Note: Descriptions are shown in the official language in which they were submitted.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
PHARMACEUTICAL COMPOSITIONS WITH ENHANCED PERMEATION
CLAIM FOR PRIORITY
This application is a continuation-in-part of U.S. Patent Application No.
15/724,234 filed
October 3, 2017 and a continuation-in-part of U.S. Patent Application No.
15/717,856, filed
September 27, 2017, which is a continuation-in-part of U.S. Patent Application
No. 15/587,376,
filed May 4, 2017, which claims priority under 35 U.S.C. 119(e) to U.S.
Patent Application
Serial No. 62/331,993 filed on May 5, 2016, and which claims priority under 35
U.S.C. 119(e)
to U.S. Patent Application Serial No. 62/735,822 filed on September 24, 2018,
each of which is
hereby incorporated by reference in its entirety.
TECHNICAL FIELD
This invention relates to pharmaceutical compositions.
BACKGROUND
Active ingredients, such as drugs or pharmaceuticals, are delivered to
patients in
deliberate fashion. Delivery of drugs or pharmaceuticals using film
transdermally or
transmucosally can require that the drug or pharmaceutical permeate or
otherwise cross a
biological membrane in an effective and efficient manner.
SUMMARY
In general a pharmaceutical composition can include a polymeric matrix, a
pharmaceutically active component in the polymeric matrix, and an adrenergic
receptor
interacter. In certain embodiments, the pharmaceutical composition can further
include a
permeation enhancer. An adrenergic receptor interacter can be an adrenergic
receptor blocker.
The permeation enhancer can also be a flavonoid, or used in combination with a
flavonoid.
In certain embodiments, the adrenergic receptor interacter can be a terpenoid,
terpene or a
C3-C22 alcohol or acid. The adrenergic receptor interacter can be a
sesquiterpene. In certain
embodiments, the adrenergic receptor interacter can include farnesol, linoleic
acid, arachidonic
1
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
acid, docosahexanoic acid, eicosapentanoic acid, or docosapentanoic acid, or
combinations
thereof
In certain embodiments, a pharmaceutical composition can include a polymeric
matrix, a
pharmaceutically active component in the polymeric matrix, and an aporphine
alkaloid
interacter.
In other embodiments, a pharmaceutical composition can include a polymeric
matrix, a
pharmaceutically active component in the polymeric matrix, and a vasodilator
interacter.
In certain embodiments, the pharmaceutical composition can be a film further
comprising
a polymeric matrix, the pharmaceutically active component being contained in
the polymeric
matrix.
In certain embodiments, the adrenergic receptor interacter can be a
phytoextract.
In certain embodiments, the permeation enhancer can be a phytoextract.
In certain embodiments, the permeation enhancer can include a phenylpropanoid.
In other embodiments, the phenylpropanoid can be eugenol.
In certain embodiments, the pharmaceutical composition can include a fungal
extract.
In certain embodiments, the pharmaceutical composition can include saturated
or
unsaturated alcohol.
In certain embodiments, the alcohol can be benzyl alcohol.
In some cases, the flavonoid, phytoextract, phenylpropanoid, eugenol, or
fungal extract
can be used as a solubilizer.
In other embodiments, the phenylpropanoid can be eugenol. In certain
embodiments, the
phenylpropanoid can be a eugenol acetate. In certain embodiments, the
phenylpropanoid can be a
cinnamic acid. In other embodiments, the phenylpropanoid can be a cinnamic
acid ester. In other
embodiments, phenylpropanoid can be a cinnamic aldehyde.
In other embodiments, the phenylpropanoid can be a hydrocinnamic acid. In
certain
embodiments, the phenylpropanoid can be chavicol. In other embodiments, the
phenylpropanoid
can be safrole.
In certain embodiments, the phytoextract can be an essential oil extract of a
clove plant.
In other examples, the phytoextract can be an essential oil extract of a leaf
of a clove plant. The
phytoextract can be an essential oil extract of a flower bud of a clove plant.
In other
embodiments, the phytoextract can be an essential oil extract of a stem of a
clove plant.
2
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
In certain embodiments, the phytoextract can be synthetic. In certain
embodiments, the
phytoextract can include 20-95% eugenol, including 40-95% eugenol, and
including 60-95%
eugenol. In certain embodiments, the phytoextract can include 80-95% eugenol.
In other embodiments, the pharmaceutically active component can be
epinephrine.
In certain embodiments, the pharmaceutically active component can be diazepam.
In certain embodiments, the pharmaceutically active component can be
alprazolam.
In certain embodiments, the polymer matrix can include a polymer. In certain
embodiments, the
polymer can include a water soluble polymer.
In certain embodiments, the polymer can be a polyethylene oxide.
In certain embodiments, the polymer can be a cellulosic polymer. In
certain
embodiments, the cellulosic polymer can be hydroxypropylmethyl cellulose,
hydroxyethyl
cellulose, hydroxyethylmethyl cellulose, hydroxypropyl cellulose,
methylcellulose,
carboxymethyl cellulose and/or sodium carboxymethylcellulose.
In certain embodiments, the polymer can include hydroxypropyl methylcellulose.
In certain embodiments, the polymer can include polyethylene oxide and/or
hydroxypropyl methylcellulose.
In certain embodiments, the polymer can include polyethylene oxide and/or
polyvinyl
pyrroli done.
In certain embodiments, the polymeric matrix can include polyethylene oxide
and/or a
polysaccharide.
In certain embodiments, the polymeric matrix can include polyethylene oxide,
hydroxypropyl methylcellulose and/or a polysaccharide.
In certain embodiments, the polymeric matrix can include polyethylene oxide, a
cellulosic polymer, polysaccharide and/or polyvinylpyrrolidone.
In certain embodiments, the polymeric matrix can include at least one polymer
selected
from the group of: pullulan, polyvinyl pyrrolidone, polyvinyl alcohol, sodium
alginate,
polyethylene glycol, xanthan gum, tragancanth gum, guar gum, acacia gum,
arabic gum,
polyacrylic acid, methylmethacrylate copolymer, carboxyvinyl copolymers,
starch, gelatin,
ethylene oxide, propylene oxide co-polymers, collagen, albumin, poly-amino
acids,
polyphosphazenes, polysaccharides, chitin, chitosan, and derivatives thereof
In certain embodiments, the pharmaceutical composition can further include a
stabilizer.
3
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Stabilizers can include antioxidants, which can prevent unwanted oxidation of
materials,
sequestrants, which can form chelate complexes and inactivating traces of
metal ions that would
otherwise act as catalysts, emulsifiers and surfactants, which can stabilize
emulsions, ultraviolet
stabilizers, which can protect materials from harmful effects of ultraviolet
radiation, UV
absorbers, chemicals absorbing ultraviolet radiation and preventing it from
penetrating the
composition, quenchers, which can dissipate the radiation energy as heat
instead of letting it
break chemical bonds, or scavengers which can eliminate free radicals formed
by ultraviolet
radiation.
In yet another aspect, the pharmaceutical composition has a suitable nontoxic,
nonionic
alkyl glycoside having a hydrophobic alkyl group joined by a linkage to a
hydrophilic saccharide
in combination with a mucosal delivery-enhancing agent selected from: (a) an
aggregation
inhibitory agent; (b) a charge-modifying agent; (c) a pH control agent; (d) a
degradative enzyme
inhibitory agent; (e) a mucolytic or mucus clearing agent; (f) a ciliostatic
agent; (g) a membrane
penetration-enhancing agent selected from: (i) a surfactant; (ii) a bile salt;
(ii) a phospholipid
.. additive, mixed micelle, liposome, or carrier; (iii) an alcohol; (iv) an
enamine; (v) a nitric oxide
donor compound; (vi) a long chain amphipathic molecule; (vii) a small
hydrophobic penetration
enhancer; (viii) sodium or a salicylic acid derivative; (ix) a glycerol ester
of acetoacetic acid; (x)
a cyclodextrin or beta-cyclodextrin derivative; (xi) a medium-chain fatty
acid; (xii) a chelating
agent; (xiii) an amino acid or salt thereof; (xiv) an N-acetylamino acid or
salt thereof; (xv) an
.. enzyme degradative to a selected membrane component; (ix) an inhibitor of
fatty acid synthesis;
(x) an inhibitor of cholesterol synthesis; and (xi) any combination of the
membrane penetration
enhancing agents recited in (i)-(x); (h) a modulatory agent of epithelial
junction physiology; (i) a
vasodilator agent; (j) a selective transport-enhancing agent; and (k) a
stabilizing delivery vehicle,
carrier, mucoadhesive, support or complex-forming species with which the
compound is
effectively combined, associated, contained, encapsulated or bound resulting
in stabilization of
the compound for enhanced mucosal delivery, wherein the formulation of the
compound with the
transmucosal delivery-enhancing agents provides for increased bioavailability
of the compound
in a blood plasma of a subject.
In general a method of making a pharmaceutical composition can include
combining an
adrenergic receptor interacter with a pharmaceutically active component and
forming a
4
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
pharmaceutical composition including the adrenergic receptor interacter and
the
pharmaceutically active component.
The pharmaceutical composition can be a chewable or gelatin based dosage form,
spray,
gum, gel, cream, tablet, liquid or film.
In general, a pharmaceutical composition can be dispensed from a device. The
device
can dispense a pharmaceutical composition in a predetermined dose as a
chewable or gelatin
based dosage form, spray, gum, gel, cream, tablet, liquid or film. A device
can include a housing
that holds an amount of a pharmaceutical composition, including a polymeric
matrix; a
pharmaceutically active component in the polymeric matrix; and an adrenergic
receptor
interacter and an opening that dispenses a predetermined amount of the
pharmaceutical
composition. The device can also dispense a pharmaceutical composition
including a
permeation enhancer including a phenylpropanoid and/or a phytoextract.
In certain embodiments, a pharmaceutical composition can include a polymeric
matrix, a
pharmaceutically active component in the polymeric matrix; and a permeation
enhancer
including a phenylpropanoid and/or a phytoextract.
In certain embodiments, a pharmaceutical composition can include a polymeric
matrix;
a pharmaceutically active component in the polymeric matrix; and an interacter
that creates
increased blood flow or enables a flushing of the tissue to modify
transmucosal uptake of the
pharmaceutically active component.
In certain embodiments, a pharmaceutical composition can include a polymeric
matrix;
a pharmaceutically active component in the polymeric matrix; and an interacter
that has a positive
or negative heat of solution which are used as aids to modify (increase or
decrease) transmucosal
uptake.
In other embodiments, a pharmaceutical composition includes a polymeric
matrix, a
pharmaceutically active component in the polymeric matrix, and an interacter,
the composition
contained in a multilayer film having at least one side where the edges are
coterminous.
In general, a method of treating a medical condition can include administering
an
effective amount of pharmaceutical composition, comprising a polymeric matrix,
a
pharmaceutically active component in the polymeric matrix, and an adrenergic
receptor
interacter. The pharmaceutically active component can be epinephrine,
diazepam, or alprazolam.
A medical condition can include a symptom of epilepsy, such as a seizure. In
certain
5
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
embodiments, the method includes administering the pharmaceutical composition
including
diazepam during an ictal state. In certain embodiments, the method includes
administering the
pharmaceutical composition including diazepam during a periictal state. In
certain embodiments,
the method includes administering the pharmaceutical composition including
diazepam during an
interictal state. The medical condition can also include anxiety, drug
withdrawal, parasomnia,
alcohol withdrawal, muscle spasms, or seizure disorder. The medical condition
can include
treatment as a sedative. The medical condition can also include sedation for a
medical
procedure.
Other aspects, embodiments, and features will be apparent from the following
description, the drawings, and the claims.
BRIEF DESCRIPTION OF THE FIGURES
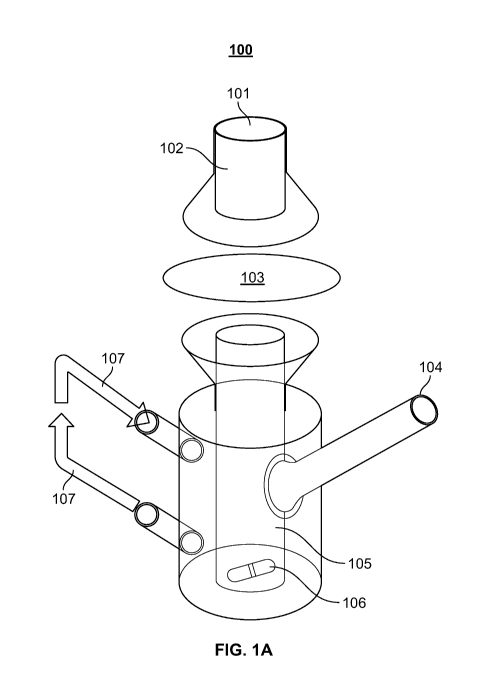
Referring to Figure 1A, a Franz diffusion cell 100 includes a donor compound
101, a
donor chamber 102, a membrane 103, sampling port 104, receptor chamber 105,
stir bar 106, and
a heater/circulator 107.
Referring to Figure 1B, a pharmaceutical composition is a film 100 comprising
a
polymeric matrix 200, the pharmaceutically active component 300 being
contained in the
polymeric matrix. The film can include a permeation enhancer 400.
Referring to Figures 2A and 2B, the graphs show the permeation of an active
material
from a composition. Referring to Figure 2A, this graph shows average amount of
active
material permeated vs. time, with 8.00 mg/mL epinephrine bitartrate and 4.4
mg/mL epinephrine
base solubilized.
Referring to Figure 2B, this graph shows average flux vs. time, with 8.00
mg/mL
bitartrate and 4.4 mg/mL epinephrine base solubilized.
Referring to Figure 3, this graph shows ex-vivo permeation of epinephrine
bitartrate as a
function of concentration. Referring to Figure 4, this graph shows permeation
of epinephrine
bitartrate as a function of solution pH. Referring to Figure 5, this graph
shows the influence of
enhancers on permeation of epinephrine, indicated as amount permeated as a
function of time.
Referring to Figure 6A and 6B, these graphs show the release of epinephrine on
polymer
platforms (6A) and the effect of enhancers on its release (6B), indicated as
amount permeated (in
6
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
ug) vs. time. Referring to Figure 7, this graph shows a pharmacokinetic model
in the male
Yucatan, miniature swine. The study compares a 0.3 mg Epipen, a 0.12 mg
Epinephrine IV and
a placebo film.
Referring to Figure 8, this graph shows the impact of no enhancer on the
concentration
profiles of a 40 mg Epinephrine film vs 0.3 mg Epipen. Referring to Figure 9,
this graph shows
the impact of Enhancer A (Labrasol) on the concentration profiles of a 40 mg
Epinephrine film
vs 0.3 mg Epipen, Referring to Figure 10, this graph shows the impact of
Enhancer L (clove oil)
on the concentration profiles of two 40 mg Epinephrine films (10-1-1) and (11-
1-1) vs. a 0.3 mg
Epipen.
Referring to Figure 11, this graph shows the impact of Enhancer L (clove oil)
and film
dimension (10-1-1 thinner, bigger film and 11-1-1 thicker, smaller film) on
the concentration
profiles of 40 mg Epinephrine films vs. a 0.3 mg Epipen.
Referring to Figure 12, this graph shows the concentration profiles for
varying doses of
Epinephrine films in a constant matrix for Enhancer L (clove oil) vs. a 0.3 mg
Epipen. Referring
to Figure 13, this graph shows the concentration profiles for varying doses of
Epinephrine films
in a constant matrix for Enhancer L (clove oil) vs. a 0.3 mg Epipen.
Referring to Figure 14, this graph shows the concentration profiles for
varying doses of
Epinephrine films in a constant matrix for Enhancer A (Labrasol) vs. a 0.3 mg
Epipen.
Referring to Figure 15, this graph shows the influence of enhancers on
permeation of
diazepam, indicated as amount permeated as a function of time.
Referring to Figure 16, this graph shows the average flux as a function of
time (diazepam
+ enhancers).
Referring to Figure 17, this graph shows the impact of Farnesol and Farnesol
in
combination with Linoleic Acid on plasma concentration profiles of 40 mg
Epinephrine Films vs.
a 0.3 mg Epipen.
Referring to Figure 18, this graph shows the impact of Farnesol on plasma
concentration
profiles of 40 mg Epinephrine Films vs. a 0.3 mg Epipen.
Referring to Figure 19, this graph shows the impact of Farnesol in combination
with
Linoleic Acid on plasma concentration profiles of 40 mg Epinephrine Films vs.
a 0.3 mg Epipen.
7
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Referring to Figure 20, this graph shows the impact of Farnesol and Farnesol
in
combination with Linoleic Acid on plasma concentration profiles of 40 mg
Epinephrine Films vs.
a 0.3 mg Epipen.
Referring to Figure 21, this graph shows the impact of Enhancer A (Labrasol)
in
combination with Enhancer L (clove oil) on the concentration profiles of a 40
mg Epinephrine
films (also shown in Fig. 22), in logarithmic view.
Referring to Figure 22, this graph shows the impact of Enhancer A (Labrasol)
in
combination with Enhancer L (clove oil) on the concentration profiles of a 40
mg Epinephrine
films vs. the average data collected from 0.3 mg Epipens.
Referring to Figure 23, this graph shows the impact of Enhancer A (Labrasol)
in
combination with Enhancer L (clove oil) on the concentration profiles of a 40
mg Epinephrine
films, shown as separate animal subjects.
Referring to Figure 24A, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam oral
disintegrating tablets
(ODT).
Referring to Figure 24B, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam
pharmaceutical composition
films.
Referring to Figure 24C, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam
pharmaceutical composition
films.
Referring to Figure 25A, this graph shows mean alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam ODTs and
alprazolam
pharmaceutical composition films.
Referring to Figure 25B, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration.
Referring to Figure 25C, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration.
Referring to Figure 26A, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam ODTs.
8
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Referring to Figure 26B, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam
pharmaceutical composition
films.
Referring to Figure 26C, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam
pharmaceutical composition
films.
Referring to Figure 27A, this graph shows mean alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam ODTs and
pharmaceutical
composition films.
Referring to Figure 27B, this graph shows mean alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam ODTs and
pharmaceutical
composition films.
Referring to Figure 27C, this graph shows the alprazolam plasma concentration
as a
function of time following sublingual administration of alprazolam ODTs and
pharmaceutical
composition films.
DETAILED DESCRIPTION
Mucosal surfaces, such as the oral mucosa, are a convenient route for
delivering drugs to
the body due to the fact that they are highly vascularized and permeable,
providing increased
bioavailability and rapid onset of action because it does not pass through the
digestive system
and thereby avoids first pass metabolism. In particular, the buccal and
sublingual tissues offer
advantageous sites for drug delivery because they are highly permeable regions
of the oral
mucosa, allowing drugs diffusing from the oral mucosa to have direct access to
systemic
circulation. This also offers increased convenience and therefore increased
compliance in
patients. For certain drugs, or pharmaceutically active components, a
permeation enhancer can
help to overcome the mucosal barrier and improve permeability. Permeation
enhancers
reversibly modulate the penetrability of the barrier layer in favor of drug
absorption. Permeation
enhancers facilitate transport of molecules through the epithelium. Absorption
profiles and their
rates can be controlled and modulated by a variety of parameters, such as but
not limited to film
size, drug loading, enhancer type/loading, polymer matrix release rate and
mucosal residence
time.
9
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
A pharmaceutical composition can be designed to deliver a pharmaceutically
active
component in a deliberate and tailored way. However, solubility and
permeability of the
pharmaceutically active component in vivo, in particular, in the mouth of a
subject, can vary
tremendously. A particular class of permeation enhancer can improve the uptake
and
bioavailability of the pharmaceutically active component in vivo. In
particular, when delivered
to the mouth via a film, the permeation enhancer can improve the permeability
of the
pharmaceutically active component through the mucosa and into the blood stream
of the subject.
The permeation enhancer can improve absorption rate and amount of the
pharmaceutically active
component by more than 5%, more than 10%, more than 20%, more than 30%, more
than 40%,
more than 50%, more than 60%, more than 70%, more than 80%, more than 90%,
more than
100%, more than 150%, about 200% or more, or less than 200%, less than 150%,
less than
100%, less than 90%, less than 80%, less than 70%, less than 60%, less than
50%, less than 40%,
less than 30%, less than 20%, less than 10%, or less than 5%, or a combination
of these ranges,
depending on the other components in the composition.
In certain embodiments, a pharmaceutical composition has a suitable nontoxic,
nonionic
alkyl glycoside having a hydrophobic alkyl group joined by a linkage to a
hydrophilic saccharide
in combination with a mucosal delivery-enhancing agent selected from: (a) an
aggregation
inhibitory agent; (b) a charge-modifying agent; (c) a pH control agent; (d) a
degradative enzyme
inhibitory agent; (e) a mucolytic or mucus clearing agent; (f) a ciliostatic
agent; (g) a membrane
penetration-enhancing agent selected from: (i) a surfactant; (ii) a bile salt;
(ii) a phospholipid
additive, mixed micelle, liposome, or carrier; (iii) an alcohol; (iv) an
enamine; (v) an NO donor
compound; (vi) a long chain amphipathic molecule; (vii) a small hydrophobic
penetration
enhancer; (viii) sodium or a salicylic acid derivative; (ix) a glycerol ester
of acetoacetic acid; (x)
a cyclodextrin or beta-cyclodextrin derivative; (xi) a medium-chain fatty
acid; (xii) a chelating
agent; (xiii) an amino acid or salt thereof; (xiv) an N-acetylamino acid or
salt thereof; (xv) an
enzyme degradative to a selected membrane component; (ix) an inhibitor of
fatty acid synthesis;
(x) an inhibitor of cholesterol synthesis; and (xi) any combination of the
membrane penetration
enhancing agents recited in (i)-(x); (h) a modulatory agent of epithelial
junction physiology; (i) a
vasodilator agent; (j) a selective transport-enhancing agent; and (k) a
stabilizing delivery vehicle,
carrier, mucoadhesive, support or complex-forming species with which the
compound is
effectively combined, associated, contained, encapsulated or bound resulting
in stabilization of
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
the compound for enhanced transmucosal delivery, wherein the formulation of
the compound
with the transmucosal delivery-enhancing agents provides for increased
bioavailability of the
compound in blood plasma of a subject. Penetration enhancers have been
described in J.
Nicolazzo, et al., I of Controlled Disease, 105 (2005) 1-15, which is
incorporated by reference
herein. There are many reasons why the oral mucosa might be an attractive site
for the delivery
of therapeutic agents into the systemic circulation. Due to the direct
drainage of blood from the
buccal epithelium into the internal jugular vein first-pass metabolism in the
liver and intestine
may be avoided. First-pass effect can be a major reason for the poor
bioavailability of some
compounds when administered orally. Additionally, the mucosa lining the oral
cavity is easily
accessible, which ensures that a dosage form can be applied to the required
site and can be
removed easily in the case of an emergency. However, like the skin, the buccal
mucosa acts as a
barrier to the absorption of xenobiotics, which can hinder the permeation of
compounds across
this tissue. Consequently, the identification of safe and effective
penetration enhancers has
become a major goal in the quest to improve oral mucosal drug delivery.
Chemical penetration enhancers are substances that control the permeation rate
of a
coadministered drug through a biological membrane. While extensive research
has focused on
obtaining an improved understanding of how penetration enhancers might alter
intestinal and
transdermal permeability, far less is known about the mechanisms involved in
buccal and
sublingual penetration enhancement.
The buccal mucosa delineates the inside lining of the cheek as well as the
area between
the gums and upper and lower lips and it has an average surface area of 100
cm2. The surface of
the buccal mucosa consists of a stratified squamous epithelium which is
separated from the
underlying connective tissue (lamina propria and submucosa) by an undulating
basement
membrane (a continuous layer of extracellular material approximately 1-2 [tm
in thickness). This
stratified squamous epithelium consists of differentiating layers of cells
which change in size,
shape, and content as they travel from the basal region to the superficial
region, where the cells
are shed. There are approximately 40-50 cell layers, resulting in a buccal
mucosa which is 500-
600 [tm thick.
Structurally the sublingual mucosa is comparable to the buccal mucosa but the
thickness
of this epithelium is 100-200 [tm. This membrane is also non-keratinised and
being relatively
thinner has been demonstrated to be more permeable than buccal mucosa. Blood
flow to the
11
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
sublingual mucosal is slower compared with the buccal mucosa and is of the
order of 1.0 ml/
The permeability of the buccal mucosa is greater than that of the skin, but
less than that
of the intestine. The differences in permeability are the result of structural
differences between
each of the tissues. The absence of organized lipid lamellae in the
intercellular spaces of the
buccal mucosa results in greater permeability of exogenous compounds, compared
to keratinized
epithelia of the skin; while the increased thickness and lack of tight
junctions results in the
buccal mucosa being less permeable than intestinal tissue.
The primary barrier properties of the buccal mucosa have been attributed to
the upper
one-third to one-quarter of the buccal epithelium. Researchers have learned
that beyond the
surface epithelium, the permeability barrier of nonkeratinized oral mucosa
could also be
attributed to contents extruded from the membrane-coating granules into the
epithelial
intercellular spaces.
The intercellular lipids of the nonkeratinized regions of the oral cavity are
of a more polar
nature than the lipids of the epidermis, palate, and gingiva, and this
difference in the chemical
nature of the lipids may contribute to the differences in permeability
observed between these
tissues. Consequently, it appears that it is not only the greater degree of
intercellular lipid
packing in the stratum corneum of keratinized epithelia that creates a more
effective barrier, but
also the chemical nature of the lipids present within that barrier.
The existence of hydrophilic and lipophilic regions in the oral mucosa has led
researchers
to postulate the existence of two routes of drug transport through the buccal
mucosa¨
paracellular (between the cells) and transcellular (across the cells).
Since drug delivery through the buccal mucosa is limited by the barrier nature
of the
epithelium and the area available for absorption, various enhancement
strategies are required in
order to deliver therapeutically relevant amounts of drug to the systemic
circulation. Various
methods, including the use of chemical penetration enhancers, prodrugs, and
physical methods
may be employed to overcome the barrier properties of the buccal mucosa.
A chemical penetration enhancer, or absorption promoter, is a substance added
to a
pharmaceutical formulation in order to increase the membrane permeation or
absorption rate of
the coadministered drug, without damaging the membrane and/or causing
toxicity. There have
been many studies investigating the effect of chemical penetration enhancers
on the delivery of
12
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
compounds across the skin, nasal mucosa, and intestine. In recent years, more
attention has been
given to the effect of these agents on the permeability of the buccal mucosa.
Since permeability
across the buccal mucosa is considered to be a passive diffusion process the
steady state flux
(Jss) should increase with increasing donor chamber concentration (CD)
according to Fick's first
law of diffusion.
Surfactants and bile salts have been shown to enhance the permeability of
various
compounds across the buccal mucosa, both in vitro and in vivo. The data
obtained from these
studies strongly suggest that the enhancement in permeability is due to an
effect of the
surfactants on the mucosal intercellular lipids.
Fatty acids have been shown to enhance the permeation of a number of drugs
through the
skin, and this has been shown by differential scanning calorimetry and Fourier
transform infrared
spectroscopy to be related to an increase in the fluidity of intercellular
lipids.
Additionally, pretreatment with ethanol has been shown to enhance the
permeability of
tritiated water and albumin across ventral tongue mucosa, and to enhance
caffeine permeability
across porcine buccal mucosa. There are also several reports of the enhancing
effect of Azone
on the permeability of compounds through oral mucosa. Further, chitosan, a
biocompatible and
biodegradable polymer, has been shown to enhance drug delivery through various
tissues,
including the intestine and nasal mucosa.
Oral transmucosal drug delivery (OTDD) is the administration of
pharmaceutically active
agents through the oral mucosa to achieve systemic effects. Permeation
pathways and predictive
models for OTDD are described, e.g. in M. Sattar, Oral transmucosal drug
delivery ¨ Current
status and future prospects, Intl Journal of Pharmaceutics, 47(2014) 498-506,
which is
incorporated by reference herein. OTDD continues to attract the attention of
academic and
industrial scientists. Despite limited characterization of the permeation
pathways in the oral
cavity compared with skin and nasal routes of delivery, recent advances in our
understanding of
the extent to which ionized molecules permeate the buccal epithelium, as well
as the emergence
of new analytical techniques to study the oral cavity, and the progressing
development of in
silico models predictive of buccal and sublingual permeation, prospects are
encouraging.
In order to deliver broader classes of drugs across the buccal mucosa,
reversible methods
of reducing the barrier potential of this tissue should be employed. This
requisite has fostered the
study of penetration enhancers that will safely alter the permeability
restrictions of the buccal
13
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
mucosa. It has been shown that buccal penetration can be improved by using
various classes of
transmucosal and transdermal penetration enhancers such as bile salts,
surfactants, fatty acids
and their derivatives, chelators, cyclodextrins and chitosan. Among these
chemicals used for the
drug permeation enhancement, bile salts are the most common.
In vitro studies on enhancing effect of bile salts on the buccal permeation of
compounds
is discussed in Sevda Senel, Drug permeation enhancement via buccal route:
possibilities and
limitations, Journal of Controlled Release 72 (2001) 133-144, which is
incorporated by
reference herein. That article also discusses recent studies on the effects of
buccal epithelial
permeability of dihydroxy bile salts, sodium glycodeoxycholate (SGDC) and
sodium
taurodeoxycholate (TDC) and tri-hydroxy bile salts, sodium glycocholate (GC)
and sodium
taurocholate (TC) at 100 mM concentration including permeability changes
correlated with the
histological effects. Fluorescein isothiocyanate (FITC), morphine sulfate were
each used as the
model compound.
Chitosan has also been shown to promote absorption of small polar molecules
and
peptide / protein drugs through nasal mucosa in animal models and human
volunteers. Other
studies have shown an enhancing effect on penetration of compounds across the
intestinal
mucosa and cultured Caco-2 cells.
The permeation enhancer can be a phytoextract. A phytoextract can be an
essential oil or
composition including essential oils extracted by distillation of the plant
material. In certain
circumstances, the phytoextract can include synthetic analogues of the
compounds extracted
from the plant material (i.e., compounds made by organic synthesis). The
phytoextract can
include a phenylpropanoid, for example, phenyl alanine, eugenol, eugenol
acetate, a cinnamic
acid, a cinnamic acid ester, a cinnamic aldehyde, a hydrocinnamic acid,
chavicol, or safrole, or a
combination thereof. The phytoextract can be an essential oil extract of a
clove plant, for
example, from the leaf, stem or flower bud of a clove plant. The clove plant
can be Syzygium
aromaticum. The phytoextract can include 20-95% eugenol, including 40-95%
eugenol,
including 60-95% eugenol, and for example, 80-95% eugenol. The extract can
also include 5%
to 15% eugenol acetate. The extract can also include caryophyllene. The
extract can also include
up to 2.1% a-humulen. Other volatile compounds included in lower
concentrations in clove
essential oil can be P-pinene, limonene, farnesol, benzaldehyde, 2-heptanone
or ethyl hexanoate.
Other permeation enhancers may be added to the composition to improve
absorption of the drug.
14
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Suitable permeation enhancers include natural or synthetic bile salts such as
sodium fusidate;
glycocholate or deoxycholate and their salts; fatty acids and derivatives such
as sodium laurate,
oleic acid, oleyl alcohol, monoolein, or palmitoylcarnitine; chelators such as
disodium EDTA,
sodium citrate and sodium laurylsulfate, azone, sodium cholate, sodium 5-
methoxysalicylate,
sorbitan laurate, glyceryl monolaurate, octoxynony1-9, laureth-9,
polysorbates, sterols, or
glycerides, such as caprylocaproyl polyoxylglycerides, e.g., Labrasol. The
permeation enhancer
can include phytoextract derivatives and/or monolignols. The permeation
enhancer can also be a
fungal extract.
Some natural products of plant origin have been known to have a vasodilatory
effect.
There are several mechanisms or modes by which plant-based products can evoke
vasodilation.
For review, see McNeill J.R. and Jurgens, T.M., Can. J. Physiol. Pharmacol.
84:803-821 (2006),
which is incorporated by reference herein. Specifically, vasorelaxant effects
of eugenol have
been reported in a number of animal studies. See, e.g., Lahlou, S., et al., J.
Cardiovasc.
Pharmacol. 43:250-57 (2004), Damiani, C.E.N., et al., Vascular Pharmacol.
40:59-66 (2003),
Nishijima, H., et al., Japanese J. Pharmacol. 79:327-334 (1998), and Hume
W.R., J. Dent Res.
62(9):1013-15 (1983), each of which is incorporated by reference herein.
Calcium channel
blockade was suggested to be responsible for vascular relaxation induced by a
plant essential oil,
or its main constituent, eugenol. See, Interaminense L.R.L. et al.,
Fundamental & Clin.
Pharmacol. 21: 497-506 (2007), which is incorporated by reference herein.
Fatty acids can be used as inactive ingredients in drug preparations or drug
vehicles.
Fatty acids can also be used as formulation ingredients due to their certain
functional effects and
their biocompatible nature. Fatty acid, both free and as part of complex
lipids, are major
metabolic fuel (storage and transport energy), essential components of all
membranes and gene
regulators. For review, see Rustan A.C. and Drevon, C.A., Fatty Acids:
Structures and
Properties, Encyclopedia of Life Sciences (2005), which is incorporated by
reference herein.
There are two families of essential fatty acids that are metabolized in the
human body: w-3 and
w-6 polyunsaturated fatty acids (PUFAs). If the first double bond is found
between the third and
the fourth carbon atom from the w carbon, they are called w-3 fatty acids. If
the first double bond
is between the sixth and seventh carbon atom, they are called w-6 fatty acids.
PUFAs are further
metabolized in the body by the addition of carbon atoms and by desaturation
(extraction of
hydrogen). Linoleic acid, which is a w-6 fatty acid, is metabolized to y-
linolenic acid, dihomo-y-
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
linolinic acid, arachidonic acid, adrenic acid, tetracosatetraenoic acid,
tetracosapentaenoic acid
and docosapentaenoic acid. a-linolenic acid, which is a w-3 fatty acid is
metabolized to
octadecatetraenoic acid, eicosatetraenoic acid, eicosapentaenoic acid (EPA),
docosapentaenoic
acid, tetracosapentaenoic acid, tetracosahexaenoic acid and docosahexaenoic
acid (DHA).
It has been reported that fatty acids, such as palmitic acid, oleic acid,
linoleic acid and
eicosapentaenoic acid, induced relaxation and hyperpolarization of porcine
coronary artery
smooth muscle cells via a mechanism involving activation of the Na+K+-APTase
pump and the
fatty acids with increasing degrees of cis-unsaturation had higher potencies.
See, Pomposiello,
S.I. et al., Hypertension 31:615-20 (1998), which is incorporated by reference
herein.
Interestingly, the pulmonary vascular response to arachidonic acid, a
metabolite of linoleic acid,
can be either vasoconstrictive or vasodilative, depending on the dose, animal
species, the mode
of arachidonic acid administration, and the tones of the pulmonary
circulation. For example,
arachidonic acid has been reported to cause cyclooxygenase-dependent and
¨independent
pulmonary vasodilation. See, Feddersen, C.O. et al., J. Appl. Physiol.
68(5):1799-808 (1990);
and see, Spannhake, E.W., et al., J .Appl. Physiol. 44:397-495 (1978) and
Wicks, T.C. et al.,
Circ. Res. 38:167-71 (1976), each of which is incorporated by reference
herein.
Many studies have reported effects of eicosapentaenoic acid (EPA) and
docosahexaenoic
acid (DHA) on vascular reactivity after being administered as ingestible
forms. Some studies
found that EPA-DHA or EPA alone suppressed the vasoconstrictive effect of
norepinephrine or
increased vasodilatory responses to acetylcholine in the forearm
microcirculation. See, Chin,
J.P.F, et al., Hypertension 21:22-8 (1993), and Tagawa, H. et al., J
Cardiovasc Pharmacol
33:633-40 (1999), each of which is incorporated by reference herein. Another
study found that
both EPA and DHA increased systemic arterial compliance and tended to reduce
pulse pressure
and total vascular resistance. See, Nestel, P. et al., Am J. Clin. Nutr.
76:326-30 (2002), which is
incorporated by reference herein. Meanwhile, a study found that DHA, but not
EPA, enhanced
vasodilator mechanisms and attenuates constrictor responses in forearm
microcirculation in
hyperlipidemic overweight men. See, Mori, T.A., et al., Circulation 102:1264-
69 (2000), which
is incorporated by reference herein. Another study found vasodilator effects
of DHA on the
rhythmic contractions of isolated human coronary arteries in vitro. See Wu, K.-
T. et al., Chinese
J. Physiol. 50(4):164-70 (2007), which is incorporated by reference herein.
16
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
The adrenergic receptors (or adrenoceptors) are a class of G protein-coupled
receptors
that are a target of catecholamines, especially norepinephrine (noradrenaline)
and epinephrine
(adrenaline). Epinephrine (adrenaline) interacts with both a- and P-
adrenoceptors, causing
vasoconstriction and vasodilation, respectively. Although a receptors are less
sensitive to
epinephrine, when activated, they override the vasodilation mediated by P-
adrenoceptors because
there are more peripheral al receptors than P-adrenoceptors. The result is
that high levels of
circulating epinephrine cause vasoconstriction. At lower levels of circulating
epinephrine, f3-
adrenoceptor stimulation dominates, producing vasodilation followed by
decrease of peripheral
vascular resistance. The al-adrenoreceptor is known for smooth muscle
contraction, mydriasis,
vasoconstriction in the skin, mucosa and abdominal vicera and sphincter
contraction of the
gastrointestinal (GI) tract and urinary bladder. The al-adrenergic receptors
are member of the Gq
protein-coupled receptor superfamily. Upon activation, a heterotrimeric G
protein, Gq, activates
phospholipase C (PLC). The mechanism of action involves interaction with
calcium channels
and changing the calcium content in a cell. For review, see Smith R. S. et
al., Journal of
Neurophysiology 102(2): 1103-14 (2009), which is incorporated by reference
herein. Many cells
possess these receptors.
al-adrenergic receptors can be a main receptor for fatty acids. For example,
saw palmetto
extract (SPE), widely used for the treatment of benign prostatic hyperplasia
(BPH), has been
reported to bind al -adrenergic, muscarinic and 1,4-dihydropyridine (1,4-DHP)
calcium channel
antagonist receptors. See, Abe M., et al., Biol. Pharm. Bull. 32(4) 646-650
(2009), and Suzuki
M. et al., Acta Pharmacologica Sinica 30:271-81 (2009), each of which is
incorporated by
reference herein. SPE includes a variety of fatty acids including lauric acid,
oleic acid, myristic
acid, palmitic acid and linoleic acid. Lauric acid and oleic acid can bind
noncompetitively to al -
adrenergic, muscarinic and 1,4-DHP calcium channel antagonist receptors.
In certain embodiments, a permeation enhancer can be an adrenergic receptor
interacter.
An adrenergic receptor interacter refers to a compound or substance that
modifies and/or
otherwise alters the action of an adrenergic receptor. For example, an
adrenergic receptor
interacter can prevent stimulation of the receptor by increasing, or
decreasing their ability to
bind. Such interacters can be provided in either short-acting or long-acting
forms. Certain short-
acting interacters can work quickly, but their effects last only a few hours.
Certain long-acting
interacters can take longer to work, but their effects can last longer. The
interacter can be
17
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
selected and/or designed based on, e.g., one or more of the desired delivery
and dose, active
pharmaceutical ingredient, permeation modifier, permeation enhancer, matrix,
and the condition
being treated. An adrenergic receptor interacter can be an adrenergic receptor
blocker. The
adrenergic receptor interacter can be a terpene (e.g. volatile unsaturated
hydrocarbons found in
the essential oils of plants, derived from units of isoprenes) or a C3-C22
alcohol or acid,
preferably a C7-C18 alcohol or acid. In certain embodiments, the adrenergic
receptor interacter
can include farnesol, linoleic acid, arachidonic acid, docosahexanoic acid,
eicosapentanoic acid,
and/or docosapentanoic acid. The acid can be a carboxylic acid, phosphoric
acid, sulfuric acid,
hydroxamic acid, or derivatives thereof. The derivative can be an ester or
amide. For example,
the adrenergic receptor interacter can be a fatty acid or fatty alcohol.
The C3-C22 alcohol or acid can be an alcohol or acid having a straight C3-C22
hydrocarbon chain, for example a C3-C22 hydrocarbon chain optionally
containing at least one
double bond, at least one triple bond, or at least one double bond and one
triple bond; said
hydrocarbon chain being optionally substituted with C1-4 alkyl, C2-4 alkenyl,
C2-4 alkynyl, C1-4
alkoxy, hydroxyl, halo, amino, nitro, cyano, C3.5 cycloalkyl, 3-5 membered
heterocycloalkyl,
monocyclic aryl, 5-6 membered heteroaryl, C1_4 alkylcarbonyloxy, C1-4
alkyloxycarbonyl, C1-4
alkylcarbonyl, or formyl; and further being optionally interrupted by -0-, -
N(Ra)-, -N(Ra)-C(0)-
0-, -0-C(0)-N(10-, -N(Ra)-C(0)-N(Rb)-, or -0-C(0)-0-. Each of le and Rb,
independently, is
hydrogen, alkyl, alkenyl, alkynyl, alkoxy, hydroxylalkyl, hydroxyl, or
haloalkyl.
Fatty acids with a higher degree of unsaturation are effective candidates to
enhance the
permeation of drugs. Unsaturated fatty acids showed higher enhancement than
saturated fatty
acids, and the enhancement increased with the number of double bonds. See, A.
Mittal, et al.
Status of Fatty Acids as Skin Penetration Enhancers ¨ A Review, Current Drug
Delivery, 2009,
6, pp. 274-279, which is incorporated by reference herein. Position of double
bond also affects
the enhancing activity of fatty acids. Differences in the physicochemical
properties of fatty acid
which originate from differences in the double bond position most likely
determine the efficacy
of these compounds as skin penetration enhancers. Skin distribution increases
as the position of
the double bond is shifted towards the hydrophilic end. It has also been
reported that fatty acid
which has a double bond at an even number position more rapidly effects the
perturbation of the
structure of both the stratum corneum and the dermis than a fatty acid which
has double bond at
an odd number position. Cis-unsaturation in the chain can tend to increase
activity.
18
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
An adrenergic receptor interacter can be a terpene. Hypotensive activity of
terpenes in
essential oils has been reported. See, Menezes I.A. et al., Z. Naturforsch.
65c:652-66 (2010),
which is incorporated by reference herein. In certain embodiments, the
permeation enhancer can
be a sesquiterpene. Sesquiterpenes are a class of terpenes that consist of
three isoprene units and
have the empirical formula C15H24. Like monoterpenes, sesquiterpenes may be
acyclic or contain
rings, including many unique combinations. Biochemical modifications such as
oxidation or
rearrangement produce the related sesquiterpenoids.
An adrenergic receptor interacter can be an unsaturated fatty acid such as
linoleic acid.
In certain embodiments, the permeation enhancer can be farnesol. Farnesol is a
15-carbon
organic compound which is an acyclic sesquiterpene alcohol, which is a natural
dephosphorylated form of farnesyl pyrophosphate. Under standard conditions, it
is a colorless
liquid. It is hydrophobic, and thus insoluble in water, but miscible with
oils. Farnesol can be
extracted from oils of plants such as citronella, neroli, cyclamen, and
tuberose. It is an
intermediate step in the biological synthesis of cholesterol from mevalonic
acid in vertebrates. It
has a delicate floral or weak citrus-lime odor and is used in perfumes and
flavors. It has been
reported that farnesol selectively kills acute myeloid leukemia blasts and
leukemic cell lines in
preference to primary hemopoietic cells. See, Rioj a A. et al., FEBS Lett 467
(2-3): 291-5 (2000),
which is incorporated by reference herein. Vasoactive properties of farnesyl
analogues have been
reported. See, Roullet, J.-B., et al., J. Clin. Invest., 1996, 97:2384-2390,
which is incorporated by
reference herein. Both Farnesol and N-acetyl-S-trans, trans-farnesyl-L-
cysteine (AFC), a
synthetic mimic of the carboxyl terminus of farnesylated proteins inhibited
vasoconstriction in
rat aortic rings.
In certain embodiments, an interacter can be an aporphine alkaloid. For
example, an
interacter can be a dicentrine.
In general, an interacter can also be a vasodilator or a therapeutic
vasodilator.
Vasodilators are drugs that open or widen blood vessels. They are typically
used to treat
hypertension, heart failure and angina, but can be used to treat other
conditions as well, including
glaucoma for example. Some vasodilators that act primarily on resistance
vessels (arterial
dilators) are used for hypertension, and heart failure, and angina; however,
reflex cardiac
stimulation makes some arterial dilators unsuitable for angina. Venous
dilators are very effective
for angina, and sometimes used for heart failure, but are not used as primary
therapy for
19
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
hypertension. Vasodilator drugs can be mixed (or balanced) vasodilators in
that they dilate both
arteries and veins and therefore can have wide application in hypertension,
heart failure and
angina. Some vasodilators, because of their mechanism of action, also have
other important
actions that can in some cases enhance their therapeutic utility or provide
some additional
therapeutic benefit. For example, some calcium channel blockers not only
dilate blood vessels,
but also depress cardiac mechanical and electrical function, which can enhance
their
antihypertensive actions and confer additional therapeutic benefit such as
blocking arrhythmias.
Vasodilator drugs can be classified based on their site of action (arterial
versus venous) or
by mechanism of action. Some drugs primarily dilate resistance vessels
(arterial dilators; e.g.,
hydralazine), while others primarily affect venous capacitance vessels (venous
dilators; e.g.,
nitroglycerine). Many vasodilator drugs have mixed arterial and venous dilator
properties (mixed
dilators; e.g., alpha-adrenoceptor antagonists, angiotensin converting enzyme
inhibitors), such as
phentolamine.
It is more common, however, to classify vasodilator drugs based on their
primary
mechanism of action. The figure to the right depicts important mechanistic
classes of vasodilator
drugs. These classes of drugs, as well as other classes that produce
vasodilation, include: alpha-
adrenoceptor antagonists (alpha-blockers); Angiotensin converting enzyme (ACE)
inhibitors;
Angiotensin receptor blockers (ARBs); beta2-adrenoceptor agonists (02-
agonists); calcium-
channel blockers (CCBs); centrally acting sympatholytics; direct acting
vasodilators; endothelin
receptor antagonists; ganglionic blockers; nitrodilators; phosphodiesterase
inhibitors; potassium-
channel openers; renin inhibitors.
In general, the active or inactive components or ingredients can be substances
or
compounds that create an increased blood flow or flushing of the tissue to
enable a modification
or difference (increase or decrease) in transmucosal uptake of the API(s),
and/or have a positive
or negative heat of solution which are used as aids to modify (increase or
decrease) transmucosal
uptake.
Sequence of Permeation Enhancer(s) and Active Pharmaceutical Ingredient(s)
The arrangement, order, or sequence of penetration enhancer(s) and active
pharmaceutical ingredient(s)(API(s)) delivered to the desired mucosal surface
can vary in order
to deliver a desired pharmacokinetic profile. For example, one can apply the
permeation
enhancer(s) first by a film, by swab, spray, gel, rinse or by a first layer of
a film then apply the
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
API(s) by single film, by swab, or by a second layer of a film. The sequence
can be reversed or
modified, for example, by applying the API(s) first by film, by swab, or by a
first layer of a film,
and then applying the permeation enhancer(s) by a film, by swab, spray, gel,
rinse or by a second
layer of a film. In another embodiment, one may apply a permeation enhancer(s)
by a film, and a
drug by a different film. For example, the permeation enhancer(s) film
positioned under a film
containing the API(s), or the film containing the API(s) positioned under a
film containing the
permeation enhancer(s), depending on the desired pharmacokinetic profile.
For example, the penetration enhancer(s) can be used as a pretreatment alone
or in
combination with at least one API to precondition the mucosa for further
absorption of the
API(s). The treatment can be followed by another treatment with neat
penetration enhancer(s) to
follow the at least one API mucosal application. The pretreatment can be
applied as a separate
treatment (film, gel, solution, swab etc.) or as a layer within a multilayered
film construction of
one or more layers. Similarly, the pretreatment may be contained within a
distinct domain of a
single film, designed to dissolve and release to the mucosa prior to release
of the secondary
domains with or without penetration enhancer(s) or API(s). The active
ingredient may then be
delivered from a second treatment, alone or in combination with additional
penetration
enhancer(s). There may also be a tertiary treatment or domain that delivers
additional
penetration enhancer(s) and/or at least one API(s) or prodrug(s), either at a
different ratio relative
to each other or relative to the overall loading of the other treatments. This
allows a custom
pharmacokinetic profile to be obtained. In this way, the product may have
single or multiple
domains, with penetration enhancer(s) and API(s) that can vary in mucosal
application order,
composition, concentration, or overall loading that leads to the desired
absorption amounts
and/or rates that achieve the intended pharmacokinetic profile and/or
pharmacodynamic effect.
The film format can be oriented such that no distinct sides, or such that the
film has at
least one side of a multiple layer film where the edges are co-terminus
(having or meeting at a
shared border or limit).
The pharmaceutical composition can be a chewable or gelatin based dosage form,
spray,
gum, gel, cream, tablet, liquid or film. The composition can include textures,
for example, at the
surface, such as microneedles or micro-protrusions. Recently, the use of
micron-scale needles in
increasing skin permeability has been shown to significantly increase
transdermal delivery,
including and especially for macromolecules. Most drug delivery studies have
emphasized solid
21
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
microneedles, which have been shown to increase skin permeability to a broad
range of
molecules and nanoparticles in vitro. In vivo studies have demonstrated
delivery of
oligonucleotides, reduction of blood glucose level by insulin, and induction
of immune responses
from protein and DNA vaccines. For such studies, needle arrays have been used
to pierce holes
.. into skin to increase transport by diffusion or iontophoresis or as drug
carriers that release drug
into the skin from a microneedle surface coating. Hollow microneedles have
also been developed
and shown to microinject insulin to diabetic rats. To address practical
applications of
microneedles, the ratio of microneedle fracture force to skin insertion force
(i.e. margin of
safety) was found to be optimal for needles with small tip radius and large
wall thickness.
Microneedles inserted into the skin of human subjects were reported as
painless. Together, these
results suggest that microneedles represent a promising technology to deliver
therapeutic
compounds into the skin for a range of possible applications. Using the tools
of the
microelectronics industry, microneedles have been fabricated with a range of
sizes, shapes and
materials. Microneedles can be, for example, polymeric, microscopic needles
that deliver
.. encapsulated drugs in a minimally invasive manner, but other suitable
materials can be used.
Applicants have found that microneedles could be used to enhance the delivery
of drugs
through the oral mucosa, particularly with the claimed compositions. The
microneedles create
micron sized pores in the oral mucosa which can enhance the delivery of drugs
across the
mucosa. Solid, hollow, or dissolving microneedles can be fabricated out of
suitable materials
including, but not limited to, metal, polymer, glass and ceramics. The
microfabrication process
can include photolithography, silicon etching, laser cutting, metal
electroplating, metal electro
polishing and molding. Microneedles could be solid which is used to pretreat
the tissue and are
removed before applying the film. The drug loaded polymer film described in
this application
can be used as the matrix material of the microneedles itself. These films can
have microneedles
or micro protrusions fabricated on their surface which will dissolve after
forming microchannels
in the mucosa through which drugs can permeate.
The term "film" can include films and sheets, in any shape, including
rectangular, square,
or other desired shape. A film can be any desired thickness and size. In
preferred embodiments,
a film can have a thickness and size such that it can be administered to a
user, for example,
placed into the oral cavity of the user. A film can have a relatively thin
thickness of from about
0.0025 mm to about 0.250 mm, or a film can have a somewhat thicker thickness
of from about
22
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
0.250 mm to about 1.0 mm. For some films, the thickness may be even larger,
i.e., greater than
about 1.0 mm or thinner, i.e., less than about 0.0025 mm. A film can be a
single layer or a film
can be multi-layered, including laminated or multiple cast films. A permeation
enhancer and
pharmaceutically active component can be combined in a single layer, each
contained in separate
layers, or can each be otherwise contained in discrete regions of the same
dosage form. In
certain embodiments, the pharmaceutically active component contained in the
polymeric matrix
can be dispersed in the matrix. In certain embodiments, the permeation
enhancer being
contained in the polymeric matrix can be dispersed in the matrix.
Oral dissolving films can fall into three main classes: fast dissolving,
moderate dissolving
and slow dissolving. Oral dissolving films can also include a combination of
any of the above
categories. Fast dissolving films can dissolve in about 1 second to about 30
seconds in the
mouth, including more than 1 second, more than 5 seconds, more than 10
seconds, more than 20
seconds, and less than 30 seconds. Moderate dissolving films can dissolve in
about 1 to about 30
minutes in the mouth including more than 1 minute, more than 5 minutes, more
than 10 minutes,
.. more than 20 minutes or less than 30 minutes, and slow dissolving films can
dissolve in more
than 30 minutes in the mouth. As a general trend, fast dissolving films can
include (or consist
of) low molecular weight hydrophilic polymers (e.g., polymers having a
molecular weight
between about 1,000 to 9,000 daltons, or polymers having a molecular weight up
to 200,000
daltons). In contrast, slow dissolving films generally include high molecular
weight polymers
(e.g., having a molecular weight in millions). Moderate dissolving films can
tend to fall in
between the fast and slow dissolving films.
It can be preferable to use films that are moderate dissolving films. Moderate
dissolving
films can dissolve rather quickly, but also have a good level of mucoadhesion.
Moderate
dissolving films can also be flexible, quickly wettable, and are typically non-
irritating to the user.
.. Such moderate dissolving films can provide a quick enough dissolution rate,
most desirably
between about 1 minute and about 20 minutes, while providing an acceptable
mucoadhesion
level such that the film is not easily removable once it is placed in the oral
cavity of the user.
This can ensure delivery of a pharmaceutically active component to a user.
A pharmaceutical composition can include one or more pharmaceutically active
components. The pharmaceutically active component can be a single
pharmaceutical component
or a combination of pharmaceutical components. The pharmaceutically active
component can be
23
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
an anti-inflammatory analgesic agent, a steroidal anti-inflammatory agent, an
antihistamine, a
local anesthetic, a bactericide, a disinfectant, a vasoconstrictor, a
hemostatic, a chemotherapeutic
drug, an antibiotic, a keratolytic, a cauterizing agent, an antiviral drug, an
antirheumatic, an
antihypertensive, a bronchodilator, an anticholinergic, an anti-anxiety drug,
an antiemetic
compound, a hormone, a peptide, a protein or a vaccine. The pharmaceutically
active component
can be the compound, pharmaceutically acceptable salt of a drug, a prodrug, a
derivative, a drug
complex or analog of a drug. The term "prodrug" refers to a biologically
inactive compound that
can be metabolized in the body to produce a biologically active drug.
In some embodiments, more than one pharmaceutically active component may be
included in the film. The pharmaceutically active components can be ace-
inhibitors, anti-anginal
drugs, anti -arrhythmi a s, anti-asthmatics, anti -chol esterol emi c s,
analgesics, anesthetics, anti -
convul s ants, anti-depressants, anti-diabetic agents, anti-diarrhea
preparations, antidotes, anti-
histamines, anti-hypertensive drugs, anti-inflammatory agents, anti-lipid
agents, anti-manics,
anti-nauseants, anti-stroke agents, anti-thyroid preparations, amphetamines,
anti-tumor drugs,
anti-viral agents, acne drugs, alkaloids, amino acid preparations, anti-
tussives, anti-uricemic
drugs, anti-viral drugs, anabolic preparations, systemic and non-systemic anti-
infective agents,
anti-neoplastics, anti-parkinsonian agents, anti-rheumatic agents, appetite
stimulants, blood
modifiers, bone metabolism regulators, cardiovascular agents, central nervous
system stimulates,
cholinesterase inhibitors, contraceptives, decongestants, dietary supplements,
dopamine receptor
agonists, endometriosis management agents, enzymes, erectile dysfunction
therapies, fertility
agents, gastrointestinal agents, homeopathic remedies, hormones, hypercalcemia
and
hypocalcemia management agents, immunomodulators, immunosuppressives, migraine
preparations, motion sickness treatments, muscle relaxants, obesity management
agents,
osteoporosis preparations, oxytocics,
parasympatholytics, parasympathomimetics,
prostaglandins, psychotherapeutic agents, respiratory agents, sedatives,
smoking cessation aids,
sympatholytics, tremor preparations, urinary tract agents, vasodilators,
laxatives, antacids, ion
exchange resins, anti-pyretics, appetite suppressants, expectorants, anti-
anxiety agents, anti-ulcer
agents, anti-inflammatory substances, coronary dilators, cerebral dilators,
peripheral
vasodilators, psycho-tropics, stimulants, anti-hypertensive drugs,
vasoconstrictors, migraine
treatments, antibiotics, tranquilizers, anti-psychotics, anti-tumor drugs,
anti-coagulants, anti-
thrombotic drugs, hypnotics, anti-emetics, anti-nauseants, anti-convulsants,
neuromuscular
24
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
drugs, hyper- and hypo-glycemic agents, thyroid and anti-thyroid preparations,
diuretics, anti-
spasmodics, uterine relaxants, anti-obesity drugs, erythropoietic drugs, anti-
asthmatics, cough
suppressants, mucolytics, DNA and genetic modifying drugs, diagnostic agents,
imaging agents,
dyes, or tracers, and combinations thereof
For example, the pharmaceutically active component can be buprenorphine,
naloxone,
acetaminophen, riluzole, clobazam, Rizatriptan, propofol, methyl salicylate,
monoglycol
salicylate, aspirin, mefenamic acid, flufenamic acid, indomethacin,
diclofenac, alclofenac,
diclofenac sodium, ibuprofen, ketoprofen, naproxen, pranoprofen, fenoprofen,
sulindac,
fenclofenac, clidanac, flurbiprofen, fentiazac, bufexamac, piroxicam,
phenylbutazone,
oxyphenbutazone, clofezone, pentazocine, mepirizole, tiaramide hydrochloride,
hydrocortisone,
predonisolone, dexarnethasone, triamcinolone acetonide, fluocinolone
acetonide, hydrocortisone
acetate, predonisolone acetate, methylpredonisolone, dexamethasone acetate,
betamethasone,
betamethasone valerate, flumetasone, fluoromethol one, beclomethasone
diproprionate,
fluocinonide, edaravone, lurasidone, esomeprazole, lumateperone, naldmedine,
doxylamine,
pyridoxine, diphenhydramine hydrochloride, diphenhydramine salicylate,
diphenhydramine,
chlorpheniramine hydrochloride, chlorpheniramine maleate isothipendyl
hydrochloride,
tripelennamine hydrochloride, promethazine hydrochloride, methdilazine
hydrochloride
dibucaine hydrochloride, dibucaine, lidocaine hydrochloride, lidocaine,
benzocaine, p-
buthylaminob enzoi c acid 2-(di e-ethyl amino) ethyl ester hydrochloride,
procaine hydrochloride,
tetracaine, tetracaine hydrochloride, chloroprocaine hydrochloride,
oxyprocaine hydrochloride,
mepivacaine, cocaine hydrochloride, piperocaine hydrochloride, dyclonine,
dyclonine
hydrochloride, thimerosal, phenol, thymol, benzalkonium chloride, benzethonium
chloride,
chlorhexidine, povidone iodide, cetylpyridinium chloride, eugenol,
trimethylammonium
bromide, naphazoline nitrate, tetrahydrozoline hydrochloride, oxymetazoline
hydrochloride,
phenylephrine hydrochloride, tramazoline hydrochloride, thrombin,
phytonadione, protamine
sulfate, aminocaproic acid, tranexamic acid, carbazochrome, carbaxochrome
sodium sulfanate,
rutin, hesperidin, sulfamine, sulfathiazole, sulfadiazine, homosulfamine,
sulfisoxazole,
sulfisomidine, sulfamethizole, nitrofurazone, penicillin, meticillin,
oxacillin, cefalotin,
cefalordin, erythromcycin, lincomycin, tetracycline, chlortetracycline,
oxytetracy cline,
metacycline, chloramphenicol, kanamycin, streptomycin, gentamicin, bacitracin,
cycloserine,
salicylic acid, podophyllum resin, podolifox, cantharidin, chloroacetic acids,
silver nitrate,
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
protease inhibitors, thymadine kinase inhibitors, sugar or glycoprotein
synthesis inhibitors,
structural protein synthesis inhibitors, attachment and adsorption inhibitors,
and nucleoside
analogues such as acyclovir, penciclovir, valacyclovir, and ganciclovir,
heparin, insulin, LHRH,
TRH, interferons, oligonuclides, calcitonin, octreotide, omeprazone,
fluoxetine, ethinylestradiol,
amiodipine, paroxetine, enalapril, lisinopril, leuprolide, prevastatin,
lovastatin, norethindrone,
ri speri done, olanzapine, albuterol, hydrochl orothi azi de,
pseudoephridrine, warfarin, teraz o sin,
cisapride, ipratropium, busprione, methylphenidate, levothyroxine, zolpidem,
levonorgestrel,
glyburide, benazepril, medroxyprogesterone, clonazepam, ondansetron, losartan,
quinapril,
nitroglycerin, midazolam versed, cetirizine, doxazosin, glipizide, vaccine
hepatitis B, salmeterol,
sumatriptan, triamcinolone acetonide, goserelin, beclomethasone, granisteron,
desogestrel,
alprazolam, estradiol, nicotine, interferon beta 1A, cromolyn, fosinopril,
digoxin, fluticasone,
bisoprolol, calcitril, captorpril, butorphanol, clonidine, premarin,
testosterone, sumatriptan,
clotrimazole, bisacodyl, dextromethorphan, nitroglycerine, nafarelin,
dinoprostone, nicotine,
bisacodyl, goserelin, or granisetron. In certain embodiments, the
pharmaceutically active
component can be epinephrine, a benzodiazepine such as diazepam or lorazepam
or alprazolam.
Epinephrine, Diazepam and Alprazolam Examples
In one example, a composition including epinephrine or its salts or esters can
have a
biodelivery profile similar to that of epinephrine administered by injection,
for example, using an
EpiPen. Epinephrine can be present in an amount of from about .01 mg to about
100 mg per
dosage, for example, at a 0.1 mg, 5 mg, 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60
mg, 70 mg, 80
mg, 90 mg or 100 mg dosage, including greater than 0.1 mg, more than 5 mg,
more than 20 mg,
more than 30 mg, more than 40 mg, more than 50 mg, more than 60 mg, more than
70 mg, more
than 80 mg, more than 90 mg, or less than 100 mg, less than 90 mg, less than
80 mg, less than 70
mg, less than 60 mg, less than 50 mg, less than 40 mg, less than 30 mg, less
than 20 mg, less than
10 mg, or less than 5 mg, or any combination thereof. In another example, a
composition
including diazepam can have a biodelivery profile similar to that of a
diazepam tablet or gel, or
better. Diazepam or its salts can be present in an amount of from about 0.5 mg
to about 100 mg
per dosage, for example, at a 0.5 mg, 1 mg, 5 mg, 10 mg, 20 mg, 30 mg, 40 mg,
50 mg, 60 mg,
70 mg, 80 mg, 90 mg or 100 mg dosage including greater than 1 mg, more than 5
mg, more than
20 mg, more than 30 mg, more than 40 mg, more than 50 mg, more than 60 mg,
more than 70
mg, more than 80 mg, more than 90 mg, or less than 100 mg, less than 90 mg,
less than 80 mg,
26
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
less than 70 mg, less than 60 mg, less than 50 mg, less than 40 mg, less than
30 mg, less than 20
mg, less than 10 mg, or less than 5 mg, or any combination thereof.
In another example, a composition (e.g., including alprazolam, diazepam or
epinephrine)
can have a suitable nontoxic, nonionic alkyl glycoside having a hydrophobic
alkyl group joined
by a linkage to a hydrophilic saccharide in combination with a mucosal
delivery-enhancing agent
selected from: (a) an aggregation inhibitory agent; (b) a charge-modifying
agent; (c) a pH control
agent; (d) a degradative enzyme inhibitory agent; (e) a mucolytic or mucus
clearing agent; (f) a
ciliostatic agent; (g) a membrane penetration-enhancing agent selected from:
(i) a surfactant; (ii)
a bile salt; (ii) a phospholipid additive, mixed micelle, liposome, or
carrier; (iii) an alcohol; (iv)
an enamine; (v) an NO donor compound; (vi) a long chain amphipathic molecule;
(vii) a
hydrophobic penetration enhancer; (viii) sodium or a salicylic acid
derivative; (ix) a glycerol
ester of acetoacetic acid; (x) a cyclodextrin or beta-cyclodextrin derivative;
(xi) a medium-chain
fatty acid; (xii) a chelating agent; (xiii) an amino acid or salt thereof;
(xiv) an N-acetylamino acid
or salt thereof; (xv) an enzyme degradative to a selected membrane component;
(ix) an inhibitor
of fatty acid synthesis; (x) an inhibitor of cholesterol synthesis; and (xi)
any combination of the
membrane penetration enhancing agents recited in (i)-(x); (h) a modulatory
agent of epithelial
junction physiology; (i) a vasodilator agent; (j) a selective transport-
enhancing agent; or (k) a
stabilizing delivery vehicle, carrier, mucoadhesive, support or complex-
forming species with
which the compound is effectively combined, associated, contained,
encapsulated or bound
resulting in stabilization of the compound for enhanced mucosal delivery,
wherein the
formulation of the compound with the transmucosal delivery-enhancing agents
provides for
increased bioavailability of the compound in a blood plasma of a subject. The
formulation can
include approximately the same active pharmaceutical ingredient (API):
enhancer ratio as in the
other examples for diazepam and alprazolam.
Treatment or Adjunctive Treatment
Status epilepticus (SE) is an epileptic seizure of greater than five minutes
or more than
one seizure within a five-minute period without the person returning to normal
between them.
Other previous definitions used a 30-minute time limit. Benzodiazepines are
some of the most
effective drugs in the treatment of acute seizures and status epilepticus. The
benzodiazepines
most commonly used to treat status epilepticus include diazepam (Valium),
lorazepam (Ativan),
or midazolam (Versed). The pharmaceutically active components in a
pharmaceutical
27
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
composition (e.g. pharmaceutical composition film) can be a treatment or
adjunctive treatment
for Angelman syndrome (AS), Benign rolandic epilepsy of childhood (BREC) and
benign
rolandic epilepsy with centro-temporal spikes (BECTS), CDKL5 disorder,
Childhood absence
epilepsy (CAE), Myoclonic-astatic epilepsy or Doose Syndrome, Dravet syndrome,
Early
Myoclonic Encephalopathy (EME), Epilepsy with generalized tonic-clonic
seizures alone
(EGTCS) or epilepsy with tonic-clonic seizures on awakening, Epilepsy with
Myoclonic-
Absences Frontal lobe epilepsy, Glutl Deficiency Syndrome, Hypothalamic
hamartoma (HH),
Infantile spasms (also called IS) or West syndrome, Juvenile absence epilepsy
(JAE), Juvenile
myoclonic epilepsy (JME), Lafora progressive myoclonus epilepsy (Lafora
disease), Landau-
syndrome, Lennox-Gastaut Syndrome (LGS), Ohtahara Syndrome (OS),
Panayiotopoulos Syndrome (PS), PCDH19 Epilepsy, Progressive myoclonic
epilepsies,
Rasmussen's syndrome Ring chromosome 20 syndrome (RC20), Reflex epilepsies,
TBCK-
related intellectual disability syndrome, Temporal lobe epilepsy and
Neurocutaneous syndromes
that can be associated with seizures including Incontinentia pigmenti,
Neurofibromatosis Type 1,
Sturge Weber Syndrome (Encephalotrigeminal Angiomatosis), and Tuberous
Sclerosis Complex.
A film and/or its components can be water-soluble, water swellable or water-
insoluble.
The term "water-soluble" can refer to substances that are at least partially
dissolvable in an
aqueous solvent, including but not limited to water. The term "water-soluble"
may not
necessarily mean that the substance is 100% dissolvable in the aqueous
solvent. The term
"water-insoluble" refers to substances that are not dissolvable in an aqueous
solvent, including
but not limited to water. A solvent can include water, or alternatively can
include other solvents
(preferably, polar solvents) by themselves or in combination with water.
The composition can include a polymeric matrix. Any desired polymeric matrix
may be
used, provided that it is orally dissolvable or erodible. The dosage should
have enough
bioadhesion to not be easily removed and it should form a gel like structure
when administered.
They can be moderate-dissolving in the oral cavity and particularly suitable
for delivery of
pharmaceutically active components, although both fast release, delayed
release, controlled
release and sustained release compositions are also among the various
embodiments
contemplated.
28
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Branched Polymers
The pharmaceutical composition film can include dendritic polymers which can
include
highly branched macromolecules with various structural architectures. The
dendritic polymers
can include dendrimers, dendronised polymers (dendrigrafted polymers), linear
dendritic
hybrids, multi-arm star polymers, or hyperbranched polymers.
Hyperbranched polymers are highly branched polymers with imperfections in
their
structure. However, they can be synthesized in a single step reaction which
can be an advantage
over other dendritic structures and are therefore suitable for bulk volume
applications. The
properties of these polymers apart from their globular structure are the
abundant functional
groups, intramolecular cavities, low viscosity and high solubility. Dendritic
polymers have been
used in several drug delivery applications. See, e.g., Dendrimers as Drug
Carriers: Applications
in Different Routes of Drug Administration. J Pharm Sci, VOL. 97, 2008, 123-
143, which is
incorporated by reference herein.
The dendritic polymers can have internal cavities which can encapsulate drugs.
The steric
hindrance caused by the highly dense polymer chains might prevent the
crystallization of the
drugs. Thus, branched polymers can provide additional advantages in
formulating crystallizable
drugs in a polymer matrix.
Examples of suitable dendritic polymers include poly(ether) based dendrons,
dendrimers
and hyperbranched polymers, poly(ester) based dendrons, dendrimers and
hyperbranched
polymers, poly(thioether) based dendrons, dendrimers and hyperbranched
polymers, poly(amino
acid) based dendrons dendrimers and hyperbranched polymers, poly(arylalkylene
ether) based
dendrons, dendrimers and hyperbranched polymers, poly(alkyleneimine) based
dendrons,
dendrimers and hyperbranched polymers, poly(amidoamine) based dendrons,
dendrimers or
hyperbranched polymers.
Other examples of hyperbranched polymers include poly(amines)s,
polycarbonates,
poly(ether ketone)s, polyurethanes, polycarbosilanes, polysiloxanes,
poly(ester amine)s,
poly(sulfone amine)s, poly(urea urethane)s or polyether polyols such as
polyglycerols.
A film can be produced by a combination of at least one polymer and a solvent,
optionally including other components. The solvent may be water, a polar
organic solvent
including, but not limited to, ethanol, isopropanol, acetone, or any
combination thereof. In some
29
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
embodiments, the solvent may be a non-polar organic solvent, such as methylene
chloride. The
film may be prepared by utilizing a selected casting or deposition method and
a controlled drying
process. For example, the film may be prepared through a controlled drying
processes, which
include application of heat and/or radiation energy to the wet film matrix to
form a visco-elastic
structure, thereby controlling the uniformity of content of the film. The
controlled drying
processes can include air alone, heat alone or heat and air together
contacting the top of the film
or bottom of the film or the substrate supporting the cast or deposited or
extruded film or
contacting more than one surface at the same time or at different times during
the drying process.
Some of such processes are described in more detail in U.S. Patent No.
8,765,167 and U.S.
Patent No. 8,652,378, which are incorporated by reference herein.
Alternatively, the films may
be extruded as described in U.S. Patent Publication No. 2005/0037055 Al, which
is incorporated
by reference herein.
A polymer included in the films may be water-soluble, water-swellable, water-
insoluble,
or a combination of one or more either water-soluble, water-swellable or water-
insoluble
polymers. The polymer may include cellulose, cellulose derivatives or gums.
Specific examples
of useful water-soluble polymers include, but are not limited to, polyethylene
oxide, pullulan,
hydroxypropylmethyl cellulose, hydroxyethyl cellulose, hydroxypropyl
cellulose, polyvinyl
pyrrolidone, carboxymethyl cellulose, polyvinyl alcohol, sodium alginate,
polyethylene glycol,
xanthan gum, tragancanth gum, guar gum, acacia gum, arabic gum, polyacrylic
acid,
methylmethacrylate copolymer, carboxyvinyl copolymers, starch, gelatin, and
combinations
thereof Specific examples of useful water-insoluble polymers include, but are
not limited to,
ethyl cellulose, hydroxypropyl ethyl cellulose, cellulose acetate phthalate,
hydroxypropyl methyl
cellulose phthalate and combinations thereof For higher dosages, it may be
desirable to
incorporate a polymer that provides a high level of viscosity as compared to
lower dosages.
As used herein the phrase "water-soluble polymer" and variants thereof refer
to a polymer
that is at least partially soluble in water, and desirably fully or
predominantly soluble in water, or
absorbs water. Polymers that absorb water are often referred to as being water-
swellable
polymers. The materials useful with the present invention may be water-soluble
or water-
swellable at room temperature and other temperatures, such as temperatures
exceeding room
temperature. Moreover, the materials may be water-soluble or water-swellable
at pressures less
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
than atmospheric pressure. In some embodiments, films formed from such water-
soluble
polymers may be sufficiently water-soluble to be dissolvable upon contact with
bodily fluids.
Other polymers useful for incorporation into the films include biodegradable
polymers,
copolymers, block polymers or combinations thereof. It is understood that the
term
"biodegradable" is intended to include materials that chemically degrade, as
opposed to materials
that physically break apart (i.e., bioerodable materials). The polymers
incorporated in the films
can also include a combination of biodegradable or bioerodable materials.
Among the known
useful polymers or polymer classes which meet the above criteria are:
poly(glycolic acid) (PGA),
poly(lactic acid) (PLA), polydioxanes, polyoxalates, poly(alpha-esters),
polyanhydrides,
polyacetates, polycaprolactones, poly(orthoesters), polyamino acids,
polyaminocarbonates,
polyurethanes, polycarbonates, polyamides, poly(alkyl cyanoacrylates), and
mixtures and
copolymers thereof Additional useful polymers include, stereopolymers of L-
and D-lactic acid,
copolymers of bis(p-carboxyphenoxy)propane acid and sebacic acid, sebacic acid
copolymers,
copolymers of caprolactone, poly(lactic acid)/poly(glycolic
acid)/polyethyleneglycol
copolymers, copolymers of polyurethane and (poly(lactic acid)õ copolymers of
alpha-amino
acids, and caproic acid, copolymers of alpha-benzyl glutamate and polyethylene
glycol,
copolymers of succinate and poly(glycols), polyphosphazene, polyhydroxy-
alkanoates or
mixtures thereof. The polymer matrix can include one, two, three, four or more
components.
Although a variety of different polymers may be used, it is desired to select
polymers that
provide mucoadhesive properties to the film, as well as a desired dissolution
and/or
disintegration rate. In particular, the time period for which it is desired to
maintain the film in
contact with the mucosal tissue depends on the type of pharmaceutically active
component
contained in the composition. Some pharmaceutically active components may only
require a few
minutes for delivery through the mucosal tissue, whereas other
pharmaceutically active
components may require up to several hours or even longer. Accordingly, in
some embodiments,
one or more water-soluble polymers, as described above, may be used to form
the film. In other
embodiments, however, it may be desirable to use combinations of water-soluble
polymers and
polymers that are water-swellable, water-insoluble and/or biodegradable, as
provided above. The
inclusion of one or more polymers that are water-swellable, water-insoluble
and/or
biodegradable may provide films with slower dissolution or disintegration
rates than films
formed from water-soluble polymers alone. As such, the film may adhere to the
mucosal tissue
31
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
for longer periods of time, such as up to several hours, which may be
desirable for delivery of
certain pharmaceutically active components.
Desirably, an individual film dosage of the pharmaceutical film can have a
suitable
thickness, and small size, which is between about 0.0625-3 inch by about
0.0625-3 inch. The
.. film size can also be greater than 0.0625 inch, greater than 0.5 inch,
greater than 1 inch, greater
than 2 inches, about 3 inches, or greater than 3 inches, less than 3 inches,
less than 2 inches, less
than 1 inch, less than 0.5 inch, less than 0.0625 inch in at least one aspect,
or greater than 0.0625
inch, greater than 0.5 inch, greater than 1 inch, greater than 2 inches, or
greater than 3 inches,
about 3 inches, less than 3 inches, less than 2 inches, less than 1 inch, less
than 0.5 inch, or less
.. than 0.0625 inch in another aspect. The aspect ratio, including thickness,
length, and width can
be optimized by a person of ordinary skill in the art based on the chemical
and physical
properties of the polymeric matrix, the active pharmaceutical ingredient,
dosage, enhancer, and
other additives involved as well as the dimensions of the desired dispensing
unit. The film
dosage should have good adhesion when placed in the buccal cavity or in the
sublingual region
of the user. Further, the film dosage should disperse and dissolve at a
moderate rate, most
desirably dispersing within about 1 minute and dissolving within about 3
minutes. In some
embodiments, the film dosage may be capable of dispersing and dissolving at a
rate of between
about 1 to about 30 minutes, for example, about 1 to about 20 minutes, or more
than 1 minute,
more than 5 minutes, more than 7 minutes, more than 10 minutes, more than 12
minutes, more
than 15 minutes, more than 20 minutes, more than 30 minutes, about 30 minutes,
or less than 30
minutes, less than 20 minutes, less than 15 minutes, less than 12 minutes,
less than 10 minutes,
less than 7 minutes, less than 5 minutes, or less than 1 minute. Sublingual
dispersion rates may
be shorter than buccal dispersion rates.
For instance, in some embodiments, the films may include polyethylene oxide
alone or in
combination with a second polymer component. The second polymer may be another
water-
soluble polymer, a water-swellable polymer, a water-insoluble polymer, a
biodegradable
polymer or any combination thereof Suitable water-soluble polymers include,
without
limitation, any of those provided above. In some embodiments, the water-
soluble polymer may
include hydrophilic cellulosic polymers, such as hydroxypropyl cellulose
and/or
hydroxypropylmethyl cellulose. In some embodiments, one or more water-
swellable, water-
insoluble and/or biodegradable polymers also may be included in the
polyethylene oxide-based
32
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
film. Any of the water-swellable, water-insoluble or biodegradable polymers
provided above
may be employed. The second polymer component may be employed in amounts of
about 0% to
about 80% by weight in the polymer component, more specifically about 30% to
about 70% by
weight, and even more specifically about 40% to about 60% by weight, including
greater than
5%, greater than 10%, greater than 15%, greater than 20%, greater than 30%,
greater than 40%,
greater than 50%, greater than 60%, and greater than 70%, about 70%, less than
70%, less than
60%, less than 50%, less than 40%, less than 30%, less than 20%, less than 10%
or less than 5%
by weight.
Additives may be included in the films. Examples of classes of additives
include
preservatives, antimicrobials, excipients, lubricants, buffering agents,
stabilizers, blowing agents,
pigments, coloring agents, fillers, bulking agents, sweetening agents,
flavoring agents,
fragrances, release modifiers, adjuvants, plasticizers, flow accelerators,
mold release agents,
polyols, granulating agents, diluents, binders, buffers, absorbents, glidants,
adhesives, anti-
adherents, acidulants, softeners, resins, demulcents, solvents, surfactants,
emulsifiers, elastomers,
anti-tacking agents, anti-static agents and mixtures thereof These additives
may be added with
the pharmaceutically active component(s).
As used herein, the term "stabilizer" means an excipient capable of preventing
aggregation or other physical degradation, as well as chemical degradation, of
the active
pharmaceutical ingredient, another excipient, or the combination thereof
Stabilizers may also be classified as antioxidants, sequestrants, pH
modifiers, emulsifiers
and/or surfactants, and UV stabilizers as discussed above and in more detail
below.
Antioxidants (i.e., pharmaceutically compatible compound(s) or composition(s)
that
decelerates, inhibits, interrupts and/or stops oxidation processes) include,
in particular, the
following substances: tocopherols and the esters thereof, sesamol of sesame
oil, coniferyl
benzoate of benzoin resin, nordihydroguaietic resin and nordihydroguaiaretic
acid (NDGA),
gallates (among others, methyl, ethyl, propyl, amyl, butyl, lauryl gallates),
butylated
hydroxyanisole (BHA/BHT, also butyl-p-cresol); ascorbic acid and salts and
esters thereof (for
example, acorbyl palmitate), erythorbinic acid (isoascorbinic acid) and salts
and esters thereof,
monothioglycerol, sodium formaldehyde sulfoxylate, sodium metabisulfite,
sodium bisulfite,
sodium sulfite, potassium metabisulfite, butylated hydroxyanisole, butylated
hydroxytoluene
(BHT), propionic acid. Typical antioxidants are tocopherol such as, for
example, a-tocopherol
33
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
and the esters thereof, butylated hydroxytoluene and butylated hydroxyanisole.
The terms
"tocopherol" also includes esters of tocopherol. A known tocopherol is a-
tocopherol. The term
"a-tocopherol" includes esters of a-tocopherol (for example, a-tocopherol
acetate).
Sequestrants (i.e., any compounds which can engage in host-guest complex
formation
with another compound, such as the active ingredient or another excipient;
also referred to as a
sequestering agent) include calcium chloride, calcium disodium ethylene
diamine tetra-acetate,
glucono delta-lactone, sodium gluconate, potassium gluconate, sodium
tripolyphosphate, sodium
hexametaphosphate, and combinations thereof. Sequestrants also include cyclic
oligosaccharides, such as cyclodextrins, cyclomannins (5 or more a-D-
mannopyranose units
linked at the 1,4 positions by a linkages), cyclogalactins (5 or more P-D-
galactopyranose units
linked at the 1,4 positions by I linkages), cycloaltrins (5 or more a-D-
altropyranose units linked
at the 1,4 positions by a linkages), and combinations thereof
pH modifiers include acids (e.g., tartaric acid, citric acid, lactic acid,
fumaric acid,
phosphoric acid, ascorbic acid, acetic acid, succininc acid, adipic acid and
maleic acid), acidic
amino acids (e.g., glutamic acid, aspartic acid, etc.), inorganic salts
(alkali metal salt, alkaline
earth metal salt, ammonium salt, etc.) of such acidic substances, a salt of
such acidic substance
with an organic base (e.g., basic amino acid such as lysine, arginine and the
like, meglumine and
the like), and a solvate (e.g., hydrate) thereof Other examples of pH
modifiers include silicified
microcrystalline cellulose, magnesium aluminometasilicate, calcium salts of
phosphoric acid
(e.g., calcium hydrogen phosphate anhydrous or hydrate, calcium, sodium or
potassium
carbonate or hydrogencarbonate and calcium lactate or mixtures thereof),
sodium and/or calcium
salts of carboxymethyl cellulose, cross-linked carboxymethylcellulose (e.g.,
croscarmellose
sodium and/or calcium), polacrilin potassium, sodium and or/calcium alginate,
docusate sodium,
magnesium calcium, aluminium or zinc stearate, magnesium palmitate and
magnesium oleate,
sodium stearyl fumarate, and combinations thereof.
Examples of emulsifiers and/or surfactants include poloxamers or pluronics,
polyethylene
glycols, polyethylene glycol monostearate, polysorbates, sodium lauryl
sulfate, polyethoxylated
and hydrogenated castor oil, alkyl polyoside, a grafted water soluble protein
on a hydrophobic
backbone, lecithin, glyceryl monostearate, glyceryl
monostearate/polyoxyethylene stearate,
ketostearyl alcohol/sodium lauryl sulfate, carbomer, phospholipids, (Cio-C20)-
alkyl and alkylene
carboxylates, alkyl ether carboxylates, fatty alcohol sulfates, fatty alcohol
ether sulfates,
34
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
alkylamide sulfates and sulfonates, fatty acid alkylamide polyglycol ether
sulfates,
alkanesulfonates and hydroxyalkanesulfonates, olefinsulfonates, acyl esters of
isethionates, a-
sulfo fatty acid esters, alkylbenzenesulfonates, alkylphenol glycol ether
sulfonates,
sulfosuccinates, sulfosuccinic monoesters and diesters, fatty alcohol ether
phosphates,
protein/fatty acid condensation products, alkyl monoglyceride sulfates and
sulfonates,
alkylglyceride ether sulfonates, fatty acid methyltaurides, fatty acid
sarcosinates,
sulforicinoleates, and acylglutamates, quaternary ammonium salts (e.g., di-
(Cio-C24)-alkyl-
dim ethyl amm onium chloride or bromide), (C io-C24)-al kyl -dim ethyl ethyl
amm onium chloride or
bromide, (C io-C24)-alkyl -trimethyl ammonium chloride
or bromide (e.g.,
cetyltrimethylammonium chloride or bromide), (C io-C24)-alkyl-dimethylbenzyl
ammonium
chloride or bromide (e.g., (C12¨C18)-alkyl-dimethylbenzylammonium chloride),
N¨(C10-C18)-
alkyl-pyridinium chloride or bromide (e.g., N¨(C12-C16)-alkyl-pyridinium
chloride or bromide),
N¨(C10-C18)-alkyl-isoquinolinium chloride, bromide or monoalkyl sulfate,
N¨(C12-C18)-alkyl-
p ol yoyl aminoformylm ethyl pyri dinium chloride, N¨(C 12-C 18)-alkyl -N-
methylmorpholinium
chloride, bromide or monoalkyl sulfate, N¨(C12-C18)-alkyl-N-ethylmorpholinium
chloride,
bromide or monoalkyl sulfate,
(C 16-C18)-al kyl -p entaox ethyl amm onium chloride,
di i sobutyl phenoxyethoxyethyl dim ethylb enzyl amm onium chloride,
salts of N,N-di -
ethyl aminoethyl stearyl amide and -ol eyl ami de with hydrochloric acid,
acetic acid, lactic acid,
citric acid, phosphoric acid, N-acylaminoethyl -N,N-di ethyl -N-m ethyl amm
onium chloride,
bromide or monoalkyl sulfate, and N-acyl aminoethyl -N,N-di ethyl -N-b
enzylammonium chloride,
bromide or monoalkyl sulfate (in the foregoing, "acyl" standing for, e.g.,
stearyl or oleyl), and
combinations thereof.
Examples of UV stabilizers include UV absorbers (e.g., benzophenones), UV
quenchers
(i.e., any compound that dissipates UV energy as heat, rather than allowing
the energy to have a
degradation effect), scavengers (i.e., any compound that eliminates free
radicals resulting from
exposure to UV radiation), and combinations thereof
In other embodiments, stabilizers include ascorbyl palmitate, ascorbic acid,
alpha
tocopherol, butylated hydroxytoluene, buthylated hydroxyani sole, cysteine HC
1, citric acid,
ethylenediamine tetra acetic acid (EDTA), methionine, sodium citrate, sodium
ascorbate, sodium
thiosulfate, sodium metabi sulfite, sodium bisulfite, propyl gallate,
glutathione, thioglycerol,
singlet oxygen quenchers, hydroxyl radical scavengers, hydroperoxide removing
agents,
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
reducing agents, metal chelators, detergents, chaotropes, and combinations
thereof. "Singlet
oxygen quenchers" include, but are not limited to, alkyl imidazoles (e.g.,
histidine, L-camosine,
histamine, imidazole 4-acetic acid), indoles (e.g., tryptophan and derivatives
thereof, such as N-
acety1-5-methoxytryptamine, N-acetylserotonin, 6-m ethoxy-1,2,3,4-tetrahydro-b
eta-carb oline),
sulfur-containing amino acids (e.g., methionine, ethionine, dj enkolic acid,
lanthionine, N-formyl
methionine, felinine, S-allyl cysteine, S-aminoethyl-L-cysteine), phenolic
compounds (e.g.,
tyrosine and derivatives thereof), aromatic acids (e.g., ascorbate, salicylic
acid, and derivatives
thereof), azide (e.g., sodium azide), tocopherol and related vitamin E
derivatives, and carotene
and related vitamin A derivatives. "Hydroxyl radical scavengers" include, but
are not limited to
azide, dimethyl sulfoxide, histidine, mannitol, sucrose, glucose, salicylate,
and L-cysteine.
"Hydroperoxide removing agents" include, but are not limited to catalase,
pyruvate, glutathione,
and glutathione peroxidases. "Reducing agents" include, but are not limited
to, cysteine and
mercaptoethylene. "Metal chelators" include, but are not limited to, EDTA,
EGTA, o-
phenanthroline, and citrate. "Detergents" include, but are not limited to, SDS
and sodium lauroyl
sarcosyl. "Chaotropes" include, but are not limited to guandinium
hydrochloride, isothiocyanate,
urea, and formamide. As discussed herein, stabilizers can be present in
0.0001%-50% by weight,
including greater than 0.0001%, greater than 0.001%, greater than 0.01%,
greater than 0.1%,
greater than 1%, greater than 5%, greater than 10%, greater than 20%, greater
than 30%, greater
than 40%, greater than 50%, less than 50%, less than 40%, less than 30%, less
than 20%, less
than 10%, less than 1%, less than 0.1%, less than 0.01%, less than 0.001%, or
less than 0.0001%
by weight.
Useful additives can include, for example, gelatin, vegetable proteins such as
sunflower
protein, soybean proteins, cotton seed proteins, peanut proteins, grape seed
proteins, whey
proteins, whey protein isolates, blood proteins, egg proteins, acrylated
proteins, water-soluble
polysaccharides such as alginates, carrageenans, guar gum, agar-agar, xanthan
gum, gellan gum,
gum arabic and related gums (gum ghatti, gum karaya, gum tragancanth), pectin,
water-soluble
derivatives of cellulose: alkylcelluloses hydroxyalkylcelluloses and
hydroxyalkylalkylcelluloses,
such as methylcellulose, hydroxym ethyl cellul ose,
hydroxyethyl cellulose,
hydroxypropyl cellulose, hydroxyethylm ethyl c ellul ose,
hydroxypropylm ethyl c ellul ose,
hydroxybutylmethylcellulose, cellulose esters and hydroxyalkylcellulose esters
such as cellulose
acetate phthalate (CAP), hydroxypropylm ethyl cellulose (HPMC);
carboxyalkylcelluloses,
36
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
carboxyalkylalkylcelluloses, carboxyalkylcellulose esters such as
carboxymethylcellulose and
their alkali metal salts; water-soluble synthetic polymers such as polyacrylic
acids and
polyacrylic acid esters, polymethacrylic acids and polymethacrylic acid
esters, polyvinylacetates,
polyvinylalcohols, polyvinylacetatephthalates (PVAP), polyvinylpyrrolidone
(PVP), PVA/vinyl
acetate copolymer, or polycrotonic acids; also suitable are phthalated
gelatin, gelatin succinate,
crosslinked gelatin, shellac, water-soluble chemical derivatives of starch,
cationically modified
acrylates and methacrylates possessing, for example, a tertiary or quaternary
amino group, such
as the diethylaminoethyl group, which may be quaternized if desired; or other
similar polymers.
The additional components can range up to about 80%, desirably about 0.005% to
50%
and more desirably within the range of 1% to 20% based on the weight of all
composition
components, including greater than 1%, greater than 5%, greater than 10%,
greater than 20%,
greater than 30%, greater than 40%, greater than 50%, greater than 60%,
greater than 70%, about
80%, greater than 80%, less than 80%, less than 70%, less than 60%, less than
50%, less than
40%, less than 30%, less than 20%, less than 10%, less than 5%, about 3%, or
less than 1%.
Other additives can include anti-tacking, flow agents and opacifiers, such as
the oxides of
magnesium aluminum, silicon, titanium, etc., desirably in a concentration
range of about 0.005%
to about 5% by weight and desirably about 0.02% to about 2% based on the
weight of all film
components, including greater than 0.02%, greater than 0.2%, greater than
0.5%, greater than
1%, greater than 1.5%, greater than 2%, greater than 4%, about 5%, greater
than 5%, less than
4%, less than 2%, less than 1%, less than 0.5%, less than 0.2%, or less than
0.02%.
In certain embodiments, the composition can include plasticizers, which can
include
polyalkylene oxides, such as polyethylene glycols, polypropylene glycols,
polyethylene-
propylene glycols, organic plasticizers with low molecular weights, such as
glycerol, glycerol
monoacetate, diacetate or triacetate, triacetin, polysorbate, cetyl alcohol,
propylene glycol, sugar
alcohols sorbitol, sodium diethylsulfosuccinate, triethyl citrate, tributyl
citrate, phytoextracts,
fatty acid esters, fatty acids, oils and the like, added in concentrations
ranging from about 0.1%
to about 40%, and desirably ranging from about 0.5% to about 20% based on the
weight of the
composition including greater than 0.5%, greater than 1%, greater than 1.5%,
greater than 2%,
greater than 4%, greater than 5%, greater than 10%, greater than 15%, about
20%, greater than
20%, less than 20%, less than 15%, less than 10%, less than 5%, less than 4%,
less than 2%, less
than 1%, or less than 0.5%. There may further be added compounds to improve
the texture
37
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
properties of the film material such as animal or vegetable fats, desirably in
their hydrogenated
form. The composition can also include compounds to improve the textural
properties of the
product. Other ingredients can include binders which contribute to the ease of
formation and
general quality of the films. Non-limiting examples of binders include
starches, natural gums,
pregelatinized starches, gelatin, polyvinylpyrrolidone, methylcellulose,
sodium
carboxymethylcellulose, ethylcellulose, polyacrylamides,
polyvinyl oxoazolidone, or
polyvinylalcohols.
Further potential additives include solubility enhancing agents, such as
substances that
form inclusion compounds with active components. Such agents may be useful in
improving the
properties of very insoluble and/or unstable actives. In general, these
substances are doughnut-
shaped molecules with hydrophobic internal cavities and hydrophilic exteriors.
Insoluble and/or
instable pharmaceutically active components may fit within the hydrophobic
cavity, thereby
producing an inclusion complex, which is soluble in water. Accordingly, the
formation of the
inclusion complex permits very insoluble and/or unstable pharmaceutically
active components to
be dissolved in water. A particularly desirable example of such agents are
cyclodextrins, which
are cyclic carbohydrates derived from starch. Other similar substances,
however, are considered
well within the scope of the present invention.
Suitable coloring agents include food, drug and cosmetic colors (FD&C), drug
and
cosmetic colors (D&C), or external drug and cosmetic colors (Ext. D&C). These
colors are dyes,
their corresponding lakes, and certain natural and derived colorants. Lakes
are dyes absorbed on
aluminum hydroxide. Other examples of coloring agents include known azo dyes,
organic or
inorganic pigments, or coloring agents of natural origin. Inorganic pigments
are preferred, such
as the oxides or iron or titanium, these oxides, being added in concentrations
ranging from about
0.001 to about 10%, and preferably about 0.5 to about 3%, including greater
than 0.001%,
greater than 0.01%, greater than 0.1%, greater than 0.5%, greater than 1%,
greater than 2%,
greater than 5%, about 10%, greater than 10%, less than 10%, less than 5%,
less than 2%, less
than 1%, less than 0.5%, less than 0.1%, less than 0.01%, or less than 0.001%,
based on the
weight of all the components.
Flavors may be chosen from natural and synthetic flavoring liquids. An
illustrative list of
such agents includes volatile oils, synthetic flavor oils, flavoring
aromatics, oils, liquids,
oleoresins or extracts derived from plants, leaves, flowers, fruits, stems and
combinations
38
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
thereof A non-limiting representative list of examples includes mint oils,
cocoa, and citrus oils
such as lemon, orange, lime and grapefruit and fruit essences including apple,
pear, peach, grape,
strawberry, raspberry, cherry, plum, pineapple, apricot or other fruit
flavors. Other useful
flavorings include aldehydes and esters such as benzaldehyde (cherry, almond),
citral i.e.,
alphacitral (lemon, lime), neral, i.e., beta-citral (lemon, lime), decanal
(orange, lemon), aldehyde
C-8 (citrus fruits), aldehyde C-9 (citrus fruits), aldehyde C-12 (citrus
fruits), tolyl aldehyde
(cherry, almond), 2,6-dimethyloctanol (green fruit), or 2-dodecenal (citrus,
mandarin),
combinations thereof and the like.
The sweeteners may be chosen from the following non-limiting list: glucose
(corn syrup),
dextrose, invert sugar, fructose, and combinations thereof, saccharin and its
various salts such as
the sodium salt; dipeptide based sweeteners such as aspartame, neotame,
advantame;
dihydrochalcone compounds, glycyrrhizin; Stevia Rebaudiana (Stevioside);
chloro derivatives of
sucrose such as sucralose; sugar alcohols such as sorbitol, mannitol, xylitol,
and the like. Also
contemplated are hydrogenated starch hydrolysates and the synthetic sweetener
3,6-dihydro-6-
methyl-1-1-1,2,3-oxathiazin-4-one-2,2-dioxide, particularly the potassium salt
(acesulfame-K),
and sodium and calcium salts thereof, and natural intensive sweeteners, such
as Lo Han Kuo.
Other sweeteners may also be used.
Anti-foaming and/or de-foaming components may also be used with the films.
These
components aid in the removal of air, such as entrapped air, from the film-
forming compositions.
Such entrapped air may lead to non-uniform films. Simethicone is one
particularly useful anti-
foaming and/or de-foaming agent. The present invention, however, is not so
limited and other
suitable anti-foam and/or de-foaming agents may be used. Simethicone and
related agents may
be employed for densification purposes. More specifically, such agents may
facilitate the
removal of voids, air, moisture, and similar undesired components, thereby
providing denser and
thus more uniform films. Agents or components which perform this function can
be referred to
as densification or densifying agents. As described above, entrapped air or
undesired components
may lead to non-uniform films.
Any other optional components described in commonly assigned U.S. Patent No.
7,425,292 and U.S. Patent No. 8,765,167, referred to above, also may be
included in the films
described herein.
39
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
The film compositions further desirably contains a buffer so as to control the
pH of the
film composition. Any desired level of buffer may be incorporated into the
film composition so
as to provide the desired pH level encountered as the pharmaceutically active
component is
released from the composition. The buffer is preferably provided in an amount
sufficient to
control the release from the film and/or the absorption into the body of the
pharmaceutically
active component. In some embodiments, the buffer may include sodium citrate,
citric acid,
bitartrate salt and combinations thereof
The pharmaceutical films described herein may be formed via any desired
process.
Suitable processes are set forth in U.S. Patent Nos. 8,652,378, 7,425,292 and
7,357,891, which
are incorporated by reference herein. In one embodiment, the film dosage
composition is formed
by first preparing a wet composition, the wet composition including a
polymeric carrier matrix
and a therapeutically effective amount of a pharmaceutically active component.
The wet
composition is cast into a film and then sufficiently dried to form a self-
supporting film
composition. The wet composition may be cast into individual dosages, or it
may be cast into a
sheet, where the sheet is then cut into individual dosages.
The pharmaceutical composition can adhere to a mucosal surface. The present
invention
finds particular use in the localized treatment of body tissues, diseases, or
wounds which may
have moist surfaces and which are susceptible to bodily fluids, such as the
mouth, the vagina,
organs, or other types of mucosal surfaces. The composition carries a
pharmaceutical, and upon
application and adherence to the mucosal surface, offers a layer of protection
and delivers the
pharmaceutical to the treatment site, the surrounding tissues, and other
bodily fluids. The
composition provides an appropriate residence time for effective drug delivery
at the treatment
site, given the control of erosion in aqueous solution or bodily fluids such
as saliva, and the slow,
natural erosion of the film concomitant or subsequent to the delivery.
The residence time of the composition depends on the erosion rate of the water
erodable
polymers used in the formulation and their respective concentrations. The
erosion rate may be
adjusted, for example, by mixing together components with different solubility
characteristics or
chemically different polymers, such as hydroxyethyl cellulose and
hydroxypropyl cellulose; by
using different molecular weight grades of the same polymer, such as mixing
low and medium
molecular weight hydroxyethyl cellulose; by using excipients or plasticizers
of various lipophilic
values or water solubility characteristics (including essentially insoluble
components); by using
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
water soluble organic and inorganic salts; by using crosslinking agents such
as glyoxal with
polymers such as hydroxyethyl cellulose for partial crosslinking; or by post-
treatment irradiation
or curing, which may alter the physical state of the film, including its
crystallinity or phase
transition, once obtained. These strategies might be employed alone or in
combination in order to
modify the erosion kinetics of the film. Upon application, the pharmaceutical
composition film
adheres to the mucosal surface and is held in place. Water absorption softens
the composition,
thereby diminishing the foreign body sensation. As the composition rests on
the mucosal surface,
delivery of the drug occurs. Residence times may be adjusted over a wide range
depending upon
the desired timing of the delivery of the chosen pharmaceutical and the
desired lifespan of the
carrier. Generally, however, the residence time is modulated between about a
few seconds to
about a few days. Preferably, the residence time for most pharmaceuticals is
adjusted from about
5 seconds to about 24 hours. More preferably, the residence time is adjusted
from about 5
seconds to about 30 minutes. In addition to providing drug delivery, once the
composition
adheres to the mucosal surface, it also provides protection to the treatment
site, acting as an
erodable bandage. Lipophilic agents can be designed to slow down erodability
to decrease
disintegration and dissolution.
It is also possible to adjust the kinetics of erodability of the composition
by adding
excipients which are sensitive to enzymes such as amylase, very soluble in
water such as water
soluble organic and inorganic salts. Suitable excipients may include the
sodium and potassium
salts of chloride, carbonate, bicarbonate, citrate, trifluoroacetate,
benzoate, phosphate, fluoride,
sulfate, or tartrate. The amount added can vary depending upon how much the
erosion kinetics is
to be altered as well as the amount and nature of the other components in the
composition.
Emulsifiers typically used in the water-based emulsions described above are,
preferably,
either obtained in situ if selected from the linoleic, palmitic, myristoleic,
lauric, stearic, cetoleic
or oleic acids and sodium or potassium hydroxide, or selected from the
laurate, palmitate,
stearate, or oleate esters of sorbitol and sorbitol anhydrides,
polyoxyethylene derivatives
including monooleate, monostearate, monopalmitate, monolaurate, fatty
alcohols, alkyl phenols,
allyl ethers, alkyl aryl ethers, sorbitan monostearate, sorbitan monooleate
and/or sorbitan
monopalmitate.
The amount of pharmaceutically active component to be used depends on the
desired
treatment strength and the composition of the layers, although preferably, the
pharmaceutical
41
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
component comprises from about 0.001% to about 99%, more preferably from about
0.003 to
about 75%, and most preferably from about 0.005% to about 50% by weight of the
composition,
including, more than 0.005%, more than 0.05%, more than 0.5%, more than 1%,
more than 5%,
more than 10%, more than 15%, more than 20%, more than 30%, about 50%, more
than 50%,
less than 50%, less than 30%, less than 20%, less than 15%, less than 10%,
less than 5%, less
than 1%, less than 0.5%, less than 0.05%, or less than 0.005%. The amounts of
other components
may vary depending on the drug or other components but typically these
components comprise
no more than 50%, preferably no more than 30%, and most preferably no more
than 15% by total
weight of the composition.
The thickness of the film may vary, depending on the thickness of each of the
layers and
the number of layers. As stated above, both the thickness and amount of layers
may be adjusted
in order to vary the erosion kinetics. Preferably, if the composition has only
two layers, the
thickness ranges from 0.005 mm to 2 mm, preferably from 0.01 to 1 mm, and more
preferably
from 0.1 to 0.5 mm, including greater than 0.1 mm, greater than 0.2 mm, about
0.5 mm, greater
than 0.5 mm, less than 0.5 mm, less than 0.2 mm, or less than 0.1 mm. The
thickness of each
layer may vary from 10 to 90% of the overall thickness of the layered
composition, and
preferably varies from 30 to 60%, including greater than 10%, greater than
20%, greater than
30%, greater than 40%, greater than 50%, greater than 70%, greater than 90%,
about 90%, less
than 90%, less than 70%, less than 50%, less than 40%, less than 30%, less
than 20%, or less
than 10%. Thus, the preferred thickness of each layer may vary from 0.01 mm to
0.9 mm, or
from 0.03 mm to 0.5 mm.
As one skilled in the art will appreciate, when systemic delivery, e.g.,
transmucosal or
transdermal delivery is desired, the treatment site may include any area in
which the film is
capable of delivery and/or maintaining a desired level of pharmaceutical in
the blood, lymph, or
other bodily fluid. Typically, such treatment sites include the oral,
esophageal, aural, ocular,
anal, nasal, and vaginal mucosal tissue, as well as, the skin. If the skin is
to be employed as the
treatment site, then usually larger areas of the skin wherein movement will
not disrupt the
adhesion of the film, such as the upper arm or thigh, are preferred.
The pharmaceutical composition can also be used as a wound dressing. By
offering a
physical, compatible, oxygen and moisture permeable, flexible barrier which
can be washed
away, the film can not only protect a wound but also deliver a pharmaceutical
in order to
42
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
promote healing, aseptic, scarification, to ease the pain or to improve
globally the condition of
the sufferer. Some of the examples given below are well suited for an
application to the skin or a
wound. As one skilled in the art will appreciate, the formulation might
require incorporating a
specific hydrophilic/hygroscopic excipient which would help in maintaining
good adhesion on
dry skin over an extended period of time. Another advantage of the present
invention when
utilized in this manner is that if one does not wish that the film be
noticeable on the skin, then no
dyes or colored substances need be used. If, on the other hand, one desires
that the film be
noticeable, a dye or colored substance may be employed.
While the pharmaceutical composition can adhere to mucosal tissues, which are
wet
tissues by nature, it can also be used on other surfaces such as skin or
wounds. The
pharmaceutical film can adhere to the skin if prior to application the skin is
wet with an aqueous-
based fluid such as water, saliva, wound drainage or perspiration. The film
can adhere to the skin
until it erodes due to contact with water by, for example, rinsing, showering,
bathing or washing.
The film may also be readily removed by peeling without significant damage to
tissue.
A Franz diffusion cell is an in vitro skin permeation assay used in
formulation
development. The Franz diffusion cell apparatus (Figure 1A) consists of two
chambers separated
by a membrane of, for example, animal or human tissue. The test product is
applied to the
membrane via the top chamber. The bottom chamber contains fluid from which
samples are
taken at regular intervals for analysis to determine the amount of active that
has permeated the
membrane. Referring to Figure 1A, a Franz diffusion cell 100 includes a donor
compound 101,
a donor chamber 102, a membrane 103, sampling port 104, receptor chamber 105,
stir bar 106,
and a heater/circulator 107.
Referring to Figure 1B, a pharmaceutical composition is a film 100 comprising
a
polymeric matrix 200, the pharmaceutically active component 300 being
contained in the
polymeric matrix. The film can include a permeation enhancer 400.
Referring to Figures 2A and 2B, the graphs show the permeation of an active
material
from a composition. The graph shows that for the Epinephrine Base ¨
solubilized in-situ vs. the
inherently soluble Epinephrine Bitartrate, no meaningful differences were
observed.
Epinephrine Bitartrate was selected for further development based on ease of
processing. Flux is
derived as slope of the amount permeated as a function of time. Steady state
flux is taken from
43
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
the plateau of flux vs time curve multiplied by the volume of receiver media
and normalized for
permeation area.
Referring to Figure 2A, this graph shows average amount of active material
permeated
vs. time, with 8.00 mg/mL epinephrine bitartrate and 4.4 mg/mL epinephrine
base solubilized.
Referring to Figure 2B, this graph shows average flux vs. time, with 8.00
mg/mL
epinephrine bitartrate and 4.4 mg/mL epinephrine base solubilized.
Referring to Figure 3, this graph shows ex-vivo permeation of epinephrine
bitartrate as a
function of concentration. The study compared concentrations of 4 mg/mL, 8
mg/mL, 16
mg/mL and 100 mg/mL. Results showed that increasing concentration resulted in
increased
permeation, and level of enhancement diminishes at higher loading.
Referring to Figure 4, this graph shows permeation of epinephrine bitartrate
as a function
of solution pH. Acidic conditions explored to promote stability. The results
compared
Epinephrine Bitartrate pH 3 buffer and Epinephrine Bitartrate pH 5 buffer, and
found that the
Epinephrine Bitartrate pH 5 buffer was slightly favorable.
Referring to Figure 5, this graph shows the influence of enhancers on
permeation of
epinephrine, indicated as amount permeated as a function of time. Multiple
enhancers were
screened, including Labrasol, capryol 90, Plurol Oleique, Labrafil, TDM, SGDC,
Gelucire 44/14
and clove oil. Significant impact on time to onset and steady state flux was
achieved, and
surprisingly enhanced permeation was achieved for clove oil and Labrasol.
Referring to Figure 6A and 6B, these graphs show the release of epinephrine on
polymer
platforms and the effect of enhancers on its release, indicated as amount
permeated (in [tg) vs.
time. Figure 6A shows the epinephrine release from different polymer
platforms. Figure 6B
shows the impact of enhancers on epinephrine release.
Referring to Figure 7, this graph shows a pharmacokinetic model in the male
Yucatan,
miniature swine. The study compares a 0.3 mg Epipen, a 0.12 mg Epinephrine IV
and a placebo
film.
Referring to Figure 8, this graphs shows the impact of no enhancer on the
concentration
profiles of a 40 mg Epinephrine film vs, a 0.3 mg Epipen.
Referring to Figure 9, this graph shows the impact of Enhancer A (Labrasol) on
the
concentration profiles of a 40 mg Epinephrine film vs, a 0.3 mg Epipen.
Referring to Figure
44
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
10, this graph shows the impact of Enhancer L (clove oil) on the concentration
profiles of two 40
mg Epinephrine films (10-1-1) and (11-1-1) vs. a 0.3 mg Epipen.
Referring to Figure 11, This graph shows the impact of Enhancer L(clove oil)
and film
dimension (10-1-1 thinner bigger film and 11-1-1 thicker smaller film) on the
concentration
profiles of 40 mg Epinephrine films vs. a 0.3 mg Epipen.
Referring to Figure 12, this graph shows the concentration profiles for
varying doses of
Epinephrine films in a constant matrix for Enhancer L (clove oil) vs. a 0.3 mg
Epipen.
Referring to Figure 13, the graph shows the concentration profiles for varying
doses of
Epinephrine films in a constant matrix for Enhancer L (clove oil) vs. a 0.3 mg
Epipen.
Referring to Figure 14, the graph shows the concentration profiles for varying
doses of
Epinephrine films in a constant matrix for Enhancer A (Labrasol) vs. a 0.3 mg
Epipen.
Referring to Figure 15, this graph shows the influence of enhancers on
permeation of
diazepam, indicated as amount permeated as a function of time.
Referring to Figure 16, this graph shows the average flux as a function of
time (diazepam
+ enhancers).
Referring to Figure 17, this graph shows the impact of Farnesol and Farnesol
in
combination with Linoleic Acid on plasma concentration profiles of 40 mg
Epinephrine Films vs.
a 0.3 mg Epipen.
Referring to Figure 18, this graph shows the impact of Farnesol and Farnesol
in
combination with Linoleic Acid on plasma concentration profiles of 40 mg
Epinephrine Films vs.
a 0.3 mg Epipen.
Referring to Figure 19, this graph shows the impact of Farnesol in combination
with
Linoleic Acid on plasma concentration profiles of 40 mg Epinephrine Films vs.
a 0.3 mg Epipen.
Referring to Figure 20, this graph shows the impact of Farnesol and Farnesol
in
combination with Linoleic Acid on plasma concentration profiles of 40 mg
Epinephrine Films vs.
a 0.3 mg Epipen.
The following examples are provided to illustrate pharmaceutical compositions,
as well
as, methods of making and using, pharmaceutical compositions and devices
described herein.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
EXAMPLE S
Example 1
Permeation Enhancers - Epinephrine
Permeation enhancement was studied using a number of permeation enhancers with
Epinephrine Bitartrate 16.00 mg/mL concentration. The results show flux
enhancement
represented in the data below. For 100% Eugenol and 100% Clove Oil, the
results showed
steady state flux reached significantly earlier along with an unexpectedly
heightened % flux
enhancement.
Average
Permeability
Donor Solution (16.00mg/mL Steady State % Flux
Epinephrine Bitartrate + enhancer) Flux enhancement Coefficient
(ug/cm2*min) (cm/s)
Epinephrine Bitartrate ,no enhancer 1.3173 N/A 1.37E-06
3% Clove Oil 8.2704 527.84 8.61E-06
3% Clove Oil Repeat 5.3776 308.24 5.60E-06
3% Eugenol 7.1311 441.35 7.43E-06
3% Eugenyl Acetate 1.8945 43.82 1.97E-06
3% B-Caryophyllene 3.5200 167.22 3.67E-06
0.3% Eugenol* 3.9735 201.65 4.14E-06
100% Eugenol 1 36.8432 2696.92 3.84E-05
0.3% Clove Oil* 3.6806 179.41 3.83E-06
100% Clove Oil 1 52.5304 3887.81 5.47E-05
3% Phenol 4.5790 247.61 4.77E-06
3% Phenol Repeat 4.1753 216.97 4.35E-06
3% Linoleic Acid 2.1788 65.40 2.27E-06
50%Clove Oil 2.5673 94.89 2.67E-06
0.3% Labrasol 3.5221 167.38 3.67E-06
3% Vanillyl Alcohol+ 6% Ethanol 1.10243 -16.31 1.15E-06
3% Safrole 2.60634 97.86 2.71E-06
3% Oleic Acid 2.06597 56.84 2.15E-06
3% Oleic Acid + 1% PEG200 2.73655 107.74 2.85E-06
3% Benzyl Alcohol 1.38455 5.11 1.44E-06
1 steady state flux reached at much earlier time point
* 0.3% Eugenol vs 0.3% Clove -similar flux rates to one another
46
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
For these examples, clove oil was obtained from clove leaf. Similar results
may be
obtained from clove oil from clove bud and/or clove stem. Based on this data,
similar
permeability enhancement results can be expected from pharmaceutical compounds
structurally
similar to epinephrine.
Example 2
Diazepam Solubility and Permeability
Diazepam was applied in the buccal area (cheek) to diffuse through the oral
mucosa and
enter directly into the bloodstream. Solubility of diazepam was also studied
using various
excipients. Figure 15 shows the influence of enhancers on permeation of
diazepam, indicated as
amount permeated indicated as ug as a function of time. Figure 16 shows the
average flux
indicated as ug/cm* min, as a function of time in minutes in a solution of
diazepam and certain
selected enhancers.
The following excipients have also been studied to improve solubility.
47
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
API
Melting Solubility
Water Point mg/gm total
EXCIPIENTS HLB Solubility C
mix
RD-0073-10 SERIES
PEG 400 S Liquid 90
Caprylic/Capric Triglyceride I Liquid <50
Propylene Glycol S Liquid <30
Glycerol Monooleate 1 I 24 71
Polysorbate 80 15 S Liquid 83
PEG 4000 S 53-59 125
PEG 32 Glyceryl Palmitostearate 11 D 50 125
Mix: PEG 400, PEG 4000, PEO N80 S 118
18 to
Poloxamer 407 23 S 52-57 110-140
11 to
Polyoxyl 50 Stearate 12 S 30-35 200
RD-0073-19 series
Benzyl Alcohol S Liquid 400
14 to
Polyoxyl 40 Hyd Castor Oil 16 S 16-26 167
12 to
Poloxamer 124 18 S 16 127
RD-0073-20 series
Clove Oil I Liquid 394
Castor Oil I Liquid <50
Light Mineral Oil I Liquid <50
Oleic Acid I Liquid <50
Polyoxyl 40 Stearate 17 S 38 83
Maisine 35-1 (GMLin0) 1 I Liquid 45
Labrasol Caprylocaproyl polyoxy1-8
glycerides 12 D Liquid 83
RD-0073-27 series
Soybean Oil I Liquid <50
RD-0073-38 series
Clove Oil FCC I Liquid 370
48
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
The following excipients can also be applied for similar enhancement
properties:
cinnamon leaf, basil, bay leaf, nutmeg, Kolliphorg TPGS, Vit E PEG Succinate,
Kolliphorg EL,
Polyoxyl 35 Castor Oil USP/NF, Menthol, N-Methyl-2-pyrrolidone, SLS (SDS),
SDBS,
Dimethyl Phthalate, Sucrose PaImitate (Sisterna PS750-C), Sucrose Stearate
(Sisterna 51370-C),
CHAPS, Octyl glucoside, Triton X 100 (Octoxyno1-9), Ethyl Maltol (flavorant
powder), Brij 58
(Ceteth-20), Vitamin E tocopherol, tocopherol acetate or tocopherol succinate,
sterols,
phytoextracts, essential oils or Cod Liver Oil.
The following results were obtained in a solution of Diazepam having a
concentration of
8.00 mg/mL.
Average Permeability
Donor Solution
Steady State
(8.00mg/mL Diazepam + Flux Coefficient
enhancer) (cm/s)
(ug/cm2*min)
100% Clove Oil 0.0008 1.63E-09
100% Benzyl Alcohol 0.0058 1.22E-08
3% Eugenyl Acetate 0.1037 2.16E-07
3% P-Caryophyllene 0.0798 1.66E-07
3% Phenol 0.2752 5.73E-07
3% Cinnamaldehyde 0.1306 2.72E-07
3% Clove Oil 0.0990 2.06E-07
3% Benzyl Alcohol 0.1438 3.00E-07
3% Labrasol 0.1251 2.61E-07
100% Cinnamaldehyde 0.0057 1.19E-08
Example 3
General Permeation Procedure ¨ Ex vivo Permeation Study Protocol
In one example, a permeation procedure is conducted as follows. A temperature
bath is
set to 37 C, and receiver media is placed in a water bath to adjust the
temperature and begin
degassing. A franz diffusion cell is obtained and prepared. The Franz
diffusion cell includes a
donor compound, a donor chamber, a membrane, sampling port, receptor chamber,
stir bar, and a
heater/circulator. A stir bar is inserted into a franz diffusion cell. Tissue
is placed over the franz
diffusion cell, and it is ensured that the tissue covers the entire area with
an overlap onto a glass
joint. The top of a diffusion cell is placed over the tissue, and the top of
the cell is clamped to
the bottom. About 5 mL of receptor media is loaded into the receiver area to
ensure that no air
49
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
bubbles are trapped in the received portion of the cell. This ensures that all
5mL can fit into the
receiver area. Stirring is begun, and temperature is allowed to equilibrate
for about 20 minutes.
Meanwhile, High Performance Liquid Chromatography (HPLC) vials are labelled by
cell
number and time point. One must then check again for air bubbles as the
solution will degas
during heating.
If testing films, one can perform the following next steps: (1) weigh films,
punch to
match diffusion area (or smaller), reweigh, record pre- and post-punching
weight; (2) wet a
donor area with approximately 100 L of phosphate buffer; (3) place film on a
donor surface, top
with 400 1..t.L of phosphate buffer, and start timers.
For solution studies, one can perform the following steps: (1) using a
micropipette,
dispense 500 [IL of the solution into each donor cell, start the timers; (2)
sample 200 [IL at the
following time points (time = 0 min, 20 min, 40 min, 60 min, 120 min, 180 min,
240 min, 300
min, 360 min), and place in labelled HPLC vials, ensure no air is trapped in
the bottom of the
vial by tapping the closed vials; (3) replace each sample time with 200 uL of
receptor media (to
maintain 5 mL); (4) When all time points completed, disassemble the cells and
dispose of all
materials properly.
Example 4
Ex vivo Permeation Evaluation
An exemplary ex vivo permeation evaluation is as follows.
1. Tissue is freshly excised and shipped (e.g. overnight) at 4 C.
2. The tissue is processed and frozen at -20 C for up to three weeks prior to
use.
3. The tissue is dermatomed to precise thickness.
4. Approximately 5 mL of receiving media is added to the receiving
compartment. The
media is selected to ensure sink conditions.
5. The tissue is placed in a franz diffusion cell, which includes a donor
compound, a
donor chamber, a membrane, sampling port, receptor chamber, stir bar, and a
heater/circulator.
6. Approximately 0.5 mL of donor solution is applied or 8 mm circular film and
wetted
with 500 1..t.L PBS buffer.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
7. Samples are taken from the receiving chamber at given intervals and
replaced with
fresh media.
Example 5
Transbuccal delivery of doxepin
The following is an exemplary permeation study on the transbuccal delivery of
doxepin.
The studies were conducted under a protocol approved by the Animal
Experimentation Ethics
Committee of the University of Barcelona (Spain) and the Committee of Animal
Experimentation of the regional autonomous government of Catalonia (Spain).
Female pigs 3-4-
months-old were used. The porcine buccal mucosa from the cheek region was
excised
immediately after the pigs were sacrificed in the animal facility at Bellvitge
Campus (University
of Barcelona, Spain) using an overdose of sodium thiopental anesthesia. The
fresh buccal tissues
were transferred from the hospital to the laboratory in containers filled with
Hank's solution. The
remaining tissue specimens were stored at -80 C in containers with a PBS
mixture containing
.. 4% albumin and 10% DMSO as cryoprotective agents.
For the permeation studies, the porcine buccal mucosa was cut to 500 +/- 50
p.m thick
sheets, which contributes to the diffusional barrier (Buccal bioadhesive drug
delivery ¨ A
promising option for orally less efficient drugs Sudhakar et al., Journal of
Controlled Release
114 (2006) 15-40), using an electric dermatome (GA 630, Aesculap, Tuttlingen,
Germany) and
.. trimmed with surgical scissors in adequate pieces. The majority of the
underlying connective
tissue was removed with a scalpel.
Membranes were then mounted in specially designed membrane holders with a
permeation orifice diameter of 9 mm (diffusion area 0.636 cm2). Using the
membrane holder,
each porcine buccal membrane was mounted between the donor (1.5 mL) and the
receptor (6
mL) compartments with the epithelium side facing the donor chamber and the
connective tissue
region facing the receiver of static Franz-type diffusion cells (Vidra Foc
Barcelona, Spain)
avoiding bubbles formation.
Infinite dose conditions were ensured by applying 100 !IL as donor solution of
a saturated
doxepin solution into the receptor chamber and sealed by Parafilmimmediately
to prevent water
.. evaporation. Prior to conducting the experiments, the diffusion cells were
incubated for 1 h in a
water bath to equalize the temperature in all cells (37 +/- C). Each cell
contained a small
51
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Teflonl coated magnetic stir bar which was used to ensure that the fluid in
the receptor
compartment remained homogenous during the experiments.
Sink conditions were ensured in all experiments by initial testing of doxepin
saturation
concentration in the receptor medium. Samples (300 [tL) were drawn via syringe
from the center
of the receptor compartment at pre-selected time intervals (0.1, 0.2, 0.3,
0.7, 1, 2, 3, 4, 5 and 6 h)
for 6 h. The removed sample volume was immediately replaced with the same
volume of fresh
receptor medium (PBS; pH 7.4) with great care to avoid trapping air beneath
the membrane.
Additional details can be found in A. Gimemo, et al. Transbuccal delivery of
doxepin: Studies
on permeation and histological evaluation, International Journal of
Pharmaceutics 477 (2014),
650-654, which is incorporated by reference herein.
Example 6
Oral transmucosal delivery
Porcine oral mucosal tissue has similar histological characteristics to human
oral mucosal
tissue (Heaney TG, Jones RS, Histological investigation of the influence of
adult porcine
alveolar mucosal connective tissues on epithelial differentiation. Arch Oral
Biol 23 (1978) 713-
717; Squier CA, and Collins P, The relationship between soft tissue
attachment, epithelial
downgrowth and surface porosity. Journal of Periodontal Research 16 (1981) 434-
440). Lesch et
al. (The Permeability of Human Oral Mucosa and Skin to Water, J Dent Res 68
(9), 1345-1349,
1989) reported that the water permeability of porcine buccal mucosa was not
significantly
different from human buccal mucosa but the floor of the mouth was more
permeable in human
tissue than in pig tissue. Comparisons between fresh porcine tissue specimens
and those stored at
-80 C revealed no significant effect on permeability as a result of freezing.
Porcine buccal
mucosal absorption has been studied for a wide range of drug molecules both in
vitro and in vivo
(see, e.g., Table 1 of M. Sattar, Oral transmucosal drug delivery ¨ current
status and future
prospects, International Journal of Pharmaceutics 471 (2014) 498-506), which
is incorporated
by reference herein. Typically, in vitro studies involve mounting excised
porcine buccal tissue in
Ussing chambers, Franz cells or similar diffusion apparatus. The in vivo
studies described in the
literature involve the application of the drug as a solution, gel or
composition to the buccal
mucosa of pigs followed by plasma sampling.
Nicolazzo et al. (The Effect of Various in Vitro Conditions on the
Permeability
Characteristics of the Buccal Mucosa, Journal of Pharmaceutical Sciences
92(12) (2002) 2399-
2410) investigated the effects of various in vitro conditions on the
permeability of porcine buccal
52
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
tissue using caffeine and oestradiol as model hydrophilic and lipophilic
molecules. Drug
permeation in the buccal mucosa was studied using modified Ussing chambers.
Comparative
permeation studies were performed through full thickness and epithelial
tissues, fresh and frozen
tissues. Tissue integrity was monitored by the absorption of the fluorescein
isothiocyanate
(FITC)-labeled dextran 20 kDa (FD20) and tissue viability was assessed using
an MTT (344,5-
dimethylthiazol-2-y1]-2,5-diphenyltetrazolium bromide) biochemical assay and
histological
evaluation. Permeability through the buccal epithelium was 1.8-fold greater
for caffeine and
16.7-fold greater for oestradiol compared with full thickness buccal tissue.
Flux values for both
compounds were comparable for fresh and frozen buccal epithelium although
histological
evaluation demonstrated signs of cellular death in frozen tissue. The tissue
appeared to remain
viable for up to 12 h postmortem using the MTT viability assay which was also
confirmed by
histological evaluation.
Kulkarni et al. investigated the relative contributions of the epithelium and
connective
tissue to the barrier properties of porcine buccal tissue. In vitro permeation
studies were
conducted with antipyrine, buspirone, bupivacaine and caffeine as model
permeants. The
permeability of the model diffusants across buccal mucosa with thickness of
250, 400, 500, 600,
and 700 p.m was determined. A bilayer membrane model was developed to
delineate the relative
contribution to the barrier function of the epithelium and the connective
tissue. The relative
contribution of the connective tissue region as a permeability barrier
significantly increased with
increasing mucosal tissue thickness. A mucosal tissue thickness of
approximately 500 p.m was
recommended by the authors for in vitro transbuccal permeation studies as the
epithelium
represented the major permeability barrier for all diffusants at this
thickness. The authors also
investigated the effects of a number of biological and experimental variables
on the permeability
of the same group of model permeants in porcine buccal mucosa (Porcine buccal
mucosa as in
vitro model: effect of biological and experimental variables, Kulkarni et al.,
J Pharm Sci. 2010
99(3):1265-77). Significantly, higher permeability of the permeants was
observed for the thinner
region behind the lip (170-220 p.m) compared with the thicker cheek (250-280
p.m) region.
Porcine buccal mucosa retained its integrity in Kreb's bicarbonate ringer
solution at 4 C for 24 h.
Heat treatment to separate the epithelium from underlying connective tissue
did not adversely
affect its permeability and integrity characteristics compared with surgical
separation.
53
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Additional details can be found at M. Sattar, Oral transmucosal drug delivery
¨ current
status and future prospects, International Journal of Pharmaceutics 471 (2014)
498-506, which
is incorporated by reference herein.
Example 7
Cryopreservation of buccal mucosa
Different areas of porcine buccal mucosa have different pattern of
permeability, there is
significantly higher permeability in the region behind the lips in comparison
to cheek region,
because in porcine buccal mucosa, the epithelium acts as a permeability
barrier, and the
thickness of the cheek epithelium is greater than that of the region behind
the lips (Harris and
Robinson, 1992). In exemplary permeation studies, the fresh or frozen porcine
buccal mucosa
from the same area was cut to 500 50 p.m thick sheets, which contributes to
the diffusional
barrier (Sudhakar et al., 2006), were obtained using an electric dermatome
(model GA 630,
Aesculap, Tuttlingen, Germany) and trimmed with surgical scissors in adequate
pieces. All
devices utilized were previously sterilized. The majority of the underlying
connective tissue was
removed with a scalpel. Membranes were then mounted in specially designed
membrane holders
with a permeation orifice diameter of 9 mm (diffusion area 0.63 cm2). Using
the membrane
holder, each porcine buccal membrane was mounted between the donor (1.5 mL)
and the
receptor (6 mL) compartments with the epithelium facing the donor chamber and
the connective
tissue region facing the receiver of static Franz-type diffusion cells (Vidra
Foc, Barcelona,
Spain) avoiding bubbles formation. Experiments were performed using PP, which
has lipophilic
characteristics (logP = 1.16; n-octanol/PBS, pH 7.4), ionisable (pKa = 9.50)
and a MW= 259.3
g/mol, as a model drug (Modamio et al., 2000).
Infinite dose conditions were ensured by applying 300 tL as a donor solution
of a
saturated solution of PP (CO = 588005 5852 g/mL at 370 1 C, n = 6), in
PBS (pH 7.4) into
the receptor chamber and sealed by Parafilm immediately to prevent water
evaporation.
Prior to conducting the experiments, the diffusion cells were incubated for 1
h in a water
bath to equilibrate the temperature in all cells (37 1 C). Each cell
contained a small Teflon
coated magnetic stir bar which was used to ensure that the fluid in the
receptor compartment
remained homogenous during the experiments. Sink conditions were ensured in
all experiments
after initial testing of PP saturation concentration in the receptor medium.
Samples (300 L) were drawn via syringe from the center of the receptor
compartment at
the following time intervals: 0.25, 0.5, 1, 2, 3, 4, 5 and 6 h. The removed
sample volume was
54
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
immediately replaced with the same volume of fresh receptor medium (PBS; pH
7.4) with great
care to avoid trapping air beneath the dermis. Cumulative amounts of the drug
( g) penetrating
the unit surface area of the mucosa membrane (cm2) were corrected for sample
removal and
plotted versus time (h). The diffusion experiments were carried out 27 times
for the fresh and 22
times for the frozen buccal mucosa.
Additional details can be found at S. Amores, An improved cryopreservation
method for
porcine buccal mucosa in ex vivo drug permeation studies using Franz diffusion
cells, European
Journal of Pharmaceutical Sciences 60 (2014) 49-54.
Example 8
Permeation of quinine across sublingual mucosa sections
Since porcine and human oral membranes are similar in composition, structure
and
permeability measurements, porcine oral mucosa is a suitable model for human
oral mucosa.
Permeability across the porcine oral mucosa is not metabolically linked
therefore it is not
important for the tissue to be viable.
To prepare the porcine membranes, porcine floor of mouth and ventral
(underside)
tongue mucosa membranes were excised by blunt dissection using a scalpel. The
excised mucosa
were cut into approximately 1 cm squares and frozen on aluminium foil at ¨20 C
until used (<2
weeks). For non-frozen ventral surface of porcine tongue, the mucosa was used
in the permeation
studies within 3 h of excision.
The permeability of the membranes to quinine was determined using all-glass
Franz
diffusion cells with a nominal receptor volume of 3.6 mL and diffusional area
of 0.2 cm2. The
cell flanges were greased with high performance vacuum grease and the
membranes mounted
between the receptor and donor compartments, with the mucosal surface
uppermost. Clamps
were used to hold the membranes into position before the receptor compartments
were filled with
degassed phosphate buffered saline (PBS), pH 7.4. Micromagnetic stirrer bars
were added to the
receptor compartments and the complete cells were placed in a water bath at 37
C. The
membranes were equilibrated with PBS applied to the donor compartments for 20
min before
being aspirated with a pipette. Aliquots of 54, of the quinine solution or 100
tL of the saturated
solutions of Q/2-HP-I3-CD complex in different vehicles were applied to each
of the donor
compartments. In the study to determine the effect of saliva on the permeation
of quinine across
the ventral surface of the tongue, 100
of sterile saliva was added to the donor compartments
before adding 5 tL of the quinine solution.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
At 2, 4, 6, 8, 10 and 12 h, the receptor phases were withdrawn from the
sampling ports
and aliquots of 1 mL samples were transferred to HPLC autosampler vials,
before being replaced
with fresh PBS stored at 37 C. Apart from the studies involving Q/2-HP-0-CD
saturated
solutions (where an infinite dose was applied at the start of the
experiments), 5 of the
respective quinine solution was reapplied to the donor phase up to 10 h. The
purpose of this was
to represent a hypothetical in-use finite dosing regimen based upon an
interval of 2 h between
doses. At least 3 replicates were carried out for each study.
Additional details can be found at C. Ong, Permeation of quinine across
sublingual
mucosa, in vitro, International Journal of Pharmaceutics 366 (2009) 58-64.
Example 9
Ex-Vivo Initial Study ¨ Form of the API
In this example, the permeation of Epinephrine Base was tested ¨ solubilized
in situ vs.
the inherently soluble Epinephrine Bitartrate and no differences were found.
Epinephrine
Bitartrate was selected for further development based on ease of processing.
Flux was derived as
slope of the amount permeated as a function of time. Steady state flux
extrapolated from the
plateau of flux vs time curve multiplied by the volume of receiver media. The
graph in Figure
2A shows average amount permeated vs. time, with 8.00 mg/mL Epinephrine
bitartrate and 4.4
mg/mL Epinephrine base solubilized. The graph in Figure 2B shows average flux
vs. time,
with 8.00 mg/mL Epinephrine bitartrate and 4.4 mg/mL Epinephrine base
solubilized.
Average Steady State
Donor Solution
Flux (uacm2*min)
Epinephrine Base (conc 4.4mg/mL) 0.512
Epinephrine Bitartrate (corm 8.00mg/m1) 0.466
Example 10
Concentration dependence on permeation/flux
In this study, ex-vivo permeation of Epinephrine Bitartrate as a function of
concentration
was studied. Figure 3 shows ex-vivo permeation of epinephrine bitartrate as a
function of
concentration. The study compared concentrations of 4 mg/mL, 8 mg/mL, 16 mg/mL
and 100
mg/mL. Results showed that increasing concentration resulted in increased
permeation, and
level of enhancement diminishes at higher loading. The study compared
concentrations of 4
mg/mL, 8 mg/mL, 16 mg/mL and 100 mg/mL.
56
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Average Steady
Donor Solution State Flux
(ugfcm2*min)
Epinephrine Bitartrate (conc 4mg/mL) 0.167
Epinephrine Bitartrate (conc Bmg/mL) 0.466
Epinephrine Bitartrate (conc 16mg/mL)* L317
Epinephrine Bitartrate (conc 100mg/mL) 2.942
Ratio of Theoretical
Donor Solution
enhancement enhancement
Epinephrine Bitartrate (4.00mg/mL) N/A N/A
Epinephrine Bitartrate (8.00mg/mL) 2.8 2
Epinephrine Bitartrate (16.00mg/mL) 7.9 4
Epinephrine Bitartrate (100.00mgfing 17.6 25
Example 11
Influence of pH
In this example, the permeation of Epinephrine Bitartrate as a function of
solution pH
was studied. In this example, acidic conditions were explored for the ability
to promote stability.
The results showed that pH 5 was slightly more favorable as compared to pH 3.
The inherent pH
of epinephrine bitartrate in solution in the concentration ranges explored is
4.5-5. No pH
adjustment with buffers was required.
Figure 4 shows permeation of epinephrine bitartrate as a function of solution
pH. Acidic
conditions were explored to promote stability. The results compared
Epinephrine Bitartrate pH
3 buffer and Epinephrine Bitartrate pH 5 buffer, and found that the
Epinephrine Bitartrate pH 5
buffer was slightly favorable.
Example 12
Influence of enhancers on permeation of epinephrine
In this example, the permeation of epinephrine to test for transmucosal
delivery was
studied as the amount permeated ( g) vs. time (in minutes). The following
enhancers were
screened for concentration effects in a solution containing 16.00 mg/mL
Epinephrine. The graph
on Figure 5 demonstrates the results of these enhancers as a function of time.
57
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Average Steady State Flux Percent
Legend Enhancer ( g/cm2*min) Enhancement
None N/A
No Enhancer 1.317
3% Labrasol
Caprylocaproyl polyoxy1-8
Enhancer A glycerides 5.208 395
3% Propylene glycol
late monocapry
Enhancer B 2.385 181
Enhancer C 3% Polyglycery1-3 oleate 1.482 112
3% Oleoyl polyoxy1-6
Enhancer D glycerides
0.281 21
3% TDM
Enhancer E 2.642 201
3% SGDC
Enhancer F 0.342 26
3% Lauroyl polyoxy1-32
Enhancer G glycerides 1.641 125
3% Ethanol
Enhancer H 0.163 12
6% Ethanol
Enhancer I 0.254 19
6% Labrasol
Caprylocaproyl polyoxy1-8
Enhancer J glycerides 4.444 337
6% Polyglycery1-3 oleate
Enhancer K 0.306 23
3% clove oil
Enhancer L 8.216 624
Enhancers were selected and designed with functionality influencing different
barriers in
the mucosa. While all tested enhancers did improve the amout permeated over
time, clove oil
and Labrasol, in particular have shown significantly and unexpectedly high
enhancement of
permeation.
58
CA 03076815 2020-03-23
WO 2019/067667 PCT/US2018/053026
set 2 (A,B,C) steady
std dev set set 2 (D,E,F)
Time average std dev
state
2 amount average flux
(min) amount set 2
flux
perm (p,g/cm2*min)
permeated average
30 0 0 0 0
45 2.5 4.33012702 0.260416667
0.4510549 1.317274
8.00mg/0.500mL 60 5.5 7.08872344 0.3125
0.3125
40mg/ strip 120 31.33333333 26.1549868
0.672743056 0.5050226
no enhancer 180 72.66666667 58.215834
1.076388889 0.8496863
240 112.1666667 80.1878004 1.028645833 0.5733604
300 160.1666667 103.254943 1.25 0.6036108
360 213.3333333 131.305306 1.384548611 0.7308311
set 1 (A,B,C)
average std dev set set 1 (A, B,C)
Time std dev
steady
amount 1 amount average flux
(min) set 1
state
permeated perm (p,g/cm2*min)
flux
(ug)
average
0 0 0 0 0
30 0.5 0.8660254 0.026041667
0.0451055 5.208333
8.00mg/0.500mL 45
2.666666667 0.28867513 0.225694444 0.0601407
40mg/ strip 60 4 1.80277564 0.138888889
0.1674245
Labrasol 120 28 16.3935963 0.625
0.3836177
180 98 53.3291665 1.822916667
0.9701176
240 238.1666667 93.0017921 3.650173611 1.136722
300 421.1666667 115.153521 4.765625 0.6675003
360 638.1666667 130.709921 5.651041667 0.4732495
set 2 (A" ) B Cl steady
std dev set set 2 (D,E,F)
Time average 2 amount average flux std dev
state
(min) amount set 2 flux
perm (p,g/mL*min)
permeated average
0 0 0 0 0
30 0.00 0 0 0
8.00mg/0.500mL 45 0.00 0 0 0
1.931424
40mg/strip 60 0.00 0 0 0
capryol 90 120 9.17
5.79511288 0.238715278 0.1509144
180 38.67 16.7655401 0.768229167
0.2864583
240 88.50 30.2654919 1.297743056
0.4018216
300 150.67 39.6936183 1.618923611
0.3269068
360 236.83 51.9358579 2.243923611
0.4999616
59
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
set 2 (A,B,C) std dev set set 2 (D,E,F) steady
Time average 2 amount average flux std
dev state
(min) amount set 2 flux
perm ( g/mL*min)
permeated average
0 0.8 1.78885438 0 0
30 10.80 13.325727 0.520833333
0.7186273
8.00mg/0.500mL 45 20.90
22.1624683 1.052083333 0.9356173 1.481771
40mg/strip 60 33.00
30.8058436 1.260416667 0.9319861
plurol oleique 120 90.70 68.1951245
1.502604167 1.005753
180 157.00 107.373763 1.7265625 1.0427891
240 239.80 140.586539 2.15625 1.2085059
300 285.60 184.236397 1.192708333
1.484335
360 353.60 221.81676 1.770833333
0.993644
set 2 (A,B,C) std dev set set 2 (D,E,F) steady
Time average std dev
state
-- 2 amount average flux
(min) amount set 2 flux
perm ( g/mL*min)
permeated average
0 0.00 0.00 0.00 0.00
20 0.00 0.00 0.00 0.00
8.00mg/0.500mL 40 3.00 2.43 0.23 0.32
2.642144
40mg/strip 60 8.83 4.86 0.46 0.35
TDM 120 41.33 15.08 0.85 0.49
180 99.75 30.17 1.52 0.79
240 179.92 48.30 2.09 0.98
300 276.92 72.35 2.53 1.19
360 382.83 102.02 2.76 1.38
set 2 (A,B,C) std dev set set 2 (D,E,F) steady
Time average 2 amount average flux std
dev state
(min) amount set 2 flux
perm ( g/mL*min)
permeated average
0 0 0 0 0
30 0.00 0 0 0
8.00mg/0.500mL 45 0.67
1.30384048 0.069444444 0.1261521 0.341797
40mg/strip 60 1.58
1.94935887 0.095486111 0.1063147
SG DC 120 7.00
9.44325156 0.141059028 0.1910856
180 16.17 22.0323626 0.238715278
0.316132
240 28.58 37.6191441 0.323350694
0.3977149
300 43.00 54.3927844 0.375434028
0.4112124
360 54.83 65.7976063 0.308159722
0.3008536
CA 03076815 2020-03-23
WO 2019/067667 PCT/US2018/053026
set 2 (A,B,C) std dev set set 2 (D,E,F) steady
Time average 2 amount average flux std
dev state
(min) amount set 2 flux
perm (p,g/mL*min)
permeated average
0 0.8 1.15108644 0 0
30 1.10 1.51657509 0.015625 0.0232924
8.00mg/0.500mL 45 1.10 1.51657509 0 0 0.28125
40mg/strip 60 1.10 1.51657509 0 0
Labrafil 120 4.00
4.89897949 0.075520833 0.0984775
180 9.10 10.9167303 0.1328125 0.1671035
240 15.20 18.7169709 0.158854167
0.2034753
300 25.70 29.8487856 0.2734375 0.2910091
360 36.80 43.3093523 0.2890625 0.3532943
set 2 (A,B,C) steady
std dev set set 2 (D,E,F)
Time average 2 amount average flux std
dev state
(min) amount set 2 flux
perm (p,g/mL*min)
permeated average
0 0 0 0 0
20 0.33 0.89442719 0.026041667
0.0637888
8.00mg/0.500mL 40 3.83 5.94138031
0.2734375 0.378725 1.629774
40mg/strip 60 11.50
17.1850225 0.598958333 0.8405853
Gelucire 44/14 120 41.58
48.5059275 0.783420139 0.8042794
180 91.92 82.5124233 1.310763889
0.9525224
240 150.17 118.949569 1.516927083
0.9914576
300 217.50 158.792947 1.753472222
1.0756081
360 275.33 189.967563 1.506076389
0.9083155
set 2 (A,B,C)
steady
Time average std dev set set 2
(D,E,F) state
(min) amount 2 amount average flux std
dev flux
permeated perm (p,g/cm2*min) set 2
average
0 0 0 0 0
20 0 0 0 0
8.00mg/0.500mL 40 28.66666667 27.360251 2.239583333 2.1375196 8.270399
40mg/strip 60 96.5
79.5424415 5.299479167 4.0954816
clove oil 120 389.6666667 278.072533
7.634548611 5.2070528
180 688.6666667 451.678628 7.786458333 4.5210426
240 1009.166667 603.252089 8.346354167 3.9590828
300 1333.5 759.653046 8.446180556
4.1168559
360 1644.333333 878.2762 8.094618056 3.2301203
61
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
set 2 (A,B,C)
steady
std dev set set 2 (D,E,F)
Time average std dev
state
2 amount average flux
(min) amount set 2 flux
perm (ug/mL*min)
permeated
average
8.00mg/0.500mL 0 0 0 0 0
40mg/strip 20 0 0 0 0
3.161892
3% Labrasol
+1%TDM 40
1.666666667 2.88675135 0.130208333 0.2255274
60 6.833333333 11.8356805 0.403645833 0.6991351
120 68.83333333 92.6907942 1.614583333 2.1084505
180 103.6666667 101.539812 0.907118056 0.3351021
240 180 130.184484 1.987847222
0.7476876
300 291.5 149.81572 2.903645833
0.5193664
360 422.8333333 164.032263 3.420138889 0.5530917
Example 13
Impact of enhancers on epinephrine release
Release profiles of epiphrine were studied to determine the impact of
enhancers (Labrasol
and clove oil) on epinephrine release. Figure 6A shows the epinephrine release
from different
polymer platforms. Figure 6B shows the impact of enhancers on epinephrine
release. The
results showed that the amount permeated leveled off after about 40 minutes to
be between
approximately 3250 and 4250 ug. The tested enhancers were shown not to
restrict the release of
epinephrine from the matrix.
62
CA 03076815 2020-03-23
WO 2019/067667 PCT/US2018/053026
Example 14
Accelerated stability
The stabilizer loading variants were tested.
Formulation 10 with Formulation 10 with
Formulation 10 0.25% Stabilizer 1%
Stabilizer
Time (weeks) @
40 C/75% R.H. EPI mg/film EPI mg/film EPI mg/film
0 39.3 37.9 38.3
2 35.2 36.8 34.7
4 38.2 36.8 36.2
8 37.7 35.6 35.1
12 36.1 35.4 35.1
Example 15
Impact of Enhancer
A pharmacokinetic model in the male Yucatan, miniature swine was studied. The
graph
on Figure 7 shows the results of a pharmacokinetic model in the male Yucatan,
miniature swine.
The study compares a 0.3 mg Epipen, a 0.12 mg Epinephrine IV and a placebo.
The impact of no enhancer is shown in Figure 8 on the concentration profiles
of a 0.3 mg
Epipen and a 40 mg Epinephrine film with no enhancer.
The impact of enhancer 3% Labrasol is shown in Figure 9, which shows the
impact of
Enhancer A (Labrasol) on the concentration profiles of a 40 mg Epinephrine
film vs. a 0.3 mg
Epipen. Figure 10 shows the impact of Enhancer L (clove oil) on the
concentration profiles of
two 40 mg Epinephrine films (10-1-1) and (11-1-1) vs. a 0.3 mg Epipen.
In addition, the influence of film dimensions and impact of clove oil (3%) is
also shown
in Figure 11. This study was carried out comparing 0.30 mg EpiPen (n=4), a 40
mg Epinephrine
Film (10-1-1) (n=5) and a 40 mg Epinephrine Film (11-1-1) (n=5). The
concentration vs. time
profiles followed subligual or intramuscular epinephrine administration to
male miniature swine.
63
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Studies were performed to vary the ratio of ephinephrine to an enhancer. These
studies
were also concentration vs. time profiles following subligual or intramuscular
epinephrine
administration to male miniature swine. Varying the ratio of Epinephrine to
clove oil (Enhancer
L) produced the results shown in Figure 12. This study was carried out
comparing 0.30 mg
EpiPen (n=4), a 40 mg Epinephrine Film (12-1-1) (n=5) and a 20 mg Epinephrine
Film (13-1-1)
(n=5).
Example 16
A varying dose was carried out in constant matrix with enhancer Labrasol (3%)
and clove
oil (3%) are shown in Figures 13 and 14 respectively. The study in Figure 13
was carried out
comparing 0.30 mg EpiPen (n=4), a 40 mg Epinephrine Film (18-1-1) (n=5) and a
30 mg
Epinephrine Film (20-1-1) (n=5). The study in Figure 14 was carried out
comparing 0.30 mg
EpiPen (n=4), a 40 mg Epinephrine Film (19-1-1) (n=5) and a 30 mg Epinephrine
Film (21-1-1)
(n=5).
These studies were also concentration vs. time profiles following subligual
or
intramuscular epinephrine administration to male miniature swine.
Example 17
A pharmacokinetic model in the male miniature swine was studied to determine
the
impact of an enhancer (farnesol) on epinephrine concentration over time. The
graph on Figure
17 shows the epinephrine plasma concentration (in ng/mL) as a function of time
(in minutes)
following sublingual or intramuscular administration of a farnesol permeation
enhancer. This
study compares a 0.3 mg Epipen (n=3), a 30 mg Epinephrine Film 31-1-1 (n=5)
and a 30 mg
Epinephrine Film 32-1-1 (n=5) each Epinephrine Film being formulated with a
farnesol
enhancer. As shown in this figure, the 31-1-1 film demonstrates enhanced
stability of
epinephrine concentration starting at about 30-40 minutes until approximately
130 minutes.
The graph in Figure 18 is taken from the same study as Figure 17, but shows
exclusively
the data points comparing the 0.3 mg Epipen against the 30 mg Epinephrine Film
31-1-1 (n=5).
The graph in Figure 19 is taken from the same study as Figure 17, but shows
exclusively
the data points comparing the 0.3 mg Epipen against the 30 mg Epinephrine Film
32-1-1 (n=5).
Example 18
Referring to Figure 20, this graph shows a pharmacokinetic model in the male
miniature
swine was studied to determine the impact of an enhancer (farnesol) on
epinephrine
64
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
concentration over time following sublingual or intramuscular administration.
The epinephrine
plasma concentration (in ng/mL) is shown as a function of time (in minutes)
following
sublingual or intramuscular administration of a farnesol permeation enhancer
in Epinephrine
films. The study compared data from three 0.3 mg Epipens against five 30 mg
Epinephrine films
(32-1-1). The data shows the Epinephrine films film having enhanced stability
of epinephrine
concentration starting at about 20-30 minutes until approximately 130 minutes.
Example 19
In one embodiment, an epinephrine pharmaceutical composition film can be made
with
the following formulation:
Formulation A
MATERIAL WT % dry WT % wet mg/Strip
EPINEPHRINE bitartrate 46.40 18.56 54.56
Hydroxypropylmethyl cellulose 11.54 4.61 13.57
Polyvinyl pyrrolidone 27.92 11.17 32.84
Glycerol monooleate 0.58 0.23 0.68
Polyethylene Oxide 1.16 0.46 1.36
Polysorbate 0.58 0.23 0.68
Phytoextract 9.98 3.99 9.97
Stabilizer 0.12 0.05 0.14
Buffer 0.58 0.23 0.68
Artifical sweetener 1.16 0.46 1.36
Linoleic acid 0.0037 0.00 0.00
Farnesol
Yellow # 5
TOTAL 100.00 40.00 115.84
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Example 20
An epinephrine pharmaceutical composition film was made with the following
formulation:
Formulation B
MATERIAL WT % dry WT % wet mg/Strip
EPINEPHRINE bitartrate 46.17 18.47 54.29
Hydroxypropylmethyl cellulose 11.48 4.59 13.50
Polyvinyl pyrrolidone 27.78 11.11 32.67
Glycerol monooleate 0.58 0.23 0.68
Polyethylene Oxide 1.15 0.46 1.35
Polysorbate 0.58 0.23 0.68
Phytoextract 9.93 3.97 9.92
Stabilizer 0.12 0.05 0.14
Buffer 0.58 0.23 0.68
Artifical sweetener 1.15 0.46 1.35
Linoleic acid 0.50 0.20 0.59
Farnesol
Yellow # 5
TOTAL 100.00 40.00 115.85
66
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Example 21
In another embodiment, pharmaceutical film compositions were made with the
following
formulation:
Formulation C
MATERIAL WT % dry WT % wet mg/Strip
EPINEPHRINE bitartrate 46.35 18.54 54.51
Hydroxypropylmethyl cellulose 11.53 4.61 13.55
Polyvinyl pyrrolidone 27.90 11.16 32.80
Glycerol monooleate 0.58 0.23 0.68
Polyethylene oxide 1.16 0.46 1.36
Polysorbate 0.58 0.23 0.68
Phytoextract 9.97 3.99 9.96
Stabilizer 0.12 0.05 0.14
Buffer 0.58 0.23 0.68
Artifical sweetener 1.16 0.46 1.36
Linoleic acid
Farnesol 0.10 0.04 0.06
Yellow # 5
TOTAL 100.00 40.00 115.78
67
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Example 22
In another embodiment, pharmaceutical film compositions were made with the
following
formulation:
Formulation D
MATERIAL WT % dry WT % wet mg/Strip
EPINEPHRINE bitartrate 46.07 18.43 54.52
Hydroxypropylmethyl cellulose 11.46 4.58 13.56
Polyvinyl pyrrolidone 27.73 11.09 32.81
Glycerol monooleate 0.57 0.23 0.68
Polyethylene oxide 1.15 0.46 1.36
Polysorbate 0.57 0.23 0.68
Phytoextract 9.91 3.96 9.96
Stabilizer 0.11 0.05 0.14
Buffer 0.57 0.23 0.68
Artificial sweetener 1.15 0.46 1.36
Linoleic acid 0.10 0.04 0.06
Farnesol 0.50 0.20 0.29
Yellow # 5 0.10 0.04 0.06
TOTAL 100.00 40.00 116.16
68
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Example 23
Referring to Figure 21, this graph shows a pharmacokinetic model (logarithmic
scale) in
the male miniature swine studied to determine the impact of an enhancer (6%
clove oil and 6%
Labrasol) on epinephrine plasma concentration over time following sublingual
or intramuscular
administration. The epinephrine plasma concentration (in ng/mL) is shown as a
function of time
(in minutes) following sublingual or intramuscular administration of a
farnesol permeation
enhancer in Epinephrine films. The data shows the Epinephrine films film
having enhanced
stability of epinephrine concentration starting at just after the 10 minute
time point through about
30 minutes, and until approximately 100 minutes.
Referring to Figure 22, this graph shows a pharmacokinetic model of the
Epinephrine
film formulation in the male miniature swine as referenced in Figure 21
compared against the
average data collected from a 0.3 mg Epipen (indicated in diamond data
points). As the data
indicates, the average plasma concentration for the 0.3 mg Epipen peaked
between 0.5 and 1
ng/mL. By contrast, the Epinephrine film formulation peaked between 4 and 4.5
ng/mL.
Example 24
Referring to Figure 23, this graph shows a pharmacokinetic model in the male
miniature
swine studied to determine the impact of an enhancer (9% clove + 3% Labrasol)
on epinephrine
concentration over time following sublingual or intramuscular administration
across 7 animal
models. The general peak concentration was achieved between 10-30 minutes.
Alprazolam Data
Example 25
Referring to Figure 24A, Figure 24B, and Figure 24B, these graphs represent
data from
a male miniature swine study comparing alprazolam plasma concentration over
time (in hours)
upon sublingual administration of oral alprazolam disintegrating tablet (ODT)
and alprazolam
pharmaceutical composition film.
Figure 24A shows mean data from alprazolam ODT (Group 1). Peak concentration
of
between 7-12 ng/mL was achieved at approximately 1-8 hours.
Figure 24B shows mean data from alprazolam pharmaceutical composition film
(Group
2). Peak concentration of between 5-17 ng/mL, including greater than 5 ng/mL,
greater than 10
ng/mL, greater than 12 ng/mL, greater than 15 ng/mL, greater than 17 ng/mL,
less than 17
69
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
ng/mL, less than 15 ng/mL, less than 12 ng/mL, less than 10 ng/mL, less than 5
ng/mL, was
achieved between between 10 minutes to 4 hours, including greater than 10
minutes, greater than
20 minutes, greater than 30 minutes, greater than 45 minutes, greater than 1
hour, greater than
1.5 hours, greater than 2 hours, greater than 2.5 hours, greater than 3 hours,
greater than 3.5
hours, or about 4 hours, less than 4 hours, less than 3.5 hours, less than 3
hours, less than 2.5
hours, less than 2 hours, less than 1.5 hours, less than 1 hour, less than 45
minutes, less than 30
minutes, or less than 20 minutes.
Figure 24C shows mean data from alprazolam pharmaceutical composition film
from
another group of male miniature swine (Group 3). Peak concentration of between
5-17 ng/mL,
including greater than 5 ng/mL, greater than 10 ng/mL, greater than 12 ng/mL,
greater than 15
ng/mL, greater than 17 ng/mL, less than 17 ng/mL, less than 15 ng/mL, less
than 12 ng/mL, less
than 10 ng/mL, less than 5 ng/mL, was achieved between 10 minutes to 4 hours,
including
greater than 10 minutes, greater than 20 minutes, greater than 30 minutes,
greater than 45
minutes, greater than 1 hour, greater than 1.5 hours, greater than 2 hours,
greater than 2.5 hours,
greater than 3 hours, greater than 3.5 hours, and about 4 hours, less than 4
hours, less than 3.5
hours, less than 3 hours, less than 2.5 hours, less than 2 hours, less than
1.5 hours, less than 1
hour, less than 45 minutes, less than 30 minutes, or less than 20 minutes.
Example 26
Referring to Figure 25 A, this graph illustrates data from a male miniature
swine study
comparing alprazolam plasma concentration over time following sublingual
administration (in
hours) of oral alprazolam disintegrating tablet (ODT) (indicated by circle
data points) and two
groups with alprazolam pharmaceutical composition film (indicated by square
and triangle data
points).
As the graph indicates, the data from the alprazolam pharmaceutical
composition film
(both groups) obtained a higher alprazolam plasma concentration of up to
approximately 15-25
mg/mL in a therapeutic window of about 30 minutes or less, including more than
10 minutes,
more than 20 minutes, about 30 minutes, more than 30 minutes, less than 30
minutes, less than
20 minutes less than 15 minutes, or less than 10 minutes.
Referring to Figure 25B, this graph indicates the individual data points from
the studies
referenced in Figure 25A.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Referring to Figure 25C, this graph indicates the individual data points from
the studies
referenced in Figure 25A from 0-1 hour.
Referring to Figure 26A, this graph indicates the individual data points for
the
alprazolam ODT referenced in Figure 25C.
Referring to Figure 26B, this graph indicates the individual data points for
the
alprazolam pharmaceutical film referenced in Figure 25C.
Referring to Figure 26C, this graph indicates the individual data points for
the
alprazolam pharmaceutical film (second group) referenced in Figure 25C.
The data from the graphs referenced above is also summarized in the following
table:
Pharmaceutical Tmax (hrs) Cmax (ng/mL) AUC ng*hr/mL
Alprazolam 2 mg (ODT) 4.25 16.18 2.90 120.61 29.63
Alprazolam 2 mg film 12-1-1 0.5 27 6.0 154.46 41.29
Alprazolam 2 mg Film 13-1-1 1.5 20.52 11.01 103.74 35.57
Alprazolam 2 mg Film 6-1-1 2.5 11.5 4.7 86.0 35.5
Alprazolam 2 mg Film 1-1-2 1 15.2 5.7 96.6 44.3
Example 27
Referring to Figure 27A, this figure illustrates mean data from a male
miniature swine
study comparing alprazolam plasma concentration over time following sublingual
administration
of oral alprazolam disintegrating tablet (ODT) (indicated by circle data
points) and two groups
with alprazolam pharmaceutical composition film (indicated by square and
triangle data points).
As the data indicates, the 0.5 mg alprazolam ODT achieved a range of peak
concentration of
about 5-6 ng/mL between 0-4 hours, including greater than 10 minutes, greater
than 20 minutes,
greater than 30 minutes, greater than 45 minutes, greater than 1 hour, greater
than 1.5 hours,
greater than 2 hours, greater than 2.5 hours, greater than 3 hours, greater
than 3.5 hours, or about
4 hours, less than 4 hours, less than 3.5 hours, less than 3 hours, less than
2.5 hours, less than 2
hours, less than 1.5 hours, less than 1 hour, less than 45 minutes, less than
30 minutes, or less
71
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
than 20 minutes. The 0.5 mg alprazolam pharmaceutical composition film
achieved peak
concentration of about 7-8 ng/mL and 6-7 ng/mL, respectively between 0-4
hours, including
greater than 10 minutes, greater than 20 minutes, greater than 30 minutes,
greater than 45
minutes, greater than 1 hour, greater than 1.5 hours, greater than 2 hours,
greater than 2.5 hours,
greater than 3 hours, greater than 3.5 hours, or about 4 hours, less than 4
hours, less than 3.5
hours, less than 3 hours, less than 2.5 hours, less than 2 hours, less than
1.5 hours, less than 1
hour, less than 45 minutes, less than 30 minutes, or less than 20 minutes.
Referring to Figure 27B, this graph illustrates mean data of alprazolam plasma
concentration over time following sublingual administration of oral alprazolam
disintegrating
tablet (ODT) (indicated by circle data points) and two groups with alprazolam
pharmaceutical
composition film (indicated by square and triangle data points) between 0-2
hours. Unlike the
ODT, the therapeutic window for the alprazolam pharmaceutical composition
films started at 10-
minutes, whereas the ODT started at approximately 17-20 minutes.
Referring to Figure 27C, this graph illustrates complete data referenced in
Fig. 27B, for
15 ODT (n=4), 0.5 mg alprazolam pharmaceutical composition film 14-1-1
(n=5), and 0.5 mg
alprazolam pharmaceutical composition film 15-1-1 (n=5).
The data from the graphs referenced above is also summarized in the following
table:
Pharmaceutical Tmax (hrs) Cmax (ng/mL) AUC ng*hr/mL
Alprazolam 0.5 mg (ODT) 1.5 5.56 1.41 34.01 14.74
Alprazolam 0.5 mg film 14-1-1 1 10.87 3.08 50.80 10.03
Alprazolam 0.5 mg Film 15-1-1 2 7.33 2.80 37.31 10.09
Example 28
In one investigation, the usability of diazepam buccal soluble film was
studied as an oral
treatment in adult patients with epilepsy. Diazepam buccal soluble film (DBSF)
is an exemplary
embodiment of the claimed subject matter. DBSF is novel dosage form of
diazepam under
development for the management of selected patients with refractory epilepsy
who require
intermittent use of diazepam to control episodes of increased seizure
activity. DBSF has the
potential to be the first orally administered treatment for this indication
and would provide an
72
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
alternative to rectal diazepam gel. DBSF is administered by placing the film
against the inner
aspect of the cheek, where it adheres, dissolves and releases drug onto the
buccal mucosa. The
DBSF dosage form is expected to be accepted by a broad range of patients with
epilepsy. A
Phase 2 pharmacokinetic study in the epilepsy monitoring unit (EMU) assessed
safety,
pharmacokinetics and usability of DBSF.
A Phase 2, multi-center, open-label, crossover study in adult subjects were
studied during
two treatment visits separated by at least three weeks. The study enrolled
male and female
subjects 17-65 years of age with a clinical diagnosis of epilepsy (tonic
clonic seizures or focal
seizures with impaired awareness) who required EMU evaluation. Subjects
received DBSF
administered in 12.5 mg during the interictal state (Treatment A) and the
ictal/periictal state
(during or within 5 min of a seizure; Treatment B). Usability endpoints for
investigator
placement of DBSF included: (1) whether successful placement was achieved, (2)
number of
attempts needed to successfully insert the film, (3) whether the subject spit
or blew out the film,
and (4) whether or not DBSF was swallowed.
The results were as follows: A total of 35 subjects received at least one dose
of DBSF.
Of those subjects, 33 subjects had usability data collected during Treatment A
and 33 subjects
had usability data collected during Treatment B. During both Treatment A and
Treatment B,
there were no unsuccessful attempts of DBSF insertion. During Treatment A, 2
(6.1%) subjects,
and during Treatment B, 1(3.0%) subject, were noted to have swallowed DBSF
during dosing. 3
(9.1%) additional subjects swallowed DBSF per the protocol which instructed
all subjects to
swallow the film if it had not completely dissolved 15 minutes after
placement. During
Treatment A, no patients spit or blew out DBSF following initial adherence to
the buccal
mucosa, while 3 (9.1%) subjects spit or blew out the film during Treatment B
following initial
placement. DBSF was generally safe and well tolerated. The most common adverse
event
possibly related to drug was somnolence reported in two (5.7 %) of 35 of
subjects. There were
no serious adverse events related to study drug, and no subject withdrew
because of an adverse
event.
This allowed the investigators to conclude that surprisingly, in the EMU
setting, DBSF
was successfully administered in both the interictal and ictal/periictal
states. This study data
supports the safe and effective use of DBSF for the management of selected
patients with
73
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
refractory epilepsy who require intermittent use of diazepam to control
episodes of increased
seizure activity. This could also support the capability for usability in the
home setting.
Example 29
Another investigation evaludated the pharmacokinetics of diazepam buccal
soluble film
.. in adult patients with epilepsy. A comparison of bioavailability with
periictal and interictal
administration was assessed.
As indicated above, dibuccal soluble film (DBSF) is an exemplary embodiment of
the
claimed subject matter. It is a novel dosage form of diazepam under
development for the
management of selected patients with refractory epilepsy who require
intermittent use of
diazepam to control episodes of increased seizure activity. In this study, the
investigators sought
to assess pharmacokinetic (PK) performance during or immediately after a
seizure. Subjects
were investigated while undergoing a clinical epilepsy monitoring unit (EMU)
evaluation and on
another PK-only visit, with the visits separated by approximately 3 weeks. The
study
investigated the diazepam maximal plasma concentration (C.), time to maximal
concentration
(T.), and partial area under the curve (partial AUC) at 2 or 4 hours in adult
subjects with
epilepsy following single doses of DBSF under interictal and ictal/periictal
conditions.
In this study, adult men and women age 17-65 years with poorly controlled
tonic clonic
seizures or focal seizures with impaired awareness (N=35) were enrolled in a
single-dose
crossover to receive DBSF 12.5 mg under both interictal (Treatment A) and
ictal/peri-ictal
.. (Treatment B) conditions in a clinical EMU setting. Plasma samples for
analysis of diazepam
were obtained pre-dose and at intervals until 2h or 4h after dosing.
Administration was classified
as interictal if there was no observed seizure activity in the preceding 4
hours and as
ictal/periictal if DBSF was given during clinically observed seizure activity
or within 5 minutes
of cessation of seizure activity. Subjects were monitored for adverse events
(AE) throughout the
study.
The results were as follows: Pharmacokinetic (PK) profiles valid for analysis
for both
treatment conditions were available for 21 subjects. Most of these subjects
had samples collected
up to 4 hours but 3 subjects were sampled only up to 2 hours. Subjects were
excluded from
analysis if both treatments were not completed (N=4), if critical PK time
points were missed as
adjudicated by an expert blinded to the concentration data (N=3), if pre-dose
diazepam
concentrations were greater than 5% of C. (N=2), or if DBSF was administered
in a manner
74
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
contrary to instructions (N=5). The table shows values for C., AUC(0,210, and
AUC(0-410
(geometric means) and the ratio of the geometric means (Treatment B/Treatment
A) with 90%
CI. Values for Cmax, AUC(0.2h), and AUC(0.4h) were surprisingly similar in
interictal and
ictal/periictal state, and median values for Tinax were not significantly
different (Table). DBSF
12.5 mg was shown to be effective, safe and well-tolerated. The most common
adverse event
possibly related to drug was somnolence reported in two (5.7 %) of 35 of
subjects. There were no
serious adverse events related to study drug, and no subject withdrew because
of an adverse
event.
These results demonstrate surprisingly that a single dose of DBSF 12.5 mg
administered
to adults with epilepsy provides exposure to diazepam under both ictal/peri-
ictal conditions that
is comparable to that obtained under inter-ictal conditions.
CA 03076815 2020-03-23
WO 2019/067667
PCT/US2018/053026
Table of Pharmacokinetic Parameters following DBSF 12.5mg in the Interictal
and
Ictal/Periictal State
2-hour Profiles; N=21
A. Interictal B. Ictal/periictal
Ratio of Geometric 90% CI (%)2
Means B/A (%)1
Geometric Mean Geometric Mean
Cinax (ng/mL) 198.76 184.40 92.77
74.5 ¨ 112.53
AUC(0_2h) (ng.h/mL) 264.57 248.06 93.76 73.86 ¨
119.02
Median Median
T(h) 0.680 0.983
4-hour Profiles; N=18
A. Interictal B. Ictal/periictal Ratio B/A 90% CI
Geometric Mean Geometric Mean
Cinax (ng/mL) 190.34 179.96 95.54
73.34-121.89
AUC(0_4h) (ng.h/mL) 483.79 433.32 89.57
69.21 -115.91
Median Median
T(h) 0.767 0.533 **
'Calculated using least-squares means according to the formula: e(Difference)
X 100.
2 90% Geometric Confidence Interval using ln-transformed data.
*Difference in Tinax not significant, p = 0.8620; ** Difference in Tinax
not significant, p=0.5708
(Wilcoxon signed-rank test)
All references cited herein are hereby incorporated by reference herein in
their entirety.
Other embodiments are within the scope of the following claims.
76