Note: Descriptions are shown in the official language in which they were submitted.
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
PYRIMIDINE DERIVATIVES AS TROPOMYOSIN RECEPTOR KINASE A (TRKA) INHIBITORS
The present invention relates to novel compounds of Formula (1) that are TrkA
inhibitors and are useful in the treatment or prevention of acute and chronic
pain but
also for other abnormal activities of TrkA beyond pain therapy, such as
inflammation
and cancer.
Background of the Invention
Based on the physical causes, pain can be divided into three types:
nociceptive,
neuropathic, and mix-type.
Nociceptive pain is the term for pain that is detected by nociceptors.
Nociceptors are
free nerve endings that terminate just below the skin, in tendons, in joints,
and in
internal organs. Nociceptive pain typically responds well to treatment with
opioids
and NSA1Ds. There are several types of nociceptive pain: somatic pain,
visceral pain,
and cutaneous pain. Visceral pain comes from the internal organs. Deep somatic
pain is initiated by stimulation of nociceptors in ligaments, tendons, bones,
blood
vessels, fascies and muscles, and is dull, aching, poorly localized pain.
Examples
include sprains and broken bones. Superficial pain is initiated by activation
of
nociceptors in the skin or other superficial tissue, and is sharp, well-
defined and
clearly located. Examples of injuries that produce superficial somatic pain
include
minor wounds and minor (first degree) burns. Nociceptive pain is usually short
in
duration and ends when the damage recovers. Examples of nociceptive pain
include
postoperative pain, sprains, bone fractures, burns, bumps, bruises, and
inflammatory
nociceptive pain. Inflammatory nociceptive pain is associated with tissue
damage
and the resulting inflammatory process.
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Neuropathic pain is produced by damage to the neurons in the peripheral and
central
nervous systems and involves sensitization of these systems. Because the
underlying etiologies are usually irreversible, most of neuropathic pain are
chronic
pain. Most people describe neuropathic pain as shooting, burning, tingling,
lancinating, electric shock qualities, numbness, and persistent allodynia. The
nomenclature of neuropathic pain is based on the site of initiating nervous
system
with the etiology; for examples, central post-stroke pain, diabetes peripheral
neuropathy, post-herpetic (or post-shingles) neuralgia, terminal cancer pain,
phantom
limb pain.
Mix-type pain is featured by the coexistence of both nociceptive and
neuropathic
pain. For example, muscle pain trigger central or peripheral neuron
sensitization
leading to chronic low back pain, migraine, and myofascial pain.
Receptor tyrosine kinases (RTKs) are a sub-family of protein kinases that play
a
critical role in cell signaling and also are involved in a variety of
processes related to
nerve activity. These include pain transmission in the spinal cord as well as
in the
peripheral nerve endings where the pain signal starts.
The tyrosine kinase receptor (Trk) family members are high affinity receptors
of the
RTK class and are activated by a group of soluble growth factors called
neurotrophins (NT). The Trk receptor family has three members ¨TrkA, TrkB and
TrkC. Among the neurotrophins are (i) nerve growth factor (NGF) which
activates
TrkA, (ii) brain-derived neurotrophic factor (BNDF) and NT-4/5 which activates
TrkB,
and (iii) NT3 which activates TrkC. Trks are widely expressed in neuronal
tissue and
are implicated in the maintenance, signaling and survival of neuronal cells
(Patapoutian, A. et al, Current Opinion In Neurobiology, 2001, 11, 272-280).
Recent
literature also indicates that activation of TrkA with NGF causes downstream
upregulation of certain ion channels which are important in increasing the
electric
signaling from the nerve endings which experience the inflammation, thus
inducing
pain (e.g. VR-1, Winston et al. Pain 2001,89,181; sodium channels, Choi et al.
Molecular and Cellular Biology 2001, 21, 2695; ASIC, Mamet et al. Journal of
Biological Chemistry 2003, 278, 48907). Binding of Nerve Growth Factor (NGF)
to
TrkA triggers TrkA-dimerization and activation of downstream signaling
pathways
linked to neuronal development and nociception (Khan, N. et al., Molecules 20,
10657-88 (2015)).
2
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Each Trk receptor contains an extra-cellular domain (ligand binding), trans-
membrane region and intra-cellular domain (including kinase domain). Upon
binding
of the ligand, the kinase domain catalyzes the auto-phosphorylation and
triggers
downstream signal transduction pathways.
Over the last 10 years many scientific reports have been published which link
Trk
signaling with induction of pain. Inhibitors of the Trk/neurotrophin pathway
have been
demonstrated to be effective in numerous pre-clinical animal models of pain.
For
example, antagonistic NGF and TrkA antibodies (e.g., RN-624) have been shown
to
be efficacious in inflammatory and neuropathic pain animal models and in human
clinical trials (Woolf, C.J. et al. (1994) Neuroscience 62, 327-331; Zahn,
P.K. et al.
(2004) J. Pain 5, 157-163; McMahon, S.B. et al., (1995) Nat. Med. 1, 774-780;
Ma,
Q.P. and Woolf, C.J. (1997) Neuroreport 8, 807-810; Shelton, D.L. et al.
(2005) Pain
116, 8-16; Delafoy, L. et al. (2003) Pain 105, 489-497; Lamb, K. et al. (2003)
Neurogastroenterol. Moth. 15, 355-361; Jaggar, S.I. et al. (1999) Br. J.
Anaesth. 83,
442-448). Additionally, literature indicates that after inflammation, BDNF
levels and
TrkB signaling is increased in the dorsal root ganglion (Cho, L. et al. Brain
Research
1997, 749, 358) and several studies have shown antibodies that decrease
signaling
through the BDNF/TrkB pathway inhibit neuronal hypersensitization and the
associated pain (Chang-Qi, L et al. Molecular Pain 2008, 4:27). A very recent
article
reports that TrkA is a validated drug target for cancer and pain (Norman, B.
H. et al.
J. Med. Chem. 60, 66-88 (2017)). In addition, pain suppression effect of TrkA
inhibitors has been shown in in-vitro models (Nwosu, L. N., et al., Ann.
Rheum. Dis.
annrheumdis-2014-207203 (2015).
doi:10.1136/annrheumdis-2014-207203;
Andrews, S. W., Array biopharma Available
at:
http://www.an-avbiopharma.com/files/6313/9810/8021/PubAttachment587.pdf). A
recent publication shows that loss-of-function TrkA variants are associated
with
congenital insensitivity to pain (Bakri, F. G. et al. Olin. Case Reports 4,
997-1000
(2016)).
There are few reports of selective Trk tyrosine kinase inhibitors that are
highly
.. selective for TrkA and TrkB. Cephalon describes CEP-751, CEP-701 (George,
D. et
al Cancer Research, 1999, 59, 2395-2401) and other indolocarbazole analogs
(W00114380) as Trk inhibitors. It was shown that the alkaloid K252a, which is
3
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
related to CEP-701/751, when injected into rats with pancreatitis could
suppress
mechanical hypersensitivity (Winston et al. Journal of Pain 2003, 4, 329).
In addition, it has been shown that tumor cell invading macrophages directly
stimulate TrkA located on peripheral pain fibers. Using various tumor models
in both
mice and rats it was demonstrated that neutralizing NGF with a monoclonal
antibody
inhibits cancer related pain to a degree similar or superior to the highest
tolerated
dose of morphine. In addition, activation of the BDNF/TrkB pathway has been
implicated in numerous studies as a modulator of various types of pain
including
inflammatory pain (Matayoshi, S., J. Physiol. 2005, 569:685-95), neuropathic
pain
(Thomson, S.W., Proc. Natl. Acad. Sci. USA 1999, 96:7714-18) and surgical pain
(Li,
C.-Q. et al., Molecular Pain, 2008, 4(28), 1-11). Because TrkA and TrkB
kinases may
serve as a mediator of NGF driven biological responses, inhibitors of TrkA
and/or
other Trk kinases may provide an effective treatment for chronic pain states.
Recent literature has also shown that overexpression, activation,
amplification and/or
mutation of Trks are associated with many cancers including neuroblastoma
(Brodeur, G.M., Nat. Rev. Cancer 2003, 3, 203-2016), ovarian cancer (Davidson.
B.,
et al., Clin. Cancer Res. 20039, 2248-2259), breast cancer (Kruettgen et al,
Brain
Pathology 2006, 16: 304-310), prostate cancer (Dionne et al, Clin. Cancer Res.
1998,4(8): 1887-1898), pancreatic cancer (Dang et al, Journal of
Gastroenterology
and Hepatology 2006, 21(5): 850-858), multiple myeloma (Hu et al, Cancer
Genetics
and Cytogenetics 2007, 178: 1-10), astrocytoma and medulloblastoma (Kruettgen
et
al, Brain Pathology 2006, 16: 304-310), glioma (Hansen et al, Journal of
Neurochemistry 2007, 28(3)221-229), lung adenocarcinoma (Perez-Pinera et al,
Molecular and Cellular Biochemistry 2007, 295(1&2), 19-26), large cell
neuroendocrine tumors (Marchetti et al, Human Mutation 2008, 29(5), 609-616),
and
colorectal cancer (Bardelli, A., Science 2003, 300, 949). In preclinical
models of
cancer, Trk inhibitors are efficacious in both inhibiting tumor growth and
stopping
tumor metastasis. In particular, non-selective small molecule inhibitors of
Trk A, B
and C and Trk/Fc chimeras were efficacious in both inhibiting tumor growth and
stopping tumor metastasis (Nakagawara, A. (2001) Cancer Letters 169:107-114;
Meyer, J. et al. (2007) Leukemia, 1-10; Pierottia, M.A. and Greco A., (2006)
Cancer
Letters 232:90-98; Eric Adriaenssens, E. et at. Cancer Res. (2008) 68(2) 346-
351)
(Truzzi et at, Journal of Investigative Dermatology 2008, 128(8): 2031-2040.
4
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Therefore, an inhibitor of the Trk family of kinases is expected to have
utility in the
treatment of cancer.
In addition, inhibition of the neurotrophinifrk pathway has been shown to be
effective
in treatment of pre-clinical models of inflammatory diseases. For example,
inhibition
of the neurotrophiniTrk pathway has been implicated in pre-clinical models of
inflammatory lung diseases including asthma (Freund-Michel, V; Frossard, N.;
Pharmacology & Therapeutics (2008), 117(1), 52-76), interstitial cystitis (Hu
Vivian Y;
et. al. The Journal of Urology (2005), 173(3), 1016-21), inflammatory bowel
diseases
including ulcerative colitis and Crohn's disease (Di Mola, F.F, et. al., Gut
(2000),
46(5), 670-678) and inflammatory skin diseases such as atopic dermatitis (Dou,
Y.-
C.; et. al. Archives of Dermatological Research (2006), 298(1), 31-37), eczema
and
psoriasis (Raychaudhuri, S.P., et. al. Journal of Investigative Dermatology
(2004),
122(3), 812-819).
Inflammation is a process by which microbes or tissue injury induce the
release of
cytokines and chemokines from various cell types producing increased blood
vessel
permeability, upregulation of endothelial receptors, and thus increased egress
of
various cells of the innate and adaptive immune system which enter surrounding
tissue and grossly produce the classical picture of inflammation, i.e.
redness,
swelling, heat and pain.
Inflammation is a localized reaction of live tissue due to an injury, which
may be
caused by various endogenous and exogenous factors. The exogenous factors
include physical, chemical, and biological factors. The endogenous factors
include
inflammatory mediators, antigens, and antibodies. Endogenous factors often
develop
under the influence of an exogenous damage. An inflammatory reaction is often
followed by an altered structure and penetrability of the cellular membrane.
Endogenous factors, such as mediators and antigens define the nature and type
of
an inflammatory reaction, especially its course in the zone of injury. In the
case
where tissue damage is limited to the creation of mediators, an acute form of
inflammation develops. If immunologic reactions are also involved in the
process,
through the interaction of antigens, antibodies, and auto-antigens, a long-
term
inflammatory process will develop. Various exogenous agents, for example,
infection,
injury, and radiation, also provide the course of inflammatory process on a
molecular
level by damaging cellular membranes which initiate biochemical reactions.
5
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
The current treatment regimens for pain conditions utilize compounds which
exploit a
very limited range of pharmacological mechanisms. One class of compounds, the
opioids, stimulates the endogenous endorphin system. Morphine is an example of
this class. Compounds of the opioid class have several drawbacks that limit
their use,
e.g. emetic and constipation effects, and negative influence on respiratory
capability.
Their use is also restricted because of their addiction liabilities. The
second major
class of analgesics, the non-steroidal anti-inflammatory analgesics of the COX-
1 or
COX-2 types, also have liabilities such as insufficient efficacy in severe
pain and
inflammatory conditions and at long term use the COX-1 inhibitors cause ulcers
of
1.0 the mucosa.
Thus, there is a need for new active agents useful for the treatment of pain
and
inflammatory conditions.
Description of the Present Invention
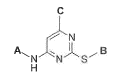
The present invention relates to compounds of Formula (1)
C
--'-''N
B
N N S-
H
wherein
A: 5-membered monocyclic aromatic heterocyclic ring or 8-10 membered
bicyclic
aromatic heterocyclic ring each containing 2-4 nitrogen atoms and optionally
one sulfur or oxygen atom, said ring optionally substituted (a) with one or
more
C1-C4 alkyl or alkenyl groups, or (b) with a 5- or 6-membered alkyl ring or
alkenyl ring or aryl ring that are optionally substituted with one or more
halogen or C1-C4 alkyl or alkenyl groups including mono- or poly-halogenated
C1-C4 alkyl or alkenyl groups
B: methylene-aryl or aryl rings that are optionally substituted with one or
more
halogen or C1-C4 alkyl or alkenyl groups including mono- or poly-halogenated
01-C4 alkyl or alkenyl groups
6
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
C: Monocyclic or bicyclic, saturated or monounsaturated or
polyunsaturated or
aromatic heterocycles having 5 ¨ 10 ring atoms among them 1 ¨ 5
heteroatoms which are preferably N, 0 and S, substituted, where appropriate,
once, twice or thrice with residues R5
R5: -OH, -SH, -01-C4 alkyl, -0-01_8 alkyl, -0-C6_14 aryl, -S-01_4 alkyl, -S-
06_14 aryl, -
S0-01_4 alkyl,-S0-06_14 aryl, -S02-01.4 alkyl, -S02-C6_14 aryl, -S03H, -0S02C1-
8
alkyl, -0S0206_14 aryl, -000H, -00001_8 alkyl, -(C0)01_8 alkyl,-000H, -
CONH2, -CONHC1_6 alkyl, -CON(C1_6 alky1)2, -NH2, -NHC1..6 alkyl, -N(01-6
alky1)2, -NH06_14 aryl, -NH-hetaryl, -N(06_14 ary1)2, -N(C1..6 alkyl)(06-14
aryl),-C16
alkyl, -02_12 alkenyl, halogen, -0H2CH2OH, -CH2CH2SH, -CH20H2SCH3, -
sulfamoyl, alkylsulfamoyl, dialkyl sulfamoyl, -sulfo, phosphono, -CN, -NO2
and/or ¨SCN
as well as pharmaceutically compatible salts, solvates, active metabolites,
tautomers
and prodrugs of these compounds.
As used herein, the following definitions shall apply unless otherwise
indicated.
The phrase "optionally substituted" is used interchangeably with the phrase
"substituted or unsubstituted" or with the term "(un)substituted". Unless
otherwise
indicated, an optionally substituted group may be a substituent at each
substitutable
position of the group, and each substitution is independent of the other.
The term "01-04 alkyl or alkenyl" designates both the unbranched and branched,
as
well as saturated and unsaturated functional aliphatic groups. In particular,
it means
methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl,
ethenyl
(vinyl), propenyl, or butenyl.
"5- or 6-membered alkyl or alkenyl ring" means cyclopentyl, cyclohexyl,
cyclopentenyl
or cyclohexenyl. The alkyl- or alkenyl ring can be substituted, where
appropriate, with
one or several halogen substituents or 01-C4 alkyl or alkenyl groups including
mono-
or poly-halogenated derivatives thereof.
"Aryl" refers to either an aromatic 5 ¨ 6 membered monocyclic carbocyclic or
heterocyclic ring system, or a 8-10 membered bicyclic carbocyclic or
heterocyclic ring
system, including heteroaromatic systems with 1 ¨ 5 hetero atoms (N, 0 or S),
preferably 1, 2 or 3 heteroatoms.
7
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
The aryl ring can be substituted, where appropriate, with one or several
halogen
substituents or 01-C4 alkyl or alkenyl groups including mono- or poly-
halogenated
derivatives thereof. The preferred aryl group is a carbocyclic ring, e.g.
phenyl, benzyl,
tolyl, xylyl or naphthyl. Examples for preferred "heteroaromatic rings" or
"heteroaryls"
are pyrazinyl, pyridinyl, furanyl, thienyl, thiazinyl, thiophenyl,
pyrimidinyl, isoxazolyl,
isothiazolyl, oxazolyl, thiazolyl, pyrazolyl, pyrrolyl, oxadiazolyl, pyridyl,
benzoimidazolyl, benzothienyl, imidazolyl, triazolyl, tetrazolyl or
benzothiazolyl.
A "5-membered monocyclic aromatic heterocyclic ring or 8-10 membered bicyclic
aromatic heterocyclic ring each containing 2-4, preferably 2 or 3, nitrogen
atoms and
optionally one sulfur or oxygen atom" comprises, for example, a imidazolyl,
pyrazolyl,
triazolyl, pyrazolo[1,5-]pyridinyl, pyrazolopyri mid inyl,
benzoimidazolyl,
imidazopyridinyl, purine or indolyl ring.
"Halogen" means fluorine, chlorine, bromine or iodine.
"Mono- or poly-halogenated alkyl or alkenyl groups" means an alkyl or alkenyl
group
substituted with one or more halogen atoms, e.g. CH2F, CHF2 or CF3.
"Monocyclic or bicyclic, saturated or monounsaturated or polyunsaturated or
aromatic heterocycles having 5 ¨ 14 ring atoms among them 1 ¨ 5 heteroatoms
which are preferably N, 0 and S, substituted, where appropriate, once, twice
or thrice
with residues W" preferably comprise the following groups: methylpiperazinyl,
thienyl,
pyridinyl, pyrimidinyl, piperazinyl, pyridyl, isoxazolyl, piperidinyl,
pyrazinyl,
morpholino, pyrrolyl, triazinyl, tetrazolyl, oxazolyl, benzo[d][1,3]dioxolyl,
indolyl,
imidazolyl, pyrazolyl, furanyl. Monocyclic or polycyclic aromatic heterocycles
denote
a ring system having 5 to 14 ring atoms (also denoted "heteroaryl" or
"hetaryl"),
preferably 5 to 10 ring atoms, where 1 or more ring atoms are an element other
than
carbon, e.g. N, 0 or S, as such or in combination. Preferred heteroaryls
contain 5 or
6 ring atoms. The heteroaryls may be substituted, where appropriate, at one or
more
ring systems. Examples of suitable heteroaryls are mentioned above.
In the sense of the invention, all residues are considered combinable with one
another unless stated otherwise in the definition of the residues. All
conceivable
subgroups thereof shall be considered disclosed.
8
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
The invention also relates to physiologically compatible salts of the
compounds of
general Formula (1). The physiologically compatible salts are obtained as
usual by
reaction of basic compounds of general Formula (1) with inorganic or organic
salts or
acids and by neutralization with inorganic or organic bases. Hydrochloric
acid,
.. sulphuric acid, nitric acid or hydrobromic acid are preferably used as
inorganic acids
and e.g. formic acid, acetic acid, propionic acid, glycolic acid, lactic acid,
amygdalic
acid, tartaric acid, malic acid, citric acid, malonic acid, maleic acid,
fumaric acid,
succinic acid, alginic acid, benzoic acid, 2-, 3- and 4-alkyloxy and acyloxy
benzoic
acids, ascorbic acid, C1-C3 alkylsulfonic acids, benzenesulfonic acid,
nicotinic acid,
isonicotinic acid and amino acids are used as organic acids. For example,
ammonia,
sodium hydroxide, and potassium hydroxide solution are used as inorganic bases
and alkylamines, pyridine, quinoline, isoquinoline, piperazine and derivatives
thereof,
and picolines, quinaldine or pyrimidine are used as organic bases. In
addition,
physiologically compatible salts of the compounds according to general Formula
(1)
can be obtained by converting the substances which have as substituents a
tertiary
amino group in a basically known manner with alkylating agents ¨such as alkyl
or
aralkyl halides ¨ into the corresponding quaternary ammonium salts.
The invention also relates to solvates of the compounds, including the
pharmaceutically acceptable salts, acids, bases and esters as well as the
active
metabolites thereof and, where appropriate, the tautomers thereof according to
general Formula (1) including prodrug formulations. Prodrug formulations here
comprise all substances which are formed by simple transformation including
hydrolysis, oxidation or reduction either enzymatically, metabolically or in
any other
way. A suitable prodrug contains e.g. a substance of general Formula (1) bound
via
an enzymatically cleavable linker (e.g. carbamate, phosphate, N-glycoside or a
disulfide group) to a dissolution-improving substance (e.g. tetraethylene
glycol,
saccharides, formic acids or glucuronic acid, etc.). Such a prodrug of a
compound
according to the invention can be applied to a patient, and this prodrug can
be
transformed into a substance of general Formula (1) so as to obtain the
desired
pharmacological effect.
The term "pharmaceutically acceptable" indicates that the substance or
composition
is compatible chemically and/or toxicologically, with the other ingredients
comprising
a formulation, and/or the mammal being treated therewith.
9
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
The present invention further provides a pharmaceutical composition, which
comprises a compound of Formula (1) or a pharmaceutically acceptable salt
thereof,
as defined hereinabove. In one embodiment, the pharmaceutical composition
includes the compound of Formula (1) together with a pharmaceutically
acceptable
excipient, diluent or carrier.
The present invention further provides a compound of Formula (1) or a
pharmaceutically acceptable salt thereof, for use in therapy.
The compounds according to the invention can be administered in different
ways,
e.g. topically, orally, parenterally, cutaneously, subcutaneously,
intravenously,
intramuscularly, rectally, or by inhalation. The oral or intravenous
administration is
preferred. The compound is given to a patient who needs a therapy for a
disease
coming under the indication spectrum of the compounds according to the
invention
over a period to be determined by a physician. The compound can be
administered
to both humans and other mammalian animals.
The dosage of the compounds according to the invention is determined by the
physician on the basis of the patient-specific parameters, such as age,
weight, sex,
severity of the disease, etc. The dosage is preferably from 0.001 mg/kg to
1000
mg/kg body weight, preferably from 0.001 to 500 mg/kg body weight and most
preferably from 0.001 to 100 mg/kg body weight.
Corresponding to the kind of administration, the medicament is suitably
formulated,
e.g. in the form of solutions or suspensions, simple tablets or dragees, hard
or soft
gelatine capsules, suppositories, ovules, preparations for injection, which
are
prepared according to common galenic methods.
The compounds according to the invention can be formulated, where appropriate,
together with further active substances and with excipients common in
pharmaceutical compositions, e.g. - depending on the preparation to be
produced -
talcum, gum arabic, lactose, starch, magnesium stearate, cocoa butter, aqueous
and
non-aqueous carriers, fatty bodies of animal or vegetable origin, paraffin
derivatives,
glycols (in particular polyethylene glycol), various plasticizers, dispersants
or
emulsifiers, pharmaceutically compatible gases (e.g. air, oxygen, carbon
dioxide,
etc.), preservatives.
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
In order to produce liquid preparations, additives and/or solvents, such as
sodium
chloride solution, DSMO, ethanol, acetone, sorbitol, glycerine, olive oil,
almond oil,
propylene glycol or ethylene glycol, can be used. For example, DMSO is an
organic
solvent and belongs to the class of sulfoxides. It is miscible with water and
various
organic solvents in any ratio. DMSO is characterized by a pharmaceutical
effect
(antiphlogistic and analgetic).
Since it was recognized that several compounds of the present invention have a
low
solubility and are (highly) hydrophobic, a solubilizing agent may be useful. A
solubilizing agent is a non-ionic surface-active substance (surfactant).
According to
the present invention, preferred solubilizing agents are selected from PEG-
hydrated
castor oil (Cremophor0-series), D-tocopherol polyethylene glycol 1000-
succinate
(Kolliphor0 TPGS), polysorbate 20 (Tween0 20), polysorbate 80 (Tween@ 80),
polyoxyethylated 12-hydroxystearic acid (Kolliphor@ HS 15; Soluto10 HS15),
sorbitan
monooleate (Span 20), poloxamer 407 (Kolliphor0 P 407), poloxamer 188
(Kolliphor0 P 188), PEG 300 caprylic acid/capric glycerides (Softigen@ 767),
PEG
400 caprylic acid/capric glycerides (Labrasol0), PEG 300 oleic acid glycerides
(Labrafil@ M-1944CS), PEG 300 linoleic acid glycerides (Labrafil@ M-2125CS),
lauroyl polyoxyglycerides (Gellusire0 44/14), n-decanoic acid ester with 1,2,3-
propane trioloctanoate (Softigen0 767), mono- or di-fatty acid esters of PEG
330,
400 or 1750.
Commercial products of PEG hydrated castor oil are for example known on the
basis
of PEG7, PEG25, PEG35, PEG40, PEG50 and PEG60: CroduretTM 25 (Croda),
CroduretTm 40 (Croda), CroduretTM 50 (Croda), CroduretTM 60 (Croda),
Cremophor0
RH 40 (BASF), Cremophor@ RH 60 (BASF), Cremophor0 RH 410 (BASF), Kolliphor
EL or Cremophor EL (BASF), Emulgin@ HRE 40 (BASF), Emulgin@ HRE 455 (BASF)
und EMAROL H40 (CISME, Italy). According to the present invention, PEG35
hydrated castor oil (Cremophore EL) is particularly suitable.
When solutions for infusion or injection are used, they are preferably aqueous
solutions or suspensions, it being possible to produce them prior to use, e.g.
from
lyophilized preparations which contain the active substance as such or
together with
a carrier, such as mannitol, lactose, glucose, albumin and the like. The ready-
made
solutions are sterilized and, where appropriate, mixed with excipients, e.g.
with
preservatives, stabilizers, emulsifiers, solubilizers, buffers and/or salts
for regulating
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
the osmotic pressure. The sterilization can be obtained by sterile filtration
using filters
having a small pore size according to which the composition can be
lyophilized,
where appropriate. Small amounts of antibiotics can also be added to ensure
the
maintenance of sterility.
Furthermore, inhalation compositions, e.g. in the form of aerosols, sprays or
as
micronized powder, are preferably produced. For this purpose, the compounds
according to the invention are either dissolved or suspended in
pharmaceutically
conventional solvents and finely divided by means of excess pressure in a
certain
volume and inhaled. The procedure is made correspondingly in the solid
substances
to be inhaled which are also finely divided by means of excess pressure and
inhaled.
Other applicators working by means of excess pressure are also included here.
The invention also relates to pharmaceutical preparations which contain a
therapeutically active amount of the active ingredients (compound according to
the
invention of Formula (1)) together with organic or inorganic solid or liquid,
pharmaceutically compatible carriers which are suited for the intended
administration
and which interact with the active ingredients without drawbacks.
The compounds of the Formula (1) are believed to be novel and are provided as
further aspects of the invention.
The ability of the compounds of the present invention to act as TrkA
inhibitors has
been demonstrated by the assays in the Examples of the present application.
Certain compounds which are inhibitors of TrkA are particularly useful in the
treatment of multiple types of pain including inflammatory pain, neuropathic
pain, and
pain associated with cancer, surgery, and bone fracture.
In one embodiment, compounds of Formula (1) are useful for treating pain,
including
chronic and acute pain in a mammal.
Acute pain, as defined by the International Association for the Study of Pain,
results
from disease, inflammation, or injury to tissues. This type of pain generally
comes on
suddenly, for example, after trauma or surgery, and may be accompanied by
anxiety
or stress. The cause can usually be diagnosed and treated, and the pain is
confined
to a given period of time and severity. In some rare instances, it can become
chronic.
12
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Chronic pain, as defined by the International Association for the Study of
Pain, is
widely believed to represent disease itself. It can be made much worse by
environmental and psychological factors. Chronic pain persists over a longer
period
than acute pain and is resistant to most medical treatments, generally over 3
months
or more. It can and often does cause severe problems for patients.
Compounds of Formula (1) are also useful for treating or preventing cancer in
a
mammal. Examples of cancer types to be treated or prevented are pancreatic
tumors, prostate tumors, lung tumors, kidney tumors, bladder tumors, liver
tumors,
lymphoma, leukemia (e.g. myeloid leukemia), oesophageal tumors, ovarian
tumors,
oral tumors, thyroid tumors, cervical tumors, head-and-neck tumors, breast
tumors,
neuroblastoma, gastric tumors, colon tumors, brain tumors (e.g. glioblastoma
or
medulloblastoma) and skin tumors (e.g. melanoma).
Compounds of Formula (1) are also useful for treating inflammation in a
mammal.
Accordingly, the present invention is further related to a method of treating
or
preventing pain in a mammal, comprising administering to said mammal one or
more
compounds of Formula (1) or a pharmaceutically acceptable salt thereof in an
amount effective to treat or prevent said pain. In one embodiment, the pain is
chronic
pain. In one embodiment, the pain is acute pain. In one embodiment, the pain
is
inflammatory pain. In one embodiment, the pain is neuropathic pain. In one
embodiment, the pain is pain associated with cancer. In one embodiment, the
pain is
pain associated with surgery. In one embodiment, the pain is pain associated
with
bone fracture. In one embodiment, the method comprises a method of treating
said
pain in a mammal. In one embodiment, the method comprises a method of
preventing said pain in a mammal.
The invention is further related to a method of treating or preventing
inflammation in a
mammal, comprising administering to said mammal one or more compounds of
Formula (1) or a pharmaceutically acceptable salt thereof in an amount
effective to
treat or prevent said inflammation. In one embodiment, the method comprises a
method of treating said inflammation in a mammal. In one embodiment, the
method
comprises a method of preventing said inflammation in a mammal.
The invention is further related to a method of treating or preventing cancer
in a
mammal, comprising administering to said mammal one or more compounds of
13
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Formula (1) or a pharmaceutically acceptable salt thereof in an amount
effective to
treat or prevent said cancer.
In one embodiment, the method comprises a method of treating said cancer in a
mammal. In one embodiment, the method comprises a method of preventing said
cancer in a mammal.
Compounds of Formula (1) may be administered alone as a sole therapy or can be
administered in addition with one or more other substances and/or treatments
that
work by the same or a different mechanism of action. Examples include anti-
inflammatory compounds, steroids (e.g., dexamethasone, cortisone and
fluticasone),
analgesics such as NSAIDs (e.g., aspirin, ibuprofen, indomethacin, and
ketoprofen),
and opioids (such as morphine), and chemotherapeutic agents. These agents may
be administered with one or more compounds of Formula (1) as part of the same
or
separate dosage forms, via the same or different routes of administration, and
on the
same or different administration schedules according to standard
pharmaceutical
practice known to one skilled in the art.
In the field of medical oncology it is normal practice to use a combination of
different
forms of treatment to treat each patient with cancer. In medical oncology the
other
component(s) of such conjoint treatment in addition to compositions of the
present
invention may be, for example, surgery, radiotherapy, chemotherapy, signal
transduction inhibitors and/or monoclonal antibodies.
Accordingly, the compounds of Formula (1) may be administered in combination
with
one or more agents selected from mitotic inhibitors, alkylating agents, anti-
metabolites, antisense DNA or RNA, intercalating antibiotics, growth factor
inhibitors,
signal transduction inhibitors, cell cycle inhibitors, enzyme inhibitors,
retinoid receptor
.. modulators, proteasome inhibitors, topoisomerase inhibitors, biological
response
modifiers, anti- hormones, angiogenesis inhibitors, cytostatic agents, anti-
androgens,
targeted antibodies, HMG-CoA reductase inhibitors, and prenyl-protein
transferase
inhibitors. These agents may be administered with one or more compounds of
Formula (1) as part of the same or separate dosage forms, via the same or
different
.. routes of administration, and on the same or different administration
schedules
according to standard pharmaceutical practice known to one skilled in the art.
14
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
As used herein, terms "treat" or "treatment" refer to therapeutic,
prophylactic,
palliative or preventative measures. Beneficial or desired clinical results
include, but
are not limited to, alleviation of symptoms, diminishment of extent of
disease,
stabilized (i.e., not worsening) state of disease, delay or slowing of disease
progression, amelioration or palliation of the disease state, and remission
(whether
partial or total), whether detectable or undetectable. "Treatment" can also
mean
prolonging survival as compared to expected survival if not receiving
treatment.
Those in need of treatment include those already with the condition or
disorder, as
well as those prone to have the condition or disorder or those in which the
condition
or disorder is to be prevented.
In one embodiment, the terms "treatment" or "treating" as used herein, mean an
alleviation, in whole or in part, of symptoms associated with a disorder or
condition as
described herein (e.g., multiple types of pain including inflammatory pain,
neuropathic pain, and pain associated with cancer, surgery, and bone
fracture), or
slowing, or halting of further progression or worsening of those symptoms.
In one embodiment, the term "preventing" as used herein means the prevention
of
the onset, recurrence or spread, in whole or in part, of the disease or
condition as
described herein (e.g., multiple types of pain including inflammatory pain,
neuropathic pain, and pain associated with cancer, surgery, and bone
fracture), or a
symptom thereof.
The terms "effective amount" and "therapeutically effective amount" refer to
an
amount of compound that, when administered to a mammal in need of such
treatment, is sufficient to (i) treat or prevent a particular disease,
condition, or
disorder, (ii) attenuate, ameliorate, or eliminate one or more symptoms of the
particular disease, condition, or disorder, or (iii) prevent or delay the
onset of one or
more symptoms of the particular disease, condition, or disorder described
herein.
The amount of a compound of Formula (1) that will correspond to such an amount
will vary depending upon factors such as the particular compound, disease
condition
and its severity, the identity (e.g., weight) of the mammal in need of
treatment, but
can nevertheless be routinely determined by one skilled in the art.
As used herein, the term "mammal" refers to a warm-blooded animal that has or
is at
risk of developing a disease described herein and includes, but is not limited
to,
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
guinea pigs, dogs, cats, horses, cows, rats, mice, hamsters, and primates,
including
humans.
Preferred compounds according to Formula (1) are:
Compound A
N-N
al
CN-N
a3
N--N
a4
a8
(with C = as defined above for Formula (1); preferably: C = methylpiperazinyl)
16
SUBSTITUTE SHEET (RULE 26)
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Compound A
N-N
bl
N-N
b2
N= N
b3
N-N
b4 ¨S),y
F F
N-N
b5
N= N vc9
b6
N-N F
b7 ¨cky
N-N
b8
N-N
b9
(with C = as defined above for Formula (1); preferably: C = methylpiperazinyl)
Particular preferred compounds according to the present invention are:
17
SUBSTITUTE SHEET (RULE 26)
CA 03076444 2020-03-19
WO 2019/101843 PCT/EP2018/082183
r
L N,?
N N N
\
N N' S =11"--N" N S
al b7
C C
N -.N N N N
,
N N S N'N'S
a4 b8
Particularly preferred
is al (N-[5-(3-fluoropheny1)-1H-pyrazol-3-y1]-6-(4-
methylpiperazin-l-y1)-2-(phenylsulfanyl)pyrimidin-4-amine). This means that in
accordance with the above mentioned Formula (1) the substituent A is (3-
.. fluorophenyI)-1H-pyrazol-3-yl, B is phenyl and C is methylpiperazinyl.
N-N 0N N' S
For the sake of completeness it is mentioned that all general definitions
given above
in in connection with Formula (1) compounds apply, of course, to each
individual
preferred compound, too. Thus, if a compound of Formula (1) is mentioned, each
of
the aforementioned preferred compounds is comprised, too.
18
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Compounds in clinical trials provide a rich source for initiating new drug
design efforts
as for instance many compounds inhibit more than one molecular target and can
have a therapeutic impact via so far unknown or ignored mechanisms. Following
this
idea, the inventors mined all kinases and corresponding inhibitors that had
entered
clinical trials and analyzed the respective protein binding sites with respect
to
selectivity determining features (a crucial element for the rational design of
selective
compounds). Based on these analyses, the target¨compound pair tropomyosin
receptor kinase A (TrkA) and tozasertib was selected as starting point for the
design
of improved TrkA inhibitors. Tozasertib (Originator: Vertex Pharma) is in
clinical trials
for aurora kinase A (AurA), but also inhibiting TrkA, which is a validated
drug target
for cancer and pain. However, the selectivity of tozasertib for TrkA is not
very high
and its use is accompanied by several severe side effects, e.g. neutropenia,
diarrhea, mucositis. In line with these side effects, the inhibition of the
AurA has
common adverse effects such as neutropenia and hematological toxicities
(Falchook,
G. S., Bastida, C. C. & Kurzrock, R. Aurora kinase inhibitors in oncology
clinical trials:
Current state of the progress. Semin. Oncol. 42, 832-848 (2015)). Thus, the
inventors decided to switch the selectivity of tozasertib, originally
developed against
AurA as cancer target, towards the pain target TrkA.
The design of improved TrkA inhibitors itself was achieved by jointly
employing a
proprietary de novo design platform and a multi-objective selection scheme
that
considers selectivity and activity aspects as predicted by the also
proprietary in silico
tools. First, selectivity-determining features in the TrkA structure were
identified
(mentioned also above) by a novel approach called 'selectivity grids', where
atom-
based interaction energy grids of several TrkA, Aurora A and B structures were
fused
into one content-rich representation of target specific sub-pockets. This step
highlighted three areas of interest for compound optimization: two favorable
hydrophobic sub-pockets for TrkA-selectivity were identified adjacent to the
gatekeeper residue and enclosed between the Asp residue of the DFG-motif and a
Phe residue of the glycine-rich loop (G-loop), respectively, and one
unfavorable
pocket for TrkA-selectivity was identified that overlaps with the cyclopropyl
moiety of
tozasertib. The identified selectivity-determining areas subsequently guided
the
compound library design where the two function groups A and B in Formula 1
were
enumerated using commercially available drug-like fragments and retrosynthetic
rules. The resulting library was subsequently prioritized using a multi-
objective
19
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
compound selection scheme that filters for selective and highly active
compounds.
Key aspects of the filtering process included I) that the final selection of
compounds
was obtained in an automated fashion based on the scoring ranks (i.e., without
any
manual selection beside synthesizability criteria) and that II) the highly
conserved
.. and large target class of kinase was considered in addition to AurA as
primary off-
target.
In detail, the multi-objective compound selection scheme included the
following
steps. Initially, binding poses of the compounds were generated via docking
calculations. Compounds were removed for reasons of either poor docking
scores,
wrong orientation, or lack of key interactions. In the next step, compounds
with an
unfavorable selectivity profile were filtered out. This was accomplished via
machine
learning-based activity prediction models (Merget, B., Turk, S., Eid, S.,
Rippmann, F.
& Fulle, S. Profiling prediction of kinase inhibitors: Toward the virtual
assay. J. Med.
Chem. 60, 474-485 (2017)) that were used I) to remove promiscuous compounds
(i.e., predicted to be active at IC50 of 500 nM on 20 kinases) and II) those
that are
predicted to be highly active (IC50 < 10 nM) on Aurora A, B, or C kinases. The
selectivity filtering was complemented by a structure-based procedure
employing the
TrkA-Aurora 'selectivity grids' for rescoring of docking solutions. The
remaining
compounds were finally prioritized for highly active compounds using two
complementary machine learning (ML) technologies. I) All compounds were
evaluated by an `MMP/ML' approach that is trained on fragment-based Matched
Molecular Pairs (MMPs) and quantifies compound activity differences (Turk, S.,
Merget, B., Rippmann, F. & Fulle, S. Coupling Matched Molecular Pairs with
Machine
Learning for virtual compound optimization, doi: 10.1021/acs.jcim.7b00298).
II) In
.. addition, compounds interacting with the Phe gatekeeper were additionally
evaluated
by a hybrid QM/ML pipeline. This pipeline is trained on high-level quantum
mechanical (QM) calculations to quantify ligand-gatekeeper interactions and
rescores the top hits in a second step with fragment molecular orbital
calculations,
taking the entire binding pocket into consideration. The final selection of
compounds
was obtained in an automated fashion based on the scoring ranks (i.e., without
any
manual selection beside synthesizability criteria). Overall, the employed
pipeline
resulted into a new compound series of TrkA inhibitors with an improved
selectivity
profile across the kinome compared to the starting compound tozasertib. The
most
preferred compound al is highly active on TrkA (pKd = 8.6), has nanomolar
cellular
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
potency (26 nM) and high selectivity in the kinome panel (Selectivity Score =
0.08 @
100 nM and < 35% ctrl as activity cut-off) (Tables 1-4). This was achieved by
removing the cyclopropylcarboxamide tail of tozasertib and modifying the amino-
5-
methylpyrazole (best hits: al and a4).
The substitution at the 5- or 6-membered alkyl ring or alkenyl or aryl ring
(e.g. via a
fluorine as present in al) seems to be an important step to achieve TrkA
versus AurA
selectivity. Analogues of tozasertib have not been employed for pain treatment
so far.
Modifying part B (i.e. N-(4-aminothiophene)cyclopropylcarboxamide) also
resulted in
two hits (b7 and b8 ) but with lower kinome selectivity compared to al.
The particular preferred compound al was subjected to a pharmacokinetic
evaluation
in mice. The formulations contained compound al in DMSO together with a
solubilizing agent of the Cremophor0 series (e.g. Cremophor0 EL) and NaCl. In
this
regard reference is made to Example 6 and Figs. 2 and 3. In the experiments it
has
been clearly shown that, despite a certain expectation that compound al may be
toxic, none of the animals showed any toxic sign. Thus, it was concluded that
at least
compound al may be a suitable candidate for forthcoming clinical trials.
The invention is further described with respect to the figures which show:
Fig. 1: General synthesis route for tozasertib analogues
The invention is further described in the following examples which are not
construed
to limit the invention.
Examples:
Example 1: Method for the preparation of tozasertib and tozasertib analogues
having improved TrkA activity
The general synthetic route for the preparation of tozasertib (a-d) and
tozasertib
analogs (B: a, e-g; A: a, h-j) is shown in Fig. 1.
21
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Reagents and conditions: (a) 3-chloroperoxybenzoic acid, DCM, rt, 3h; (b) N-(4-
mercaptophenyl)cyclopropanecarboxamide, TEA, CH3CN, 80 C, 3-10h; (c) 5-methyl-
1H-pyrazol-3-amine, DIPEA, DMF, 95 C, 16h; (d) amine (1-methylpiperazine or
morpholine), DMF, DIPEA, 90 C, 6-12h; (e) corresponding thiol, TEA, CH3CN, 80
C, 3-10h; (f) 5-methyl-1H-pyrazol-3-amine, DIPEA, dioxane, 95 C, 3-6h, (g)
amine
(1-methylpiperazine or morpholine), DMF, DIPEA, 90 C, 6-12h, (h) thiophenol,
TEA,
THF, 50 C; (i) corresponding amine, DIPEA, dioxane, 95 C, 3-6h, (j) amine (1-
methylpiperazine or morpholine), DMF, DIPEA, 90 C, 6-12h.
The structures and batch purities (90%) of the compounds have been confirmed
by
using standard analytical methods (LC/MS and NMR).
Analytical data (NMR and LC/MS)
Tozasertib: N44-({4-115-methyl-1 H-pyrazol-3-y0amino]-6-(4-methylpiperazin-1-
yl)pyrimidin-2-yl}sulfanyOphenyl]cyclopropanecarboxamide
1H NMR (400 MHz, DMSO-d6) 6 0.8 (4H, CH2), 1.8 (1H, C(=0)CH), 2.0 (3H, ArCH3),
2.2 (3H, NCH3), 2.3 (4H, N(CH2)0H2), 3.3 (4H, ArN(CH2)CH2), 5.4 (1H, ArNHAr),
6.0
(1H, ArH), 7.4 (2H, ArH), 7.7 (2H, ArH), 9.2 (1H, ArH), 10.4 (1H, ArNHCO),
11.7 (1H,
ArH). LCMS purity 100%.
Reference 2: N-(5-methy1-1H-pyrazol-3-y1)-6-(4-methylpiperazin-1 -y1)-2-
(phenylsulfany1)-pyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 52.0 (3H, ArCH3), 2.2 (3H, NCH3), 2.3 (4H,
N(CH2)CH2), 3.3 (4H, ArN(CH2)0H2), 5.5 (1H, ArNHAr), 6.1 (1H, ArH), 7.5 (3H,
ArH), 7.6 (2H, ArH), 9.2 (1H, ArH), 11.7 (1H, ArH). LCMS purity 100%.
.. al: N-15-(3-fluorophenyI)-1 H-pyrazol-3-y11-6-(4-methylpiperazin-l-y1)-2-
(phenylsulfanyOpyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.2 (3H, NCH3), 2.3 (4H, N(CH2)CH2), 3.4 (4H,
ArN(CH2)CH2), 6.2 (2H, ArNHAr, ArH), 7.2 (1H, ArH), 7.4 (6H, ArH), 7.6 (2H,
ArH),
9.5 (1H, ArH), 9.3 (1H, ArH), 12.7 (1H, ArH). LCMS purity 100%.
a3: 6-(4-methylpiperazin-1 -y1)-2-(phenylsulfany1)-N-{pyrazolo[1,5-a]pyridin-2-
yOpyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.2 (3H, NCH3), 2.3 (4H, N(CH2)0H2), 3.4 (4H,
ArN(CH2)CH2), 5.8 (1H, ArH), 6.1 (1H, ArNHAr), 6.6 (1H, ArH), 7.1 (1H, ArH),
7.5
22
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
(2H, ArH), 7.6 (3H, ArH), 8.4 (1H, ArH), 9.8 (1H, ArH). LCMS purity 100%. LCMS
purity 98.5%.
a4: N-(5-cyclopenty1-1 H-pyrazol-3-y1)-6-(4-methylpiperazin-1 -y1)-2-
(phenylsuffanyOpyrimidin-4-amine
s 1H NMR (400 MHz, DMSO-d6) 6 1.4 (2H, cyclopentyl-H), 1.6 (4H, cyclopentyl-
H), 1.9
(2H, cyclopentyl-H), 2.2 (3H, NCH3), 2.3 (4H, N(CH2)CH2), 2.9 (1H, 2H,
cyclopentyl-
H), 5.6 (1H, ArNHAr), 6.3 (1H, ArH), 7.4 (3H, ArH), 7.6 (2H, ArH), 9.3 (1H,
ArH), 11.9
(1H, ArH). LCMS purity 100%.
a8: N-(5-tert-buty1-1H-pyrazol-3-y1)-6-(4-methylpiperazin-1-y1)-2-
(phenylsulfany0
pyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 1.2 (9H, CH3), 2.2 (3H, NCH3), 2.3 (4H,
N(CH2)CH2),
5.7 (1H, ArNHAr), 6.5 (1H, ArH), 7.5(5H, ArH), 9.2 (1H, ArH), 11.9 (1H, ArH).
LCMS
purity 100%.
bl : 21(2,5-dimethylphenyOsulfanyli-N-(5-methyl-1 H-pyrazol-3-y0-6-(4-
methylpiperazin-1 -y0pyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 62.0 (3H, ArCH3), 2.2 (6H, NCH3, ArCH3), 2.4 (3H,
ArCH3), 2.6 (4H, N(CH2)CH2), 3.4 (4H, ArN(CH2)CH2), 5.4 (1H, ArNHAr), 6.0 (1H,
ArH), 7.2 (2H, ArH), 7.4 (1H, ArH), 9.2 (1H, ArH), 11.7 (1H, ArH). LCMS purity
95.8%.
b2: 24(2,5-dimethylphenyOmethylkulfany0-N-(5-methyl-1 H-pyrazol-3-0-6-(4-
methylpiperazin-1 -yl)pyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.1 (3H, ArCH3), 2.2 (6H, 2xArCH3), 2.3 (3H,
NCH3),
2.4 (4H, N(CH2)CH2), 3.5 (4H, ArN(CH2)CH2), 4.3 (2H, CH2), 5.9 (111, ArNHAr),
6.4
(1H, ArH), 7.0 (1H, ArH), 7.1 (1H, ArH), 7.2 (1H, ArH), 9.2 (1H, ArH), 11.9
(1H, ArH).
LCMS purity 91.8%.
b3: 2-{[(3,4-dimethylphenyOmethylisulfanyli-N-(5-methyl-1H-pyrazol-3-y1)-6-(4-
methylpiperazin-1 -yOpyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 62.2 (12H, 3xArCH3, NCH3), 2.4 (4H, N(CH2)CH2),
3.5 (4H, ArN(CH2)CH2), 4.3 (2H, CH2), 5.9 (1H, ArNHAr), 6.5 (1H, ArH), 7.0
(1H,
ArH), 7.1 (1H, ArH), 7.2 (1H, ArH), 9.2 (1H, ArH), 11.9 (1H, ArH). LCMS purity
98.1%.
b4: N-(5-methyl-1 H-pyrazol-3-y1)-2-113-methylphenyOsulfany11-6-(4-
methylpiperazin-1-
yOpyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.0 (3H, ArCH3), 2.2 (3H, NCH3), 2.3 (7H, ArCH3,
N(CH2)CH2), 3.3 (4H, ArN(CH2)CH2), 5.5 (1H, ArNHAr), 6.1 (1H, ArH), 7.3 (5H,
ArH),
9.2 (1H, ArH), 11.7(1H, ArH). LCMS purity 91.1%.
23
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
b5: N-(5-methy1-1 H-pyrazol-3-y1)-6-(4-methylpiperazin-1-y1)-2-0-
(trifluoromethyl)
phenylisulfanyl}pyrimidin-4-amine
NMR (400 MHz, DMSO-d6) 6 2.0 (3H, ArCH3), 2.1 (3H, NCH3), 2.3 (4H,
N(CH2)CH2), 3.3 (4H, ArN(CH2)CH2), 5.5 (1H, ArNHAr), 6.2 (1H, ArH), 7.7 (1H,
ArH),
7.8 (2H, ArH), 7.9 (1H, ArH), 9.3 (1H, ArH), 11.8 (1H, ArH). LCMS purity
95.8%.
b6: N-(5-methy1-1 H-pyrazo1-3-y1)-6-(4-methylpiperazin-1-y1)-2-[(naphthalen-1-
ylmethyl)sulfanyl]pyrimidin-4-a mine
1H NMR (400 MHz, DMSO-d6) 6 2.1 (3H, ArCH3), 2.2 (3H, NCH3), 2.3 (4H,
N(CH2)CH2), 3.5 (4H, ArN(CH2)CH2), 4.8 (2H, CH2), 5.9 (1H, ArNHAr), 6.5 (1H,
ArH),
7.3 (1H, ArH), 7.6 (2H, ArH), 7.7 (1H, ArH), 7.8 (1H, ArH), 8.0 (1H, ArH), 8.2
(1H,
ArH), 9.3 (1H, ArH), 11.9 (1H, ArH). LCMS purity 100%.
b7: 2-{[(4-fluorophenyl)methyl]sulfany1}-N-(5-methyl-1 H-pyrazol-3-y1)-6-(4-
methylpiperazin-1 -yl)pyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.2 (6H, ArCH3, NCH3), 2.3 (4H, N(CH2)CH2), 3.5
(4H, ArN(CH2)CH2), 4.3 (2H, CH2), 5.9 (1H, ArNHAr), 6.5 (1H, ArH), 7.1 (2H,
ArH),
7.4 (2H, ArH), 9.2 (1H, ArH), 11.9 (1H, ArH). LCMS purity 95.6%.
b8: N-(5-methy1-1 H-pyrazol-3-y1)-2-{1(4-methylphenyl)methylisulfany1}-6-(4-
methylpiperazin-l-Apyrimidin-4-amine
1H NMR (400 MHz, DMSO-d6) 6 2.1 (9H, 2xArCH3, NCH3), 2.3 (4H, N(CH2)0H2), 3.4
(4H, ArN(CH2)CH2), 4.3 (2H, CH2), 5.8 (1H, ArNHAr), 6.4 (1H, ArH), 7.1 (2H,
ArH),
7.2 (2H, ArH), 9.2 (1H, ArH), 11.9 (1H, ArH). LCMS purity 92.8%.
b9: 2-[(2,4-dimethylphenyl)sulfany1]-N-(5-methyl-1 H-pyrazol-3-y1)-6-(4-
methylpiperazin-1 -yOpyrimidin-4-a mine
1H NMR (400 MHz, DMSO-d6) 6 2.0 (3H, ArCH3), 2.2 (3H, NCH3), 2.3 (10H,
N(CH2)CH2 & ArCH3), 3.3 (4H, ArN(CH2)CH2), 5.4 (1H, ArNHAr), 6.0 (1H, ArH),
7.1
(1H, ArH), 7.2 (1H, ArH) , 7.4 (1H, ArH), 9.2 (1H, ArH), 11.7 (1H, ArH). LCMS
purity
95.4%.
Example 2: Primary screen
Compounds were initially tested in a primary screen against TrkA (at 10 and
100 nM)
and AurA (at 100 nM and 1 pM) using the KINOMEscan technology from DiscoverX
Corporation, Fremont, CA, USA (https://www.discoverx.com/services/druq-
discoverv-
development-services/kinase-profilinalkinomescan).
24
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Results were reported as A of control ( /0 Ctrl. = (test compound signal -
positive
control signal) I (DMSO signal - positive control signal). KINOMEscan is an
active
site-directed competition binding assay that quantitatively measures the
ability of a
compound to compete with an immobilized, active-site directed ligand.
Table 1: Results of primary screens of biological assay.
ID % Ctrl. TrkA % Ctrl. AurA
@10 nM @100 nM @100 nM @1 pM
Tozasertib 31 5.2 0.4
Reference 2 58 10 15 0.95
al 11 1.9 97 92
a3 93 29 87 76
a4 15 0.95 49 4.5
a8 69 18 100 79
b1 80 11 7.3 0.5
b2 93 31 54 9.4
b3 53 11 46 4.5
b4 50 6.7 2.6 0.1
b5 90 32 36 2.4
b6 74 35 63 7.4
b7 9.3 0.5 15 0.85
b8 35 5.3 30 1.3
b9 77 24 51 3.8
'Compound "Reference 2": Tozasertib without cyclopropylcarboxamide tail
N-N
S
Reference 2 has compared to tozasertib an improved selectivity towards AurA
(i.e.
AApKd = 1; Table 2) pointing to the contribution of removing the
cyclopropylcarboxamide tail in addition to the modifications in compound part
A.
25
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Example 3: Kd measurement
Kd measurements were done via the KdELECT product service from DiscoverX
Corporation (Freemont, CA, USA) which also employs the KINOMEscan technology.
Here, inhibitor binding constants (Kd values) are calculated from duplicate 11-
point
dose-response curves.
Table 2: Experimental Kd / pKd values of top hits.
ID 1 TrkA AurA TrkA AurA
ApKd
[nNI] [AI] [pKdi [pl(d]
Tozasertib 3.4 0.47 8.5 9.3 -0.8
Reference 30 41 7.5 7.4 0.1
2
al 2.6 4500 8.6 5.3 3.3
a4 1.5 93 8.8 7.0 1.8
b7 1.1 28 9.0 7.6 1.4
b8 3.8 53 8.4 7.3 1.1
i
Example 4: Selectivity profiling
Selectivity profiling was done over a panel of 97 kinases at 100 nM
concentration
(scanEDGE assay panel from Discover X, which also uses the KINOMEscan
technology, whereat KIT(D816V) and KIT (V559D, T670I) were replaced by TrkB
and
TrkC). The best compound (al) was additionally profiled at 1 pM concentration
using
the same panel. The employed profiling panel consisted of the following
kinases:
ABL1(T3151)-phosphorylated, ABL1-nonphosphorylated, ABL1-phosphorylated,
ACVR1B, ADCK3, AKT1, AKT2, ALK, AURKA, AURKB, AXL, BMPR2, BRAF, BRAF
zo (V600E), BTK, CDK11, CDK2, CDK3, CDK7, CDK9, CHEK1, CSF1R, CSNK1D,
CSNK1G2, DCAMKL1, DYRK1B, EGFR, EGFR (L858R), EPHA2, ERBB2, ERBB4,
ERK1, FAK, FGFR2, FGFR3, FLT3, GSK3B, 1GF1R, IKK-alpha, IKK-beta, INSR,
JAK2 (JH1domain-catalytic), JAK3 (JH1domain-catalytic), JNK1, JNK2, JNK3, KIT,
LKB1, MAP3K4, MAPKAPK2, MARK3, MEK1, MEK2, MET, MKNK1, MKNK2, MLK1,
p38-alpha, p38-beta, PAK1, PAK2, PAK4, PCTK1, PDGFRA, PDGFRB, PDPK1,
PIK3C2B, PIK3CA, P1K3CG, PIM1, PIM2, PIM3, PKAC-alpha, PLK1, PLK3, PLK4,
26
CA 03076444 2020-03-19
WO 2019/101843 PCT/EP2018/082183
PRKCE, RAF1, RET, RIOK2, ROCK2, RSK2 (Kin.Dom.1-N-terminal), SNARK, SRC,
SRPK3, TGFBR1, T1E2, TRKA, TRKB, TRKC, TSSK1B, TYK2 (JH1 domain-
catalytic), ULK2, VEGFR2, YANK3, ZAP70
Table 3: Experimental profiling data @ 100 nM screening concentration.
Selectivity
ID List of inhibited kinases
Hits Score
TrkA, ABL1, AurA, AurB, AXL, CK1d, FLT3, JAK2,
Tozasertib 10 0.109 PLK4, RET
al 7 0.076 TrkA, TrkB, TrkC, FLT3, KIT, PDGFRb, RET
__
TrkA, TrkB, TrkC, ABL1, AurA, AXL, FGFR2,
a4 19 0.207 FGFR3, FLT3, FMS, INSR, JAK2, JAK3, KIT,
MAP2K2, MET, PDGFRb, RET, SRC
TrkA, TrkB, TrkC, ABL1, ALK, AurA, FLT3, FMS,
b7 18 0.196 JAK2, JAK3, KDR, KIT, NuaK2, PDGFRb, PLK4,
RET, SRC, TYK2
TrkA, TrkB, TrkC, ABL1, AurA, AXL, BTK, FGFR3,
b8 17 0.185 FLT3, JAK2, JAK3, KDR, KIT, PLK4, RET,
SRC,
TYK2
Kinase panel: N = 92 non-mutant kinases; activity cut-off: <35% ctrl @ 100 nM
Selectivity Score = Number of inhibited kinases / Number of tested kinases
Experimental profiling data @ 1 pM screening concentration for al: Selectivity
Score = 0.207
Example 5: Cellular Assay
The two compounds al and b7 were additionally tested in a functional assay
using
the PathHunter0 technology (DiscoverX Corporation, Freemont, CA, USA;
https.11www.discoverx.com/technoloqies-pla fforms/enzyme-fraqment-
complementation-technology/cell-based-efc-assays/protein-protein-
interactions/kinases-cell-based-(rtk-ctk)). In this assay, the full-length
human TrkA
receptor (i.e. including ligand-binding and kinase domains) is expressed as a
C-
terminal (intracellular) fusion with a small peptide epitope, called ProlinkTM
(PK).
Furthermore, a SH2 domain containing protein (i.e. Shcl) is co-expressed with
a
larger Enzyme Acceptor (EA) fragment. Activation of the TrkA receptor by
adding the
agonist ligand (13-NGF), leads to the auto-phosphorylation of the kinase and
subsequent binding of the Shcl -EA protein. That recruitment results in an
active B-
galactosidase enzyme that is detected by addition of a chemiluminescent
substrate.
27
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Compounds were tested for antagonism using EC80 concentration (i.e. 0.03
pg/ml)
of 13-NGF. Data was normalized to the maximal and minimal response observed in
the presence of EC80 agonist ligand (13-NGF) and vehicle (DMSO, final
concentration
1%), respectively.
Table 4: Compound activity in cell-based assay.
ID Cellular assay [RC50]
al 26 nM
b7 23 nM
Example 6: Pharmacokinetic evaluation of compound al after intravenous
administration in mice
Compound al is highly active on TrkA, most importantly from a design
perspective,
has a 10,000-fold improved selectivity against the selected off-target AurA.
Nanomolar cellular potency against TrkA underlines the potential value of
compound
al as an advanced hit compound for the treatment of acute and chronic pain or
other
conditions associated with abnormal TrkA activity such as inflammation and
cancer.
Compound al was synthesized under contract by the company Enamine Ltd.
according to Example 1. Dimethylsulfoxide (DMSO) from J.T. Baker (Germany),
Cremophor EL (Polyoxyethyleneglycerol triricinoleate 35 castor oil) from BASF
(Germany), and Saline solution 0.9% from B. Braun (Germany) were used in this
study. Two types of formulations were prepared: a formulation with a high dose
of
compound al (9 mM) and a formulation with a low dose of compound al (1.8 mM);
c.f. Tables 5 and 6. First, a 100 mM stock solution of compound al in DMSO was
prepared by dissolving 18.46 mg of compound al (molecular weight of 461.56
g/mol)
in 400 pl DMSO at room temperature. The formulation with the high dose of al
was
prepared using 9% al-DMSO stock solution, 10% Cremophor EL, and 81% saline.
The formulation with the low dose of al contained 1.8% al-DMSO stock solution,
5%
Cremophor EL, and 93.2% saline; c.f. Fig. 2. Both formulations were checked in
28
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
serial dilutions in order to make sure that compound al did not precipitate
after
intravenous injection.
Table 5: Preparation of test solutions for i.v. injection in mice
al_low al_high vehicle_low vehicle_high saline
al-DMSO stock solution 1.8% 9.0%
--
(100 mM)
DMSO 1.8% 9.0%
Cremophor EL 5.0% 10.0% 5.0% ____ 10.0%
NaCI 0.9% 93.2% 81.0% 93.2% 81.0%
100%
Experiments were carried out in accordance with the EU Directive 86/609/EEC on
the protection of animals used for scientific purposes. Female adult C57131/6
mice (6-
8 weeks old, body weight 20 g) were treated with 5 mg/kg (low dose) and 25
mg/kg
1.0 (high dose) compound al or with vehicle (formulations without compound
al) or with
saline by intravenous (i.v.) injections for 1 hour using a 1 ml syringe (29-G,
Terumo);
c.f. Table 6. After injection, the mice were returned to their home cages, and
after 1
hour, the mice were anaesthetized with isoflurane. Terminal blood samples were
collected using a 1 ml syringe (20-G needle, B. Braun). The blood was
immediately
transferred into a 1.3 ml heparin-coated tube (Sarstedt). Plasma was collected
by
centrifugation at 1500g for 15 minutes. The supernatant was placed in a 1.5 ml
tube
on dry ice and stored at -20 C.
The quantification of compound al in murine plasma samples was performed via
liquid chromatography tandem mass spectrometry (LC-MS-MS).
29
CA 03076444 2020-03-19
WO 2019/101843
PCT/EP2018/082183
Table 6: Pharmacokinetic evaluation of compound al after i.v. injection in
mice
Animal No. Test Item Injection Toxicity
Plasma Conc.
(C57BI/6 mice) Volume
of Compound
al
1 al low (5 150 pl None
407 nM
mg/kg)
2 al low (5 150 pl None
272 nM
mg/kg)
3 al high (25 150 pl None
805 nM
mg/kg)
4 al high (25 150 pl None
920 nM
mg/kg)
vehicle_ low 150 pl None n.d.
6 vehicle_low 150 pl None
n.d.
7 vehicle_high 150 pl None
n.d.
8 vehicle_high 150 pl , None
n.d.
9 , saline 150 pl None
n.d.
saline 150 pl None n.d.
5 n.d. = not detectable (below detection limit)
Results: The LC-MS-MS analysis confirmed an average concentration of compound
al in plasma of mice which were treated for 1 hour with 5 mg/kg (low dose) and
25
mg/kg (high dose) of 340 nM and 863 nM, respectively. Surprisingly, none of
the
10 animals showed any obvious signs of toxicity (Table 6).