Note: Descriptions are shown in the official language in which they were submitted.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
1
METHODS FOR PROTECTING PORCINE FETUSES FROM INFECTION WITH VIRUS
REFERENCE TO SEQUENCE LISTING SUBMITTED ELECTRONICALLY
[0001] The official copy of the sequence listing is submitted electronically
via EFS-Web
as an ASCII-formatted sequence listing with a file named "SEQ LISTING
18054W0", created
on March 27, 2018 and having a size of 175.5 kilobytes, and is filed
concurrently with the
specification. The sequence listing contained in this ASCII-formatted document
is part of the
specification and is herein incorporated by reference in its entirety.
FIELD OF THE INVENTION
[0002] The present invention relates to methods for protecting porcine fetuses
from
infection with Porcine Reproductive and Respiratory Syndrome Virus (PRRSV).
BACKGROUND OF THE INVENTION
[0003] Porcine reproductive and respiratory syndrome (PRRS) is the most
economically
important disease of swine in North America, Europe and Asia, costing North
American
producers approximately $600 million annually (Holtkamp et al., 2013).
Clinical disease
syndromes caused by infection with porcine reproductive and respiratory
syndrome virus
(PRRSV) were first reported in the United States in 1987 (Keffaber, 1989) and
later in Europe in
1990 (Wensvoort et al., 1991). Infection with PRRSV results in respiratory
disease including
cough and fever, reproductive failure during late gestation, and reduced
growth performance.
The virus also participates in a variety of polymicrobial disease syndrome
interactions while
maintaining a life-long subclinical infection (Rowland et al., 2012).Losses
are the result of
respiratory disease in young pigs, poor growth performance, reproductive
failure, and in utero
infection (Keffaber, 1989).
[0004] The reproductive form of the disease accounts for an estimated 45% of
losses, the
result of abortions, dead fetuses, and respiratory disease in newborns. In its
severest form,
reproductive PRRS can result in 90% mortality of fetuses/neonates, along with
increased
mortality for the dams. The reproductive form of PRRS occurs following the
infection of
pregnant gilts or sows at about 90 days of the 114 day gestation period
(Christianson et al.,
1993; Rowland, 2010). After an initial phase of replication in maternal
macrophages, the virus
crosses the placenta and begins to productively infect fetuses. The virus
initially infects only a
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
2
small number of fetuses, followed by horizontal transmission of virus from
fetus to fetus
(Wilkinson et al., 2016). The exact mechanism of how the virus crosses the
placenta remains
unknown, but could be similar to the infected "Trojan Horse" macrophage,
previously described
for lactate dehydrogenase-elevating virus (LDV) (Cafruny, 1996). Unlike the
alveolar
macrophages in adult animals, the primary site of PRRSV replication in the
fetus is the thymus
(Rowland, 2003). Since the pig fetus becomes immunocompetent at about 70 days
of gestation,
PRRSV infection occurs in a fetal immune environment containing functional B
and T cells
(Rowland, 2003; Rowland, 2010).
[0005] Pigs that survive in utero infection become continuous sources of virus
in
downstream production phases, resulting in endemically infected herds
(Rowland, et al., 2003).
The severest form of reproductive disease is associated with a group of highly
virulent isolates
referred to as atypical PRRSV (Halbur et al., 1997; Mengeling et al., 1998).
Interestingly, many
of the atypical PRRSV isolates emerged from PRRS-vaccinated farms (Key et al.,
2001). In
2006, an atypical virus, called high pathogenic PRRSV (HP-PRRSV), appeared in
China and
continues to decimate pig populations in that country (Tian et al., 2007).
Since the standard
commercial breeding facility contains about 5,000 sows, an outbreak of high
mortality
reproductive PRRS can have a devastating impact. To ensure sustainability of
pork production
and food security, solutions for the control of reproductive PRRS remain a
priority. Vaccines
have been unable to control the disease, largely because of genetic diversity
within the structural
proteins of the virus (Shi et al., 2010). In practice, intensive biosecurity
measures provide the
only means of protecting the reproductive herd.
[0006] Porcine reproductive and respiratory syndrome virus (PRRSV) belongs to
the
family Arterividae along with murine lactate dehydrogenase-elevating virus,
simian
hemorrhagic fever virus, and equine arteritis virus. Structurally, the
arteriviruses resemble
togaviruses, but similar to coronaviruses, replicate via a nested 3'-co-
terminal set of subgenomic
mRNAs, which possess a common leader and a poly-A tail. The arteriviruses
share important
properties related to viral pathogenesis, including a tropism for macrophages
and the capacity to
cause severe disease and persistent infection (Plagemann, 1996). Molecular
comparisons
between North American and European viruses place all PRRSV isolates into one
of two
genotypes, Type 2 or Type 1, respectively. Even though the two genotypes
possess only about
70% identity at the nucleotide level (Nelsen et al., 1999), both share a
tropism for CD163-
positive cells, establish long-term infections, and produce similar clinical
signs.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
3
[0007] CD163 is a 130 kDa type 1 membrane protein composed of nine scavenger
receptor cysteine-rich (SRCR) domains and two spacer domains along with a
transmembrane
domain and a short cytoplasmic tail (Fabriek et al., 2005). Porcine CD163
contains 17 exons that
code for a peptide signal sequence followed by nine SRCR domains, two linker
domains (also
referred to as proline serine threonine (PST) domains, located after SRCR 6
and SRCR 9), and a
cytoplasmic domain followed by a short cytoplasmic tail. Surface expression of
CD163 is
restricted to cells of the monocyte-macrophage lineage. In addition to
functioning as a virus
receptor, CD163 exhibits several important functions related to maintaining
normal homeostasis.
For instance, following infection or tissue damage, CD163 functions as a
scavenger molecule,
removing haptoglobin-hemoglobin complexes from the blood (Kristiansen et al.,
2001). The
resulting heme degradation products regulate the associated inflammatory
response (Fabriek et
al., 2005). HbHp scavenging is a major function of CD163 and locates to SRCR 3
(Madsen et
al., 2004). Metabolites released by macrophages following HbHp degradation
include bilirubin,
CO, and free iron. One important function of CD163 the prevention of oxidative
toxicity that
results from free hemoglobin (Kristiansen et al., 2001; Soares et al., 2009).
[0008] Other important functions of C163 include erythroblast adhesion
(SRCR2), being
a TWEAK (tumor necrosis factor-like weak inducer of apoptosis) receptor (SRCR1-
4 & 6-9),
being a bacterial receptor (SRCR5), and being an African Swine Virus receptor
(Sanchez-Torres
et al. 2003). CD163 also has a potential role as an immune-modulator
(discussed in Van Gorp et
al. 2010).
[0009] CD163 was first described as a receptor for PRRSV by Calvert et. al.
(2007).
Transfection of non-permissive cell lines with CD163 cDNAs from a variety of
species,
including simian, human, canine, and mouse, can make cells permissive for
PRRSV infection
(Calvert et al., 2007). In addition to CD163, a second receptor protein, CD169
(also known as
sialoadhesin or SIGLEC1), was identified as being a primary PRRSV receptor
involved in
forming the initial interaction with the GP5-matrix (M) heterodimer, the major
protein on the
surface of the virion (Delputte et al., 2002). In this model, the subsequent
interaction between
CD163 and the GP2, 3, 4 heterotrimer in an endosomal compartment mediates
uncoating and the
release of the viral genome into the cytoplasm (Van Breedam et al., 2010,
Allende et al., 1999).
A previous model describing PRRSV infection of alveolar macrophages identified
SIGLEC1
(CD169) as the primary viral receptor on the surface of macrophages; however,
previous work
using SIGLEC1-1- pigs showed no difference in virus replication compared to
wild type pigs
(Prather et al., 2013). These results supported previous in vitro studies
showing that PRRSV-
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
4
resistant cell lines lacking surface CD169 and CD163 supported virus
replication after
transfection with a CD163 plasmid (Welch et al., 2010).
[0010] Many characteristics of both PRRSV pathogenesis (especially at the
molecular
level) and epizootiology are poorly understood, thus making control efforts
difficult. Currently,
producers often vaccinate swine against PRRSV with modified-live attenuated
strains or killed
virus vaccines, however, current vaccines often do not provide satisfactory
protection. This is
due to both the strain variation and inadequate stimulation of the immune
system. In addition to
concerns about the efficacy of the available PRRSV vaccines, there is strong
evidence that the
modified-live vaccine currently in use can persist in individual pigs and
swine herds and
accumulate mutations (Mengeling et al. 1999), as has been demonstrated with
virulent field
isolates following experimental infection of pigs (Rowland et al., 1999).
Furthermore, it has
been shown that vaccine virus is shed in the semen of vaccinated boars
(Christopher-Hennings
et al., 1997). As an alternative to vaccination, some experts are advocating a
"test and removal"
strategy in breeding herds (Dee et al., 1998). Successful use of this strategy
depends on removal
of all pigs that are either acutely or persistently infected with PRRSV,
followed by strict controls
to prevent reintroduction of the virus. The difficulty, and much of the
expense, associated with
this strategy is that there is little known about the pathogenesis of
persistent PRRSV infection
and thus there are no reliable techniques to identify persistently infected
pigs.
[0011] As can be seen, a need exists in the art for the development of
strategies to induce
PRRSV resistance in animals. There is also a particular need for techniques
for protecting
fetuses from PRRSV infection while in utero and for preventing transmission of
PRRSV from
mother to fetus.
BRIEF SUMMARY OF THE INVENTION
[0012] A method for protecting a porcine fetus from infection with porcine
reproductive
and respiratory syndrome virus (PRRSV) is provided. The method comprises
breeding a female
porcine animal with a male porcine animal. The female porcine animal comprises
modified
chromosomal sequences in both alleles of its CD163 gene, wherein the modified
chromosomal
sequences reduce the susceptibility of the female porcine animal to infection
by PRRSV, as
compared to the susceptibility to infection by PRRSV of a female porcine
animal that does not
comprise any modified chromosomal sequences in the alleles of its CD163 gene.
The male
porcine animal comprises at least one wild-type CD163 allele.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
[0013] Other objects and features will be in part apparent and in part pointed
out
hereinafter.
BRIEF DESCRIPTION OF THE DRAWINGS
[0014] FIG. 1. Targeting vectors and CRISPRs used to modify CD163. Panel A
depicts
wild type exons 7, 8 and 9 of the CD163 gene that was targeted for
modification using
CRISPRs. Panel B shows the targeting vector designed to replace pig exon 7
(pig domain
SRCR5 of CD163) with DNA that encodes human SRCR8 of CD163L. This targeting
vector
was used in transfections with drug selection by G418. PCR primers for the
long range, left arm
and right arm assay are labelled with arrows for 1230, 3752, 8791, 7765 and
7775. Panel C
depicts a targeting vector identical to the one shown in panel B, but wherein
the Neo cassette
was removed. This targeting vector was used to target CD163 in cells that were
already
neomycin resistant. Primers used in small deletions assays are illustrated
with arrows and
labeled GCD163F and GCD163R. Panel D emphasizes the exons targeted by CRISPRs.
Location of CRISPRs 10, 131, 256 and 282 are represented by the downward
facing arrows on
exon 7. The CRISPR numbers represent the number of base pairs from the intron-
exon junction
of intron 6 and exon 7.
[0015] FIG. 2. Targeting vector and CRISPRs used to modify CD1D. Panel A
depicts
wild type exons 3, 4, 5, 6 and 7 of the CD1D gene that was targeted for
modification by
CRISPRs. Panel B shows the targeting vector designed to replace exon 3 with
the selectable
marker Neo. This targeting vector was used in combination with CRISPRs to
modify CD1D.
PCR primers for the long range, left arm and right arm assay are labeled with
arrows for 3991,
4363, 7373 and 12806. Panel C depicts the exons targeted by CRISPRs. Locations
of CRISPRs
4800, 5350, 5620 and 5626 are represented by the downward facing arrows on
exon 3. Primers
used in small deletions assays are illustrated with arrows and labelled GCD1DF
and GCD1DR.
[0016] FIG. 3. Generation of CD163 and CD1D knockout pigs by CRISPR/Cas9 and
SCNT. A) Targeted deletion of CD163 in somatic cells after transfection with
CRISPR/Cas9
and donor DNA. A wild-type (WT) genotype results in a 6545 base pair (bp)
band. Lanes 1-6
represent six different colonies from a single transfection with CRISPR 10
with Cas9 and donor
DNA containing Neo. Lanes 1, 4, and 5 show a large homozygous deletion of 1500-
2000 bp.
Lane 2 represents a smaller homozygous deletion. Lanes 3 and 6 represent
either a WT allele
and a small deletion or a biallelic modification of both alleles. The exact
modifications of each
colony were only determined by sequencing for colonies used for SCNT. The
faint WT band in
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
6
some of the lanes may represent cross-contamination of fetal fibroblasts from
a neighboring WT
colony. NTC = no template control. B) Targeted deletion of CD1D in somatic
cells after
transfection with CRISPR/Cas9 and donor DNA. A WT genotype results in an 8729
bp band.
Lanes 1-4 represent colonies with a 500-2000 bp deletion of CD1D. Lane 4
appears to be a WT
colony. NTC = no template control. C) Image of CD163 knockout pig produced by
SCNT
during the study. This male piglet contains a homozygous 1506 bp deletion of
CD163. D) Image
of CD1D pigs produced during the study. These piglets contain a 1653 bp
deletion of CD1D. E)
Genotype of two SCNT litters containing the 1506 bp deletion of CD163. Lanes 1-
3 (litter 63)
and lanes 1-4 (litter 64) represent the genotype for each piglet from each
litter. Sow indicates
the recipient female of the SCNT embryos, and WT represents a WT control. NTC
= no
template control. F) Genotype of two SCNT litters containing the 1653 bp
deletion of CD1D.
Lanes 1-7 (litter 158) and lanes 1-4 (litter 159) represent the genotype for
each piglet.
[0017] FIG. 4. Effect of CRISPR/Cas9 system in porcine embryos. A) Frequency
of
blastocyst formation after injection of different concentrations of
CRISPR/Cas9 system into
zygotes. Toxicity of the CRISPR/Cas9 system was lowest at 10 ng/p.l. B) The
CRISPR/Cas9
system can successfully disrupt expression of eGFP in blastocysts when
introduced into
zygotes. Original magnification X4. C) Types of mutations on eGFP generated
using the
CRISPR/Cas9 system: WT genotype (SEQ ID NO:16), #1 (SEQ ID NO:17), #2 (SEQ ID
NO:18), and #3 (SEQ ID NO:19).
[0018] FIG. 5. Effect of CRISPR/Cas9 system in targeting CD163 in porcine
embryos.
A) Examples of mutations generated on CD163 by the CRISPR/Cas9 system: WT
genotype
(SEQ ID NO:20), #1-1 (SEQ ID NO:21), #1-4 (SEQ ID NO:22), and #2-2 (SEQ ID
NO:23). All
the embryos examined by DNA sequencing showed mutation on the CD163 (18/18).
CRISPR
131 is highlighted in bold. B) Sequencing read of a homozygous deletion caused
by the
CRISPR/Cas9 system. The image represents # 1-4 from panel A carrying a 2 bp
deletion of
CD163.
[0019] FIG. 6. Effect of CRISPR/Cas9 system when introduced with two types of
CRISPRs. A) PCR amplification of CD163 in blastocysts injected with CRISPR/
Cas9 as
zygotes. Lanes 1,3,6, and 12 show the designed deletion between two different
CRISPRs. B)
PCR amplification of CD1D in blastocysts injected with CRISPR/Cas9 as zygotes.
CD1D had a
lower frequency of deletion as determined by gel electrophoresis when compared
to CD163
(3/23); lanes 1,8, and 15 show obvious deletions in CD1D. C) CRISPR/Cas9
system
successfully targeted two genes when the system was provided with two CRISPRs
targeting
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
7
CD163 and eGFP. The modifications of CD163 and eGFP are shown: CD163 WT (SEQ
ID
NO:24), CD163 #1 (SEQ ID NO:25), CD163 #2 (SEQ ID NO:26), CD163 #3 (SEQ ID
NO:27),
eGFP WT (SEQ ID NO:28), eGFP #1-1 (SEQ ID NO:29), eGFP #1-2 (SEQ ID NO: 30),
eGFP
#2 (SEQ ID NO:31), and eGFP #3 (SEQ ID NO:32).
[0020] FIG. 7. CD163 knockout pigs generated by CRISPR/Cas9 system injected
into
zygotes. A) PCR amplification of CD163 from the knockout pigs; a clear sign of
deletion was
detected in litters 67-2 and 67-4. B) Image of CD163 knockout pigs with a
surrogate. All the
animals are healthy and show no signs of abnormalities. C) Genotype of CD163
knockout
pigs. Wild-type (WT) sequence is shown as SEQ ID NO: 33. Two animals (from
litters 67-
1 (SEQ ID NO:34) and 67-3 (SEQ ID NO:37)) are carrying a homozygous deletion
or
insertion in CD163. The other two animals (from litters 67-2 and 67-4) are
carrying a biallelic
modification of CD163: #67-2 Al (SEQ ID NO:35), #67-2 A2 (SEQ ID NO:36), #67-4
Al (SEQ
ID NO:38), and #67-4 a2 (SEQ ID NO:39). The deletion was caused by introducing
two
different CRISPRs with Cas9 system. No animals from the zygote injection for
CD163
showed a mosaic genotype.
[0021] FIG. 8. CD1D knockout pigs generated by CRISPR/Cas9 system injected
into
zygotes. A) PCR amplification of CD1D from knockout pigs; 166-1 shows a mosaic
genotype
for CD1D. 166-2, 166-3, and 166-4 do not show a change in size for the
amplicon, but
sequencing of the amplicon revealed modifications. WT FF = wild-type fetal
fibroblasts. B)
PCR amplification of the long-range assay showed a clear deletion of one
allele in piglets 166-1
and 166-2. C) Image of CD1D knockout pigs with surrogate. D) Sequence data of
CD1D knock
out pigs; WT (SEQ ID NO:40), #166-1.1 (SEQ ID NO: 41), #166-1.2 (SEQ ID
NO:42), #166-2
(SEQ ID NO:43), #166-3.1 (SEQ ID NO:44), #166-3.2 (SEQID NO:45), and #166-4
(SEQ ID
NO:46). The atg start codon in exon 3 is shown in bold and also lower case.
[0022] FIG. 9. Clinical signs during acute PRRSV infection. Results for daily
assessment for the presence of respiratory signs and fever for CD163 +/+ (n=6)
and CD163 ¨/¨
(n=3).
[0023] FIG. 10. Lung histopathology during acute PRRSV infection.
Representative
photomicrographs of H and E stained tissues from wild-type and knockout pigs.
The left panel
shows edema and infiltration of mononuclear cells. The right panel from a
knockout pig shows
lung architecture of a normal lung.
[0024] FIG. 11. Viremia in the various genotypes. Note that the CD163-/-
piglet data lies
along the X axis.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
8
[0025] FIG. 12. Antibody production in null, wild type and uncharacterized
allele pigs.
[0026] FIG. 13. Cell surface expression of CD163 in individual pigs. Lines
appearing
towards the right in the uncharacterized A, uncharacterized B, and CD163 +/+
panels represent
the CD163 antibody while the lines appearing towards the left-hand sides of
these panels are the
no antibody controls (background). Note that in the CD163-/- animals, the
CD163 staining
overlaps with the background control, and that the CD163 staining in the
uncharacterized alleles
is roughly half way between the WT level and the background (also note that
this is a log scale,
thus less than ¨10%).
[0027] FIG. 14. Level of CD169 on alveolar macrophages from three
representative pigs
and the no antibody control (FITC labelled anti-CD169).
[0028] FIG. 15. Viremia in the various genotypes. Note that the A43 amino acid
piglet
data lies along the X-axis.
[0029] FIG. 16. Genomic Sequence of wild type CD163 exons 7-10 used as a
reference
sequence (SEQ ID NO: 47). The sequence includes 3000 bp upstream of exon 7 to
the last base
of exon 10. The underlined regions show the locations of exons 7, 8,9, and 10,
respectively.
[0030] FIG. 17. Diagram of CD163 modifications illustrating several CD163
chromosomal modifications, the predicted protein product for each
modification, and relative
macrophage expression for each modification, as measured by the level of
surface CD163 on
porcine alveolar macrophages (PAMs). Black regions indicate introns and white
regions indicate
exons. The hatched region indicates the hCD163L1 exon 11 mimic, the homolog of
porcine
exon 7. The grey region indicates the synthesized intron with PGK Neo
construct.
[0031] FIG. 18. Diagram of the porcine CD163 protein and gene sequence. A)
CD163
protein SRCR (ovals) and PST (squares) domains along with the corresponding
gene exons. B)
Comparison of the porcine CD163 SRCR 5 (SEQ ID NO: 120) with the human CD163L1
SRCR
8 (SEQ ID NO: 121) homolog.
[0032] FIG. 19. Representative results for surface expression of CD163 and
CD169 on
PAMs from wild-type and CD163-modified pigs. Panels A¨E show results for the
CD163
modifications as illustrated in FIG. 17. Pooled data for d7(1467) and d7(1280)
are shown in
panel D.
[0033] FIG. 20. Serum haptoglobin levels in wild-type and CD163-modified pigs.
[0034] FIG. 21. Relative permissiveness of wild-type and HL11m PAMs to
infection
with Type 2 PRRSV isolates.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
9
[0035] FIG. 22. Infection of CD163 modified pigs with Type 1 and Type 2 PRRSV
isolates.
[0036] FIG. 23. Virus load for WT and CD163-modified pigs infected with Type 2
viruses.
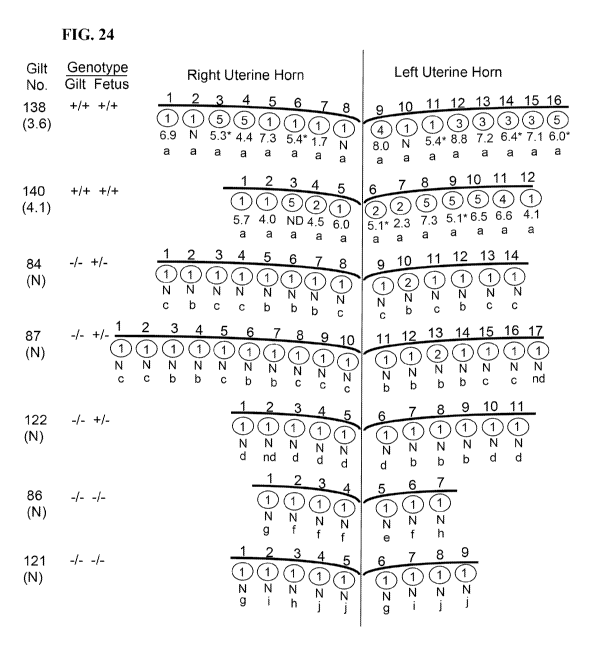
[0037] FIG. 24. Fetal outcomes following maternal infection with PRRSV. The
numbers
on the left identify each dam ("Dam No."; see Table 16 below). Below each dam
number in
parenthesis is the result for PRRS PCR in serum, measured as logio templates
per reaction. "N"
is negative for PRRSV nucleic acid (Ct>39). Fetuses are identified by number
and relative
position within each uterine horn. Asterisks identify fetal PCR samples
obtained from abdominal
fluid. The number below each fetus is the result for PRRS PCR in fetal serum
(logio templates
per reaction). The number within each circle refers to the presence of
anatomical pathology: 1)
normal fetus; 2) small fetus; 3) placenta changes such as detached placenta
and/or necrosis; 4)
meconium stained fetus; 5) fetus is dead and necrotic. Lower case letters
identify the genotype
of the individual fetuses (see Table 16). Key: a, A/A; b, C/A; c, B/A; d, E/A;
e, B/C; f, B/D; g,
D/C; h, D/D; i, E/C; j, E/D; ND not determined because the fetus was necrotic;
nd, genotype was
not determined.
DETAILED DESCRIPTION OF THE INVENTION
[0038] The present invention is directed to methods for protecting porcine
fetuses from
infection with porcine reproductive and respiratory syndrome virus (PRRSV).
Pigs having
inactivating mutations in both alleles of the CD163 gene are resistant to
infection with PRRSV.
It has now unexpectedly been found that CD163-positive fetuses (e.g., fetuses
that have one or
two wild-type CD163 alleles) can be protected from PRRSV infection while in
utero so long as
the dam possesses inactivating mutations in both alleles of her CD163 gene.
Thus, for example,
dams having inactivating mutations in both alleles of the CD163 gene can be
mated males
having two wild-type CD163 alleles, and the resulting heterozygous fetuses
will be protected
from PRRSV infection.
Definitions
[0039] When introducing elements of the present invention or the preferred
embodiments(s) thereof, the articles "a", "an", "the", and "said" are intended
to mean that there
are one or more of the elements.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
[0040] The term "and/or" means any one of the items, any combination of the
items, or
all of the items with which this term is associated.
[0041] The term "breeding" as used herein refers to the union of male and
female
gametes so that fertilization occurs. Such a union may be brought about by
mating (copulation)
or by in vitro or in vivo artificial methods. Such artificial methods include,
but are not limited to,
artificial insemination, surgical assisted artificial insemination, in vitro
fertilization,
intracytoplasmic sperm injection, zona drilling, in vitro culture of
fertilized oocytes, ovary
transfer, and ovary splitting. The term "breeding" as used herein also
includes transferring of a
fertilized oocyte into the reproductive tract of a female animal.
[0042] The terms "comprising", "including", and "having" are intended to be
inclusive
and mean that there may be additional elements other than the listed elements.
[0043] The term "CRISPR" stands for "clustered regularly interspaced short
palindromic
repeats." CRISPR systems include Type I, Type II, and Type III CRISPR systems.
[0044] The term "Cas" refers to "CRISPR associated protein." Cas proteins
include but
are not limited to Cas9 family member proteins, Cas6 family member proteins
(e.g., Csy4 and
Cas6), and Cas5 family member proteins.
[0045] The term "Cas9" can generally refer to a polypeptide with at least
about 5%,
10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% sequence identity and/or
sequence
similarity to a wild-type Cas9 polypeptide (e.g., Cas9 from S. pyogenes).
Illustrative Cas9
sequences are provided by SEQ ID NOs. 1-256 and 795-1346 of U.S. Patent
Publication No.
2016/0046963. SEQ ID NOs. 1-256 and 795-1346 of U.S. Patent Publication No.
2016/0046963 are hereby incorporated herein by reference. "Cas9" can refer to
can refer to a
polypeptide with at most about 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%,
90%, or
100% sequence identity and/or sequence similarity to a wild type Cas9
polypeptide (e.g., from S.
pyogenes). "Cas9" can refer to the wild-type or a modified form of the Cas9
protein that can
comprise an amino acid change such as a deletion, insertion, substitution,
variant, mutation,
fusion, chimera, or any combination thereof.
[0046] The term "Cas5" can generally refer to can refer to a polypeptide with
at least
about 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% sequence
identity and/or
sequence similarity to a wild type illustrative Cas5 polypeptide (e.g., Cas5
from D. vulgaris).
Illustrative Cas5 sequences are provided in Figure 42 of U.S. Patent
Publication No.
2016/0046963. Figure 42 of U.S. Patent Publication No. 2016/0046963 is hereby
incorporated
herein by reference. "Cas5" can generally refer to can refer to a polypeptide
with at most about
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
11
5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% sequence identity
and/or
sequence similarity to a wild-type Cas5 polypeptide (e.g., a Cas5 from D.
vulgaris). "Cas5" can
refer to the wild-type or a modified form of the Cas5 protein that can
comprise an amino acid
change such as a deletion, insertion, substitution, variant, mutation, fusion,
chimera, or any
combination thereof.
[0047] The term "Cas6" can generally refer to can refer to a polypeptide with
at least
about 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% sequence
identity and/or
sequence similarity to a wild type illustrative Cas6 polypeptide (e.g., a Cas6
from T
thermophilus). Illustrative Cas6 sequences are provided in Figure 41 of U.S.
Patent Publication
No. 2016/0046963. Figure 41 of U.S. Patent Publication No. 2016/0046963 is
hereby
incorporated herein by reference. "Cas6" can generally refer to can refer to a
polypeptide with at
most about 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% sequence
identity
and/or sequence similarity to a wild-type Cas6 polypeptide (e.g., from T
thermophilus). "Cas6"
can refer to the wildtype or a modified form of the Cas6 protein that can
comprise an amino acid
change such as a deletion, insertion, substitution, variant, mutation, fusion,
chimera, or any
combination thereof.
[0048] The terms "CRISPR/Cas9" or "CRISPR/Cas9 system" refer to a programmable
nuclease system for genetic engineering that includes a Cas9 protein, or
derivative thereof, and
one or more non-coding RNAs that can provide the function of a CRISPR RNA
(crRNA) and
trans-activating RNA (tracrRNA) for the Cas9. The crRNA and tracrRNA can be
used
individually or can be combined to produce a "guide RNA" (gRNA). The crRNA or
gRNA
provide sequence that is complementary to the genomic target.
[0049] References herein to a deletion in a nucleotide sequence from
nucleotide x to
nucleotide y mean that all of the nucleotides in the range have been deleted,
including x and y.
Thus, for example, the phrase "an 11 base pair deletion from nucleotide 3,137
to nucleotide
3,147 as compared to SEQ ID NO: 47" means that each of nucleotides 3,317
through 3,147 have
been deleted, including nucleotides 3,317 and 3,147.
[0050] "Resistance" of an animal to a disease is a characteristic of an
animal, wherein
the animal avoids the disease symptoms that are the outcome of animal-pathogen
interactions,
such as interactions between a porcine animal and PRRSV. That is, pathogens
are prevented
from causing animal diseases and the associated disease symptoms, or
alternatively, a reduction
of the incidence and/or severity of clinical signs or reduction of clinical
symptoms. One of skill
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
12
in the art will appreciate that the methods disclosed herein can be used with
other compositions
and methods available in the art for protecting animals from pathogen attack.
[0051] As used herein, "gene editing," "gene edited", "genetically edited" and
"gene
editing effectors" refer to the use of homing technology with naturally
occurring or artificially
engineered nucleases, also referred to as "molecular scissors," "homing
endonucleases," or
"targeting endonucleases." The nucleases create specific double-stranded
chromosomal breaks
(DSBs) at desired locations in the genome, which in some cases harnesses the
cell's endogenous
mechanisms to repair the induced break by natural processes of homologous
recombination
(HR) and/or nonhomologous end-joining (NHEJ). Gene editing effectors include
Zinc Finger
Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs),
Clustered
Regularly Interspaced Short Palindromic Repeats (CRISPR) systems (e.g., the
CRISPR/Cas9
system), and meganucleases (e.g., meganucleases re-engineered as homing
endonucleases). The
terms also include the use of transgenic procedures and techniques, including,
for example,
where the change is a deletion or relatively small insertion (typically less
than 20nt) and/or does
not introduce DNA from a foreign species. The term also encompasses progeny
animals such as
those created by sexual crosses or asexual propagation from the initial gene
edited animal.
[0052] The terms "genome engineering," "genetic engineering," "genetically
engineered," "genetically altered," "genetic alteration," "genome
modification," "genome
modification," and "genomically modified" can refer to altering the genome by
deleting,
inserting, mutating, or substituting specific nucleic acid sequences. The
altering can be gene or
location specific. Genome engineering can use nucleases to cut a nucleic acid
thereby generating
a site for the alteration. Engineering of non-genomic nucleic acid is also
contemplated. A protein
containing a nuclease domain can bind and cleave a target nucleic acid by
forming a complex
with a nucleic acid-targeting nucleic acid. In one example, the cleavage can
introduce double
stranded breaks in the target nucleic acid. A nucleic acid can be repaired
e.g. by endogenous
non-homologous end joining (NHEJ) machinery. In a further example, a piece of
nucleic acid
can be inserted. Modifications of nucleic acid-targeting nucleic acids and
site-directed
polypeptides can introduce new functions to be used for genome engineering.
[0053] As used herein "homing DNA technology," "homing technology" and "homing
endonuclease" include any mechanisms that allow a specified molecule to be
targeted to a
specified DNA sequence including Zinc Finger (ZF) proteins, Transcription
Activator-Like
Effectors (TALEs) meganucleases, and CRISPR systems (e.g., the CRISPR/Cas9
system).
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
13
[0054] The terms "increased resistance" and "reduced susceptibility" herein
mean, but
are not limited to, a statistically significant reduction of the incidence
and/or severity of clinical
signs or clinical symptoms which are associated with infection by pathogen.
For example,
"increased resistance" or "reduced susceptibility" can refer to a
statistically significant reduction
of the incidence and/or severity of clinical signs or clinical symptoms which
are associated with
infection by PRRSV in an animal comprising a modified chromosomal sequence in
a CD163
gene protein as compared to a control animal having an unmodified chromosomal
sequence. The
term "statistically significant reduction of clinical symptoms" means, but is
not limited to, the
frequency in the incidence of at least one clinical symptom in the modified
group of subjects is
at least 10%, preferably at least 20%, more preferably at least 30%, even more
preferably at least
50%, and even more preferably at least 70% lower than in the non-modified
control group after
the challenge with the infectious agent.
[0055] "Knock-out" means disruption of the structure or regulatory mechanism
of a
gene. Knock-outs may be generated through homologous recombination of
targeting vectors,
replacement vectors, or hit-and-run vectors or random insertion of a gene trap
vector resulting in
complete, partial or conditional loss of gene function.
[0056] As used herein, the term "mutation" includes alterations in the
nucleotide
sequence of a polynucleotide, such as for example a gene or coding DNA
sequence (CDS),
compared to the wild-type sequence. The term includes, without limitation,
substitutions,
insertions, frameshifts, deletions, inversions, translocations, duplications,
splice-donor site
mutations, point-mutations and the like.
[0057] Herein, "reduction of the incidence and/or severity of clinical signs"
or
"reduction of clinical symptoms" means, but is not limited to, reducing the
number of infected
subjects in a group, reducing or eliminating the number of subjects exhibiting
clinical signs of
infection, or reducing the severity of any clinical signs that are present in
one or more subjects,
in comparison to wild-type infection. For example, these terms encompass any
clinical signs of
infection, lung pathology, viremia, antibody production, reduction of pathogen
load, pathogen
shedding, reduction in pathogen transmission, or reduction of any clinical
sign symptomatic of
PRRSV. Preferably these clinical signs are reduced in one or more animals of
the invention by at
least 10% in comparison to subjects not having a modification in the CD163
gene and that
become infected. More preferably clinical signs are reduced in subjects of the
invention by at
least 20%, preferably by at least 30%, more preferably by at least 40%, and
even more
preferably by at least 50%.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
14
[0058] A "TALE DNA binding domain" or "TALE" is a polypeptide comprising one
or
more TALE repeat domains/units. The repeat domains are involved in binding of
the TALE to
its cognate target DNA sequence. A single "repeat unit" (also referred to as a
"repeat") is
typically 33-35 amino acids in length and exhibits at least some sequence
homology with other
TALE repeat sequences within a naturally occurring TALE protein. Zinc finger
and TALE
binding domains can be "engineered" to bind to a predetermined nucleotide
sequence, for
example via engineering (altering one or more amino acids) of the recognition
helix region of
naturally occurring zinc finger or TALE proteins. Therefore, engineered DNA
binding proteins
(zinc fingers or TALEs) are proteins that are non-naturally occurring. Non-
limiting examples of
methods for engineering DNA-binding proteins are design and selection. A
designed DNA
binding protein is a protein not occurring in nature whose design/composition
results principally
from rational criteria. Rational criteria for design include application of
substitution rules and
computerized algorithms for processing information in a database storing
information of existing
ZFP and/or TALE designs and binding data. See, for example, U.S. Pat. Nos.
6,140,081;
6,453,242; and 6,534,261; see also WO 98/53058; WO 98/53059; WO 98/53060; WO
02/016536 and WO 03/016496 and U.S. Publication No. 20110301073.
[0059] A "zinc finger DNA binding protein" (or binding domain) is a protein,
or a
domain within a larger protein, that binds DNA in a sequence-specific manner
through one or
more zinc fingers, which are regions of amino acid sequence within the binding
domain whose
structure is stabilized through coordination of a zinc ion. The term zinc
finger DNA binding
protein is often abbreviated as zinc finger protein or ZFP.
[0060] A "selected" zinc finger protein or TALE is a protein not found in
nature whose
production results primarily from an empirical process such as phage display,
interaction trap or
hybrid selection. See e.g., U.S. Pat. No. 5,789,538; U.S. Pat. No. 5,925,523;
U.S. Pat. No.
6,007,988; U.S. Pat. No. 6,013,453; U.S. Pat. No. 6,200,759; WO 95/19431; WO
96/06166; WO
98/53057; WO 98/54311; WO 00/27878; WO 01/60970 WO 01/88197, WO 02/099084 and
U.S.
Publication No. 20110301073.
[0061] Various other terms are defined hereinbelow.
Methods for Protecting Porcine Fetuses from PRRSV Infection
[0062] A method for protecting a porcine fetus from infection with porcine
reproductive
and respiratory syndrome virus (PRRSV) is provided. The method comprises
breeding a female
porcine animal with a male porcine animal. The female porcine animal comprises
modified
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
chromosomal sequences in both alleles of its CD163 gene, wherein the modified
chromosomal
sequences reduce the susceptibility of the female porcine animal to infection
by PRRSV, as
compared to the susceptibility to infection by PRRSV of a female porcine
animal that does not
comprise any modified chromosomal sequences in the alleles of its CD163 gene.
The male
porcine animal comprises at least one wild-type CD163 allele.
[0063] In the methods described herein, the modified chromosomal sequences can
be
sequences that are altered such that a CD163 protein function as it relates to
PRRSV infection is
impaired, reduced, or eliminated. Thus, the female porcine animals used in the
methods
described herein can be referred to as a "knock-out" animal.
[0064] The male porcine animal can comprise two wild-type CD163 alleles.
[0065] The term "wild-type CD163 allele" as used herein means that the
sequence of the
CD163 allele is a sequence as found in nature, or that the sequence of the
CD163 allele contains
one or more mutations (e.g., insertions, deletions, or substitutions) that do
not substantially
impair CD163 activity. Thus, wild-type CD163 alleles can contain polymorphisms
and/or
mutations, so long as those polymorphisms or mutations do not substantially
impair CD163
activity.
[0066] Using the methods described herein, the fetuses will be protected from
both Type
1 and Type 2 PRRSV viruses, including various Type 1 and Type 2 PRRSV
isolates.
[0067] Thus, in the methods described herein, the modified chromosomal
sequences can
reduce the susceptibility of the female porcine animal to a Type 1 PRRSV
virus, a Type 2
PRRSV, or to both Type 1 and Type 2 PRRSV viruses.
[0068] The modified chromosomal sequences can reduce the susceptibility of the
female
porcine animal to a PRRSV isolate selected from the group consisting of NVSL
97-7895, KS06-
72109, P129, VR2332, C090, AZ25, MLV-ResPRRS, KS62-06274, KS483 (SD23983),
C084,
SD13-15, Lelystad, 03-1059, 03-1060, SD01-08, 4353PZ, and combinations
thereof.
[0069] The female porcine animal can comprise a genetically edited female
porcine
animal.
[0070] The genetically edited female porcine animal can be an animal that has
been
edited using a homing endonuclease. The homing endonuclease can be a naturally
occurring
endonuclease but is preferably a rationally designed, non-naturally occurring
homing
endonuclease that has a DNA recognition sequence that has been designed so
that the
endonuclease targets a chromosomal sequence in gene encoding a CD163 protein.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
16
[0071] Thus, the homing endonuclease can be a designed homing endonuclease.
The
homing endonuclease can comprise, for example, a Clustered Regularly
Interspaced Short
Palindromic Repeats (CRISPR) system, a Transcription Activator-Like Effector
Nuclease
(TALEN), a Zinc Finger Nuclease (ZFN), a recombinase fusion protein, a
meganuclease, or a
combination of any thereof.
[0072] The homing nuclease preferably comprises a CRISPR system. Examples of
CRISPR systems that can be used to create the female porcine animals for use
in the methods
described herein include, but are not limited to CRISPR/Cas9, CRISPR/Cas5, and
CRISPR/Cas6. The use of CRISPR systems to generate genetically edited animals
is discussed
further hereinbelow.
[0073] In any of the methods described herein, the female porcine animal can
comprise
the same modified chromosomal sequence in both alleles of the CD163 gene.
[0074] Alternatively, the female porcine animal can comprise a first modified
chromosomal sequence in a first allele of the CD163 gene and a second modified
chromosomal
sequence in a second allele of the CD163 gene, the first and second modified
chromosomal
sequences being different from each other.
[0075] In any of the methods described herein, each allele of the CD163 gene
of the
female porcine animal can comprise an insertion, a deletion, or a combination
thereof.
[0076] For example, at least one allele of the CD163 gene of the female
porcine animal
can comprise a deletion.
[0077] At least one allele of the CD163 gene can comprise an in-frame
deletion.
[0078] At least one allele of the CD163 gene of the female porcine animal can
comprise
an insertion.
[0079] In the methods described herein, the modified chromosomal sequences
preferably
cause CD163 protein production or activity to be reduced, as compared to CD163
protein
production or activity in a female porcine animal that lacks the modified
chromosomal
sequences.
[0080] Preferably, the modified chromosomal sequences result in production of
substantially no functional CD163 protein by the female porcine animal. By
"substantially no
functional CD163 protein," it is meant that the level of CD163 protein in the
animal, offspring,
or cell is undetectable, or if detectable, is at least about 90% lower,
preferably at least about 95%
lower, more preferably at least about 98%, lower, and even more preferably at
least about 99%
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
17
lower than the level observed in an animal, offspring, or cell that does not
comprise the modified
chromosomal sequences.
[0081] For example, in any of the methods described herein, the female porcine
animal
does not produce CD163 protein.
[0082] In any of the methods described herein, each allele of the CD163 gene
of the
female porcine animal can comprise a modification in exon 7, a modification in
exon 8, a
modification in an intron that is contiguous with exon 7 or exon 8, or a
combination of any
thereof.
[0083] For example, one or both alleles of the CD163 gene of the female
porcine animal
can comprise a modification in exon 7 of the CD163 gene.
[0084] The modification in exon 7 can comprise a deletion.
[0085] In any of the methods described herein wherein an allele of the CD163
gene of
the female porcine animal comprises a deletion, the deletion can comprise an
in-frame deletion.
[0086] The modification in exon 7 can comprise an insertion.
[0087] In any of the methods described herein, the modified chromosomal
sequences in
one or both alleles of the CD163 gene of the female porcine animal can result
in a miscoding.
[0088] Where a modified chromosomal sequence results in a miscoding in an
allele of
the CD163 gene, the miscoding can result in a premature stop codon downstream
of the
miscoding in the allele of the CD163 gene.
[0089] In any of the methods described herein, at least one of the alleles of
the CD163
gene in the female porcine animal comprises a modification selected from the
group consisting
of: an 11 base pair deletion from nucleotide 3,137 to nucleotide 3,147 as
compared to reference
sequence SEQ ID NO: 47; a 2 base pair insertion between nucleotides 3,149 and
3,150 as
compared to reference sequence SEQ ID NO: 47, with a 377 base pair deletion
from nucleotide
2,573 to nucleotide 2,949 as compared to reference sequence SEQ ID NO: 47 on
the same allele;
a 124 base pair deletion from nucleotide 3,024 to nucleotide 3,147 as compared
to reference
sequence SEQ ID NO: 47; a 123 base pair deletion from nucleotide 3,024 to
nucleotide 3,146 as
compared to reference sequence SEQ ID NO: 47; a 1 base pair insertion between
nucleotides
3,147 and 3,148 as compared to reference sequence SEQ ID NO: 47; a 130 base
pair deletion
from nucleotide 3,030 to nucleotide 3,159 as compared to reference sequence
SEQ ID NO: 47; a
132 base pair deletion from nucleotide 3,030 to nucleotide 3,161 as compared
to reference
sequence SEQ ID NO: 47; a 1506 base pair deletion from nucleotide 1,525 to
nucleotide 3,030
as compared to reference sequence SEQ ID NO: 47; a 7 base pair insertion
between nucleotide
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
18
3,148 and nucleotide 3,149 as compared to reference sequence SEQ ID NO: 47; a
1280 base pair
deletion from nucleotide 2,818 to nucleotide 4,097 as compared to reference
sequence SEQ ID
NO: 47; a 1373 base pair deletion from nucleotide 2,724 to nucleotide 4,096 as
compared to
reference sequence SEQ ID NO: 47; a 1467 base pair deletion from nucleotide
2,431 to
nucleotide 3,897 as compared to reference sequence SEQ ID NO: 47; a 1930 base
pair deletion
from nucleotide 488 to nucleotide 2,417 as compared to reference sequence SEQ
ID NO: 47,
wherein the deleted sequence is replaced with a 12 base pair insertion
beginning at nucleotide
488, and wherein there is a further 129 base pair deletion in exon 7 from
nucleotide 3,044 to
nucleotide 3,172 as compared to reference sequence SEQ ID NO: 47; a 28 base
pair deletion
from nucleotide 3,145 to nucleotide 3,172 as compared to reference sequence
SEQ ID NO: 47; a
1387 base pair deletion from nucleotide 3,145 to nucleotide 4,531 as compared
to reference
sequence SEQ ID NO: 47; a 1382 base pair deletion from nucleotide 3,113 to
nucleotide 4,494
as compared to reference sequence SEQ ID NO: 47, wherein the deleted sequence
is replaced
with an 11 base pair insertion beginning at nucleotide 3,113; a 1720 base pair
deletion from
nucleotide 2,440 to nucleotide 4,160 as compared to reference sequence SEQ ID
NO: 47; a 452
base pair deletion from nucleotide 3,015 to nucleotide 3,466 as compared to
reference sequence
SEQ ID NO: 47; and combinations of any thereof
[0090] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 11 base pair deletion from nucleotide 3,137 to nucleotide 3,147
as compared to
reference sequence SEQ ID NO: 47.
[0091] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 2 base pair insertion between nucleotides 3,149 and 3,150 as
compared to
reference sequence SEQ ID NO: 47, with the 377 base pair deletion from
nucleotide 2,573 to
nucleotide 2,949 as compared to reference sequence SEQ ID NO: 47 on the same
allele.
[0092] Where the modification comprises the 2 base pair insertion between
nucleotides
3,149 and 3,150 as compared to reference sequence SEQ ID NO: 47, with the 377
base pair
deletion from nucleotide 2,573 to nucleotide 2,949 as compared to reference
sequence SEQ ID
NO: 47 on the same allele, and the 2 base pair insertion can comprise the
dinucleotide AG.
[0093] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 124 base pair deletion from nucleotide 3,024 to nucleotide 3,147
as compared to
reference sequence SEQ ID NO: 47.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
19
[0094] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 123 base pair deletion from nucleotide 3,024 to nucleotide 3,146
as compared to
reference sequence SEQ ID NO: 47.
[0095] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1 base pair insertion between nucleotides 3,147 and 3,148 as
compared to
reference sequence SEQ ID NO: 47.
[0096] Where the modification comprises the 1 base pair insertion between
nucleotides
3,147 and 3,148 as compared to reference sequence SEQ ID NO: 47, the 1 base
pair insertion
can comprise a single adenine residue.
[0097] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 130 base pair deletion from nucleotide 3,030 to nucleotide 3,159
as compared to
reference sequence SEQ ID NO: 47.
[0098] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 132 base pair deletion from nucleotide 3,030 to nucleotide 3,161
as compared to
reference sequence SEQ ID NO: 47.
[0099] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1506 base pair deletion from nucleotide 1,525 to nucleotide 3,030
as compared to
reference sequence SEQ ID NO: 47.
[00100] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 7 base pair insertion between nucleotide 3,148 and nucleotide
3,149 as compared
to reference sequence SEQ ID NO: 47.
[00101] Where the modification comprises the 7 base pair insertion between
nucleotide 3,148 and nucleotide 3,149 as compared to reference sequence SEQ ID
NO: 47, the 7
base pair insertion can comprise the sequence TACTACT (SEQ ID NO: 115).
[00102] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the1280 base pair deletion from nucleotide 2,818 to nucleotide 4,097
as compared to
reference sequence SEQ ID NO: 47.
[00103] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1373 base pair deletion from nucleotide 2,724 to nucleotide 4,096
as compared to
reference sequence SEQ ID NO: 47.
[00104] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1467 base pair deletion from nucleotide 2,431 to nucleotide 3,897
as compared to
reference sequence SEQ ID NO: 47.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
[00105] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1930 base pair deletion from nucleotide 488 to nucleotide 2,417
as compared to
reference sequence SEQ ID NO: 47, wherein the deleted sequence is replaced
with a 12 base
pair insertion beginning at nucleotide 488, and wherein there is a further 129
base pair deletion
in exon 7 from nucleotide 3,044 to nucleotide 3,172 as compared to reference
sequence SEQ ID
NO: 47.
[00106] Where the modification comprises the 1930 base pair deletion from
nucleotide 488 to nucleotide 2,417 as compared to reference sequence SEQ ID
NO: 47, wherein
the deleted sequence is replaced with a 12 base pair insertion beginning at
nucleotide 488, and
wherein there is a further 129 base pair deletion in exon 7 from nucleotide
3,044 to nucleotide
3,172 as compared to reference sequence SEQ ID NO: 47, the 12 base pair
insertion can
comprise the sequence TGTGGAGAATTC (SEQ ID NO: 116).
[00107] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 28 base pair deletion from nucleotide 3,145 to nucleotide 3,172
as compared to
reference sequence SEQ ID NO: 47.
[00108] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1387 base pair deletion from nucleotide 3,145 to nucleotide 4,531
as compared to
reference sequence SEQ ID NO: 47.
[00109] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1382 base pair deletion from nucleotide 3,113 to nucleotide 4,494
as compared to
reference sequence SEQ ID NO: 47, wherein the deleted sequence is replaced
with an 11 base
pair insertion beginning at nucleotide 3,113.
[00110] Where the modification comprises the 1382 base pair deletion from
nucleotide 3,113 to nucleotide 4,494 as compared to reference sequence SEQ ID
NO: 47,
wherein the deleted sequence is replaced with an 11 base pair insertion
beginning at nucleotide
3,113, the 11 base pair insertion can comprises the sequence AGCCAGCGTGC (SEQ
ID NO:
117).
[00111] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 1720 base pair deletion from nucleotide 2,440 to nucleotide 4,160
as compared to
reference sequence SEQ ID NO: 47.
[00112] At least one of the alleles of the CD163 gene in the female porcine
animal can
comprise the 452 base pair deletion from nucleotide 3,015 to nucleotide 3,466
as compared to
reference sequence SEQ ID NO: 47.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
21
[00113] For example, at least one of the alleles of the CD163 gene in the
female
porcine animal comprises a modification selected from the group consisting of:
the 7 base pair
insertion between nucleotide 3,148 and nucleotide 3,149 as compared to
reference sequence
SEQ ID NO: 47; the 2 base pair insertion between nucleotides 3,149 and 3,150
as compared to
reference sequence SEQ ID NO: 47, with the 377 base pair deletion from
nucleotide 2,573 to
nucleotide 2,949 as compared to reference sequence SEQ ID NO: 47 on the same
allele; the 11
base pair deletion from nucleotide 3,137 to nucleotide 3,147 as compared to
reference sequence
SEQ ID NO: 47; the 1382 base pair deletion from nucleotide 3,113 to nucleotide
4,494 as
compared to reference sequence SEQ ID NO: 47, wherein the deleted sequence is
replaced with
the 11 base pair insertion beginning at nucleotide 3,113; and combinations of
any thereof
[00114] The CD163 gene in the female porcine animal can comprise any
combination
of the modified chromosomal sequences described herein.
[00115] For example, the female porcine animal can comprise the 7 base pair
insertion
between nucleotide 3,148 and nucleotide 3,149 as compared to reference
sequence SEQ ID NO:
47 in one allele of the CD163 gene; and the 2 base pair insertion between
nucleotides 3,149 and
3,150 as compared to reference sequence SEQ ID NO: 47, with the 377 base pair
deletion from
nucleotide 2,573 to nucleotide 2,949 as compared to reference sequence SEQ ID
NO: 47, in the
other allele of the CD163 gene.
[00116] The female porcine animal can comprise the 1382 base pair deletion
from
nucleotide 3,113 to nucleotide 4,494 as compared to reference sequence SEQ ID
NO: 47,
wherein the deleted sequence is replaced with the 11 base pair insertion
beginning at nucleotide
3,113, in one allele of the CD163 gene; and the 2 base pair insertion between
nucleotides 3,149
and 3,150 as compared to reference sequence SEQ ID NO: 47, with the 377 base
pair deletion
from nucleotide 2,573 to nucleotide 2,949 as compared to reference sequence
SEQ ID NO: 47,
in the other allele of the CD163 gene.
[00117] The female porcine animal can comprise the 7 base pair insertion
between
nucleotide 3,148 and nucleotide 3,149 as compared to reference sequence SEQ ID
NO: 47 in
one allele of the CD163 gene; and the 11 base pair deletion from nucleotide
3,137 to nucleotide
3,147 as compared to reference sequence SEQ ID NO: 47 in the other allele of
the CD163 gene.
[00118] The female porcine animal can comprise the 1382 base pair deletion
from
nucleotide 3,113 to nucleotide 4,494 as compared to reference sequence SEQ ID
NO: 47,
wherein the deleted sequence is replaced with the 11 base pair insertion
beginning at nucleotide
3,113, in one allele of the CD163 gene; and the 11 base pair deletion from
nucleotide 3,137 to
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
22
nucleotide 3,147 as compared to reference sequence SEQ ID NO: 47 in the other
allele of the
CD163 gene.
[00119] In any of the methods described herein, the alleles of the CD163 gene
of the
female porcine animal can comprise a chromosomal sequence having at least 80%
sequence
identity to SEQ ID NO: 47 in the regions of said chromosomal sequence outside
of the insertion
or deletion.
[00120] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 85% sequence identity to SEQ ID NO: 47 in
the regions
of said chromosomal sequence outside of the insertion or deletion.
[00121] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 90% sequence identity to SEQ ID NO: 47 in
the regions
of said chromosomal sequence outside of the insertion or deletion.
[00122] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 95% sequence identity to SEQ ID NO: 47 in
the regions
of said chromosomal sequence outside of the insertion or deletion.
[00123] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 98% sequence identity to SEQ ID NO: 47 in
the regions
of said chromosomal sequence outside of the insertion or deletion.
[00124] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 99% sequence identity to SEQ ID NO: 47 in
the regions
of said chromosomal sequence outside of the insertion or deletion.
[00125] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having at least 99.9% sequence identity to SEQ ID NO: 47
in the
regions of said chromosomal sequence outside of the insertion or deletion.
[00126] The alleles of the CD163 gene of the female porcine animal can
comprise a
chromosomal sequence having 100% sequence identity to SEQ ID NO: 47 in the
regions of said
chromosomal sequence outside of the insertion or deletion.
[00127] In any of the methods described herein, the female porcine animal can
comprise a chromosomal sequence comprising SEQ ID NO: 98, 99, 100, 101, 102,
103, 104,
105, 106, 107, 108, 109, 110, 111, 112, 113, 114, or 119 in one or both
alleles of the CD163
gene.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
23
[00128] For example, the female porcine animal can comprise a chromosomal
sequence comprising SEQ ID NO: 99, 102, 103, or 113 in one or both alleles of
the CD163
gene.
[00129] The alleles of the CD163 gene in the female porcine animal can
comprise any
combination of chromosomal sequences comprising SEQ ID NO: 98, 99, 100, 101,
102, 103,
104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, or 119. Thus, the
female porcine animal
can comprise a chromosomal sequence comprising any one of SEQ ID NO: 98, 99,
100, 101,
102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, or 119 in one
allele of the
CD163 gene and a chromosomal sequence comprising any one of SEQ ID NO: 98, 99,
100, 101,
102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, or 119 in the
other allele of the
CD163 gene.
[00130] For example, the female porcine animal can comprise a chromosomal
sequence comprising SEQ ID NO: 99 in one allele of the CD163 gene, and a
chromosomal
sequence comprising SEQ ID NO: 103 in the other allele of the CD163 gene.
[00131] The female porcine animal can comprise a chromosomal sequence
comprising SEQ ID NO: 113 in one allele of the CD163 gene, and a chromosomal
sequence
comprising SEQ ID NO: 99 in the other allele of the CD163 gene.
[00132] The female porcine animal can comprise a chromosomal sequence
comprising SEQ ID NO: 99 in one allele of the CD163 gene, and a chromosomal
sequence
comprising SEQ ID NO: 102 in the other allele of the CD163 gene.
[00133] The female porcine animal can comprise a chromosomal sequence
comprising SEQ ID NO: 113 in one allele of the CD163 gene, and a chromosomal
sequence
comprising SEQ ID NO: 102 in the other allele of the CD163 gene.
[00134] In any of the methods described herein, the breeding will produce one
or
more fetuses that comprise a modified chromosomal sequence in a single allele
of the CD163
gene. Since the female porcine animal used in the methods described herein
comprises modified
chromosomal sequences in both alleles of its CD163 gene, breeding the female
porcine animal
with a male porcine animal comprising at least one wild-type CD163 allele will
produce fetuses
that have inherited a CD163 allele comprising the modified chromosomal
sequence from the
female porcine animal and a wild-type CD163 allele from the male porcine
animal. Thus, the
breeding will produce fetuses that are heterozygous for the modified CD163
chromosomal
sequence. Where the male animal comprises two wild-type CD163 alleles, all of
the fetuses
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
24
produced as a result of the breeding will be heterozygous for the modified
CD163 chromosomal
sequence.
[00135] The fetuses produced by the breeding will have reduced susceptibility
to
infection by PRRSV while in utero, as compared to fetuses in utero in a wild-
type female
porcine animal.
[00136] In any of the methods described herein, the breeding can comprise
mating
(copulation) of the female porcine animal with the male porcine animal.
[00137] In any of the methods described herein, the breeding can comprises
artificial
insemination of the female animal with sperm obtained from the male animal.
[00138] In any of the methods described herein, the breeding can comprise
transferring a fertilized oocyte into the reproductive tract of the female
porcine animal.
[00139] In methods where the breeding comprises transferring a fertilized
oocyte into
the reproductive tract of the female porcine animal, the fertilized oocyte can
be generated by in
vitro fertilization of the oocyte with sperm obtained from the male porcine
animal.
[00140] The in vitro fertilization can comprise intracytoplasmic injection of
an oocyte
with sperm obtained from the male porcine animal.
[00141] Where the breeding comprises transferring a fertilized oocyte into the
reproductive tract of the female porcine animal, the oocyte can be an oocyte
derived from the
porcine female animal, such that the oocyte comprises modified chromosomal
sequences in both
alleles of its CD163 gene. Alternatively, the oocyte can be an oocyte derived
from a different
female porcine animal comprising modified chromosomal sequences in both
alleles of its
CD163 gene, wherein the modified chromosomal sequences reduce the
susceptibility of the
animal to infection by PRRSV, as compared to the susceptibility to infection
by PRRSV of a
female porcine animal that does not comprise any modified chromosomal
sequences in the
alleles of its CD163 gene. Thus, for example, any oocyte having modified
chromosomal
sequences in both alleles of its CD163 gene (e.g., knockouts of both alleles
of the C163 gene)
can be used.
[00142] However, where the breeding comprises transferring a fertilized oocyte
into
the reproductive tract of the female porcine animal, the oocyte need not
comprise any modified
chromosomal sequences in the alleles of its CD163 gene. An oocyte containing
two wild-type
CD163 alleles can be used. The oocyte containing two wild-type CD163 alleles
can be fertilized
with sperm obtained from a male porcine animal, wherein the sperm comprises
two wild-type
CD163 alleles, to create a fertilized oocyte containing two wild-type CD163
alleles. If such a
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
fertilized oocyte (containing two wild-type CD163 alleles) is transferred into
the reproductive
tract of the female porcine animal comprising the modified chromosomal
sequences in both
alleles of its CD163 gene, the resulting fetus will be protected from PRRSV
infection.
[00143] Alternatively, where the breeding comprises transferring a fertilized
oocyte
into the reproductive tract of the female porcine animal, the fertilized
oocyte can comprise a
modified chromosomal sequence in a single allele of its CD163 gene. Such
fertilized oocytes
will also produce fetuses that are protected from PRRSV infection upon
transfer of the fertilized
oocyte into the reproductive tract of the female porcine animal comprising the
modified
chromosomal sequences in both alleles of its CD163 gene.
[00144] Thus, where the breeding comprises transferring a fertilized oocyte
into the
reproductive tract of the female porcine animal, the fertilized oocyte can
comprise modified
chromosomal sequences in both alleles of its CD163 gene (e.g., knockouts of
both alleles of its
CD163 gene), can comprise a modified chromosomal sequence in only one allele
of its CD163
gene, or can comprise only wild-type CD163 alleles.
Affinity Tags
[00145] An "affinity tag" can be either a peptide affinity tag or a nucleic
acid affinity
tag. The term "affinity tag" generally refers to a protein or nucleic acid
sequence that can be
bound to a molecule (e.g., bound by a small molecule, protein, or covalent
bond). An affinity tag
can be a non-native sequence. A peptide affinity tag can comprise a peptide. A
peptide affinity
tag can be one that is able to be part of a split system (e.g., two inactive
peptide fragments can
combine together in trans to form an active affinity tag). A nucleic acid
affinity tag can comprise
a nucleic acid. A nucleic acid affinity tag can be a sequence that can
selectively bind to a known
nucleic acid sequence (e.g. through hybridization). A nucleic acid affinity
tag can be a sequence
that can selectively bind to a protein. An affinity tag can be fused to a
native protein. An affinity
tag can be fused to a nucleotide sequence.
[00146] Sometimes, one, two, or a plurality of affinity tags can be
fused to a native
protein or nucleotide sequence. An affinity tag can be introduced into a
nucleic acid-targeting
nucleic acid using methods of in vitro or in vivo transcription. Nucleic acid
affinity tags can
include, for example, a chemical tag, an RNA-binding protein binding sequence,
a DNA-binding
protein binding sequence, a sequence hybridizable to an affinity-tagged
polynucleotide, a
synthetic RNA aptamer, or a synthetic DNA aptamer. Examples of chemical
nucleic acid
affinity tags can include, but are not limited to, ribo-nucleotriphosphates
containing biotin,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
26
fluorescent dyes, and digoxeginin. Examples of protein-binding nucleic acid
affinity tags can
include, but are not limited to, the MS2 binding sequence, the UlA binding
sequence, stem-
loop binding protein sequences, the boxB sequence, the eIF4A sequence, or any
sequence
recognized by an RNA binding protein. Examples of nucleic acid affinity-tagged
oligonucleotides can include, but are not limited to, biotinylated
oligonucleotides, 2, 4-
dinitrophenyl oligonucleotides, fluorescein oligonucleotides, and primary
amine-conjugated
oligonucleotides.
[00147] A nucleic acid affinity tag can be an RNA aptamer. Aptamers can
include,
aptamers that bind to theophylline, streptavidin, dextran B512, adenosine,
guanosine,
guanine/xanthine, 7-methyl-GTP, amino acid aptamers such as aptamers that bind
to arginine,
citrulline, valine, tryptophan, cyanocobalamine, N-methylmesoporphyrin IX,
flavin, NAD, and
antibiotic aptamers such as aptamers that bind to tobramycin, neomycin,
lividomycin,
kanamycin, streptomycin, viomycin, and chloramphenicol.
[00148] A nucleic acid affinity tag can comprise an RNA sequence that can be
bound
by a site-directed polypeptide. The site-directed polypeptide can be
conditionally enzymatically
inactive. The RNA sequence can comprise a sequence that can be bound by a
member of Type I,
Type II, and/or Type III CRISPR systems. The RNA sequence can be bound by a
RAMP family
member protein. The RNA sequence can be bound by a Cas9 family member protein,
a Cas6
family member protein (e.g., Csy4, Cas6). The RNA sequence can be bound by a
Cas5 family
member protein (e.g., Cas5). For example, Csy4 can bind to a specific RNA
hairpin sequence
with high affinity (Kd ¨50 pM) and can cleave RNA at a site 3' to the hairpin.
[00149] A nucleic acid affinity tag can comprise a DNA sequence that can be
bound
by a site-directed polypeptide. The site-directed polypeptide can be
conditionally enzymatically
inactive. The DNA sequence can comprise a sequence that can be bound by a
member of the
Type I, Type II and/or Type III CRISPR system. The DNA sequence can be bound
by an
Argonaut protein. The DNA sequence can be bound by a protein containing a zinc
finger
domain, a TALE domain, or any other DNA-binding domain.
[00150] A nucleic acid affinity tag can comprise a ribozyme sequence. Suitable
ribozymes can include peptidyl transferase 23 SrRNA, RnaseP, Group I introns,
Group II
introns, GIR1 branching ribozyme, Leadzyme, hairpin ribozymes, hammerhead
ribozymes,
HDV ribozymes, CPEB3 ribozymes, VS ribozymes, glmS ribozyme, CoTC ribozyme,
and
synthetic ribozymes.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
27
[00151] Peptide affinity tags can comprise tags that can be used for tracking
or
purification (e.g., a fluorescent protein such as green fluorescent protein
(GFP), YFP, RFP, CFP,
mCherry, tdTomato; a His tag, (e.g., a 6XHis tag); a hemagglutinin (HA) tag; a
FLAG tag; a
Myc tag; a GST tag; a MBP tag; a chitin binding protein tag; a calmodulin tag;
a V5 tag; a
streptavidin binding tag; and the like).
[00152] Both nucleic acid and peptide affinity tags can comprise small
molecule tags
such as biotin, or digitoxin, and fluorescent label tags, such as for example,
fluoroscein,
rhodamin, Alexa fluor dyes, Cyanine3 dye, Cyanine5 dye.
[00153] Nucleic acid affinity tags can be located 5' to a nucleic acid (e.g.,
a nucleic
acid- targeting nucleic acid). Nucleic acid affinity tags can be located 3' to
a nucleic acid.
Nucleic acid affinity tags can be located 5' and 3' to a nucleic acid. Nucleic
acid affinity tags
can be located within a nucleic acid. Peptide affinity tags can be located N-
terminal to a
polypeptide sequence. Peptide affinity tags can be located C-terminal to a
polypeptide sequence.
Peptide affinity tags can be located N-terminal and C-terminal to a
polypeptide sequence. A
plurality of affinity tags can be fused to a nucleic acid and/or a polypeptide
sequence.
Capture Agents
[00154] As used herein, "capture agent" can generally refer to an agent that
can purify
a polypeptide and/or a nucleic acid. A capture agent can be a biologically
active molecule or
material (e.g. any biological substance found in nature or synthetic, and
includes but is not
limited to cells, viruses, subcellular particles, proteins, including more
specifically antibodies,
immunoglobulins, antigens, lipoproteins, glycoproteins, peptides,
polypeptides, protein
complexes, (strept)avidin-biotin complexes, ligands, receptors, or small
molecules, aptamers,
nucleic acids, DNA, RNA, peptidic nucleic acids, oligosaccharides,
polysaccharides,
lipopolysccharides, cellular metabolites, haptens, pharmacologically active
substances,
alkaloids, steroids, vitamins, amino acids, and sugars). In some embodiments,
the capture agent
can comprise an affinity tag. In some embodiments, a capture agent can
preferentially bind to a
target polypeptide or nucleic acid of interest. Capture agents can be free
floating in a mixture.
Capture agents can be bound to a particle (e.g. a bead, a microbead, a
nanoparticle). Capture
agents can be bound to a solid or semisolid surface. In some instances,
capture agents are
irreversibly bound to a target. In other instances, capture agents are
reversibly bound to a target
(e.g., if a target can be eluted, or by use of a chemical such as imidazole).
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
28
DNA-Binding Polypeptides
[00155] Site-specific integration can be accomplished by using factors
that are
capable of recognizing and binding to particular nucleotide sequences, for
example, in the
genome of a host organism. For instance, many proteins comprise polypeptide
domains that are
capable of recognizing and binding to DNA in a site-specific manner. A DNA
sequence that is
recognized by a DNA-binding polypeptide may be referred to as a "target"
sequence.
Polypeptide domains that are capable of recognizing and binding to DNA in a
site-specific
manner generally fold correctly and function independently to bind DNA in a
site-specific
manner, even when expressed in a polypeptide other than the protein from which
the domain
was originally isolated. Similarly, target sequences for recognition and
binding by DNA-binding
polypeptides are generally able to be recognized and bound by such
polypeptides, even when
present in large DNA structures (e.g., a chromosome), particularly when the
site where the target
sequence is located is one known to be accessible to soluble cellular proteins
(e.g., a gene).
[00156] While DNA-binding polypeptides identified from proteins that exist in
nature
typically bind to a discrete nucleotide sequence or motif (e.g., a consensus
recognition
sequence), methods exist and are known in the art for modifying many such DNA-
binding
polypeptides to recognize a different nucleotide sequence or motif. DNA-
binding polypeptides
include, for example and without limitation: zinc finger DNA-binding domains;
leucine zippers;
UPA DNA-binding domains; GAL4; TAL; LexA; Tet repressors; Lad; and steroid
hormone
receptors.
[00157] For example, the DNA-binding polypeptide can be a zinc finger.
Individual
zinc finger motifs can be designed to target and bind specifically to any of a
large range of DNA
sites. Canonical Cys2His2 (as well as non-canonical Cys3His) zinc finger
polypeptides bind DNA
by inserting an a-helix into the major groove of the target DNA double helix.
Recognition of
DNA by a zinc finger is modular; each finger contacts primarily three
consecutive base pairs in
the target, and a few key residues in the polypeptide mediate recognition. By
including multiple
zinc finger DNA-binding domains in a targeting endonuclease, the DNA-binding
specificity of
the targeting endonuclease may be further increased (and hence the specificity
of any gene
regulatory effects conferred thereby may also be increased). See, e.g., Urnov
et al. (2005) Nature
435:646-51. Thus, one or more zinc finger DNA-binding polypeptides may be
engineered and
utilized such that a targeting endonuclease introduced into a host cell
interacts with a DNA
sequence that is unique within the genome of the host cell.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
29
[00158] Preferably, the zinc finger protein is non-naturally occurring
in that it is
engineered to bind to a target site of choice. See, for example, Beerli et al.
(2002) Nature
Biotechnol. 20:135-141; Pabo et al. (2001) Ann. Rev. Biochem. 70:313-340;
Isalan et al. (2001)
Nature Biotechnol. 19:656-660; Segal et al. (2001) Curr. Opin. Biotechnol.
12:632-637; Choo et
al. (2000) Curr. Opin. Struct. Biol. 10:411-416; U.S. Pat. Nos. 6,453,242;
6,534,261; 6,599,692;
6,503,717; 6,689,558; 7,030,215; 6,794,136; 7,067,317; 7,262,054; 7,070,934;
7,361,635;
7,253,273; and U.S. Patent Publication Nos. 2005/0064474; 2007/0218528;
2005/0267061.
[00159] An engineered zinc finger binding domain can have a novel binding
specificity, compared to a naturally-occurring zinc finger protein.
Engineering methods include,
but are not limited to, rational design and various types of selection.
Rational design includes,
for example, using databases comprising triplet (or quadruplet) nucleotide
sequences and
individual zinc finger amino acid sequences, in which each triplet or
quadruplet nucleotide
sequence is associated with one or more amino acid sequences of zinc fingers
which bind the
particular triplet or quadruplet sequence. See, for example, U.S. Pat. Nos.
6,453,242 and
6,534,261.
[00160] Exemplary selection methods, including phage display and two-hybrid
systems, are disclosed in U.S. Pat. Nos. 5,789,538; 5,925,523; 6,007,988;
6,013,453; 6,410,248;
6,140,466; 6,200,759; and 6,242,568; as well as WO 98/37186; WO 98/53057; WO
00/27878;
WO 01/88197 and GB 2,338,237. In addition, enhancement of binding specificity
for zinc finger
binding domains has been described, for example, in WO 02/077227.
[00161] In addition, as disclosed in these and other references, zinc
finger domains
and/or multi-fingered zinc finger proteins may be linked together using any
suitable linker
sequences, including for example, linkers of 5 or more amino acids in length.
See, also, U.S. Pat.
Nos. 6,479,626; 6,903,185; and 7,153,949 for exemplary linker sequences 6 or
more amino
acids in length. The proteins described herein may include any combination of
suitable linkers
between the individual zinc fingers of the protein.
[00162] Selection of target sites: ZFPs and methods for design and
construction of
fusion proteins (and polynucleotides encoding same) are known to those of
skill in the art and
described in detail in U.S. Pat. Nos. 6,140,0815; 789,538; 6,453,242;
6,534,261; 5,925,523;
6,007,988; 6,013,453; 6,200,759; WO 95/19431; WO 96/06166; WO 98/53057; WO
98/54311;
WO 00/27878; WO 01/60970 WO 01/88197; WO 02/099084; WO 98/53058; WO 98/53059;
WO 98/53060; WO 02/016536 and WO 03/016496.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
[00163] In addition, as disclosed in these and other references, zinc
finger domains
and/or multi-fingered zinc finger proteins may be linked together using any
suitable linker
sequences, including for example, linkers of 5 or more amino acids in length.
See, also, U.S. Pat.
Nos. 6,479,626; 6,903,185; and 7,153,949 for exemplary linker sequences 6 or
more amino
acids in length. The proteins described herein may include any combination of
suitable linkers
between the individual zinc fingers of the protein.
[00164] Where a porcine female animal for use in the methods described
herein has
been genetically edited using a zinc-finger nuclease, the female animal can be
created using a
process comprising introducing into an embryo or cell at least one RNA
molecule encoding a
targeted zinc finger nuclease and, optionally, at least one accessory
polynucleotide. The method
further comprises incubating the embryo or cell to allow expression of the
zinc finger nuclease,
wherein a double-stranded break introduced into the targeted chromosomal
sequence by the zinc
finger nuclease is repaired by an error-prone non-homologous end-joining DNA
repair process
or a homology-directed DNA repair process. The method of editing chromosomal
sequences
encoding a protein associated with germline development using targeted zinc
finger nuclease
technology is rapid, precise, and highly efficient.
[00165] Alternatively, the DNA-binding polypeptide is a DNA-binding domain
from
GAL4. GAL4 is a modular transactivator in Saccharomyces cerevisiae, but it
also operates as a
transactivator in many other organisms. See, e.g., Sadowski et al. (1988)
Nature 335:563-4. In
this regulatory system, the expression of genes encoding enzymes of the
galactose metabolic
pathway in S. cerevisiae is stringently regulated by the available carbon
source. Johnston (1987)
Microbiol. Rev. 51:458-76. Transcriptional control of these metabolic enzymes
is mediated by
the interaction between the positive regulatory protein, GAL4, and a 17 bp
symmetrical DNA
sequence to which GAL4 specifically binds (the upstream activation sequence
(UAS)).
[00166] Native GAL4 consists of 881 amino acid residues, with a molecular
weight of
99 kDa. GAL4 comprises functionally autonomous domains, the combined
activities of which
account for activity of GAL4 in vivo. Ma and Ptashne (1987) Cell 48:847-53);
Brent and
Ptashne (1985) Cell 43(3 Pt 2):729-36. The N-terminal 65 amino acids of GAL4
comprise the
GAL4 DNA-binding domain. Keegan et al. (1986) Science 231:699-704; Johnston
(1987)
Nature 328:353-5. Sequence-specific binding requires the presence of a
divalent cation
coordinated by six Cys residues present in the DNA binding domain. The
coordinated cation-
containing domain interacts with and recognizes a conserved CCG triplet at
each end of the 17
bp UAS via direct contacts with the major groove of the DNA helix. Marmorstein
et al. (1992)
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
31
Nature 356:408-14. The DNA-binding function of the protein positions C-
terminal
transcriptional activating domains in the vicinity of the promoter, such that
the activating
domains can direct transcription.
[00167] Additional DNA-binding polypeptides that can be used include, for
example
and without limitation, a binding sequence from a AVRBS3-inducible gene; a
consensus binding
sequence from a AVRBS3-inducible gene or synthetic binding sequence engineered
therefrom
(e.g., UPA DNA-binding domain); TAL; LexA (see, e.g., Brent & Ptashne (1985),
supra); LacR
(see, e.g., Labow et al. (1990) Mol. Cell. Biol. 10:3343-56; Baim et al.
(1991) Proc. Natl. Acad.
Sci. USA 88(12):5072-6); a steroid hormone receptor (Ellliston et al. (1990)
J. Biol. Chem.
265:11517-121); the Tet repressor (U.S. Pat. No. 6,271,341) and a mutated Tet
repressor that
binds to a tet operator sequence in the presence, but not the absence, of
tetracycline (Tc); the
DNA-binding domain of NF-kappaB; and components of the regulatory system
described in
Wang et al. (1994) Proc. Natl. Acad. Sci. USA 91(17):8180-4, which utilizes a
fusion of GAL4,
a hormone receptor, and VP16.
[00168] The DNA-binding domain of one or more of the nucleases used in the
methods and compositions described herein can comprise a naturally occurring
or engineered
(non-naturally occurring) TAL effector DNA binding domain. See, e.g., U.S.
Patent Publication
No. 2011/0301073.
[00169] Alternatively, the nuclease can comprise a CRISPR system. For example,
the
nuclease can comprise a CRISPR/Cas system.
[00170] The (CRISPR-associated) system evolved in bacteria and archaea as an
adaptive immune system to defend against viral attack. Upon exposure to a
virus, short segments
of viral DNA are integrated into the CRISPR locus. RNA is transcribed from a
portion of the
CRISPR locus that includes the viral sequence. That RNA, which contains
sequence
complementary to the viral genome, mediates targeting of a Cas protein (e.g.,
Cas9 protein) to
the sequence in the viral genome. The Cas protein cleaves and thereby silences
the viral target.
Recently, the CRISPR/Cas system has been adapted for genome editing in
eukaryotic cells. The
introduction of site-specific double strand breaks (DSBs) enables target
sequence alteration
through one of two endogenous DNA repair mechanisms¨ either non-homologous end-
joining
(NHEJ) or homology-directed repair (HDR). The CRISPR/Cas system has also been
used for
gene regulation including transcription repression and activation without
altering the target
sequence. Targeted gene regulation based on the CRISPR/Cas system can, for
example, use an
enzymatically inactive Cas9 (also known as a catalytically dead Cas9).
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
32
[00171] CRISPR/Cas systems include a CRISPR (clustered regularly interspaced
short palindromic repeats) locus, which encodes RNA components of the system,
and a Cas
(CRISPR-associated) locus, which encodes proteins (Jansen et al., 2002. Mol.
Microbiol. 43:
1565-1575; Makarova et al., 2002. Nucleic Acids Res. 30: 482-496; Makarova et
al., 2006. Biol.
Direct 1: 7; Haft et al., 2005. PLoS Comput. Biol. 1: e60). CRISPR loci in
microbial hosts
contain a combination of Cas genes as well as non-coding RNA elements capable
of
programming the specificity of the CRISPR-mediated nucleic acid cleavage.
[00172] The Type II CRISPR is one of the most well characterized systems and
carries out targeted DNA double-strand break in nature in four sequential
steps. First, two non-
coding RNAs, the pre-crRNA array and tracrRNA, are transcribed from the CRISPR
locus.
Second, tracrRNA hybridizes to the repeat regions of the pre-crRNA and
mediates the
processing of pre-crRNA into mature crRNAs containing individual spacer
sequences. Third, the
mature crRNA:tracrRNA complex directs Cas9 to the target DNA via Watson-Crick
base-
pairing between the spacer on the crRNA and the protospacer on the target DNA
next to the
protospacer adjacent motif (PAM), an additional requirement for target
recognition. Finally,
Cas9 mediates cleavage of target DNA to create a double-stranded break within
the protospacer.
[00173] For use of the CRISPR/Cas system to create targeted insertions and
deletions,
the two non-coding RNAs (crRNA and the TracrRNA) can be replaced by a single
RNA
referred to as a guide RNA (gRNA). Activity of the CRISPR/Cas system comprises
of three
steps: (i) insertion of exogenous DNA sequences into the CRISPR array to
prevent future
attacks, in a process called "adaptation," (ii) expression of the relevant
proteins, as well as
expression and processing of the array, followed by (iii) RNA-mediated
interference with the
foreign nucleic acid. In the bacterial cell, several Cas proteins are involved
with the natural
function of the CRISPR/Cas system and serve roles in functions such as
insertion of the foreign
DNA etc.
[00174] The Cas protein can be a "functional derivative" of a naturally
occurring Cas
protein. A "functional derivative" of a native sequence polypeptide is a
compound having a
qualitative biological property in common with a native sequence polypeptide.
"Functional
derivatives" include, but are not limited to, fragments of a native sequence
and derivatives of a
native sequence polypeptide and its fragments, provided that they have a
biological activity in
common with a corresponding native sequence polypeptide. A biological activity
contemplated
herein is the ability of the functional derivative to hydrolyze a DNA
substrate into fragments.
The term "derivative" encompasses both amino acid sequence variants of
polypeptide, covalent
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
33
modifications, and fusions thereof Suitable derivatives of a Cas polypeptide
or a fragment
thereof include but are not limited to mutants, fusions, covalent
modifications of Cas protein or a
fragment thereof. Cas protein, which includes Cas protein or a fragment
thereof, as well as
derivatives of Cas protein or a fragment thereof, may be obtainable from a
cell or synthesized
chemically or by a combination of these two procedures. The cell may be a cell
that naturally
produces Cas protein, or a cell that naturally produces Cas protein and is
genetically engineered
to produce the endogenous Cas protein at a higher expression level or to
produce a Cas protein
from an exogenously introduced nucleic acid, which nucleic acid encodes a Cas
that is same or
different from the endogenous Cas. In some case, the cell does not naturally
produce Cas protein
and is genetically engineered to produce a Cas protein.
[00175] Where a porcine female animal for use in the methods described herein
has
been genetically edited using a CRISPR system, a CRISPR/Cas9 system can be
used to generate
the porcine female animal. To use Cas9 to edit genomic sequences, the protein
can be delivered
directly to a cell. Alternatively, an mRNA that encodes Cas9 can be delivered
to a cell, or a gene
that provides for expression of an mRNA that encodes Cas9 can be delivered to
a cell. In
addition, either target specific crRNA and a tracrRNA can be delivered
directly to a cell or target
specific gRNA(s) can be to a cell (these RNAs can alternatively be produced by
a gene
constructed to express these RNAs). Selection of target sites and designed of
crRNA/gRNA are
well known in the art. A discussion of construction and cloning of gRNAs can
be found at
http://www.genome-engineering.org/crispr/wp-content/uploads/2014/05/CRISPR-
Reagent-
Description-Rev20140509.pdf.
[00176] A DNA-binding polypeptide can specifically recognize and bind to a
target
nucleotide sequence comprised within a genomic nucleic acid of a host
organism. Any number
of discrete instances of the target nucleotide sequence may be found in the
host genome in some
examples. The target nucleotide sequence may be rare within the genome of the
organism (e.g.,
fewer than about 10, about 9, about 8, about 7, about 6, about 5, about 4,
about 3, about 2, or
about 1 copy(ies) of the target sequence may exist in the genome). For
example, the target
nucleotide sequence may be located at a unique site within the genome of the
organism. Target
nucleotide sequences may be, for example and without limitation, randomly
dispersed
throughout the genome with respect to one another; located in different
linkage groups in the
genome; located in the same linkage group; located on different chromosomes;
located on the
same chromosome; located in the genome at sites that are expressed under
similar conditions in
the organism (e.g., under the control of the same, or substantially
functionally identical,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
34
regulatory factors); and located closely to one another in the genome (e.g.,
target sequences may
be comprised within nucleic acids integrated as concatemers at genomic loci).
Targeting Endonucleases
[00177] A DNA-binding polypeptide that specifically recognizes and binds to a
target
nucleotide sequence can be comprised within a chimeric polypeptide, so as to
confer specific
binding to the target sequence upon the chimeric polypeptide. In examples,
such a chimeric
polypeptide may comprise, for example and without limitation, nuclease,
recombinase, and/or
ligase polypeptides, as these polypeptides are described above. Chimeric
polypeptides
comprising a DNA-binding polypeptide and a nuclease, recombinase, and/or
ligase polypeptide
may also comprise other functional polypeptide motifs and/or domains, such as
for example and
without limitation: a spacer sequence positioned between the functional
polypeptides in the
chimeric protein; a leader peptide; a peptide that targets the fusion protein
to an organelle (e.g.,
the nucleus); polypeptides that are cleaved by a cellular enzyme; peptide tags
(e.g., Myc, His,
etc.); and other amino acid sequences that do not interfere with the function
of the chimeric
polypeptide.
[00178] Functional polypeptides (e.g., DNA-binding polypeptides and nuclease
polypeptides) in a chimeric polypeptide may be operatively linked. Functional
polypeptides of a
chimeric polypeptide can be operatively linked by their expression from a
single polynucleotide
encoding at least the functional polypeptides ligated to each other in-frame,
so as to create a
chimeric gene encoding a chimeric protein. Alternatively, the functional
polypeptides of a
chimeric polypeptide can be operatively linked by other means, such as by
cross-linkage of
independently expressed polypeptides.
[00179] A DNA-binding polypeptide, or guide RNA that specifically recognizes
and
binds to a target nucleotide sequence can be comprised within a natural
isolated protein (or
mutant thereof), wherein the natural isolated protein or mutant thereof also
comprises a nuclease
polypeptide (and may also comprise a recombinase and/or ligase polypeptide).
Examples of such
isolated proteins include TALENs, recombinases (e.g., Cre, Hin, Tre, and FLP
recombinase),
RNA-guided CRISPR/Cas9, and meganucleases.
[00180] As used herein, the term "targeting endonuclease" refers to natural or
engineered isolated proteins and mutants thereof that comprise a DNA-binding
polypeptide or
guide RNA and a nuclease polypeptide, as well as to chimeric polypeptides
comprising a DNA-
binding polypeptide or guide RNA and a nuclease. Any targeting endonuclease
comprising a
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
DNA-binding polypeptide or guide RNA that specifically recognizes and binds to
a target
nucleotide sequence comprised within a CD163 locus (e.g., either because the
target sequence is
comprised within the native sequence at the locus, or because the target
sequence has been
introduced into the locus, for example, by recombination) can be used.
[00181] Some examples of suitable chimeric polypeptides include,
without limitation,
combinations of the following polypeptides: zinc finger DNA-binding
polypeptides; a FokI
nuclease polypeptide; TALE domains; leucine zippers; transcription factor DNA-
binding motifs;
and DNA recognition and/or cleavage domains isolated from, for example and
without
limitation, a TALEN, a recombinase (e.g., Cre, Hin, RecA, Tre, and FLP
recombinases), RNA-
guided CRISPR/Cas9, a meganuclease; and others known to those in the art.
Particular examples
include a chimeric protein comprising a site-specific DNA binding polypeptide
and a nuclease
polypeptide. Chimeric polypeptides may be engineered by methods known to those
of skill in
the art to alter the recognition sequence of a DNA-binding polypeptide
comprised within the
chimeric polypeptide, so as to target the chimeric polypeptide to a particular
nucleotide
sequence of interest.
[00182] The chimeric polypeptide can comprise a DNA-binding domain (e.g., zinc
finger, TAL-effector domain, etc.) and a nuclease (cleavage) domain. The
cleavage domain may
be heterologous to the DNA-binding domain, for example a zinc finger DNA-
binding domain
and a cleavage domain from a nuclease or a TALEN DNA-binding domain and a
cleavage
domain, or meganuclease DNA-binding domain and cleavage domain from a
different nuclease.
Heterologous cleavage domains can be obtained from any endonuclease or
exonuclease.
Exemplary endonucleases from which a cleavage domain can be derived include,
but are not
limited to, restriction endonucleases and homing endonucleases. See, for
example, 2002-2003
Catalogue, New England Biolabs, Beverly, Mass.; and Belfort et al. (1997)
Nucleic Acids Res.
25:3379-3388. Additional enzymes which cleave DNA are known (e.g., 51
Nuclease; mung
bean nuclease; pancreatic DNAse I; micrococcal nuclease; yeast HO
endonuclease; see also
Linn et al. (eds.) Nucleases, Cold Spring Harbor Laboratory Press, 1993). One
or more of these
enzymes (or functional fragments thereof) can be used as a source of cleavage
domains and
cleavage half-domains.
[00183] Similarly, a cleavage half-domain can be derived from any nuclease or
portion thereof, as set forth above, that requires dimerization for cleavage
activity. In general,
two fusion proteins are required for cleavage if the fusion proteins comprise
cleavage half-
domains. Alternatively, a single protein comprising two cleavage half-domains
can be used. The
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
36
two cleavage half-domains can be derived from the same endonuclease (or
functional fragments
thereof), or each cleavage half-domain can be derived from a different
endonuclease (or
functional fragments thereof). In addition, the target sites for the two
fusion proteins are
preferably disposed, with respect to each other, such that binding of the two
fusion proteins to
their respective target sites places the cleavage half-domains in a spatial
orientation to each other
that allows the cleavage half-domains to form a functional cleavage domain,
e.g., by dimerizing.
Thus, the near edges of the target sites can be separated by 5-8 nucleotides
or by 15-18
nucleotides. However any integral number of nucleotides, or nucleotide pairs,
can intervene
between two target sites (e.g., from 2 to 50 nucleotide pairs or more). In
general, the site of
cleavage lies between the target sites.
[00184] Restriction endonucleases (restriction enzymes) are present in many
species
and are capable of sequence-specific binding to DNA (at a recognition site),
and cleaving DNA
at or near the site of binding, for example, such that one or more exogenous
sequences
(donors/transgenes) are integrated at or near the binding (target) sites.
Certain restriction
enzymes (e.g., Type ITS) cleave DNA at sites removed from the recognition site
and have
separable binding and cleavage domains. For example, the Type ITS enzyme Fok I
catalyses
double-stranded cleavage of DNA, at 9 nucleotides from its recognition site on
one strand and
13 nucleotides from its recognition site on the other. See, for example, U.S.
Pat. Nos. 5,356,802;
5,436,150 and 5,487,994; as well as Li et al. (1992) Proc. Natl. Acad. Sci.
USA 89:4275-4279;
Li et al. (1993) Proc. Natl. Acad. Sci. USA 90:2764-2768; Kim et al. (1994a)
Proc. Natl. Acad.
Sci. USA 91:883-887; Kim et al. (1994b) J. Biol. Chem. 269:31,978-31,982.
Thus, fusion
proteins can comprise the cleavage domain (or cleavage half-domain) from at
least one Type ITS
restriction enzyme and one or more zinc finger binding domains, which may or
may not be
engineered.
[00185] An exemplary Type ITS restriction enzyme, whose cleavage domain is
separable from the binding domain, is Fok I. This particular enzyme is active
as a dimer.
Bitinaite et al. (1998) Proc. Natl. Acad. Sci. USA 95: 10,570-10,575.
Accordingly, for the
purposes of the present disclosure, the portion of the Fok I enzyme used in
the disclosed fusion
proteins is considered a cleavage half-domain. Thus, for targeted double-
stranded cleavage
and/or targeted replacement of cellular sequences using zinc finger-Fok I
fusions, two fusion
proteins, each comprising a FokI cleavage half-domain, can be used to
reconstitute a
catalytically active cleavage domain. Alternatively, a single polypeptide
molecule containing a
DNA binding domain and two Fok I cleavage half-domains can also be used.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
37
[00186] A cleavage domain or cleavage half-domain can be any portion of a
protein
that retains cleavage activity, or that retains the ability to multimerize
(e.g., dimerize) to form a
functional cleavage domain.
[00187] Exemplary Type ITS restriction enzymes are described in U.S. Patent
Publication No. 2007/0134796. Additional restriction enzymes also contain
separable binding
and cleavage domains, and these are contemplated by the present disclosure.
See, for example,
Roberts et al. (2003) Nucleic Acids Res. 31:418-420.
[00188] The cleavage domain can comprise one or more engineered cleavage half-
domain (also referred to as dimerization domain mutants) that minimize or
prevent
homodimerization, as described, for example, in U.S. Patent Publication Nos.
2005/0064474;
2006/0188987 and 2008/0131962.
[00189] Alternatively, nucleases may be assembled in vivo at the nucleic acid
target
site using so-called "split-enzyme" technology (see e.g. U.S. Patent
Publication No.
20090068164). Components of such split enzymes may be expressed either on
separate
expression constructs, or can be linked in one open reading frame where the
individual
components are separated, for example, by a self-cleaving 2A peptide or IRES
sequence.
Components may be individual zinc finger binding domains or domains of a
meganuclease
nucleic acid binding domain.
Zinc Finger Nucleases
[00190] A chimeric polypeptide can comprise a custom-designed zinc finger
nuclease
(ZFN) that may be designed to deliver a targeted site-specific double-strand
DNA break into
which an exogenous nucleic acid, or donor DNA, may be integrated (see US
Patent publication
2010/0257638). ZFNs are chimeric polypeptides containing a non-specific
cleavage domain
from a restriction endonuclease (for example, FokI) and a zinc finger DNA-
binding domain
polypeptide. See, e.g., Huang et al. (1996) J. Protein Chem. 15:481-9; Kim et
al. (1997a) Proc.
Natl. Acad. Sci. USA 94:3616-20; Kim et al. (1996) Proc. Natl. Acad. Sci. USA
93:1156-60;
Kim et al. (1994) Proc Natl. Acad. Sci. USA 91:883-7; Kim et al. (1997b) Proc.
Natl. Acad. Sci.
USA 94:12875-9; Kim et al. (1997c) Gene 203:43-9; Kim et al. (1998) Biol.
Chem. 379:489-95;
Nahon and Raveh (1998) Nucleic Acids Res. 26:1233-9; Smith et al. (1999)
Nucleic Acids Res.
27:674-81. The ZFNs can comprise non-canonical zinc finger DNA binding domains
(see US
Patent publication 2008/0182332). The FokI restriction endonuclease must
dimerize via the
nuclease domain in order to cleave DNA and introduce a double-strand break.
Consequently,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
38
ZFNs containing a nuclease domain from such an endonuclease also require
dimerization of the
nuclease domain in order to cleave target DNA. Mani et al. (2005) Biochem.
Biophys. Res.
Commun. 334:1191-7; Smith et al. (2000) Nucleic Acids Res. 28:3361-9.
Dimerization of the
ZFN can be facilitated by two adjacent, oppositely oriented DNA-binding sites.
Id.
[00191] A method for the site-specific integration of an exogenous nucleic
acid into at
least one CD163 locus of a host can comprise introducing into a cell of the
host a ZFN, wherein
the ZFN recognizes and binds to a target nucleotide sequence, wherein the
target nucleotide
sequence is comprised within at least one CD163 locus of the host. In certain
examples, the
target nucleotide sequence is not comprised within the genome of the host at
any other position
than the at least one CD163 locus. For example, a DNA-binding polypeptide of
the ZFN may be
engineered to recognize and bind to a target nucleotide sequence identified
within the at least
one CD163 locus (e.g., by sequencing the CD163 locus). A method for the site-
specific
integration of an exogenous nucleic acid into at least one CD163 performance
locus of a host
that comprises introducing into a cell of the host a ZFN may also comprise
introducing into the
cell an exogenous nucleic acid, wherein recombination of the exogenous nucleic
acid into a
nucleic acid of the host comprising the at least one CD163 locus is
facilitated by site-specific
recognition and binding of the ZFN to the target sequence (and subsequent
cleavage of the
nucleic acid comprising the CD163 locus).
Optional Exogenous Nucleic Acids for Integration at a CD163 Locus
[00192] Exogenous nucleic acids for integration at a CD163 locus include: an
exogenous nucleic acid for site-specific integration in at least one CD163
locus, for example and
without limitation, an ORF; a nucleic acid comprising a nucleotide sequence
encoding a
targeting endonuclease; and a vector comprising at least one of either or both
of the foregoing.
Thus, particular nucleic acids include nucleotide sequences encoding a
polypeptide, structural
nucleotide sequences, and/or DNA-binding polypeptide recognition and binding
sites.
Optional Exogenous Nucleic Acid Molecules for Site-Specific Integration
[00193] As noted above, insertion of an exogenous sequence (also called a
"donor
sequence" or "donor" or "transgene") is provided, for example for expression
of a polypeptide,
correction of a mutant gene or for increased expression of a wild-type gene.
It will be readily
apparent that the donor sequence is typically not identical to the genomic
sequence where it is
placed. A donor sequence can contain a non-homologous sequence flanked by two
regions of
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
39
homology to allow for efficient homology-directed repair (HDR) at the location
of interest.
Additionally, donor sequences can comprise a vector molecule containing
sequences that are not
homologous to the region of interest in cellular chromatin. A donor molecule
can contain
several, discontinuous regions of homology to cellular chromatin. For example,
for targeted
insertion of sequences not normally present in a region of interest, said
sequences can be present
in a donor nucleic acid molecule and flanked by regions of homology to
sequence in the region
of interest.
[00194] The donor polynucleotide can be DNA or RNA, single-stranded or double-
stranded and can be introduced into a cell in linear or circular form. See
e.g., U.S. Patent
Publication Nos. 2010/0047805, 2011/0281361, 2011/0207221, and 2013/0326645.
If
introduced in linear form, the ends of the donor sequence can be protected
(e.g. from
exonucleolytic degradation) by methods known to those of skill in the art. For
example, one or
more dideoxynucleotide residues are added to the 3' terminus of a linear
molecule and/or self-
complementary oligonucleotides are ligated to one or both ends. See, for
example, Chang et al.
(1987) Proc. Natl. Acad. Sci. USA 84:4959-4963; Nehls et al. (1996) Science
272:886-889.
Additional methods for protecting exogenous polynucleotides from degradation
include, but are
not limited to, addition of terminal amino group(s) and the use of modified
internucleotide
linkages such as, for example, phosphorothioates, phosphoramidates, and 0-
methyl ribose or
deoxyribose residues.
[00195] A polynucleotide can be introduced into a cell as part of a vector
molecule
having additional sequences such as, for example, replication origins,
promoters and genes
encoding antibiotic resistance. Moreover, donor polynucleotides can be
introduced as naked
nucleic acid, as nucleic acid complexed with an agent such as a liposome or
poloxamer, or can
be delivered by viruses (e.g., adenovirus, AAV, herpesvirus, retrovirus,
lentivirus and integrase
defective lentivirus (IDLV)).
[00196] The donor is generally integrated so that its expression is
driven by the
endogenous promoter at the integration site, namely the promoter that drives
expression of the
endogenous gene into which the donor is integrated (e.g., CD163). However, it
will be apparent
that the donor may comprise a promoter and/or enhancer, for example a
constitutive promoter or
an inducible or tissue specific promoter.
[00197] Furthermore, although not required for expression, exogenous sequences
may
also include transcriptional or translational regulatory sequences, for
example, promoters,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
enhancers, insulators, internal ribosome entry sites, sequences encoding 2A
peptides and/or
polyadenylation signals.
[00198] Exogenous nucleic acids that may be integrated in a site-specific
manner into
at least one CD163 locus, so as to modify the CD163 locus include, for example
and without
limitation, nucleic acids comprising a nucleotide sequence encoding a
polypeptide of interest;
nucleic acids comprising an agronomic gene; nucleic acids comprising a
nucleotide sequence
encoding an RNAi molecule; or nucleic acids that disrupt the CD163 gene.
[00199] An exogenous nucleic acid can be integrated at a CD163 locus, so as to
modify the CD163 locus, wherein the nucleic acid comprises a nucleotide
sequence encoding a
polypeptide of interest, such that the nucleotide sequence is expressed in the
host from the
CD163 locus. In some examples, the polypeptide of interest (e.g., a foreign
protein) is expressed
from a nucleotide sequence encoding the polypeptide of interest in commercial
quantities. In
such examples, the polypeptide of interest may be extracted from the host
cell, tissue, or
biomass.
Nucleic Acid Molecules Comprising a Nucleotide Sequence Encoding a Targeting
Endonuclease
[00200] A nucleotide sequence encoding a targeting endonuclease can be
engineered
by manipulation (e.g., ligation) of native nucleotide sequences encoding
polypeptides comprised
within the targeting endonuclease. For example, the nucleotide sequence of a
gene encoding a
protein comprising a DNA-binding polypeptide may be inspected to identify the
nucleotide
sequence of the gene that corresponds to the DNA-binding polypeptide, and that
nucleotide
sequence may be used as an element of a nucleotide sequence encoding a
targeting endonuclease
comprising the DNA-binding polypeptide. Alternatively, the amino acid sequence
of a targeting
endonuclease may be used to deduce a nucleotide sequence encoding the
targeting
endonuclease, for example, according to the degeneracy of the genetic code.
[00201] In exemplary nucleic acid molecules comprising a nucleotide sequence
encoding a targeting endonuclease, the last codon of a first polynucleotide
sequence encoding a
nuclease polypeptide, and the first codon of a second polynucleotide sequence
encoding a DNA-
binding polypeptide, may be separated by any number of nucleotide triplets,
e.g., without coding
for an intron or a "STOP." Likewise, the last codon of a nucleotide sequence
encoding a first
polynucleotide sequence encoding a DNA-binding polypeptide, and the first
codon of a second
polynucleotide sequence encoding a nuclease polypeptide, may be separated by
any number of
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
41
nucleotide triplets. The last codon (i.e., most 3' in the nucleic acid
sequence) of a first
polynucleotide sequence encoding a nuclease polypeptide, and a second
polynucleotide
sequence encoding a DNA-binding polypeptide, can be fused in phase-register
with the first
codon of a further polynucleotide coding sequence directly contiguous thereto,
or separated
therefrom by no more than a short peptide sequence, such as that encoded by a
synthetic
nucleotide linker (e.g., a nucleotide linker that may have been used to
achieve the fusion).
Examples of such further polynucleotide sequences include, for example and
without limitation,
tags, targeting peptides, and enzymatic cleavage sites. Likewise, the first
codon of the most 5'
(in the nucleic acid sequence) of the first and second polynucleotide
sequences may be fused in
phase-register with the last codon of a further polynucleotide coding sequence
directly
contiguous thereto, or separated therefrom by no more than a short peptide
sequence.
[00202] A sequence separating polynucleotide sequences encoding functional
polypeptides in a targeting endonuclease (e.g., a DNA-binding polypeptide and
a nuclease
polypeptide) may, for example, consist of any sequence, such that the amino
acid sequence
encoded is not likely to significantly alter the translation of the targeting
endonuclease. Due to
the autonomous nature of known nuclease polypeptides and known DNA-binding
polypeptides,
intervening sequences will not interfere with the respective functions of
these structures.
Other Knockout Methods
[00203] Various other techniques known in the art can be used to inactivate
genes to
make knock-out animals and/or to introduce nucleic acid constructs into
animals to produce
founder animals and to make animal lines, in which the knockout or nucleic
acid construct is
integrated into the genome. Such techniques include, without limitation,
pronuclear
microinjection (U.S. Pat. No. 4,873,191), retrovirus mediated gene transfer
into germ lines (Van
der Putten et al. (1985) Proc. Natl. Acad. Sci. USA 82, 6148-1652), gene
targeting into
embryonic stem cells (Thompson et al. (1989) Cell 56, 313-321),
electroporation of embryos
(Lo (1983) Mol. Cell. Biol. 3, 1803-1814), sperm-mediated gene transfer
(Lavitrano et al. (2002)
Proc. Natl. Acad. Sci. USA 99, 14230-14235; Lavitrano et al. (2006) Reprod.
Fert. Develop. 18,
19-23), and in vitro transformation of somatic cells, such as cumulus or
mammary cells, or adult,
fetal, or embryonic stem cells, followed by nuclear transplantation (Wilmut et
al. (1997) Nature
385, 810-813; and Wakayama et al. (1998) Nature 394, 369-374). Pronuclear
microinjection,
sperm mediated gene transfer, and somatic cell nuclear transfer are
particularly useful
techniques. An animal that is genomically modified is an animal wherein all of
its cells have the
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
42
modification, including its germ line cells. When methods are used that
produce an animal that
is mosaic in its modification, the animals may be inbred and progeny that are
genomically
modified may be selected. Cloning, for instance, may be used to make a mosaic
animal if its
cells are modified at the blastocyst state, or genomic modification can take
place when a single-
cell is modified. Animals that are modified so they do not sexually mature can
be homozygous
or heterozygous for the modification, depending on the specific approach that
is used. If a
particular gene is inactivated by a knock out modification, homozygosity would
normally be
required. If a particular gene is inactivated by an RNA interference or
dominant negative
strategy, then heterozygosity is often adequate.
[00204] Typically, in embryo/zygote microinjection, a nucleic acid construct
or
mRNA is introduced into a fertilized egg; one or two cell fertilized eggs are
used as the nuclear
structure containing the genetic material from the sperm head and the egg are
visible within the
protoplasm. Pronuclear staged fertilized eggs can be obtained in vitro or in
vivo (i.e., surgically
recovered from the oviduct of donor animals). In vitro fertilized eggs can be
produced as
follows. For example, swine ovaries can be collected at an abattoir, and
maintained at 22-28 C.
during transport. Ovaries can be washed and isolated for follicular
aspiration, and follicles
ranging from 4-8 mm can be aspirated into 50 mL conical centrifuge tubes using
18 gauge
needles and under vacuum. Follicular fluid and aspirated oocytes can be rinsed
through pre-
filters with commercial TL-HEPES (Minitube, Verona, Wis.). Oocytes surrounded
by a compact
cumulus mass can be selected and placed into TCM-199 00CYTE MATURATION MEDIUM
(Minitube, Verona, Wis.) supplemented with 0.1 mg/mL cysteine, 10 ng/mL
epidermal growth
factor, 10% porcine follicular fluid, 50 tM 2-mercaptoethanol, 0.5 mg/ml cAMP,
10 IU/mL
each of pregnant mare serum gonadotropin (PMSG) and human chorionic
gonadotropin (hCG)
for approximately 22 hours in humidified air at 38.7 C. and 5% CO2.
Subsequently, the oocytes
can be moved to fresh TCM-199 maturation medium, which will not contain cAMP,
PMSG or
hCG and incubated for an additional 22 hours. Matured oocytes can be stripped
of their cumulus
cells by vortexing in 0.1% hyaluronidase for 1 minute.
[00205] For swine, mature oocytes can be fertilized in 500 11.1 Minitube
PORCPRO
IVF MEDIUM SYSTEM (Minitube, Verona, Wis.) in Minitube 5-well fertilization
dishes. In
preparation for in vitro fertilization (IVF), freshly-collected or frozen boar
semen can be washed
and resuspended in PORCPRO IVF Medium to 400,000 sperm. Sperm concentrations
can be
analyzed by computer assisted semen analysis (SPERMVISION, Minitube, Verona,
Wis.). Final
in vitro insemination can be performed in a 10 11.1 volume at a final
concentration of
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
43
approximately 40 motile sperm/oocyte, depending on boar. All fertilizing
oocytes can be
incubated at 38.7 C. in 5.0% CO2 atmosphere for six hours. Six hours post-
insemination,
presumptive zygotes can be washed twice in NCSU-23 and moved to 0.5 mL of the
same
medium. This system can produce 20-30% blastocysts routinely across most boars
with a 10-
30% polyspermic insemination rate.
[00206] Linearized nucleic acid constructs or mRNA can be injected into one of
the
pronuclei or into the cytoplasm. Then the injected eggs can be transferred to
a recipient female
(e.g., into the oviducts of a recipient female) and allowed to develop in the
recipient female to
produce the transgenic or gene edited animals. In particular, in vitro
fertilized embryos can be
centrifuged at 15,000 x g for 5 minutes to sediment lipids allowing
visualization of the
pronucleus. The embryos can be injected with using an Eppendorf FEMTOJET
injector and can
be cultured until blastocyst formation. Rates of embryo cleavage and
blastocyst formation and
quality can be recorded.
[00207] Embryos can be surgically transferred into uteri of asynchronous
recipients.
Typically, 100-200 (e.g., 150-200) embryos can be deposited into the ampulla-
isthmus junction
of the oviduct using a 5.5-inch TOMCAT catheter. After surgery, real-time
ultrasound
examination of pregnancy can be performed.
[00208] In somatic cell nuclear transfer, a transgenic or gene edited cell
such as an
embryonic blastomere, fetal fibroblast, adult ear fibroblast, or granulosa
cell that includes a
nucleic acid construct described above, can be introduced into an enucleated
oocyte to establish
a combined cell. Oocytes can be enucleated by partial zona dissection near the
polar body and
then pressing out cytoplasm at the dissection area. Typically, an injection
pipette with a sharp
beveled tip is used to inject the transgenic or gene edited cell into an
enucleated oocyte arrested
at meiosis 2. In some conventions, oocytes arrested at meiosis-2 are termed
eggs. After
producing a porcine or bovine embryo (e.g., by fusing and activating the
oocyte), the embryo is
transferred to the oviducts of a recipient female, about 20 to 24 hours after
activation. See, for
example, Cibelli et al. (1998) Science 280, 1256-1258 and U.S. Pat. Nos.
6,548,741, 7,547,816,
7,989,657, or 6,211,429. For pigs, recipient females can be checked for
pregnancy
approximately 20-21 days after transfer of the embryos.
[00209] Standard breeding techniques can be used to create animals that are
homozygous for the inactivated gene from the initial heterozygous founder
animals.
Homozygosity may not be required, however. Gene edited pigs described herein
can be bred
with other pigs of interest.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
44
[00210] Once gene edited animals have been generated, inactivation of an
endogenous
nucleic acid can be assessed using standard techniques. Initial screening can
be accomplished by
Southern blot analysis to determine whether or not inactivation has taken
place. For a
description of Southern analysis, see sections 9.37-9.52 of Sambrook et al.,
1989, Molecular
Cloning, A Laboratory Manual, second edition, Cold Spring Harbor Press,
Plainview; N.Y.
Polymerase chain reaction (PCR) techniques also can be used in the initial
screening PCR refers
to a procedure or technique in which target nucleic acids are amplified.
Generally, sequence
information from the ends of the region of interest or beyond is employed to
design
oligonucleotide primers that are identical or similar in sequence to opposite
strands of the
template to be amplified. PCR can be used to amplify specific sequences from
DNA as well as
RNA, including sequences from total genomic DNA or total cellular RNA. Primers
typically are
14 to 40 nucleotides in length, but can range from 10 nucleotides to hundreds
of nucleotides in
length. PCR is described in, for example PCR Primer: A Laboratory Manual, ed.
Dieffenbach
and Dveksler, Cold Spring Harbor Laboratory Press, 1995. Nucleic acids also
can be amplified
by ligase chain reaction, strand displacement amplification, self-sustained
sequence replication,
or nucleic acid sequence-based amplified. See, for example, Lewis (1992)
Genetic Engineering
News 12,1; Guatelli et al. (1990) Proc. Natl. Acad. Sci. USA 87:1874; and
Weiss (1991) Science
254:1292. At the blastocyst stage, embryos can be individually processed for
analysis by PCR,
Southern hybridization and splinkerette PCR (see, e.g., Dupuy et al. Proc Natl
Acad Sci USA
(2002) 99:4495).
Interfering RNAs
[00211] A variety of interfering RNA (RNAi) systems are known. Double-stranded
RNA (dsRNA) induces sequence-specific degradation of homologous gene
transcripts. RNA-
induced silencing complex (RISC) metabolizes dsRNA to small 21-23-nucleotide
small
interfering RNAs (siRNAs). RISC contains a double stranded RNAse (dsRNAse,
e.g., Dicer)
and ssRNAse (e.g., Argonaut 2 or Ago2). RISC utilizes antisense strand as a
guide to find a
cleavable target. Both siRNAs and microRNAs (miRNAs) are known. A method of
inactivating
a gene in a genetically edited animal comprises inducing RNA interference
against a target gene
and/or nucleic acid such that expression of the target gene and/or nucleic
acid is reduced.
[00212] For example the exogenous nucleic acid sequence can induce RNA
interference against a nucleic acid encoding a polypeptide. For example,
double-stranded small
interfering RNA (siRNA) or small hairpin RNA (shRNA) homologous to a target
DNA can be
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
used to reduce expression of that DNA. Constructs for siRNA can be produced as
described, for
example, in Fire et al. (1998) Nature 391:806; Romano and Masino (1992) Mol.
Microbiol.
6:3343; Cogoni et al. (1996) EMBO J. 15:3153; Cogoni and Masino (1999) Nature
399:166;
Misquitta and Paterson (1999) Proc. Natl. Acad. Sci. USA 96:1451; and
Kennerdell and
Carthew (1998) Cell 95:1017. Constructs for shRNA can be produced as described
by McIntyre
and Fanning (2006) BMC Biotechnology 6:1. In general, shRNAs are transcribed
as a single-
stranded RNA molecule containing complementary regions, which can anneal and
form short
hairpins.
[00213] The probability of finding a single, individual functional siRNA or
miRNA
directed to a specific gene is high. The predictability of a specific sequence
of siRNA, for
instance, is about 50% but a number of interfering RNAs may be made with good
confidence
that at least one of them will be effective.
[00214] In vitro cells, in vivo cells, or a genetically edited animal such as
a livestock
animal that express an RNAi directed against a gene encoding CD163 can be
used. The RNAi
may be, for instance, selected from the group consisting of siRNA, shRNA,
dsRNA, RISC and
miRNA.
Inducible Systems
[00215] An inducible system may be used to inactivate a CD163 gene. Various
inducible systems are known that allow spatial and temporal control of
inactivation of a gene.
Several have been proven to be functional in vivo in porcine animals.
[00216] An example of an inducible system is the tetracycline (tet)-on
promoter
system, which can be used to regulate transcription of the nucleic acid. In
this system, a mutated
Tet repressor (TetR) is fused to the activation domain of herpes simplex virus
VP 16 trans-
activator protein to create a tetracycline-controlled transcriptional
activator (tTA), which is
regulated by tet or doxycycline (dox). In the absence of antibiotic,
transcription is minimal,
while in the presence of tet or dox, transcription is induced. Alternative
inducible systems
include the ecdysone or rapamycin systems. Ecdysone is an insect molting
hormone whose
production is controlled by a heterodimer of the ecdysone receptor and the
product of the
ultraspiracle gene (USP). Expression is induced by treatment with ecdysone or
an analog of
ecdysone such as muristerone A. The agent that is administered to the animal
to trigger the
inducible system is referred to as an induction agent.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
46
[00217] The tetracycline-inducible system and the Cre/loxP recombinase system
(either constitutive or inducible) are among the more commonly used inducible
systems. The
tetracycline-inducible system involves a tetracycline-controlled
transactivator (tTA)/reverse tTA
(rtTA). A method to use these systems in vivo involves generating two lines of
genetically edited
animals. One animal line expresses the activator (tTA, rtTA, or Cre
recombinase) under the
control of a selected promoter. Another line of animals expresses the
acceptor, in which the
expression of the gene of interest (or the gene to be altered) is under the
control of the target
sequence for the tTA/rtTA transactivators (or is flanked by loxP sequences).
Mating the two of
animals provides control of gene expression.
[00218] The tetracycline-dependent regulatory systems (tet systems) rely on
two
components, i.e., a tetracycline-controlled transactivator (tTA or rtTA) and a
tTA/rtTA-
dependent promoter that controls expression of a downstream cDNA, in a
tetracycline-
dependent manner. In the absence of tetracycline or its derivatives (such as
doxycycline), tTA
binds to tet0 sequences, allowing transcriptional activation of the tTA-
dependent promoter.
However, in the presence of doxycycline, tTA cannot interact with its target
and transcription
does not occur. The tet system that uses tTA is termed tet-OFF, because
tetracycline or
doxycycline allows transcriptional down-regulation. Administration of
tetracycline or its
derivatives allows temporal control of transgene expression in vivo. rtTA is a
variant of tTA that
is not functional in the absence of doxycycline but requires the presence of
the ligand for
transactivation. This tet system is therefore termed tet-ON. The tet systems
have been used in
vivo for the inducible expression of several transgenes, encoding, e.g.,
reporter genes,
oncogenes, or proteins involved in a signaling cascade.
[00219] The Cre/lox system uses the Cre recombinase, which catalyzes site-
specific
recombination by crossover between two distant Cre recognition sequences,
i.e., loxP sites. A
DNA sequence introduced between the two loxP sequences (termed foxed DNA) is
excised by
Cre-mediated recombination. Control of Cre expression in a transgenic and/or
gene edited
animal, using either spatial control (with a tissue- or cell-specific
promoter), or temporal control
(with an inducible system), results in control of DNA excision between the two
loxP sites. One
application is for conditional gene inactivation (conditional knockout).
Another approach is for
protein over-expression, wherein a foxed stop codon is inserted between the
promoter sequence
and the DNA of interest. Genetically edited animals do not express the
transgene until Cre is
expressed, leading to excision of the foxed stop codon. This system has been
applied to tissue-
specific oncogenesis and controlled antigene receptor expression in B
lymphocytes. Inducible
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
47
Cre recombinases have also been developed. The inducible Cre recombinase is
activated only by
administration of an exogenous ligand. The inducible Cre recombinases are
fusion proteins
containing the original Cre recombinase and a specific ligand-binding domain.
The functional
activity of the Cre recombinase is dependent on an external ligand that is
able to bind to this
specific domain in the fusion protein.
[00220] In vitro cells, in vivo cells, or a genetically edited animal such as
a livestock
animal that comprises a CD163 gene under control of an inducible system can be
used. The
chromosomal modification of an animal may be genomic or mosaic. The inducible
system may
be, for instance, selected from the group consisting of Tet-On, Tet-Off, Cre-
lox, and Hifl alpha.
Vectors and Nucleic Acids
[00221] A variety of nucleic acids may be introduced into cells for knockout
purposes,
for inactivation of a gene, to obtain expression of a gene, or for other
purposes. As used herein,
the term nucleic acid includes DNA, RNA, and nucleic acid analogs, and nucleic
acids that are
double-stranded or single-stranded (i.e., a sense or an antisense single
strand). Nucleic acid
analogs can be modified at the base moiety, sugar moiety, or phosphate
backbone to improve,
for example, stability, hybridization, or solubility of the nucleic acid.
Modifications at the base
moiety include deoxyuridine for deoxythymidine, and 5-methyl-2'-deoxycytidine
and 5-bromo-
2'-doxycytidine for deoxycytidine. Modifications of the sugar moiety include
modification of
the 2' hydroxyl of the ribose sugar to form 2'-0-methyl or 2'-0-ally1 sugars.
The deoxyribose
phosphate backbone can be modified to produce morpholino nucleic acids, in
which each base
moiety is linked to a six membered, morpholino ring, or peptide nucleic acids,
in which the
deoxyphosphate backbone is replaced by a pseudopeptide backbone and the four
bases are
retained. See, Summerton and Weller (1997) Antisense Nucleic Acid Drug Dev.
7(3):187; and
Hyrup et al. (1996) Bioorgan. Med. Chem. 4:5. In addition, the deoxyphosphate
backbone can
be replaced with, for example, a phosphorothioate or phosphorodithioate
backbone, a
phosphoroamidite, or an alkyl phosphotriester backbone.
[00222] The target nucleic acid sequence can be operably linked to a
regulatory region
such as a promoter. Regulatory regions can be porcine regulatory regions or
can be from other
species. As used herein, operably linked refers to positioning of a regulatory
region relative to a
nucleic acid sequence in such a way as to permit or facilitate transcription
of the target nucleic
acid.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
48
[00223] Any type of promoter can be operably linked to a target nucleic acid
sequence. Examples of promoters include, without limitation, tissue-specific
promoters,
constitutive promoters, inducible promoters, and promoters responsive or
unresponsive to a
particular stimulus. Suitable tissue specific promoters can result in
preferential expression of a
nucleic acid transcript in beta cells and include, for example, the human
insulin promoter. Other
tissue specific promoters can result in preferential expression in, for
example, hepatocytes or
heart tissue and can include the albumin or alpha-myosin heavy chain
promoters, respectively. A
promoter that facilitates the expression of a nucleic acid molecule without
significant tissue or
temporal-specificity can be used (i.e., a constitutive promoter). For example,
a beta-actin
promoter such as the chicken beta-actin gene promoter, ubiquitin promoter,
miniCAGs
promoter, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter, or 3-
phosphoglycerate kinase (PGK) promoter can be used, as well as viral promoters
such as the
herpes simplex virus thymidine kinase (HSV-TK) promoter, the 5V40 promoter, or
a
cytomegalovirus (CMV) promoter. For example, a fusion of the chicken beta
actin gene
promoter and the CMV enhancer can be used as a promoter. See, for example, Xu
et al. (2001)
Hum. Gene Ther. 12:563; and Kiwaki et al. (1996) Hum. Gene Ther. 7:821.
[00224] Additional regulatory regions that may be useful in nucleic acid
constructs,
include, but are not limited to, polyadenylation sequences, translation
control sequences (e.g., an
internal ribosome entry segment, IRES), enhancers, inducible elements, or
introns. Such
regulatory regions may not be necessary, although they may increase expression
by affecting
transcription, stability of the mRNA, translational efficiency, or the like.
Such regulatory regions
can be included in a nucleic acid construct as desired to obtain optimal
expression of the nucleic
acids in the cell(s). Sufficient expression, however, can sometimes be
obtained without such
additional elements.
[00225] A nucleic acid construct may be used that encodes signal peptides or
selectable markers. Signal peptides can be used such that an encoded
polypeptide is directed to a
particular cellular location (e.g., the cell surface). Non-limiting examples
of selectable markers
include puromycin, ganciclovir, adenosine deaminase (ADA), aminoglycoside
phosphotransferase (neo, G418, APH), dihydrofolate reductase (DHFR),
hygromycin-B-
phosphotransferase, thymidine kinase (TK), and xanthin-guanine
phosphoribosyltransferase
(XGPRT). Such markers are useful for selecting stable transformants in
culture. Other selectable
markers include fluorescent polypeptides, such as green fluorescent protein or
yellow
fluorescent protein.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
49
[00226] A sequence encoding a selectable marker can be flanked by recognition
sequences for a recombinase such as, e.g., Cre or Flp. For example, the
selectable marker can be
flanked by loxP recognition sites (34-bp recognition sites recognized by the
Cre recombinase) or
FRT recognition sites such that the selectable marker can be excised from the
construct. See,
Orban, et al., Proc. Natl. Acad. Sci. (1992) 89:6861, for a review of Cre/lox
technology, and
Brand and Dymecki, Dev. Cell (2004) 6:7. A transposon containing a Cre- or Flp-
activatable
transgene interrupted by a selectable marker gene also can be used to obtain
animals with
conditional expression of a transgene. For example, a promoter driving
expression of the
marker/transgene can be either ubiquitous or tissue-specific, which would
result in the
ubiquitous or tissue-specific expression of the marker in FO animals (e.g.,
pigs). Tissue specific
activation of the transgene can be accomplished, for example, by crossing a
pig that ubiquitously
expresses a marker-interrupted transgene to a pig expressing Cre or Flp in a
tissue-specific
manner, or by crossing a pig that expresses a marker-interrupted transgene in
a tissue-specific
manner to a pig that ubiquitously expresses Cre or Flp recombinase. Controlled
expression of
the transgene or controlled excision of the marker allows expression of the
transgene.
[00227] The exogenous nucleic acid can encode a polypeptide. A nucleic acid
sequence encoding a polypeptide can include a tag sequence that encodes a
"tag" designed to
facilitate subsequent manipulation of the encoded polypeptide (e.g., to
facilitate localization or
detection). Tag sequences can be inserted in the nucleic acid sequence
encoding the polypeptide
such that the encoded tag is located at either the carboxyl or amino terminus
of the polypeptide.
Non-limiting examples of encoded tags include glutathione S-transferase (GST)
and FLAGTmtag
(Kodak, New Haven, Conn.).
[00228] Nucleic acid constructs can be methylated using an SssI CpG methylase
(New
England Biolabs, Ipswich, Mass.). In general, the nucleic acid construct can
be incubated with
S-adenosylmethionine and SssI CpG-methylase in buffer at 37 C.
Hypermethylation can be
confirmed by incubating the construct with one unit of HinPlI endonuclease for
1 hour at 37 C.
and assaying by agarose gel electrophoresis.
[00229] Nucleic acid constructs can be introduced into embryonic, fetal, or
adult
animal cells of any type, including, for example, germ cells such as an oocyte
or an egg, a
progenitor cell, an adult or embryonic stem cell, a primordial germ cell, a
kidney cell such as a
PK-15 cell, an islet cell, a beta cell, a liver cell, or a fibroblast such as
a dermal fibroblast, using
a variety of techniques. Non-limiting examples of techniques include the use
of transposon
systems, recombinant viruses that can infect cells, or liposomes or other non-
viral methods such
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
as electroporation, microinjection, or calcium phosphate precipitation, that
are capable of
delivering nucleic acids to cells.
[00230] In transposon systems, the transcriptional unit of a nucleic
acid construct, i.e.,
the regulatory region operably linked to an exogenous nucleic acid sequence,
is flanked by an
inverted repeat of a transposon. Several transposon systems, including, for
example, Sleeping
Beauty (see, U.S. Pat. No. 6,613,752 and U.S. Publication No. 2005/0003542);
Frog Prince
(Miskey et al. (2003) Nucleic Acids Res. 31:6873); To12 (Kawakami (2007)
Genome Biology
8(Supp1.1):57; Minos (Pavlopoulos et al. (2007) Genome Biology 8(Supp1.1):52);
Hsmarl
(Miskey et al. (2007)) Mol Cell Biol. 27:4589); and Passport have been
developed to introduce
nucleic acids into cells, including mice, human, and pig cells. The Sleeping
Beauty transposon is
particularly useful. A transposase can be delivered as a protein, encoded on
the same nucleic
acid construct as the exogenous nucleic acid, can be introduced on a separate
nucleic acid
construct, or provided as an mRNA (e.g., an in vitro-transcribed and capped
mRNA).
[00231] Insulator elements also can be included in a nucleic acid construct to
maintain
expression of the exogenous nucleic acid and to inhibit the unwanted
transcription of host genes.
See, for example, U.S. Publication No. 2004/0203158. Typically, an insulator
element flanks
each side of the transcriptional unit and is internal to the inverted repeat
of the transposon. Non-
limiting examples of insulator elements include the matrix attachment region-
(MAR) type
insulator elements and border-type insulator elements. See, for example, U.S.
Pat. Nos.
6,395,549, 5,731,178, 6,100,448, and 5,610,053, and U.S. Publication No.
2004/0203158.
[00232] Nucleic acids can be incorporated into vectors. A vector is a broad
term that
includes any specific DNA segment that is designed to move from a carrier into
a target DNA. A
vector may be referred to as an expression vector, or a vector system, which
is a set of
components needed to bring about DNA insertion into a genome or other targeted
DNA
sequence such as an episome, plasmid, or even virus/phage DNA segment. Vector
systems such
as viral vectors (e.g., retroviruses, adeno-associated virus and integrating
phage viruses), and
non-viral vectors (e.g., transposons) used for gene delivery in animals have
two basic
components: 1) a vector comprised of DNA (or RNA that is reverse transcribed
into a cDNA)
and 2) a transposase, recombinase, or other integrase enzyme that recognizes
both the vector and
a DNA target sequence and inserts the vector into the target DNA sequence.
Vectors most often
contain one or more expression cassettes that comprise one or more expression
control
sequences, wherein an expression control sequence is a DNA sequence that
controls and
regulates the transcription and/or translation of another DNA sequence or
mRNA, respectively.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
51
[00233] Many different types of vectors are known. For example, plasmids and
viral
vectors, e.g., retroviral vectors, are known. Mammalian expression plasmids
typically have an
origin of replication, a suitable promoter and optional enhancer, necessary
ribosome binding
sites, a polyadenylation site, splice donor and acceptor sites,
transcriptional termination
sequences, and 5' flanking non-transcribed sequences. Examples of vectors
include: plasmids
(which may also be a carrier of another type of vector), adenovirus, adeno-
associated virus
(AAV), lentivirus (e.g., modified HIV-1, Sly or Fly), retrovirus (e.g., ASV,
ALV or MoMLV),
and transposons (e.g., Sleeping Beauty, P-elements, To1-2, Frog Prince,
piggyBac).
[00234] As used herein, the term nucleic acid refers to both RNA and DNA,
including, for example, cDNA, genomic DNA, synthetic (e.g., chemically
synthesized) DNA, as
well as naturally occurring and chemically modified nucleic acids, e.g.,
synthetic bases or
alternative backbones. A nucleic acid molecule can be double-stranded or
single-stranded (i.e., a
sense or an antisense single strand).
[00235] Having described the invention in detail, it will be apparent that
modifications
and variations are possible without departing from the scope of the invention
defined in the
appended claims.
EXAMPLES
[00236] The following non-limiting examples are provided to further illustrate
the
present invention.
Example 1: Use of the CRISPR/Cas9 System to Produce Genetically Engineered
Pigs from
In Vitro-Derived Oocytes and Embryos
[00237] Recent reports describing homing endonucleases, such as zinc-finger
nucleases (ZFNs), transcription activator-like effector nucleases (TALENs),
and components in
the clustered regularly interspaced short palindromic repeat (CRISPR)/ CRISPR-
associated
(Cas9) system suggest that genetic engineering (GE) in pigs might now be more
efficient.
Targeted homing endonucleases can induce double-strand breaks (DSBs) at
specific locations in
the genome and cause either random mutations through nonhomologous end joining
(NHEJ) or
stimulation of homologous recombination (HR) if donor DNA is provided.
Targeted
modification of the genome through HR can be achieved with homing
endonucleases if donor
DNA is provided along with the targeted nuclease. After introducing specific
modifications in
somatic cells, these cells were used to produce GE pigs for various purposes
via SCNT. Thus,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
52
homing endonucleases are a useful tool in generating GE pigs. Among the
different homing
endonucleases, the CRISPR/Cas9 system, adapted from prokaryotes where it is
used as a
defense mechanism, appears to be an effective approach. In nature, the Cas9
system requires
three components, an RNA (-20 bases) that contains a region that is
complementary to the target
sequence (cis- repressed RNA [crRNA]), an RNA that contains a region that is
complementary
to the crRNA (trans-activating crRNA [tracrRNA]), and Cas9, the enzymatic
protein component
in this complex. A single guide RNA (gRNA) can be constructed to serve the
roles of the base-
paired crRNA and tracrRNA. The gRNA/protein complex can scan the genome and
catalyze a
DSB at regions that are complementary to the crRNA/gRNA. Unlike other designed
nucleases,
only a short oligomer needs to be designed to construct the reagents required
to target a gene of
interest whereas a series of cloning steps are required to assemble ZFNs and
TALENs.
[00238] Unlike current standard methods for gene disruption, the use of
designed
nucleases offers the opportunity to use zygotes as starting material for GE.
Standard methods for
gene disruption in livestock involve HR in cultured cells and subsequent
reconstruction of
embryos by somatic cell nuclear transfer (SCNT). Because cloned animals
produced through
SCNT sometimes show signs of developmental defects, progeny of the SCNT/GE
founders are
typically used for research to avoid confounding SCNT anomalies and phenotype
that could
occur if founder animals are used for experiments. Considering the longer
gestation period and
higher housing costs of pigs compared to rodents, there are time and cost
benefits to the reduced
need for breeding. A recent report demonstrated that direct injection of ZFNs
and TALENs into
porcine zygotes could disrupt an endogenous gene and produce piglets with the
desired
mutations. However, only about 10% of piglets showed biallelic modification of
the target gene,
and some presented mosaic genotypes. A recent article demonstrated that
CRISPR/ Cas9 system
could induce mutations in developing embryos and produce GE pigs at a higher
efficiency than
ZFNs or TALENs. However, GE pigs produced from the CRISPR/ Cas9 system also
possessed
mosaic genotypes. In addition, all the above-mentioned studies used in vivo
derived zygotes for
the experiments, which require intensive labor and numerous sows to obtain a
sufficient number
of zygotes.
[00239] The present example describes an efficient approach to use the CRISPR/
Cas9
system in generating GE pigs via both injection of in vitro derived zygotes
and modification of
somatic cells followed by SCNT. Two endogenous genes (CD163 and CD1D) and one
transgene
(eGFP) were targeted, and only in vitro derived oocytes or zygotes were used
for SCNT or RNA
injections, respectively. CD163 appears to be required for productive
infection by porcine
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
53
reproductive and respiratory syndrome virus, a virus known to cause a
significant economic loss
to swine industry. CD1D is considered a nonclassical major histocompatibility
complex protein
and is involved in presentation of lipid antigens to invariant natural killer
T cells. Pigs deficient
in these genes were designed to be models for agriculture and biomedicine. The
eGFP transgene
was used as a target for preliminary proof-of-concept experiments and
optimizations of methods.
MATERIALS AND METHODS
[00240] Chemical and Reagents. Unless otherwise stated, all of the chemicals
used in
this study were purchased from Sigma.
Design of gRNAs to build specific CRISPRs
[00241] Guide RNAs were designed to regions within exon 7 of CD 163 that were
unique to the wild type CD 163 and not present in the domain swap targeting
vector (described
below), so that the CRISPR would result in DSB within wild type CD 163 but not
in the domain
swap targeting vector. There were only four locations in which the targeting
vector would
introduce a single nucleotide polymorphism (SNP) that would alter an S.
pyogenes (Spy)
protospacer adjacent motif (PAM). All four targets were selected including:
(SEQ ID NO:1) GGAAACCCAGGCTGGTTGGAgGG (CRISPR 10),
(SEQ ID NO:2) GGAACTACAGTGCGGCACTGtGG (CRISPR 131),
(SEQ ID NO:3) CAGTAGCACCCCGCCCTGACgGG (CRISPR 256) and
(SEQ ID NO:4) TGTAGCCACAGCAGGGACGTcGG (CRISPR 282).
The PAM can be identified by the bold font in each gRNA.
[00242] For CD 1D mutations, the search for CRISPR targets was arbitrarily
limited to
the coding strand within the first 1000 bp of the primary transcript. However,
RepeatMasker
[26] ("Pig" repeat library) identified a repetitive element beginning at base
943 of the primary
transcript. The search for CRISPR targets was then limited to the first 942 bp
of the primary
transcript. The search was further limited to the first 873 bp of the primary
transcript since the
last Spy PAM is located at base 873. The first target (CRISPR 4800) was
selected because it
overlapped with the start codon located at base 42 in primary transcript
(CCAGCCTCGCCCAGCGACATgGG (SEQ ID NO:5)). Two additional targets (CRISPRs
5620 and 5626) were selected because they were the most distal to the first
selection within the
arbitrarily selected region (CTTTCATTTATCTGAACTCAgGG (SEQ ID NO:6)) and
TTATCTGAACTCAGGGTCCCcGG (SEQ ID NO:7)). These targets overlap. In relation to
the
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
54
start codon, the most proximal Spy PAMs were located in simple sequence that
contained
extensively homopolymeric sequence as determined by visual appraisal. The
forth target
(CRISPR 5350) was selected because, in relation to the first target selection,
it was the most
proximal target that did not contain extensive homopolymeric regions
(CAGCTGCAGCATATATTTAAgGG (SEQ ID NO:8)). Specificity of the designed crRNAs
was confirmed by searching for similar porcine sequences in GenBank. The
oligonucleotides
(Table 1) were annealed and cloned into the p330X vector which contains two
expression
cassettes, a human codon-optimized S. pyogenes (hSpy) Cas9 and the chimeric
guide RNA.
P330X was digested with Bbsl (New England Biolabs) following the Zhang
laboratory protocol
(http://www.addgene.org/crisprizhang/).
[00243] To target eGFP, two specific gRNAs targeting the eGFP coding sequence
were designed within the first 60 bp of the eGFP start codon. Both eGFP1 and
eGFP2 gRNA
were on the antisense strand and eGFP1 directly targeted the start codon. The
eGFP1 gRNA
sequence was CTCCTCGCCCTTGCTCACCAtGG (SEQ ID NO:9) and the eGFP2 gRNA
sequence was GACCAGGATGGGCACCACCCcGG (SEQ ID NO:10).
Table 1. Designed crRNAs. Primer 1 and primer 2 were annealed following the
Zhang
protocol.
Primer Sequence (5' ¨ 3') SEQ ID NO.
CD163 101 CACCGGAAACCCAGGCTGGTTGGA 48
CD163 102 AAACTCCAACCAGCCTGGGTTTCC 49
CD163 131 1 CACCGGAACTACAGTGCGGCACTG 50
CD163 131 2 AAACCAGTGCCGCACTGTAGTTCC 51
CD163 256 1 CACCGCAGTAGCACCCCGCCCTGAC 52
CD163 256 2 AAACGTCAGGGCGGGGTGCTACTGC 53
CD163 282 1 CACCGTGTAGCCACAGCAGGGACGT 54
CD163 282 2 AAACACGTCCCTGCTGTGGCTACAC 55
CD1D 4800 1 CACCGCCAGCCTCGCCCAGCGACAT 56
CD 1D 4800 2 AAACATGTCGCTGGGCGAGGCTGGC 57
CD1D 5350 1 CACCGCAGCTGCAGCATATATTTAA 58
CD 1D 5350 2 AAACTTAAATATATGCTGCAGCTGC 59
CD1D 5620 1 CACCGCTTTCATTTATCTGAACTCA 60
CD1D 5620 2 AAACTGAGTTCAGATAAATGAAAGC 61
CD1D 5626 1 CACCGTTATCTGAACTCAGGGTCCC 62
CD 1D 5626 2 AAACGGGACCCTGAGTTCAGATAAC 63
eGFP 11 CACCGCTCCTCGCCCTTGCTCACCA 64
eGFP 12 AAACTGGTGAGCAAGGGCGAGGAGC 65
eGFP 21 CACCGGACCAGGATGGGCACCACCC 66
eGFP 22 AAACGGGTGGTGCCCATCCTGGTCC 67
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
Synthesis of Donor DNA for CD163 and CD1D Genes
[00244] Both porcine CD163 and CD1D were amplified by PCR from DNA isolated
from the fetal fibroblasts that would be used for later transfections to
ensure an isogenic match
between the targeting vector and the transfected cell line. Briefly, LA taq
(Clontech) using the
forward primer CTCTCCCTCACTCTAACCTACTT (SEQ ID NO:11), and the reverse primer
TATTTCTCTCACATGGCCAGTC (SEQ ID NO:12) were used to amplify a 9538 bp fragment
of CD163. The fragment was DNA sequence validated and used to build the domain-
swap
targeting vector (Fig. 1). This vector included 33 point mutations within exon
7 so that it would
encode the same amino acid sequence as human CD163L from exon 11. The
replacement exon
was 315 bp. In addition, the subsequent intron was replaced with a modified
myostatin intron B
that housed a selectable marker gene that could be removed with Cre-
recombinase (Cre) and had
previously demonstrated normal splicing when harboring the retained loxP site
(Wells,
unpublished results). The long arm of the construct was 3469 bp and included
the domain swap
DS exon. The short arm was 1578 bp and included exons 7 and 8 (Fig. 1, panel
B). This plasmid
was used to attempt to replace the coding region of exon 7 in the first
transfection experiments
and allowed for selection of targeting events via the selectable marker
(G418). If targeting were
to occur, the marker could be deleted by Cre-recombinase. The CD163 DS-
targeting vector was
then modified for use with cell lines that already contained a SIGLEC1 gene
disrupted with Neo
that could not be Cre deleted. In this targeting vector, the Neo cassette,
loxP and myostatin
intron B, were removed, and only the DS exon remained with the WT long and
short arm (Fig.
1, panel C).
[00245] The genomic sequence for porcine CD1D was amplified with LA taq using
the forward primer CTCTCCCTCACTCTAACCTACTT(SEQ ID NO:13) and reverse primer
GACTGGCCATGTGAGAGAAATA (SEQ ID NO:14), resulting in an 8729 bp fragment. The
fragment was DNA sequenced and used to build the targeting vector shown in
Fig. 2. The Neo
cassette is under the control of a phosphoglycerol kinase (PGK) promoter and
flanked with loxP
sequences, which were introduced for selection. The long arm of the construct
was 4832 bp and
the short arm was 3563 bp, and included exons 6 and 7. If successful HR
occurred, exons 3, 4,
and 5 would be removed and replaced with the Neo cassette. If NHEJ repair
occurred
incorrectly, then exon 3 would be disrupted.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
56
Fetal Fibroblast Collection
[00246] Porcine fetal tissue was collected on Day 35 of gestation to create
cell lines.
Two wild-type (WT) male and female fetal fibroblast cell lines were
established from a large
white domestic cross. Male and female fetal fibroblasts that had previously
been modified to
contain a Neo cassette (SIGLEC1-/- genetics) were also used in these studies.
Fetal fibroblasts
were collected as described with minor modifications; minced tissue from each
fetus was
digested in 20 ml of digestion media (Dulbecco-modified Eagle medium [DMEM]
containing L-
glutamine and 1 g/L D-glucose [Cellgro] supplemented with 200 units/ml
collagenase and 25
Kunitz units/ml DNAseI) for 5 hours at 38.5 C. After digestion, fetal
fibroblast cells were
washed and cultured with DMEM, 15% fetal bovine serum (FBS), and 40 g/ml
gentamicin.
After overnight culture, the cells were trypsinized and frozen at ¨80 C in
aliquots in FBS with
10% dimethyl sulfoxide and stored in liquid nitrogen.
Cell Transfection and Genotyping
[00247] Transfection conditions were essentially as previously reported. The
donor
DNA was always used at a constant amount of 1 [tg with varying amounts of
CRISPR/Cas9
plasmid (listed below). Donor DNA was linearized with MLUI (CD163) (NEB) or
AFLII
(CD1D) (NEB) prior to transfection. The gender of the established cell lines
was determined by
PCR as described previously prior to transfection. Both male and female cell
lines were
transfected, and genome modification data was analyzed together between the
transfections.
Fetal fibroblast cell lines of similar passage number (2-4) were cultured for
2 days and grown to
75%-85% confluency in DMEM containing L-glutamine and 1 g/L D-glucose
(Cellgro)
supplemented with 15% FBS, 2.5 ng/ml basic fibroblast growth factor, and 10
mg/ml
gentamicin. Fibroblast cells were washed with phosphate-buffered saline (PBS)
(Life
Technologies) and trypsinized. As soon as cells detached, the cells were
rinsed with an
electroporation medium (75% cytosalts [120 mM KC1, 0.15 mM CaCl2, 10 mM
K2HPO4, pH
7.6, 5 Mm MgCl2]) and 25% Opti-MEM (LifeTechnologies). Cell concentration was
quantified
by using a hemocytometer. Cells were pelleted at 600 X g for 5 minutes and
resuspended at a
concentration of 1 X 106 in electroporation medium. Each electroporation used
200 11.1 of cells in
2 mm gap cuvettes with three (1 msec) square-wave pulses administered through
a BTX ECM
2001 at 250 V. After the electroporation, cells were resuspended in DMEM
described above. For
selection, 600 g/ml G418 (Life Technologies) was added 24 hours after
transfection, and the
medium was changed on Day 7. Colonies were picked on Day 14 after
transfection. Fetal
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
57
fibroblasts were plated at 10,000 cells/plate if G418 selection was used and
at 50 cells/plate if no
G418 selection was used. Fetal fibroblast colonies were collected by applying
10 mm autoclaved
cloning cylinders sealed around each colony by autoclaved vacuum grease.
Colonies were rinsed
with PBS and harvested via trypsin; then resuspended in DMEM culture medium. A
part (1/3) of
the resuspended colony was transferred to a 96-well PCR plate, and the
remaining (2/3) cells
were cultured in a well of a 24-well plate. The cell pellets were resuspended
in 6 .1 of lysis
buffer (40 mM Tris, pH 8.9, 0.9% Triton X-100, 0.4 mg/ml proteinase K [NEB]),
incubated at
65 C for 30 minutes for cell lysis, followed by 85 C for 10 minutes to
inactivate the proteinase
K.
PCR Screening for DS and Large and Small Deletions
[00248] Detection of HR-directed repair. Long-range PCRs were used to identify
mutations on either CD163 or CD1D. Three different PCR assays were used to
identify HR
events: PCR amplification of regions spanning from the CD163 or CD1D sequences
in the
donor DNA to the endogenous CD163 or CD1D sequences on either the right or
left side and a
long-range PCR that amplified large regions of CD163 or CD1D encompassing the
designed
donor DNAs. An increase in the size of a PCR product, either 1.8 kb (CD1D) or
3.5 kb
(CD163), arising from the addition of exogenous Neo sequences, was considered
evidence for
HR-directed repair of the genes. All the PCR conditions included an initial
denaturation of 95 C
for 2 minutes followed by 33 cycles of 30 seconds at 94 C, 30 seconds at 50 C,
and 7-10
minutes at 68 C. LA taq was used for all the assays following the
manufacturers'
recommendations. Primers are shown in Table 2.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
58
Table 2. Primers used to identify HR directed repair of CD163 and CD1D
SEQ
ID
Primer Sequence (5' ¨ 3') NO.
CD163 Long Range Assay Primer 1230F TTGTTGGAAGGCTCACTGTCCTTG 68
CD163 Long Range Assay Primer 7775 R ACAACTAAGGTGGGGCAAAG 69
CD163 Left Arm Assay Primer 1230 F TTGTTGGAAGGCTCACTGTCCTTG 70
CD163 Left Arm Assay Primer 8491 R GGAGCTCAACATTCTTGGGTCCT 71
CD163 Right Arm Assay Primer 3752 F GGCAAAATTTTCATGCTGAGGTG 72
CD163 Right Arm Assay Primer 7765 R GCACATCACTTCGGGTTACAGTG 73
CD1D Long Range Assay Primer F 3991 F CCCAAGTATCTTCAGTTCTGCAG 74
CD1D Long Range Assay Primer R 12806 R
TACAGGTAGGAGAGCCTGTTTTG 75
CD1D Left Arm Assay Primer F 3991 F CCCAAGTATCTTCAGTTCTGCAG 76
CD1D Left Arm Assay Primer 7373 R CTCAAAAGGATGTAAACCCTGGA 77
CD1D Right Arm Assay Primer 4363 F TGTTGATGTGGTTTGTTTGCCC 78
CD1D Right Arm Assay Primer 12806 R TACAGGTAGGAGAGCCTGTTTTG 79
[00249] Small deletions assay (NHEJ) . Small deletions were determined by PCR
amplification of CD163 or CD1D flanking a projected cutting site introduced by
the
CRISPR/Cas9 system. The size of the amplicons was 435 bp and 1244 bp for CD163
and
CD1D, respectively. Lysates from both embryos and fetal fibroblasts were PCR
amplified with
LA taq. PCR conditions of the assays were an initial denaturation of 95 C for
2 minutes
followed by 33 cycles of 30 seconds at 94 C, 30 seconds at 56 C, and 1 minute
at 72 C. For
genotyping of the transfected cells, insertions and deletions (INDELs) were
identified by
separating PCR amplicons by agarose gel electrophoresis. For embryo
genotyping, the resulting
PCR products were subsequently DNA sequenced to identify small deletions using
forward
primers used in the PCR. Primer information is shown in Table 3.
Table 3. Primers used to identify mutations through NHEJ on CD163 and CD1D
Primer Sequence (5' ¨ 3') SEQ ID NO.
GCD I 63F GGAGGTCTAGAATCGGCTAAGCC 80
GCD 163R GGCTACATGTCCCGTCAGGG 81
GCD IDF GCAGGCCACTAGGCAGATGAA 82
GCD IDR GAGCTGACACCCAAGAAGTTCCT 83
eGFP1 GGCTCTAGAGCCTCTGCTAACC 84
eGFP2 GGACTTGAAGAAGTCGTGCTGC 85
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
59
Somatic Cell Nuclear Transfer (SCNT)
[00250] To produce SCNT embryos, either sow-derived oocytes (ART, Inc.) or
gilt-
derived oocytes from a local slaughter house were used. The sow-derived
oocytes were shipped
overnight in maturation medium (TCM-199 with 2.9 mM Hepes, 5 pg/m1 insulin, 10
ng/ml
epidermal growth factor [EGF], 0.5 tg/m1 porcine follicle-stimulating hormone
[p-FSH], 0.91
mM pyruvate, 0.5 mM cysteine, 10% porcine follicular fluid, and 25 ng/ml
gentamicin) and
transferred into fresh medium after 24 hours. After 40-42 hours of maturation,
cumulus cells
were removed from the oocytes by vortexing in the presence of 0.1%
hyaluronidase. The gilt-
derived oocytes were matured as described below for in vitro fertilization
(IVF). During
manipulation, oocytes were placed in the manipulation medium (TCM-199 [Life
Technologies]
with 0.6 mM NaHCO3, 2.9 mM Hepes, 30 mM NaCl, 10 ng/ml gentamicin, and 3 mg/ml
BSA,
with osmolarity of 305 mOsm) supplemented with 7.0 pg/m1 cytochalasin B. The
polar body
along with a portion of the adjacent cytoplasm, presumably containing the
metaphase II plate,
was removed, and a donor cell was placed in the perivitelline space by using a
thin glass
capillary. The reconstructed embryos were then fused in a fusion medium (0.3 M
mannitol, 0.1
mM CaCl2, 0.1 mM MgCl2, and 0.5 mM Hepes) with two DC pulses (1-second
interval) at 1.2
kV/cm for 30 seconds using a BTX Electro Cell Manipulator (Harvard Apparatus).
After fusion,
fused embryos were fully activated with 2001..LM thimerosal for 10 minutes in
the dark and 8
mM dithiothreitol for 30 minutes. Embryos were then incubated in modified
porcine zygote
medium PZM3-MU1 with 0.5 pJV1 Scriptaid (S7817; Sigma-Aldrich), a histone
deacetylase
inhibitor, for 14-16 hours, as described previously.
In Vitro Fertilization (IVF)
[00251] For IVF, ovaries from prepubertal gilts were obtained from an abattoir
(Farmland Foods Inc.). Immature oocytes were aspirated from medium size (3¨ 6
mm) follicles
using an 18-gauge hypodermic needle attached to a 10 ml syringe. Oocytes with
evenly dark
cytoplasm and intact surrounding cumulus cells were then selected for
maturation. Around 50
cumulus oocyte complexes were place in a well containing 50011.1 of maturation
medium, TCM-
199 (Invitrogen) with 3.05 mM glucose, 0.91 mM sodium pyruvate, 0.57 mM
cysteine, 10 ng/ml
EGF, 0.5 pg/m1 luteinizing hormone (LH), 0.5 pg/m1FSH, 10 ng/ml gentamicin
(APP Pharm),
and 0.1% polyvinyl alcohol for 42-44 hours at 38.5 C, 5% CO2, in humidified
air. At the end of
the maturation, the surrounding cumulus cells were removed from the oocytes by
vortexing for 3
minutes in the presence of 0.1% hyaluronidase. Then, in vitro matured oocytes
were placed in
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
50 11.1 droplets of IVF medium (modified Tris-buffered medium containing 113.1
mM NaCl, 3
mM KC1, 7.5 mM CaCl2, 11 mM glucose, 20 mM Tris, 2 mM caffeine, 5 mM sodium
pyruvate,
and 2 mg/ml bovine serum albumin [BSA]) in groups of 25-30 oocytes. One
10011.1 frozen
semen pellet was thawed in 3 ml of Dulbecco PBS supplemented with 0.1% BSA.
Either frozen
WT or fresh eGFP semen was washed in 60% Percoll for 20 minutes at 650 3 g and
in modified
Tris-buffered medium for 10 minutes by centrifugation. In some cases, freshly
collected semen
heterozygous for a previously described eGFP transgene was washed three times
in PBS. The
semen pellet was then resuspended with IVF medium to 0.5 X 106 cells/ml. Fifty
microliters of
the semen suspension was introduced into the droplets with oocytes. The
gametes were
coincubated for 5 hours at 38.5 C in an atmosphere of 5% CO2 in air. After
fertilization, the
embryos were incubated in PZM3-MU1 at 38.5 C and 5% CO2 in air.
Embryo Transfer
[00252] Embryos generated to produce GE CD163 or CD1D pigs were transferred
into surrogates either on Day 1 (SCNT) or 6 (zygote injected) after first
standing estrus. For Day
6 transfer, zygotes were cultured for five additional days in PZM3-MU1 in the
presence of 10
ng/ml ps48 (Stemgent, Inc.). The embryos were surgically transferred into the
ampullary-isthmic
junction of the oviduct of the surrogate.
In Vitro Synthesis of RNA for CRISPR/Cas9 System
[00253] Template DNA for in vitro transcription was amplified using PCR (Table
4).
CRISPR/Cas9 plasmid used for cell transfection experiments served as the
template for the
PCR. In order to express the Cas9 in the zygotes, the mMES SAGE mMACHINE Ultra
Kit
(Ambion) was used to produce mRNA of Cas9. Then a poly A signal was added to
the Cas9
mRNA using a Poly (A) tailing kit (Ambion). CRISPR guide RNAs were produced by
MEGAshortscript (Ambion). The quality of the synthesized RNAs were visualized
on a 1.5%
agarose gel and then diluted to a final concentration of 10 ng/p1 (both gRNA
and Cas9) and
distributed into 3 11.1 aliquots.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
61
Table 4. Primers used to amplify templates for in vitro transcription.
Primers Sequence (5' ¨ 3') SEQ ID
NO.
Cas9 F: TAATACGACTCACTATAGGGAGAATGGACTATAAGGACCACGAC 86
R: GCGAGCTCTAGGAATTCTTAC 87
eGFP 1 F: TTAATACGACTCACTATAGGCTCCTCGCCCTTGCTCACCA 88
R: AAAAGCACCGACTCGGTGCC 89
CD 163 F: TTAATACGACTCACTATAGGAAACCCAGGCTGGTTGGA 90
R: AAAAGCACCGACTCGGTGCC 91
CD 163 F: TTAATACGACTCACTATAGGAACTACAGTGCGGCACTG 92
131 R: AAAAGCACCGACTCGGTGCC 93
CD 1D F: TTAATACGACTCACTATAGGCCAGCCTCGCCCAGCGACAT 94
4800 R: AAAAGCACCGACTCGGTGCC 95
CD 1D F: TTAATACGACTCACTATAGGCAGCTGCAGCATATATTTAA 96
5350 R: AAAAGCACCGACTCGGTGCC 97
Microinjection of Designed CRISPR/Cas9 System in Zygotes
[00254] Messenger RNA coding for Cas9 and gRNA was injected into the cytoplasm
of fertilized oocytes at 14 hours postfertilization (presumptive zygotes)
using a FemtoJet
microinjector (Eppendorf). Microinjection was performed in manipulation medium
on the
heated stage of a Nikon inverted microscope (Nikon Corporation; Tokyo, Japan).
Injected
zygotes were then transferred into the PZM3-MU1 with 10 ng/ml ps48 until
further use.
Statistical Analysis
[00255] The number of colonies with a modified genome was classified as 1, and
the
colonies without a modification of the genome were classified as 0.
Differences were
determined by using PROC GLM (SAS) with a P-value of 0.05 being considered as
significant.
Means were calculated as least-square means. Data are presented as numerical
means SEM.
RESULTS
CRISPR/Cas9-Mediated Knockout of CD163 and CD1D in Somatic Cells
[00256] Efficiency of four different CRISPRs plasmids (guides 10, 131, 256,
and 282)
targeting CD163 was tested at an amount of 2 pg/11.1 of donor DNA (Table 5).
CRISPR 282
resulted in significantly more average colony formation than CRISPR 10 and 256
treatments (P
<0.05). From the long-range PCR assay described above, large deletions were
found ranging
from 503 bp to as much as 1506 bp instead of a DS through HR as was originally
intended (Fig.
3, panel A). This was not expected because previous reports with other DNA-
editing systems
showed much smaller deletions of 6-333 bp using ZFN in pigs. CRISPR 10 and a
mix of all four
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
62
CRISPRs resulted in a higher number of colonies with a modified genome than
CRISPR 256
and 282 (Table 5, P < 0.002). Transfection with CRISPR 10 and a plasmid
containing Neo but
no homology to CD163 resulted in no colonies presenting the large deletion.
Interestingly, one
monoallelic deletion was also detected when the donor DNA was introduced
without any
CRISPR. This assay likely represents an underestimation of the mutation rate
because any
potential small deletions by sequencing which could not be detected on an
agarose gel in the
transfected somatic cells were not screened for.
0
Table 5. Efficiency of four different CRISPR plasmids (guides 10, 131, 256,
and 282) targeting CD163. Four different CRISPRs were tested at t..)
o
,-,
an amount of 2 1.ig to 11.ig Donor DNA (shown in Fig. 1).
,z
o
(...)
cio
o
-4
Average
Total Total No. of No. of
Percent Colonies
No. of No. of Colonies/pi Colonies
Colony with a Modified
Treatment* Colonies Plates atet NHEJ with
HR Genomet Reps
10+Donor DNA 76 102 0.75bc 11 11:
15.79a 4
131+Donor DNA 102 51 2.00ab 11 0
10.78ab 3
256+Donor DNA 43 49 0.88' 2 0
4.65bc 3 P
0
282+Donor DNA 109 46 2.37a 3 0
2.75bc 3
mix of 4+Donor DNA 111 55 2.02ab 20 0
18.02a 3
Donor DNA 48 52 0.92bc 1 0
2.08bc 3
0
+ Neo (no CD163) 26 20 1.3111a 0 0
0.00' 1 ,
,
0
,
* Mix of 4 + Donor DNA represents an equal mixing of 0.5m of each CRISPR with
1 jig of Donor DNA. The Donor DNA treatment served as
the no CRISPR control and the 10 + Neo treatment illustrates that the large
deletions observed in the CRISPR treatments were present only when
the CD163 Donor DNA was also present.
t ANOVA was performed comparing the average number of colonies/plate to
estimate CRISPR toxicity and on the percent colonies with a
modified genome. P-values were 0.025 and 0.0002, respectively. n/a = There
were no replicates for this treatment so no statistical analysis was
performed.
1-d
n
The one colony with HR represents a partial HR event.
a-c Superscript letters indicate a significant difference between treatments
for both average number of colonies/plate and percent colonies with a cp
t..)
modified genome (P <0.05).
,-,
cio
O-
t..)
-4
,z
.6.
.6.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
64
[00257] The initial goal was to obtain a domain swap (DS)-targeting event by
HR for
CD163, but CRISPRs did not increase the efficiency of targeting CD163. It
should be noted that
various combinations of this targeting vector had been used to modify CD163 by
HR by
traditional transfections and resulted in 0 targeting events after screening
3399 colonies
(Whitworth and Prather, unpublished results). Two pigs were obtained with a
full DS resulting
from HR that contained all 33 of the mutations that were attempted to be
introduced by
transfection with CRISPR 10 and the DS-targeting vector as donor DNA.
[00258] Next, the efficiency of CRISPR/Cas9-induced mutations without drug
selection was tested; the fetal fibroblast cell line used in this study
already had an integration of
the Neo resistant cassette and a knockout of SIGLEC1. Whether the ratio of
CRISPR/Cas9 and
donor DNA would increase genome modification or result in a toxic effect at a
high
concentration was also tested. CRISPR 131 was selected for this trial because
in the previous
experiment, it resulted in a high number of total colonies and an increased
percentage of colonies
possessing a modified genome. Increasing amounts of CRISPR 131 DNA from 3:1 to
20:1 did
not have a significant effect on fetal fibroblast survivability. The percent
of colonies with a
genome modified by NHEJ was not significantly different between the various
CRISPR
concentrations but had the highest number of NHEJ at a 10:1 ratio (Table 6, P
= 0.33). Even at
the highest ratio of CRISPR DNA to donor DNA (20:1), HR was not observed.
Table 6. Efficiency of CRISPR/Cas9-induced mutations without drug selection.
Four different ratios of Donor DNA to CRISPR 131
DNA were compared in a previously modified cell line without the use of G418
selection. 0
Number Percent
cio
Number Mean of Colonies
Colony Percent
Donor DNA: Number of Number of Colonies with
with Colonies
CRISPR Ratio Plates Colonies Colonies/Plate NHEJ NHEJ
HR with HR Reps
1:0 30 79 2.6 1 1.3a
0 0.0 2
1:3 30 84 2.8 1 1.2a
0 0.0 2
1:5 27 76 2.8 2 2.6a
0 0.0 2
1:10 32 63 2.0 5 7.9a
0 0.0 2
1:20 35 77 2.2 3 3.9a
0 0.0 2
a Significant difference between treatments for percent colonies with NHEJ
repair (P>0.05).
There was not a significant difference in the number of genome modified
colonies with increasing concentration of CRISPR
(P>0.33).
1-d
cio
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
66
[00259] Based on this experience, targeted disruption of CD1D in somatic cells
was
attempted. Four different CRISPRs were designed and tested in both male and
female cells.
Modifications of CD1D could be detected from three of the applied CRISPRs, but
use of
CRISPR 5350 did not result in modification of CD1D with a deletion large
enough to detect by
agarose gel electrophoresis (Table 7). Interestingly, no genetic changes were
obtained through
HR although donor DNA was provided. However, large deletions similar to the
CD163 knockout
experiments were observed (Fig. 3, panel B). No targeted modification of CD1D
with a large
deletion was detected when CRISPR/Cas9 was not used with the donor DNA.
Modification of
CD1D from CRISPR/Cas9-guided targeting was 4/121 and 3/28 in male and female
colonies of
cells, respectively. Only INDELs detectable by agarose gel electrophoresis
were included in the
transfection data.
Table 7. Four different CRISPRS were tested at an amount of 21.ig to 11.ig
Donor DNA (shown
in Fig. 2). The Donor DNA treatment served as the no CRISPR control.
Total Number
Gender Treatment of Colonies INDEL Efficiency (%)
4800 +Donor
male DNA 29 2 6.9
5350+Donor
male DNA 20 0 0
5620+Donor
male DNA 43 1 2.33
5626+Donor
male DNA 29 2 6.9
male Donor DNA 28 0 0
4800 +Donor
female DNA 2 0 0
5350+Donor
female DNA 8 0 0
5620+Donor
female DNA 10 0 0
5626+Donor
female DNA 8 3 37.5
female Donor DNA 7 0 0
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
67
Production of CD163 and CD1D Pigs through SCNT Using the GE Cells
[00260] The cells presenting modification of CD163 or CD1D were used for SCNT
to
produce CD163 and CD1D knockout pigs (Fig. 3). Seven embryo transfers (CD163
Table 8), six
embryo transfers (CD163-No Neo), and five embryo transfers (CD1D) into
recipient gilts were
performed with SCNT embryos from male and female fetal fibroblasts transfected
with CRISPR/
Cas9 systems. Six (CD163), two (CD163-No Neo), and four (CD1D) (Table 9) of
the recipient
gilts remained pregnant to term resulting in pregnancy rates of 85.7%. 33.3%,
and 80%,
respectively. Of the CD163 recipients, five delivered healthy piglets by
caesarean section. One
(0044) farrowed naturally. Litter size ranged from one to eight. Four pigs
were euthanized
because of failure to thrive after birth. One piglet was euthanized due to a
severe cleft palate. All
the remaining piglets appear healthy (Fig. 3, panel C). Two litters of male
piglets resulting from
fetal fibroblasts transfected with CRISPR 10 and donor DNA described in Fig.
3, panel B had a
30 bp deletion in exon 7 adjacent to CRISPR 10 and an additional 1476 bp
deletion of the
preceding intron, thus removing the intron 6/exon 7 junction of CD163 (Fig. 3,
panel E). The
genotypes and predicted translations are summarized in Table 10. One male
piglet and one
female litter (4 piglets) were obtained from the CD163-No Neo transfection of
previously
modified SIGLEC1 cells. All five piglets were double knockouts for SIGLEC1 and
CD163. The
male piglet had a biallelic modification of CD163 with a 28 bp deletion in
exon 7 on one allele
and a 1387 bp deletion on the other allele that included a partial deletion of
exon 7 and complete
deletion of exon 8 and the proceeding intron, thus removing the intron exon
junction. The female
piglets had a biallelic mutation of CD163, including a 1382 bp deletion with a
11 bp insertion on
one allele and a 1720 bp deletion of CD163 on the other allele. A summary of
the CD163
modifications and the predicted translations can be found in Table 10. A
summary of the CD1D
modifications and predicted translations by CRISPR modification can be found
in Table 11.
Briefly, one female and two male litters were born, resulting in 13 piglets.
One piglet died
immediately afterbirth. Twelve of the 13 piglets contained either a biallelic
or homozygous
deletion of CD1D (Fig. 3, panel F). One piglet was WT.
Table 8. Embryo Transfer data for CD 163.
# Embryos Oocyte Day of
0
t..)
Pig ID Line* Gender Transferred Source t
Estrus Piglet Result =
,-,
0047 CD163 CRISPR NT Male 240 ART 2
4 live piglets (2 euthanized after birth)
o
0015 CD163 CRISPR NT Male 267 ART 1
3 live piglets (all healthy) c,.)
oe
o
7 live piglets (1 born dead, 1 euthanized
0044 CD163 CRISPR NT Male 206 ART 1
after birth)
0053 CD163 CRISPR NT Male 224 ART 2
1 male piglet (euthanized at day 13)
008 CD 163 CRISPR NT Male 226 ART 1
0 piglets
0094 CD163 CRISPR NT Female 193 MU 2
8 live piglets (1 euthanized due to FTT)
9 live piglets (2 euthanized at day 0, 2 due
0086 CD 163 CRISPR NT Female 213 MU 1
to FTT)
CRISPR Injected CD163
P
0082 10/131 Male/Female 50 Blast MU 5
0 piglets .
CRISPR Injected CD163
.
00
.
0083 10/131 Male 46 Blast MU 5
4 live piglets
099 CD163 CRISPR NT-no Neo Male 156 ART 1
1 live piglet, 1 dead piglet 2
o
,
,
0128 CD163 CRISPR NT-no Neo Male 196 ART 2
0 piglets .
0100 CD163 CRISPR NT-no Neo Male 261 MU 3
0 piglets
0134 CD 163 CRISPR NT-no Neo Male/Female 181 MU 1
0 piglets
200889 CD 163 CRISPR NT-no Neo Female 202 ART 1
4 live piglets
0135 CD163 CRISPR NT-no Neo Female 169 ART 2
0 piglets
*The CD163 CRISPR NT line represents embryos created by NT with a fetal
fibroblast line modified by transfection. CRISPR injected embryos were
IVF embryos injected at the 1 cell stage with CD163 guide RNA with CAS9 RNA.
CD163 CRISPR NT-no Neo fetal line represents embryos created 1-d
by NT with a previously modified fetal fibroblast that was already Neo
resistant line modified by transfection without the use of a selectable
marker. n
1-i
1- MU refers to gilt oocytes that were aspirated and matured at the University
of Missouri as described in the IVF se4ction of the Materials and
cp
Methods. ART refers to sow oocytes that were purchased and matured as
described in the SCNT section of the Materials and Methods. t..)
o
,-,
cio
O-
t..)
-.1
.6.
.6.
Table 9. Embryo transfer data for CD1D.
0
# Embryos Oocyte Day of
Pig ID Line* Gender Transferred Sourcet Estrus
Result
200888 CD1D CRISPR NT Male 201 ART 2
7 live piglets
061 CD1D CRISPR NT Male 239 ART 0
4 live piglets cio
0164 CD1D CRISPR NT Female 199 MU 2
0 piglets
0156 CD1D CRISPR NT Female 204 MU 2
0 piglets
CD1D Injected
0165 4800/5350 Male/Female 55 Blast MU 6
4 piglets (1 female, 3 male)
CD1D Injected
0127 4800/5350 Male/Female 55 Blast MU 6
0 piglets
0121 CD1D CRISPR NT Female 212 ART 1
2 live piglets
* CD1D CRISPR NT line represents embryos created by NT with a fetal fibroblast
line modified by transfection. CRISPR injected embryos
were IVF embryos injected at the 1 cell stage with CD1D guide RNA with CAS9
RNA.
t MU refers to gilt oocytes that were aspirated and matured at the University
of Missouri as described in the IVF se4ction of the Materials and
Methods. ART refers to sow oocytes that were purchased and matured as
described in the SCNT section of the Materials and Methods.
1-d
cio
Table 10. Genotype and Translational Prediction for CD163 modified pigs. Some
pigs contain a biallelic type of modification, but only have
0
one allele described and another modified allele that was not amplified by
PCR.
,--
8 (5
=C-
:)-
*
.,.') 5 0
0 0
u 0 Z Z o
sm o
oe
;-, o
. g d sm 0 a ' (21 rn
u
.1 Z 'PI )4 g E ' .CA 4 r`i
4.-' ,t 4:' 8 'c:i 4 w
(z)
63 & 7 NHEJ biallelic 1506 bp deletion 30 bp
deletion in exon 7 KO or CD163 A422- No Deletion from
nt 1,525 to nt 98
64 527
3,030
Other allele Uncharacterized, unamplifiable
65 3 NHEJ Biallelic 7 bp insertion Insertion
into exon 7 KO Yes (491) Insertion between nt 3,148 &
99
3,149 a
65 2 NHEJ Biallelic 503 bp deletion Partial
deletion of exon 7 and 8 KO Yes (491) ** **
Other allele Uncharacterized
P
65 2 NHEJ Biallelic 1280 bp deletion
Complete deletion of exons 7 and 8 CD 163 A422-6" No Deletion from nt
2,818 to nt 100 0
µ,.
4,097
e,
e,
N,
IV
1373 bp deletion Complete deletion of exons 7 and
8 A422-631 No Deletion from nt 2,724 to nt 101
CD163
--A 1,;
4,096
e,
**
**
66 1 NHEJ Homozygous 2015 bp insertion
Insertion of targeting vector
,
1-
e,
backbone into exon 7
'
e,
67-1 1 NHEJ Biallelic 11 bp deletion Deletion
in exon 7 KO Yes (485) Deletion from nt 3,137 to nt
102 N,
3,147
2 bp insertion, 377 bp Insertion in exon 7
2 bp insertion between nt 3,149 103
deletion in intron 6
& nt 3,150b with a 377 bp
deletion from nt 2,573 to nt
2,949
67-2 1 NHEJ Biallelic 124 bp deletion Deletion
in exon 7 KO Yes (464) Deletion from nt 3,024 to nt
104
3,147
,-i
A429-470
123 bp deletion Deletion in exon 7 CD 163
No Deletion from nt 3,024 to nt 105
3,146
ci)
n.)
67-3 1 NHEJ Biallelic 1 bp insertion Insertion
into exon 7 KO Yes (489) Insertion between nt 3,147 &
106 o
1--,
3,148'
oe
-C;
n.)
--.1
Other allele Uncharacterized, unamplifiable
o
.6.
67-4 1 NHEJ Biallelic 130 bp deletion Deletion
in exon 7 KO Yes (462) Deletion from nt 3,030 to
nt 107 .6.
3,159
A430-474
132 bp deletion Deletion in exon 7 CD 163
No Deletion from nt 3,030 to nt 108 0
3,161
n.)
o
1¨,
68 & 6 NHEJ Biallelic 1467 bp deletion
Complete deletion of exons 7 and 8 CD 163A422-631 No Deletion from nt
2,431 to nt 109 ):1
o
69
3,897 c,.)
oe
o
--.1
Other allele Uncharacterized, unamplifiable
68 & 2 NHEJ Biallelic 129 bp deletion, 1930 bp Deletion in exon 7
CD 163A435-478 No Deletion from nt 488 to nt 110
69 intron 6 deletion
2,417 in exon 6, deleted
sequence is replaced with a 12
bp insertion' starting at nt 488,
& an additional 129 bp deletion
from nt 3,044 to nt 3,172
other allele Uncharacterized, unamplifiable
65 & 3 WT Wild type pigs created from a
SEQ ID NO: 47 47
69 mixed colony
Q
70 2 NHEJ On SIGLEC1-/- 28 bp deletion Deletion in exon 7 KO
Yes (528) Deletion from nt 3,145 to nt 111
µ,.
Biallelic
3,172 .
.
N)
N,
1387 bp deletion Partial deletion in exon 7 and
all of KO No Deletion from nt 3,145 to nt 112 IV
exon 8
4,531
"
I-I
0
I
73 4 NHEJ On SIGLEC1-/- 1382 bp deletion
Partial deletion in exon 7 and all of KO No Deletion from nt 3,113 to
nt 113 ,
,
Biallelic +11 bp insertion exon 8
4,494, deleted sequence .
N,
replaced with an 11 bp
insertione starting at nt 3,113
114
1720 bp deletion Complete deletion of exons 7 and
8 CD 163A422-631 Deletion from nt 2,440 to nt
4,160
*KO, knock-out
** Not included because piglets were euthanized.
t SEQ ID NOs. in this column refer to the SEQ ID NOs. for the sequences that
show the INDELs in relation to SEQ ID NO: 47. Iv
a The inserted sequence was TACTACT (SEQ ID NO: 115)
n
,-i
b The inserted sequence was AG.
c The inserted sequence was a single adenine (A) residue.
cp
N
d The inserted sequence was TGTGGAGAATTC (SEQ ID NO:116).
o
1¨,
oe
e The inserted sequence was AGCCAGCGTGC (SEQ ID NO: 117).
-1
n.)
--.1
.6.
.6.
Table 11. Genotype and Translational Prediction for CD1D modified pigs
0
Number
t..)
Repair
Protein =
Litter of Type Size of INDEL Description
,-,
,z
Mechanism
Translation
Piglets
o
(...)
158,
cio
o
11 NHEJ homozygous 1653 bp deletion Deletion of
exon 3,4 and 5 KO* -4
159
Deletion of exon 5 and 72 bp of exon
167 2 NHEJ homozygous
1265 bp deletion 6
KO
166-1 1 NHEJ biallelic 24 bp deletion Removal of
start codon in exon 3 KO
27 bp deletion Disruption
of start codon in exon 3
362 bp deletion + 5 bp Deletion of
exon 3
6 bp insertion + 2 bp Addition of
6 bp before start codon in
166-2 1 NHEJ biallelic
P
mismatch exon 3
CD1D"I+ o
.
Removal of start codon in exon 3 and deletion of exons
g
"0
1598 bp deletion 4,5
--.1 N)
"
t.)
Addition of G/T in exon 3 before
166-3 1 NHEJ biallelic
.
,
1 bp insertion start codon
in exon 3 CD1D+I+ '8
,
Addition of A in exon 3 before start
166-4 1 NHEJ homozygous 1 bp insertion
codon in exon 3
CD1D+I+
*KO, knock-out
Iv
n
,-i
cp
t..)
=
oe
7a3
t..)
-4
,z
.6.
.6.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
73
Efficiency of CRISPR/Cas9 System in Porcine Zygotes
[00261] Based on targeted disruption of CD163 and CD1D in somatic cells
using the CRISPR/Cas9 system, this approach was applied to porcine
embryogenesis.
First, the effectiveness of the CRISPR/Cas9 system in developing embryos was
tested.
CRISPR/Cas9 system targeting eGFP was introduced into zygotes fertilized with
semen
from a boar heterozygous for the eGFP transgene. After the injection,
subsequent
embryos expressing eGFP were monitored. Various concentrations of the
CRISPR/Cas9
system were tested and cytotoxicity of the delivered CRISPR/Cas9 system was
observed
(Fig. 4, panel A); embryo development after CRISPR/Cas9 injection was lower
compared to control. However, all the concentrations of CRISPR/Cas9 that were
examined were effective in generating modification of eGFP because no embryos
with
eGFP expression were found in the CRISPR/Cas9-injected group (Fig. 4, panel
B); of the
noninjected control embryos 67.7% were green, indicating expression of eGFP.
When
individual blastocysts were genotyped, it was possible to identify small
mutations near
the CRISPR binding sites (Fig. 4, panel C). Based on the toxicity and
effectiveness, 10
ng/p.1 of gRNA and Cas9 mRNA were used for the following experiments.
[00262] When CRISPR/Cas9 components designed to target CD163 were
introduced into presumptive zygotes, targeted editing of the genes in the
subsequent
blastocysts was observed. When individual blastocysts were genotyped for
mutation of
CD163, specific mutations were found in all the embryos (100% GE efficiency).
More
importantly, while embryos could be found with homozygous or biallelic
modifications
(8/18 and 3/18, respectively) (Fig. 5), mosaic (monoallelic modifications)
genotypes
were also detected (4/18 embryos). Some embryos (8/10) from the pool were
injected
with 2 ng/p,1 Cas9 and 10 ng/p,1 CRISPR and no difference was found in the
efficiency of
mutagenesis. Next, based on the in vitro results, two CRISPRs representing
different
gRNA were introduced to disrupt CD163 or CD1D during embryogenesis to induce a
specific deletion of the target genes. As a result, it was possible to
successfully induce a
designed deletion of CD163 and CD1D by introducing two guides. A designed
deletion
is defined as a deletion that removes the genomic sequence between the two
guides
introduced. Among the embryos that received two CRISPRs targeting CD163, all
but
one embryo resulted in a targeted modification of CD163. In addition, 5/13
embryos
were found to have a designed deletion on CD163 (Fig. 6, panel A) and 10/13
embryos
appeared to have modification of CD163 in either homozygous or biallelic
fashion.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
74
Targeting CD1D with two CRISPRs was also effective because all the embryos
(23/23)
showed a modification of CD1D. However, the designed deletion of CD1D could
only
be found in two embryos (2/23) (Fig. 6, panel B). Five of twenty-three embryos
possessing mosaic genotypes were also found, but the rest of embryos had
either
homozygous or biallelic modification of CD1D. Finally, whether multiple genes
can be
targeted by the CRISPR/Cas9 system within the same embryo was tested. For this
purpose, targeting both CD163 and eGFP was performed in the zygotes that were
fertilized with heterozygous eGFP semen. When blastocysts from the injected
embryos
were genotyped for CD163 and eGFP, it was found that found that CD163 and eGFP
were successfully targeted during embryogenesis. Sequencing results
demonstrated that
multiple genes can be targeted by introducing multiple CRISPRs with Cas9 (Fig.
6, panel
C).
Production of CD163 and CD1D Mutants from CRISPR/ Cas9-Injected Zygotes
[00263] Based on the success from the previous in vitro study, some
CRISPR/Cas9-injected zygotes were produced and 46-55 blastocysts were
transferred
per recipient (because this number has been shown to be effective in producing
pigs from
the in vitro derived embryos). Four embryo transfers were performed, two each
for
CD163 and CD1D, and a pregnancy for each modification was obtained. Four
healthy
piglets were produced carrying modifications on CD163 (Table 8). All the
piglets, litter
67 from recipient sow ID 0083 showed either homozygous or biallelic
modification of
CD163 (Fig. 7). Two piglets showed the designed deletion of CD163 by the two
CRISPRs delivered. All the piglets were healthy. For CD1D, one pregnancy also
produced four piglets (litter 166 from recipient sow identification no. 0165):
one female
and three males (Table 9). One piglet (166-1) did carry a mosaic mutation of
CD1D,
including a 362 bp deletion that completely removed exon 3 that contains the
start codon
(Fig. 8). One piglet contained a 6 bp insertion with a 2 bp mismatch on one
allele with a
large deletion on the other allele. Two additional piglets had a biallelic
single bp
insertion. There were no mosaic mutations detected for CD163.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
DISCUSSION
[00264] An increase in efficiency of GE pig production can have a wide
impact by providing more GE pigs for agriculture and biomedicine. The data
described
above show that by using the CRISPR/Cas9 system, GE pigs with specific
mutations can
be produced at a high efficiency. The CRISPR/Cas9 system was successfully
applied to
edit genes in both somatic cells and in preimplantation embryos.
[00265] When the CRISPR/Cas9 system was introduced into somatic cells, it
successfully induced targeted disruption of the target genes by NHEJ but did
not increase
the ability to target by HR. Targeting efficiency of individual CRISPR/Cas9 in
somatic
cells was variable, which indicated that the design of the guide can affect
the targeting
efficiency. Specifically, it was not possible to find targeted modification of
CD1D when
CRISPR 5350 and Cas9 were introduced into somatic cells. This suggests that it
could be
beneficial to design multiple gRNAs and validate their efficiencies prior to
producing
pigs. A reason for the lack of HR-directed repair with the presence of donor
DNA is still
unclear. After screening 886 colonies (both CD163 and CD1D) transfected with
CRISPR
and donor DNA, only one colony had evidence for a partial UR event. The
results
demonstrated that the CRISPR/Cas9 system worked with introduced donor DNA to
cause unexpected large deletions on the target genes but did not increase UR
efficiency
for these two particular targeting vectors. However, a specific mechanism for
the large
deletion observation is not known. Previous reports from our group suggested
that a
donor DNA can be effectively used with a ZFN to induce HR-directed repair.
Similarly,
an increase in the targeting efficiency was seen when donor DNA was used with
CRISPR/ Cas9 system, but complete UR directed repair was not observed. In a
previous
study using ZFN, it was observed that targeted modification can occur through
a
combination of HR and NHEJ because a partial recombination was found of the
introduced donor DNA after induced DSBs by the ZFN. One explanation might be
that
UR and NHEJ pathways are not independent but can act together to complete the
repair
process after DSBs induced by homing endonucleases. Higher concentrations of
CRISPRs might improve targeting efficiency in somatic cells although no
statistical
difference was found in these experimental results. This may suggest that
CRISPR is a
limiting factor in CRISPR/Cas9 system, but further validation is needed.
Targeted cells
were successfully used to produce GE pigs through SCNT, indicating the
application of
CRISPR/Cas9 does not affect the ability of the cells to be cloned. A few
piglets were
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
76
euthanized because of health issues; however, this is not uncommon in SCNT-
derived
piglets.
[00266] When the CRISPR/Cas9 system was introduced into developing
embryos by zygote injection, nearly 100% of embryos and pigs contained an
INDEL in
the targeted gene, demonstrating that the technology is very effective during
embryogenesis. The efficiency observed during this study surpasses frequencies
reported
in other studies utilizing homing endonucleases during embryogenesis. A
decrease in the
number of embryos reaching the blastocyst stage suggested that the
concentration of
CRISPR/Cas9 introduced in this study may be toxic to embryos. Further
optimization of
the delivery system may increase survivability of embryos and thus improve the
overall
efficiency of the process. The nearly 100% mutagenesis rate observed here was
different
from a previous report in CRISPR/Cas9-mediated knockout in pigs; however, the
difference in efficiency between the studies could be a combination of the
guide and
target that was selected. In the present study, lower concentrations of
CRISPR/Cas9 (10
ng/p1 each) were effective in generating mutations in developing embryos and
producing
GE pigs. The concentration is lower than previously reported in pig zygotes
(125 ng/p,1
of Cas9 and 12.5 ng/p1 of CRISPR). The lower concentration of CRISPR/Cas9
components could be beneficial to developing embryos because introducing
excess
amounts of nucleic acid into developing embryos can be toxic. Some mosaic
genotypes
were seen in CRISPR/Cas9-injected embryos from the in vitro assays; however,
only one
piglet produced through the approach had a mosaic genotype. Potentially, an
injection
with CRISPR/Cas9 components may be more effective than introduction of other
homing endonucleases because the mosaic genotype was considered to be a main
hurdle
of using the CRISPR/Cas9 system in zygotes. Another benefit of using the
CRISPR/Cas9
system demonstrated by the present results is that no CD163 knockout pigs
produced
from IVF- derived zygotes injected with CRISPR/Cas9 system were lost, whereas
a few
piglets resulting from SCNT were euthanized after a few days. This suggests
that the
technology could not only bypass the need of SCNT in generating knockout pigs
but
could also overcome the common health issues associated with SCNT. Now that
injection of CRISPR/Cas9 mRNA into zygotes has been optimized, future
experiments
will include coinjection of donor DNA as well.
[00267] The present study demonstrates that introducing two CRISPRs with
Cas9 in zygotes can induce chromosomal deletions in developing embryos and
produce
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
77
pigs with an intended deletion, that is, specific deletion between the two
CRISPR guides.
This designed deletion can be beneficial because it is possible to specify the
size of the
deletion rather than relying on random events caused by NHEJ. Specifically, if
there is
insertion/deletion of nucleotides in a multiple of three caused by a homing
endonuclease,
the mutation may rather result in a hypomorphic mutation because no frame
shift would
occur. However, by introducing two CRISPRs, it is possible to cause larger
deletions that
will have a higher chance of generating non-functional protein. Interestingly,
CD1D
CRISPRs were designed across a greater area in the genome than CD163; there
was a
124 bp distance between CD163 CRISPR 10 and 131 while there was a distance of
550
bp between CRISPR 4800 and 5350 for CD1D. The longer distance between CRISPRs
was not very effective in generating a deletion as shown in the study.
However, because
the present study included only limited number of observations and there is a
need to
consider the efficacy of individual CRISPRs, which is not addressed here,
further study
is need to verify the relationship between the distance between CRISPRs and
probability
of causing intended deletions.
[00268] The CRISPR/Cas9 system was also effective in targeting two genes
simultaneously within the same embryo with the only extra step being the
introduction of
one additional CRISPR with crRNA. This illustrates the ease of disrupting
multiples
genes compared to other homing endonucleases. These results suggest that this
technology may be used to target gene clusters or gene families that may have
a
compensatory effect, thus proving difficult to determine the role of
individual genes
unless all the genes are disrupted. The results demonstrate that CRISPR/Cas9
technology
can be applied in generating GE pigs by increasing the efficiency of gene
targeting in
somatic cells and by direct zygote injection.
Example 2: Increased resistance to PRRSV in swine having a modified
chromosomal sequence in a gene encoding a CD163 protein
[00269] Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) has
ravaged the swine industry over the last quarter of a century. Speculation
about the mode
of viral entry has included both SIGLEC1 and CD163. While knockout of SIGLEC1
did
not affect the response to a viral challenge, it is shown in the present
example that
CD163 null animals show no clinical signs of infection, lung pathology,
viremia or
antibody production that are all hallmarks of PRRSV infection. Not only has a
PRRSV
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
78
entry mediator been confirmed; but if similarly created animals were allowed
to enter the
food supply, then a strategy to prevent significant economic losses and animal
suffering
has been described.
MATERIALS AND METHODS
Genotyping
[00270] Genotyping was based on both DNA sequencing and mRNA
sequencing. The sire's genotype had an 11 bp deletion in one allele that when
translated
predicted 45 amino acids into domain 5, resulting in a premature stop codon at
amino
acid 64. In the other allele there was a 2 bp addition in exon 7 and 377 bp
deletion in
intron before exon 7, that when translated predicted the first 49 amino acids
of domain 5,
resulting in a premature stop code at amino acid 85. One sow had a 7 bp
addition in one
allele that when translated predicted the first 48 amino acids of domain 5,
resulting in a
premature stop codon at amino acid 70. The other allele was uncharacterized
(A), as
there was no band from exon 7 by either PCR or long range 6.3 kb PCR. The
other 3
sows were clones and had a 129 bp deletion in exon 7 that is predicted to
result in a
deletion of 43 amino acids from domain 5. The other allele was uncharacterized
(B).
Growth of PRRSV in culture and production of virus inoculum for the infection
of pigs
are covered under approved IBC application 973
[00271] A type strain of PRRSV, isolate NVSL 97-7895 (GenBank
AF325691 2001.-02-11), was grown as described in approved 113C protocol 973.
This
laboratory isolate has been used in experimental studies for about 20 years
(Ladinig et
al., 2015). A. second isolate was used for the 2' trial, K.S06-72109 as
described
previously (Prather et al., 2013).
Infection of pigs with .PRRSV
[00272] A standardized infection protocol for PRRSV was used for the
infection of pigs. Three week old piglets were inoculated with approximately
104
TOD50 of PRRS virus which was administered by intramuscular (TM) and
intranasal
(fN) routes. Pigs were monitored daily and those exhibiting symptoms of
illness are
treated according to the recommendations of the CMG veterinarians. Pigs that
show
severe distress and are in danger of succumbing -to infection are humanely
euthanized
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
79
and samples collected. Staff and veterinarians were blind to the genetic
status of the pigs
to eliminate bias in evaluation or treatment PRRSV is present in body fluids
during
infection; therefore, blood samples were collected and stored at ¨80 C until
measured to
determine the amount or degree of viremia in each pig At the end of the
experiment,
pigs were weighed and humanely euthanized, and tissues collected and fixed in
10%
buffered formalin, embedded in paraffin, and processed for histopathology by a
board-
certified pathologist.
Phenotype Scoring of the Challenged pigs
[00273] The phenotype of the pigs was blindly scored daily as follows: What
is the attitude of the pig? Attitude Score: 0: BAR, 1: QAR, 2: Slightly
depressed, 3:
Depressed, 4: Moribund. What is the body condition of the pig? Body Condition
Score:
1: Emaciated, 2: Thin, 3: Ideal, 4: Fat, 5: Overfat/Obese. What is the rectal
temperature
of the pig? Normal Body Temperature 101.6-103.6 F (Fever considered > 104 F).
Is
there any lameness (grade)? What limb? Evaluate limbs for joint swelling and
hoof
lesions (check bottom and sides of hoof). Lameness Score: 1: No lameness, 2:
Slightly
uneven when walking, appears stiff in some joints but no lameness, 3: Mild
lameness,
slight limp while walking, 4: Moderate lameness, obvious limp including toe
touching
lame, 5: Severe lameness, non-weight bearing on limb, needs encouragement to
stand/walk. Is there any respiratory difficulty (grade)? Is there open mouth
breathing? Is
there any nasal discharge (discharge color, discharge amount:
mild/moderate/severe)?
Have you noticed the animal coughing? Is there any ocular discharge?
Respiratory Score:
0: Normal, 1: mild dyspnea and/or tachypnea when stressed (when handled), 2:
mild
dyspnea and/or tachypnea when at rest, 3: moderate dyspnea and/or tachypnea
when
stressed (when handled), 4: moderate dyspnea and/or tachypnea when at rest, 5:
severe
dyspnea and/or tachypnea when stressed (when handled), 6: severe dyspnea
and/or
tachypnea when at rest. Is there evidence of diarrhea (grade) or vomiting? Is
there any
blood or mucus? Diarrhea Score: 0: no feces noted, 1: normal stool, 2: soft
stool but
formed (soft serve yogurt consistency, creates cow patty), 3: liquid diarrhea
of brown/tan
coloration with particulate fecal material, 4: liquid diarrhea of brown/tan
coloration
without particulate fecal material, 5: liquid diarrhea appearing similar to
water.
1002741 This scoring system was developed by Dr. Megan Niederwerder at
KSU and is based on the following publications (Halbur et al., 1995; Merck;
Miao et al.,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
2009; Patience and Thacker, 1989; Winckler and Willen, 2001). Scores and
temperatures
were analyzed by using ANOVA separated based on genotypes as treatments.
Measurement qf PRRSV viremia
[002751 Viremia was determined via two approaches. Virus titration was
performed by adding serial 1:10 dilutions of serum to confluent MARC-145 cells
in a 96
well-plate. Serum was diluted in Eagle's minimum essential medium supplemented
with
8% fetal bovine serum, penicillin; streptomycin, and amphotericin B as
previously
described (Prather et at, 2013). The cells were examined after 4 days of
incubation for
the presence of a cytopathic effect by using microscope. The highest dilution
showing a
cytopathic effect was scored as the titration endpoint. Total RNA was isolated
from
serum by using the Life Technologies MagMAX-96 viral RNA isolation kit for
measuring viral nucleic acid. The reverse transcription polymerase chain
reaction was
performed by using the EL-PR:RSV MPX 4.0 kit from 717etracore on a CFX-96 real-
time
PCR system (Bio-Rad) according to the manufacturer's instructions. Each
reaction (25
pi) contained RNA from 5.8 il of serum. The standard curve was constructed by
preparing serial dilutions of an RNA control supplied in the kit (Tetracore).
The number
of templates per PCR are reported.
SIGLEC7.1 and CL)/63 staining of PAM cells
[002761 Porcine alveolar macrophages (PAMs) were collected by excising the
lungs and filling them with ¨100 ml cold phosphate buffered saline. After
recovering the
phosphate buffered saline wash cells were pelleted and resuspended in 5 ml
cold
phosphate buffered saline and stored on ice. Approximately 107 PAMs were
incubated in
5 ml of the various antibodies (anti-porcine CI) 169 (clone 3B I ill 1; AbD
Serotec); anti
porcine CD163 (clone 2A10/11; AbD Serotec)) diluted in phosphate buffered
saline with
5% fetal bovine serum and 0,1% sodium azide for 30 minutes on ice. Cells were
washed
and resuspended in 1/100 dilution of fluorescein isothiocyanate (FITC)-
conjugated to
goat anti-rnouse IgG (life Technologies) diluted in staining buffer and
incubated for 30
minutes on ice. At least 104 cells were analyzed by using a FACSCalibur flow
cytometer
and Cell Quest software (Becton Dickinson).
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
81
Measurement of PRRSV-spectfic 1g
[00277] To measure PRRSV-specific 1g recombinant PRRSV N protein was
expressed in bacteria (Trible et al., 2012) and conjugated to magnetic Luminex
beads by
using a kit (Luminex Corporation). The N protein-coupled beads were diluted in
phosphate buffered saline containing 10% goat serum to 2,500 beads/50 ul and
placed
into the wells of a 96-well round-bottomed polystyrene plate. Serum was
diluted 1:400 in
phosphate buffered saline containing 10% goat serum and 50 pl was added in
duplicate
wells and incubated for 30 minutes with gentle shaking at room temperature.
Next the
plate was washed (3X) with phosphate buffered saline containing 10% goat serum
and
50 pl of biotin-SP-conjugated affinity-purified goat anti-swine secondary
antibody (IgG,
Jackson ImmunoResearch) or biotin-labeled affinity purified goat anti-swine
IgM (KPL)
diluted to 2 p.g/rril in phosphate buffered saline containing 10% goat serum
was added.
The plates were washed (3X) after 30 minutes of incubation and then 50 pi of
streptavidin-conjugated phycoerythrin (2 pginal (Moss. Inc.) in phosphate
buffered saline
containing 10% goat serum) was added. The plates were washed 30 minutes later
and
microspheres were resuspended in 100 pl of phosphate buffered saline
containing 10%
goat serum an analyzed by using the MAGPIX and the Luminex xPONENT 4.2
software. Mean fluorescence intensity (MF1) is reported.
RESULTS
[00278] Mutations in CD 163 were created by using the CRISPR/Cas9
technology as described above in Example 1. Several founder animals were
produced
from zygote injection and from somatic cell nuclear transfer. Some of these
founders
were mated creating offspring to study. A single founder male was mated to
females with
two genotypes. The founder male (67-1) possessed an 11 bp deletion in exon 7
on one
allele and a 2 bp addition in exon 7 (and 377 bp deletion in the preceding
intron) of the
other allele and was predicted to be a null animal (CD 163-1). One founder
female (65-1)
had a 7 bp addition in exon 7 in one allele and an uncharacterized
corresponding allele
and was thus predicted to be heterozygous for the knockout (CD/63-/?). A
second
founder female genotype (3 animals that were clones) contained an as yet
uncharacterized allele and an allele with a 129 bp deletion in exon 7. This
deletion is
predicted to result in a deletion of 43 amino acids in domain 5. Matings
between these
animals resulted in all piglets inheriting a null allele from the boar and
either the 43
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
82
amino acid deletion or one of the uncharacterized alleles from the sows. In
addition to
the wild type piglets that served as positive controls for the viral
challenge, this produced
4 additional genotypes (Table 12).
Table 12. Genotypes tested for resistance to PRRSV challenge (NVSL and KS06
strains)
Alleles Resistance to PRRSV Challenge as Measured by
Viremia
Paternal Maternal NVSL KS06
Null Null Resistant N/A
Null A43 Amino Acids N/A Resistant
Null Uncharacterized A Susceptible N/A
Null Uncharacterized B Susceptible Susceptible
Wild Type Wild Type Susceptible Susceptible
[00279] At weaning, gene edited piglets and wild type age-matched piglets
were transported to Kansas State University for a PRRSV challenge. A PRRSV
challenge was conducted as previously described (Prather et al., 2013).
Piglets, at three
weeks of age, were brought into the challenge facility and maintained as a
single group.
All experiments were initiated after approval of institutional animal use and
biosafety
committees. After acclimation, the pigs were challenged with a PRRSV isolate,
NVSL
97-7895 (Ladinig et al., 2015), propagated on MARC-145 cells (Kim et al.,
1993). Pigs
were challenged with approximately 105 TCID50 of virus. One-half of the
inoculum was
delivered intramuscularly and the remaining delivered intranasally. All
infected pigs
were maintained as a single group, which allowed the continuous exposure of
virus from
infected pen mates. Blood samples were collected at various days up to 35 days
after
infection and at termination, day 35. Pigs were necropsied and tissues fixed
in 10%
buffered formalin, embedded in paraffin and processed for histopathology.
PRRSV
associated clinical signs recorded during the course of the infection included
respiratory
distress, inappetence, lethargy and fever. The results for clinical signs over
the study
period are summarized in Fig 9. As expected, the wild-type Wild Type
(CD163+I+) pigs
showed early signs of PRRSV infection, which peaked at between days 5 and 14
and
persisted in the group during the remainder of the study. The percentage of
febrile pigs
peaked on about day 10. In contrast, Null (CD 163-1-) piglets showed no
evidence of
clinical signs over the entire study period. The respiratory signs during
acute PRRSV
infection are reflected in significant histopathological changes in the lung
(Table 9). The
infection of the wild type pigs showed hi stopathology consistent with PRRS
including
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
83
interstitial edema with the infiltration of mononuclear cells (Fig. 10). In
contrast there
was no evidence for pulmonary changes in the Null (CD163-I-) pigs. The sample
size for
the various genotypes is small; nevertheless the mean scores were 3.85 (n=7)
for the wild
type, 1.75 (n=4) for the uncharacterized A, 1.33 (n=3) for the uncharacterized
B, and 0
(n=3) and for the null (CD163-/-).
Table 13. Microscopic Lung evaluation
Pig Genotype Description Score
41 Wild Type 100% congestion. Multifocal areas of edema. Infiltration
3
of moderate numbers of lymphocytes and macrophages
42 Wild Type 100% congestion. Multifocal areas of edema. Infiltration
3
of moderate numbers of lymphocytes and macrophages
47 Wild Type 75% multifocal infiltration with mononuclear cells and
2
mild edema
50 Wild Type 75% multifocal infiltration of mononuclear cells within
3
alveolar spaces and around small blood vessels
perivascular edema
51 Wild Type 25% atelectasis with moderate infiltration of 1
mononuclear cells
52 Wild Type 10% of alveolar spaces collapsed with infiltration of
1
small numbers of mononuclear cells
56 Wild Type 100% diffuse moderate interstitial infiltration of 4
mononuclear cells. Interalveolar septae moderately
thickened by hemorrhage and edema.
45 Uncharacterized A 75% multifocal infiltrates of mononuclear cells, .. 3
especially around bronchi, blood vessels, subpleural
spaces, and interalveolar septae.
49 Uncharacterized A 75% multifocal moderate to large infiltration of .. 2
mononuclear cells. Some vessels with mild edema.
53 Uncharacterized A 10% multifocal small infiltration of mononuclear cells
1
57 Uncharacterized A 15% infiltration of mononuclear cells 1
46 Uncharacterized B Moderate interstitial pneumonia 2
48 Uncharacterized B Perivascular edema and infiltration of mononuclear
cells 2
around small and medium sized vessels and around
interalveolar septae
54 Uncharacterized B No changes 0
40 Null No changes 0
43 Null No changes 0
55 Null No changes 0
[00280] Peak clinical signs correlated with the levels of PRRSV in the blood.
The measurement of viral nucleic acid was performed by isolation of total RNA
from
serum followed by amplification of PRRSV RNA by using a commercial reverse
transcriptase real-time PRRSV PCR test (Tetracore, Rockville, MD). A standard
curve
was generated by preparing serial dilutions of a PRRSV RNA control, supplied
in the
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
84
RT-PCR kit and results were standardized as the number templates per 50 11.1
PCR
reaction. The PRRSV isolate followed the course for PRRSV viremia in the wild
type
CD163+I+ pigs (Fig. 11). Viremia was apparent at day four, reached a peak at
day 11
and declined until the end of the study. In contrast viral RNA was not
detected in the
CD163-1- pigs at any time point during the study period. Consistent with the
viremia,
antibody production by the null and uncharacterized allele pigs was detectable
by 14 and
increased to day 28. There was no antibody production in the null animals
(Fig. 12).
Together, these data show that wild type pigs support PRRSV replication with
the
production of clinical signs consistent with PRRS. In contrast, the knockout
pigs
produced no viremia and no clinical signs, even though pigs were inoculated
and
constantly exposed to infected pen mates.
[00281] At the end of the study, porcine alveolar macrophages were removed
by lung lavage and stained for surface expression of SIGLEC1 (CD169, clone
3B11/11)
and CD163 (clone 2A10/11), as described previously (Prather et al., 2013).
Relatively
high levels of CD163 expression were detected on CD163+I+ wild type animals
(Fig.
13). In contrast, CD 163-1- pigs showed only background levels of anti-CD163
staining,
thus confirming the knockout phenotype. Expression levels for another
macrophage
marker CD169 were similar for both wild type and knockout pigs (Fig. 14).
Other
macrophage surface markers, including MHC II and CD172 were the same for both
genotypes (data not shown).
[00282] While the sample size was small the wild type pigs tended to gain less
weight over the course of the experiment (average daily gain 0.81 kg 0.33,
n=7) versus
the pigs of the other three genotypes (uncharacterized A 1.32 kg 0.17, n=4;
uncharacterized B 1.20 kg 0.16, n=3; null 1.21 kg 0.16, n=3).
[00283] In a second trial 6 wild type, 6 A43 amino acids, and 6 pigs with an
uncharacterized allele (B) were challenged as described above, except KS06-
72109 was
used to inoculate the piglets. Similar to the NVSL data the wild type and
uncharacterized
B piglets developed viremia. However, in the A43 amino acid pigs the K506 did
not
result in viremia (Fig. 15; Table 7).
IMPLICATIONS AND CONCLUSION
[00284] The most clinically relevant disease to the swine industry is PRRS.
While vaccination programs have been successful to prevent or ameliorate most
swine
CA 03096022 2020-10-02
WO 2019/203807
PCT/US2018/027944
pathogens, the PRRSV has proven to be more of a challenge. Here CD163 is
identified
as an entry mediator for this viral strain. The founder boar was created by
injection of
CRISPR/Cas9 into zygotes (Whitworth et al., 2014) and thus there is no
transgene.
Additionally one of the alleles from the sow (also created by using
CRISPR/Cas9) does
not contain a transgene. Thus piglet #40 carries a 7 bp addition in one allele
and a 11 bp
deletion in the other allele, but no transgene. These virus-resistance alleles
of CD163
represent minor genome edits considering that the swine genome is about 2.8
billion bp
(Groenen et al., 2012). If similarly created animals were introduced into the
food supply,
significant economic losses could be prevented.
Example 3: Increased resistance to genotype 1 porcine reproductive and PRRS
viruses in swine with CD163 SRCR domain 5 replaced with human CD163-like
homology SRCR domain 8
[00285] CD163 is considered the principal receptor for porcine reproductive
and respiratory syndrome virus (PRRSV). In this study, pigs were genetically
edited
(GE) to possess one of the following genotypes: complete knock out (KO) of
CD163,
deletions within CD163 scavenger receptor cysteine-rich (SRCR) domain 5, or
replacement (domain swap) of SRCR domain 5 with a synthesized exon encoding a
homolog of human CD163-like (hCD163L1) SRCR 8 domain. Immunophenotyping of
porcine alveolar macrophages (PAMs) showed that pigs with the KO or SRCR
domain 5
deletions did not express CD163 and PAMs did not support PRRSV infection. PAMs
from pigs that possessed the hCD163L1 domain 8 homolog expressed CD163 and
supported the replication of Type 2, but not Type 1 genotype viruses.
Infection of
CD163-modified pigs with representative Type 1 and Type 2 viruses produced
similar
results. Even though Type 1 and Type 2 viruses are considered genetically and
phenotypically similar at several levels, including the requirement of CD163
as a
receptor, the results demonstrate a distinct difference between PRRSV
genotypes in the
recognition of the CD163 molecule.
MATERIALS AND METHODS
Genomic modifications of the porcine CD163 gene
[00286] Experiments involving animals and viruses were performed in
accordance with the Federation of Animal Science Societies Guide for the Care
and Use
of Agricultural Animals in Research and Teaching, the USDA Animal Welfare Act
and
Animal Welfare Regulations, and were approved by the Kansas State University
and
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
86
University of Missouri Institutional Animal Care and Use Committees and
Institutional
Biosafety Committees. Mutations in CD163 used in this study were created using
the
CRISPR/Cas9 technology as described hereinabove in the preceding examples. The
mutations are diagrammed in Fig. 17. The diagrammed genomic region shown in
Fig. 17
covers the sequence from intron 6 to intron 8 of the porcine CD163 gene. The
introns
and exons diagrammed in Fig. 17 are not drawn to scale. The predicted protein
product is
illustrated to the right of each genomic structure. Relative macrophage
expression, as
measured by the level of surface CD163 on PAMs, is shown on the far right of
Fig. 17.
The black regions indicate introns; the white regions indicate exons; the
hatched region
indicates hCD163L1 exon 11 mimic, the homolog of porcine exon 7; and the gray
region
indicates a synthesized intron with the PGK Neo construct as shown in Fig. 17.
[00287] The CD163 gene construct KO-d7(11) shown in Fig. 17 possesses
an
11 base pair deletion in exon 7 from nucleotide 3,137 to nucleotide 3,147. The
CD163
gene construct KO-17(2), possesses a 2 base pair insertion in exon 7 between
nucleotides
3,149 and 3,150 as well as a 377 base pair deletion in the intron upstream of
exon 7,
from nucleotide 2,573 to nucleotide 2,949. These edits are predicted to cause
frameshift
mutations and premature stop codons, resulting in only partial translation of
SRCR 5 and
the KO phenotype. Three other mutations produced deletions in exon 7. The
first,
d7(129), has a 129 base pair deletion in exon 7 from nucleotide 3,044 to
nucleotide
3,172. The d7(129) construct also has a deletion from nucleotide 488 to
nucleotide 2,417
in exon 6, wherein the deleted sequence is replaced with a 12 bp insertion.
The other two
deletion constructs, d7(1467) and d7(1280), have complete deletions of exons 7
and 8 as
illustrated in Fig. 17. d7(1467) has a 1467 base pair deletion from nucleotide
2,431 to
nucleotide 3,897, and d7(1280) has a 1280 base pair deletion from nucleotide
2,818 to
nucleotide 4,097. For these deletion constructs the other CD163 exons remained
intact.
[00288] The last construct shown in Fig. 17, HL11m, was produced using a
targeting event that deleted exon 7 and replaced it with a synthesized exon
that encoded a
homolog of SRCR 8 of the human CD163-like 1 protein (hCD163L1 domain 8 is
encoded by hCD163L1 exon 11). The SRCR 8 peptide sequence was created by
making
33 nucleotide changes in the porcine exon 7 sequence. A neomycin cassette was
included
in the synthesized exon to enable screening for the modification. SEQ ID NO:
118
provides the nucleotide sequence for the HL 11m construct in the region
corresponding to
the same region in reference sequence SEQ ID NO: 47.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
87
[00289] A diagram of the porcine CD163 protein and gene is provided Fig. 18.
The CD163 protein SCRC (ovals) and PST (squares) domains along with the
corresponding gene exons are shown in panel A of Fig. 18. A peptide sequence
comparison for porcine CD163 SRCR 5 (SEQ ID NO: 120) and human CD163 SRCR 8
homolog (SEQ ID NO: 121) is shown in panel B of Fig. 18. The figure is based
on
GenBank accession numbers AJ311716 (pig CD163) and GQ397482 (hCD163-L1).
Viruses
[00290] The panel of viruses used in this example is listed in Table
14. Isolates
were propagated and titrated on MARC-145 cells (Kim et al., 1993). For
titration, each
virus was serially diluted 1:10 in MEM supplemented with 7% FBS, Pen-Strep (80
Units/ml and 801.tg/ml, respectively), 3 1.tg/m1FUNGIZONE (amphotericin B),
and 25
mM HEPES. Diluted samples were added in quadruplicate to confluent MARC-145
cells
in a 96 well plate to a final volume of 200 pi per well and incubated for four
days at
37 C in 5% CO2. The titration endpoint was identified as the last well with a
cytopathic
effect (CPE). The 50% tissue culture infectious dose (TCID50/m1) was
calculated using a
method as previously described (Reed and Muench 1938).
Table 14. PRRSV isolates.
Year GenBank
Virus Genotype
Isolated No.
NVSL 97-7895 2 1997 AY545985
K506-72109 2 2006 KM252867
P129 2 1995 AF494042
VR2332 2 1992 AY150564
C090 2 2010 KM035799
AZ25 2 2010 KM035800
MLV-ResPRRS 2 NA* AF066183
K562-06274 2 2006 KM035798
K5483 (5D23983) 2 1992 JX258843
C084 2 2010 KM035802
5D13-15 1 2013 NA
Lelystad 1 1991 M96262
03-1059 1 2003 NA
03-1060 1 2003 NA
SD01-08 1 2001 DQ489311
4353PZ 1 2003 NA
*NA, Not available
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
88
Infection of alveolar macrophages
[00291] The preparation and infection of macrophages were performed as
previously described (Gaudreault, et al., 2009 and Patton, et al., 2008).
Lungs were
removed from euthanized pigs and lavaged by pouring 100 ml of cold phosphate
buffered saline (PBS) into the trachea. The tracheas were clamped and the
lungs gently
massaged. The alveolar contents were poured into 50 ml centrifuge tubes and
stored on
ice. Porcine alveolar macrophages (PAMs) were sedimented by centrifugation at
1200 x g for 10 minutes at 4 C. The pellets were re-suspended and washed once
in cold
sterile PBS. The cell pellets were re-suspended in freezing medium containing
45%
RPMI 1640, 45% fetal bovine serum (FBS), and 10% dimethylsulfoxide (DMSO) and
stored in liquid nitrogen until use. Frozen cells were thawed on ice, counted
and adjusted
to 5x105 cells/ml in media (RPMI 1640 supplemented with 10% FBS, PenStrep, and
FUNGIZONE; RPMI-FBS). Approximately 103 PAMs per well were added to 96 well
plates and incubated overnight at 37 C in 5% CO2. The cells were gently washed
to
remove non-adherent cells. Serial 1:10 dilutions of virus were added to
triplicate wells.
After incubation overnight, the cells were washed with PBS and fixed for 10
minutes
with 80% acetone. After drying, wells were stained with PRRSV N-protein
specific
SDOW-17 mAb (Rural Technologies Inc.) diluted 1:1000 in PBS with 1% fish
gelatin
(PBS-FG; Sigma Aldrich). After a 30 minute incubation at 37 C, the cells were
washed
with PBS and stained with ALEXAFLUOR 488-labeled anti-mouse IgG (Thermofisher
Scientific) diluted 1:200 in PBS-FG. Plates were incubated for 30 minutes in
the dark at
37 C, washed with PBS, and viewed under a fluorescence microscope. The 50%
tissue
culture infectious dose (TCID50)/m1 was calculated according to a method as
previously
described (Reed and Muench 1938).
Measurement of CD 169 and CD163 surface expression on PAMs
[00292] Staining for surface expression of CD169 and CD163 was performed
as described previously (Prather et al., 2013). Approximately 1X106 PAMs were
placed
in 12 mm x 75 mm polystyrene flow cytometry (FACS) tubes and incubated for 15
minutes at room temp in 1 ml of PBS with10% normal mouse serum to block Fc
receptors. Cells were pelleted by centrifugation and re-suspended in 5 Ill of
FITC-
conjugated mouse anti-porcine CD169 mAb (clone 3B11/11; AbD Serotec) and 5 Ill
of
PE-conjugated mouse anti-porcine CD163 mAb (Clone: 2A10/11, AbD Serotec).
After
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
89
30 minutes incubation the cells were washed twice with PBS containing 1%
bovine
serum albumin (BSA Fraction V; Hyclone) and immediately analyzed on a BD LSR
Fortessa flow cytometer (BD Biosciences) with FCS Express 5 software (De Novo
Software). A minimum of 10,000 cells were analyzed for each sample.
Measurement of PRRS viremia
[00293] RNA was isolated from 50 IA of serum using Ambion's MagMAX 96
Viral Isolation Kit (Applied Biosystems) according to the manufacturer's
instructions.
PRRSV RNA was quantified using EZ-PRRSV MPX 4.0 Real Time RT-PCR Target-
Specific Reagents (Tetracore) performed according to the manufacturer's
instructions.
Each plate contained Tetracore Quantification Standards and Control Sets
designed for
use with the RT-PCR reagents. PCR was carried out on a CFX96 Touch Real-Time
PCR
Detection System (Bio-Rad) in a 96-well format using the recommended cycling
parameters. The PCR assay results were reported as logio PRRSV RNA copy number
per
50 IA reaction volume, which approximates the number of copies per ml of
serum. The
area under the curve (AUC) for viremia over time was calculated using GraphPad
Prism version 6.00 for Windows.
Measurement of PRRSV antibody
[00294] The microsphere fluorescent immunoassay (FMIA) for the detection
of antibodies against the PRRSV nucleocapsid (N) protein was performed as
described
previously (Stephenson et al., 2015). Recombinant PRRSV N protein was coupled
to
carboxylated Luminex MAGPLEX polystyrene microsphere beads according to the
manufacturer's directions. For FMIA, approximately 2500 antigen-coated beads,
suspended in 50 [iL PBS with 10% goat serum (PBS-GS), were placed in each well
of a
96-well polystyrene round bottom plate. Sera were diluted 1:400 in PBS-GS and
50 IA
added to each well. The plate was wrapped in foil and incubated for 30 minutes
at room
temperature with gentle shaking. The plate was placed on a magnet and beads
were
washed three times with 19011.1 of PBS-GS. For the detection of IgG, 50 11.1
of biotin-SP-
conjugated affinity purified goat anti-swine secondary antibody (IgG, Jackson
ImmunoResearch) was diluted to 2 l.g/m1 in PBS-GS and 100 IA added to each
well. The
plate was incubated at room temperature for 30 minutes and washed three times
followed
by the addition of 5011.1 of streptavidin-conju,gated phycoerythrin (2 gg/m1
in PBS-GS;
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
SAPE). After 30 minutes, the microspheres were washed, resuspended in 100 .1
of PBS-
GS, and analyzed using a MAGPIX instrument (LUMINEX) and LUMINEX xPONENT
4.2 software. The mean fluorescence intensity (MFI) was calculated on a
minimum of
100 microsphere beads.
Measurement of haptoglobin (HP)
[00295] The amount of Hp in serum was measured using a porcine-specific Hp
ELISA kit (Genway Biotech Inc.) and steps performed according to the
manufacturer's
instructions. Serum samples were diluted 1:10,000 in lx diluent solution and
pipetted in
duplicate on a pre-coated anti-pig Hp 96 well ELISA plate, incubated at room
temperature for 15 minutes, then washed three times. Anti-Hp-horseradish
peroxidase
(HRP) conjugate was added to each well and incubated in the dark at room
temperature
for 15 minutes. The plate was washed and 100 11.1 chromogen-substrate solution
added to
each well. After incubating in the dark for 10 minutes, 100 .1 of stop
solution was added
to each well. The plate was read at 450 nm on a Fluostar Omega filter-based
microplate
reader (BMG Labtech).
RESULTS
Phenotypic properties of PAMs from CD] 63-modified pigs
[00296] The forward and side scatter properties of cells in the lung lavage
material were used to gate on the mononuclear subpopulation of cells.
Representative
CD169 and CD163 staining results for the different chromosomal modifications
shown
in Fig. 17 are presented in Fig. 19. In the representative example presented
in panel A of
Fig. 19, greater than 91% of PAMs from the WT pigs were positive for both
CD169 and
CD163. Results for 12 WT pigs used in this study showed a mean of 85 +/-8% of
double-positive cells. As shown in panel B of Fig. 19, PAMs from the CD163 KO
pigs
showed no evidence of CD163, but retained normal surface levels of CD169.
Although it
was predicted that the CD163 polypeptides derived from the d7(1467) and
d7(1280)
deletion genotypes should produce modified CD163 polypeptides anchored to the
PAM
surface, immunostaining results showed no surface expression of CD163 (see
Fig. 19,
panel D). Since MAb 2A10 recognizes an epitope located in the first three SRCR
domains, the absence of detection was not the result of the deletion of an
immunoreactive epitope. The d7(129) genotype was predicted to possess a 43
amino acid
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
91
deletion in SRCR 5 (see Fig. 17). In the example presented in panel C of Fig.
19, only
2.4% of cells fell in the double-positive quadrant. The analysis of PAMs from
nine
d7(129) pigs used in this study showed percentages of double-positive cells
ranging from
0% to 3.6% (mean = 0.9%). The surface expression of CD169 remained similar to
WT
PAMs. For the purpose of this study, pigs possessing the KO, d7(1467),
d7(1280), and
d7(129) genotypes were all categorized as possessing a CD163-null phenotype.
[00297] The CD163 modification containing the hCD163L1 domain 8 peptide
sequence HL11m, showed dual expression of CD163+ and CD169+ on PAMs (panel E
of
Fig. 19). However, in all of the HL11m pigs analyzed in this study, the
surface
expression of CD163 was markedly reduced compared to the WT PAMs. The levels
of
CD163 fell on a continuum of expression, ranging from no detectable CD163 to
pigs
possessing moderate levels of CD163. In the example shown in panel E of Fig.
19,
approximately 60% of cells were in the double-positive quadrant while 40% of
cells
stained for only CD169. The analysis of PAMs from a total 24 HL11m pigs showed
38
+/¨ 12% of PAM cells were positive only for CD169 and 54+/-14% were double-
positive (CD169+CD163+).
Circulating haptoglobin levels in WT and CD 163-modified pigs
[00298] As a scavenging molecule, CD163 is responsible for removing HbHp
complexes from the blood (Fabriek, et al., 2005; Kristiansen et al., 2001; and
Madsen et
al., 2004). The level of Hp in serum provides a convenient method for
determining the
overall functional properties of CD163-expressing macrophages. Hp levels in
sera from
WT, HL11m and CD163-null pigs were measured at three to four weeks of age,
just prior
to infection with PRRSV. The results, presented in Fig. 20, showed that sera
from WT
pigs had the lowest amounts of Hb (mean A450=23+/¨ 0.18, n=10). The mean and
standard deviation for each group were WT, 0.23+/¨ 0.18, n=10; HL11m, 1.63+/¨
0.8,
n=11; and 2.06 +/¨ 0.57, n=9, for the null group. The null group was composed
of
genotypes that did not express CD163 (CD163 null phenotype pigs). Hp
measurements
were made on a single ELISA plate. Groups with the same letter were not
significantly
different (p>0.05, Kruskal-Wallis one-way ANOVA with Dunnett's post-test). The
mean
A450 value was for WT pigs was significantly different from that of the HL11m
and
CD163-null pigs (p< 0.05). Although the mean A450 value was lower for the
HL11m
group compared to the CD163-null group (A450 = 1.6+/-0.8 versus 2.1+/-0.6),
the
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
92
difference was not statistically significant. Since the interaction between
HbHp and
CD163 occurs through SRCR 3 (Madsen et al., 2004), increased circulating Hp in
the
HL11m pigs compared to WT pigs was likely not a consequence of a reduced
affinity of
CD163 for Hb/Hp, but the result of reduced numbers of CD163+ macrophages along
with
reduced CD163 expression on the remaining macrophages (see panel E of Fig.
19).
Infection of PAMs with Type 1 and Type 2 viruses
[00299] The permissiveness of the CD163-modified pigs for PRRSV was
initially evaluated by infecting PAM cells in vitro with a panel of six Type 1
and nine
Type 2 PRRSV isolates (see Table 14 for the list of viruses). The viruses in
the panel
represent different genotypes, as well as differences in nucleotide and
peptide sequences,
pathogenesis, and years of isolation. The data presented in Table 15 show the
results
form experiments using PAMs from three pigs for each CD 163 genotype group.
The
viruses listed correspond to the PRRSV isolates listed in Table 14. The
results are shown
as mean +/¨ standard deviation of the percent of PAMs infected. The CD163-null
PAMs
were from pigs expressing the d7 ( 129) allele (see Figs. 17 and 19 for CD163
gene
constructs and CD163 expression on PAMs, respectively).
Table 15. Infection of PAMs from wild-type and GE pigs with different PRRSV
isolates
Genotype/Phenotype (% Infection)
Type 1 WT (%) HL11m Null
13-15 56 +/-9 0 0
Lelystad 62 +/-15 0 0
03-1059 50 +/-18 0 0
03-1060 61 +/-12 0 0
01-08 64 +/-20 0 0
4353-PZ 62+/-15 0 0
Type 2 WT (%) HL11m Null
NVSL 97 59 +/-15 8 +/-08 0
KS-06 56 +/-20 12 +/-09 0
P129 64 +/-11 8 +/-06 0
VR2332 54+/-05 6+/-03 0
CO 10-90 43 +/-18 8 +/-08 0
CO 10-84 51 +/-22 7 +/-04 0
MLV-ResP 55 +/-12 3 +/-01 0
K562 49 +/-03 10 +/-11 0
K5483 55 +/-23 6 +/-03 0
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
93
[00300] As expected, the WT PAMs were infected by all viruses. In contrast,
the CD163-null phenotype pigs were negative for infection by all viruses. A
marked
difference was observed in the response of PAMs from the HL11m pigs. None of
the
Type 1 viruses were able to infect the HL11m PAMs; whereas, all viruses in the
Type 2
panel infected the HL11m PAMs, albeit at much lower percentages compared to
the WT
PAMs.
[00301] Permissiveness was also evaluated by comparing virus titration
endpoints between WT and HL11m PAMs for the same Type 2 viruses. Results are
shown for two WT and two HL11m pigs (Fig. 21). The logioTCID50 values were
calculated based on the infection of macrophage cultures with the same virus
sample.
Infection results represent two different pigs from each genotype. Viruses
used for
infection are listed in Table 14. The logioTCID50 values for PAMs from the
HL11m pigs
were 1-3 logs lower compared to WT PAMs infected with the same virus. The only
exception was infection with a modified-live virus vaccine strain. When taken
altogether,
the results suggest that PAMs from HL11m pigs possess a reduced susceptibility
or
permissiveness to infection with Type 2 viruses.
Infection of CD163-modified pigs with Type 1 and Type 2 viruses
[00302] WT (circles), HL11m (squares), and CD163-null (triangles) pigs were
infected with representative Type 1 (SD13-15) (Fig. 22, panel A, left graph)
and Type 2
(NVSL 97-7895) (Fig. 22, panel A, right graph) viruses. The null phenotype
pigs were
derived from the KO and d(I567) alleles (see Fig. 17). Pigs from the three
genotypes
inoculated with the same virus were co-mingled in one pen, which allowed for
the
continuous exposure of CD163-modified pigs to virus shed from WT pen mates.
The
number of pigs infected with representative Type 1 virus were: WT (n=4), HL11m
(n=5),
and Null (n=3); and Type 2 virus: WT (n=4), HL11m (n=4), and Null (n=3). As
shown in
Fig. 22, the CD163-null pigs infected with either the Type 1 or Type 2 virus
were
negative for viremia at all time points and did not seroconvert. As expected,
the WT pigs
were productively infected possessing mean viremia levels approaching 106
templates
per 50 tl PCR reaction at 7 days after infection for both viruses. By 14 days,
all WT pigs
had seroconverted (see Fig. 22, panel B). Consistent with the PAM infection
results
(Table 15), the five HL11m pigs infected with the Type 1 virus showed no
evidence of
viremia or PRRSV antibody. All HL11m pigs infected with the Type 2 isolate,
NVSL,
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
94
supported infection and seroconverted (Fig. 22, panel B). The presence of a
reduced
permissive of the HL11m pigs was unclear. Mean viremia for three of the four
HL11m
pigs were similar to the WT pigs. However, for one HL11m pig, #101 (open
squares in
Fig. 22, panel A right graph), viremia was greatly reduced compared to the
other pigs in
HL11m genotype group. An explanation for the 3 to 4 log reduction in viremia
for Pig
#101 was not clear, but suggested that some HL11m pigs may be less permissive
for
PRRSV, an observation supporting the in vitro PAM infection results (Table
15). Since
all pigs were inoculated with the same amount of virus and remained co-mingled
with
the WT pigs, the lower viremia in Pig #101 was not the result of receiving a
lower
amount of virus or less exposure to virus. Flow cytometry of macrophages
showed that
CD163 expression for Pig #101 was comparable to the other HL11m pigs (data not
shown). There was no difference in the sequence in the exon 11 mimic sequence.
[00303] Additional virus infection trials were conducted using two viruses,
NVSL 97-7895 and KS06-72109. Results are shown in Fig. 23. Pigs were followed
for
35 days after infection and data reported as the area under the curve (AUC)
for viremia
measurements taken at 3, 7, 11, 14, 21, 28 and 35 days after infection. As
shown in Fig.
23, for NVSL, the mean AUC value for the seven WT pigs infected with NVSL was
168
+/¨ 8 versus 165 +/¨ 15 for the seven HL11m pigs. For K506, the mean AUC
values for
the six WT and six HL11m pigs were 156 +/¨ 9 and 163 +/-13, respectively. For
both
viruses, there was no statistically significant difference between the WT and
HL11m pigs
(p> 0.05). When taken altogether, the results showed that the HL11m pigs
failed to
support infection with Type 1 PRRSV, but retained permissiveness for infection
with
Type 2 viruses. Even though there was a reduction in the PRRSV permissiveness
of
PAMs from HL11m pigs infected in vitro with the Type 2 isolates, this
difference did not
translate to the pig. For the results shown in Fig. 23, virus load was
determined by
calculating the area under the curve (AUC) for each pig over a 35 day
infection period.
The AUC calculation was performed using logio PCR viremia measurements taken
at 0,
4, 7, 10, 14, 21, 28 and 35 days after infection. The horizontal lines show
mean and
standard deviation. Key: WT = wild-type pigs, HL11 = HL11m genotype pigs; Null
=
CD163-null genotype.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
DISCUSSION
[00304] CD163 is a macrophage surface protein important for scavenging
excess Hb from the blood and modulating inflammation in response to tissue
damage. It
also functions as a virus receptor. CD163 participates in both pro- and anti-
inflammatory
responses (Van Gorp et al., 2010). CD163-positive macrophages are placed
within the
alternatively activated M2 group of macrophages, which are generally described
as
highly phagocytic and anti-inflammatory. M2 macrophages participate in the
cleanup
and repair after mechanical tissue damage or infection (Stein et al., 1992).
In an anti-
inflammatory capacity, CD163 expression is upregulated by anti-inflammatory
proteins,
such as IL-10 (Sulahian, et al., 2002). During inflammation, CD163 decreases
inflammation by reducing oxidative through the removal of circulating heme
from the
blood. Heme degradation products, such as bilverdin, bilirubin, and carbon
monoxide are
potent anti-inflammatory molecules (Soares and Bach, 2009 and Jeney et al.,
2002). In a
pro-inflammatory capacity, the crosslinking of CD163 on the macrophage surface
by
anti-CD163 antibody or bacteria results in the localized release of pro-
inflammatory
cytokines, including IL-6, GM-CSF, TNFa and IL-10 (Van den Heuvel et al., 1999
and
Fabriek et al., 2009).
[00305] GE pigs that lack CD163 fail to support the replication of a Type 2
PRRSV isolate (Whitworth et al., 2016). In this study, in vitro infection
trials
demonstrate the resistance of CD163 null phenotype macrophages to an extensive
panel
of Type 1 and Type 2 PRRSV isolates, further extending resistance to
potentially include
all PRRSV isolates (Table 15). Resistance of the CD163-null phenotype
macrophages to
Type 1 and Type 2 viruses was confirmed in vivo (Fig. 22 and Fig. 23). Based
on these
results, the contribution of other PRRSV receptors previously described in the
literature
(Zhang and Yoo, 2015) can be ruled out. For example, Shanmukhappa et al.
(2007)
showed that non-permissive BHK cells transfected with a CD151 plasmid acquired
the
ability to support PRRSV replication, and incubation with a polyclonal anti-
CD151
antibody was shown to significantly reduce the infection of MARC-145 cells. In
addition, a simian cell line, SJPL, originally developed for use in
propagating swine
influenza viruses, was previously shown to support PRRSV replication (Provost,
et al.,
2012). Important properties of the SJPL cell line included the presence of
CD151 and the
absence of sialoadhesin and CD163. When taken together, these data provided
convincing evidence that the presence of CD151 alone is sufficient to support
PRRSV
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
96
replication. The results from this study showing the absence of PRRSV
infection in
macrophages and pigs possessing a CD163 null phenotype indicates that CD151 as
an
alternative receptor for PRRSV is not biologically relevant.
[00306] The viral proteins GP2a and GP4, which form part of the GP2a, GP3,
GP4 heterotrimer complex on the PRRSV surface, can be co-precipitated with
CD163 in
pull-down assays from cells transfected with GP2 and GP4 plasmids (Das, et
al., 2009).
Presumably, GP2 and GP4 form an interaction with one or more of the CD163 SRCR
domains. In vitro infectivity assays incorporating a porcine CD163 cDNA
backbone
containing a domain swap between porcine SRCR 5 and the homolog from hCD163-L1
SRCR 8 further localized the region utilized by Type 1 viruses to SRCR 5 (Van
Gorp, et
al., 2010). It is interesting to speculate that the stable interaction between
GP2/GP4 and
CD163 occurs through SRCR 5. Additional viral glycoproteins, such as GP3 and
GP5,
may further stabilize the virus-receptor complex or may function as co-
receptor
molecules. The requirement for SRCR 5 was investigated in this study by
infecting
macrophages and pigs possessing the HL11m allele, which recreated the CD163L1
SRCR 8 domain swap by making 33 bp substitutions in porcine exon 7. The HL11m
allele also included a neomycin cassette for selection of cells positive for
the genetic
alteration (Fig. 17). The HL11m pigs expressed CD163 on PAMs, albeit at
reduced
levels compared to WT PAMs (Fig. 19, compare panels A and E). Reduced
expression
was likely due to the presence of the neomycin cassette, which was located
between the
exon 11 mimic and the following intron. HL11m pigs were not permissive for
infection
with a Type 1 virus, confirming the importance of SRCR 5. However, HL11m
macrophages and HL11m pigs did support infection with Type 2 viruses. Based on
virus
titration and percent infection results, the PAMs from the HL11m pigs showed
an overall
decrease in permissiveness for virus compared to the WT macrophages (Table 15
and
Fig. 17). Decreased permissiveness may be due to reduced levels of CD163 on
the
HL11m macrophages, combined with a reduced affinity of virus for the modified
CD163
protein. Assuming that Type 2 viruses possesses a requirement of SRCR 5 and
that Li
SRCR 8 can function as a suitable substitute, the lower affinity may be
explained by the
difference in peptide sequences between human SRCR 8 and porcine SRCR 5 (see
Fig.
18, panel B). However, the reduced permissiveness of PAMs did not translate to
the pig.
Mean viremia for the HL11m pigs was not significantly different when compared
to WT
pigs (Fig. 23). In addition to PAMs, PRRSV infection of intravascular, septal
and
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
97
lymphoid tissue macrophages contribute to viremia (Lawson et al., 1997 and
Morgan et
al., 2014). The potential contributions of these and other CD163-positive
cells
populations in maintaining the overall virus load in HL 11m pigs deserves
further study.
[00307] Even though CD163 plasmids possessing deletions of SRCR domains
are stably expressed in HEK cells (Van Gorp et al., 2010), the deletion of
exons 7 and 8
in d7(1467) and d7(1280) resulted in a lack of detectable surface expression
of CD163
(Fig. 19, panel D). Since the 2A10 mAB used for flow cytometry recognizes the
three N-
terminal SRCR domains (Van Gorp et al., 2010), and possibly the 7th and 8th
domains
(Sanchez, et al., 1999), the absence of detection was not due to the removal
of a 2A10
epitope in the mutated proteins. While a small amount of CD163 expression
could be
detected on PAMs from some of the d7(129) pigs (see Fig. 19, panel C), the
quantity of
expressed protein was not sufficient to support PRRSV infection in PAMs or
pigs. The
absence of CD163 expression in the exon 7 and 8 deletion mutants is not fully
understood, but is likely the result of mRNA and/or protein degradation.
[00308] In 2003, CD163 was identified as a receptor for African swine fever
virus (ASFV; Sanchez-Torres et al., 2003). This conclusion was based on the
observation
that infected macrophages possess a mature CD163-positive phenotype, and anti-
CD163
antibodies, such as 2A10, block ASFV infection of macrophages in vitro. It
remains to
be determined if CD163-null pigs are resistant to ASFV infection.
[00309] Cell culture models incorporating modifications to the PRRSV
receptor have provided valuable insight into the mechanisms of PRRSV entry,
replication and pathogenesis. One unique aspect of this study was the conduct
of parallel
experiment in vivo using receptor-modified pigs. This research has important
impacts on
the feasibility of developing preventative cures for one of the most serious
diseases to
ever face the global swine industry.
Example 4: Knockout of maternal CD163 protects fetuses from infection with
porcine reproductive and respiratory syndrome virus (PRRSV).
[00310] Examples 1 to 3 above demonstrate that pigs having a complete
knockout (KO) of the CD163 gene lack CD163 expression on macrophages and fail
to
support PRRSV infection (see also Whitworth et al., 2016; Wells et al., 2017).
Since
CD163 expression is a dominant trait and inherited in a classic Mendelian
fashion,
offspring possessing normal CD163 expression and function can be derived by
crossing
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
98
a KO CD163-1- female pig with a wildtype (WT) CD163 +I+ male. For this study,
CD163
KO gilts were bred with WT boars, producing heterozygous, CD163 +1- fetuses in
order to
determine whether the presence of the CD163 KO genotype of the dam would be
sufficient to protect fetuses following maternal infection with PRRSV. In this
study,
CD163-positive fetuses, recovered between 109 days of gestation or 20 days
after
maternal infection, were completely protected from PRRSV in dams possessing a
complete knockout of the CD163 receptor. The results demonstrate a practical
means to
eliminate PRRSV-associated reproductive disease, a major source of economic
hardship
to agriculture.
Materials and Methods
[00311] CD163 gene editing. The CRISPR/Cas9 methods used to generate all
of the KO alleles are described in detail in Examples 1-3 above. Wild-type
animals and
knockout, or heterozygous animals generated as described in Examples 1-3 and
having
the alleles described in Table 16 were used in the experiments described in
this Example.
Each of these alleles is described in Examples 1-3 and in PCT Publication No.
WO
2017/023570, which is incorporated herein by reference in its entirety. The
specific edits
for alleles B, D and E are also described in Whitworth et al., 2014, and the
specific edit
in Allele C (2 bp insertion) is described in Whitworth et al., 2016. All of
the alleles
described in Table 16 were identified based on DNA sequencing. The knockout
genotype
was confirmed by the absence of CD163 expression, which was measured by
staining
alveolar macrophages with anti-CD163 mAb, 2A10, as described above in Example
2.
Table 16. CD163 alleles.
Allele Description In Reference to SEQ ID NO: 47 SEQ ID
NO.
SEQ ID NO:47 (partial WT CD163 sequence
A Wild Type 47
including exon 7)
Knockout (7 bp 7 base pair insertion between nucleotide
insertion in exon 7) 3,148 and nucleotide 3,149 as compared to 99
reference sequence SEQ ID NO: 47
Knockout (2 bp 2 base pair insertion between nucleotides
insertion in exon 7) 3,149 and 3,150 as compared to reference
sequence SEQ ID NO: 47, with a 377 base
103
pair deletion from nucleotide 2,573 to
nucleotide 2,949 as compared to reference
sequence SEQ ID NO: 47 on the same allele
Knockout (11 bp 11 base pair deletion from nucleotide 3,137 to
deletion in exon 7) nucleotide 3,147 as compared to reference
102
sequence SEQ ID NO: 47.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
99
Knockout (1382 bp 1382 base pair deletion from nucleotide 3,113
deletion that included to nucleotide 4,494 as compared to reference
part of exon 7 and 8 sequence SEQ ID NO: 47, wherein the 113
with an 11 bp deleted sequence is replaced with an 11 base
insertion in exon 7) pair insertion beginning at nucleotide 3,113
[00312] PRRSV infection. The PRRSV strain used in this study, NVSL 97-
7895 (NVSL), is a laboratory strain isolated in 1997 from a herd in Southeast
Iowa, USA
that was experiencing a PRRS abortion storm (Halbur et al., 1997). The virus,
maintained as a low passage isolate, was propagated and titered on MARC-145
cells. At
89 to 91 days of gestation, gilts were inoculated with 105 TCID50 of virus
diluted in 5 ml
of culture medium. One half of the inoculum was administered by intramuscular
injection and the remainder was administered intranasally. All gilts were
maintained in
an environment that allowed for the continuous exposure to virus shed by
infected pen
mates. Blood samples were taken from the gilts prior to infection, seven days
post-
inoculation (dpi), and at the time of euthanasia. PRRSV nucleic acid was
measured by
isolation of total RNA from serum followed by reverse transcriptase real-time
PRRSV
PCR (Tetracore, Rockville, MD). A standard curve was generated using the
quantification standards supplied in the RT-PCR kit. Results are reported as
logio
templates per 25 pi reaction, which approximates the number of viral RNA
templates per
ml of blood.
RESULTS
[00313] A
detailed description of the knockout alleles used in this study is
shown in Table 16 above. Each knockout allele possessed a mutation in exon 7
that was
predicted to result in a codon frameshift followed by a premature stop codon
in the
mRNA. The matings between WT and CD163 KO parents are summarized in Table 17.
The first group of three dams, which served as positive infection controls,
were CD163+I+
dams carrying CD 163+1+ fetuses (++/++ group). A second group (¨ ¨1+¨) were CD
163-/-
dams carrying CD 163+/- fetuses. In this group, the CD163-1- dams are unable
to support
PRRS replication, while the CD163+/- fetuses retain susceptibility to PRRS
infection.
And finally, a third group (¨ ¨/¨ ¨) consisted of CD 163-/- dams carrying CD
163-/¨
fetuses. For the last group, both dams and fetuses should be resistant to
infection.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
100
Table 17. CD163 parental and fetal genotypes.
CD163 Genotype Collection of Fetuses
Parents*' Day of Day of No. of
Gilt No. Male Dam Fetus Infection*2 Gestation*3 Fetuses
138 +1+ (A/A) +/+ (A/A) +/+ 91 106 16
139*4 / (A/A) +/+ (A/A) +/+ 91 106 14
140 +/-k (A/A) +/+ (A/A) +/+ 91 106 12
84 +/+ (A/A) ¨/¨(B/C) +/¨ 89 109 14
87 +/+ (A/A) ¨/¨(B/C) +/¨ 89 109 17
122 +/+ (A/A) ¨/¨(E/C) +/¨ 89 109 11
86 ¨/¨ (C/D) ¨/¨ (B/D) ¨/¨ 90 109 7
121 ¨/¨(C/D) ¨/¨(B/D) ¨/¨ 90 109 9
*1 CD163 alleles are identified in Table 1
*2 Gestation day when dams were infected
*3 Gestation day when fetuses were removed
PRRSV-infected dam aborted at 106 days of gestation
[00314] Clinical signs in the infected wild-type (WT) dams included lethargy
and transient inappetence. The KO dams showed no clinical signs. During the
study
period, one WT dam, No. 139, aborted on day 106 of gestation (15 dpi). PRRSV
nucleic
acid, measured at 7 dpi, showed a viremia level for Dam No. 139 of 5.5 logio
templates
per reaction, demonstrating the presence of a productive PRRSV infection.
Between 15
and 20 dpi, all remaining dams were euthanized and uterine horns immediately
removed.
Beginning at the tip of each horn, fetuses and placentas were removed and
assessed for
the presence of anatomic pathology. A blood sample was obtained from each
fetus. If
blood was not obtainable, a sample of fluid was collected from the abdominal
cavity.
The number of fetuses recovered from each dam is listed in Table 17. For the
CD163
WT group ++/++) (including the dam that aborted), the number of fetuses were
16, 14
and 12 (mean=14.0). The CD163 KO dams carrying the CD163+/- fetuses (¨ ¨/+¨
group)
yielded 14, 17 andll fetuses (mean= 13.6). For the CD163 KO dams carrying
CD163
KO fetuses (¨ ¨/¨ ¨ group), the numbers of fetuses were 7 and 9. The results
for fetal
viremia and gross pathology are summarized in FIG. 24 and Table 18.
CA 03096022 2020-10-02
WO 2019/203807
PCT/US2018/027944
101
Table 18. Summary of fetal infection and pathology."
No.
Dam Fetus Total No. Dam Infected
Gilt No. Genotype Genotype Fetuses Viremia*2 Fetuses Pathology*3
138 +/+ +/+ 16 3.6 13 (80%) 8 (50%)
139*4 +/+ +/+ 14 5.5 ND ND
140 +/+ +/+ 12 4.1 11(92) 8(72)
84 ¨/¨ +/¨ 14 N 0 (0) 1 (07)
87 ¨/¨ +/¨ 17 N 0 (0) 1 (06)
122 ¨/¨ +/¨ 11 N 0 (0) 0 (0)
86 ¨/¨ ¨/¨ 7 N 0 (0) 0 (0)
121 ¨/¨ ¨/¨ 9 N 0 (0) 0 (0)
*1 The table is a combined summary of the data presented in Table 17 and Fig.
24
*2 Viremia shown as logio virus nucleic templates per PCR
*3 Fetuses showing pathology as described for Fig. 24
*4 Gilt aborted prior to recovery of fetuses
[00315] FIG. 24 depicts each of the fetal outcomes following maternal
infection with PRRSV. The numbers on the left identify each dam. Below each
dam in
parenthesis is the result for PRRS PCR in serum, measured as logio templates
per
reaction. "N" is negative for PRRSV nucleic acid (Ct>39). Fetuses are
identified by
number and relative position within each uterine horn. Asterisks identify
fetal PCR
samples obtained from abdominal fluid. The number below each fetus is the
result for
PRRS PCR in fetal serum (logio templates per reaction). The number within each
circle
refers to the presence of anatomical pathology: 1) normal fetus; 2) small
fetus; 3)
placenta changes such as detached placenta and/or necrosis; 4) meconium
stained fetus;
5) fetus is dead and necrotic. Lower case letters identify the genotype of the
individual
fetuses (see Table 17). Key: a, A/A; b, C/A; c, B/A; d, E/A; e, B/C; f, B/D;
g, D/C; h,
D/D; i, E/C; j, E/D; ND not determined because the fetus was necrotic; nd,
genotype was
not determined.
[00316] At the anatomic level, 50% and 72% of fetuses derived from the two
CD163 WT (++/++) dams, No. 138 and No. 140, showed some degree of pathology,
including smaller than normal fetuses (11% of all fetuses), fetuses with
detached or
necrotic placentas (14%), meconium staining (7%), and fetuses that were dead
and
necrotic (25%). The pathology observations are typical of reproductive PRRS.
The same
litters showed a high rate of PRRSV infection, with 92% of the fetuses testing
positive
for the presence of PRRSV nucleic acid. The PCR results for the fetuses from
the WT
dams illustrate two important properties of fetal PRRSV infection. First,
there was a
wide variation between fetuses in the concentration of virus detected in
serum, the result
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
102
of fetuses becoming infected at different times. Secondly, the level of
viremia was not
always correlated with pathology. For example, Fetus No. 5 from Dam No. 138
possessed a high level of viremia (7.3 logio templates per reaction) and yet
the fetus
appeared unaffected. The reason for the discrepancy between viremia and the
pathology
is unclear. One possibility is that fetal pathology is the result of tissue
damage that
occurs on the maternal side and not related to the level of fetal viremia. In
the field, these
normal, but infected newborn piglets can function as "supershedders," which
facilitate
the rapid dissemination of PRRSV throughout a production system. For the ¨ ¨1+
¨group
(dams No. 84, 87 and 122), all fetuses appeared normal, with the minor
exception of two
fetuses that were smaller than the other littermates. The smaller than normal
size is likely
a consequence of crowding within the uterine horn that decreases the surface
area of the
placenta, thus restricting the growth of the developing fetus. All dams and
fetuses in the
¨ ¨1+ ¨ group were negative for the presence of PRRSV nucleic acid. For the
last group,
¨ ¨/¨ ¨, there was no visible pathology, and all dams (No. 86 and 121) and
fetuses were
negative for PRRSV nucleic acid.
[00317] The results from this study clearly demonstrate that the absence of
CD163 in the dam is sufficient to protect the PRRSV-susceptible fetus.
Although
CD163-positive offspring derived from CD163 KO dams are susceptible to virus
immediately after birth, the protection from PRRSV in utero provides a means
to
eliminate a major source of economic loss and animal suffering.
[00318] Examples disclosed herein are provided by way of exemplification
and are not intended to limit the scope of the invention.
[00319] In view of the above, it will be seen that the several objects of the
invention are achieved and other advantageous results attained.
[00320] As various changes could be made in the above products and methods
without departing from the scope of the invention, it is intended that all
matter contained
in the above description and shown in the accompanying drawing[s] shall be
interpreted
as illustrative and not in a limiting sense.
REFERENCES
Allende R, et al., 1999. North American and European porcine reproductive and
respiratory syndrome viruses differ in non-structural protein coding regions.
J. Gen.
Virol. 80: 307-315.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
103
Bauer BK, et al., Arginine supplementation in vitro increases porcine embryo
development and affects mRNA transcript expression. Reprod Fertil Dev 2011;
23:107.
Beaton BP, et al., Compound transgenics: recombinase- mediated gene stacking.
In:
Pinkert CA (ed.). Transgenic Animal Technology: A Laboratory Handbook, 3rd ed.
Waltham, MA: Elsevier; 2014:565-578.
Benfield DA, et al., 1992. Characterization of swine infertility and
respiratory syndrome
(SIRS) virus (Isolate ATCC VR-2332). J Vet Diag Invest 4:127-133.
Boddicker, et al., 2014. Genome-wide association and genomic prediction for
host
response to porcine reproductive and respiratory syndrome virus infection.
Genetics,
selection, evolution: GSE 46,18.
Borg NA, et al., CD id-lipid- antigen recognition by the semi-invariant NKT T-
cell
receptor. Nature 2007; 448:44-49.
Brinster RL, et al., Factors affecting the efficiency of introducing foreign
DNA into mice
by microinjecting eggs. Proc Nat! Acad Sci U S A 1985; 82:4438-4442.
Cafruny, WA, et al, Trojan Horse macrophages: studies with the murine lactate
dehydrogenase-elevating virus and implications for sexually transmitted virus
infection.
J. Gen. Virol. 77(12): 3005-3012 (1996).
Calvert JG, et al., 2007. CD163 expression confers susceptibility to porcine
reproductive
and respiratory syndrome viruses. J Virol. 81:7371-7379.
Carter DB, et al., Phenotyping of transgenic cloned piglets. Cloning Stem
Cells 2002;
4:131-145.
Cong L, et al., Multiplex genome engineering using CRISPR/Cas systems. Science
2013;
339:819-823.
Christianson, W. T. et al. Experimental reproduction of swine infertility and
respiratory
syndrome in pregnant sows. American Journal of Veterinary Research 53,485-488
(1993).
Christopher-Hennings, et al., Effects of a modified-live virus vaccine against
porcine
reproductive and respiratory syndrome in boars, American J. Veterinary
Research 58(1),
40-45 (1997).
Dai Y, et al., Targeted disruption of the alphal,3-galactosyltransferase gene
in cloned
pigs. Nat Biotechnol 2002; 20:251-255.
Das PB, et al., 2009. The minor envelope glycoproteins GP2a and GP4 of porcine
reproductive and respiratory syndrome virus interact with the receptor CD163.
J Virol.
84:1731-40.
Dee et al., Elimination of porcine reproductive and respiratory syndrome virus
using a
test and removal process, The Veterinary Record 143,474-476 (1998).
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
104
Delputte PL, et al., 2002. Involvement of the matrix protein in attachment of
porcine
reproductive and respiratory syndrome virus to a heparin-like receptor on
porcine
alveolar macrophages. J Virol. 76:4312-4320.
Etzerodt, A., et al., 2013. Plasma clearance of hemoglobin and haptoglobin in
mice and
effect of CD163 gene targeting disruption. Antioxidants & redox signaling 18,
2254-
2263.
Etzerodt, A., et al., 2013. CD163 and inflammation: biological, diagnostic,
and
therapeutic aspects. Antioxidants & redox signaling 18, 2352-2363.
Fabriek BO, et al., 2005. The macrophage scavenger receptor CD163.
Immunobiology.
210:153-160.
Fabriek BO, et al., 2009. The macrophage scavenger receptor CD163 functions as
an
innate immune sensor for bacteria. Blood. 113:887-892.
Gaj T, et al., ZFN, TALEN, and CRISPR/Cas- based methods for genome
engineering.
Trends Biotechnol 2013; 31: 397-405.
Gaudreault N, et al., 2009. Factors affecting the permissiveness of porcine
alveolar
macrophages for porcine reproductive and respiratory syndrome virus. Archiv
Virol.
154:133-136.
Graversen, J.H., et al., 2012. Targeting the hemoglobin scavenger receptor
CD163 in
macrophages highly increases the anti-inflammatory potency of dexamethasone.
Molecular therapy : the journal of the American Society of Gene Therapy 20,
1550-1558.
Groenen, M.A., et al., 2012. Analyses of pig genomes provide insight into
porcine
demography and evolution. Nature 491, 393-398.
Hai T, et al., One-step generation of knockout pigs by zygote injection of
CRISPR/Cas
system. Cell Res 2014; 24: 372-375.
Halbur, P.G., et al., 1995. Comparison of the pathogenicity of two US porcine
reproductive and respiratory syndrome virus isolates with that of the Lelystad
virus.
Veterinary pathology 32, 648-660.
Halbur, P. & Bush, E. B. Update on abortion storms and sow mortality. Journal
of Swine
Health and Production 5, 73 (1997).
Hammer RE, et al., Production of transgenic rabbits, sheep and pigs by
microinjection.
Nature 1985; 315:680-683.
Hauschild J, et al., Efficient generation of a biallelic knockout in pigs
using zinc-finger
nucleases. Proc Nat! Acad Sci US A 2011; 108:12013-12017.
Holtkamp, D.J., et al., 2013. Assessment of the economic impact of porcine
reproductive
and respiratory syndrome virus on United States pork producers. Journal of
Swine Health
and Production 21, 72-84.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
105
Hwang WY, et al., Efficient genome editing in zebrafish using a CRISPR-Cas
system.
Nat Biotechnol 2013; 31:227-229.
Jeney V, et al., 2002. Pro-oxidant and cytotoxic effects of circulating heme.
Blood.
100:879-887.
Keffaber, K.K., 1989. Reproductive failure of unknown etiology. American
Association
of Swine Practitioners Newsletter 1, 1-10.
Key, K. F. et al. Genetic variation and phylogenetic analyses of the ORF5 gene
of acute
porcine reproductive and respiratory syndrome virus isolates. Veterinary
Microbiology
83, 249-263 (2001).
Kim, H. S., et al.,1993. Enhanced replication of porcine reproductive and
respiratory
syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch
Virol 133, 477-483.
Kristiansen M, et al., 2001. Identification of the haemoglobin scavenger
receptor. Nature.
409:198-201.
Kwon DN, et al., Production of biallelic CMP-Neu5Ac hydroxylase knock-out
pigs. Sci
Rep 2013; 3:1981.
Ladinig, A., et al., 2015. Pathogenicity of three type 2 porcine reproductive
and
respiratory syndrome virus strains in experimentally inoculated pregnant
gilts. Virus Res
203, 24-35.
Lai L, et al., Production of alpha-1,3-galactosyltransferase knockout pigs by
nuclear
transfer cloning. Science 2002; 295:1089-1092.
Lai L, et al., Creating genetically modified pigs by using nuclear transfer.
Reprod Biol
Endocrinol 2003; 1:82.
Lai L, et al., Production of cloned pigs by using somatic cells as donors.
Cloning Stem
Cells 2003; 5:233-241.
Lawson SR, et al.,1997. Porcine reproductive and respiratory syndrome virus
infection of
gnotobiotic pigs: sites of virus replication and co-localization with MAC-387
staining at
21 days post-infection. Virus Res. 51:105-113.
Lee K, et al., Engraftment of human iPS cells and allogeneic porcine cells
into pigs with
inactivated RAG2 and accompanying severe combined immunodeficiency. Proc Nat!
Acad Sci U S A 2014; 111:7260-7265.
Lee K, et al., Piglets produced from cloned blastocysts cultured in vitro with
GM-CSF.
Mol Reprod Dev 2013; 80: 145-154.
Li D, et al., Heritable gene targeting in the mouse and rat using a CRISPR-Cas
system.
Nat Biotechnol 2013; 31:681-683.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
106
Lillico SG, et al., Live pigs produced from genome edited zygotes. Sci Rep
2013;
3 :2847.
Machaty Z, et al., Complete activation of porcine oocytes induced by the
sulfhydryl
reagent, thimerosal. Biol Reprod 1997; 57:1123-1127.
Madsen M, et al., 2004. Molecular characterization of the haptoglobin-
hemoglobin
receptor CD163. Ligand binding properties of the scavenger receptor cysteine-
rich
domain region. J Biol Chem. 279:51561-51567.
Mengeling, W. L. et al. Clinical consequences of exposing pregnant gilts to
strains of
porcine reproductive and respiratory syndrome (PRRS) virus isolated from field
cases of
"atypical" PRRS. American Journal of Veterinary Research. 59,1540-1544 (1998).
Merck, The Merck Veterinary Manual.
http://www.merckmanuals.com/vet/appendixes/reference guides/normal rectal
temperat
ure ranges.html.
Miao, Y.L., et al., 2009. Centrosome abnormalities during porcine oocyte
aging.
Environmental and molecular mutagenesis 50,666-671.
Morgan SB, et al., 2014. Pathology and Virus Distribution in the Lung and
Lymphoid
Tissues of Pigs Experimentally Inoculated with Three Distinct Type 1 PRRS
Virus
Isolates of Varying Pathogenicity. Transbound Emerg Dis. Nov 10. pp 1-11.
Nelsen CJ, et al., 1999. Porcine reproductive and respiratory syndrome virus
comparison:
divergent evolution on two continents. J. Virol. 73,270-280.
Neumann EJ, et al., Assessment of the economic impact of porcine reproductive
and
respiratory syndrome on swine production in the United States. J Am Vet Med
Assoc
2005; 227:385-392.
Niu Y, et al., Generation of gene-modified Cynomolgus monkey via Cas9/RNA-
mediated gene targeting in one-cell embryos. Cell 2014; 156:836-843.
Patience, J.F., et al.,1989. Swine Nutrition Guide, in: Center, P.S. (Ed.),
University of
Saskatchewan, Saskatoon, CA, pp. 149-171.
Patton JP, et al., 2008. Modulation of CD163 receptor expression and
replication of
porcine reproductive and respiratory syndrome virus in porcine macrophages.
Virus Res.
140: 161-171.
Plagemann, P. G. W. Lactate dehydrogenase-elevating virus and related viruses.
In
Fields Virology, 3rd Ed, Fields, B. ed. Lippincott- Raven, Philadelphia, pp
1105-1120
(1996).
Prather RS, et al., An intact sialoadhesin (Sn/SIGLEC1/CD169) is not required
for
attachment/ internalization of the porcine reproductive and respiratory
syndrome virus. J
Virol 2013; 87:9538-9546.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
107
Prather, R.S., et al., 2013. An intact sialoadhesin (Sn/SIGLEC1/CD169) is not
required
for attachment/internalization of the porcine reproductive and respiratory
syndrome
virus. J Virol 87, 9538-9546.
Provost C, et al., 2012. Identification of a new cell line permissive to
porcine
reproductive and respiratory syndrome virus infection and replication which is
phenotypically distinct from MARC-145 cell line. Virol J. 9:267.
Reed JL, et al., 1938. A simple method of estimating fifty percent endpoints.
The
American Journal of Hygiene 27:493-497.
Ross JW, et al., Optimization of square-wave electroporation for transfection
of porcine
fetal fibroblasts. Transgenic Res 2010; 19:611-620.
Rowland, R.R.R., et al., The evolution of porcine reproductive and respiratory
syndrome
virus: quasispecies and emergence of a virus subpopulation during infection of
pigs with
VR-2332, Virology 259, 262-266 (1999).
Rowland, R. R.R. et al. Lymphotropism of porcine reproductive and respiratory
syndrome virus replication during persistent infection of pigs originally
exposed to virus
in utero. Veterinary Microbiology 96, 219-235 (2003).
Rowland, R. R.R. The interaction between PRRSV and the late gestation pig
fetus. Virus
Research 154, 114-122 (2010).
Rowland, R.R., et al., 2012. Control of porcine reproductive and respiratory
syndrome
(PRRS) through genetic improvements in disease resistance and tolerance.
Frontiers in
genetics 3, 260.
Sanchez C, et al., 1999. The porcine 2A10 antigen is homologous to human CD163
and
related to macrophage differentiation. Journal of Immunology 162:5230-5237.
Sanchez-Torres C, et al., 2003. Expression of porcine CD163 on
monocytes/macrophages correlates with permissiveness to African swine fever
infection.
Arch Virol. 148:2307-2323.
Schaer, C.A., et al., 2006a. Constitutive endocytosis of CD163 mediates
hemoglobin-
heme uptake and determines the noninflammatory and protective transcriptional
response
of macrophages to hemoglobin. Circulation research 99, 943-950.
Schaer, D.J., et al., 2006b. CD163 is the macrophage scavenger receptor for
native and
chemically modified hemoglobins in the absence of haptoglobin. Blood 107, 373-
380.
Schaer, D.J., et al., 2006c. Hemophagocytic macrophages constitute a major
compartment of heme oxygenase expression in sepsis. European journal of
haematology
77, 432-436.
Shanmukhappa, K, et al., 2007. Role of CD151, A tetraspanin, in porcine
reproductive
and respiratory syndrome virus infection. Virol J. 4:62.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
108
Shimozawa N, et al., Abnormalities in cloned mice are not transmitted to the
progeny.
Genesis 2002; 34:203-207.
Shi, M. et al. Phylogeny-based evolutionary, demographical, and geographical
dissection
of North American type 2 porcine reproductive and respiratory syndrome
viruses.
Journal of Virology 84, 8700-8711 (2010).
Smit, AFA, et al., RepeatMasker Open-3Ø 1996-2010. Current Version: open-
4Ø5
(RMLib: 20140131 and Dfam: 1.2). http://www.repeatmasker.org>. CD163:
accessed
July 25, 2014; CD1D: accessed August 27, 2013.
Soares MP, et al., 2009. Heme oxygenase-1: from biology to therapeutic
potential.
Trends Mol Med. 15:50-58.
Stein M, et al.,1992. Interleukin 4 potently enhances murine macrophage
mannose
receptor activity: a marker of alternative immunologic macrophage activation.
J Exp
Med. 176:287-292.
Stephenson R, et al., 2015. Multiplex serology for common viral infections in
feral pigs
in Hawaii between 2007 and 2010. Accepted for publication. J Wildlife Dis
51:239-
2343.
Sulahian TH1, et al., 2002. 2002. Human monocytes express CD163, which is
upregulated by IL-10 and identical to p155. Cytokine. 2000 12:1312-1321.
Terns MP, et al., CRISPR-based adaptive immune systems. Curr Opin Microbiol
2011;
14:321-327.
Tian, K. et al. Emergence of fatal PRRSV variants: unparalleled outbreaks of
atypical
PRRS in China and molecular dissection of the unique hallmark. PLoS One. 2,
e526
(2007).
Trible, B.R., et al., 2012. Recognition of the different structural forms of
the capsid
protein determines the outcome following infection with porcine circovirus
type 2. J
Virol 86, 13508-13514.
U.S. Department of Agriculture and U.S. Department of Health and Human
Services.
Dietary Guidelines for Americans, 2010. 7th Edition,
Van Breedam W, et al., Porcine reproductive and respiratory syndrome virus
entry into
the porcine macrophage. J Gen Virol 2010; 91:1659-1667.
Van Breedam, W., et al., 2010. Porcine reproductive and respiratory syndrome
virus
entry into the porcine macrophage. J Gen Virol 91, 1659-1667.
Van den Heuvel MM, et al., 1999. Regulation of CD 163 on human macrophages:
cross-
linking of CD163 induces signaling and activation. J Leukoc Biol. 66:858-866.
van den Hoff MJ, et al., Electroporation in 'intracellular' buffer increases
cell survival.
Nucleic Acids Res 1992; 20:2902.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
109
Van Gorp, H., et al., 2010. Scavenger receptor CD163, a Jack-of-all-trades and
potential
target for cell-directed therapy. Mol Immunol 47,1650-1660.
Van Gorp H, et al., 2010. Identification of the CD163 protein domains involved
in
infection of the porcine reproductive and respiratory syndrome virus. J Virol.
84:3101-
3105.
Walters EM, et al., Advancing swine models for human health and diseases. Mo
Med
2013; 110:212-215.
Wang H, et al., One-step generation of mice carrying mutations in multiple
genes by
CRISPR/Cas-mediated genome engineering. Cell 2013; 153: 910-918.
Welch SK, et al., 2010. A brief review of CD163 and its role in PRRSV
infection. Virus
Res. 154:98-103.
Wells, K.D., et al., 2014. Use of the CRISPR/Cas9 system to produce
genetically
engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod 91,78.
Wells, K.D. et al. Substitution of porcine CD163 SRCR domain 5 with a CD163-
like
homolog confers resistance of pigs to genotypel but not genotype 2 porcine
reproductive
and respiratory syndrome (PRRS) viruses. Journal of Virology In press (2017).
Wensvoort, G., et al., 1991. Mystery swine disease in The Netherlands: the
isolation of
Lelystad virus. Veterinary Quarterly 13,121-130.
Whitworth KM, et al., 2016. CD163 facilitates both entry and replication of
porcine
reproductive and respiratory syndrome virus. Nature Biotech. 34:20-22.
Whitworth KM, et al., Method of oocyte activation affects cloning efficiency
in pigs.
Mol Reprod Dev 2009;76:490-500.
Whitworth et al. Use of the CRISPR/Cas9 system to produce genetically
engineered pigs
from in vitro-derived oocytes and embryos, Biology of Reproduction 91(3):1-13
(2014).
Whitworth KM, et al., Activation method does not alter abnormal placental gene
expression and development in cloned pigs. Mol Reprod Dev 2010; 77:1016-1030.
Whitworth KM, et al., Scriptaid corrects gene expression of a few aberrantly
reprogrammed transcripts in nuclear transfer pig blastocyst stage embryos.
Cell
Reprogram 2011; 13:191-204.
Whitworth, K.M., et al., 2014. Use of the CRISPR/Cas9 system to produce
genetically
engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod 91,78.
Whyte JJ, et al., Genetic modifications of pigs for medicine and agriculture.
Mol Reprod
Dev 2011; 78:879-891.
Whyte JJ, et al., Gene targeting with zinc finger nucleases to produce cloned
eGFP
knockout pigs. Mol Reprod Dev 2011; 78:2.
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
110
Wiedenheft B, et al., RNA-guided genetic silencing systems in bacteria and
archaea.
Nature 2012; 482:331-338.
Wilkinson et al. Genome-wide analysis of the transcriptional response to
porcine
reproductive and respiratory syndrome virus infection at the maternal/fetal
interface and
in the fetus. BMC Genomics 17,383 (2016).
Whitworth, K. M. et al. Gene-edited pigs are protected from porcine
reproductive and
respiratory syndrome virus. Nature Biotechnology 34,20-22 (2016).
Winckler, C., et al., 2001. The reliability and repeatability of a lameness
scoring system
for use as an indicator of welfare in dairy cattle. Acta Agricultura
Scandinavica Section
A. Animal Science, 103-107.
Yang D, et al., Generation of PPARgamma mono-allelic knockout pigs via zinc-
finger
nucleases and nuclear transfer cloning. Cell Res 2011; 21:979-982.
Yoshioka K, et al., Birth of piglets derived from porcine zygotes cultured in
a chemically
defined medium. Biol Reprod 2002; 66:112-119.
Zhang Q, et al., 2015. PRRS virus receptors and their role for pathogenesis.
Vet
Microbiol. 177:229-241.
Zhao J, et al., Histone deacetylase inhibitors improve in vitro and in vivo
developmental
competence of somatic cell nuclear transfer porcine embryos. Cell Reprogram
2010;
12:75-83.
Zhao J, et al., Significant improvement in cloning efficiency of an inbred
miniature pig
by histone deacetylase inhibitor treatment after somatic cell nuclear
transfer. Biol Reprod
2009; 81:525-530.
TABLE OF SEQUENCES
SEQ TYPE DESCRIPTION
SEQ ID NO:1 nucleotide CRISPR 10
SEQ ID NO:2 nucleotide CRISPR 131
SEQ ID NO:3 nucleotide CRISPR 256
SEQ ID NO:4 nucleotide CRISPR 282
SEQ ID NO:5 nucleotide CRISPR 4800
SEQ ID NO:6 nucleotide CRISPR 5620
SEQ ID NO:7 nucleotide CRISPR 5626
SEQ ID NO:8 nucleotide CRISPR 5350
SEQ ID NO:9 nucleotide eGFP1
SEQ ID NO:10 nucleotide eGFP2
SEQ ID NO:11 nucleotide forward primer 9538 fragment
SEQ ID NO:12 nucleotide reverse primer 9538 fragment
CA 03096022 2020-10-02
WO 2019/203807 PCT/US2018/027944
111
SEQ ID NO:13 nucleotide forward primer 8729 fragment
SEQ ID NO:14 nucleotide forward primer 8729 fragment
SEQ ID NO:15 nucleotide WILD TYPE CD163
SEQ ID NO:16 nucleotide Fig. 4, panel C WT
SEQ ID NO:17 nucleotide Fig. 4, panel C #1
SEQ ID NO:18 nucleotide Fig. 4, panel C #2
SEQ ID NO:19 nucleotide Fig. 4, panel C #3
SEQ ID NO:20 nucleotide Fig. 5, panel A WT
SEQ ID NO:21 nucleotide Fig. 5, panel A #1-1
SEQ ID NO:22 nucleotide Fig. 5, panel A #1-4
SEQ ID NO:23 nucleotide Fig. 5, panel A #2-2
SEQ ID NO:24 nucleotide Fig. 6, panel C CD163 WT
SEQ ID NO:25 nucleotide Fig. 6, panel C CD163 #1
SEQ ID NO:26 nucleotide Fig. 6, panel C CD163 #2
SEQ ID NO:27 nucleotide Fig. 6, panel C CD163 #3
SEQ ID NO:28 nucleotide Fig. 6, panel C eGFP WT
SEQ ID NO:29 nucleotide Fig. 6, panel C eGFP #1-1
SEQ ID NO: 30 nucleotide Fig. 6, panel C eGFP #1-2
SEQ ID NO:31 nucleotide Fig. 6, panel C eGFP #2
SEQ ID NO:32 nucleotide Fig.6, panel C eGFP #3
SEQ ID NO:33 nucleotide Fig. 7, panel C WT
SEQ ID NO:34 nucleotide Fig. 7, panel C #67-1
SEQ ID NO:35 nucleotide Fig. 7, panel C #67-2 al
SEQ ID NO:36 nucleotide Fig. 7, panel C #67-2 a2
SEQ ID NO:37 nucleotide Fig. 7, panel C #67-3
SEQ ID NO:38 nucleotide Fig. 7, panel C #67-4 al
SEQ ID NO:39 nucleotide Fig. 7, panel C #67-4 a2
SEQ ID NO:40 nucleotide Fig. 8, panel D WT
SEQ ID NO:41 nucleotide Fig. 8, panel D #166-1.1
SEQ ID NO:42 nucleotide Fig. 8, panel D #166-1.2
SEQ ID NO:43 nucleotide Fig. 8, panel D #166-2
SEQ ID NO:44 nucleotide Fig. 8, panel D #166-3.1
SEQ ID NO:45 nucleotide Fig. 8, panel D #166-3.2
SEQ ID NO:46 nucleotide Fig. 8, panel D #166-4
CA 03096022 2020-10-02
WO 2019/203807
PCT/US2018/027944
112
SEQ ID NO:47 nucleotide Fig. 16 WT CD163 partial
SEQ ID NOs. 48-67 nucleotide Primer sequences (Table 1)
SEQ ID NOs. 68-79 nucleotide Primer sequences (Table 2)
SEQ ID NOs. 80-85 nucleotide Primer sequences (Table 3)
SEQ ID NOs. 86-97 nucleotide Primer sequences (Table 4)
SEQ ID NO: 98 nucleotide Allele with 1506 bp deletion
SEQ ID NO: 99 nucleotide Allele with 7 bp insertion
SEQ ID NO: 100 nucleotide Allele with 1280 bp deletion
SEQ ID NO: 101 nucleotide Allele with 1373 bp deletion
SEQ ID NO: 102 nucleotide Allele with 11 bp deletion
SEQ ID NO: 103 nucleotide Allele with 2 bp insertion &
377 bp deletion
SEQ ID NO: 104 nucleotide Allele with 124 bp deletion
SEQ ID NO: 105 nucleotide Allele with 123 bp deletion
SEQ ID NO: 106 nucleotide Allele with 1 bp insertion
SEQ ID NO: 107 nucleotide Allele with 130 bp deletion
SEQ ID NO: 108 nucleotide Allele with 132 bp deletion
SEQ ID NO: 109 nucleotide Allele with 1467 bp deletion
SEQ ID NO: 110 nucleotide Allele with 1930 bp deletion
in exon 6,129 bp deletion in
exon 7, and 12 bp insertion
SEQ ID NO: 111 nucleotide Allele with 28 bp deletion
SEQ ID NO: 112 nucleotide Allele with 1387 bp deletion
SEQ ID NO: 113 nucleotide Allele with 1382 bp deletion
&11 bp insertion
SEQ ID NO: 114 nucleotide Allele with 1720 bp deletion
SEQ ID NO: 115 nucleotide Inserted sequence for SEQ ID
NO: 99
SEQ ID NO: 116 nucleotide Inserted sequence for SEQ ID
NO: 110
SEQ ID NO: 117 nucleotide Inserted sequence for SEQ ID
NO: 113
SEQ ID NO: 118 nucleotide Domain swap sequence
SEQ ID NO: 119 nucleotide Allele with 452 bp deletion
SEQ ID NO: 120 peptide Porcine CD163 SRCR 5
SEQ ID NO: 121 peptide Human CD163L1 SRCR 8
homolog