Note: Descriptions are shown in the official language in which they were submitted.
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
Title: Calibration Free In-Vivo Measurement of Analytes Using Electrochemical
Sensors
[0001] CROSS-REFERENCE TO RELATED APPLICATIONS: This application claims the
benefit of priority to United States Provisional Patent Application Serial
Number 62/578,665
entitled "Calibration Free In-Vivo Measurement of Analytes Using
Electrochemical
Sensors," filed October 30, 2017, the contents of which are hereby
incorporated by reference.
[0002] STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH OR
DEVELOPMENT: This invention was made with government support under grant
number
W911NF-09-0001 U.S. Army Research Office awarded by the U.S. Army Research
Office.
The government has certain rights in the invention.
[0003] Background of the Invention
[0004] Various types of electrochemical sensors for the detection of target
molecules are
known. Many such sensors produce a current output, wherein the magnitude of
the current
output changes in response to binding of the target molecule. Electrochemical
aptamer-based
(EA-B) sensors use redox-reporter-modified, electrode-bound aptamers, wherein
binding-
induced conformational changes in the aptamer result in measurable changes in
electron
flows between the redox reporter and electrode. EA-B sensors have provided the
art with a
versatile platform for the measurement of target analytes in complex samples.
EA-B sensors
have even been demonstrated to work in vivo for the real-time detection of
drugs and other
target species in flowing whole blood. However, despite the great potential of
this platform,
E-AB sensors suffer from certain shortcomings that have limited their clinical
deployment.
[0005] Specifically, EA-B sensors, like all complex devices, suffer from
inconsistencies in
fabrication, wherein different numbers of recognition elements are present on
individual
sensors, even when manufactured in the same batch. This sensor-to-sensor
physical
variability means that outputs obtained from different sensors of the same
design may vary
significantly. Furthermore, when deployed in complex sample environments, such
as whole
blood, even the best E-AB's are subject to drift, wherein non-specific
interactions between
the aptamers and complex mixture of molecules in the sample result in variable
readings over
time. These factors dictate that calibration steps or signal correction
measures must be
performed in order to interpret sensor outputs. In the case of sensors
implanted in vivo,
1
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
where calibration is not practical or is often impossible, the sources of
error described above
present a serious obstacle to clinical implementation.
[0006] Accordingly, there remains a need in the art for novel electrochemical
sensor system
and methods of operating such sensors that enable calibration-free
measurement.
Furthermore, there remains a need in the art for a means of accurately
measuring analytes in
vivo without being confounded by signal drift. There also remains a need in
the art for
improving the performance and efficiency of existing sensing platforms.
[0007] Summary of the Invention.
[0008] Prior art electrochemical sensing methods have relied on measurements
of current
output (i.e. absolute current values), such as SWV peaks, to determine target
concentration,
which such measurements are strongly affected by sensor-to-sensor variability
and sensor
drift. The inventors of the present disclosure have advantageously discovered
that certain
aspects of the signal output kinetics of electrochemical sensors,
specifically, current decay
kinetics, are insensitive to sensor-to-sensor variability and sensor drift.
Like absolute
currents, these current decay kinetic parameters are responsive to target
binding in a
concentration-dependent manner, but unlike absolute currents, they are stable
across sensors
of a given class, and are stable over time, providing a means of avoiding the
variability
observed in absolute currents.
[0009] The inventors of the present disclosure have advantageously developed
novel
methods of operating electrochemical sensors and interpreting sensor outputs,
enabling
signals generated by different sensors or at different times to be accurately
correlated with
target concentration in the sample. The methods of the invention enable
sensors within a
class (e.g., employing the same recognition element) to be effectively
calibrated a priori and
to be deployed for extended, calibration-free measurement of target species in
complex
environments, such as whole blood in vivo.
[0010] In a first aspect, the scope of the invention encompasses methods of
obtaining and
interpreting sensor outputs to provide accurate measurements of target analyte
concentration.
In another aspect, the scope of the invention encompasses sensor systems that
may be
operated with drift-free or calibration-free measurement. In another aspect,
the scope of the
2
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
invention encompasses electrochemical sensors of a given class, wherein the
sensor's outputs
is correlated with target concentration by a relationship that is stable
across all sensors of the
class. In another aspect, the scope of the invention encompasses computer
programs,
software, and operations which enable the acquisition and interpretation of
electrochemical
sensor outputs to measure target concentration.
[0011] Brief Description of the Drawings.
[0012] Fig. 1A and 1B. Fig. 1A depicts the basic operation of an E-AB sensing
platform,
wherein an aptamer (102) bound to an electrode substrate (101) is modified
with a methylene
blue redox reporter (103). In the absence of target, electron flow (104)
between the
methylene blue and substrate is slow. When target (105) binds to the aptamer,
the resulting
conformation change alters the proximity of the reporter (103) to the
substrate (101),
increasing the rate of electron transfer (104). Fig. 1B depicts a
representative square wave
voltammetry trace for this type of E-AB sensor wherein peak current is higher
in the
presence than in the absence of target.
[0013] Fig. 2A and 2B. Fig. 2A depicts SWV titration curves for a set six of
aminoglycoside-detecting sensors using tobramycin as a target. The absolute
peak currents
generated using SWV depend not only on the concentration of the target
molecule, but also
on the number of redox-reporter-modified probes on the sensor surface. Due to
variations in
the active area of the working electrode and the density of the aptamer probes
packed onto it,
these absolute peak currents can vary significantly from sensor to sensor,
leading in turn to
large variations in raw (uncalibrated) sensor output. Fig. 2B depicts sensor
outputs of Fig.
2A when normalized by performing a calibration step.
[0014] Fig. 3. Fig. 3 depicts a current transient log-log plot of average
current vs. time
measured by chronoamperometry of aminoglycoside-binding E-AB sensors in
flowing whole
blood. The current decay is well described as the sum of two exponential
phases. From left
to right, a first phase, to the left of the dotted vertical line, is the decay
of double layer
charging current, wherein the sensors are insensitive to target. To the right
of the vertical
line, current transients are highly target dependent. In transients obtained
in the absence of
target, a slower exponential decay of faradaic current is observed. In the
presence of target
(here tobramycin), the average current lifetime is substantially reduced. The
illustrated
3
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
transients were recorded by stepping the potential from -0.1 V to -0.3 V (all
potentials
reported versus Ag/AgC1) and sampling the resultant current every 10 .is. The
solid lines are
multi exponential fits of the experimental data.
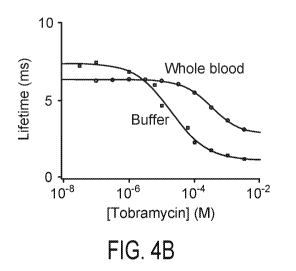
[0015] Fig. 4A, 4B, and 4C. Fig. 4A depicts average chronoamperometry-measured
current transients for aminoglycoside-binding E-AB sensors (with tobramycin as
the target)
with decaying current transients with lifetimes that decrease with increasing
target
concentration. The solid lines represent mono-exponential fits of the two
current transients.
Fig. 4B depicts the relationship between current lifetime and target
concentration in buffer
and whole blood, established for a set of aminoglycoside-binding E-AB sensors.
The
differences in lifetimes between the two sample types are likely due to
changes in electrolyte
composition and viscosity that affect electron transfer from methylene blue.
Fig. 4C depicts
tobramycin concentration measured by five sensors of the same class as used to
generate the
standard curve of Fig. 4B, but which were not used to generate the standard
curve. Using
these independent sensors to estimate the concentration of tobramycin in
flowing whole
blood by the curve generated for the sensor class, accurate and precise
measurements of
tobramycin concentration were obtained over a broad range of concentrations
without the
need to calibrate individual sensors. Measured concentration values are within
10% of the
actual (spiked) concentration of tobramycin over the range from 1 tM to 1 mM
when
challenged in undiluted whole blood. In Fig. 4B and Fig. 4C the error bars
(which are so
small as to be difficult to see in Fig. 4B) represent the standard deviation.
[0016] Fig. 5A and 5B. Fig. 5A depicts E-AB sensor output in flowing whole
blood (here
lacking target), where the the average peak current recorded from SWV drifts
significantly
over the course of a few hours (uncorrected signal). Kinetic differential
measurements
correction techniques applied to the measurements provide a corrected signal.
Fig. 4B
depicts average current amplitude and average current lifetime for the same
sensors deployed
in whole blood (lacking target) over time. The amplitude of the
chronoamperometric current
decays drift quite significantly over the test period. However, in contrast,
the current lifetimes
are stable across time and do not drift over the course of the test period.
[0017] Fig. 6A, 6B, 6C, and 6D. Fig. 6A depicts sensor placement in the
jugular vein of a
live, anesthetized rat. Sensors were encased in a 22-gauge catheter for
structural support, and
placed inside the external jugular vein at a depth of 2 cm. An infusion line
was implanted in
4
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
the opposite side to carry out drug infusions. Fig. 6B depicts real-time non-
linear regression
analysis of the current transients generated by chronoamperometry to extract
current lifetimes
and convert them to target concentration in real time. The trace is the
rolling average of 20
points. The dotted box depicts the timeframe during which tobramycin was
infused. Fig. 6C
depicts the lifetime vs. time trace (rolling average of 20 points) at 300 ms
per time point,
wherein the time resolution of this approach is sufficient to monitor not only
the injection of
the drug but also the subsequent few tens of seconds "mixing" phase associated
with the drug
homogenizing within the circulatory system. This panel corresponds to the
zoomed area
marked in dashes from Fig. 6B. Fig. 6D depicts the average concentration vs.
time, wherein
the unprecedented time resolution enables measurement of the minute-scale
distribution
phase of the drug with more than 1,000 experimental points, producing in turn
ultra-high
precision estimates of the associated pharmacokinetic parameters.
[0018] Detailed Description of the Invention.
[0019] Operating Principal. The inventions disclosed herein were derived by
extensive
study of electrochemical sensor operations and the discovery that certain
outputs are
insensitive to sensor fabrication variability and signal drift. In standard
electrochemical
sensor implementation, current flows are altered by target binding and are is
assessed by
voltammetric methods, including cyclic voltammetry, alternating current
voltammetry, and
square wave voltammetry. These methods measure peak currents, which are
related to the
fraction of redox-reporter-modified recognition elements that are bound, but
are also highly
dependent on the total number of recognition elements (e.g., aptamers) present
and active on
the sensor.
[0020] The inventors of the present disclosure have developed an alternative
approach for
monitoring changes in electron transfer induced by target binding. Rather than
measuring
voltammetric peak currents, which are indirectly related to binding-dependent
electron
transfer kinetics, electron transfer kinetics can be measured directly, and,
advantageously, the
inventors of the present disclosure have discovered that these values are
insensitive to the
sources of error that affect peak current measurements. The inventors of the
present
disclosure have determined that the electron transfer kinetics measured via
chronoamperometric current decays are largely determined by target binding
and, unlike peak
currents, are independent of the number of active recognition units on each
sensor.
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
Accordingly, the relationship between target concentration and electron
transfer kinetics can
be determined for a selected sensor configuration and this relationship will
be stable and
applicable to all like sensors operating under similar conditions.
Furthermore, this predictive
relationship also stable for an individual sensor operating for long periods
of time.
[0021] This novel operating principal of the invention obviates the need to
calibrate
individual sensors, and provide a means for drift-free operation in
challenging environments,
such as in vivo. The measurements can also be obtained on extremely fine time
scales,
enabling resolution of biological processes by electrochemical sensors at time
scales of
milliseconds.
[0022] The various elements of the invention are described next.
[0023] Electrochemical Sensors. The various embodiments of the invention are
directed to
methods of using electrochemical sensors. As used herein, an electrochemical
sensor is any
sensor that is capable of measuring the concentration of a target species in a
sample, wherein
binding of the target species to a recognition element of the sensor induces
measurable
changes in current output by the sensing element, such that the output of the
sensor can be
used to estimate the concentration of the target in the sample.
[0024] EAB Sensors. In a first implementation, the electrochemical sensors
utilized in the
methods of the invention comprises E-AB sensors. Any E-AB sensor design or
configuration
known in the art may be used. In an E-AB sensor, the recognition element
comprises an
aptamer, as known in the art. The aptamer may comprise a DNA aptamer, RNA
aptamer, or
an aptamer comprising non-natural nucleic acids, as well as hybrids of the
foregoing.
Variants of the E-AB concept wherein the recognition element is other than a
nucleic acid,
for example sensors using proteins, chemical species, and other molecules, are
also within the
scope of the invention.
[0025] In an E-AB sensor, one or more selected portions of the working
electrode are
functionalized with the aptamer. The aptamer may be conjugated to or otherwise
associated
with the electrode surface by any appropriate chemistry, for example by
covalent bonding,
chemisorption, or adsorption. Alkane thiol monolayers may be used to conjugate
aptamers to
the electrode surface, being particularly suitable for gold electrode
surfaces.
6
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0026] Each aptamer is functionalized with one or more redox reporters.
Binding of the
target species causes the aptamer to change its configuration, such that the
position of (or the
accessibility to the electrode of) the one or more redox reporters is
detectably altered. The
redox species may comprise any composition of matter that interacts with the
electrode such
that a change in its accessibility to or proximity to the electrode causes a
change in the
electron transfer kinetics. Exemplary redox species include methylene blue,
ferrocene,
viologen, anthraquinone or any other quinones, ethidium bromide, daunomycin,
organo-
metallic redox labels, for example porphyrin complexes or crown ether cycles
or linear
ethers, ruthenium, bis-pyridine, tris-pyridine, bis-imidizole,
ethylenetetracetic acid-metal
complexes, cytochrome c, plastocyanin, and cytochrome c'.
[0027] In some implementations, the E-AB sensor is a signal-on type sensor,
such that target
binding enhances the signal, and in other implementations the E-AB sensor may
comprise a
signal-off configuration. In one embodiment, the E-AB is a dual-strand sensor,
wherein the
redox species is present on a separate strand, a portion of which is
complementary to or
otherwise capable of reversibly binding to a portion of the aptamer. In the
presence of the
target species, the redox species' strand is liberated from the aptamer,
allowing the target
species to bind to the aptamer and the redox species to come into contact or
proximity to the
electrode.
[0028] Sensor Components. The electrochemical sensor will comprise one or more
working electrodes, to which a plurality of recognition elements are bound
(for example, at
densities of 0.1x1011 to lx1013molecules/cm2). The working electrode may
comprise any
suitable electrode material for electrochemical sensing, including, for
example: any metallic
surface that forms a bond with thiols or amines; gold; any gold-coated metal,
(such as
titanium, tungsten, platinum, carbon, aluminum, copper, etc); bare palladium
electrodes,
carbon electrodes, etc.
[0029] The working electrode may be configured in any desired shape or size.
For example,
paddle-shaped electrodes, rectangular electrodes, wire electrodes, electrode
arrays, screen-
printed electrodes, and other configurations may be used. For in vivo
measurements, a thin
wire configuration is advantageous, as the low profile wire may be inserted
into veins,
arteries, tissue or organs and will not impede blood flow in blood vessels or
cause substantial
damage in tissues. For example, a wire having a diameter of 1-500 [Am, for
example, 100
7
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[Am, may be used.
[0030] The electrochemical sensing systems of the invention further comprise
an auxiliary
or counter electrode, for example, a platinum auxiliary electrode. The
electrochemical
sensing element may be used with a reference electrode, for example an Ag/AgC1
electrode,
or other reference electrode known in the art. The electrochemical sensor of
the invention
may be configured in a two-electrode or a three-electrode system,
appropriately configured
for performing chronoamperometric measurements. The electrode-containing cell
system
may comprise a mixing chamber or other vessel wherein the electrodes are
present and are in
contact with the sample.
[0031] The sensor and electrode system may comprise an assembly for obtaining
faradic
current measurements when deployed in the sample or exposed to the sample. The
assembly
may comprise a housing. For example, for placement in the body of a living
organism, the
housing may comprise a needle, catheter, or other implantable structure. For
ex vivo
applications, the housing may comprise a well, microfluidic vessel, or other
structure, such as
found in a lab-on-chip device.
[0032] The electrochemical sensors of the invention will be in functional
connection with
appropriate components for performing chronoamperometry measurements. The
chronoamperometry components may comprise two or more devices in electrical
and/or
network connection with one another, or may comprise a single integrated
device.
[0033] A first component for performing chronoamperometry measurements
comprises a
device or combination of devices that can deliver excitation voltage pulses of
a desired
magnitude, frequency, and waveform to the sensing element. Chronoamperometry
components may include potientiostats or other voltage sources and voltage
controllers for
imposing voltage steps on the working electrode.
[0034] A second component for performing chronoamperometry measurements
comprises a
device or combination of devices that can acquire time-resolved faradic
current outputs from
the sensing element. These components will comprise circuitry for reading
sensor outputs
and storing such outputs or routing the outputs to other devices, including
components as
analog-to-digital converters, amplifiers, and storage media. Resolution at
very fine time
8
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
scales will be necessary for measuring the current decay kinetics in most
sensor systems, for
example, time scales of microseconds to milliseconds.
[0035] Other Sensor Types. The scope of the invention is not limited to E-AB
sensors.
The scope of the invention further encompasses any electrochemical sensor
wherein target
binding to recognition elements creates measurable changes in electron
transfer rates
measurable by the sensing element. In one aspect, sensors that employ non-
mediated
electrochemical sensing may be used, including the use of direct electron
transfer and redox
equilibrium as a way to generate a signal. These biochemical sensors include,
for example,
sensors that detect the consumption or generation of species belonging to
redox couples.
Other sensors may employ a mediated electrochemical analysis, i.e. using a
redox species
mediator for electron transfer and establishing redox equilibrium. Examples of
mediated and
non-mediated sensors can be found in Sander et at. 2015, A Review of
Nonmediated and
Mediated Approaches. Environ. Sci. Technol. 49:5862-5878.
[0036] Additional sensor types include chemically modified electrodes,
immunosensors,
oligopeptide-based sensors, and sensors that utilize, organelles (e.g.
chloroplasts,
mitochondria), animal and vegetable tissues, microorganisms, enzymes, tissue
slices,
peptides, and antibodies.
[0037] Additional sensor types include sensors that measure changes in
electron transfer
from solution-phase redox reporters, for example, ferrocyanate/ferricyanate
redox coupled
with a 17mer peptide that specifically recognizes cyclic AMP, as described in
Katayama et
al. 2000, The Design of Cyclic AMP¨Recognizing Oligopeptides and Evaluation of
Its
Capability for Cyclic AMP Recognition Using an Electrochemical System. Anal
Chem.
2000;72(19):4671-4.
[0038] Another sensor architecture that may be used is a polymeric sensor,
such as a sensors
using cationic polythiophene bearing a ferrocene substituent as a mediator in
an aptamer
system as described in Le Floch, 2006, Label-Free Electrochemical Detection of
Protein
Based on a Ferrocene-Bearing Cationic Polythiophene and Aptamer. Anal Chem.
2006;78(13):4727-31. doi: 10.1021/ac0521955.
[0039] Another sensor type that may be used is a sensor based on electron
transfer changes
9
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
due to target binding-induced displacement of ligands, for example,
hexamethylphosphoramide with samarium (II) iodide as described in Prasad,
2004, The Role
of Ligand Displacement in Sm(II)¨HMPA-Based Reductions. J Am Chem Soc.
2004;126(22):6891-4.
[0040] Another exemplary sensor type is based on changes in a redox reporter's
reorganizational energy, for example, ferrocenoyl-peptides as described in
Plumb, 2003,
Interaction of a Ferrocenoyl-Modified Peptide with Papain: Toward Protein-
Sensitive
Electrochemical Probes. Bioconj Chem. 2003;14(3):601-6. doi:
10.1021/bc0256446; or
trinuclear ruthenium clusters, for example, as described in Feld 2012,
Trinuclear Ruthenium
Clusters as Bivalent Electrochemical Probes for Ligand¨Receptor Binding
Interactions.
Langmuir. 2012;28(1):939-49. doi: 10.1021/1a202882k.
[0041] Another sensor type that may be used is a sensor based on sterically
induced changes
in the efficiency with which a scaffold-attached redox reporter approaches an
underlying
electrode surface, for example, duplex DNA, quadruplex DNA, and DNA
nanoswitches as
described in Ge 2010, A Robust Electronic Switch Made of Immobilized
Duplex/Quadruplex
DNA. Angew Chem Int Ed. 2010;49(51):9965-7. doi: 10.1002/anie.201004946, or
DNA-
containing a small molecule recognition element as described in Cash 2009, An
Electrochemical Sensor for the Detection of Protein¨Small Molecule
Interactions Directly in
Serum and Other Complex Matrices. J Am Chem Soc. 2009;131(20):6955-7. doi:
10.1021/ja9011595.
[0042] Sensor Class. The various methods of the invention are based on the
discovery
that certain sensor outputs, i.e. current decay kinetics, are stable among
sensors of a given
type. A class, as used herein, refers to a plurality of sensors having one or
more shared
characteristics. The sensor characteristics may comprise various factors,
including: the
configuration and materials of the electrodes, the type of sample to be
analyzed, the type of
recognition element, the type of redox reporter(s) and placement thereof, the
packing density
of the recognition elements on the working electrode, the electrode
functionalization
chemistry, and other sensor parameters that affect sensor output. Another
class parameter
may be the sensor's manufacturing lot, wherein sensors in a class are those
fabricated in the
same allotment.
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0043] In one embodiment, a class of sensors comprises a plurality of sensors
having
substantially identical sensor architecture, identical recognition elements
and redox reporter
elements, identical chemistries for attachment of the recognition elements to
the working
electrode, identical manufacturing methods, and similar recognition element
packing
densities (e.g., packing densities that vary by 1-20% among sensors within the
batch in terms
of moles of probe per square centimeter).
[0044] Target Species. The sensors employed in the methods of the invention
are directed
to the detection of a target species. The target species may comprise any
inorganic or organic
molecule, for example: a small molecule drug, a metabolite, a hormone, a
peptide, a protein,
a carbohydrate, a nucleic acid, a lipid, a hormone, metabolite, growth factor,
neurotransmitter, or a nutrient. The target may comprise a pollutant or
contaminant. The
target may comprise a toxin. The target may comprise a pathogen-induced or
pathogen-
derived factor, or a virus or cell. In some embodiments, the target species
comprises a drug
having significant side effects, such as a chemotherapeutic drug, or a drug
having a narrow
therapeutic index, wherein accurate measurement of blood level is critical to
ensure safe
dosing or minimal side effects.
[0045] Operating Conditions. Sensors are utilized under selected operating
conditions,
which encompass various aspects of the detection process. Operating conditions
may
encompass any combination of factors that affect the operation and output of
the sensor.
[0046] In a first aspect, the operating conditions encompass the sample type
to be analyzed.
The target species of the invention are assessed in a sample. The sample will
comprise a
liquid. The sample may comprise whole blood, serum, saliva, urine, sweat,
interstitial fluid,
spinal fluid, cerebral fluid, tissue exudates, macerated tissue samples, cell
solutions,
intracellular compartments, water, wash water, wastewater, groundwater, food,
beverages, or
other biological and environmental samples. In some embodiments, the sample is
derived
from a subject, for example a human patient or a non-human animal such as a
veterinary
subject or test animal. In one embodiment, the sample comprises flowing whole
blood, i.e.,
blood sampled by a sensing system comprising a sensor implanted in the living
body (e.g., in
the circulatory system) of a subject. In an alternative embodiment, the method
is used to
measure target species in a gas, wherein the gas has equilibrated with the
liquid sample.
11
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0047] In one embodiment, the sample is processed prior to measurement.
Examples of
processing include filtering, dilution, buffering, centrifugation, and the
application of other
materials or processes to the sample prior to analysis. In some embodiments,
the sample is
not processed prior to performing measurements, for example, the sample being
undiluted,
unfiltered, or unconcentrated.
[0048] In a second aspect, the operating conditions encompass the assay
conditions. The
general assay conditions will refer to reaction conditions for the assay, such
as sample
volume, temperature, pH, etc.
[0049] In a third aspect, the operating conditions may be defined by
operational parameters
used to obtain sensor measurements. For example, shape and frequency of the
applied
voltage waveform, voltage step values, and sampling intervals are some of the
variables that
constitute operational parameters.
[0050] Measurement of Target Concentration by Current Decays. In a first
aspect, the
invention encompasses a general method of measuring the concentration of a
target species in
a sample by the use of an electrochemical sensor. The method encompasses the
following
general steps:
(A)a mathematical relationship between a selected measure of current decay and
target
concentration is determined for a selected class of electrochemical sensors
operating
under a selected set of operating conditions;
(B) an electrochemical sensor of the selected class is deployed in a sample of
unknown
target concentration, under the selected operating conditions, and a value of
the
selected measure of current decay is acquired; and
(C) applying the relationship between current decay and concentration
established in step
(A), the current decay value observed in step (B) is used to determine the
concentration of the target species in the sample.
[0051] For example, in one embodiment, the invention encompasses a method of
measuring
the concentration of a target species in a sample by the use of an
electrochemical sensor, the
12
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
method comprising the steps of:
deploying an electrochemical sensor such that it is exposed to a sample,
wherein the
output of the electrochemical sensor is a faradic current that varies in a
concentration-
dependent manner to the concentration of a target species in the sample;
applying one or more excitation pulses to the electrochemical sensor, wherein
a time-
dependent faradic current output is generated by each pulse;
acquiring time-resolved faradic current data following each of the one or more
excitation pulses;
by the acquired time-resolved faradic current data, calculating the value of a
selected
measure of current decay;
by the calculated value of a measure of current decay, calculating the
concentration of
the target species by application of a mathematical relationship between
selected
measure of current decay and the concentration of the target species in the
sample.
[0052] Measure of Current Decay. The inventors of the present disclosure have
advantageously determined that the decay rate of current transients generated
in response to
an excitation stimulus are related to the concentration of the target species
and are stable
across sensors of the same class. The methods of the invention thus rely on
the measurement
of current decay, the relationship between current decay rate and target
concentration, and the
remarkable stability of this relationship among like sensors and over time.
The lifetime of
chronoamperometric decays depend only on the relative populations of the bound
and
unbound recognition elements (e.g. aptamers), not the absolute numbers of
bound and
unbound recognition elements, and thus provides a way to measure target
concentration that
is independent of factors that may vary significantly between individual
sensors of the same
class.
[0053] For example, in the case of an electrochemical sensor comprising an
electrode
substrate functionalized with a plurality of recognition elements, wherein
each recognition
13
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
element is functionalized with one or more redox reporters, electrical
excitation of the redox
reporters will induce a temporary flow of current between the redox reporters
and an
electrode substrate (or between the electrode substrate and the redox
reporters, depending on
the configuration of the system). Upon stepping the voltage of a working
electrode, such as
the substrate of an E-AB sensor, the working electrode becomes either a
stronger reducing
agent (in the case of stepping to a more negative potential) or a stronger
oxidizing agent (in
the case of stepping to a more positive potential). Within appropriate ranges
(e.g., near or
above the redox potential of the redox reporter), this step in voltage will
induce a faradaic
current flow between redox reporters of the sensor's recognition elements and
the electrode
substrate. As current flows, the pool of electrons mobilized by the excitation
becomes
depleted, and the current will decay exponentially (or multi-exponentially) at
rates and
amplitudes dependent upon the binding status of the recognition elements of
the sensor,
wherein target binding induces faster or slower transfer of current, for
example, by changing
the proximity of the redox reporter to the working electrode. Accordingly, the
ratio of target-
bound recognition elements to recognition elements not bound by the target,
which ratio is
proportional to the concentration of the target species in the sample, will
determine the
observed current decay rates and amplitudes for the sensor as a whole.
[0054] As used herein, "current" will refer to the flow of electrons measured
by a sensor
that has been deployed in a sample. For example, current may comprise flow of
electrons
from redox reporters to an electrode, or may comprise the flow of electrons
from an electrode
to redox reporters. "Current decay," as used herein, means the behavior of
current transients
measured by the sensor over time in response to the application of an
excitation stimulus to
the sensor or sample.
[0055] The measurement of current decay may be achieved using
chronoamperometric
methods, as known in the art. Decay parameters may be measured by applying an
excitation
to the sensor and/or sample and measuring the response of current over a
period of time
following the excitation. The measurement of decay parameters thus requires a
sensor or
sample environment in connection with components that (1) deliver excitation
pulses of a
desired voltage, frequency, and waveform; and (2) measure the current response
over time
scales corresponding to the duration of the current transients.
[0056] The excitation pulse may be stimulus of any kind that induces a current
transient, for
14
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
example, a stepping of the sensor electrode's potential to values where redox
reporters will
be substantially (e.g., fully) oxidized or reduced. Appropriate excitation
waveforms may be
selected, as known in the art. For example, voltage steps in the range of +/-
0.1 V to 0.5 V
may be utilized, at repetition rates of 1 to 10,000 Hz, for example 5 Hz, 10
Hz, 20 Hz, 50 Hz,
100 Hz, and intermediate values between 1 and 10,000 Hz.
[0057] Acquisition of time-resolved current measurements at time scales of
microseconds to
milliseconds will typically be required. Typical current transients have a
duration in the
range of 10-100 ms, and may be resolved by sampling at shorter time intervals,
for example,
every 1 [is, 2 [is, 3 [is, 5 [is, or 10 [is.
[0058] Once time-resolved current data has been acquired, these data may then
be analyzed
to derive any number of mathematical parameters that describe the kinetics of
the current
decay. The selected measure of decay may be any parameter of the current
transient which
varies in a concentration-dependent manner to the concentration of the target
species in the
sample. The selected measure of current decay can be assessed as any measure
of decay
kinetics, for example, a rate constant, lifetime, half-life, or any other
quantification of current
decay. For example, in one embodiment, as described below, the measure of
decay is
derived by fitting the entire decay curve to a function and employing the
lifetime (inverse
rate constant) or half-life for that decay curve. In another embodiment, as
described below,
the decay is derived by fitting the time-resolved data to a function that
derives the sum of two
or more exponentials and employing the relative amplitudes, or like measures,
of the
exponential components. .
[0059] A selected mathematical analysis is applied to the acquired, time-
resolved current
data to derive the selected measure of decay. Any regression analysis may be
applied to
derive the selected measure of current decay, to resolve the coincident
electron transfer of
bound and unbound recognition elements. In one embodiment, the measure of
current decay
is derived using a monoexponential fit to the sensor current trace. In one
embodiment, the
measure of current decay is derived using a multiexponential fit. In one
embodiment, the
measure of current decay is derived using a biexponential fit. In one
embodiment, the
measure of current decay is derived using a triexponential fit. A current
decay value may be
assessed by a single data point, or by the averaging of multiple data points.
For example, a
sampled decay value may comprise an average value observed over a selected
number of
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
excitation-decay cycles ranging between 1-100 cycles, for example, 5, 10, 20,
50, 75, or 100
cycles or intermediate values thereof.
[0060] In some sensor systems, the kinetics of interconversion between the
bound and
unbound states of the recognition element are faster than the electron
transfer events
measured by the sensor. Thus, the observed current transients reflect a
population-weighted
average of the bound and unbound states. This may be analyzed by approximating
the
current decay lifetime using a mono-exponential fit, for example as in Fig.
4A. The lifetime
of this "best fit" mono-exponential is monotonically related to the
concentration of the target.
In other words, current decay traces which are best described as the sum of
two exponential
processes (unbound decay and bound decay) can be fit with a monoexponential
curve, the
parameters of which (e.g., decay constant, half-life, etc.) provide a means of
determining
target concentration.
[0061] In one embodiment, the selected measure of decay is derived by fitting
the time-
resolved current data to a function that derives the sum of two exponents and
using the
relative amplitude of the exponential components as a measure of target
binding. For
example, as the observed current decay is a combination of bound and unbound
decays, a
biexponential fit may be used to describe the decay curve, with a more rapid
phase
representing target-bound decays and a slower phase representing unbound
decay. The
relative amplitude of either decay, i.e., its amplitude relative to the total
amplitude of the two
phases together or the ratio of the amplitudes of the two phases, may be
utilized as a measure
of the proportion of target-bound recognition elements. In alternative
implementations, the
decays may be described by three or more exponentials, e.g., a triexponential
fit, etc.
[0062] For example, exemplary methods of utilizing exponential fits are
described in
Kamman et al., C., Multi-exponential relaxation analysis with MR imaging and
NMR
spectroscopy using fat-water systems, In Magnetic Resonance Imaging, Volume 5,
Issue 5,
1987, Pages 381-392.
[0063] For example, in one implementation, chronoamperometric current decays
from the
sensors are plotted in log-i(current)-log-t(time) plots, and non-linear
regression analysis is
performed using the following equations:
For mono-exponential systems, by Equation 1:
16
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
(Equation 1) i = i0i * exp(7:) + C
For bi-exponential systems, by Equation 2:
(Equation 2) : i = i0i * exp(7:) + i02 * exp(72t) + C
For tri-exponential systems, by Equation 3:
(Equation 3) i = i0i * exp(7:) + i02 * exp(7t2) + iO3 * exp(73t) + C
wherein, for Equations 1-3, t is time following the excitation, T is the time
constant, i is
current, and C is constant background current, if any.
The relative amplitudes are calculated as the ratio of one selected amplitude
over the sum of
all the amplitudes. For example, for a biexponential fit:
Equation 4: Relative Amplitude 1 ¨ 101 and
(i01+i02) ;
Equation 5: Relative Amplitude 2 ¨ 102 (i01+i02)
[0064] Fig. 3 provides an illustration of current decay dynamics for an
electrochemical
sensor. In this example, pulsed voltage is applied to an E-AB sensor
comprising methylene
blue redox reporters and the current is measured following the pulse. Two
curves are
presented, a first depicting current dynamics for a sample comprising no
target, and a second
depicting current dynamics for a sample comprising a saturating concentration
of target (i.e.,
under which all recognition elements aptamers of the sensor are bound by
target). Upon
application of a step in voltage at a sufficiently negative potential, the
complete reduction of
all methylene blue reporters is achieved. The flow of these electrons is
measured over time
following the end of the pulse. In a first phase, typically about 0.1 ms after
application of the
17
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
pulse in aqueous solutions, the decay is rapid. This phase is attributed to
charging of the
electrical double layer formed on the electrode surface at this potential bias
(i.e., the
migration of aqueous ions that has time-scales of microseconds). The double
layer charging
decay is largely insensitive to target concentration in the sample and is
generally not useful
for predicting target concentration. Following this phase, a second phase is
observed,
wherein the decay kinetics are strongly related to target binding,
corresponding to the
faradaic reduction of the reporter, e.g., methylene blue to leucomethylene
blue. In Fig. 2, for
the target-saturated recognition elements, a fast exponential decay in current
is observed,
with a lifetime of 100 30 .is. For the recognition elements in a target-free
sample, a slower
decay is observed with a lifetime of 6.5 0.5 ms. The ¨ 5 fold decrease in
lifetime (when
comparing saturated samples to samples devoid of target) corresponds to a
change in the
proximity of the redox reporter to the electrode surface and reflects a
population of target-
bound aptamers that transfers electrons more rapidly than the target-free
aptamer.
[0065] Class Calibration. In one aspect, the scope of the invention is
directed to a method
of calibrating a class of sensors, by means of a representative subset of
sensors in the class.
The purpose of the calibration step is to generate a standard curve for the
interpretation of
sensor outputs for all sensors in the class. Accordingly, the calibration of
sensors of a given
class is performed using representative sensors of the same design, assaying
representative
samples, under operating conditions similar to and/or representative of those
in which the
sensors will be deployed. Once such calibration has been performed using the
representative
subset, other sensors from the class may be used to perform calibration-free
measurement of
the target species under similar operating conditions.
[0065] Class calibration is achieved by establishing a "decay-concentration
relationship" for
the selected sensor type in representative samples under the selected
operating conditions.
The decay-concentration relationship is a predictive relationship between
target concentration
in a sample and the observed current decay value resulting from a step in
voltage at the
working electrode. The decay-concentration relationship may be calculated
using any
appropriate regression analysis. As signal output is a combined sum of bound
and unbound
decays, models which estimate a monotonic function based on the sum of two
exponential
decay curves will be well suited to the calculation step. For example, if
current lifetime is the
selected measure of current decay, a relationship between current lifetime and
concentration
is determined. In another embodiment, calibration data is fitted to extract
multiple
18
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
exponential processes, and relative amplitudes calculated for each exponential
phase serve as
the measure of decay, such that a relationship between relative amplitude and
concentration
is established.
[0066] The calibration measurements used to establish the relationship in Step
(A) are
performed using a set of standards of various and known target concentrations.
Measurements should be made across a range of target concentrations within the
dynamic
range of the sensor, i.e., from zero target to saturating target levels, for
example by the use of
spiked samples. Any number of data points may be generated, for example 2-
1,000 data
points may be sampled to generate the calibration data. For example, 25-100,
e.g., 50-75
data points may be used in the calibration.
[0067] The class calibration process will employ a representative set of
sensors. The
representative set will comprise a sufficient number of sensors to provide a
calibration curve
that is accurately predictive for other sensors in the class. The number of
sensors in the
representative set may be established, for example, by methods known in the
art for
determining a sample mean based on sub-sample values. A representative set may
comprise,
for example 1, 3, 5, 10, 20, 50, 100, or more sensors, and values intermediate
thereto.
[0068] Fig. 3B illustrates a calibration curve of the invention. For five E-AB
aminoglycoside-detecting sensors, current lifetime was assessed across a range
of
concentrations, in both whole blood and buffer. A "best fit" mono-exponential
was
monotonically related to the concentration of the target by a non-linear
regression of lifetime
versus concentration to a Langmuir isotherm. These results illustrate the
effects of different
operating conditions on sensor output, wherein the blood and buffer
calibration curves are
different.
[0069] Sensors of a given class, operated under similar operating conditions,
will have
sufficiently similar decay outputs to enable the prediction of target using a
standard curve
generated for all sensors of that class. Sensors of the same class may have
decay outputs
that, when exposed to a like sample comprising a given target concentration,
will have values
of a selected current decay measure that diverge by less than 20%, less than
15%, less than
10%, less than 5%, or less than 1% depending on the stringency of
manufacturing, signal-to-
noise inherent in the system, and other variables.
19
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0070] In one embodiment, the scope of the invention encompasses a class-
calibrated
sensor. A class-calibrated sensor means a sensor of a selected class, e.g.,
having a selected
recognition element, selected redox reporter, selected electrode attachment
chemistry,
selected electrode configuration, and other parameters that define sensor
performance,
wherein a decay-concentration relationship between a selected measure of
current decay and
the concentration of a selected target is known for sensors of the class. In
one embodiment,
the class calibrated sensor is an aptamer-based electrochemical sensor.
[0071] Calibration-Free Measurement. In one aspect, the scope of the invention
comprises a method of calibration free-measurement of target species
concentration.
Calibration-free operation, as used herein, means the operation of an
electrochemical sensor
of a selected class wherein the sensor output (for example, current decay
lifetime or relative
amplitudes) is directly converted to a target concentration value by use of a
calibration curve
specific for the selected class. In calibration-free operation, the sensor is
not necessarily
calibrated prior to, during or after measurement.
[0072] Drift-Free Operation. In one aspect, the scope of the invention
encompasses the
drift -free operation of an electrochemical sensor, wherein the sensor is
operated for an
extended period of time, e.g. hours, days, months, or longer. As depicted in
Fig. 5A, E-AB
sensors deployed directly in flowing whole blood will have outputs that drift
considerably
when sensor output is measured as an amplitude by square wave voltammetry.
This severe
baseline drift is eliminated current lifetime is measured instead, as in Fig.
5B, wherein
current lifetimes are stable for a period of 8 hours, while absolute sensor
output drifts
considerably during the test period.
[0073] Subsecond Temporal Resolution. The methods of the invention enable
calibration-
free, sub-second-resolved measurement of molecules directly in vivo. As set
forth in the
Examples, real-time resolution of target concentration in the blood or other
body
compartment is enabled by the methods of the invention, for example with time
scales of
100-500 ms. The unprecedented temporal resolution of this approach enables the
measurement of rapidly fluctuating target dynamics during physiological
events, such as drug
uptake and distribution, hormone and neurotransmitter release, and other
physiological events
taking place over short time scales. The fine time-scale resolutions enabled
by
chronoamperometry make the methods of the invention especially well suited to
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
pharmacokinetic analysis. In one embodiment, the invention encompasses the
real-time
calculation of pharmacokinetic parameters by the calibration-free measurement
of drugs,
metabolites, excretion products, or other species involved in drug metabolism.
[0074] Sensor Deployment and Operation. The inherent stability of electron
transfer
kinetics across sensors of a class operated under similar operating conditions
enables the use
of electrochemical sensors in various contexts. In various implementations,
the methods of
the invention enable sensors to make accurate measurements when deployed in
contexts
where calibration is impossible, burdensome, or expensive. The calibration-
free and drift-
free methods of the invention are especially amenable to in vivo measurements.
For example,
the sensing element or housing of a sensing system may be inserted, implanted,
or otherwise
placed within the body of a living organism. The sensor element of a sensing
system may be
implanted in the circulatory system, subcutaneously, intraperitoneally, within
an organ, or in
other body compartments, wherein the the sensing element is exposed to in vivo
fluids, e.g.,
interstitial fluid, blood, for example, flowing whole blood. The implanted
sensing system of
the invention may comprise an implanted sensing element in connection with
components
external to the body (e.g. by leads, wires, or wireless communication means)
wherein the
external components perform pulse generation, data acquisition or processing.
Alternatively,
the one or more ancillary components to the sensing element, or even the
entire sensing
system of the invention, may be implanted in the body, with communication (
by, for
example, leads, wires, or wireless communication devices) to external devices
for data
collection.
[0075] In one embodiment, methods of the invention are performed in a feedback
controlled
dosing system, as known in the art, wherein a drug is administered to maintain
blood
concentrations within a therapeutic index or safety index. For example, in
such methods and
systems, the methods of the invention are applied using an electrochemical
sensing element
implanted in a subject to measure the concentration of a target species,
wherein the target
species is a drug, metabolite of a drug, or biomarker indicating that the drug
should be
administered. When the detected levels of the target species indicate that the
subject is in
need of administration of a drug or other agent, an implanted pump or other
agent delivery
means administers the drug or agent metered at a dosage to maintain the
concentration of
drug or agent within a desired range.
21
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0076] In other contexts, the sensors of the invention are utilized for long-
term and/or
continuous monitoring of environmental or industrial sites, for example, in
rivers, seas, water
treatment plants, industrial facilities, food processing facilities, etc.
[0077] The sensors of the invention may also be used in ex-vivo diagnostic
applications. In
one embodiment, the method of the invention comprises the steps of withdrawing
a sample
from a living organism, and measuring the concentration of the target species
in the sample
by calibration-free measurement. In one embodiment, the sensors of the
invention are
employed in point-of-care testing systems. For example, in one embodiment, the
sample is a
blood sample, for example, a self-withdrawn pin-prick or finger-prick blood
sample, or a
urine, sweat, or saliva sample. In such embodiments, the electrochemical
sensor may be
deployed in a housing such as a well, slide, lab-on-chip, microfluidic
chamber, or other
device.
[0078] Chronoamperometry with Calibration. The scope of the invention is not
limited
to calibration-free chronoamperometry. In one implementation, the accuracy of
concentration measurements attained by electrochemical sensors performing
chronoamperometry is enhanced by the performance of one or more calibration
steps, i.e.
wherein a deployed sensor is exposed to one more samples of known target
concentration in
order to check for deviation between observed and expected concentration
values and to
correct for any such deviations. This embodiment still entrains the improved
time resolution
chronoamperometry affords relative to established electrochemical methods for
interrogating
sensors in this class.
[0079] In another embodiment, the decay-concentration relationship for a
deployed sensor is
established by calibration. The deployed sensor may be exposed to one more
samples of
known target concentration in order to establish the decay-concentration
relationship for the
sensor under its current operating conditions.
[0080] Computer Processes and Programs. The scope of the invention encompasses
various embodiments comprising software, computer programs, and programmed
devices. In
one embodiment, the scope of the invention encompasses non-transitory computer-
readable
recording media having stored thereon data and/or an encoding program that
causes a
computer to execute series of operations. The computer may comprise any
general purpose
22
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
computer, processor, embedded processor, mobile device, or other computing
device. The
computer may encompass hardware elements comprising input devices such as
keyboards,
mouse, touchscreen and other inputs. The computer may encompass hardware
elements for
the output, storage, or display of data, including graphical user interfaces,
displays, and
storage devices.
[0081] In one embodiment, the invention comprises a non-transitory computer-
readable
recording media having stored thereon data and/or an encoding program that
causes a
computer to execute series of operations, wherein the data and/or series of
operations causes
the operation of an electrochemical sensing system to effect the methods of
the invention. In
various embodiments, the non-transitory computer-readable recording media may
effect
operations such as: controlling a potentiostat or equivalent device to deliver
a series of
stepped voltage pulses to a deployed electrochemical sensor; controlling a
data collection
device to record sensor current outputs following the delivery of voltage
pulses; controlling a
processor to perform calculations that derive one or more selected measures of
current decay
from sensor output data; storing a calibration curve that relates a selected
measure of current
decay to target concentration; controlling a processor to calculate target
concentration based
on sensor outputs and a stored calibration curve; storing instructions for the
performance of
the methods of the invention; and other operations of the invention.
[0082] In one embodiment, the scope of the invention encompasses a device
programmed to
perform the operations of the invention, for example, a device comprising or
in connection
with (e.g. network connection) the aforementioned non-transitory computer
readable
medium.
[0083] Kits and Systems. The scope of the invention extends to collections of
components
configured to perform the analyte concentration measurements of the invention.
In various
embodiments, the scope of the invention encompasses collections comprising two
or more
items for the performance of the methods of the invention, the two more
components being
selected from the group consisting of: electrochemical sensors of a class
wherein the
relationship between a measure of decay acquired by the sensor under selected
operating
conditions and target concentration is known; sensor housings for deployment
of the sensors
to a selected sample type; potentiostat and controllers thereof which may
deliver electrical
signals to generate current transients; data acquisition and computer readable
storage media
23
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
and/or processing means for calculating a selected measure of current decay
from sensor
current data; computer readable storage media and/or processing means for
calculating target
concentration from current decay data; and instructions for performing the
methods of the
invention.
[0084] Exemplary Embodiments.
[0085] In one embodiment, the invention is a method of measuring the
concentration of a
target species in a sample by the use of an electrochemical sensor, the method
comprising the
steps of:
deploying an electrochemical sensor such that it is exposed to a sample,
wherein the
output of the electrochemical sensor is a faradic current that varies in a
concentration-
dependent manner to the concentration of a target species in the sample;
applying one or more excitation pulses to the electrochemical sensor, wherein
a
faradic current output is generated by each pulse;
acquiring time-resolved faradic current data following each of the one or more
excitation pulses;
by the acquired time-resolved faradic current data, calculating the value of a
selected
measure of current decay;
by the calculated value of a measure of current decay, calculating the
concentration of
the target species by application of a mathematical relationship between
selected
measure of current decay and the concentration of the target species in the
sample.
[0086] In one embodiment, the electrochemical sensor comprises an electrode
functionalized with a plurality of recognition elements that undergoes a
conformational
change upon target binding, wherein each recognition element is functionalized
with one or
more redox reporters. In one embodiment, the recognition element comprises an
aptamer.
[0087] In one embodiment, the sample is selected from the group consisting of
whole blood,
serum, saliva, urine, sweat, interstitial fluid, spinal fluid, cerebral fluid,
tissue exudates,
macerated tissue samples, cell solutions, intracellular compartments, water,
wash water,
wastewater, groundwater, food, and beverages. In one embodiment, the sample is
not
24
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
processed prior to measurement.
[0088] In one embodiment, the target species is selected from the group
consisting of a
small molecule drug, a metabolite, a hormone, a peptide, a protein, a
carbohydrate, a nucleic
acid, a lipid, a hormone, a metabolite, a growth factor, a neurotransmitter, a
nutrient, and a
pollutant, a pathogen-induced or pathogen-derived factor, a pathogen, or a
cell.
[0089] In one embodiment the selected measure of target decay is selected from
a decay
constant, an average lifetime, a half-life, and a relative amplitude. In one
embodiment, the
selected measure of current decay is derived from an exponential fit of the
time-resolved
current data. In one embodiment, the selected measure of current decay is
derived from a
monoexponential fit of the time-resolved current data. In one embodiment, the
selected
measure of current decay is derived from a biexponential fit of the time-
resolved current data.
[0090] In one embodiment, the mathematical relationship between the selected
measure of
current decay and target concentration has been derived for sensors of the
same class as the
deployed electrochemical sensor.
[0091] In one embodiment no calibration step is performed prior to or after
the
measurement. In one embodiment, repeated measurements are obtained over an
extended
period of time.
[0092] In one embodiment, the electrochemical sensor is deployed in vivo.
In one
embodiment, the electrochemical sensor is deployed in a human subject. In one
embodiment,
the electrochemical sensor is deployed in a non-human animal. In one
embodiment, the
electrochemical sensor is deployed ex vivo. In one embodiment, the ex vivo
deployment is in
a point-of-care system.
[0093] In one embodiment, the electrochemical sensor is configured such that
when it is
deployed in a sample, the output of the sensor is a faradic current which
varies in a
concentration dependent manner to the concentration of a target species in a
sample; and
wherein a stable mathematical relationship relating a measure of faradic
current decay and
target concentration is known for sensors of a class to which the
electrochemical sensor
belongs.
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
[0094] In one embodiment, the electrochemical sensor comprises an electrode
functionalized with a plurality of recognition elements which undergo a
conformational
change upon target binding and wherein each recognition element is
functionalized with one
or more redox reporters. In one embodiment, the sensors of the class comprise
sensors
having the same recognition element type, same redox reporter type, and same
attachment
chemistry for conjugation to the electrode. In one embodiment, the recognition
element
comprises an aptamer.
[0095] In one embodiment, invention is a sensing system comprising an
electrochemical
sensor, wherein the electrochemical sensor is configured such that when it is
deployed in a
sample, the output of the sensor is a faradic current which varies in a
concentration dependent
manner to the concentration of a target species in a sample;
hardware components comprising devices for the application of excitation
pulses to the
electrochemical sensor and the acquisition of time-resolved faradic current
decay from the
electrochemical sensor following the application of each pulse; and
non-transitory computer-readable medium having stored thereon data and a
computer
program which enable performance of the methods described herein by the
electrochemical
sensor and hardware components. In one embodiment of the system, the
electrochemical
sensor comprises an electrode functionalized with a plurality of recognition
elements which
undergo a conformational change upon target binding and wherein each
recognition element
is functionalized with one or more redox reporters. In one embodiment, the
recognition
element comprises an aptamer. In one embodiment, the electrochemical sensing
system is
programmed to perform chronoamperometric current decay analyses to derive the
concentration of target species in the sample, by the methods disclosed
herein. In one
embodiment, the sensor element of the sensing system is implanted in the body
of an animal
and is in wireless or wired communication with ex vivo hardware components. In
one
embodiment, one or more ancillary hardware elements for delivery of voltage
pulses,
collection of time-resolved current data, and calculation of target
concentration is implanted
with the sensor. In one embodiment, the sensor and the ancillary elements,
including
elements for the delivery of voltage pulses and elements for the collection
and analysis of
time resolved current data, in addition to a power supply (e.g., battery) are
implanted in the
body of an animal. In one embodiment, the system further comprises a drug
delivery means
for administering a pharmaceutical agent to the animal in response to
threshold levels of the
26
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
target species being detected by the methods of the invention, or a means for
issuing an alert
that a pharmaceutical administration is necessary. In one embodiment, the drug
delivery
means is a pump. In one embodiment, the means for issuing an alert that a
pharmaceutical
administration is necessary comprises a wearable or mobile device in wired or
wireless
connection with implanted sensor system components.
[0096] In one embodiment, the invention comprises a non-transitory storage
medium having
thereon computer readable instructions for operating a chronoamperometric
electrochemical
sensing system to perform chronoamperometric current decay analyses to derive
the
concentration of a target species in a sample, by the systems and methods
disclosed herein.
[0097] EXAMPLES. Example 1. Real-time, sub-second-resolved measurement of
specific
molecules directly in the living body using chronoamperometrically
interrogated E-AB
sensors.
[0098] Electrochemical, aptamer-based sensors provide a modular approach to
the
continuous, real-time measurement of specific molecular targets irrespective
of their
chemical reactivity. The platform, which consists of an aptamer "probe"
modified with a
redox-active "reporter" and attached to an interrogating electrode (Fig. 1A),
signals via a
binding-induced conformational change that alters electron transfer from the
reporter, leading
in turn to an easily measurable electrochemical output (Fig. 1B). Because
their signaling
mechanism mimics the conformation-linked signal transduction employed by
naturally
occurring receptors in the body, E-AB sensors are particularly insensitive to
non-specific
binding and easily support continuous, extended measurements directly in
flowing, undiluted
blood serum. And while E-AB sensors often exhibit significant drift when
challenged in
undiluted whole blood, the inventors of the present disclosures have recently
shown that,
when combined with protective membranes, improved surface passivation
chemistries and/or
active drift correction mechanisms, E-AB sensors support the continuous, real-
time
measurement of specific molecules in whole blood and even in situ in the
living body over
the course of hours.
[0099] E-AB signaling is driven by binding-induced changes in the electron
transfer kinetics
of the aptamer-bound redox reporter. Previously cyclic current, alternating
current, or square
wave voltammetry has been used to "read out" this change by observation of
peak currents.
The most commonly employed of these, square wave voltammetry (SWV) achieves
this
27
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
conversion by subjecting the sensor to a series of potential pulses and
sampling the resultant
faradaic currents after a delay defined by the square wave frequency. The
magnitude of the
observed current is thus dependent on the electron transfer rate (which
defines how much the
current has decayed by the time it is measured), which, in turn, depends
quantitatively on the
concentration of the target. Specifically, when an E-AB sensor is interrogated
by SWV the
resultant peak current rises or falls monotonically (depending on the square-
wave frequency)
with rising target concentration (Fig. 1B). The relative magnitude of this
binding-induced
change (i.e., the signal gain) is dependent on the aptamer employed in the
sensor and can be
maximized by optimizing the square wave frequency and amplitude.
[0100] Although SWV has proven a particularly convenient and reliable means of
converting binding-induced changes in electron transfer kinetics into an
easily measurable
output, the approach is not without limitations. First, the peak currents
(measured in amperes)
produced by SWV are dependent not only on the presence or absence of target
but also on the
number of redox-reporter-modified aptamers on the sensor's surface, which can
fluctuate
significantly from device-to-device due to variations in fabrication (Fig.
2A). Previously, this
this variation was addressed by calibrating each device in a reference sample
of known
(typically zero) target concentration prior to use, which, while effective
(Fig. 2B), increases
complexity. Second, while SWV-interrogated E-AB sensors are selective enough
to deploy
directly in undiluted blood serum, they exhibit significant drift when
deployed either in vitro
or in vivo in whole blood a problem that, has previously been overcome using a
variety of
drift correction and drift-avoidance mechanisms. Finally, because of the time
required to scan
the necessary several hundred millivolt potential window of SWV, its time-
resolution is
limited to several seconds. Herein is demonstrated that by replacing
voltammetry, which
measures changes in electron transfer rates indirectly, with
chronoamperometry, which
measures them directly, these limitations are overcome to achieve the
calibration-free, sub-
second-resolved measurement of specific molecules in situ in the living body.
[0101] Results and Discussion. Unlike SWV, which converts changes in electron
transfer
rates into a change in peak current, thus indirectly reporting on transfer
kinetics,
chronoamperometry measures electron transfer kinetics directly. It does so by
determining
the lifetimes of current transients generated in response to a stepping of the
electrode's
potential to values where the redox reporter will either be fully oxidized or
reduced. For E-
AB sensors binding to the aminoglycoside antibiotics a sufficiently negative
potential was
28
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
applied to drive the complete reduction of all methylene blue reporters and
measured the
resultant current (Fig. 3). Current decay traces were observed that are best
described as the
sum of two exponential processes. In the absence of their target, for example,
these sensors
exhibit one rapid exponential phase with a lifetime of 100 30 [is
(throughout this
manuscript, errors represent the standard deviation from 5 independently
fabricated sensors)
and a slower one with lifetime of 6.5 0.5 ms. The more rapid phase is
attributed to charging
of the double layer formed on the electrode surface at this potential bias
(i.e., the migration of
aqueous ions that has time-scales of [is) which remains insensitive to changes
in target
concentration. The slower phase, in contrast, corresponds to the faradaic
reduction of
methylene blue to leucomethylene blue. Upon the addition of saturating target
concentrations, the second phase becomes more rapid with a lifetime of 1.20
0.01 ms. This
¨ 5 fold decrease in lifetime (when comparing with a sample devoid of target)
agrees with a
change in the proximity of the redox reporter to the electrode surface and
presumably reflects
a population of target-bound aptamers that transfers electrons more rapidly
than the target-
free aptamer.
[0102] In theory, if the conformation dynamics of aminoglycoside-binding
aptamers obey a
two-state model, the relative amplitudes of the exponential phases (which
reflect the
populations of bound and unbound aptamers) would change monotonically with
target
concentration. The two-state model assumes, however, that the two exponential
phases can
be measured independently. This is not the case here, however, because the
lifetimes of the
two exponential phases are quite similar at any given concentration of target,
rendering it
difficult to extract their amplitudes with sufficient precision. In other
words, when the
kinetics of interconversion between the bound and unbound states of the
aptamer are faster
than the electron transfer event, such as is the case here, the conformational
equilibrium
cannot be sampled as discrete static populations. Instead, the measured
lifetimes reflect a
population-weighted average of the bound and unbound states. This limitation
is overcome
by approximating the current decay lifetime using a mono-exponential fit (Fig.
4A).
[0103] The monotonic relationship between the chronoamperometric lifetime of
an E-AB
sensor and the concentration of its target provides a calibration-free
approach to performing
E-AB measurements. That is, unlike absolute SWV peak currents, which depend on
the total
number of aptamers on the sensor, the lifetime of chronoamperometric decays
depend only
on the relative populations of the bound and unbound aptamer. Thus, once
established for a
29
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
given type of sensor, the lifetime-concentration relationship can be used to
determine target
concentrations without the need to calibrate each individual sensor. The
lifetime-
concentration was established relationship for aminoglycoside-detecting E-AB
sensors when
being used for the detection of tobramycin in vitro in flowing whole blood.
Specifically, a
non-linear regression of lifetime versus concentration to a Langmuir isotherm
was performed
(red line in Fig. 4B) and solved for concentration. A batch of five new E-AB
sensors (i.e.,
sensors not in the initial training set) was challenged with tobramycin in
whole blood, and the
previously-determined Langmuir isotherm was used to convert the observed
chronoamperometric lifetimes into estimated concentrations (Fig. 4C). This
successfully
determined concentrations of the drug with precision and accuracy of better
than 10% over
the range from 1 p.M to 1000 M.
[0104] In addition to being calibration-free, the use of
chronoamperometrically-determined
current decay lifetimes as a means of defining target concentration is also
resistant to drift.
Again, while SWV-interrogated E-AB sensors are selective enough to perform
well in
undiluted blood serum, they often exhibit severe baseline drift when deployed
directly in
flowing whole blood (Fig. 5A). Previously this has been corrected using square
wave
voltammetry approaches that involve measurements taken at multiple frequencies
(Fig. 5A).
Chronoamperometric lifetime measurements, in contrast, are inherently
resistant to such drift;
as noted above, while the total amplitude of the current transient drifts
significantly
(presumably due to surface reorganization of the monolayer22), the lifetime of
its exponential
decay is independent of its amplitude and thus is largely drift-free (Fig.
5B).
[0105] The drift-resistance of chronoamperometrically-interrogated E-AB
sensors is
sufficient to support continuous, real-time measurements directly in situ in
the blood of live
animals. Aminoglycoside-binding E-AB were fabricated sensors on 75 p.m-
diameter gold
micro-wires, encased in 22-gauge catheters for structural support, and
deployed directly in
the jugular veins of live rats (Fig. 6A). The plasma pharmacokinetics of the
antibiotic
tobramycin were monitored following an intravenous administration of 30 mg/kg
by
performing continuous chronoamperometric measurements for a total of two hours
(Fig. 6B).
Continuous measurements were achieved by pulsing the potential of the E-AB
sensors
serially between -0.1 V and -0.3 V, holding each pulse with a duration of only
100 ms. Then,
non-linear regression analysis was performed of the current transients
generated at -0.3 V to
extract current decay lifetimes in real time, and these lifetimes were
converted to
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
concentration using the previously determined Langmuir isotherm (Fig. 6C). At
a time
resolution of 300 ms (limited by aptamer-target binding kinetics, data
acquisition and
computation time) the chronoamperometric measurements resolve not only the
duration of
the drug infusion but also the time it takes for the drug to "mix" and reach
homogeneity in
the bloodstream after the end of the infusion (Fig. 6C).
[0106] The sub-second time resolution of chronoamperometrically interrogated E-
AB
sensors measurement of drug pharmacokinetics with unprecedented precision. The
distribution, a, and elimination, /3, lifetimes of tobramycin were determined
by fitting the in
vivo data to a two-compartment pharmacokinetic model. The 300-ms time
resolution of
chronoamperometrically-interrogated E-AB sensors supports the determination of
the
distribution phase of the drug, a = 3.74 0.04 min, with more than one
thousand
measurement points and a calculated standard error from the fit of ¨1%.
Similarly, the
elimination phase of the drug was determined as /3 = 69 2 min with fourteen
thousand
measurement points, bringing down the standard error of the fit to only 3%,
with much of this
small deviation likely arising due to metabolic fluctuations in the animal
over the course of
the experiment (i.e., /3 is not truly a constant). This precision is an order
of magnitude
improved over previous in-vivo E-AB measurements achieved using square wave
voltammetry, which in turn was a large improvement over prior measurements
using blood
draws and ex-vivo analysis.
[0107] Herein chronoamperometric interrogation of E-AB sensors was used to
achieve the
calibration-free, sub-second-resolved measurement of specific small molecules
directly in
vivo. The unprecedented temporal resolution of this approach suggests that it
could improve
our understanding of rapidly fluctuating physiological events, such as drug
uptake, hormone
and neurotransmitter release, and the movement of drugs and metabolites within
the central
nervous system. The ability to perform the calibration-free measurement of
specific
molecules in the body in real-time could also enhance the efficiency and
accuracy with which
drugs are dosed, in applications ranging from therapeutic drug monitoring to
long-term
feedback-controlled drug delivery.
[0108] E-AB sensors are not the only class of biosensors that relies on
binding-induced
changes in electron transfer kinetics for the detection of analytes. Other
examples include
sensors that measure changes in electron transfer from solution-phase redox
reporters,
electron transfer changes due to binding-induced displacement of ligands,
changes in the
31
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
reporter's reorganizational energy, or sterically induced changes in the
efficiency with which
a scaffold-attached redox reporter approaches an underlying electrode surface.
From this
perspective, we postulate that the ability of chronoamperometry to measure
electron transfer
kinetics directly may also prove of value in the interrogation of these other
platforms.
[0109] Methods. E-AB sensors were fabricated as follows: segments of pure gold
(12 cm
in length), platinum (11.5 cm) and silver (11 cm) wire, were cut to make
sensors. The
insulation at both ends of these wires, about 2 cm, was removed using a
surgical blade to
allow electrical contact. These were then soldered each to one of the three
ends of a
connector cable using 60% tin/40% lead rosin-core solder (0.8 mm diameter) and
then
attached together by applying heat to shrinkable tubing around the body of the
wires, except
for a small window of about 5 mm at the edge of each wire. The wires were
attached in a
layered fashion, with the gold wire being insulated alone first, then both
gold and platinum
wires together, and finally all three wires together. The purpose of this
three-layer-thick
insulation was to give mechanical strength to the body of the malleable probe.
To prevent
electrical shorts between wires, different lengths were used for each wire as
described above.
The sensor window (i.e., the region devoid of insulation) in the gold wire was
cut to
approximately 3 mm in length. The silver wire was employed as a reference
electrode by first
immersing it in bleach overnight to form a silver chloride film. To increase
surface area of
the gold working electrodes (to obtain larger peak currents) the sensor
surface was roughened
electrochemically via immersion in 0.5 M sulfuric acid followed by stepping
the potential
between Einitial= 0.0 V to Ehigh= 2.0 V vs Ag/AgC1, back and forth, for 16,000
pulses. Each
potential step was of 20 ms duration with no "quiet time." To fabricate
sensors an aliquot of
the DNA construct was reduced for 30 min at room temperature with a 1000-fold
molar
excess of tris(2-carboxyethyl)phosphine. A freshly roughened probe was then
rinsed in
deionized water before being immersed in a solution of the reduced DNA
construct at 200
nM in PBS for 1 h at room temperature. Following this the sensor was immersed
overnight at
4 C for 12 h in 20 mM 6-mercapto-1-hexanol in PBS to coat the remaining gold
surface.
After this the sensor was rinsed with deionized water and stored in PBS.
[0110] For the SWV measurements, the sensors were interrogated from 0.0 V to -
0.5 V
versus Ag/AgC1, using an amplitude of 50 mV, potential step sizes of 1-5 mV,
and varying
frequencies from 10 Hz to 500 Hz. All SWV measurements were performed using a
three-
electrode setup and with a CH Instruments(TM) electrochemical workstation
(Austin, TX,
32
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
Model 660D) using commercial Ag/AgC1 reference electrodes filled with
saturated KC1
solution and platinum counter electrodes. For chronoamperometry, the potential
of the
sensors was serially stepped from -0.1 V to -0.3 V, each step for a duration
of 100 ms.
Current sampling was carried out every 10 [is for in vitro measurements, and
every 100 [is
for in vivo measurements (to reduce the number of experimental points and
speed data
acquisition). All chronoamperometric measurements were performed using the
three-
electrode E-AB sensor described above and recorded with a GAMRY(TM) Reference
600+
Potentiostat/Galvanostat/ZRA (Warminster, PA).
[0111] To study the behavior of chronoamperometric current decays, to measure
aptamer
affinity and to correlate signal gain to target concentration, sensors were
interrogated by
either square wave voltammetry or chronoamperometry first in flowing PBS and
next in
flowing heparinized bovine blood with increasing concentrations of the
corresponding target.
These experiments were carried out in a closed flow system intended to mimic
the type of
blood transport found in veins. Blood flow was achieved using a magnetic gear
pump, setting
flow rates to 1-10 mL min-1 as measured by a flow meter. To construct the
binding curves
(titrations of aptamer with target), stock solutions of tobramycin were
prepared fresh prior to
measurements in PBS buffer or blood, respectively. The sensor challenge to
demonstrate
calibration-free behavior was performed by challenging a fresh batch of
aminoglycoside-
binding E-AB sensors against stock solutions made from a tobramycin reference
standard.
[0112] In-vivo measurements were performed in anaesthetized rats, wherein
either a silastic
catheter was inserted for infusions or the E-AB sensor was placed for
measurements in the
jugular vein. All in vivo measurements were performed using a three-electrode
setup in
which the reference electrode was a silver wire coated with a silver chloride
film as described
above, and the counter electrode was a platinum wire. Recordings were taken
for up to 3 h,
with sampling rates of one point every 300 milliseconds. To obtain
pharmacokinetic profiles
from our real-time data non-linear regression analysis was performed using a
two-
compartment model to fit intravenous injections. The equation employed in the
regressions
was the following:
Cp = Ae-tia ¨ Be tli6
where Cp is the measured plasma concentration, A and B are contributions of
each
pharmacokinetic compartment to the maximum concentration A + B = CA/mx, a is
the first-
33
CA 03081401 2020-04-29
WO 2019/089465 PCT/US2018/058020
order time constant of drug distribution and /3 is the drug's elimination time
constant. During
the regression analysis, all variables were floating such that the best fit
was determined by
minimizing the squared errors.
[0113] All patents, patent applications, and publications cited in this
specification are herein
incorporated by reference to the same extent as if each independent patent
application, or
publication was specifically and individually indicated to be incorporated by
reference. The
disclosed embodiments are presented for purposes of illustration and not
limitation. While
the invention has been described with reference to the described embodiments
thereof, it will
be appreciated by those of skill in the art that modifications can be made to
the structure and
elements of the invention without departing from the spirit and scope of the
invention as a
whole.
34