Note: Descriptions are shown in the official language in which they were submitted.
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
METHODS AND COMPOSITIONS FOR THE ANALYSIS OF CANCER
BIOMARKERS
BACKGROUND
[0001] Molecular tests can detect residual disease after a treatment. The
presence of residual
disease indicates that the treatment did not completely eliminate a tumor,
where treatment
may include surgery, radiotherapy, chemotherapy, endocrine therapy, or
targeted molecular
therapy.
[0002] Following surgical treatments, positive surgical margins are defined as
tumor cells on
the surface of an excised tissue specimen. Since the surface of the excised
specimen is
topologically equivalent to the wall of the incision, tumor cells on the
surface of the incision
indicate the presence of residual tumor in a patient after surgical treatment.
[0003] Following medical treatments, Pathologic Complete Response (pCR) is
defined as the
absence of residual tumor in tissue from patients who were previously
diagnosed with
invasive cancer. pCR is used as a primary endpoint to determine the success of
emerging
breast cancer treatments in the neoadjuvant setting. Innovative clinical trial
designs have
validated pathologic complete response (pCR) as a surrogate endpoint, and are
now
validating pCR as a therapeutic endpoint.
SUMMARY
[0004] Described herein are methods and compositions that are useful for an
improved RNA-
based test suitable for analysis of tumor margins from surgical samples for
residual disease,
or for analysis of residual disease in post-treatment cancer patients from
other samples.
[0005] In some aspects, the disclosure provides a method of distinguishing a
cancer from
adjacent healthy tissue, said method comprising: (a) obtaining a specimen from
a human
subject, (b) detecting a presence of a set of markers in said specimen by
performing an
amplification reaction in a plurality of polynucleotides from said specimen,
wherein said set
of markers is selected from the group consisting essentially of: Matrix
Metallopeptidase 11
(WP 11), integrin binding sialoprotein (IBSP), and collagen type X alpha 1
chain
(COL10A1); and (c) distinguishing said cancer when a threshold level of said
set of markers
is detected. In some instances, said plurality of polynucleotides comprise
RNA, cDNA, or
DNA. In some instances, the detecting comprises using a DNA-intercalating dye
or a
fluorescent probe, such as a TaqMan probe. In some instances said
amplification reaction is a
-1-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
PCR reaction, such as a qPCR reaction or an RTqPCR reaction. In some
instances, said
method can distinguish said cancer in at least lOng of said plurality of
polynucleotides from
specimen. In some instances, said method can distinguish said cancer in at
least 250 cells of
said specimen. In some instances, said amplification reaction uses at least
one primer
sequence that has at least 90% identity to SEQ ID NO: 1- SEQ ID NO: 356, for
example to
convert RNA into cDNA and/or to amplify a cDNA. In some instances, the
specimen is a
frozen specimen, a fresh specimen, or a fixed specimen. In some instances, the
specimen is a
biopsy specimen, such as a liquid biopsy, a solid tissue biopsy, or a surgical
excision. In some
instances, said specimen is obtained by imprint cytology, with for example a
touch-
preparation. In some instances said specimen is obtained by scrape
preparation, a nipple
aspiration, or a ductal lavage. In some instances said cancer is breast
cancer, including, but
not-limited to, invasive adenocarcinoma, invasive ductal breast cancer, and
invasive lobular
breast cancer. In some instances, said method distinguishes said breast cancer
from adjacent
healthy tissue with greater than 90% accuracy, greater than 90% sensitivity,
or greater than
90% specificity. In some instances, said method quantitates an amount of said
cancer. In
some instances said method further comprises outputting a percentage of said
plurality of
polynucleotides expressing said markers from said specimen. In some instances,
the method
further comprises comparing said set of markers from said specimen to said set
of markers
from said control specimen, such as a second specimen from said human subject
or a
synthetic nucleotide control. In some instances, the method further comprises
performing a
second assay to distinguish said cancer, such as an immunohistochemistry
assay. In some
instances, said threshold level of said WP 11 is 1,000 copies per microliter,
said threshold
level of said IBSP is 25 copies per microliter, and said threshold level of
said COL10A1 is
700 copies per microliter. In some instances, said set of markers is selected
from the group
consisting of: Matrix Metallopeptidase 11 (WPM, integrin binding sialoprotein
(IBSP),
and collagen type X alpha 1 chain (COL10A1). In some aspects, said
amplification reaction
can be a singleplex reaction or a multiplex reaction.
[0006] In some aspects, the disclosure provides a kit comprising, at least one
primer
sequence that has at least 90% identity to any one of SEQ ID NO: 1- SEQ ID NO:
356, and a
buffer system. In some instances said buffer system is a PCR buffer system. In
some
instances, the kits further comprise a DNA-intercalating dye, a fluorescent
probe, such as a
TaqMan compatible probe. In some instances the kit also comprises a negative
control
sample, a positive control sample, or a synthetic nucleotide control.
-2-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
[0007] In some aspects, the disclosure provides isolated nucleic acid
comprising a primer
sequence that has at least 90%, at least 95%, or at least 99% identity to SEQ
ID NO: 1- SEQ
ID NO: 356.
[0008] In some aspects the disclosure provides a method of identifying a
biomarker for a
cancer comprising: (a) analyzing, by a computer system, a cohort of biomarkers
from a
population of subjects afflicted with a cancer; (b) applying, by said computer
system, a first
filter to said cohort of said biomarkers to identify a first subset of
biomarkers from said
cohort that has at least a 3-fold higher expression level in said cancer as
compared to a
healthy control biomarker; (c) applying, by said computer system, a second
filter to said first
subset of biomarkers to identify a second subset of biomarkers that have a
false discovery
rate for said cancer that is less than 0.000001; and (d) applying, by said
computer system, a
correlation based filter selection to said second subset of biomarkers to
identify the
biomarkers that classify the largest number of different types of said cancer.
In some aspects,
said correlation based filter is an anti-correlation based method. In some
aspects, the method
further comprises using the identified biomarkers as features input into a
machine learning
algorithm that distinguishes clinical specimens based on predefined
attributes. In some
aspects said cancer is breast cancer, including, but not-limited to invasive
adenocarcinoma,
invasive ductal breast cancer, and invasive lobular breast cancer. In some
aspects, said one or
more biomarkers identify said cancer with greater than 90% accuracy, greater
than 90%
sensitivity, or greater than 90% specificity. In some aspects, said one or
more biomarkers are
therapeutic targets. In some aspects, said false discovery rate is ap-value
for said cancer that
is less than 0.0000001.
[0009]
Additional aspects and advantages of the present disclosure will become
readily
apparent to those skilled in this art from the following detailed description,
wherein only
illustrative embodiments of the present disclosure are shown and described. As
will be
realized, the present disclosure is capable of other and different
embodiments, and its several
details are capable of modifications in various obvious respects, all without
departing from
the disclosure. Accordingly, the drawings and description are to be regarded
as illustrative in
nature, and not as restrictive.
INCORPORATION BY REFERENCE
[0010] All publications, patents, and patent applications mentioned in this
specification are
herein incorporated by reference to the same extent as if each individual
publication, patent,
-3-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
or patent application was specifically and individually indicated to be
incorporated by
reference.
BRIEF DESCRIPTION OF THE DRAWINGS
[0011] The novel features of the invention are set forth with particularity
in the appended
claims. A better understanding of the features and advantages of the present
invention will be
obtained by reference to the following detailed description that sets forth
illustrative
embodiments, in which the principles of the invention are utilized, and the
accompanying
drawings (also "Figure" and "FIG." herein), of which:
[0012] FIGURE 1 is a diagram illustrating positive versus clear surgical
margins.
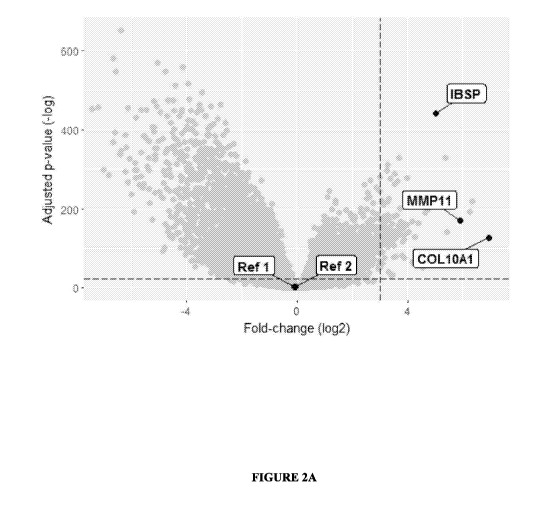
[0013] FIGURE 2A is a Volcano plot of 20,253 mRNAs in 1,014 samples. RNA Seq
was
used to analyze 1,014 samples from early-stage tumors and healthy samples from
adjacent
tissue. Selected genes had the highest Correlation-based Feature Selection
scores among
genes that passed p-value threshold (dashed horizontal line) and fold-change
threshold
(dashed vertical line).
[0014] FIGURE 2B panels (a-c) are cumulative frequency plots of 1,536 patient
samples
that show that a 3-gene set (WP11,COL10A1,IBSP) is overexpressed in samples
from
early-stage tumors and adjacent healthy tissue. The genes have comparable
distributions on
RNA Seq samples (a), a subset of samples that were also analyzed by microarray
(b), and a
subset that were also analyzed by RTqPCR (c). These results confirm that
expression is not
platform-specific. Panels (c-e) are 2D-Density maps illustrating the advantage
of a
multianalyte test over a single biomarker. Separation of tumor and healthy
improves as we
progress from RNA Seq to Microarray to custom RTqPCR.
[0015] FIGURE 3 is a chart showing a Principal Component Analysis (PCA) of all
available
microarray probes shows a clear demarcation between tumor (left dots) and
healthy samples
(right dots).
[0016] FIGURE 4 depicts receiver-operator characteristic (ROC) curves of
classifiers for a
3-gene set including WP11,COL10A1,IBSP. ROC curves show the tradeoff between
sensitivity and specificity over all possible thresholds. The solid dark line
shows performance
of the 3-gene test on 939 cross-validated RNA Seq samples.
[0017] FIGURE 5 illustrates an error plot of a 3-gene set (WP11,COL10A1,IBSP)
in 939
RNA Seq samples. In contrast to ROC plots, which show the tradeoff between
sensitivity and
specificity, error plots set the threshold based on the tradeoff between Type
I and Type II
errors. Type I errors (False Positives) trigger unnecessary re-excisions. Type
II errors (False
-4-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
Negatives) indicate positive margins that were not detected. We use these
tradeoffs to guide
our threshold selection.
[0018] FIGURE 6 is a graph illustrating that negative controls to detect over-
fitting
demonstrate that predictive models were correctly cross-validated (n = 939 RNA
Seq
samples).
[0019] FIGURE 7A and FIGURE 7B depict charts showing analytic validation of
qPCR
assays for using clinical-grade reagents. FIGURE 7A panel a depicts
amplification plots of
20 microliter qPCR reactions. 12 concentrations of synthetic cDNA template 1.1
million
copies per microliter to 0 copies per microliter), including 10-fold dilutions
for 6 high
concentrations (5 technical replicates) and 2-fold dilutions for 5 low
concentrations (7
technical replicates). One concentration point overlapped in the high and low
concentration
series. Each primer pair includes 24 replicates of no-template controls. Error
bars at each
cycle represent 95% CI of technical replicates.
[0020] FIGURE 7A panel b depict fluorescence versus cycle plots to determine
Ct for
WP11. A 4-parameter linear model was fitted to 5 technical replicates
(circles). The
maximum of the second derivative was used to define the Ct (CtD2).
[0021] FIGURE 7B panel c depicts threshold cycle versus template dilution
plots to
calculate linear range. The linear range is defined as the range of
concentrations where CtD2
fit a straight line with R-squared >0.995. Red lines indicate 95% Confidence
Intervals
calculated from 200 bootstraps. FIGURE 7B panel d depicts melt plots confirm
to specificity
of the primers. Increasing temperature denatures PCR amplicons, which
decreases
fluorescence. A single peak of the negative first derivative confirms the
presence of a single
amplicon. The peak corresponds to the expected melting temperature (dashed
line).
[0022] FIGURE 7A and FIGURE 7B panels e-h depict charts showing analytic
validation
of qPCR assays for IBSP RNA as for WP11 . All assays used clinical-grade
reagents. Panel
e depicts amplification plots of 20 microliter qPCR reactions. 12
concentrations of synthetic
cDNA template (1.1M to 0 copies per microliter), including 10-fold dilutions
for 6 high
concentrations (5 technical replicates) and 2-fold dilutions for 5 low
concentrations (7
technical replicates). One concentration point overlapped in the high and low
concentration
series. Each primer pair includes 24 replicates of no-template controls. Error
bars at each
cycle represent 95% Confidence Intervals of technical replicates.
[0023] FIGURE 7A Panel f depicts fluorescence versus cycle plots to determine
Ct for IBSP
A 4-parameter linear model was fitted to 5 technical replicates (circles). The
maximum of the
second derivative was used to define the Ct (CtD2). FIGURE 7B panel g depicts
threshold
-5-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
cycle versus template dilution plots to calculate linear range. The linear
range is defined as
the range of concentrations where CtD2 fit a straight line with R-squared
>0.995. Red lines
indicate 95% Confidence Intervals calculated from 200 bootstraps.
[0024] FIGURE 7B panel h depict melt plots that demonstrate the specificity of
the primers.
Increasing temperature denatures PCR amplicons, which decreases fluorescence.
A single
peak of the negative first derivative confirms the presence of a single
amplicon. The peak
corresponds to the expected melting temperature (dashed line).
[0025] FIGURE 7A and FIGURE 7B panels 1-1 depict analytic validation of qPCR
assays
for COL10A/ RNA as for WP11. All assays use clinical-grade reagents. FIGURE 7A
panel i depict amplification plots of 20 microliter qPCR reactions. 12
concentrations of
synthetic cDNA template (1.1M to 0 copies per microliter), including 10-fold
dilutions for 6
high concentrations (5 technical replicates) and 2-fold dilutions for 5 low
concentrations (7
technical replicates). One concentration point overlapped in the high and low
concentration
series. Each primer pair included 24 replicates of no-template controls. Error
bars at each
cycle represent 95% Confidence Intervals of technical replicates. FIGURE 7A
Panel j
depicts fluorescence versus cycle plots to determine Ct for COL10A1. A 4-
parameter linear
model was used to fit all 5 technical replicates (circles). The maximum of the
second
derivative (green curve) was used to define the Ct (CtD2). FIGURE 7A panel k
depicts
threshold cycle versus template dilution plots to calculate linear range. The
linear range is
defined as the range of concentrations where CtD2 fit a straight line with R-
squared >0.995.
Red lines indicate 95% Confidence Intervals calculated from 200 bootstraps.
Panel 1 depicts
melt plots confirm to specificity of the primers. Increasing temperature
denatures PCR
amplicons, which decreases fluorescence (black line). A single peak of the
negative first
derivative (red line) confirms the presence of a single amplicon. The peak
corresponds to the
expected melting temperature (dashed line).
[0026] FIGURE 8A, FIGURE 8B, and FIGURE 8C are graphs depicting absolute
quantification (RT-qPCR) of the 3 RNAs in the 3-gene set (WP11, COL10A1, IBSP)
in 22
patient samples using Tukey Boxplots. Tukey Boxplots: the thick center line
represents the
mean, boxes show the interquartile range (Q1-Q3). Cumulative Frequency plots
show the
distribution of expression in tumor and healthy samples. Panel b depicts
absolute
quantification (RTqPCR) of RNAs in 22 patient samples using density plots.
Density plots
illustrate the advantage of combining multiple biomarkers. Copy numbers are
adjusted for
tumor percent because each tumor specimen contains a differing amount of
healthy cells.
-6-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[0027] FIGURE 9 is a graph depicting a Receiver-Operator Characteristic (ROC)
Curve of
the 3-Gene Classifier. ROC curves show the tradeoff between sensitivity and
specificity over
all possible thresholds. The 3-gene classifier uses Random Forest to
distinguish between
tumor and adjacent healthy tissue. Performance estimates are based on 5-fold
cross validation
of 22 samples that were analyzed with the disclosed RTqPCR assays.
[0028] FIGURE 10 depicts a plot showing Generalized Linear Model (glm) (dashed
line)
sample discrimination using IBSP RNA in 22 patient samples, analyzed by the
disclosed
RTqPCR assay. The disclosed RTqPCR assays can resolve a greater difference in
analytes
than RNA Seq. The disclosed assays perform so well that a simple linear model
can correctly
classify 100% of the analyzed samples using a single biomarker. In contrast,
RNA Seq
required a complex combination of 3 biomarkers, and still did not achieve 100%
accuracy.
[0029] FIGURE 11 depicts a plot showing Generalized Linear Model (glm) (dashed
line)
sample discrimination using WP 11 RNA in 22 patient samples, analyzed by the
disclosed
RTqPCR assay.
[0030] FIGURE 12 shows a chart depicting a Tumor Probability Score calculated
using the
3-gene classifier described in EXAMPLE 1. The 3-gene classifier uses the
Random Forest
algorithm to calculate a Tumor Probability Score (T) from zero to one. Panel a
shows the T
score for RNA Seq samples from 901 tumors (black) and 113 adjacent healthy
samples
(grey). Panel b shows the T score for RTqPCR samples from 11 tumors (black)
and 11
adjacent healthy samples (grey).
[0031] FIGURE 13 shows a computer system that is programmed or otherwise
configured to
implement methods provided herein.
DETAILED DESCRIPTION
I. Overview of Pathologic Complete Response
[0032] While various embodiments of the invention have been shown and
described herein, it
will be obvious to those skilled in the art that such embodiments are provided
by way of
example only. Numerous variations, changes, and substitutions may occur to
those skilled in
the art without departing from the invention. It should be understood that
various alternatives
to the embodiments of the invention described herein may be employed.
[0033] pCR has quickly become the primary endpoint for ¨50% of enrolling phase
II rectal
cancer trials, and 45% of phase III preoperative breast cancer trials.
Unpublished results from
the I-SPY 2 TRIAL of high-risk breast cancer patients indicate that pCR was
statistically
associated with 3-year outcomes on pooled patients across all treatment arms.
After 3 years,
-7-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
patients who achieved pCR had a 6% recurrence risk (event-free survival),
compared to 24%
recurrence risk for those who did not achieve pCR.
[0034] Improving surrogate endpoints will help to replace treatment regimens
with ones that
are more effective, less toxic, and that improve survival. However, existing
technologies are
subjective, qualitative, and underpowered because they are based on visual
analysis of a
limited number of tissue sections. Moreover, pCR is labor intensive and
currently only
provided by specialty clinical centers as part of research protocols.
Pathology labs routinely
examine 3-5 microscopic tissue sections. If therapeutic response is ultimately
verified as a
therapeutic goal, busy pathology practices will be overwhelmed by requests to
examine
thousands of sections from hundreds of thousands of U.S. patients with
invasive breast
cancer. Described herein is a quantitative molecular analysis of residual
tumor for
identifying improved treatment regimens and complete excision of malignant
tissue from
patients.
II. Overview of Positive Surgical Margins
[0035] Most U.S. breast cancer patients are treated with breast conservation
surgery
(lumpectomy), where the goal is to remove the entire tumor, bounded by a thin
margin of
healthy tissue (FIG.1a). Positive margins are defined as malignant cells that
touch the cut
surface of a specimen (FIG.1b), indicating residual tumor in the bed of the
incision. Positive
margins increase the risk of recurrence and disease-specific mortality. As an
example, in a
cohort of 1,043 consecutive patients, positive margins were the strongest risk
factor of
disease-specific mortality among patients with early-stage breast tumors: the
10-year risk of
death from breast cancer was 3.9x higher for patients with positive margins,
relative to
patients with negative margins (95% CI: 1.4-11.5, p = 0.011). See, e.g., Meric
F, Mirza NQ,
Vlastos G, Buchholz TA, Kuerer HM, Babiera GV, Singletary SE, Ross MI, Ames
FC, Feig
BW, Krishnamurthy S, Perkins GH, McNeese MD, Strom EA, Valero V, Hunt KK.
Positive
surgical margins and ipsilateral breast tumor recurrence predict disease-
specific survival after
breast-conserving therapy, Cancer, 2003 Feb 15;97(4):926-33.
[0036] Patients with positive margins have a higher risk of recurrence (HR:
2.52, 95%CI:
1.04-6.09) than patients with 10 positive lymph nodes (HR: 2.32, 95% CI: 1.29-
4.14). These
findings hold, even under modern treatment protocols that include localized
radiation,
endocrine therapy, targeted molecular therapy, and the option of systemic
chemotherapy.
Detecting and treating positive margins is important because the risk of
recurrence typically
cannot be mitigated by additional chemotherapy or a radiation boost. Obtaining
clear margins
-8-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
is a canon of surgical oncology, and is codified in clinical guidelines (ASCO
and NCCN) and
consensus statements (SSO and ASRO).
[0037] There is a need to improve the evaluation of surgical margins.
Histopathology has
been the best way to examine tumors for over a century, but it is not an ideal
way to hunt for
residual disease on the surface of a specimen. A retrospective analysis of
1,201 lumpectomy
margins from Harvard's Brigham and Women's Hospital found that when microscopy
was
used to detect positive margins, it had a 51% sensitivity, 69.5% specificity,
19% false
negative rate, and 65% false positive rate. See, e.g., Tang R, Coopey SB,
Specht MC, Lei L,
Gadd MA, Hughes KS, Brachtel EF, Smith BL. Lumpectomy specimen margins are not
reliable in predicting residual disease in breast conserving surgery. Am J
Surg. 2015
Jul;210(1):93-8. These results were consistent with a prospective, randomized-
control trial at
Yale, where microscopy of the primary specimen had a false negative rate of
20%.
Undersampling is likely to be a primary culprit; microscopic sections only
sample a small
portion of a specimen's surface. Some pathologists therefore conclude that
margin analysis is
the weak link in breast cancer care.
[0038] Many have tried to reduce reexcisions by testing margins during an
operation, but
these technologies have failed to reach a clinical impact. This is primarily
due to the
preliminary nature of rapid intraoperative test results -- surgeons use them
to predict post-
operative test results. Since the relevant reference-standard has a 51%
sensitivity and 70%
specificity, test discordance has created an insurmountable barrier for
adoption¨even a
perfect intraoperative test cannot predict which margins pathology will call
positive.
Accordingly, we describe herein a method using nucleic acid tests to improve
post-operative
testing.
[0039] Improved testing has the potential to reduce Type I & II Errors. Type I
errors are
known as false positives. False positives have proven a significant barrier in
the adoption of
analysis of tumor margins by microscopy/histology; in a previous study of
lumpectomy
margin analysis by Tang et at. only 149 (32%) of 462 positive microscopy
results actually
had residual tumor along the margin. See, e.g., Tang R, Coopey SB, Specht MC,
Lei L, Gadd
MA, Hughes KS, Brachtel EF, Smith BL. Lumpectomy specimen margins are not
reliable in
predicting residual disease in breast conserving surgery. Am J Surg. 2015
Jul;210(1):93-8.
[0040] The 313 false positives triggered an alarming number of unnecessary
surgeries. Type
II errors are known as false negatives; in the same Tang et al. study, false
negatives also
presented a problem as traditional microscopy only detected 149 (51%) of the
293 margins
that contained residual disease. The 144 patients with false negative results
had a high risk of
-9-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
recurrence and mortality, which could have been mitigated by surgical
excision. Improving
post-operative testing could reduce reexcisions and improve long-term
outcomes.
[0041] Clinical utility involves a balance between Type I and II errors. The
clinical
consequence of Type II errors (False Negatives) is that undetected positive
margins place
patients at high risk of recurrence. Some estimate that microscopy has a Type
II error rate of
19% (patients who have positive margins but test negative). Assuming RNA Seq
performance is a reasonable indicator of clinical performance, a Type II error
rate <5%
represents a 75-100% improvement over existing methods. However, exclusive
focus on
Type II errors would be insufficient; high Type I errors (False Positives)
would result in
overtreatment. Surgeons may even avoid using a test with high Type I errors
(False Positives)
because it would trigger unnecessary reexcisions. Some estimates have placed
Type I errors
(False Positives) as 65% using existing microscopy methods. Reducing Type I
errors from
65% to 5% would reduce unnecessary surgical reexcisions >90%.
III. Overview of Ductal Lavage
[0042] There is an urgent need to improve breast cancer screening and
evaluation. Current
screening tests have rates of false negative results, which fail to detect
potentially lethal
tumors. Current screening tests also have high rates of false positive
results, which lead to
invasive biopsies in patients who do not have breast cancer. Error rates of
existing tests are
not uniform. For example, it is not clear from current evidence whether the
tradeoff is
beneficial for screening mammography in women less than 50 years old. In the
U.S., only
0.5% of women who are screened have cancer, but approximately 10% of women who
undergo breast cancer screening require additional tests. On a population
level, the false
positive rate of breast cancer screening is therefore approximately 9.5%.
[0043] Mammography is the most widely used screening modality for the
detection of breast
cancer. There is conflicting evidence about whether screening mammography
decreases
breast cancer mortality. The evidence is strongest for women aged 50 to 69
years. However,
screening in all age groups is also associated with harms. Harms can include
unnecessary
invasive procedures for patients who do not have breast cancer, and
overdiagnosis, which is
the detection of tumors that are not clinically significant. The error rates
for mammography in
women less than 50 are so high relative to the incidence of invasive breast
cancers that the
benefit of mammography is uncertain for women between 40 to 49 years old. In
2014, the
Canadian National Breast Screening Study completed 25 years of follow-up and
found no
survival benefit associated with screening mammograms for women of all ages.
While it is
-10-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
debatable how these findings should be applied to individual patients, it is
clear that
screening technologies are insufficient.
[0044] Alternative imaging technologies sometimes provide benefit for high-
risk
populations, or as adjuncts to mammography, but are not recommended as primary
screening
tools for the general population. This group of technologies includes
molecular breast
imaging, ultrasound, and magnetic resonance imaging.
[0045] In the past, patients were advised to perform breast self-exams, but
subsequent studies
found that breast self-exams have no mortality benefit. Breast exams performed
by clinicians
(Clinical Breast Exams, CBE) have not been evaluated as an independent
screening test. This
leaves patients with poor options for early cancer detection, and limited
options to determine
whether a suspicious screening result warrants an invasive diagnostic
procedure.
IV. Overview of a Molecular Test for Complete Response
[0046] Described herein is a method for analysis of residual tumor cells. The
method and kits
disclosed herein can identify improved treatment regimens. Accordingly,
disclosed herein
are post-operative devices and methods for obtaining and analyzing gene
expression from
cells from patient samples (e.g. from an excisional surgical biopsy) for
residual disease. A
panel of one to three cDNAs can serve as biomarkers to distinguish invasive
breast cancer
from adjacent healthy tissue with an accuracy of 96-100%. When cross-validated
on 939
RNA Seq samples, the disclosed 3-gene test had a 96% Accuracy, 96%
Sensitivity, and 94%
Specificity. On an independent test set of 75 RNA Seq samples, the 3-gene test
had a 97%
Accuracy, 98% Sensitivity, 96% Specificity, 98% Positive Predictive Value, and
96 %
Negative Predictive Value. We used The Cancer Genome Atlas (TCGA) project from
the
National Cancer Institute for biomarker discovery to identify a cohort of
biomarkers from a
population of subjects afflicted with a cancer. In contrast to many freely
available datasets,
the Biospecimen Core maintains rigorous protocols and quality controls that
increase our
confidence in pre-analytical variables. mRNA was profiled by RNA Seq (n=1,218)
and
microarray (n=132). Subsets from the cohort of biomarkers were identified in
subsequent
analysis and informed a selection of biomarkers that correctly identified a
cancer with high
sensitivity and specificity.
V. Overview of a Molecular Test for Positive Surgical Margins
[0047] mRNAs are promising biomarkers because changes in cell and tissue
morphology
necessarily involve changes in gene activity and are therefore ideally
situated to improve
margin analysis. Moreover, we can now catalog tumor mRNAs across the genome.
Finally,
-11-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
clinical labs routinely perform sensitive nucleic acid tests, positioning this
qPCR assay for
rapid adoption.
[0048] Prosignag (PAM50 gene expression test) has 510K clearance from the FDA
as a
prognostic test for the risk of recurrence, in conjunction with clinical
factors. However, by
design, half of the 50 mRNAs in PAM50 are expressed at lower levels in tumors
than in
healthy tissues, and PAM50 is only valid when at least 50% of the sample is
tumor. The
PAM50 strategy of using genes that are downregulated in tumors could therefore
not be used
to detect rare tumor cells. Since our clinical indication involves detecting
tumor cells in a
population of healthy cells, we validated tumor-specific mRNAs with high
expression in
tumors.
[0049] Described herein is a method for analysis of residual tumor cells. The
method and kits
disclosed herein can identify complete excision of malignant tissue from
patients.
Accordingly, disclosed herein are post-operative devices and methods for
obtaining and
analyzing gene expression from cells from patient samples (e.g. on the surface
of surgical
specimens) for residual disease. Nucleic acid tests for residual tumor cells
provide a powerful
solution to address positive surgical margins when combined with methods to
acquire
samples from the surface of a surgical sample.
VI. Overview of Molecular Test for Breast Cancer Screening
Described herein is a method for analysis of rare tumor cells. The method and
kits disclosed
herein can identify rare cancer cells, even when those tumor cells are not
found in the context
of healthy tissue. Accordingly, disclosed herein are screening devices and
methods for
obtaining and analyzing gene expression from cells from patient samples (e.g.
nipple
aspirates from ductal lavage) for disease. Disclosed herein are also adjuvant
devices and
methods to determine whether a screening test result warrants further
investigation.
VII. Definitions
[0050] As used in the specification and in the claims, the singular form "a,"
"an," and "the"
include plural referents unless the context clearly dictates otherwise.
[0051] The term "subject" or "patient" can include human or non-human animals.
Thus, the
methods and described herein are applicable to both human and veterinary
disease and animal
models. Preferred subjects are "patients," e.g., living humans that are
receiving medical care
for a disease or condition (e.g., cancer). This includes persons with no
defined illness who are
being investigated for signs of pathology. The methods described herein are
particularly
useful for the evaluation of patients having or suspected of having breast
adenocarcinomas.
-12-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[0052] Biomarkers broadly refer to any characteristics that are objectively
measured and
evaluated as indicators of normal biological processes, pathogenic processes,
or pharmacologic
responses to therapeutic intervention. Unless otherwise noted, the term
biomarker as used
herein specifically refers to biomarkers that have biophysical properties,
which allow their
measurements in biological samples (e.g., plasma, serum, lavage, biopsy).
Unless otherwise
noted, the term biomarker is used interchangeably with "molecule biomarker" or
"molecular
markers." Examples of biomarkers include nucleic acid biomarkers (e.g.,
oligonucleotides or
polynucleotides), peptides or protein biomarkers, lipids, and
lipopolysaccharide markers.
[0053] The term "polynucleotide" or "nucleic acid" as used herein refers to a
polymeric form
of nucleotides of any length, either ribonucleotides or deoxyribonucleotides,
that comprise
purine and/or pyrimidine bases, or other naturally modified nucleotide bases.
Polynucleotides
of the embodiments of the invention include sequences of deoxyribonucleic acid
(DNA),
ribonucleic acid (RNA), or DNA copies of ribonucleic acid (cDNA), all of which
may be
isolated from natural sources, recombinantly produced, or artificially
synthesized. The
polynucleotides and nucleic acids may exist as single-stranded or double-
stranded.
[0054] The term "primer" as used herein refers to an oligonucleotide which is
capable of acting
as a point of initiation of synthesis when placed under conditions in which
synthesis of a primer
extension product, which is complementary to a nucleic acid strand, is
induced, i.e., in the
presence of nucleotides and an inducing agent such as a DNA polymerase and at
a suitable
temperature and pH. The primer may be either single-stranded or double-
stranded and must be
sufficiently long to prime the synthesis of the desired extension product in
the presence of the
inducing agent. The exact length of the primer will depend upon many factors,
including
temperature, source of primer and the method used. For example, for diagnostic
applications,
depending on the complexity of the target sequence, the oligonucleotide primer
typically
contains 15-35 or more nucleotides, although it may vary for certain
biomarkers or
applications.
[0055] "Biological sample" as used herein is a sample of biological tissue or
chemical fluid
that is suspected of containing a biomarker or an analyte of interest. The
sample may be an ex
vivo sample or in vivo sample. Samples include, for example, tissue biopsies,
e.g., from the
breast or any other tissue suspected to be affected by, for instance, a
metastasis of a cancer. The
biopsy can be a liquid biopsy or a solid tissue biopsy. The sample can be a
surgical excision
from a tissue margin or another area suspected to be affected. A sample may be
suspended or
dissolved in, e.g., buffers, extractants, solvents, and the like. The terms
sample and specimen
can be used interchangeably herein.
-13-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[0056] Ranges can be expressed herein as from "about" one particular value,
and/or to
"about" another particular value. When such a range is expressed, another
embodiment
includes from the one particular value and/or to the other particular value.
Similarly, when
values are expressed as approximations, by use of the antecedent "about," it
will be
understood that the particular value forms another embodiment. It will be
further understood
that the endpoints of each of the ranges are significant both in relation to
the other endpoint,
and independently of the other endpoint. The term "about" as used herein
refers to a range
that is 15% plus or minus from a stated numerical value within the context of
the particular
usage. For example, about 10 would include a range from 8.5 to 11.5.
VIII. Samples
[0057] Methods for detecting molecules (e.g., nucleic acids, proteins, etc.)
in a subject in
order to detect, diagnose, monitor, or evaluate the presence of residual
cancer are described in
this disclosure. In some cases, the molecules are circulating molecules. In
some cases, the
molecules are expressed in the cytoplasm of blood, endothelial, or organ
cells. In some cases,
the molecules are expressed on the surface of blood, endothelial, or organ
cells.
[0058] The methods, kits, and systems disclosed herein can be used to classify
one or more
samples from one or more subjects. A sample can be any material containing
tissues, cells,
nucleic acids, genes, gene fragments, expression products, polypeptides,
exosomes, gene
expression products, or gene expression product fragments of a subject to be
tested. A sample
can include but is not limited to, tissue, cells, or biological material from
cells or derived
from cells of an individual. The sample can be a heterogeneous or homogeneous
population
of cells or tissues. The sample can be a fluid that is acellular or depleted
of cells (e.g.,
serum). In some cases, the sample is from a single patient. In some cases, the
method
comprises analyzing multiple samples at once, e.g., via massively parallel
multiplex
expression analysis on protein arrays or the like.
[0059] The sample may be obtained using any suitable method. The sample may be
obtained
by a minimally-invasive method, e.g., venipuncture or ductal lavage. The
sample obtained by
venipuncture may comprise whole blood or a component thereof (e.g. serum,
white blood
cells). Ductal lavage may be performed by e.g. the method described in
U520020058887A1,
which is incorporated by reference herein. Alternatively, the sample may be
obtained an
invasive method, such as by biopsy. Biopsies could include core biopsies,
punch biopsies,
incisional biopsies and excisional biopsies. A sample obtained by surgical
excision may
comprise a subsection of an excised tissue chunk (e.g. a representative cross-
section of
tissue). A sample obtained by surgical excision may comprise a cell-
dissociated or
-14-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
homogenized chunk of some or all of the excised tissue. A sample obtained by
surgical
excision may comprise a surface sample of excised tissue. A surface sample of
excised tissue
may comprise a "touch prep" sample which reflects the population of cells
along the margins
of the excised tissue (e.g. tumor).
[0060] In some embodiments, obtaining a sample comprises directly isolating a
sample from
a patient. In some embodiments, obtaining a sample comprises obtaining a
sample
previously isolated from a patient. In some embodiments, obtaining a sample
comprises
obtaining polynucleotides isolated from a sample previously isolated from a
patient.
[0061] The cellular specimen may be obtained using imprint cytology
acquisition strategies,
one form of which is a 'touch prep' or similar method. A 'touch prep' is known
as a type of
imprint cytology. Generally, the term 'touch prep' refers to both the process
of preparing the
slide, rapid staining the slide, and analyzing the slide under a microscope.
The 'touch prep'
method may involve smearing or spreading the obtained cellular specimen onto a
slide or a
plurality of slides. The 'touch prep' method may involve pressing the slide to
the biological
sample. The 'touch prep' method may involve pressing the slide to the excised
tissue. The
'touch prep' method may involve pressing the slide to a tissue on or within
the subject. The
'touch prep' method may involve pressing the slide to an area, wall or margin
surrounding a
tissue or biological sample on or within the subject. The 'touch prep' method
may involve
pressing the slide to an area, wall or margin surrounding a site where a
tissue was excised.
Touch prep may be performed in, e.g. less than about 60 minutes, less than
about 55 minutes,
less than about 50 minutes, less than about 45 minutes, about less than 40
minutes, about less
than 35 minutes, about less than 30 minutes, about less than 25 minutes, less
than about 20
minutes, less than about 15 minutes, less than about 10 minutes, less than
about 5 minutes,
less than about 3 minutes, less than about 2 minutes, less than about 1
minute, less than about
30 seconds, less than about 10 seconds, less than about 5 seconds, less than
about 2 seconds,
or less than about 1 second. The 'touch prep' method may be performed in a few
seconds per
slide. The 'touch prep' method may be performed by a surgeon, a nurse, an
assistant, a
cytopathologist, a person with no medical training or the subject. The 'touch
prep' method
may be operated manually. The 'touch prep' method may be operated
automatically by a
machine. The 'touch prep' method may be performed intraoperatively to detect
or rule out
malignant cells along the surgical margin (e.g. during a breast lumpectomy).
During the
'touch prep' method, the excised tissue may be pressed against a sample
collection unit
which is a glass slide coated with poly-Lysine, or other surface. The cellular
specimen
obtained by a touch prep method may be used to determine the presence or
absence of
-15-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
malignant cells along the margin of excised tissue. In some cases, the surface
comprises
sample collection unit. In some cases, the sample is then applied to a sample
input unit of a
device.In some embodiments, the touch prep sample may be obtained according to
the
methodology described in US20040030263A1, which is incorporated by reference
herein.
[0062] In some embodiment, the samples comprise tissue samples and are
prepared by tumor
dissociation/homogenization. In some embodiments, this is accomplished using
the Miltenyi
Biotec Tumor Dissociation Kit in combination with a gentleMACS Tissue
Dissociator to
homogenize tissue samples in a sterile environment. The Tissue Dissociator
uses disposable
Miltenyi M tubes with rotor-stators that are built into the tube lids. Frozen
samples may be
used to achieve more comsistent yields. Tissue is added to cell lysis in
buffer directly in the
disruptor tube. After dissociation and lysis, RNA is isolated using an RNA
isolation kit, such
as Qiagen RNeasy Mini Kit. This method can isolate high-quality RNA from both
tumor and
adipose-based tissues. Larger specimens may be divided into smaller pieces
depending on
maximum tissue input. If tissue dissociation alone does not collect enough
high-quality RNA
for RTqPCR, samples may be pre-incubated with enzymatic treatments (e.g.
Collagenases).
Enzymatic treatments may be applied during mechanical dissociation, which
others have
validated for the GentleMACS Tissue Dissociator.
[0063] In some embodiments, the methods or compositions herein are capable of
detecting
breast cancer in a sample from a cancer patient, detecting residual breast
cancer in a sample
from a cancer patient (e.g. post-chemotherapy/radiation/surgery) , or
distinguishing between
breast cancer and surrounding healthy breast tissue. In some embodiments, the
detection is
based on a minimal amount of polynucleotides or nucleic acids isolated from a
sample.
[0064] In some embodiments the minimal amount of polynucleotides or nucleic
acids
isolated from the sample is at least 10 ng, 50 ng, 10Ong, 200 ng, 500ng, 1 mg,
2 mg, 3 mg, 4
mg, 5 mg, 10 mg, 15 mg, 20 mg, 50 mg, or 500ng. In some embodiments, the
methods or
compositions herein are capable of detecting residual cancer in a sample from
a patient, or
distinguishing between cancer and surrounding healthy tissue, based on a
minimal weight of
tissue sample used to isolate polynucleotides or nucleic acids. In some
embodiments, the
minimal amount of tissue sample is at least 100 ng, 200 ng, 500 ng, lmg, 2 mg,
3 mg, 4 mg, 5
mg, 10 mg, 15 mg, 20 mg, 50 mg, 100 mg, 200 mg, 300 mg, or 500 mg.
IX. Biomarkers
[0065] The term "biomarker" as used herein refers to a measurable indicator of
some
biological state or condition. In some instances, a biomarker can be a
substance found in a
subject, a quantity of the substance, or some other indicator. For example, a
biomarker can
-16-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
be the amount of a protein and/or other gene expression products in a sample.
In some
embodiments, a biomarker is a total level of protein in a sample. In some
embodiments, a
biomarker is a total level of a particular type of nucleic acid (e.g. RNA,
cDNA) in a sample.
In some embodiments, a biomarker is a therapeutic target, or an indicator of
response to
therapy.
[0066] The methods, compositions and systems as described here also relate to
the use of
biomarker panels for purposes of research, identification, diagnosis,
classification, treatment
or to otherwise characterize the status of cancer in a patient. Sets of
biomarkers useful for
classifying biological samples are provided, as well as methods of obtaining
such sets of
biomarkers. Often, the pattern of levels of biomarkers in a panel (also known
as a signature)
is determined from a control sample or population and then used to evaluate
the signature of
the same panel of biomarkers in an experimental sample or population, such as
by a measure
of similarity between the sample signature and the reference signature.
[0067] In some embodiments, the panels of biomarkers described herein are
useful for the
detection of breast cancer (e.g. detection of positive surgical margins on a
biopsy sample,
detection of residual disease in a cancer patient post-
radiation/chemotherapy/surgery, or
detection of disease in a patient suspected of having cancer). In some
embodiments the
breast cancer is invasive adenocarcinoma, invasive ductal breast cancer,
invasive lobular
breast cancer, or a combination thereof In some embodiments, the breast cancer
is HER2
positive, ER (estrogen receptor) positive, or PR (progesterone receptor)
positive, or a
combination thereof In some embodiments, the breast cancer is HER2 negative,
ER
(estrogen receptor) negative, or PR (progesterone receptor) negative, or a
combination
thereof.
[0068] In some embodiments, the methods herein comprise measuring expression
levels of
genes selected from the group consisting essentially of Matrix
Metallopeptidase 11
(WP 11), integrin binding sialoprotein (IBSP), and collagen type X alpha 1
chain
(COL10A1). In some embodiments, the methods herein comprise measuring
expression
levels of genes selected from the group consisting of Matrix Metallopeptidase
11 (WP 11),
integrin binding sialoprotein (IBSP), and collagen type X alpha 1 chain
(COL10A1). In some
embodiments the methods herein comprise measuring expression levels of genes
selected
from the group consisting essentially of Matrix Metallopeptidase 11 (WP 11)
and integrin
binding sialoprotein (IBSP). In some embodiments the methods herein comprise
measuring
expression levels of genes selected from the group consisting of Matrix
Metallopeptidase 11
(WP 11) and integrin binding sialoprotein (IBSP).
-17-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[0069] The biomarkers that form the basis for the 3-gene test described herein
(WPII,
IBSP, and COL10A _I) particularly useful in that their expression is higher
(upregulated) in
cancerous tissues than in normal tissues. As a result, the fraction of a
sample that must
contain cancerous cells for the sample to be labeled as positive is much lower
than for a test
that depends on genes that have decreased expression (downregulated) in
cancerous tissue.
In some embodiments, the methods, compositions and systems as described here
also relate
to the use of a biomarker test of research, identification, diagnosis,
classification, treatment or
to otherwise characterize the status of cancer in a patient, wherein at least
one of Matrix
Metallopeptidase 11 (WP 11), integrin binding sialoprotein (IBSP), and
collagen type X
alpha 1 chain (COL10A1) are higher in said cancer than in healthy tissue. In
some
embodiments, at least two of Matrix Metallopeptidase 11 (WP I 1), integrin
binding
sialoprotein (IBSP), and collagen type X alpha 1 chain (COL10A1) are higher in
said cancer
than in healthy tissue. In some embodiments, the levels of each of Matrix
Metallopeptidase
11 (WP I 1), integrin binding sialoprotein (IBSP), and collagen type X alpha 1
chain
(COL10A1) are higher in said cancer than in healthy tissue.
X. Biomarker Expression Profiles
[0070] The methods, kits, and systems disclosed herein may comprise
specifically detecting,
profiling, or quantitating biomolecules (e.g., nucleic acids, DNA, RNA,
polypeptides, etc.)
that are within the biological samples to determine an expression profile. In
some instances,
genomic expression products, including RNA, or polypeptides, may be isolated
from the
biological samples. In some cases, nucleic acids, DNA, RNA, polypeptides may
be isolated
from a cell-free source. In some cases, nucleic acids, DNA, RNA, polypeptides
may be
isolated from cells derived from the cancer patient. In some cases, the
molecules detected are
derived from molecules endogenously present in the sample via an enzymatic
process (e.g.,
cDNA derived from reverse transcription of RNA from the biological sample
followed by
amplification).
[0071] Expression profiles are preferably measured at the nucleic acid level,
meaning that
levels of mRNA or nucleic acid derived therefrom (e.g., cDNA or RNA) are
measured. An
expression profile refers to the expression levels of a plurality of genes in
a sample. A
nucleic acid derived from mRNA means a nucleic acid synthesized using mRNA as
a
template. Methods of isolation and amplification of mRNA are described in,
e.g., Chapter 3
of Laboratory Techniques in Biochemistry and Molecular Biology: Hybridization
With
Nucleic Acid Probes, Part I. Theory and Nucleic Acid Preparation, (P. Tijssen,
ed.) Elsevier,
N.Y. (1993). If mRNA or a nucleic acid therefrom is amplified, the
amplification is
-18-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
performed under conditions that approximately preserve the relative
proportions of mRNA in
the original samples, such that the levels of the amplified nucleic acids can
be used to
establish phenotypic associations representative of the mRNAs.
[0072] In some embodiments, expression levels are determined by direct
detection of nucleic
acids. Such methods include e.g. gel or capillary electrophoresis, wherein
specifically
amplified DNA is detected by its intrinsic fluorescence/absorbance, or by
complexing with a
suitable absorbent or fluorescent DNA-binding dye. Such methods can be used
alongside
PCR or RT-PCR with forward and reverse primers against specific genes to
detect levels of
genes within nucleic acids isolated from a sample.
[0073] In other methods, expression levels are determined by NanoStringTM
assay.
NanoStringTM based assays are described in the U.S. Patent Nos. 8,415, 102,
8,519,115, and
7,919,237, which are herein incorporated by reference in their entirety.
NanoString's
NCOUNTER technology is a variation on the DNA microarray. It uses molecular
"barcodes"
and microscopic imaging to detect and count up to several hundred unique
transcripts in one
hybridization reaction. Each color-coded barcode is attached to a single
target-specific probe
corresponding to a target of interest. The protocol typically includes
hybridization
(employing two ¨50 base probes per mRNA that hybridize in solution; the
reporter probe
carries the signal, while the capture probe allows the complex to be
immobilized for data
collection); purification and immobilization (after hybridization, the excess
probes are
removed and the probe/target complexes are aligned and immobilized in the
cartridge); and
data collection (sample cartridges are placed in a digital analyzer instrument
for data
collection; color codes on the surface of the cartridge are counted and
tabulated for each
target molecule). The protocol is carried out with a prep station, which is an
automated
fluidic instrument that immobilizes code set complexes for data collection,
and a digital
analyzer, which derives data by counting fluorescent barcodes. Code set
complexes are
custom-made or pre-designed sets of color-coded probes pre -mixed with a set
of system
controls. Probes for the barcode-based assay can be designed according to
desired variables
such as melting temperature (Tm) and specificity for the template mRNA/cDNA to
be
detected.
[0074] In other methods, expression levels are determined by so-called "real
time
amplification" methods also known as quantitative PCR (qPCR) or Taqman. The
basis for
this method of monitoring the formation of amplification product formed during
a PCR
reaction with a template using oligonucleotide probes/oligos specific for a
region of the
template to be detected. In some embodiments, qPCR or Taqman are used
immediately
-19-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
following a reverse-transcriptase reaction performed on isolated cellular
mRNA; this variety
serves to quantitate the levels of individual mRNAs during qPCR.
[0075] Taqman uses a dual-labeled fluorogenic oligonucleotide probe. The dual
labeled
fluorogenic probe used in such assays is typically a short (ca. 20-25 bases)
polynucleotide
that is labeled with two different fluorescent dyes. The 5' terminus of the
probe is typically
attached to a reporter dye and the 3' terminus is attached to a quenching dye.
Regardless of
labelling or not, the qPCR probe is designed to have at least substantial
sequence
complementarity with a site on the target mRNA or nucleic acid derived from.
Upstream and
downstream PCR primers that bind to flanking regions of the locus are also
added to the
reaction mixture. When the probe is intact, energy transfer between the two
fluorophores
occurs and the quencher quenches emission from the reporter. During the
extension phase of
PCR, the probe is cleaved by the 5' nuclease activity of a nucleic acid
polymerase such as
Taq polymerase, thereby releasing the reporter from the polynucleotide-
quencher and
resulting in an increase of reporter emission intensity which can be measured
by an
appropriate detector. The recorded values can then be used to calculate the
increase in
normalized reporter emission intensity on a continuous basis and ultimately
quantify the
amount of the mRNA being amplified. mRNA levels can also be measured without
amplification by hybridization to a probe, for example, using a branched
nucleic acid probe,
such as a QuantiGeneg Reagent System from Panomics. This format of test is
particularly
useful for the multiplex detection of multiple genes from a single sample
reaction, as each
fluorophore/quencher pair attached to an individual probe may be spectrally
orthogonal to the
other probes used in the reaction such that multiple probes (each directed
against a different
gene product) can be detected during the amplification/detection reaction.
[0076] qPCR can also be performed without a dual-labeled fluorogenic probe by
using a
fluorescent dye (e.g. SYBR Green) specific for dsDNA that reflects the
accumulation of
dsDNA amplified specific upstream and downstream oligonucleotide primers. The
increase
in fluorescence during the amplification reaction is followed on a continuous
basis and can be
used to quantify the amount of mRNA being amplified.
[0077] For qPCR or Taqman, the levels of particular genes may be expressed
relative to one
or more internal control gene measured from the same sample using the same
detection
methodology. Internal control genes may include so-called "housekeeping" genes
(e.g.
ACTB, B2M, UBC, GAPD and HPRT1). In some embodiments, the one or more internal
control gene is TTC5, C2orf44, or Chr3.
-20-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[0078] In some embodiments, for qPCR or Taqman detection, a "pre-
amplification" step is
performed on cDNA transcribed from cellular RNA prior to the quantitatively
monitored
PCR reaction. This serves to increase signal in conditions where the natural
level of the
RNA/cDNA to be detected is very low. Suitable methods for pre-amplification
include but
are not limited LM-PCR, PCR with random oligonucleotide primers (e.g. random
hexamer
PCR), PCR with poly-A specific primers, and any combination thereof.
[0079] In some embodiments, for qPCR or Taqman detection, an RT-PCR step is
first
performed to generate cDNA from cellular RNA. Such amplification by RT-PCR can
either
be general (e.g. amplification with partially/fully degenerate oligonucleotide
primers) or
targeted (e.g. amplification with oligonucleotide primers directed against
specific genes
which are to be analyzed at a later step).
[0080] In other methods, expression levels are determined by sequencing, such
as by RNA
sequencing or by DNA sequencing (e.g., of cDNA generated from reverse-
transcribing RNA
(e.g., mRNA) from a sample). Sequencing may also be general (e.g. with
amplification using
partially/fully degenerate oligonucleotide primers) or targeted (e.g. with
amplification using
oligonucleotide primers directed against specific genes which are to be
analyzed at a later
step). Sequencing may be performed by any available method or technique.
Sequencing
methods may include: Next Generation sequencing, high-throughput sequencing,
pyrosequencing, classic Sanger sequencing methods, sequencing-by-ligation,
sequencing by
synthesis, sequencing-by-hybridization, RNA- Seq (Illumina), Digital Gene
Expression
(Helicos), next generation sequencing, single molecule sequencing by synthesis
(SMSS)
(Helicos), Ion Torrent Sequencing Machine (Life Technologies/Thermo-Fisher),
massively-
parallel sequencing, clonal single molecule Array (Solexa), shotgun
sequencing, Maxim-
Gilbert sequencing, primer walking, and any other sequencing methods known in
the art.
[0081] Measuring gene expression levels may comprise reverse transcribing RNA
(e.g.,
mRNA) within a sample in order to produce cDNA. The cDNA may then be measured
using
any of the methods described herein (e.g., qPCR, sequencing, etc.).
[0082] Alternatively, or additionally, expression levels of genes can be
determined at the
protein level, meaning that levels of proteins encoded by the genes discussed
above are
measured. Several methods and devices are well known for determining levels of
proteins
including immunoassays such as sandwich, competitive, or non-competitive assay
formats, to
generate a signal that is related to the presence or amount of a protein
analyte of interest.
Immunoassays such as, but not limited to, lateral flow, enzyme-linked
immunoassays
(ELISA), radioimmunoassays (RIAs), and competitive binding assays may be
utilized.
-21-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
Numerous formats for antibody arrays have been described proposed employing
antibodies.
Other ligands having specificity for a particular protein target can also be
used, such as
synthetic antibodies.
XI: Sensitivity, Specificity, Accuracy and other measures of performance
[0083] The methods provided herein can detect the presence of residual
disease, such as a
positive margin on a surgical cancer biopsy or presence of disease (e.g. of in
a sample from a
cancer patient with a high degree of accuracy, sensitivity, and/or
specificity. In some cases,
the accuracy (e.g., for detecting residual disease, or distinguishing between
residual disease
and surrounding healthy tissue) is at least about 75%, 80%, 85%, 90%, 91%,
92%, 93%,
94%, 95%, 96%, 97% 98% or 99%. In some cases, the sensitivity (e.g., for
detecting residual
disease, or distinguishing between residual disease and surrounding healthy
tissue) is at least
about 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% 98% or 99%. In
some
cases, the specificity (e.g., for detecting residual disease, or
distinguishing between residual
disease and surrounding healthy tissue) is at least about 75%, 80%, 85%, 90%,
91%, 92%,
93%, 94%, 95%, 96%, 97% 98% or 99%.. In some cases, the positive predictive
value (e.g.,
for detecting residual disease, or distinguishing between residual disease and
surrounding
healthy tissue) of the method at least 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%,
95%,
96%, 97% 98% or 99%.. The AUC after thresholding in any of the methods
provided herein
may be at least about 0.7, 0.75, 0.8, 0.85, 0.9, 0.91, 0.92, 0.93, 0.94, 0.95.
0.96, 0.97, 0.98,
0.99, 0.995, or 0.999. In some embodiments, the methods disclosed herein have
a positive
predictive value of at least about 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or
99%. In
some embodiments, the methods disclosed herein have a negative predictive
value of at least
about 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99%.
XII. Clinical Applications
[0084] The methods, compositions, systems and kits provided herein can be used
to detect,
diagnose, predict or monitor a condition of a pregnant patient. In some
instances, the
methods, compositions, systems and kits described herein provide information
to a medical
practitioner that can be useful in making a therapeutic decision. Therapeutic
decisions can
include decisions to: continue with a particular therapy, modify a particular
therapy, alter the
dosage of a particular therapy, stop or terminate a particular therapy,
altering the frequency of
a therapy, introduce a new therapy, introduce a new therapy to be used in
combination with a
current therapy, or any combination of the above. In some cases, the methods
provided herein
can be applied in an experimental setting, e.g., a clinical trial. In some
embodiments, the
guidance of a test result herein (e.g. presence of residual disease) may be
used to determine
-22-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
the end of a course of therapy (e.g. standard chemotherapy regimens). In some
embodiments,
the guidance of a test result herein (e.g. presence of residual disease) may
be used to indicate
the location of a further tumor excision to be performed on the patient (e.g.
in the case where
the test is used in combination with touch prep multiple touch prep samples
derived as
described above to indicate where surgical margins have been insufficient in
an excised
sample).
XIII. Monitoring a Condition of a Patient
[0085] Provided herein are methods, systems, kits and compositions for
monitoring a
condition of a cancer patient (e.g. presence of residual disease). Often, the
monitoring is
conducted by serial testing, such as serial non-invasive tests, serial
minimally-invasive tests
(e.g., blood draws, ductal lavage), or some combination thereof.
[0086] In some instances, the cancer patient is monitored as needed using the
methods
described herein. Alternatively the cancer patient can be monitored weekly,
monthly, or at
any pre-specified intervals. In some instances, the cancer patient is
monitored at least once
every 24 hours. In some instances the cancer patient is monitored at least
once every 1 day to
30 days. In some instances the cancer patient is monitored at least once every
at least 1 day.
In some instances the cancer patient is monitored at least once every at most
30 days. In some
instances the cancer patient is monitored at least once every 1 day to 5 days,
1 day to 10 days,
1 day to 15 days, 1 day to 20 days, 1 day to 25 days, 1 day to 30 days, 5 days
to 10 days, 5
days to 15 days, 5 days to 20 days, 5 days to 25 days, 5 days to 30 days, 10
days to 15 days,
days to 20 days, 10 days to 25 days, 10 days to 30 days, 15 days to 20 days,
15 days to 25
days, 15 days to 30 days, 20 days to 25 days, 20 days to 30 days, or 25 days
to 30 days. In
some instances the cancer patient is monitored at least once every 1, 2, 3, 4,
5, 6, 7, 8, 9, 10,
11,12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 28, 29, 30 or 31 days. In some
instances, the cancer
patient is monitored at least once every 1, 2, 3, or 6 months.
XIV. Sequences and Embodiments of Combinations of Sequences
[0087] The primers disclosed herein, such as a pair of primers as described
herein,
specifically a forward primer ("F") and a reverse primer ("R") for both
strands to be detected,
can be in a composition in amounts effective to permit detection of native,
mutant, reference,
or control sequences. Detection of native, mutant, reference, or control
sequences is
accomplished using any of the methods described herein or known by one of
ordinary skill in
the art in the art for detecting a specific nucleic acid molecule in a sample.
The primers
disclosed herein may be provided as part of a kit. A kit can also comprise
buffers, nucleotide
-23-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
bases and other compositions to be used in hybridization and/or amplification
reactions. In
other cases, the primers described herein may be part of a device.
[0088] In some embodiments a panel of nucleic acids is detected in a sample
from a patient.
A panel of one to three cDNAs can serve as biomarkers to distinguish invasive
breast cancer
from adjacent healthy tissue. A panel of one to three cDNAs can serve to
residual breast
cancer post-chemotherapy, post-radiation treatment, or post-surgical excision
of tumor(s).
Such cDNA panels may comprise IBSP, MMP11, and/or COL10A1 cDNA. A panel may
comprise two or three genes selected from IBSP, MMP11, and COL10A1, which can
be
amplified using the primers disclosed herein. In some embodiments, the
relative levels of
cDNA panels may be assessed relative to the cDNA levels of a reference gene
panel. Such
reference gene panel may comprise TTC5 and/or C2orf44, which can be amplified
using the
primers disclosed herein. In some cases, the single genes or gene panels are
compared to a
negative control for genomic DNA, for example, chr3 gDNA, which can be
amplified using
the primers disclosed herein.
XV. Primers
[0089] Exemplary forward primers for IBSP cDNA
TABLE 1
SEQ ID NO: F primers Sequence
Ref Code:
SEQ ID NO: 1 A21 CACAGGGTATACAGGGTTAGCTG
SEQ ID NO: 2 A27 ATGAAAAATTTGCATCGAAGAG
SEQ ID NO: 3 A6 TCAAAATAGAGGATTCTGAAGA
SEQ ID NO: 4 A19 CAATCTGTGCCACTCACTGC
SEQ ID NO: 5 A32 ACTGCCTTGAGCCTGCTTC
SEQ ID NO: 6 A30 AGAGGAGGAGGAAGAAGAG
SEQ ID NO: 7 Al2 TGAGTGAGTGAGAGGGCAGA
SEQ ID NO: 8 A35 AGTGAGTGAGAGGGCAGAGG
SEQ ID NO: 9 A22 TGCTTTAATTTTGCTCAGCATT
SEQ ID NO: 10 A23 TTGGGAATGGCCTGTGCTTTCTCA
SEQ ID NO: 11 A36 AAGCAATCACCAAAATGAAGAC
SEQ ID NO: 12 A8 TGAAGAAAATGGG
SEQ ID NO: 13 A16 ACAGGGTTAGCTGCAATCCA
SEQ ID NO: 14 AS GTCTTTAAGTACAGGCCACGAT
-24-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 15 A7 ATTATCTTTACAAGCATGCCTA
SEQ ID NO: 16 A13 GAGACTTCAAATGAAGGAGAAA
SEQ ID NO: 17 A26 ACAATGAAGAATCGAATGAAGA
SEQ ID NO: 18 A25 TGAAGACTCTGAGGCTGAGAAT
SEQ ID NO: 19 A4 ACCACACTTTCTGCTACAACAC
SEQ ID NO: 20 A20 TGGGCTATGGAGAGGACGCCAC
SEQ ID NO: 21 A24 CAACACTGGGCTATGGAGAGG
SEQ ID NO: 22 Al GCCTGGCACAGGGTATACAGGG
SEQ ID NO: 23 Al7 GAGTGAGAGGGCAGAGGAAA
SEQ ID NO: 24 A29 CTTTTATCCTCATTTAAAACGA
SEQ ID NO: 25 A38 TTAGCTGCAATCCAGCTTCCCAAGAAG
SEQ ID NO: 26 A14 CTCAATCTGTGCCACTCACTGC
SEQ ID NO: 27 Al 1 CTGCTTCCTCACTCCAGGAC
SEQ ID NO: 28 A18 CAAGCATGCCTACTTTTATCCTC
SEQ ID NO: 29 A37 CTTGAGCCTGCTTCCTCACT
SEQ ID NO: 30 A39 GTCTTTAAGTACAGGCCACGA
SEQ ID NO: 31 A9 ACAACACTGGGCTATGGAGAGG
SEQ ID NO: 32 A10 GAGTGAGTGAGAGGGCAGAGGA
SEQ ID NO: 33 A40 AATACTCAATCTGTGCCACTCA
SEQ ID NO: 34 A31 CTGCCTTGAGCCTGCTTCCTCA
SEQ ID NO: 35 A3 GTGAGAGGGCAGAGGAAATAC
SEQ ID NO: 36 A28 CTCCAGGACTGCCAGAGG
SEQ ID NO: 37 A34 TTTCCAGTTCAG
SEQ ID NO: 38 A15 GGCAGTAGTGACTCATCCGAAG
SEQ ID NO: 39 A2 GTCTTTAAGTACAGGCCACGA
SEQ ID NO: 40 A33 AAAATGGAGATGACAGTTCAGA
[0090] Exemplary reverse primers for IBSP cDNA
TABLE 2
SEQ ID NO: R primers Sequence
Ref Code:
SEQ ID NO: 41 B1 TTCTGCCTCTGTGCTGTTGGTA
-25-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 42 B2 CTGGTGCCGTTTATGCCTTGTT
SEQ ID NO: 43 B3 AGCTTTATTTGTTATATCCCCAGC
SEQ ID NO: 44 B4 GATGCAAATTTTTCAT
SEQ ID NO: 45 B5 TCCTCTTCTTCTTCATCACTTTCC
SEQ ID NO: 46 B6 AGAAAGCACAGGCCATTCC
SEQ ID NO: 47 B7 TGAGAAAGCACAGGCCATTCCC
SEQ ID NO: 48 B8 ATTTTGACTCTTCGATGCAAAT
SEQ ID NO: 49 B9 GGTGCCGTTTATGCCTTGTT
SEQ ID NO: 50 B10 CTTCTTGGGAAGCTGGATTGCA
SEQ ID NO: 51 B11 CTGAACTGGAAATCGTTTTAAA
SEQ ID NO: 52 B12 GCTAACCCTGTATACCCTGTGC
SEQ ID NO: 53 B13 GAACTGTCATCTCCATTTTCTT
SEQ ID NO: 54 B14 CATCTCCATTTTCTTCGGATG
SEQ ID NO: 55 B15 GCCGTTTATGCCTTGTTCGT
SEQ ID NO: 56 B16 GCCATTCCCAAAATGCTGAG
SEQ ID NO: 57 B17 TCCTCTTCCTCCTCTTCTTCTT
SEQ ID NO: 58 B18 CAGTCTTCATTTTGGTGATTGC
SEQ ID NO: 59 B19 CTTCATCTTCATTCGATTCTTC
SEQ ID NO: 60 B20 TCCCCTTCTTCTCCATTGTCTC
SEQ ID NO: 61 B21 GCCCAGTGTTGTAGCAGAAAGT
SEQ ID NO: 62 B22 GCAGTCCTGGAGTGAGGAAG
SEQ ID NO: 63 B23 AAAATGCTGAGCAAAATTAAAG
SEQ ID NO: 64 B24 CGTTTTCATCCACTTCTGCTTC
SEQ ID NO: 65 B25 CAGTCCTGGAGTGAGGAAGC
SEQ ID NO: 66 B26 GTGGTATTCTCAGCCTCAGAGT
SEQ ID NO: 67 B27 GCTTTCTTCGTTTTCATTTCCT
SEQ ID NO: 68 B28 GTACTTAAAGAC
SEQ ID NO: 69 B29 CCCATTTTCTTCAGAATCCTCT
SEQ ID NO: 70 B30 TCTCCATTTTCTTCGGATGAG
SEQ ID NO: 71 B31 ATTGTTTTCTCCTTCATTTGAAGTCTC
SEQ ID NO: 72 B32 CTTCTGCTTCGCTTTCTTCG
SEQ ID NO: 73 B33 CTTCTGAACTGTCATCTCCATTTTC
-26-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 74 B34 TGTAAAGATAATATCGTGGCCT
SEQ ID NO: 75 B35 TCCATTTTCTTCGGATGAGTC
SEQ ID NO: 76 B36 CATCACTTTCCTTCTCTTTTGT
SEQ ID NO: 77 B37 AGTCCTGGAGTGAGGAAGCA
SEQ ID NO: 78 B38 TGAGGATAAAAGTAGGCATGCT
SEQ ID NO: 79 B39 CGGATGAGTCACTACTGCC
SEQ ID NO: 80 B40 TCTTACCCTCTGGCAGTCCT
SEQ ID NO: 81 B41 CTGAGAAAGCACAGGCCATT
SEQ ID NO: 82 B42 CTCTTCTTCCTCCTCCTCTTCT
SEQ ID NO: 83 B43 CTCCGCTGCTGCCGTTGCCGTT
SEQ ID NO: 84 B44 CAGGCGTGGCGTCCTCTCCATA
SEQ ID NO: 85 B45 CATCTCCATTTTCTTCGGATG
[0091] Exemplary forward primers for WP11 cDNA
TABLE 3
SEQ ID NO: F primers Sequence
Ref Code:
SEQ ID NO: 86 Cl CGAGGCGAGCTCTTTTTCT
SEQ ID NO: 87 C2 CCTGGACTATCGGGGATGACCAGG
SEQ ID NO: 88 C3 ATAAGGGGCGGCGGCCCGGAGC
SEQ ID NO: 89 C4 GCCCCCAGGCTGGGATAGACAC
SEQ ID NO: 90 C5 TTGGTTCTTCCAAGGTGAGG
SEQ ID NO: 91 C6 AGGGCCGTGCTGACATCATGAT
SEQ ID NO: 92 C7 GTACTGGCATGGGGACGACCTG
SEQ ID NO: 93 C8 CCGTTTGATGGGCCTGGGGGCA
SEQ ID NO: 94 C9 TACCCAGCATTGGCCTCTC
SEQ ID NO: 95 C10 GTGAGGCCTCCTTTGACGCGGT
SEQ ID NO: 96 C11 CTGAGTCTCAGCCCAGATGACT
SEQ ID NO: 97 C12 ACCTCCAGGACCCCAGCCCTGG
SEQ ID NO: 98 C13 GCTGCAGCCCGGCTACCCAGCA
SEQ ID NO: 99 C14 TAGGTGCCTGCATCTGTCTG
SEQ ID NO: 100 C15 GATCCTTCGGTTCCCATGGCAG
-27-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 101 C16 TGTGGCGCCTCCGTGGGGGCCA
SEQ ID NO: 102 C17 AGACGATGGCAGAGGCCCTAAA
SEQ ID NO: 103 C18 CTCCACCATCCGAGGCGAGCTC
SEQ ID NO: 104 C19 ATCCGAGGCGAGCTCTTTTT
SEQ ID NO: 105 C20 TCCTGGCCCATGCCTTCTTCCC
SEQ ID NO: 106 C21 CAATGAGATTGCACCGCTGGAG
SEQ ID NO: 107 C22 CAGCCAAGGCCCTGATGTCCGC
SEQ ID NO: 108 C23 GTGAGGCCTCCTTTGACGCGGT
SEQ ID NO: 109 C24 CACATTTGGTTCTTCCAAG
SEQ ID NO: 110 C25 T GC C T TC GAGGAT GC C CAGGGC
SEQ ID NO: 111 C26 CGCGCCCTCCTGCCCCCGATGC
SEQ ID NO: 112 C27 GTTTCCACCCCAGCACC
SEQ ID NO: 113 C28 CTTCTACACCTTTCGCTACCCA
SEQ ID NO: 114 C29 GACTGCCCAGCCCTGTGGACGC
SEQ ID NO: 115 C30 C GGGGC GGAT GGC T C C GGC C GC
SEQ ID NO: 116 C31 TGCTGCTGCTGCTCCAGCCGCC
SEQ ID NO: 117 C32 GCACAGACCTGCTGCAGGTGGC
SEQ ID NO: 118 C33 TTTTTCTTCAAAGCGGGCTTTG
SEQ ID NO: 119 C34 CACATTTGGTTCTTCCAAG
SEQ ID NO: 120 C35 CGACTTCGCCAG
SEQ ID NO: 121 C36 CTGTGGACGCTGCCTTC
SEQ ID NO: 122 C37 CTACCCAGCATTGGCCTC
SEQ ID NO: 123 C38 TTGGCCTCTCGCCACTGGCAGG
SEQ ID NO: 124 C39 TGTGGCGCCTCCGTGGGGGCCA
SEQ ID NO: 125 C40 TGGCCAGCCCTGGCCCACTGTC
SEQ ID NO: 126 C41 CTCACCTTTACTGAGGTGCACG
SEQ ID NO: 127 C42 CAAGACTCACCGAGAAGGGGAT
SEQ ID NO: 128 C43 AGCCCATGAATTTGGCCACGTG
SEQ ID NO: 129 C44 TTTTTCTTCAAAGCGGGCTTTG
SEQ ID NO: 130 C45 GGCGAGCTCTTTTTCTTCAA
SEQ ID NO: 131 C46 GGCCCAGCAAGCCCAGCAGCCC
SEQ ID NO: 132 C47 GTCCACTTCGACTATGATGAGA
-28-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 133 C48 CCAGACGCCCCGCCAGATGCCT
SEQ ID NO: 134 C49 GCCGCTGCTGGCCCGGGCTCTGCCGCCG
SEQ ID NO: 135 C50 GACTGCCCAGCCCTGTGGACGC
SEQ ID NO: 136 C51 TGCCTTCGAGGATGCCCAGGGC
SEQ ID NO: 137 C52 CTACCCAGCATTGGCCTCTC
SEQ ID NO: 138 C53 CTGTGGACGCTGCCTTC
SEQ ID NO: 139 C54 CCAGACGCCCCGCCAGATGCCT
SEQ ID NO: 140 C55 GCTGCAGCCCGGCTACCCAGCA
SEQ ID NO: 141 C56 TTGGTGCAGGAGCAGGTGCGGC
SEQ ID NO: 142 C57 GGCCACTGACTGGAGAGG
SEQ ID NO: 143 C58 CTCCACCATCCGAGGCGAGCTC
SEQ ID NO: 144 C59 GCAGGGGCGTTCAACACCTATA
SEQ ID NO: 145 C60 CTGGGGCTGCAGCACACAACAG
SEQ ID NO: 146 C61 GGTATGGAGCGATGTGACGCCA
SEQ ID NO: 147 C62 CTGGCTCCGCAGCGCGGCCGCG
SEQ ID NO: 148 C63 TTGGCCTCTCGCCACTGGCAGG
SEQ ID NO: 149 C64 AGGCGAGCTCTTTTTCTTCA
SEQ ID NO: 150 C65 GCGAGCTCTTTTTCTTCAA
[0092] Exemplary reverse primers for WP11 cDNA
TABLE 4
SEQ ID NO: R primers Sequence
Ref Code:
SEQ ID NO: 151 D1 CTTGGAAGAACCAAATGTGGCC
SEQ ID NO: 152 D2 CCTGCCTCGGAAGAAGTAGA
SEQ ID NO: 153 D3 CTGTAGGTGAGGTCCGTCTTCT
SEQ ID NO: 154 D4 GCGGACATCAGGGCCTTGGCTG
SEQ ID NO: 155 D5 CGGGCACGCCACAGCGGGGAGG
SEQ ID NO: 156 D6 CACCCCTCTCCAGTCAGTG
SEQ ID NO: 157 D7 CATCAGCATCCTGGAAGGCAGC
SEQ ID NO: 158 D8 CCAGACCAAGGCAGCATGGACC
SEQ ID NO: 159 D9 GGTGGAAACGCCAGTAGTCC
-29-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 160 D10 TGGCTTTTCACCGTCGTACA
SEQ ID NO: 161 D1 1 GACTGGCTTTTCACCGTCGTAC
SEQ ID NO: 162 D12 CATCTGGCGGGGCGTCTGG
SEQ ID NO: 163 D13 CTGTTGTGTGCTGCAGCCCCAG
SEQ ID NO: 164 D14 ACATCCCCTTCTCGGTGAGTCT
SEQ ID NO: 165 D15 CTGGGCATCCTCGAAGGCAGCG
SEQ ID NO: 166 D16 TAGTCCCTGCCTCGGAAGAAGT
SEQ ID NO: 167 D17 CCTGGTCATCCCCGATAGTCCA
SEQ ID NO: 168 D18 GACAGTGGGCCAGGGCTGGCCA
SEQ ID NO: 169 D19 GGCTGCATGCCAGGGCTGTGGC
SEQ ID NO: 170 D20 CTCCAGCGGTGCAATCTCATTG
SEQ ID NO: 171 D21 AGAATACCCCTCCCCATTTG
SEQ ID NO: 172 D22 GCCACCTGCAGCAGGTCTGTGC
SEQ ID NO: 173 D23 GGGTGGAAACGCCAGTAGT
SEQ ID NO: 174 D24 GGTGCTGGGGTGGAAACGCCAG
SEQ ID NO: 175 D25 CCCCCACGGAGGCGCCACACAA
SEQ ID NO: 176 D26 ACCCAGTACTGAGCAC
SEQ ID NO: 177 D27 GGGAACCTCACCAGGCCCAGCT
SEQ ID NO: 178 D28 GACTGGCTTTTCACCGTCGTAC
SEQ ID NO: 179 D29 GTCGATCTCAGAGGGCACCCCT
SEQ ID NO: 180 D30 GGGGCTTCCTGCGTGGCAGGGG
SEQ ID NO: 181 D31 GATGCCCCCAGGCCCATCAAAC
SEQ ID NO: 182 D32 AGATCTTGTTCTTCTCGGGACC
SEQ ID NO: 183 D33 CCAAGGTGAGGGGCCTGGTGAG
SEQ ID NO: 184 D34 GGTGCTGGGGTGGAAACGCCAG
SEQ ID NO: 185 D35 CC TGAGGCTGCTGGCAGGCCGG
SEQ ID NO: 186 D36 GGCAGGTCGTCCCCATGCCAGTAC
SEQ ID NO: 187 D37 AGTCATCTGGGCTGAGACTCAG
SEQ ID NO: 188 D38 AGATCTTGTTCTTCTCGGGACC
SEQ ID NO: 189 D39 GAACCTCTTCTGTCGGTTGCGG
SEQ ID NO: 190 D40 TGGGTAGCGAAAGGTGTAGAAG
SEQ ID NO: 191 D41 GACTGGCTTTTCACCGTCGT
-30-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 192 D42 TACTTCTTCCGAGGCAGG
SEQ ID NO: 193 D43 CTCGCCTCGGATGGTGGAGACC
SEQ ID NO: 194 D44 CGGTGAGGGGTGCGGGGCCCAG
SEQ ID NO: 195 D45 GCACTCAGCCCATCAGATGGGT
SEQ ID NO: 196 D46 GTGTCTATCCCAGCCTGGGGGC
SEQ ID NO: 197 D47 ACCCAGTACTGAGCAC
SEQ ID NO: 198 D48 GCGCAGGAAGTAGGCATAG
SEQ ID NO: 199 D49 GCACGGGACTGTCTACACGCCG
SEQ ID NO: 200 D50 CGGTGAGGGGTGCGGGGCCCAG
SEQ ID NO: 201 D51 CTACCCAGCATTGGCCTC
SEQ ID NO: 202 D52 TGGGTAGCCGGGCTGCAGCTGG
SEQ ID NO: 203 D53 GTCGATCTCAGAGGGCACCCCT
SEQ ID NO: 204 D54 CTCCAGTCAGTGGCCCTGCGGG
SEQ ID NO: 205 D55 CCCCTCCTCTCGGCATGGAGGT
SEQ ID NO: 206 D56 CTCCAGTCAGTGGCCCTGCGGG
SEQ ID NO: 207 D57 GGGAACCTCACCAGGCCCAGCT
SEQ ID NO: 208 D58 GGTGGGCGTC
SEQ ID NO: 209 D59 TATAGGTGTTGAACGCCCCTGC
SEQ ID NO: 210 D60 TAGTCCCTGCCTCGGAAGAAGT
SEQ ID NO: 211 D61 GCACGGGACTGTCTACACGCCG
SEQ ID NO: 212 D62 GGTCTCATCATAGTCGAAGTGG
SEQ ID NO: 213 D63 GTCAAACTTCCAGTAGAGGCG
SEQ ID NO: 214 D64 CCTCACCTTGGAAGAACCAA
SEQ ID NO: 215 D65 GCCAGTGGCGAGAGGCCAATGC
SEQ ID NO: 216 D66 TCCACAGGGCTGGGCAGTCCCT
SEQ ID NO: 217 D67 CACGTGGCCAAATTCATGGGCT
SEQ ID NO: 218 D68 CCAGGGCTGGGGTCCTGGAGGT
SEQ ID NO: 219 D69 CAGGTGCCGGGCTACTGGGCAG
SEQ ID NO: 220 D70 AGCCCGCTTTGAAGAAAAAGAG
SEQ ID NO: 221 D71 CATCAGCATCCTGGAAGGCAGC
SEQ ID NO: 222 D72 GCGTCAAAGGAGGCCTCACAGG
SEQ ID NO: 223 D73 ATGGACCGGGAACCTCAC
-31-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 224 D74 CCAGACCAAGGCAGCATGGACC
SEQ ID NO: 225 D75 CCCAGCGCCCGCCAGAAAGCAC
SEQ ID NO: 226 D76 ATGGACCGGGAACCTCAC
SEQ ID NO: 227 D77 TGGGGAAGAAGGCATGGGCCAG
[0093] Exemplary forward primers for COL10A1 cDNA
TABLE 5
F primers
SEQ ID NO: Ref Code: Sequence
SEQ ID NO: 228 El CTTCTGCACTGCTCATCTGG
SEQ ID NO: 229 E2 AGCTTCAGAAAGCTGCCAAG
SEQ ID NO: 230 E3 GCCACAAATACCCTTTTTGC
SEQ ID NO: 231 E4 CCCAACACCAAGACACAGTTC
SEQ ID NO: 232 E5 TGCTGCCACAAATACCCTTT
SEQ ID NO: 233 E6 ACTCCCAGCACGCAG
SEQ ID NO: 234 E7 TACCCCACCCTACAAAATGC
SEQ ID NO: 235 E8 GGCAGAGGAAGCTTCAGAAAGC
SEQ ID NO: 236 E9 ACGATACCAAATGCCCACAG
SEQ ID NO: 237 E10 ACTCCCAGCACGCAG
SEQ ID NO: 238 El 1 GGCAGAGGAAGCTTCAGAAA
SEQ ID NO: 239 E12 TGCCAAGGCACCATCTCCAGGA
SEQ ID NO: 240 E13 GGCAGAGGAAGCTTCAGAAA
SEQ ID NO: 241 E14 CACCTTCTGCACTGCTCATCTG
SEQ ID NO: 242 EIS GGCAGAGGAAGCTTCAGAAAGC
SEQ ID NO: 243 E16 AGCTTCAGAAAGCTGCCAAG
SEQ ID NO: 244 E17 GGCAGAGGAAGCTTCAGAAA
SEQ ID NO: 245 E18 ACGATACCAAATGCCCACAG
SEQ ID NO: 246 E19 TGCCAAGGCACCATCTCCAGGA
SEQ ID NO: 247 E20 CACCTTCTGCACTGCTCATCTG
SEQ ID NO: 248 E21 CAAGGCACCATCTCCAGGAA
[0094] Exemplary reverse primers for COL10A1 cDNA
-32-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
TABLE 6
SEQ ID NO: R primers Sequence
Ref Code:
SEQ ID NO: 249 Fl CTTTACTCTTTATGGTGTAGGG
SEQ ID NO: 250 F2 CGTTGCTGCTCACTTTTCAG
SEQ ID NO: 251 F3 GTGGGCATTTGGTATCGTTC
SEQ ID NO: 252 F4 AGCAGCAAAAAGGGTATTTGTGG
SEQ ID NO: 253 F5 GGACTTCCGTAGCCTGGTTTTC
SEQ ID NO: 254 F6 GTACCTTGCTCTCCTCTTACTG
SEQ ID NO: 255 F7 AATGAAGAACTGTGTCTTGGTG
SEQ ID NO: 256 F8 GTGCCCTCGAGGTCCAGCAGGG
SEQ ID NO: 257 F9 ATGGTCCCGGTGGTCCTGGCAA
SEQ ID NO: 258 F10 ACACCTGGTTTCCCTACAGCTG
SEQ ID NO: 259 F 11 CAGATGGATTCTGCGTGCT
SEQ ID NO: 260 F12 TTTTATGCCTGTGGGCATTT
SEQ ID NO: 261 F13 GGCAGCATATTCTCAGATGGA
SEQ ID NO: 262 F14 GCTCTCCTCTTACTGCTATAC
SEQ ID NO: 263 F15 AAGTTCAAGGATACTAGCAGCA
SEQ ID NO: 264 F16 TTTTATGCCTGTGGGCATTT
SEQ ID NO: 265 F17 CTGGTGGTCCAGAAGGACCTGG
SEQ ID NO: 266 F18 GTGGGCATTTGGTATCGTTC
SEQ ID NO: 267 F19 AAGGGTATTTGTGGCAGCA
SEQ ID NO: 268 F20 CCCTGGCTCTCCTTGGAGTCCA
SEQ ID NO: 269 F21 AAAAGGGTATTTGTGGCAGCAT
SEQ ID NO: 270 F22 TTGGGTAGTGGGCCTTTTATGC
SEQ ID NO: 271 F23 GTGGGCATTTGGTATCGTTC
SEQ ID NO: 272 F24 TGGTTTTCCTGGGAGTCCTGGC
SEQ ID NO: 273 F25 GTAGCCTGGTTTTCCTGGTG
SEQ ID NO: 274 F26 AGCGTAAAACACTCCATGAACC
SEQ ID NO: 275 F27 TGGGCATTTGGTATCGTTCAG
SEQ ID NO: 276 F28 ATTCTCAGATGGATT
SEQ ID NO: 277 F29 GTACCTTGCTCTCCTCTTACTG
-33-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 278 F30 CCTGGTGGACCAGGAGTACCTT
SEQ ID NO: 279 F31 CTGTGGGCATTTGGTATCGTTC
[0095] Exemplary forward primers for TTC5 cDNA
TABLE 7
SEQ ID NO: F primers Sequence
Ref Code:
SEQ ID NO: 280 G1 GCTTCCCATTCACCACTAGC
SEQ ID NO: 281 G2 TCTCCACTCGAACACTGGA
SEQ ID NO: 282 G3 GGCCAGAACCCTAAGATCTCC
SEQ ID NO: 283 G4 CTGGAGCTCAAGCCACTGAG
SEQ ID NO: 284 G5 AGGCTTCCCATTCACCACTA
SEQ ID NO: 285 G6 GTAGCCATTCCTGAGCCC
SEQ ID NO: 286 G7 TTGTGCTGAATTCGGTG
SEQ ID NO: 287 G8 TCCACTCGAACACTGGAAAA
SEQ ID NO: 288 G9 TCTCCACTCGAACACTGGAAAA
SEQ ID NO: 289 G10 TCTCCACTCGAACACTGGAA
SEQ ID NO: 290 Gil GCTTCACCGAATTCAGCACA
SEQ ID NO: 291 G12 CACCGAATTCAGCACAAAGG
SEQ ID NO: 292 G13 CCCTTTACTTCTCTACTGGCC
SEQ ID NO: 293 G14 CTGAGTACGCTTCAGCCTG
[0096] Exemplary reverse primers for TTC5 cDNA
TABLE 8
R primers
SEQ ID NO: Ref Code: Sequence
SEQ ID NO: 294 H1 TTCAGATGAAGGTCAGGATTGC
SEQ ID NO: 295 H2 GAGGCTTCCCATTCACCACT
SEQ ID NO: 296 H3 TCAGATGAAGGTCAGGATTGCT
SEQ ID NO: 297 H4 TCTGTAGCCATTCCTGAGC
SEQ ID NO: 298 H5 TCAGATGAAGGTCAGGATTGCT
SEQ ID NO: 299 H6 GCTTTTCTGTCAACTTTCTCTGC
-34-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
SEQ ID NO: 300 H7 TGGACCTTGCTATGCAGTGA
SEQ ID NO: 301 H8 TTGGCCTGGTAGATTCAGATG
SEQ ID NO: 302 H9 CTGTAGCCATTCCTGAGCCC
SEQ ID NO: 303 H10 TTCACCGAATTCAGCACAAA
SEQ ID NO: 304 H11 GCTTCACCGAATTCAGCACA
SEQ ID NO: 305 H12 TCACCGAATTCAGCACAAAG
SEQ ID NO: 306 H13 TGTACACCATCACTGCATAGC
SEQ ID NO: 307 H14 CCCATTCACCACTAGCAGGAG
SEQ ID NO: 308 H15 TTGGCCTGGTAGATTCAGATG
SEQ ID NO: 309 H16 CACCGAATTCAGCACAAAGG
SEQ ID NO: 310 H17 CTGTAGCCATTCCTGAGC
[0097] Exemplary forward primers for C2orf44 cDNA
TABLE 9
SEQ ID NO: F primers Sequence
Ref Code:
SEQ ID NO: 311 Ii GAAGCACCGCTCTTTTTTCA
SEQ ID NO: 312 12 TTTACCATCACAGAGAAGCAC
SEQ ID NO: 313 13 CTGAGCCTTAGTTTACCA
SEQ ID NO: 314 14 GCCATAGAAGCTCCATTAGCAC
SEQ ID NO: 315 IS CCGTCTGCATAATGGGAAGA
SEQ ID NO: 316 16 ATGTAGCATTTCAATGAGAGAA
SEQ ID NO: 317 17 CAAACCGTCTGCATAATGGG
SEQ ID NO: 318 18 GCCAAAAGTCTGCTGAACT
SEQ ID NO: 319 19 AGAGAAGCACCGCTCTTTTT
SEQ ID NO: 320 110 ATTTCAATGAGAGAAAGGCCAAA
SEQ ID NO: 321 Iii TCTGGTAAATGATGTGAACATA
SEQ ID NO: 322 112 CAGAGAAGCACCGCTCTT
SEQ ID NO: 323 113 GGTGCTTCTCTGTGATGGTAA
SEQ ID NO: 324 114 ATGTAGCATTTCAATGAG
[0098] Exemplary reverse primers for C2orf44 cDNA
-35-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
TABLE 10
SEQ ID NO: R primers Sequence
Ref Code:
SEQ ID NO: 325 J1 CAGAGAAGCACCGCTCTTTTT
SEQ ID NO: 326 J2 TGTGGCTGTGAAGGTTAACG
SEQ ID NO: 327 J3 ACCGTCTGCATAATGGG
SEQ ID NO: 328 J4 ACCGTCTGCATAATGGGA
SEQ ID NO: 329 J5 TTTCCTAACCCAGCTCCATC
SEQ ID NO: 330 J6 GTGTATCCACTCTCTCAAGAT
SEQ ID NO: 331 J7 CAAGATCTTCCTTATGTTC
SEQ ID NO: 332 J8 ACCGTCTGCATAATGGGAAG
SEQ ID NO: 333 J9 CAAGATCTTCCTTATGTTCA
SEQ ID NO: 334 J10 TATGTTCACATCATTTA
SEQ ID NO: 335 J11 TATGTTCACATCATTTACC
SEQ ID NO: 336 J12 GCTGAACTGTACTGAGCCTTAG
SEQ ID NO: 337 J13 AACCGTCTGCATAATGGGAAGA
SEQ ID NO: 338 J14 GAAATCCTCTTCAGTGTATC
[0099] Exemplary forward primers for Chr3 gDNA
TABLE 11
SEQ ID NO: F primers Sequence
Ref Code:
SEQ ID NO: 339 K1 TTGGTGGTTTGTATGGGT
SEQ ID NO: 340 K2 GGCTTGCCTCCGAATTCTAT
SEQ ID NO: 341 K3 GGTGGTTTGTATGGGTCAA
SEQ ID NO: 342 K4 TTGGTGGTTTGTATGGGTCA
SEQ ID NO: 343 K5 GCTTGCCTCCGAATTCTATG
SEQ ID NO: 344 K6 TGGTACGTGCCTCAGAACAG
SEQ ID NO: 345 K7 AATCCTGCTCACCTTTCTGAG
SEQ ID NO: 346 K8 GCTTGCCTCCGAATTCTATG
SEQ ID NO: 347 K9 TAGTCCCAGGAGGTGGTACG
[00100] Exemplary reverse primers for chr3 gDNA
-36-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
TABLE 12
SEQ ID NO: R primers Sequence
Ref Code:
SEQ ID NO: 348 Li AACGTTTACTAGCCCCACCA
SEQ ID NO: 349 L2 AGGTGGTCTCTGGAGGGTCT
SEQ ID NO: 350 L3 CCTCTGCATAATCCACTGTCTG
SEQ ID NO: 351 L4 AGGTGGTCTCTGGAGGGTCT
SEQ ID NO: 352 L5 GGTGGTCTCTGGAGGGTC
SEQ ID NO: 353 L6 GCCACTTGGCTCCTAACAGA
SEQ ID NO: 354 L7 AGCCACTTGGCTCCTAAC
SEQ ID NO: 355 L8 CCACTTGGCTCCTAACAG
SEQ ID NO: 356 L9 AACGTTTACTAGCCCCACC
XVI. Exemplary Forward/Reverse primer combinations for RTqPCR measurement of
IBSP cDNA
[00101]
TABLE 13
Combination IBSP Primer Combinations
1 A3+B16
2 Al8+B33
3 A2+B45
4 Al6+B5
A24+B15
6 A21+B32
7 A9+B14
8 A24+B9
9 A2+B30
A2+B35
11 Al9+B22
12 A32+B25
13 Al7+B37
14 Al2+B40
-37-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
15 Al 1+B41
16 A37+B6
17 A39+B45
18 Al0+B7
19 A40+B23
20 A31+B18
21 A28+B18
22 A36+B29
23 A22+B8
24 A23+B4
25 A27+B11
26 A6+B38
27 A8+B34
28 A8+B28
29 A5+B42
30 A7+B13
31 A29+B39
32 A34+B39
33 Al5+B10
34 A33+B12
35 A30+B44
36 A30+B21
37 A30+B26
38 A30+B19
39 A30+B31
40 Al3+B20
41 A26+B43
42 A25+B1
43 A4+B2
44 A20+B24
45 Al +B27
46 A38+B17
-38-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
47 A38+B36
48 A38+B3
[00102] Exemplary Forward/Reverse primer combinations for RTqPCR/qPCR
measurement ofWP11 cDNA
TABLE 14
Combination MMP11 Primer Combinations
1 C19+D10
2 C57+D48
3 C14+D21
4 C37+D41
C27+D63
6 C36+D76
7 C37+D76
8 C27+D51
9 C53+D73
C5+D23
11 C52+D64
12 C9+D9
13 Cl+D2
14 C64+D6
C45+D33
16 C65+D42
17 C3+D3
18 C46+D75
19 C3O+D39
C62+D45
21 C26+D5
22 C31+D35
23 C49+D30
24 C49+D69
C49+D19
-39-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
26 C49+D55
27 C49+D58
28 C3+D77
29 C46+D78
30 C3O+D79
31 C62+D80
32 C26+D81
33 C31+D82
34 C49+D83
35 C15+D17
36 C56+D62
37 C17+D14
38 C61+D77
39 C41+D31
40 C6+D36
41 C35+D36
42 C7+D20
43 C8+D46
44 C2O+D68
45 C42+D18
46 C47+D59
47 C2+D37
48 C2+D40
49 C2+D4
50 C2+D13
51 C2+D67
52 C2+D22
53 C32+D1
54 C43+D15
55 C6O+D66
56 C22+D65
57 C28+D52
-40-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
58 Cl 1+D25
59 C59+D70
60 C4O+D43
61 C12+D72
62 C4+D12
63 C21+D12
64 C54+D7
65 C23+D29
66 C58+D54
67 C33+D49
68 C39+D34
69 C13+D60
70 C63+D38
71 C5O+D8
72 C25+D57
73 C34+D50
74 C34+D28
75 C34+D26
76 C48+D71
77 C1O+D53
78 C18+D56
79 C44+D61
80 C16+D24
81 C55+D16
82 C38+D32
83 C29+D74
84 C51+D27
85 C24+D44
86 C66+D44
87 C67+D44
[00103] Exemplary Forward/Reverse primer combinations for RTqPCR/qPCR
measurement of COL10A/ cDNA
-41-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
TABLE 15
Combination COL10A1 For/Rev Primer Combinations
1 E21+F27
2 E4+F25
3 E7+F2
4 El8+F29
E21+F4
6 E9+F6
7 E3+F23
8 E13+F 1 1
9 El6+F13
E5+F19
11 E2+F16
12 Ell+F18
13 El7+F12
14 El+F3
E2O+Fl
16 E8+F7
17 El9+F22
18 E1O+F31
19 El 0+F26
E1O+F15
21 El 0+F21
22 El 0+F28
23 El4+F24
24 El5+F10
El2+F9
26 E6+F20
27 E6+F5
28 E6+F17
29 E6+F8
E6+F30
-42-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
31 E6+F14
[00104] Exemplary Forward/Reverse Primer Combinations for RTqPCR/qPCR
measurement of TTC5 cDNA
TABLE 16
Combination TTC5 Primer Combinations
1 Gll+H2
2 G14+H13
3 G12+H14
4 G3+H1
G13+H6
6 G4+H3
7 G3+H5
8 G14+H11
9 G8+H4
G1O+H7
11 Gl+H12
12 G5+H10
13 G9+H17
14 G2+H9
G6+H15
16 G7+H8
[00105] Exemplary Forward/Reverse Primer Combinations for RTqPCR/qPCR
measurement of C2orf44 cDNA
TABLE 17
Combination C2orf44 Primer Combinations
1 17+J12
2 113+J2
3 15+J1
4 I4+J5
5 I9+J8
-43-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
6 I12+J4
7 I1O+J3
8 1+J7
9 114+J10
I6+J11
11 I8+J9
12 I2+J6
13 11+J13
14 13+J14
[00106] Exemplary Forward/Reverse Primer Combinations for
RTqPCR/qPCR/RTPCR of Chr3 gDNA
TABLE 18
Combination gDNA chr3 Primer Combinations
1 K7+L3
2 K5+L2
3 K9+Ll
4 K4+L6
5 Kl+L7
6 K2+L4
7 K3+L8
8 K8+L5
9 K6+L9
[00107] In some embodiments, the nucleic acids disclosed herein may be
used a
biomarker. For example, a portion of the cDNA sequence ofWP11,IBSP, or COL10A1
may be used as a biomarker to detect cancer.
[00108] In some embodiments, the sequence of an WP11 cDNA is according to:
[00109] ATAAGGGGCGGCGGCCCGGAGCGGCCCAGCAAGCCCAGCAGCCCC
GGGGCGGATGGCTCCGGCCGCCTGGCTCCGCAGCGCGGCCGCGCGCGCCCTCCT
GCCCCCGATGCTGCTGCTGCTGCTCCAGCCGCCGCCGCTGCTGGCCCGGGCTCTG
CCGCCGGACGCCCACCACCTCCATGCCGAGAGGAGGGGGCCACAGCCCTGGCAT
GCAGCCCTGCCCAGTAGCCCGGCACCTGCCCCTGCCACGCAGGAAGCCCCCCGG
-44-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
CCTGCCAGCAGCCTCAGGCCTCCCCGCTGTGGCGTGCCCGACCCATCTGATGGGC
TGAGTGCCCGCAACCGACAGAAGAGGTTCGTGCTTTCTGGCGGGCGCTGGGAGA
AGACGGACCTCACCTACAGGATCCTTCGGTTCCCATGGCAGTTGGTGCAGGAGCA
GGTGCGGCAGACGATGGCAGAGGCCCTAAAGGTATGGAGCGATGTGACGCCACT
CACCTTTACTGAGGTGCACGAGGGCCGTGCTGACATCATGATCGACTTCGCCAGG
TACTGGCATGGGGACGACCTGCCGTTTGATGGGCCTGGGGGCATCCTGGCCCATG
CCTTCTTCCCCAAGACTCACCGAGAAGGGGATGTCCACTTCGACTATGATGAGAC
CTGGACTATCGGGGATGACCAGGGCACAGACCTGCTGCAGGTGGCAGCCCATGA
ATTTGGCCACGTGCTGGGGCTGCAGCACACAACAGCAGCCAAGGCCCTGATGTC
CGCCTTCTACACCTTTCGCTACCCACTGAGTCTCAGCCCAGATGACTGCAGGGGC
GTTCAACACCTATATGGCCAGCCCTGGCCCACTGTCACCTCCAGGACCCCAGCCC
TGGGCCCCCAGGCTGGGATAGACACCAATGAGATTGCACCGCTGGAGCCAGACG
CCCCGCCAGATGCCTGTGAGGCCTCCTTTGACGCGGTCTCCACCATCCGAGGCGA
GCTCTTTTTCTTCAAAGCGGGCTTTGTGTGGCGCCTCCGTGGGGGCCAGCTGCAG
CCCGGCTACCCAGCATTGGCCTCTCGCCACTGGCAGGGACTGCCCAGCCCTGTGG
ACGCTGCCTTCGAGGATGCCCAGGGCCACATTTGGTTCTTCCAAGGTGCTCAGTA
CTGGGTGTACGACGGTGAAAAGCCAGTCCTGGGCCCCGCACCCCTCACCGAGCT
GGGCCTGGTGAGGTTCCCGGTCCATGCTGCCTTGGTCTGGGGTCCCGAGAAGAAC
AAGATCTACTTCTTCCGAGGCAGGGACTACTGGCGTTTCCACCCCAGCACCCGGC
GTGTAGACAGTCCCGTGCCCCGCAGGGCCACTGACTGGAGAGGGGTGCCCTCTG
AGATCGACGCTGCCTTCCAGGATGCTGATGGCTATGCCTACTTCCTGCGCGGCCG
CCTCTACTGGAAGTTTGACCCTGTGAAGGTGAAGGCTCTGGAAGGCTTCCCCCGT
CTCGTGGGTCCTGACTTCTTTGGCTGTGCCGAGCCTGCCAACACTTTCCTCTGACC
ATGGCTTGGATGCCCTCAGGGGTGCTGACCCCTGCCAGGCCACGAATATCAGGCT
AGAGACCCATGGCCATCTTTGTGGCTGTGGGCACCAGGCATGGGACTGAGCCCA
TGTCTCCTCAGGGGGATGGGGTGGGGTACAACCACCATGACAACTGCCGGGAGG
GCCACGCAGGTCGTGGTCACCTGCCAGCGACTGTCTCAGACTGGGCAGGGAGGC
TTTGGCATGACTTAAGAGGAAGGGCAGTCTTGGGCCCGCTATGCAGGTCCTGGCA
AACCTGGCTGCCCTGTCTCCATCCCTGTCCCTCAGGGTAGCACCATGGCAGGACT
GGGGGAACTGGAGTGTCCTTGCTGTATCCCTGTTGTGAGGTTCCTTCCAGGGGCT
GGCACTGAAGCAAGGGTGCTGGGGCCCCATGGCCTTCAGCCCTGGCTGAGCAAC
TGGGCTGTAGGGCAGGGCCACTTCCTGAGGTCAGGTCTTGGTAGGTGCCTGCATC
TGTCTGCCTTCTGGCTGACAATCCTGGAAATCTGTTCTCCAGAATCCAGGCCAAA
AAGTTCACAGTCAAATGGGGAGGGGTATTCTTCATGCAGGAGACCCCAGGCCCT
-45-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
GGAGGCTGCAACATACCTCAATCCTGTCCCAGGCCGGATCCTCCTGAAGCCCTTT
TCGCAGCACTGCTATCCTCCAAAGCCATTGTAAATGTGTGTACAGTGTGTATAAA
CCTTCTTCTTCTTTTTTTTTTTTTAAACTGAGGATTGTCATTAAACACAGTTGTTTT
CTAAAAAAAAAAAAAAAA (SEQ ID NO: 357)
[00110] In some embodiments, the sequence of an IBSP cDNA is according to:
[00111] GAGTGAGTGAGAGGGCAGAGGAAATACTCAATCTGTGCCACTCAC
TGCCTTGAGCCTGCTTCCTCACTCCAGGACTGCCAGAGGAAGCAATCACCAAAAT
GAAGACTGCTTTAATTTTGCTCAGCATTTTGGGAATGGCCTGTGCTTTCTCAATGA
AAAATTTGCATCGAAGAGTCAAAATAGAGGATTCTGAAGAAAATGGGGTCTTTA
AGTACAGGCCACGATATTATCTTTACAAGCATGCCTACTTTTATCCTCATTTAAAA
CGATTTCCAGTTCAGGGCAGTAGTGACTCATCCGAAGAAAATGGAGATGACAGT
TCAGAAGAGGAGGAGGAAGAAGAGGAGACTTCAAATGAAGGAGAAAACAATGA
AGAATCGAATGAAGATGAAGACTCTGAGGCTGAGAATACCACACTTTCTGCTAC
AACACTGGGCTATGGAGAGGACGCCACGCCTGGCACAGGGTATACAGGGTTAGC
TGCAATCCAGCTTCCCAAGAAGGCTGGGGATATAACAAATAAAGCTACAAAAGA
GAAGGAAAGTGATGAAGAAGAAGAGGAGGAAGAGGAAGGAAATGAAAACGAA
GAAAGCGAAGCAGAAGTGGATGAAAACGAACAAGGCATAAACGGCACCAGTAC
CAACAGCACAGAGGCAGAAAACGGCAACGGCAGCAGCGGAGGAGACAATGGAG
AAGAAGGGGAAGAAGAAAGTGTCACTGGAGCCAATGCAGAAGACACCACAGAG
ACCGGAAGGCAGGGCAAGGGCACCTCGAAGACAACAACCTCTCCAAATGGTGG
GTTTGAACCTACAACCCCACCACAAGTCTATAGAACCACTTCCCCACCTTTTGGG
AAAACCACCACCGTTGAATACGAGGGGGAGTACGAATACACGGGCGCCAATGAA
TACGACAATGGATATGAAATCTATGAAAGTGAGAACGGGGAACCTCGTGGGGAC
AATTACCGAGCCTATGAAGATGAGTACAGCTACTTTAAAGGACAAGGCTACGAT
GGCTATGATGGTCAGAATTACTACCACCACCAGTGAAGCTCCAGCCTGGGATGA
ATTCATCCATTCTGGCTTTGCATCCGGCTACCATTTTCGAAGTTCAACTCAGGAAG
GTGCAATATAACAAATGTGCATATTATAATGAGGAATGGTACTACCGTTCCAGAT
TTTCTGTAATTGCTTCTGCAAAGTAATAGGCTTCTTGTCCCTTTTTTTTCTGGCATG
TTATGGAATGATCATTGTAAATCAGGACCATTTATCAAGCAGTACACCAACTCAT
AAGATCAAATTTCATTGAATGGTTTGAGGTTGTAGCTCTATAAATAGTAGTTTTT
AACATGCCTGTAGTATTGCTAACTGCAAAAACATACTCTTTGTACAAGAAGTGCT
TCTAAGAATTTCATTGACATTAATGACACTGTATACAATAAATGTGTAGTTTCTTA
ATCGCACTACCTATGCAACACTGTGTATTAGGTTTATCATCCTCATGTATTTTTAT
-46-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
GTGACCTGTATGTATATTC TAATCTACGAGTTTTATCACAAATAAAAATGCAATC
CTTCAAATGTGTTATAATTAAAAAA (SEQ ID NO:358)
[00112] In some embodiments, the sequence of a COL10A1 cDNA is according
to:
[00113] CAC CUUCUGCACUGCUCAUCUGGGCAGAGGAAGCUUCAGAAAGC
UGC C AAGGCAC CAUCUC C AGGAACUC C C AGCAC GCAGAAUCCAUCUGAGAAUA
UGCUGC CAC AAAUAC CCUUUUUGCUGCUAGUAUCCUUGAACUUGGUUCAUGGA
GUGUUUUACGCUGAACGAUACCAAAUGCC CACAGGCAUAAAAGGCC CACUACC
CAAC AC C AAGACAC AGUUCUUCAUUC CCUACACCAUAAAGAGUAAAGGUAUAG
CAGUAAGAGGAGAGCAAGGUACUC CUGGUC C AC C AGGC C CUGCUGGAC CUC GA
GGGCAC C C AGGUC CUUCUGGAC CAC CAGGAAAAC C AGGCUAC GGAAGUC CUGG
ACUC CAAGGAGAGCCAGGGUUGC CAGGAC CAC CGGGAC CAUCAGCUGUAGGGA
AACCAGGUGUGC CAGGACUCC CAGGAAAACCAGGAGAGAGAGGACCAUAUGG
AC C AAAAGGAGAUGUUGGAC CAGCUGGCCUACCAGGACC C C GGGGC C C AC C AG
GAC C AC CUGGAAUC CCUGGACCGGCUGGAAUUUCUGUGCCAGGAAAACCUGGA
CAACAGGGAC C CAC AGGAGC CCCAGGAC CCAGGGGCUUUC CUGGAGAAAAGGG
UGCAC CAGGAGUCC CUGGUAUGAAUGGACAGAAAGGGGAAAUGGGAUAUGGU
GCUC CUGGUC GUCCAGGUGAGAGGGGUCUUCCAGGCCCUCAGGGUCCCACAGG
AC C AUCUGGC CCUCCUGGAGUGGGAAAAAGAGGUGAAAAUGGGGUUCCAGGA
CAGC CAGGCAUCAAAGGUGAUAGAGGUUUUC CGGGAGAAAUGGGAC CAAUUG
GC C CAC C AGGUC C CCAAGGCC CUC CUGGGGAACGAGGGCCAGAAGGCAUUGGA
AAGCCAGGAGCUGCUGGAGCC CCAGGC CAGC CAGGGAUUCCAGGAACAAAAGG
UCUC CCUGGGGCUC CAGGAAUAGCUGGGCCC CCAGGGCCUCCUGGCUUUGGGA
AACCAGGCUUGCCAGGCCUGAAGGGAGAAAGAGGAC CUGCUGGCCUUCCUGGG
GGUCCAGGUGC CAAAGGGGAACAAGGGCCAGCAGGUCUUCCUGGGAAGCCAGG
UCUGACUGGAC CCC CUGGGAAUAUGGGACCC CAAGGAC CAAAAGGCAUC CC GG
GUAGCCAUGGUCUCC CAGGC CCUAAAGGUGAGACAGGGC CAGCUGGGC CUGC A
GGAUAC CCUGGGGCUAAGGGUGAAAGGGGUUCC CCUGGGUCAGAUGGAAAAC
CAGGGUACCCAGGAAAACCAGGUCUCGAUGGUCCUAAGGGUAACC CAGGGUUA
CCAGGUCCAAAAGGUGAUCCUGGAGUUGGAGGACCUCCUGGUCUC CCAGGCC C
UGUGGGC CCAGCAGGAGCAAAGGGAAUGCC CGGACACAAUGGAGAGGCUGGC C
CAAGAGGUGCC CCUGGAAUAC CAGGUACUAGAGGC CCUAUUGGGC CAC C AGGC
AUUCCAGGAUUC CCUGGGUCUAAAGGGGAUC CAGGAAGUCC CGGUC CUC CUGG
CC CAGCUGGCAUAGCAACUAAGGGCCUCAAUGGACC CAC C GGGC C AC C AGGGC
CUC CAGGUC CAAGAGGCCACUCUGGAGAGCCUGGUCUUCCAGGGCC CCCUGGG
-47-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
C CUC CAGGC C C AC C AGGUCAAGCAGUC AUGC CUGAGGGUUUUAUAAAGGCAGG
CCAAAGGCC CAGUCUUUCUGGGACC CCUCUUGUUAGUGCCAACCAGGGGGUAA
CAGGAAUGCCUGUGUCUGCUUUUACUGUUAUUCUCUCCAAAGCUUAC CCAGCA
AUAGGAACUCC CAUAC CAUUUGAUAAAAUUUUGUAUAACAGGCAACAGCAUU
AUGACC CAAGGACUGGAAUCUUUACUUGUCAGAUAC CAGGAAUAUACUAUUU
UUCAUAC CAC GUGCAUGUGAAAGGGACUCAUGUUUGGGUAGGCCUGUAUAAG
AAUGGC AC C C CUGUAAUGUAC AC CUAUGAUGAAUACAC C AAAGGCUAC CUGGA
UCAGGCUUCAGGGAGUGC CAUCAUCGAUCUCACAGAAAAUGACCAGGUGUGGC
UCCAGCUUC CCAAUGCC GAGUCAAAUGGC CUAUACUC CUCUGAGUAUGUC C AC
UCCUCUUUCUCAGGAUUCCUAGUGGCUCCAAUGUGAGUACACACAGAGCUAAU
CUAAAUCUUGUGCUAGAAAAAGCAUUCUCUAACUCUACCC CAC CCUACAAAAU
GCAUAUGGAGGUAGGCUGAAAAGAAUGUAAUUUUUAUUUUCUGAAAUACAGA
UUUGAGCUAUCAGACCAACAAAC CUUC CCC CUGAAAAGUGAGCAGCAAC GUAA
AAACGUAUGUGAAGCCUCUCUUGAAUUUCUAGUUAGCAAUCUUAAGGCUCUU
UAAGGUUUUCUCCAAUAUUAAAAAAUAUCACCAAAGAAGUCCUGCUAUGUUA
AAAACAAACAACAAAAAACAAACAACAAAAAAAAAAUUAAAAAAAAAAACAG
AAAUAGAGCUCUAAGUUAUGUGAAAUUUGAUUUGAGAAACUCGGCAUUUCCU
UUUUAAAAAAGCCUGUUUCUAACUAUGAAUAUGAGAACUUCUAGGAAACAUC
CAGGAGGUAUCAUAUAACUUUGUAGAACUUAAAUACUUGAAUAUUCAAAUUU
AAAAGACACUGUAUCCC CUAAAAUAUUUCUGAUGGUGC ACUACUCUGAGGC CU
GUAUGGC CCCUUUCAUCAAUAUCUAUUCAAAUAUACAGGUGCAUAUAUACUU
GUUAAAGCUCUUAUAUAAAAAAGCCCCAAAAUAUUGAAGUUCAUCUGAAAUG
CAAGGUGCUUUCAUCAAUGAACCUUUUCAAACUUUUCUAUGAUUGCAGAGAA
GCUUUUUAUAUAC CCAGCAUAACUUGGAAACAGGUAUCUGAC CUAUUCUUAU
UUAGUUAACACAAGUGUGAUUAAUUUGAUUUCUUUAAUUCCUUAUUGAAUCU
UAUGUGAUAUGAUUUUCUGGAUUUACAGAACAUUAGCACAUGUACCUUGUGC
CUCCCAUUCAAGUGAAGUUAUAAUUUACACUGAGGGUUUCAAAAUUCGACUA
GAAGUGGAGAUAUAUUAUUUAUUUAUGCACUGUACUGUAUUUUUAUAUUGCU
GUUUAAAACUUUUAAGCUGUGCCUCACUUAUUAAAGCACAAAAUGUUUUACC
UACUC CUUAUUUACGAC GCAAUAAAAUAACAUCAAUAGAUUUUUAGGCUGAA
UUAAUUUGAAAGCAGCAAUUUGCUGUUCUCAACCAUUCUUUCAAGGCUUUUC
AUUGUUCAAAGUUAAUAAAAAAGUAGGACAAUAAAGUGAAAAAAAAAAAAAA
AAAA (SEQ ID NO:359)
XVII. Computer Systems
-48-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00114] While various embodiments of the invention have been shown and
described
herein, it will be obvious to those skilled in the art that such embodiments
are provided by
way of example only. Numerous variations, changes, and substitutions may occur
to those
skilled in the art without departing from the invention. It should be
understood that various
alternatives to the embodiments of the invention described herein may be
employed.
Computer systems
[00115] The present disclosure provides computer systems that are
programmed to
implement methods of the disclosure. FIG. 13 shows a computer system 1301 that
is
programmed or otherwise configured to identify biomarkers for a cancer, such
as a breast
cancer. The computer system 1301 can regulate various aspects of the analysis
of the present
disclosure, such as, for example, it can analyze a cohort of biomarkers from a
population of
subjects afflicted with a cancer; it can identify a first subset from said
cohort of said
biomarkers that has at least a 3-fold higher expression level in said cancer
as compared to
tissue samples that do not contain cancer, such a healthy control biomarker;
it can identify a
second subset from said first subset of said biomarkers that have a false
discovery rate of less
than a 10-6 it can use at least one biomarker from said second subset of said
biomarkers as
input for a machine learning algorithm such as correlation feature selection
(CFS); and it can
further output one or more biomarkers that identify said cancer. The computer
system 1301
can be an electronic device of a user or a computer system that is remotely
located with
respect to the electronic device. The electronic device can be a mobile
electronic device.
[00116] The computer system 1301 includes a central processing unit (CPU, also
"processor" and "computer processor" herein) 1305, which can be a single core
or multi core
processor, or a plurality of processors for parallel processing. The computer
system 1301
also includes memory or memory location 1310 (e.g., random-access memory, read-
only
memory, flash memory), electronic storage unit 1315 (e.g., hard disk),
communication
interface 1320 (e.g., network adapter) for communicating with one or more
other systems,
and peripheral devices 1325, such as cache, other memory, data storage and/or
electronic
display adapters. The memory 1310, storage unit 1315, interface 1320 and
peripheral devices
1325 are in communication with the CPU 1305 through a communication bus (solid
lines),
such as a motherboard. The storage unit 1315 can be a data storage unit (or
data repository)
for storing data. The computer system 1301 can be operatively coupled to a
computer
network ("network") 1330 with the aid of the communication interface 1320. The
network
1330 can be the Internet, an interne and/or extranet, or an intranet and/or
extranet that is in
communication with the Internet. The network 1330 in some cases is a
telecommunication
-49-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
and/or data network. The network 1330 can include one or more computer
servers, which
can enable distributed computing, such as cloud computing. The network 1330,
in some
cases with the aid of the computer system 1301, can implement a peer-to-peer
network,
which may enable devices coupled to the computer system 1301 to behave as a
client or a
server. The system can train a number of classifiers that identify breast
cancer.
[00117] The CPU 1305 can execute a sequence of machine-readable instructions,
which
can be embodied in a program or software. The instructions may be stored in a
memory
location, such as the memory 1310. The instructions can be directed to the CPU
1305, which
can subsequently program or otherwise configure the CPU 1305 to implement
methods of the
present disclosure. Examples of operations performed by the CPU 1305 can
include fetch,
decode, execute, and writeback.
[00118] The CPU 1305 can be part of a circuit, such as an integrated circuit.
One or more
other components of the system 1301 can be included in the circuit. In some
cases, the
circuit is an application specific integrated circuit (ASIC).
[00119] The storage unit 1315 can store files, such as drivers, libraries
and saved
programs. The storage unit 1315 can store user data, e.g., user preferences
and user
programs. The computer system 1301 in some cases can include one or more
additional data
storage units that are external to the computer system 1301, such as located
on a remote
server that is in communication with the computer system 1301 through an
intranet or the
Internet.
[00120] The computer system 1301 can communicate with one or more remote
computer
systems through the network 1330. For instance, the computer system 1301 can
communicate with a remote computer system of a user (e.g., it can access
electronic data
from the TCGA project). Examples of remote computer systems include personal
computers
(e.g., portable PC), slate or tablet PC's (e.g., Apple iPad, Samsung Galaxy
Tab),
telephones, Smart phones (e.g., Apple iPhone, Android-enabled device,
Blackberry ), or
personal digital assistants. The user can access the computer system 1301 via
the network
1330.
[00121] Methods as described herein can be implemented by way of machine
(e.g.,
computer processor) executable code stored on an electronic storage location
of the computer
system 1301, such as, for example, on the memory 1310 or electronic storage
unit 1315. The
machine executable or machine readable code can be provided in the form of
software.
During use, the code can be executed by the processor 1305. In some cases, the
code can be
retrieved from the storage unit 1315 and stored on the memory 1310 for ready
access by the
-50-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
processor 1305. In some situations, the electronic storage unit 1315 can be
precluded, and
machine-executable instructions are stored on memory 1310.
[00122] The code can be pre-compiled and configured for use with a machine
having a
processer adapted to execute the code, or can be compiled during runtime. The
code can be
supplied in a programming language that can be selected to enable the code to
execute in a
pre-compiled or as-compiled fashion.
[00123] Aspects of the systems and methods provided herein, such as the
computer system
1301, can be embodied in programming. Various aspects of the technology may be
thought
of as "products" or "articles of manufacture" typically in the form of machine
(or processor)
executable code and/or associated data that is carried on or embodied in a
type of machine
readable medium. Machine-executable code can be stored on an electronic
storage unit, such
as memory (e.g., read-only memory, random-access memory, flash memory) or a
hard disk.
"Storage" type media can include any or all of the tangible memory of the
computers,
processors or the like, or associated modules thereof, such as various
semiconductor
memories, tape drives, disk drives and the like, which may provide non-
transitory storage at
any time for the software programming. All or portions of the software may at
times be
communicated through the Internet or various other telecommunication networks.
Such
communications, for example, may enable loading of the software from one
computer or
processor into another, for example, from a management server or host computer
into the
computer platform of an application server. Thus, another type of media that
may bear the
software elements includes optical, electrical and electromagnetic waves, such
as used across
physical interfaces between local devices, through wired and optical landline
networks and
over various air-links. The physical elements that carry such waves, such as
wired or
wireless links, optical links or the like, also may be considered as media
bearing the
software. As used herein, unless restricted to non-transitory, tangible
"storage" media, terms
such as computer or machine "readable medium" refer to any medium that
participates in
providing instructions to a processor for execution.
[00124] Hence, a machine-readable medium, such as computer-executable code,
may take
many forms, including but not limited to, a tangible storage medium, a carrier
wave medium
or physical transmission medium. Non-volatile storage media include, for
example, optical
or magnetic disks, such as any of the storage devices in any computer(s) or
the like, such as
may be used to implement the databases, etc. shown in the drawings. Volatile
storage media
include dynamic memory, such as main memory of such a computer platform.
Tangible
transmission media include coaxial cables; copper wire and fiber optics,
including the wires
-51-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
that comprise a bus within a computer system. Carrier-wave transmission media
may take
the form of electric or electromagnetic signals, or acoustic or light waves
such as those
generated during radio frequency (RF) and infrared (IR) data communications.
Common
forms of computer-readable media therefore include for example: a floppy disk,
a flexible
disk, hard disk, magnetic tape, any other magnetic medium, a CD-ROM, DVD or
DVD-
ROM, any other optical medium, punch cards paper tape, any other physical
storage medium
with patterns of holes, a RAM, a ROM, a PROM and EPROM, a FLASH-EPROM, any
other
memory chip or cartridge, a carrier wave transporting data or instructions,
cables or links
transporting such a carrier wave, or any other medium from which a computer
may read
programming code and/or data. Many of these forms of computer readable media
may be
involved in carrying one or more sequences of one or more instructions to a
processor for
execution.
[00125] The computer system 1301 can include or be in communication with
an
electronic display 1335 that comprises a user interface (UI) 1340 for
providing, for example,
an output listing one or more biomarkers that identify a cancer, such as
breast cancer.
Examples of UI's include, without limitation, a graphical user interface (GUI)
and web-based
user interface.
[00126] Methods and systems of the present disclosure can be implemented by
way of one
or more algorithms. An algorithm can be implemented by way of software upon
execution
by the central processing unit 1305.
XIII. Kits
[00127] In one aspect, the invention provides kits comprising any of the
primers and
reagents for detecting the 3-gene panel of biomarkers described in this
application. The kits
may comprise at least one primer sequence that has at least 90% identity to
any one of SEQ
ID NO: 1- SEQ ID NO: 356, and a buffer solution/system. In some embodiments,
the kit
comprises at least one forward primer that has at least 90% identity to any
one of SEQ ID
NO:1-40, SEQ ID NO:56-150, or SEQ ID NO:228-248 and at least one reverse
primer that
has at least 90% identity to any one of SEQ ID NO:41-85, SEQ ID NO: 151-227,
or SEQ ID
NO:249-279. In some embodiments, the kit comprises at least one forward
reference primer
that has at least 90% identity to any one of SEQ ID NO:280-293 or SEQ ID
NO:311-324 and
at least one reverse reference primer that has at least 90% identity to any
one of SEQ ID NO:
294-310 or SEQ ID NO: 325-338. In some embodiments, the kit comprises at least
one
forward positive control primer that has at least 90% identity to any one of
SEQ ID NO:339-
347 and at least one reverse positive control primer that has at least 90%
identity to any one
-52-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
of SEQ ID NO: 348-356. In some embodiments, the kit comprises at least one
forward and
reverse primer sequence for each of IBSP, MMP11, and COL10A that has at least
90%
identity to any of the primer combinations in Table 13, Table 14, and Table
15. In some
embodiments, the kit comprises at least one forward and reverse primer
sequence for each of
TTC5 and C2orf44 that has at least 90% identity to any of the primer
combinations in Table
16 and Table 17. In some embodiments, the kit comprises at least one forward
and reverse
primer sequence for chr3 gDNA that has at least 90% identity to any of the
primer
combinations in Table 18.
[00128] In some instances, the kits further comprise a DNA-intercalating
dye or a
fluorescent probe, such as a TaqMan compatible probe. A TaqMan compatible
probe may
comprise a short oligonucleotide sequence designed to hybridize to the desired
gene, in
combination with a 5'-fluorophore and a 3'-quencher attached to either end of
the
oligonucleotide. In some instances the kit also comprises a negative control
sample, a
positive control sample, or a using a synthetic nucleotide control.
[00129] The kits can further comprise a set of reagents for a polymerase
chain reaction.
Such reagents for a polymerase chain reaction include a suitable thermostable
DNA
polymerase (e.g. Taq polymerase, which may be a hot-start polymerase to
improve fidelity)
solution, a solution of 4 dNTPs (e.g. dATP, dTTP, dGTP, dCTP), a buffer
solution, DNAse-
free water, and/or solutions of PCR stabilizers/enhancers. Buffers are
prepared at the pH
optimum for the enzyme and may additionally comprise salts such as KC1, NaCl,
and/or
MgCl2, reducing agents such as DTT or B-me, detergents such as triton-x or
tween-20,
and/or glycerol as useful for function of the enzyme. Stabilizers/additives
may include agents
such as DMSO, betaine monohydrate, formamide, MgCl2, glycerol, BSA, tween-20,
Tetramethyl ammonium chloride, and/or 7-deaza-2'-deoxyguanosine. For qPCR
applications, a polymerase chain reaction kit may include suitable fluorescent
DNA-binding
dyes such as SYBR Green, ethidium bromide, or EvaGreen.
[00130] In some examples, the set of reagents can be for a reverse-
transcriptase
polymerase chain reaction. Reagents for a reverse-transcriptase polymerase
chain reaction
include a suitable reverse transcriptase (such as Maloney murine leukemia
virus, M-MLV,
reverse transcriptase) solution, solution of 4 dNTPs (e.g. dATP, dTTP, dGTP,
dCTP), a
buffer solution, an RNAse inhibitor solution, and/or RNAse-free water. In the
case of
solutions for reverse-transcriptase polymerase chain reaction, all the
reagents (e.g. dNTPs,
water, buffer) are certified RNAse free to prevent template degradation.
-53-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00131] The kit can further comprise written instructions for a use
thereof. Such
instructions may include instructions for isolating/preparing the sample,
operating
instrumentation (e.g. qPCR instrumentation), and/or data interpretation
[00132] The kit can further comprise components for touch-prep. Such
components
include poly-D-lysine coated glass slides, an RNA isolation kit, and/or spin
columns (suitable
for isolation/purification of RNA) and collection tubes. A minimal RNA
isolation kit may
comprise a solution RNAse-free sample disruption buffer, solutions of RNA
isolation
reagents (e.g. Trizol or phenol/chloroform or phenol/chloroform/isoamyl
alcohol mixtures),
RNAse-free DNase, and/or a solution of an RNAse inhibitor.
[00133] The kit can further comprise components for tumor/tissue
dissociation. Such
components include a) solutions of enzymes for extracellular matrix (ECM) or
other protein
degradation such as collagenase, trypsin, elastase, hyaluronidase, and/or
papain; b) solutions
for lysis of red blood cells from tissue (e.g. a hypotonic lysis buffer); c) a
tissue dissociator
(e.g. Miltenyi gentleMACS Octo Tissue Dissociator); d) a stabilization buffer
(e.g. containing
protease, DNAse, and/or RNAse inhibitors); e) a lysis buffer (a buffered
solution, optionally
hypotonic, containing ionic or nonionic detergents such as Triton X-100, tween-
20, beta-octyl
glucoside, and/or SDS).
[00134] In some embodiments, the kit is a kit for the detection of
positive surgical
margins. Such a kit includes components such as instructions, primers or
primer
combinations outlined above (e.g. forward and reverse primers for each target
gene, forward
and reverse primers for a reference gene, and forward and reverse primers for
a gDNA
control gene), touch prep components as described above, reagents for
polymerase chain
reaction, and reagents for reverse-polymerase chain reaction. In some
embodiments such a
kit consists of as instructions, touch prep components as described above,
reagents for
polymerase chain reaction, and reagents for reverse-polymerase chain reaction.
[00135] In some embodiments, the kit is a kit for detection of molecular
complete
response (mCR). Such a kit includes components such as instructions, primers
or primer
combinations outlined above (e.g. forward and reverse primers for each target
gene, forward
and reverse primers for a reference gene, and forward and reverse primers for
a gDNA
control gene), components for tumor/tissue dissociation as described above, an
RNA isolation
kit, and/or spin columns suitable for isolation/purification of RNA.
[00136] While preferred embodiments of the present invention have been shown
and
described herein, it will be obvious to those skilled in the art that such
embodiments are
provided by way of example only. It is not intended that the invention be
limited by the
-54-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
specific examples provided within the specification. While the invention has
been described
with reference to the aforementioned specification, the descriptions and
illustrations of the
embodiments herein are not meant to be construed in a limiting sense. Numerous
variations,
changes, and substitutions will now occur to those skilled in the art without
departing from
the invention. Furthermore, it shall be understood that all aspects of the
invention are not
limited to the specific depictions, configurations or relative proportions set
forth herein which
depend upon a variety of conditions and variables. It should be understood
that various
alternatives to the embodiments of the invention described herein may be
employed in
practicing the invention. It is therefore contemplated that the invention
shall also cover any
such alternatives, modifications, variations or equivalents. It is intended
that the following
claims define the scope of the invention and that methods and structures
within the scope of
these claims and their equivalents be covered thereby.
EXAMPLES
[00137] Example 1. A 3-gene test for residual disease/pathologic complete
response (pCR)
[00138] General Methods
[00139] Data from the TCGA project was analyzed to inform the
identification of a
cohort of biomarkers from a population of subjects afflicted with a cancer. In
addition,
lumpectomies were performed on small, early-stage tumors. cDNA from these
samples was
prepared from clinical samples and q-PCR performed according to standard
protocols. A
variety of standard protocols and kits for cDNA preparation/q-PCR are known to
those of
skill in the art. Exemplary protocols include, those from ThermoFisher (e.g.
Manual for
Power SYBR Green RNA-to-CTTml-Step Kit Part Number 4391003 Rev. D; Manual for
EXPRESS One-Step; SuperScript qRT-PCR Kits, Rev. Date: 28 June 2010 Manual
part no.
A10327), BioRad (e.g. Manual for iTaqTrn Universal SYBR Green One-Step Kit
10032048
Rev B), NEB (e.g. Luna Universal One-Step RT-qPCR Kit Protocol (E3005)),
Qiagen (e.g.
QuantiFast SYBR Green RT-PCR Handbook ver 07/2011, US 5,994,056, and US
6,171,785),
Roche (e.g. Transcriptor One-Step RT-PCR Kit; FastStart Universal SYBR Green
Master
(Rox), each of which are specifically incorporated by reference herein.
[00140] Inclusion/Exclusion Criteria.
[00141] Since genomic signatures may evolve during metastasis, AJCC TNM
staging (<T3)
was used to restrict samples to female patients who would be eligible for a
lumpectomy (see
TABLE 19). One sample was excluded for failing quality control, and 8 samples
were
-55-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
excluded for having clinical data. Inclusion/exclusion criteria preserved the
racial and ethnic
representation of the U.S. population, except for the only available American
Indian/ Alaskan
Native (APAN) subject, who did not satisfy our inclusion criteria. 1,014 early-
stage tumors
were divided (T1-T2) into a training set with 939 samples and an independent
test with 75
samples. The early-stage tumor sets (Cross-validation Set and Independent Set
#1) also
included healthy samples from patient with late-stage tumors. This test was
designed to detect
early-stage tumors, but the analysis also included 175 late-stage tumors (T3-
T4) as a second
independent test set. In sum, novel biomarkers for cancer were identified when
a computer
system was used to analyze a cohort of biomarkers from the aforementioned
population of
subjects afflicted with a cancer. The method identified a first subset of
biomarkers that had at
least a 3-fold higher expression level in said cancer as compared to a healthy
control
biomarker; and a second subset from said first subset of said biomarkers that
provided a false
discovery rate for said cancer that was less than 0.000001. The markers
identified were used
to train a machine learning algorithm and were experimentally validated.
1001421 The method identified a 3-gene set of markers from a plurality of
biomarkers from 939
RNA Seq samples. The method was tested on two independent RNA Seq test sets
(TABLE 19A
and TABLE 19B). The selected 3-gene set of markers correctly classified 96.2%
of 939 samples
in the Cross-validation Set (early stage, AJCC 1NN4 Tumor Stages T1-12). Since
these results
were unexpected, we tested whether the performance estimates from cross-
validation were
inflated by potential modeling errors (e.g. overfitting). First, a suite of
negative controls did not
detect any modeling errors in the cross validation. Second, the classifier was
trained on all 939
samples in the cross-validation set, and tested on a hold-out set of 75
samples. The 3-gene
Random Forest test correctly classified 97.3% of 75 early-stage samples in one
independent test
set, and correctly classified 94.3% of 175 late-stage samples in a second
independent test set.
Performance was not significantly affected by race, ethnicity, tumor stage, or
ER/PR/Her2 status.
By definition, overfit models have higher performance from resampling
estimates like cross
validation than on independent validation sets. In this case, cross validation
estimates and
performance on the independent validation set were within the 95% confidence
intervals. These
results therefore firmly exclude overfitting.
TABLE 19A
Cross validation Early-Stage Independent Test Set
#1:
Available Sample Correctly P-value Sample Correctly P-value
samples Size classified (Fisher's) Size
classified (Fisher's)
-56-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
TABLE 19A
Cross validation Early-Stage Independent Test Set
#1:
Available Sample Correctly P-value Sample Correctly P-value
samples Size classified (Fisher's) Size classified
(Fisher's)
Samnle Size 1210 939 903 75 73
Gender
Female 1197 939 903 75 73
Male 13
Sample Type
Tumor 1089 851 820 50 49
Healthy 114 88 83 25 24
Metastatic 7
Race 0.6605
0.4843
AI or AN1 1
Asian 62 48 47 4 4
Black or AA' 187 147 140 11 10
White 861 672 648 54 53
NA/NE2 99 72 68 6 6
Ethnicity 1.0000
1.0000
Hispanic or Latino 39 30 29 3 3
(96.7%) (100.0%)
Not Hispanic or 201 154 148 18 18
Latino (96.1%) (100.0%)
NA/NE/Unknown2 970 755 726 54 52
(96.2%) (963%)
Tumor Pathology 0.3293
1.0000
Ti 281 263 251 17 17
T2 632 588 569 33 32
T3 % T4 172
TX (Unknown) 4
Estrogen 0.6678
1.0000
Positive 801 621 597 34 33
Negative 238 190 183 14 14
Indeterminate 2 1 1
Not Evaluated 48 39 39 2 2
Progesterone 0.1206
1.0000
Positive 694 544 522 28 27
Negative 342 263 255 20 20
Indeterminate 4 4 3 (75.0%)
Not Evaluated 49 40 40 2 2
Her2/Neu Status 0.9653
0.1400
Positive 162 127 123 9 9
Negative 560 438 420 27 27
Equivocal 178 150 145 5 4(80.0%)
-57-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
TABLE 19A
Cross validation Early-Stage Independent Test Set
#1:
Available Sample Correctly P-value Sample Correctly P-value
samples Size classified (Fisher's) Size classified
(Fisher's)
Not Evaluated 12 7 7 2 2
NA/NE2 177 129 125 7 7
Triple Negative 0.3751
0.2580
Triple Negative 116 94 89 6 6
Not Triple 973 757 731 44 43
1
American Indian (AI), Alaska Native (AN), African American (AA)
2
Not Available (NA), Not Evaluated (NE)
TABLE 19B
Cross validation Early-Stage Independent Test Set
#2:
(T1-T2) Late-Stage (T3-T4)
Available Sample Correctly P-value Sample Correctly P-value
samples Size classified (Fisher's) Size classified
(Fisher's)
Samule Size 1210 939 903 175 165
Gender
Female 1197 939 903 175 165
Male 13
Sample Type
Tumor 1089 851 820 175 165
Healthy 114 88 83
Metastatic 7
Race 0.6605
0.6840
AI or AN1 1
Asian 62 48 47 10 9 (90.0%)
Black or AA1 187 147 140 26 24
White 861 672 648 118 112
NA/NE2 99 72 68 21 20
Ethnicity 1.0000
Hispanic or Latino 39 30 29
(96.7%)
Not Hispanic or 201 154 148 175 165
Latino (96.1%) (94.3%)
NA/NE/Unknown2 970 755 726
(96.2%)
Tumor Pathology 0.3293
1.0000
Ti 281 263 251
T2 632 588 569
T3 % T4 172 171 161
TX (Unknown) 4 4 4
-58-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
TABLE 19B
Cross validation Early-Stage Independent Test Set
#2:
(T1-T2) Late-Stage (T3-T4)
Available Sample Correctly P-value Sample Correctly P-value
samples Size classified (Fisher's) Size classified
(Fisher's)
Estrogen 0.6678
0.1347
Positive 801 621 597 134 129
Negative 238 190 183 34 30
Indeterminate 2 1 1 1 1
Not Evaluated 48 39 39 6 5 (83.3%)
Progesterone 0.1206
0.1052
Positive 694 544 522 112 108
Negative 342 263 255 57 52
Indeterminate 4 4 3 (75.0%)
Not Evaluated 49 40 40 6 5 (83.3%)
Her2/Neu Status 0.9653
0.1186
Positive 162 127 123 23 23
Negative 560 438 420 90 86
Equivocal 178 150 145 21 20
Not Evaluated 12 7 7 3 2 (66.7%)
NA/NE2 177 129 125 38 34
Triple Negative 0.3751
0.2288
Triple Negative 116 94 89 16 14
Not Triple 973 757 731 159 151
[00143] Biomarker Discovery.
[00144] A subset of biomarkers that had a large mean difference between
groups, with
two clearly separated distributions, was first identified using a computer
system. In addition,
to detect tumor cells in a population of healthy cells, additional biomarkers
that had a higher
level of expression in tumors than healthy samples were selected. To identify
such
biomarkers, genes with a 10g2(fold-change) = +3 and genes with a False
Discovery Rate
(adjusted p-value) of p < 10-6 were identified. The method identified a first
subset of
biomarkers that were overexpressed in tumors (FIG.2A). Subsequently,
Correlation-based
Feature Selection (CF S) was applied in the first subset from the broader
cohort of biomarkers
to identify genes with at least a 3-fold higher expression level in the
selected cancer as
compared to a healthy control biomarker to select the top genes that
contributed the most
unique information.
[00145] In addition, expression of many disease-associated genes is highly
correlated.
Thus, the identification of genes that contribute the most unique information
informed the
selection of relevant markers. For instance, estrogen signaling is the classic
example in breast
-59-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
tumors, where multiple ER-responsive genes can be the strongest biomarkers in
a given
tumor. But these genes would only help identify ER+ tumors, and would miss
every tumor
that is not ER+. Selection of highly expressed genes with CFS, or another
suitable method,
allows the identification of a subset of genes that not only contribute
information, but that
contribute the most unique information relative to the other selected genes.
Using CFS, we
selected panels of 200, 100, 20, 10, 5, 4, 3, 2, and 1 gene panels. Six
machine learning
algorithms were tested and performed similarly in identifying gene panels. The
6 algorithms
were the support vector algorithm SMO, Naive Bayes, J48 Decision Tree, Lazy-
IBk, the
Multilayer Perceptron neural network, and Random Forest. The Random Forest
ensemble
machine learning method was used in the remaining of the experiments. There
are at least 9
published classifiers for breast cancer that use gene expression, including
OncoTypeDX
and PAM50. Principal Component Analysis (PCA) (FIG.3) suggests a rationale for
why the
disclosed 3-gene set of markers had higher performance than existing breast
cancer disease
classifiers. Existing classifiers attempt to identify subgroups among the
cluster of tumor
samples. This leads to the strongest performance being focused on
distinguishing the two
most prominent groups, such as tumor and healthy (FIG. 3).
[00146] The 3-gene test had an accuracy of 94-97% when analyzed on 3 sets
of RNA
Seq. samples (TABLE 20). PAM50 could not be used for margin analysis because
it requires
samples with >50% tumor.
[00147] TABLE 20: Comparison of 3-gene test with PAM50 on RNA Seq samples
TABLE 20
Accuracy Sensitivity Specificity Sample Size
3-Gene Test
Cross-validated Set (Early-stage tumors) 96.2% 96.4% 94.4%
939
Independent Test Set #1 (Early-stage tumors) 97.3% 98.0% 96.0% 75
Independent Test Set #2 (Late-stage tumors) 94.3% 94.3% NA 175
Microscopy (Early-stage tumors) 64.9% 50.9% 69.5% 1,201
PAM50 97.3% 98.8% 83.0% 995
[00148] Example 2. Test performance and validation
[00149] 1,014 RNA Seq samples were divided from early stage breast tumors
and
adjacent healthy tissue into a Cross-validation set of 939 samples and an
Independent Test
Set of 75 samples (TABLE 19). The 3-gene test correctly classified 96.2% of
the samples in
the Cross-validation set (TABLE 20). The Area Under the Receiver Operator
Characteristic
Curve (AUC ROC) was 0.990 (95% CI: 0.997-1.000) (FIG.4). The 3-gene test has
equivalent
-60-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
performance on the early-stage Independent Test Set: 97.3% Accuracy, 0.998 AUC
ROC
(95% Cl: 0.992-1.000), 98.0% Sensitivity, 96.0% Specificity, 98.0% Positive
Predictive
Value, and 96.0% Negative Predictive Value.
[00150] To validate the test in other tumor samples, T3 and T4 (later
stage) samples,
were also tested. 175 late-stage primary tumors were used as a second
independent test set. In
this analysis, the classifier correctly detects 94.3% of late-stage tumors. In
all tests sets, the 3-
gene test performed equally well regardless of racial groups or clinical
subtypes (ER , PR,
Her2 ) (see TABLE 19).
[00151] 10-Fold Cross validation (CV).
[00152] Our classifier combines a 3-gene set of markers including WP 11 ,
IBSP , and
COL10A 1, using the Random Forest machine learning algorithm. We used ten-fold
CV to
estimate performance with RNA Seq data. For ten-fold CV, each of the 939
samples was
used once (and only once) in 1 of 10 independent validation sets. The first
iteration used
subsets 2-10 (S2.10) as a training set (Ti), while subset 1 (Si) was withheld
as the validation
set (VI). This was repeated on a total of 10 independent validation sets.
[00153] Negative Controls for Cross Validation.
[00154] After analyzing test performance, a panel of four separate
negative controls
was created to demonstrate that the modelling was performed correctly.
[00155] Negative Control I: Randomized, Fictitious Class Labels to Detect
Overfitting
[00156] Biomarker selection workflow was performed on a dataset with
randomized
class labels to detect overfitting. Using the existing classification of
samples in the dataset as
either Tumor or Healthy, markers were randomly assigned to a fictitious class
or to a gene
expression class (Class A or B). The workflow was repeated in the same manner
used to
develop the 3-gene test, this time trying to distinguish Class A from B. The 3
best genes were
selected in each of 10 cross validation folds, and used Random Forest to train
a classifier for
each fold. Subsequently, 10 independent test sets were used to determine
performance of the
models. By performing the disclosed workflow on a dataset with randomized
class labels,
the strategy detects overfitting. FIG.6, Negative Control I (Random Class)
clearly shows no
evidence of overfitting: 0.51 Area Under the ROC Curve, 51.9% Accuracy, 51.6%
Sensitivity, 52.1% Specificity.
[00157] Negative Control II: Randomly Selected Genes
[00158] 3 randomly selected genes were modeled in each of 10 cross
validation folds
(FIG.6): 0.733 AUC ROC, 72.6% Accuracy, 73.8% Sensitivity, 61.8% Specificity,
however
-61-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
randomly selected genes perform much worse than our 3-Gene Test. This data
demonstrates
that randomly selected genes do not provide an adequate set of biomarkers.
[00159] Negative Control III: Reversed Selection Criteria
[00160] Poorly performing genes were selected by reversing the disclosed
selection
criteria: i.e., genes with poor p-values and small differences between tumor
and healthy
samples were selected in the reversed selection criteria (FIG.6): 0.653 AUC
ROC, 61.9%
Accuracy, 61.5% Sensitivity, and 65.4% Specificity. The ROC plot confirms that
poor genes
provide less information than other disclosed genes.
[00161] Negative Control IV: Comparison to Null Model
[00162] We applied a Null Model to our dataset and compared the performance
of our
3-gene panel to it. The Null Model consistently guesses that each sample is a
member of the
most prevalent class; in this case, that each sample is tumor (FIG.6, dashed
line). The 3-gene
test (FIG.6, dark line) has a p-value smaller than 5 x 1011 compared to the
Null Model. While
this provides a performance benchmark, it also models the diagnostic
performance of a
treatment strategy. Some well-intentioned surgeons proposed taking additional
tissues
(routine second margins) from all patients. Like routine second margins, the
Null Model
assumes that all patients have positive margins. FIG.4 shows how the
sensitivity and
specificity of the Null Model (dashed line) compares to microscopy.
[00163] Example 3. Assay development for qPCR test
[00164] Primer Design.
[00165] Hundreds of primers were designed for use in in-silico evaluations.
Approximately 40 primer pairs were synthesized and tested empirically using
synthetic
cDNA template. For all experiments, we used clinical-grade reagents and
pipettes that are
certified to 1S08655 standards. qPCR reagents were manufactured in cGMP
conditions under
IS09001 management in a facility that is IS013485-registered.
[00166] The disclosed one-step RTqPCR assay uses targeted primers to
reverse
transcribe RNA into cDNA, followed by qPCR amplification of cDNA and detection
using a
DNA-intercalating dye. Synthetic templates were utilized to optimize the
concentration of
each primer (titrations of primer concentrations) and annealing temperatures
(temperature
gradients). Some RNA primers were designed to span exon junctions. For exon-
spanning
primers, genomic DNA from HeLa cells was used to verify that RNA
quantification is not
impacted by the presence of genomic DNA. For each primer pair, a synthetic
template was
used to determine performance parameters: 10-fold dilutions (5 technical
replicates of 6
-62-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
concentrations), 2-fold dilutions (7 technical replicates of 6
concentrations), and 24 no-
template controls. Lastly, pooled RNA from 3 invasive breast tumors was used
to test each
primer pair. The testing evaluated 3 tumor-specific genes (IBSP, WP 1 1, and
COL10A1), 2
reference genes (C2orf44 and TTC5), and a control to detect genomic DNA, chr3
gDNA. For
each primer pair, 2 negative controls were included and 3 technical replicates
were
conducted. All RNA experiments also included a positive control for each
primer pair.
Altogether, assay development and validation involved greater than 3,700
reactions of 20-
microliter reactions.
[00167] Absolute Quantification of 22 Clinical Samples.
[00168] The disclosed qPCR assays were used to analyze RNA from 22
clinical
samples (11 pairs of invasive breast adenocarcinomas and adjacent healthy
samples).
Specificity, prevision, sensitivity, linearity, and PCR efficiency were
determined for the top
qPCR primer combinations, which were all designed to span exon junctions
(TABLES 1-6).
Performance criteria were found to satisfy MIQE and CLSI guidelines.
[00169] TABLE 21: Analytical Validation of qPCR primers
Parameter Criteria IBSP WP 11
COL10A1 CTDNEP 1 Method
Specificity. Melt curve. 100% 100% 100% 100%
% of area under the
melt curve (negative
first derivative)
corresponding to the
expected melting
temperature
Specificity. Blank 44.5 50 39.7 42.9
Limit of the blank =
Detection, reported in
(N*0.95)=0.5, where
number of PCR cycles. N = 90
technical
replicates of no
template controls
Precision (Standard Standard 0.027 0.012 0.013 0.009
Standard deviation
Deviation,) Deviation < (SD) of
technical
0.167 replicates
with
10,700
copies/microliters
-63-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
Sensitivity. Limit of <0.34 <0.67 <0.34 <0.34
LoD = (mean of no
detection, reported in template
controls
number of copies per minus
standard
microliter deviation of
no
template controls) -
cf3*SD,
Linearity (R2) R2> 0.95 0.999 0.999 0.999 0.999
Maximum of the linear R2 > 0.98 1x106 1x106 1x106 1x106
range, reported in
copies per microliter
Minimum of the linear R2 > 0.98 0.34 0.67 0.34 0.34
range, reported in
copies per microliter
PCR efficiency (6) 90-110% 90% 91% 94% 95%
[00170] FIGURE 7A and FIGURE 7B depict charts showing analytic validation
of
qPCR assays for using clinical-grade reagents. FIGURE 7A panel a depicts
amplification
plots of 20 microliter qPCR reactions. 12 concentrations of synthetic cDNA
template (1.1
million to 0 copies per microliter), including 10-fold dilutions for 6 high
concentrations (5
technical replicates) and 2-fold dilutions for 5 low concentrations (7
technical replicates).
One concentration point overlapped in the high and low concentration series.
Each primer
pair includes 24 replicates of no-template controls. Error bars at each cycle
represent 95% CI
of technical replicates.
[00171] FIGURE 7A panel b depict fluorescence versus cycle plots to
determine Ct
forWP11. A 4-parameter linear model was fitted to 5 technical replicates
(circles). The
maximum of the second derivative was used to define the Ct (CtD2).
[00172] FIGURE 7B panel c depicts threshold cycle versus template dilution
plots to
calculate linear range. The linear range is defined as the range of
concentrations where CtD2
fit a straight line with R-squared >0.995. Red lines indicate 95% Confidence
Intervals
calculated from 200 bootstraps. FIGURE 7B panel d depicts melt plots confirm
to specificity
of the primers. Increasing temperature denatures PCR amplicons, which
decreases
-64-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
fluorescence. A single peak of the negative first derivative confirms the
presence of a single
amplicon. The peak corresponds to the expected melting temperature (dashed
line).
[00173] FIGURE 7A and FIGURE 7B panels e-h depict charts showing analytic
validation of qPCR assays for IBSP RNA as for WP 11. All assays used clinical-
grade
reagents. Panel e depicts amplification plots of 20 microliter qPCR reactions.
12
concentrations of synthetic cDNA template (1.1M to 0 copies per microliter),
including 10-
fold dilutions for 6 high concentrations (5 technical replicates) and 2-fold
dilutions for 5 low
concentrations (7 technical replicates). One concentration point overlapped in
the high and
low concentration series. Each primer pair includes 24 replicates of no-
template controls.
Error bars at each cycle represent 95% Confidence Intervals of technical
replicates.
[00174] FIGURE 7A Panel f depicts fluorescence versus cycle plots to
determine Ct
for IBSP. A 4-parameter linear model was fitted to 5 technical replicates
(circles). The
maximum of the second derivative was used to define the Ct (CtD2). FIGURE 7B
panel g
depicts threshold cycle versus template dilution plots to calculate linear
range. The linear
range is defined as the range of concentrations where CtD2 fit a straight line
with R-squared
>0.995. Red lines indicate 95% Confidence Intervals calculated from 200
bootstraps.
[00175] Absolute quantification (qPCR) of RNA from 22 clinical samples
confirms
that biomarker expression is substantially higher in tumors (TABLE 21). We
present
adjusted copy numbers for all tumor results from this experiment, including
TABLE 21 and
FIG.8. If a tumor sample did not contain 100% tumor (e.g. 95% tumor), the
estimated
number of copies was adjusted to the equivalent of 100% tumor. The mean number
of RNA
copies was 34 to 176 times higher in tumor than healthy. On average,
expression was 72 to
189 times higher when we compared each tumor to paired healthy tissue from the
same
patient. One advantage of multi-analyte modeling is that all genes do not need
to be elevated
in every patient (otherwise the test would only require 1 gene). In each pair
of samples, at
least one of the tumor biomarkers was markedly elevated. On average, the best
of the 3 genes
was 273 times higher in the tumor sample than the paired healthy sample.
[00176] TABLE 22. Absolute quantification (RTqPCR) of RNA from 22 tumors
and healthy tissues.
TABLE 22
WP I I IBSP COL10A1
Mean number of copies
Tumor 13,472 269 291
-65-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
Healthy 76 3 12
Mean fold-change
176 86 34
Mean per-patient fold-change
155 189 72
[00177] qPCR Test Performance
A panel of 3 biomarkers correctly classified 100% of the samples as tumor or
healthy using
Random Forest (EXAMPLE 4), Generalized Linear Models of the Binomial Family
(EXAMPLE 5), and Regularized Discriminant Analysis (EXAMPLE 6). FIG.9 shows
the
ROC curve for a 3-gene test using Random Forest. In addition, IBSP RNA and WP
I I RNA
can unexpectedly be used in combination in a Generalized Linear Model of the
Binomial
Family to correctly classify 100% of samples (EXAMPLE 7). We further
demonstrated that
IBSP RNA (FIG.10) and WP I I RNA (FIG.11), correctly classified 100% of
samples when
used individually in a Generalized Linear Model of the Binomial Family
(EXAMPLE 7).
Additionally, when COL10A1 RNA was used as an individual biomarker in a
Generalized
Linear Model of the Binomial Family, the disclosed qPCR assay correctly
classified 77.3% of
samples as tumor of healthy.
[00178] Example 4. Performance of the 3-gene test using the Random Forest
(RF)
machine learning algorithm, as determined by 5-fold cross validation.
[00179] 22 samples were analyzed using the disclosed RTqPCR assay as
follows:
[00180] 2 classes of samples were analyzed: RNA from 11 tumor samples and
11
healthy samples were analyzed using the disclosed clinical-grade RTqPCR
assays.
Resampling was used to estimate performance and statistical parameters of a
test generated
using Random Forest. Five-fold cross validation showed that the 3-gene RF test
had an
accuracy of 100%, as shown in TABLE 24.
TABLE 23
Reference
Prediction Tumor Healthy
Tumor 11 0
Healthy 0 11
-66-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
[00181] The following parameters were used or determined in the analysis:
TABLE 24
ROC Sens Spec Accuracy Kappa
1 1 1 1 1
> confusionMatrix(data = CMcvRF, CMcvRF$Sample Type)
Cross-Validated (5-fold) Confusion Matrix
[00182] Example 5. Performance of the 3-gene test using a Generalized
Linear
Model (Om).
[00183] 22 samples were analyzed using the disclosed RTqPCR assay as
follows:
[00184] 2 classes of samples were analyzed: RNA from 11 tumor samples and
11
healthy samples were analyzed using the disclosed clinical-grade RTqPCR
assays.
Resampling was used to estimate performance and statistical parameters of a
test generated
using a Generalized Linear Model in the binomial family. Five-fold cross
validation showed
that the 3-gene glm test had an accuracy of 100%, as shown in TABLE 26.
TABLE 25
Reference
Prediction Tumor Healthy
Tumor 11 0
Healthy 0 11
[00185] The following describes R output for this analysis:
TABLE 26
> CMcvGLM 3 log
ROC Sens Spec Accuracy Kappa
1 1 1 1 1
> summary(CMcvGLM 3 log)
Call:
NULL
Deviance Residuals:
Min 1Q Median 3Q Max
-1.067e-05 -2.110e-08 0.000e+00 2.110e-08 1.037e-05
-67-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
Coefficients:
Estimate Std. Error z value
(Intercept) 8.358e+01 2.403e+05
TB SP -1.763e+01 7.812e+04
MMP11 -4.382e+00 5.577e+04
COL10A1 7.443e-01 4.136e+04
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 3.0498e+01 on 21 degrees of freedom
Residual deviance: 3.4098e-10 on 18 degrees of freedom
AIC: 8
Number of Fisher Scoring iterations: 25
> confusionMatrix(data = CMcvGLM 3 log CMcvGLM 3 log$Sample Type)
_ _
Cross-Validated (5 fold) Confusion Matrix
[00186] Example 6. Performance of the 3-gene test using a Regularized
Discriminant Analysis (RDA).
[00187] 22 samples were analyzed using the disclosed RTqPCR assay as
follows:
[00188] 2 classes of samples were analyzed: RNA from 11 tumor samples and
11
healthy samples were analyzed using the disclosed clinical-grade RTqPCR
assays.
Resampling was used to estimate performance and statistical parameters of a
test generated
using Regularized Discriminant Analysis (RDA). Five-fold cross validation
showed that the
3-gene RDA test had an accuracy of 100%, as shown in TABLE 27.
TABLE 26
Reference
Prediction Tumor Healthy
Tumor 11 0
Healthy 0 11
[00189] The following describes R output for this analysis:
TABLE 27
> CMcvRDA
Regularized Discriminant Analysis
-68-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
gamma lambda ROC Sens Spec Accuracy Kappa
0.0 0.0 1 1.0000000 1 1.00 1.0000000
0.0 0.5 1 0.6000000 1 0.82 0.6000000
0.0 1.0 1 0.5333333 1 0.77 0.5321678
0.5 0.0 1 1.0000000 1 1.00 1.0000000
0.5 0.5 1 0.5333333 1 0.77 0.5321678
0.5 1.0 1 0.6333333 1 0.82 0.6321678
1.0 0.0 1 1.0000000 1 1.00 1.0000000
1.0 0.5 1 0.6333333 1 0.82 0.6321678
1.0 1.0 1 0.6333333 1 0.82 0.6321678
> confusionMatrix(data = CMcvRDA, CMcvRDA$Sample Type)
[00190] Accuracy was used to select the optimal model using the largest
value. The
final values used for the model were gamma = 0 and lambda = 0.
[00191] Example 7. Performance of the 2-gene test using a Generalized
Linear
Model (glm).
[00192] 22 samples were analyzed using the disclosed RTqPCR assay as
follows:
[00193] 2 classes of samples were analyzed: RNA from 11 tumor samples and
11
healthy samples were analyzed using the disclosed clinical-grade RTqPCR
assays. The two
genes were IBSP and WP 1 1 . Resampling was used to estimate performance and
statistical
parameters of a test generated using a Generalized Linear Model (glm). Five-
fold cross
validation showed that the 2-gene glm test had an accuracy of 100%, as shown
in TABLE
29.
TABLE 28
Reference
Prediction Tumor Healthy
Tumor 11 0
Healthy 0 11
[00194] The following describes R output for this analysis:
TABLE 29
-69-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
> CMcvGLM 2 genes
Generalized Linear Model
ROC Sens Spec Accuracy Kappa
1 1 1 1 1
> summary(CMcvGLM 2 genes)
Call:
NULL
Deviance Residuals:
Min 1Q Median 3Q Max
-3.200e-05 -2.100e-08 7.230e-07 4.130e-06 2.197e-05
Coefficients:
Estimate Std. Error z value Pr(>1z1)
(Intercept) 2.810e+01 2.988e+04 0.001 0.999
TB SP -4.418e-01 7.770e+02 -0.001 1.000
MIVIP11 -5.536e-03 1.426e+01 0.000 1.000
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 3.0498e+01 on 21 degrees of freedom
Residual deviance: 2.2434e-09 on 19 degrees of freedom
AIC: 6
Number of Fisher Scoring iterations: 25
> confusionMatrix(data = CMcvGLM 2 genes, CMcvGLM 2 genes$Sample Type)
[00195] Example 8. Analysis of Validation Data for 3-gene test
[00196] Data from 1,211 samples using 3 different technologies was used to
validate a
3-gene test. Cross-validation was performed using 939 RNA Seq samples. Since
cross-
validation uses independent test sets, one can mathematically prove that it is
a reliable
estimate of performance. This can be confirmed with a suite of negative
controls. Similar
expression patterns have been confirmed with RNA Seq and microarray,
confirming that the
signals are not platform specific. Independent test sets were then analyzed to
determined
performance on early and late stage tumors, specifically, test sets 1 (75
early-stage samples)
and independent test set 2 (175 late-stage samples).
[00197] The results of these analyses were extremely surprising in light
of the current
beliefs about breast cancer biology. To further investigate these surprising
results, we used
clinical-grade reagents to develop and validate RTqPCR assays for the selected
mRNAs. We
-70-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
analyzed 22 samples with our assays. This represents the third independent set
of samples.
Using the disclosed RTqPCR assays, the selected biomarkers clearly demarcate
tumor and
healthy, and even provides more separation between tumor and healthy than RNA
Seq.
[00198] We used predictive models to combine the biomarkers and set
clinically
actionable thresholds. We discovered that multiple predictive models can
achieve excellent
performance. For example, when all 3 biomarkers are detected using the
disclosed RTqPCR
assays, Random Forest, Regularized Discriminant Analysis, and Generalized
Linear Models
of the Binomial Family can correctly classify 100% of the samples as tumor or
healthy. In
addition, we unexpectedly discovered that when IBSP RNA (FIG.10) and MMP11 RNA
(FIG.11) are detected using the disclosed assays, they can be used
individually to correctly
classify 100% of samples. These experiments firmly establish that the signal
is tumor-specific
and reproducible across three detection technologies.
[00199] Example 9. Evaluation of surgical margins
[00200] As demonstrated herein, the markers and methods can be used to
improve the
evaluation of surgical margins. In this case, cells are collected from the
surface of a surgical
specimen and the disclosed assays are used to detect the disclosed markers. A
number of
methods can be used to collect cells from the surface of a surgical specimen.
As non-limiting
examples, cells can be collected using a surface with a functionalized
surface, such as a poly-
lysine coated touch imprint cytology slide. Cells could also be collected
using a membrane,
such as a nitrocellulose membrane. In addition, cells could be collected using
a sharp or blunt
instrument, such as scrape preparations, which are routinely performed for
pathologic
examination.
[00201] The markers and methods could be used to screen patients for
invasive breast
cancer. Specimens can be collected using nipple aspirates or ductal lavage,
where the
mammary ducts and glands are flushed with fluid and aspirated, sometimes
following brief
hormonal stimulation. Existing screening methods suffer from poor sensitivity
or specificity,
and often exposure patients to radiation. Ductal lavage is the preferred
screening method for
some surgeons, because it directly samples the ducts and glands that give rise
to epithelial
tumors like adenocarcinomas. However, the analysis of rare tumor cells is not
ideal.
Microscopic detection of tumors has the best performance when the tumor is
analyzed in the
context of its surrounding healthy tissue. In fact, the name histo-pathology
derives from the
Greek histos, meaning tissue. When cells are scraped or flushed into a
suspension, they are no
longer in the context of surrounding tissue. Ductal lavage is therefore a
promising screening
-71-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
strategy that is currently limited by the microscopic analysis required to
detect rare or isolated
breast cancer cells. Molecular analysis is particularly well suited to solve
this problem
because it does not rely on visual analysis, and does not require tumor to be
evaluated in the
context of healthy tissue. The disclosed markers and methods could therefore
be used as a
screening tool to determine whether there are invasive cancer cells present in
screened
patients.
[00202] The markers and methods could be used to detect or diagnose
invasive
adenocarcinoma from biopsies of the breast. Biopsies could include core
biopsies, punch
biopsies, incisional biopsies and excisional biopsies. In many biopsy samples,
the procedure
did not collect a sufficient amount of cells, or the tissue architecture has
been disrupted,
making it challenging to reach a definitive histopathologic or cytological
diagnosis. These
challenging cases are prime examples of the advantage of molecular analysis.
Molecular
analysis does not require abundant tissue, and does not require intact tissue
structures in order
to detect the disclosed signatures of invasive cancer.
[00203] Example 10. Identifying pre-cancerous lesions
[00204] The disclosed markers and methods can be used to establish a new
diagnostic
paradigm for pre-cancerous lesions. Lesions like ductal carcinoma in situ
(DCIS) and lobular
carcinoma in situ (LCIS) are currently considered pre-cancerous lesions or
risk factors for
invasive cancer. In only some cases do they develop into invasive cancer, but
there is
currently no way to identify which lesions have invasive potential. Moreover,
precursor
lesions are only analyzed by a few microscopic sections. The current
diagnostic paradigm for
precancerous lesions is based on whether a pathologist happens to observe
cells that penetrate
the basement membrane on the few slides that they examine. There is therefore
thought to be
a subset of pre-cancerous lesions with undiagnosed invasive potential. The
disclosed markers
and methods provide a molecular analysis of invasiveness that could identify
those
precancerous lesions with invasive potential. In addition, the disclosed
methods can be
performed on a more representative portion of the specimen than 3 microscopic
sections. As
non-limiting examples, tissue or biopsy specimens can be morcellated, digested
enzymatically, and/or chemically lysed to release the disclosed biomarkers,
which can then
be detected using the disclosed methods. In this way, the disclosed biomarkers
represent a
strategy to stratify patients by their risk for developing invasive cancer.
[00205] Pathologic Complete Response (pCR) is the absence of residual
cancer in a
solid tissue specimen, obtained from a patient who was previously diagnosed
with invasive
-72-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
cancer. pCR is used as a surrogate endpoint for solid tumor neoadjuvant
therapies. However,
FDA guidance acknowledges that there is an "uncertain relationship between pCR
and long-
term outcome," and emphasizes the possibility "that a neoadjuvant trial could
fail to
demonstrate a significant difference in pCR rates and result in abandoned
development of a
drug that is, in fact, active in the adjuvant or metastatic setting." A 2016
analysis found that
pCR is the primary endpoint of ¨50% of enrolling phase II rectal cancer
trials, and 45% of
phase III preoperative breast cancer trials. However, there are reasons to
continually improve
the metrics and technologies that serve as surrogates for long-term outcomes.
Hormonal
therapies exemplify treatments that substantially improve survival, with only
minimal
impacts on pCR. Conversely, pertuzumab was approved by the FDA following a
phase II
randomized clinical trial that demonstrated an improvement in pCR. Yet, to
date, there have
been no data that suggest pertuzumab improves event-free survival, disease-
free survival, or
overall survival in the neoadjuvant setting. These cautionary notes underscore
the importance
of efforts to vigorously improve the detection of minimal residual disease and
the need to
develop a molecular complete response (mCR) assay.
[00206] Histopathology has been the best way to examine tumors for over a
century,
but it is not ideal to hunt for minimal residual disease (MRD). While FDA
guidance
documents emphasize the importance of compressive sectioning, sampling by
pathology is
woefully underpowered to provide a statistically meaningful analysis of the
specimen (e.g. in
practice, only a few sections are used to hunt for elusive residual tumor).
[00207] Detecting metastases to lymph nodes exemplifies the challenges of
detecting
breast cancer MRD using microscopy. Donald Weaver wrote that "It is quite
clear that the
more sections we evaluate from SLNs the more metastases we identify; however,
it is
impractical to expect the practicing pathologist to mount, stain, and
microscopically examine
every section through the SLN paraffin blocks." Nevertheless, "when we fail to
examine the
entire node, we as pathologists miss metastases that are present."
[00208] Older recommendations of sectioning lymph nodes in intervals is no
longer
considered appropriate because thicker intervals (3-4mm intervals) mean less
metastases are
detected. Examining a greater number of thinner sections detects more
metastases. When thin
sectioning was adopted in the United States between 1995 to 1999, node
positive Stage II
breast cancer increased from 60 to 80 cases per 100,000 population-based
individuals in the
SEER national cancer database.
-73-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00209] EMBODIMENTS
[00210] Embodiment 1. A method of distinguishing a cancer from adjacent
healthy
tissue, said method comprising: a) obtaining a specimen from a human subject,
b) collecting a
sample from said specimen, c) detecting a presence of a set of markers in said
sample by
performing an amplification reaction in a plurality of polynucleotides from
said sample,
wherein said set of markers is selected from the group consisting essentially
of: Matrix
Metallopeptidase 11 (WP 1 1), integrin binding sialoprotein (IBSP), and
collagen type X
alpha 1 chain (COL10A1); and d) distinguishing said cancer when a threshold
level of said set
of markers is detected.
[00211] Embodiment 2. The method of Embodiment 1, wherein said
amplification
reaction is a PCR reaction.
[00212] Embodiment 3. The method of Embodiment 1, wherein said PCR
reaction is a
qPCR reaction.
[00213] Embodiment 4. The method of Embodiment 1, wherein said PCR
reaction is a
RTqPCR reaction.
[00214] Embodiment 5. The method of Embodiment 1, wherein said method can
distinguish said cancer in at least lOng of said plurality of polynucleotides
from sample.
[00215] Embodiment 6. The method of Embodiment 1, wherein said method can
distinguish said cancer in at least 100 mg of said sample.
[00216] Embodiment 7. The method of Embodiment 1, wherein said
amplification
reaction uses at least one primer sequence that has at least 90% identity to
SEQ ID NO: 1-
SEQ ID NO: 356.
[00217] Embodiment 8. The method of Embodiment 1, wherein said sample is
frozen.
[00218] Embodiment 9. The method of Embodiment 1, wherein said sample is a
biopsy sample.
[00219] Embodiment 10. The method of Embodiment 9, wherein said biopsy is
a
liquid biopsy.
[00220] Embodiment 11. The method of Embodiment 9, wherein said biopsy is
a solid
tissue biopsy.
[00221] Embodiment 12. The method of Embodiment 1[00209], wherein said
cancer is
breast cancer.
[00222] Embodiment 13. The method of Embodiment 12, wherein said breast
cancer is
invasive breast cancer.
-74-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00223] Embodiment 14. The method of Embodiment [00221], wherein said
method
distinguishes said breast cancer from adjacent healthy tissue with greater
than 96% accuracy.
[00224] Embodiment 15. The method of Embodiment [00221], wherein said
method
distinguishes said breast cancer from adjacent healthy tissue with greater
than 96%
sensitivity.
[00225] Embodiment 16. The method of Embodiment [00221], wherein said
method
distinguishes said breast cancer from adjacent healthy tissue with greater
than 94%
specificity.
[00226] Embodiment 17. The method of Embodiment 1, wherein said cancer is
a
urothelial carcinoma.
[00227] Embodiment 18. The method of Embodiment [00209], further
comprising
outputting a percentage of said plurality of polynucleotides expressing said
markers from said
sample.
[00228] Embodiment 19. The method of Embodiment [00209], further
comprising
comparing said set of markers from said sample to said set of markers from
said a control
sample.
[00229] Embodiment 20. The method of Embodiment [00227], wherein said
control
sample is a second sample from said human subject.
[00230] Embodiment 21. The method of Embodiment 21, further comprising
performing a second assay to distinguish said cancer.
[00231] Embodiment 22. The method of Embodiment 21, wherein said second
assay
is an immunohistochemistry assay.
[00232] Embodiment 23. The method of Embodiment [00209], wherein said
threshold
level of said WP 11 is 1,000 copies.
[00233] Embodiment 24. The method of Embodiment 1, wherein said threshold
level
of said IBSP is 25 copies.
[00234] Embodiment 25. The method of Embodiment [00209], wherein said
threshold
level of said COL10A1 is 700 copies.
[00235] Embodiment 26. The method of Embodiment [00209], wherein said set
of
markers is selected from the group consisting of: Matrix Metallopeptidase 11
(WP 1 1),
integrin binding sialoprotein (IBSP), and collagen type X alpha 1 chain
(COL10A1).
[00236] Embodiment 27. A kit comprising, at least one primer sequence that
has at
least 90% identity to SEQ ID NO: 1- SEQ ID NO: 356 and a buffer system.
-75-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00237] Embodiment 28. The kit of claim [00235], wherein said buffer
system is a
PCR buffer system.
[00238] Embodiment 29. Isolated nucleic acid comprising a primer sequence
that has
at least 90% identity to SEQ ID NO: 1- SEQ ID NO: 356.
[00239] Embodiment 30. A method of identifying a biomarker for a cancer
comprising:
(a) analyzing, by a computer system, a cohort of biomarkers from a
population of
subjects afflicted with a cancer;
(b) identifying, by said computer system, a first subset from said cohort
of said
biomarkers that has at least a 3-fold higher expression level in said cancer
as
compared to a healthy control biomarker;
(c) identifying, by a computer system, a second subset from said first
subset of said
biomarkers that provides a false discovery rate for said cancer that is less
than a
10-6 rate;
(d) instructing said computer system to use at least one biomarker from
said
second subset of said biomarkers as a training set of a machine learning
algorithm; and
(e) outputting one or more biomarkers that identify said cancer within a
95%
confidence interval.
[00240] Embodiment 31. The method of Embodiment 30, wherein said cancer is
breast
cancer.
[00241] Embodiment 32. The method of Embodiment 31, wherein said breast
cancer is
invasive breast cancer.
[00242] Embodiment 33. The method of Embodiment 30, wherein said one or
more
biomarkers identify said cancer with greater than 96% accuracy.
[00243] Embodiment 34. The method of Embodiment 30, wherein said one or
more
biomarkers identify said cancer with greater than 96% sensitivity.
[00244] Embodiment 35. The method of Embodiment 30, wherein said one or
more
biomarkers identify said cancer with greater than 94% specificity.
[00245] Embodiment 36. The method of Embodiment 30, wherein said training
set
comprises one or more markers selected from the group consisting essentially
of: Matrix
Metallopeptidase 11 integrin binding sialoprotein (IBSP), and collagen
type X
alpha 1 chain (COL10A1).
-76-
CA 03103572 2020-12-11
WO 2019/245587 PCT/US2018/039163
[00246] Embodiment 37. A method of diagnosing a cancer in a human subject,
said
method comprising:
(a) obtaining a sample from said human subject,
(b) detecting whether one or more markers are present in said sample by
performing an
amplification reaction in a plurality of polynucleotides from said sample and
detecting
the presence of said one or more markers, wherein said one or more markers are
selected from the group consisting of: Matrix Metallopeptidase 11 (WP I 1),
integrin
binding sialoprotein (IBSP), and collagen type X alpha 1 chain (COL10A1); and
(c) distinguishing said cancer when a threshold level of one or more said
markers is
detected.
[00247] Embodiment 38. A method of detecting Matrix Metallopeptidase 11
(WP 11)
in a human subject, said method comprising:
(a) obtaining a sample from said human subject; and
(b) detecting whether WP I I is present in said sample by performing an
amplification
reaction in a plurality of polynucleotides from said sample and detecting the
presence
of an WP I I transcript.
[00248] Embodiment 39. A method of detecting integrin binding sialoprotein
(IBSP) in
a human subject, said method comprising:
(c) obtaining a sample from said human subject; and
(d) detecting whether IBSP is present in said sample by performing an
amplification
reaction in a plurality of polynucleotides from said sample and detecting the
presence
of an IBSP transcript.
[00249] Embodiment 40. A method of detecting collagen type X alpha 1 chain
(COL10A1) in a human subject, said method comprising:
(e) obtaining a sample from said human subject; and
(f) detecting whether COL 10AI is present in said sample by performing an
amplification
reaction in a plurality of polynucleotides from said sample and detecting the
presence
of an COL10A/ transcript.
[00250] While preferred embodiments of the present invention have been
shown and
described herein, it will be obvious to those skilled in the art that such
embodiments are
provided by way of example only. Numerous variations, changes, and
substitutions will now
occur to those skilled in the art without departing from the invention. It
should be understood
that various alternatives to the embodiments of the invention described herein
may be
employed in practicing the invention. It is intended that the following claims
define the
-77-
CA 03103572 2020-12-11
WO 2019/245587
PCT/US2018/039163
scope of the invention and that methods and structures within the scope of
these claims and
their equivalents be covered thereby.
-78-