAGATACGAAACGGGA
GCGGCGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTG
GACCTATGTACAGAATGCAGCTGCTGAGCTGCATCGCCCTGAGCCTGGCCCTGGTGAC
CAACAGCGCACCCACGTCCTCTAGCACCAAGAAGACCCAGTTACAGTTGGAGCATCTAC
TTTTAGACCTGCAAATGATTTTGAACGGCATCAACAACTACAAGAATCCTAAACTTACTC
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
28
GCATGCTTACCTTCAAATTTTACATGCCCAAGAAGGCCACCGAACTGAAGCACTTGCAA
TGTCTGGAGGAAGAACTCAAGCCGCTGGAGGAAGTTCTCAACCTCGCGCAGTCCAAGA
ATTTCCACCTCCGGCCAAGAGACCTGATCAGTAACATTAATGTGATAGTGCTGGAGCTG
AAGGGAAGCGAGACTACATTTATGTGCGAGTACGCCGATGAAACCGCTACAATCGTCGA
GTTCCTGAATAGATGGATCACATTTTGCCAGTCAATTATCTCTACTCTGACATGATAACT
CGAGTCTAGAGGGCCCGTTTAAACCCGCTGATCAGCCTCGACTGTGCCTTCTAGTTGCC
AGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGGAAGGTGCCACTCCC
ACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGAGTAGGTGTCATTCT
ATTCTGGGGGGTGGGGTGGGGCAGGACAGCAAGGGGGAGGATTGGGAAGACAATAGC
AGGCATGCTGGGGATGCGGTGGGCTCTATGGCTTCTACTGGGCGGTTTTATGGACAGC
AAGCGAACCGGAATTGCCAGCTGGGGCGCCCTCTGGTAAGGTTGGGAAGCCCTGCAAA
GTAAACTGGATGGCTTTCTCGCCGCCAAGGATCTGATGGCGCAGGGGATCAAGCTCTG
ATCAAGAGACAGGATGAGGATCGTTTCGCATGGCCAAAGAAGACAATATTGAAATGCAA
GGTACCGTTCTTGAAACGTTGCCTAATACCATGTTCCGCGTAGAGTTAGAAAACGGTCA
CGTGGTTACTGCACACATCTCCGGTAAAATGCGCAAAAACTACATCCGCATCCTGACGG
GCGACAAAGTGACTGTTGAACTGACCCCGTACGACCTGAGCAAAGGCCGCATTGTCTT
CCGTAGTCGCTGATAAATTATTAACGCTTACAATTTCCTGATGCGGTATTTTCTCCTTAC
GCATCTGTGCGGTATTTCACACCGCATACAGGTGGCACTTTTCGGGGAAATGTGCGCG
GAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATA
ACCCTGATAAATGCTTCAATAATAGCACGTGCTAAAACTTCATTTTTAATTTAAAAGGATC
TAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTC
CACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTG
CGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCC
GGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATAC
CAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCAC
CGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAG
TCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGG
GCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACT
GAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCG
GACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCA
GGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCG
TCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCG
GCCTTTTTACGGTTCCTGGGCTTTTGCTGGCCTTTTGCTCACATGTTCTT.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
29
In a fourth embodiment the plasmid of the invention has the sequence SEQ ID NO
29: full (non-annotated) plasmid sequence
GACTCTTCGCGATGTACGGGCCAGATATACGCGTTGACATTGATTATTGACTAG
TTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGT
TACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCATTG
ACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAA
TGGGTGGACTATTTACGGTAAACTGCCCACTTGGCAGTACATCAAGTGTATCATATGCC
AAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCGCCTGGCATTATGCCCAGT
ACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATTAGTCATCGCTATTA
CCATGGTGATGCGGTTTTGGCAGTACATCAATGGGCGTGGATAGCGGTTTGACTCACG
GGGATTTCCAAGTCTCCACCCCATTGACGTCAATGGGAGTTTGTTTTGGCACCAAAATC
AACGGGACTTTCCAAAATGTCGTAACAACTCCGCCCCATTGACGCAAATGGGCGGTAG
GCGTGTACGGTGGGAGGTCTATATAAGCAGAGCTCTCTGGCTAACTAGAGAACCCACT
GCTTACTGGCTTATCGAAATTAATACGACTCACTATAGGGAGACCCAAGCTGGCTAGCG
TTTAAACTTAAGCTTGGTACCGAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTCTGC
AGCTCGCATCTCTCCTTCACGCGCCCGCCGCCCTACCTGAGGCCGCCATCCACGCCGG
TTGAGTCGCGTTCTGCCGCCTCCCGCCTGTGGTGCCTCCTGAACTGCGTCCGCCGTCT
AGGTAAGTTTAAAGCTCAGGTCGAGACCGGGCCTTTGTCCGGCGCTCCCTTGGAGCCT
ACCTAGACTCAGCCGGCTCTCCACGCTTTGCCTGACCCTGCTTGCTCAACTCTAGGTAA
GTTAATGAGACAGATAGAAACTGGTCTTGTAGAAACAGAGTAGTCGCCTGCTTTTCTGC
CAGGTGCTGACTTCTCTCCCCTGGGCTTTTTTCTTTTTCTCAGGTTGAAAAGAAGAAGAC
GAAGAAGACGAAGAAGACAAACCGTCGTCGACTGCCATGCGCCGCTGATTAACGCCGC
CACCATGGCCCACCGACGCAGATCCAGAAGCTGCCGTGAGGACCAGAAGCCCGTGAT
GGATGATCAGAGGGACCTTATCTCTAACAATGAACAACTGCCAATGCTCGGCAGACGGC
CTGGGGCCCCGGAGAGCAAGTGCAGCAGAGGAGCCTTGTACACGGGGTTCTCCATTTT
AGTGACTCTCCTTCTCGCCGGCCAAGCTACCACCGCCTACTTTCTGTACCAACAGCAAG
GCAGACTAGACAAACTGACAATCACAAGCCAGAACCTTCAGCTGGAGTCTCTGCGGATG
AAGCTGCCCGCTTTGTGGATGAGATTGCTTCCTCTACTTGCTCTCCTGGCGCTCTGGGG
ACCTGACCCCGAGCAAGAGTTTGTTAATCAGCACCTGTGTGGGAGTCATCTGGTGGAG
GCACTCTATTTAGTGTGCGGAGAGAGGGGCTTCTTCTACACTCCAAAGACCAGACGGG
AGGCCGAAGACCTTCAAGTGGGGCAAGTAGAACTGGGTGGCGGACCCGGTGCCGGGA
GCCTTCAGCCGCTCGCCCTGGAGGGCTCTCTTCAGAAACGCGGCATCGTGGAGCAGTG
TTGCACATCCATTTGCTCACTCTACCAGCTGGAGAACTACTGCAACGGAAGCGGAGTGA
AGCAGACGTTGAATTTTGATTTGTTGAAGTTGGCGGGGGATGTGGAGAGCAATCCGGG
GCCGATGCCCCCTAGTGGCCTCAGACTTTTGTTATTGTTATTACCGCTTTTATGGCTCTT
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
GGTGCTGACACCGGGCCGTCCGGCTGCTGGCTTGTCGACTTGTAAGACAATTGATATG
GAATTGGTGAAACGAAAACGGATTGAGGCCATCCGAGGACAGATTTTGAGCAAGCTGC
GGCTTGCCTCGCCACCCTCGCAAGGGGAAGTCCCACCCGGACCTCTACCAGAAGCAGT
CCTAGCGCTGTACAACAGTACAAGAGATAGAGTGGCCGGGGAATCCGCAGAACCAGAG
5 CCTGAGCCTGAAGCCGATTATTATGCAAAGGAAGTGACTAGGGTCCTGATGGTCGAGA
CCCATAACGAAATCTACGACAAATTCAAACAAAGTACCCACTCTATCTACATGTTCTTCA
ACACCAGTGAGCTAAGAGAAGCCGTGCCCGAACCTGTGCTTCTTTCCCGCGCAGAACT
CCGCCTCTTGAGACTCAAATTGAAAGTTGAACAACACGTAGAGCTTTACCAGAAATACTC
TAATAATTCATGGCGATATCTTTCTAATCGTCTCCTCGCCCCATCTGACAGCCCTGAATG
10 GCTCTCCTTCGACGTTACGGGAGTTGTGCGCCAGTGGCTCAGCAGAGGCGGAGAGATA
GAGGGCTTTCGGCTGAGCGCACATAGCTCTAGCGACTCAAGGGACAACACATTGCAAG
TGGATATTAACGGTTTTACAACTGGACGGAGAGGGGACCTGGCGACCATCCACGGCAT
GAATAGACCTTTCCTGCTGCTGATGGCTACTCCCCTGGAGAGGGCACAGCACTTACAGT
CTTCCAGACACCGGCGCGCCCTGGATACAAACTACTGCTTCAGCTCCACCGAAAAGAA
15 CTGTTGCGTGCGGCAGCTGTACATTGACTTCAGAAAGGATCTGGGCTGGAAGTGGATT
CATGAGCCCAAGGGGTATCATGCCAACTTCTGTCTTGGGCCATGCCCATACATCTGGTC
ACTGGATACCCAGTACTCCAAAGTTCTGGCCTTGTACAATCAACACAACCCTGGAGCTT
CCGCCGCTCCTTGCTGTGTGCCCCAAGCCCTAGAGCCCCTGCCCATCGTTTATTATGTC
GGACGCAAGCCCAAAGTAGAACAGCTATCAAATATGATCGTGAGAAGCTGCAAGTGTAG
20 CTGATAAACGCGTCGAGCATGCATCTAGGGCGGCCAATTCCGCCCCTCTCCCCCCCAC
CCCTCTCCCTCCCCCCCCCCTAACGTTACTGGCCGAAGCCGCTTGGAATAAGGCCGGT
GTGCGTTTGTCTATATGTTATTTTCCACCATATTGCCGTCTTTTGGCAATGTGAGGGCCC
GGAAACCTGGCCCTGTCTTCTTGACGAGCATTCCTAGGGGTCTTTCCCCTCTCGCCAAA
GGAATGCAAGGTCTGTTGAATGTCGTGAAGGAAGCAGTTCCTCTGGAAGCTTCTTGAAG
25 ACAAACAACGTCTGTAGCGACCCTTTGTAGACAGCGGAACCCCCCACCTGGCGATAGA
TGCCTCTGCGGCCAAAAGCCACGTGTATAAGATACACCTGCAAAGGCGGCACAACCCC
AGTGCCACGTTGTGAGTTGGATAGTTGTGGAAAGAGTCAAATGGCTCTCCTCAAGCGTA
TTCAACAAGGGGCTGAAGGATGCCCAGAAGGTACCCCATTGTATGGGATCTGATCTGG
GGCCTCGGTGCACATGCTTTACATGTGTTTAGTCGAGGTTAAAAAACGTCTAGGCCCCC
30 CGAACCACGGGGACGTGGTTTTCCTTTGAAAAACACGATGATAATATGATGCACAGCTC
AGCACTGCTCTGTTGCCTGGTCCTCCTGACTGGGGTGAGGGCCAGCCCAGGCCAGGG
CACCCAGTCTGAGAACAGCTGCACCCACTTCCCAGGCAACCTGCCTAACATGCTTCGA
GATCTCCGAGATGCCTTCAGCAGAGTGAAGACTTTCTTTCAAATGAAGGATCAGCTGGA
CAACTTGTTGTTAAAGGAGTCCTTGCTGGAGGACTTTAAGGGTTACCTGGGTTGCCAAG
CCTTGTCTGAGATGATCCAGTTTTACCTGGAGGAGGTGATGCCCCAAGCTGAGAACCAA
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
31
GACCCAGACATCAAGGCGCATGTGAACTCCCTGGGGGAGAACCTGAAGACCCTCAGGC
TGAGGCTACGGCGCTGTCATCGATTTCTTCCCTGTGAAAACAAGAGCAAGGCCGTGGA
GCAGGTGAAGAATGCCTTTAATAAGCTCCAAGAGAAAGGCATCTACAAAGCCATGAGTG
AGTTTGACATCTTCATCAACTACATAGAAGCCTACATGACAATGAAGATACGAAACGGGA
GCGGCGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTG
GACCTATGTACAGAATGCAGCTGCTGAGCTGCATCGCCCTGAGCCTGGCCCTGGTGAC
CAACAGCGCACCCACGTCCTCTAGCACCAAGAAGACCCAGTTACAGTTGGAGCATCTAC
TTTTAGACCTGCAAATGATTTTGAACGGCATCAACAACTACAAGAATCCTAAACTTACTC
GCATGCTTACCTTCAAATTTTACATGCCCAAGAAGGCCACCGAACTGAAGCACTTGCAA
TGTCTGGAGGAAGAACTCAAGCCGCTGGAGGAAGTTCTCAACCTCGCGCAGTCCAAGA
ATTTCCACCTCCGGCCAAGAGACCTGATCAGTAACATTAATGTGATAGTGCTGGAGCTG
AAGGGAAGCGAGACTACATTTATGTGCGAGTACGCCGATGAAACCGCTACAATCGTCGA
GTTCCTGAATAGATGGATCACATTTTGCCAGTCAATTATCTCTACTCTGACATGATAACT
CGAGTCTAGAGGGCCCGTTTAAACCCGCTGATCAGCCTCGACTGTGCCTTCTAGTTGCC
AGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGGAAGGTGCCACTCCC
ACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGAGTAGGTGTCATTCT
ATTCTGGGGGGTGGGGTGGGGCAGGACAGCAAGGGGGAGGATTGGGAAGACAATAGC
AGGCATGCTGGGGATGCGGTGGGCTCTATGGCTTCTACTGGGCGGTTTTATGGACAGC
AAGCGAACCGGAATTGCCAGCTGGGGCGCCCTCTGGTAAGGTTGGGAAGCCCTGCAAA
GTAAACTGGATGGCTTTCTCGCCGCCAAGGATCTGATGGCGCAGGGGATCAAGCTCTG
ATCAAGAGACAGGATGAGGATCGTTTCGCATGGCCAAAGAAGACAATATTGAAATGCAA
GGTACCGTTCTTGAAACGTTGCCTAATACCATGTTCCGCGTAGAGTTAGAAAACGGTCA
CGTGGTTACTGCACACATCTCCGGTAAAATGCGCAAAAACTACATCCGCATCCTGACGG
GCGACAAAGTGACTGTTGAACTGACCCCGTACGACCTGAGCAAAGGCCGCATTGTCTT
CCGTAGTCGCTGATAAATTATTAACGCTTACAATTTCCTGATGCGGTATTTTCTCCTTAC
GCATCTGTGCGGTATTTCACACCGCATACAGGTGGCACTTTTCGGGGAAATGTGCGCG
GAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATA
ACCCTGATAAATGCTTCAATAATAGCACGTGCTAAAACTTCATTTTTAATTTAAAAGGATC
TAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTC
CACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTG
CGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCC
GGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATAC
CAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCAC
CGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAG
TCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGG
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
32
GCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACT
GAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCG
GACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCA
GGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCG
TCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCG
GCCTTTTTACGGTTCCTGGGCTTTTGCTGGCCTTTTGCTCACATGTTCTT
The term "GLP-1/GLP-1 peptide/GLP-1R aponist peptide" as used herein refers to
GLP-1 molecules/peptides/proteins/variants/agonists herein are molecules
having GLP-1R
agonist function meaning that they are agonists of the GLP-1 receptor. This
class of drugs is
normally used for the treatment of diabetes, in particular type 2 diabetes.
The amino acid
sequence of mature "human GLP-1" is: HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRG (SEQ
ID NO: 21).
The term "GLP-1 analogue" as used herein refers to a peptide or a compound,
which is a variant of GLP-1 (SEQ ID NO: 15). The terms "GLP-1 analog" and
"analogue" may
be used interchangeably herein.
GLP-1 analogues may be described by reference to i) the number of the amino
acid
residue in human GLP-1 (SEQ ID NO: 15) which corresponds to the amino acid
residue
which is modified (i.e. the corresponding position in GLP-1 (SEQ ID NO: 15)),
and to ii) the
actual modification.
The term GLP-1 Derivatives refer to derivatives of GLP-1 analogues. The term
"derivative" as used herein in the context of a GLP-1 analogue means a
chemically modified
GLP-1 analogue in which one or more substituents have been covalently attached
to the
GLP-1 analogue. The term "substituent" as used herein, means a chemical moiety
or
group/side group conjugated to the GLP-1 protein/agonist/analogue. The
derivative may
comprise one or more modifications selected from amides, carbohydrates, alkyl
groups, acyl
groups, esters and the like.
In some embodiments the substituent is covalently attached via an amino acid
residue in said polypeptide e.g. at one of the amino acid positions selected
from the group
consisting of position 22, 23, 27, 34, 35, and 36.
In some embodiments the GLP-1 derivative comprises a substituent comprising a
lipophilic moiety. The term "lipophilic moiety" as used herein, means an
aliphatic or cyclic
hydrocarbon moiety with more than 6 and less than 30 carbon atoms, wherein
said
hydrocarbon moiety may comprise additional substituents.
Examples of GLP-1 agonists include (but are not limited to) exenatide,
liraglutide,
lixisentide, albiglutide, dulaglutide, taspoglutide, and semaglutide. DNA
immuno-therapy
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
33
vaccines using the plasmids herein may be combined initially with a parallel
GLP-1R agonist
treatment in treatment of e.g. recent onset T1D patients. GLP-1 co-
administration may be
chronic or temporary and include oral routes in addition to parenteral routes.
Liraglutide: (SEQ ID NO 22):
0
11-12--1-1AEGT FTSDYSSYLEGOAANõ)¨EF I AWLVR G R
01
0
Semaglutide (SEQ ID NO 23):
0 G T F TS 0 VSSY L EGOAAN,JL0
E F IAINUMGR G ¨
0
0 0
0 0
0 0 0
Pharmaceutical compositions herein are preferably aqueous formulations
comprising at least 50% water, more preferably at least 60% water, more
preferably at least
75% water, more preferably at least 90% water, more preferably at least 95%
water, and
most preferably at least 99% water. The pharmaceutical compositions herein may
alternatively be dry formulations, such as lyophilized formulations, intended
for reconstitution,
inhalation, intranasal instillation, intradermal administration, etc.
Pharmaceutical formulations herein are preferably administered without the use
of
methods for enhancing transformation, such as electroporation. In one
embodiment,
pharmaceutical formulations are intended for parenteral administration, e.g.
subcutaneous
administration, intradermal administration, intravenous administration,
intramuscular
administration, etc. In another embodiment, pharmaceutical compositions herein
may
furthermore be administered topically, orally, rectally, or by inhalation.
Pharmaceutical compositions herein are preferably without addition of any
condensation agents or other excipients that may induce local reactions.
Formulations herein
preferably contain free-radical scavengers (e.g. 1`)/0 ethanol) and/or
chelators such as e.g.
divalent cation scavengers (e.g. EDTA [CAS #60-00-4], EGTA [CAS #67-42-5], or
DPTA
[CAS #67-43-6]) in order to enhance stability of aqueous plasmid DNA.
Pharmaceutical
compositions herein may furthermore be in the form of a saline solution and/or
a buffer
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
34
solution or comprise a saline solution and/or comprise a buffer solution (e.g.
PBS ¨
phosphate buffered saline, TRIS buffer, or equivalent pharmaceutically
acceptable buffers).
Pharmaceutical formulations herein are preferably free from any adjuvants as
well other
typical vaccine ingredients such as e.g. aluminium hydroxide, phenol,
sorbitol, silicone, etc.
Administration: The DNA immuno-therapy vaccine herein may be administered to a
T1D patient, or a patient in risk of developing Ti D. The vaccine may be
administered e.g on
a daily basis, every second day, twice a week, once a week, twice monthly,
once a month,
every second month, four times a year, or once a year ¨ frequency may be
adjusted
according to general or individual needs. The immuno-therapy herein may be
chronic. The
duration of therapy may be e.g. one month, two months, three months, 6 months,
one year,
two years, three years, five years, six years, seven years, eight years, nine
years, or 10
years.
Embodiments
The following embodiments illustrate the invention and are not to be
understood in
any limiting way. It is understood that all embodiments can be combined in all
possible ways.
1. A plasmid which encodes:
i. an antigen;
ii. TGF-6; and
iii. IL-10.
2. The plasmid according to embodiment 1, which said antigen is an insulin
antigen.
3. A plasmid which co-expresses/encodes (preferably from a single operon): (i)
an
antigen, such as e.g. an insulin antigen; (ii) TGF-6/TGF-61 (such as in a
constitutively active
form); and (iii) IL-10.
4. The plasmid according to any of the preceding embodiments, wherein said
insulin
antigen is selected from the group consisting of: proinsulin, secretion-
incapable pre-
proinsulin, or a functional or immuno-dominant peptide fragment thereof.
5. The plasmid according to any of the preceding embodiments, wherein said
insulin
antigen is selected from the group consisting of: proinsulin, pre-proinsulin,
and a functional or
immuno-dominant peptide fragment thereof.
6. The plasmid according to any of the preceding embodiments, wherein said
insulin
antigen is endosomally targeted insulin.
7. The plasmid according to any of the preceding embodiments, wherein said
plasmid expresses the insulin antigen and TGF-6 in a ratio of about 1:1.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
8. The plasmid according to any of the preceding embodiments, wherein said
plasmid expresses insulin antigen and TGF-[3 in an amount of at least 200 fold
lower than IL-
10.
9. The plasmid according to any of the preceding embodiments, wherein said
5 plasmid expresses insulin antigen and TGF-[3 in an amount of at least 2
fold lower than IL-10.
10. The plasmid according to any of the preceding embodiments, wherein said
plasmid furthermore co-expresses Interleukin-2 (IL-2).
11. The plasmid according to any of the preceding embodiments, wherein said
plasmid expresses an excess of IL-10 and IL-2 over the antigen (e.g. insulin)
and TGF-[3.
10 12. The plasmid according to any of the preceding embodiments, wherein
said
plasmid expresses IL-10 and IL-2 at least about one fold, two fold, five fold
or at least about
one hundred fold over TGF-[3 and insulin antigen (ratio of IL-10+IL-2 to
insulin+TGF13 may
be at least 1:1, or 2:1, or 5:1 or 100:1).
13. The plasmid according to any of the preceding embodiments, wherein said
15 plasmid expresses IL-10 and IL-2 at least about one hundred fold, two
hundred fold, five
hundred fold or at least about one thousand fold over TGF-[3 and insulin
antigen (ratio of IL-
10+IL-2 to insulin+TGF13 may be at least 100:1, or 200:1, or 500:1 or 1000:1).
14. The plasmid according to any of the preceding embodiments, wherein said
plasmid expresses IL-10 and IL-2 in a ratio of about 1:1 ¨ 100:1, such as e.g.
1:1 - 50:1, such
20 as e.g. 1:1 ¨25:1, such as e.g. 1:1 - 10:1, alternatively 1:1 ¨ 5:1,
alternatively 1:1 ¨ 3:1,
alternatively 1:1 ¨ 2:1. Alternatively, the ratio between expressed IL-10 and
expressed IL-2
may be about 1:1, 1:0.9, 1:0.8, 1:0,7, 1:0.6, 1:0.5, 1:0.4; 1:0.3, 1:0.2, or
1:0.1.
15. The plasmid according to any of the preceding embodiments, wherein said
plasmid comprises: (i) an FMDV 2A element separating the insulin antigen
encoding
25 sequence and the TGF-[3 encoding sequence, (ii) an EMCV IRES element
separating the
TGF-[3 encoding sequence and the IL-10 encoding sequence, and (iii) a 2A
element
separating the IL-10 encoding sequence and the IL-2 encoding sequence.
16. The plasmid according to any of the preceding embodiments, wherein said
plasmid comprises:
30 (I) a 2A element (such as an FMDV 2A or a P 2A element) separating
the
insulin antigen encoding sequence and the TGF-[3 encoding sequence,
(ii) an EMCV IRES element (alternatively a bi-directional
promoter) separating
the TGF-[3 encoding sequence and the IL-10 encoding sequence
(preferably, three alanine amino acids are encoded immediately N-
35 terminal to the IL-10 gene), and
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
36
(iii) a 2A element (such as a P 2A element) separating the IL-10
encoding
sequence and the IL-2 encoding sequence.
17. The plasmid according to any of the preceding embodiments, wherein the TGF-
13 encoding sequence encodes constitutively active TGF-13, preferably
constitutively active
human TGF-131.
18. The plasmid according to any of the preceding embodiments, wherein said
plasmid comprises: (i) an endosomally targeted pre-pro-insulin encoding
sequence, (ii) an
FMDV 2A element, (iii) a TGF-13 encoding sequence, (iv) an EMCV IRES element,
(v) an IL-
encoding sequence, (vi) a P 2A element, (vii) an IL-2 encoding sequence,
(viii) a
10 polyadenylation /termination element, (ix) a selection gene, (x) an
origin of replication, (xi) a
eukaryotic promoter element, (xii) a eukaryotic translational start sequence,
(xiii) an
endosomal sorting sequence, and (xiv) optionally an intron.19. The plasmid
according to any
of the preceding embodiments, wherein said plasmid comprises the following
elements:
(i) a promoter (such as a CMV IE promoter),
(ii) an intron (located within the noncoding leader sequence), and
(iii) a eukaryotic translational start sequence (such as a Kozak element),
(iv) an endosomally targeted antigen encoding sequence (such as an
endosomally targeted human secretion-defective pre-pro-insulin encoding
sequence),
(v) an FMDV 2A element preferably separating the antigen encoding sequence
and the TGF-13 encoding sequence,
(vi) a TGF-13 encoding sequence (such as a constitutively active
human TGF-13
encoding sequence, preferably a constitutively active human TGF-131
encoding sequence),
(vii) an EMCV IRES element (or alternatively a bi-directional eukaryotic
promoter), wherein said EMCV IRES element separates the TGF-13 encoding
sequence and the IL-10 encoding sequences,
(viii) an IL-10 encoding sequence (such as a human IL-10 encoding sequence
with a three alanine amino acid N-terminal addition),
(ix) a 2A element, such as a P 2A element, wherein said 2A element
separates
the IL-10 encoding sequence and the IL-2 encoding sequence,
(x) an IL-2 encoding sequence (such as a human IL-2 encoding sequence),
(xi) a termination element (such as a bGH_PA termination element),
(xii) a selection gene (such as a kanamycin encoding sequence or a wt infA
encoding sequence),
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
37
(xiii) an origin of replication (such as a prokaryotic origin of
replication, such as
e.g. pUC on).
20. The plasmid according to embodiment 18, wherein the elements (i)-(xiii)
are
arranged by order of expression.
21. The plasmid according to any of the preceding embodiments, wherein the DNA
sequence of the plasmid is as set forth SEQ ID NO 24, or essentially as set
forth in SEQ ID
NO 24.
22. The plasmid according to embodiment 21, wherein a few minor modifications,
resulting in e.g. one, two, three, or four amino acid substitution in one or
more of the antigen
and/or the cytokines are made to SEQ ID NO 24 herein.
23. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:26 or a modification of SEQ ID
NO:26 resulting in
e.g. one, two, three or four amino acid substitutions in one or more of the
antigen and/or
cytokines, or a modification of SEQ ID NO:26 which results in expression of
the same
polypeptide sequences as from SEQ ID NO:26.
24. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:26 or a modification of SEQ ID
NO:26 having less
than 100 bases which are different than SEQ ID NO:26.
25. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:28 or a modification of SEQ ID
NO:28 resulting in
e.g. one, two, three or four amino acid substitutions in one or more of the
antigen and/or
cytokines, or a modification of SEQ ID NO:28 which results in expression of
the same
polypeptide sequences as from SEQ ID NO:28.
26. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:28 or a modification of SEQ ID
NO:28 having less
than 100 bases which are different than SEQ ID NO:28.
27. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:29 or a modification of SEQ ID
NO:29 resulting in
e.g. one, two, three or four amino acid substitutions in one or more of the
antigen and/or
cytokines, or a modification of SEQ ID NO:29 which results in expression of
the same
polypeptide sequences as from SEQ ID NO:29.
28. The plasmid according to any of embodiments 1-20, wherein the DNA sequence
of the plasmid is as set forth in SEQ ID NO:29 or a modification of SEQ ID
NO:29 having less
than 100 bases which are different than SEQ ID NO:29.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
38
29. The plasmid according to any of embodiments 1-20, wherein said plasmid
comprises a TGF-6 gene comprising SEQ ID NO:25 or SEQ ID NO:25 having less
than 10
base substitutions.
30. The plasmid according to any of the preceding embodiments for use in
delaying
or preventing type I diabetes.
31. The plasmid according to any of the preceding embodiments for intra-
muscular,
intradermal, intranasal, or subcutaneous administration.
32. The plasmid according to embodiment 31 for subcutaneous administration.
33. The plasmid according to embodiment 31 for intra-muscular injection.
34. The plasmid according to any of the preceding embodiments for use in
treating a
medical condition in a subject, such as e.g. type I diabetes, early-onset type
I diabetes, or
increased risk of developing type I diabetes (including type 1,5 diabetes type
of conditions).
35. A DNA immuno-therapy vaccine comprising a plasmid according to any of the
preceding embodiments.
36. The DNA immuno-therapy vaccine according to embodiment 35 for use in
delaying or preventing type I diabetes.
37. The DNA immuno-therapy vaccine according to any of embodiments 35-36 for
intra-muscular, intradermal, intranasal, or subcutaneous administration.
38. The DNA immuno-therapy vaccine according to embodiment 37 for
subcutaneous administration.
39.The DNA immuno-therapy vaccine according to embodiment 37 for intra-
muscular administration.
40. The DNA immuno-therapy vaccine according to any of embodiments 35-39 used
in association with, or in parallel with other types of medical treatments
such as e.g. beta
cell/beta stem cell therapy, beta cell/beta stem cell grafting, etc. to
prolong the survival and
efficacy of engrafted cells.
41. A pharmaceutical composition comprising the DNA immuno-therapy vaccine
according to any of embodiments 34-39, or a plasmid according to any of
embodiments 1-34,
wherein said pharmaceutical composition comprises a saline solution and/or a
buffer and/or
a chelator.
42. A pharmaceutical composition comprising the DNA immuno-therapy vaccine
according to any of embodiments 35-40, or a plasmid according to any of
embodiments 1-34,
wherein said pharmaceutical composition comprises a saline solution and/or a
buffer and/or
a chelator and/or ethanol.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
39
43. The pharmaceutical composition according to any of embodiments 41-42,
wherein the volume/volume percentage of ethanol is less than 5%, less than 4%,
less than
3%, less than 2%, or less than 1%.
44. The pharmaceutical composition according to any of embodiments 41-43,
wherein said composition does not comprise any virus, lipid co-packing agent,
or
condensation agent.
45. The pharmaceutical composition according to any of embodiments 41-44,
wherein said composition further comprises a GLP-1R agonist.
46. The pharmaceutical composition according to any of embodiments 41-44,
wherein said composition furthermore comprises a GLP-1 analogue/GLP-1R
agonist.
47. The pharmaceutical composition according to any of embodiments 45-46
wherein said GLP-1 analogue or said GLP-1R agonist is selected from
liraglutide,
semaglutide or a mixture thereof.
48. A kit comprising a pharmaceutical composition according to any of
embodiments
41-47 and a pharmaceutical composition comprising a GLP-1 analogue/GLP-1R
agonist (e.g.
liraglutide and/or semaglutide).
49. A method of producing a plasmid according to any of embodiments 1-34,
wherein said method comprises (i) incubating a host cell, such as a host cell
of bacterial
origin such as e.g. E. coli) transfected with said plasmid under suitable
conditions and (ii)
recovering/purifying said plasmid.
50. The method according to embodiment 49, wherein said host cell is a E. coli
infA
thermosensitive strain.
51. A method of delaying the onset of Type-1 diabetes (Ti D) or symptoms
thereof in
a patient at risk of developing Ti D, or recently diagnosed with Ti D, said
method comprising
administering a DNA immuno-therapy vaccine comprising the plasmid according to
any of
embodiments 1-31, optionally in combination with a GLP-1 analogue/GLP-1R
agonist.
52. A method of preserving beta cell function and/or endogenous insulin
production
in an individual, said method comprising administering a DNA immuno-therapy
vaccine
comprising the plasmid according to any of embodiments 1-34, optionally in
combination with
a GLP-1 analogue/GLP-1R agonist.
53. A method of treating a diabetic individual comprising administering a
vaccine
comprising the plasmid according to any of embodiments 1-34, optionally in
combination with
a GLP-1 analogue/GLP-1R agonist (e.g. liraglutide and/or semaglutide).
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
54. A vaccine for preventing or delaying the onset of Type-1 diabetes (Ti D)
symptoms in a patient at risk of developing, or recently diagnosed with, T1D
said vaccine
comprising the plasmid according to any of embodiments 1-34.
55. A method of reducing the dosage of insulin in an individual having Type-1
5 diabetes (Ti D), or a person at risk of developing Ti D, said method
comprising administering
a DNA immuno-therapy vaccine comprising a plasmid according to any of
embodiments 1-
33, optionally in combination with a GLP-1 analogue/GLP-1R agonist (e.g.
liraglutide and/or
semaglutide).
10 EXAMPLES
Non Obese Diabetic mice (NOD mouse model of type 1 diabetes): Immune function
in autoimmunity relies on a complex network of cellular interactions that
cannot be
adequately evaluated in vitro.
Disease suppression and/or treatment evaluations herein were carried out in
the
15 NOD mouse model, this model is a polygenic spontaneous onset model where
most mice
develop elevated blood glucose concentrations (BGV, blood glucose value,
determined from
tail-vein needlestick and handheld meter) between 12 and 30 weeks of age.
Incidence and
progression of disease is unpredictable, with total incidence ranging from 60%
to 95% at 30
weeks of age (WoA) and progression from diagnosis (two sequential BGV readings
of >250)
20 .. to terminal (two sequential BGV readings of 600 or higher) ranging from
2 days to 4 weeks.
Replication of elevated BGVs on sequential readings are necessary as mice are
allowed
food and water ad libitum which results in moderate BGV variability beyond
that caused by
immuno-pathology.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
41
An example of a plasmid nucleotide sequence herein:
SEQ ID NO 24: full (non-annotated) plasmid sequence (6,401 base pairs)
GACTCTTCGCGATGTACGGGCCAGATATACGCGTTGACATTGATTATTGACTAG
TTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGT
TACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCATTG
ACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAA
TGGGTGGACTATTTACGGTAAACTGCCCACTTGGCAGTACATCAAGTGTATCATATGCC
AAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCGCCTGGCATTATGCCCAGT
ACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATTAGTCATCGCTATTA
CCATGGTGATGCGGTTTTGGCAGTACATCAATGGGCGTGGATAGCGGTTTGACTCACG
GGGATTTCCAAGTCTCCACCCCATTGACGTCAATGGGAGTTTGTTTTGGCACCAAAATC
AACGGGACTTTCCAAAATGTCGTAACAACTCCGCCCCATTGACGCAAATGGGCGGTAG
GCGTGTACGGTGGGAGGTCTATATAAGCAGAGCTCTCTGGCTAACTAGAGAACCCACT
GCTTACTGGCTTATCGAAATTAATACGACTCACTATAGGGAGACCCAAGCTGGCTAGCG
TTTAAACTTAAGCTTGGTACCGAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTCTGC
ACTGCAGCTCGCATCTCTCCTTCACGCGCCCGCCGCCCTACCTGAGGCCGCCATCCAC
GCCGGTTGAGTCGCGTTCTGCCGCCTCCCGCCTGTGGTGCCTCCTGAACTGCGTCCGC
CGTCTAGGTAAGTTTAAAGCTCAGGTCGAGACCGGGCCTTTGTCCGGCGCTCCCTTGG
AGCCTACCTAGACTCAGCCGGCTCTCCACGCTTTGCCTGACCCTGCTTGCTCAACTCTA
GGTAAGTTAATGAGACAGATAGAAACTGGTCTTGTAGAAACAGAGTAGTCGCCTGCTTT
TCTGCCAGGTGCTGACTTCTCTCCCCTGGGCTTTTTTCTTTTTCTCAGGTTGAAAAGAAG
AAGACGAAGAAGACGAAGAAGACAAACCGTCGTCGACTGCCATGCGCCGCTGATTAAC
GCCGCCACCATGGCCCACCGACGCAGATCCAGAAGCTGCCGTGAGGACCAGAAGCCC
GTGATGGATGATCAGAGGGACCTTATCTCTAACAATGAACAACTGCCAATGCTCGGCAG
ACGGCCTGGGGCCCCGGAGAGCAAGTGCAGCAGAGGAGCCTTGTACACGGGGTTCTC
CATTTTAGTGACTCTCCTTCTCGCCGGCCAAGCTACCACCGCCTACTTTCTGTACCAACA
GCAAGGCAGACTAGACAAACTGACAATCACAAGCCAGAACCTTCAGCTGGAGTCTCTGC
GGATGAAGCTGCCCGCTTTGTGGATGAGATTGCTTCCTCTACTTGCTCTCCTGGCGCTC
TGGGGACCTGACCCCGAGCAAGAGTTTGTTAATCAGCACCTGTGTGGGAGTCATCTGG
TGGAGGCACTCTATTTAGTGTGCGGAGAGAGGGGCTTCTTCTACACTCCAAAGACCAGA
CGGGAGGCCGAAGACCTTCAAGTGGGGCAAGTAGAACTGGGTGGCGGACCCGGTGCC
GGGAGCCTTCAGCCGCTCGCCCTGGAGGGCTCTCTTCAGAAACGCGGCATCGTGGAG
CAGTGTTGCACATCCATTTGCTCACTCTACCAGCTGGAGAACTACTGCAACGGAAGCGG
AGTGAAGCAGACGTTGAATTTTGATTTGTTGAAGTTGGCGGGGGATGTGGAGAGCAATC
CGGGGCCGATGCCCCCTAGTGGCCTCAGACTTTTGTTATTGTTATTACCGCTTTTATGG
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
42
CTCTTGGTGCTGACACCGGGCCGTCCGGCTGCTGGCTTGTCGACTTGTAAGACAATTG
ATATGGAATTGGTGAAACGAAAACGGATTGAGGCCATCCGAGGACAGATTTTGAGCAAG
CTGCGGCTTGCCTCGCCACCCTCGCAAGGGGAAGTCCCACCCGGACCTCTACCAGAAG
CAGTCCTAGCGCTGTACAACAGTACAAGAGATAGAGTGGCCGGGGAATCCGCAGAACC
AGAGCCTGAGCCTGAAGCCGATTATTATGCAAAGGAAGTGACTAGGGTCCTGATGGTC
GAGACCCATAACGAAATCTACGACAAATTCAAACAAAGTACCCACTCTATCTACATGTTC
TTCAACACCAGTGAGCTAAGAGAAGCCGTGCCCGAACCTGTGCTTCTTTCCCGCGCAG
AACTCCGCCTCTTGAGACTCAAATTGAAAGTTGAACAACACGTAGAGCTTTACCAGAAAT
ACTCTAATAATTCATGGCGATATCTTTCTAATCGTCTCCTCGCCCCATCTGACAGCCCTG
AATGGCTCTCCTTCGACGTTACGGGAGTTGTGCGCCAGTGGCTCAGCAGAGGCGGAGA
GATAGAGGGCTTTCGGCTGAGCGCACATGTATCTGTGGACTCAAGGGACAACACATTG
CAAGTGGATATTAACGGTTTTACAACTGGACGGAGAGGGGACCTGGCGACCATCCACG
GCATGAATAGACCTTTCCTGCTGCTGATGGCTACTCCCCTGGAGAGGGCACAGCACTTA
CAGTCTTCCAGACACCGGCGCGCCCTGGATACAAACTACTGCTTCAGCTCCACCGAAAA
GAACTGTTGCGTGCGGCAGCTGTACATTGACTTCAGAAAGGATCTGGGCTGGAAGTGG
ATTCATGAGCCCAAGGGGTATCATGCCAACTTCTGTCTTGGGCCATGCCCATACATCTG
GTCACTGGATACCCAGTACTCCAAAGTTCTGGCCTTGTACAATCAACACAACCCTGGAG
CTTCCGCCGCTCCTTGCTGTGTGCCCCAAGCCCTAGAGCCCCTGCCCATCGTTTATTAT
GTCGGACGCAAGCCCAAAGTAGAACAGCTATCAAATATGATCGTGAGAAGCTGCAAGTG
TAGCTGATAAACGCGTCGAGCATGCATCTAGGGCGGCCAATTCCGCCCCTCTCCCCCC
CACCCCTCTCCCTCCCCCCCCCCTAACGTTACTGGCCGAAGCCGCTTGGAATAAGGCC
GGTGTGCGTTTGTCTATATGTTATTTTCCACCATATTGCCGTCTTTTGGCAATGTGAGGG
CCCGGAAACCTGGCCCTGTCTTCTTGACGAGCATTCCTAGGGGTCTTTCCCCTCTCGCC
AAAGGAATGCAAGGTCTGTTGAATGTCGTGAAGGAAGCAGTTCCTCTGGAAGCTTCTTG
AAGACAAACAACGTCTGTAGCGACCCTTTGTAGACAGCGGAACCCCCCACCTGGCGAT
AGATGCCTCTGCGGCCAAAAGCCACGTGTATAAGATACACCTGCAAAGGCGGCACAAC
CCCAGTGCCACGTTGTGAGTTGGATAGTTGTGGAAAGAGTCAAATGGCTCTCCTCAAGC
GTATTCAACAAGGGGCTGAAGGATGCCCAGAAGGTACCCCATTGTATGGGATCTGATCT
GGGGCCTCGGTGCACATGCTTTACATGTGTTTAGTCGAGGTTAAAAAACGTCTAGGCCC
CCCGAACCACGGGGACGTGGTTTTCCTTTGAAAAACACGATGATAATATGGCTGCCGCT
CATTCTAGTGCCCTTCTTTGCTGCCTGGTCCTGCTCACCGGGGTGCGAGCTAGCCCTG
GACAAGGGACACAATCCGAAAACTCGTGCACCCACTTCCCGGGCAACCTCCCTAACAT
GCTGAGGGACCTCCGTGATGCCTTCAGTAGAGTGAAGACGTTCTTCCAAATGAAAGATC
AGTTAGATAACCTGCTCCTGAAGGAGTCACTCTTAGAAGACTTCAAAGGATACCTCGGC
TGCCAAGCACTTAGCGAGATGATTCAATTCTACTTAGAAGAAGTCATGCCTCAAGCTGA
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
43
GAATCAAGACCCCGACATCAAAGCTCATGTGAATTCTTTGGGAGAAAATTTGAAGACTTT
GCGGCTGCGGCTGCGGAGATGTCACCGCTTTCTGCCCTGTGAGAACAAATCAAAAGCG
GTCGAGCAAGTTAAGAATGCCTTCAATAAGCTACAAGAGAAGGGCATCTACAAAGCAAT
GAGCGAGTTTGATATCTTTATCAATTACATTGAAGCCTACATGACAATGAAGATTAGGAA
TGCCGCGGGGAGCGGCGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGA
GGAGAACCCTGGACCTATGTACAGAATGCAGCTGCTGAGCTGCATCGCCCTGAGCCTG
GCCCTGGTGACCAACAGCGCACCCACGTCCTCTAGCACCAAGAAGACCCAGTTACAGT
TGGAGCATCTACTTTTAGACCTGCAAATGATTTTGAACGGCATCAACAACTACAAGAATC
CTAAACTTACTCGCATGCTTACCTTCAAATTTTACATGCCCAAGAAGGCCACCGAACTGA
AGCACTTGCAATGTCTGGAGGAAGAACTCAAGCCGCTGGAGGAAGTTCTCAACCTCGC
GCAGTCCAAGAATTTCCACCTCCGGCCAAGAGACCTGATCAGTAACATTAATGTGATAG
TGCTGGAGCTGAAGGGAAGCGAGACTACATTTATGTGCGAGTACGCCGATGAAACCGC
TACAATCGTCGAGTTCCTGAATAGATGGATCACATTTTGCCAGTCAATTATCTCTACTCT
GACATGATAACTCGAGGTCTAGAGGGCCCGTTTAAACCCGCTGATCAGCCTCGACTGT
GCCTTCTAGTTGCCAGCCATCTGTTGTTTGCCCCTCCCCCGTGCCTTCCTTGACCCTGG
AAGGTGCCACTCCCACTGTCCTTTCCTAATAAAATGAGGAAATTGCATCGCATTGTCTGA
GTAGGTGTCATTCTATTCTGGGGGGTGGGGTGGGGCAGGACAGCAAGGGGGAGGATT
GGGAAGACAATAGCAGGCATGCTGGGGATGCGGTGGGCTCTATGGCTTCTACTGGGC
GGTTTTATGGACAGCAAGCGAACCGGAATTGCCAGCTGGGGCGCCCTCTGGTAAGGTT
GGGAAGCCCTGCAAAGTAAACTGGATGGCTTTCTCGCCGCCAAGGATCTGATGGCGCA
GGGGATCAAGCTCTGATCAAGAGACAGGATGAGGATCGTTTCGCATGGCCAAAGAAGA
CAATATTGAAATGCAAGGTACCGTTCTTGAAACGTTGCCTAATACCATGTTCCGCGTAGA
GTTAGAAAACGGTCACGTGGTTACTGCACACATCTCCGGTAAAATGCGCAAAAACTACA
TCCGCATCCTGACGGGCGACAAAGTGACTGTTGAACTGACCCCGTACGACCTGAGCAA
AGGCCGCATTGTCTTCCGTAGTCGCTGATAAATTATTAACGCTTACAATTTCCTGATGCG
GTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATACAGGTGGCACTTTTCGG
GGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGC
TCATGAGACAATAACCCTGATAAATGCTTCAATAATAGCACGTGCTAAAACTTCATTTTTA
ATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGT
GAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGA
TCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGT
GGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCA
GAGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAG
AACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGC
CAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAG
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
44
GCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACG
ACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCG
AAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCA
CGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCA
CCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAA
AACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGGCTTTTGCTGGCCTTTTGCTCACAT
GTTCTT
Example 1 - Antigen encoding plasmids compared to antigen + IL-10 encoding
plasmids:
It has been suggested in the prior art that depletion of immuno-stimulatory
CpG
sequences in the plasmid back bone would be required for effective DNA immuno-
therapy
treatment of Ti D. This experiment was thus modelled after previously
published experiments
(2008 J lmmunol. 181(12):8298-307).
NOD mice were given eight once weekly doses of plasmid beginning at week 9
(age): either empty vector (pVAX1, 50 ug) was given, or pVAX1-proinsulin Ag
(not
endosomally targeted, not preproinsulin), or CpG depleted pVAX1-proinsulin Ag,
or a
bicistronic construct pVAX1-1L10-IRES-proinsulin Ag in equimolar ratios.
All administrations were intramuscular in the left quadriceps under isoflurane
anaesthesia and contained only plasmid in PBS + EDTA. BGVs were assessed in
all mice on
a weekly basis and incidence of type 1 diabetes was scored based on two BGV
readings
over 250 mg/d1. Mice were evaluated until 30 weeks of age or a BGV of 600 were
reached,
followed by sacrifice.
The results from this experiment (table 1) demonstrate that A) CpG depletion
is
neither necessary nor beneficial for efficacy, B) the inclusion of immuno-
modulatory
cytokines significantly increases efficacy, and C) the plasmid backbone (empty
vector) is
equivalent to untreated groups.
CA 03042321 2019-04-30
WO 2018/083111
PCT/EP2017/077949
Table 1: T1D incidence in NOD mice at 30 weeks of age.
Plasmid T1DDisease incidence at 30 weeks of
age
Historical untreated colony incidence 77.8%
pVAX1 (empty vector negative control) 23/29 = 79.3%
CpG depleted pVAX1-proinsulin Ag 24/29 = 82.7%
(antigen+modified vector)
pVAX1-proinsulin Ag 18/30 = 60%
(antigen)
pVAX1-1L10-IRES-proinsulin Ag 10/26 = 38.5%
(antigen+I L-10)
Example 2 ¨ Expressed protein products resulting from plasmids encoding
antigen, IL-10, IL-2 and TGF-I3
5 Multi-cistronic plasmids were created to co-express TGF-13, IL-10, and
optionally IL-
2. Freestyle293 cells were transiently transfected and cultured in serum-free
media.
Supernatants were collected and subjected to ELISA quantification after 72
hours.
The results in table 2 below shows that: A) expression of multiple independent
cytokines is achieved from a single vector, B) significant amounts of each
cytokine are
10 produced and in the expected ratios, C) minor sequence changes
significantly improve IL-10
expression from the first generation IL10/proinsulin plasmid, and D) neither
the plasmid
backbone (empty vector) or endosomal targeting of antigen (11Ag) induces
cytokine
production or dysregulation.
15 Table 2: ELISA quantification of expressed protein products.
Plasmid Active TGF-b1 Interleukin-10
Interleukin-2
(ng/ml) (ng/ml) (ng/ml)
pVAX1 (empty vector) <0.0035 <0.0027 <0.0009
pVAX1-1L10/Proinsulin <0.0035 85.3 <0.0009
(antigen+I L-10)
pVAX1-I lAg/TGF13/1L10/ 7.35 1,238.8 <0.0009
(antigen+TGF[3+I L-10)
pVAX1-I lAg/TGF13/1L10/1L2 2.39 1,259.5
777.0
(antigen+TGF[3+I L-10+IL-2)
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
46
Example 3 ¨ Impact of TGF43 and IL-2 on disease suppression
Multi-cistronic plasmids were evaluated for disease prevention in NOD mice as
in
Example 1, with the exception that dosing was continued once weekly until
sacrifice (onset of
diabetes) or week 30. One mouse from each group (initial n=24) was sent out
for full
necropsy after 10 weeks of dosing - including pathology on 10 standard highly
perfused
tissues, complete blood count, and clinical chemistry. Other than minor muscle
disruption
and regrowth due to mechanical trauma at the injection site, there were no
deviations from
un-dosed animals.
The results in table 3 below shows that: A) addition of TGFI3 significantly
increases
efficacy, B) the inclusion of Interleukin-2 may increase efficacy and does not
induce
pathology, C) chronic dosing with plasmids expressing IL-10 and antigen
increases efficacy
in disease prevention, and D) chronic dosing with plasmids expressing TGF13,
IL-10 and IL-2
increases efficacy without resulting in any safety signals.
Table 3: T1D incidence in NOD mice.
Plasmid Disease incidence at 30 weeks of
age
Historical untreated colony incidence 77.8%
Untreated (negative control) 18/21 = 85.7%
pVAX1-Ag/I L10 5/23 = 21.7%
(antigen+IL-10)
pVAX1-I lAg/TGF13/1L10 2/23 = 8.7%
(antigen+TGFr3+IL-10)
pVAX1-11Ag/TGF13/1L10/IL2 1/23 = 4.3%
(antigen+TGF[3+1L-10+1L-2)
Example 4 Evaluation of RES elements, introns as well as subcutaneous
administration
Multi-cistronic plasmids were evaluated for disease prevention in NOD mice as
in
Example 3, except that dosing began earlier (at week 5) in order to better
mimic chronic
pediatric administration. In addition to validating the pVAX1-11Ag/TGF13/IL10
and pVAX1-
11Ag/TGF13/1L10/1L2 plasmids containing introns, other control groups were
examined.
Specifically, a different IRES segment (CrPV [from Cricket Paralysis Virus] as
opposed to the
EMCV [from EncephaloMyoCarditis Virus]) was evaluated for expected increases
in efficacy,
as was a deletion of the intron segment to assess its necessity. Due to
obvious lack of
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
47
efficacy compared to the parental plasmid (pVAX1-11Ag/TGF13/1L10/IL2) the CrPV
and intron-
free (n.i. = no intron) groups were terminated early. In addition, the cohort
of mice utilized in
this experiment experienced more rapid progression of disease than previous
cohorts, with
time from diagnosis to sacrifice averaging 1.25 weeks rather than 2.75 from
previous
experiments. Finally, a subcutaneous administration group was added. This
group was
dosed with the triple cytokine plasmid (pVAX1-11Ag/TGF13/1L10/IL2) with once
weekly injection
in the s.c. space in the scruff of the neck without anaesthesia.
The results in table 4 show that: A) EMCV IRES elements provide significantly
better
efficacy than the CrPV IRES, B) the inclusion of an intron (in this plasmid
located within the
CD74 endosomal targeting region) significantly increases efficacy, C) while
the inclusion of
IL-2 provides minimal benefit in mild disease settings its presence
significantly increases the
efficacy and robustness of treatment in aggressive disease settings, and D)
subcutaneous
dosing, which is ineffective in most DNA vaccine applications, here shows
modest efficacy
and a significant delay of disease even without optimization.
Table 4: T1D incidence in NOD mice.
Treatment type Diabetic/total % diabetic
Historical control 80% @ 30 Weeks
Untreated 15/21 71.4% @ 30 Weeks
Empty vector control i.m. 13/21 61.9% @ 30 Weeks
pVAX1-11Ag/TGF13/1L10/IL2 10/24 41.6% @22 Weeks
(no lntron) i.m.
pVAX1-11Ag/TGF13/1L10/IL2 7/22 31.8% @22 Weeks
(CrPv IRES instead of EMCV IRES) i.m.
pVAX1-11Ag/TGF13/IL10 i.m. (no IL-2) 12/42 28.6%@ 30 Weeks
pVAX1-11Ag/TGF13/1L10/IL2 i.m. 1/42 2.4% @30 Weeks
pVAX1-11Ag/TGF13/1L10/IL2 s.c. 12/42 28.6% @ 30 Weeks
Example 5 Comparison of commercial antibiotic free selection with antibiotic
selection systems
An alternate plasmid backbone was evaluated with the object of removing
kanamycin resistance to comply with European Medicines Agency guidance. The
same
insert (11Ag/TGF13/1L10/1L2, including intron) was cloned into the Nature
Technology
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
48
NTC9385R "nanoplasmid" backbone. The resultant plasmid was evaluated in NOD
mice as
in Example 3, except that treatment began on week 11 (late start) and
terminated early due
to failure of the NTC9385R-based plasmid.
The results in table 5 below show that: A) changes to selection system of the
plasmid backbone surprisingly induce significant changes to effectiveness of
the plasmids,
and B) a late start to treatment results in early conversions. Data from
other, related
experiments indicates that dosing with these tolerogenic DNA vaccine plasmids
requires two
to four weeks to have efficacy, such that a late start to treatment results in
several early
cases of diabetes before the treatment becomes efficacious.
Table 5: T1D disease incidence in NOD mice.
Plasmid Disease incidence at 30 weeks of
age
Historical untreated colony incidence 77.8%
Untreated (negative control) 16/21 = 76.2%
pVAX1-11Ag/TGF13/1L10/1L2 with intron 5/21 = 23.8%
(kanamycin resistant)
pNTC9385R-11Ag/TGF13/1L10/1L2 with intron 13/21 = 61.9%
(commercial antibiotic free selection system)
Example 6 Disease suppression efficacy with plasmids with and without
antigen
To determine the role of the encoded antigen in the function of the plasmid
two
experiments were performed (Examples 6 and 7). An alternate plasmid was
evaluated with
the object of removing the antigen (pre-proinsulin) encoding region while
retaining the CD74
targeting domain and all three secreted cytokines. The resultant plasmid was
evaluated in
NOD mice as in Example 3, except that treatment began on week 11 (late start).
This experiment demonstrates that the antigen portion is required for full
efficacy
and that it is not merely cytokine production driving the function of the
plasmid. This is one of
two criteria needed to demonstrate antigen-specificity of the treatment.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
49
Table 6: T1D incidence in NOD mice.
Plasmid Disease incidence at 30 weeks of
age
Historical untreated colony incidence 77.8%
pVAX1-11Ag/TGF13/1L10/1L2 2/22 = 9.1%
(antigen+cytokines)
pVAX1-11/TGF13/1L10/IL2 15/28 = 53.5%
(no antigen+cytokines)
Example 7 Impact of the antigen immuno-therapy herein on efficiency of
unrelated antigen vaccines
To determine the role of the encoded antigen in the function of the plasmid
two
experiments were performed (Examples 6 and 7). NOD mice were either sham
treated with
PBS injection or treated with pVAX1-11Ag/TGF13/1L10/1L2 plasmid as in Example
3. Following
four doses (i.e. at 13 weeks of age) each mouse was immunized i.p. with 50 g
of an
irrelevant antigen (Chicken Ovalbumin, OVA) in 100 I of a 1:1 alum
suspension. Sham or
plasmid treatments were continued once weekly until sacrifice three weeks (21
days) post-
immunization at which time serum was collected. Class-switched (total IgG and
IgG2a)
antibodies against the ovalbumin antigen were determined via commercial ELISA
kits. No
significant differences were observed between plasmid and sham treated groups
in their total
anti-OVA IgG levels, nor did either group produce anti-OVA IgG2a.
The results in table 7 below show that while the plasmid suppresses immune
responses related to the targeted disease, it does not suppress immune
reactivity toward
unrelated antigens (i.e. any antigens not encoded by the plasmid). This is the
second of two
criteria needed to demonstrate antigen-specificity of the treatment. As
treatment of pediatric
patients will involve concomitant administration of standard childhood
vaccinations this is a
significant advantage over systemic / generic immunosuppression via agents
such as
methotrexate or cyclosporine A.
Table 7: Response to irrelevant antigen in NOD mice that have received DNA
immu no-therapy vaccination against Ti D.
Treatment # samples Mean ug anti-OVA IgG / mL serum Error
Plasmid treated 8 7.517 +/- 0.967
PBS (Sham) treated 5 8.954 +/- 1.227
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
These values result in a non-significant p value of 0.377 and a confidence
interval of -1.99 to
4.87. These results indicate that treatment with the immunomodulatory plasmid
does not
impact immune response to other antigens not encoded by the plasmid, and
therefore does
not result in broad or systemic immuno suppression.
5
Example 8 Individual protein products expressed from the plasmid
The TaV 2A element resulted in unexpected IL-10+IL-2 fusion products herein
(data
not shown) and other separation strategies were therefore evaluated. Initial
separation
technologies included upstream extensions of the TaV 2A sequence (leading to
rapid
10 degradation and lack of secreted IL-10) and also a carboxypeptidase
cleavage site (which
induced death of transfected cell lines). Further separation strategies
evaluated were GSG-
TaV 2A, a furin cleavage site, a furin site followed by TaV 2A, P 2A, and E 2A
(equine rhinitis
virus A).
Freestyle293 cells were transiently transfected and cultured in serum-free
media.
15 Both cell pellets and supernatants were collected and subjected to semi-
quantitative
multicolor Western blotting after 72 hours.
The results in table 8 below show that: A) unexpectedly, proteolytic cleavage
sites
fail to function between IL-10 and IL-2 genes, B) GSG tags (decoupler
sequences) between
IL-10 IL-2 are preferable to extended insulator sequences, C) P 2A is
preferable to either
20 TaV 2A or E 2A, and D) 2A sequences may have significant and unexpected
effects on the
degradation and secretion of expressed upstream proteins such as IL-10.
Table 8: Separation of expressed IL-10 and IL-2 protein products.
Plasmid Cellular Secreted Cellular Secreted Cellular Secreted
Interleukin Interleukin Interleukin- Interleukin- Fused Fused
-10 -10 2 2 product
product
GSG-TaV ++++ ++ - +++ ++ -
2A
Furin + - - - ++++ ++
cleavage
site
Furin/TaV +++ ++ - ++ ++ +
2A
P 2A ++ ++++ - +++ + -
E 2A +++ ++ - ++ ++ -
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
51
Example 9 Comparison of commercial selection system with a heat sensitive
selection system provided herein as well as comparison between plasmids
encoding
IL-2 and plasmids not encoding IL-2 (subcutaneous administration)
Plasmid backbones were created and evaluated with the object of removing
kanamycin resistance to comply with European Medicines Agency guidance. The
corrected
insert (11Ag/GSG-FMDV 2A/TGF13/EMCV IRES/IL10/GSG-P 2A/1L2, including an
intron in the
upstream noncoding region) was cloned into either a retrofitted/minimally
modified pVAX1
vector containing the Nature Technology "RNA-OUT" selection marker or an
equivalent
minimally modified pVAX1 vector encoding wt infA ("pNN") as backbones.
Additionally,
plasmids either containing an additional SV40 enhancer element or defiecient
in IL-2 were
produced. The resultant plasmids were evaluated in NOD mice as in Example 3,
except that
administration was s.c. either once weekly or three times weekly (preferred).
The results shown in table 9+10 below show that: A) the commercially available
exchange of RNA-OUT for the Kanamycin antibiotic resistance in the pVAX1
backbone still
unexpectedly underperforms, B) the infA complementation antibiotic-free
selection system
performs equivalently to the parental pVAX1 vector, C) Interleukin-2 is
required for optimal
efficacy, D) addition of the SV40 enhancer element does not improve efficacy,
and E) the
corrected triple cytokine insert retains full functionality.
Table 9: T1D incidence in NOD mice.
Plasmid, administered 3x weekly (optimal) Disease incidence at 30 weeks
of age
Historical untreated colony incidence 78.9%
Untreated (negative control) 12/15 = 80%
pNN empty vector 12/16 = 75%
(negative control with heat sensitive selection but
no protein encoding sequences)
pVAX1-I lAg/FMDV/TGF13/1L10/TaV2A/1 L2 1/16 = 6.3%
(kanamycin selection and protein encoding
sequences)
pNN-11Ag/FMDV/TGF13/11_10/P2A/1 L2 1/23 = 4.3%
(temperature selective system and protein
encoding sequences)
pVAX1-RNA-OUT-I lAg/FMDV/TGF13/11_10/P2A/1 L2 9/23 = 39.1%
(commercial selective system and protein
encoding sequences)
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
52
Table 10: T1D incidence in NOD mice.
Plasmid, administered lx weekly (sub-optimal) Disease incidence at 27 weeks of
age
Historical untreated colony incidence 78.9%
Untreated (negative control) 12/15 = 80%
pNN-11Ag/FMDV/TGF8/1L10/P2A/1 L2 16/37 = 43.2%
(temperature selective system and protein
encoding sequences)
pNN-SV40e-IlAg/FMDV/TGFWIL10/P2A/IL2 20/37 = 54%
(temperature selective system and protein
encoding sequences as well as an enhancer)
pNN-11Ag/FMDV/TGF8/1L10 (1L-2 deficient) 25/40 = 62.5%
(temperature selective system and protein
encoding sequences - except IL-2)
pVAX1-RNA-OUT-I lAg/FMDV/TGF8/1L10/P2A/1 L2 27/38 = 71%
(commercial selective system and protein
encoding sequences)
Example 10 Examination of durability of tolerance effect following plasmid
withdrawal
In the previous experiment (represented in Table 9), the pNN-
IlAg/FMDV/TGFWIL10/P2A/IL2 group was not sacrificed at 30 weeks of age but
ceased
dosing with plasmid. Blood glucose values were followed for an additional ten
(10) weeks, to
a total of 40 weeks of age, to assess whether the plasmid had induced a
durable state of
tolerance or whether continued dosing was necessary for efficacy.
The results shown in table 11 below indicate that continued dosing is required
for
durability of tolerance, as a stable disease-free state to 30 weeks of age
rapidly deteriorated
following discontinuation of dosing. This indicates a beneficial safety
profile, as any adverse
events that might be encountered with plasmid dosing would also be expected to
cease with
dosing.
Table 11: T1D incidence in NOD mice following cessation of plasmid dosing.
Disease incidence to 30 weeks of age
Disease incidence to 40 weeks of age
1/23 = 4.3% 9/23 = 39.1%
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
53
Example 11 Examination of plasmid stability and durability on injection
A key issue with plasmid administration is degradation on administration. In
the case
of injection, shear forces encountered by large and viscous plasmid molecules
passing
through a thin needle under pressure lead to breakage of the covalently closed
circular
structure of the plasmid - rendering it linear and both subject to reduced
transfection ability
and rapid destruction. Most plasmids have 5 to 15% degradation to linear forms
on injection
through needles of sizes acceptable for clinical use, which leads to either
reduced efficacy or
a need for larger initial doses to compensate for the loss. Several types of
sequence
structures which may lead to plasmid unwinding and susceptibility to shear
degradation were
intentionally minimized in the plasmids disclosed, with the intention of
increasing robustness
and reliability with injection protocols. In order to assess shear degradation
of plasmid, which
can vary with viscosity and therefore concentration, the human lead plasmid
was
resuspended in Tris EDTA buffer to concentrations of 5, 7, and 9 mg/ml and
passed three
times though a G30 needle (expelled, re-drawn into syringe, then re-expelled)
and one (1)
microgram samples were run on an agarose gel against reference samples which
were not
passaged through the injection process.
The results shown in Figure 3 indicate, surprisingly, that the plasmid is not
noticeably degraded by three injection passages at any tested concentration or
viscosity.
Plasmid degradation would be visualized as both a smearing of smaller bands
(in between
the main supercoiled band at 6 Kb and the small process impurity band at the
bottom of the
gel or roughly 600 bp). Such linearization / degradation smears are not seen
for any sample
passaged through the injection process. This robust physical stability on
dosing is highly
desirable and both greater than anticipated or previously reported in
literature.
Example 12 Verification of plasmid retention with infA complementation
system
In order to verify that the infA-based plasmid retention selection system
functioned
as desired, the plasmid transformed bacteria were grown through 100 passages
(roughly 36
doublings/generations per passage, for a total of 3,600 generations of
potential drift or
plasmid loss examined). Passages 1-100 were generated at 11 per week, 2
passages per
weekday at 37 C and one each weekend at 30 C. All were performed in liquid
animal-free LB
media (Teknova soy-tone) supplemented with 15 micrograms/ml naladixic acid
(selecting for
DH5a base strain, not for plasmid presence). Glycerol stocks were generated
from each
passage and retained until all 100 passages were obtained for concurrent
processing.
CA 03042321 2019-04-30
WO 2018/083111 PCT/EP2017/077949
54
Scrapes of the glycerol stocks were used to inoculate 5 ml overnight cultures
which
were processed via supplier instructions on Qiagen miniprep kits using a
vacuum manifold
(either 16 or 32 cultures per run, due to gel size constraints). No attempt
was made to collect
0D600 readings for cell input normalization and all preps were done based on
standard
volumes. One microliter of each miniprep was subjected to Pstl/Xhol digestion
to resolve
backbone (approximately 2.4 Kbp) from insert (approximately 4 Kbp), without
correction for
plasmid concentration resulting from each miniprep. Each gel was run with
flanking Tridye 2-
Log ladders (NEB https://vvww.neb.com/products/n3200-2-log-dna-ladder-01-100-
kb), a first
sample lane of undigested plasmid, and visualized with SybrSafe dye. In the
images of the
gels, despite lack of control for nucleic acid quantity, all digest lanes show
both the presence
and expected digestion pattern for plasmid (seen on the images for passages 1-
16, 17-48,
49-80, and 81-100).
As an additional confirmation, glycerol stocks for passages 1-100 were also
streaked onto 50 sector antibiotic-free and animal-free LB agar plates and
incubated
overnight at 30 C. No attempt to control for streak inoculum was made. As
shown in Figure
4, all glycerol stock representative streaks resulted in noticeable growth and
thus plasmid
retention.
Example 13 Suitability for scale up with infA complementation system
In order to verify that the infA-based plasmid retention selection system
functioned
as desired at production scale, the plasmid transformed bacteria were used in
a 50 L pilot
fed-batch fermentor run with a specific yield enhancing temperature shift
step. Minimal
medium with the addition of yeast extract was utilized, reducing the doubling
rate to 0.88 /
hour. The fed-batch was initiated at 17h00 post inoculation and the regulation
of dissolved
oxygen at 30% was realized by successive increase of the p02 cascade
parameters (stirring
at 32h15 , pressure at 40h30, then air flow at 45h40). The biomass increase
rate lessened
immediately following the shift to 42 C, as anticipated. The amount of plasmid
DNA produced
was estimated at 1.03 0.17 g/L using a small scale plasmid extraction
procedure mimicking
immediate post-lysis yield.