Note: Descriptions are shown in the official language in which they were submitted.
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
METHODS, APPARATUSES, AND SYSTEMS FOR ANALYZING MICROORGANISM
STRAINS IN COMPLEX HETEROGENEOUS COMMUNITIES, DETERMINING
FUNCTIONAL RELATIONSHIPS AND INTERACTIONS THEREOF, AND
DIAGNOSTICS AND BlOSTATE MANAGEMENT BASED THEREON
[0001] This application claims priority to and benefit of U.S. Provisional
Patent Application
No. 62/439,804, filed on December 28, 2016; this application also claims
priority to and benefit
of U.S. Provisional Patent Application No. 62/560,174, filed on September 18,
2017; the entirety
of the aforementioned application(s) are hereby expressly incorporated by
reference for all
purposes.
[0002] This application may contain material that is subject to copyright,
mask work, and/or
other intellectual property protection. The respective owners of such
intellectual property have
no objection to the facsimile reproduction of the disclosure by anyone as it
appears in published
Patent Office file/records, but otherwise reserve all rights.
BACKGROUND
[0003] Microorganisms coexist in nature as communities and engage in a variety
of
interactions, resulting in both collaboration and competition between
individual community
members. Advances in microbial ecology have revealed high levels of species
diversity and
complexity in most communities. Microorganisms are ubiquitous in the
environment, inhabiting
a wide array of ecosystems within the biosphere. Individual microorganisms and
their respective
communities play unique roles in environments such as marine sites (both deep
sea and marine
surfaces), soil, and animal tissues, including human tissue.
SUMMARY
[0004] This disclosure is directed to methods, apparatuses, and systems for
analyzing
microorganism strains in complex heterogeneous communities, determining
functional
relationships and interactions thereof, and diagnostics and biostate
management based thereon.
Methods for diagnostics, analytics, and treatments of states and state
aberrations and state
deviations, including treatments comprising synthetic microbial ensembles, are
also disclosed.
[0005] In one aspect of the disclosure, a diagnostic method is disclosed. The
method can
comprise obtaining at least two samples sharing at least one common
environmental parameter
1
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
(such as sample type, sample location, sample time, etc.). At least one of the
at least two samples
can be defined as being in a first state, and at least one of the at least two
samples can be defined
as being in a second state, the second state different from the first state.
For example, in one
embodiment one of the at least two states is a healthy state or a state
associated with a healthy
sample source (e.g., a sample source having one or more desirable
characteristics or metadata),
while the other state is an unhealthy/sick state or a state associated with an
unhealthy/sickly
sample source (e.g., a sample source having one or more undesirable
characteristics or metadata,
in some instances, especially when compared to the corresponding
characteristic(s) or metadata
of a healthy sample source). For each sample, the presence of one or more
microorganism types
in the sample is detected and a number of each detected microorganism type of
the one or more
microorganism types in each sample is determined.
100061 Unique first markers in each sample, and quantity thereof, are
measured, each unique
first marker being a marker of a microorganism strain of a detected
microorganism type. The
absolute cell count of each microorganism strain in each sample is determined,
based on the
number of each microorganism type and the number/respective number of the
unique first
markers. Then, at least one unique second marker for each microorganism strain
is measured,
and an activity level for that microorganism strain is determined (e.g., based
on the unique
second marker exceeding a specified activity threshold). Depending on the
implementation, the
activity level can be numerical, relative, and/or binary (e.g.,
active/inactive). The absolute cell
count of each microorganism strain is filtered by the determined activity to
provide a set or list of
active microorganisms strains and their respective absolute cell counts for
each of the at least
two samples. The filtered absolute cell counts of active microorganisms
strains for the at least
one sample from the first state and the at least one sample from the second
state can be compared
or processed to define or determine a baseline state (e.g., a healthy state or
normal state). The
baseline state can be defined or characterized by the presence or absence of
specified taxonomic
groups and/or strains. In some embodiments, the method includes or further
comprises obtaining
at least one further sample, the further sample having an unknown state. Then,
for the at least one
further sample, the presence of one or more microorganism types is detected
and a number of
each detected microorganism type of the one or more microorganism types is
determined.
Unique first markers, and quantity thereof, are determined, each unique first
marker being a
marker of a microorganism strain of a detected microorganism type. The
absolute cell count of
2
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
each microorganism strain is determined from the number of each microorganism
type and the
number of the unique first markers. At least one unique second marker is used,
for each
microorganism strain based on a specified threshold, to determine an activity
level for that
microorganism strain. The absolute cell count of each microorganism strain is
filtered by the
determined activity to provide a set of active microorganisms strains and
their respective
absolute cell counts. The set of active microorganisms strains and their
respective absolute cell
counts for the at least one further sample is then compared to the baseline
state to determine a
state of the at least one further sample (e.g., healthy or unhealthy, normal
or abnormal, etc.). The
determined state of the at least one further sample is then output and/or
displayed (e.g., on a
display screen or graphic interface).
[0007] According to some further embodiments, the determined state of the at
least one further
sample corresponds to a state of an environment associated with the at least
one further sample.
Depending on the implementation, the environment associated with the at least
one further
sample can include a geospatial environment, such as a field or pasture, a
feed environment or
source (e.g., grain silo), a target animal and/or herd, etc. Treatments can be
identified or
determined for the environment associated with the at least one further
sample. In embodiments
where the baseline is healthy or the like, the treatment can be configured to
shift the state of the
environment toward the baseline. In some embodiments, the treatment can be
configured to shift
the state of the environment toward a state associated with desired goal or
favorable outcome.
The treatment can include a synthetic ensemble (especially a synthetic
ensemble formed
according to the methods of the disclosure), a chemical/biological treatment
or medicine, a
treatment regime, a combination of two or more of the preceding treatments,
and/or the like. In
some embodiments, the baseline state can be updated based on the at least one
further sample.
[0008] In another aspect of the disclosure, an analytic method is disclosed.
Such a method can
comprise obtaining at least two sample sets, each sample set including a
plurality of samples. In
some implementations, at least one sample set of the at least two sample sets
can be defined as
being in a first state, and at least one sample set of the at least two sample
sets can be defined as
being in a second state, wherein the first state is different from the second
state, and the range of
the sample in the sample set corresponds to the range of the state
corresponding to the sample
set. In other implementations, samples within the sample set are defined as
being in respective
states, or the state determination or definition is made post-analysis. The
method then includes
3
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
detecting a plurality of microorganism types in each sample, determining an
absolute number of
cells of each detected microorganism type of the plurality of microorganism
types in each
sample, and measuring unique first markers in each sample, and quantity
thereof, each unique
first marker being a marker of a microorganism strain of a detected
microorganism type. The
method includes then determining the absolute cell count of each microorganism
strain present in
each sample based on the number of each detected microorganism types in that
sample and the
number of unique first markers and quantity thereof in that sample and
measuring at least one
unique second marker for each microorganism strain to determine active
microorganism strains
in each sample. A set of active microorganisms strains and their respective
absolute cell counts is
then generated for each sample of the at least two sample sets. The method
includes analyzing
the active microorganisms strains and respective absolute cell counts for each
sample of the at
least two sample sets and/or respective samples to define a baseline state.
The baseline state can
be, in some embodiments, defined and/or characterized by the presence or
absence of specified
taxonomic groups and/or strains.
[0009] Then, at least one further sample having an unknown state is obtained.
For the at least
one further sample, the method further includes: (1) detecting the presence of
one or more
microorganism types; (2) determining a number of each detected microorganism
type; (3)
measuring unique first markers, and quantity thereof, each unique first marker
being a marker of
a microorganism strain of a detected microorganism type; (4) determining the
absolute cell count
of each microorganism strain from the number of each microorganism type and
the number of
the unique first markers; (5) measuring at least one unique second marker for
each
microorganism strain based on a specified threshold to determine an activity
level for that
microorganism strain; and (6) filtering the absolute cell count of each
microorganism strain by
the determined activity to provide a set of active microorganisms strains and
their respective
absolute cell counts. The set of active microorganisms strains and their
respective absolute cell
counts for the at least one further sample is compared to the baseline state
to determine a state
associated with the at least one further sample, and the determined state
associated with the at
least one further sample is displayed or output
10010] The method can further comprise selecting a plurality of active
microorganism strains
based on the baseline state and the determined state associated with the at
least one further
sample, and combining the selected plurality of active microorganism strains
with a carrier
4
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
medium to form a synthetic ensemble of active microorganisms configured to be
introduced to
an environment associated with the at least one further sample and modify a
state of the
environment associated with associated with the at least one further sample.
[0011] According to some embodiments, a method for identifying active
microorganisms from
a plurality of samples, analyzing identified microorganisms with at least one
metadata, and
creating an ensemble of microorganisms based on the analysis is disclosed.
Ensembles can be
used in treatments for disorders or undesirable states, and/or for biostate
shifting (e.g., shifting
from a disease state to a healthy or baseline state; or shifting from a
baseline or normal state to a
productive or enhanced state). Embodiments of the method include determining
the absolute cell
count of one or more active microorganism strains in a sample, wherein the one
or more active
microorganism strains is present in a microbial community in the sample. The
one or more
microorganism strains can be a subtaxon of a microorganism type. Samples used
in the methods
provided herein can be of any environmental origin. For example, in one
embodiment, the
sample is from animal, soil (e.g., bulk soil or rhizosphere), air, saltwater,
freshwater, wastewater
sludge, sediment, oil, plant, an agricultural product, plant, food or beverage
(e.g., cheese, beer,
wine, bread, or other fermented food) or an extreme environment. In another
embodiment, the
animal sample is a blood, tissue, tooth, perspiration, fingernail, skin, hair,
feces, urine, semen,
mucus, saliva, gastrointestinal tract, rumen, muscle, brain, tissue, or organ
sample. In one
embodiment, a method for determining the absolute cell count of one or more
active
microorganism strains is provided. The methods can also be used for defining
states/biostates
and/or analytics for determining the state of a sample (and corresponding
sample source).
[0012] According to some embodiments, a method of forming a bioensemble of
active
microorganism strains configured to alter a property in and/or biostate of a
target biological
environment is provided. Such methods can comprise obtaining at least two
samples (or sample
sets) sharing at least one common environmental parameter (such as sample
type, sample time,
sample location, sample source type, etc.) and detecting the presence of a
plurality of
microorganism types in each sample. Then the absolute number of cells of each
detected
microorganism type of the plurality of microorganism types in each sample is
determined (e.g.,
by way of non-limiting example, the dyeing procedures, cell sorting/FACS,
etc., as discussed
herein), and measuring a number of unique first markers in each sample, and
quantity thereof,
each unique first marker being a marker of a microorganism strain of a
detected microorganism
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
type. The absolute cell count of each microorganism strain present in each
sample is determined
based on the number of each detected microorganism types in that sample and
the number of
unique first markers and quantity thereof in that sample. At least one unique
second marker,
indicative of activity (e.g., metabolic activity) is measured for each
microorganism strain to
determine active microorganism strains in each sample, and a set or list of
active microorganisms
strains and their respective absolute cell counts for each of the at least two
samples is generated.
The active microorganisms strains and respective absolute cell counts for each
of the at least two
samples with at least one measured metadata for each of the at least two
samples are analyzed to
identify relationships between each active microorganism strain and at least
one measured
metadata, measured metadata for each sample, and/or measured metadata for a or
the sample
set(s). Based on the analysis, a plurality of active microorganism strains are
selected and
combined with a carrier medium to form a bioensemble of active microorganisms,
the
bioensemble of active microorganisms configured to alter at least one property
(that corresponds
to the at least one metadata) of a target biological environment when the
bioensemble is
introduced into that target biological environment. Depending on the
embodiment, the metadata
can be the or a environmental parameter, and can be the same or relatively
similar across
samples or sample sets, have different values across different samples or
sample sets. For
example, the metadata for dairy cows could include feed and milk output, and
the feed metadata
value could be the same (i.e., the cows are fed the same feed) while the milk
output/composition
could vary (i.e., the sample from one cow or set of samples from a particular
herd of cows has an
average milk output/composition that is different from milk output/composition
corresponding to
a sample from a second cow or sample set for a separate herd of cows). In some
embodiments, a
one sample set can be utilized to define a biostate, such as a baseline state.
[0013] According to some embodiments of the disclosure, diagnostic methods and
methods for
analyzing microbial communities are provided. Such methods can comprise
obtaining at least
two samples (or data for at least two samples), each sample including a
heterogeneous microbial
community, and detecting the presence of a plurality of microorganism types in
each sample. An
absolute number of cells of each detected microorganism type of the plurality
of microorganism
types in each sample is then determined (e.g., via FACS or other methods as
discussed herein). A
number of unique first markers in each sample, and quantity thereof, are
measured, each unique
first marker being a marker of a microorganism strain of a detected
microorganism type. A value
6
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
(activity, concentration, expression, etc.) of one or more unique second
markers is measured, a
unique second marker indicative of activity (e.g., metabolic activity) of a
particular
microorganism strain of a detected microorganism type, and the activity of
each detected
microorganism strain is determined based on the measured value of the one or
more unique
second markers (e.g., based on the value exceeding a specified set threshold).
The respective
ratios of each active detected microorganism strain in each sample are
determined (e.g., based on
the respective absolute cell counts, values, etc.). Then each of the active
detected microorganism
strains (or a subset thereof) of the at least two samples are analyzed to
identifying relationships
and the strengths thereof between each active detected microorganism strain
and the other active
detected microorganism strains, and between each active detected microorganism
strain and at
least one measured metadata. The identified relationships are then displayed
or otherwise output,
and can be utilized for defining a biostate and/or generation of a
bioensemble. In some
embodiments, only relationships that exceed a certain strength or weight are
displayed. As
detailed throughout the disclosure, biostates or states based on the disclosed
analytics can be
defined for purposes of analytics and treatment, and bioensembles can be
configured such that,
when introduced into a target environment, a bioensemble can change or alter a
biostate or
property of the target environment, an in particular, a property related to
the measured metadata.
[0014] According to some embodiments of the disclosure, methods comprise
detecting the
presence of a plurality of microorganism types in a plurality of samples and
determining the
absolute number of cells of each of the detected microorganism types in each
sample. A number
of unique first markers in each sample, and quantity thereof, can be measured,
a unique first
marker being a marker of a microorganism strain. A value or level of one or
more unique second
markers is measured, a unique second marker being indicative of metabolic
activity of a
particular microorganism strain. Based on measured value or level, an activity
of each of the
detected microorganism strains for each sample is determined or defined (e.g.,
based on the
measured value or level exceeding a specified threshold). A weighted or cell-
adjusted value of
each active detected microorganism strain in the sample is determined (the
weighted or cell-
adjusted value is not relative abundance). In some implementations, the
weighted or cell-adjusted
value is the absolute cell count for a strain relative to the sum of all
absolute cell counts for all
strains.
7
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
100151 Each of the detected active microorganism strains of each sample (or
sample sets) is
analyzed. The analysis can include identifying relationship and the strengths
thereof between
each detected active microorganism strain having a weighted value and every
other active
microorganism strain having a weighted value, and each active microorganism
strain having a
weighted value and one or more measured metadata.
10016.1 The identified relationships (an in some embodiments, related data
such as weighted
values and strengths) can be used to define a biostate, such as a baseline
state, and/or can then be
displayed or otherwise output, and can be utilized for generation of a
synthetic ensemble and/or
for biostate management. In some embodiments, the identified relationships for
each metadata
are displayed or output. In some embodiments, the displayed or output
relationships identify or
are configured to facilitate identification of a state or states, and/or one
or more microbial strains
responsible for a disease or deviation from a baseline state. In some
embodiments, the displayed
or output relationships identify or are configured to facilitate
identification of one or more
microbial strains to modify a biostate and/or treat a disease or disorder.
100171 In some embodiments, only relationships that exceed a certain strength
or weight (e.g.,
exceeding a specified threshold or base value) are displayed or output. As
detailed throughout
the disclosure, synthetic ensembles can be configured such that, when
introduced into a target
environment, a synthetic ensemble can modify a biostate and/or change or alter
a property of the
target environment, in particular, a property that is related to the measured
metadata. In some
implementations, the above method can be used to form a synthetic ensemble of
active
microorganism strains configured to modify a biostate or alter a property in a
biological
environment, and is based on two or more sample sets each having a plurality
of environmental
parameters, at least one parameter of the plurality of environmental
parameters being a common
environmental parameter that is similar between the two or more sample sets
and at least one
environmental parameter being a different environmental parameter that is
different between
each of the two or more sample sets. In some implementations, each sample set
includes at least
one sample comprising a heterogeneous microbial community obtained from a
biological sample
source. In some implementations, at least one of the active microorganism
strains is a subtaxon
of one or more microorganism types.
8
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[0018] In some embodiments of the disclosure, the one or more microorganism
types are one
or more bacteria (e.g., mycoplasma, coccus, bacillus, rickettsia, spirillum),
fungi (e.g.,
filamentous fungi, yeast), nematodes, protozoans, archaea, algae,
dinoflagellates, viruses (e.g.,
bacteriophages), viroids and/or a combination thereof. In one embodiment, the
one or more
microorganism strains is one or more bacteria (e.g., mycoplasma, coccus,
bacillus, rickettsia,
spirillum), fungi (e.g., filamentous fungi, yeast), nematodes, protozoans,
archaea, algae,
dinoflagellates, viruses (e.g., bacteriophages), viroids and/or a combination
thereof. In a further
embodiment, the one or more microorganism strains is one or more fungal
species or fungal sub-
species. In a further embodiment, the one or more microorganism strains is one
or more
bacterial species or bacterial sub-species. In even a further embodiment, the
sample is a ruminal
sample. In some embodiments, the ruminal sample is from cattle. In some
embodiments, the
sample is a gastrointestinal sample. In some embodiments, the gastrointestinal
sample is from a
pig or chicken.
[0019] In some embodiments, the methods include determining the absolute cell
count of one
or more active microorganism strains in a sample, the presence of one or more
microorganism
types in the sample is detected and the absolute number of each of the one or
more
microorganism types in the sample is determined. Such embodiments can be used
to determine a
biostate or deviation from a previously-defined baseline state A number of
unique first markers
is measured along with the relative quantity of each of the unique first
markers. As described
herein, a unique first marker is a marker of a unique microorganism strain.
Activity can then be
assessed, e.g., at the protein or RNA level, by measuring the level of
expression of one or more
unique second markers. The unique second marker can be the same or different
from the first
unique marker, and is a marker of activity of an organism strain. Based on the
level of
expression of one or more of the unique second markers, a determination is
made which (if any)
one or more microorganism strains are active. In one embodiment, a
microorganism strain is
considered active if it expresses the second unique marker at threshold level,
or at a percentage
above a threshold level. The absolute cell count of the one or more active
microorganism strains
is determined based upon the quantity of the one or more first markers of the
one or more active
microorganism strains and the absolute number of the microorganism types from
which the one
or more microorganism strains is a subtaxon.
9
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[0020] In one embodiment, determining the number of each of the one or more
organism types
in the sample comprises subjecting the sample or a portion thereof to nucleic
acid sequencing,
centrifugation, optical microscopy, fluorescence microscopy, staining, mass
spectrometry,
microfluidics, quantitative polymerase chain reaction (qPCR) or flow
cytometry.
[0021] In one embodiment, measuring the number of first unique markers in the
sample
comprises measuring the number of unique genomic DNA markers. In another
embodiment,
measuring the number of first unique markers in the sample comprises measuring
the number of
unique RNA markers. In another embodiment, measuring the number of unique
first markers in
the sample comprises measuring the number of unique protein markers. In
another embodiment,
measuring the number of unique first markers in the sample comprises measuring
the number of
unique metabolite markers. In a further embodiment, measuring the number of
unique
metabolite markers in the sample comprises measuring the number of unique
carbohydrate
markers, unique lipid markers or a combination thereof.
100221 In another embodiment, measuring the number of unique first markers,
and quantity
thereof, comprises subjecting genomic DNA from the sample to a high throughput
sequencing
reaction. The measurement of a unique first marker in one embodiment,
comprises a marker
specific reaction, e.g., with primers specific for the unique first marker. In
another embodiment,
a metagenomic approach.
[0023] In one embodiment, measuring the level of expression of one or more
unique second
markers comprises subjecting RNA (e.g., miRNA, tRNA, rRNA, and/or mRNA) in the
sample to
expression analysis. In a further embodiment, the gene expression analysis
comprises a
sequencing reaction. In yet another embodiment, the RNA expression analysis
comprises a
quantitative polymerase chain reaction (qPCR), metatranscriptome sequencing,
and/or
transcriptome sequencing.
[0024] In some embodiments, measuring the number of second unique markers in
the sample
comprises measuring the number of unique protein markers. In some embodiments,
measuring
the number of unique second markers in the sample comprises measuring the
number of unique
metabolite markers. In some embodiments, measuring the number of unique
metabolite markers
in the sample comprises measuring the number of unique carbohydrate markers.
In some
embodiments, measuring the number of unique metabolite markers in the sample
comprises
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
measuring the number of unique lipid markers. In some embodiments, the
absolute cell count of
the one or more microorganism strains is measured in a plurality of samples.
The absolute cell
counts of the plurality of samples can be used to define a state or biostate,
such as a baseline
state, and/or can be used to determine if sample sources deviate from a
predefined biostate, such
as a baseline state. In further embodiments, the plurality of samples is
obtained from the same
environment or a similar environment. In some embodiments, the plurality of
samples are
obtained at a plurality of time points. For example, in biostate management, a
plurality of
samples can be obtained for a particular environment or target, such as an
animal, over a course
of time to monitor and manage the biostate of the animal, and provide
treatments, supplements,
etc., to move the target toward or keep the target at a baseline state or
other desired biostate.
[0025] In some embodiments, measuring the level of one or more unique second
markers
comprises subjecting the sample or a portion thereof to mass spectrometry
analysis. In some
embodiments, measuring the level of expression of one more unique second
markers comprises
subjecting the sample or a portion thereof to metaribosome profiling and/or
ribosome profiling.
[0026] In another aspect of the disclosure, a method for determining the
absolute cell count of
one or more active microorganism strains is determined in a plurality of
samples, and the
absolute cell count levels are related to one or more metadata (e.g.,
environmental) parameters.
Relating the absolute cell count levels to one or more metadata parameters
comprises in one
embodiment, a co-occurrence measurement, a mutual information measurement, a
linkage
analysis, and/or the like. The one or more metadata parameters in one
embodiment, is the
presence of a second active microorganism strain. Accordingly, the absolute
cell count values
are used in one embodiment of this method to determine the co-occurrence of
the one or more
active microorganism strains in a microbial community with an environmental
parameter. In
another embodiment, the absolute cell count levels of the one or more active
microorganism
strains is related to an environmental parameter such as feed conditions, pH,
nutrients or
temperature of the environment from which the microbial community is obtained.
[0027] In this aspect, the absolute cell count of one or more active
microorganism strains is
related to one or more environmental parameters. The environmental parameter
can be a
parameter of the sample itself, e.g., pH, temperature, amount of protein in
the sample, the
presence of other microbes in the community. In one embodiment, the parameter
is a particular
11
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
genomic sequence of the host from which the sample is obtained (e.g., a
particular genetic
mutation). Alternatively, the environmental parameter is a parameter that
affects a change in the
identity of a microbial community (i.e., where the "identity" of a microbial
community is
characterized by the type of microorganism strains and/or number of particular
microorganism
strains in a community), or is affected by a change in the identity of a
microbial community. For
example, an environmental parameter in one embodiment, is the food intake of
an animal or the
amount of milk (or the protein or fat content of the milk) produced by a
lactating ruminant. In
some embodiments described herein, an environmental parameter is referred to
as a metadata
parameter.
100281 In one embodiment, determining the co-occurrence of one or more active
microorganism strains in the sample comprises creating matrices populated with
linkages
denoting one or more environmental parameters and active microorganism strain
associations.
[0029] In one embodiment, determining the co-occurrence of one or more active
organism
strains and a metadata parameter comprises a network and/or cluster analysis
method to measure
connectivity of strains within a network, wherein the network is a collection
of two or more
samples that share a common or similar environmental parameter. In some
embodiments, the
network analysis and/or network analysis methods comprise one or more of graph
theory, species
community rules, Eigenvectors/ modularity matrix, Gambit of the Group, and/or
network
measures. In some implementations, network measures include one or more of
observation
matrices, time-aggregated networks, hierarchical cluster analysis, node-level
metrics and/or
network level metrics. In some embodiments, node-level metrics include one or
more of: degree,
strength, betweenness centrality, Eigenvector centrality, page rank, and/or
reach. In some
embodiments, network level metrics include one or more of density,
homophilylassortativity,
and/or transitivity
[0030] In some embodiments, network analysis comprises linkage analysis,
modularity
analysis, robustness measures, betweenness measures, connectivity measures,
transitivity
measures, centrality measures or a combination thereof. In another embodiment,
the cluster
analysis method comprises building a connectivity model, subspace model,
distribution model,
density model, or a centroid model. In another embodiment, the network
analysis comprises
predictive modeling of network through link mining and prediction, collective
classification,
12
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
link-based clustering, relational similarity, or a combination thereof. In
another embodiment, the
network analysis comprises mutual information, maximal information coefficient
calculations, or
other nonparametric methods between variables to establish connectivity. In
another
embodiment, the network analysis comprises differential equation based
modeling of
populations. In another embodiment, the network analysis comprises Lotka-
Volterra modeling.
100311 Based on the analysis, strain relationships can be displayed or
otherwise output, and/or
one or more active relevant strains are identified for including in a
microbial ensemble.
BRIEF DESCRIPTION OF THE FIGURES
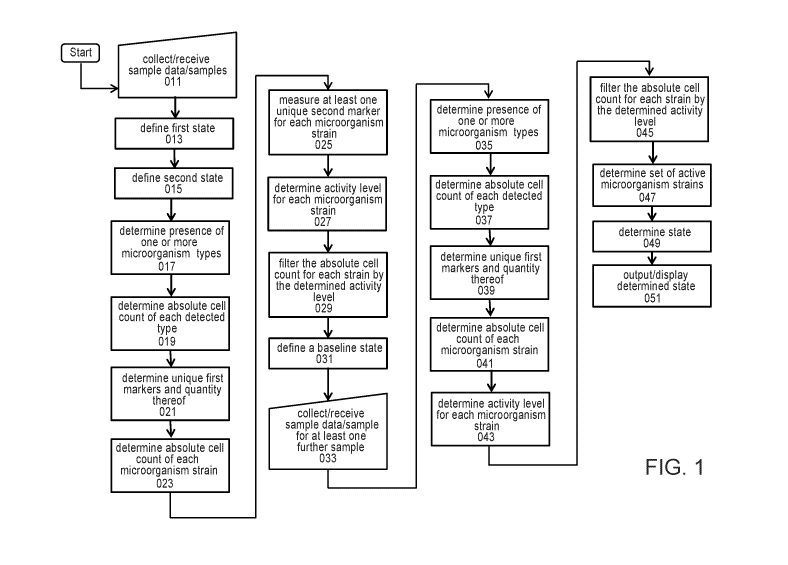
100321 FIG. 1 shows an exemplary high-level process flow state determination
and
diagnostics, according to some embodiments.
100331 FIG. IA shows an exemplary high-level process flow for screening and
analyzing
microorganism strains from complex heterogeneous communities, predicting
functional
relationships and interactions thereof, and selecting and synthesizing
microbial ensembles based
thereon, according to some embodiments.
100341 FIG. 1B shows a general process flow for determining the absolute cell
count of one or
more active microorganism strains, according to some embodiments.
100351 FIG. 1C shows a process flow for microbial community analysis,
typelstrain-metadata
relationship determination, display, and bioensemble generation, according to
some
embodiments.
100361 FIG. 1D illustrates exemplary visual output of analyzed strains and
relationships,
according to some embodiments.
100371 FIG. 1E illustrates MIC Score Distribution for Rumen Bacteria and Milk
Fat
Efficiency, according to some embodiments.
100381 FIG. 1F illustrates MIC Score Distribution for Rumen Fungi and Milk Fat
Efficiency,
according to some embodiments.
100391 FIG. 1G illustrates MIC Score Distribution for Rumen Bacteria and Dairy
Efficiency,
according to some embodiments.
13
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
100401 FIG. 1H illustrates MIC Score Distribution for Rumen Fungi and Dairy
Efficiency,
according to some embodiments.
100411 FIG. 2 shows a general process flow determining the co-occurrence of
one or more
active microorganism strains in a sample or sample with one or more metadata
(environmental)
parameters, according to some embodiments.
10042.1 FIG. 3A is a schematic diagram that illustrates an exemplary microbe
interaction
analysis and selection system 300, according to some embodiments, and FIG. 3B
is example
process flow for use with such a system. Systems and processes to determine
multi-dimensional
interspecies interactions and dependencies within natural microbial
communities, identify active
microbes, and select a plurality of active microbes to form an ensemble,
aggregate or other
synthetic grouping of microorganisms that will alter specified parameter(s)
and/or related
measures, is described with respect to FIGs. 3A and 3B.
100431 FIGs. 3C and 3D provides exemplary data illustrating some aspects of
the disclosure.
100441 FIG. 4 shows the non-linearity of pounds of milk fat produced over the
course of an
experiment to determine rumen microbial community constituents that impact the
production of
milk fat in dairy cows.
100451 FIG. 5 shows the correlation of the absolute cell count with activity
filter of target
strain Ascus_713 to pounds (lbs) of milk fat produced.
100461 FIG. 6 shows the absolute cell count with activity filter of target
strain Ascus_7 and the
pounds (lbs) of milk fat produced over the course of an experiment.
100471 FIG. 7 shows the correlation of the relative quantity or abundance with
no activity filter
of target strain Ascus_3038 to pounds (lbs) of milk fat produced.
100481 FIG. 8 shows the results of a field trial in which dairy cows were
administered a
microbial ensemble prepared according to the disclosed methods; FIG. 8A shows
the average
number of pounds of milk fat produced over time; FIG. 8B shows the average
number of pounds
of milk protein produced over time; and FIG. 8C shows the average number of
pounds of energy
corrected milk (ECM) produced over time.
100491 FIG. 9 shows the results of a bird study based on an embodiment of the
disclosure.
14
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
100501 FIG. 10 shows results of a horse study based on an embodiment of the
disclosure.
[0051] FIG. 11 shows an overview of example diagnostic platform workflow
according to
some embodiments of the disclosure.
[0052] FIGs. 12a-d illustrates an embodiment of the disclosure relating to
equine state
identification and microbial insights.
[0053] FIGs. 13a-b and 14a-c illustrates example embodiments of the disclosure
relating to
dairy state identification and microbial insights.
DETAILED DESCRIPTION
[0054] Microbial communities are central to environmental processes in many
different types
ecosystems as well and the Earth's biogeochemistry, e.g., by cycling nutrients
and fixing carbon
(Falkowski et al. (1998) Science 281, pp. 237-240, incorporated by reference
herein in its
entirety for all purposes). However, because of community complexity and the
lack of
culturability of most of the members of any given microbial community, the
molecular and
ecological details as well as influencing factors of these processes are still
poorly understood.
[0055] Microbial communities differ in qualitative and quantitative
composition and each
microbial community is unique, and its composition depends on the given
ecosystem and/or
environment in which it resides. The absolute cell count of microbial
community members is
subject to changes of the environment in which the community resides, as well
as the
physiological and metabolic changes caused by the microorganisms (e.g., cell
division, protein
expression, etc.). Changes in environmental parameters and/or the quantity of
one active
microorganism within a community can have far-reaching effects on the other
microorganisms of
the community and on the ecosystem and/or environment in which the community
is found. To
understand, predict, and react to changes in these microbial communities, it
is necessary to
identify the active microorganisms in a sample, and the number of the active
microorganisms in
the respective community. However, to date, the vast majority of studies of
microbial community
members have focused on the proportions of microorganisms in the particular
microbial
community, rather than absolute cell count (Segata et al. (2013). Molecular
Systems Biology 9,
p. 666, incorporated by reference herein in its entirety for all purposes).
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[0056] Although microbial community compositions can be readily determined for
example,
via the use of high throughput sequencing approaches, a deeper understanding
of how the
respective communities are assembled and maintained is needed.
[0057] Microorganism communities are involved in critical processes such as
biogeochemical
cycling of essential elements, e.g., the cycling of carbon, oxygen, nitrogen,
sulfur, phosphorus
and various metals; and the respective community's structures, interactions
and dynamics are
critical to the biosphere's existence (Zhou et at (2015). mBio 6(1):e02288-14.
Doi:10.1128/mBio.02288-14, herein incorporated by reference in its entirety
for all purposes).
Such communities are highly heterogeneous and almost always include complex
mixtures of
bacteria, viruses, archaea, and other micro-eukaryotes such as fungi. The
levels of microbe
community heterogeneity in human environments such as the gut and vagina have
been linked to
diseases such as inflammatory bowel disease and bacterial vaginosis (Nature
(2012). Vo. 486, p.
207, herein incorporated by reference in its entirety for all purposes).
Notably however, even
healthy individuals differ remarkably in the microbes that occupy tissues in
such environments
(Nature (2012). Vo. 486, p. 207).
[0058] As many microbes may be unculturable or otherwise difficult/expensive
to culture,
cultivation-independent approaches such as nucleic acid sequencing have
advanced the
understanding of the diversity of various microbial communities. Amplification
and sequencing
of the small subunit ribosomal RNA (SSU rRNA or 16s rRNA) gene was the
foundational
approach to the study of microbial diversity in a community, based in part on
the gene's
universal presence and relatively uniform rate of evolution. Advances in high-
throughput
methods have led to metagenomics analysis, where entire genomes of microbes
are sequenced.
Such methods do not require a priori knowledge of the community, enabling the
discovery of
new microorganism strains. Metagenomics, metatranscriptomics,
metaproteomics and
metabolomics all enable probing of a community to discern structure and
function.
[0059] The ability to not only catalog the microorganisms in a community but
to decipher
which members are active, the number of those organisms, and co-occurrence of
a microbial
community member(s) with each other and with environmental parameter(s), for
example, the
co-occurrence of two microbes in a community in response to certain changes in
the
community's environment, would allow for the understanding of the importance
of the
16
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
respective environmental factor (e.g., climate, nutrients present,
environmental pH) has on the
identity of microbes within a microbial community (and their respective
numbers), as well as the
importance of certain community members have on the environment in which the
community
resides. The present disclosure addresses these and other needs.
[0060] As used in this specification, the singular forms "a," "an" and "the"
include plural
referents unless the context clearly dictates otherwise. Thus, for example,
the term "an organism
type" is intended to mean a single organism type or multiple organism types.
For another
example, the term "an environmental parameter" can mean a single environmental
parameter or
multiple environmental parameters, such that the indefinite article "a" or
"an" does not exclude
the possibility that more than one of environmental parameter is present,
unless the context
clearly requires that there is one and only one environmental parameter.
100611 Reference throughout this specification to "one embodiment", "an
embodiment", "one
aspect", or "an aspect", "one implementation", or "an implementation" means
that a particular
feature, structure or characteristic described in connection with the
embodiment is included in at
least one embodiment of the present disclosure. Thus, the appearances of the
phrases "in one
embodiment" or "in an embodiment" in various places throughout this
specification are not
necessarily all referring to the same embodiment. Furthermore, the particular
features,
structures, or characteristics can be combined in any suitable manner in one
or more
embodiments.
[0062] As used herein, in particular embodiments, the terms "about" or
"approximately" when
preceding a numerical value indicates the value plus or minus a range of 10%.
Where a range of
values is provided, it is understood that each intervening value, to the tenth
of the unit of the
lower limit unless the context clearly dictates otherwise, between the upper
and lower limit of
that range and any other stated or intervening value in that stated range is
encompassed within
the disclosure. That the upper and lower limits of these smaller ranges can
independently be
included in the smaller ranges is also encompassed within the disclosure,
subject to any
specifically excluded limit in the stated range. Where the stated range
includes one or both of the
limits, ranges excluding either or both of those included limits are also
included in the disclosure.
[0063] As used herein, "isolate," "isolated," "isolated microbe," and like
terms, are intended to
mean that the one or more microorganisms has been separated from at least one
of the materials
17
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
with which it is associated in a particular environment (for example soil,
water, animal tissue).
Thus, an "isolated microbe" does not exist in its naturally occurring
environment; rather, it is
through the various techniques described herein that the microbe has been
removed from its
natural setting and placed into a non-naturally occurring state of existence.
Thus, the isolated
strain may exist as, for example, a biologically pure culture, or as spores
(or other forms of the
strain) in association with an acceptable carrier.
[0064] As used herein, "bioreactive modificator" refers to a composition, such
as microbial
ensemble comprising one or more active microbes, identified by methods,
systems, and/or
apparatuses of the present disclosure and that does not naturally exist in a
naturally occurring
environment, and/or at ratios, percentages, and/or amounts that are not
consistently found
naturally and/or that do not exist in a nature. For example, a bioreactive
modificator such as
microbial ensemble (also synthetic ensemble or bioensemble), or bioreactive
modificators
aggregate could be formed from identified or generated compounds/compositions,
and/or one or
more isolated microbe strains, along with an appropriate medium or carrier.
Bioreactive
modificators can be applied or administered to a target, such as a target
environment, population,
individual, animal, and/or the like.
[0065] In some embodiments, bioreactive modificators, such as microbial
ensembles according
to the disclosure are selected from and/or based on sets, subsets, and/or
groupings of active,
interrelated individual microbial species, or strains of a species. The
relationships and networks,
as identified by methods of the disclosure, are grouped, associated, and/or
linked based on
carrying out one or more a common functions, or can be described as
participating in, or leading
to, and/or associated with, a recognizable parameter, such as a phenotypic
trait of interest (e.g.,
increased milk production in a ruminant). In some implementations, groups from
which the
microbial ensemble is selected and/or upon which a bioreactive modificator is
selected, and/or
the bioreactive modificator, such as a microbial ensemble itself, can include
two or more species,
strains of species, or strains of different species, of microbes. In some
instances, the microbes
coexist can within the groups, bioreactive modificator, and/or microbial
ensemble symbiotically.
[0066] In certain aspects of the disclosure, bioreactive modificators and/or
microbial
ensembles are or are based on one or more isolated microbes that exist as
isolated and
biologically pure cultures. It will be appreciated that an isolated and
biologically pure culture of
18
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
a particular microbe, denotes that said culture is substantially free (within
scientific reason) of
other living organisms and contains only the individual microbe in question.
The culture can
contain varying concentrations of said microbe. The present disclosure notes
that isolated and
biologically pure microbes often "necessarily differ from less pure or impure
materials." See,
e.g. In re Bergstrom, 427 F.2d 1394, (CCPA 1970)(discussing purified
prostaglandins), see also,
In re Bergy, 596 F.2d 952 (CCPA 1979)(discussing purified microbes), see also,
Parke-Davis &
Co. v. H.K. Mulford & Co., 189 F. 95 (S.D.N.Y. 1911) (Learned Hand discussing
purified
adrenaline), aff d in part, rev'd in part, 196 F. 496 (2d Cir. 1912), each of
which are incorporated
herein by reference in their entireties. Furthermore, in some aspects,
implementation of the
disclosure can require certain quantitative measures of the concentration, or
purity limitations,
that must be achieved for an isolated and biologically pure microbial culture
to be used in the
disclosed microbial ensembles. The presence of these purity values, in certain
embodiments, is a
further attribute that distinguishes the microbes identified by the presently
disclosed method
from those microbes existing in a natural state. See, e.g., Merck & Co. v.
Olin Mathieson
Chemical Corp., 253 F.2d 156 (4th Cir. 1958) (discussing purity limitations
for vitamin B12
produced by microbes), incorporated herein by reference.
[0067] As used herein, "carrier", "acceptable carrier", or "pharmaceutical
carrier" refers to a
diluent, adjuvant, excipient, or vehicle with which is used with or in the
microbial ensemble.
Such carriers can be sterile liquids, such as water and oils, including those
of petroleum, animal,
vegetable, or synthetic origin; such as peanut oil, soybean oil, mineral oil,
sesame oil, and the
like. Water or aqueous solution saline solutions and aqueous dextrose and
glycerol solutions are
preferably employed as carriers, in some embodiments as injectable solutions.
Alternatively, the
carrier can be a solid dosage form carrier, including but not limited to one
or more of a binder
(for compressed pills), a glidant, an encapsulating agent, a flavorant, and a
colorant. The choice
of carrier can be selected with regard to the intended route of administration
and standard
pharmaceutical practice. See Hardee and Baggo (1998. Development and
Formulation of
Veterinary Dosage Forms. 2nd Ed. CRC Press. 504 pg.); E.W. Martin (1970.
Remington's
Pharmaceutical Sciences. 17th Ed. Mack Pub. Co.); and Blaser et al. (US
Publication
US20110280840A1), each of which is herein expressly incorporated by reference
in their
entirety.
19
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[0068] The terms "microorganism" and "microbe" are used interchangeably herein
and refer to
any microorganism that is of the domain Bacteria, Eukarya or Archaea.
Microorganism types
include without limitation, bacteria (e.g., mycoplasma, coccus, bacillus,
rickettsia, spirillum),
fungi (e.g., filamentous fungi, yeast), nematodes, protozoans, archaea, algae,
dinoflagellates,
viruses (e.g., bacteriophages), viroids and/or a combination thereof. Organism
strains are
subtaxons of organism types, and can be for example, a species, sub-species,
subtype, genetic
variant, pathovar or serovar of a particular microorganism.
100691 The term "marker" or "unique marker" as used herein is an indicator of
unique
microorganism type, microorganism strain or activity of a microorganism
strain. A marker can
be measured in biological samples and includes without limitation, a nucleic
acid-based marker
such as a ribosomal RNA gene, a peptide- or protein-based marker, and/or a
metabolite or other
small molecule marker.
100701 The term "metabolite" as used herein is an intermediate or product of
metabolism. A
metabolite in one embodiment is a small molecule. Metabolites have various
functions, including
in fuel, structural, signaling, stimulatory and inhibitory effects on enzymes,
as a cofactor to an
enzyme, in defense, and in interactions with other organisms (such as
pigments, odorants and
pheromones). A primary metabolite is directly involved in normal growth,
development and
reproduction. A secondary metabolite is not directly involved in these
processes but usually has
an important ecological function. Examples of metabolites include but are not
limited to
antibiotics and pigments such as resins and terpenes, etc. Some antibiotics
use primary
metabolites as precursors, such as actinomycin which is created from the
primary metabolite,
tryptophan. Metabolites, as used herein, include small, hydrophilic
carbohydrates; large,
hydrophobic lipids and complex natural compounds.
[0071] Embodiments of the disclosure include diagnostic methods. As
illustrated in FIG. 1,
such a method can include obtaining at least two samples or data therefor
(011), the at least two
samples sharing at least one common environmental parameter (such as sample
type, sample
location, sample time, etc.). At least one of the at least two samples can be
defined as being in a
first state (013), and at least one of the at least two samples can be defined
as being in a second
state (015), the second state different from the first state. For example, in
one embodiment one of
the at least two states is a healthy state or a state associated with a
healthy sample source (e.g., a
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
sample source having one or more desirable characteristics or metadata), while
the other state is
an unhealthy/sick state or a state associated with an unhealthy/sickly sample
source (e.g., a
sample source having one or more undesirable characteristics or metadata, in
some instances,
especially when compared to the corresponding characteristic(s) or metadata of
a healthy sample
source). For each sample, the presence of one or more microorganism types in
the sample is
detected (017) and a number of each detected microorganism type of the one or
more
microorganism types in each sample is determined (019).
100721 Unique first markers in each sample, and quantity thereof, are then
measured (021),
each unique first marker being a marker of a microorganism strain of a
detected microorganism
type. The absolute cell count of each microorganism strain in each sample is
determined (023),
based on the number of each microorganism type and the number/respective
number of the
unique first markers. Then, at least one unique second marker for each
microorganism strain is
measured (025), and an activity level for that microorganism strain is
determined (027), e.g.,
based on the unique second marker exceeding a specified activity threshold.
Depending on the
implementation, the activity level can be numerical, relative, and/or binary
(e.g., active/inactive).
The absolute cell count of each microorganism strain is filtered by the
determined activity (029)
to provide a set or list of active microorganisms strains and their respective
absolute cell counts
for each of the at least two samples. The filtered absolute cell counts of
active microorganisms
strains for the at least one sample from the first state and the at least one
sample from the second
state can be compared or processed to define or determine a baseline state
(031), e.g., a healthy
state or normal state. The baseline state can be defined or characterized by
the presence or
absence of specified taxonomic groups and/or strains. In some embodiments, the
method
includes or further comprises obtaining at least one further sample (033), the
at least one further
sample having an unknown state. Then, for the at least one further sample, the
presence of one or
more microorganism types is detected (035) and a number of each detected
microorganism type
of the one or more microorganism types is determined (037). Unique first
markers, and quantity
thereof, are determined (039), each unique first marker being a marker of a
microorganism strain
of a detected microorganism type. The absolute cell count of each
microorganism strain is
determined (041) from the number of each microorganism type and the number of
the unique
first markers. At least one unique second marker is used, for each
microorganism strain based on
a specified threshold, to determine an activity level for that microorganism
strain (043). The
21
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
absolute cell count of each microorganism strain is filtered by the determined
activity level (045)
to provide a set or list of active microorganism strains and their respective
absolute cell counts
(047). The set of active microorganisms strains and their respective absolute
cell counts for the at
least one further sample is then compared to the baseline state to determine a
state of the at least
one further sample (049), e.g., healthy or unhealthy, normal or abnormal, etc.
The determined
state of the at least one further sample is then output and/or displayed
(051), e.g., on a display
screen or graphic interface.
100731 According to some further embodiments, the determined state of the at
least one further
sample corresponds to a state of an environment associated with the at least
one further sample.
Depending on the implementation, the environment associated with the at least
one further
sample can include a geospatial environment, such as a field or pasture, a
feed environment or
source (e.g., grain silo), a target animal and/or herd, etc. Treatments can be
identified or
determined for the environment associated with the at least one further
sample. In embodiments
where the baseline is healthy or the like, the treatment can be configured to
shift the state of the
environment toward the baseline. In some embodiments, the treatment can be
configured to shift
the state of the environment toward a state associated with desired goal or
favorable outcome.
The treatment can include a synthetic ensemble (especially a synthetic
ensemble formed
according to the methods of the disclosure), a chemical/biological treatment
or medicine, a
treatment regime, a combination of two or more of the preceding treatments,
and/or the like. In
some embodiments, the baseline state can be updated based on the at least one
further sample.
[0074] In another aspect of the disclosure, an analytical method is disclosed.
Such a method
can comprise obtaining at least two sample sets, each sample set including a
plurality of samples.
In some implementations, at least one sample set of the at least two sample
sets can be defined as
being in a first state, and at least one sample set of the at least two sample
sets can be defined as
being in a second state, wherein the first state is different from the second
state, and the range of
the sample in the sample set corresponds to the range of the state
corresponding to the sample
set. In other implementations, samples within the sample set are defined as
being in respective
states, or the state determination or definition is made post-analysis. The
method then includes
detecting a plurality of microorganism types in each sample, determining an
absolute number of
cells of each detected microorganism type of the plurality of microorganism
types in each
sample, and measuring unique first markers in each sample, and quantity
thereof, each unique
22
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
first marker being a marker of a microorganism strain of a detected
microorganism type. In some
embodiments, measuring unique first markers, and quantity thereof, includes at
least one of:
subjecting genomic DNA from each sample to a high throughput sequencing
reaction; and/or
subjecting genomic DNA from each sample to metagenome sequencing. The unique
first
markers can include at least one of an mRNA marker, an siRNA marker, a
ribosomal RNA
marker, a sigma factor, a transcription factor, a nucleoside associated
protein, and/or a metabolic
enzyme. In some embodiments, measuring unique first markers includes at least
one of
measuring unique genomic DNA markers in each sample, measuring unique RNA
markers in
each sample, and/or measuring unique protein markers in each sample. In some
embodiments,
measuring unique first markers includes measuring unique metabolite markers in
each sample,
which can include at least one of measuring unique lipid markers in each
sample and/or
measuring unique carbohydrate markers in each sample.
100751 The method includes then determining the absolute cell count of each
microorganism
strain present in each sample based on the number of each detected
microorganism types in that
sample and the number of unique first markers and quantity thereof in that
sample and measuring
at least one unique second marker for each microorganism strain to determine
active
microorganism strains in each sample. In some embodiments, measuring at least
one unique
second marker for each microorganism strain includes measuring a level of
expression of the at
least one unique second marker. In some embodiments, measuring the level of
expression of the
at least one unique second marker includes at least one of: subjecting sample
mRNA to gene
expression analysis; subjecting each sample or a portion thereof to mass
spectrometry analysis;
and/or subjecting each sample or a portion thereof to metaribosome profiling
or ribosome
profiling.
[0076] A set of active microorganisms strains and their respective absolute
cell counts is then
generated for each sample of the at least two sample sets. The method includes
analyzing the
active microorganisms strains and respective absolute cell counts for each
sample of the at least
two sample sets and/or respective samples to define a baseline state. The
baseline state can be, in
some embodiments, defined and/or characterized by the presence or absence of
specified
taxonomic groups and/or strains.
23
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[0077] Then, at least one further sample having an unknown state is obtained.
For the at least
one further sample, the method further includes: (a) detecting the presence of
one or more
microorganism types; (b) determining a number of each detected microorganism
type; (c)
measuring unique first markers, and quantity thereof, each unique first marker
being a marker of
a microorganism strain of a detected microorganism type; (d) determining the
absolute cell count
of each microorganism strain from the number of each microorganism type and
the number of
the unique first markers; (e) measuring at least one unique second marker for
each
microorganism strain based on a specified threshold to determine an activity
level for that
microorganism strain; and (f) filtering the absolute cell count of each
microorganism strain by
the determined activity to provide a set of active microorganisms strains and
their respective
absolute cell counts. The set of active microorganisms strains and their
respective absolute cell
counts for the at least one further sample is compared to the baseline state
to determine a state
associated with the at least one further sample, and the determined state
associated with the at
least one further sample is displayed or output. While generally discussed as
a singular state, it
should be understood that for some embodiments and applications, a baseline
state or biostate
can refer to multiple states and/or biostates associated with a particular
microbiome, and multiple
states can also be utilized in characterizing, identifying, and/or treating
particular indications,
whether on an individual or herd level.
[0078] The method can further comprise selecting a plurality of active
microorganism strains
based on the baseline state and the determined state associated with the at
least one further
sample, and combining the selected plurality of active microorganism strains
with a carrier
medium to form a synthetic ensemble of active microorganisms configured to be
introduced to
an environment associated with the at least one further sample and modify a
state of the
environment associated with associated with the at least one further sample.
[0079] In one aspect of the disclosure, a method for identifying relationships
between a
plurality of microorganism strains and one or more metadata and/or parameters
is disclosed. As
illustrated in FIG. 1A, samples and/or sample data for at least two samples is
received from at
least two sample sources 101, and for each sample, the presence of one or more
microorganism
types is determined 103. The number (cell count) of each detected
microorganism type of the one
or more microorganism types in each sample is determined 105, and a number of
unique first
markers in each sample, and quantity thereof is determined 107, each unique
first marker being a
24
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
marker of a microorganism strain. The number of each microorganism type and
the number of
the first markers is integrated to yield the absolute cell count of each
microorganism strain
present in each sample 109, and an activity level for each microorganism
strain in each sample is
determined 111 based on a measure of at least one unique second marker for
each
microorganism strain exceeding a specified threshold, a microorganism strain
being identified as
active if the measure of at least one unique second marker for that strain
exceeds the
corresponding threshold. The absolute cell count of each microorganism strain
is then filtered by
the determined activity to provide a set or list of active microorganisms
strains and their
respective absolute cell counts for each of the at least two samples 113. A
network analysis of
the set or list of filtered absolute cell counts of active microorganisms
strains for each of the at
least two samples with at least one measured metadata or additional active
microorganism strain
is conducted 115, the network analysis including determining maximal
information coefficient
scores between each active microorganism strain and every other active
microorganism strain
and determining maximal information coefficient scores between each active
microorganism
strain and the at least one measured metadata or additional active
microorganism strain. The
active microorganism strains can then be categorized based on function,
predicted function
and/or chemistry 117, and a plurality of active microorganism strains
identified and output based
on the categorization 119. In some embodiments, the method further comprises
assembling an
active microorganism ensemble from the identified plurality of microorganism
strains 121, the
microorganism ensemble configured to, when applied to a target, alter a
property corresponding
to the at least one measured metadata. The method can further comprise
identifying at least one
pathogen based on the output plurality of identified active microorganism
strains (see Example 4
for additional detail). In some embodiments, the plurality of active
microorganism strains can be
utilized to assemble an active microorganism ensemble that is configured to,
when applied to a
target, address the at least one identified pathogen and/or treat a symptom
associated with the at
least one identified pathogen.
100801 In one aspect of the disclosure, a method for determining the absolute
cell count of one
or more active microorganism strains in a sample or plurality of samples is
provided, wherein the
one or more active microorganism strains are present in a microbial community
in the sample.
The one or more microorganism strains is a subtaxon of one or more organism
types (see method
1000 at FIG. 1B). For each sample, the presence of one or more microorganism
types in the
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
sample is detected (1001). The absolute number of each of the one or more
organism types in
the sample is determined (1002). The number of unique first markers is
measured along with the
quantity of each of the unique first markers (1003). As described herein, a
unique first marker is
a marker of a unique microorganism strain. Activity is then assessed at the
protein and/or RNA
level by measuring the level of expression of one or more unique second
markers (1004). The
unique second marker can be the same or different as the first unique marker,
and is a marker of
activity of an organism strain. Based on the level of expression of one or
more of the unique
second markers, a determination is made which (if any) microorganism strains
are active (1005).
A microorganism strain is considered active if it expresses the second unique
marker at a
particular level, or above a threshold level (1005), for example, at least
about 10%, at least about
20%, at least about 30% or at least about 40% above a threshold level (it is
to be understood that
the various thresholds can be determined based on the particular application
and/or
implementation, for example, thresholds can vary by sample source(s), such as
a particular
species, sample origin location, metadata of interest, environment, etc.). The
absolute cell count
of the one or more active microorganism strains can be determined based upon
the quantity of
the one or more first markers of the one or more active microorganism strains
and the absolute
number of the organism types from which the one or more microorganism strains
is a subtaxon.
[0081] Some embodiments of the disclosure can be configured for analyzing
microbial
communities. As illustrated by FIG. 1C, data for two or more samples (and/or
sample sets) are
obtained (1051), each sample including a heterogeneous microbial community,
and a plurality of
microorganism types is detected in each sample (1053). An absolute number of
cells of each
detected microorganism type of the plurality of microorganism types in each
sample is then
determined (1055), e.g., via FACS or other methods as discussed herein. Unique
first markers in
each sample, and quantity thereof, are measured (1057), each unique first
marker being a marker
of a microorganism strain of a detected microorganism type. A value (activity,
concentration,
expression, etc.) of one or more unique second markers is measured (1059), a
unique second
marker indicative of activity (e.g., metabolic activity) of a particular
microorganism strain of a
detected microorganism type, and the activity of each detected microorganism
strain is
determined (1061), based on the measured value of the one or more unique
second markers (e.g.,
based on the value exceeding a specified set threshold). The respective ratios
of each active
detected microorganism strain in each sample are determined (1063), e.g.,
based on the
26
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
respective absolute cell counts, values, etc. Then each of the active detected
microorganism
strains (or a subset thereof) of the at least two samples are analyzed to
identify a biostate, such as
a baseline state, and/or relationships and the strengths thereof (1065)
between and among each
active detected microorganism strain and the other active detected
microorganism strains, and
between each active detected microorganism strain and at least one measured
metadata. The
identified biostate and/or relationships are then displayed or otherwise
output (1067), e.g., on a
graphical display/interface (e.g., FIG. 1D), and can be utilized for biostate
management and/or
generation of a bioensemble (1069). In some embodiments, the display/output of
relationships
can be limited such that only relationships that exceed a certain strength or
weight are displayed
(1066a, 1066b).
100821 Microbial ensembles according to the disclosure can be selected from
sets, subsets,
and/or groupings of active, interrelated individual microbial species, or
strains of a species. The
relationships and networks, as identified by methods of the disclosure, are
grouped and/or linked
based on carrying out one or more a common functions, or can be described as
participating in,
or leading to, or associated with, a recognizable parameter, such as a
phenotypic trait of interest
(e.g. increased milk production in a ruminant). In FIG. 1D, the Louvain
community detection
method was used to identify groups associated with dairy cow-relevant metadata
parameters.
Each node represents a specific rumen microorganism strain or a metadata
parameter. The links
between nodes represent significant relationships. Unconnected nodes are
irrelevant
microoganisms. Each colored "bubble" represents a group detected by the
Louvain analysis. This
grouping allows for prediction of the functionality of strains based on the
groups they fall into.
10083.1 Some embodiments of the disclosure are configured to leverage mutual
information to
rank the importance of native microbial strains residing in the
gastrointestinal tract of the animal
to specific animal traits. The maximal information coefficient (MIC) is
calculated for all
microorganisms and the desired animal trait. Relationships are scored on a
scale of 0 to 1, with 1
representing a strong relationship between the microbial strain and animal
trait and 0
representing no relationship. A cut-off based on this score is used to define
useful and non-useful
microorganisms with respect to the improvement of specific traits. FIGs. 1E
and 1F depict
examples of MIC score distributions for rumen microbial strains that share a
relationship with
milk fat efficiency. Here, the point where the curve shifts from exponential
to linear (-0.45-0.5
27
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
for bacteria, and -0.3 for fungi) represents the cut off between useful and
non-useful
microorganism strains. FIGS. 1G and 1H depict examples of MIC score
distributions for rumen
microbial strains that share a relationship with dairy efficiency. The point
where the curve shifts
from exponential to linear (-0.45-0.5 for bacteria, and -0.25 for fungi)
represents the cut off
between useful and non-useful microorganism strains.
[0084] As provided in FIG. 2, in another aspect of the disclosure, the
absolute cell count of one
or more active microorganisms is determined in a plurality of samples, and the
absolute cell
count is related to a metadata (environmental parameter) (2001-2008). A
plurality of samples
are subjected to analysis for the absolute cell count of one or more active
microorganism strains,
wherein the one or more active microorganism strains is considered active if
an activity
measurement is at a threshold level or above a threshold level in at least one
of the plurality of
samples (2001-2006). The absolute cell count of the one or more active
microorganism strains is
then related to a metadata parameter of the particular implementation and/or
application (2008).
100851 In one embodiment, the plurality of samples is collected over time from
the same
environmental source (e.g., the same animal over a time course). In another
embodiment, the
plurality of samples is from a plurality of environmental sources (e.g.,
different animals). In one
embodiment, the environmental parameter is the absolute cell count of a second
active
microorganism strain. In a further embodiment, the absolute cell count values
of the one or more
active microorganism strains is used to determine the co-occurrence of the one
or more active
microorganism strains, with a second active microorganism strain of the
microbial community.
In a further embodiment, a second environmental parameter is related to the
absolute cell count
of the one or more active microorganism strains and/or the absolute cell count
of the second
environmental strain.
[0086] Aspects of the disclosed embodiments are discussed throughout the
disclosure.
[0087] The samples for use with the methods provided herein importantly can be
of any type
that includes a microbial community. For example, samples for use with the
methods provided
herein encompass without limitation, an animal sample (e.g., mammal, reptile,
bird), soil, air,
water (e.g., marine, freshwater, wastewater sludge), sediment, oil, plant,
agricultural product,
plant, soil (e.g., rhizosphere), food (e.g. cheese, beer, wine, bread), and
extreme environmental
sample (e.g., acid mine drainage, hydrothermal systems). In the case of marine
or freshwater
28
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
samples, the sample can be from the surface of the body of water, or any depth
of the body
water, e.g., a deep sea sample. The water sample, in one embodiment, is an
ocean, river or lake
sample.
10088.1 The animal sample in one embodiment is a body fluid. In another
embodiment, the
animal sample is a tissue sample. Non-limiting animal samples include tooth,
perspiration,
fingernail, skin, hair, feces, urine, semen, mucus, saliva, gastrointestinal
tract. The animal
sample can be, for example, a human, primate, bovine, porcine, canine, feline,
rodent (e.g.,
mouse or rat), or bird sample. In one embodiment, the bird sample comprises a
sample from one
or more chickens. In another embodiment, the sample is a human sample. The
human
microbiome comprises the collection of microorganisms found on the surface and
deep layers of
skin, in mammary glands, saliva, oral mucosa, conjunctiva and gastrointestinal
tract. The
microorganisms found in the microbiome include bacteria, fungi, protozoa,
viruses and archaea.
Different parts of the body exhibit varying diversity of microorganisms. The
quantity and type
of microorganisms may signal a healthy or diseased state for an individual.
The number of
bacteria taxa are in the thousands, and viruses may be as abundant. The
bacterial composition
for a given site on a body varies from person to person, not only in type, but
also in abundance or
quantity.
[0089] In another embodiment, the sample is a ruminal sample. Ruminants such
as cattle rely
upon diverse microbial communities to digest their feed. These animals have
evolved to use feed
with poor nutritive value by having a modified upper digestive tract
(reticulorumen or rumen)
where feed is held while it is fermented by a community of anaerobic microbes.
The rumen
microbial community is very dense, with about 3 x 1010 microbial cells per
milliliter. Anaerobic
fermenting microbes dominate in the rumen. The rumen microbial community
includes
members of all three domains of life: Bacteria, Archasa, and Eukarya. Ruminal
fermentation
products are required by their respective hosts for body maintenance and
growth, as well as milk
production (van Houtert (1993). Anim. Feed Sci. Technol. 43, pp. 189-225;
Bauman et a/.
(2011). Annu. Rev. Nutr. 31, pp. 299-319; each incorporated by reference in
its entirety for all
purposes). Moreover, milk yield and composition has been reported to be
associated with
ruminal microbial communities (Sandri et al. (2014). Animal 8, pp. 572-579;
Palmonari et al.
(2010). J. Dairy Sci. 93, pp. 279-287; each incorporated by reference in its
entirety for all
purposes). Ruminal samples, in one embodiment, are collected via the process
described in
29
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Jewell et al. (2015). App!. Environ. Microbiol. 81, pp. 4697-4710,
incorporated by reference
herein in its entirety for all purposes.
[0090] In another embodiment, the sample is a soil sample (e.g., bulk soil or
rhizosphere
sample). It has been estimated that 1 gram of soil contains tens of thousands
of bacterial taxa,
and up to 1 billion bacteria cells as well as about 200 million fungal hyphae
(Wagg et al. (2010).
Proc Natl. Acad. Sci. USA 111, pp. 5266-5270, incorporated by reference in its
entirety for all
purposes). Bacteria, actinomycetes, fungi, algae, protozoa and viruses are all
found in soil. Soil
microorganism community diversity has been implicated in the structure and
fertility of the soil
microenvironment, nutrient acquisition by plants, plant diversity and growth,
as well as the
cycling of resources between above- and below-ground communities. Accordingly,
assessing
the microbial contents of a soil sample over time and the co-occurrence of
active microorganisms
(as well as the number of the active microorganisms) provides insight into
microorganisms
associated with an environmental metadata parameter such as nutrient
acquisition and/or plant
diversity.
[0091] The soil sample in one embodiment is a rhizosphere sample, i.e., the
narrow region of
soil that is directly influenced by root secretions and associated soil
microorganisms. The
rhizosphere is a densely populated area in which elevated microbial activities
have been
observed and plant roots interact with soil microorganisms through the
exchange of nutrients and
growth factors (San Miguel et al. (2014). App!. Microbiol. Biotechnol. DOI
10.1007/s00253-
014-5545-6, incorporated by reference in its entirety for all purposes). As
plants secrete many
compounds into the rhizosphere, analysis of the organism types in the
rhizosphere may be useful
in determining features of the plants which grow therein.
[0092] In another embodiment, the sample is a marine or freshwater sample.
Ocean water
contains up to one million microorganisms per milliliter and several thousand
microbial types.
These numbers may be an order of magnitude higher in coastal waters with their
higher
productivity and higher load of organic matter and nutrients. Marine
microorganisms are crucial
for the functioning of marine ecosystems; maintaining the balance between
produced and fixed
carbon dioxide; production of more than 50% of the oxygen on Earth through
marine
phototrophic microorganisms such as Cyanobacteria, diatoms and pico- and
nanophytoplankton;
providing novel bioactive compounds and metabolic pathways; ensuring a
sustainable supply of
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
seafood products by occupying the critical bottom trophic level in marine
foodwebs. Organisms
found in the marine environment include viruses, bacteria, archaea and some
eukarya. Marine
viruses may play a significant role in controlling populations of marine
bacteria through viral
lysis. Marine bacteria are important as a food source for other small
microorganisms as well as
being producers of organic matter. Archaea found throughout the water column
in the ocean are
pelagic Archaea and their abundance rivals that of marine bacteria.
100931 In another embodiment, the sample comprises a sample from an extreme
environment,
i.e., an environment that harbors conditions that are detrimental to most life
on Earth. Organisms
that thrive in extreme environments are called extremophiles. Though the
domain Archaea
contains well-known examples of extremophiles, the domain bacteria can also
have
representatives of these microorganisms. Extremophiles include: acidophiles
which grow at pH
levels of 3 or below; alkaliphiles which grow at pH levels of 9 or above;
anaerobes such as
Spinolorkus Cinzia which does not require oxygen for growth; cryptoendoliths
which live in
microscopic spaces within rocks, fissures, aquifers and faults filled with
groundwater in the deep
subsurface; halophiles which grow in about at least 0.2M concentration of
salt;
hyperthermophiles which thrive at high temperatures (about 80-122 C) such as
found in
hydrothermal systems; hypoliths which live underneath rocks in cold deserts;
lithoautotrophs
such as Nitrosomonas ettropaea which derive energy from reduced mineral
compounds like
pyrites and are active in geochemical cycling; metallotolerant organisms which
tolerate high
levels of dissolved heavy metals such as copper, cadmium, arsenic and zinc;
oligotrophs which
grow in nutritionally limited environments; osmophiles which grow in
environments with a high
sugar concentration; piezophiles (or barophiles) which thrive at high
pressures such as found
deep in the ocean or underground; psychrophiles/cryophiles which survive, grow
and/or
reproduce at temperatures of about -15 C or lower; radioresistant organisms
which are resistant
to high levels of ionizing radiation; thermophiles which thrive at
temperatures between 45-122
C; xerophiles which can grow in extremely dry conditions. Polyextremophiles
are organisms
that qualify as extremophiles under more than one category and include
thermoacidophiles
(prefer temperatures of 70-80 C and pH between 2 and 3). The Crenarchaeota
group of Archaea
includes the thermoacidophiles.
10094] The sample can include microorganisms from one or more domains. For
example, in
one embodiment, the sample comprises a heterogeneous population of bacteria
and/or fungi (also
31
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
referred to herein as bacterial or fungal strains). Additional applications of
teaching of the
disclosure include use in foods, especially fermented foods and microbial
foods, e.g., breads,
cheese, wine, beer, kimchi, kombucha, chocolates, etc.
[0095] In the methods provided herein for determining the presence and
absolute cell count of
one or more microorganisms in a sample, for example the absolute cell count of
one or more
microorganisms in a plurality of samples collected from the same or different
environments,
and/or over multiple time points, the one or more microorganisms can be of any
type. For
example, the one or more microorganisms can be from the domain Bacteria,
Archaea, Eukarya or
a combination thereof. Bacteria and Archaea are prokaryotic, having a very
simple cell structure
with no internal organelles. Bacteria can be classified into gram positive/no
outer membrane,
gram negative/outer membrane present and ungrouped phyla. Archaea constitute a
domain or
kingdom of single-celled microorganisms. Although visually similar to
bacteria, archaea possess
genes and several metabolic pathways that are more closely related to those of
eukaryotes,
notably the enzymes involved in transcription and translation. Other aspects
of archaeal
biochemistry are unique, such as the presence of ether lipids in their cell
membranes. The
Archaea are divided into four recognized phyla: Thaumarchaeota, Aigarchaeota,
Crenarchaeota
and Korarchaeota.
[0096] The domain of Eukarya comprises eukaryotic organisms, which are defined
by
membrane-bound organelles, such as the nucleus. Protozoa are unicellular
eukaryotic organisms.
All multicellular organisms are eukaryotes, including animals, plants and
fungi. The eukaryotes
have been classified into four kingdoms: Protista, Plantae, Fungi and
Animalia. However,
several alternative classifications exist. Another classification divides
Eukarya into six
kingdoms: Excavata (various flagellate protozoa); amoebozoa (lobose amoeboids
and slime
filamentous fungi); Opisthokonta (animals, fungi, choanoflagellates); Rhizaria
(Foraminifera,
Radiolaria, and various other amoeboid protozoa); Chromalveolata
(Stramenopiles (brown algae,
diatoms), Haptophyta, Cryptophyta (or cryptomonads),
and Al veolata);
Archaeplastida/Primoplantae (Land plants, green algae, red algae, and
glaucophytes).
[0097] Within the domain of Eukarya, fungi are microorganisms that are
predominant in
microbial communities. Fungi include microorganisms such as yeasts and
filamentous fungi as
well as the familiar mushrooms. Fungal cells have cell walls that contain
glucans and chitin, a
32
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
unique feature of these organisms. The fungi form a single group of related
organisms, named
the Eutnycota that share a common ancestor. The kingdom Fungi has been
estimated at 1.5
million to 5 million species, with about 5% of these having been formally
classified. The cells of
most fungi grow as tubular, elongated, and filamentous structures called
hyphae, which may
contain multiple nuclei. Some species grow as unicellular yeasts that
reproduce by budding or
binary fission. The major phyla (sometimes called divisions) of fungi have
been classified
mainly on the basis of characteristics of their sexual reproductive
structures. Currently, seven
phyla are proposed: Microsporidia,
Chytridiomycota, Blastocladiomycota,
Neocallimastigomycota, Glomeromycota, Ascomycota, and Basidiomycota.
100981 Microorganisms for detection and quantification by the methods
described herein can
also be viruses. A virus is a small infectious agent that replicates only
inside the living cells of
other organisms. Viruses can infect all types of life forms in the domains of
Eukarya, Bacteria
and Archaea. Virus particles (known as virions) consist of two or three parts:
(i) the genetic
material which can be either DNA or RNA; (ii) a protein coat that protects
these genes; and in
some cases (iii) an envelope of lipids that surrounds the protein coat when
they are outside a cell.
Seven orders have been established for viruses: the Caudovirales,
Herpesvirales,
Ligamenvirales, Mononegavirales, Nidovirales, Picornavirales, and Tymovirales.
Viral
genomes may be single-stranded (ss) or double-stranded (ds), RNA or DNA, and
may or may not
use reverse transcriptase (RT). In addition, ssRNA viruses may be either sense
(+) or antisense
(¨). This classification places viruses into seven groups: I: dsDNA viruses
(such as
Adenoviruses. Herpesviruses, Poxviruses); (+) ssDNA viruses (such as
Parvoviruses);
dsRNA viruses (such as Reoviruses); IV: (+)ssRNA viruses (such as
Picornaviruses,
Togaviruses); V: (¨)ssRNA viruses (such as Orthomyxoviruses, Rhabdoviruses);
VI: (+)ssRNA-
RT viruses with DNA intermediate in life-cycle (such as Retroviruses); VII:
dsDNA-RT viruses
(such as Hepadnaviruses).
[0099] Microorganisms for detection and quantification by the methods
described herein can
also be viroids. Viroids are the smallest infectious pathogens known,
consisting solely of short
strands of circular, single-stranded RNA without protein coats. They are
mostly plant pathogens,
some of which are of economical importance. Viroid genomes are extremely small
in size,
ranging from about 246 to about 467 nucleobases.
33
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[001001 According to the methods provided herein, a sample is processed to
detect the presence
of one or more microorganism types in the sample (FIG. 1B, 1001; FIG. 2,
2001). The absolute
number of one or more microorganism organism type in the sample is determined
(FIG. 1B,
1002; FIG. 2, 2002). The determination of the presence of the one or more
organism types and
the absolute number of at least one organism type can be conducted in parallel
or serially. For
example, in the case of a sample comprising a microbial community comprising
bacteria (i.e.,
one microorganism type) and fungi (i.e., a second microorganism type), the
user in one
embodiment detects the presence of one or both of the organism types in the
sample (FIG. 1B,
1001; FIG. 2, 2001). The user, in a further embodiment, determines the
absolute number of at
least one organism type in the sample ¨ in the case of this example, the
number of bacteria, fungi
or combination thereof, in the sample (FIG. 1B, 1002; FIG. 2, 2002).
[00101] In one embodiment, the sample, or a portion thereof is subjected to
flow cytometry
(FC) analysis to detect the presence and/or number of one or more
microorganism types (FIG.
1B, 1001, 1002; FIG. 2, 2001, 2002). In one flow cytometer embodiment,
individual microbial
cells pass through an illumination zone, at a rate of at least about 300 *s-1,
or at least about 500
*s-1, or at least about 1000 *s-1. However, it should be recognized that this
rate can vary
depending on the type of instrument is employed. Detectors which are gated
electronically
measure the magnitude of a pulse representing the extent of light scattered.
The magnitudes of
these pulses are sorted electronically into "bins" or "channels," permitting
the display of
histograms of the number of cells possessing a certain quantitative property
(e.g., cell staining
property, diameter, cell membrane) versus the channel number. Such analysis
allows for the
determination of the number of cells in each "bin" which in embodiments
described herein is an
"microorganism type" bin, e.g., a bacteria, fungi, nematode, protozoan,
archaea, algae,
dinoflagellate, virus, viroid, etc.
[00102] In one embodiment, a sample is stained with one or more fluorescent
dyes wherein a
fluorescent dye is specific to a particular microorganism type, to enable
detection via a flow
cytometer or some other detection and quantification method that harnesses
fluorescence, such as
fluorescence microscopy. The method can provide quantification of the number
of cells and/or
cell volume of a given organism type in a sample. In a further embodiment, as
described herein,
flow cytometry is harnessed to determine the presence and quantity of a unique
first marker
and/or unique second marker of the organism type, such as enzyme expression,
cell surface
34
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
protein expression, etc. Two- or three-variable histograms or contour plots
of, for example, light
scattering versus fluorescence from a cell membrane stain (versus fluorescence
from a protein
stain or DNA stain) can also be generated, and thus an impression may be
gained of the
distribution of a variety of properties of interest among the cells in the
population as a whole. A
number of displays of such multiparameter flow cytometric data are in common
use and are
amenable for use with the methods described herein.
1001031 In one embodiment of processing the sample to detect the presence and
number of one
or more microorganism types, a microscopy assay is employed (FIG. 1B, 1001,
1002). In one
embodiment, the microscopy is optical microscopy, where visible light and a
system of lenses
are used to magnify images of small samples. Digital images can be captured by
a charge-couple
device (CCD) camera. Other microscopic techniques include, but are not limited
to, scanning
electron microscopy and transmission electron microscopy. Microorganism types
are visualized
and quantified according to the aspects provided herein.
(001041 In another embodiment of the disclosure, in order to detect the
presence and number of
one or more microorganism types, each sample, or a portion thereof is
subjected to fluorescence
microscopy. Different fluorescent dyes can be used to directly stain cells in
samples and to
quantify total cell counts using an epifluorescence microscope as well as flow
cytometry,
described above. Useful dyes to quantify microorganisms include but are not
limited to acridine
orange (AO), 4,6-di-amino-2 phenylindole (DAPI) and 5-cyano-2,3 Dytolyl
Tetrazolium
Chloride (CTC). Viable cells can be estimated by a viability staining method
such as the
LIVE/DEAD Bacterial Viability Kit (Bac-LightTM) which contains two nucleic
acid stains: the
green-fluorescent SYTO 9TM dye penetrates all membranes and the red-
fluorescent propidium
iodide (PI) dye penetrates cells with damaged membranes. Therefore, cells with
compromised
membranes will stain red, whereas cells with undamaged membranes will stain
green.
Fluorescent in situ hybridization (FISH) extends epifluorescence microscopy,
allowing for the
fast detection and enumeration of specific organisms. FISH uses fluorescent
labelled
oligonucleotides probes (usually 15-25 basepairs) which bind specifically to
organism DNA in
the sample, allowing the visualization of the cells using an epifluorescence
or confocal laser
scanning microscope (CLSM). Catalyzed reporter deposition fluorescence in situ
hybridization
(CARD-FISH) improves upon the FISH method by using oligonucleotide probes
labelled with a
horse radish peroxidase (HRP) to amplify the intensity of the signal obtained
from the
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
microorganisms being studied. FISH can be combined with other techniques to
characterize
microorganism communities. One combined technique is high affinity peptide
nucleic acid
(PNA)-FISH, where the probe has an enhanced capability to penetrate through
the Extracellular
Polymeric Substance (EPS) matrix. Another example is LIVE/DEAD-FISH which
combines the
cell viability kit with FISH and has been used to assess the efficiency of
disinfection in drinking
water distribution systems.
[00105] In another embodiment, each sample, or a portion thereof is subjected
to Raman micro-
spectroscopy in order to determine the presence of a microorganism type and
the absolute
number of at least one microorganism type (FIG. 1B, 1001-1002; FIG. 2, 2001-
2002). Raman
micro-spectroscopy is a non-destructive and label-free technology capable of
detecting and
measuring a single cell Raman spectrum (SCRS). A typical SCRS provides an
intrinsic
biochemical "fingerprint" of a single cell. A SCRS contains rich information
of the
biomolecules within it, including nucleic acids, proteins, carbohydrates and
lipids, which enables
characterization of different cell species, physiological changes and cell
phenotypes. Raman
microscopy examines the scattering of laser light by the chemical bonds of
different cell
biomarkers. A SCRS is a sum of the spectra of all the biomolecules in one
single cell, indicating
a cell's phenotypic profile. Cellular phenotypes, as a consequence of gene
expression, usually
reflect genotypes. Thus, under identical growth conditions, different
microorganism types give
distinct SCRS corresponding to differences in their genotypes and can thus be
identified by their
Raman spectra.
[00106] In yet another embodiment, the sample, or a portion thereof is
subjected to
centrifugation in order to determine the presence of a microorganism type and
the number of at
least one microorganism type (FIG. 1B, 1001-1002; FIG. 2, 2001-2002). This
process sediments
a heterogeneous mixture by using the centrifugal force created by a
centrifuge. More dense
components of the mixture migrate away from the axis of the centrifuge, while
less dense
components of the mixture migrate towards the axis. Centrifugation can allow
fractionation of
samples into cytoplasmic, membrane and extracellular portions. It can also be
used to determine
localization information for biological molecules of interest. Additionally,
centrifugation can be
used to fractionate total microbial community DNA. Different prokaryotic
groups differ in their
guanine-plus-cytosine (G+C) content of DNA, so density-gradient centrifugation
based on G+C
content is a method to differentiate organism types and the number of cells
associated with each
36
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
type. The technique generates a fractionated profile of the entire community
DNA and indicates
abundance of DNA as a function of G+C content. The total community DNA is
physically
separated into highly purified fractions, each representing a different G+C
content that can be
analyzed by additional molecular techniques such as denaturing gradient gel
electrophoresis
(DGGE)/amplified ribosomal DNA restriction analysis (ARDRA) (see discussion
herein) to
assess total microbial community diversity and the presence/quantity of one or
more
microorganism types.
1001071 In another embodiment, the sample, or a portion thereof is subjected
to staining in order
to determine the presence of a microorganism type and the number of at least
one microorganism
type (FIG. 1B, 1001-1002; FIG. 2, 2001-2002). Stains and dyes can be used to
visualize
biological tissues, cells or organelles within cells. Staining can be used in
conjunction with
microscopy, flow cytometry or gel electrophoresis to visualize or mark cells
or biological
molecules that are unique to different microorganism types. In vivo staining
is the process of
dyeing living tissues, whereas in vitro staining involves dyeing cells or
structures that have been
removed from their biological context. Examples of specific staining
techniques for use with the
methods described herein include, but are not limited to: gram staining to
determine gram status
of bacteria, endospore staining to identify the presence of endospores, Ziehl-
Neelsen staining,
haematoxylin and eosin staining to examine thin sections of tissue,
papanicolaou staining to
examine cell samples from various bodily secretions, periodic acid-Schiff
staining of
carbohydrates, Masson's trichome employing a three-color staining protocol to
distinguish cells
from the surrounding connective tissue, Romanowsky stains (or common variants
that include
Wright's stain, Jenner's stain, May-Grunwald stain, Leishman stain and Giemsa
stain) to examine
blood or bone marrow samples, silver staining to reveal proteins and DNA,
Sudan staining for
lipids and Conklin's staining to detect true endospores. Common biological
stains include
acridine orange for cell cycle determination; bismarck brown for acid mucins;
carmine for
glycogen; carmine alum for nuclei; Coomassie blue for proteins; Cresyl violet
for the acidic
components of the neuronal cytoplasm; Crystal violet for cell walls; DAPI for
nuclei; eosin for
cytoplasmic material, cell membranes, some extracellular structures and red
blood cells;
ethidium bromide for DNA; acid fuchsine for collagen, smooth muscle or
mitochondria;
haematoxylin for nuclei; Hoechst stains for DNA; iodine for starch; malachite
green for bacteria
in the Gimenez staining technique and for spores; methyl green for chromatin;
methylene blue
37
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
for animal cells; neutral red for Nissl substance; Nile blue for nuclei; Nile
red for lipohilic
entities; osmium tetroxide for lipids; rhodamine is used in fluorescence
microscopy; safranin for
nuclei. Stains are also used in transmission electron microscopy to enhance
contrast and include
phosphotungstic acid, osmium tetroxide, ruthenium tetroxide, ammonium
molybdate, cadmium
iodide, carbohydrazide, ferric chloride, hexamine, indium trichloride,
lanthanum nitrate, lead
acetate, lead citrate, lead(11) nitrate, periodic acid, phosphomolybdic acid,
potassium
ferricyanide, potassium ferrocyanide, ruthenium red, silver nitrate, silver
proteinate, sodium
chloroaurate, thallium nitrate, thiosemicarbazide, uranyl acetate, uranyl
nitrate, and vanadyl
sulfate.
1001081 In another embodiment, the sample, or a portion thereof is subjected
to mass
spectrometry (MS) in order to determine the presence of a microorganism type
and the number
of at least one microorganism type (FIG. 1B, 1001-1002; FIG. 2, 2001-2002).
MS, as discussed
below, can also be used to detect the presence and expression of one or more
unique markers in a
sample (FIG. 1B, 1003-1004; FIG. 2, 2003-2004). MS is used for example, to
detect the
presence and quantity of protein and/or peptide markers unique to
microorganism types and
therefore to provide an assessment of the number of the respective
microorganism type in the
sample. Quantification can be either with stable isotope labelling or label-
free. De novo
sequencing of peptides can also occur directly from MS/MS spectra or sequence
tagging
(produce a short tag that can be matched against a database). MS can also
reveal post-
translational modifications of proteins and identify metabolites. MS can be
used in conjunction
with chromatographic and other separation techniques (such as gas
chromatography, liquid
chromatography, capillary electrophoresis, ion mobility) to enhance mass
resolution and
determination.
[001091 In another embodiment, the sample, or a portion thereof is subjected
to lipid analysis in
order to determine the presence of a microorganism type and the number of at
least one
microorganism type (FIG. 1B, 1001-1002; FIG. 2, 2001-2002). Fatty acids are
present in a
relatively constant proportion of the cell biomass, and signature fatty acids
exist in microbial
cells that can differentiate microorganism types within a community. In one
embodiment, fatty
acids are extracted by saponification followed by derivatization to give the
respective fatty acid
methyl esters (FAMEs), which are then analyzed by gas chromatography. The FAME
profile in
38
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
one embodiment is then compared to a reference FAME database to identify the
fatty acids and
their corresponding microbial signatures by multivariate statistical analyses.
1001101 In the aspects of the methods provided herein, the number of unique
first makers in the
sample, or portion thereof (e.g., sample aliquot) is measured, as well as the
quantity of each of
the unique first markers (FIG. 1B, 1003; FIG. 2, 2003). A unique marker is a
marker of a
microorganism strain. It should be understood that depending on the unique
marker being
probed for and measured, the entire sample need not be analyzed. For example,
if the unique
marker is unique to bacterial strains, then the fungal portion of the sample
need not be analyzed.
As described above, in some embodiments, measuring the absolute cell count of
one or more
organism types in a sample comprises separating the sample by organism type,
e.g., via flow
cytometry.
[00111] Any marker that is unique to an organism strain can be employed
herein. For example,
markers can include, but are not limited to, small subunit ribosomal RNA genes
(16S/18S
rDNA), large subunit ribosomal RNA genes (23S/25S/28S rDNA), intercalary 5.8S
gene,
cytochrome c oxidase, beta-tubulin, elongation factor, RNA polymerase and
internal transcribed
spacer (ITS).
[00112] Ribosomal RNA genes (rDNA), especially the small subunit ribosomal RNA
genes,
i.e., 18S rRNA genes (18S rDNA) in the case of eukaryotes and 16S rRNA (16S
rDNA) in the
case of prokaryotes, have been the predominant target for the assessment of
organism types and
strains in a microbial community. However, the large subunit ribosomal RNA
genes, 28S
rDNAs, have been also targeted. rDNAs are suitable for taxonomic
identification because: (i)
they are ubiquitous in all known organisms; (ii) they possess both conserved
and variable
regions; (iii) there is an exponentially expanding database of their sequences
available for
comparison. In community analysis of samples, the conserved regions serve as
annealing sites
for the corresponding universal PCR and/or sequencing primers, whereas the
variable regions
can be used for phylogenetic differentiation. In addition, the high copy
number of rDNA in the
cells facilitates detection from environmental samples.
[00113] The internal transcribed spacer (ITS), located between the 18S rDNA
and 28S rDNA,
has also been targeted. The ITS is transcribed but spliced away before
assembly of the
ribosomes. The ITS region is composed of two highly variable spacers, ITS1 and
ITS2, and the
39
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
intercalary 5.8S gene. This rDNA operon occurs in multiple copies in genomes.
Because the
ITS region does not code for ribosome components, it is highly variable.
1.001141 In one embodiment, the unique RNA marker can be an mRNA marker, an
siRNA
marker or a ribosomal RNA marker.
1.001151 Protein-coding functional genes can also be used herein as a unique
first marker. Such
markers include but are not limited to: the recombinase A gene family
(bacterial RecA, archaea
RadA and RadB, eukaryotic Rad51 and Rad57, phage UvsX); RNA polymerase II
subunit
(RpoB) gene, which is responsible for transcription initiation and elongation;
chaperonins.
Candidate marker genes have also been identified for bacteria plus archaea:
ribosomal protein S2
(rpsB), ribosomal protein S10 (rpsJ), ribosomal protein Li (rplA), translation
elongation factor
EF-2, translation initiation factor IF-2, metalloendopeptidase, ribosomal
protein L22, ffh signal
recognition particle protein, ribosomal protein L4/Lie (rp1D), ribosomal
protein L2 (rp1B),
ribosomal protein S9 (rpsI), ribosomal protein L3 (rp1C), phenylalanyl-tRNA
synthetase beta
subunit, ribosomal protein Ll 4b/L23e (rp1N), ribosomal protein S5, ribosomal
protein S19
(rpsS), ribosomal protein S7, ribosomal protein L16/L10E (rp1P), ribosomal
protein S13 (rpsM),
phenylalanyl-tRNA synthetase a subunit, ribosomal protein L15, ribosomal
protein L25/L23,
ribosomal protein L6 (rp1F), ribosomal protein LI1 (rp1K), ribosomal protein
L5 (rplE),
ribosomal protein S12/S23, ribosomal protein L29, ribosomal protein S3 (rpsC),
ribosomal
protein S 11 (rpsK), ribosomal protein L10, ribosomal protein S8, tRNA
pseudouridine synthase
B, ribosomal protein Ll8P/L5E, ribosomal protein S 1 5P/S13e, Porphobilinogen
deaminase,
ribosomal protein S17, ribosomal protein L13 (rp1M),
phosphoribosylformylglycinamidine
cyclo-ligase (rpsE), ribonuclease HII and ribosomal protein L24. Other
candidate marker genes
for bacteria include: transcription elongation protein NusA (nusA), rpoB DNA-
directed RNA
polymerase subunit beta (rpoB), GTP-binding protein EngA, rpoC DNA-directed
RNA
polymerase subunit beta', priA primosome assembly protein, transcription-
repair coupling factor,
CTP synthase (pyrG), secY preprotein translocase subunit SecY, GTP-binding
protein
Obg/CgtA, DNA polymerase I, rpsF 30S ribosomal protein S6, poA DNA-directed
RNA
polymerase subunit alpha, peptide chain release factor 1, rpll 505 ribosomal
protein L9,
polyribonucleotide nucleotidyltransferase, tsf elongation factor Ts (tsf),
rplQ 505 ribosomal
protein L17, tRNA (guanine-N(1)-)-methyltransferase (rp1S), rplY probable 505
ribosomal
protein L25, DNA repair protein RadA, glucose-inhibited division protein A,
ribosome-binding
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
factor A, DNA mismatch repair protein MutL, smpB SsrA-binding protein (smpB),
N-
acetylgl ucosaminy I transferase, S-adenosyl-
methyltransferase MraW, UDP-N-
acetylmuramoylalanine--D-glutamate ligase, rp1S 50S ribosomal protein L19,
rp1T 50S
ribosomal protein L20 (rp1T), ruvA Holliday junction DNA helicase, ruvB
Holliday junction
DNA helicase B, serS seryl-tRNA synthetase, rplU 50S ribosomal protein L21,
rpsR 30S
ribosomal protein S18, DNA mismatch repair protein MutS, rpsT 30S ribosomal
protein S20,
DNA repair protein RecN, frr ribosome recycling factor (frr), recombination
protein RecR,
protein of unknown function UPF0054, miaA tRNA isopentenyltransferase, GTP-
binding protein
YchF, chromosomal replication initiator protein DnaA, dephospho-CoA kinase,
16S rRNA
processing protein RimM, ATP-cone domain protein, 1-deoxy-D-xylulose 5-
phosphate
reductoisomerase, 2C-methyl-D-erythritol 2,4-
cyclodiphosphate synthase, fatty
acid/phospholipid synthesis protein PlsX, tRNA(Ile)-lysidine synthetase, dnaG
DNA primase
(dnaG), ruvC Holliday junction resolvase, rpsP 30S ribosomal protein S16,
Recombinase A
recA, riboflavin biosynthesis protein RibF, glycyl-tRNA synthetase beta
subunit, trmU tRNA (5-
methylaminomethy1-2-thiouridylate)-methyltransferase, rpm! 50S ribosomal
protein L35, hemE
uroporphyrinogen decarboxylase, Rod shape-determining protein, rpmA 50S
ribosomal protein
L27 (rpmA), peptidyl-tRNA hydrolase, translation initiation factor IF-3
(infC), UDP-N-
acetylmuramyl-tripeptide synthetase, rpmF 50S ribosomal protein L32, rpIL 50S
ribosomal
protein L7/L12 (rpIL), leuS leucyl-tRNA synthetase, ligA NAD-dependent DNA
ligase, cell
division protein FtsA, GTP-binding protein TypA, ATP-dependent Clp protease,
ATP-binding
subunit ClpX, DNA replication and repair protein RecF and UDP-N-
acetyl enolpyruvoy lglucosamine reductase.
[00116] Phospholipid fatty acids (PLFAs) can also be used as unique first
markers according to
the methods described herein. Because PLFAs are rapidly synthesized during
microbial growth,
are not found in storage molecules and degrade rapidly during cell death, it
provides an accurate
census of the current living community. All cells contain fatty acids (FAs)
that can be extracted
and esterified to form fatty acid methyl esters (FAMEs). When the FAMEs are
analyzed using
gas chromatography¨mass spectrometry, the resulting profile constitutes a
'fingerprint' of the
microorganisms in the sample. The chemical compositions of membranes for
organisms in the
domains Bacteria and Eukarya are comprised of fatty acids linked to the
glycerol by an ester-
type bond (phospholipid fatty acids (PLFAs)). In contrast, the membrane lipids
of Archaea are
41
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
composed of long and branched hydrocarbons that are joined to glycerol by an
ether-type bond
(phospholipid ether lipids (PLELs)). This is one of the most widely used non-
genetic criteria to
distinguish the three domains. In this context, the phospholipids derived from
microbial cell
membranes, characterized by different acyl chains, are excellent signature
molecules, because
such lipid structural diversity can be linked to specific microbial taxa.
100117.1 As provided herein, in order to determine whether an organism strain
is active, the level
of expression of one or more unique second markers, which can be the same or
different as the
first marker, is measured (FIG. 1B, 1004; FIG. 2, 2004). Unique first markers
are described
above. The unique second marker is a marker of microorganism activity. For
example, in one
embodiment, the mRNA or protein expression of any of the first markers
described above is
considered a unique second marker for the purposes of this disclosure.
1001181 In one embodiment, if the level of expression of the second marker is
above a threshold
level (e.g., a control level) or at a threshold level, the microorganism is
considered to be active
(FIG. 1B, 1005; FIG. 2, 2005). Activity is determined in one embodiment, if
the level of
expression of the second marker is altered by at least about 5%, at least
about 10%, at least about
15%, at least about 20%, at least about 25%, or at least about 30%, as
compared to a threshold
level, which in some embodiments, is a control level.
[00119] Second unique markers are measured, in one embodiment, at the protein,
RNA or
metabolite level. A unique second marker is the same or different as the first
unique marker.
[00120] As provided above, a number of unique first markers and unique second
markers can be
detected according to the methods described herein. Moreover, the detection
and quantification
of a unique first marker can be carried out according to methods known to
those of ordinary skill
in the art in light of the disclosure (FIG. 1B, 1003-1004, FIG. 2, 2003-2004).
[00121] Nucleic acid sequencing (e.g., gDNA, cDNA, rRNA, mRNA) in one
embodiment is
used to determine absolute cell count of a unique first marker and/or unique
second marker.
Sequencing platforms include, but are not limited to, Sanger sequencing and
high-throughput
sequencing methods available from Roche/454 Life Sciences, Illumina/Solexa,
Pacific
Biosciences, Ton Torrent and Nanopore. The sequencing can be amplicon
sequencing of
particular DNA or RNA sequences or whole metagenome/transcriptome shotgun
sequencing.
42
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00122] Traditional Sanger sequencing (Sanger et al. (1977) DNA sequencing
with chain-
terminating inhibitors. Proc Natl. Acad. Sci. USA, 74, pp. 5463-5467,
incorporated by reference
herein in its entirety) relies on the selective incorporation of chain-
terminating
dideoxynucleotides by DNA polymerase during in vitro DNA replication and is
amenable for use
with the methods described herein.
[00123] In another embodiment, the sample, or a portion thereof is subjected
to extraction of
nucleic acids, amplification of DNA of interest (such as the rRNA gene) with
suitable primers
and the construction of clone libraries using sequencing vectors. Selected
clones are then
sequenced by Sanger sequencing and the nucleotide sequence of the DNA of
interest is retrieved,
allowing calculation of the number of unique microorganism strains in a
sample.
[00124] 454 pyrosequencing from Roche/454 Life Sciences yields long reads and
can be
harnessed in the methods described herein (Margulies et a/. (2005) Nature,
437, pp. 376-380;
U.S. Patents Nos. 6,274,320; 6,258,568; 6,210,891, each of which is herein
incorporated in its
entirety for all purposes). Nucleic acid to be sequenced (e.g., amplicons or
nebulized
genomic/metagenomic DNA) have specific adapters affixed on either end by PCR
or by ligation.
The DNA with adapters is fixed to tiny beads (ideally, one bead will have one
DNA fragment)
that are suspended in a water-in-oil emulsion. An emulsion PCR step is then
performed to make
multiple copies of each DNA fragment, resulting in a set of beads in which
each bead contains
many cloned copies of the same DNA fragment Each bead is then placed into a
well of a fiber-
optic chip that also contains enzymes necessary for the sequencing-by-
synthesis reactions. The
addition of bases (such as A, C, G, or T) trigger pyrophosphate release, which
produces flashes
of light that are recorded to infer the sequence of the DNA fragments in each
well. About 1
million reads per run with reads up to 1,000 bases in length can be achieved.
Paired-end
sequencing can be done, which produces pairs of reads, each of which begins at
one end of a
given DNA fragment. A molecular barcode can be created and placed between the
adapter
sequence and the sequence of interest in multiplex reactions, allowing each
sequence to be
assigned to a sample bioinformatically.
[00125] IlluminalSolexa sequencing produces average read lengths of about 25
basepairs (bp) to
about 300 bp (Bennett et al. (2005) Pharmacogenomics, 6:373-382; Lange et al.
(2014). BMC
Genomics 15, p. 63; Fadrosh etal. (2014) Microbiome 2, p. 6; Caporaso et al.
(2012) 1SME J, 6,
43
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
p. 1621-1624; Bentley et al. (2008) Accurate whole human genome sequencing
using reversible
terminator chemistry. Nature, 456:53-59). This sequencing technology is also
sequencing-by-
synthesis but employs reversible dye terminators and a flow cell with a field
of oligos attached.
DNA fragments to be sequenced have specific adapters on either end and are
washed over a flow
cell filled with specific oligonucleotides that hybridize to the ends of the
fragments. Each
fragment is then replicated to make a cluster of identical fragments.
Reversible dye-terminator
nucleotides are then washed over the flow cell and given time to attach. The
excess nucleotides
are washed away, the flow cell is imaged, and the reversible terminators can
be removed so that
the process can repeat and nucleotides can continue to be added in subsequent
cycles. Paired-
end reads that are 300 bases in length each can be achieved. An Illumina
platform can produce 4
billion fragments in a paired-end fashion with 125 bases for each read in a
single run. Barcodes
can also be used for sample multiplexing, but indexing primers are used.
1001261 The SOLiD (Sequencing by Oligonucleotide Ligation and Detection, Life
Technologies) process is a "sequencing-by-ligation" approach, and can be used
with the methods
described herein for detecting the presence and quantity of a first marker
and/or a second marker
(FIG. 1B, 1003-1004; FIG. 2, 2003-2004) (Peckham et cd. SOLiDTM Sequencing and
2-Base
Encoding. San Diego, CA: American Society of Human Genetics, 2007; Mitra et
al. (2013)
Analysis of the intestinal microbiota using SOLiD 16S rRNA gene sequencing and
SOLiD
shotgun sequencing. BMC Genomics, 14(Suppl 5): S16; Mardis (2008) Next-
generation DNA
sequencing methods. Annu Rev Genomics Hum Genet, 9:387-402; each incorporated
by
reference herein in its entirety). A library of DNA fragments is prepared from
the sample to be
sequenced, and are used to prepare clonal bead populations, where only one
species of fragment
will be present on the surface of each magnetic bead. The fragments attached
to the magnetic
beads will have a universal P1 adapter sequence so that the starting sequence
of every fragment
is both known and identical. Primers hybridize to the PI adapter sequence
within the library
template. A set of four fluorescently labelled di-base probes compete for
ligation to the
sequencing primer. Specificity of the di-base probe is achieved by
interrogating every 1st and
2nd base in each ligation reaction. Multiple cycles of ligation, detection and
cleavage are
performed with the number of cycles determining the eventual read length. The
SOLiD platform
can produce up to 3 billion reads per run with reads that are 75 bases long.
Paired-end
sequencing is available and can be used herein, but with the second read in
the pair being only 35
44
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
bases long. Multiplexing of samples is possible through a system akin to the
one used by
Illumina, with a separate indexing run.
1001271 The Ion Torrent system, like 454 sequencing, is amenable for use with
the methods
described herein for detecting the presence and quantity of a first marker
and/or a second marker
(FIG. 1B, 1003-1004; FIG. 2, 2003-2004). It uses a plate of microwells
containing beads to
which DNA fragments are attached. It differs from all of the other systems,
however, in the
manner in which base incorporation is detected. When a base is added to a
growing DNA strand,
a proton is released, which slightly alters the surrounding pH. Microdetectors
sensitive to pH are
associated with the wells on the plate, and they record when these changes
occur. The different
bases (A, C, G, T) are washed sequentially through the wells, allowing the
sequence from each
well to be inferred. The Ion Proton platform can produce up to 50 million
reads per run that have
read lengths of 200 bases. The Personal Genome Machine platform has longer
reads at 400
bases. Bidirectional sequencing is available. Multiplexing is possible through
the standard in-
line molecular barcode sequencing.
[00128] Pacific Biosciences (PacBio) SMRT sequencing uses a single-molecule,
real-time
sequencing approach and in one embodiment, is used with the methods described
herein for
detecting the presence and quantity of a first marker and/or a second marker
(FIG. 1B, 1003-
1004; FIG. 2, 2003-2004). The PacBio sequencing system involves no
amplification step,
setting it apart from the other major next-generation sequencing systems. In
one embodiment,
the sequencing is performed on a chip containing many zero-mode waveguide
(ZMW) detectors.
DNA polymerases are attached to the ZMW detectors and phospholinked dye-
labeled nucleotide
incorporation is imaged in real time as DNA strands are synthesized. The
PacBio system yields
very long read lengths (averaging around 4,600 bases) and a very high number
of reads per run
(about 47,000). The typical "paired-end" approach is not used with PacBio,
since reads are
typically long enough that fragments, through CCS, can be covered multiple
times without
having to sequence from each end independently. Multiplexing with PacBio does
not involve an
independent read, but rather follows the standard "in-line" barcoding model.
[00129] In one embodiment, where the first unique marker is the ITS genomic
region,
automated ribosomal intergenic spacer analysis (ARTSA) is used in one
embodiment to
determine the number and identity of microorganism strains in a sample (FIG.
1B, 1003, FIG. 2,
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
2003) (Ranjard et al. (2003). Environmental Microbiology 5, pp. 1111-1120,
incorporated by
reference in its entirety for all purposes). The ITS region has significant
heterogeneity in both
length and nucleotide sequence. The use of a fluorescence-labeled forward
primer and an
automatic DNA sequencer permits high resolution of separation and high
throughput The
inclusion of an internal standard in each sample provides accuracy in sizing
general fragments.
100130.1 In another embodiment, fragment length polymorphism (RFLP) of PCR-
amplified
rDNA fragments, otherwise known as amplified ribosomal DNA restriction
analysis (ARDRA),
is used to characterize unique first markers and the quantity of the same in
samples (FIG. 1B,
1003, FIG. 2, 2003) (for additional detail, see Massol-Deya et al. (1995).
Mol. Microb. Ecol.
Manual. 3.3.2, pp. 1-18, the entirety of which is herein incorporated by
reference for all
purposes). rDNA fragments are generated by PCR using general primers, digested
with
restriction enzymes, electrophoresed in agarose or acrylamide gels, and
stained with ethidium
bromide or silver nitrate.
1001311 One fingerprinting technique used in detecting the presence and
relative quantities of a
unique first marker is single-stranded-conformation polymorphism (SSCP) (see
Lee et al.
(1996). App! Environ Microbiol 62, pp. 3112-3120; Scheinert et al. (1996). J.
Microbiol.
Methods 26, pp. 103-117; Schwieger and Tebbe (1998). App!. Environ. Microbiol.
64, pp. 4870-
4876, each of which is incorporated by reference herein in its entirety). In
this technique, DNA
fragments such as PCR products obtained with primers specific for the 16S rRNA
gene, are
denatured and directly electrophoresed on a non-denaturing gel. Separation is
based on
differences in size and in the folded conformation of single-stranded DNA,
which influences the
electrophoretic mobility. Reannealing of DNA strands during electrophoresis
can be prevented
by a number of strategies, including the use of one phosphorylated primer in
the PCR followed
by specific digestion of the phosphorylated strands with lambda exonuclease
and the use of one
biotinylated primer to perform magnetic separation of one single strand after
denaturation. To
assess the identity of the predominant populations in a given microbial
community, in one
embodiment, bands are excised and sequenced, or SSCP-patterns can be
hybridized with specific
probes. Electrophoretic conditions, such as gel matrix, temperature, and
addition of glycerol to
the gel, can influence the separation.
46
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00132] In addition to sequencing based methods, other methods for quantifying
expression
(e.g., gene, protein expression) of a second marker are amenable for use with
the methods
provided herein for determining the level of expression of one or more second
markers (FIG. 1B,
1004; FIG. 2, 2004). For example, quantitative RT-PCR, microarray analysis,
linear
amplification techniques such as nucleic acid sequence based amplification
(NASBA) are all
amenable for use with the methods described herein, and can be carried out
according to methods
known to those of ordinary skill in the art in light of this disclosure.
1001331 In another embodiment, the sample, or a portion thereof is subjected
to a quantitative
polymerase chain reaction (PCR) for detecting the presence and quantity of a
first marker and/or
a second marker (FIG. 1B, 1003-1004; FIG. 2, 2003-2004). Specific
microorganism strains
activity is measured by reverse transcription of transcribed ribosomal and/or
messenger RNA
(rRNA and mRNA) into complementary DNA (cDNA), followed by PCR (RT-PCR).
[00134] In another embodiment, the sample, or a portion thereof is subjected
to PCR-based
fingerprinting techniques to detect the presence and quantity of a first
marker and/or a second
marker (FIG. 1B, 1003-1004; FIG. 2, 2003-2004). PCR products can be separated
by
electrophoresis based on the nucleotide composition. Sequence variation among
the different
DNA molecules influences the melting behavior, and therefore molecules with
different
sequences will stop migrating at different positions in the gel. Thus
electrophoretic profiles can
be defined by the position and the relative intensity of different bands or
peaks and can be
translated to numerical data for calculation of diversity indices. Bands can
also be excised from
the gel and subsequently sequenced to reveal the phylogenetic affiliation of
the community
members. Electrophoresis methods can include, but are not limited to:
denaturing gradient gel
electrophoresis (DOGE), temperature gradient gel electrophoresis (TGGE),
single-stranded-
conformation polymorphism (SSCP), restriction fragment length polymorphism
analysis (RFLP)
or amplified ribosomal DNA restriction analysis (ARDRA), terminal restriction
fragment length
polymorphism analysis (T-RFLP), automated ribosomal intergenic spacer analysis
(ARISA),
randomly amplified polymorphic DNA (RAPD), DNA amplification fingerprinting
(DAF) and
Bb-PEG electrophoresis.
[00135] In another embodiment, the sample, or a portion thereof is subjected
to a chip-based
platform such as microarray or microfluidics to determine the quantity of a
unique first marker
47
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
and/or presence/quantity of a unique second marker (FIG. 1B, 1003-1004, FIG.
2, 2003-2004).
The PCR products are amplified from total DNA in the sample and directly
hybridized to known
molecular probes affixed to microarrays. After the fluorescently labeled PCR
amplicons are
hybridized to the probes, positive signals are scored by the use of confocal
laser scanning
microscopy. The microarray technique allows samples to be rapidly evaluated
with replication,
which is a significant advantage in microbial community analyses. The
hybridization signal
intensity on microarrays can be directly proportional to the quantity of the
target organism. The
universal high-density 16S microarray (e.g., PHYLOCHIP) contains about 30,000
probes of
16SrRNA gene targeted to several cultured microbial species and "candidate
divisions". These
probes target all 121 demarcated prokaryotic orders and allow simultaneous
detection of 8,741
bacterial and archaeal taxa. Another microarray in use for profiling microbial
communities is the
Functional Gene Array (FGA). Unlike PHYLOCHPs, FGAs are designed primarily to
detect
specific metabolic groups of bacteria. Thus, FGA not only reveal the community
structure, but
they also shed light on the in situ community metabolic potential. FGA contain
probes from
genes with known biological functions, so they are useful in linking microbial
community
composition to ecosystem functions. An FGA termed GEOCHIP contains >24,000
probes from
all known metabolic genes involved in various biogeochemical, ecological, and
environmental
processes such as ammonia oxidation, methane oxidation, and nitrogen fixation.
[00136] A protein expression assay, in one embodiment, is used with the
methods described
herein for determining the level of expression of one or more second markers
(FIG. 1B, 1004;
FIG. 2, 2004). For example, in one embodiment, mass spectrometry or an
immunoassay such as
an enzyme-linked immunosorbant assay (ELISA) is utilized to quantify the level
of expression of
one or more unique second markers, wherein the one or more unique second
markers is a protein.
[00137] In one embodiment, the sample, or a portion thereof is subjected to
Bromodeoxyuridine
(BrdU) incorporation to determine the level of a second unique marker (FIG.
1B, 1004; FIG. 2,
2004). BrdU, a synthetic nucleoside analog of thymidine, can be incorporated
into newly
synthesized DNA of replicating cells. Antibodies specific for BRdU can then be
used for
detection of the base analog. Thus BrdU incorporation identifies cells that
are actively
replicating their DNA, a measure of activity of a microorganism according to
one embodiment of
the methods described herein. BrdU incorporation can be used in combination
with FISH to
provide the identity and activity of targeted cells.
48
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00138] In one embodiment, the sample, or a portion thereof is subjected to
microautoradiography (MAR) combined with FISH to determine the level of a
second unique
marker (FIG. 1B, 1004; FIG. 2, 2004). MAR-FISH is based on the incorporation
of radioactive
substrate into cells, detection of the active cells using autoradiography and
identification of the
cells using FISH. The detection and identification of active cells at single-
cell resolution is
performed with a microscope. MAR-FISH provides information on total cells,
probe targeted
cells and the percentage of cells that incorporate a given radiolabelled
substance. The method
provides an assessment of the in situ function of targeted microorganisms and
is an effective
approach to study the in vivo physiology of microorganisms. A technique
developed for
quantification of cell-specific substrate uptake in combination with MAR-FISH
is known as
quantitative MAR (QMAR).
1001391 In one embodiment, the sample, or a portion thereof is subjected to
stable isotope
Raman spectroscopy combined with FISH (Raman-FISH) to determine the level of a
second
unique marker (FIG. 1B, 1004; FIG. 2, 2004). This technique combines stable
isotope probing,
Raman spectroscopy and FISH to link metabolic processes with particular
organisms. The
proportion of stable isotope incorporation by cells affects the light scatter,
resulting in
measurable peak shifts for labelled cellular components, including protein and
mRNA
components. Raman spectroscopy can be used to identify whether a cell
synthesizes compounds
including, but not limited to: oil (such as alkanes), lipids (such as
triacylglycerols (TAG)),
specific proteins (such as heme proteins, metalloproteins), cytochrome (such
as P450,
cytochrome c), chlorophyll, chromophores (such as pigments for light
harvesting carotenoids and
rhodopsins), organic polymers (such as polyhydroxyalkanoates (PHA),
polyhydroxybutyrate
(PHB)), hopanoids, steroids, starch, sulfide, sulfate and secondary
metabolites (such as vitamin
B12).
[00140] In one embodiment, the sample, or a portion thereof is subjected to
DNA/RNA stable
isotope probing (SIP) to determine the level of a second unique marker (FIG.
1B, 1004; FIG. 2,
2004). SIP enables determination of the microbial diversity associated with
specific metabolic
pathways and has been generally applied to study microorganisms involved in
the utilization of
carbon and nitrogen compounds. The substrate of interest is labelled with
stable isotopes (such
as '3C or '5N) and added to the sample. Only microorganisms able to metabolize
the substrate
will incorporate it into their cells. Subsequently, '3C-DNA and 15N-DNA can be
isolated by
49
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
density gradient centrifugation and used for metagenomic analysis. RNA-based
SIP can be a
responsive biomarker for use in SIP studies, since RNA itself is a reflection
of cellular activity.
[00141] In one embodiment, the sample, or a portion thereof is subjected to
isotope array to
determine the level of a second unique marker (FIG. 1B, 1004; FIG. 2, 2004).
Isotope arrays
allow for functional and phylogenetic screening of active microbial
communities in a high-
throughput fashion. The technique uses a combination of SIP for monitoring the
substrate
uptake profiles and microarray technology for determining the taxonomic
identities of active
microbial communities. Samples are incubated with a "C-labeled substrate,
which during the
course of growth becomes incorporated into microbial biomass. The "C-labeled
rRNA is
separated from unlabeled rRNA and then labeled with fluorochromes. Fluorescent
labeled rRNA
is hybridized to a phylogenetic microarray followed by scanning for
radioactive and fluorescent
signals. The technique thus allows simultaneous study of microbial community
composition and
specific substrate consumption by metabolically active microorganisms of
complex microbial
communities.
[00142] In one embodiment, the sample, or a portion thereof is subjected to a
metabolomics
assay to determine the level of a second unique marker (FIG. 1B, 1004; FIG. 2,
2004).
Metabolomics studies the metabolome which represents the collection of all
metabolites, the end
products of cellular processes, in a biological cell, tissue, organ or
organism. This methodology
can be used to monitor the presence of microorganisms and/or microbial
mediated processes
since it allows associating specific metabolite profiles with different
microorganisms. Profiles of
intracellular and extracellular metabolites associated with microbial activity
can be obtained
using techniques such as gas chromatography-mass spectrometry (GC-MS). The
complex
mixture of a metabolomic sample can be separated by such techniques as gas
chromatography,
high performance liquid chromatography and capillary electrophoresis.
Detection of metabolites
can be by mass spectrometry, nuclear magnetic resonance (NMR) spectroscopy,
ion-mobility
spectrometry, electrochemical detection (coupled to HPLC) and radiolabel (when
combined with
thin-layer chromatography).
[00143] According to the embodiments described herein, the presence and
respective number of
one or more active microorganism strains in a sample are determined (FIG. 1B,
1006; FIG. 2,
2006). For example, strain identity information obtained from assaying the
number and presence
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
of first markers is analyzed to determine how many occurrences of a unique
first marker are
present, thereby representing a unique microorganism strain (e.g., by counting
the number of
sequence reads in a sequencing assay). This value can be represented in one
embodiment as a
percentage of total sequence reads of the first maker to give a percentage of
unique
microorganism strains of a particular microorganism type. In a further
embodiment, this
percentage is multiplied by the number of microorganism types (obtained at
step 1002 or 2002,
see FIG. 1B and FIG. 2) to give the absolute cell count of the one or more
microorganism strains
in a sample and a given volume.
[00144] The one or more microorganism strains are considered active, as
described above, if the
level of second unique marker expression is at a threshold level, higher than
a threshold value,
e.g., higher than at least about 5%, at least about 10%, at least about 20% or
at least about 30%
over a control level.
[00145] In another aspect of the disclosure, a method for determining the
absolute cell count of
one or more microorganism strains is determined in a plurality of samples
(FIG. 2, see in
particular, 2007). For a microorganism strain to be classified as active, it
need only be active in
one of the samples. The samples can be taken over multiple time points from
the same source, or
can be from different environmental sources (e.g., different animals).
[00146] The absolute cell count values over samples are used in one embodiment
to relate the
one or more active microorganism strains, with an environmental parameter
(FIG. 2, 2008). In
one embodiment, the environmental parameter is the presence of a second active
microorganism
strain. Relating the one or more active microorganism strains to the
environmental parameter, in
one embodiment, is carried out by determining the co-occurrence of the strain
and parameter by
network analysis and/or graph theory.
[00147] In one embodiment, determining the co-occurrence of one or more active
microorganism strains with an environmental parameter comprises a network
and/or cluster
analysis method to measure connectivity of strains or a strain with an
environmental parameter
within a network, wherein the network is a collection of two or more samples
that share a
common or similar environmental parameter. Examples of measurement of
independence are
provided and discussed herein, and additional details can be understood by
configuring the
teachings and methods of: Blomqvist "On a measure of dependence between two
random
51
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
variables" The Annals of Mathematical Statistics (1950): 593-600; Hollander et
al.
"Nonparametric statistical methods - Wiley series in probability and
statistics Texts and
references section" (1999); and/or Blum et al. "Distribution free tests of
independence based on
the sample distribution function" The Annals of Mathematical Statistics
(1961): 485-498; the
entirety of each of the aforementioned publications being herein expressly
incorporated by
reference for all purposes.
[00148] In another embodiment, correlation methods including Pearson
correlation, Spearman
correlation, Kendall correlation, Canonical Correlation Analysis, Likelihood
ratio tests (e.g., by
adapting the teachings and methods detailed in Wilks, S.S. "On the
Independence of k Sets of
Normally Distributed Statistical Variables" Econometric& Vol. 3, No. 3, July
1935, pp 309-326,
the entirety of which is herein expressly incorporated by reference for all
purposes), and
canonical correlation analysis are used establish connectivity between
variables. Multivariate
extensions of these methods, Maximal correlation (see, e.g., Alfred Renyi "On
measures of
dependence" Acta mathematica hungarica 10.3-4 (1959): 441-451, herein
expressly incorporated
by reference in its entirety), or both (MAC) can be used when appropriate,
depending on the
number of variables being compared. Some embodiments utilize Maximal
Correlation Analysis
and/or other multivariate correlation measures configured for discovering
multi-dimensional
patterns (for example, by adapting the methods and teachings of "Multivariate
Maximal
Correlation Analysis," Nguyen et al., Proceedings of the 31st International
Conference on
Machine Learning, Beijing, China, 2014, which is herein expressly incorporated
by reference in
its entirety for all purposes). In some embodiments, network metrics and
analysis, such as
discussed by Farine et al, in "Constructing, Conducting and Interpreting
Animal Social Network
Analysis" Journal of Animal Ecology, 2015, 84, pp. 1144-1163. doi:10.1111/1365-
2656.12418
(the entirety of which is herein expressly incorporated by reference for all
purposes) can be
utilized and configured for the disclosure.
[00149] In some embodiments, network analysis comprises nonparametric
approaches (e.g., by
adapting the teaching and methods detailed in Taskinen et al. "Multivariate
nonparametric tests
52
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
of independence." Journal of the American Statistical Association 100.471
(2005): 916-925; and
Gieser et al. "A Nonparametric Test of Independence Between Two Vectors."
Journal of the
American Statistical Association, Vol. 92, No. 438, June, 1977, pp 561-567;
entirety of each of
being herein expressly incorporated by reference for all purposes), including
mutual information
Maximal Information Coefficient, Maximal Information Entropy (MW; e.g., by
adapting the
teachings and methods of Zhang Ya-hong et al. "Detecting Multivariable
Correlation with
Maximal Information Entropy[J]" Journal of Electronics & Information
Technology, 2015-01
(37(1): 123-129), the entirety of which is herein expressly incorporated by
reference for all
purposes), Kernel Canonical Correlation Analysis (KCCA; e.g., by adapting the
teachings and
methods detailed in Bach et al. "Kernel Independent Component Analysis"
Journal of Machine
Learning Research 3 (2002) 1-48, the entirety of which is herein expressly
incorporated by
reference for all purposes), Alternating Conditional Expectation or
backfitting algorithms (ACE;
e.g., by adapting the teaching and methods detailed in Breiman et al.
"Estimating Optimal
Transformations for Multiple Regression and Correlation: Rejoinder." Journal
of the American
Statistical Association 80, no. 391 (1985): 614-19, doi:10.2307/2288477, the
entirety of which is
herein expressly incorporated by reference for all purposes), Distance
correlation measure (dcor;
e.g., by adapting the teaching and methods detailed in Szekely et al.
"Measuring and Testing
Dependence by Correlation of Distances" The Annals of Statistics, 2007, Vol.
35, No. 6, 2769-
2794, doi:10.1214/009053607000000505, the entirety of which is herein
expressly incorporated
by reference for all purposes), Brownian distance covariance (dcov; e.g., by
adapting the
teaching and methods detailed in Szekely et al. "Brownian Distance Covariance"
The Annals of
Applied Statistics, 2009, Vol. 3, No. 4, 1236-1265, Doi:10.1214/09-AOAS312,
the entirety of
which is herein expressly incorporated by reference for all purposes), Hilbert-
Schmidt
Independence Criterion (HSCI / CHSI; e.g., by adapting the teachings and
methods detailed in
Gretton et al. "A Kernal Two-Sample Test" Journal of Machine Learning Research
13 (2012)
723-773, and Poczos et al. "Copula-based Kernel Dependency Measures" Carnegie
Mellow
University, Research Showcase@CMU, Proceedings of the 29th International
Conference on
53
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Machine Learning, each of which is herein expressly incorporated by reference
in their entireties
for all purposes), Randomized Dependence Coefficient (RDC; e.g., by adapting
the teaching and
methods detailed in Lopez-Paz et al. "The Randomized Dependence Coefficient"
Advances in
Neural Information Processing Systems (2013), the entirety of which is herein
expressly
incorporated by reference for all purposes) to establish connectivity between
variables. In some
embodiments, one or more of these methods can be coupled to bagging or
boosting methods, or k
nearest neighbor estimators (e.g., by adapting the teaching and methods
detailed in: Breiman,
"Arcing Classifiers" The Annals of Statistics, 1998, Vol. 26, No. 3, 801-849;
Liu, "Modified
Bagging of Maximal Information Coefficient for Genome-wide Identification"
Int. J. Data
Mining and Bioinformatics, Vol. 14, No. 3, 2016, pp. 229-257; and/or Gao et
al. "Efficient
Estimation of Mutual Information for Strongly Dependent Variables" Proceedings
of the 18th
International Conference on Artificial Intelligence and Statistics (AISTATS),
2015, San Diego,
CA, JMLR: W&CP Volume 38; each of which is herein expressly incorporated by
reference in
its entirety for all purposes).
[00150] In some embodiments, the network analysis comprises node-level
analysis, including
degree, strength, betweenness centrality, eigenvector centrality, page rank,
and reach. In another
embodiment, the network analysis comprises network level metrics, including
density,
homophily or assortativity, transitivity, linkage analysis, modularity
analysis, robustness
measures, betweenness measures, connectivity measures, transitivity measures,
centrality
measures or a combination thereof. In others embodiments, species community
rules (see, e.g.,
Connor et al. "The Assembly of Species Communities: Chance or Competition?"
Ecology, Vol.
60, No. 6 (Dec., 1979), pp. 1132-1140, the entirety of which is herein
incorporated by reference
for all purposes) are applied to the network, which can include leveraging
Gambit of the Group
assumptions (e.g., by applying the methods and teachings of Franks et al.
"Sampling Animal
Association Networks with the Gambit of the Group' Behav Ecol Sociobiol (2010)
64:493,
doi:10.1007/x00265-0098-0865-8, the entirety of which is herein expressly
incorporated by
reference for all purposes). In some embodiments, eigenvectors/modularity
matrix analysis
54
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
methods can be used, e.g., by configuring the teachings and methods as
discussed by Mark EJ
Newman in "Finding community structure in networks using the eigenvectors of
matrices"
Physical Review E 74.3 (2006): 036104, the entirety of which is herein
expressly incorporated
by reference for all purposes.
[00151] In some embodiments, time-aggregated networks or time-ordered networks
are utilized.
In another embodiment, the cluster analysis method comprises building or
constructing an
observation matrix, connectivity model, subspace model, distribution model,
density model, or a
centroid model, using community detection in graphs, and/or using community
detection
algorithms such as, by way of non-limiting example, the Louvain, Bron-
Kerbosch, Girvan-
Newman, Clauset-Newman-Moore, Pons-Latapy, and Wakita-Tsurumi algorithms.
[00152] In some embodiments, the cluster analysis method is a heuristic method
based on
modularity optimization. In a further embodiment, the cluster analysis method
is the Louvain
method (see, e.g., the method described by Blondel et al. (2008) Fast
unfolding of communities
in large networks. Journal of Statistical Mechanics: Theory and Experiment,
Volume 2008,
October 2008, incorporated by reference herein in its entirety for all
purposes, and which can be
adapted for use in the methods disclosed herein).
[00153] In other embodiments, the network analysis comprises predictive
modeling of network
through link mining and prediction, collective classification, link-based
clustering, hierarchical
cluster analysis, relational similarity, or a combination thereof. In another
embodiment, the
network analysis comprises differential equation based modeling of
populations. In another
embodiment, the network analysis comprises Lotka-Volterra modeling.
[00154] In some embodiments, relating the one or more active microorganism
strains to an
environmental parameter (e.g., determining the co-occurrence) in the sample
comprises creating
matrices populated with linkages denoting environmental parameter and
microorganism strain
associations.
[00155] In some embodiments, the multiple sample data obtained at step 2007
(e.g., over two or
more samples which can be collected at two or more time points where each time
point
corresponds to an individual sample) is compiled. In a further embodiment, the
number of cells
of each of the one or more microorganism strains in each sample is stored in
an association
matrix (which can be in some embodiments, a quantity matrix). In one
embodiment, the
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
association matrix is used to identify associations between active
microorganism strains in a
specific time point sample using rule mining approaches weighted with
association (e.g.,
quantity) data. Filters are applied in one embodiment to remove insignificant
rules.
[00156] In some embodiments, the absolute cell count of one or more, or two or
more active
microorganism strains is related to one or more environmental parameters (FIG.
2, 2008), e.g.,
via co-occurrence determination. Environmental parameters can be selected
depending on the
sample(s) to be analyzed and are not restricted by the methods described
herein. The
environmental parameter can be a parameter of the sample itself, e.g., pH,
temperature, amount
of protein in the sample. Alternatively, the environmental parameter is a
parameter that affects a
change in the identity of a microbial community (i.e., where the "identity" of
a microbial
community is characterized by the type of microorganism strains and/or number
of particular
microorganism strains in a community), or is affected by a change in the
identity of a microbial
community. For example, an environmental parameter in one embodiment, is the
food intake of
an animal or the amount of milk (or the protein or fat content of the milk)
produced by a
lactating ruminant. In one embodiment, the environmental parameter is the
presence, activity
andlor quantity of a second microorganism strain in the microbial community,
present in the
same sample. In some embodiments described herein, an environmental parameter
is referred to
as a metadata parameter, and vice-versa.
[00157] Other examples of metadata parameters include but are not limited to
genetic
information from the host from which the sample was obtained (e.g., DNA
mutation
information), sample pH, sample temperature, expression of a particular
protein or mRNA,
nutrient conditions (e.g., level and/or identity of one or more nutrients) of
the surrounding
environment/ecosystem), susceptibility or resistance to disease, onset or
progression of disease,
susceptibility or resistance of the sample to toxins, efficacy of xenobiotic
compounds
(pharmaceutical drugs), biosynthesis of natural products, or a combination
thereof.
[00158] For example, according to one embodiment, microorganism strain number
changes are
calculated over multiple samples according to the method of FIG. 2 (i.e., at
2001-2007). Strain
number changes of one or more active strains over time is compiled (e.g., one
or more strains
that have initially been identified as active according to step 2006), and the
directionality of
change is noted (i.e., negative values denoting decreases, positive values
denoting increases).
56
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
The number of cells over time is represented as a network, with microorganism
strains
representing nodes and the quantity weighted rules representing edges. Markov
chains and
random walks are leveraged to determine connectivity between nodes and to
define clusters.
Clusters in one embodiment are filtered using metadata in order to identify
clusters associated
with desirable metadata (FIG. 2, 2008).
[00159] In a further embodiment, microorganism strains are ranked according to
importance by
integrating cell number changes over time and strains present in target
clusters, with the highest
changes in cell number ranking the highest.
[00160] Network and/or cluster analysis method in one embodiment, is used to
measure
connectivity of the one or more strains within a network, wherein the network
is a collection of
two or more samples that share a common or similar environmental parameter. In
one
embodiment, network analysis comprises linkage analysis, modularity analysis,
robustness
measures, betweenness measures, connectivity measures, transitivity measures,
centrality
measures or a combination thereof In another embodiment, network analysis
comprises
predictive modeling of network through link mining and prediction, social
network theory,
collective classification, link-based clustering, relational similarity, or a
combination thereof In
another embodiment, network analysis comprises mutual information, maximal
information
coefficient calculations, or other nonparametric methods between variables to
establish
connectivity. In another embodiment, network analysis comprises differential
equation based
modeling of populations. In yet another embodiment, network analysis comprises
Lotka-
Vol terra modeling.
[00161] Cluster analysis method comprises building a connectivity model,
subspace model,
distribution model, density model, or a centroid model.
[00162] Network and cluster based analysis, for example, to carry out method
step 2008 of FIG.
2, can be carried out via a processor, component and/or module. As used
herein, a component
and/or module can be, for example, any assembly, instructions and/or set of
operatively-coupled
electrical components, and can include, for example, a memory, a processor,
electrical traces,
optical connectors, software (executing in hardware) and/or the like.
[00163] FIG. 3A is a schematic diagram that illustrates a microbe analysis,
screening and
selection platform and system 300, according to an embodiment. A platform
according to the
57
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
disclosure can include systems and processes to determine multi-dimensional
interspecies
interactions and dependencies within natural microbial communities, and an
example is
described with respect to FIG. 3A. FIG. 3A is an architectural diagram, and
therefore certain
aspects are omitted to improve the clarity of the description, though these
aspects should be
apparent to one of skill when viewed in the context of the disclosure.
[00164] As shown in FIG. 3A, the microbe screening and selection platform and
system 300 can
include one or more processors 310, a database 319, a memory 320, a
communications interface
390, an input/output interface configured to interact with user input devices
396 and peripheral
devices 397 (including but not limited to data collection and analysis device,
such as FACs,
selection/incubation/formulation devices, and/or additional databases/data
sources, remote data
collection devices (e.g., devices that can collect metadata environmental
data, such as sample
characteristics, temperature, weather, etc., including mobile smart phones
running apps to collect
such information as well as other mobile or stationary devices), a network
interface configured
to receive and transmit data over communications network 392 (e.g., LAN, WAN,
and/or the
Internet) to clients 393b (which can include user interfaces and/or displays,
such as graphical
displays) and users 393a; a data collection component 330, an absolute count
component 335, a
sample relation component 340, an activity component 345, a network analysis
component 350,
a strain selection/microbial ensemble generation component 355, and a
biostateldiagnostics
component 360. In some embodiments, the microbe screening system 300 can be a
single
physical device. In other embodiments, the microbe screening system 300 can
include multiple
physical devices (e.g., operatively coupled by a network), each of which can
include one or
multiple components and/or modules shown in FIG. 3A. In some embodiments, the
screening
system can be utilized for diagnostics and therapeutics, e.g., by adapting the
teaching and
methods detailed in U.S. Pat. App. Pub. Nos. 2016/0110515, 2016/0230217, and
2016/0224749,
each of which is herein expressly incorporated by reference in its entirety
for all purposes.
[00165] Each component or module in the microbe screening system 300 can be
operatively
coupled to each remaining component and/or module. Each component and/or
module in the
microbe screening system 300 can be any combination of hardware and/or
software (stored
and/or executing in hardware) capable of performing one or more specific
functions associated
with that component and/or module.
58
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00166] The memory 320 can be, for example, a random-access memory (RAM)
(e.g., a
dynamic RAM, a static RAM), a flash memory, a removable memory, a hard drive,
a database
and/or so forth. In some embodiments, the memory 320 can include, for example,
a database
(e.g., as in 319), process, application, virtual machine, and/or some other
software components,
programs and/or modules (stored and/or executing in hardware) or hardware
components/modules configured to execute a microbe screening process and/or
one or more
associated methods for microbe screening and ensemble generation (e.g., via
the data collection
component 330, the absolute count component 335, the sample relation component
340, the
activity component 345, the network analysis component 350, the strain
selection/microbial
ensemble generation component 355 (and/or similar modules)). In such
embodiments,
instructions of executing the microbe screening and/or ensemble generation
process and/or the
associated methods can be stored within the memory 320 and executed at the
processor 310. In
some embodiments, data collected via the data collection component 330 can be
stored in a
database 319 and/or in the memory 320.
[00167] The processor 310 can be configured to control, for example, the
operations of the
communications interface 390, write data into and read data from the memory
320, and execute
the instructions stored within the memory 320. The processor 310 can also be
configured to
execute and/or control, for example, the operations of the data collection
component 330, the
absolute count component 335, the sample relation component 340, the activity
component, and
the network analysis component 350, as described in further detail herein. In
some
embodiments, under the control of the processor(s) 310 and based on the
methods or processes
stored within the memory 320, the data collection component 330, absolute
count component
335, sample relation component 340, activity component 345, network analysis
component 350,
and strain selection/ensemble generation component 355 can be configured to
execute a microbe
screening, selection and synthetic ensemble generation process, as described
in further detail
herein.
[00168] The communications interface 390 can include and/or be configured to
manage one or
multiple ports of the microbe screening system 300 (e.g., via input out
interface(s) 395). In some
instances, for example, the communications interface 390 (e.g., a Network
Interface Card (NIC))
can include one or more line cards, each of which can include one or more
ports (operatively)
coupled to devices (e.g., peripheral devices 397 and/or user input devices
396). A port included
59
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
in the communications interface 390 can be any entity that can actively
communicate with a
coupled device or over a network 392 (e.g., communicate with end-user devices
393b, host
devices, servers, etc.). In some embodiments, such a port need not necessarily
be a hardware
port, but can be a virtual port or a port defined by software. The
communication network 392
can be any network or combination of networks capable of transmitting
information (e.g., data
and/or signals) and can include, for example, a telephone network, an Ethernet
network, a fiber-
optic network, a wireless network, and/or a cellular network. The
communication can be over a
network such as, for example, a Wi-Fi or wireless local area network ("WLAN")
connection, a
wireless wide area network ("WWAN") connection, and/or a cellular connection.
A network
connection can be a wired connection such as, for example, an Ethernet
connection, a digital
subscription line ("DSL") connection, a broadband coaxial connection, and/or a
fiber-optic
connection. For example, the microbe screening system 300 can be a host device
configured to
be accessed by one or more compute devices 393b via a network 392. In such a
manner, the
compute devices can provide information to and/or receive information from the
microbe
screening system 300 via the network 392. Such information can be, for
example, information
for the microbe screening system 300 to collect, relate, determine, analyze
and/or generate
ensembles of active, network-analyzed microbes, as described in further detail
herein. Similarly,
the compute devices can be configured to retrieve and/or request determined
information from
the microbe screening system 300.
[00169] In some embodiments, the communications interface 390 can include
and/or be
configured to include input/output interfaces 395. The input/output interfaces
can accept,
communicate, and/or connect to user input devices, peripheral devices,
cryptographic processor
devices, and/or the like. In some instances, one output device can be a video
display, which can
include, for example, a Cathode Ray Tube (CRT) or Liquid Crystal Display
(LCD), LED, or
plasma based monitor with an interface (e.g., Digital Visual Interface (DVT)
circuitry and cable)
that accepts signals from a video interface. In such embodiments, the
communications interface
390 can be configured to, among other functions, receive data and/or
information, and send
microbe screening modifications, commands, and/or instructions.
[00170] The data collection component 330 can be any hardware and/or software
component
and/or module (stored in a memory such as the memory 320 and/or executing in
hardware such
as the processor 310) configured to collect, process, and/or normalize data
for analysis on multi-
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
dimensional interspecies interactions and dependencies within natural
microbial communities
performed by the absolute count component 335, sample relation component 340,
activity
component 345, network analysis component 350, and/or strain
selection/ensemble generation
component 355. In some embodiments, the data collection component 330 can be
configured to
determine absolute cell count of one or more active organism strains in a
given volume of a
sample. Based on the absolute cell count of one more active microorganism
strains, the data
collection component 330 can identify active strains within absolute cell
count datasets using
marker sequences. The data collection component 330 can continuously collect
data for a period
of time to represent the dynamics of microbial populations within a sample.
The data collection
component 330 can compile temporal data and store the number of cells of each
active organism
strain in a quantity matrix in a memory such as the memory 320.
1001711 The sample relation component 340 and the network analysis component
350 can be
configured to collectively determine multi-dimensional interspecies
interactions and
dependencies within natural microbial communities. The sample relation
component 340 can be
any hardware and/or software component (stored in a memory such as the memory
320 and/or
executing in hardware such as the processor 310) configured to relate a
metadata parameter
(environmental parameter, e.g., via co-occurrence) to presence of one or more
active
microorganism strains. In some embodiments, the sample relation component 340
can relate the
one or more active organism strains to one or more environmental parameters.
[001721 The network analysis component 350 can be any hardware and/or software
component
(stored in a memory such as the memory 320 and/or executing in hardware such
as the processor
310) configured to determine co-occurrence of one or more active microorganism
strains in a
sample to an environmental (metadata) parameter. In some embodiments, based on
the data
collected by the data collection component 330, and the relation between the
one or more active
microorganism strains to one or more environmental parameters determined by
the sample
relation component 340, the network analysis component 350 can create matrices
populated with
linkages denoting environmental parameters and microorganism strain
associations, the absolute
cell count of the one or more active microorganism strains and the level of
expression of the one
or more unique second markers to represent one or more networks of a
heterogeneous population
of microorganism strains. For example, the network analysis can use an
association (quantity
and/or abundance) matrix to identify associations between an active
microorganism strain and a
61
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
metadata parameter (e.g., the associations of two or more active microorganism
strains) in a
sample using rule mining approaches weighted with quantity data. In some
embodiments, the
network analysis component 350 can apply filters to select and/or remove
rules. The network
analysis component 350 can calculate cell number changes of active strains
over time, noting
directionality of change (i.e., negative values denoting decreases, positive
values denoting
increases). The network analysis component 350 can represent matrix as a
network, with
microorganism strains representing nodes and the quantity weighted rules
representing edges.
The network analysis component 350 can use leverage markov chains and random
walks to
determine connectivity between nodes and to define clusters. In some
embodiments, the network
analysis component 350 can filter clusters using metadata in order to identify
clusters associated
with desirable metadata. In some embodiments, the network analysis component
350 can rank
target microorganism strains by integrating cell number changes over time and
strains present in
target clusters, with highest changes in cell number ranking the highest.
[00173] In some embodiments, the network analysis includes linkage analysis,
modularity
analysis, robustness measures, betweenness measures, connectivity measures,
transitivity
measures, centrality measures or a combination thereof. In another embodiment,
a cluster
analysis method can be used including building a connectivity model, subspace
model,
distribution model, density model, or a centroid model. In another embodiment,
the network
analysis includes predictive modeling of network through link mining and
prediction, collective
classification, link-based clustering, relational similarity, or a combination
thereof. In another
embodiment, the network analysis comprises mutual information, maximal
information
coefficient calculations, or other nonparametric methods between variables to
establish
connectivity. In another embodiment, the network analysis includes
differential equation based
modeling of populations. In another embodiment, the network analysis includes
Lotka-Volterra
modeling.
[00174] FIG 3B shows an exemplary logic flow according to one embodiment of
the disclosure.
To begin, a plurality of samples and/or sample sets are collected and/or
received 3001. It is to be
understood that as used herein, "sample" can refer to one or more samples, a
sample set, a
plurality of samples (e.g., from particular population), such that when two or
more different
samples are discussed, that is for ease of understanding, and each sample can
include a plurality
62
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
of sub sample (e.g., when a first sample and second sample are discussed, the
first sample can
include 2, 3, 4, 5 or more sub samples, collected from a first population, and
the second sample
can include 2, 3, 4, 5 or more sub samples collected from a second population,
or alternatively,
collected from the first population but at a different point in time, such as
one week or one month
after collection of the first sub-sample). When sub-samples are collected,
individual collection
indicia and parameters for each sub-sample can be monitored and stored,
including
environmental parameters, qualitative and/or quantitative observations,
population member
identity (e.g., so when sample are collected from the same population at two
or more different
time, the sub-samples are paired by identify, so subsample at time 1 from
animal 1 is linked to a
subsample collected from that same animal at time 2, and so forth).
1001751 For each sample, sample set, and/or subsample, the cells are stained
based on the target
organism type 3002, each sample/subsample or portion thereof is weighed and
serially diluted
3003, and processed 3004 to determine the number of cells of each
microorganism type in each
sample/subsample. In one exemplary implementation, a cell sorter can be used
to count
individual bacterial and fungal cells from samples, such as from an
environmental sample. As
part of the disclosure, specific dyes were developed to enable counting of
microorganisms that
previously were not countable according to the traditional methods. Following
the methods of
the disclosure, specific dyes are used to stain cell walls (e.g., for bacteria
and/or fungi), and
discrete populations of target cells can be counted from a greater population
based on cellular
characteristics using lasers. In one specific example, environmental samples
are prepared and
diluted into isotonic buffer solution and stained with dyes: (a) for bacteria,
the following dyes
can be used to stain - DNA: Sybr Green, Respiration: 5-cyano-2,3-
ditolyltetrazolium chloride
and/or CTC, Cell wall : Malachite Green and/or Crystal Violet; (b) for fungi,
the following dyes
can be used to stain - Cell wall: Calcofluor White, Congo Red, Trypan Blue,
Direct Yellow 96,
Direct Yellow 11, Direct Black 19, Direct Orange 10, Direct Red 23, Direct Red
81, Direct
Green 1, Direct Violet 51, Wheat Germ Agglutinin - WGA, Reactive Yellow 2,
Reactive Yellow
42, Reactive Black 5, Reactive Orange 16, Reactive Red 23, Reactive Green 19,
and/or Reactive
Violet 5.
100176.1 In the development of this disclosure, it was advantageously
discovered that although
direct and reactive dyes are typically associated with the staining of
cellulose-based materials
(i.e., cotton, flax, and viscose rayon), they can also be used to stain chitin
and chitosan because
63
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
of the presence of 13-(1-4)-linked N-acetylglucosamine chains, and 13-(1¨>4)-
linked D-
glucosamine and N-acetyl-D-glucosamine chains, respectively. When these
subunits assemble
into a chain, a flat, fiber-like structure very similar to cellulose chains is
formed. Direct dyes
adhere to chitin and/or chitosan molecules via Van der Waals forces between
the dye and the
fiber molecule. The more surface area contact between the two, the stronger
the interaction.
Reactive dyes, on the other hand, form a covalent bond to the chitin and/or
chitosan.
[00177] Each dyed sample is loaded onto the FACs 3004 for counting. The sample
can be run
through a microfluidic chip with a specific size nozzle (e.g., 100 pm,
selected depending on the
implementation and application) that generates a stream of individual droplets
(e.g.,
approximately 1/10th of a microliter (0.1 !IL)). These variables (nozzle size,
droplet formation)
can be optimized for each target microorganism type. Ideally, encapsulated in
each droplet is one
cell, or "event," and when each droplet is hit by a laser, anything that is
dyed is excited and emits
a different wavelength of light The FACs optically detects each emission, and
can plot them as
events (e.g., on a 2D graph). A typical graph consists of one axis for size of
event (determined by
"forward scatter"), and the other for intensity of fluorescence. "Gates" can
be drawn around
discrete population on these graphs, and the events in these gates can be
counted.
[00178] FIG. 3C shows example data from fungi stained with Direct Yellow;
includes yeast
monoculture 3005a (positive control, left), E. coli 3005b (negative control,
middle), and
environmental sample 3005c (experimental, right). In the figure, "back
scatter" (BSC-A)
measures complexity of event, while FITC measures intensity of fluorescent
emission from
Direct Yellow. Each dot represents one event, and density of events is
indicated by color change
from green to red. Gate B indicates general area in which targeted events, in
this case fungi
stained with Direct Yellow, are expected to be found.
[00179] Returning to FIG. 3B, beginning with the two or more samples 3001
collected from one
or more sources (including samples collected from an individual animal or
single geographical
location over time; from two or more groups differing in geography, breed,
performance, diet,
disease, etc.; from one or more groups that experience a physiological
perturbation or event;
and/or the like) the samples can be analyzed to establish absolute counts
using flow cytometry,
including staining 3002, as discussed above. Samples are weighed and serially
diluted 3003, and
processed using a FACs 3004. Output from the FACs is then processed to
determine the absolute
64
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
number of the desired organism type in each sample 3005. The following code
fragment shows
an exemplary methodology for such processing, according to one embodiment:
# User defined variables
# volume = volume of sample measured by FACs
# dilution = dilution factor
# beads_num = counting bead factor
# total_volume = total volume of sample (if applicable) in mL
# Note on total_volume: This is can be directly measured (i.e.
# rumen evacuation to measure entire volume content of the rumen),
# or via a stable tracer (i.e. use of an undigestible marker dosed
# in a known quantity in order to backcalculate volume of small
# intestine.)
Read FACsoutput as x
for i in range(len(x)):
holder = x[i]
for j in range(len(holder)):
beads = holder[-1]
if beads == 0:
temp =
(((holder[j]/beads_num)*(51300/volume))*1000)*diluti on*100*tota I volume
mule.append(temp)
else:
temp = (((holder[Wholded-
1])*(51300/volume))*1000)*dilution*100*total_volume
mule.append(temp)
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
organism type_l = mule[column_location]
call = sample_names[i]
cell count = [call, organism type 1]
savetxt(output_file,cell_count)
output_fi le. close()
[00180] The total nucleic acids are isolated from each sample 3006. The
nucleic acid sample
elutate is split into two parts (typically, two equal parts), and each part is
enzymatically purified
to obtain either purified DNA 3006a or purified RNA 3006b. Purified RNA is
stabilized through
an enzymatic conversion to cDNA 3006c. Sequencing libraries (e.g., ILLUMINA
sequencing
libraries) are prepared for both the purified DNA and purified cDNA using PCR
to attach the
appropriate barcodes and adapter regions, and to amplify the marker region
appropriate for
measuring the desired organism type 3007. Library quality can be assessed and
quantified, and
all libraries can then be pooled and sequenced.
[00181] Raw sequencing reads are quality trimmed and merged 3008. Processed
reads are
dereplicated and clustered to generate a set or list of all of the unique
strains present in the
plurality of samples 3009. This set or list can be used for taxonomic
identification of each strain
present in the plurality of samples 3010. Sequencing libraries derived from
DNA samples can be
identified, and sequencing reads from the identified DNA libraries are mapped
back to the set or
list of dereplicated strains in order to identity which strains are present in
each sample, and
quantify the number of reads for each strain in each sample 3011. The
quantified read list is then
integrated with the absolute cell count of target microorganism type in order
to determine the
absolute number or cell count of each strain 3013. The following code fragment
shows an
exemplary methodology for such processing, according to one embodiment:
# User defined variables
# input = quantified count output from sequence analysis
# count = calculated absolute cell count of organism type
# taxonomy = predicted taxonomy of each strain
66
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Read absolute cell count file as counts
Read taxonomy file as tax
ncols= len(counts)
num_samples = ncols/2
tax_level =
tax_level.append(unique(taxonomyrkingdomlvalues.ravel()))
tax_level.append(unique(taxonomyrphyluml.values.ravel()))
tax_level.append(unique(taxonomy['class'].values.ravel()))
tax_level.append(unique(taxonomyrorderl.values.ravel()))
tax_level.append(unique(taxonomyrfamilylvalues.ravel()))
tax_level.append(unique(taxonomyrgenus/values.ravel()))
tax_level.append(unique(taxonomyrspeciegvalues.ravel()))
tax_counts = merge(left=counts,right=tax)
# Species level analysis
tax_counts.to_csv('species.txf)
# Only pull DNA samples
data_mule = loadcsv('species.txt', usecols=xrange(2,ncols,2))
data_mule_normalized = data_mule/sum(data_mule)
data_mule_with_counts = data_mule_normalized*counts
Repeat for every taxonomic level
100182.1 Sequencing libraries derived from cDNA samples are identified 3014.
Sequencing
reads from the identified cDNA libraries are then mapped back to the list of
dereplicated strains
67
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
in order to determine which strains are active in each sample. If the number
of reads is below a
specified or designated threshold 3015, the strain is deemed or identified as
inactive and is
removed from subsequent analysis 3015a. If the number of reads exceeds the
threshold 3015, the
strain is deemed or identified as active and remains in the analysis 3015b.
Inactive strains are
then filtered from the output 3013 to generate a set or list of active strains
and respective
absolute numbers/cell counts for each sample 3016. The following code fragment
shows an
exemplary methodology for such processing, according to one embodiment:
# continued using variables from above
# Only pull RNA samples
active_data_mule = loadcsv(species.csvi, usecols=xrange(3,ncols+1,2))
threshold = percentile(active_data_mule, 70)
for i in range(len(active_data_mule)):
if data_mule activity >= threshold
multiplier[i] = 1
else
multiplier[i] =0
acti ve_data_mul e_with_counts = multi plier*data_mule_with_counts
Repeat for every taxonomic level
[00183] Qualitative and quantitative metadata (e.g., environmental parameters,
etc.) is
identified, retrieved, and/or collected for each sample 3017 (set of samples,
subsamples, etc.) and
stored 3018 in a database (e.g., 319). Appropriate metadata can be identified,
and the database is
queried to pull identified and/or relevant metadata for each sample being
analyzed 3019,
depending on the application/implementation. The subset of metadata is then
merged with the set
or list of active strains and their corresponding absolute numbers/cell counts
to create a large
species and metadata by sample matrix 3020.
68
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1.001841 The maximal information coefficient (MIC) is then calculated between
strains and
metadata 3021a, and between strains 3021b. Results are pooled to create a set
or list of all
relationships and their corresponding MIC scores 3022. If the relationship
scores below a given
threshold 3023, the relationship is deemed/identified as irrelevant 3023b. If
the relationship is
above a given threshold 3023, the relationship deemed/identified as relevant
3023a, and is
further subject to network analysis 3024. The following code fragment shows an
exemplary
methodology for such analysis, according to one embodiment:
Read total list of relationships file as links
threshold = 0.8
for i in range(len(links)):
if links >= threshold
multiplier[i] = 1
else
multiplier[i] =0
end if
1 inks_temp = multipl ier* links
final_links = links temp[links temp != 0]
savetxt(output_filefinal_links)
output file.close()
[00185] Based on the output of the network analysis, a biostate is defined
and/or active strains
are selected 3025 for preparing products (e.g., ensembles, aggregates, and/or
other synthetic
groupings) containing the selected strains. The output of the network analysis
can also be used to
inform diagnostics and/or the selection of strains for further product
composition testing.
1.001861 The use of thresholds is discussed above for analyses and
determinations. Thresholds
can be, depending on the implementation and application: (1) empirically
determined (e.g., based
69
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
on distribution levels, setting a cutoff at a number that removes a specified
or significant portion
of low level reads); (2) any non-zero value; (3) percentage/percentile based;
(4) only strains
whose normalized second marker (i.e., activity) reads is greater than
normalized first marker
(cell count) reads; (5) 1og2 fold change between activity and quantity or cell
count; (6)
normalized second marker (activity) reads is greater than mean second marker
(activity) reads
for entire sample (and/or sample set); and/or any magnitude threshold
described above in
addition to a statistical threshold (i.e., significance testing). The
following example provides
thresholding detail for distributions of RNA-based second marker measurements
with respect to
DNA-based first marker measurements, according to one embodiment.
1001871 The small intestine contents of one male Cobb500 was collected and
subjected to
analysis according to the disclosure. Briefly, the total number of bacterial
cells in the sample was
determined using FACs (e.g., 3004). Total nucleic acids were isolated (e.g.,
3006) from the fixed
small intestine sample. DNA (first marker) and cDNA (second marker) sequencing
libraries were
prepared (e.g., 3007), and loaded onto an ILLUMINA MISEQ. Raw sequencing reads
from each
library were quality filtered, dereplicated, clustered, and quantified (e.g.,
3008). The quantified
strain lists from both the DNA-based and cDNA-based libraries were integrated
with the cell
count data to establish the absolute number of cells of each strain within the
sample (e.g., 3013).
Although cDNA is not necessarily a direct measurement of strain quantity
(i.e., highly active
strains may have many copies of the same RNA molecule), the cDNA-based library
was
integrated with cell counting data in this example to maintain the same
normalization procedure
used for the DNA library.
[00188] After analysis, 702 strains (46 unique) were identified in the cDNA-
based library and
1140 strains were identified in the DNA-based library. If using 0 as the
activity threshold (i.e.
keeping any nonzero value), 57% of strains within this sample that had a DNA-
based first
marker were also associated with a cDNA-based second marker. These strains are
identified
as/deemed the active portion of the microbial community, and only these
strains continue into
subsequent analysis. If the threshold is made more stringent and only strains
whose second
marker value exceed the first marker value are considered active, only 289
strains (25%) meet
the threshold. The strains that meet this threshold correspond to those above
the DNA (first
marker) line in FIG. 3D.
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1001891 The disclosure includes a variety of methods identifying a plurality
of active microbe
strains that influence each other as well as one or more parameters or
metadata, and selecting
identified microbes for use in a microbial ensemble that includes a select
subset of a microbial
community of individual microbial species, or strains of a species, that are
linked in carrying out
or influence a common function, or can be described as participating in, or
leading to, or
associated with, a recognizable parameter, such as a phenotypic trait of
interest (e.g. increased
milk production in a ruminant). The disclosure also includes a variety of
systems and apparatuses
that perform and/or facilitate the methods.
1001901 In some embodiments, the method, comprises: obtaining at least two
samples sharing at
least one common characteristic (such as sample geolocation, sample type,
sample source,
sample source individual, sample target animal, sample time, breed, diet,
temperature, etc.) and
having a least one different characteristic (such as sample
geolocation/temporal location, sample
type, sample source, sample source individual, sample target animal, sample
time, breed, diet,
temperature, etc., different from the common characteristic). For each sample,
detecting the
presence of one or more microorganism types, determining a number of each
detected
microorganism type of the one or more microorganism types in each sample; and
measuring a
number of unique first markers in each sample, and quantity thereof, each
unique first marker
being a marker of a microorganism strain. This is followed by integrating the
number of each
microorganism type and the number of the first markers to yield the absolute
cell count of each
microorganism strain present in each sample; measuring at least one unique
second marker for
each microorganism strain based on a specified threshold to determine an
activity level for that
microorganism strain in each sample; filtering the absolute cell count by the
determined activity
to provide a set or list of active microorganisms strains and their respective
absolute cell counts
for each of the at least two samples; comparing the filtered absolute cell
counts of active
microorganisms strains for each of the at least two samples with each other
and with at least one
measured metadata for each of the at least two samples and categorizing the
active
microorganism strains into one of at least two groups, at least three groups,
at least four groups,
at least five groups, at least six groups, at least seven groups, at least
eight groups, at least nine
groups, at least 10 groups, at least 15 groups, at least 20 groups, at least
25 groups, at least 50
groups, at least 75 groups, or at least 100 groups, based on predicted
function and/or chemistry.
For example, the comparison can be network analysis that identifies the ties
between the
71
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
respective microbial strains and between each microbial strain and metadata,
and/or between the
metadata and the microbial strains. At least one microorganism can be selected
from the at least
two groups, and combined to form an ensemble of microorganisms configured to
alter a property
corresponding to the at least one metadata (e.g., a property in a target, such
as milk production in
a cow or cow population). Forming the ensemble can include isolating the
microorganism strain
or each microorganism strain, selecting a previously isolated microorganism
strain based on the
analysis, and/or incubating/growing specific microorganism strains based on
the analysis, and
combining the strains, including at particular amounts/counts and/or ratios
and/or
media/carrier(s) based on the application, to form the microbial ensemble. The
ensemble can
include an appropriate medium, carrier, and/or pharmaceutical carrier that
enables delivery of the
microorganisms in the ensemble in such a way that they can influence the
recipient (e.g.,
increase milk production).
[00191] Measurement of the number of unique first markers can include
measuring the number
of unique genomic DNA markers in each sample, measuring the number of unique
RNA markers
in each sample, measuring the number of unique protein markers in each sample,
and/or
measuring the number of unique metabolite markers in each sample (including
measuring the
number of unique lipid markers in each sample and/or measuring the number of
unique
carbohydrate markers in each sample).
[00192] In some embodiments, measuring the number of unique first markers, and
quantity
thereof, includes subjecting genomic DNA from each sample to a high throughput
sequencing
reaction and/or subjecting genomic DNA from each sample to metagenome
sequencing. The
unique first markers can include at least one of an mRNA marker, an siRNA
marker, and/or a
ribosomal RNA marker. The unique first markers can additionally or
alternatively include at
least one of a sigma factor, a transcription factor, nucleoside associated
protein, and/or metabolic
enzyme.
[00193] In some embodiments, measuring the at least one unique second marker
includes
measuring a level of expression of the at least one unique second marker in
each sample, and can
include subjecting mRNA in the sample to gene expression analysis. The gene
expression
analysis can include a sequencing reaction, a quantitative polymerase chain
reaction (qPCR),
metatranscriptome sequencing, and/or transcriptome sequencing.
72
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00194] In some embodiments, measuring the level of expression of the at least
one unique
second marker includes subjecting each sample or a portion thereof to mass
spectrometry
analysis and/or subjecting each sample or a portion thereof to metaribosome
profiling, or
ribosome profiling. The one or more microorganism types includes bacteria,
archaea, fungi,
protozoa, plant, other eukaryote, viruses, viroids, or a combination thereof,
and the one or more
microorganism strains includes one or more bacterial strains, archaeal
strains, fungal strains,
protozoa strains, plant strains, other eukaryote strains, viral strains,
viroid strains, or a
combination thereof. The one or more microorganism strains can be one or more
fungal species
or sub-species, and/or the one or more microorganism strains can be one or
more bacterial
species or sub-species.
[00195] In some embodiments, determining the number of each of the one or more
microorganism types in each sample includes subjecting each sample or a
portion thereof to
sequencing, centrifugation, optical microscopy, fluorescent microscopy,
staining, mass
spectrometry, microfluidics, quantitative polymerase chain reaction (qPCR),
gel electrophoresis,
and/or flow cytometry.
[00196] Unique first markers can include a phylogenetic marker comprising a 5S
ribosomal
subunit gene, a 16S ribosomal subunit gene, a 23S ribosomal subunit gene, a
5.8S ribosomal
subunit gene, a 18S ribosomal subunit gene, a 28S ribosomal subunit gene, a
cytochrome c
oxidase subunit gene, a fi-tubulin gene, an elongation factor gene, an RNA
polymerase subunit
gene, an internal transcribed spacer (ITS), or a combination thereof.
Measuring the number of
unique markers, and quantity thereof, can include subjecting genomic DNA from
each sample to
a high throughput sequencing reaction, subjecting genomic DNA to genomic
sequencing, and/or
subjecting genomic DNA to amplicon sequencing.
[00197] In some embodiments, the at least one different characteristic
includes: a collection
time at which each of the at least two samples was collected, such that the
collection time for a
first sample is different from the collection time of a second sample, a
collection location (either
geographical location difference and/or individual sample target/animal
collection differences) at
which each of the at least two samples was collected, such that the collection
location for a first
sample is different from the collection location of a second sample. The at
least one common
characteristic can include a sample source type, such that the sample source
type for a first
73
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
sample is the same as the sample source type of a second sample. The sample
source type can be
one of animal type, organ type, soil type, water type, sediment type, oil
type, plant type,
agricultural product type, bulk soil type, soil rhizosphere type, plant part
type, and/or the like. In
some embodiments, the at least one common characteristic includes that each of
the at least two
samples are gastrointestinal samples, which can be, in some implementations,
ruminal samples.
In some implementations, the common/different characteristics provided herein
can be, instead,
different/common characteristics between certain samples. In some embodiments,
the at least
one common characteristic includes animal sample source type, each sample
having a further
common characteristic such that each sample is a tissue sample, a blood
sample, a tooth sample,
a perspiration sample, a fingernail sample, a skin sample, a hair sample, a
feces sample, a urine
sample, a semen sample, a mucus sample, a saliva sample, a muscle sample, a
brain sample, or
an organ sample.
1001981 In some embodiments, the above method can further comprise obtaining
at least one
further sample from a target, based on the at least one measured metadata,
wherein the at least
one further sample from the target shares at least one common characteristic
with the at least two
samples. Then, for the at least one further sample from the target, detecting
the presence of one
or more microorganism types, determining a number of each detected
microorganism type of the
one or more microorganism types, measuring a number of unique first markers
and quantity
thereof, integrating the number of each microorganism type and the number of
the first markers
to yield the absolute cell count of each microorganism strain present,
measuring at least one
unique second marker for each microorganism strain to determine an activity
level for that
microorganism strain, filtering the absolute cell count by the determined
activity to provide a set
or list of active microorganisms strains and their respective absolute cell
counts for the at least
one further sample from the target. In such embodiments, the selection of the
at least one
microorganism strain from the at least two groups is based on the set or list
of active
microorganisms strain(s) and the/their respective absolute cell counts for the
at least one further
sample from the target such that the formed ensemble is configured to alter a
property of the
target that corresponds to the at least one metadata. For example, using such
an implementation,
a microbial ensemble could be identified from samples taken from Holstein
cows, and a target
sample taken from a Jersey cow or water buffalo, where the analysis identified
the same,
74
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
substantially similar, or similar network relationships between the same or
similar
microorganism strains from the original sample and the target sample(s).
[00199] In some embodiments, comparing the filtered absolute cell counts of
active
microorganisms strains for each of the at least two samples with at least one
measured metadata
or additional active microorganism strain for each of the at least two samples
includes
determining the co-occurrence of the one or more active microorganism strains
in each sample
with the at least one measured metadata or additional active microorganism
strain. The at least
one measured metadata can include one or more parameters, wherein the one or
more parameters
is at least one of sample pH, sample temperature, abundance of a fat,
abundance of a protein,
abundance of a carbohydrate, abundance of a mineral, abundance of a vitamin,
abundance of a
natural product, abundance of a specified compound, bodyweight of the sample
source, feed
intake of the sample source, weight gain of the sample source, feed efficiency
of the sample
source, presence or absence of one or more pathogens, physical
characteristic(s) or
measurement(s) of the sample source, production characteristics of the sample
source, or a
combination thereof. Parameters can also include abundance of whey protein,
abundance of
casein protein, and/or abundance of fats in milk produced by the sample
source.
[00200] In some embodiments, determining the co-occurrence of the one or more
active
microorganism strains and the at least one measured metadata or additional
active
microorganism strain in each sample can include creating matrices populated
with linkages
denoting metadata and microorganism strain associations in two or more sample
sets, the
absolute cell count of the one or more active microorganism strains and the
measure of the one
or more unique second markers to represent one or more networks of a
heterogeneous microbial
community or communities. Determining the co-occurrence of the one or more
active
microorganism strains and the at least one measured metadata or additional
active
microorganism strain and categorizing the active microorganism strains can
include network
analysis and/or cluster analysis to measure connectivity of each microorganism
strain within a
network, the network representing a collection of the at least two samples
that share a common
characteristic, measured metadata, and/or related environmental parameter. The
network analysis
and/or cluster analysis can include linkage analysis, modularity analysis,
robustness measures,
betweenness measures, connectivity measures, transitivity measures, centrality
measures, or a
combination thereof. The cluster analysis can include building a connectivity
model, subspace
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
model, distribution model, density model, and/or a centroid model. Network
analysis can, in
some implementations, include predictive modeling of network(s) through link
mining and
prediction, collective classification, link-based clustering, relational
similarity, a combination
thereof, and/or the like. The network analysis can comprise differential
equation based modeling
of populations and/or Lotka-Volterra modeling. The analysis can be a heuristic
method. In some
embodiments, the analysis can be the Louvain method. The network analysis can
include
nonparametric methods to establish connectivity between variables, and/or
mutual information
and/or maximal information coefficient calculations between variables to
establish connectivity.
1002011 For some embodiments, the method for forming an ensemble of active
microorganism
strains configured to alter a property or characteristic in an environment
based on two or more
sample sets that share at least one common or related environmental parameter
between the two
or more sample sets and that have at least one different environmental
parameter between the
two or more sample sets, each sample set comprising at least one sample
including a
heterogeneous microbial community, wherein the one or more microorganism
strains is a
subtaxon of one or more organism types, comprises: detecting the presence of a
plurality of
microorganism types in each sample; determining the absolute number of cells
of each of the
detected microorganism types in each sample; and measuring the number of
unique first markers
in each sample, and quantity thereof, wherein a unique first marker is a
marker of a
microorganism strain. Then, at the protein or RNA level, measuring the level
of expression of
one or more unique second markers, wherein a unique second marker is a marker
of activity of a
microorganism strain, determining activity of the detected microorganism
strains for each sample
based on the level of expression of the one or more unique second markers
exceeding a specified
threshold, calculating the absolute cell count of each detected active
microorganism strains in
each sample based upon the quantity of the one or more first markers and the
absolute number of
cells of the microorganism types from which the one or more microorganism
strains is a
subtaxon, wherein the one or more active microorganism strains expresses the
second unique
marker above the specified threshold. The co-occurrence of the active
microorganism strains in
the samples with at least one environmental parameter is then determined based
on maximal
information coefficient network analysis to measure connectivity of each
microorganism strain
within a network, wherein the network is the collection of the at least two or
more sample sets
with at least one common or related environmental parameter. A plurality of
active
76
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
microorganism strains from the one or more active microorganism strains is
selected based on
the network analysis, and an ensemble of active microorganism strains is
formed from the
selected plurality of active microorganism strains, the ensemble of active
microorganism strains
configured to selectively alter a property or characteristic of an environment
when the ensemble
of active microorganism strains is introduced into that environment For some
implementations,
at least one measured indicia of at least one common or related environmental
factor for a first
sample set is different from a measured indicia of the at least one common or
related
environmental factor for a second sample set. For example, if the
samples/sample sets are from
cows, the first sample set can be from cows fed on a grass diet, while the
second sample set can
be from cows fed on a corn diet While one sample set could be a single sample,
it could
alternatively be a plurality of samples, and a measured indicia of at least
one common or related
environmental factor for each sample within a sample set is substantially
similar (e.g., samples in
one set all taken from a herd on grass feed), and an average measured indicia
for one sample set
is different from the average measured indicia from another sample set (first
sample set is from a
herd on grass feed, and the second sample set is samples from a herd on corn
feed). There may
be additional difference and similarities that are taken into account in the
analysis, such as
differing breeds, differing diets, differing performance, differing age,
differing feed additives,
differing growth stage, differing physiological characteristics, differing
state of health, differing
elevations, differing environmental temperatures, differing season, different
antibiotics, etc.
While in some embodiments each sample set comprises a plurality of samples,
and a first sample
set is collected from a first population and a second sample set is collected
from a second
population, in additional or alternative embodiments, each sample set
comprises a plurality of
samples, and a first sample set is collected from a first population at a
first time and a second
sample set is collected from the first population at a second time different
from the first time. For
example, the first sample set could be taken at a first time from a herd of
cattle while they were
being feed on grass, and a second sample set could be taken at a second time
(e.g., 2 months
later), where the herd had been switched over to corn feed right after the
first sample set was
taken. In such embodiments, the samples can be collected and the analysis
performed on the
population, and/or can include specific reference to individual animals so
that the changes that
happened to individual animals over the time period could be identified, and a
finer level of data
granularity provided. In some embodiments, a method for forming a synthetic
ensemble of active
77
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
microorganism strains configured to alter a property in a biological
environment, based on two
or more samples (or sample sets, each set comprising at least one sample),
each having a
plurality of environmental parameters (and/or metadata), at least one
parameter of the plurality of
environmental parameters being a common environmental parameter that is
similar between the
two or more samples or sample sets and at least one environmental parameter
being a different
environmental parameter that is different between each of the two or more
samples or sample
sets, each sample set including at least one sample comprising a heterogeneous
microbial
community obtained from a biological sample source, at least one of the active
microorganism
strains being a subtaxon of one or more organism types, comprises: detecting
the presence of a
plurality of microorganism types in each sample; determining the absolute
number of cells of
each of the detected microorganism types in each sample; measuring the number
of unique first
markers in each sample, and quantity thereof, a unique first marker being a
marker of a
microorganism strain; measuring the level (e.g., level of expression) of one
or more unique
second markers, wherein a unique second marker is a marker of activity of a
microorganism
strain; determining activity of each of the detected microorganism strains for
each sample based
on the level (e.g., level of expression) of the one or more unique second
markers exceeding a
specified threshold to identify one or more active microorganism strains;
calculating the absolute
cell count of each detected active microorganism strain in each sample from
the quantity
(relative quantity, proportional quantity, percentage quantity, etc.) of each
of the one or more
unique first markers and the absolute number of cells of the respective or
corresponding
microorganism types from which the one or more microorganism strains is a
subtaxon (wherein
the calculating is mathematical function such as multiplication, dot operator,
and/or other
operation), the one or more active microorganism strains having or expressing
one or more
unique second markers above the specified threshold; analyzing the active
microorganism strains
of the two or more sample sets, the analyzing including conducting
nonparametric network
analysis of each of the active microorganism strains for each of the two or
more sample sets, the
at least one common environmental parameter, and the at least one different
environmental
parameter, the nonparametric network analysis including determining the
maximal information
coefficient score between each active microorganism strain and every other
active
microorganism strain and determining the maximal information coefficient score
between each
active microorganism strain and the at least one different environmental
parameter; selecting a
78
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
plurality of active microorganism strains from the one or more active
microorganism strains
based on the nonparametric network analysis; and forming a synthetic ensemble
of active
microorganism strains comprising the selected plurality of active
microorganism strains and a
microbial carrier medium, the ensemble of active microorganism strains
configured to selectively
alter a property of a biological environment when the synthetic ensemble of
active
microorganism strains is introduced into that biological environment.
Depending on the
embodiment or implementation, the at least two samples or sample sets can
comprise three
samples, four samples, five samples, six samples, seven samples, eight
samples, nine samples,
ten samples, eleven samples, twelve samples, thirteen samples, fourteen
samples, fifteen
samples, sixteen samples, seventeen samples, eighteen samples, nineteen
samples, twenty
samples, twenty one samples, twenty two samples, twenty three samples, twenty
four samples,
twenty five samples, twenty six samples, twenty seven samples, twenty eight
samples, twenty
nine samples, thirty samples, thirty five samples, forty samples, forty five
samples, fifty samples,
sixty samples, seventy samples, eighty samples, ninety samples, one hundred
samples, one
hundred fifty samples, two hundred samples, three hundred samples, four
hundred samples, five
hundred samples, six hundred samples, and/or the like. The total number of
samples can,
depending on the embodiment/implementation, can be less than 5, from 5 to 10,
10 to 15, 15 to
20, 20 to 30, 30 to 40, 40 to 50, 50 to 60, 60 to 70, 70 to 80, 80 to 90, 90
to 100, less than 100,
more than 100, less than 200 more than 200, less than 300, more than 300, less
than 400, more
than 400, less than 500, more than 500, less than 1000, more than 1000, less
than 5000, less than
10000, less than 20000, and so forth.
[00202] In some embodiments, at least one common or related environmental
factor includes
nutrient information, dietary information, animal characteristics, infection
information, health
status, and/or the like.
[00203] The at least one measured indicia can include sample pH, sample
temperature,
abundance of a fat, abundance of a protein, abundance of a carbohydrate,
abundance of a
mineral, abundance of a vitamin, abundance of a natural product, abundance of
a specified
compound, bodyweight of the sample source, feed intake of the sample source,
weight gain of
the sample source, feed efficiency of the sample source, presence or absence
of one or more
pathogens, physical characteristic(s) or measurement(s) of the sample source,
production
characteristics of the sample source, abundance of whey protein in milk
produced by the sample
79
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
source, abundance of casein protein produced by the sample source, and/or
abundance of fats in
milk produced by the sample source, or a combination thereof.
[00204] Measuring the number of unique first markers in each sample can,
depending on the
embodiment, comprise measuring the number of unique genomic DNA markers,
measuring the
number of unique RNA markers, and/or measuring the number of unique protein
markers. The
plurality of microorganism types can include one or more bacteria, archaea,
fungi, protozoa,
plant, other eukaryote, virus, viroid, or a combination thereof.
[00205] In some embodiments, determining the absolute number of each of the
microorganism
types in each sample includes subjecting the sample or a portion thereof to
sequencing,
centrifugation, optical microscopy, fluorescent microscopy, staining, mass
spectrometry,
microfluidics, quantitative polymerase chain reaction (qPCR), gel
electrophoresis and/or flow
cytometry. In some embodiments, one or more active microorganism strains is a
subtaxon of one
or more microbe types selected from one or more bacteria, archaea, fungi,
protozoa, plant, other
eukaryote, virus, viroid, or a combination thereof. In some embodiments, one
or more active
microorganism strains is one or more bacterial strains, archaeal strains,
fungal strains, protozoa
strains, plant strains, other eukaryote strains, viral strains, viroid
strains, or a combination
thereof. In some embodiments, one or more active microorganism strains is one
or more bacterial
species or subspecies. In some embodiments, one or more active microorganism
strains is one or
more fungal species or subspecies.
[00206] In some embodiments, at least one unique first marker comprises a
phylogenetic
marker comprising a 5S ribosomal subunit gene, a 16S ribosomal subunit gene, a
23S ribosomal
subunit gene, a 5.8S ribosomal subunit gene, a 18S ribosomal subunit gene, a
28S ribosomal
subunit gene, a cytochrome c oxidase subunit gene, a beta-tubulin gene, an
elongation factor
gene, an RNA polymerase subunit gene, an internal transcribed spacer (ITS), or
a combination
thereof.
[00207] In some embodiments, measuring the number of unique first markers, and
quantity
thereof, comprises subjecting genomic DNA from each sample to a high
throughput sequencing
reaction, and/or subjecting genomic DNA from each sample to metagenome
sequencing. In some
implementations, unique first markers can include an mRNA marker, an siRNA
marker, and/or a
ribosomal RNA marker. In some implementations, unique first markers can
include a sigma
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
factor, a transcription factor, nucleoside associated protein, metabolic
enzyme, or a combination
thereof.
[00208] In some embodiments, measuring the level of expression of one or more
unique second
markers comprises subjecting mRNA in each sample to gene expression analysis,
and in some
implementations, gene expression analysis comprises a sequencing reaction. In
some
implementations, the gene expression analysis comprises a quantitative
polymerase chain
reaction (qPCR), metatranscriptome sequencing, and/or transcriptome
sequencing.
[00209] In some embodiments, measuring the level of expression of one or more
unique second
markers includes subjecting each sample or a portion thereof to mass
spectrometry analysis,
metaribosome profiling, and/or ribosome profiling.
1002101 In some embodiments, measuring the level of expression of the at least
one or more
unique second markers includes subjecting each sample or a portion thereof to
metaribosome
profiling or ribosome profiling (Ribo-Seq) (see, e.g., Ingolia, N.T., S.
Ghaemmaghami, J.R
Newman, and J.S. Weissman, 2009, "Genome-wide analysis in vivo of translation
with
nucleotide resolution using ribosome profiling' Science 324:218-223; Ingolia,
N.T., 2014,
"Ribosome profiling: new views of translation, from single codons to genome
scale" Nat. Rev.
Genet. 15:205-213; each of which is incorporated by reference in it entirety
for all purposes).
Ribo-seq is a molecular technique that can be used to determine in vivo
protein synthesis at the
genome-scale. This method directly measures which transcripts are being
actively translated via
footprinting ribosomes as they bind and interact with mRNA. The bound mRNA
regions are then
processed and subjected to high-throughput sequencing reactions. Ribo-seq has
been shown to
have a strong correlation with quantitative proteomics (see, e.g., Li, G.W.,
D. Burkhardt, C.
Gross, and J.S. Weissman. 2014 "Quantifying absolute protein synthesis rates
reveals principles
underlying allocation of cellular resources" Cell 157:624-635, the entirety of
which is herein
expressly incorporated by reference).
[00211] The source type for the samples can be one of animal, soil, air,
saltwater, freshwater,
wastewater sludge, sediment, oil, plant, an agricultural product, bulk soil,
soil rhizosphere, plant
81
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
part, vegetable, an extreme environment, or a combination thereof. In some
implementations,
each sample is a digestive tract and/or ruminal sample. In some
implementations, samples can be
tissue samples, blood samples, tooth samples, perspiration samples, fingernail
samples, skin
samples, hair samples, feces samples, urine samples, semen samples, mucus
samples, saliva
samples, muscle samples, brain samples, tissue samples, and/or organ samples.
[00212] Depending on the implementation, a microbial ensemble of the
disclosure can comprise
two or more substantially pure microbes or microbe strains, a mixture of
desired
microbes/microbe strains, and can also include any additional components that
can be
administered to a target, e.g., for restoring microbiota to an animal.
Microbial ensembles made
according to the disclosure can be administered with an agent to allow the
microbes to survive a
target environment (e.g., the gastrointestinal tract of an animal, where the
ensemble is configured
to resist low pH and to grow in the gastrointestinal environment). In some
embodiments,
microbial ensembles can include one or more agents that increase the number
and/or activity of
one or more desired microbes or microbe strains, said strains being present or
absent from the
microbes/strains included in the ensemble. Non-limiting examples of such
agents include
fructooligosaccharides (e.g., oligofructose, inulin, inulin-type fructans),
galactooligosaccharides,
amino acids, alcohols, and mixtures thereof (see Ramirez-Farias et al. 2008.
Br. J. Nutr. 4:1-10
and Pool-Zobel and Sauer 2007. J. Nutr. 137:2580-2584 and supplemental, each
of which is
herein incorporated by reference in their entireties for all purposes).
[00213] Microbial strains identified by the methods of the disclosure can be
cultured/grown
prior to inclusion in an ensemble. Media can be used for such growth, and can
include any
medium suitable to support growth of a microbe, including, by way of non-
limiting example,
natural or artificial including gastrin supplemental agar, LB media, blood
serum, and/or tissue
culture gels. It should be appreciated that the media can be used alone or in
combination with
one or more other media. It can also be used with or without the addition of
exogenous nutrients.
The medium can be modified or enriched with additional compounds or
components, for
example, a component which may assist in the interaction and/or selection of
specific groups of
microorganisms and/or strains thereof. For example, antibiotics (such as
penicillin) or sterilants
(for example, quaternary ammonium salts and oxidizing agents) could be present
and/or the
physical conditions (such as salinity, nutrients (for example organic and
inorganic minerals (such
82
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
as phosphorus, nitrogenous salts, ammonia, potassium and micronutrients such
as cobalt and
magnesium), pH, and/or temperature) could be modified.
1.002141 As discussed above, systems and apparatuses can be configured
according to the
disclosure, and in some embodiments, can comprise a processor and memory, the
memory
storing processor-readable/issuable instructions to perform the method(s). In
one embodiment, a
system and/or apparatus are configured to perform the method. Also disclosed
are processor-
implementations of the methods, as discussed with reference for FIG 3A. For
example, a
processor-implemented method, can comprise: receiving sample data from at
least two samples
sharing at least one common characteristic and having a least one different
characteristic; for
each sample, determining the presence of one or more microorganism types in
each sample;
determining a number of cells of each detected microorganism type of the one
or more
microorganism types in each sample; determining a number of unique first
markers in each
sample, and quantity thereof, each unique first marker being a marker of a
microorganism strain;
integrating, via one or more processors, the number of each microorganism type
and the number
of the first markers to yield the absolute cell count of each microorganism
strain present in each
sample; determining an activity level for each microorganism strain in each
sample based on a
measure of at least one unique second marker for each microorganism strain
exceeding a
specified threshold, a microorganism strain being identified as active if the
measure of at least
one unique second marker for that strain exceeds the corresponding threshold;
filtering the
absolute cell count of each microorganism strain by the determined activity to
provide a list of
active microorganisms strains and their respective absolute cell counts for
each of the at least
two samples; analyzing via one or more processors the filtered absolute counts
of active
microorganisms strains for each of the at least two samples with at least one
measured metadata
or additional active microorganism strain for each of the at least two samples
and categorizing
the active microorganism strains based on function, predicted function, and/or
chemistry;
identifying a plurality of active microorganism strains based on the
categorization; and
outputting the identified plurality of active microorganism strains for
assembling an active
microorganism ensemble configured to, when applied to a target, alter a
property of the target
corresponding to the at least one measured metadata. In some embodiments, the
output can be
utilized in the generation, synthesis, evaluation, and/or testing of synthetic
and/or transgenic
microbes and microbe strains. Some embodiments can include a processor-
readable non-
83
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
transitory computer readable medium that stores instructions for performing
and/or facilitating
execution of the method(s). In some embodiments, analysis and screening
methods, apparatuses,
and systems according to the disclosure can be used for identifying
problematic microorganisms
and strains, such as pathogens, as discussed in Example 4 below. In such
situations, a known
symptom metadata, such as lesion score, would be used in the network analysis
of the samples.
[00215] The state and phenotype of a host can be inherently linked to the
composition of the
microbiome residing within the host. Measurements of these compositions can be
learned in
relation to host data to identify biomarkers to accurately predict patient
outcomes and state shifts.
Diagnostic tools used to determine states can utilize readily obtained samples
and are applied and
analyzed in short periods of time, thus, in some embodiments, making them a
candidates for the
replacement of methods that rely on cultivation.
1002161 There are a variety of methods that can be utilized for the
measurement of the
genotype/phenotype of the microbiome, including but not limited to
metabolomics, amplicon
metagenomics, metagenomics, metatranscriptomics, and/or proteomics. However,
each
measurement is resolved in tables where rows represent samples and columns
represent the items
of measure. For example, amplicon metagenomics resolves in a table of samples
in the rows and
OTUs (i.e., microbes) in the columns where the table is populated by the
measurement in that
sample. In some instances, the measured variable is called a feature where the
table has the
dimensions of samples by features. The table of measurements can be referred
to as the target
data while external data about each sample is referred to as labels. The label
data can be ordered
match to the target rows and contains at least 1 column(s).
[00217] According to some embodiments, the first step in some diagnostic
methods involves
preprocessing target datasets. A variety of possible normalization methods can
be used in
measurement-specific cases and even more for measurement/model-specific cases.
In such cases,
tables may contain gross outliers where one sample is skewed by an abundant
feature not found
in other samples. Samples that contain gross outliers can cause models to
perform poorly.
Disclosed herein are a variety of methods to address outliers, such as scaling
datasets to
minimize their effects, or removing them entirely (see e.g., Iglewicz 1983;
Art et al. 1982;
Janssen et al. 1995; Girman 1994; McLachlan and Peel 2004; the entirety of
each being herein
expressly incorporated by reference for all purposes). Outliers can also be
produced from sparse
data where many values are missing, which can be common in biological
measurements. This
84
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
can be corrected through matrix completion, decomposition, and/or other
methodologies that
allows missing values to be approximated (see, e.g., Keshavan et al. 2009;
Kapur et al. 2016;
Mazumder et al. 2010, the entirety of each being herein expressly incorporated
by reference for
all purposes). In cases where absolute quantities are unknown, scaling can be
performed in
compositions, e.g., using centered log-ratio transforms and inverse log-ratio
transforms (see, e.g.,
Morton et al. 2017). In some cases the signal pertaining to a specific set of
labels can depleted of
non-relevant features through feature selections (see, e.g., Baraniuk 2007).
Feature selection
leverages measures of relationships, such as MIC and Hoffding. However, there
are many
methods for feature extraction whereby features deemed irrelevant are either
removed or lowly
weighted, and features of high importance are highly weighted.
[00218] According to some embodiments, machine learning can be utilized as
part of the
disclosed methods, in particular, it can be used both to determine mechanisms
in target data
related to labels, or discover biomarkers in target data related to labels.
Machine learning can be
sub-grouped into supervised machine learning and unsupervised machine learning
methods.
Supervised machine learning directly integrates labels into the modeling
process both for
development and validation of the model. Unsupervised machine learning
describes the class of
machine learning where labels are not known or incorporated and data is
analyzed based purely
on target data characteristics.
[00219] Unsupervised machine learning incorporates many methods for measuring
the inherent
structure of the target data between samples or features. The main goal of
most unsupervised
machine learning methods, such as Manifold learning (Criminisi et al. 2012),
Clustering (Kluger
et al. 2003), and Decompositions (Bouwmans et al. 2015), is to determine the
number of inherent
labels in the data. The most common use of these methods in diagnostic tools
is in
dimensionality reduction where samples in the target data can be viewed in a
lower dimension
that can be visualized (i.e. 1-3 dimensions). In the microbiome the most
common dimensionality
methods used are Principal Coordinates Analysis (PCoA) on differing distance
matrices
(Lozupone et al. 2011), Principal Component Analysis (Jolliffe 1986), and
Linear Discriminant
Analysis (Ye et al. 2005). Furthermore, in all but the case of PCoA
dimensionality reduction
techniques can be used as a preprocessing step to supervised machine learning.
[00220] While supervised machine learning is a broad classification of
methods, particular
methods disclosed herein are especially useful for the microbiome-related
analyses of the
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
disclosure, including but not limited to the following. Within the class of
supervised machine
learning, and the category of predictive models two sub categories exist
between regression and
classification. Regression describes the instance where labels are continuous.
Classification can
be binary in the case of two label possibilities or multi-class where several
possible labels exist.
In any manifestation classification each label must occur more than once in
any given column.
1002211 In some embodiments, target data is preprocessed as necessary to
maximize model
optimization and labels data is processed to contain no missing entries. Each
column in the labels
data is then separated and evaluated as either being continuous regression,
binary classification
or multi-class classification. Depending on the subclass a method is
determined commonly using
but not limited to Random Forests (Breiman 2001), Nearest Neighbors (Indyk and
Motwani
1998), Neural Networks (supervised) (Moller 1993), Support Vector Machines
(Smola and
Scholkopf 2004), or a Gaussian Process (Neumann et al. 2009). The model is
cross-validated
through splitting the target and label data into a training dataset (for
example, 80%) and the test
dataset (for example, 20%). This is done iteratively (folds) shuffling
features and samples in each
iteration. On each iteration a metric of model performance is calculated
between the predictions
from the training data to the target data (Taylor 2001). The performance
metric is used to both
tune the model's parameters also called hype-parameter tuning and to validate
the model's
prediction power.
[00222] Following the production of models with high prediction power the
development of
automated prediction platforms are produced as well as high-throughput
biomarker probes. In
some embodiments, the whole community of measurements is utilized to give
accurate results
where the input measurement is used to produce predictions. The predictive
model developed is
used to predict labels from new data after being trained on the entire known
dataset. The
predictions can be produced with an associated confidence and probability
distributions. This can
be done in an automated function from input sample to prediction
visualization. In some
embodiments feature selection or the model reveals a small sub group or a
single feature that has
high prediction power. In such an embodiment, a high-throughput probe can be
developed to
quickly identify the feature in relation to the prediction. For example, in
the case of amplicon
metagenomics, single microbes or a small community of microbes can directly
determine state
and predict patient outcomes. A high-throughput probe can be a real time PCR
primer that can
reveal the abundance or presence of specific features.
86
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00223] It is intended that the systems and methods described herein can be
performed by
software (stored in memory and/or executed on hardware), hardware, or a
combination thereof.
Hardware components and/or modules can include, for example, a general-purpose
processor, a
field programmable gate array (FPGA), and/or an application specific
integrated circuit (AS1C).
Software components and/or modules (executed on hardware) can be expressed in
a variety of
software languages (e.g., computer code), including Unix utilities, C, C++,
JavaTm, JavaScript
(e.g., ECMAScript 6), Ruby, SQL, SAS , the R programming language/software
environment,
Visual BasicTM, and other object-oriented, procedural, or other programming
language and
development tools. Examples of computer code include, but are not limited to,
micro-code or
micro-instructions, machine instructions, such as produced by a compiler, code
used to produce a
web service, and files containing higher-level instructions that are executed
by a computer using
an interpreter. Additional examples of computer code include, but are not
limited to, control
signals, encrypted code, and compressed code.
[00224] Some embodiments described herein relate to devices with a non-
transitory computer-
readable medium (also can be referred to as a non-transitory processor-
readable medium or
memory) having instructions or computer code thereon for performing various
computer-
implemented operations. The computer-readable medium (or processor-readable
medium) is
non-transitory in the sense that it does not include transitory propagating
signals per se (e.g., a
propagating electromagnetic wave carrying information on a transmission medium
such as space
or a cable). The media and computer code (also can be referred to as code) may
be those
designed and constructed for the specific purpose or purposes. Examples of non-
transitory
computer-readable media include, but are not limited to: magnetic storage
media such as hard
disks, floppy disks, and magnetic tape; optical storage media such as Compact
Disc/Digital
Video Discs (CD/DVDs), Compact Disc-Read Only Memories (CD-ROMs), and
holographic
devices; magneto-optical storage media such as optical disks; carrier wave
signal processing
components and/or modules; and hardware devices that are specially configured
to store and
execute program code, such as Application-Specific Integrated Circuits
(AS1Cs), Programmable
Logic Devices (PLDs), Read-Only Memory (ROM) and Random-Access Memory (RAM)
devices. Other embodiments described herein relate to a computer program
product, which can
include, for example, the instructions and/or computer code discussed herein.
87
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00225] While various embodiments of FIG. 3A have been described above, it
should be
understood that they have been presented by way of example only, and not
limitation. Where
methods and steps described above indicate certain events occurring in certain
order, the
ordering of certain steps can be modified. Additionally, certain of the steps
can be performed
concurrently in a parallel process when possible, as well as performed
sequentially as described
above. Although various embodiments have been described as having particular
features and/or
combinations of components, other embodiments are possible having any
combination or sub-
combination of any features and/or components from any of the embodiments
described herein.
Furthermore, although various embodiments are described as having a particular
entity
associated with a particular compute device, in other embodiments different
entities can be
associated with other and/or different compute devices.
EXPERIMENTAL DATA AND EXAMPLES
[00226] The present disclosure is further illustrated by reference to the
following Experimental
Data and Examples. However, it should be noted that these Experimental Data
and Examples,
like the embodiments described above, are illustrative and are not to be
construed as restricting
the scope of the disclosure in any way.
E pie 1
1002271 Reference is made to steps provided at FIG. 2.
[00228] 2000: Cells from a cow rumen sample are sheared off matrix. This can
be done via
blending or mixing the sample vigorously through sonication or vortexing
followed by
differential centrifugation for matrix removal from cells. Centrifugation can
include a gradient
centrifugation step using Nycodenz or Percoll.
[00229] 2001: Organisms are stained using fluorescent dyes that target
specific organism types.
Flow cytometry is used to discriminate different populations based on staining
properties and
size.
[00230] 2002: The absolute number of organisms in the sample is determined by,
for example,
flow cytometry. This step yields information about how many organism types
(such as bacteria,
archaea, fungi, viruses or protists) are in a given volume.
88
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00231] 2003: A cow rumen sample is obtained and cells adhered to matrix are
directly lysed
via bead beating. Total nucleic acids are purified. Total purified nucleic
acids are treated with
RNAse to obtain purified genomic DNA (gDNA). qPCR is used to simultaneously
amplify
specific markers from the bulk gDNA and to attach sequencing adapters and
barcodes to each
marker. The qPCR reaction is stopped at the beginning of exponential
amplification to minimize
PCR-related bias. Samples are pooled and multiplexed sequencing is performed
on the pooled
samples using an Illumina Miseq.
1002321 2004: Cells from a cow rumen sample adhered to matrix are directly
lysed via bead
beating. Total nucleic acids are purified using a column-based approach. Total
purified nucleic
acids are treated with DNAse to obtain purified RNA. Total RNA is converted to
cDNA using
reverse transcriptase. qPCR is used to simultaneously amplify specific markers
from the bulk
cDNA and to attach sequencing adapters and barcodes to each marker. The qPCR
reaction is
stopped at the beginning of exponential amplification to minimize PCR-related
bias. Samples
are pooled and multiplexed sequencing is performed on the pooled samples using
an Illumina
Miseq.
[00233] 2005: Sequencing output (fastq files) is processed by removing low
quality base pairs
and truncated reads. DNA-based datasets are analyzed using a customized UPARSE
pipeline,
and sequencing reads are matched to existing database entries to identify
strains within the
population. Unique sequences are added to the database. RNA-based datasets are
analyzed using
a customized UPARSE pipeline. Active strains are identified using an updated
database.
[00234] 2006: Using strain identity data obtained in the previous step (2005),
the number of
reads representing each strain is determined and represented as a percentage
of total reads. The
percentage is multiplied by the counts of cells (2002) to calculate the
absolute cell count of each
organism type in a sample and a given volume. Active strains are identified
within absolute cell
count datasets using the marker sequences present in the RNA-based datasets
along with an
appropriate threshold. Strains that do not meet the threshold are removed from
analysis.
[00235] 2007: Repeat 2003-2006 to establish time courses representing the
dynamics of
microbial populations within multiple cow rumens. Compile temporal data and
store the number
of cells of each active organism strain and metadata for each sample in a
quantity or abundance
matrix. Use quantity matrix to identify associations between active strains in
a specific time
89
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
point sample using rule mining approaches weighted with quantity data. Apply
filters to remove
insignificant rules.
[00236] 2008: Calculate cell number changes of active strains over time,
noting directionality of
change (i.e., negative values denoting decreases, positive values denoting
increases). Represent
matrix as a network, with organism strains representing nodes and the quantity
weighted rules
representing edges. Leverage markov chains and random walks to determine
connectivity
between nodes and to define clusters. Filter clusters using metadata in order
to identify clusters
associated with desirable metadata (environmental parameter(s)). Rank target
organism strains
by integrating cell number changes over time and strains present in target
clusters, with highest
changes in cell number ranking the highest.
Example 2
Experimental Design and Materials and Methods
100237l Objective: Determine rumen microbial community constituents that
impact the
production of milk fat in dairy cows.
1002381 Animals: Eight lactating, ruminally cannulated, Holstein cows were
housed in
individual tie-stalls for use in the experiment. Cows were fed twice daily,
milked twice a day,
and had continuous access to fresh water. One cow (cow 1) was removed from the
study after
the first dietary Milk Fat Depression due to complications arising from an
abortion prior to the
experiment.
1002391 Experimental Design and Treatment: The experiment used a crossover
design with
2 groups and 1 experimental period. The experimental period lasted 38 days: 10
days for the
covariateiwash-out period and 28 days for data collection and sampling. The
data collection
period consisted of 10 days of dietary Milk Fat Depression (MFD) and 18 days
of recovery.
After the first experimental period, all cows underwent a 10-day wash out
period prior to the
beginning of period 2.
[00240] Dietary MFD was induced with a total mixed ration (TMR) low in fiber
(29% NDF)
with high starch degradability (70% degradable) and high polyunsaturated fatty
acid levels
(PUFA, 3.7%). The Recovery phase included two diets variable in starch
degradability. Four
cows were randomly assigned to the recovery diet high in fiber (37% NDF), low
in PUFA
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
(2.6%), and high in starch degradability (70% degradable). The remaining four
cows were fed a
recovery diet high in fiber (37% NDF), low in PUFA (2.6%), but low in starch
degradability
(35%).
[00241] During the 10-day covariate and 10-day wash out periods, cows were fed
the high fiber,
low PUFA, and low starch degradability diet.
[00242] Samples and Measurements: Milk yield, dry matter intake, and feed
efficiency were
measured daily for each animal throughout the covariate, wash out, and sample
collection
periods. TMR samples were measured for nutrient composition. During the
collection period,
milk samples were collected and analyzed every 3 days. Samples were analyzed
for milk
component concentrations (milk fat, milk protein, lactose, milk urea nitrogen,
somatic cell
counts, and solids) and fatty acid compositions.
1002431 Rumen samples were collected and analyzed for microbial community
composition and
activity every 3 days during the collection period. The rumen was intensively
sampled 0, 2, 4, 6,
8, 10, 12, 14, 16, 18, 20, and 22 hours after feeding during day 0, day 7, and
day 10 of the dietary
MFD. Similarly, the rumen was intensively sampled 0, 2, 4, 6, 8, 10, 12, 14,
16, 18, 20, and 22
hours after feeding on day 16 and day 28 during the recovery period. Rumen
contents were
analyzed for pH, acetate concentration, butyrate concentration, propionate
concentration, isoacid
concentration, and long chain and CLA isomer concentrations.
[00244] Rumen Sample Preparation and Sequencing: After collection, rumen
samples were
centrifuged at 4,000 rpm in a swing bucket centrifuge for 20 minutes at 4 C.
The supernatant
was decanted, and an aliquot of each rumen content sample (1-2mg) was added to
a sterile
1.7mL tube prefilled with 0.1 mm glass beads. A second aliquot was collected
and stored in an
empty, sterile 1.7 mL tube for cell counting.
[00245] Rumen samples with glass beads (1' aliquot) were homogenized with bead
beating to
lyse microorganisms. DNA and RNA was extracted and purified from each sample
and prepared
for sequencing on an Illumina Miseq. Samples were sequenced using paired-end
chemistry, with
300 base pairs sequenced on each end of the library. Rumen samples in empty
tubes (2nd aliquot)
were stained and put through a flow cytometer to quantify the number of cells
of each
microorganism type in each sample.
91
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00246] Sequencing Read Processing and Data Analysis: Sequencing reads were
quality
trimmed and processed to identify bacterial species present in the rumen based
on a marker gene.
Count datasets and activity datasets were integrated with the sequencing reads
to determine the
absolute cell numbers of active microbial species within the rumen microbial
community.
Production characteristics of the cow over time, including pounds of milk
produced, were linked
to the distribution of active microorganisms within each sample over the
course of the
experiment using mutual information. Maximal information coefficient (MIC)
scores were
calculated between pounds of milk fat produced and the absolute cell count of
each active
microorganism. Microorganisms were ranked by MIC score, and microorganisms
with the
highest MIC scores were selected as the target species most relevant to pounds
of milk produced.
[00247] Tests cases to determine the impact of count data, activity data, and
count and activity
on the final output were run by omitting the appropriate datasets from the
sequencing analysis.
To assess the impact of using a linear correlation rather than the MIC on
target selection,
Pearson's coefficients were also calculated for pounds of milk fat produced as
compared to the
relative abundance of all microorganisms and the absolute cell count of active
microorganisms.
Results and Discussion
[00248] Relative Abundances vs. Absolute cell counts
[00249] The top 15 target species were identified for the dataset that
included cell count data
(absolute cell count, Table 2) and for the dataset that did not include cell
count data (relative
abundance, Table 1) based on MIC scores. Activity data was not used in this
analysis in order to
isolate the effect of cell count data on final target selection. Ultimately,
the top 8 targets were the
same between the two datasets. Of the remaining 7, 5 strains were present on
both lists in
varying order. Despite the differences in rank for these 5 strains, the
calculated M1C score for
each strain was the identical between the two lists. The two strains present
on the absolute cell
count list but not the relative abundance list, ascus_111 and ascus_288, were
rank 91 and rank
16, respectively, on the relative abundance list. The two strains present on
the relative abundance
list but not the absolute cell count list, ascus_102 and ascus_252, were rank
50 and rank 19,
respectively, on the absolute cell count list. These 4 strains did have
different MIC scores on
each list, thus explaining their shift in rank and subsequent impact on the
other strains in the list.
92
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1002501 Table 1: Top 15 Target Strains using Relative Abundance with no
Activity Filter
Target
Strain MIC Nearest Taxonomy
d:Bacteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.8756),o:Clostridiales(0.
5860),CRuminococcaceae(0.3217),g
ascus_7 0.97384 :Ruminococcus(0.0605)
d:Bacteria(1.0000),p:Firmicutes(0.8349),c:Clostridia(0.52510:Clostridiales(0.27
14),f:Ruminococcaceae(0.1062),g
ascus_82 0.97173 :Saccharofermentans(0.0073)
ascus_209 0.95251 d:Bacteria(1.0000),p:TM7(0.9991),g:TM7_genera
jncertae_sedis(0.8645)
d:Bacteria(1.0000),p:Firmicutes(0.8349),c:Clostridia(0.52510:Clostridiales(0.27
14),f:Ruminococcaceae(0.1242),g
ascus_126 0.91477 :Saccharofermentans(0.0073)
ascus_1366 0.89713 d:Bacteria(1.0000),p:TM7(0.9445),g:TM7_genera
jncertae_sedis(0.0986)
d:Bacteria(0.940 I ),p:Bacteroidetes{0.4304),c:Bacteroidia(0.055 I
Lo:Bacteroidales(0.0198),f:Prevotellaceae{0.006
ascus_1780 0.89466 7),w Prevotella(0.0052)
d:
8acteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.8823),o:Clostridiales(0.62
67),f:Ruminococcaceae(0.2792),g
ascus...64 0.89453 :Ruminococcus(0.0605)
ascus_299 0.88979
d:8acteria(1.0000),p:TM7(0.9963),g:TM7_genera_incertae_sedis(0.5795)
d:8acteria(1.0000),p:Firmicutes(0.9628),c:Clostridia(0.8317},o:Clostridiales(0.
4636},f:Ruminococcaceae(0.2367),g
ascus.. 1.02 0.87095 :Saccharofermentans(0.0283)
d:Bacteria(0.8663),9:8acteroidetes(0.2483),c:Bacteroidia(0.0365),o:Bacteroidale
s(0.0179),f:Porphyromonadacea
ascus...1801 0.87038 e(0.0059),g:Butyriamonas(0.0047)
ascus...295 0.86724
d:8acteria(1.0000),p:SR1(0.9990),g:SRI...genera..incertae..sedis(0.9793)
ascus_1139 0.8598
d:Bacteria(1.0000),p:TM7(0.99S1),g:TM7_genera_incertae_sedis(0.4747)
ascus..127 0.84082 d:Bacteria(1.0000),p:M47(0.9992),g:TM7..genera
Jacertae..sedis(0.8035)
ascus_341 0.8348
d:Bacteria(1.0000),p:TM7(0.9992),g:TIV17..genera...incertae_sedis(0.8035)
d:Bacteria(1.0000),p:Firmicutes(0.9986),c:Clostridia(0.9022),o:Clostridiales(0.
7491),fiLachnospiraceae(0.3642),g:
ascus_252 0.82891 Lachnospiracea_incertae_sedis(0.0859)
[002511 Table 2: Top 15 Target Strains using Absolute cell count with no
Activity Filter
Target
Strain MIC Nearest Taxonomy
d:Bacteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.87560:Clostridiales(0.58
60},f:Ruminococcaceae(0.3217),g
ascus_7 0.97384 :Ruminococcus(0.0605)
d:8acteria(1.0000),p:Firmicutes(0.8349},c:Clostridia(0.5251),o:Clostridiales(0.
2714),fiRuminococcaceae(0.1062),g
ascus_82 0.97173 :Saccharofermentans(0.0073)
ascus_209 0.95251 d:Bacteria(1.0000),p:TM7(0.9991),g:TM7_genera
jncertae_sedis(0.8645)
d:8acteria(1.0000),p:Firmicutes(0.8349),c:Clostridia(0.5251},o:Clostridiales(0.
2714},f:Ruminococcaceae(0.1242),g
ascus_126 0.91701 :Saccharofermentans(0.0073)
ascus_1366 0.89713 d:Bacteria(1.0000),p:TM7(0.9445),g:TM7_genera
_incertae_sedis(0.0986)
d:8acteria(0.9401),p:Bacteroidetes(0.4304},c:Bacteroidia(0.0551),o:Bacteroidale
s(0.0198),f:Prevotellaceae(0.006
ascus_1780 0.89466 7),w Prevotella(0.0052)
d:Bacteria(1.0000),9:Firmicutes(0.9922),c:Clostridia(0.8823),o:Clostridiales(0.
6267),f:Ruminococcaceae(0.2792),g
a5cu5...64 0.89453 :Ruminococcus(0.0605)
ascus_299 0.88979
d:8acteria(1.0000),p:TM7(0.9963),g:TM7_genera_incertae_sedis(0.5795)
d:Bacteria(0.8663),9:8acteroidetes(0.2483),c:Bacteroidia(0.03651,o:Bacteroidale
s(0.0179),EPorphyromonadacea
ascus_1801 0.87038 e(0.0059),g:Butyricimonas(0.0047)
ascus_295 0.86724
d:Bacteria(1.0000),p:SR1(0.9990),g:SRl_genera_incertae_sedis(0.9793)
ascus_1139 0.8598
d:Bacteria(1.0000),p:TM7(0.99S1),g:TM7_genera_incertae_sedis(0.4747)
ascus..127 0.84082
d:Bactena(1.0000),p:TM7(0.9992),g:TM7..generaincertae_sedis(0.8035)
a5cu5..341 I 0.8348
d:Bacteria(1.0000),p:TM7(0.9992),g:TIV17..genera...incertae_sedis(0.8035)
d:Bactena(1.0000),p:Firmicutes(0.7947),c:Clostridia(0.4637),o:Clostridiales(0.2
335),fiRuminococcaceae(0.1062),g
ascus_111 0.83358 :Papillibacter(0.0098)
93
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
d:Bacteria(0.7925),9:3acteroidetes(0.2030},c:Bacteroidia(0.0327),o:Bacteroidale
s(0.0160),f:Porphyromonadacea
ascus_288 0.82833 e(0.0050),g:Butyricimonas(0.0042)
[00252] Integration of cell count data did not always affect the final MIC
score assigned to each
strain. This may be attributed to the fact that although the microbial
population did shift within
the rumen daily and over the course of the 38-day experiment, it was always
within 107-108 cells
per milliliter. Much larger shifts in population numbers would undoubtedly
have a broader
impact on final MIC scores.
[00253] Inactive Species vs. Active Species
[00254] In order to assess the impact of filtering strains based on activity
data, target species
were identified from a dataset that leveraged relative abundance with (Table
3) and without
(Table 1) activity data as well as a dataset that leveraged absolute cell
counts with (Table 4) and
without (Table 2) activity data.
[00255] For the relative abundance case, ascus_126, ascus_1366, ascus_l 780,
ascus_299,
ascus_1139, ascus_127, ascus_341, and ascus_252 were deemed target strains
prior to applying
activity data. These eight strains (53% of the initial top 15 targets) fell
below rank 15 after
integrating activity data. A similar trend was observed for the absolute cell
count case.
Ascus_126, ascus_1366, ascus_1780, ascus_299, ascus_1139, ascus_i 27, and
ascus_341 (46%
of the initial top 15 targets) fell below rank 15 after activity dataset
integration.
[00256] The activity datasets had a much more severe effect on target rank and
selection than
the cell count datasets. When integrating these datasets together, if a sample
is found to be
inactive it is essentially changed to a "0" and not considered to be part of
the analysis. Because of
this, the distribution of points within a sample can become heavily altered or
skewed after
integration, which in turn greatly impacts the final MIC score and thus the
rank order of target
microorganisms.
[00257] Table 3: Top 15 Target Strains using Relative Abundance with Activity
Filter
Target
Strain MIC Nearest Taxonomy
d:Bacteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.8756),o:Clostridiales(0.
5860),CRuminococcaceae(0.3217),g
ascus_7 0.97384 :Ruminococcus(0.0605)
d:Bacteria(1.0000),p:Firmicutes(0.8349),c:Clostridia(0 5253.0:Clostridiales(0
273.4},f:Ruminococcateae(0.1062),g
ascus_82 0.93391 :Saccharofermenkcns(0.0073)
ascus..1.02 0.87095
d:Bacteria(1.0000),p:Firmicutes(0.9628),c:Clostridia(0.8317},o:Clostridiales(0.
4636},f:Ruminococcaceae(0.2367),g
94
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
:5accharofermentan5(0.0283)
ascus_209 0.84421
d:Bacteria(1.0000),p:M47(0.9991),g:TM7_genera_incertae_sedis(0.8645)
d:Bacteria(0.8663),p:Sacteroidetesi0.2483),c:Bacteroidia(0.0365),o.8acteroidale
s(0.0179),f:Porphyromonadacea
ascus_1801 0.82398 e(0.0059),g.Butyricimonasi0.00471
d:8acteria(1.0000),p:Spirochaetes(0.9445},c:Spirochaetes(0.8623),o:Spirochaetal
es(0.5044),f:Spirochaetaceae(0.
ascus_372 0.81735 3217{,g:Spirochaeta(0.0190)
d:Bacteria(1.0000),9:Firmicutes(0.9080),c:Clostridia(0.7704),o:Clostridiales(0.
4230),f:Ruminococcaceae(0.1942),g
ascus_26 0.81081 :Clostridium_IV(0.0144{
d:Sacteria(1.0000),p:Spirochaetesi0.9445),c:Spirochaetes(0.8623),o:Spirochaetal
es(0.5044),Bpirochaetaceae(0.
ascus_180 0.80702 3217),g:Spirochaeta(0.0237)
d:Bacteria(1.0000),p:Firmicutes(0.7036{,c:Clostridia{0.4024),o:Clostridiales(0.
1956),f:Ruminococcaceae(0.0883).g
ascus_32 0.7846 :Hydrogenoanaerobacterium(0.0144)
d:Bacteria(0.7925),p:8acteroidetes(0.2030},c:Bactesoidia(0.0327{,o:8acteroidale
s(0.0160),f:Posphyromonadacea
ascus_288 0.78229 e(0.0050),g:8utysicimonas(0.0042)
d:Bacteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.8823),o:Clostridiales(0.
6267),f:Ruminococcaceae(0.2792),g
ascus_64 0.77514 :Ruminococcus(0.0605)
ascus_295 0.76639
d:Bacteria(1.0000),p:SR1(0.9990{,g:SRl_genera_incertae_sedis(0.9793}
d:Bacteria(1.0000),p:Firmicutes(0.6126),c:Clostridia(0.2851),o:Clostridiales(0.
1324),f:Clostridiaceae_1(0.0208),g:
ascus_546 0.76114 Clostridium_sensu_stricto(0.0066)
d:Bacteria(1.0000),p:Firmicutes(0.9922{,c:Clostridia(0.8756{,o:Clostridiales(0.
5860),CRuminoccoccaceae(0.3642),g
ascus_233 0.75779 :Ruminococcus(0.0478)
d:8acteria(1.0000),p:Firmicutes(0.7947),c:Clostridia(0.4637},o:Clostridiales(0.
2335},f:Ruminococcaceae(0.0883),g
ascus_651 0.74837 :Clostridium_IV(0.0069{
[00258] Table 4: Top 15 Target Strains using Absolute cell count with Activity
Filter
Target
Strain NIIC Nearest Taxonomy
d:Bacteria(1.0000),9:Firmicutes(0.9922),c:Clostridia(0.8756),o:Clostridiales(0.
5860),f:Ruminococcaceae(0.3217),g
ascus_7 0.97384 :Ruminococcus(0.0605)
d:Sacteria(1.0000),p:Firmicutesi0.83491,c:Clostridia10.5251),o:Clostridiales10.
2714),f:Ruminococcaceae(0.1062),g
ascus_82 0.93391 :Saccharofermentans(0.0073)
ascus_209 0.84421
d:8acteria(1.0000),p:TM7(0.9991),g:TM7_genera_incertae.. sedis(0.8645)
d:Sacteria(0.8663),p:Bacteraidetes(0.2483),c:Bacteroidia(0.0365{,o:Bacteroidale
s(0.0179),f:Porphyromonadacea
ascus_1801 0.82398 e(0.0059),g:Butyriamonas(0.0047)
d:Bacteria(1.0000).p:Spirochaetes(0.9445),c:Spirochaetes(0.8623),0:Spirochaetal
es(0.5044).VSpirochaetaceae(0.
ascus_372 0.81735 3217),g:Spirochaeta10.0190)
d:Bacteria(1.0000),p:Firmicutes(0.9080{,c:Clostridia(0.7704},o:Clostridiales(0.
4230},f:Ruminococcaceae(0.1942),g
ascus_26 0.81081 :Clostridium_l V(0.01.44)
d:Bacteria(1.0000),p:Firmicutes(0.9628),c:Clostridia(0.8317),o:Clostridiales(0.
4636),f:Ruminococcaceae(0.2367),g
ascus_102 0.81048 :Saccharofermentans(0.0283)
d:Bacteria(1.0000),p:Firmicutes(0.7947{,c:Clostridia(0.4637),o:Clostridiales(0.
2335),CRuminococcaceae(0.1062),g
ascus_111 0.79035 :Papillibacter10.0098)
d:Bacteria(0.7925{,p:Bacteroidetes{0.2030),c:Bacteroidia(0.0327{,0.8acteroidale
s(0.0160),UPorphyromonadacea
ascus_288 0.78229 e(0.0050),g.3utyricimonas(0.0042{
d:Bacteria(1.0000),9:Firmicutes(0.9922),c:Clostridia(0.8823),o:Clostridiales(0.
6267),f:Ruminococcaceae(0.2792),g
ascu5_64 0.77514 :Ruminococcus(0.0605)
ascus_295 0.76639 d:Bacteria(1.0000{,p:5111(0.9990),8:911_genera
jncertae_sedis(0.9793)
d:8acteria(1.0000),p:Pirmicutes(0.6126),c:Clostridia(0.2851},o:Clostridiales(0.
1324},f:Clostridiaceae_1(0.0208{,g:
ascus_546 0.76114 Ciostridium_sensu_stricto(0.0066)
d:Sacteria(1.0000),p:Firmicutesi0.7036{,c:Clostridia(0.4024),o:Clostridialesi0.
1956{,i:Ruminococcaceae(0.0883),g
ascus_32 0.75068 :1-1ydrogenoanaerobacterium(0.0144)
d:Bacteria(1.0000).p:Firmicutes(0.7947{,c:Clostridia{0.4637),o:Clostridiales{0.
2335),f:Ruminococcaceae(0.0883).g
ascus_651 0.74837 :Clostridium_IV(0.0069)
d:Bacteria(1.0000),p:Firmicutes(0.9922{,c:Clostridia(0.8756},o:Clostridiales(0.
5860},f:Ruminococcaceae(0.3642),g
ascus_233 0.74409 :Ruminococcus(0.0478{
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
100259] Relative Abundances and Inactive vs. Absolute cell counts and Active
[00260] Ultimately, the method defined here leverages both cell count data and
activity data to
identify microorganisms highly linked to relevant metadata characteristics.
Within the top 15
targets selected using both methods (Table 4, Table 1), only 7 strains were
found on both lists.
Eight strains (53%) were unique to the absolute cell count and activity list
The top 3 targets on
both lists matched in both strain as well as in rank. However, two of the
three did not have the
same MIC score on both lists, suggesting that they were influenced by activity
dataset integration
but not enough to upset their rank order.
[00261] Linear Correlations vs. Nonparametric Approaches
[00262] Pearson's coefficients and MIC scores were calculated between pounds
of milk fat
produced and the absolute cell count of active microorganisms within each
sample (Table 5).
Strains were ranked either by MIC (Table 5a) or Pearson coefficient (Table 5b)
to select target
strains most relevant to milk fat production. Both MIC score and Pearson
coefficient are reported
in each case. Six strains were found on both lists, meaning nine (60%) unique
strains were
identified using the MIC approach. The rank order of strains between lists did
not match¨the
top 3 target strains identified by each method were also unique.
[002631 Like Pearson coefficients, the MIC score is reported over a range of 0
to 1, with 1
suggesting a very tight relationship between the two variables. Here, the top
15 targets exhibited
MK scores ranging from 0.97 to 0.74. The Pearson coefficients for the
correlation test case,
however, ranged from 0.53 to 0.45¨substantially lower than the mutual
information test case.
This discrepancy may be due to the differences inherent to each analysis
method. While
correlations are a linear estimate that measures the dispersion of points
around a line, mutual
information leverages probability distributions and measures the similarity
between two
distributions. Over the course of the experiment, the pounds of milk fat
produced changed
nonlinearly (FIG. 4). This particular function may be better represented and
approximated by
mutual information than correlations. To investigate this, the top target
strains identified using
correlation and mutual information, Ascus_713 (Fig. 5) and Ascus_7 (Fig. 6)
respectively, were
plotted to determine how well each method predicted relationships between the
strains and milk
96
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
fat. If two variables exhibit strong correlation, they are represented by a
line with little to no
dispersion of points when plotted against each other. In Fig. 5, Ascus...713
correlates weakly with
milk fat, as indicated by the broad spread of points. Mutual information,
again, measures how
similar two distributions of points are. When Ascus...7 is plotted with milk
fat (Fig. 6), it is
apparent that the two point distributions are very similar.
[002641 The Present Method in Entirety I.'s. Conventional Approaches
1002651 The conventional approach of analyzing microbial communities relies on
the use of
relative abundance data with no incorporation of activity information, and
ultimately ends with a
simple correlation of microbial species to metadata (see, e.g., U.S. Patent
No. 9,206,680, which
is herein incorporated by reference in its entirety for all purposes). Here,
we have shown how the
incorporation of each dataset incrementally influences the final list of
targets. When applied in
its entirety, the method described herein selected a completely different set
of targets when
compared to the conventional method (Tables 5a and Sc). Ascus....3038, the top
target strain
selected using the conventional approach, was plotted against milk fat to
visualize the strength of
the correlation (Fig. 7). Like the previous example, Ascus....3038 also
exhibited a weak
correlation to milk fat.
100266i Table 5: Top 15 Target Strains using Mutual Information or
Correlations
1002671 Table 5a. MC using Absolute cell count with Activity Filter
Target
Strain MEC Pearson Coefficient Nearest Taxonomy
d:Bacteria(1.0000),p:Firrnicutes(0.9922),c:Clostridia(0.8756),o:Clostridiales(0
.5860),
ascus...7 0.97384 0.25282502
f:Ftuminococcaceae(0.3217},e:Ruminococcus(0.0605)
d:Bacteria(1.0000),p:Firmicutes(0.8349),c.Clostridia(0.5251),o:Clostridiales(0.
2714 ),
ascus_82 0.93391 0.42776647
f:Ruminococcaceae(0.10621,8:Saccharafermentans(0.0073)
ascus...209 0.84421 0.3036308
d:Bacieria(1.0000),p:TM7(0.9991),g:TM7...genera..incertae...sedis(0.8645)
d:Bacteria(0.8663),p:Bacteroidetes(0.2483),c:Bacteroidia(0.0365),a:Bacteroidale
s(0.
ascus...1801 0.82398 0.5182261
0179),f:Porphyromorsadaceae(0.0059),g:Butyricirnonas(0.0047)
d:Bacteria(1.0000),p:Spirochaetes(0.9445).c.Spirochaetes(0.8623),o:Spirochaetal
es(
ascus_372 0.81735 0.34172258 0
.5044),f:Spirochaetaceae(0.3217),g:Spirochaeta(0.0190)
d:Bacteria(1.0000),p:Firrnicutes(0.9080),c:Clostridia(0.7704),o:Clostridiales(0
.4230),
a5cus..26 0.81081 0.5300298
f:Ruminococcaceae(0.1942},g:Clostridium..1V(0.0144)
d:Bacteria(1.0000),p:Firmicutes(0.9628),c:Clostridia(0.8317),o:Clostridiales(0.
4636),
ascus..102 0.81048 0.35456932
f:Ruminococcaceae(0.2367),g:Saccharoferrnentans(0.0283)
d:Bacteria(1.0000),p:Firmiattes(0.7947),vCiostridia(0.4637),o.Clostridiales(0.2
335),
ascus_111 1 0.79035 0.45881805
f:Ruminacoccaceae(0.1062),g:Papillibacter(0.0098)
d:Bacteria(0.7925),p:Bacteroidetes(0.2030},c:Bacteroidia(0.0327),o:Bacteroidaie
s(0.
ascus_288 0.78229 0.46522045
0160),I:Porphyrornonadaceae(0.0050),g.3utyricimonas(0.0042{
d:Bacteria(1.0000),p:Firrnicutes(0.9922),c:Clostridia(0.8823),o:Clostrodiales(0
.6267),
ascus_64 0.77514 0.45417055
f:Ftuminococcaceae(0.2792},e:Ruminococcus(0.0605)
ascus_295 0.76639 0.24972263
d:Bacteria(1.0000),p:SR1(0.9990),g5Rl_genera jncertae_sedis(0.9793)
97
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
d:Bacteria(1.0000),p:Firrnicutes(0.6126),c:Clostridia(0.2851),o:Clostridiales(0
.1324),
ascus...546 0.76114 0.23819838
f:Clostridiaceae_1(0.0208},g:Clostridium_sensu_stricto(0.0066)
d:Bacteria(1.0000),p:Firmicutes(0.7036),c:Clastridia(0.4024),o:Clostridiales(0.
19561,
ascus_32 0.75068 0.5179697
f:Ruminococcaceae(0.08831,8:Hydrogenoanaerobacterium(0.0144)
d:Bacteria(1.0000),p:Firmicutes(0.7947).c:Clostridia(0.4637)syClostridiales(0.2
335{,
ascus_651 0.74837 0.27656645
f:Rurninococcaceae(0.08834:Clostridium_IV(0.0069{
d:Bacteria(1.0000),p:Firrnicutes(0.9922),c:Clostridia(0.8756),o:Clostridiales(0
.5860),
ascus..233 0.74409 0.36095098
f:Rurninococcaceae(0.3642},g:Ruminococcus(0.0478)
1002681 Table 5b. Correlation using Absolute cell count with Activity Filter
Target Strain NIIC Pearson Coefficient Nearest Taxonomy
d:Bacteria(1.0000),p:Firmicutes(0.8349),c:Clostridia(0.5251).o:Clostridiales(0.
2714).
ascus_713 0.71066 0.5305876
f:Rurninococuceae(0.1062{.g:Saccharofermentans(0.0073)
d:Bacteria(1.0000},p:Firrnicutes(0.9080),c:Clostridia(0.7704),o:Clostridiales(0
.4230),
ascus_26 0.81081 0.5300298
f:Rurninococcaceae(0.1942},g:Clostridiurn_IV(0.0144)
d:Bacteria(0.8663),p:Bacteroidetes(0.2483),c:Bacteroidia(0.0365),o:Bacteroidale
s(0.
ascus..1801 0.82398 0.5182261
0179),f:Porphyrornonadaceae(0.0059),g:Butyricirnonas(0.0047)
d:Bacteria(1.000,p:Firmicutes(0.7036),c:Clostridia(0.4024),o:Clastridiales(0.19
56),
ascus_32 0.75068 0.5179697
f:Ruminococcaceae(0.0883),g:Hydrogenoanaerobacterium(0.0144)
d:Bacteria(1.0000},p:Firrnicutes(0.9922),caostridia(0.8756),o:Clostridiales(0.5
860),
ascus_119 0.6974 0.4968678
f:Ruminococcaceae(0.3217),g:Ruminococcus(0.0478)
d:Bacteria(1.0000),p:Actinobacteria(0.1810),c:Actinobacteria(0.0365),o:Actinorn
yce
ascus...13899 0.64556 0.48739454
tales(0.0179),f:Propionibacteriaceae(0.0075),g:Microlunatus(0.0058)
d:Bacteria(1.0000),p:Firmicutes(0.8349),c:Clastridia(0.5251),o:Clostridiales(0.
2714),
ascus...906 0.49256 0.48418677
f:Rurninococcaceae(0.1242),g:Papillibacter(0.0098)
d:Bacteria(1.0000),p:Bacteroidetes(0.9991),c8acteroidia(0.9088),o:Bacteroidales
(0.
ascus_221 0.44006 0.47305903 7898
),f:Prevotellaceae(0.3217),g:Prevotella(0. 0986)
d:Bacteria(1.0000),p:Firrnicutes(0.7036),c:Clostridia(0.2851),o:Clostridiales(0
.1324),
ascus_1039 0.65629 0.46932846
f:Rurninococcaceae(0.0329),g:Clostridiurn_IV(0.0069)
d:Bacteria(0.7925),p:Bacteroidetes(0.2030),c:Bacteroidia(0.0327),o:Bacteroidale
s(0.
ascus_288 0.78229 0.46522045
0160),f:Porphyrornonadaceae(0.0050),g:Butyricirnonas(0.0042)
d:Bacteria(1.000,p:Firmicutes(0.9981),c:Clostridia(0.9088),o:Clostridiales(0.78
98),
ascus_589 0.40868 0.4651165
flachnospiraceae10.598Ã4:Clostridium_XiVa10.3698
d:Bacteria(1.0000},p:Firrnicutes(0.6126),caostridia(0.3426),o:Clostridiales(0.1
618),
ascus_41 0.67227 0.46499047
f:Ruminococcaceae(0.0703),g:Hydrogenoanaerobacteriurn(0.0098)
d:Bacteria(1.0000),p:Firrnicutes(0.7947),c:Clostridia(0.4637),o:Clostridiales(0
.2335),
ascus...111 0.79035 0.45881805
f:Ruminococcaceae(0.1062),8:Papillibacter(0.0098)
d:Bacteria(1.0000),p:Firmicutes(0.6126),c:Clastridia(0.3426),o:Clostridiales(0.
1618),
ascus_205 0.72441 0.45684373
f:Peptococcaceae_2(0.0449),g:Pelotomaculurn(0.0069)
d:Bacteria(1.0000),p:Firmicutes(0.9922),c:Clostridia(0.8823).o:Clostridiales(0.
6267).
ascus_64 0.77514 0.45417055
f:Rurninococuceae(0.2792{.g:Ruminococcus(0.0605)
[002691 Table 5c. Correlation using Relative Abundance with no Activity Filter
Target
Strain MC Pearson Coefficient Nearest Taxonomy
d:Bacteria(1.0000),p:Firrnicutes(0.9945),c:Clostridia(0.8623),o:Clostridiales(0
.5044),
ascus_3038 0.56239 0.6007549
f:Lachnospiraceae(0.2367),g:Clostridiurn_XiVa{0.0350)
d:Bacteria(1.0000),p:Firrnicutes(0.7947),c:Clostridia(0.3426),o:Clostridiales(0
.1618),
ascus...1555 0.66965 0.59716415 f:
Ruminococcaceae(0.0449),e:Clostridium_l V(0.0073)
d:Bacteria(1.0000),p:Firmicutes(0.7036),ctlastridia(0.2851),o:Clostridiales(0.1
3241,
ascus_1039 0.68563 0.59292555
f:Ruminococcaceae(0.03291,8:Clostridiurn_l V(0.00691
d:Bacteria(1.0000),p:Firmicutes(0.8897).c:Clostridia(0.7091),o:Ciostridiales(0.
3851),
ascus_1424 0.55509 0.57589555
f:Rurninococcaceae(0.1422),g:Papillibacter(0.0144)
d:Bacteria(1.0000),p:Firrnicutes(0.8349),c:Clostridia(0.5251),o:Clostridiales(0
.2714),
ascus_378 0.77519 . 0.5671971
f:Rurninococcaceae(0.1062),g:Saccharofermentans(0.0073)
d:Bacteria(1.0000),p:Firmicutes(0.7036),c:Clostridia(0.3426),o:Clostridiales(0.
1618),
ascus_407 0.69783 0.56279755
f:Clostridiaceae..1(0.0329),g:Clostridiurn_sensu_stricto(0.0069)
98
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
d:Bacteria(1.0000),p:Firrnicutes(0.9945),c:Clostridia(0.8756),o:Clostridiales(0
.5860),
ascus_1584 0.5193 0.5619939 f:Lachnospiraceae(0.3217),g:Coprococcus(0.0605)
d:Bacteria(1.0000),p:Firmicutes(0.6126),c.Clastridia(0.2851),o:Clostridiales(0.
13241,
ascus_760 0.61363 0.55807924
t:Clastridiaceae_1(0.0208),g:Clostridium sensu_stricto(0.0066)
d:Bacteria(1.0000),p:"Bacteroidetes"(0.9-
992),c:"Bacteroidia"(0.8690),o:"Bacteroida
ascus_1184 0.70593 0.5578006
les"(0.5452),f:Bacteroidaceae(0.1062),g.3acteroides(0.0237)
d:Bacteria(1.0000),p:Firrnicutes(0.9939),c:Clostridia(0.7704),o:Clostridiales(0
.4230),
ascus_7394 0.6269 0.5557023
f:Lachnospiraceae(0.1422),g:Clostridium_XlVa(0.0350)
d:Bacteria(1.0000),p:Firmicutes(0.9992),c:Clostridia(0.9351
),oklastridiales(0.8605),
ascus_1360 0.57343 0.5535785
filachnospiraceae(0.7052),g:Clostrichurn_Ma(0.2649)
d:Bacteria(1.0000),p:"Bacteraidetes"(0.9991),c:"Bacteroidia"(0.8955),a:"Bactero
ida
ascus_3175 0.53565 0.54864305
les"(0.7083),f:"Prevotellaceae"(0.1942),g:Prevatella(0.0605)
d:Bacteria(1.0000),p:"Spirochaetes"(0.9445},c:Spirochaetes(0.8623),o:Spirochaet
aie
ascus_2581 0.68361 0.5454486 50.5044),
f:Spirochaetaceae(0.3217),g:Spirochaeta(0.0237)
d:Bacteria(1.0000),p:Firrnicutes(0.6126),c:Clostridia(0.2851),o:Clostrediales(0
.1324),
ascus_531 0.71315 0.5400517
f:Clostridiaceae_1(0.0208},g:Clostridium_sensu_stricto(0.0066)
d:Bacteria(1.0000),p:"Spirochaetes"(0.9263),c:Spirochaetes(0.8317),o:Spirochaet
ale
ascus_1858 0.65165 0.5393882
s(0.4636),f:Spirochaetaceae(0.2792),g:Spirochaeta(0.0237)
Example 3,
Increase total Milk Fat, Milk Protein, and Energy-Corrected Milk (ECM) in Cows
[00270] Example 3 shows a specific implementation with the aim to increase the
total amount
of milk fat and milk protein produced by a lactating ruminant, and the
calculated ECM. As used
herein, ECM represents the amount of energy in milk based upon milk volume,
milk fat, and
milk protein. ECM adjusts the milk components to 3.5% fat and 3.2% protein,
thus equalizing
animal performance and allowing for comparison of production at the individual
animal and herd
levels over time. An equation used to calculate ECM, as related to the present
disclosure, is:
ECM = (0.327 x milk pounds) + (12.95 x fat pounds) + (7.2 x protein pounds)
100271] Application of the methodologies presented herein, utilizing the
disclosed methods to
identify active interrelated microbes/microbe strains and generating microbial
ensembles
therefrom, demonstrate an increase in the total amount of milk fat and milk
protein produced by
a lactating ruminant. These increases were realized without the need for
further addition of
hormones.
[00272] In this example, a microbial ensemble comprising two isolated
microbes, Ascusb X
and Ascusf Y, identified and generated according to the above disclosure, was
administered to
Holstein cows in mid-stage lactation over a period of five weeks. The cows
were randomly
assigned into 2 groups of 8, wherein one of the groups was a control group
that received a buffer
lacking a microbial ensemble. The second group, the experimental group, was
administered a
microbial ensemble comprising Ascusb_X and Ascusf Y once per day for five
weeks. Each of
99
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
the cows were housed in individual pens and were given free access to feed and
water. The diet
was a high milk yield diet. Cows were fed ad libitum and the feed was weighed
at the end of the
day, and prior day refusals were weighed and discarded. Weighing was performed
with a PS-
2000 scale from Salter Brecknell (Fairmont, MN).
[00273] Cows were cannulated such that a cannula extended into the rumen of
the cows. Cows
were further provided at least 10 days of recovery post cannulation prior to
administering control
dosages or experimental dosages.
[00274] Administration to the control group consisted of 20 ml of a neutral
buffered saline,
while administration to the experimental group consisted of approximately 109
cells suspended in
20 mL of neutral buffered saline. The control group received 20 ml of the
saline once per day,
while the experimental group received 20 ml of the saline further comprising
109 microbial cells
of the described microbial ensemble.
[00275] The rumen of every cow was sampled on days 0, 7, 14, 21, and 35,
wherein day 0 was
the day prior to microbial administration. Note that the experimental and
control administrations
were performed after the rumen was sampled on that day. Daily sampling of the
rumen,
beginning on day 0, with a pH meter from Hanna Instruments (Woonsocket, RI)
was inserted
into the collected rumen fluid for recordings. Rumen sampling included both
particulate and
fluid sampling from the center, dorsal, ventral, anterior, and posterior
regions of the rumen
through the cannula, and all five samples were pooled into 15ml conical vials
containing 1.5m1
of stop solution (95% ethanol, 5% phenol). A fecal sample was also collected
on each sampling
day, wherein feces were collected from the rectum with the use of a palpation
sleeve. Cows were
weighed at the time of each sampling.
[00276] Fecal samples were placed in a 2 ounce vial, stored frozen, and
analyzed to determine
values for apparent neutral detergent fibers (NDF) digestibility, apparent
starch digestibility, and
apparent protein digestibility. Rumen sampling consisted of sampling both
fluid and particulate
portions of the rumen, each of which was stored in a 15m1 conical tube. Cells
were fixed with a
10% stop solution (5% phenol/95% ethanol mixture) and kept at 4 C and shipped
to Ascus
Biosciences (San Diego, California) on ice.
100
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1002771 The milk yield was measured twice per day, once in the morning and
once at night.
Milk composition (% fats and % proteins, etc.) was measured twice per day,
once in the morning
and once at night Milk samples were further analyzed with near-infrared
spectroscopy for
protein fats, solids, analysis for milk urea nitrogen (MUN), and somatic cell
counts (SCC) at the
Tulare Dairy Herd Improvement Association (DH1A) (Tulare, California). Feed
intake of
individual cows and rumen pH were determined once per day.
1002781 A sample of the total mixed ration (TMR) was collected the final day
of the adaptation
period, and then successively collected once per week. Sampling was performed
with the
quartering method, wherein the samples were stored in vacuum sealed bags which
were shipped
to Cumberland Valley Analytical Services (Hagerstown, MD) and analyzed with
the N1R1
package. The final day of administration of buffer and/or microbial
bioensemble was on day 35,
however all other measurements and samplings continued as described until day
46.
1002791 FIG. 8A demonstrates that cows that received the microbial ensemble
based on the
disclosed methods exhibited a 20.9% increase in the average production of milk
fat versus cows
that were administered the buffered solution alone. FIG. 8B demonstrates that
cows that were
administered the microbial ensemble exhibited a 20.7% increase in the average
production of
milk protein versus cows that were administered the buffered solution alone.
FIG. 8C
demonstrates that cows that were administered the microbial ensemble exhibited
a 19.4%
increase in the average production of energy corrected milk. The increases
seen in FIG. 8A-C
became less pronounced after the administration of the ensemble ceased, as
depicted by the
vertical line intersecting the data points.
Example -I
Detection of Clostridium perfringens as causative agent for lesion formation
in broiler
chickens
[002801 160 male Cobb 500s were challenged with various levels of Clostridium
petfringens
(Table 6a). They were raised for 21 days, sacrificed, and lesion scored to
quantify the
progression of necrotic enteritis and the impact of C. petfringens.
[00281] Table 6a
101
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
NE No. of
Number of
Challenge Birds/ No. of Birds/
Treatment
(YIN) Treatment Description Pen Pens
Treatment
1N Non-Challenged 20 2 40
Challenged with half typical dose
(1.25 ml/bird; 2.0 ¨ 9.0 X108 20 2 40
cfu/ml)
Challenged with typical dose (2.5
3 20 2 40
ml/bird; 2.0 ¨9.0 X108cfu/m1)
Challenged with twice the typical
4 Y dose (5.0 ml/bird; 2.0 ¨9.0 X108 20 2 40
cfu/ml)
Total 8 160
1002821 Experimental Design
100283] Birds were housed within an environmentally controlled facility in
wooden floor pens
(¨ 4' x 4' minus 2.25 sq. ft for feeder space) providing floor space & bird
density of [-0.69
ft2/bird], temperature, lighting, feeder and water. Birds were placed in clean
pens containing an
appropriate depth of wood shavings to provide a comfortable environment for
the chicks.
Additional shavings were added to pens if they become too damp for comfortable
conditions for
the test birds during the study. Lighting was via incandescent lights and a
commercial lighting
program was used as follows.
[00284] Table 6b
Approximate Hours
Approximate of Continuous Light -tight intensity
Bird Age (days) per 24 hr period (foot candles)
0 ¨ 4 24 1.0 ¨ 1.3
5-10 10 1.0 ¨ 1.3
11 ¨ 18 12 0.2 ¨ 0.3
19 ¨ end 16 0.2 ¨ 0.3
[00285] Environmental conditions for the birds (i.e. bird density,
temperature, lighting, feeder
and water space) were similar for all treatment groups. In order to prevent
bird migration and
bacterial spread from pen to pen, each pen had a solid (plastic) divider for
approximately 24
inches in height between pens.
[00286] Vaccinations and Therapeutic Medication:
102
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1002871 Birds were vaccinated for Mareks at the hatchery. Upon receipt (study
day 0), birds
were vaccinated for Newcastle and Infectious Bronchitis by spray application.
Documentation
of vaccine manufacturer, lot number and expiration date were provided with the
final report.
100288.1 Water:
1002891 Water was provided ad libitum throughout the study via one Plasson
drinker per pen.
Drinkers were checked twice daily and cleaned as needed to assure a clean and
constant water
supply to the birds.
100290.1 Feed:
1002911 Feed was provided ad libitum throughout the study via one hanging, -17-
inch diameter
tube feeder per pen. A chick feeder tray was placed in each pen for
approximately the first 4
days. Birds were placed on their respective treatment diets upon receipt (day
0) according to the
Experimental Design. Feed added and removed from pens from day 0 to study end
were
weighed and recorded.
100292] Daily observations:
1002931 The test facility, pens and birds were observed at least twice daily
for general flock
condition, lighting, water, feed, ventilation and unanticipated events. If
abnormal conditions or
abnormal behavior was noted at any of the twice-daily observations they were
documented and
documentation included with the study records. The minimum-maximum
temperatures of the
test facility were recorded once daily.
1002941 Pen Cards:
1002951 There were 2 cards attached to each pen. One card identified the pen
number and the
second denoted the treatment number.
1002961 Animal 1kindling:
1002971 The animals were kept under ideal conditions for livability. The
animals were handled
in such a manner as to reduce injuries and unnecessary stress. Humane measures
were strictly
enforced.
1002981 Veterinaty Care, Intervention and Euthanasia:
103
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1002991 Birds that developed clinically significant concurrent disease
unrelated to the test
procedures were, at the discretion of the Study Investigator, or a designee,
removed from the
study and euthanized in accordance with site SOPs. In addition, moribund or
injured birds were
also euthanized upon authority of a Site Veterinarian or a qualified
technician. The reasons for
any withdrawal were documented. If an animal died, or was removed and
euthanized for
humane reasons, it was recorded on the mortality sheet for the pen and a
necropsy performed and
filed to document the reason for removal.
1003001 If euthanasia was deemed necessary by the Study Investigator, animals
were euthanized
by cervical dislocation.
1003011 Mortality and Culls:
1003021 Starting on study day 0, any bird that was found dead or was removed
and sacrificed
was weighed and necropsied. Cull birds that were unable to reach feed or water
were sacrificed,
weighed and documented. The weight and probable cause of death and necropsy
findings were
recorded on the pen mortality record.
1003031 Body Weights and Feed Intake:
1003041 Birds were weighed, by pen and individually, on approximately days 14
and 21. The
feed remaining in each pen was weighed and recorded on study days 14 and 21.
The feed intake
during days 14-21 was calculated.
1003051 Weight Gains and Feed Conversion:
1003061 Average bird weight, on a pen and individual basis, on each weigh day
were
summarized. The average feed conversion was calculated on study day 21 (i.e.
days 0-21) using
the total feed consumption for the pen divided by the total weight of
surviving birds. Adjusted
feed conversion was calculated using the total feed consumption in a pen
divided by the total
weight of surviving birds and weight of birds that died or were removed from
that pen.
1003071 CLOSTRIDIUM PERFRINGENS CHALLENGE
1003081 Method of Administration:
1003091 Clostridium perfringens (CL-15, Type A, a and 132 toxins) cultures in
this study were
administered via the feed. Feed from each pen's feeder was used to mix with
the culture. Prior
104
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
to placing the cultures in the pens the treatment feed was removed from the
birds for
approximately 4 ¨ 8 hours. For each pen of birds, a fixed amount based on
study design of the
broth culture at a concentration of approximately 2.0 ¨ 9.0 X108 cfu/ml was
mixed with a fixed
amount of feed (-25g/bird) in the feeder tray and all challenged pens were
treated the same.
Most of the culture-feed was consumed within 1 ¨ 2 hours. So that birds in all
treatments are
treated similar, the groups that are not challenged also had the feed removed
during the same
time period as the challenged groups.
[00310] Clostridium Challenge:
100311] The Clostridium petfringens culture (CL-15) was grown ¨5 hrs at ¨37 C
in Fluid
Thioglycollate medium containing starch. CL-15 is a field strain of
Clostridium petfringens
from a broiler outbreak in Colorado. A fresh broth culture was prepared and
used each day. For
each pen of birds, a fixed amount of the overnight broth culture was mixed
with a fixed amount
of treatment feed in the feeder tray (see administration). The amount of feed,
volume and
quantitation of culture inoculum, and number of days dosed were documented in
the final report
and all pens will be treated the same. Birds received the C. perfringens
culture for one day
(Study day 17).
[00312] DATA COLLE'CIED:
- intestinal content for analysis with the Ascus platform methods according to
the
disclosure.
- Bird weights, by pen and individually and feed efficiency, by pen,
on approximately
days 14 and 21.
- Feed amounts added and removed from each pen from day 0 to study
end.
- Mortality: sex, weight and probable cause of death day 0 to study
end.
- Removed birds: reason for culling, sex and weight day 0 to study
end.
- Daily observation of facility and birds, daily facility temperature.
- Lesion scores 5 birds / pen on approximate day 21
[00313] Lesion Scoring:
105
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1003141 Four days following the last C. perfringens culture administration,
five birds were
randomly selected from each pen by first bird caught, sacrificed and
intestinal lesions scored for
necrotic enteritis. Lesions scored as follows:
- 0 =normal: no NE lesions, small intestine has normal elasticity (rolls back
to
normal position after being opened)
- 1 -mild: small intestinal wall is thin and flaccid (remains flat when opened
and
doesn't roll back into normal position after being opened); excess mucus
covering
mucus membrane
- 2 =moderate: noticeable reddening and swelling of the intestinal wall; minor
ulceration and necrosis of the intestine membrane; excess mucus
- 3 =severe: extensive area(s) of necrosis and ulceration of the small
intestinal
membrane; significant hemorrhage; layer of fibrin and necrotic debris on the
mucus
membrane (Turkish towel appearance)
- 4 =dead or moribund: bird that would likely die within 24 hours and
has NE lesion
score of 2 or more
1003151 RESULTS
1003161 The results were analyzed using the methods disclosed above (e.g., as
discussed with
reference to FIGs. 1A, 1B, and 2, as well as throughout the specification) as
well as the
conventional correlation approach (as discussed above). Strain-level microbial
abundance and
activity were determined for the small intestine content of each bird, and
these profiles were
analyzed with respect to two different bird characteristics: individual lesion
score, and average
lesion score of the pen.
1003171 37 birds were used in the individual lesion score analysis - although
40 birds were
scored, only 37 had sufficient intestinal material for analysis. The same
sequencing reads and
same sequencing analysis pipeline was used for both the Ascus approach of the
disclosure and
the conventional approach. However, the Ascus approach also integrated
activity information, as
well as cell count information for each sample, as detailed earlier.
1003181 The Ascus mutual information approach was used to score the
relationships between
the abundance of the active strains and the individual lesion scores of the 37
broilers. Pearson
106
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
correlations were calculated between the strains and individual lesion scores
of the 37 broilers
for the conventional approach. The causative strain, C. petfringens, was
confirmed via global
alignment search against the list of organisms identified from the pool of
samples. The rank of
this specific strain was then identified on the output of each analysis
method. The Ascus
approach identified the C. perfringens administered in the experiment as the
number one strain
linked to individual lesion score. The conventional approach identified this
strain as the 26th
highest strain linked to individual lesion score. Since C. petfringens was
successfully identified
as the causative agent using the disclosed methods / approach, the first
marker and/or second
marker representing the pathogenic strain can be used as an indicator of a
pathogenic and/or
undesirable state in future samples. The abundance of the marker can also be
used as an indicator
of the severity of a pathogenic state.
1003191 102 birds were used in the average lesion score analysis. As in the
previous case, the
same sequencing reads and same sequencing analysis pipeline was used for both
the Ascus
approach and the conventional approach. Again, the Ascus approach also
integrated activity
information, as well as cell count information for each sample.
[00320] The Ascus mutual information approach was used to score the
relationships between
the abundance of the active strains and the average lesion score of each pen.
Pearson correlations
were calculated between the strains and average lesion score of each pen for
the conventional
approach. The causative strain, C. petfringens, was confirmed via global
alignment search
against the list of organisms identified from the pool of samples. The rank of
this specific strain
was then identified on the output of each analysis method. The Ascus approach
identified the C.
pedkingens administered in the experiment as the 4th highest strain linked to
average lesion
score of the pen. The conventional approach identified C petfringens as the
15th highest strain
linked to average lesion score of the pen. Average lesion score of the pen is
a less accurate
measurement than individual lesion score due to the variable levels of C
petfringens infection
being masked by the bulk/average measurement. The drop in rank when comparing
the
individual lesion score analysis to the average pen lesion score analysis was
expected. The
collected metadata is provided below
[00321] Table 7
107
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
====00i.k== ' .**µ.. .W=.#04A.W*4*===.**kc'i.=*=...'.
wz*z=..:".i,=1=' .t6:::': L'Z'.:ssf$
i:.................... :i ii........:.:.:.:.:.:.:.:.:.............i
i............:.:.:.:.:.:.:.:............. i............................
2122 2 3.4
4124 1 1.4 , I 3:0 :;&5,1:i :i :i 49320=:i :i434.,18fa:5'1:41330
:i :11t4.F.44.43,4t .
323:3 2 3. :..i.= ::: :.:: := :=:=:=:=:==
::: :::0,,,,,i......,,,, :: :: =-,==,..,,,,,-..,
.1,3:1: a ...,.....1i . .
a 1.1 -.... 2 1.4$
4 .,
.$.
21?i4, 2
23.3.0 24 $ 21.11 '
, 2.41 4,
. .
2220 3.41. 214-2$ 2 2 4i
4124 1.4 i 213141 , 2..4
2118, 4.4
,.. 2 .4 ...
$128 3.4$
4154 4 4.a =k
.11.17 1.4 i 4 ...................... t ....
$3128 3.4i 2 2 ....
4144 1.4 i 415'3'4 .4:
t
.
2.1.54
a ......
4. ...
a.As
- ................................... +- .. -4 - 4 4
4524 4 2.4 i a 112' __
- "
'
4.. 2.4 4
4
4420 , 4 4.1.. , 4i'.,.
, - 4 2.2 t
09:21 .4 = - =
= =
4445 4 4 4.21
43124 4 $ 4:4=432:1
"2.11
45.24, za :.,,,x,..:', 4µ
. ...
45.32., 4 . 4
....: ....t
2-z..,... :
:=*.=t'.4 4
4524 4
840:8 4 a.2 .................. ..4 ^4 ,
.µ,
_
4480 4 4.4 ,;42 4 3.3.
*93.3. 4 2.8 :cµ'a; a 88
444:::: 4 2.2 5 wat A. II .....
'
4544 a 2.1 =?, 41nt 4
,..%4'...'F:: 4 ;4:2 i 4
,n24:1 4. 4...4.:
:,:..q:i.f. 4 4 4i 1
3352::: 4 ,=,.z,t '.4.,
IEIIIIIIII IIIIIIIIIIIIII 21721 ,
2.04 A:
1 12'
13]
.41 .?'...,: 1M
4441 2.i 7 1 q 0
2142 :21 ..2.2 1
. . ___________________________
2144 5 3-41 43:34; 1. 5?
21.45 : .4 i 3. 41.55; 1 5 t
3.549 4 al: 4 41 t -.......a, .. 1. .i..
33 ..........................................................
45:37 1 4.5 i 1 ;
4155, 1
1. ` 3.
.
4545 4== at .. &= ::,.I a
4544 1 4.51. 5 4- 4. ...
811433 2 4;4 z: 0
8:443. 1 1.
04 $ $ _____________________
413715; 2
t 44221 :S.. 2.2 4
4841 a .. :3
- .a, :
...7:,..::4a, I :,..:.i. :-.. ,s,,:r.n.31 -3 2.2
2..,,
;.I.Al.
.4,t :3, =:'..03.',31 3 .,, .....: ....
...,:,...,n,, I .41 4004:1 :.:k 4.4_
4534. A 54! 43.4:4; :.*S
s.4.$3.i= 1 4.P,, i
4524 1 "t
4.5., 4 4n:'.3t;
. t :1 2,2 _ 4
4022 I. v.4 0..kx..., ..............
$1...s, , .., .3. ii,a.a. 3., 8.8
.i __________________________________________________________
11$::$ ;k .;,..4i: ::$$:,...$81 ''' LI 4
Example 5
Selection of an ensemble of active microorganism strains to shift the
composition of the
gastrointestinal rnicrobiome of broiler chickens towards a more productive
state
108
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1003221 96 male Cobb 500s were raised for 21 days. Weight and feed intake were
determined
for individual birds, and cecum scrapings were collected after sacrifice. The
cecum samples were
processed using the methods of the present disclosure to identify an ensemble
of microorganisms
that will enhance feed efficiency when administered to broiler chickens in a
production setting.
100323] EXPERIMENTAL DESIGN
1003241 120 Cobb 500 chicks were divided and placed into pens based on dietary
treatment.
The birds were placed in floor pens by treatment from 0-14D. The test facility
was divided into 1
block of 2 pens and 48 blocks of 2 individual cages each. Treatments were
assigned to the
pens/cages using a complete randomized block design; pens/cages retained their
treatments
throughout the study. The treatments were identified by numeric codes. Birds
were assigned to
the cages/pens randomly. Specific treatment groups were as follows in Table 9.
1003251 Table 9
No. of No. of No. of No. of No.
Treatment
Treatment . . Strain Birds/ Floor Bi rds/ Cages Birds/
Description
Floor Pen Pens/Trt Cage /Trt Treatment
1
0.042% Cobb 60 1 1 48 48(D14)
Salinomycin 500 60 (DO)
No Cobb 48 (D14)
60 1 1 48
Sal inomycin 500 60 (DO)
1003261 Housing:
1003271 Assignment of treatments to cages/pens was conducted using a computer
program. The
computer-generated assignment were as follows:
1003281 Birds were housed in an environmentally controlled facility in a large
concrete floor
pen (4' x 8') constructed of solid plastic (4' tall) with clean litter. At day
14, 96 birds were
moved into cages within the same environmentally controlled facility. Each
cage was
24"x1 8"x24".
1003291 Lighting was via incandescent lights and a commercial lighting program
was used.
Hours of continuous light for every 24-hour period were as follows in Table
10.
[00330] Table 10
109
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Approximate Hours
Approximate of Continuous -Light Intensity
Bird Age (days) Light (foot candles)
per 24 hr period
0-6 23 1.0 - 1.3
7-21 16 0.2 - 0.3
100331] Environmental conditions for the birds (i.e. 0.53 ft), temperature,
lighting, feeder and
water space) were similar for all treatment groups.
1003321 In order to prevent bird migration, each pen was checked to assure no
openings greater
than 1 inch existed for approximately 14 inches in height between pens.
1003331 Vaccinations:
1003341 Birds were vaccinated for Mareks at the hatchery. Upon receipt (study
day 0), birds
were vaccinated for Newcastle and Infectious Bronchitis by spray application.
Documentation of
vaccine manufacturer, lot number and expiration date were provided with the
final report.
1003351 Water:
1003361 Water was provided ad libitum throughout the study. The floor pen
water was via
automatic bell drinkers. The battery cage water was via one nipple waterer.
Drinkers were
checked twice daily and cleaned as needed to assure a clean water supply to
birds at all times.
1003371 Feed:
1003381 Feed was provided ad libitum throughout the study. The floor pen feed
was via
hanging, -17-inch diameter tube feeders. The battery cage feed was via one
feeder trough,
9"x4". A chick feeder tray was placed in each floor pen for approximately the
first 4 days.
1003391 Daily observations:
1003401 The test facility, pens and birds were observed at least twice daily
for general flock
condition, lighting, water, feed, ventilation and unanticipated events. The
minimum-maximum
temperature of the test facility was recorded once daily.
100341] Mortality and Culls:
110
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00342] Starting on study day 0, any bird that was found dead or was removed
and sacrificed
was necropsied. Cull birds that are unable to reach feed or water were
sacrificed and necropsied.
The probable cause of death and necropsy findings were recorded on the pen
mortality record.
[00343] Body Weights and Feed Intake:
[00344] ¨96 birds were weighed individually each day. Feed remaining in each
cage was
weighed and recorded daily from 14-21 days. The feed intake for each cage was
determined for
each day.
[00345] Weight Gains and Feed Conversion:
[00346] Body weight gain on a cage basis and an average body weight gain on a
treatment basis
were determined from 14-21 days. Feed conversion was calculated for each day
and overall for
the period 14-21D using the total feed consumption for the cage divided by
bird weight.
Average treatment feed conversion was determined for the period 14-21 days by
averaging the
individual feed conversions from each cage within the treatment.
1003471 Veterinary (are, Intervention and Euthanasia:
[00348] Animals that developed significant concurrent disease, are injured and
whose condition
may affect the outcome of the study were removed from the study and euthanized
at the time that
determination is made. Six days post challenge all birds in cages were removed
and lesion
scored.
[00349] Data Collected:
1003501 Bird weights and feed conversion, individually each day from days 14-
21.
[00351] Feed amounts added and removed from floor pen and cage from day 0 to
study end.
[00352] Mortality: probable cause of death day 0 to study end.
[003531 Removed birds: reason for culling day 0 to study end.
100354] Daily observation of facility and birds, daily facility temperature.
100355] Cecum content from each bird on day 21.
[00356] RESULTS
111
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00357] The results were analyzed using the methods disclosed above (e.g., as
discussed with
reference to FIGs. 1A, 1B, and 2, as well as throughout the specification).
Strain-level microbial
abundance and activity were determined for the cecal content of each bird. A
total of 22,461
unique strains were detected across all 96 broiler cecum samples. The absolute
cell counts of
each strain was filtered by the activity threshold to create a list of active
microorganism strains
and their respective absolute cell counts. On average, only 48.3% of the
strains were considered
active in each broiler at the time of sacrifice. After filtering, the profiles
of active microorganism
in each bird were integrated with various bird metadata, including feed
efficiency, final body
weight, and presence/absence of salinomycin in the diet, in order to select an
ensemble that
improves performance of all of these traits.
[00358] The mutual information approach of the present disclosure was used to
score the
relationships between the absolute cell counts of the active strains and
performance
measurements, as well as relationships between two different active strains,
for all 96 birds.
After applying a threshold, 4039 metadata-strain relationships were deemed
significant, and
8842 strain-strain relationships were deemed significant. These links,
weighted by MIC score,
were then used as edges (with the metadata and strains as nodes) to create a
network for
subsequent community detection analysis. A Louvain method community detection
algorithm
was applied to the network to categorize the nodes into subgroups.
[003591 The Louvain method optimizes network modularity by first removing a
node from its
current subgroup, and placing into neighboring subgroups. If modularity of the
node's neighbors
has improved, the node is reassigned to the new subgroup. If multiple groups
have improved
modularity, the subgroup with the most positive change is selected. This step
is repeated for
every node in the network until no new assignments are made. The next step
involves the
creation of a new, coarse-grained network, i.e. the discovered subgroups
become the new nodes.
The edges between nodes are defined by the sum of all of the lower-level nodes
within each
subgroup. From here, the first and second steps are repeated until no more
modularity-optimizing
changes can be made. Both local (i.e. groups made in the iterative steps) and
global (i.e. final
grouping) maximas can be investigated to resolve sub-groups that occur within
the total
microbial community, as well as identify potential hierarchies that may exist.
[00360] Modularity:
112
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1 ic,ki õ
Q = E [Ai, -2,3
0 kei, ej)
GM - Zrfl
[00361] Where A is the matrix of metadata-strain and strain-strain
relationships; ki=fylij is the
total link weight attached to node i; and m = 1/2 EtjAtj. The Kronecker delta
ii(ci,ci) is 1 when
nodes i and j are assigned to the same community, and 0 otherwise.
[00362] Computing change in modularity when moving nodes:
Ein +ki,in (Etot +ki) 2 rtot )2 21
AQ
2m 2m. 2m 2rn 2m
[00363] AQ is the gain in modularity in subgroup C. Lo is the sum of the
weights of the link in
C, Lot is the sum of the weights of the links incident to nodes in C, IQ is
the sum of weights of
links incident to node i, kiin is the sum of weights of links from / to nodes
in C, and m is the sum
of the weights of all links in the network.
[00364] Five different subgroups were detected in the chicken microbial
community using the
Louvain community detection method. Although a vast amount of microbial
diversity exists in
nature, there is far less functional diversity. Similarities and overlaps in
metabolic capability
create redundancies. Microorganism strains responding to the same
environmental stimuli or
nutrients are likely to trend similarly¨this is captured by the methods of the
present disclosure,
and these microorganisms will ultimately be grouped together. The resulting
categorization and
hierarchy reveal predictions of the functionality of strains based on the
groups they fall into after
community-detection analysis. This categorization can also be used to define a
more successful /
productive state. Once established, this state can be used to define and
describe the state of future
samples.
[00365] After the categorization of strains is completed, microorganism
strains are cultured
from the samples. Due to the technical difficulties associated with isolating
and growing axenic
cultures from heterogeneous microbial communities, only a small fraction of
strains passing both
the activity and relationship thresholds of the methods of the present
disclosure will ever be
propagated axenically in a laboratory setting. After cultivation is completed,
the ensemble of
microorganism strains is selected based on whether or not an axenic culture
exists, and which
1 1 3
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
subgroups the strains were categorized into. Ensembles are created to contain
as much functional
diversity possible¨that is, strains are selected such that a diverse range of
subgroups are
represented in the ensemble. These ensembles are then tested in efficacy and
field studies to
determine the effectiveness of the ensemble of strains as a product, and if
the ensemble of strains
demonstrates a contribution to production, the ensemble of strains could be
produced and
distributed as a product.
Example 6
Using small sample sizes to identify active microorganism strains
[003661 As detailed below, as few as two samples can be effective to identify
active
microorganism strains. In particular, the below experiment show that the
methods of the
disclosure properly identify C. perfringens as an active microorganism strain
and causative agent
of intestinal lesions and necrotic enteritis for all comparisons, including in
a 2 sample
comparison.
1003671 EXPERIMENTAL DESIGN
1003681 Birds housed within an environmentally controlled facility in concrete
floor pens (-4' x
4' minus 2.25 sq ft of feeder space) providing floor space & bird density of 1-
0.55 ft2/bird (day
0); ¨ 0.69 ft2/bird (day 21 after lesion scores)], temperature, humidity,
lighting, feeder and water
space will be similar for all test groups. Birds placed in clean pens
containing an appropriate
depth of clean wood shavings to provide a comfortable environment for the
chicks. Additional
shavings added to pens in order to maintain bird comfort. Lighting via
incandescent lights and a
commercial lighting program used as follows.
1003691 Table 11
Approximate Hours
of Continuous Light
Approximate ¨Light Intensity
per 24 hr period
Bird Age days) (foot candles)
0-4 24 1.0 ¨ 1.3
5-10 10 1.0 ¨ 1.3
11 -- 18 12 0.2 ¨ 0.3
19¨ end 16 0.2 ¨ 0.3
114
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1003701 Environmental conditions for the birds (i.e., bird density,
temperature, lighting, feeder
and water space) were similar for all treatment groups. In order to prevent
bird migration and
bacterial spread from pen to pen, each pen had a solid (plastic) divider of
approximately 24
inches in height between pens.
1003711 Vaccinations and Therapeutic Medication:
100372.1 Birds were vaccinated for Mareks at the hatchery. Upon receipt (study
day 0), birds
were vaccinated for Newcastle and Infectious Bronchitis by spray application.
Documentation
of vaccine manufacturer, lot number and expiration date were provided with the
final report.
1003731 Water:
100374.1 Water was provided ad libitum throughout the study via one Plasson
drinker per pen.
Drinkers were checked twice daily and cleaned as needed to assure a clean and
constant water
supply to the birds.
100375] Feed:
1003761 Feed was provided ad libitum throughout the study via one hanging, -47-
inch diameter
tube feeder per pen. A chick feeder tray was placed in each pen for
approximately the first 4
days. Birds were placed on their respective treatment diets upon receipt (day
0) according to the
Experimental Design. Feed added and removed from pens from day 0 to study end
were
weighed and recorded.
1003771 Daily observations:
1003781 The test facility, pens and birds were observed at least twice daily
for general flock
condition, lighting, water, feed, ventilation and unanticipated events. If
abnormal conditions or
abnormal behavior is noted at any of the twice-daily observations they were
documented, and the
documentation was included with the study records. The minimum-maximum
temperature of the
test facility were recorded once daily.
[00379] Pen Cards:
100380] There were 2 cards attached to each pen. One card identified the pen
number and the
second denoted the treatment number.
1003811 Animal Handling:
115
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1003821 The animals were kept under ideal conditions for livability. The
animals were handled
in such a manner as to reduce injuries and unnecessary stress. Humane measures
were strictly
enforced.
100383] Veterinary Care, Intervention and Euthanasia:
1003841 Birds that develop clinically significant concurrent disease unrelated
to the test
procedures may, at the discretion of the Study Investigator, or a designee, be
removed from the
study and euthanized in accordance with site SOPs. In addition, moribund or
injured birds may
also be euthanized upon authority of a Site Veterinarian or a qualified
technician. The reasons for
withdrawal were documented. If an animal dies, or is removed and euthanized
for humane
reasons, it was recorded on the mortality sheet for the pen and a necropsy was
performed and
filed to document the reason for removal.
1003851 If euthanasia was deemed necessary by the Study Investigator, animals
were euthanized
by cervical dislocation.
[00386] Mortality and Culls:
1003871 Starting on study day 0, any bird that was found dead or was removed
and sacrificed
was weighed and necropsied. Cull birds that were unable to reach feed or water
were sacrificed,
weighed and documented. The weight and probable cause of death and necropsy
findings were
recorded on the pen mortality record.
1003881 CLOSTRIDIUM PERFRINGENS CHALLENGE
[003891 Method of Administration:
1003901 Clostridium perfringens (CL-15, Type A, a and 132 toxins) cultures in
this study were
administered via the feed. Feed from each pen's feeder was used to mix with
the culture. Prior
to placing the cultures in the pens the treatment feed was removed from the
birds for
approximately 4 ¨ 8 hours. For each pen of birds, a fixed amount based on
study design of the
broth culture at a concentration of approximately 2.0 ¨ 9.0 X108 cfulml was
mixed with a fixed
amount of feed (-25g/bird) in the feeder tray and all challenged pens were
treated the same.
Most of the culture-feed was consumed within 1 ¨ 2 hours. So that birds in all
treatments were
treated similarly, the groups that are not challenged also had the feed
removed during the same
time period as the challenged groups.
116
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1003911 Clostridium Challenge:
[003921 The Clostridium petfringens culture (CL-15) was grown ¨5 hrs at ¨37 C
in Fluid
Thioglycollate medium containing starch. CL-15 is a field strain of
Clostridium petfringens
from a broiler outbreak in Colorado. A fresh broth culture was prepared and
used each day. For
each pen of birds, a fixed amount of the overnight broth culture was mixed
with a fixed amount
of treatment feed in the feeder tray. The amount of feed, volume and
quantitation of culture
inoculum, and number of days dosed were documented in the final report and all
pens will be
treated the same. Birds will receive the C. petfringens culture for one day
(Study day 17).
[00393] DATA COLLECTED
[00394] Intestinal content for analysis with the methods of the present
application
100395] Bird weights, by pen and individually, and feed efficiency, by pen, on
approximately
days 14 and 21.
[00396] Feed amounts added and removed from each pen from day 0 to study end.
100397] Mortality: sex, weight and probable cause of death day 0 to study end.
[00398] Removed birds: reason for culling, sex and weight day 0 to study end.
[00399] Daily observation of facility and birds, daily facility temperature.
[00400] Lesion score 5 birds / pen on approximate day 21
[00401] Samples collected from 48 lesion scored birds
[00402] Lesion Scoring:
[00403] Four days following the last C. petfringens culture administration,
five birds were
randomly selected from each pen by first bird caught, sacrificed and
intestinal lesions scored for
necrotic enteritis. Lesions scored as follows:
[00404] 0 =normal: no NE lesions, small intestine has normal elasticity (rolls
back to normal
position after being opened)
[00405] 1 =mild: small intestinal wall is thin and flaccid (remains flat when
opened and doesn't
roil back into normal position after being opened); excess mucus covering
mucus membrane
117
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1004061 2 =moderate: noticeable reddening and swelling of the intestinal wall;
minor ulceration
and necrosis of the intestine membrane; excess mucus
1004071 3 =severe: extensive area(s) of necrosis and ulceration of the small
intestinal
membrane; significant hemorrhage; layer of fibrin and necrotic debris on the
mucus membrane
(Turkish towel appearance)
100408.1 4 -dead or moribund: bird that would likely die within 24 hours and
has NE lesion
score of 2 or more
100409.1 RESULTS
1004101 The results were analyzed using the methods of the present
application. Strain-level
microbial absolute cell count and activity were determined for the small
intestine content of all
48 birds. The methods of the present application integrated activity
information, as well as
absolute cell count information for each sample.
1004111 The mutual information approach of the present application was used to
score the
relationships between the absolute cell count of the active strains and the
individual lesion scores
of 10 randomly selected broilers. One sample was randomly removed from the
dataset, and the
analysis was repeated. This was repeated until only two broiler samples were
compared.
1004121 The causative strain, C. petfringens, was confirmed via global
alignment search against
the list of organisms identified from the pool of samples. Its rank (with a
rank position of 1 being
the strain most implicated in causing lesion scores) against all strains
analyzed are presented in
Table 12:
Table 12
Number of Samples Rank
9
8
7 (2 tied for 1)
6 I (3 tied for I)
5 I (3 tied for I)
4 I (3 tied for I)
3 1 (25 tied for 1)
118
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
2 1 (31 lied for 1)
[00413] Table 12 illustrates that C. pelfringens was properly identified as an
active
microorganism strain and causative agent of lesion scores for all comparisons,
including the 2
sample comparison, using the disclosed methods. As the sample number was
reduced, the
number of false positives (i.e., other strains also being identified as
causative agents) increased
beginning at the 7-sample comparison where two strains, including C.
perfringens, tied for a
rank of 1. This trend continued down to the 2 sample comparison, where 31
strains, including C.
petfringens, tied for the number 1 rank.
[00414] Generally, while using additional samples can reduce the noise/number
of false
positives, further analysis and processing of the resulting strains can be
used to identify C.
pedringens as the causative strain, including from a total of 31 identified
strains. Depending on
the embodiment, configuration, and application, methods of the disclosure can
be practiced with
small numbers of samples, and the number of samples utilized can vary
depending on the sample
source, sample type, metadata, complexity of the target microbiome, and so
forth.
Example 7
Platform for diagnostics - broilers infected with Clostridium pedringens
[00415] This study illustrates an example of the disclosure used to provide
diagnostics. The
objective of the study was to determine the difference in microbial
compositions in broilers
during necrotic enteritis when challenged with various levels of Clostridium
petfringens.
Additional details regarding Clostridium petfringens can be found in Al-
Sheikhly et al. "The
interaction of Clostridium peifringens and its toxins in the production of
necrotic enteritis of
chickens" Avian diseases (1977): 256-263, the entirety of which is herein
expressly incorporated
by reference for all purposes.
[00416] This study utilized 160 Cobb 500 broiler chickens over 21 study days.
The Cobb 500
commercial production broiler chickens were all male and were ¨ 1 day of age
upon receipt (Day
0); Cobb 500 chickens were from Siloam Springs North. Chickens were separated
into four
treatments with twenty birds per pen and two pens per treatment.
119
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00417] The study utilized a feed additive, Phytase 2500 from Nutra Blend,
LLC; Lot Number:
06115A07. Phytase 2500 occurred was commercially available at a concentration
of 2,500
FTU/g with an inclusion level of 0.02%, and is stored in a secured and
temperature-monitored
dry area. The method of administration was via feed over a duration of 21
days.
[00418] The basal feed and treatment diets were sampled in duplicate (-300 g
sample size). One
sample of the basal and each treatment diet was submitted to the sponsor for
assay.
[00419] Experimental Design
[00420] Test Groups
[00421] The test facility was divided into 2 blocks of 4 pens. Treatments were
assigned to the
pens/cages using a completely randomized block design. Specific treatment
groups were
designed as depicted in Table 13.
1004221 Table 13: Experimental design for treatments 1-4.
NE No. of
No. of Birds/
Treatment Challenge Treatment Description No. Birds/ Pen
Pens Treatmen
(Y/N)
1 N Non-Challenged 20 2 40
2 V Challenged with half typical 20 2 40
dose (1.25 mVbird; 2.0-
9.0X108 cfu/m1)
3 V Challenged with typical dose 20 2 40
(2.5 ml/bird; 2.0-9.0X108
cfil/m1)
Challenged with twice the 20 2 40
typical dose (5 ml/bird; 2.0-
9.0X108 cfu/m1)
Total 80 8 160
[004231 Housing
[00424] Assignment of treatments to cages/pens were conducted using a computer
program.
The computer-generated assignment was as follows in Table 14.
[00425] Table 14: Computer selection of treatments to pens.
120
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Block Treatment 1 Treatment 2 Treatment 3
Treatment 4
81 4 1 3 2
132 7 5 8 6
[00426] Birds were housed in an environmentally control facility in wooden
floor pens (-4' x 4'
minus 2.25 sq. ft for feeder space) providing floor space and bird density of
¨0.69 ft2/bird and
temperature, lighting, feeder and water space was similar for all test groups.
Birds were placed in
clean pens containing an appropriate depth of wood shavings to provide a
comfortable
environment for the chicks. Additional shavings were added to pens if they
became too damp for
comfortable conditions for the test birds during the study. Lighting was via
incandescent lights
and a commercial lighting program was used as noted in Table 15.
1004271 Table 15: Lighting programing for incandescent bird lighting
Approximate Bird Age (Days) Approximate Hours of Approximate Light
Intensity
Continuous Light per 24 Hour (Foot Candles)
Period
0-6 23 1.0-1.3
7-21 16 0.2-0.3
[00428] In order to prevent bird migration and bacterial spread from pen to
pen, each pen had a
solid (plastic) divider for approximately 24 inches in height between pens.
[00429] Vaccinations
[00430] Birds were vaccinated for Mareks at the hatchery. Birds were
vaccinated for Newcastle
and infectious bronchitis by spray application on study day 0. No other
vaccinations, except
those in the experimental design, were administered during the study. Records
of the
vaccinations (vaccine source, type, lot number, and expiration date) were
maintained with the
study records. No vaccinations or medications other than those disclosed
herein were utilized.
[00431] Water
121
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00432] Water was provided ad libitum throughout the study via one Plasson
drinker per pen.
Drinkers were checked twice daily and cleaned as needed to assure a clean
water supply to birds
at all times.
1004331 Feed
[00434] Feed was proved ad libitum throughout the study via one hanging, -17-
inch diameter
tube feeder per pen. A chick feeder tray was placed in each floor pen for
approximately the first
4 days. Birds were placed on their respective treatment diets upon receipt
(day 0), according to
the Experimental Design. Feed added and removed from pens from day 0 to study
end were
weighed and recorded.
1004351 Daily Observations
[00436] The test facility, pens, and birds were observed at least twice daily
for general flock
condition, lighting, water, feed, ventilation, and unanticipated events. If
abnormal conditions or
abnormal behavior was noted at any of the twice-daily observations they were
noted in the study
records. The minimum-maximum temperature of the test facility was recorded
once daily.
1004371 Pen Cards
1004381 There were 2 cards attached to each pen. One card identifies the pen
number and the
second will include the treatment number.
1004391 Animal Handling
1004401 Animals were kept under ideal conditions for livability. The animals
were handled in
such a manner as to reduce injuries and unnecessary stress. Humane measures
were strictly
enforced.
1004411 Veterinary Care, Intervention, and Euthanasia
[00442] Birds that developed clinically significant concurrent disease
unrelated to the test
procedures were, at the discretion of the investigator or designee, removed
from the study and
euthanized in accordance with site standard operating procedures. In addition,
moribund or
injured birds may also be euthanized upon authority of a site veterinarian or
a qualified
technician. Any reasons for withdrawal were documented. In an animal died, or
was removed
and euthanized for humane reasons, it was recorded on the mortality sheet for
the pen and a
122
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
necropsy performed, and was filed to document the reason for removal. If
euthanasia was
deemed necessary, animals were euthanized via cervical dislocation.
100443] Mortality and Culls
[00444] Starting on study day 0, any bird that was found dead was removed
weighed and
necropsied. Birds that are unable to reach feed or water were sacrificed and
necropsied. The
weight and probable cause of death and necropsy findings were recorded on the
pen mortality
record.
[00445] Body Weight and Feed Intake
[00446] Birds were weighed by pen and individually on approximately days 14
and 21. The
feed remaining in each pen was weighed and recorded on study days 14 and 21.
The feed intake
during days 14-21 were calculated.
[00447] Weight Gain and Feed Conversion
[00448] Average bird weight, on a pen and individual basis, on each weigh day
was
summarized. The average feed conversion was calculated on study day 21 using
the total feed
consumption for the pen divided by the total weight of surviving birds.
Adjusted feed conversion
was calculated using the total feed consumption in a pen divided by the total
weight of surviving
birds and weight of birds that died or were removed from that pen.
100449] Digesta Collection
[00450] On day 21, each bird was euthanized by cervical dislocation to collect
the following
using the described procedures, gloves were changed between each bird.
[00451] immediately place the contents of one cecum in a 1.5-ml tube prefilled
with 150 pl stop
solution.
[00452] Immediately place the contents of the small intestine into a 1.5-ml
tube prefilled with
150 pi stop solution.
[004531 Dissect the gizzard out of the GI tract, remove the contents with
forceps, and place in a
1.5-ml tube prefilled with 150 pl stop solution.
123
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00454] Dissect the crop out of the GI tract, remove the contents with
forceps/scrape out
mucosa] lining, and place in a 1.5-ml tube prefilled with 150 pl stop
solution.
[00455] Store all samples at 4 C until shipment
100456] Scales
[00457] Scales used in weighing of feed and feed additives were licensed
and/or certified by the
State of Colorado. At each use the scales were checked using standard weights
according to CQR
standard operating procedures.
100458] Clostridium perfringens Challenge
100459] Method of Administration
[00460] The Clostridium petfringens culture was obtained from Microbial
Research, Inc.
Administration of the C. petfringens (CL-15, Type A, a and 32 toxins) cultures
in this study
were via the feed. Feed from each pen's feeder was used to mix with the
culture. Prior to placing
the cultures in the pens, the treatment feed was removed from the birds for
approximately 4-8
hours. For each pen of birds, a fixed amount based on study design of the
broth culture at a
concentration of approximately 2.0 ¨ 9.0 X 108 cfu/m1 was mixed with a fixed
amount of feed
(-25g/bird) in the feeder tray and all challenged pens were treated the same.
Most of the culture-
feed was consumed within 1-2 hours. So that birds in all treatments are
treated similar, the
groups that are not challenged also had the feed removed during the same time
period as the
challenged groups.
[00461] Clostridium Challenge
[00462] The C. petfringens culture (CL-15) was grown for ¨5 hours at ¨37 C in
fluid
thioglycollate medium containing starch. CL-15 is a field strain of C.
petfringens from a broiler
outbreak in Colorado. A fresh broth culture was prepared and used each day.
For each pen of
birds, a fixed amount of the overnight broth culture was mixed with a fixed
amount of treatment
feed in the feeder tray (see administration). The amount of feed, volume, and
quantitation of
culture inoculum, and number of days dosed was documented in the final report,
and all pens
124
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
were treated the same. Birds received the C. perfringens culture for one day
(day 17).
Quantitation was conducted by Microbial Research, Inc on the culture and
results were
documented in the final report. There was no target mortality for this study.
[00463] Lesion Scoring
[00464] Four days following the last C. perfringens culture administration,
five birds were
randomly selected from each pen by first bird caught, sacrificed, and
intestinal lesions scored for
necrotic enteritis. Lesions were scored as follows:
[00465] 0 = normal: No NE lesions, small intestine has normal elasticity
(rolls back to normal
position after being opened).
[00466] 1 = mild: Small intestinal wall is thin and flaccid (remains flat when
opened and doesn't
roll back into normal position after being opened); excess mucus covering
mucus membrane.
[00467] 2 = moderate: Noticeable reddening and swelling of the intestinal
wall; minor
ulceration and necrosis of the intestinal membrane; excess mucus.
[00468] 3 = severe: Extensive area(s) of necrosis and ulceration of the small
intestinal
membrane; significant hemorrhage; layer of fibrin and necrotic debris on the
mucus membrane
(Turkish towel appearance).
[00469] 4 = dead or moribund: Bird that would likely die within 24 hours and
has NE lesion
score of 2 or more.
[00470] Dispositions
[00471] Excess Test Articles
[00472] An accounting was maintained of the test articles received and used
for this study.
Excess test articles were dispositioned or returned to the sponsor.
Documentation was provided
with the study records.
[00473] Feed
[00474] An accounting was maintained of all diets. The amount mixed, used and
discarded was
documented. Unused feed was disposed of either by salvage sale and/or placing
into a dumpster
125
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
for commercial transport to a local landfill for burial. Disposition was
documented in the study
records.
1004751 Test Animals
[00476] An accounting was maintained for birds received for the study.
Disposal of mortalities
and birds sacrificed during the study and at study end was discarded to the
landfill at study end.
Documentation of disposition was provided with the study records. No food
products derived
from animals enrolled in this study entered the human food chain.
[00477] Sample Analysis
[00478] A portion of each digesta sample was stained and put through a flow
cytometer to
quantify the number of cells of each microorganism type in each sample. A
separate portion of
the same digesta sample was homogenized with bead beating to lyse
microorganisms. DNA and
RNA was extracted and purified from each sample and prepared for sequencing on
an Illumina
Miseq. Samples were sequenced using paired-end chemistry, with 300 base pairs
sequenced on
each end of the library. The sequencing reads were used to quantify the number
of cells of each
active, microbial member present in each bird after C. perfringens infection.
[00479] Necrotic enteritis, the severe necrosis of intestinal mucosa, is
caused by toxins
generated by C. perfringens. Thus, to assess the ability of the platform as a
diagnostic for
disease, presence and activity of C. perfringens was analyzed in context of
lesion scores for each
bird sampled. All organs were analyzed¨the results indicated that the small
intestine, however,
was the best predictor of C. petfrigens infection. This is expected, as the
small intestine is the
primary location of pathogen establishment
[00480] The results are presented in FIG. 9 and Table 16. C. perfrigens was
detected in all but
one bird that scored 1 or higher during lesion scoring. The amount of C.
perfrigens present
tended to correlate with the lesion score measured for each bird¨the more C
pedilgens present,
the more likely the bird was scored as a "4". Multiple birds that scored "0"
for lesion scores did
have C perfringens present in their GI tract. Despite this presence, activity
analysis revealed that
C perfringens was not active in these birds. These results indicate that the
disclosed methods
and systems are able to detect the quantity and activity of C. perfringens in
birds with necrotic
126
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
enteritis. This information can be used as a diagnostic to predict the
causative agent of a necrotic
enteritis outbreak in broiler chickens, as well as the severity of the disease
in individual, sick
birds.
[004811 Table 16: Lesion score and C. po:fringens abundance for each bird in
the trial
Bird number Lesion score perfringensAbundance
1 1 1.08
2 1 0.10
3 0 0.16
4 3 0.26
1 0.03
6 1 0.19
7 2 8.41
8 0 0.11
9 3 0.06
4 33.80
11 3 8.10
12 1 0.08
13 2 0.05
14 4 0.45
1 0.06
16 0 0.08
17 3 0.06
18 1 0.02
19 0 0.08
0 0.00
21 2 0.01
22 0 0.00
23 0 0.00
24 3 0.00
3 0.38
26 3 0.25
27 0 0.19
28 3 28.79
29 4 0.24
3 5.23
31 3 1.88
32 4 5.49
33 1 0.04
34 1 0.65
127
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
35 0 3.00
36 3 0.28
37 3 0.07
38 0 0.02
39 3 0.07
40 2 3.31
41 1 74.61
42 0 0.06
43 3 0.07
44 1 1.12
45 4 28.81
46 2 0.03
47 3 0.05
48 0 0.09
49 4 43,83
50 4 89 78
51 4 88.00
52 4 77.19
53 4 86.00
54 4 65.65
55 4 43 96
56 4 57 81
57 4 64.08
100482]
Example 8
Shifts in rumen microbial composition after administration of a microbial
composition
[00483] The methods of the disclosure were applied to increase the total
amount of milk fat and
milk protein produced by a lactating ruminant, and the calculated ECM.
[00484] The methodologies of the disclosure presented herein¨based on
utilizing the disclosed
isolated microbes, ensembles, and compositions comprising the same¨demonstrate
an increase
in the total amount of milk fat and milk protein produced by a lactating
ruminant. These
increases were realized without the need for further addition of hormones.
[00485] In this example, a microbial ensemble comprising two isolated
microbes, a bacterium
and a fungus, identified and synthesized by the disclosed methods, was
administered to Holstein
cows in mid-stage lactation over a period of five weeks.
128
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00486] The cows were randomly assigned into 2 groups of 8, wherein one of the
groups was a
control group that received a buffer lacking a microbial ensemble. The second
group, the
experimental group, was administered the microbial ensemble once per day for
five weeks. Each
of the cows were housed in individual pens and were given free access to feed
and water. The
diet was a high milk yield diet. Cows were fed ad libitum and the feed was
weighed at the end of
each day, and prior day refusals were weighed and discarded. Weighing was
performed with a
PS-2000 scale from Salter Brecknell (Fairmont, MN).
[00487] Cows were cannulated such that a cannula extended into the rumen of
the cows. Cows
were further provided at least 10 days of recovery post cannulation prior to
administering control
dosages or experimental dosages.
[00488] Each administration consisted of 20 ml of a neutral buffered saline,
and each
administration consisted of approximately 109 cells suspended in the saline.
The control group
received 20 ml of the saline once per day, while the experimental group
received 20 ml of the
saline further comprising 109 microbial cells of the described microbial
ensemble.
[00489] The rumen of every cow was sampled on days 0, 7, 14, 21, and 35,
wherein day 0 was
the day prior to microbial administration. Note that the experimental and
control administrations
were performed after the rumen was sampled on that day. Daily sampling of the
rumen,
beginning on day 0, with a pH meter from Hanna Instruments (Woonsocket, RI)
was inserted
into the collected rumen fluid for recordings. Rumen sampling included both
particulate and
fluid sampling from the center, dorsal, ventral, anterior, and posterior
regions of the rumen
through the cannula, and all five samples were pooled into 15ml conical vials
containing 1.5m1
of stop solution (95% ethanol, 5% phenol) and stored at 4 C and shipped to
Ascus Biosciences
(Vista, California) on ice.
[00490] A portion of each rumen sample was stained and put through a flow
cytometer to
quantify the number of cells of each microorganism type in each sample. A
separate portion of
the same rumen sample was homogenized with bead beating to lyse
microorganisms. DNA and
RNA was extracted and purified from each sample and prepared for sequencing on
an 11lumina
Miseq. Samples were sequenced using paired-end chemistry, with 300 base pairs
sequenced on
each end of the library. The sequencing reads were used to quantify the number
of cells of each
129
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
active, microbial member present in each animal rumen in the control and
experimental groups
over the course of the experiment.
[00491] Both the bacterium and fungus colonized the rumen, and were active in
the rumen after
¨3-5 days of daily administration, depending on the animal. This colonization
was observed in
the experimental group, but not in the control group. The rumen is a dynamic
environment,
where the chemistry of the cumulative rumen microbial population is highly
intertwined. The
artificial addition of the microbial ensemble could have effects on the
overall structure of the
community. To assess this potential impact, the entire microbial community was
analyzed over
the course of the experiment to identify higher level taxonomic shifts in
microbial community
population.
[00492] Distinct trends were not observed in the fungal populations over time,
aside from the
higher cell numbers of fungus administered in the experimental animals. The
bacterial
populations, however, did change more predictably. To assess high level trends
across individual
animals over time, percent compositions of the microbial populations were
calculated and
compared. Only genera composing greater than 1% of the community were
analyzed. Percent
composition of genera containing known fiber-degrading bacteria, including
Ruminococcus,
were found to increase in experimental animals as compared to control animals.
Volatile fatty
acid-producing genera, including Clostridial cluster XlVa, Clostridium,
Pseudobutyrivibrio,
Butyricimonas, and Lachnospira were also found at higher abundances in the
experimental
animals. The biggest shift was observed in the genera Prevotella. Members of
this genus have
been shown to be involved in the digestion of cellobiose, pectin, and various
other structural
carbohydrates within the rumen. Prevotella sp. Have also been implicated in
the conversion of
plant lignins into beneficial antioxidants (prevotella source).
[00493] To more directly measure quantitative changes in the rumen over time,
cell count data
was integrated with sequencing data to identify bulk changes in the population
at the cell level.
Fold changes in cell numbers were determined by dividing the average number of
cells of each
genera in the experimental group by the average number of cells of each genera
in the control
group. The cell count analysis captured many genera that fell under the
threshold in the previous
analysis Promicromonospora, Rhodopirellula, Olivibacter, Victivallis,
Nocardia, Lentisphaera,
Eubacteiru, Pedobacter, Butyricimonas, Mogibacterium, and Desulfovibrio were
all found to be
130
CA 03048247 2019-06-21
WO 2018/126033
PCT/US2017/068753
at least 10 fold higher on average in the experimental animals. Prevotella,
Lachnospira,
Butyrieicoceus, Clostridium 1N7Va, Roseburia, Clostridiunt_sensu_stricto, and
Pseudobutyrivibrio were found to be -1.5 times higher in the experimental
animals.
(00494) Table 17 - Family level Analysis:
Taxonomy Control (%) Variation Experimental (%)
Variation
Prevotellaceae 15.27 6.43 18.62 5.63
Ruminococcaceae 16.40 5.14 17.84 6.44
Lachnospiraceae 23.85 7.63 24.58 6.96
[00495] Table 18 - Genus level Analysis:
Taxonomy Control (%) Variation Experimental (I%)
Variation
Prevotella 16.14 5.98 19.14 5.27
Clostridium_XlVa 12.41 5.35 12.83 4.81
Lachnospiracea...incertae_sedis 3.68 1.68 3.93 1.33
Ruminococcus 3.70 2.21 3.82 1.82
Clostridium_IV 3.02 1.87 3.51 1.74
Fibrobacter 2.10 1.72 2.06 1.33
Butyricimonas 1.68 1.35 1.83 2.38
Clostridium...sensu_stricto 1.52 0.65 1.81 0.53
Pseudobutyrivibrio 1.00 0.64 1.42 1.03
Citrobacter 0.71 1.86 1.95 3.00
Selenomonas 1.04 0.83 1.34 0.86 .
Hydrogenoanaerobacterium 1.03 1.08 1.11 0.78
[00496] Table 19 - Fold changes in cells:
Genus Fold change
(experimental/control)
Promicromonospora 22619.50
Rhodopirellula 643.31
Olivibacter 394.01
Victivallis 83.97
Nocardia 73.81 .
Lentisphaera 57.70
Eubacterium 50.19
Pedobacter 26.15
Butyricimonas 15.47
Mogibacterium 15.23
Desulfovibrio 13.55 .
Anaeroplasma 8.84
Sharpea 8.78
131
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Erysipelotrichaceae...incertae..sedis 5.71
Saccharofermentans 5.09
Parabacteroides 4.16
Papillibacter 3.63
Citrobacter 2.95
Lachnospiracea_incertae_sedis 2.27
Prevotella 1.60
Butyricicoccus 1.95
Clostridium_XlVa 1.47
Roseburia 1.44
Pseudobutyrivibrio 1.43
Clostridium_sensu_stricto 1.29
Selenomonas 1.25
Oisendia 1.04
100497]
Example 9
Determining the equine fecal microbiota in horses with colic and site-matched
healthy control
horses
[00498] Horses are often diagnosed with colic, and common intestinal disorder
that causes
severe abdominal pain to the animal. The source of colic is highly variable.
It can be caused by
blockages due to ingestion of indigestible objects, gas, or torsion of the
digestive track. Some
colics are linked to abnormalities in the microbial populations residing in
the animal's
gastrointestinal tract. In most cases, it is very difficult to diagnose the
exact cause of colic,
particularly in chronic cases. Here, the feces of twenty horses were analyzed
with disclosed
methods to diagnose animals with microbial-based colic.
[00499] Over the course of two months, twenty horses (ten control, ten
experimental) were
assayed. Animals were sampled in pairs. For each colic horse, a control hose
living on the same
farm was sampled. The control horse had a similar travel history as the colic
horse, and did not
receive antimicrobials nor have an episode of colic in the previous 6 months.
[00500] The owner of each horse completed a signed consent form and survey.
Each horse
received a physical examination that measured heart rate, respiratory rate,
temperature, mucous
membrane color, capillary refill time, and gastrointestinal borborygmi. Any
other abnormalities
found on examination were reported. Blood was collected for complete blood
count and
132
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
chemistry panel, and fecal samples were collected by inserting the swab 4 to 6
cm into the
rectum of the animal. The swab was gently rubbed against the inner walls of
the rectum to
collect cells and fecal material. The swab was then fully immersed into a tube
prefi Iled with
stop solution, and then immediately transferred to a new, sterile 1.7mL tube.
Excess swab
stick was removed prior to closing the tube. This was repeated to generate a
duplicate
sample. For colic horses, feces were also collected during rectal palpation.
Feces were stored
in a 50mL conical pre-filled with 15mL stop solution. Stomach fluids and
contents were
collected when possible.
[00501] All samples were stored at 4 C/on ice during transit. Swabs were
stored at -20 C
upon return to the lab, and remained at -20 C until shipped.
1005021 Data collection included:
= Age, breed, predominant use
= Blood Test results
= DievFeeding/supplement Regime
= Housing Type
= Travel History
= Deworming history
= Treatment
= Medication and Medical history (esp. if colic is reoccurring), to include
any episodes of
anesthesia
= Any additional information about horse symptoms/behavior, pathogen tests
(Salmonella, Clostridium)
= Final diagnosis
[00503] All fecal samples were analyzed using the methods of the disclosure.
[00504] Weese et al. ("Changes in the faecal microbiota of mares precede the
development of
post partum colic" Equine veterinary journal 47.6 2015: 641-649, herein
expressly incorporated
by reference in its entirety for all purposes) identified that mares tended to
develop an episode of
colic due to large colon volvulus when they had a higher relative abundance of
Proteobacteria in
their feces as compared to control horses that did not colic. Large colon
volvulus is one of the
133
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
most severe forms of colic, and may be prevented if the animal's diet and
management is
changed prior to progression of the colic to a more severe state. In many
cases, early detection is
not possible, and horses with large colon volvulus undergo invasive surgeries
or are put down
when the colic relapses.
[00505] The analysis revealed, and as corroborated by veterinary diagnosis,
that only a few of
the ten horses had a microbial-caused colic. One horse, in particular, was
diagnosed with colic
due to large colon volvulus. As can be seen in FIG. 10, Colic Horse 3 did have
elevated levels of
highly active proteobacteria (pink bar) as compared to all of the other
horses. Further analysis
showed that this proteobacteria is a distant relative of Helicobacier equorum.
Although previous
studies have not been able to link this species to pathogenicity (see, e.g.,
Moyaert et al.
"Helicobacter equorum: prevalence and significance for horses and humans" FEMS
Immunology
& Medical Microbiology 57.1 (2009): 14-16, the entirety of which is herein
expressly
incorporated by reference for all purposes), the results here indicate that it
does play a role in the
development in large colon volvulus colic. Thus, although horses are afflicted
by a wide variety
of colics, the disclosed methods are able to diagnose animals with microbial-
based colic. FIG. 10
illustrates relative abundance of the active microorganisms in horse feces at
the phylum level.
Proteobacteria are represented by a light pink color. Colic Horse 3, the horse
diagnosed with
large colon volvulus colic, is denoted by the red rectangle.
[00506] FIG. 11 provides an overview summary of an example diagnostic platform
workflow,
according to some embodiments.
Example 10
Equine State Identification and Microbial Insights
[00507] The objective of the study was to produce biomarkers and possible
biological
mechanisms in and differentiate multiple states of colic (i.e. bacterial vs.
non-bacterial equine
colic). A total of 60 patients were sampled at multiple times, 30 of the
patients were identified as
having a form of colic. The other 30 patients were identified as healthy with
no other diagnosed
conditions.
100508] Sample Processing: Fecal samples were taken from each sampling
point and
immediately added to a 15 ml conical tube prefilled with stabilization
solution and stored at 4 C.
The solution was mixed via inversion several times and stored at 4 C
immediately after. Fecal
134
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
samples were centrifuged at 4,000 rpm for 15 min, the supernatant was decanted
and 0.5 mL was
aliquoted for Total RNA and DNA extraction using the PowerViral Environmental
RNA/DNA
Isolation Kit (Mo Bio Laboratories, Inc., Carlsbad, CA, USA). Decanted
supernatant was flash
frozen in liquid nitrogen for downstream metabolomics processing.
[00509] 16S rRNA. The 16S rRNA gene was amplified using 27F and 534R
modified for
Illumina sequencing, and the ITS region was amplified using ITS5 and ITS4
modified for
Illumina sequencing following standard protocols Q5 High-Fidelity DNA
Polymerase (New
England Biolabs, Inc., Ipswich, MA, USA). Following amplification, PCR
products were
verified with a standard agarose gel electrophoresis and purified using AMPure
XP bead
(Beckman Coulter, Brea, CA, USA). The purified amplicon library was quantified
and
sequenced on the MiSeq Platform (Illumina, San Diego, CA, USA) according to
standard
protocols (see, e.g., Flores et al. 2014). Raw fastq read were de-multiplexed
on the MiSeq
Platform (Illumina, San Diego, CA, USA). All total cell counts were performed
on an SH800S
Cell Sorter (Sony, San Jose, CA, USA). All raw sequencing data was trimmed of
adapter
sequences and phred33 quality filtered at a cutoff of 20 using Trim Galore
(see, e.g., Krueger
2015). All remaining sequences were then filtered for PhiX, low-complexity
reads and cross-talk
(see, e.g., Edgar 2016). 16S taxonomic sequence clustering and classification
was performed
with the USEARCH's UNOISE and SINTAX (v10Ø240) (see, e.g., Edgar and
Flyvbjerg 2015;
Edgar 2016) with the RDP 16S rRNA database (see, e.g., Cole et al. 2014) in
conjunction with
the target sequences for DY20 and 21.
[00510] Activity Measurement. cDNA synthesis was performed on RNA samples
after
DNase I treatment (New England Biolabs, Inc., Ipswich, MA, USA). Random Primer
Mix (New
England Biolabs, Inc., Ipswich, MA, USA), Superscript IV Reverse
Transcriptase (Thermo
Fisher Scientific, Waltham, MA, USA), and Rnasin (Promega, Madison, WI, USA)
were used
for cDNA synthesis following manufacturers protocols. The 16S rRNA gene was
amplified
using 27F and 534R modified for Illumina sequencing, and the ITS region was
amplified using
ITS5 and ITS4 modified for Illumina sequencing following standard protocols.
Following
amplification, PCR products were verified and purified using AMPure XP beads
(Beckman
Coulter, Brea, CA, USA). The purified amplicon library was quantified with
Qubit DNA HS
kit (Thermo Fisher Scientific, Waltham, MA, USA) and sequenced on the MiSeq
Platform
135
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
(IIlumina, San Diego, CA, USA) according to standard protocols. Raw fastq
reads were de-
multiplexed on the MiSeq Platform (IIlumina, San Diego, CA, USA).
[00511]
Cell Staining and Counting. A small aliquot of each sample was separated into
a
new 1.7mL tube and weighed. 1 mL of sterile PBS was added to each sample, and
bead beat
without beads to separate cells from fibrous rumen content. Samples were then
centrifuged to
remove large debris. An aliquot of the supernatant was diluted in PBS, and
then strained.
Counting beads were added to each tube (Spherotech ACFP-70-10). Dyed samples
were then
processed on a Sony SH800 cell sorter (Sony, San Jose, CA, USA), and number of
fungal and
bacterial cells per gram of original sample was determined.
1005121
Biomarker Identification. Absolute cell counts were used to produce absolute
cell counts and inactive OTUs were filtered through cDNA sequencing
normalization. Sample
output was processed in a OTU table and preprocessed through matrix
completion. Following
completion the data was learned with respect to health state (bacterial colic
vs. non-bacterial
colic or Healthy) with a ROC greater than 0.9 in a ten fold validation. Data
was visualized in
PCoA dimensionality reduction. Furthermore, common pathogenic biomarkers were
screened
from the OTU table. Finally compositional composites were compared between
health states.
[00513]
Case Study. New samples for screening were submitted and run through the
platform using the methods of the disclosure. The Random Forests machine
learning model
produced distributions based on predicted health states (FIG. 12a). Common
pathogenic
biomarkers revealed no highly abundant markers (FIG. 12b). PCoA revealed the
sample fell
within a colic distribution (FIG. 12c). Finally the compositional composite
between samples
states compared to the submitted sample revealed the sample submitted matched
colic
compositions (FIG. 12d). The sample and subsequent analysis suggests that the
horse it was
derived from was in a colic state at the time of sampling.
Example 11
Dairy State Identification and Microbial Insights
[00514] The
objective of the study was to produce biomarkers and possible biological
mechanisms in the dairy rumen related to production and other important
external factors. A total
of 5,000 samples were collected from varying climates, geographies, breeds,
feed systems, and
health states. Furthermore, several healthy states were sampled primarily
driven by diet type (i.e.
TMR, pTMR, grazing) in contrast to several general unhealthy states (i.e. Milk
Fat Depression).
136
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00515] Sample Processing. Fecal samples were taken from each sampling
point and
immediately added to a 15 ml conical tube prefilled with stabilization
solution and stored at 4 C.
The solution was mixed via inversion several times and stored at 4 C
immediately after. Fecal
samples were centrifuged at 4,000 rpm for 15 min, the supernatant was decanted
and 0.5 mL was
aliquoted for Total RNA and DNA extraction using the PowerViral Environmental
RNA/DNA
Isolation Kit (Mo Bio Laboratories, Inc., Carlsbad, CA, USA). Decanted
supernatant was flash
frozen in liquid nitrogen for downstream metabolomics processing.
[00516] 16S rRNA. The 16S rRNA gene was amplified using 27F and 534R
modified for
Illumina sequencing, and the ITS region was amplified using ITS5 and ITS4
modified for
Illumina sequencing following standard protocols Q50 High-Fidelity DNA
Polymerase (New
England Biolabs, Inc., Ipswich, MA, USA). Following amplification, PCR
products were
verified with a standard agarose gel electrophoresis and purified using AMPure
XP bead
(Beckman Coulter, Brea, CA, USA). The purified amplicon library was quantified
and
sequenced on the MiSeq Platform (Illumina, San Diego, CA, USA) according to
standard
protocols (see, e.g., Flores et al. 2014). Raw fastq read were de-multiplexed
on the MiSeq
Platform (Illumina, San Diego, CA, USA). All total cell counts were performed
on an SH800S
Cell Sorter (Sony, San Jose, CA, USA). All raw sequencing data was trimmed of
adapter
sequences and phred33 quality filtered at a cutoff of 20 using Trim Galore
(see, e.g., Krueger
2015). All remaining sequences were then filtered for PhiX, low-complexity
reads and cross-talk
(see, e.g., Edgar 2016). 16S taxonomic sequence clustering and classification
was performed
with the USEARCH's UNOISE and SINTAX (v10Ø240) (see, e.g., Edgar and
Flyvbjerg 2015;
Edgar 2016) with the RDP 16S rRNA database (see, e.g., Cole et al. 2014) in
conjunction with
the target sequences for DY20 and 21.
[00517] Activity Measurement. cDNA synthesis was performed on RNA samples
after
DNase I treatment (New England Biolabs, Inc., Ipswich, MA, USA). Random Primer
Mix (New
England Biolabs, Inc., Ipswich, MA, USA), Superscript IV Reverse
Transcriptase (Thermo
Fisher Scientific, Waltham, MA, USA), and Rnasin (Promega, Madison, WI, USA)
were used
for cDNA synthesis following manufacturers protocols. The 16S rRNA gene was
amplified
137
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
using 27F and 534R modified for Illumina sequencing, and the ITS region was
amplified using
ITS5 and ITS4 modified for Illumina sequencing following standard protocols.
Following
amplification, PCR products were verified and purified using AMPure XP beads
(Beckman
Coulter, Brea, CA, USA). The purified amplicon library was quantified with
Qubit DNA HS kit
(Thermo Fisher Scientific, Waltham, MA, USA) and sequenced on the MiSeq
Platform
(Illumina, San Diego, CA, USA) according to standard protocols. Raw fastq
reads were de-
multiplexed on the MiSeq Platform (Illumina, San Diego, CA, USA).
1005181
Cell Staining and Counting. A small aliquot of each sample was separated into
a
new 1.7mL tube and weighed. 1 nIL of sterile PBS was added to each sample, and
bead beat
without beads to separate cells from fibrous rumen content. Samples were then
centrifuged to
remove large debris. An aliquot of the supernatant was diluted in PBS, and
then strained.
Counting beads were added to each tube (Spherotech ACFP-70-10). Dyed samples
were then
processed on a Sony SH800 cell sorter (Sony, San Jose, CA, USA), and number of
fungal and
bacterial cells per gram of original sample was determined.
[00519]
Biomarker and predictive model building. Absolute cell counts were used to
produce absolute cell counts and inactive OTUs were filtered through cDNA
sequencing
normalization. Data was then completed through matrix completion. Data was
visualized in
PCoA dimensionality reduction. This revealed several tightly clustered healthy
states with TMR
based diet on the left and pTMR based diet on the right and a large dispersed
group of unhealthy
states below (FIG. 13b). Animal data was first learned with respect to the
microbial compositions
through partial-least squares regression. The model produced was accurate with
an R-squared
above 0.9 and a mean squared error less than I. This allowed compositions to
be predicted based
off nutritional, geographical, and climate input Through the manipulation of
these data forecasts
of microbial compositions could be produced. Random Forests machine learning
was used to
predict nutritional, geographical, and climate data from microbial
compositions with an ROC
greater than 0.9 in a ten fold validation. Both of these methods could be used
in tandem where
either sample metadata or sample microbial compositions can be learned and
predicted. This is
fit to the many healthy and unhealthy states where by any state can be
predictively optimized.
1005201
Case study. A sample was submitted from a rumen sample of a healthy dairy cow
on a pIMR diet. The rumen sample was analyzed using the described method, and
sequenced on
an Illumina Miseq. The PCoA dimensionality reduction placed the sample in the
healthy
138
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
distribution (FIG. 13a). Furthermore, the optimization of NDF and pH of the
rumen in silico
placed the rumen composition in a more productive state on a pTMR diet (FIG.
13b), suggesting
that alterations to these two variables via feed changes or feed additives
will make the sampled
animal's microbial composition match that of the closest most productive
state. The microbial
compositions could also be learned to predict the external factors not
measured for the
identification of possible mis-managements in health (FIG. 14a), diet (FIG.
14b), and climate
(FIG. 14c).
[00521] While generally discussed as a singular state, it should be understood
that for some
embodiments and applications, a state (e.g., baseline state) or biostate can
refer to multiple states
and/or biostates associated with a particular microbiome, and multiple states
can also be utilized
in defining a baseline, defining particular state, characterizing samples,
identifying potential
problems, and/or treating particular indications, whether on an individual or
group (e.g., herd)
level. For example, with the colic examples above, there can be multiple
causes of colic, and
such are reflected in the microbiome. In some embodiments, a comparison
according to the
disclosure can utilize the following states: control (healthy), microbial
colic, and non-microbial
colic (and in some embodiments, multiple different states/substates).
[00522] Additional Example Embodiments
[00523] Embodiment Al is a method, comprising: obtaining at least two samples
sharing at
least one common characteristic and having at least one different
characteristic; for each sample,
detecting the presence of one or more microorganism types in each sample;
determining a
number of each detected microorganism type of the one or more microorganism
types in each
sample; measuring a number of unique first markers in each sample, and
quantity thereof, each
unique first marker being a marker of a microorganism strain; integrating the
number of each
microorganism type and the number of the first markers to yield the absolute
cell count of each
microorganism strain present in each sample; measuring at least one unique
second marker for
each microorganism strain based on a specified threshold to determine an
activity level for that
microorganism strain in each sample; filtering the absolute cell count by the
determined activity
to provide a list of active microorganisms strains and their respective
absolute cell counts for
each of the at least two samples; comparing the filtered absolute cell counts
of active
microorganisms strains for each of the at least two samples with at least one
measured metadata
or additional active microorganism strain for each of the at least two samples
and categorizing
139
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
the active microorganism strains into at least two groups based on predicted
function and/or
chemistry; selecting at least one microorganism strain from the at least two
groups; and
combining the selected at least one microorganism strain from the at least two
groups to form a
ensemble of microorganisms configured to alter a property corresponding to the
at least one
metadata.
100524] Embodiment A2 is a method according to embodiment Al, wherein
measuring the
number of unique first markers includes measuring the number of unique genomic
DNA markers
in each sample. Embodiment A3 is a method according to embodiment Al, wherein
measuring
the number of unique first markers includes measuring the number of unique RNA
markers in
each sample. Embodiment A4 is a method according to embodiment Al, wherein
measuring the
number of unique first markers includes measuring the number of unique protein
markers in each
sample. Embodiment A5 is a method according to embodiment Al, wherein
measuring the
number of unique first markers includes measuring the number of unique
metabolite markers in
each sample. Embodiment A6 is a method according to embodiment A5, wherein
measuring the
number of unique metabolite markers includes measuring the number of unique
lipid markers in
each sample. Embodiment A7 is a method according to embodiment A5, wherein
measuring the
number of unique metabolite markers includes measuring the number of unique
carbohydrate
markers in each sample. Embodiment A8 is a method according to embodiment Al,
wherein
measuring the number of unique first markers, and quantity thereof, includes
subjecting genomic
DNA from each sample to a high throughput sequencing reaction. Embodiment A9
is a method
according to embodiment Al, wherein measuring the number of unique first
markers, and
quantity thereof, includes subjecting genomic DNA from each sample to
metagenome
sequencing. Embodiment A10 is a method according to embodiment Al, wherein the
unique first
markers include at least one of an mRNA marker, an siRNA marker, and/or a
ribosomal RNA
marker. Embodiment All is a method according to embodiment Al, wherein the
unique first
markers include at least one of a sigma factor, a transcription factor,
nucleoside associated
protein, and/or metabolic enzyme.
1005251 Embodiment Al2 is a method according to any one of embodiments Al-All,
wherein
measuring the at least one unique second marker includes measuring a level of
expression of the
at least one unique second marker in each sample. Embodiment A13 is a method
according to
embodiment Al2, wherein measuring the level of expression of the at least one
unique second
140
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
marker includes subjecting inRNA in the sample to gene expression analysis.
Embodiment A14
is a method according to embodiment A13, wherein the gene expression analysis
includes a
sequencing reaction. Embodiment A15 is a method according to embodiment A13,
wherein the
gene expression analysis includes a quantitative polymerase chain reaction
(qPCR),
metatranscriptome sequencing, andlor transcriptome sequencing. Embodiment A16
is a method
according to embodiment Al2, wherein measuring the level of expression of the
at least one
unique second marker includes subjecting each sample or a portion thereof to
mass spectrometry
analysis. Embodiment A17 is a method according to embodiment Al2, wherein
measuring the
level of expression of the at least one unique second marker includes
subjecting each sample or a
portion thereof to metaribosome profiling, or ribosome profiling.
[00526] Embodiment A18 is a method according to any one of embodiments Al -
A17, wherein
the one or more microorganism types includes bacteria, archaea, fungi,
protozoa, plant, other
eukaryote, viruses, viroids, or a combination thereof. Embodiment A19 is a
method according to
any one of embodiments A1-A18, wherein the one or more microorganism strains
is one or more
bacterial strains, archaeal strains, fungal strains, protozoa strains, plant
strains, other eukaryote
strains, viral strains, viroid strains, or a combination thereof. Embodiment
A20 is a method
according to embodiment A19, wherein the one or more microorganism strains is
one or more
fungal species or sub-species; and/or wherein the one or more microorganism
strains is one or
more bacterial species or sub-species.
[00527] Embodiment A21 is a method according to any one of embodiments A 1 -
A20, wherein
determining the number of each of the one or more microorganism types in each
sample includes
subjecting each sample or a portion thereof to sequencing, centrifugation,
optical microscopy,
fluorescent microscopy, staining, mass spectrometry, microfluidics,
quantitative polymerase
chain reaction (qPCR), gel electrophoresis, and/or flow cytometry.
[00528] Embodiment A22 is a method according to embodiment Al, wherein the
unique first
markers include a phylogenetic marker comprising a 5S ribosomal subunit gene,
a 16S ribosomal
subunit gene, a 23S ribosomal subunit gene, a 5.8S ribosomal subunit gene, a
18S ribosomal
subunit gene, a 28S ribosomal subunit gene, a cytochrome c oxidase subunit
gene, a I3-tubulin
gene, an elongation factor gene, an RNA polymerase subunit gene, an internal
transcribed spacer
(ITS), or a combination thereof.
141
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00529] Embodiment A22a is a method according to embodiment Al, wherein the
unique first
marker does not include a phylogenetic marker. Embodiment A22b is a method
according to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 5S ribosomal subunit gene. Embodiment A22c is a method according
to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 16S ribosomal subunit gene. Embodiment A22d is a method according
to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 235 ribosomal subunit gene. Embodiment A22e is a method according
to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 5.8S ribosomal subunit gene. Embodiment A22f is a method
according to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 18S ribosomal subunit gene. Embodiment A22g is a method according
to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a 28S ribosomal subunit gene. Embodiment A22h is a method according
to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a cytochrome c oxidase subunit gene. Embodiment A22i is a method
according to
embodiment Al, wherein the unique first marker does not include a phylogenetic
marker
comprising a [3-tubulin gene. Embodiment A22j is a method according to
embodiment Al,
wherein the unique first marker does not include a phylogenetic marker
comprising an
elongation factor gene. Embodiment A22k is a method according to embodiment
Al, wherein
the unique first marker does not include a phylogenetic marker comprising an
RNA polymerase
subunit gene. Embodiment A221 is a method according to embodiment Al, wherein
the unique
first marker does not include a phylogenetic marker comprising an internal
transcribed spacer
(ITS).
[00530] Embodiment A23 is a method according to embodiment A22, wherein
measuring the
number of unique markers, and quantity thereof, includes subjecting genomic
DNA from each
sample to a high throughput sequencing reaction. Embodiment A24 is a method
according to
embodiment A22, wherein measuring the number of unique markers, and quantity
thereof,
comprises subjecting genomic DNA to genomic sequencing. Embodiment A25 is a
method
according to embodiment A22, wherein measuring the number of unique markers,
and quantity
thereof, comprises subjecting genomic DNA to amplicon sequencing.
142
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[005311 Embodiment A26 is a method according to any one of embodiments A 1 -
A25, wherein
the at least one different characteristic includes a collection time at which
each of the at least two
samples was collected, such that the collection time for a first sample is
different from the
collection time of a second sample.
[005321 Embodiment A27 is a method according to any one of embodiments A 1 -
A25, wherein
the at least one different characteristic includes a collection location at
which each of the at least
two samples was collected, such that the collection location for a first
sample is different from
the collection location of a second sample.
1005331 Embodiment A28 is a method according to any one of embodiments A1-A27,
wherein
the at least one common characteristic includes a sample source type, such
that the sample source
type for a first sample is the same as the sample source type of a second
sample. Embodiment
A29 is a method according to embodiment A28, wherein the sample source type is
one of animal
type, organ type, soil type, water type, sediment type, oil type, plant type,
agricultural product
type, bulk soil type, soil rhizosphere type, or plant part type.
1005341 Embodiment A30 is a method according to any one of embodiments A1-A27,
wherein
the at least one common characteristic includes that each of the at least two
samples is a
gastrointestinal sample.
1005351 Embodiment A31 is a method according to any one of embodiments A1-A27,
wherein
the at least one common characteristic includes an animal sample source type,
each sample
having a further common characteristic such that each sample is a tissue
sample, a blood sample,
a tooth sample, a perspiration sample, a fingernail sample, a skin sample, a
hair sample, a feces
sample, a urine sample, a semen sample, a mucus sample, a saliva sample, a
muscle sample, a
brain sample, or an organ sample.
[00536] Embodiment A32 is a method according to any one of embodiments AI-A31,
further
comprising: obtaining at least one further sample from a target, based on the
at least one
measured metadata, wherein the at least one further sample from the target
shares at least one
common characteristic with the at least two samples; and for the at least one
further sample from
the target, detecting the presence of one or more microorganism types,
determining a number of
each detected microorganism type of the one or more microorganism types,
measuring a number
of unique first markers and quantity thereof, integrating the number of each
microorganism type
143
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
and the number of the first markers to yield the absolute cell count of each
microorganism strain
present, measuring at least one unique second marker for each microorganism
strain to determine
an activity level for that microorganism strain, filtering the absolute cell
count by the determined
activity to provide a list of active microorganisms strains and their
respective absolute cell counts
for the at least one further sample from the target; wherein the selection of
the at least one
microorganism strain from each of the at least two groups is based on the list
of active
microorganisms strains and their respective absolute cell counts for the at
least one further
sample from the target such that the formed ensemble is configured to alter a
property of the
target that corresponds to the at least one metadata.
1005371 Embodiment A33 is a method according to any one of embodiments A1-A32,
wherein
comparing the filtered absolute cell counts of active microorganisms strains
for each of the at
least two samples with at least one measured metadata or additional active
microorganism strain
for each of the at least two samples includes determining the co-occurrence of
the one or more
active microorganism strains in each sample with the at least one measured
metadata or
additional active microorganism strain. Embodiment A34 is a method according
to embodiment
A33, wherein the at least one measured metadata includes one or more
parameters, wherein the
one or more parameters is at least one of sample pH, sample temperature,
abundance of a fat,
abundance of a protein, abundance of a carbohydrate, abundance of a mineral,
abundance of a
vitamin, abundance of a natural product, abundance of a specified compound,
bodyweight of the
sample source, feed intake of the sample source, weight gain of the sample
source, feed
efficiency of the sample source, presence or absence of one or more pathogens,
physical
characteristic(s) or measurement(s) of the sample source, production
characteristics of the
sample source, or a combination thereof. Embodiment A35 is a method according
to
embodiment A34, wherein the one or more parameters is at least one of
abundance of whey
protein, abundance of casein protein, and/or abundance of fats in milk.
[00538] Embodiment A36 is a method according to any one of embodiments A33-
A35, wherein
determining the co-occurrence of the one or more active microorganism strains
and the at least
one measured metadata in each sample includes creating matrices populated with
linkages
denoting metadata and microorganism strain associations, the absolute cell
count of the one or
more active microorganism strains and the measure of the one more unique
second markers to
represent one or more networks of a heterogeneous microbial community or
communities.
144
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
Embodiment A37 is a method according to embodiment A36, wherein the at least
one measured
metadata comprises a presence, activity and/or quantity of a second
microorganism strain.
[00539] Embodiment A38 is a method according to any one of embodiments A33-
A37, wherein
determining the co-occurrence of the one or more active microorganism strains
and the at least
one measured metadata and categorizing the active microorganism strains
includes network
analysis and/or cluster analysis to measure connectivity of each microorganism
strain within a
network, wherein the network represents a collection of the at least two
samples that share a
common characteristic, measured metadata, and/or related environmental
parameter.
Embodiment A39 is a method according to embodiment A38, wherein the at least
one measured
metadata comprises a presence, activity and/or quantity of a second
microorganism strain.
Embodiment A40 is a method according to embodiment A38 or A39, wherein the
network
analysis and/or cluster analysis includes linkage analysis, modularity
analysis, robustness
measures, betweenness measures, connectivity measures, transitivity measures,
centrality
measures, or a combination thereof. Embodiment A41 is a method according to
any one of
embodiments A38-A40, wherein the cluster analysis includes building a
connectivity model,
subspace model, distribution model, density model, or a centroid model.
[00540] Embodiment A42 is a method according to embodiment A38 or embodiment
A39,
wherein the network analysis includes predictive modeling of network through
link mining and
prediction, collective classification, link-based clustering, relational
similarity, or a combination
thereof. Embodiment A43 is a method according to embodiment A38 or embodiment
3A9,
wherein the network analysis comprises differential equation based modeling of
populations.
Embodiment A44 is a method according to embodiment A43, wherein the network
analysis
comprises Lotka-Volterra modeling. Embodiment A45 is a method according to
embodiment
A38 or embodiment A39, wherein the cluster analysis is a heuristic method.
Embodiment A46 is
a method according to embodiment A45, wherein the heuristic method is the
Louvain method.
[00541] Embodiment A47 is a method according to embodiment A38 or embodiment
A39,
where the network analysis includes nonparametric methods to establish
connectivity between
variables. Embodiment A48 is a method according to embodiment A38 or
embodiment A39,
wherein the network analysis includes mutual information and/or maximal
information
coefficient calculations between variables to establish connectivity.
145
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
11005421 Embodiment A49 is a method for forming an ensemble of active
microorganism strains
configured to alter a property or characteristic in an environment based on
two or more sample
sets that share at least one common or related environmental parameter between
the two or more
sample sets and that have at least one different environmental parameter
between the two or
more sample sets, each sample set comprising at least one sample including a
heterogeneous
microbial community, wherein the one or more microorganism strains is a
subtaxon of one or
more organism types, comprising: detecting the presence of a plurality of
microorganism types in
each sample; determining the absolute number of cells of each of the detected
microorganism
types in each sample; measuring the number of unique first markers in each
sample, and quantity
thereof, wherein a unique first marker is a marker of a microorganism strain;
at the protein or
RNA level, measuring the level of expression of one or more unique second
markers, wherein a
unique second marker is a marker of activity of a microorganism strain;
determining activity of
the detected microorganism strains for each sample based on the level of
expression of the one or
more unique second markers exceeding a specified threshold; calculating the
absolute cell count
of each detected active microorganism strain in each sample based upon the
quantity of the one
or more first markers and the absolute number of cells of the microorganism
types from which
the one or more microorganism strains is a subtaxon, wherein the one or more
active
microorganism strains expresses the second unique marker above the specified
threshold;
determining the co-occurrence of the active microorganism strains in the
samples with at least
one environmental parameter or additional active microorganism strain based on
maximal
information coefficient network analysis to measure connectivity of each
microorganism strain
within a network, wherein the network is the collection of the at least two or
more sample sets
with at least one common or related environmental parameter; selecting a
plurality of active
microorganism strains from the one or more active microorganism strains based
on the network
analysis; and forming an ensemble of active microorganism strains from the
selected plurality of
active microorganism strains, the ensemble of active microorganism strains
configured to
selectively alter a property or characteristic of an environment when the
ensemble of active
microorganism strains is introduced into that environment
100543] Embodiment A50 is a method according to embodiment A49, wherein the at
least one
environmental parameter comprises a presence, activity and/or quantity of a
second
microorganism stain. Embodiment A51 is a method according to embodiment A49 or
146
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
embodiment A50, wherein at least one measured indicia of at least one common
or related
environmental factor for a first sample set is different from a measured
indicia of the at least one
common or related environmental factor for a second sample set.
[00544] Embodiment A52 is a method according to embodiment A49 or embodiment
A50,
wherein each sample set comprises a plurality of samples, and a measured
indicia of at least one
common or related environmental factor for each sample within a sample set is
substantially
similar, and an average measured indicia for one sample set is different from
the average
measured indicia from another sample set Embodiment A53 is a method according
to
embodiment A49 or embodiment A50, wherein each sample set comprises a
plurality of samples,
and a first sample set is collected from a first population and a second
sample set is collected
from a second population. Embodiment A54 is a method according to embodiment
A49 or A50,
wherein each sample set comprises a plurality of samples, and a first sample
set is collected from
a first population at a first time and a second sample set is collected from
the first population at a
second time different from the first time. Embodiment A55 is a method
according to any one of
embodiments A49-A54, wherein at least one common or related environmental
factor includes
nutrient information.
[00545] Embodiment A56 is a method according to any one of embodiments A49-
A54, wherein
at least one common or related environmental factor includes dietary
information. Embodiment
A57 is a method of any one of embodiments A49-A54, wherein at least one common
or related
environmental factor includes animal characteristics. Embodiment A58 is a
method according to
any one of embodiments A49-A54, wherein at least one common or related
environmental factor
includes infection information or health status.
[00546] Embodiment A59 is a method according to embodiment A51, wherein at
least one
measured indicia is sample pH, sample temperature, abundance of a fat,
abundance of a protein,
abundance of a carbohydrate, abundance of a mineral, abundance of a vitamin,
abundance of a
natural product, abundance of a specified compound, bodyweight of the sample
source, feed
intake of the sample source, weight gain of the sample source, feed efficiency
of the sample
source, presence or absence of one or more pathogens, physical
characteristic(s) or
measurement(s) of the sample source, production characteristics of the sample
source, or a
combination thereof.
147
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00547] Embodiment A60 is a method according to embodiment A49 or embodiment
A50,
wherein the at least one parameter is at least one of abundance of whey
protein, abundance of
casein protein, and/or abundance of fats in milk. Embodiment A61 is a method
according to any
one of embodiments A49-A60, wherein measuring the number of unique first
markers in each
sample comprises measuring the number of unique genomic DNA markers.
Embodiment A62 is
a method according to any one of embodiments A49-A60, wherein measuring the
number of
unique first markers in the sample comprises measuring the number of unique
RNA markers.
Embodiment A63 is a method according to any one of embodiments A49-A60,
wherein
measuring the number of unique first markers in the sample comprises measuring
the number of
unique protein markers.
[00548] Embodiment A64 is a method according to any one of embodiments A49-
A63, wherein
the plurality of microorganism types includes one or more bacteria, archaea,
fungi, protozoa,
plant, other eukaryote, virus, viroid, or a combination thereof. Embodiment
A65 is a method
according to any one of embodiments A49-A64, wherein determining the absolute
cell number
of each of the microorganism types in each sample includes subjecting the
sample or a portion
thereof to sequencing, centrifugation, optical microscopy, fluorescent
microscopy, staining, mass
spectrometry, microfluidics, quantitative polymerase chain reaction (qPCR),
gel electrophoresis
and/or flow cytometry. Embodiment A66 is a method according to any one of
embodiments
A49-A65, wherein one or more active microorganism strains is a subtaxon of one
or more
microbe types selected from one or more bacteria, archaea, fungi, protozoa,
plant, other
eukaryote, virus, viroid, or a combination thereof.
[00549] Embodiment A67 is a method according to any one of embodiments A49-
A65, wherein
one or more active microorganism strains is one or more bacterial strains,
archaeal strains, fungal
strains, protozoa strains, plant strains, other eukaryote strains, viral
strains, viroid strains, or a
combination thereof. Embodiment A68 is a method according to any one of
embodiments A49-
A67, wherein one or more active microorganism strains is one or more fungal
species, fungal
subspecies, bacterial species and/or bacterial subspecies. Embodiment A69 is a
method
according to any one of embodiments A49-A68, wherein at least one unique first
marker
comprises a phylogenetic marker comprising a 5S ribosomal subunit gene, a 16S
ribosomal
subunit gene, a 23S ribosomal subunit gene, a 5.8S ribosomal subunit gene, a
18S ribosomal
subunit gene, a 28S ribosomal subunit gene, a cytochrome c oxidase subunit
gene, a beta-tubulin
148
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
gene, an elongation factor gene, an RNA polymerase subunit gene, an internal
transcribed spacer
(ITS), or a combination thereof.
[00550] Embodiment A70 is a method according to embodiment A49 or embodiment
A50,
wherein measuring the number of unique first markers, and quantity thereof,
comprises
subjecting genomic DNA from each sample to a high throughput sequencing
reaction.
Embodiment A71 is a method according to embodiment A49 or A50, wherein
measuring the
number of unique first markers, and quantity thereof, comprises subjecting
genomic DNA from
each sample to metagenome sequencing. Embodiment A72 is a method according to
embodiment A49 or A50, wherein a unique first marker comprises an inRNA
marker, an siRNA
marker, or a ribosomal RNA marker. Embodiment A73 is a method according to
embodiment
A49 or embodiment A50, wherein a unique first marker comprises a sigma factor,
a transcription
factor, nucleoside associated protein, metabolic enzyme, or a combination
thereof.
[00551] Embodiment A74 is a method according to any one of embodiments A49-
A73, wherein
measuring the level of expression of one or more unique second markers
comprises subjecting
mRNA in the sample to gene expression analysis. Embodiment A75 is a method
according to
embodiment A74, wherein the gene expression analysis comprises a sequencing
reaction.
Embodiment A76 is a method according to embodiment A74, wherein the gene
expression
analysis comprises a quantitative polymerase chain reaction (qPCR),
metatranscriptome
sequencing, and/or transcriptome sequencing.
[00552] Embodiment A77 is a method according to any one of embodiments A49-A68
and
embodiments A74-A76, wherein measuring the level of expression of one or more
unique second
markers includes subjecting each sample or a portion thereof to mass
spectrometry analysis.
Embodiment A78 is a method according to any one of embodiments A49-A68 and
embodiments
A74-A76, wherein measuring the level of expression of one or more unique
second markers
comprises subjecting the sample or a portion thereof to metaribosome
profiling, and/or ribosome
profiling.
[00553] Embodiment A79 is a method according to any one of embodiments A49-
A78, wherein
the source type for the samples is one of animal, soil, air, saltwater,
freshwater, wastewater
sludge, sediment, oil, plant, an agricultural product, bulk soil, soil
rhizosphere, plant part,
vegetable, an extreme environment, or a combination thereof.
149
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1005541 Embodiment A80 is a method according to any one of embodiments A49-
A78, wherein
each sample is a gastrointestinal sample. Embodiment A81 is a method according
to any one of
embodiments A49-A78, wherein each sample is one of a tissue sample, blood
sample, tooth
sample, perspiration sample, fingernail sample, skin sample, hair sample,
feces sample, urine
sample, semen sample, mucus sample, saliva sample, muscle sample, brain
sample, or organ
sample.
1005551 Embodiment A82 is a processor-implemented method, comprising:
receiving sample
data from at least two samples sharing at least one common characteristic and
having a least one
different characteristic; for each sample, determining the presence of one or
more microorganism
types in each sample; determining a number of each detected microorganism type
of the one or
more microorganism types in each sample; determining a number of unique first
markers in each
sample, and quantity thereof, each unique first marker being a marker of a
microorganism strain;
integrating, via a processor, the number of each microorganism type and the
number of the first
markers to yield the absolute cell count of each microorganism strain present
in each sample;
determining an activity level for each microorganism strain in each sample
based on a measure
of at least one unique second marker for each microorganism strain exceeding a
specified
threshold, a microorganism strain being identified as active if the measure of
at least one unique
second marker for that strain exceeds the corresponding threshold; filtering
the absolute cell
count of each microorganism strain by the determined activity to provide a
list of active
microorganisms strains and their respective absolute cell counts for each of
the at least two
samples; conducting a network analysis, via at least one processor, of the
filtered absolute cell
counts of active microorganisms strains for each of the at least two samples
with at least one
measured metadata or additional active microorganism strain for each of the at
least two
samples, the network analysis including determining maximal information
coefficient scores
between each active microorganism strain and every other active microorganism
strain and
determining maximal information coefficient scores between each active
microorganism strain
and the respective at least one measured metadata or additional active
microorganism strain;
categorizing the active microorganism strains based on predicted function
and/or chemistry;
identifying a plurality of active microorganism strains based on the
categorization; and
outputting the identified plurality of active microorganism strains.
150
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00556] Embodiment A83 is the processor-implemented method of embodiment A82,
further
comprising: assembling an active microorganism ensemble configured to, when
applied to a
target, alter a property corresponding to the at least one measured metadata.
Embodiment A84 is
the processor-implemented method of embodiment A82, wherein the output
plurality of active
microorganism strains is used to assemble an active microorganism ensemble
configured to,
when applied to a target, alter a property corresponding to the at least one
measured metadata.
Embodiment A85 is the processor-implemented method of embodiment A82, further
comprising:
identifying at least one pathogen based on the output plurality of identified
active microorganism
strains. Embodiment A86 is a processor-implemented method of any one of
embodiments A82-
A85, wherein the output plurality of active microorganism strains is further
used to assemble an
active microorganism ensemble configured to, when applied to a target, target
the at least one
identified pathogen and treat and/or prevent a symptom associated with the at
least one identified
pathogen.
[00557] Embodiment A87 is a method of forming an active microorganism
bioensemble of
active microorganism strains configured to alter a property in a target
biological environment,
comprising: obtaining at least two samples sharing at least one common
characteristic and
having at least one different characteristic; for each sample, detecting the
presence of one or
more microorganism types in each sample; determining a number of each detected
microorganism type of the one or more microorganism types in each sample;
measuring a
number of unique first markers in each sample, and quantity thereof, each
unique first marker
being a marker of a microorganism strain; integrating the number of each
microorganism type
and the number of the first markers to yield the absolute cell count of each
microorganism strain
present in each sample; measuring at least one unique second marker for each
microorganism
strain based on a specified threshold to determine an activity level for that
microorganism strain
in each sample; filtering the absolute cell count by the determined activity
to provide a list of
active microorganisms strains and their respective absolute cell counts for
each of the at least
two samples; comparing the filtered absolute cell counts of active
microorganisms strains for
each of the at least two samples with at least one measured metadata for each
of the at least two
samples, the comparison including determining the co-occurrence of the active
microorganism
strains in each sample with the at least one measured metadata, determining
the co-occurrence of
the active microorganism strains and the at least one measured metadata in
each sample
151
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
including creating matrices populated with linkages denoting metadata and
microorganism strain
relationships, the absolute cell count of the active microorganism strains,
and the measure of the
unique second markers, to represent one or more heterogeneous microbial
community networks;
grouping the active microorganism strains into at least two groups according
to predicted
function and/or chemistry based on at least one of nonparametric network
analysis and cluster
analysis identifying connectivity of each active microorganism strain and
measured metadata
within an active heterogeneous microbial community network; selecting at least
one
microorganism strain from each of the at least two groups; and combining the
selected
microorganism strains and with a carrier medium to form a bioensemble of
active
microorganisms configured to alter a property corresponding to the at least
one metadata of
target biological environment when the bioensemble is introduced into that
target biological
environment
[005581 Embodiment A88 is the method according to embodiment A87, further
comprising:
obtaining at least one further sample, based on the at least one measured
metadata, wherein the at
least one further sample shares at least one characteristic with the at least
two samples; and for
the at least one further sample, detecting the presence of one or more
microorganism types,
determining a number of each detected microorganism type of the one or more
microorganism
types, measuring a number of unique first markers and quantity thereof,
integrating the number
of each microorganism type and the number of the first markers to yield the
absolute cell count
of each microorganism strain present, measuring at least one unique second
marker for each
microorganism strain to determine an activity level for that microorganism
strain, filtering the
absolute cell count by the determined activity to provide a list of active
microorganisms strains
and their respective absolute cell counts for the at least one further sample;
wherein comparing
the filtered absolute cell counts of active microorganisms strains comprises
comparing the
filtered absolute cell counts of active microorganism strains for each of the
at least two samples
and the at least one further sample with the at least one measured metadata,
such that the
selection of the active microorganism strains is at least partially based on
the list of active
microorganisms strains and their respective absolute cell counts for the at
least one further
sample.
1005591 Embodiment A89 is a method for forming a synthetic ensemble of active
microorganism strains configured to alter a property in a biological
environment, based on two
152
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
or more sample sets each having a plurality of environmental parameters, at
least one parameter
of the plurality of environmental parameters being a common environmental
parameter that is
similar between the two or more sample sets and at least one environmental
parameter being a
different environmental parameter that is different between each of the two or
more sample sets,
each sample set including at least one sample comprising a heterogeneous
microbial community
obtained from a biological sample source, at least one of the active
microorganism strains being
a subtaxon of one or more organism types, the method comprising: detecting the
presence of a
plurality of microorganism types in each sample; determining the absolute
number of cells of
each of the detected microorganism types in each sample; measuring the number
of unique first
markers in each sample, and quantity thereof, a unique first marker being a
marker of a
microorganism strain; measuring the level of expression of one or more unique
RNA markers,
wherein a unique RNA marker is a marker of activity of a microorganism strain;
determining
activity of each of the detected microorganism strains for each sample based
on the level of
expression of the one or more unique RNA markers exceeding a specified
threshold; calculating
the absolute cell count of each detected active microorganism strain in each
sample based upon
the quantity of the one or more first markers and the absolute number of cells
of the
microorganism types from which the one or more microorganism strains is a
subtaxon, the one or
more active microorganism strains expressing one or more unique RNA markers
above the
specified threshold; analyzing the active microorganism strains of the two or
more sample sets,
the analyzing including conducting nonparametric network analysis of each of
the active
microorganism strains for each of the two or more sample sets, the at least
one common
environmental parameter, and the at least one different environmental
parameter, the
nonparametric network analysis including (1) determining the maximal
information coefficient
score between each active microorganism strain and every other active
microorganism strain and
(2) determining the maximal information coefficient score between each active
microorganism
strain and the at least one different environmental parameter; selecting a
plurality of active
microorganism strains from the one or more active microorganism strains based
on the
nonparametric network analysis; and forming a synthetic ensemble of active
microorganism
strains comprising the selected plurality of active microorganism strains and
a microbial carrier
medium, the ensemble of active microorganism strains configured to selectively
alter a property
153
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
of a biological environment when the synthetic ensemble of active
microorganism strains is
introduced into that biological environment
[00560] Embodiment A90 is a method of forming an active microorganism
bioensemble
configured to alter a property in a target biological environment, comprising:
obtaining at least
two samples sharing at least one common environmental parameter and having at
least one
different environmental parameter; for each sample, detecting the presence of
one or more
microorganism types in each sample; determining a number of each detected
microorganism type
of the one or more microorganism types in each sample; measuring a number of
unique first
markers in each sample, and quantity thereof, each unique first marker being a
marker of a
microorganism strain of a detected microorganism type; determining the
absolute cell count of
each microorganism strain present in each sample based on the number of each
detected
microorganism type and the proportional/relative number of the corresponding
or related unique
first markers for that microorganism type; measuring at least one unique
second marker for each
microorganism strain based on a specified threshold to determine an activity
level for that
microorganism strain in each sample; filtering the absolute cell count of each
microorganism
strain by the determined activity to provide a list of active microorganisms
strains and their
respective absolute cell counts for each of the at least two samples;
comparing the filtered
absolute cell counts of active microorganisms strains for each of the at least
two samples with at
least one measured metadata for each of the at least two samples, the
comparison including
determining the co-occurrence of the active microorganism strains in each
sample with the at
least one measured metadata, determining the co-occurrence of the active
microorganism strains
and the at least one measured metadata in each sample including creating
matrices populated
with linkages denoting metadata and microorganism strain relationships, the
absolute cell count
of the active microorganism strains, and the measure of the unique second
markers, to represent
one or more heterogeneous microbial community networks; grouping the active
microorganism
strains into at least two groups according to predicted function and/or
chemistry based on at least
one of nonparametric network analysis and cluster analysis identifying
connectivity of each
active microorganism strain and measured metadata within an active
heterogeneous microbial
community network; selecting at least one microorganism strain from each of
the at least two
groups; and combining the selected microorganism strains and with a carrier
medium to form a
synthetic bioensemble of active microorganisms configured to alter a property
corresponding to
154
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
the at least one metadata of target biological environment when the
bioensemble is introduced
into that target biological environment.
1005611 While the disclosure has been communicated with reference to the
specific
embodiments thereof it should be understood by those skilled in the art that
various changes may
be made and equivalents may be substituted without departing from the true
spirit and scope of
the disclosure. In addition, many modifications may be made to adopt a
particular situation,
material, composition of matter, process, process step or steps, to the
objective spirit and scope
of the described embodiments and disclosure. All such modifications are
intended to be within
the scope of the disclosure. Patents, patent applications, patent application
publications, journal
articles and protocols referenced herein are incorporated by reference in
their entireties, for all
purposes, including the following PCT application publications:
WO/2016/210251,
WO/2017/120495, and WO/2017/181203.
1005621 While various embodiments have been described and illustrated herein,
those of skill in
the art will readily envision a variety of other ways and/or structures for
performing the function
and/or obtaining the results and/or one or more of the advantages described
herein, and each of
such variations and/or modifications is deemed to be within the scope of the
disclosure. More
generally, those skilled in the art will readily appreciate that parameters,
dimensions, materials,
and configurations described herein are provided as illustrative examples, and
that the actual
parameters, dimensions, materials, and/or configurations will depend upon the
specific
application(s) or implementation(s) for which the disclosed teachings is/are
used. Those skilled
in the art will recognize, or be able to ascertain using no more than routine
experimentation,
equivalents to the specific embodiments described herein. It is, therefore, to
be understood that
the foregoing embodiments are presented by way of example only and that,
within the scope of
the appended claims and equivalents thereto; embodiments can be practiced
otherwise than as
specifically described and claimed. Embodiments of the present disclosure are
directed to each
individual feature, system, article, material, kit, and/or method described
herein. In addition, any
combination of two or more such features, systems, articles, materials, kits,
and/or methods, if
such features, systems, articles, materials, kits, and/or methods are not
mutually inconsistent, is
included within the scope of the present disclosure.
155
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00563] The above-described embodiments can be implemented in any of numerous
ways. For
example, the embodiments can be implemented using hardware, software, or a
combination
thereof. When implemented in software, the software code can be executed on
any suitable
processor or collection of processors, whether provided in a single computer
or distributed
among multiple computers.
[00564] Further, it should be appreciated that the disclosed methods can be
used in conjunction
with a computer, which can be embodied in any of a number of forms, such as a
rack-mounted
computer, a desktop computer, a laptop computer, or a tablet computer.
Additionally, a computer
can be embedded in a device not generally regarded as a computer but with
suitable processing
capabilities, including a tablet, Personal Digital Assistant (PDA), a smart
phone or any other
suitable portable or fixed electronic device.
1005651 Also, a computer can have one or more input and output devices,
including one or more
displays. These devices can be used, among other things, to present a user
interface. Examples of
output devices that can be used to provide a user interface include printers
or display screens for
visual presentation of output and speakers or other sound generating devices
for audible
presentation of output. Examples of input devices that can be used for a user
interface include
keyboards, and pointing devices, such as mice, touch pads, and digitizing
tablets. As another
example, a computer can receive input information through speech recognition
or in other
audible format.
[00566] Such computers can be interconnected by one or more networks in any
suitable form,
including a local area network or a wide area network, such as an enterprise
network, and
intelligent network (IN) or the Internet. Such networks can be based on any
suitable technology
and can operate according to any suitable protocol and can include wireless
networks, wired
networks or fiber optic networks.
[00567] Various methods and processes outlined herein (and/or portions
thereof) can be coded
as software that is executable on one or more processors that employ any one
of a variety of
operating systems or platforms. Additionally, such software can be written
using any of a
number of suitable programming languages and/or programming or scripting
tools, and also can
be compiled as executable machine language code or intermediate code that is
executed on a
framework or virtual machine.
156
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00568] In this respect, various disclosed concepts can be embodied as a
computer readable
storage medium (or multiple computer readable storage media) (e.g., a computer
memory, one or
more floppy discs, compact discs, optical discs, magnetic tapes, flash
memories, circuit
configurations in Field Programmable Gate Arrays or other semiconductor
devices, or other non-
transitory medium or tangible computer storage medium) encoded with one or
more programs
that, when executed on one or more computers or other processors, perform
methods that
implement the various embodiments of the disclosure discussed above. The
computer readable
medium or media can be transportable, such that the program or programs stored
thereon can be
loaded onto one or more different computers or other processors to implement
various aspects of
the present disclosure as discussed above.
[00569] The terms "program" or "software" are used herein in a generic sense
to refer to any
type of computer code or set of computer-executable instructions that can be
employed to
program a computer or other processor to implement various aspects of
embodiments as
discussed above. Additionally, it should be appreciated that according to one
aspect, one or more
computer programs that when executed perform methods of the present disclosure
need not
reside on a single computer or processor, but can be distributed in a modular
fashion amongst a
number of different computers or processors to implement various aspects of
the present
disclosure.
[00570] Computer-executable instructions can be in many forms, such as program
modules,
executed by one or more computers or other devices. Generally, program modules
include
routines, programs, objects, components, data structures, etc. that perform
particular tasks or
implement particular abstract data types. Typically the functionality of the
program modules can
be combined or distributed as desired in various embodiments.
[00571] Also, data structures can be stored in computer-readable media in any
suitable form.
For simplicity of illustration, data structures can be shown to have fields
that are related through
location in the data structure. Such relationships can likewise be achieved by
assigning storage
for the fields with locations in a computer-readable medium that convey
relationship between the
fields. However, any suitable mechanism can be used to establish a
relationship between
information in fields of a data structure, including through the use of
pointers, tags or other
mechanisms that establish relationship between data elements.
157
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
[00572] Also, various disclosed concepts can be embodied as one or more
methods, of which
examples have been provided. The acts performed as part of the method can be
ordered in any
suitable way. Accordingly, embodiments can be constructed in which acts are
performed in an
order different than illustrated, which can include performing some acts
simultaneously, even
though shown as sequential acts in illustrative embodiments.
[00573] All definitions, as defined and used herein, should be understood to
control over
dictionary definitions, definitions in documents incorporated by reference,
and/or ordinary
meanings of the defined terms.
[00574] Flow diagrams are used herein. The use of flow diagrams is not meant
to be limiting
with respect to the order of operations performed. The herein described
subject matter sometimes
illustrates different components contained within, or connected with,
different other components.
It is to be understood that such depicted architectures are merely exemplary,
and that in fact
many other architectures can be implemented which achieve the same
functionality. In a
conceptual sense, any arrangement of components to achieve the same
functionality is
effectively "associated" such that the desired functionality is achieved.
Hence, any two
components herein combined to achieve a particular functionality can be seen
as "associated
with" each other such that the desired functionality is achieved, irrespective
of architectures or
intermedia components. Likewise, any two components so associated can also be
viewed as
being "operably connected," or "operably coupled," to each other to achieve
the desired
functionality, and any two components capable of being so associated can also
be viewed as
being "operably couplable," to each other to achieve the desired
functionality. Specific examples
of operably couplable include but are not limited to physically mateable
and/or physically
interacting components and/or wirelessly interactable and/or wirelessly
interacting components
and/or logically interacting and/or logically interactable components.
[00575] The indefinite articles "a" and "an," as used herein in the
specification and in the
claims, unless clearly indicated to the contrary, should be understood to mean
"at least one."
[00576] The phrase "and/or," as used herein in the specification and in the
claims, should be
understood to mean "either or both" of the elements so conjoined, i.e.,
elements that are
conjunctively present in some cases and disjunctively present in other cases.
Multiple elements
listed with "and/or" should be construed in the same fashion, i.e., "one or
more" of the elements
158
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
so conjoined. Other elements can optionally be present other than the elements
specifically
identified by the "and/or" clause, whether related or unrelated to those
elements specifically
identified. Thus, as a non-limiting example, a reference to "A and/or B", when
used in
conjunction with open-ended language such as "comprising" can refer, in one
embodiment, to A
only (optionally including elements other than B); in another embodiment, to B
only (optionally
including elements other than A); in yet another embodiment, to both A and B
(optionally
including other elements); etc.
[00577] As used herein in the specification and in the claims, "or" should be
understood to have
the same meaning as "and/or" as defined above. For example, when separating
items in a list,
"or" or "and/or" shall be interpreted as being inclusive, i.e., the inclusion
of at least one, but also
including more than one, of a number or list of elements, and, optionally,
additional unlisted
items. Only terms clearly indicated to the contrary, such as "only one of' or
"exactly one of," or,
when used in the claims, "consisting of," will refer to the inclusion of
exactly one element of a
number or list of elements. In general, the term "or" as used herein shall
only be interpreted as
indicating exclusive alternatives (i.e. "one or the other but not both") when
preceded by terms of
exclusivity, such as "either," "one of," "only one of," or "exactly one of"
"Consisting essentially
of," when used in the claims, shall have its ordinary meaning as used in the
field of patent law.
[00578] As used herein in the specification and in the claims, the phrase "at
least one," in
reference to a list of one or more elements, should be understood to mean at
least one element
selected from any one or more of the elements in the list of elements, but not
necessarily
including at least one of each and every element specifically listed within
the list of elements and
not excluding any combinations of elements in the list of elements. This
definition also allows
that elements can optionally be present other than the elements specifically
identified within the
list of elements to which the phrase "at least one" refers, whether related or
unrelated to those
elements specifically identified. Thus, as a non-limiting example, "at least
one of A and B" (or,
equivalently, "at least one of A or B," or, equivalently "at least one of A
and/or B") can refer, in
one embodiment, to at least one, optionally including more than one, A, with
no B present (and
optionally including elements other than B); in another embodiment, to at
least one, optionally
including more than one, B, with no A present (and optionally including
elements other than A);
in yet another embodiment, to at least one, optionally including more than
one, A, and at least
one, optionally including more than one, B (and optionally including other
elements); etc.
159
CA 03048247 2019-06-21
WO 2018/126033 PCT/US2017/068753
1.005791 In the claims, as well as in the specification above, all
transitional phrases such as
"comprising," "including," "carrying," "having," "containing," "involving,"
"holding,"
"composed of," and the like are to be understood to be open-ended, i.e., to
mean including but
not limited to. Only the transitional phrases "consisting of' and "consisting
essentially of' shall
be closed or semi-closed transitional phrases, respectively, as set forth in
the United States Patent
Office Manual of Patent Examining Procedures, Section 2111.03.
160