Note: Descriptions are shown in the official language in which they were submitted.
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
GENE THERAPY METHODS AND COMPOSITIONS USING AUXOTROPHIC
REGULATABLE CELLS
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] The present international application claiming priority to
provisional application U.S.
62/669,848 filed May 10, 2018, which is hereby incorporated by reference in
its entirety.
SEQUENCE LISTING
[0002] The present application is being filed along with a Sequence Listing
in electronic fonnat.
The Sequence Listing file, entitled 2158_1001PCT_SL.txt, was created on May
10, 2019, and is
1,970 bytes in size. The information in electronic format of the Sequence
Listing is incorporated
herein by reference in its entirety.
FIELD OF THE DISCLOSURE
[0003] The disclosure herein relates to gene therapy methods, compositions
and kits with
improved efficacy and safety.
BACKGROUND
[0004] Cell therapies have been shown to provide promising treatments. Yet,
reintroduction of
modified cells into a human host carries risks including immune reactions,
malignant
transformation, or overproduction or lack of control of transgenes.
[0005] Several approaches of genetic engineering enable the control over
functions of human
cells like cell signaling, proliferation or apoptosis (See, Bonifant, et al.
Mol. Ther. - Oncolytics 3,
16011(2016); Sockolosky et al., (2018). Science (80-. ). 359, 1037-1042 Tey,
(2014) Clin. Transl.
Immtmol. 3, el7; each of which is hereby incorporated by reference in its
entirety) and made it
possible to control even severe side effects of cell therapies (Bonifant et
al., 2016). Despite these
advances, other applications have been prevented from gaining widespread
application, e.g. the use
of engineered pluripotent cells for regenerative medicine (See, Ben-David and
Benvenisty, 2011,
Nat. Rev. Cancer 11, 268-277.; Lee et al., 2013, Nat. Med. 19, 998-1004;
Porteus, M. (2011) Mol.
Ther. 19,439-441; each of which is hereby incorporated by reference in its
entirety), due to the fact
that control systems that rely on the introduction of a genetically encoded
control mechanism into
the cell have multiple limitations (Tey, 2014).
[0006] Two of the major problems that can arise are "leakiness", i.e. low-
level activity of the
mechanism in the absence of its trigger (see, Ando et al. (2015) Stem Cell
Reports 5, 597-608,
-1-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
which is hereby incorporated by reference in its entirety), and the lack of
removal of the entire cell
population upon activation of the mechanism (see, Garin et al. (2001) Blood
97, 122-129; Di Stasi
et al. (2011) N Engl J Med 365, 1673-1683; Wu etal. (2014) N Engl J Med 365,
1673-1683;
Yagyu et al. (2015) Mol. 'Ther. 23, 1475-1485; each of which is hereby
incorporated by reference
in its entirety), due to several escape mechanisms from external control. For
example, the transgene
that is introduced by viral transduction can be silenced from expression by
the cell (see, Sulkowski
etal. (2018) Switch. Int. J. Mol. Sci. 19, 197, which is hereby incorporated
by reference in its
entirety) or the cell can develop resistance towards the effector mechanism
(See, Yagyu et al.
(2015) Mol. Ther. 23, 1475-1485, which are hereby incorporated by reference in
its entirety).
Another concern is the mutation of the transgene in cell types with genetic
instability, e.g. cell lines
that are cultured for prolonged periods of time or tumor cell lines (Merkle et
al. (2017) Nature 545,
229-233; D'Antonio etal. (2018) Cell Rep. 24, 883-894; each of which is hereby
incorporated by
reference in its entirety). Moreover, primary cell populations often retain
their functionality for
only limited time in ex vivo culture and many types cannot be purified by
clonal isolation.
[00071 Existing modes of safety switches also have a number of risks, such
as (1) transgene
insertion into a tumor suppressor leading to oncogenic transformation of the
cell line, and (2)
transgene insertion into an epigenetically silenced region leading to lack of
expression and thus
efficacy, or subsequent epigenetic silencing of the transgene after insertion.
Genome instability is a
common phenotype in oncogenic transformation of a cell. Further, a point
mutation or genetic loss
of an exogenous suicide switch would be quickly selected for and amplified. A
safety switch based
on targeting a signaling pathway of the cell depends on the physiology of the
cell. For example, a
cell that is in "pro-survival" mode may express caspase inhibitors, preventing
cell death upon
suicide switch induction.
[00081 An especially attractive application of gene therapy involves the
treatment of disorders
that are either caused by an insufficiency of a gene product or that are
treatable by increased
expression of a gene product, for example a therapeutic protein, antibody or
RNA.
(0009] Recent advances allow the precise modification of the genome of human
cells. This
genetic engineering enables a wide range of applications, but also requires
new methods to control
cell behavior. An alternative control system for cells is auxotrophy that can
be engineered by
targeting a gene in metabolism. This concept has been explored for
microorganisms (see, Steidler et
al. (2003) Nat. Biotechnol. 21, 785-789, which is hereby incorporated by
reference in its entirety)
and has been broadly used as a near universal research tool by yeast
geneticists. It would be
particularly powerful in mammalian cells if it is created by knockout of a
gene instead of by
introduction of a complex control mechanism, and if the auxotrophy is towards
a non-toxic
-2-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
compound that is part of the cell's endogenous metabolism. This could be
achieved by disruption of
an essential gene in a metabolic pathway, allowing the cell to function only
if the product of that
pathway is externally supplied and taken up by the cell from its environment.
Furthermore, if the
respective gene is also involved in the activation of a cytotoxic agent, the
gene knockout (KO)
would render the cells resistant to that drug, thereby enabling the depletion
of non-modified cells
and purification of the engineered cells in a cell population. Several
monogenic inborn errors of
metabolism can be treated by supply of a metabolite and can therefore be seen
as models of human
auxotrophy.
SUMMARY OF THE DISCLOSURE
[0010] Disclosed herein, in some embodiments, are donor templates
comprising (a) one or more
nucleotide sequences homologous to a fragment of an auxotrophy-inducing locus,
or homologous
to the complement of said auxotrophy-inducing locus, and (b) a transgene
encoding a therapeutic
factor, optionally linked to an expression control sequence. In some
instances, the donor template is
single stranded. In some instances, the donor template is double stranded. In
some instances, the
donor template is a plasmid or DNA fragment or vector. In some instances, the
donor template is a
plasmid comprising elements necessary for replication, optionally comprising a
promoter and a 3'
UTR. Disclosed herein, in some embodiments, are vectors comprising (a) one or
more nucleotide
sequences homologous to a fragment of the auxotrophy-inducing locus, or
homologous to the
complement of said auxotrophy-inducing locus, and (b) a transgene encoding a
therapeutic factor.
In some instances, the vector is a viral vector. In some instances, the vector
is selected from the
group consisting of retroviral, lentiviral, adenoviral, adeno-associated viral
and herpes simplex viral
vectors. In some instances, the vector further comprises genes necessary for
replication of the viral
vector. In some instances, the transgene flanked on both sides by nucleotide
sequences homologous
to a fragment of the auxotrophy-inducing locus or the complement thereof. In
some instances, the
auxotrophy-inducing locus is a gene encoding a protein that is involved in
synthesis, recycling or
salvage of an auxotrophic factor. In some instances, the auxotrophy-inducing
locus is within a gene
in Table 1 or within a region that controls expression of a gene in Table 1.
In some instances, the
auxotrophy-inducing locus is within a gene encoding uridine monophosphate
synthetase. In some
instances, the auxotrophy-inducing locus is within a gene encoding
holocarboxylase synthetase. In
some instances, the nucleotide sequence homologous to a fragment of the
auxotrophy-inducing
locus is 98% identical to at least 200 consecutive nucleotides of the
auxotrophy-inducing locus. In
some instances, the nucleotide sequence homologous to a fragment of the
auxotrophy-inducing
locus is 98% identical to at least 200 consecutive nucleotides of human
uridine monophosphate
-3-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
synthetase or holocarboxylase sy-nthetase or any of the genes in Table 1. In
some instances, the
donor template or vector further comprises an expression control sequence
operably linked to said
transgene. In some instances, the expression control sequence is a tissue-
specific expression control
sequence. In some instances, the expression control sequence is a promoter or
enhancer. In some
instances, the expression control sequence is an inducible promoter. In some
instances, the
expression control sequence is a constitutive promoter. In some instances, the
expression control
sequence is a posttranscriptional regulatory sequence. In some instances, the
expression control
sequence is a microRNA. In some instances, the donor template or vector
further comprises a
marker gene. In some instances, the marker gene comprises at least a fragment
of NGFR or EGFR,
at least a fragment of CD20 or CD19, Myc, HA, FLAG, GFP, an antibiotic
resistance gene. In
some instances, the transgene is selected from the group consisting of
hormones, cytokines,
chemokines, interferons, interleukins, interleukin-binding proteins, enzymes,
antibodies, Fe fusion
proteins, growth factors, transcription factors, blood factors, vaccines,
structural proteins, ligand
proteins, receptors, cell surface antigens, receptor antagonists, and co-
stimulating factors, structural
proteins, cell surface antigens, ion channels an epigenetic modifier or an RNA
editing protein. In
some instances, the transgene encodes a T cell antigen receptor. In some
instances, the transgene
encodes an RNA, optionally a regulatory microRNA.
[0011] Disclosed herein, in some embodiments, are nuclease systems for
targeting integration of
a transgene to an auxotrophy-inducing locus comprising a cas9 protein, and a
guide RNA specific
for an auxotrophy-inducing locus. Disclosed herein, in some embodiments, are
nuclease system for
targeting integration of a transgene to an auxotrophy-inducing locus
comprising a meganuclease
specific for said auxotrophy-inducing locus. In some instances, the
meganuclease is a ZFN or
TALEN. In some instances, the nuclease system further comprises a donor
template or vector
disclosed herein.
[0012] Disclosed herein, in some embodiments, are modified host cell ex
vivo, comprising a
transgene encoding a therapeutic factor integrated at an auxotrophy-inducing
locus, wherein said
modified host cell is auxotrophic for an auxotrophic factor and capable of
expressing the
therapeutic factor. In some instances, the modified host cell is a mammalian
cell. In some instances,
the modified host cell is a human cell. In some instances, the modified host
cell is an embryonic
stem cell, a stem cell, a progenitor cell, a pluripotent stem cell, an induced
pluripotent stem (iPS)
cell, a somatic stem cell, a differentiated cell, a mesenchymal stem cell, a
neural stein cell, a
hematopoietic stein cell or a hematopoietic progenitor cell, an adipose stein
cell, a keratinocyte, a
skeletal stem cell, a muscle stem cell, a fibroblast, an NK cell, a B-cell, a
T cell or a peripheral
-4-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
blood mononuclear cell (PBMC). In some instances, the modified host cell is
derived from cells
from a subject to be treated with the modified host cells.
[0013] Disclosed herein, in some embodiments, are methods of producing a
modified
mammalian host cell comprising (a) introducing into said mammalian host cell
one or more
nuclease systems that targets and cleaves DNA at the auxotrophy-inducing
locus, or a nucleic acid
encoding one or more components of said one or more nuclease systems, and (b)
a donor template
or vector disclosed herein. In some instances, the methods further comprising
introducing a second
nuclease or second guide RNA to target and cleave DNA at a second genomic
locus, or a nucleic
acid encoding said second nuclease or second guide RNA, and optionally (b) a
second donor
template or vector.
[0014] Disclosed herein, in some embodiments, are methods of targeting
integration of a
transgene to an auxotrophy-inducing locus in a mammalian cell ex vivo
comprising contacting said
mammalian cell with a donor template or vector disclosed herein, and a
nuclease. In some
instances, the nuclease is a ZFN. In some instances, the nuclease is a TALEN.
100151 Disclosed herein, in some embodiments, are methods of producing a
modified
mammalian host cell comprising introducing into said mammalian host cell with:
(a) a Cas9
polypeptide, or a nucleic acid encoding said Cas9 polypeptide, (b) a guide RNA
specific to an
auxotrophy-inducing locus, or a nucleic acid encoding said guide RNA, and (c)
a donor template or
vector disclosed herein. The methods further comprising introducing into said
mammalian host cell
with (a) a second guide RNA specific to a second auxotrophy-inducing locus, or
a nucleic acid
encoding said guide RNA, and optionally (b) a second donor template or vector.
100161 Disclosed herein, in some embodiments, are methods of targeting
integration of a
transgene to an auxotrophy-inducing locus in a mammalian cell ex vivo
comprising contacting said
mammalian cell with a donor template or vector disclosed herein, a cas9
polypeptide, and a guide
RNA. In some instances, the guide RNA is a chimeric RNA. In some instances,
the guide RNA
comprises two hybridized RNAs. In some instances, the methods produce one or
more single
stranded breaks within the auxotrophy-inducing locus. In some instances, the
methods produce a
double stranded break within the auxotrophy-inducing locus. In some instances,
the auxotrophy-
inducing locus is modified by homologous recombination using said donor
template or vector. In
some instances, the steps (a) and (b) are carried out before or after
expanding said cells, and
optionally culturing said cells. In some instances, the methods further
comprising (c) selecting cells
that contain the transgene integrated into the auxotrophy-inducing locus. In
some instances, the
selecting comprises (i) selecting cells that require the auxotrophic factor to
survive and optionally
(ii) selecting cells that comprise the transgene integrated into the
auxotrophy-inducing locus. In
-5-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
some instances, the auxotrophy-inducing locus is a gene encoding uridine
monophosphate
synthetase and the cells are selected by contacting with 5-F0A.
[0017] Disclosed herein, in some embodiments, are sterile composition
containing said donor
template or vector, or said nuclease system, and sterile water or a
pharmaceutically acceptable
excipient. Disclosed herein, in some embodiments, are sterile composition
comprising the modified
mammalian host cell and sterile water or a pharmaceutically acceptable
excipient. Disclosed herein,
in some embodiments, are kit containing said donor template or vector or
nuclease system or
modified host cell, or a combination thereof, of any of the preceding claims,
optionally with a
container or vial.
[0018] Disclosed herein, in some embodiments, are methods of expressing a
therapeutic factor
in a subject comprising (a) administering the modified host cells, (b)
optionally administering a
conditioning regime to permit modified cells to engraft, and (c) administering
the auxotrophic
factor. In some instances, the modified host cells and auxotrophic factor are
administered
concurrently. In some instances, the modified host cells and auxotrophic
factor are administered
sequentially. In some instances, administration of said auxotrophic factor is
continued regularly for
a period of time sufficient to promote expression of the therapeutic factor.
In some instances,
administration of said auxotrophic factor is decreased to decrease expression
of the therapeutic
factor. In some instances, administration of said auxotrophic factor is
increased to increase
expression of the therapeutic factor. In some instances, administration of
said auxotrophic factor is
discontinued to create conditions that result in growth inhibition or death of
the modified host cells.
In some instances, administration of said auxotrophic factor is temporarily
interrupted to create
conditions that result in growth inhibition of the modified host cells. In
some instances,
administration of said auxotrophic factor is continued for a period of time
sufficient to exert a
therapeutic effect in a subject. In some instances; the modified host cell is
regenerative. In some
instances, the administration of the modified host cell comprises localized
delivery. In some
instances, the administration of the auxotrophic factor comprises systemic
delivery. In some
instances, the host cell prior to modification is derived from the subject to
be treated.
10019) Disclosed herein, in some embodiments, are methods of treating a
subject with a disease,
a disorder, or a condition comprising administering to the subject (a) said
modified host cells and
(b) said auxotrophic factor in an amount sufficient to produce expression of a
therapeutic amount of
the therapeutic factor. In some instances, the disease; the disorder, or the
condition is selected from
the group consisting of cancer, Parkinson's disease, graft versus host disease
(GvHD), autoimmune
conditions, hyperproliferative disorder or condition, malignant
transformation, liver conditions,
genetic conditions including inherited genetic defects, juvenile onset
diabetes mellitus and ocular
-6-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
compartment conditions. In some instances, the disease, the disorder, or the
condition affects at
least one system of the body selected from the group consisting of muscular,
skeletal, circulatory,
nervous, lymphatic, respiratory endocrine, digestive, excretory, and
reproductive systems.
[0020] Disclosed herein, in some embodiments, are uses of a modified host
cell disclosed herein
for treatment of a disease, disorder or condition. Disclosed herein, in some
embodiments, are the
modified host cell disclosed herein for use in administration to humans, or
for use in treating a
disease, a disorder or a condition.
[0021] Disclosed herein, in some embodiments, are auxotrophic factor for
use in administration
to a human that has received a modified human host cell.
[0022] Disclosed herein, in some embodiments, are methods of alleviating or
treating a disease
or disorder in an subject in need thereof, the method comprising administering
to the subject: (a) a
composition comprising modified host cell comprising a transgene encoding a
protein integrated at
an auxotrophy-inducmg locus, wherein the modified host cell is auxotrophic for
an auxotrophic
factor: and (b) the auxotrophic factor in an amount sufficient to produce
therapeutic expression of
the protein. In some instances, the auxotrophy-inducing locus is within a gene
encoding uridine
monophosphate synthetase (UMPS). In some instances, the auxotrophic factor is
uridine. In some
instances, the auxotrophy-inducing locus is within a gene encoding
holocarboxylase synthetase
(HLCS). In some instances, the auxotrophic factor is biotin. In some
instances, the protein is an
enzyme. In some instances, the protein is an antibody. In some instances, the
modified host cell is
an embryonic stem cell, a stem cell, a progenitor cell, a pluripotent stem
cell, an induced
pluripotent stem (iPS) cell, a somatic stem cell, a differentiated cell, a
mesenchymal stem cell, a
neural stem cell, a hematopoietic stem cell or a hematopoietic progenitor
cell, an adipose stem cell,
a keratinocyte, a skeletal stem cell, a muscle stem cell, a fibroblast, an NK
cell, a B-cell, a T cell or
a peripheral blood mononuclear cell (PBMC). In some instances, the modified
host cell is a
mammalian cell. In some instances, the mammalian cell is a human cell. In some
instances, the
modified host cell is derived from the subject to be treated with the modified
host cell. In some
instances, the composition and the auxotrophic factor are administered
concurrently. In some
instances, the composition and the auxotrophic factor are administered
sequentially. In some
instances, the composition is administered before the auxotrophic factor. In
some instances, the
composition and the auxotrophic factor are administered concurrently. In some
instances,
administration of the auxotrophic factor is continued regularly for a period
of time sufficient to
promote therapeutic expression of the protein. In some instances,
administration of the auxotrophic
factor is decreased to decrease expression of the protein. In some instances,
administration of the
auxotrophic factor is increased to increase expression of the protein. In some
instances,
-7-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
discontinued administration of the auxotrophic factor induces growth
inhibition or cell death of the
modified host cell. In some instances, administration of the auxotrophic
factor is continued for a
period of time sufficient to exert a therapeutic effect in the subject. In
some instances, the modified
host cell is regenerative. In some instances, the administration of the
composition comprises
localized delivery. In some instances, the administration of the auxotrophic
factor comprises
systemic delivery. In some instances, the disease is a lysosomal storage
disease (LSD). In some
instances, the LSD is Gaucher's Disease (Type 1/2/3), MPS2 (Hunter's) disease,
Pompe disease,
Fabry disease, Krabbe disease, Hypophosphatasia, Niemann-Pick disease type
A/B, MPS I,
MPS3A, MPS3B, MPS3C, MPS3, MPS4, MPS6, MPS7, Phenylketonuria, MLD, Sandhoff
disease,
Tay-Sachs disease, or Battens disease. In some instances, the enzyme is
Glucocerebrosidase,
Idursulfase, Alglucosidase alfa, Agalsidase alfa/beta, Galactosylceramidase,
Asfotase alfa, Acid
Sphingomyelinase, Laronidase, heparan N-sulfatase, alpha-N-
acetylglucosatninidase, heparan-a-
glucosaminide N-acetyltransferase. N-acetylglucosamine 6-sulfatase, Elosulfase
alfa, Glasulfate, B-
Glucoronidase, Phenylalanine hydroxylase, Arylsulphatase A, Hexosaminidase-B,
Hexosaminidase-A, or tripeptidyl peptidase I. In some instances, the disease
is Friedreich's ataxia,
Hereditary angioedema, or Spinal muscular atrophy. In some instances, the
protein is frataxin, Cl
esterase inhibitor (which may also be referred to as HAEGAARDA subcutaneous
injection) or
SMN 1.
(00231 Various embodiments described herein provide a method of reducing the
size of a tumor
or reducing a rate of growth of a tumor in a subject, the method comprising:
administering to the
subject a modified human host cell as described herein.
INCORPORATION BY REFERENCE
[00241 All publications, patents, and patent applications mentioned in this
specification are
herein incorporated by reference to the same extent as if each individual
publication, patent, or
patent application was specifically and individually indicated to be
incorporated by reference.
BRIEF DESCRIPTION OF THE DRAWINGS
100251 The features of the subject matter encompassed by the present
disclosure are set forth
with particularity in the appended claims. A better understanding of the
features and advantages of
the present disclosure will be obtained by reference to the following detailed
description that sets
forth illustrative embodiments, in which the principles of the subject matter
encompassed by the
disclosure herein are utilized, and the accompanying drawings of which:
-8-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0026] FIG. I A and FIG. 1B exemplify the effect of serum on optimal recovery
post-
electroporation. FIG. IA is an exemplary schematic of assay used to determine
optimal
electroporation recovery conditions. Following electroporation, cells were
supplied with/without
serum, 5-fluoroorotic acid (5-F0A), or an exogenous uracil source (uridine).
FIG. 1B illustrates
cell counts by CytoFLEX flow cytometer (Beckman Coulter) after 4 days of
recovery post
electroporation in indicated media conditions. The figure shows cells
administered serum, mock
edited cells treated with/without 5-FOA with no serum, and uridine
monophosphate synthetase
(UMPS) knockout cells treated with/without 5-FOA without serum.
100271 FIG. 2A-FIG. 2F exemplifies that maintenance and growth of UMPS InDel
containing
cells requires an exogenous uracil source. FIG. 2A is an exemplary schematic
of the procedure used
to assay for growth of UMPS or mock edited T cells following electroporation
and recovery. FIG.
2B illustrates tracking of indels by decomposition (TIDE) analysis of UMPS
InDels in indicated
culture conditions. TIDE analysis was performed on sanger sequencing of UMPS
locus with
oligonucleotides UMPS-0-1 and UMPS-O-2. FIG. 2C illustrates percentage of
alleles containing
frameshift InDels analyzed by TIDE performed on day 8. FIG. 2D illustrates
predicted absolute
numbers of cells at day 8 containing alleles identified by TIDE. FIG. 2E
illustrates time course of
cell counts with/without UMP. FIG. 2F illustrates time course of cell counts
with/without uridine.
[0028] FIG. 3A-FIG. 3C exemplifies that 5-FOA is less toxic in MPS targeted
cell lines. FIG.
3A is an exemplary schematic of 5-FOA selection procedure. FIG. 3B and FIG. 3C
illustrate cell
counts after 4 days of 5-FOA selection in indicated culture conditions. In
FIG. 3B and FIG. 3C, the
mock results are represented by the left bar for each culture condition, and
the results for UMPS-7
are shown by the right bar for each culture condition.
[0029] FIG. 4A-FIG. 4D exemplifies that 5-FOA selected, UMPS targeted cell
lines exhibit
optimuin growth only in the presence of an exogenous uracil source. FIG. 4A is
an exemplary
schematic of protocol for the demonstration of uracil auxotrophy. Cell
cultures were split following
4-day selection in 5-FOA into test media and grown for 4 further days before
cell counting. FIG.
4B-FIG. 4D illustrate cell counts of 5-FOA selected cells in exogenous uracil
(UMP or uridine)
containing or deficient media.
[0030] FIG. 5A exemplifies InDel quantification performed at the UMPS locus by
the ICE
analysis. FIG. 5B exemplifies proliferation of T cells after mock treatment,
CCR5 knockout or
UMPS knockout. FIG. 5C illustrates proliferation of T cells with MPS knockout
with or without
UMP or Uridine. FIG. 5D illustrates InDel frequency on day 8 after UMPS
knockout with different
culture conditions. FIG. 5E illustrates the frequency of InDels that are
predicted to lead to a
frameshift.
-9-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0031] FIG. 6A exemplifies DNA donor constructs for targeting of the MPS
locus. FIG. 6B
illustrates expression of surface markers after targeting of K562 cells. FIG.
6C exemplifies
targeting approach to integrate Nanoluciferase and green fluorescent protein
(GFP) into the HBB
locus. FIG. 6D illustrates expression of the 3 integrated markers in K562
cells before cell sorting.
FIG. 6E illustrates K562 cell growth and cell counts on day 8 when cultured in
the presence of
different Uridine concentrations. FIG. 6F illustrates selection of UMPSK"
cells during culture
with 5-FOA. FIG. 6G illustrates proliferation of UMPS K0 cells in the presence
of 5-F0A.
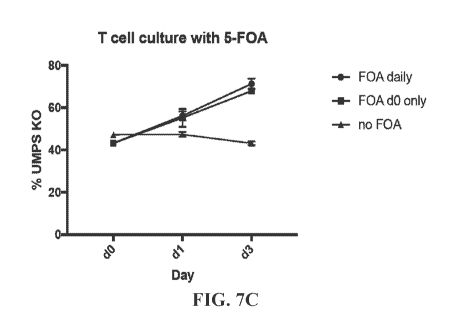
[0032] FIG. 7A exemplifies surface marker expression after IMPS targeting of T
cells. FIG. 7B
illustrates auxotrophic growth of UMPS K or wild-type (WT) T cells. FIG. 7C
illustrates that 5-
FOA selects for T cells with UMPS knockout.
100331 Groups were compared by statistical tests as indicated using Prism 7
(GraphPad).
Asterisks indicate levels of statistical significance: * = p<0.05, ** =
p<0.01, *** = p<0.001, and
**** p<0.0001.
DETAILED DESCRIPTION
I. Introduction
100341 Recent advances allow the precise modification of the genome of human
cells. This
genetic engineering enables a wide range of applications, but also requires
new methods to control
cell behavior. An alternative control system for cells is auxotrophy that can
be engineered by
targeting a gene in metabolism. The approach described herein of genetically
engineering
auxotrophy by disruption of a central gene of metabolism is an alternative
paradigm to create an
external control mechanism over cell function which has not been explored for
human cells. By
disrupting a key gene in pyiimidine metabolism, a passive containment system
was created
(Steidler et al., 2003), which is an addition and alternative to the already
existing toolbox of
systems for human cells that circumvents their previously mentioned
limitations. It enables the
control over growth of human cells through the addition or withdrawal of the
non-toxic substance
uridine. Auxotrophy has previously been engineered in microorganisms, e.g.
towards an unnatural
substance by introduction of an engineered gene circuit (see, Kato, Y. (2015)
An engineered
bacterium auxotrophic for an unnatural amino acid: a novel biological
containment system. Peed 3,
el247, which is hereby incorporated by reference in its entirety) or towards
pyrimidines by
knockout of a bacterial gene (see, Steidler et al. (2003) Nat. Biotechnol. 21,
785-789, which is
hereby incorporated by reference in its entirety). The latter concept is
appealing, since it relies on
the knockout of a gene instead of the introduction of complex expression
cassettes, which impedes
the cell from reversing the genetic modification or the development of
resistance mechanisms, and
-10-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
therefore addresses this challenge of alternative systems. The fact that
pyrimidine nucleosides and
nucleotides play an essential role in a wide array of cellular processes,
including DNA and RNA
synthesis, energy transfer, signal transduction and protein modification (see,
van Kuilenburg,
A.B.P. and Meinsma, R. (2016). Biochem. Biophys. Acta - Mol. Basis Dis. 1862,
1504-1512,
which is hereby incorporated by reference in its entirety) makes their
synthesis pathway a
theoretically appealing target.
[0035] Human cells are naturally auxotrophic for certain compounds like amino
acids that they
have to acquire, either from external sources or symbiotic organisms (See,
Murray, P.J. (2016).
Nat. Immunol. 17, 132-139, which is hereby incorporated by reference in its
entirety).
Additionally, auxotrophy is a natural mechanism to modulate the function of
immune cells, e.g. by
differential supply or depletion of the metabolite that the cells are
auxotrophic for (See, Grohmann
et al., (2017). Cytokine Growth Factor Rev. 35, 37-45, which is hereby
incorporated by reference
in its entirety). Cellular auxotrophy also plays an important role in
mechanisms of defense against
malignant growth, e.g. in the case of macrophages that inhibit tumor growth by
scavenging arginine
(Murray, 2016). In addition, several malignant cell types have been shown to
be auxotrophic for
certain metabolites (see, Fung, M.K.L. and Chan, G.C.F. (2017). J. Hematol.
Oncol. 10, 144, which
is hereby incorporated by reference in its entirety), which is exploited by
the therapeutic depletion
of asparagine for the treatment of leukemia patients (See, Hill et al.,
(1967). JAMA 202, 882).
100361 In addition to the previously developed containment strategies for
microorganisms, the
approach described herein using gene editing based on Cas9 ribonucleoprotein
(RNP)/rAAV6
allows for highly efficient engineering of a primary and therapeutically
relevant human cell type.
Auxotrophy and resistance to 5-FOA are inherent to all cells with complete
disruption of the IMPS
gene, but to show proof-of- concept, the identification of the populations was
facilitated with bi-
allelic knockout by targeted integration of selection markers. The recent
development of methods
that allow the efficient targeted modification of primary human cells (Bak et
al. (2017).
[0037] Multiplexed genetic engineering of human hematopoietic stem and
progenitor cells using
CRISPR/Cas9 and AAV6. Elife 6, e27873; Bak, et al. (2018). Nat. Protoc. 13,
358-376; Porteus,
M.H. and Baltimore, D. (2003). Science (80-.). 300, 763-763; each of which is
hereby
incorporated by reference in its entirety) together with the establishment of
metabolic auxotrophy
lays the foundation for further development of therapeutic approaches in
settings where the use of
human cells is necessary, e.g. in the use of stem cells or stein-cell derived
tissues or other
autologous somatic cells with specific effector functions and reduced
immunogenicity. Notably,
constructs and reagents have been used that would facilitate expedited
clinical translation, e.g.
-11-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
selection markers tNGFR and tEGFR in the targeting constructs, which avoid
immunogenicity, and
uridine supplied in the in vivo model using its FDA-approved prodrug.
100381 Engineered mechanisms to control cell function have the additional
challenge of
selecting an entirely pure population of cells that express the proteins
mediating the control
mechanism. The possibility of selecting the engineered cells by rendering them
resistant to a
cytotoxic agent is particularly appealing since it can substantially increase
efficiency by allowing
the creation of a highly pure population of cells that can be controlled using
a non-toxic substance,
and the removal of a gene crucial for the function of a vital metabolic
pathway prevents cells from
developing escape mechanisms. Therefore, this method offers several advantages
over existing
control mechanisms in settings where genetic instability and the risk of
malignant transfonnation
play a role and where even small numbers of cells that escape their
containment can have disastrous
effects, e.g. in the use of somatic or pluripotent stem cells.
100391 This concept has been explored for microorganisms (Steidler et al.,
2003) and has been
broadly used as a near universal research tool by yeast geneticists. It would
be particularly powerful
in mammalian cells if it is created by knockout of a gene instead of by
introduction of a complex
control mechanism, and if the auxotrophy is towards a non-toxic compound that
is part of the cell's
endogenous metabolism. This could be achieved by disruption of an essential
gene in a metabolic
pathway, allowing the cell to function only if the product of that pathway is
externally supplied and
taken up by the cell from its environment. Furthermore, if the respective gene
is also involved in
the activation of a cytotoxic agent, the gene knockout (KO) would render the
cells resistant to that
drug, thereby enabling the depletion of non-modified cells and purification of
the engineered cells
in a cell population. Several monogenic inborn errors of metabolism can be
treated by supply of a
metabolite and can therefore be seen as models of human auxotrophy.
[00401 In certain embodiments, auxotrophy is introduced to human cells by
disrupting IMPS in
the de novo pyrimidine synthesis pathway through genome editing. This makes
the cell's function
dependent on the presence of exogenous uridine. Furthermore, this abolishes
the cell's ability to
metabolize 5-fluoroorotic acid into 5-F1.1, which enables the depletion of
remaining cells with intact
UMPS alleles. The ability to use a metabolite to influence the function of
human cells by
genetically engineered auxotrophy and to deplete other cells provides for the
development of this
approach for a range of applications where a pure population of controllable
cells is necessary.
100411 One example is hereditary orotic aciduria, in which mutations in the
UMPS gene lead to
a dysfunction that can be treated by supplementation with high doses of
uridine (See, Fallon et al
(1964). N. Engl. J. Med. 270, 878-881, which is hereby incorporated by
reference in its entirety).
Transferring this concept to a cell type of interest, genetic engineering is
used to knock out the
-12-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
UMPS gene in human cells which makes the cells auxotrophic to uridine and
resistant to 5-
fluoroorotic acid (5-F0A). We show that UMPS' cell lines and primary cells
survive and
proliferate only in the presence of uridine in vitro, and that LIMPS
engineered cell proliferation is
inhibited without supplementation of uridine in vivo. Furthermore, the cells
can be selected from a
mixed population by culturing in the presence of 5-F0A.
[0042] in certain embodiments, auxotrophy is introduced to human cells by
disrupting (IMPS in
the de-novo pyrimidine synthesis pathway through genome editing. This makes
the cell's function
dependent on the presence of exogenous uridine. Furthermore, this abolishes
the cell's ability to
metabolize 5-fluoroorotic acid into 5-FU, which enables the depletion of
remaining cells with intact
(IMPS alleles. The ability to use a metabolite to influence the function of
human cells by
genetically engineered auxotrophy and to deplete other cells provides for the
development of this
approach fora range of applications where a pure population of controllable
cells is necessary.
[0043] One example of an auxotrophy is hereditary orotic aciduria, in which
mutations in the
LIMPS gene lead to a dysfunction that can be treated by supplementation with
high doses of uridine
(Fallon et al., 1964). Transferring this concept to a cell type of interest,
genetic engineering was
used to knock out the (IMPS gene in human cells which makes the cells
auxotrophic to uridine and
resistant to 5- fluoroorotic acid (5-F0A). UMPS cell lines and primary cells
are shown herein to
survive and proliferate only in the presence of uridine in vitro, and that
LIMPS engineered cell
proliferation is inhibited without supplementation of uridine in vivo.
Furthermore, the cells can be
selected from a mixed population by culturing in the presence of 5-F0A.
II. Compositions and Methods of Use of Certain Embodiments
[0044] Disclosed herein are some embodiments of methods and compositions for
use in gene
therapy. In some instances, the methods comprise delivery of a transgene,
encoding a therapeutic
factor, to host cells in a manner that renders the modified host cell
auxotrophic, and that can
provide improved efficacy, potency, and/or safety of gene therapy through
transgene expression.
Delivery of the transgene to a specific auxotrophy-inducing locus creates an
auxotrophic cell, for
example, through disruption or knockout of a gene or downregulation of a
gene's activity, that is
now dependent on continuous administration of an auxotrophic factor for growth
and reproduction.
In some instances, the methods comprise nuclease systems targeting the
auxotrophy-inducing
locus, donor templates or vectors for inserting the transgene, kits, and
methods of using such
systems, templates or vectors to produce modified cells that are auxotrophic
and capable of
expressing the introduced transgene.
[0045] .. Also disclosed herein, in some embodiments, are methods,
compositions and kits for use
of the modified host cells, including pharmaceutical compositions, therapeutic
methods, and
-13-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
methods of administration of auxotrophic factors to control ¨ increase,
decrease or cease - the
growth and reproduction of the modified cells and to control the expression of
the transgene and to
control levels of the therapeutic factor.
[0046] In some instances, delivery of the transgene to the desired locus
can be accomplished
through methods such as homologous recombination. As used herein, "homologous
recombination
(HR)" refers to insertion of a nucleotide sequence during repair of double-
strand breaks in DNA via
homology-directed repair mechanisms. This process uses a "donor" molecule or
"donor template"
with homology to nucleotide sequence in the region of the break as a template
for repairing a
double-strand break. The presence of a double-stranded break facilitates
integration of the donor
sequence. The donor sequence may be physically integrated or used as a
template for repair of the
break via homologous recombination, resulting in the introduction of all or
part of the nucleotide
sequence. This process is used by a number of different gene editing platforms
that create the
double-strand break, such as meganucleases, such as zinc finger nucleases
(ZFNs), transcription
activator-like effector nucleases (TALENs), and the CRISPR-Cas9 gene editing
systems.
[0047] In some embodiments, genes are delivered to two or more loci, for
example, for the
expression of multiple therapeutic factors, or for the introduction of a
second gene that acts as a
synthetic regulator or that acts to bias the modified cells towards a certain
lineage (e.g. by
expressing a transcription factor from the second locus). In some embodiments,
genes are delivered
to two or more auxotrophy-inducing loci. For example, a different gene or a
second copy of the
same gene is delivered to a second auxotrophy-inducing locus.
[0048] In some embodiments, the cell is auxotrophic because the cell no
longer has the ability to
produce the auxotrophic factor. As used herein, a "cell", "modified cell" or
"modified host cell"
refers to a population of cells descended from the same cell, with each cell
of the population having
a similar genetic make-up and retaining the same modification.
[0049] In some embodiments, the auxotrophic factor comprises one or two or
more nutrients,
enzymes, altered pH, altered temperature, non-organic molecules, non-essential
amino acids, or
altered concentrations of a moiety (compared to normal physiologic
concentrations), or
combinations thereof. All references to auxotrophic factor herein contemplate
administration of
multiple factors. In any of the embodiments described herein, the auxotrophic
factor is a nutrient or
enzyme that is neither toxic nor bioavailable in the subject in concentrations
sufficient to sustain
the modified host cell, and it is to be understood that any references to
"auxotrophic factor"
throughout this application may include reference to a nutrient or enzyme.
[0050] In some instances, if the modified cell is not continuously supplied
with the auxotrophic
factor, the cell ceases proliferation or dies. In some instances, the modified
cell provides a safety
-14-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
switch that decreases the risks associated with other cell-based therapies
that include oncogenic
transformation.
[0051] The methods and compositions disclosed herein provide a number of
advantages, for
example: consistent results and conditions due to integrating into the same
locus rather than random
integration such as with lentivectors; constant expression of transgene
because areas with native
promoters or enhancers or areas that are silenced are avoided; a consistent
copy number of
integration, 1 or 2 copies, rather than a Poisson distribution; and limited
chance of oncogenic
transformation. In some instances, the modified cells of the present
disclosure are less
heterogeneous than a product engineered by lentivector or other viral vector.
[0052] In some embodiments, disclosed herein, are counter selection methods
to generate a
population of cells which are 100% auxotrophic, limiting the probability of
reversion to a non-
auxotrophic state. Current safety switches rely on inserting a transgene, and
modified cells can
escape through mutation of the transgene or epigenetic silencing of its
expression (see, e.g., Wu et
Mol Ther Methods Clin Dev. 1:14053 (2014), which is hereby incorporated by
reference in its
entirety). Thus, the combination of transgene insertion with creation of an
auxotrophic mechanism
is generally safer in the long term.
[0053] In some embodiments, reducing the auxotrophic factor administration to
low levels may
cause the modified cells to enter a quiescent state rather than being killed,
permitting temporary
interruption and re-starting of therapy with cells already present in the
host. This would be an
advantage compared to having to re-edit host cells and re-introduce modified
host cells.
[0054] In some embodiments, ceasing auxotrophic factor administration will
result in death of
the modified cells when that is desired, for example if aberrant proliferation
or oncogenic
transformation has been detected, or if cessation of treatment is desired.
[0055] In some embodiments, increasing auxotrophic factor administration
increases growth and
reproduction of the modified cells and results in increased expression of the
transgene, and thus
increased levels of the therapeutic factor. In some instances, the auxotrophic
factor administration
provides a means for controlling dosage of the gene product.
[0056] An auxotrophy-based safety mechanism circumvents many of the risks to
patients
associated with current cell therapies. By supplementing a patient with a
defined auxotrophic factor
during the course of the therapy and removing the factor upon therapy
cessation or some other
safety-based indication, cell growth is physically limited. In some instances,
if the auxotrophic
factor is no longer available to the cell, then the cell stops dividing and
does not have a self-evident
mechanism for the development of resistance. By manipulating levels of the
auxotrophic factor, the
growth rate of cells in vivo is controlled. Multiple cell lines may be
controlled independently in
-15-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
vivo by using separate auxotrophies. Location specific growth may be
controlled by localized
nutrient release, such as exogenously grown pancreatic B cells administered
within a biocompatible
device that releases a nutrient and prevents cell escape. For example, the
methods and compositions
disclosed herein may be used in conjunction with chimeric antigen receptor
(CAR)-T cell
technology, to allow more defined control over the activity of CAR-T cells in
vivo. In some
instances, the compositions disclosed herein are used to inhibit or reduce
tumor growth. For
example, withdrawal of the auxotrophic factor (e.g. uridine or biotin) may
lead to tumor regression.
[0057] A considerable number of disorders are either caused by an
insufficiency of a gene
product or are treatable by increased expression of a therapeutic factor, e.g.
protein, peptide,
antibody, or RNA. In some embodiments, disclosed herein, are compositions
comprising modified
host cell comprising a transgene encoding a therapeutic factor of interest
integrated at an
auxotrophy-inducing locus, wherein the modified host cell is auxotrophic for
an auxotrophic factor.
Further disclosed herein, in some embodiments, are methods of using the
compositions of the
current disclosure to treat conditions in an individual in need thereof by
providing the auxotrophic
factor in an amount sufficient to produce therapeutic expression of the
factor.
Exemplary therapeutic factors
[00581 The following embodiments provide conditions to be treated by producing
a therapeutic
factor in an auxotrophic host cell.
[0059] Clotting disorders, for example, are fairly common genetic disorders
where factors in the
clotting cascade are absent or have reduced function due to a mutation. These
include hemophilia A
(factor VIII deficiency), hemophilia B (factor IX deficiency), or hemophilia C
(factor XI
deficiency).
[0060] Alpha-1 antitrypsin (A lAT) deficiency is an autosomal recessive
disease caused by
defective production of alpha 1-antitrypsin which leads to inadequate A lAT
levels in the blood and
lungs. It can be associated with the development of chronic obstructive
pulmonary disease (COPD)
and liver disorders.
[0061] Type I diabetes is a disorder in which immune-mediated destruction
of pancreatic beta
cells results in a profound deficiency of insulin production. Complications
include ischemic heart
disease (angina and myocardial infarction), stroke and peripheral vascular
disease, diabetic
retinopathy, diabetic neuropathy, and diabetic nephropathy, which may result
in chronic kidney
disease requiring dialysis.
[0062] Antibodies are secreted protein products used for neutralization or
clearance of target
proteins that cause disease as well as highly selective killing of certain
types of cells (e.g. cancer
cells, certain immune cells in autoimmune diseases, cells infected with virus
such as human
-16-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
immunodeficiency virus (HIV), RSV, Flu, Ebola, CMV, and others). Antibody
therapy has been
widely applied to many human conditions including oncology, rheumatology,
transplant, and ocular
disease. In some instances, the therapeutic factor encoded by the compositions
disclosed herein is
an antibody used to prevent or treat conditions such as cancer, infectious
diseases and autoimmune
diseases. In certain embodiments, the cancer is treated by reducing the rate
of growth of a tumor or
by reducing the size of a tumor in the subject.
100631 Monoclonal antibodies approved by the FDA for therapeutic use include
Adalimumab.
Bezlotoxumab, Avelumab, Dupilumab, Durvalumab, Ocrelizumab, Brodalumab,
Reslizumab,
Olaratumab, Daratttmumab, Elotuzumab, Necitumumab, Infliximab, Obiltoxaximab,
Atezolizumab, Secukinumab, Mepolizumab, Nivolumab, Alirocumab, Idaruciztunab,
Evolocumab,
Dinutuximab, Bevacizumab, Pembrolizumab, Ramucirumab, Vedolizumab, Siltuximab,
Alemtuzumab, Trastuzumab emtansine, Pertuzumab, Infliximab, Obinutuzumab,
Brentuximab,
Raxibacumab, Belimumab, 1pilimumab, Denosumab, Denosumab, Ofattunumab,
Besilesomab,
Tocilizumab, Canakinumab, Golimumab, Ustekinumab, Certolizumab pegol,
Catumaxomab,
Eculizumab, Ranibizumab, Panitumumab, Natalizumab, Catumaxomab, Bevacizumab,
Omalizumab, Cetuximab, Efalizumab, Ibritumomab tiuxetan, Fanolesomab,
Adalimtunab,
Tosittimomab, Alemturtunab, Trastuzumab, Gemtuzumab ozogamicin, lnfliximab,
Palivizumab,
Necitumumab, Basiliximab, Rituximab, Votumumab, Sulesomab, Arcittunomab,
Imiciromab,
Capromab, Nofetumomab, Abciximab, Satumomab, and Muromonab-CD3. Bispecific
antibody
approved by the FDA for therapeutic use includes Blinatumomab. In some
embodiments, the
antibody is used to prevent or treat HIV or other infectious diseases.
Antibodies for use in treatment
of HIV include human monoclonal antibody (mAb) VRC-FI1VMAB060-00-AB (VRC01);
mAb
VRC-HIVMAB080-00-AB (VRCOlLS); mAb VRC-HIVMAB075-00-AB (VRC07-523LS); mAb
F105; mAb C2F5; mAb C2G12; mAb C4E10; antibody UB-421 (targeting the HIV-1
receptor on
the CD4 molecule (domain 1) of T-lymphocytes and monocytes); Ccr5mab004 (Human
Monoclonal IgG4 antibody to Ccr5); mAb PGDM1400; mAb PGT121 (recombinant human
TgGI
monoclonal antibodies that target a V1V2 (PGDM1400) and a V3 glycan-dependent
(PGT121)
epitope region of the HIV envelope protein); KD-247 (a humanized monoclonal
antibody); PRO
140 (a monoclonal CCR5 antibody); mAb 3BNC117; and PG9 (anti-HIV-1 gp120
monoclonal
antibody).
[00641 Therapeutic RNAs include antisense, siRNAs, aptamers, microRNA
mimics/anti-miRs
and synthetic mRNA, and some of these can be expressed by transgenes.
100651 LSDs are inherited metabolic diseases that are characterized by an
abnomial build-up of
various toxic materials in the body's cells as a result of enzyme
deficiencies. There are nearly 50 of
-17-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
these disorders altogether, and they affect different parts of the body,
including the skeleton, brain,
skin, heart, and central nervous system. Common examples include
Sphingolipidoses, Farber
disease (ASAH1 deficiency), Krabbe disease (galactosylceramidase or GALC
deficiency),
Galactosialidosis, Gangliosidoses, Alpha-galactosidase, Fain), disease (a-
galactosidase
deficiency¨GLA, or agalsidase alpha/beta), Schindler disease (alpha-NAGA
deficiency), GM!
gangliosidosis, GM2 gangliosidoses (beta-hexosaminidase deficiency), Sandhoff
disease
(hexosaminidase-B deficiency), Tay-Sachs disease (hexosaminidase-A
deficiency), Gaucher's
disease Type 1/2/3 (glucocerebrosidase deficiency-gene name: GBA), Wolman
disease (LAL
deficiency), Niemann-Pick disease type A/B (sphingomyelin phosphodiesterase
1deficiency--
SMPD1 or acid sphingomyelinase), Sulfatidosis, Metachromatic leukodystrophy,
Hurler syndrome
(alpha-L iduronidase deficiency--IDUA), Hunter syndrome or MPS2 (iduronate-2-
sulfatase
deficiency-idursulfase or IDS), Sanfilippo syndrome, Morquio, Maroteaux-Lamy
syndrome, Sly
syndrome (0-glucuronidase deficiency), Mucolipidosis, I-cell disease,
Lipidosis, = Neuronal ceroid
lipofuscinoses, Batten disease (tripeptidyl peptidase-1 deficiency), Pompe
(alglucosidase alpha
deficiency), hypophosphatasia (asfotase alpha deficiency), MPS1 (laronidase
deficiency), MPS3A
(heparin N-sulfatase deficiency), MPS3B (alpha-N-acetylglucosaminidase
deficiency), MPS3C
(heparin-a-glucosaminide N-acetyltransferase deficiency), MPS3D (N-
acet3,71glucosamine 6-
sulfatase deficiency), MPS4 (elosulfase alpha deficiency), MPS6 (glasulfate
deficiency), MPS7 (B-
glucoronidase deficiency), phenylketonuria (phenylalanine hydroxylase
deficiency), and MLD
(arylsulphatase A deficiency). Collectively LSDs have an incidence in the
population of about 1 in
7000 births and have severe effects including early death. While clinical
trials are in progress on
possible treatments for some of these diseases, there is currently no approved
treatment for many
LSDs. Current treatment options for some but not all LSDs include enzyme
replacement therapy
(ERT). ERT is a medical treatment which replaces an enzyme that is deficient
or absent in the
body. In some instances, this is done by giving the patient an intravenous
(IV) infusion of a
solution containing the enzyme.
100661 Disclosed herein, in some embodiments, are methods of treating a LSD in
an individual
in need thereof, the method comprising providing to the individual enzyme
replacement therapy
using the compositions disclosed herein. In some instances, the method
comprises a modified host
cell ex vivo, comprising a transgene encoding an enzyme integrated at an
auxotrophy-inducing
locus, wherein said modified host cell is auxotrophic for an auxotrophic
factor and capable of
expressing the enzyme that is deficient in the individual, thereby treating
the LSD in the individual.
In some instances, the auxotrophy-inducing locus is within a gene in Table 1
or within a region that
controls expression of a gene in Table 1. In some instances, the auxotrophy-
inducing locus is
-18-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
within a gene encoding uridine monophosphate synthetase (UMPS). In some
instances, the
auxotrophic factor is uridine. In some instances, the auxotrophy-inducing
locus is within a gene
encoding holocarboxylase synthetase (HLCS). In some instances, the auxotrophic
factor is biotin.
In some instances, the auxotrophy-inducing locus is within a gene encoding
asparagine synthetase.
In some instances, the auxotrophic factor is asparagine. In some instances,
the auxotrophy-inducing
locus is within a gene encoding aspartate transaminase. In some instances, the
auxotrophic factor is
aspartate. In some instances, the auxotrophy-inducing locus is within a gene
encoding alanine
transaminase. In some instances, the auxotrophic factor is alanine. In some
instances, the
auxotrophy-inducing locus is within a gene encoding cystathionine beta
syrithase. In some
instances, the auxotrophic factor is cysteine. In some instances, the
auxotrophy-inducing locus is
within a gene encoding cystathionine gamma-lyase. In some instances, the
auxotrophic factor is
cysteine. In some instances, the auxotrophy-inducing locus is within a gene
encoding glutamine
synthetase. In some instances, the auxotrophic factor is glutamine. In some
instances, the
auxotrophy-inducing locus is within a gene encoding serine
hydroxymethyltransferase. In some
instances, the auxotrophic factor is serine or glycine. In some instances, the
auxotrophy-inducing
locus is within a gene encoding glycine syrithase. In some instances, the
auxotrophic factor is
glycine. In some instances, the auxotrophy-inducing locus is within a gene
encoding phosphoserine
transaminase. In some instances, the auxotrophic factor is serine. In some
instances, the
auxotrophy-inducing locus is within a gene encoding phosphoserine phospha ase.
In some
instances, the auxotrophic factor is serine. In some instances, the auxotrophy-
inducing locus is
within a gene encoding phenylalanine hydroxylase. In some instances, the
auxotrophic factor is
tyrosine. In some instances, the auxotrophy-inducing locus is within a gene
encoding
argininosuccinate synthetase. In some instances, the auxotrophic factor is
arginine. In some
instances, the auxotrophy-inducing locus is within a gene encoding
argininosuccinate lyase. In
some instances, the auxotrophic factor is arginine. In some instances, the
auxotrophy-inducing
locus is within a gene encoding dihydrofolate reductase. In some instances,
the auxotrophic factor
is folate or tetrahydrofolate.
100671 Further disclosed herein, in some embodiments, are methods of
treating a disease or
disorder in an individual in need thereof, the method comprising providing to
the individual protein
replacement therapy using the compositions disclosed herein. In some
instances, the method
comprises a modified host cell ex vivo, comprising a transgene encoding a
protein integrated at an
auxotrophy-inducing locus, wherein said modified host cell is auxotrophic for
an auxotrophic factor
and capable of expressing the protein that is deficient in the individual,
thereby treating the disease
or disorder in the individual. In some instances, the auxotrophy-inducing
locus is within a gene in
-19-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
Table 1 or within a region that controls expression of a gene in Table 1. In
some instances, the
auxotrophy-inducing locus is within a gene encoding uridine monophosphate
synthetase (UMPS).
In some instances, the auxotrophic factor is uridine. In some instances, the
auxotrophy-inducing
locus is within a gene encoding holocarboxylase synthetase (HLCS). In some
instances, the
auxotrophic factor is biotin. In some instances, the disease is Friedreich's
ataxia, and the protein is
frataxin. In some instances, the disease is hereditary angioedema and the
protein is Cl esterase
inhibitor (e.g., HAEGAARDA subcutaneous injection). In some instances, the
disease is spinal
muscular atrophy and the protein is SMNI.
III. Compositions and Methods for Making Modified Cells
A. Cells
[0068] Disclosed herein, in some embodiments, are compositions comprising
modified host
cells, preferably human cells, that are genetically engineered to be
auxotrophic (through insertion
of a transgene encoding a therapeutic factor at an auxotrophy-inducing locus)
and are capable of
expressing the therapeutic factor. Animal cells, mammalian cells, preferably
human cells, modified
ex vivo, in vitro, or in vivo are contemplated. Also included are cells of
other primates: mammals,
including commercially relevant mammals, such as cattle, pigs, horses, sheep,
cats, dogs, mice,
rats; birds, including commercially relevant birds such as poultry, chickens,
ducks, geese, and/or
turkeys.
[0069] In some embodiments, the cell is an embryonic stem cell, a stem
cell, a progenitor cell, a
pluripotent stem cell, an induced pluripotent stem (iPS) cell, a somatic stem
cell, a differentiated
cell, a mesenchymal stem cell or a mesenchymal stromal cell, a neural stein
cell, a hematopoietic
stem cell or a hematopoietic progenitor cell, an adipose stem cell, a
keratinocyte, a skeletal stem
cell, a muscle stem cell, a fibroblast, an NK cell, a B-cell, a T cell, or a
peripheral blood
mononuclear cell (PBMC). For example, the cell may be engineered to express a
CAR, thereby
creating a CAR-T cell. In some embodiments, the cell lines are T cells that
are genetically
engineered to be auxotrophic. Engineered auxotrophic T cells may be
administered to a patient with
cancer along with an auxotrophic factor. Upon destruction of the cancer, the
auxotrophic nutrient
may be removed, which results in the elimination of the engineered auxotrophic
T cells. In some
embodiments, the cell lines are pluripotent stem cells that are genetically
engineered to be
auxotrophic. Engineered auxotrophic pluripotent stem cells may be administered
to a patient along
with an auxotrophic factor. Upon conversion of an engineered auxotrophic
pluripotent stem cell to
a cancerous cell, the auxotrophic factor may be removed, which results in the
elimination of the
cancerous cell and the engineered auxotrophic pluripotent stem cells
-20-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[00701 To prevent immune rejection of the modified cells when administered
to a subject, the
cells to be modified are preferably derived from the subject's own cells.
Thus, preferably the
mammalian cells are from the subject to be treated with the modified cells. In
some instances, the
mammalian cells are modified to be autologous cell. In some instances, the
mammalian cells are
further modified to be allogeneic cell. In some instances, modified T cells
can be further modified
to be allogeneic, for example, by inactivating the T cell receptor locus. In
some instances, modified
cells can further be modified to be allogeneic, for example, by deleting B2M
to remove ME-IC class
I on the surface of the cell, or by deleting B2M and then adding back an HLA-G-
B2M fusion to the
surface to prevent NK cell rejection of cells that do not have MI-IC Class I
on their surface.
100711 The cell lines may include stein cells that were maintained and
differentiated using the
techniques below as shown in U.S. 8,945,862, which is hereby incorporated by
reference in its
entirety. In some embodiments, the stem cell is not a human embryonic stem
cell. Furthermore, the
cell lines may include stem cells made by the techniques disclosed in WO
2003/046141 or Chung
et al. (Cell Stem Cell, February 2008, Vol. 2, pages 113-117); each of which
are hereby
incorporated by reference in its entirety.
100721 For example, the cells may be stem cells isolated from the subject
for use in a
regenerative medical treatment in any of epithelium, cartilage, bone, smooth
muscle, striated
muscle, neural epithelium, stratified squamous epithelium, and ganglia.
Disease that results from
the death or dysfunction of one or a few cell types, such as Parkinson's
disease and juvenile onset
diabetes, are also commonly treated using stem cells (See, Thomson et al.,
Science, 282:1145-1147,
1998, which is hereby incorporated by reference in its entirety).
100731 In some embodiments, cells are harvested from the subject and modified
according to the
methods disclosed herein, which can include selecting certain cell types,
optionally expanding the
cells and optionally culturing the cells, and which can additionally include
selecting cells that
contain the transgene integrated into the auxotrophy-inducing locus.
B. Donor templates or vectors for inserting the transgene
100741 In some embodiments, the compositions disclosed herein comprise donor
templates or
vectors for inserting the transgene into the auxotrophy-inducing locus.
100751 In some embodiments, the donor template comprises (a) one or more
nucleotide
sequences homologous to a fragment of the auxotrophy-inducing locus, or
homologous to the
complement of said auxotrophy-inducing locus, and (b) a transgene encoding a
therapeutic factor,
optionally linked to an expression control sequence. For example, after a
nuclease system is used to
cleave DNA, introduction of a donor template can take advantage of homology-
directed repair
mechanisms to insert the transgene sequence during their repair of the break
in the DNA. In some
-21-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
instances, the donor template comprises a region that is homologous to
nucleotide sequence in the
region of the break so that the donor template hybridizes to the region
adjacent to the break and is
used as a template for repairing the break.
[0076] In some embodiments, the transgene is flanked on both sides by
nucleotide sequences
homologous to a fragment of the auxotrophy-inducing locus or the complement
thereof.
[0077] In some instances, the donor template is single stranded, double
stranded, a plasmid or a
DNA fragment.
[0078] In some instances, plasmids comprise elements necessary for
replication, including a
promoter and optionally a 3' UTR.
[0079] Further disclosed herein are vectors comprising (a) one or more
nucleotide sequences
homologous to a fragment of the auxotrophy-inducing locus, or homologous to
the complement of
said auxotrophy-inducing locus, and (b) a transgene encoding a therapeutic
factor.
[0080] The vector can be a viral vector, such as a retroviral, lentiviral
(both integration
competent and integration defective lentiviral vectors), adenoviral, adeno-
associated viral or herpes
simplex viral vector. Viral vectors may further comprise genes necessary for
replication of the viral
vector.
[0081] In some embodiments, the targeting construct comprises: (1) a viral
vector backbone,
e.g. an AAV backbone, to generate virus; (2) arms of homology to the target
site of at least 200 bp
but ideally 400 bp on each side to assure high levels of reproducible
targeting to the site (see,
Porteus, Annual Review of Pharmacology and Toxicology, Vol. 56:163-190 (2016);
which is
hereby incorporated by reference in its entirety): (3) a transgene encoding a
therapeutic factor and
capable of expressing the therapeutic factor; (4) an expression control
sequence operably linked to
the transgene; and optionally (5) an additional marker gene to allow for
enrichment and/or
monitoring of the modified host cells.
[0082] Suitable marker genes are known in the art and include Myc, HA, FLAG,
GFP, truncated
NGFR, truncated EGFR, truncated CD20, truncated CD19, as well as antibiotic
resistance genes.
[0083] Any AAV known in the art can be used. In some embodiments the primary
AAV
serotype is AAV6.
[0084] In any of the preceding embodiments, the donor template or vector
comprises a
nucleotide sequence homologous to a fragment of the auxotrophy-inducing locus,
optionally any of
the genes in Table 1 below, wherein the nucleotide sequence is at least 85,
88, 90, 92, 95, 98, or
99% identical to at least 200, 250, 300, 350, or 400 consecutive nucleotides
of the auxotrophy-
inducing locus; up to 400 nucleotides is usually sufficient to assure accurate
recombination. Any
combination of the foregoing parameters is envisioned, e.g. at least 85%
identical to at least 200
-22-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
consecutive nucleotides, or at least 88% identical to at least 200 consecutive
nucleotides, or at least
90% identical to at least 200 consecutive nucleotides, or at least 92%
identical to at least 200
consecutive nucleotides, or at least 95% identical to at least 200 consecutive
nucleotides, or at least
98% identical to at least 200 consecutive nucleotides, or at least 99%
identical to at least 200
consecutive nucleotides, or at least 85% identical to at least 250 consecutive
nucleotides, or at least
88% identical to at least 250 consecutive nucleotides, or at least 90%
identical to at least 250
consecutive nucleotides, or at least 92% identical to at least 250 consecutive
nucleotides, or at least
95% identical to at least 250 consecutive nucleotides, or at least 98%
identical to at least 250
consecutive nucleotides, or at least 99% identical to at least 250 consecutive
nucleotides, or at least
85% identical to at least 300 consecutive nucleotides, or at least 88%
identical to at least 300
consecutive nucleotides, or at least 90% identical to at least 300 consecutive
nucleotides, or at least
92% identical to at least 300 consecutive nucleotides, or at least 95%
identical to at least 300
consecutive nucleotides, or at least 98% identical to at least 300 consecutive
nucleotides, or at least
99% identical to at least 300 consecutive nucleotides, or at least 85%
identical to at least 350
consecutive nucleotides, or at least 88% identical to at least 350 consecutive
nucleotides, or at least
90% identical to at least 350 consecutive nucleotides, or at least 92%
identical to at least 350
consecutive nucleotides, or at least 95% identical to at least 350 consecutive
nucleotides, or at least
98% identical to at least 350 consecutive nucleotides, or at least 99%
identical to at least 350
consecutive nucleotides, or at least 85% identical to at least 400 consecutive
nucleotides, or at least
88% identical to at least 400 consecutive nucleotides, or at least 90%
identical to at least 400
consecutive nucleotides, or at least 92% identical to at least 400 consecutive
nucleotides, or at least
95% identical to at least 400 consecutive nucleotides, or at least 98%
identical to at least 400
consecutive nucleotides, or at least 99% identical to at least 400 consecutive
nucleotides.
[00851 The disclosure herein also contemplates a system for targeting
integration of a transgene
to an auxotrophy-inducing locus comprising said donor template or vector, a
cas9 protein, and a
guide RNA.
100861 The disclosure herein further contemplates a system for targeting
integration of a
transgene to an auxotrophy-inducing locus comprising said donor template or
vector and a
meganuclease specific for said auxotrophy-inducing locus. The meganuclease can
be, for example,
a ZFN or TALEN.
[00871 The inserted construct can also include other safety switches, such
as a standard suicide
gene into the locus (e.g. iCasp9) in circumstances where rapid removal of
cells might be required
due to acute toxicity. The present disclosure provides a robust safety switch
so that any engineered
-23-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
cell transplanted into a body can be eliminated by removal of an auxotrophic
factor. This is
especially important if the engineered cell has transformed into a cancerous
cell.
[0088] In some
instances, the donor poly-nucleotide or vector optionally further comprises an
expression control sequence operably linked to said transgene. In some
embodiments, the
expression control sequence is a promoter or enhancer, an inducible promoter,
a constitutive
promoter, a tissue-specific promoter or expression control sequence, a
posttranscriptional
regulatory sequence or a microRNA.
C. Nuclease systems
[0089] In some
embodiments, the compositions disclosed herein comprise nuclease systems
targeting the auxotrophy-inducing locus. For example, the present disclosure
contemplates (a) a
meganuclease that targets and cleaves DNA at said auxotrophy-inducing locus,
or (b) a
polynucleotide that encodes said meganuclease, including a vector system for
expressing said
meganuclease. As one example, the meganuclease is a TALEN that is a fusion
protein comprising
(i) a Transcription Activator Like Effector (TALE) DNA binding domain that
binds to the
auxotrophy-inducing locus, wherein the TALE DNA binding protein comprises a
plurality of
TALE repeat units, each TALE repeat unit comprising an amino acid sequence
that binds to a
nucleotide in a target sequence in the auxotrophy-inducing locus, and (ii) a
DNA cleavage domain.
[0090] Also disclosed herein are CRISPR/Cas or CRISPR/Cpfl system that targets
and cleaves
DNA at said auxotrophy-inducing locus that comprises (a) a Cas (e.g. Cas9) or
Cpfl polypeptide or
a nucleic acid encoding said polypeptide, and (b) a guide RNA that hybridizes
specifically to said
auxotrophy-inducing locus, or a nucleic acid encoding said guide RNA. In
nature, the Cas9 system
is composed of a cas9 polypeptide, a crRNA, and a trans-activating crRNA
(tracrRNA). As used
herein, "cas9 polypeptide" refers to a naturally occurring cas9 polypeptide or
a modified cas9
polypeptide that retains the ability to cleave at least one strand of DNA. The
modified cas9
polypeptide can, for example, be at least 75%, 80%, 85%, 90%, or 95% identical
to a naturally
occurring Cas9 polypeptide. Cas9 polypeptides from different bacterial species
can be used: S.
pyogenes is commonly sold commercially. The cas9 polypeptide normally creates
double-strand
breaks but can be converted into a nickase that cleaves only a single strand
of DNA (i.e. produces a
"single stranded break") by introducing an inactivating mutation into the HNH
or RuvC domain.
Similarly, the naturally occurring tracrRNA and crRNA can be modified as long
as they continue to
hybridize and retain the ability to target the desired DNA, and the ability to
bind the cas9. The
guide RNA can be a chimeric RNA, in which the two RNAs are fused together,
e.g. with an
artificial loop, or the guide RNA can comprise two hybridized RNAs. The
meganuclease or
CRISPR/Cas or CRISPR/Cpfl system can produce a double stranded break or one or
more single
-24-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
stranded breaks within the auxotrophy-inducing locus, for example, to produce
a cleaved end that
includes an overhang.
[0091] In some instances, the nuclease systems described herein, further
comprises a donor
template as described herein.
[0092] Various methods are known in the art for editing nucleic acid, for
example to cause a
gene knockout or expression of a gene to be downregulated. For example,
various nuclease
systems, such as zinc finger nucleases (ZFN), transcription activator-like
effector nucleases
(TALEN), meganucleases, or combinations thereof are known in the art to be
used to edit nucleic
acid and may be used in the present disclosure. Meganucleases are modified
versions of naturally
occurring restriction enzymes that typically have extended or fused DNA
recognition sequences.
[0093] The CRISPR/Cas system is detailed in, for example WO 2013/176772, WO
2014/093635 and WO 2014/089290; each of which is hereby incorporated by
reference in its
entirety. Its use in T cells is suggested in WO 2014/191518, which is hereby
incorporated by
reference in its entirety. CRISPR engineering of T cells is discussed in EP
3004349, which is
hereby incorporated by reference in its entirety.
100941 The time-limiting factor for generation of mutant (knock-out, knock-
in, or gene replaced)
cell lines was the clone screening and selection before development of the
CRISPR/Cas9 platform.
The term "CRISPR/Cas9 nuclease system" as used herein, refers to a genetic
engineering tool that
includes a guide RNA (gRNA) sequence with a binding site for Cas9 and a
targeting sequence
specific for the area to be modified. The Cas9 binds the gRNA to form a
ribonucleoprotein that
binds and cleaves the target area. CRISPR/Cas9 permits easy multiplexing of
multiple gene edits.
In some embodiments, the gRNA comprises the nucleic acid sequence of SEQ ID
NO: I.
100951 In addition to the CRISPR/Cas 9 platform (which is a type II CRISPR/Cas
system),
alternative systems exist including type 1 CR1SPR/Cas systems, type 111
CR1SPR/Cas systems, and
type V CRISPR/Cas systems. Various CRISPR/Cas9 systems have been disclosed,
including
Streptococcus pyogenes Cas9 (SpCas9), Streptococcus thermophilus Cas9
(StCas9),
Campylobacter jejuni Cas9 (CjCas9) and Neisseria cinerea Cas9 (NcCas9) to name
a few.
Alternatives to the Cas system include the Francisella novicida Cpfl (FnCpfl),
Acidaminococcus
sp. Cpfl (AsCpfl), and Lachno.spiraceae bacterium ND2006 Cpfl (LbCpfl)
systems. Any of the
above CRISPR systems may be used in methods to generate the cell lines
disclosed herein. For
example, the CRISPR system used may be the CRISPR/Cas9 system, such as the S.
pyogenes
CRISPR/Cas9 system.
-25-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
IV. Methods of Creating the Modified Host Cells
[0096] In some embodiments, the auxotrophy-inducing locus is within a
target gene selected
from those disclosed in Table 1, or the region controlling expression of that
gene. In some
embodiments, the target gene is selected from IMPS (creating a cell line
auxotrophic for uracil)
and holocarboxylase synthetase (creating a cell line auxotrophic for biotin).
In some embodiments,
the auxotrophic factor is selected from biotin, alanine, aspartate,
asparagine, glutamate, serine,
uracil and cholesterol.
[0097] Further disclosed herein are methods of using said nuclease systems
to produce the
modified host cells described herein, comprising introducing into the cell (a)
the components of one
or more nuclease systems that target and cleave DNA at an auxotrophy-inducing
locus, e.g.
meganuclease such as ZFN or TALEN, or CRISPR/Cas nuclease such as CRTSPR/Cas9,
and (b) a
donor template or vector as described herein. Each component can be introduced
into the cell
directly or can be expressed in the cell by introducing a nucleic acid
encoding the components of
said one or more nuclease systems. The methods can also comprise introducing a
second nuclease
system, e.g. a second meganuclease or second CRISPR/Cas nuclease that targets
and cleaves DNA
at a second locus, or a second guide RNA that targets DNA at a second locus,
or a nucleic acid that
encodes any of the foregoing, and (b) a second donor template or vector. The
second donor
template or vector can contain a different transgene, or a second copy of the
same transgene, which
will then be integrated at the second locus according to such methods.
(0098) Such methods will target integration of the transgene encoding the
therapeutic factor to
an auxotrophy-inducing locus in a host cell ex vivo.
[0099] Such methods can further comprise (a) introducing a donor template
or vector into the
cell, optionally after expanding said cells, or optionally before expanding
said cells, and (b)
optionally culturing the cell.
[0100] In some embodiments, the disclosure herein contemplates a method of
producing a
modified mammalian host cell comprising introducing into a mammalian cell: (a)
a Cas9
polypeptide, or a nucleic acid encoding said Cas9 polypeptide, (b) a guide RNA
specific to an
auxotrophy-inducing locus, or a nucleic acid encoding said guide RNA, and (c)
a donor template or
vector as described herein. The methods can also comprise introducing (a) a
second guide RNA
specific to a second auxotrophy-inducing locus and (b) a second donor template
or vector. In such
methods, the guide RNA can be a chimeric RNA or two hybridized RN As.
[0101] In any of these methods, the nuclease can produce one or more single
stranded breaks
within the auxotrophy-inducing locus, or a double stranded break within the
auxotrophy-inducing
-26-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
locus. In these methods, the attxotrophy-inducing locus is modified by
homologous recombination
with said donor template or vector to result in insertion of the transgene
into the locus.
[0102] The methods can further comprise (c) selecting cells that contain
the transgene integrated
into the auxotrophy-inducing locus. The selecting steps can include (i)
selecting cells that require
the auxotrophic factor to survive and optionally (ii) selecting cells that
comprise the transgene
integrated into the auxotrophy-inducing locus.
[0103] In some embodiments, the auxotrophy-inducing locus is a gene encoding
uridine
monophosphate synthetase and the cells are selected by contacting them with 5-
F0A. The (MPS
gene is required to metabolize 5-FOA into 5-FUMP, which is toxic to cells due
to its incorporation
into RNA/DNA. Thus, cells which have a disruption in the UMPS gene will
survive 5-FOA
treatment. The resulting cells will all be auxotrophic, although not all cells
may contain the
transgene. Subsequent positive selection for the transgene will isolate only
modified host cells that
are auxotrophic and that are also capable of expressing the transgene.
[0104] In some embodiments, the disclosure herein provides a method of
creating a modified
human host cell comprising the steps of: (a) obtaining a pool of cells, (b)
using a nuclease to
introduce a transgene to the auxotrophy-inducing locus, for example by
knocking out or
downregulating expression of a gene, and (c) screening for auxotrophy, and (d)
screening for the
presence of the transgene.
[0105] The screening step may be carried out by culturing the cells with or
without one of the
auxotrophic factors disclosed in Table 1.
[0106] Techniques for insertion of transgenes, including large transgenes,
capable of expressing
functional factors, antibodies and cell surface receptors are known in the art
(See, e.g. Bak and
Porteus, Cell Rep. 2017 Jul 18; 20(3): 750-756 (integration of EGFR); Kanojia
et al., Stem Cells.
2015 Oct;33(10):2985-94 (expression of anti-Her2 antibody); Eyquem et al.,
Nature. 2017 Mar
2;543(7643):113-117 (site-specific integration of a CAR); O'Connell et al.,
2010 PLoS ONE 5(8):
el2009 (expression of human IL-7); Tuszynski et al., Nat Med. 2005
May;11(5):551-5 (expression
of NGF in fibroblasts); Sessa et al., Lancet. 2016 Jul 30;388(10043):476-87
(expression of
arylsulfatase A in ex vivo gene therapy to treat MLD): Rocca et al., Science
Translational Medicine
25 Oct 2017: Vol. 9, Issue 413, eaaj2347 (expression of frataxin); Bak and
Porteus, Cell Reports,
Vol. 20, Issue 3, 18 July 2017, Pages 750-756 (integrating large transgene
cassettes into a single
locus), Dever et al., Nature 17 November 2016: 539, 384-389 (adding tNGFR into
hematopoietic
stem cells (HSC) and HSPCs to select and enrich for modified cells); each of
which is hereby
incorporated by reference in its entirety.
-27-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
A. Auxotrophy-inducing locus and auxotrophic factor
101071 In some embodiments, disruption of a single gene causes the desired
auxotrophy. In
alternative embodiments, disruption of multiple genes produces the desired
auxotrophy.
[0108] In some embodiments, the auxotrophy-inducing locus is a gene
encoding a protein that
produces an auxotrophic factor, which includes proteins upstream in the
pathway for producing the
auxotrophic factor.
[0109] In some embodiments described herein, the auxotrophy-inducing locus
is the gene
encoding uridine monophosphate synthetase (UMPS) (and the corresponding
auxotrophic factor is
uracil), or the gene encoding holocarboxylase synthetase (and the
corresponding auxotrophic factor
is biotin). In some embodiments, auxotrophy-inducing loci are selected from
the following genes in
Table 1. The genes of Table 1. were collated by selecting S. cerevisiae genes
with a phenotype
annotated as "Auxotrophy" downloaded with "Chemical" data from the yeast
phenotype ontology
database on the Saccharomyces genome database (SOD) (See, Cherry et al. 2012,
Nucleic Acids
Res. 40:D700-D705, which is hereby incorporated by reference in its entirety).
These genes were
converted into human homologues using the YeastMinet database or, in
alternative embodiments,
the Saccharocyces Genome Database (SGD). The genes are identified by their
ENSEMBL gene
symbol and ENSG identifier, which are found in the ENSEMBL database
(www.ensembl.org). The
first five zeroes of the ENSG identifiers (e.g., ENSG00000) have been removed.
Table 1. Auxotrophy-inducing loci
Gene ENSG(s) Au xot rophic factor Gene ENSG(s) Au xot rophie
factor
AACS 081760 lysine HSD1.7B 1 2 149084 ergosterol
A ADAT 109576 histidine HSD17B3 130948 ergosterol
AASDHPPT 149313 lysine HSD17B7 _ 132196 _ ergosterol
AASS 008311 lysine HSDI7B7P2 099251 ergosterol
ACAT1 075239 ergosterol HSDL I 103160 ergosterol
ACCS 110455 histidine HSDL2 119471 _ ergosterol
ACC SL 205126 histidine IBA57 181873 glutatnic acid
ACO1 122729 leucine IDO I 131203 nicotinic acid
ACO2 100412 leucine I1)02 188676 nicotinic acid
ACSS3 111058 lysine IL4I 1 104951 0.1mM beta-alanine
ADSL 239900 adenine ILVBL 105135 'aline. isoleucine
ADSS 035687 adenine IP6K1 _ 176095 asinine
ADSSL 1 185100 adenine (P6K2 068745 arginine
ALAD 148218 cy stet ne IP6K3 161896 arginine
ALASI 023330 cysteine IPMK 151151 arginine
ALAS2 158578 cysteine IREB2 136381 leucine
ALDH1A I 165092 pantothenic acid ISCA1 135070 lysine
ALDH1A2 128918 pantothenic acid ISCA1PI 217416 Mine
ALDH IA3 184254 pantothenic acid ISCA2 165898 lysine
ALDH 1BI 137124 pantothenic acid KAThAI 186625 ethanolarnine
ALDH2 111275 pantothenic acid K ATN ALI 102781
ethanolamine
AMD1 123505 0.25M4 spermine KATNAL2 167216 ethanolamine
A SL 126522 arginine KDM1B 165097 0.1mM beta-alani no
ASS I 130707 arginine KDSR 119537 lysine
ATF4 128272 tnethionine KMO 117009 nicotinic acid
-28-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
ATF5 169136 methionine KYNU 115919 nicotinic acid
AZIN I 155096 0.25mM putrescine LGSN 146166 glutamine
281289;
AZIN2 142920 0.25mM put tesci tie LSS 160285 . ergosterol
BcAT i 060982 val i tie, leucine MARS 166986 methionine
BCAT2 105552 . valine, leucine MAR S2 247626 met hioni ne .
CAD 084774 unicil MAX 125952 methionine
CBS 160200 cysteine IvIITF 187098 glutamate( 1-)
CBSL 274276 cv stet ne MLX 108788 glutamate( I.-)
CCBL I 171097 histidine 'WM S 19 155229 methionine
CCBL2 137944 histidine MPC1 060762 . vali ne, leucine
CCS 173992 methionine MPC IL 238205 'aline. leucine
CEBPA 245848 . methionine MP! 178802 D-mannose .
CEBPB 172216 methionine MSMO I 052802 ergosterol
CEBPD 221869 methionine IvITHFD1 100714 adenine
CEBPE 092067 methionine NM-1FD IL 120254 adenine
CEBPG 153879 methionine MTHFD2 065911 adenine
CI-125H 138135 ergosterol MTHFD21., 163738 adenine
COQ6 119723 nicotinic acid IVITHFR 177000 methionine
CPS1 021826 . arginine MIRR 124275 methionine .
01-1. 116761 cysteine MVI( 110921 ergosterol
CYP51 A I 001630 ergosteml IvIYB 118513 adenine
DECR 1 104325 ergosterol MYB LI 185697 adenine
DHFR 228716 dThIP MYBL2 101057 adenine
DHFR LI 178700 (MP NAGS 161653 arginine
DHODH 102967 ttracil ODC I 115758 0.25mM putrescine
DI-1167 100612 _ lvsine arc 036473 arginine .
DI-IRS7B 109016 lysine PA.ICS 128050 adenine
DHRS7C 184544 Iv si ne PAOX 148832 O. I inM beta-
alanine
DPYD 188641 uraci I PAPSS I 138801 methionine
DLIT 128951 dThIP PAPSS2 198682 methionine
F:rFoli 171503 thiamine( I 4-) PDHB 168291 try ptophan
FAXDC2 170271 et:g9stem1 PDX1 139515 adenine ...
079459;
FDFT1 284967 ergosterol PFAS 178921 . adenine
FDPS 160752 ergosterol PIN1 127445 galactose
FDXR 161513 tuna PLCB1 182621 omithine
HI 091483 arginine PLCB2 137841, ontithine
FPGS 136877 methionine PLCB3 149782 omithine
G6PD 160211 met hio ni tie PLCB4 101333 omithine
GCAT 100116 cvsteine PLCD I 187091 ornithine
GCH1 131979 5-fortny het rahvdro fblicac id PLCD3 161714 .
ornithine
GCLC 001084 glutathione PLCD4 115556 omithine
GFPT I 198380 Dilucosamine PLCE I 138193 omithine
GFPT2 131459 D-glucosatni tie PLCG1 124181, ontithine
GLRX5 182512 glutamic acid PLCG2 197943 omithine
GLUL 135821 glutamine PLCH1 114805 omithine
276429;
GAPS 163655 ..Kuanine PLCH2 149527 omithine
GPT 167701 . hist icing: PLCL I. 115896 omithine .
154822;
GPT2 166123 histidine PLCL2 284017 omithine
GSX2 180613 adenine PLCZ I 139151 ornithine
H6P1) 049239 methionine PM20D1 162877 leucine
HAAO 162882 nicotinic acid PPAT 128059 adenine
'I-R.(7S 159267 biotin PSAT I. 135069 seri ne
256269;
HMBS 281702 heme PSPH 146733 seine
HMGCL 117305 lysine PYCR I 183010 proline .
-29-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
HMGCLL1 146151 lysine PYCR2 143811 prohne
HMGCS I 112972 ergosterol 104524 proline
HMGCS2 134240 . ergosterol QPRT 103485 Nicotinic acid
=
HOXA1 105991 adenine RDH8 80511 Lvsine
HOXA10 253293 adenine RPUSD2 166133 riboflavin
HOXA I 1 005073 adenine SC!) 99194 oleic acid
HOXA13 106031 adenine SCD5 145284 oleic acid
HOXA2 105996 adenine SLC25A I 9 125454 -dna ini ne
144741;
HOXA3 105997 adenine SLC25A26 282739 biotin
HOXA4 197576 adenine SLC25A34 162461 . leucine
HOXA5 106004 adenine 5LC25A35 125434 !endue
HOXA6 106006 adenine SLC7A 10 130876 L-arginine
HOX A7 122592 adenine SLC7A 11 151012 L-argi nine
HOXA9 078399 adenine SLC7A 13 164893 L-argi nine
HOXB1 120094 adenine SLC7A5 103257 L-arginine
HOXBI3 159184 adenine SLC7A6 103064 L-argi Ili ne
HOXB2 173917 adenine SLC7A7 155465 . L-arginine
HOXB3 120093 adenine SLC7A8 092068 L-argi nine
HOXB4 182742 adenine SLC7A9 021488 L-argi nine
HOX B5 120075 adenine SMOX 088826 0.1mM beta-alani
tie
HOXB6 108511 adenine SMS 102172 0.25mM spenuine
HOXB7 260027 adenine SNA PC4 165684 adenine
HOXB8 120068 adenine SOD! 142168 methionine
HOXB9 170689 adenine SOD3 109610 . methionine
HOXCIO 180818 adenine SQLE 104549 ergosterol
HOXC I 1 123388 adenine SRM 116649 0.25mM spermine
HOX Cl2 123407 adenine TAT 198650 histid i tie
HOXC13 123364 adenine TFE3 068323 glutamate(1-)
HOXC4 198353 adenine TFEB I 1 2561 glutamate(1 -)
HOXC5 172789 adenine TFEC 105967 glutamate(1-)
HOXC6 197757 adenine THNSL1 185875 . threonine
HOXC8 037965 adenine THNSL2 144115 threonine
HOXC9 180806 adenine TKT 163931 tryp9phan
HOX DI 128645 adenine TKTL I 007350 t ry ptophim
HOXDIO 128710 adenine TKTL2 151005 tiyptophan
HOXD I 1 128713 adenine ti MPS 114491 mei!
HOXD12 170178 adenine UROD 126088 home _
HOXD13 128714 adenine UROS 188690 . benne
HOXD3 128652 adenine U SF I 158773 glutamate(1-)
HOXD4 170166 . adenine USF2 105698 glutamate(1-) .
HOX D8 175879 adenine VPS33A 139719 tnethioni tie
HOXD9 128709 adenine VF'S33B 184056 methionine
HRSP12 132541 isoleucirie VP S36 136100 ellianolartiine
HSDI1B 1 117594 lvsine VPS4A 132612 ethanolamine
HSDIIB IL 167733 lysine VPS4B 119541 ethatiolainine
101101 CCBL1 may also be referred to as KYATI . CCBL2 may also be referred to
as KYAT3.
DHFRL I may also be referred to as DHFR2. PYCRL may also be referred to as
PYCR3. HRSP12
may also be referred to as RI DA.
101111 The auxotrophic factor may be one or two or more nutrients, enzymes,
altered pH,
altered temperature, non-organic molecules, non-essential amino acids, or
altered concentrations of
a moiety (compared to normal physiologic concentrations), or combinations
thereof All references
to auxotrophic factor herein contemplate administration of multiple factors.
Any factor is suitable
-30-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
as long as it is not toxic to the subject and is not bioavailable or present
in a sufficient concentration
in an untreated subject to sustain growth and reproduction of the modified
host cell.
[0112] For example, the auxotrophic factor may be a nutrient that is a
substance required for
proliferation or that functions as a cofactor in metabolism of the modified
host cell. Various
auxotrophic factors are disclosed in Table 1. In certain embodiments, the
auxotrophic factor is
selected from biotin, alanine, aspartate, asparagine, glutamate, serine, wadi,
valine and cholesterol.
Biotin, also known as vitamin B7, is necessary for cell growth. In some
instances, valine is needed
for the proliferation and maintenance of hematopoietic stem cells. In some
instances, the
compositions disclosed herein are used to express the enzymes in HSCs that
relieve the need for
valine supplementation and thereby give those cells a selective advantage when
valine is removed
from the diet compared to the unmodified cells.
B. Transgene
[0113] Therapeutic entities encoded by the genome of the modified host cell
may cause
therapeutic effects, such as molecule trafficking, inducing cell death,
recruitment of additional
cells, or cell growth. In some embodiments, the therapeutic effect is
expression of a therapeutic
protein. In some embodiments, the therapeutic effect is induced cell death,
including cell death of a
tumor cell.
C. Control of transgene expression
[0114] In some instances, the transgene is optionally linked to one or more
expression control
sequences, including the gene's endogenous promoter, or heterologous
constitutive or inducible
promoters, enhancers, tissue-specific promoters, or post-transcriptional
regulatory sequences. For
example, one can use tissue-specific promoters (transcriptional targeting) to
drive transgene
expression or one can include regulatory sequences (microRNA (miRNA) target
sites) in the RNA
to avoid expression in certain tissues (post-transcriptional targeting). In
some instances, the
expression control sequence functions to express the therapeutic transgene
following the same
expression pattern as in normal individuals (physiological expression) (See
Toscano et al., Gene
Therapy (2011) 18, 117-127 (2011), incorporated herein by reference in its
entirety for its
references to promoters and regulatory sequences).
[0115] Constitutive mammalian promoters include, but are not limited to,
the promoters for the
following genes: hypoxanthine phosphoribosyl transferase (HPTR), adenosine
deaminase, pyruvate
kinase, a-actin promoter and other constitutive promoters. Exemplary viral
promoters which
function constitutively in eukaryotic cells include, for example, promoters
from the simian virus,
papilloma virus, adenovirus, human immunodeficiency virus (HIV), Rous sarcoma
virus,
cytomegalovirus, the long terminal repeats (LTR) of Moloney leukemia virus and
other
-31-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
retroviruses, and the thymidine kinase promoter of herpes simplex virus.
Commonly used
promoters including the CMV (cytomegalovirus) promoter/enhancer, EF la
(elongation factor la),
SV40 (simian virus 40), chicken I3-actin and CAG (CMV, chicken (-actin, rabbit
13-globin),
Ubiquitin C and PGK, all of which provide constitutively active, high-level
gene expression in
most cell types. Other constitutive promoters are known to those of ordinary
skill in the art.
[0116] inducible promoters are activated in the presence of an inducing
agent. For example, the
metallothionein promoter is activated to increase transcription and
translation in the presence of
certain metal ions. Other inducible promoters include alcohol-regulated,
tetracycline-regulated,
steroid-regulated, metal-regulated, nutrient-regulated promoters, and
temperature-regulated
promoters.
[0117] For liver-specific targeting: Natural and chimeric promoters and
enhancers have been
incorporated into viral and non-viral vectors to target expression of factor
Vila, factor VIII or factor
IX to hepatocytes. Promoter regions from liver-specific genes such as albumin
and human al
antitrypsin (hAAT) are good examples of natural promoters. Alternatively,
chimeric promoters
have been developed to increase specificity and/or vectors efficiency. Good
examples are the
(ApoE)4/hAAT chimeric promoter/enhancer, harboring four copies of a liver-
specific ApoE/hAAT
enhancer/promoter combination and the DC! 72 chimeric promoter, consisting in
one copy the
hAAT promoter and two copies of the a(1)-microglobulin enhancer.
[0118] For muscle-specific targeting: Natural (creatine kinase promoter-
MCK, desmin) and
synthetic (a-myosin heavy chain enhancer-/MCK enhancer-promoter (MI-ICK7))
promoters have
been included in viral and non-viral vectors to achieve efficient and specific
muscle expression.
[0119] For endothelium-specific targeting, both natural (NW, FLT-1 and ICAM-2)
and
synthetic promoters have been used to drive endothelium-specific expression.
[0120] For myeloid cell targeting, a synthetic chimeric promoter that
contains binding sites for
myeloid transcription factors CAAT box enhancer-binding family proteins
(C/EBPs) and PU.1,
which are highly expressed during granulocytic differentiation, has been
reported to direct
transgene expression primarily in myeloid cells (See, Santilli et al., Mol
Ther. 2011 Jan;19(1):122-
32, which is hereby incorporated by reference in its entirety. CD68 may also
be used for myeloid
targeting.
101211 Examples of tissue-specific vectors for gene therapy of genetic
diseases are shown in
Table 2.
Table 2. Tissue-specific vectors
Promoter Vector type Target
cell/tissue
WAS proximal promoter HIV- I -based vectors Hematopoietic
cells
CD4 mini-promoter/enhancer IVILV-based vectors T cells
-3
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
MIN based and HIV-1-based
CD2 locus control region vectors T cells
CD4 minimal promoter and proximal enhancer and silencer HIV-1-based
vectors T cells
(1)4 mini-promoter/enhancer H I -based vectors T cells
GATA-1 enhancer HS2 within the LTR SFCM retroviral vector Erythroid
linage
Anky tin-1 and a-spectfin promoters combined or not with HS-
40, ARE and intron 8 enhancers HIV-1-based vectors Ely throid
lAnkyrin- I promoter/13-00bn] HS-40 enhancer HIV-1-based
vectors Erythroid linage
1GATA-1 enhancer HS1 to HS2 within the retroviral LTR SFCM retroviral
vector Erythroid linage
'Hybrid tomegalovirus (CMV) enhancer/D.-actin promoter Sleeping Beauty
transposon ErVihroid linage
IMCH II-specific HLA-DR promoter HIV-1-based vectors APCs
Fascin promoter (pFascin) Plasmid APCs
Dectin-2 gene promoter HIV-I-based vectors APCs
5' untranslated region from the DC-STAMP HIV-1-based vectors APCs
Heavy chain intronic enhancer (Ell) and matrix attachment
regions HIV-1-based vectors B cells
CD19 promoter MLV based vectors B cells
Hybrid immunoglobulin promoter (Igk promoter, int ronic
Enhancer and 3' enhancer front Ig genes) H I -based vectors
B cells
CD68L promoter and first intron MLV-based vectors Megakaryocvtes
Glycoprotein Iba promoter HIV-1-based vectors Megakaiyocytes
Apolipoprotein E (Apo E) enhancer/alpha1-antitrypsin (hAAT)
promoter (ApoE/hAAT) MIN based vectors Hepatocytes
HAAT promoter/Apo E locus control region Plasmid Hepatocytes
Albumin promoter H I -based vectors Hepatocytes
HAAT promoter/four copies of the Apo E enhancer AAV-2-based
vectors Hepatocvtes
Albumin and hAAT promoters/al-microglobulin and
prothrombin enhancers Plasmid Hepatocvtes
HAAT promoter/Apo E. locus control region AAV8
Hepatocytes
MAT promoter/four copies of the Apo E enhancer AAV2/8
Hepatocytes
TBG promoter (thyroid hormone-binding globulin promoter
and al-microglobulin/bikunin enhancer) AAV Hepatocytes
DC172 promoter (a1-antitrypsin promoter and al-
microglobulin enhancer) Adenovirus. plasmid Hepatocvtes
kLSP-IVS, ApoE/hAAT and liver-fatty acid-binding
protein promoters AAV I õAAV2, AAV6, AAV8 Hepatocytes
RU486-responsive promoter Adenovirus Hepatocytes
Creatine kinase promoter Adenovirus Muscle
Creatine kinase promoter AAV6 Muscle
,Synthetic muscle-specificpromoter C5-12 AV-1. Muscle
Creatine kinase promoter AAV2/6 Muscle
Hybrid enhancer/promoter regions of a-myosin and creatine
kinase (MI-1CM) AAV6 Muscle
Hybrid enhancer/promoter regions of a-myosin and creatine
kinase AAV2/8 Muscle
Synthetic muscle-specific promoter C5-12 HIV-1-based vectors Muscle
Cardiac troponin-I proximal promoter HIV-1-based vectors Cantomyocyles
E-selectin and KDR promoters MIN-based vectors Endothelial cell
Prepro-endothelin-1 promoter MLV-based vectors Endothelial cell
KDR promoterthypoxia-responsive element MIN-based vectors Endothelial
cell
Flt-1 promoter Adenovims Endothelial cell
Flt-1. promoter Adenovims Endothelial cell
ICAM-2 promoter Plasmid Endothelial cell
Synthetic endothelial promoter HIV-1-based vectors Endothelial
cell
Endothelin-1 gene promoter Sleeping Beauty transposon Endothelial
cell
Amylase promoter Adenovirus Pancreas
Insulin and human pdx-1 promoters Adenovirus Pancreas
TRE-regulated insulin promoter Plasmid Pancreas
Enolase promoter Herpesvirus Neurons
-33-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
Enolase promoter Adenovintses Neurons
TRE-regulated synapsin promoter Adenoviruses Neurons
Synapsin 1 promoter Adenoviruses Neurons
PDGE-11 promoter/Cs/IV enhancer Plasmid Neurons
PDGF3 synapsin. tubulin-a and ca2 /calmodulin-PK2
promoters combined with CivIV enhancer HIV-1-based vectors Neurons
Phosphate-activated glutaminase and vesicular glutamate Glutainatergic
transporter-1 promoters Heivesvirus neurons
Glutamic acid decarbox-ylase-67 promoter Herpesvinis GABAergic neuron
Cmecholaminergic
Tyrosine hydroxylase promoter Herpesvitus neurons
Neurofilamem heavy gene promoter Herpesvirus Neurons
Human red psi's promoter Recombinant AAV Cone cells
Keratin-18 promoter Adenovints Epithelial cells
keratin-14 (K14) promoter Lentiviral vectors Epithelial cells
Keratin-5 promoter HIV-1-based vectors Epithelial
cells
101221 Examples of physiologically regulated vectors for gene therapy of
genetic diseases are
shown in Table 3.
Table 3. Physiologically regulated vectors
Promoter Vector type Target cell/tissue
WAS proximal promoter (1600 bp) HIV-1-based vectors IHematopoietic cells
WAS proximal promoter (500 bp) HIV-1-based vectors Hematopoietic cells
WAS proximal promoter (170 bp) HIV-1-based vectors iffematopoietic cells
WAS proximal promoter (500 bp)AVAS
alternative promoter (386 bp) HIV-1-based vectors Hematopoietic cells
CD4OL promoter and regulators' sequences Human
artificial chromosome (H.AC) ;Activated T cells
CD4OL promoter HIV-1-based vectors iActivated T
cells
P-Globitt promoter/LCR HIV-1-based vectors :Ei-ythroid
linage
13-Globin and 0-globin promoters combined or
not with HS-40, GATA-1, ARE, and intron 8
enhancers HIV-1-based vectors Etythroid
linage
13-Globin, LCR HS4. H53, HS2 and a truncated
0-globin intron 2 HIV-1-based vectors lErythroid
linage
promoter/LCR/cHS4 HIV-I -based vectors lEty
throid linage
HSFE/LCR/D-globin promoter MSCV renoviral vector iElythroid
linage
hategtin an promoter (nucleotides ¨889 to +35) MLV-based vectors
IMegakaryocytes
Dystrophin promoter and regulatory sequences HAC IMuscle
Endoglin promoter Plasmid Endothelial
cells
RPE65 promoter AAV2/4 IRetinal pigmented
epithelium
TRE-regulated synapsin promoter Adenoviruses Neurons
[0123] Tissue-specific and/or physiologically regulated expression can also
be pursued by
modifying mRNA stability and/or translation efficiency (post-transcriptional
targeting) of the
transgenes. Alternatively, the incorporation of miRNA target recognition sites
(miRTs) into the
expressed mRNA has been used to recruit the endogenous host cell machinery to
block transgene
expression (detargeting) in specific tissues or cell types. miRNAs are
noncoding RNAs,
approximately 22 nucleotides, that are fully or partially complementary to the
3' UTR region of
particular mRNA, referred to as miRTs. Binding of a miRNA to its particular
miRTs promotes
translational attenuation/inactivation and/or degradation. Regulation of
expression through
-34-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
miRNAs is described in Geisler and Fechner, World J Exp Med. 2016 May 20,
6(2): 37-54; Brown
and Naldini, Nat Rev Genet. 2009 Aug, 10(8):578-85; Gentner and Naldini,
Tissue Antigens. 2012
Nov, 80(5):393-403, each of which is hereby incorporated by reference in its
entirety. Engineering
miRTs-vector recognized by a specific miRNA cell type has been shown to be an
effective way for
knocking down the expression of a therapeutic gene in undesired cell types
(See, Toscano et al.,
supra., which is hereby incorporated by reference in its entirety).
D. Pharmaceutical compositions
[0124] Disclosed herein, in some embodiments, are methods, compositions and
kits for use of
the modified cells, including pharmaceutical compositions, therapeutic
methods, and methods of
administration of auxotrophic factors to control ¨ increase, decrease or cease
- the growth and
reproduction of the modified cells and to control the expression of the
therapeutic factor by the
transgene.
[0125] The modified mammalian host cell may be administered to the subject
separately from
the auxotrophic factor or in combination with the auxotrophic factor. Although
the descriptions of
pharmaceutical compositions provided herein are principally directed to
pharmaceutical
compositions which are suitable for administration to humans, it will be
understood by the skilled
artisan that such compositions are generally suitable for administration to
any animals.
[0126] Subjects to which administration of the pharmaceutical compositions
is contemplated
include, but are not limited to, humans and/or other primates; mammals,
including commercially
relevant mammals such as cattle, pigs, horses, sheep, cats, dogs, mice, rats,
birds, including
commercially relevant birds such as poultry, chickens, ducks, geese, and/or
turkeys. In some
embodiments, compositions are administered to humans, human patients, or
subjects.
[0127] In some instances, the pharmaceutical compositions described herein
is used in a method
of treating a disease, a disorder, or a condition in a subject, the method
including: (i) generating a
cell line which is auxotrophic for a nutrient, an enzyme, an altered pH, an
altered temperature, an
altered concentration of a moiety, and/or a niche environment, such that the
nutrient, enzyme,
altered pH, altered temperature, and niche environment is not present in the
subject; (ii) contacting
the subject with the resulting auxotrophic cell line of step (i); (iii)
contacting the subject of (ii) with
the auxotrophic factor which is selected from the nutrient, enzyme, moiety
that alters pH and/or
temperature, and a cellular niche environment in the subject, such that the
auxotrophic factor
activates the auxotrophic system or element resulting in the growth of the
cell line and/or the
expression of one or more therapeutic entities for the subject.
[0128] The pharmaceutical compositions of the disclosure herein may also be
used in a method
of treating a disease, a disorder, or a condition in a subject, comprising (a)
administering to the
-35-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
subject a modified host cell according to the disclosure herein, and (b)
administering the
auxotrophic factor to the subject in an amount sufficient to promote growth of
the modified host
cell.
[0129] Compositions comprising a nutrient auxotrophic factor may also be
used for
administration to a human comprising a modified host cell of the disclosure
herein.
V. Formulations
A. Cellular Engineering Formulations
[0130] The modified host cell is genetically engineered to insert the
transgene encoding the
therapeutic factor into the auxotrophy-inducing locus. Delivery of Cas9
protein/gRNA
ribonucleoprotein complexes (Cas9 RNPs) targeting the desired locus may be
performed by
liposome-mediated transfection, electroporation, or nuclear localization. In
some embodiments, the
modified host cell is in contact with a medium containing serum following
electroporation. In some
embodiments, the modified host cell is in contact with a medium containing
reduced serum or
containing no serum following electroporation.
B. Therapeutic Formulations
101311 The modified host cell or auxotrophic factor of the disclosure
herein may be formulated
using one or more excipients to: (1) increase stability; (2) alter the
biodistribution (e.g., target the
cell line to specific tissues or cell types); (3) alter the release profile of
an encoded therapeutic
factor; and/or (4) improve uptake of the auxotrophic factor.
[0132] Formulations of the present disclosure can include, without
limitation, saline, liposomes,
lipid nanoparticles, polymers, peptides, proteins, and combinations thereof.
[0133] Formulations of the pharmaceutical compositions described herein may be
prepared by
any method known or hereafter developed in the art of pharmacology. As used
herein the term
"pharmaceutical composition" refers to compositions including at least one
active ingredient and
optionally one or more pharmaceutically acceptable excipients. Pharmaceutical
compositions of the
present disclosure may be sterile.
101341 in general, such preparatory methods include the step of associating
the active ingredient
with an excipient and/or one or more other accessory ingredients. As used
herein, the phrase
"active ingredient- generally refers to either (a) a modified host cell or
donor template including a
transgene capable of expressing a therapeutic factor inserted into an
auxotrophy-inducing locus, or
(b) the corresponding auxotrophic factor. or (c) the nuclease system for
targeting cleavage within
the auxotrophy-inducing locus.
[0135] Formulations of the modified host cell or the auxotrophic factor and
pharmaceutical
compositions described herein may be prepared by a variety of methods known in
the art.
-36-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0136] A pharmaceutical composition in accordance with the present disclosure
may be
prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a
plurality of single unit
doses. As used herein, a "unit dose" refers to a discrete amount of the
pharmaceutical composition
including a predetermined amount of the active ingredient.
101371 Relative amounts of the active ingredient (e.g. the modified host
cell or auxotrophic
factor), a pharmaceutically acceptable excipient, and/or any additional
ingredients in a
pharmaceutical composition in accordance with the present disclosure may vary,
depending upon
the identity, size, and/or condition of the subject being treated and further
depending upon the route
by which the composition is to be administered. For example, the composition
may include
between 0.1% and 99% (w/w) of the active ingredient. By way of example, the
composition may
include between 0.1% and 100%, e.g., between 0.5 and 50%, between 1-30%,
between 5-80%, or at
least 80% (w/w) active ingredient.
C. Excipients and Diluents
[0138] In some embodiments, a pharmaceutically acceptable excipient may be at
least 95%, at
least 96%, at least 97%, at least 98%, at least 99%, or 100% pure. In some
embodiments, an
excipient is approved for use for humans and for veterinary use. In some
embodiments, an
excipient may be approved by United States Food and Drug Administration. In
some embodiments,
an excipient may be of pharmaceutical grade. In some embodiments, an excipient
may meet the
standards of the United States Pharmacopoeia (USP), the European Pharmacopoeia
(EP), the
British Pharmacopoeia, and/or the International Pharmacopoeia.
[0139] Excipients, as used herein, include, but are not limited to, any and
all solvents, dispersion
media, diluents, or other liquid vehicles, dispersion or suspension aids,
surface active agents,
isotonic agents, thickening or emulsifying agents, preservatives, and the
like, as suited to the
particular dosage form desired. Various excipients for formulating
pharmaceutical compositions
and techniques for preparing the composition are known in the art (see
Remington: The Science
and Practice of Pharmacy, 21st Edition, A. R. Gennaro, Lippincott, Williams &
Wilkins, Baltimore,
MD, 2006; incorporated herein by reference in its entirety). The use of a
conventional excipient
medium may be contemplated within the scope of the present disclosure, except
insofar as any
conventional excipient medium may be incompatible with a substance or its
derivatives, such as by
producing any undesirable biological effect or otherwise interacting in a
deleterious manner with
any other component(s) of the pharmaceutical composition.
[0140] Exemplary diluents include, but are not limited to, calcium
carbonate, sodium carbonate,
calcium phosphate, dicalcium phosphate, calcium sulfate, calcium hydrogen
phosphate, sodium
-37-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
phosphate lactose, sucrose, cellulose, microcrystalline cellulose, kaolin,
mannitol, sorbitol, inositol,
sodium chloride, dry starch, cornstarch, powdered sugar, etc., and/or
combinations thereof.
D. Inactive Ingredients
[0141] In some embodiments, fornmlations may include at least one inactive
ingredient. As used
herein, the term "inactive ingredient" refers to one or more agents that do
not contribute to the
activity of the active ingredient of the pharmaceutical composition included
in formulations. In
some embodiments, all, none or some of the inactive ingredients which may be
used in the
formulations of the present disclosure may be approved by the U.S. Food and
Drug Administration
(FDA).
E. Pharmaceutically acceptable salts
[0142] The auxotrophic factor may be administered as a pharmaceutically
acceptable salt
thereof. As used herein, "pharmaceutically acceptable salts" refers to
derivatives of the disclosed
compounds such that the parent compound is modified by converting an existing
acid or base
moiety to its salt form (e.g., by reacting the free base group with a suitable
organic acid). Examples
of pharmaceutically acceptable salts include, but are not limited to, mineral
or organic acid salts of
basic residues such as amines; alkali or organic salts of acidic residues such
as carboxylic acids;
and the like. Representative acid addition salts include acetate; acetic acid,
adipate, alginate,
ascorbate, aspartate, benzenesulfonate, benzene sulfonic acid, benzoate,
bisulfate, borate, butyrate,
camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate,
dodecyl sulfate,
ethanesulfonate, fumarate, glucoheptonate, glycerophosphate, hemisulfate,
heptonate, hexanoate,
hydrobromide, hydrochloride, hydroiodide, 2-hydroxy-ethanesulfonate,
lactobionate, lactate,
laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-
naphthalenesulfonate,
nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate,
persulfate, 3-phenylpropionate,
phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate,
tartrate, thiocyanate,
toluenesulfonate, undecanoate, valerate salts, and the like. Representative
alkali or alkaline earth
metal salts include sodium, lithium, potassium, calcium, magnesium, and the
like, as well as
nontoxic ammonium, quaternary ammonium; and amine cations, including, but not
limited to
ammonium, tetramethylanunonium, tetraethylammonium, methylamine,
dimethylamine,
trimethylamine, triethylamine, ethylamine, and the like. The pharmaceutically
acceptable salts of
the present disclosure include the conventional non-toxic salts of the parent
compound formed, for
example, from non-toxic inorganic or organic acids.
VI. Dosing and Administration
[0143] The modified host cells or auxotrophic factors of the present
disclosure included in the
pharmaceutical compositions described above may be administered by any
delivery route, systemic
-38-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
delivery or local delivery, which results in a therapeutically effective
outcome. These include, but
are not limited to, enteral (into the intestine), gastroenteral, epidural
(into the dura mater), oral (by
way of the mouth), transdermal, intracerebral (into the cerebrum),
intracerebroventricular (into the
cerebral ventricles), epicutaneous (application onto the skin), intradermal
(into the skin itself),
subcutaneous (under the skin), nasal administration (through the nose),
intravenous (into a vein),
intravenous bolus, intravenous drip, intra-arterial (into an artery),
intramuscular (into a muscle),
intracardiac (into the heart), intraosseous infusion (into the bone marrow),
intrathecal (into the
spinal canal), intraparenchymal (into brain tissue), intraperitoneal (infusion
or injection into the
peritoneum), intravesical infusion, intravitreal, (through the eye),
intracavernous injection (into a
pathologic cavity), intracavitaty (into the base of the penis), intravaginal
administration,
intrauterine, extra-amniotic administration, transdermal (diffusion through
the intact skin for
systemic distribution), transmucosal (diffusion through a mucous membrane),
transvaginal,
insufflation (snorting), sublingual, sublabial, enema, eye drops (onto the
conjunctiva), or in ear
drops, auricular (in or by way of the ear), buccal (directed toward the
cheek), conjunctival,
cutaneous, dental (to a tooth or teeth), electro-osmosis, endocervical,
endosinusial, endotracheal,
extracorporeal, hemodialysis, infiltration, interstitial, intra-abdominal,
intra-amniotic, intra-
articular, intrabiliary, intrabronchial, intrabursal, intracartilaginous
(within a cartilage), intracaudal
(within the cauda equine), intracisternal (within the cistema magna
cerebellomedularis),
intracorneal (within the cornea), dental intracornal, intracoronary (within
the coronary arteries),
intracorpoms cavemosum (within the dilatable spaces of the corpoms cavernosa
of the penis),
intradiscal (within a disc), intraductal (within a duct of a gland),
intraduodenal (within the
duodenum), intradural (within or beneath the dura), intraepidermal (to the
epidermis),
intraesophageal (to the esophagus), intragastric (within the stomach),
intragingival (within the
gingivae), intraileal (within the distal portion of the small intestine),
intralesional (within or
introduced directly to a localized lesion), intraluminal (within a lumen of a
tube), intralymphatic
(within the lymph), intrarnedullary (within the marrow cavity of a bone),
intrarneningeal (within the
meninges), intramyocardial (within the myocardium), intraocular (within the
eye), intraovarian
(within the ovary), intrapericardial (within the pericardium), intrapleural
(within the pleura),
intraprostatic (within the prostate gland), intrapulmonary (within the lungs
or its bronchi), intrasinal
(within the nasal or periorbital sinuses), intraspinal (within the vertebral
column), intrasynovial
(within the synovial cavity of a joint), intratendinous (within a tendon),
intratesticular (within the
testicle), intrathecal (within the cerebrospinal fluid at any level of the
cerebrospinal axis),
intrathoracic (within the thorax), intratubular (within the tubules of an
organ), intratumor (within a
tumor), intratympanic (within the aurus media), intravascular (within a vessel
or vessels),
-39-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
intraventricular (within a ventricle), iontophoresis (by means of electric
current where ions of
soluble salts migrate into the tissues of the body), irrigation (to bathe or
flush open wounds or body
cavities), laryngeal (directly upon the larynx), nasogastric (through the nose
and into the stomach),
occlusive dressing technique (topical route administration which is then
covered by a dressing
which occludes the area), ophthalmic (to the external eye), oropharyngeal
(directly to the mouth
and pharynx), parenteral, percutaneous, periarticular, peridural, perineural,
periodontal, rectal,
respiratoiy (within the respiratory tract by inhaling orally or nasally for
local or systemic effect),
retrobulbar (behind the pons or behind the eyeball), soft tissue,
subarachnoid, subconjunctival,
submucosal, topical, transplacental (through or across the placenta),
transtracheal (through the wall
of the trachea), transtympanic (across or through the tympanic cavity).
ureteral (to the ureter),
urethral (to the urethra), vaginal, caudal block, diagnostic, nerve block,
biliary perfusion, cardiac
perfusion, photopheresis, and spinal.
A. Parenteral and injectable administration
101441 In some embodiments, the modified host cells may be administered
parenterally.
101451 Injectable preparations, for example, sterile injectable aqueous or
oleaginous suspensions
may be formulated according to the known art using suitable dispersing agents,
wetting agents,
and/or suspending agents. Sterile injectable preparations may be sterile
injectable solutions,
suspensions, and/or emulsions in nontoxic parenterally acceptable diluents
and/or solvents, for
example, as a solution in 1,3-butanediol. Among the acceptable vehicles and
solvents that may be
employed are water, Ringer's solution, U.S.P., and isotonic sodium chloride
solution. Sterile, fixed
oils are conventionally employed as a solvent or suspending medium. For this
purpose, any bland
fixed oil can be employed including synthetic mono- or diglycerides. Fatty
acids such as oleic acid
can be used in the preparation of injectables.
101461 Injectable formulations may be sterilized, for example, by
filtration through a bacterial-
retaining filter, and/or by incorporating sterilizing agents in the form of
sterile solid compositions
which can be dissolved or dispersed in sterile water or other sterile
injectable medium prior to use.
101471 In order to prolong the effect of active ingredients, it is often
desirable to slow the
absorption of active ingredients from subcutaneous or intramuscular
injections. This may be
accomplished by the use of liquid suspensions of crystalline or amorphous
material with poor water
solubility. The rate of absorption of active ingredients depends upon the rate
of dissolution which,
in turn, may depend upon crystal size and crystalline form. Alternatively,
delayed absorption of a
parenterally administered drug form is accomplished by dissolving or
suspending the drug in an oil
vehicle. Injectable depot forms are made by forming microencapsule matrices of
the drug in
biodegradable polymers such as polylactide-polyglycolide. Depending upon the
ratio of drug to
-40-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
polymer and the nature of the particular polymer employed, the rate of drug
release can be
controlled. Examples of other biodegradable polymers include poly(orthoesters)
and
poly(anhydrides). Depot injectable formulations are prepared by entrapping the
drug in liposomes
or microemulsions which are compatible with body tissues.
B. Depot administration
101481 As described herein, in some embodiments, pharmaceutical
compositions including the
modified host cell of the present disclosure are formulated in depots for
extended release.
Generally, specific organs or tissues ("target tissues") are targeted for
administration. In some
embodiments, localized release is affected via utilization of a biocompatible
device. For example,
the biocompatible device may restrict diffusion of the cell line in the
subject.
[0149] In some aspects of the disclosure herein, pharmaceutical
compositions including the
modified host cell of the present disclosure are spatially retained within or
proximal to target
tissues. Provided are methods of providing pharmaceutical compositions
including the modified
host cell or the auxotrophic factor, to target tissues of mammalian subjects
by contacting target
tissues (which include one or more target cells) with pharmaceutical
compositions including the
modified host cell or the auxotrophic factor, under conditions such that they
are substantially
retained in target tissues, meaning that at least 10, 20, 30, 40, 50, 60, 70,
80, 85, 90, 95, 96, 97, 98,
99, 99.9, 99.99, or greater than 99.99% of the composition is retained in the
target tissues. For
example, at least 1%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 85%, 90%,
95%, 96%,
97%, 98%, 99%, 99.9%, 99.99% or greater than 99.99% of pharmaceutical
compositions including
the modified host cell or the auxotrophic factor administered to subjects are
present at a period of
time following administration.
101501 Certain aspects of the present disclosure are directed to methods of
providing
pharmaceutical compositions including the modified host cell or the
auxotrophic factor of the
present disclosure to target tissues of mammalian subjects, by contacting
target tissues with
pharmaceutical compositions including the modified host cell under conditions
such that they are
substantially retained in such target tissues. Pharmaceutical compositions
including the modified
host cell include enough active ingredient such that the effect of interest is
produced in at least one
target cell. In some embodiments, pharmaceutical compositions including the
modified host cell
generally include one or more cell penetration agents, although "naked"
formulations (such as
without cell penetration agents or other agents) are also contemplated, with
or without
pharmaceutically acceptable excipients.
-41-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
C. Therapeutic methods
101511 The present disclosure additionally provides a method of delivering
to a subject,
including a mammalian subject, any of the above-described modified host cells
or auxotrophic
factors including as part of a pharmaceutical composition or formulation.
D. Dose and Regimen
[0152] The present disclosure provides methods of administering modified
host cells or
auxotrophic factors in accordance with the disclosure to a subject in need
thereof. The
pharmaceutical compositions including the modified host cell or the
auxotrophic factor, and
compositions of the present disclosure may be administered to a subject using
any amount and any
route of administration effective for preventing, treating, managing, or
diagnosing diseases,
disorders and/or conditions. The exact amount required will vary from subject
to subject,
depending on the species, age, and general condition of the subject, the
severity of the disease, the
particular composition, its mode of administration, its mode of activity, and
the like. The subject
may be a human, a mammal, or an animal. The specific therapeutically
effective, prophylactically
effective, or appropriate diagnostic dose level for any particular individual
will depend upon a
variety of factors including the disorder being treated and the severity of
the disorder; the activity
of the specific payload employed; the specific composition employed; the age,
body weight,
general health, sex and diet of the patient; the time of administration, route
of administration, and
rate of excretion of the auxotrophic factor; the duration of the treatment;
drugs used in combination
or coincidental with the specific modified host cell or auxotrophic factor
employed; and like factors
well known in the medical arts.
[0153] In certain embodiments, modified host cell or the auxotrophic factor
pharmaceutical
compositions in accordance with the present disclosure may be administered at
dosage levels
sufficient to deliver from about 0.0001 mg/kg to about 100 mg/kg, from about
0.001 mg/kg to
about 0.05 mg/kg, from about 0.005 mg/kg to about 0.05 mg/kg, from about 0.001
mg/kg to about
0.005 mg/kg, from about 0.05 mg/kg to about 0.5 mg/kg, from about 0.01 mg/kg
to about 50
mg/kg, from about 0.1 mg/kg to about 40 mg/kg, from about 0.5 mg/kg to about
30 mg/kg, from
about 0.01 mg/kg to about 10 mg/kg, from about 0.1 mg/kg to about 10 mg/kg, or
from about 1
mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a
day, to obtain the
desired therapeutic, diagnostic, or prophylactic, effect.
[0154] In certain embodiments, modified host cell or auxotrophic factor
pharmaceutical
compositions in accordance with the present disclosure may be administered at
about 10 to about
600 I/site, 50 to about 500 1/site, 100 to about 400 1/site, 120 to about
300 I/site, 140 to about
-42-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
200 1/site, about 160 I/site. As non-limiting examples, the modified host
cell or auxotrophic
factor may be administered at 50 1/site and/or 150 I/site.
[0155] The desired dosage of the modified host cell or auxotrophic factor
of the present
disclosure may be delivered only once, three times a day, two times a day,
once a day, every other
day, every third day, every week, every two weeks, every three weeks, or every
four weeks. In
certain embodiments, the desired dosage may be delivered using multiple
administrations (e.g.,
two, three, four, five, six, seven, eight, nine, ten, eleven, twelve,
thirteen, fourteen, or more
administrations).
101561 The desired dosage of the modified host cells of the present
disclosure may be
administered one time or multiple times. The auxotrophic factor is
administered regularly with a set
frequency over a period of time, or continuously as a "continuous flow". A
total daily dose, an
amount given or prescribed in 24-hour period, may be administered by any of
these methods, or as
a combination of these methods.
[0157] In some embodiments, delivery of the modified host cell or
auxotrophic factor of the
present disclosure to a subject provides a therapeutic effect for at least 1
month, 2 months, 3
months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months,
11 months, 1
year, 13 months, 14 months, 15 months, 16 months, 17 months, 18 months, 19
months, 20 months,
20 months, 21 months, 22 months, 23 months, 2 years, 3 years, 4 years, 5
years, 6 years, 7 years, 8
years, 9 years, 10 years or more than 10 years.
[0158] The modified host cells may be used in combination with one or more
other therapeutic,
prophylactic, research or diagnostic agents, or medical procedures, either
sequentially or
concurrently. In general, each agent will be administered at a dose and/or on
a time schedule
determined for that agent. In some embodiments, the present disclosure
encompasses the delivery
of pharmaceutical, prophylactic, research, or diagnostic compositions in
combination with agents
that may improve their bioavailability, , reduce and/or modify their
metabolism, inhibit their
excretion, and/or modify their distribution within the body.
[0159] For example, the modified host cell or auxotrophic factor is
administered as a
biocompatible device that restricts diffusion in the subject to increase
bioavailability in the area
targeted for treatment. The modified host cell or auxotrophic factor may also
be administered by
local delivery.
101601 The disclosure herein contemplates methods of expressing a therapeutic
factor in a
subject comprising (a) administering said modified cells, (b) optionally
administering a
conditioning regime to permit modified cells to engraft, and (c) administering
said auxotrophic
factor.
-43-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0161] The term "conditioning regime" refers to a course of therapy that a
patient undergoes
before stem cell transplantation. For example, before hematopoietic stem cell
transplantation, a
patient may undergo myeloablative therapy, non-myeloablative therapy or
reduced intensity
conditioning to prevent rejection of the stem cell transplant even if the stem
cell originated from the
same patient. The conditioning regime may involve administration of cytotoxic
agents. The
conditioning regime may also include immtmosuppression, antibodies, and
irradiation. Other
possible conditioning regiments include antibody mediated conditioning (see
e.g., Czechowicz et
al., 318(5854) Science 1296-9 (2007); Palchaudari et al., 34(7) Nature
Biotechnology 738-745
(2016); Chhabra etal., 10:8(351) Science Translational Medicine 351ra105
(2016)) and CAR-T
mediated conditioning (see, e.g., Arai etal., 26(5) Molecular Therapy 1181-
1197 (2018); each of
which is hereby incorporated by reference in its entirety). Conditioning needs
to be used create
space in the brain for microglia derived from engineered HSCs to migrate into
to deliver the protein
of interest (recent gene therapy trials for ALD and MLD). The conditioning
regimen is also
designed to create niche "space" to allow the transplanted cells to have a
place in the body to
engraft and proliferate. In hematopoietic stem cell transplantation, for
example, the conditioning
regimen creates niche space in the bone marrow for the transplanted
hematopoietic stem cells to
engraft into. Without a conditioning regimen the transplanted hematopoietic
stem cells cannot
engraft. In some embodiments, the cell lines are T cells that are genetically
engineered to be
auxotrophic. Engineered auxotrophic T cells may be used as CAR T cells to act
as a living drug and
administered to a patient along with an auxotrophic factor to condition the
patient for a
hematopoietic stem cell transplant. Prior to the delivery of the donor
hematopoietic stem cells, the
auxotrophic factor may be removed, which results in the elimination of the
engineered auxotrophic
T cells. In some embodiments, the cell lines are allogenic T cells that are
genetically engineered to
be auxotrophic. Engineered auxotrophic allogenic T cells may be administered
to a patient along
with an auxotrophic factor to provide a therapeutic effect. Upon the patient
developing graft-
versus-host disease (GvHD), the auxotrophic factor may be removed, which
results in the
elimination of the engineered auxotrophic allogenic T cells which have become
alloreactive.
[0162] In some embodiments, administration of said auxotrophic factor is
continued regularly
for a period of time sufficient to express the therapeutic factor, and
preferably for a period of time
sufficient for the therapeutic factor to exert a therapeutic effect. In some
embodiments,
administration of said auxotrophic factor is decreased to decrease expression
of the therapeutic
factor. In some embodiments, administration of said auxotrophic factor is
increased to increase
expression of the therapeutic factor. In some embodiments, administration of
said auxotrophic
factor is discontinued to create conditions that result in growth inhibition
or death of the modified
-44-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
cells. In some embodiments, administration of said auxotrophic factor is
temporarily interrupted to
create conditions that result in growth inhibition of the modified cells.
[0163] The disclosure herein also contemplates a method of treating a
subject with a disease, a
disorder, or a condition comprising administering to the subject (a) said
modified mammalian host
cells and (b) said auxotrophic factor in an amount sufficient to produce
expression of a therapeutic
amount of the therapeutic factor.
[0164] Use of a modified mammalian host cell according to the present
disclosure for treatment
of a disease, disorder or condition is also encompassed by the disclosure.
101651 Certain embodiments provide the disease, the disorder, or the
condition as selected from
the group consisting of cancer. Parkinson's disease, graft versus host disease
(GvHD), autoimmune
conditions, hyperproliferative disorder or condition, malignant
transformation, liver conditions,
genetic conditions including inherited genetic defects, juvenile onset
diabetes mellitus and ocular
compartment conditions.
[0166] In certain embodiments, the disease, the disorder, or the condition
affects at least one
system of the body selected from the group consisting of muscular, skeletal,
circulatory, nervous,
lymphatic, respiratory endocrine, digestive, excretory, and reproductive
systems. Conditions that
affect more than one cell type in the subject may be treated with more than
one modified host cell
with each cell line activated by a different auxotrophic factor. In some
cases, a subject may be
administered more than one auxotrophic factor.
[0167] Certain embodiments provide the cell line as regenerative. In an
aspect of the present
disclosure, the subject may be contacted with more than one modified host cell
and/or with one or
more auxotrophic factor. Certain embodiments provide localized release of the
auxotrophic factor,
e.g. nutrient or the enzyme. Alternative embodiments provide systemic
delivery. For example,
localized release is affected via utilization of a biocompatible device. In an
aspect of the present
disclosure, the biocompatible device may restrict diffusion of the cell line
in the subject. Certain
embodiments of the method provide removing the auxotrophic factor to deplete
therapeutic effects
of the modified host cell in the subject or to induce cell death in the
modified host cell. Certain
embodiments of the method provide the therapeutic effects as including at
least one selected from
the group consisting of molecule trafficking, inducing cell death, cell death,
and recruiting of
additional cells. Certain embodiments of the method provide that the
unmodified host cells are
derived from the same subject prior to treatment of the subject with the
modified host cells.
[0168] The present disclosure contemplates kits comprising such compositions
or components
of such compositions, optionally with a container or vial.
-45-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
VII. Definitions
101691 The term "about" in relation to a numerical value x means, for example,
x+10%.
[0170] The term "active ingredient" generally refers to the ingredient in a
composition that is
involved in exerting a therapeutic effect. As used herein, it generally refers
to (a) the modified host
cell or donor template including a transgene as described herein, (b) the
corresponding auxotrophic
factor as described herein, or (c) the nuclease system for targeting cleavage
within the auxotrophy-
inducing locus.
[0171] The term "altered concentration" as used herein, refers to an
increase in concentration of
an auxotrophic factor compared to the concentration of the auxotrophic factor
in the subject prior to
administration of the pharmaceutical compositions described herein.
[0172] The term "altered pH" as used herein, refers to a change in pH induced
in a subject
compared to the pH in the subject prior to administration of the
pharmaceutical composition
described herein.
[0173] The term "altered temperature" as used herein refers to a change in
temperature induced
in a subject compared to the temperature in the subject prior to
administration of the
pharmaceutical composition as described herein.
[0174] The term "auxotrophy" or "auxotrophic" as used herein, refers to a
condition of a cell
that requires the exogenous administration of an auxotrophic factor to sustain
growth and
reproduction of the cell.
[0175] The term "auxotrophy-inducing locus" as used herein refers to a region
of a chromosome
in a cell that, when disrupted, causes the cell to be auxotrophic. For
example, a cell can be rendered
auxotrophic by disrupting a gene encoding an enzyme involved in synthesis,
recycling or salvage of
an auxotrophic factor (either directly or upstream through synthesizing
intermediates used to make
the auxotrophic factor), or by disrupting an expression control sequence that
regulates the gene's
expression.
[0176] The term "bioavailability" as used herein, refers to systemic
availability of a given
amount of the modified host cell or auxotrophic factor administered to a
subject.
[0177] The term "Cas9" as used herein, refers to CRISPR-associated protein 9,
which is an
endonuclease for use in genome editing.
101781 The term "comprising" means "including" as well as "consisting" e.g.
a composition
"comprising" X may consist exclusively of X or may include something
additional e.g. X + Y.
[0179] The term "conditioning regime" refers to a course of therapy that a
patient undergoes
before stem cell transplantation.
-46-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0180] The term "continuous flow" as used herein, refers to a dose of
therapeutic administered
continuously for a period of time in a single route/single point of contact,
i.e., continuous
administration event.
[0181] The term "CRISPR" as used herein, refers to clustered regularly
interspaced short
palindromic repeats of DNA that deploy an enzyme that cuts the RNA nucleotides
of an invading
cell.
[0182] The term "CRISPR/Cas9 nuclease system" as used herein, refers to a
genetic engineering
tool that includes a guide RNA (gRNA) sequence with a binding site for Cas9
and a targeting
sequence specific for the site to be cleaved in the target DNA. The Cas9 binds
the gRNA to form a
ribonucleoprotein complex that binds and cleaves the target site.
[0183] The term "expanding" when used in the context of cells refers to
increasing the number
of cells through generation of progeny.
[0184] The term "expression control sequence" refers to a nucleotide sequence
capable of
regulating or controlling expression of a nucleotide sequence of interest.
Examples include a
promoter, enhancer, transcription factor binding site, miRNA binding site.
101851 The term "homologous recombination" (FIR) refers to insertion of a
nucleotide sequence
during repair of breaks in DNA via homology-directed repair mechanisms. This
process uses a
"donor" molecule or "donor template" with homology to nucleotide sequence in
the region of the
break as a template for repairing the break. The inserted nucleotide sequence
can be a single base
change in the genome or the insertion of large sequence of DNA.
[0186] The term "homologous" or "homology," when used in the context of two or
more
nucleotide sequences, refers to a degree of base pairing or hybridization that
is sufficient to
specifically bind the two nucleotide sequences together in a cell under
physiologic conditions.
Homology can also be described by calculating the percentage of nucleotides
that would undergo
Watson-Crick base pairing with the complemental), sequence, e.g. at least 70%
identity, preferably
at least 75 /0, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or
higher
identity over a specified number of bases. With respect to donor templates,
for example, the
homology may be over 200-400 bases. With respect to guide sequences, for
example, the homology
may be over 15-20 bases.
101871 The term "operatively linked" refers to functional linkage between a
nucleic acid
expression control sequence (such as a promoter, enhancer, signal sequence, or
array of
transcription factor binding sites) and a second nucleic acid sequence,
wherein the expression
control sequence affects transcription and/or translation of the second
nucleic acid sequence.
-47-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0188] The term "pharmaceutical composition" as used herein, refers to a
composition including
at least one active ingredient and optionally one or more pharmaceutically
acceptable excipients.
[0189] The term "pharmaceutically acceptable salt" as used herein, refers
to derivatives of the
disclosed compounds such that the parent compound is modified by converting an
existing acid or
base moiety to its salt form (e.g., by reacting the free base group with a
suitable organic acid). All
references herein to compounds or components include the pharmaceutically
acceptable salt
thereof.
[0190] The term "regenerative" as used herein, refers to renewal or
restoration of an organ or
system of the subject.
[0191] The term "therapeutic factor" refers to a product encoded by the
inserted transgene that
treats and/or alleviates symptoms of the disease, disorder, or condition of
the subject.
101921 The term "therapeutic amount" refers to an amount of therapeutic factor
sufficient to
exert a "therapeutic effect", which means an alleviation or amelioration of
symptoms of the disease,
disorder or condition.
[0193] The term "unit dose" as used herein, refers to a discrete amount of
the pharmaceutical
composition including a predetermined amount of the active ingredient.
EXAMPLES
Example 1. General T cell culture methods
101941 K562 cells (acquired from ATCC) and Nalm6 cells (kindly provided by C.
Mackall)
were cultured in RPMI 1640 (HyClone) supplemented with 10% bovine growth
serum, 2mM L-
glutamine and 100 U/ml Penicillin and 100 U/ml Streptomycin. T cells were
either used fresh after
isolation from buffy coats obtained from healthy donors. T cells were isolated
through a Ficoll
density gradient centrifugation followed by magnetic enrichment using the Pan
T Cell Isolation Kit
(Miltenyi Biotec).
[0195] Cells were cryopreserved in BAMBANKERTm medium. After thawing cells
were
cultured at 37 C, 5% CO2 in X-Vivo 15 (Lonza) supplemented with or without 5%
human senun
(Sigma-Aldrich) and 100 human recombinant 1L-2 (Peprotech) and 10 ng/ml human
recombinant
IL-7 (BD Biosciences). UMP or Uridine was added at 250 jag/ml. 5-FOA was added
at 100 p.g/m1
to lmg/ml. During culture, medium was refreshed every 2 days.
[0196] T cells were activated using immobilized Anti-CD3 (clone OKT3, Tonbo
Biosciences)
and soluble anti-CD28 (clone CD28.2, Tonbo Biosciences) for three days before
electroporation.
[0197] 1.4 million activated T cells were resuspended in electroporation
solution, mixed with
the pre-complexed RNP, and electroporated using a 4D-NUCLEOFECTORTm system
(Lonza)
-48-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
using program EO-115. The RNP consisted of Cas9 protein (Alt-Re CRISPR/Cas9
system based
on S. pyogenes, IDT) at 300 tg/m1 and sgRNA using a sgRNA:Cas9 molar ratio of
2.5.
[0198] Gcnomic DNA was harvested using QUICKEXTRACTTm DNA Extraction Kit
(Epicentre). Cells were counted on an automated cell counter using Trypan blue
staining or on a
CytoFLEX flow cytometer (Beckman Coulter) with automatic plate reader using
COUNTBRIGHTThi beads (ThermoFisher) as a reference for normalizing the values.
Alternatively,
cells were analyzed after staining with fluorochrome-labelled antibodies
(Biolegend) on an
ACCURITM C6 flow cytometer (BD Biosciences), which also measures volumes, or a
FACS
ARIATM II SORP cell sorter (BD Biosciences). Data was analyzed using Excel
(Microsoft) and
FlowJo software (Tree Star).
[0199] Sanger sequencing of the UMPS locus was performed using UMPS-O-1 and
bl4PS-0-2,
with the region amplified using PHUSIONTm Hot Start Flex 2x Master Mix (New
England Biolabs,
Inc.). Sanger sequencing traces were analyzed by TIDE analysis (see, Brinkman
et al, 2014,
Nucleic Acids Res. 42(22):e168), which is hereby incorporated by reference in
its entirety) to
identify insertions and deletions (InDels) after editing. InDel quantification
was performed on the
sequences using the TIDE online tool (www.deskgen.com/landing/tide.html) (See,
M. Sadelain, N.
Engl. J. Med. 365, 1735-7 (2011), which is hereby incorporated by reference in
its entirety.
[0200] gRNA sequences (including protospacer adjacent motifs, also referred to
as PAMs):
[0201] UMPS-7
[0202] GCC CCG CAG AUC GAU GUA GAG UUU UAG AGC UAG AAA UAG CAA GUU
AAA AUA AGG CUA GUC CGU UAU CAA CUU GAA AAA GUG GCA CCG AGU COG
UGC UUU U (SEQ ID NO: I)
102031 Sequencing oligonucleotides for UMPS locus TIDE analysis:
[0204] UMPS-0-1: CCCGGGGAAACCCACGGGTGC (SEQ ID NO: 2)
[0205] UMPS-0-2: AGGGTCGGTCTGCCTGCTTGGCT (SEQ ID NO: 3)
[0206] After the initial screening, sgRNA "UMPS-7," which showed the highest
frequency of
InDels was chosen for further analysis,
Example 2. UMPS editing bv Cas9-sgRNA electroporation in human T cells
[0207] T Cells were thawed and cultured, followed by activation and
subsequent electroporation
with Cas9-UMPS-7 sgRNA RNP as described above. Following electroporation,
cells were allowed
to recover in medium with or without serum, 5-FOA or an exogenous uracil
source (FIG. 1A). Cell
survival following electroporation was markedly increased when serum was
included in the media
(FIG. 1B), and thus a four-day recovery period in medium with serum, uridine,
and UMP was
performed in all subsequent experiments. Cell counts post-electroporation are
shown in Table 4.
-49-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
Table 4. Cell counts
Sample intact cells (absolute)
Semm 30217
Mock +120A 580
Mock 901
UMPS KO + FOA 395
LIMPS KO 560
Example 3. Growth of a mixed LIMPS edited population and maintenance of LIMPS
mutations
102081 T Cells were electroporated and edited as in Example 2 and allowed to
recover for a 4-
day period in medium with serum, uridine, and UMP. On day 4, cells were
shifted to UMP, uridine,
or uracil source media. This experiment did not feature a selection step and
thus the resulting
population of cells was a heterogeneous mix of wild-type (WT), heterozygous
mutant and
homozygous mutant cells. The growth of homozygous (IMPS mutant cells was
observed to be
dependent on an exogenous uracil source ¨ as these should be auxotrophic (FIG.
2A). When UMPS
is targeted, InDels were observed to be generated in about 50% of cells (as
assayed by TIDE
analysis (See, Brinkman et al, 2014, Nucleic Acids Res. 42(22):e168), which is
hereby incorporated
by reference in its entirety).
102091 When the exogenous uracil source was removed, the InDel frequency in
the population
was reduced after three days of growth (Day 7 = four days of recovely and
three days in test
media). This was consistent with the model showing that any homozygous
auxotrophic UMPS
mutant cells would be outcompeted in the population by non-auxotrophic
heterozygous mutants
and WT cells still present after editing ¨ resulting in a reduced apparent
InDel frequency (see. FIG.
2B). The percentages of alleles with InDels are shown in Table 5.
Table 5. Alleles with InDels
Condition Percent of alleles
(without 5-F0A)
no metabolites 57.9
with LIMP 71.1
with Uridine 77.0
102101 The optimal growth of the heterogeneous UMPS edited population was
observed to be
dependent on the presence of an exogenous source of uracil (FIG. 2C-FIG. 2F).
The percent of
alleles with a frameshift InDel is shown in FIG. 2C, and the values are shown
in Table 6.
Table 6. Alleles with frameshift InDels
Percent of alleles (without 5-F0A)
no metabolites 14.3
with UMP 46.1
with Uridine 52.5
-50-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0211] FIG. 2D compares the predicted absolute numbers of cells at day 8
containing alleles
identified by TIDE. The values are shown in Table 7.
Table 7. Predicted viable cell counts
noLCondition Cells with frameshift InDel Cells with In-
frame InDel Cells without InDel
metabolites 365000 1110000 1073550
with UMP 1670000 908000 1049070
;with Uridine 1660000 777000 729100
[0212] FIG. 2E shows the time course (eight days) of cell counts with/without
UMP. The values
are shown in Table 8.
Table 8. Cell density [cells per ml]
Treatment Metabolite Day 0 Day 1 . Day 2 Day 4 Day 6
Day 8
Mock no
metabolites 5.00E+05 9.67E+05 2.35E+06 3.44E+06 4.15E+06 3.71E+06
CCR5 knockout no metabolites 5.00E+05 8.35E+05 2.29E+06 3.42E+06 3.90E+06
_3.91E+06
LIMPS knockout no metabolites 5.00E+05 8.08E1-05 1.59E+06 2.51E+06 2.21E+06
2.55E+06
Mock with UMP 5.00E+05
9.83E+05 2.01E+06 3.80E+06 4.18E+06 3.90E+06
CCR5 knockout with UMP 5.00E+05 I
.02E+06 1.80E+06 3.32E+061 3.80E+06 4.03E+06
UMPS knockout with UMP 5.00E+05
8.74E+05 1.86E+06 3.47E+0613.88E+06 3.63E+06
[0213] FIG. 2F shows the time course (eight days) of cell counts
with/without uridine. The
values are shown in Table 9.
Table 9. Cell density [cells per ml]
Treatment Metabolite Day 0 Day I Day 2 Day 4
Day 6 Day 8
Mock no metabolites 5.00E+05 9.67E+05 2.35E+06 3.44E+06 4.15E+06
3.71E+06
CCR5 knockout no metabolites 5.00E+05 8.35E+05 2.29E+06 3.42E+06 3.90E+06
3.91E+06
UMPS knockout no metabolites 5.00E+05 8.08E+05 1.59E+06 2.51E+06 2.21E+06
2.55E+06
Mock with Uridine 5.00E+05 9.78E+05 1.98E+06 3.90E+06 4.91E+06
4.09E+06
CCR5 knockout IN ith Uridine 5.00E+05 9.67E+05 1.71E+06 3.70E+06 3.92E+06
3.96E+06
UMPS knockout with Uridine 5.00E+05 7.69E+05 1.59E+06 3.43E+06 3.79E+06
3.17E+06
102141 UMP and uridine rescued the growth of an UMPS edited culture to the
same level as
mock edited cells. This rescue of growth is dependent on UMPS editing and is
not seen in mock
cells treated with an exogenous uracil source, indicating that edited (IMPS
makes human T cells
specifically dependent on uracil supplementation for optimal cell growth.
102151 It is worth reiterating the UMPS edited population contained
unedited or heterozygous
cells that are not expected to be auxotrophic, and thus complete lack of
growth of UMPS edited
cells in uracil deficient media is not expected.
Example 4. 5-FOA treatment selects for UMPS tar2eted cells
[0216] 5-FOA selects for uracil auxotrophic cells in other organisms (e.g.
Boeke et al. 1984,
Mol. Gen. Genet. 197(2):345-6), which is hereby incorporated by reference in
its entirety). To
investigate the potential utility of 5-FOA for the selection of uracil
auxotrophs among human cells,
the UMPS gene was targeted in human T cells by Cas9-gRNA complex
electroporation followed by
-51-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
recovery (as shown in Example 2) followed by an assay of resistance to 5-FOA
treatment (FIG.
3A). Cells were grown in 5-FOA and a variety of combinations of serum and wadi
sources for 4
days before cell counting was performed.
[0217] Table 10 compares cell counts for cell populations grown with or
without serum.
Table 10. Cell counts
Average number of cells per volume unit
Culture condition Substrates Mock UMPS-7
Wilh Serum UMP + Und 6307L71 72181.87
With Serum No UMP/Uridine 13403.28 54282.95
No Serum UMP Urid 49125.44 72385.14
No Serum No UMP/Uridine 13947.04 56895.21
102181 Serum, while important for the recovery of cells post
electroporation, had no effect on
the viability of cells in 5-FOA (FIG. 3B). The cell counts for additional
samples grown in 5-FOA
without serum are shown in FIG. 3C and Table 11.
Table 11. Cell counts
Average of cells per volume unit
Substrates Mock U MPS-7
UM? 24770.99 58299.26
No UMP/Uridine 12279.07 52156.98
Uridine 53052.43 77755.72
No UMP/Uridine 16467.39 67438.73
[0219] Uridine and UMP improved the survival of both mock treated and (IMPS
targeted cells
in 5-FOA compared to control . This is likely through a competition-based
mechanism (uridine can
reverse 5-fluorouracil toxicity in htunans (see, van Groeningen et al. 1992,
Semin. Oncol. 19(2
Suppl 3):148-54, which is hereby incorporated by reference in its entirety))
(FIG. 3B and FIG. 3C).
In all cases. UMPS targeted cells exhibited increased survival compared to
mock targeted cells.
This data indicated that 5-FOA can be used for the selection of uracil
auxotrophic cells in a human
cell culture.
Example 5. 5-FOA selected UMIn targeted cells exhibit uracil auxotrophv
[0220] To assay whether or not the cells selected for by 5-FOA treatment were
uracil
auxotrophs, mock or (IMPS targeted T cells were exposed to 5-FOA as shown in
Example 4.
Following 4 days of 5-FOA selection, the population of cells was split into an
uracil containing
media (UMP, uridine or both) and an uracil deficient media. A growth assay was
subsequently
performed by cell counting after following 4 days incubation in test media
(Day 8) (FIG. 4A). In all
cases, cell growth in the mock targeted cell cultures was negligible and
independent of uracil
source supplementation ¨ indicating successful killing of non-UMPS targeted
cells during the 5-
FOA selection step (FIG. 4B-FIG. 4D). In the UMPS targeted population, in all
conditions cell
-52-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
growth was stimulated by the addition of uracil and poor cell growth was
observed in its absence
(FIG. 4B-FIG. 4D).
[0221] FIG. 4B compares the cell counts in culture on Day 8 for samples
without serum. The
values are shown in Table 12.
Table 12. Cell counts on Day 8
No UMP/Uridine UMP Urid
Replicate 1 I 2 3 4 1 1 2 1 3 4
Mock 893 1365 223 512 1061 1185 4161 292
UMPS knockout 10268 10585 4318 4352 13908 13526 8045 6190
[0222] FIG. 4C compares the cell counts in cultures supplemented with UMP and
without
serum. The values are shown in Table 13.
Table 13. Cells counts on Day 8
No UMP UMP
Replicate 1 Replicate 2 Replicate 1 Replicate
2
Mock 1116 409 1421 490
UMPS knockout 7847 4100 9978 6392
[0223] FIG. 4D
compares the cell counts in cultures supplemented with uridine and without
serum. The values are shown in Table 14.
Table 14. Cells counts on Day 8
No Uridine Uridine
Replicate 1 Replicate 2 Replicate 1 Replicate 2
Mock 1386 431 1249 687
UMPS knockout 7795 3945 12006 5629
[0224] Taken together, the results of Examples 1-5 indicate that editing of
the (IMPS locus by
Cas9 in human T cells generates cells that are dependent on an exogenous
uracil source for optimal
cell growth. These results demonstrate that engineered human auxotrophy can be
used as a
mechanism for controlling the proliferation of T cells or some other cell
therapy. In addition, 5-
FOA selection of UMPS edited cells provides a useful mechanism for selection
of a true
auxotrophic population of T cells.
Example 6. Culturine stem cells
[0225] In order
to evaluate another cell type with potential therapeutic relevance, IMPS was
engineered in human pluripotent cells. The modified host cells that are the
subject matter of the
disclosure herein may include stem cells that were maintained and
differentiated using the
techniques below as shown in U.S. 8,945,862, which is hereby incorporated by
reference in its
entirety.
-53-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0226] Undifferentiated hESCs (H9 line from WICELLC, passages 35 to 45) were
grown on an
inactivated mouse embryonic fibroblast (MEF) feeder layer (Stem Cells, 2007.
25(2): p. 392-401,
which is hereby incorporated by reference in its entirety). Briefly, the cell
was maintained at an
undifferentiated stage on irradiated low-passage MEF feeder layers on 0.1%
gelatin-coated plates.
The medium was changed daily. The medium consists of Dulbecco's Modified Eagle
Medium
(DMEM)/F-12, 20% knockout serum replacement, 0.1 mM nonessential amino acids,
2 mM L-
glutamine, 0.1 mM13-mercaptoethanol, and 4 ng/ml rhFGF-2 (R&D Systems Inc.,
Minneapolis).
The undifferentiated hESCs were treated by 1 mg/ml collagenase type IV in
DMEM/F12 and
scraped mechanically on the day of passage. Prior to differentiation, hESCs
were seeded onto
MATRIGELC protein mixture (Corning, Inc.)-coated plates in conditioned medium
(CM) prepared
from MEF as follows (Nat Biotechnol, 2001. 19(10): p. 971-4, which is hereby
incorporated by
reference in its entirety). MEF cells were harvested and irradiated with 50 Gy
and were cultured
with hES medium without basic fibroblast growth factor (bFGF). CM was
collected daily and
supplemented with an additional 4 ng/ml of bFGF before feeding hES cells.
Example 7. In vitro differentiation of human embryonic stem cell (ESC)-
endothelial cells
(ECs)
102271 To induce hESC differentiation, undifferentiated hESCs were cultured
in differentiation
medium containing Iscove's Modified Dulbecco's Medium (IMDM) and 15% defined
fetal bovine
serum (FBS) (Hyclone, Logan, Utah), 0.1 mM nonessential amino acids, 2 mM L-
glutamine, 450
gM monothioglycerol (Sigma, St. Louis, Mo.), 50 U/ml penicillin, and 50 g/m1
streptomycin,
either in ultra-low attachment plates for the formation of suspended embryoid
bodies (EBs) as
previously described (see, Proc Nat! Acad Sci USA, 2002. 99(7): p. 4391-6 and
Stem Cells, 2007.
25(2): p. 392-401; each of which is hereby incorporated by reference in its
entirety). Briefly, hESCs
cultured on MATRIGELC protein mixture (Corning, Inc.) coated plate with
conditioned media
were treated by 2 mg/ml dispase (Invitrogen, Carlsbad, Calif.) for 15 minutes
at 37 C. to loosen the
colonies. The colonies were then scraped off and transferred into ultra-low-
attachment plates
(Corning incorporated. Corning, N.Y.) for embiyoid body formation.
Example 8. Selection of auxotrophic modified host cells
[0228] The (MPS locus was disrupted in the hESCs by electroporation of Cas9
RNP and
selection of a clone with InDels in exon 1 as evaluated by amplification and
Sanger sequencing of
the genomic locus. For gene editing, hESCs were treated with 10 gm ROCK
inhibitor (Y-27632)
for 24 hours before electroporation. Cells at 70-80% confluence were harvested
with
ACCUTASEC solution (Life Technologies). 500,000 cells were used per reaction
with a SpCas
concentration of 150 gg/mL (Integrated DNA Technologies) and a Cas9:sgRNA
molar ratio of 1:3
-54-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
and electroporation performed in P3 Primary Cell solution (Lonza) in 16-well
NUCLEOCUVETTETm Strips in the 4D NUCLEOFECTOR system (Lonza). Immediately
after
cicctroporation, cells were transferred into one well of a MATRIGEL protein
mixture (Corning,
Inc.)-coated 24 well plate containing 500 I of mTeSRTm media (STEMCELL
Technologies) with
M Y-27632. Media was changed 24 hours after editing and Y-27632 was removed 48
hours
after.
[0229] Sanger sequencing compared the hESC population before editing, the
bulk population
after RNP electroporation, and the genotype of the selected clone. Results
showed a deletion of
10bp around the sgRNA target region. The lack of a sequence trace in this
region indicated both
alleles had been modified.
[0230] An auxotrophy assay was performed over four days with different
concentrations of
uridine. Microscope photos of wells were taken on day 4 after seeding UMPS""
hESCs at
similar densities and culturing in the presence of different uridine
concentrations. The photos
showed that cells proliferated in the presence of 2.5-250 g/m1 but showed no
proliferation without
added uridine. Quantification of viable cells on day 4 after seeding to
evaluate the effect of
different uridine concentrations is shown in Table 15.
Table 15. Viable cell counts
Uridine Replicate 1 Replicate 2 Replicate 3
None 0 0 0
2.5 pc/nil 31040 38065 45189
25 pg/inl 31810 39635 36283
250 pglinl 19147 31050 33955
192311 Kill curves with different concentrations of supplement versus
control were generated to
demonstrate that an exogenously supplied version of the product of the knocked-
out gene rescues
the auxotrophic phenotype of the cell line.
[0232] To assess resistance to 5-F0A, the (IMPS-KO hESCs were genetically
engineered to
express GFP from an expression cassette integrated into a safe-harbor locus
for easier identification
in co-culture with UMPS-WT cells.
[0233] A clone that showed bright and stable expression of GFP was selected.
These
UMPSKc" hESCs were mixed with UMPSwrAvr cells that were not expressing GFP and
followed
up by fluorescence-activated cell sorting (FACS) analysis in the presence of
different
concentrations of 5-F0A. Table 16 provides counts of viable GFP+ and GFP-
cells after culture
with different 5-FOA concentrations.
Table 16. Viable cell counts
GFP+ GFP-
None 133875 121125
-55-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
0.25 1Lem1 142820 5180
2.5 jig/m1 11812.5 687.5
25 jig/ml 8455.98 334.02
[0234] Similar to the previous cell types, enrichment for GFP+ cells over
time was observed.
54.8% of the cells were GFP+ in the group without 5-F0A, and 95.0% of the
cells were GFP+ in
the groups with 5-F0A. In this cell type, UMPS-WT cells were sensitive to all
tested 5-FOA
concentrations, and UMPS-K0 cells tolerated the concentration of 0.25 tig/m1
well, while showing
impaired proliferation at higher concentrations as shown in Table 16.
[0235] In conclusion, these results confirm that a key pathway of metabolism
may be
engineered efficiently to create auxotrophy in a range of human cells from
leukemia cell lines to
pluripotent cell lines and primary immune cells. Gene targeting of both UMPS
alleles may be used
to create and purify a cell population with homozygous knockout or enrich
those cells using 5-
FOA.Cell lines with multiple knockouts and mutations may be also generated to
provide rapid
multiplexed genome engineering and selection (e.g. 5 auxotrophic mutations and
5 antibiotics).
Example 9. In vivo analysis
[0236] In vitro validated auxotrophic knockout cell lines also may be analyzed
in vivo. These
cell lines are constrained by toxicity and bioavailability of the auxotrophic
factor in humans. The
gene knockout cell lines are engineered from human T cells or any other
lymphocyte. Conditional
in vitro growth by the cell line is demonstrated in the presence of the
auxotrophic factor, and not in
the absence of the auxotrophic factor. The modified mammalian host cells
confirmed to be
auxotrophic for the factor and capable of expressing the transgene may be
administered in a mouse
model. Only mice consuming the auxotrophic factor supplement sustain growth of
human
lymphocytes. Further, cell growth stops in vivo upon removal of nutrient from
the mouse food
source.
Example 10. Creatine auxotrophy in human cells throueh genetic eneineering
[0237] Bioinformatics tools (crisportefor.net) were used to identify
possible sgRNA target sites
in exon 1 of the UMPS gene for spCas9. Putative off-target (0T) effects were
predicted using
COSMID (crispubme.gatech.edu0 (See, Majzner et a1. Cancer Cell. 31, 476-485
(2017), which is
hereby incorporated by reference in its entirety). Potential off-target sites
in the human genome
(hg38) were identified using the web-based bioinformatics program COSMID
(crispr.bme.gatech.edu) with up to 3 mismatches or lbp deletion/insertion with
1 mismatch allowed
in the 19 PAM proximal bases. The sgRNAs were ranked by number of highly-
similar off-target
sites (COSMID score <1) and then ranked by number of OT sites with higher
scores. Primers for
amplifying all sites were also designed by the COSMID program. All sites were
amplified by locus
specific PCR, barcoded via a second round of PCR, pooled at equimolar amounts
and sequenced
-56-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
using an Illumina MiSeq using 250bp paired end reads as previously described
in Porteus, NI. Mol.
Ther. 19, 439-441(2011), which is hereby incorporated by reference in its
entirety. The resulting
data was analyzed using the custom script indelQuantificationFromFastqPaired-
1Ø1.p1(10)(
https://github.com/piyuranjan/NucleaseIndelActivityScript/blob/master/indelQuan
tificationFromFa
stqPaired-1Ø1.p1).
102381 The 3 sgRNAs with the lowest number of OT sites were identified and
used for an in
vitro screening of activity. These sgRNAs are shown in Table 17.
Table 17. sgRNAs with fewest OT sites
Name Target sequence + PAM SEQ
ID NO. COSMID total OT sites MIT Specificity Score
U4.IPS-3 CCCCGCAGATCGATGTAGAT GGG 4, 1 96
MPS-7 GCCCCGCAGATCGATGTAGA TGG 5 6 94
t.,TAiPS-6 GGCGGTCGCTCGTGCAGCTT TGG 6 3 94
[02391 sgRNAs were acquired with chemical modifications from Synthego
Corporation. The
sgRNAs were complexed with Cas9 protein (IDT) at a molar ration of 2.5:1
(sgRNA:protein) and
electroporated into activated T cells using a 4D- NUCLEOFECTORTm system
(Lonza). 4 days
later, cells were harvested, and genomic DNA extracted using QUICKEXTRACTTm
DNA
Extraction Kit (Epicentre) according to the manufacturer's protocol. The sgRNA
target site was
amplified with specific primers (Table 18) and the amplicon sequenced by
Sanger sequencing
(MCLab, South San Francisco).
Table 18. Primers
Name Sequence SEQ ID NO.
LIMPS TIDE Fwd CCCGGGGAAACCCACGGGTGC _ 2
LIMPS TIDE Rev AGGGTCGGTCTGCCTGC,TTGGCT 3
102401 InDel quantification was performed on the sequences using the
interference of CRISPR
edits (ICE) and 10E-D online tools (ice.synthego.com) (FIG. 5A). Results are
shown in Table 19.
Table 19. InDel quantification
ICE InDels CYO ICE-D InDels CYO
UMPS-3 45 43
UMPS-6 12 11
LIMPS-7 39 93
[0241] sgRNA "UMPS-7" was chosen for further experiments. This sgRNA led to
the creation
of a high proportion of large (greater than 30 bp) deletions that were
detectable by inference of
CRISPR edits - discordance (ICE-D) but not by conventional ICE or TIDE
analysis
(www.deskgen.comtlanding/tide.html).
102421 To evaluate whether the (IMPS knockout leads to differential cell
proliferation if
cultured without the addition of Uridine or Uridine monophosphate (UMP), the
cell counts in
-57-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
culture were followed over time by automatic cell counting with Tiypan blue
staining. (IMPS
knockout led to lower cell counts from day 2 after electroporation, compared
to cells that were
mock electroporated or electroporated using Cas9 targeting a different genomic
locus (i.e., CCR3)
(FIG. 5B). The cell counts are shown in Table 20.
Table 20. InDel quantification
Days Mock, no metabolites CCR5 knockout, no metabolites UMPS knockout, no
metabolites
0 500000 500000 500000
967000 835000 808000
2 2350000 2290000 1590000
4 3440000 3420000 2510000
6 4150000 39()0000 2210000
8 3710000 3910000 2550000
102431 In contrast, cell proliferation was not impaired after UMPS knockout
if UMP or Uridine
were supplemented at high concentrations (250 pg/m1 each) (FIG. 5C). The
number of viable cells
per ml is shown in Table 21.
Table 21. Number of viable cells per ml
Day U MPS
knockout, no metabolites UMPS knockout, with UMP UMPS knockout, with Uridine
0 500000 500000 500000
1 808000 874000 769000
2 1590000 1860000 1590000
4 2510000 3470000 3430000
6 2210000 388000Q_ 3790000
8 2550000 3630000 3170000
102441 To confirm the results on the genomic level, genomic DNA was harvested
at the end of
the experiment and InDels were quantified (FIG. 5D-FIG. 5E). FIG. 5D compares
the frequency of
InDels in different culture conditions for cells not exposed to 5-F0A.
Percentages are shown in
Table 22.
Table 22. Percentages of overall InDel frequency
Culture condition Percent (%)
no metabolites 57.9
with UMP 71.1
with Uridine 77.0
192451 FIG. 5E compares the frequency of frameshift InDels in different
culture conditions for
cells not exposed to 5-FOA. Percentages are shown in Table 23.
Table 23. Percentages of fraineshift InDel frequency
Culture condition Percent
no metabolites 14.3
with UMP 46.1
with Uridine 52.5
-58-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0246] Overall InDel frequency was slightly reduced after culture without
Uridine or UMP, but
when quantifying InDels that would lead to a frameshift (not multiples of +3/-
3), there was a
reduction of InDels in the cell population without the metabolite addition.
This confirms that cells
with UMPS knockout due to a frameshift InDel in exon I have a disadvantage in
survival and
proliferation compared to UMPS wild-type cells or cells with InDel in exon 1
that preserved the
reading frame.
[0247] Next, gene targeting constructs were generated that allow the
integration of 2 different
markers into the UMPS locus, thereby disrupting gene expression and enabling
the identification of
the cells with bi-allelic gene knockout through co-expression of tEGFR and
tNGFR (FIG. 6A),
using the approach described in Bak et al.õ Elife 28:6 (2017), which is hereby
incorporated by
reference in its entirety. The constructs were cloned by Gibson assembly using
standard molecular
biology methods with a plasmid backbone that is flanked by the AAV2 inverted
terminal repeats
(1TRs).
[0248] For targeting of stein cells and primary human cells, the constructs
were packaged in
recombinant adeno-associated virus type 6 (rAAV6) to deliver the DNA after
creation of the
double-strand break, thereby stimulating homologous recombination to integrate
the transgenes.
Transfer plasmids for the production of rAAV6 were created by cloning the
transgene and
surrounding aims homologous to the targeted genomic region into the backbone
of pAAV-MCS
plasmid (Agilent Technologies) adjacent to the flanking inverted terminal
repeats (ITR) by Gibson
assembly (NEBUILDER HiFi DNA Assembly Master Mix, New England Biolabs Inc.).
The
homology arms were amplified by PCR from healthy donor genomic DNA. For the
expression of
surface markers, we used the tNGFR and tEGFR (See, Teixeira et al. Cum. Opin.
Biotechnol. 55,
87-94 (2019); Chen et al. Sci. Transl. Med. 3 (2011); each of which is hereby
incorporated by
reference in its entirety). For transcription termination, the poly-
adenylation sequence from bovine
growth hormone (bGH) was used.
[0249] Production of AAV was performed in HEK293T cells by co-transfection of
the transfer
plasmid with the pdgm6 packaging plasmid and purified by lodixanol gradient
centrifugation. The
HEK293 cells were co-transfected with polyethyleneimine with the pDGM6 helper
plasmid and the
respective transfer plasmid carrying the transgene between homology arms
flanked by the AAV2
ITRs. After 48 hours the cells were detached, separated from the supernatant
and lysed. The
suspension was treated with Benzonase (Sigma Aldrich) and debris pelleted. The
crude AAV
extract was purified on an Iodixanol density gradient and then subjected to 2
cycles of dialysis
against PBS and one cycle against PBS with 5% sorbitol in 1 x 104 molecular
weight cut off
(MWCO) SLIDE-A-LYZERTm G2 Dialysis Cassettes (Thermo Fisher Scientific). The
AAV titer
-59-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
was determined by extraction of genomic DNA by QUICKEXTRACTTm DNA Extraction
Kit
(Epicentre) and measuring the absolute concentration of ITR copy numbers by
droplet digital PCR
(Bio-rad) according to the manufacturers protocol using previously reported
primer and probe sets
(See, Jaen et al., Mol. Ther. Methods Clin. Dev. 6, 1-7 (2017, which is hereby
incorporated by
reference in its entirety.).
[0250]
Targeting with these donor constructs used as plasmids was first tested in the
myeloid
leukemia cell line K562 (ATCCS CCL-243Tm). The cells were electroporated with
2 pg of each
plasmid on a SF Cell Line 4D NUCLEOFECTORTm system (Lonza) following the
manufacturer's
protocol. When targeting the 2 markers into the UMPS locus, a small but stable
population of cells
that showed co-expression of both markers was identified (FIG. 6B).
[0251] Magnetic
bead enrichment was used to sequentially enrich for the cells expressing the
surface markers EGFR and NGFR. For magnetic separation, cells expressing both
tNGFR and
tEGFR were enriched by sequential magnetic bead sorting using antibodies
against NGFR and
EGFR with PE and APC as fluorochromes (Biolegend), the Anti-phycoerythrin (PE
MultiSort kit
(Miltenyi) and anti-APC MicroBeads (Miltenyi) on LS or MS columns (Miltenyi).
FACS sorting
was performed on an FACS ARIATM II SORP cell sorter (BD Biosciences).
[0252] To make identification easier, a second editing step was performed in
which an
expression cassette with firefly luciferase and TurboGFP was targeted into a
safe harbor locus
(HBB) (FIG. 6C). The K562 cells were suspended in 20 ul SF cell line solution
with 6 pg Cas9
protein (IDT) and 3.2 pg sgRNA (Trilink) and electroporated. After
resuspension in K562 cell
medium (RPMI with 10% BGS and supplemented with GLUTAMAXTm and
Penicillin/Streptomycin), the cells were transduced with rAAV carrying the
expression cassette.
This resulted in a cell population expressing all 3 markers (tNGFR, tEGFR and
GFP) that were
sorted by flow cytometry. Results of the flow cytometiy are shown in FIG. 6D.
The percent of
GFP+ cells in each group is shown in FIG. 6F.
[0253] The sorted IJMPSI")"/GFP+ cell population were subjected to assays
evaluating their
auxotrophy and their resistance to 5-F0A. The cells were split into samples of
equal numbers and
cultured in the presence of different concentrations of Uridine or without.
With supplementation of
high concentrations of Uridine (250 gimp the cells expanded rapidly. Cell
growth was inhibited at
a lower concentration (25 pg/ml) while cell numbers declined with a lower
concentration or no
Uridine (FIG. 6E). The number of cells per ml is shown in Table 24.
Table 24. Number of cells per ml from Day 1 to Day 8
250 ug/mlUridine 25 u /m1 Uridine 2.5 u ml Uridine No Uridine
Day 1 83.41 109.11 64.28 60.92 69.81
58.10 57.83 49.52 40.56 131.21 103.97 18.04
Day 2 130.60 80.43 39.92 150.58 73.78 N/A 99.62
40.41 31.70 97.77 28.14 29.30
Day 4 520.75 356.31 142.97 305.15 114.37 71.23 124.37 33.19 20.39 89.31
24.21 13.69
-60-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
1 Day 6 1 474.67 1 460.12 1 205.32 1 146.01 1 56.14 1 43.75 1 46.59 1 10.77 1
8.05 1 35.06 1 3.98 1 4.99 1
Day 8 631.12 629.35 318.17 242.61 46.45 39.19 28.68 2.06
1.85 15.82 0.54 0.48
[0254] The same experiment was performed with Na1m6 cells and a similar
dependency on the
uridine concentration in the culture was observed that was not visible for
cells with intact (IMPS.
Table 25 provides data of growth curves of U4PSK" Nalm6 cells cultured with
different uridine
concentrations. No difference was observed between the groups receiving
uridine supplement
treatment and those not for wild-type cells.
Table 25. Cell counts of UMPSK " Nalm6 cells
___________________________________________ Day
0 1 2 3 4 5 6 7
17980 33784 59524 167715 303894 1163678 3511660 7293447
UMPS-WT, 250 pg/m1uridine 24390 40000 58737 160160 213547 1115507 3058542
N/A
19505 39683 70175 194185 311891 1173622 2708995 5746352.
UN1PS-WT, 25 p.g/m1uridinc 36496 40000 68027 174292 376249 1216000 3792593
7585185
27548 31847 70922 214190 447240 1413856 3792593 6320988
20052 35088 67340 156019 244368 923391 2017336 3869992
UMPS-WT, 2.5 ug/ml uridine 19661 34130 38314 153257 225750 11 19256 3269476
6117085
21142 28986 62598 137812 338624 1148582 3269476 6538953
13496 15343 43860 86496 252167 1048734 3058542 6772487
UN1PS-WT, no Uridine 14582
23529 31974 93077 278867 1138455 3511660 7585185
14140 18994 45045 111810 316049 1223111 3511660 6538953
20704 37175 101781 210805 551249 2072099 3792593 7585185
250 g/m1uricline 24510
35461 106667 265340 551249 2343280 4514991 6320988
29762 30769 112045 257649 564374 1844675 4122383 7901235
14984 21277 69085 142944 388585 1624178 3646724 7293447
25 pg/mi undine 18952
25189 68027 176018 324708 1360329 3386243 5746352
14400 19688 71301 189125 395062 1360329 3792593 6538953
20121 42194 69808 70287 80625 193482 380782 755497
2.5 pg/ml uridim 29155
38023 88300 70547 81737 188952 383866 796763
28090 40650 87146 78663 79012 193913 377748 793429
10258 13661 13222 11234 14667 24413 27139 40429
No undinc 12284 16244 13126 11518
15131 21683 27762 46573
12427 15772 15585 11729 16441 25738 29410 43139
102551 Significantly greater growth was observed in the groups supplemented
with uridine,
especially the groups supplemented with 25 lg/m1 and 250 1.ig/m1 uridine.
[0256] To determine the resistance of UMPS knockout cells to 5-FOA the
purified
umpsicoxo/GFp+ K562 cells were mixed at an equal ratio with UMPSwilwT/GFP-
negative K562
cells. The cells were cultured in the presence of Uridine and different
concentrations of 5-FOA
(Fig. 6F). Table 26 provides the percentages of GFP-positive (+) cells under
different culture
conditions.
Table 26. Percentages of GFP+ cells
1000 uWm15-FOA 100 uvfml 5-FOA 10 ugind 5-FOA No 5-FOA
Day 1 51.9 1 49.4 52.6 48.9 45.4 48.5 38.1
37.6
Day 2 65.0 : 58.9 67.1 64.2 54.7 56.8 34.6
34.9
i
Day 3 72.9 1 66.9 79.5 71.4 65.6 64.0 33.6
31.7
-61-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
DM' 4 82.0 777 85.2 74,4 64.8 69.7 32.4 31.5
Day 6 92.6 90.5 92.1 86.2 79.1 76.5 31.2 29.2
Day 8 90. I 75.7 92.8 82.8 85.0 79.8 24.8 26.0
[0257] FIG. 66 shows the growth curve for GFP+ cells at different amounts of 5-
F0A. The
values are show in Table 27.
Table 27. Number of GFP+ cells per .1
1000 ug/m15-FOA 100 ug/ml 5-FOA 10 ug/ml 5-FOA No 5-FOA
Day 1 75925.72 115272.35 87013.04 80080.81
Day 2 1 79080.77 106377.73 163245.74 135616.84
Day 4 1 151010.55 376993.53 569281.05 304640.45
Day 6 217794.10 501940.62 550780.31 282520.65
Day 8 339282.35 693093.53 719624.13 203799.66
[0258] At all concentrations that were used, the fraction of UPS""" cells
increased over
time. Cells with UMP knockouts proliferated well at the concentrations 10 and
100 1.ig/m1 of 5-
FOA, while the highest concentration slowed their cell growth down.
Example 11. UMPS editine creates auxotronhv in T cells and allows for
selection with 5-FOA
102591 T cells were isolated from buffy coats that were acquired from the
Stanford Blood Center
(Palo Alto, CA) using Ficoll density gradients and MACS negative selection
(Miltenyi T cell
enrichment kit). The T cells were cultured in X-VIV015 medium supplemented
with 5% human
serum (Sigma) and 100IU/m1 IL-2.
102601 Before electroporation, T cells were activated for 3 days with Anti-
CD3/-CD28 beads
(STEMCELL Technologies), also referred to as Dynabeads in the art, and IL-2
(100IU/m1).
Activation beads were removed by magnetic immobilization before
electroporation. K562 cells and
Nalm6 cells were kept in logarithmic growth phase before electroporation.
sgRNAs were acquired
from Synthego with 2'-0-methyl-3=-phosphorothioate modifications at the three
terminal
nucleotides of both ends (See. Bonifant, et al. Mol. Ther. - Oncolytics. 3,
16011 (2016), which is
hereby incorporated by reference in its entirety).
102611 The two selection markers, tEGFR and tNGFR, were targeted into the UMPS
locus in
primary human T cells after isolation of CD3+ T cells from healthy donors and
activation of the
cells.
[0262] Large-scale sgRNAs were acquired high-performance liquid chromatography
(HPLC)-
purified. High-fidelity (HiFi) Cas9 protein was purchased from 1DT. The sgRNAs
were complexed
with HiFi spCas9 protein (IDT) at a molar ratio of 2.5:1 (sgRNA : protein) and
electroporated into
the cell lines or activated T cells using a 4D-NUCLEOFECTORTm System (Lonza)
in 16-cuvette
strips.
-62-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
[0263] For targeting of transgenes into specific loci of the genome, cells
were edited as
described, resuspended directly after electroporation in 80111 of medium, then
incubated with
rAAV6 for transduction at multiplicities of infection (MOI) of 5000 vg/cell.
After 8-12 hours, the
suspension was diluted with medium to reach a cell concentration of 0.5-1E6
cells per ml. For
targeting of the HBB locus, a previously characterized sgRNA with the target
sequence
CTTGCCCCACAGGGCAGTAA (SEQ ID NO:?) was used (See, Teixeira et al., Curr. Opin.
Biotechnol. 55, 87-94 (2019), which is hereby incorporated by reference in its
entirety, ).Cas9 and
sgRNA were complexed to an RNP and mixed with the T cells resuspended in P3
buffer and
electroporated in the 4D NUCLEOFECTORTm system (Lonza) using program EO-115.
Human T
cells are known in the art to allow high editing frequencies at low toxicity
as described in Bak et
al., 2018, to create a population of cells with a bi-allelic UMPS knockout
using RNP/rAAV6 gene
targeting methods. Cells expressing both markers were simultaneously
expressed. The following
cell counts per electroporation, electroporation solutions and programs were
used: 2E5 K562 cells
in SF cell line solution using program FF420, 2E5 Nalm6 cells in SF-cell line
solution and
program CV-104 and 1E6 activated T cells in P3 solution. For controls edited
at the CCR5 locus
the genomic target sequence of the sgRNA was GCAGCATAGTGAGCCCAGAA (SEQ ID NO:
8). After electroporation, the cells were resuspended in medium and rAAV
added.
[0264] Three days after targeting, a population of EGFR+/NGFR+ cells was
identified and
expanded by co-culturing with Anti-CD3/-CD28 magnetic beads in the presence of
high Uridine
concentrations. The population of EGFR+/NGFR+ cells was differentiated from
cells that received
AAV alone due to brighter expression indicating stable integration as opposed
to episomal
expression from AAV.
102651 After expansion, the EGFR+/NGFR+ population was sorted using flow
cytometry to get
a population of T cells with bi-allelic UMPS knockout. Results are shown in
FIG. 7A.
102661 These T cells were also subjected to an auxotrophy assay and the
possibility to select
these cells with 5-FOA was tested. When culturing the cells in the presence of
Anti-CD3/-CD28
beads and different concentrations of Uridine, cells proliferated only in the
presence of Uridine,
which confirmed their auxotrophic cell growth. Higher Uridine concentrations
led to higher
proliferation rates. Auxotrophic growth of UMPS KO or wild-type (WT) T cells
is shown in in
FIG. 7B and Table 28.
Table 28. Viable cells per ml
UMPS KO WT
250 344/miuridine 319120 345862
348022 609575 412493 468354
25 uglnd uridine _ 282368 268684 304864 384503 410116 338547
2.5 tighnluddine 226596 217594
224448 486192 362626 364194
No widine 46037 45351 52771 424742
414301 393938
-63-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
102671 Table 29 show the relative viability of the cell population on Day
4.
Table 29. Viable cells per ml
LIMPS KO ________________________________________ WT
250 ng/tril Uridine 85.66 85.29 84.38 86.05 86.97
87.62
25 ftgind Uridine 82.16 81.57 83.09 83.91 83.40
79.66
2.5 lighnl Uridine 78.78 79.28 79 74 84.64 82.19
80.45
No thidine 46.06 48.32 50.46 83.78 84.15
82.96
102681 To evaluate the possibility to select for UMPSK(3" cells with 5-F0A,
the sorted cells
were mixed with wild-type cells, which were labeled with different tracking
dyes, and cultured in
the presence or absence of 5-F0A. In part of the samples, 5-FOA was only added
on the first day
(Day 0) while in another group it was supplemented daily. Table 30 and FIG. 7C
show the percent
(%) of the UMPS-KO T cells (labelled with eFluor670) over time when culturing
with or without 5-
FOA.
Table 30. Percent of UMPS"/" cells in mixed cell population
5-FOA daily 5-FOA Day 0 only
I no 5-FOA
Day 0 43.2 43.2 47.2
Day 1 58.5 1 55.4 1 54.5 59.2 1 55.6 I 50.8 46.5 1 48.0 49.3 1 46.5 47.5 1
46.4
Day 3 74.0 70.7 0.2 67.2 68.6 67.8 43.4 42.9 43.1 43.8 44.2 41.7
102691 Groups were compared for statistically significant differences using
an unpaired t test.
No statistical significance was observed between the groups treated with 5-
F0A, while there was a
significant increase in the percent of UMPS-KO T cells in the treated groups
compared to the
untreated group.
102701 In both 5-FOA treated groups, the fraction of cells with (IMPS knockout
increased over
time, indicating their increased resistance to the compound compared to wild-
type cells, and that a
one-time treatment with 5-FOA was sufficient to lead to an enrichment of
modified cells over
several days. The data in Table 30 illustrates 5-FOA selects for T cells with
UMPS knockout.
102711 In fact, FACS analysis of a culture of a mixed population of UMPS
knockout and wild-
type T cells with 5-F0A. (IMPS-KO T cells were labeled with eFluor670, and
wild-type cells were
labeled with carboxyfluorescein succinimidyl ester (CFSE). Results showed that
on Day 0 in the
group treated with 5-FOA only on the first day (Data 43.7% of the cells were
UMPS-KO T cells,
while 56.0% were observed to the wild-type cells. On Day 0 in the control
group not treated with 5-
FOA, 47.7% of the cells were UMPS-KO T cells, and 52.1% were wild-type cells.
On Day 3 in the
group with 5-FOA supplemented daily, 74.0% of the cells were UMPS-KO T cells,
while 25.8%
were wild-type cells. On Day 3 in the control group not treated with 5-F0A,
43.1% of the cells
were UMPS-K0 T cells, and 56.4% were wild-type cells.
-64-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
Example 12. Cellular theranv
102721 Pluripotent stem cells are genetically engineered to make them
dependent on externally
supplied factors. These cells are injected into immunodeficient NSG mice as
teratoma-fonning
assays to evaluate the safety system, which prevents teratoma formation
through withdrawal of the
externally supplied compound. Cell lines used are iPSCs: iLiF3, iSB7-M3
(source: Nakauchi Lab at
Stanford University), and hES: H9.
Example 13. Teratoma-formin2 assay in 2astrocnemius muscle
[0273] To determine whether the safety switch can eradicate teratomas that
originate from
pluripotent cells, iPSCs or ES cells that were genetically modified (or
control cells) were
transplanted into mice. The cells expressed luciferase for in vivo detection.
1x106 UMPS-
engineered hESCs were resuspended in a 100 I of MATRIGEL protein mixture
(Corning, Inc.)
and PBS mixture and injected into the gastrocnemius muscle of the right hind
leg of anesthetized
=NSG mice. The mice were followed up for tumor formation by tumor size
measurement and by
bioluminescence imaging. After establishment of tumors, whether withdrawal of
Uridine triacetate
(UTA) led to tumor regression was tested. At the endpoint (tumor sizes above
1.7cm or impairment
of mouse activity, otherwise 24 weeks) tumor was explanted and fixated for
histological analysis.
Example 14. K562 xenograft model
[0274] For the K562 xenograft assay, 6 to 12 weeks old male NOD SCID gamma
mouse (NSG)
mice were transplanted with lx106 K562 cells resuspended in MATRIGELO protein
mixture
(Corning, Inc.) 1:1 diluted with PBS under anesthesia. All animals were kept
and handled
according to institutional guidelines and the experimental protocol was
approved by Stanford
University's Administrative Panel on Laboratory Animal Care.
102751 The growth of UMPS'Kaw engineered cells was analyzed in vivo after
transplantation
into a model organism by supplying the animal with high doses of uridine.
Uridine has been used in
humans for the treatment of hereditary orotic aciduria and for toxicity from
fluoropyrimidine
overdoses (see, van Groeningen, et al. Ann. Oncol. 4,317-320 (1993); Becroft,
et al. J. Pediatr. 75,
885-91 (1969); each of which are hereby incorporated by reference in its
entirety), but it is poorly
absorbed in the gastrointestinal tract and broken down in the liver (See,
Gasser, et al. Science. 213,
777-8 (1981); each of which are hereby incorporated by reference in its
entirety). Its bioavailability
can be increased by administration as the prodrug uridine triacetate (UTA,
PN401), which has FDA
approval for the above-mentioned indications (See, Weinberg et al., PLoS One.
6, el4709 (2011);
Ison et al., Clin. Cancer Res. 22,4545-9 (2016); each of which is hereby
incorporated by reference
in its entirety). In humans and mouse models, this can effectively increase
uridine serum levels by
greater than 10-fold (See, Garcia et al., Brain Res. 1066,164-171 (2005); FDA,
"XURIDEN -
-65-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
Highlights of prescribing information." (2015), (available at
https://www.accessdata.fda.gov/drugsatfda_docs/labe1/2015/208169s0001b1.pdf);
each of which is
hereby incorporated by reference in its entirety.
102761 The previously engineered IIMP5K011(c) K562 cell line expressing
firefly luciferase
(FLuc) was used in a xenograft model in NSG mice. Control K562 cells with wild-
type LIMPS
were engineered by targeting an expression cassette with FLuc and GFP into a
safe-harbor locus, in
order to establish comparable xenograft models for both UMPS genotypes in
which the tumors can
be monitored by bioluminescence imaging. Cas9 RNP is targeted to exon 1 of the
HBB locus with
a guide RNA and a DNA donor template transduced by rAAV6 which carries a FLuc-
2A-GFP-
polyA cassette under control of the SFFV promoter. FACS analysis was performed
four days after
targeting of K562 cells to evaluate GFP expression before sorting of the GFP+
population. In a
control group administered the AAV only, 1.61% of the cells were GFP+, and
13.4% of the cells.
102771 Mice were fed with either regular mouse food or with a custom food
which had been
enriched with 8% (w/w) UTA, an amount that had previously been shown to
increase serum levels
in mice while being well tolerated (See, Garcia et al., 2005). UTA was
acquired from Accela
ChemBio Inc. and added to make the 8% (w/w) to Teklad mouse food (Envigo) and
the food
irradiated before use. Control food was the standard mouse food Teklad 2018
(irradiated).
[0278] Alternatively, the food was supplemented with uridine monophosphate.
These cells may
be implanted into the inununocompromised mice in a local (hind leg) or
systemically through an
intravenous (iv) injection.
[0279] UMPSK". K562 cells or control cells were transplanted
subcutaneously and observed
weekly with bioluminescence imaging. Luminescence imaging of K562 cells was
performed 5
minutes after intraperitoneal (ip) injection of 125 mg/kg D-Luciferin
(PerkinElmer) on an IVIS
Spectrum imaging system (PerkinElmer). The localized growth that has been
described for K562
cells after subcutaneous xeno-transplantation was observed (See, Sontakke, et
al. Stem Cells Int.
2016, 1625015 (2016), which is hereby incorporated by reference in its
entirety). Mice were
euthanized when they got moribund or if longest tumor diameter exceeded 1.75
cm. Except for one
mouse with engraftment failure, an increase in tumor burden in UMPS wild-type
cells with both
normal or UTA supplemented food was observed. In contrast, luminescence of
UMPSKc" K562-
derived tumors were observed to only increased in the mice fed with 8 /h UTA,
while tumor
burdens were observed to remain stable in the majority of mice that received
food without UTA.
102801 Auxotrophic cell proliferation of the (84PSKa4( -engineered hES cells
in vivo was also
analyzed. Except for one mouse with failure to form a teratoma, masses were
observed in all the
mice fed with supplemented UTA, after injection of the pluripotent cells into
the hind legs of NSG
-66-
CA 03098874 2020-10-29
WO 2019/217803 PCT/US2019/031699
mice. When euthanizing the mice 7 weeks after cell injection, large teratomas
that had formed in
the region of injection in mice fed with UTA were extracted, while in mice on
normal food the
teratomas were visible but significantly smaller and weighed less as shown in
Table 31. Bone
marrow is analyzed at the time that the animal dies or is sacrificed (latest
16 weeks after injection).
Table 31 shows quantification results of teratoma weights (p<0.05 by unpaired
t-test comparing all
mice between groups, p<0.01 when censoring the mouse without engraftment).
Groups were
compared by statistical tests as indicated using Prism 7 (GraphPad).
Table 31. Teratoma weight
Mouse No. 1 2 3 I 4 I 5
Weight [g]
No UTA 603 3H 468 91 174
With LITA 3108 33 2923 1545 937
[0281] The in vivo results were consistent with the previous in vitro
results, which had shown
reduced but not completely abrogated proliferation of UMPS"" cells at the
uridine concentration
of 2.5 pg/m1 (= 10 nmol/ml). This concentration corresponds to serum uridine
levels of mice, which
are reported in the literature to range from 8 to 11.8 nmol/ml (See, Karle, et
al. Anal. Biochem.
109, 41-46 (1980), which is hereby incorporated by reference in its entirety).
[0282] Overall, these results are evidence a metabolic auxotrophy can be
engineered to add a
control mechanism over cell proliferation of human cells both in vitro and in
vivo.
Example 15. GvHD model
[0283] Whether the safety system can prevent the side effects of xeno-GvHD is
determined in a
mouse model. Genetically modified human T cells or control T cells are
transplanted into irradiated
immunocompromised mice and mice are supplied with UTA or not. Mice are
evaluated for weight
loss or other signs of GvHD and sacrificed upon establishment of disease
(latest 16 weeks). Cells
are followed by bioluminescence imaging and blood draws.
Example 16. Enzyme replacement therapy in Ivsosomal storaee disease (LSD)
102841 Pluripotent stem cells are genetically engineered to encode for an
enzyme of interest
integrated at MPS locus to make them dependent on externally supplied uridine.
Individuals in
need of enzyme replacement therapy for the specific enzyme to treat a LSD are
administered
compositions comprising these cells along with uridine, to promote expression
of the enzyme that
is deficient in the individual. The dosing and timing of the administration of
uridine is adjusted
based on the desired expression of the enzyme.
[0285] In some instances, cells are genetically engineered to encode for an
enzyme of interest at
HLCs locus to make them dependent on externally supplied biotin.
-67-
CA 03098874 2020-10-29
WO 2019/217803
PCT/US2019/031699
[0286] While embodiments of the present disclosure have been shown and
described herein, it
will be obvious to those skilled in the art that such embodiments are provided
by way of example
only. Numerous variations, changes, and substitutions will now occur to those
skilled in the art
without departing from the present disclosure. It should be understood that
various alternatives to
the embodiments of the disclosure described herein may be employed in
practicing the subject
matter of the disclosure. It is intended that the following claims define the
scope of the disclosure
herein and that methods and structures within the scope of these claims and
their equivalents be
covered thereby.
-68-