Note: Descriptions are shown in the official language in which they were submitted.
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
METHODS AND COMPOSITIONS FOR DELIVERING MYCOPHENOLIC
ACID ACTIVE AGENTS TO NON-HUMAN MAMMALS
BACKGROUND
Technical Field
Autoimmune diseases represent a heterogeneous family of chronic
diseases. The hallmarks of such diseases include activation and/or
proliferation of
lymphocytes, development of autoantibodies, and dysregulation of the immune
system
leading to chronic inflammation and tissue damage. In the veterinary context,
autoimmune diseases represent a category of diseases with few viable treatment
options.
Description of the Related Art
Mycophenolate mofetil has been recognized as a treatment for
autoimmune diseases and other conditions in both human and veterinary
subjects.
However, current methods and compositions for delivery of mycophenolate
mofetil and
sodium mycophenolate can produce significant side effects in veterinary
subjects,
including, for example, gastrointestinal intolerance related to mucosal
ulceration, and
erosion and necrosis of the stomach and the small and large intestines. See,
e.g., Arns,
W., "Noninfectious Gastrointestinal (GI) Complications of Mycophenolic Acid
Therapy:
A Consequence of Local GI Toxicity?," Transplantation Proceedings 39:88-93
(2007).
Moreover, currently available formulations have uneven pharmacodynamic
activity in
vivo and require frequent dosing to sustain desired activity.
BRIEF SUMMARY
The present disclosure provides compositions and methods for providing
a mycophenolic acid (WA) active agent to a canine subject in a controlled-
release
manner. Disclosed compositions and methods possess pharmacodynamic,
pharmacokinetic, safety, and/or convenience advantages over immediate-release
WA-containing (e.g., mycophenolate mofetil) formulations. For example,
presently
1
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
disclosed formulations and methods provide controlled-release profiles of a
MPA active
agent that attain an average plasma [MPA] concentration over about 8 hours in
a canine
subject such that 2.5, 4, and 8 hours following a first dose of the
formulation, lymphocyte
proliferation is reduced as compared to a pre-dose amount. In certain
embodiments, the
canine subject achieves an average plasma [MPA] of about 250 ng/ml to about
3000
ng/ml over about 8 hours following a first dose of the controlled-release
formulation,
whereupon at 2.5 hours, 4 hours, and 8 hours following the first dose, a
percentage of
proliferating lymphocytes in a whole blood sample from the subject is reduced
as
compared to the percentage of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the first dose, as
determined using
monoclonal antibody Ki-67.
Further, administration according to the instant disclosure produces a
systemic effect such that 24 hours following a second (or more) once-daily
dose, and
prior to an eighth dose, the amount of proliferating lymphocytes in a whole
blood sample
from the canine is lower than the amount of proliferating lymphocytes in whole
blood
obtained from the canine 15 or fewer minutes prior to the dose of the first
day.
In some embodiments, lymphocyte proliferation according to the
disclosed controlled-release methods and compositions is reduced by a greater
degree
and/or for a longer period of time as compared to the reduction attained using
an
immediate-release formulation comprising a MPA active agent. In certain
embodiments,
(i) the percentage of proliferating lymphocytes in a whole blood sample
obtained from
the subject at 4 hours (and/or at 8 hours) following administration of a first
dose of
controlled-release formulation of the present disclosure is lower than (ii)
the percentage
of proliferating lymphocytes in a whole blood sample obtained from a reference
canine
subject that received an immediate-release formulation comprising a MPA active
agent.
Moreover, administration of the instantly disclosed formulations to a
canine subject surprisingly provides a reduction (i.e., in the number, in the
severity, or
both) of adverse gastrointestinal events in the subject as compared to an
immediate-release formulation comprising a MPA active agent. This occurs even
when
2
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
the amount of MPA active agent administered using a controlled-release
formulation
exceeds that administered in the immediate-release form.
In certain embodiments, administering comprises administering a single
dose for each of 10 or more (optionally 15 or more) consecutive days, wherein
over the
10 days (optionally 15 or more days), the subject exhibits no adverse
gastrointestinal
events, wherein an adverse gastrointestinal event comprises emesis, diarrhea,
soft stool,
or any combination thereof.
Accordingly, the presently disclosed methods and compositions have
utility in, for example, treating an autoimmune disease or disorder in a
canine subject,
wherein the autoimmune disease or disorder is characterized by aberrant
proliferation
and/or activation of lymphocytes, as described herein. Autoimmune diseases and
disorders treatable using the presently disclosed methods and compositions
include
atopic dermatitis, arthritis, myasthenia gravis, celiac disease, diabetes
mellitus type 1,
Grave's disease, inflammatory bowel disease, multiple sclerosis, psoriasis,
rheumatoid
arthritis, systemic lupus erythematosus, Behcef s disease, pemphigus vulgaris,
refractory
incomplete systemic lupus erythematosus, lupus nephritis, immunoglobulin A
nephropathy, small vessel vasculitides, scleroderma (systemic sclerosis or
SSc),
idiopathic thrombocytopenic purpura (ITP), psoriasis, apernicious anemia,
vitiligo,
autoimmune hemolytic disease, glomerulonephritis, immune cytopenias,
meningoencephalomyelitis, subepidermal blistering autoimmune disease,
immunobullous diseases, cutaneous vasculitis, recurrent erythema multiforme,
erythema
nodosum, lichen planus, cutaneous Crohn's disease, sarcoidosis, hepatitis,
pyoderma
gangrenosum, and any combination thereof
Also provided herein are methods of administering presently disclosed
controlled-release formulations that comprise a MPA active agent to a canine
subject that
has undergone, is undergoing, or will undergo an organ transplant and/or an
artificial
implant.
In any of the herein disclosed embodiments, the MPA active agent
comprises mycophenolate sodium. In any of the herein disclosed embodiments,
the
canine subject receives a controlled-release formulation in a fed state, or in
a fasted state,
3
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
as described herein. In certain embodiments, a controlled-release formulation
is
administered orally.
Controlled-release formulations comprising a MPA active agent, and kits
comprising the formulations, are also provided.
In another aspect, the present disclosure provides uses of the
controlled-release compositions to suppress lymphocyte proliferation in a
canine subject.
In a further aspect, methods are provided for manufacturing a medicament
comprising a controlled-release formulation of the present disclosure.
Further aspects, embodiments, features, and advantages of the disclosure,
as well as the structure and operation of the certain embodiments, are
described in detail
below with reference to accompanying drawings.
BRIEF DESCRIPTION OF THE SEVERAL VIEWS OF THE DRAWINGS
FIG. 1 shows a schematic example of a particulate subunit of a
controlled-release formulation of the present disclosure.
FIG. 2 shows a representation of an embodiment of a further particulate
subunit of a controlled-release formulation in accordance with an embodiment
hereof.
FIG. 3 shows a representation of an embodiment of a controlled-release
formulation according of the present disclosure, in the form of a capsule,
comprising
particulate subunits in accordance with the present disclosure.
FIG. 4 shows the release of mycophenolate sodium in biphasic media (pH
1.2 for 2 hours, followed by incubation at pH 6.8) from two embodiments of a
particulate
subunit in accordance with the present disclosure.
FIG. 5 shows the release of mycophenolate sodium from additional
embodiments of a particulate subunit of a controlled-release formulation in
accordance
with the present description. Release was measured during incubation in media
at pH
6.8.
FIG. 6 shows the release of mycophenolate sodium from yet another
embodiment of a particulate subunit in accordance with the present disclosure,
where the
4
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
particulate subunit was exposed to a 2-hour incubation in acidic media (pH
1.2), followed
by pH 6.8 media.
FIG. 7 shows release of mycophenolate sodium from another embodiment
of a particulate subunit in accordance with the present disclosure.
FIG. 8 shows release of mycophenolate sodium from yet another
embodiment of a particulate subunit in accordance with the present disclosure
that was
exposed to a 2-hour incubation in acidic media (pH 1.2), followed pH 6.8
media.
FIG. 9 shows release of mycophenolate sodium from an embodiment of a
particulate subunit according to the present disclosure, where the particulate
subunit
comprises a bead with a solvent-based coating and where the particulate
subunit was
exposed to a 2-hour incubation in acidic media (pH 1.2), followed by 12 hours
in pH 6.8
media.
FIG. 10 shows relative levels of serum mycopenolic acid ([MPA] =
ng/mL MPA) measured at the indicated timepoints following administration of
either an
immediate release mycophenolate mofetil solution (120 mg MMF BID, each dose
containing ; circles) to fasted canines or a controlled-release composition of
the present
disclosure to fed canines (270mg "OKV-1001" containing 252mg MPA; squares).
FIG. 11 provides another view of the data shown in FIG. 10 ("Groups" 2
and 3) and further provides mean serum levels (ng/mL) of MPA in fasted canines
following administration of the controlled-release composition ("Group 4").
FIG. 12 shows mean serum levels (ng/mL) of the MPA metabolite acyl
MPA glucoronide (AcMPAG) measured in the indicated canine treatment groups.
FIG. 13 shows mean serum levels (ng/mL) of the MPA metabolite MPA
glucoronide (MPAG) measured in the indicated canine treatment groups.
FIG. 14 provides a schematic diagram showing the design of a
2-treatment, 2-period, sequential adaptive cross-over study by the inventors
of the
present disclosure.
FIG. 15 shows Day 1 serum MPA concentrations over time from the
canine "Period 1" and "Period 2" treatment groups depicted in FIG. 14.
5
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
FIG. 16 shows Day 5 serum MPA concentrations over time from the
canine "Period 1" and "Period 2" treatment groups depicted in FIG. 14.
FIGS. 17-26 relate to a 15-day open-label study investigating
pharmacodynamic (PD) activity of a controlled-release composition of the
present
disclosure ("OKV-1001"). FIG. 17 shows the design of the 15-day study. In the
depicted
study arm, male beagle dogs received a "base dose" of the controlled-release
composition
(270 mg, containing 252 mg MPA; QD); a "low dose" once-daily (180 mg,
containing
168 mg MPA; QD); or a "low dose" twice-daily (180 mg, containing 168 mg MPA;
BID). In each treatment group, n=5 dogs. In a parallel study (not shown), dogs
received
an immediate-release mycophenolate mofetil formulation (120 mg; ¨10 mg/kg for
approximately 12 kg dogs) MMF, BID; n=5) or placebo (n=1).
FIG. 18 illustrates the data capture timeline for the open-label study
shown in Figure 17. PD endpoints were collected at Days 1, 8, and 15. On each
endpoint
collection day, a total of four (4) sampling timepoints were used, as shown in
Table 17
herein.
FIG. 19 shows the percentage of lymphocytes expressing the Ki-67
antigen, a marker associated with proliferation, in whole blood samples taken
from male
beagle dogs prior to, or 2.5 hours, 4 hours, or 8 hours following oral
administration of a
controlled-release formulation of the present disclosure (containing 252 mg
MPA, QD)
.. during a 15-day pharmacodynamics study. Ki-67 expression was determined
using
monoclonal antibody Ki-67 in samples taken at Days 1, 8 and 15 of the study.
The
indicated percentages on the Y-axis are with reference to baseline (i.e.,
percent of
baseline Ki67+ cells (+/- SD)) such that lower bars represent a reduction in
expression as
compared to baseline.
FIG. 20 shows the overall percentage of lymphocytes expressing the
Ki-67 antigen in whole blood samples taken from male beagle dogs prior to, or
2 hours, 4
hours, or 8 hours following a single oral administration of a controlled-
release
formulation of the present disclosure.
FIG. 21 shows the percentage of lymphocytes expressing the Ki-67
antigen in whole blood samples taken from male beagle dogs prior to, or 2.5
hours, 4
6
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
hours, or 8 hours following oral administration of a controlled-release
formulation (180
mg, containing 168 mg MPA, QD) of the present disclosure. Samples were taken
at Days
1, 8, and 15 of the 15-day study. The indicated percentages are with reference
to
baseline.
FIG. 22 shows the percentage of lymphocytes expressing the Ki-67
antigen in whole blood samples taken from male beagle dogs prior to, or 0.75
hours, 4
hours, or 8 hours following oral administration of an immediate-release
mycophenolate
mofetil formulation (120 mg MMF; BID). Samples were taken at Days 1, 8, and 15
of the
15-day study. The indicated percentages are with reference to baseline.
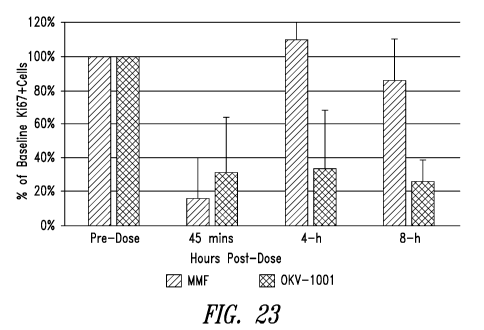
FIG. 23 shows a comparison of single-dose (Day 1) pharmacodynamics of
an immediate-release mycophenolate mofetil formulation (120 mg MMF), BID) and
of a
controlled-release formulation of the present disclosure (270 mg, containing
252 mg
MPA; QD). Ki-67 expression was measured in whole blood samples taken from male
beagle dogs at the indicated timepoints. The indicated percentages are with
reference to
baseline.
FIG. 24 shows a comparison of pharmacodynamics of an
immediate-release mycophenolate mofetil formulation (120 mg MMF, BID) and a
controlled-release formulation of the present disclosure (270 mg, containing
252 mg
MPA; QD) on days 8 and 15 of the 15-day study. Ki-67 expression was measured
in
whole blood samples taken from male beagle dogs at the indicated timepoints.
The
indicated percentages are with reference to baseline.
FIG. 25 shows an eight-hour pharmacokinetic profile (mean plasma MPA
concentration) in male beagle dogs administered a controlled-release
formulation of the
present disclosure (270 mg, containing 252 mg MPA) measured following 8 days
of
administration (QD) in the presently disclosed 15-day study. N=5 male beagle
dogs.
FIG. 26 shows eight-hour pharmacokinetic profiles (mean plasma MPA
concentration) in male beagle dogs administered a controlled-release
formulation of the
present disclosure (180 mg, containing 168 mg MPA; administered BID or QD)
measured following 8 days of administration in the presently disclosed 15-day
study.
N=5 male beagle dogs per treatment group.
7
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
FIG. 27 compares 8-hour pharmacokinetic profiles (mean plasma MPA
concentration) of male beagle dogs administered a controlled-release
formulation of the
present disclosure (270 mg, containing 252 mg MPA; QD) on Day 8 of the 15-day
study
with average pharmacokinetic profiles from "Group A" and "Group C" treatment
groups
on Day 5 of the 5-day repeat dosing study illustrated in Figure 14.
DETAILED DESCRIPTION
The present disclosure provides methods and compositions for controlled
delivery of mycophenolic acid active agents, including mycophenolate sodium,
in
veterinary subjects. The methods and compositions disclosed herein are useful
for,
among other applications, suppressing lymphocyte proliferation (e.g., reducing
the
number of proliferating lymphocytes) in a veterinary subject, and for treating
autoimmune diseases, blood disorders associated with aberrant lymphocyte
proliferation
and/or activation, and immune rejection related to transplant or graft
procedures.
The published patents, patent applications, websites, company names, and
scientific literature referred to herein are hereby incorporated by reference
in their
entireties to the same extent as if each was specifically and individually
indicated to be
incorporated by reference. Any conflict between any reference cited herein and
the
specific teachings of this specification shall be resolved in favor of the
latter. Likewise,
any conflict between an art-understood definition of a word or phrase and a
definition of
the word or phrase as specifically taught in this specification shall be
resolved in favor of
this specification.
Definitions
Prior to setting forth this disclosure in more detail, it may be helpful to an
understanding thereof to provide definitions of certain terms to be used
herein.
Additional definitions are set forth throughout this disclosure.
Technical and scientific terms used herein have the meaning commonly
understood by one of skill in the art to which the present application
pertains, unless
8
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
otherwise defined. Reference is made herein to various methodologies and
materials
known to those of ordinary skill in the art.
In the present description, any concentration range, percentage range,
ratio range, or integer range is to be understood to include the value of any
integer within
the recited range and, when appropriate, fractions thereof (such as one tenth
and one
hundredth of an integer), unless otherwise indicated. Also, any number range
recited
herein relating to any physical feature, such as polymer subunits, size or
thickness, are to
be understood to include any integer within the recited range, unless
otherwise indicated.
As used herein, the term "about" means 20% of the indicated range, value, or
structure,
unless otherwise indicated. It should be understood that the terms "a" and
"an" as used
herein refer to "one or more" of the enumerated components. The use of the
alternative
(e.g., "or") should be understood to mean either one, both, or any combination
thereof of
the alternatives. As used herein, the terms "include," "have" and "comprise"
are used
synonymously, which terms and variants thereof are intended to be construed as
non-limiting.
"Optional" or "optionally" means that the subsequently described
element, component, event, or circumstance may or may not occur, and that the
description includes instances in which the element, component, event, or
circumstance
occurs and instances in which they do not.
The term "consisting essentially of' is not equivalent to "comprising" and
refers to the specified materials or steps of a claim, or to those that do not
materially
affect the basic characteristics of a claimed subject matter.
The terms "gastrointestinal tract," "GI tract," and "GI" may be used
interchangeably herein and refer to an organ system in veterinary subjects
which takes in
.. food, digests the food to extract and absorb energy and nutrients, and
expels the
remaining waste. The GI tract is commonly considered to comprise two subparts:
the
upper GI tract (also "upper GI" herein) includes the buccal cavity, pharynx,
esophagus,
stomach, and duodenum, and the lower GI tract (also "lower GI" herein)
includes the
small and large intestines, the jejeunum, the ileum, the colon, the cecum, the
rectum, the
anal canal, and the anus.
9
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
The terms "MPA active agent" and "mycophenolic acid active agent" may
be used interchangeably herein and refer to MPA or a MPA-based ingredient
(e.g., of a
veterinary composition of the present disclosure) that exerts a physiological
or
pharmacodynamic effect on a subject. MPA active agents comprise MPA and
pharmaceutically acceptable salts, esters, prodrugs, homologs, hydrates or
solvates
thereof. In certain embodiments, a MPA active agent comprises mycophenolate
sodium.
In certain embodiments, a MPA active agent comprises M_MF.
As used herein, "bioavailability" refers to the fraction of a drug that is
absorbed and therefore available to produce a physiological effect.
Bioavailability may
be measured by quantifying the AUC, by, for example, plotting serum
concentration over
time plots using labeled drugs and mass spectroscopy. Bioavailability can be
measured
in terms of "absolute bioavailablity" or "relative bioavailablity."
Absolute bioavailability (Fabs) relates to bioavailability when
administered in a non-intravenous dosage form (e.g., oral tablet) compared
with the same
drug administered intravenously. Absolute bioavailability may be determined by
comparing the AUC of the non-i.v. and i. v. forms, and correcting for the
respective doses:
Fabs¨(AUCnon-intravenous/AUCintravenous) * (DOSeintravenous/DOSenon-
intravenous)
Relative bioavailability (Frei)compares the bioavailability of two different
dosage forms of a drug. The relative AUCs for each dosage form are compared
and
relative doses are used to normalize the calculation:
Fret (AUCdosageVAUCdosageB)* (DoseB/DoseA)
Pharmacodynamics ("PD"), as used herein, refers to the biochemical or
physiological effect or effects of a drug on a subject. PD may be described in
the context
of a dose-response relationship or a concentration-response relationship, and
may
encompass a range of desirable, undesirable, or neutral effects through
mechanisms such
as stimulating or depressing action through receptor agonism and downstream
effects,
blocking or antagonizing action (e.g., of a signaling pathway, or catalytic
activity of an
enzyme, or the like), stabilizing action, exchanging, replacing, or
accumulating
substances (e.g., glycogen storage), conferring a direct beneficial chemical
reaction, or
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
conferring a direct harmful chemical reaction (e.g., cytotoxicity,
mutagenesis, or
irritation). PD values described herein with respect to MPA compositions and
related
methods include, for example, adverse effects on a canine subject administered
an MPA
active agent (e.g., adverse gastrointestinal events such as diarrhea, soft
stools, emesis, or
the like), autoantibody levels or activity, cytokine release rate or levels,
inflammation,
and B or T lymphocyte count(s) or functionality(ies), including proliferation,
e.g., as
determined by the presence of the proliferation-associated marker antigen Ki-
67. Other
markers and assays for determining proliferation of lymphocytes are known in
the art and
are contemplated herein. Suppression of lymphocyte proliferation may be
measured by a
reduction in the percentage of proliferating lymphocytes in a whole blood
sample, and
can be reported as a reduction relative to a baseline level, as a raw
percentage, or by other
accepted means. As set forth in additional detail herein, Ki-67 expression can
be
measured using monoclonal antibody Ki-67 ("mAb-Ki-67").
As used herein, a "canine" refers to any member of the family Canidae,
and includes domestic dogs (including any breed or any variant thereof, as
well as any
combination of two or more breeds or variants, and combinations thereof),
wolves, foxes,
jackals, and coyotes.
"Autoimmune disease" and "autoimmune disorder" may be used
interchangeably herein and refer to conditions in which the immune system of a
subject
recognizes the subject's own cell(s) or tissue(s) as antigenic and produces an
inflammatory response against the subject's cell(s) or tissue(s).
In some embodiments, an autoimmune disease or disorder is associated
with aberrant lymphocyte proliferation and/or activation. Aberrant
proliferation of
lymphocytes includes, for example, increased proliferation (e.g., an increase
in the
overall percentage of lymphocytes in a population that are proliferating; an
increase in
the rate of proliferation of a single lymphocyte or of a population of
lymphocytes; or an
increase in the speed of one or more cycles of cell division by a lymphocyte
or population
of lymphocytes, relative to a normal baseline; proliferation induced exposure
to healthy
cells or tissues, rather than, for example, a mitogen, a cancer antigen, or an
antigen
associated with an infection) and proliferation that results in abnormal
and/or
11
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
dysfunctional lymphocytes, including lymphocytes with a decreased native
functionality
and lymphocytes with an acquired undesirable functionality, such as
autoreactivity.
Aberrant activation of lymphocytes refers to one or more aberration in a
functionality of a
lymphocyte that typically occurs following contact with an antigen or mitogen,
such as
the production of lymphokines, the enlargement of cytoplasm, the synthesis of
macromolecules (e.g., antibodies), and differentiation into memory and
effector cell
types.
In certain embodiments, compositions and methods according to the
present disclosure are useful to treat an (i.e., one or more) autoimmune
disease, such as,
for example, atopic dermatitis, rheumatoid arthritis, celiac disease, diabetes
mellitus type
1, Grave's disease, inflammatory bowel disease, multiple sclerosis, psoriasis,
systemic
lupus erythematosus, Behcet's disease, pemphigus vulgaris, refractory
incomplete
systemic lupus erythematosus, lupus nephritis, immunoglobulin A nephropathy,
small
vessel vasculitides, scleroderma (systemic sclerosis or SSc), idiopathic
thrombocytopenic purpura (ITP), myasthenia gravis, psoriasis, pernicious
anemia,
vitiligo, autoimmune hemolytic disease, glomerulonephritis, immune cytopenias,
meningoencephalomyelitis, subepidermal blistering autoimmune disease,
immunobullous diseases, cutaneous vasculitis, recurrent erythema multiforme,
erythema
nodosum, lichen planus, cutaneous Crohn's disease, sarcoidosis, immune
reactions
associated with veterinary transplant or implant procedures (e.g., tissue
transplants,
grafts, and device implants), including host-versus-graft disease (HvGD) and
other
forms of implant rejection, hepatitis, and pyoderma gangrenosum. Blood
disorders or
diseases treatable according to the presently disclosed methods and
compositions
include, but are not limited to, aplastic anemia, immune mediated hemolytic
anemia, and
immune-mediated thrombocytopenia.
In some embodiments, the presently disclosed compositions and methods
are useful in providing immunosuppression (e.g., by suppressing proliferation
and/or
activation of lymphocytes) in a canine subject that has undergone, is
undergoing, or will
undergo an organ transplant and/or an artificial implant (e.g., a corneal
implant, an
artificial implant or replacement of a joint, a ligament, a bone, or the
like).
12
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
"Treat," "treatment," and "ameliorate," as used herein, refer to the
prevention, lessening of the likelihood of, or medical management of a
disease, disorder,
or condition of a subject (e.g., a canine having a an autoimmune disease or
disorder
associated with aberrant lymphocyte proliferation, or a surgical graft or
transplant). A
canine subject according to the presently disclosed methods and compositions
may, but
need not necessarily, be evaluated, diagnosed, or treated by a veterinarian or
other
veterinary care professional). In general, a dose or treatment regimen
comprising a
controlled-release veterinary composition of the present disclosure is
administered to the
canine subject in an amount sufficient to elicit a therapeutic or prophylactic
benefit.
Therapeutic or prophylactic/preventive benefits include, but are not limited
to: improved
clinical outcome; lessening or alleviation of symptoms associated with a
disease; reduced
frequency of occurrence of symptoms; improved quality of life; longer disease-
free
status; diminishment of extent of disease, stabilization of disease state;
delay of disease
onset, progression; remission; survival; prolonged survival; or any
combination thereof
A "therapeutically effective amount" or "effective amount" of a MPA
active agent or a controlled-release formulation of the present disclosure
refers to an
amount sufficient to result in a therapeutic effect, including: improved
clinical outcome;
lessening or alleviation of symptoms associated with a disease; decreased
occurrence of
symptoms; improved quality of life; longer disease-free status; diminishment
of extent of
disease; stabilization of disease state; delay of disease progression;
remission; survival;
or prolonged survival in a statistically significant manner. For example, a
therapeutically
effective amount of a MPA active agent according to the compositions and
methods of
the present disclosure may be an amount sufficient to reduce or delay
proliferation and/or
activation of B or T lymphocytes, to prevent, reduce, or ameliorate an
inflammatory
response in a canine subject; to treat an autoimmune disease or disorder; or
to prevent,
reduce the severity of, or delay the onset of a rejection occurring in the
course of a cell,
organ, or tissue transplant or graft. When referring to an individual active
ingredient,
administered alone, a therapeutically effective amount refers to the effects
of that
ingredient alone. When referring to a combination, a therapeutically effective
amount
13
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
refers to the combined amounts of active ingredients that result in a
therapeutic effect,
whether administered sequentially, contemporaneously, or simultaneously.
As used herein, "modulating" means reducing, raising, hastening,
delaying, or preventing an occurrence, or increasing or decreasing the
intensity or
efficiency of the occurrence being modulated, through either direct or
indirect means.
The term "controlled-release" is used to describe products that alter the
timing and/or the rate of release of the drug substance in a way that deviates
from
immediate-release following administration. A controlled-release dosage form
is a
formulation in which the drug-release characteristics of time, course, and/or
location are
chosen to accomplish therapeutic or convenience objectives not offered by
immediate-release dosage forms such as immediate-release tablets or
suspensions or
other promptly dissolving or releasing dosage forms. Controlled-release oral
drug
formulations include, for example, extended-release formulations (which allow
a
reduction in dosage frequency as compared to the same drug presented as an
immediate-release (conventional) dosage form, e.g., sustained-release and long-
acting
formulations); delayed-release formulations (which release an identifiable
portion or
portions of drug at a time other than promptly after administration, e.g.,
enteric-coated
aspirin and other NSAID products); targeted-release formulations (which
release the
drug at or near the intended physiologic site of action, and may have either
immediate- or
.. extended-release characteristics); and orally disintegrating tablets (ODT),
which
disintegrate rapidly in the saliva after oral administration. The terms
"controlled-release," "modified-release," "sustained-release," "extended-
release,"
"long-acting," "targeted-release," and "delayed-release" may be used
interchangeably
herein to refer to the release of an administered MPA active agent in a way
that deviates
from immediate release following administration. As used herein, an "immediate
release" dosage refers to any dosage form that is formulated to release or
make available
the active ingredient immediately upon administration. A controlled-release
formulation
according to the present disclosure may, in certain embodiments, be formulated
or
administered to achieve one or more of the following characteristics: release
of a MPA
active agent at or within a certain time following administration; release of
a MPA active
14
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
agent under specific physiological conditions (e.g., pH, temperature); release
of a MPA
active agent within a particular part of the body based on known, estimated,
or predicted
digestive, circulatory, or metabolic rates; release of a MPA active agent
with, upon, or
following administration with another reagent; in a predetermined amount;
release of a
MPA active agent for a predetermined amount of time; release of a MPA active
agent
according to particular release profile; or any combination thereof.
Certain embodiments of the presently disclosed controlled-release
formulations comprise a means for controlling release of the MPA active agent
so that the
MPA active agent is released from the formulation in vivo such that a canine
subject
administered the MPA active agent at a single dose achieves a desired PK
effect and a
desired PD effect, as described herein. Various means for controlling in vivo
release of a
MPA active agent are known and described further herein, and include, for
example,
cellulose polymers, acrylate polymers, cellulose acetates, cellulose acetate
butyrates,
ethyl celluloses, hydroxypropyl methyl celluloses, methyl cellulose polymers,
ethyl
celluloses, hydroxypropyl methyl celluloses, methyl cellulose polymers,
EUDRAGIT
polymers for controlled release, poly(vinyl acrylate) (PVA) polymers (e.g.,
KOLLIDON series), poly(vinyl acrylate) (PVA) polymers, or any combination
thereof.
Means for controlling release of a MPA active agent also comprise layers
comprising one
or more of the herein-described materials (optionally combined with one or
more other
materials), which layers can be present in any number, type, and thickness as
appropriate
to achieve a desired controlled-release. Examples of such layers include
controlled-release layers, protective layers, and seal coat layers, which are
described
herein.
The terms "pharmaceutically acceptable excipient or carrier" or
"physiologically acceptable excipient or carrier," as used herein, refer to
non-active
biologically compatible vehicles, which are described in greater detail
herein, that are
suitable for administration to a human or other non-human mammalian (e.g.,
canine)
subject and generally recognized as safe or not causing a serious adverse
event. In certain
embodiments, a pharmaceutically acceptable carrier includes food items or
liquids to be
administered to the subject. For example, a controlled-release formulation of
the present
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
disclosure can be sprinkled on, sprayed on, or otherwise added to, or combined
with, food
(including "treats") or water to be consumed by a canine subject. In certain
embodiments, a MPA composition of the present disclosure may be carried by
(i.e.,
contained within, combined with, or coated on) a food item such as a dry dog
food, a
treat, a bone, or the like. Feeding regimes useful for practicing such
embodiments are
described herein.
As used herein, "statistically significant" refers to a p value of 0.050 or
less when calculated using the Students t-test and indicates that it is
statistically unlikely
that a particular event or result being measured has arisen by chance.
MPA, MPA Active Agents, and Pharmacology Thereof
Mycophenolic acid (Ci7H2006; "MPA") is a nonnucleoside,
noncompetitive, reversible inhibitor of the enzyme inosine 5'-monophosphate
dehydrogenase (IMPDH), which catalyzes the synthesis of xanthine monophosphate
(XMP) from inosine-5'-monophosphate (IMP). IMP4XMP is the rate-limiting step
in
the de novo synthesis of guanine nucleotides required for nucleic acid
synthesis,
proliferation, and differentiation cells, including B and T lymphocytes. By
inhibiting
IMPDH activity, MPA acts as an immunosuppressive agent. See, e.g., Arns, W.,
Transplantation Proceedings 39:88-93 (2007), the disclosure and methods of
which are
herein incorporated by reference in their entirety. MPA has the following
basic structure
shown in Formula I:
OH 0
HO
0
0
0
Formula I
IMPDH has two isoenzymes, IMPDH1 and IMPDH2. The former is
expressed in most cell types, while the latter predominates in activated
lymphocytes (see,
16
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
e.g., Winnicki et al., Pharmacogenomics 10(1):70-6 (2009)). MPA inhibits
IMPDH2
up to 4-to 5-fold more than IMPDHL and therefore has a more potent cytostatic
effect on
activated lymphocytes than on other cells.
MPA has been prepared for use in humans as an adjunctive
immunosuppressant as a mycophenolate mofetil ester (MMF; approved for human
use in
the U.S. as CellCeptg) and as Na.MPA (Myforticg).
Adverse drug reactions (>1% of human patients) associated with
mycophenolate therapy (i.e., any single-dose or multi-dose therapeutic regimen
involving use of MPA or an active agent thereof, as defined herein) include
diarrhea,
nausea, loose stools, emesis, joint pain, infections, leukopenia, and anemia.
Mycophenolate sodium is also commonly associated with fatigue, headache, cough
and/or breathing issues. Intravenous (IV) administration of MMF is also
commonly
associated with thrombophlebitis and thrombosis. Adverse effects associated
with MMF
use (0.1-1% of subjects) include esophagitis, gastritis, diarrhea, loose
stools, emesis,
gastrointestinal tract hemorrhage, and/or invasive cytomegalovirus (CMV)
infection.
Less frequently, pulmonary fibrosis or various neoplasia occur, such as, for
example,
melanoma, lymphoma, and other malignancies, which MMF-related neoplasias can
occur at frequencies of 1 in 20 to 1 in 200, depending on the type, with
neoplasia in the
skin being the most common site. Cases of pure red cell aplasia (PRCA) have
also been
reported.
Compositions and methods according to the present disclosure may be
described in pharmacological terms, including pharmacokinetics ("PK") and
pharmacodynamics ("PD"). As is understood in the art, pharmacokinetics relate
to the
fate¨e.g., the concentration, metabolism, distribution, absorption, half-life,
or
excretion¨of a drug administered to an organism. Non-limiting measures of PK
include
Cmax (the maximum serum concentration of a drug in a specified compartment or
test area
of the body), Tmax (the time at which the C. is observed), Cmm (minimum or
trough
concentration), Tim, (time at which Calla is observed), T112 (half-life of the
drug or
metabolite, i.e., the time taken for the drug concentration to fall to one
half of its original
value, which may be calculated using one or more points along the terminal
phase of the
17
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
elimination), elimination rate constant "k" (the slope calculated using one or
more
concentrations in the log domain the terminal phase), and AUC ("area under the
curve";
the definite integral in a plot of concentration of a drug in blood plasma
overtime). AUC
represents the total drug exposure over time in a given dose or dosing
regimen, and may
be computed starting at the time of administration and ending when the plasma
concentration is minimal, or may be measured at chosen points in time and
calculated
therefrom. It will be appreciated that certain PK values, such as Cmax and
Ci,õõ, may be
reported with respect to particular timeframes herein, e.g., a particular time
window, or a
particular number of hours, following administration of a controlled-release
formulation.
PK values described herein with respect to MPA compositions and related
methods include, for example, [WA] (concentration of mycophenolic acid drug),
[WAG], and [Acyl-MPAG]. Serum or plasma concentrations of a drug or metabolite
may be reported in any appropriate unit, such as, for example, ng/mL, mg/kg,
pg/mL,
pg/L, and so on. Concentrations over time may be reported in any appropriate
unit, such
as, for example, pg*h/L or ng*h/mL. The AUC may be used to report the
concentration
over a given time interval (AUC) or unbound by a particular time interval
(AUCinf).
Other measures of MPA PK include, for example, drug:metabolite ratios,
e.g., drug:metabolite ratios obtainable following administration of a MPA-
containing
agent (e.g., an immediate-release formulation or a controlled-release
formulation of the
present disclosure).
Moreover, presently disclosed methods and compositions may, in some
embodiments, possess desired MPA pharmacodynamics. For example, in some
embodiments, presently disclosed methods and compositions of the present
disclosure
may be used for e.g., reducing lymphocyte proliferation, providing
immunosuppression,
modulating an inflammatory response in a canine subject, and providing MPA-
based
therapies with improved safety profiles.
Formulations and Kits
In certain aspects, controlled-release formulations are provided, wherein
the formulations comprise: about 3 mg to about 2.2 g of a MPA active agent;
and
18
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
(ii) a means for controlling release of the MPA active agent so that the MPA
active agent
is released from the formulation in vivo such that a canine subject
administered the MPA
active agent at a single dose of about 21 mg/kg achieves a plasma [MPA] Cmax
of up to
about 3000 ng/ml (e.g., about 1000, 1500, 2000, 2500, or about 3000 ng/ml)
over about 8
.. hours following the single dose, and a plasma [MPA] Cmin of no less than
about 250
ng/mL from about 2.5 to about 8 hours following the first dose, and the
subject achieves
an average plasma [MPA] of 250 ng/ml to about 2000 ng/ml for about 8 hours
following
the single dose, and whereupon at 2.5 hours, 4 hours, and 8 hours following
the single
dose, a percentage of proliferating lymphocytes in a whole blood sample from
the subject
is reduced as compared to the percentage of proliferating lymphocytes in a
whole blood
sample obtained from the canine 15 or fewer minutes prior to the single dose,
as
determined using monoclonal antibody Ki-67.
In some embodiments, the total amount of the MPA active agent present
in the controlled-release formulation is about 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, 16,
17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35,
36, 37, 38, 39, 40,
41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59,
60, 61, 62, 63, 64,
65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 81, 82,
84, 85, 86, 87, 88,
89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 105, 110, 115, 120, 125, 130,
145, 150, 155,
160, 165, 170, 175, 180, 185, 190, 195, 200, 205, 210, 215, 220, 225, 230,
235, 240, 245,
250, 255, 260, 265, 270, 275, 280, 285, 290, 295, 300, 305, 310, 315, 320,
325, 330, 335,
340, 345, 350, 355, 360, 365, 370, 375, 380, 385, 390, 395, 400, 405, 410,
415, 420, 425,
430, 435, 440, 445, 450, 455, 460, 465, 470, 475, 480, 485, 490, 495, 500,
505, 510, 515,
520, 525, 530, 535, 540, 545, 550, 555, 560, 565, 570, 575, 580, 585, 590,
595, 600, 605,
610, 615, 620, 625, 630, 635, 640, 645, 650, 655, 660, 665, 670, 675, 680,
685, 690, 695,
.. 700, 705, 710, 715, 720, 725, 730, 735, 740, 745, 750, 755, 760, 765, 770,
775, 780, 785,
790, 795, 800, 805, 810, 815, 820, 825, 830, 835, 840, 845, 850, 855, 860,
865, 870, 875,
880, 885, 890, 895, 900, 905, 910, 915, 920, 925, 930, 935, 940, 945, 950,
955, 960, 965,
970, 975, 980, 985, 990, 995, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700,
1800,
1900, 2000, 2100, or 2200 mg.
19
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
In some embodiments, the MPA active agent comprises mycophenolate
sodium. In particular embodiments, the formulation comprises about 180 mg of
mycophenolate sodium. In other embodiments, the formulation comprises about
270 mg
of mycophenolate sodium.
Any suitable delivery system may be used to deliver a formulation of the
instant disclosure, such as, for example: (i) a multiparticulate drug delivery
system
(MDDS) containing a plurality of particulate subunits that each comprise the
MPA active
agent; (ii) a tablet; (iii) a suspension; (iv) a dragee; (v) a minitablet; or
(vi) any
combination thereof For example, as shown in FIG. 3, a capsule 300 includes a
plurality
of subunits 100, 200, dispersed or mixed within the capsule 300.
The term "multi-particulate drug delivery system," also abbreviated
herein as "MDDS," refers to a multi-unit drug dosage form comprising a
plurality of
discrete particulate subunits (e.g., granules, beads, microspheres, spheroids,
pellets, and
minitablets) that contain or otherwise carry the drug to be delivered. MDDS of
the
present disclosure are controlled-release oral compositions that release a MPA
active
agent in vivo along a desired release profile, and include, for example,
tablets (including
minitablets), capsules, dragees, sachets, and suspensions that comprise
particulate
subunits. A "particulate subunit," as described herein, refers to a drug-
containing subunit
of a MDDS, and can take the form of, for example, a bead, a granule, a
microsphere, a
spheroid, a pellet, or the like, as described further herein.
In certain embodiments, a formulation comprises a MDDS that is
prepared for delivery via a capsule, a sachet, a tablet, or any combination
thereof.
In certain embodiments, a MDDS comprises a single type of a particulate
subunit. In
other embodiments, a MDDS comprises multiple types of the particulate
subunits.
For example, a MDDS may comprise a mixture of particulate subunits having
different
release characteristics so as to achieve a desired drug release profile in a
canine subject.
Thus, in certain embodiments, a plurality of particulate subunits having cores
of different
sizes, or the presence or absence of a protective layer (described herein), or
other
characteristics may be present in a MDDS of the present disclosure. Without
wishing to
be bound by theory, this flexibility advantageously permits selecting or
calibrating a
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
MDDS for a specific canine subject (e.g., an individual canine) or for a
population or
sub-population of canine subjects (e.g., a breed or species of canine) as may
be warranted
by characteristics such as the specific size, activity, level, responsiveness
to a pending or
prior treatment, general health, metabolic rate or function, and other
specific
characteristics of the canine subject. Particulate subunits include, for
example, beads,
granules, microspheres, spheroids, pellets, and minitablets.
In certain embodiments, a particulate subunit (e.g., FIG. 1, 100) includes a
core and a MPA active agent. In further embodiments, a core (e.g., FIG. 1,
102) of a
particulate subunit 100 comprises a solid support core, such as, for example,
a sugar
bead, a sugar sphere, a nonpareil bead, a microcrystalline cellulose bead, a
silica bead, a
calcium carbonate bead, a tartaric acid bead, a mannitol bead, a lactose bead,
a starch
bead, or another pharmaceutically acceptable core onto which an MPA active
agent and
other layers described herein can be disposed. In some embodiments, a core 102
comprises an active agent layer (e.g., FIG. 1, 104) disposed over at least a
portion of the
core 102 (e.g., disposed over a portion, or all, of the core 102). The MPA
active agent
may be disposed over core 102 using methods known in the art, such as, for
example,
spray coating, extrusion, suspension layering, dry powder layering, spray
granulation,
direct pelletizing, dip coating, layering, painting, deposition methods, and
the like (see,
e.g., methods outlined by Glatt GmbH, Binzen, Germany, www.glatt.com).
In alternative embodiments, as exemplified in FIG. 2, a core 202 is an
extruded core, in which the MPA active agent is contained. Extruded cores can
be
prepared as described, for example, in U.S. Patent Nos. 4,808,413 and
5,049,394 (the
disclosures of each of which are incorporated by reference herein in their
entireties), and
may include a binder-plasticizer (e.g., a non-lipophilic binder-plasticizer
(such as
microcrystalline cellulose)), an excipient (e.g., a starch-based excipient) or
a binder.
Additional exemplary extruded cores can be prepared as described in Missaghi
et at.,
"Investigation of Venlafaxine HC1 Release from Extruded and Spheronized Beads
Coated with Ethylcellulose Using Organic or Aqueous Coating Systems,"
Controlled
Release Society Annual Meeting July 2008, the disclosure of which is
incorporated by
reference herein in its entirety for all purposes.
21
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
For example, in certain embodiments, extruded core 202 includes a MPA
active agent (e.g., mycophenolate sodium) from about 50 wt% to about 90 wt%,
an
extrusion/spheronization aid, such as microcrystalline cellulose, from about
10 wt% to
about 30 wt%, or from about 15 wt% to 20wt%. As used herein, the amount of a
substance in a composition may be described "by weight," by "percent weight,"
or
"wt%," meaning the weight of a substance relative to the weight of an
individual
composition (e.g., a single particulate subunit) rather than relative to the
total weight of
the MDDS comprising the plurality of the particulate subunits). In certain
embodiments,
an extruded core 202 further comprises one or more of: a binder (e.g.,
hydroxypropyl
cellulose, hydroxyl propyl methyl cellulose, pregelatinized starch, ethyl
cellulose or poly
vinyl pyrrolidone) from about 1 wt% to about 10 wt%, preferably from about 2
wt% to
about 5 wt%; a release excipient, such as, for example, hydroxpropyl methyl
cellulose
(HPMC), hydroxypropyl cellulose (HPC), acrylic polymers, hydroxyethyl
cellulose
(HEC), ethyl cellulose (EC), which can be incorporated into the beads or
applied as
coating; a filler, such as, for example, lactose, maltodextrin, mannitol,
sorbitol, dicalcium
phosphate, and the like; and a superdisintegrant, such as, for example,
crosslinked
poly(vinyl pyrrolidone) (PVP), sodium starch glycolate, or sodium
croscarmellose, and
the like.
In embodiments having an extruded core (e.g., extruded core 202), at least
a portion of the MPA active agent can be dispersed, dissolved, mixed in, or
otherwise
distributed throughout the core. For example, the MPA active agent may be co-
dissolved
with the various polymers and other excipients for producing the extruded
cores, and then
passed through an extruder to form the desired size beads, prior to drying.
Methods
suitable for making extruded cores according to the presently disclosed
particulate
subunits are described in, for example, US Patent No. 5,049,394, which methods
are
incorporated herein by reference.
It will be appreciated that an extruded core 202 of the present disclosure
may also have an active layer (i.e., of MPA active agent) disposed partially
or fully
thereover.
22
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
In certain embodiments, and without wishing to be bound by theory, the
size of the core 102, 202 of a particulate subunit can be important to ensure
sufficient
delivery of a MPA active agent to a canine subject. For example, in certain
instances it
has been reported that canines do not readily pass objects above certain
dimensions in the
lower GI. Accordingly, in any of the herein disclosed embodiments, a
particulate subunit
core can have a diameter of about 0.5 mm to about 10 mm. For example, in some
embodiments, a diameter of a core (e.g., 102, 202) is from about 0.5 mm to
about 9 mm,
about 1 mm to about 8 mm, about 1 mm to about 7 mm, about 1 mm to about 6 mm,
about
1 mm to about 5 mm, about 2 mm to about 5 mm, about 2 mm to about 4 mm, or
about 1
mm, about 2 mm, about 3 mm, about 4 mm, about 5 mm, about 6 mm, about 7 mm,
about
8 mm, about 9 mm, or about 10 mm in diameter. In certain embodiments, the
diameter of
the core is about 5 mm or less, or is about 2 mm to about 4 mm. For example,
in some
canine subjects, a core diameter of less than about 5 mm allows the
particulate subunit,
once freed or dispersed from an administered MDDS, to move readily through the
stomach of a canine subject, in particular, through a canine stomach and into
the upper
and then lower gastrointestinal tract for delivery of the MPA active agent.
For some
canine subjects, particulate subunit with cores having a diameter of greater
than about 5
mm may remain in the stomach for an undesirably long period of time, thereby
impacting
the targeted delivery of the MPA active agent.
In addition, the crushing strength of the stomach of certain canines can be
significantly higher than the crushing strength of a human stomach (about 1.5N
(human)
vs. about 3.2N (canine); (see, e.g., Kamba et al., Int. I Pharmaceutics 228(1-
2):209-217
(2001)). Thus, in particular embodiments, smaller-sized cores, such as cores
having
diameters of less than about 5 mm, may prevent a particulate subunit from
being crushed
in the stomach, which may cause premature (and therefore less effective and
possibly
adverse) release of a MPA active agent in the stomach of the canine subject.
In some embodiments, particulate subunits (100, 200) of the present
disclosure include a controlled-release layer 108, 208 disposed over at least
a portion of
core 102 or 202. As used herein, a "controlled-release layer" refers to a
layer of material
that provides release of a MPA active agent over a pre-determined time or
period of time,
23
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
or at a pre-determined rate, or otherwise along a release profile that does
not include
immediate release of a MPA active agent following administration. Non-limiting
examples of materials suitable for forming a controlled-release layer include
various
polymers, such as cellulose polymers or acrylate polymers, cellulose acetates,
cellulose
acetate butyrates, ethyl cellulose, hydroxypropyl methyl cellulose, methyl
cellulose
polymers, EUDRAGIT polymers for modified release (e.g., one or both of
EUDRAGIT RS100 and RL100, which if both present can be in any ratio), and
poly(vinyl acrylate) (PVA) polymers (e.g., KOLLIDON series).
In certain embodiments, a controlled-release layer comprises from about
15 wt% to about 35 wt% of the composition. In certain embodiments, a
controlled-release layer may comprise ethyl cellulose, such as in the form of
an aqueous
ethyl cellulose rate-controlling polymer. A controlled-release layer may be
applied in
any way that provides an appropriate rate controlling membrane. For example, a
powder
coating may be used as a deposition vehicle for the controlled-release layer.
Any suitable
dispersion product may be used, such as, for example, Surelease (Colorcon,
Harleysville,
PA, USA) or other products and materials known in the art.
Any desired polymer ratio, using any polymer blend, may be employed
using known techniques to produce a composition having a desired release
profile.
Accordingly, in certain embodiments, a controlled-release layer 108, 208
includes a polymer as described herein at about 5 wt% to about 50 wt%, or
about 10 wt%
to about 40 wt%, or about 20 wt% to about 30 wt%, or about 5 wt%, about 10
wt%, about
15 wt%, about 20 wt%, about 21 wt%, about 22 wt%, about 23 wt%, about 24 wt%,
about
wt%, about 26 wt%, about 27 wt%, about 28 wt%, about 29 wt%, about 30 wt%,
about
31 wt%, about 32 wt%, about 33 wt%, about 34 wt%, about 35 wt%, about 36 wt%,
about
25 37 wt%, about 38 wt%, about 39 wt%, or about 40 wt%. In certain
embodiments, the
polymer may be an ethyl cellulose polymer. In still further embodiments,
controlled-release layer 108, 208 may include a soluble component to modulate
the
permeability thereof Release of a MPA active agent may be further adjusted by
varying
the thickness of one or more polymer layers utilized to form a controlled -
release layer
24
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
(i.e., by varying the weight of the polymer layer) or by adding pore forming-
agents to
affect the permeability of the controlled-release layer.
The controlled-release layer can be applied as a single layer. In specific
embodiments, a controlled-release layer includes multiple layers, optionally
(in the case
of spherical, circular, round, or ovular subunits) concentrically disposed on
one another.
In embodiments where an active agent is disposed over at least a portion of
core 102 as
active layer 104, the controlled -release layer 108 can be disposed over at
least a portion
of the active layer 104. In embodiments where core 202 is an extruded core
comprising
at least a portion of the active agent, a controlled-release layer 208 is
disposed over at
.. least a portion of core 202.
In any of the embodiments described herein, a particulate subunit 100,
200 may further include a protective layer 110, 210 disposed over at least a
portion of
controlled-release layer 108, 208. As used herein, "protective layer" refers
to a layer of
material that provides protection from degradation or dissolution to an
ingested
composition (e.g., a particulate subunit of the present disclosure) as it
travels through the
stomach. In certain embodiments, the protective layer 110, 210 is selected or
designed to
delay release of at least a portion of the MPA active agent until the
formulation reaches
the a desired site within the canine, such as the lower GI tract. For example,
one or more
polymers of the Eudragit L series (Evonik, Essen, DE), such as L100, may be
used to
form a protective layer. In certain embodiments, the protective layer
comprises from
about 8wt% to about 15wt% of the composition.
Where utilized, a protective layer 110, 210 may be a pH sensitive layer
that can maintain integrity at the pH of stomach acid (e.g., roughly pH 1.2 to
pH 4.5 in
canines), but at least partially degrades once it reaches the small or large
intestine (having
.. a pH of about 4 to about 8 in canines). In certain embodiments, protective
layer 110, 210
dissolves at a pH above about 6Ø It will be understood that the pH
sensitivity of the
protective layer, as well as the overall strength and release characteristics
of the
particulate subunit or formulation, may vary in accordance with the
physiological
characteristics of the canine subject to be treated (e.g., large canine versus
small canine).
Examples of suitable materials for forming protective layer 110, 210 include
enteric
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
polymers, such as methacrylate-based polymers including EUDRAGIT L or
EUDRAGIT S polymers, cellulose acetate phthalate, cellulose acetate
succinate,
HPMC phthalate, HPMC acetate succinate, sodium alginate, zein, polyvinyl
acetate
phthalate (PVAP), shellac, methacrylic aid-ethyl acrylate copolymer (Kollicoat
MAE),
and mixtures thereof. In further embodiments, controlled-release layer 108,
208 and
protective layer 110, 210 can be designed so as to provide a timed release,
rather than a
pH-dependent release, of the MPA active agent, so that they enable the
compositions to
pass through the stomach intact and release (at least a portion of) the MPA
active agent in
the small and/or large intestine, as desired.
In further embodiments, particulate subunits of the present disclosure may
include a seal coat 106, 206. As shown in the exemplary embodiment of FIG. 1,
in a
particulate subunit 100, seal coat 106 separates core 102 and active agent
layer 104 from
controlled-release layer 108. In the exemplary embodiment represented in FIG.
2, a
particulate subunit 200 includes a seal coat 206 that separates controlled-
release layer
208 from core 202, which is an extruded core containing at least some (i.e.,
all or less
than all) of the MPA active agent. In certain embodiments, a seal coat 106,
206 may be
useful to separate, partially or fully, a MPA active agent from controlled-
release layer
108, 208 so as to reduce or eliminate interactions and degradation of the
controlled-release layer or of the active agent. However, in embodiments where
a
non-aqueous coating method is used to apply the active agent layer 104 to core
102,
degradation may be less of a concern and seal coat 106 may therefore be
excluded or
reduced in thickness. Exemplary compositions for use in seal coat 106, 206
include
various cellulose polymers, including hydroxypropyl methylcellulose,
poly(vinyl
alcohol) (Opadry AMB, Kollicoat), hydroxypropyl methylcellulose, methyl
cellulose,
hydroxyethylcellulose, Opadry series, and the like.
In any of the embodiments described herein, a controlled-release
formulation (and/or a particulate subunit thereof) may further comprise a
buffering agent
or buffer to protect a MPA active agent from degradation by gastric acid.
Accordingly, a
buffer can be added to core 102, 202 or to active agent layer 104 surrounding
core 102. In
the case of extruded cores 202, the buffer may be added to core 202 or added
to a layer
26
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
206, 208, 210 surrounding core 202 to provide buffering and to maintain the
integrity and
activity of the MPA active agent. Exemplary buffers for use in the
formulations and
particulate subunites described herein include, but are not limited to,
phosphate buffers,
citrate buffers and acetate buffers.
In any of the embodiments described herein, a particulate subunit may
further comprise a buffer to affect stability or release of the MPA active
agent under
certain pH conditions.
Illustrative materials, amounts, ratios, and constructions of
controlled-release formulations are described in the Examples, and the design
and dosing
of the instant controlled-release formulations will more generally be
understood by
persons of ordinary skill in light of this disclosure.
Kits are also provided herein that comprise a controlled-release
formulation of the instant disclosure, and optionally further comprise
instructions for
administering the formulation to a canine subject. In certain embodiments, the
kit further
comprises a companion delivery piece. A companion delivery piece can be, for
example,
an irrigation syringe, a syringe, a tube, a transdermal patch, a mixing flask
for producing
a solution containing the formulation; or a food item to be provided to the
veterinary
subject with the formulation. In certain embodiments, a food item is of a
recommended
meal size for the veterinary subject. As described herein, a food item may be
useful in
accompanying the formulation for oral ingestion as a separate item or as a
coating, filling,
or mixture with the formulation. A controlled-release formulation (e.g.,
powder or
microbeads) may also be mixed with water or another liquid so that the
formulation is
ingested when the canine subject takes a drink. If the subject is not drinking
or resists
drinking, the irrigation syringe may be useful to deliver the formulation to
the subject.
In related embodiments, the kit comprises a sealed package housing
individually sealed unit dosage forms comprising the formulation, e.g., MDD S,
capsules,
tablets, or the like, along with instructions for use and an optional
companion delivery
piece.
27
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Methods of Suppressing Lymphocyte Proliferation
Controlled-release MPA formulations, including the formulations
described herein, may be useful for suppressing lymphocyte proliferation and
in treating
diseases and conditions in which such suppression may be desired. Moreover,
the
present disclosure provides, for the first time, a pharmacokinetic "window" of
MPA
which advantageous pharmacodynamic effects are achieved in a canine subject
using
controlled-release formulations.
In certain aspects, a method for suppressing lymphocyte proliferation in a
canine subject comprises administering to the subject a controlled-release
formulation
comprising a MPA active agent such that the subject achieves an average plasma
[MPA]
of about 250 ng/mL to about 3000 ng/mL (e.g., about 250, 300, 350, 400, 450,
500, 550,
600, 650, 700, 750, 800, 850, 900, 950, 1000, 1100, 1200, 1300, 1400, 1500,
1600, 1700,
1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, or
about 3000
ng/mL) over about 8 hours following a first dose of the controlled-release
formulation,
whereupon at 2.5 hours, 4 hours, and 8 hours following the first dose, a
percentage of
proliferating lymphocytes in a whole blood sample from the subject is reduced
as
compared to the percentage of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the first dose, as
determined using
monoclonal antibody Ki-67.
In certain embodiments, the subject achieves an average plasma [MPA] of
about 250 ng/mL to about 2500 ng/mL for about 8 hours following a first dose.
In certain embodiments, the subject achieves an average plasma [MPA] of
about 350 ng/mL to about 2000 ng/mL for about 8 hours following a first dose.
It will be
understood that the pharmacokinetics of a controlled-release MPA formulation
may
depend on whether the canine subject receiving the formulation is in a fed
state, or is in a
fasted state, as described herein. In certain embodiments, a canine subject is
administered a first dose of the controlled-release formulation when in a fed
state,
wherein the subject achieves an average plasma [MPA] of about 500 ng/mL to
about
2500 ng/mL for about 8 hours following a first dose. In other embodiments, the
canine
subject is administered a first dose of the controlled-release formulation
when in a fasted
28
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
state, wherein the subject achieves an average plasma [MPA] of about 500 ng/mL
to
about 3000 ng/mL for about 8 hours following the first dose.
In certain embodiments, a fasted state may comprise a state in which the
canine subject was fed no later than about 1, 2, 3, 4, 5, 6, 7, 8, 9,10, 11,
12, 14, 16, 18, 20,
24, or more hours prior to administration, and then optionally not fed again
until about 1,
2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 16, 18, or 24 hours following
administration. A fed
state may comprise a state in which the canine subject has been fed
immediately prior to,
or no more than 1 hour prior to, administration.
In particular embodiments, the subject achieves an average plasma [MPA]
of about 500 ng/mL to about 1700 ng/mL for about 8 hours following a first
dose.
In particular embodiments, the subject achieves an average plasma [MPA]
of about 650 ng/mL to about 1500 ng/mL for about 8 hours following a first
dose.
In other embodiments, subject achieves an average plasma [MPA] of
about 250 ng/mL to about 600 ng/mL for about 8 hours following a first dose.
In particular embodiments, the subject achieves a plasma [MPA] Cmax of
about 2000 ng/mL over about 8 hours following a first dose, and a plasma [MPA]
Cmin
of no less than about 500 ng/mL from about 2.5 to about 8 hours following the
first dose.
In some embodiments, the subject achieves a plasma [MPA] Cmax of
about 2500 ng/mL over about 8 hours following a first dose, and a plasma [MPA]
Cmin
.. of no less than about 250 ng/mL from about 2.5 to about 8 hours following
the first dose.
In certain embodiments, the subject achieves a plasma [MPA] Cmax of
about 1500 ng/mL over about 8 hours following a first dose, and a plasma [MPA]
Cmin
of no less than about 600 ng/mL from about 2.5 to about 8 hours following the
first dose.
In other embodiments, the subject achieves a plasma [MPA] Cmax of
about 700 ng/mL over about 8 hours following a first dose, and a plasma [MPA]
Cmin of
no less than about 250 ng/mL from about 2.5 to about 8 hours following the
first dose
In still other embodiments, the subject achieves a plasma [MPA] Cmax of
about 600 ng/mL from about 1 to about 8 hours following a first dose, and a
plasma
[MPA] Cmin of no less than about 250 ng/mL from about 2.5 to about 8 hours
following
.. the first dose.
29
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Without wishing to be bound by theory, the presently disclosed PK
profiles are believed to provide a desirable MPA PD in a canine subject
administered a
controlled-release formulation. In some embodiments, at 2.5 hours following a
first
dose, the percentage of proliferating lymphocytes in a whole blood sample from
the
subject is reduced by at least about 35%, 40%, 45%, 50%, 55%, 60 %, 65%, 70%,
75%,
80%, or more, relative to the percentage of proliferating lymphocytes in a
whole blood
sample obtained from the subject 15 or fewer minutes prior to the first dose.
In certain embodiments, at 4 hours following a first dose, the percentage
of proliferating lymphocytes in a whole blood sample from the subject is
reduced by at
least about 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, or more,
relative to
the percentage of proliferating lymphocytes in a whole blood sample obtained
from the
subject 15 or fewer minutes prior to the first dose.
In certain embodiments, at 8 hours following a first dose, the percentage
of proliferating lymphocytes in a whole blood sample from the subject is
reduced by at
least about 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, or more,
relative to
the amount of proliferating lymphocytes in a whole blood sample obtained from
the
subject 15 or fewer minutes prior to the first dose.
It will be understood that the reduction in the percentage of proliferating
lymphocytes may be achieved at 2.5, 4, or 8 hours following a first dose, or
may be
achieved at any two or all three of the indicated timepoints (i.e., at 2.5 and
4, at 2.5 and 8,
at 4 and 8, or at 2.5, at 4, and at 8 hours following a first dose).
The pharmacodynamic effects achieved by the present disclosure
advantageously allow for once-daily administration of a controlled-release
formulation,
which may in some embodiments be easier, less stressful, and more convenient
for both
the canine subject and the owner or care provider. For once-daily dosing, a
dose may be
administered at any time, though generally in the morning or evening.
Both single-day and multi-day administrations of a controlled-release
MPA formulation are contemplated in the presently disclosed methods. For multi-
day
administrations, doses are generally administered at around the same time on
each day;
e.g., about 24 hours following the previous dose.
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
In some embodiments, administering comprises administering a single
dose of the controlled-release composition per day for 2, 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, or more, consecutive days. Advantageously, and quite surprisingly,
administering a once-daily dose according of the presently disclosed methods
for
multiple (i.e., two or more) consecutive days provides a systemic effect.
In certain embodiments, administering comprises administering a single
dose for two or more consecutive days (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, or
more), whereupon 24 hours after the dose of the second day, and prior to any
subsequent
dose, the percentage of proliferating lymphocytes in a whole blood sample from
the
subject is reduced by about 35%, about 40%, about 45%, about 50%, about 55%,
about
60%, about 65%, about 70%, about 75%, or more, relative to the amount of
proliferating
lymphocytes in a whole blood sample obtained from the subject 15 or fewer
minutes
prior to the dose of the first day.
In further embodiments, administering is performed for 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 12, 13, 14, 15, or more days, and whereupon 24 hours after the dose
of each of
the second, third, fourth, fifth, sixth, seven, eighth, ninth, tenth,
eleventh, twelfth,
thirteenth, fourteenth, and fifteenth days, and prior to any subsequent dose,
the
percentage of proliferating lymphocytes in a whole blood sample from the
subject is
reduced by about 35%, about 40%, about 45%, about 50%, about 55%, about 60%,
about
65%, about 70%, about 75%, or more, relative to the amount of proliferating
lymphocytes in a whole blood sample obtained from the subject 15 or fewer
minutes
prior to the dose of the first day.
In some embodiments, administering comprises administering a single
dose for seven or more consecutive days, whereupon 24 hours following the
seventh
dose, and prior to any subsequent dose, the percentage of proliferating
lymphocytes in a
whole blood sample from the canine subject is reduced (e.g., by any amount,
including
and up to at least about 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, or
more) as compared to the amount of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the dose of the first
day.
31
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
In further embodiments, administering is performed for 8, 9, 10, 11, 12,
13, 14, 15, or more days, whereupon 24 hours after the dose of each of the
eighth, ninth,
tenth, eleventh, twelfth, thirteenth, fourteenth, and fifteenth days, and
prior to any
subsequent dose, the percentage of proliferating lymphocytes in a whole blood
sample
from the subject is reduced by about 35%, about 40%, about 45%, about 50%,
about
55%, about 60%, about 65%, about 70%, about 75%, or more, relative to the
amount of
proliferating lymphocytes in a whole blood sample obtained from the subject 15
or fewer
minutes prior to the dose of the first day.
As illustrated in the Examples and described further herein, the presently
disclosed methods are also advantageous in that a single dose of a controlled-
release
formulation has improved activity, both in degree and duration, over immediate-
release
(IR) formulations that comprise an MPA active agent (e.g., mycophenolate
mofetil),
including when the IR formulation is administered twice daily. In some
embodiments, (i)
the percentage of proliferating lymphocytes in a whole blood sample obtained
from the
subject at 4 hours following administration of a single dose is lower than
(ii) the
percentage of proliferating lymphocytes in a whole blood sample obtained from
a
reference canine subject that received an immediate-release formulation
comprising a
MPA active agent. In further embodiments, (i) the amount of proliferating
lymphocytes
in a whole blood sample obtained from the subject at 8 hours following
administration of
a single dose is lower than (ii) the amount of proliferating lymphocytes in a
whole blood
sample obtained from a reference canine subject that received an immediate-
release
formulation comprising a MPA active agent.
A "reference canine subject," as referred to herein, is a comparator canine
of a similar or same age, size, gender, breed, and disease state as the canine
subject
receiving the controlled-release formulation. A reference canine subject
receives, in one
or more doses of the immediate-release formulation, a total daily intake of an
MPA active
agent that is present in in a standard-of-care treatment such as CellCept
(Genentech)
(e.g., 10 mg/kg BID; MPA dose-equivalent of approximately 7.4 mg/kg for each
dose for
an approximately 10kg canine). In some embodiments, a reference canine subject
receives a total daily intake of a MPA active agent that is about 40%, 45%,
50%, 55%,
32
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
60%, 65%, 70%, or up to about 100% of the amount present in the controlled-
release
formulation administered to the canine subject. For example, in some
embodiments, a
reference male beagle dog weighing approximately 12 kg may receive two daily
administrations of an immediate-release formulation, each administration
containing
about 88.5 mg MPA and totaling about 177 mg MPA, while a male beagle dog
weighing
approximately 12kg may receive a once-daily administration of a controlled-
release
formulation of the instant disclosure containing about 168mg MPA or about 252
mg
MPA, or any amount therebetween.
Furthermore, the disclosed methods and compositions advantageously
.. possess improved safety profiles over immediate-release formulations. In
some
embodiments, administering comprises administering the dose for 10 or more
days,
wherein over the 10 days, the subject exhibits no adverse gastrointestinal
events, wherein
an adverse gastrointestinal event comprises emesis, diarrhea, soft stool, or
any
combination thereof. In further embodiments, administering comprises
administering
.. the dose for 15 or more days, and wherein over the 15 days, the canine
subject exhibits no
adverse gastrointestinal events.
In some embodiments, administering comprises administering a dose for
10 or more days, wherein over the 10 days, the subject exhibits a reduced
number, a
reduced severity, or both, of an adverse gastrointestinal event as compared to
a reference
canine subject that received an immediate-release formulation comprising a MPA
active
agent, wherein an adverse gastrointestinal event comprises emesis, diarrhea,
soft stool, or
any combination thereof
In further embodiments, administering comprises administering a dose
for 15 or more days, wherein over the 15 days, the canine subject exhibits a
reduced
number, a reduced severity, or both, of an adverse gastrointestinal event as
compared to
the reference canine subject.
In any of the embodiments described herein, administration can comprise
oral administration.
In some embodiments, a method comprises use of any one or more of a
.. controlled-release formulation as described herein, such as, for example,
(i) a
33
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
multiparticulate drug delivery system (MDDS) containing a plurality of
particulate
subunits that each comprise the MPA active agent; (ii) a tablet; (iii) a
suspension; (iv) a
dragee; or (v) any combination thereof In particular embodiments, a controlled-
release
formulation comprises a MDDS comprising a plurality of particulate subunits
each
having a diameter of than about 5 mm.
In certain embodiments, a formulation comprises a MDDS that is
prepared for delivery to the subject via a capsule, a sachet, a tablet, or any
combination
thereof. In further embodiments, each of the plurality of particulate subunits
of the
MDDS comprises a core, wherein: (i) the MPA active agent is contained in an
active
layer disposed over at least a portion of the core; or (ii) the MPA active
agent is contained
within the core.
In still further embodiments, each particulate subunit of the plurality
further comprises a controlled-release layer, wherein the controlled-release
layer is
disposed over the MPA active layer of the core of (i), or is disposed over at
least a portion
of the core of (ii).
In yet other embodiments, each particulate subunit of the plurality
comprises a seal coat layer, wherein the seal coat layer is disposed between
the MPA
active layer of the core of (i) and the controlled-release layer, or disposed
between the
core of (ii) and the controlled-release layer.
In further embodiments, each particulate subunit of the plurality further
comprises a protective layer disposed over the controlled-release layer.
In any of the herein disclosed embodiments, a first dose comprises the
MPA active agent at about 3 mg/kg to about 35 mg/kg.
In further embodiments, a first dose comprises the MPA active agent at
about 15 mg/kg to about 30 mg/kg.
In some embodiments of the presently disclosed methods, a first dose
comprises from about 3mg to about 2200 mg of the MPA active agent.
In particular embodiments, a first dose comprises about 168 mg of the
MPA active agent. In some embodiments, a first dose comprises about 252 mg of
the
MPA active agent. In any of the presently disclosed embodiments, the MPA
active agent
34
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
can comprise mycophenolate sodium. In some embodiments, the controlled-release
composition is administered to the subject with food, and may in any
embodiment be
administered in a fed state or a fasted state.
In certain embodiments, the subject: (i) has, or is suspected of having, an
autoimmune disease or disorder associated with aberrant lymphocyte
proliferation;
(ii) has undergone, is undergoing, or will undergo an organ transplant and/or
an artificial
implant; or (iii) both of (i) and (ii).
In some embodiments, the canine subject has or is suspected of having an
autoimmune disease or disorder comprising atopic dermatitis, arthritis,
myasthenia
gravis, celiac disease, diabetes mellitus type 1, Grave's disease,
inflammatory bowel
disease, multiple sclerosis, psoriasis, rheumatoid arthritis, systemic lupus
erythematosus,
Behcef s disease, pemphigus vulgaris, refractory incomplete systemic lupus
erythematosus, lupus nephritis, immunoglobulin A nephropathy, small vessel
vasculitides, scleroderma (systemic sclerosis or SSc), idiopathic
thrombocytopenic
purpura (ITP), psoriasis, apernicious anemia, vitiligo, autoimmune hemolytic
disease,
glomerulonephritis, immune cytopenias, meningoencephalomyelitis, subepidermal
blistering autoimmune disease, immunobullous diseases, cutaneous vasculitis,
recurrent
erythema multiforme, erythema nodosum, lichen planus, cutaneous Crohn's
disease,
sarcoidosis, hepatitis, pyoderma gangrenosum, or any combination thereof.
Treatment Methods
Also provided herein are methods for treating an autoimmune disease or
disorder in a canine subject, the autoimmune disease or disorder being
characterized by
aberrant proliferation of lymphocytes, wherein the method comprises
administering to
the subject a single dose of a controlled-release composition comprising a MPA
active
agent, such that the subject achieves an average plasma [MPA] of about 250
ng/mL to
about 3000 ng/ml (e.g., about 250, 300, 350, 400, 450, 500, 550, 600, 650,
700, 750, 800,
850, 900, 950, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900,
2000, 2100,
2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, or about 3000 ng/mL)over about
8
hours following the single dose, and whereupon at 2.5 hours, 4 hours, and 8
hours
following the single dose, the percentage of proliferating lymphocytes in a
whole blood
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
sample from the subject is reduced by at least about 35%, 40%, 45%, 50%, 55%,
60%,
65%, 70%, 75%, 80%, or more, as compared to the percentage of proliferating
lymphocytes in a whole blood sample obtained from the subject 15 or fewer
minutes
prior to the single dose, as determined using monoclonal antibody Ki-67.
In some embodiments, the subject achieves an average plasma [MPA] of
about 250 ng/mL to about 2500 ng/mL for about 8 hours following a first dose.
In some embodiments, the subject achieves an average plasma [MPA] of
about 350 ng/mL to about 2000 ng/mL for about 8 hours following a first dose.
In further embodiments, the subject achieves an average plasma [MPA] of
.. about 500 ng/mL to about 1700 ng/mL for about 8 hours following a first
dose.
In still further embodiments, the subject achieves an average plasma
[MPA] of about 650 ng/mL to about 1500 ng/mL for about 8 hours following a
first dose.
In some embodiments, the subject achieves a plasma [MPA] Cmax of
about 2500 ng/mL over about 8 hours following a first dose, and a plasma [MPA]
Cmin
.. of no less than about 250 ng/mL from about 2.5 to about 8 hours following a
first dose.
In further embodiments, the subject achieves a plasma [MPA] Cmax of
about 2000 ng/mL over about 8 hours following a single dose, and a plasma
[MPA] Cmin
of no less than about 500 ng/mL from about 2.5 to about 8 hours following the
single
dose.
In further embodiments, the subject achieves a plasma [MPA] Cmax of
about 1500 ng/mL over about 8 hours following a single dose, and a plasma
[MPA] Cmin
of no less than about 600 ng/mL from about 2.5 to about 8 hours following the
single
dose.
In other embodiments, the subject achieves a plasma [MPA] Cmax of
about 700 ng/mL over about 8 hours following a single dose, and a plasma [MPA]
Cmin
of no less than about 250 ng/mL from about 2.5 to about 8 hours following the
single
dose.
In further embodiments, the subject achieves a plasma [MPA] Cmax of
about 600 ng/mL from about 1 to about 8 hours following the first dose, and a
plasma
36
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
[MPA] Cmin of no less than about 250 ng/mL from about 2.5 to about 8 hours
following
the first dose.
In some embodiments, the administering comprises administering a single
dose for 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or more, consecutive
days.
In certain embodiments, administering comprises administering a single
dose for two or more consecutive days (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, or
more), whereupon 24 hours after the dose of the second day, and prior to any
subsequent
dose, the percentage of proliferating lymphocytes in a whole blood sample from
the
subject is reduced by about 35%, about 40%, about 45%, about 50%, about 55%,
about
.. 60%, about 65%, about 70%, about 75%, or more, relative to the amount of
proliferating
lymphocytes in a whole blood sample obtained from the subject 15 or fewer
minutes
prior to the dose of the first day.
In further embodiments, the administering is performed for 2, 3, 4, 5, 6, 7,
8,9, 10, 11, 12, 13, 14, 15, or more days, and whereupon 24 hours after the
dose of each
of the second, third, fourth, fifth, sixth, seven, eighth, ninth, tenth,
eleventh, twelfth,
thirteenth, fourteenth, and fifteenth days, and prior to any subsequent dose,
the
percentage of proliferating lymphocytes in a whole blood sample from the
subject is
reduced by about 35%, about 40%, about 45%, about 50%, about 55%, about 60%,
about
65%, about 70%, about 75%, or more, relative to the amount of proliferating
lymphocytes in a whole blood sample obtained from the subject 15 or fewer
minutes
prior to the dose of the first day.
In further embodiments, the administering comprises administering a
single dose for seven or more consecutive days, whereupon 24 hours after the
dose of the
seventh day, and prior to any subsequent dose, the percentage of proliferating
lymphocytes in a whole blood sample from the subject is reduced by about 35%,
about
40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%, about
75%,
or more, relative to the amount of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the dose of the first
day.
In some embodiments, the administering comprises administering a single
dose for 10 or more days, wherein over the 10 days, the subject exhibits no
adverse
37
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
gastrointestinal events, wherein an adverse gastrointestinal event comprises
emesis,
diarrhea, soft stool, or any combination thereof.
In particular embodiments, the autoimmune disease or disorder comprises
atopic dermatitis, arthritis, myasthenia gravis, celiac disease, diabetes
mellitus type 1,
Grave's disease, inflammatory bowel disease, multiple sclerosis, psoriasis,
rheumatoid
arthritis, systemic lupus erythematosus, Behcef s disease, pemphigus vulgaris,
refractory
incomplete systemic lupus erythematosus, lupus nephritis, immunoglobulin A
nephropathy, small vessel vasculitides, scleroderma (systemic sclerosis or
SSc),
idiopathic thrombocytopenic purpura (ITP), psoriasis, apernicious anemia,
vitiligo,
autoimmune hemolytic disease, glomerulonephritis, immune cytopenias,
meningoencephalomyelitis, subepidermal blistering autoimmune disease,
immunobullous diseases, cutaneous vasculitis, recurrent erythema multiforme,
erythema
nodosum, lichen planus, cutaneous Crohn's disease, sarcoidosis, hepatitis,
pyoderma
gangrenosum, or any combination thereof
In another aspect, methods are provided for providing a MPA active agent
to a canine subject in relation to an organ transplant or artificial implant
procedure to, for
example, suppress or treat an immune response against the transplanted organ
and/or
artificial implant. In some embodiments, a method comprises administering to a
canine
subject that has undergone, is undergoing, or will undergo an organ transplant
and/or
artificial implant, a controlled-release composition comprising a MPA active
agent, at a
dose of about 3 mg/kg to about 35 mg/kg of the MPA active agent, wherein the
subject
receives a single dose of the controlled-release composition per day, such
that the subject
achieves a plasma [WA] Cmax of about 3000 ng/mL over about 8 hours following a
single dose, and a plasma [WA] Cmin of no less than about 250 ng/mL from about
2.5
to about 8 hours following the single dose, and the subject achieves an
average plasma
[WA] of 250 ng/mL to about 2500 ng/ml for about 8 hours following the single
dose,
and at 2.5 hours, 4 hours, and 8 hours following the single dose, the
percentage of
proliferating lymphocytes in a whole blood sample from the subject is reduced
by at least
about 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, or more, as compared
to
the percentage of proliferating lymphocytes in a whole blood sample obtained
from the
38
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
subject 15 or fewer minutes prior to the single dose, as determined using
monoclonal
antibody Ki-67.
In some embodiments, the subject achieves a plasma [MPA] Cmax of
about 2500 ng/mL over about 8 hours following the first dose, and a plasma
[MPA] Cmin
of no less than about 250 ng/mL from about 2.5 to about 8 hours following the
single
dose.
In some embodiments, the subject achieves a plasma [MPA] Cmax of
about 2000 ng/mL over about 8 hours following the single dose, and a plasma
[MPA]
Cmin of no less than about 500 ng/mL from about 2.5 to about 8 hours following
the
single dose.
In certain embodiments, the subject achieves a plasma [MPA] Cmax of
about 1500 ng/mL over about 8 hours following the single dose, and a plasma
[MPA]
Cmin of no less than about 600 ng/mL from about 2.5 to about 8 hours following
the
single dose.
In particular embodiments, the subject achieves a plasma [MPA] Cmax of
about 700ng/mL over about 8 hours following the single dose, and a plasma
[MPA]
Cmin of no less than about 250 ng/mL from about 1 to about 8 hours following
the single
dose.
In some embodiments, the administering comprises administering the
dose for 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or more, consecutive
days.
In certain embodiments, the administering comprises administering the
single dose for seven or more consecutive days, whereupon 24 hours after the
dose of the
seventh day, and prior to any subsequent dose, the percentage of proliferating
lymphocytes in a whole blood sample from the canine subject is reduced by
about 35%,
about 40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%,
about
75%, or more, relative to the amount of proliferating lymphocytes in a whole
blood
sample obtained from the subject 15 or fewer minutes prior to the dose of the
first day.
In further embodiments, the administering comprises administering the
dose for 10 or more days, wherein over the 10 days, the subject exhibits no
adverse
39
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
gastrointestinal events, wherein an adverse gastrointestinal event comprises
emesis,
diarrhea, soft stool, or any combination thereof.
In some embodiments, following the administration, the canine subject
exhibits a reduction in the severity of a symptom associated with the
autoimmune disease
or disorder. Such symptoms include, for example, rash, elevated levels of
autoantibodies
and/or inflammatory cytokine levels, fever, fatigue, stiffness, pain,
blisters, itchiness,
discharge, eczema, swelling, hair loss, loss of appetite, asthma, and foul
odor.
Uses
In another aspect, the present disclosure provides uses of a
controlled-release MPA formulation for suppressing lymphocyte proliferation
and/or
activation in a canine subject.
In some embodiments, a use comprises administering to the subject a
controlled-release formulation comprising a mycophenolic acid (MPA) active
agent such
that the subject achieves an average plasma [MPA] of about 250 ng/mL to about
3000
ng/mL (e.g., about 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800,
850, 900,
950, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100,
2200,
2300, 2400, 2500, 2600, 2700, 2800, 2900, or about 3000 ng/mL)over about 8
hours
following a first dose of the controlled-release formulation, wherein the
subject achieves
a plasma [MPA] Cmax of about 3000 ng/mL, optionally about 2500 ng/mL,
optionally
about 2000 ng/mL, optionally about 1500 mL, optionally about 700 ng/mL,
optionally
about 600 ng/mL, optionally about 500 ng/mL, over about 8 hours following the
first
dose, and a plasma [MPA] Cmin of no less than about 250 ng/mL from about 2.5
to about
8 hours following the first dose, whereupon at 2.5 hours, 4 hours, and 8 hours
following
the first dose, a percentage of proliferating lymphocytes in a whole blood
sample from
the subject is reduced as compared to the percentage of proliferating
lymphocytes in a
whole blood sample obtained from the subject 15 or fewer minutes prior to the
first dose,
as determined using monoclonal antibody Ki-67.
In further embodiments, the subject achieves an average of plasma [MPA]
of: (i) about 350 ng/ml to about 2000 ng/mL over 8 hours following the first
dose; (ii)
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
about 500 ng/ml to about 1700 ng/mL over 8 hours following the first dose; or
(iii) about
650 ng/ml to about 1500 ng/mL over 8 hours following the first dose.
In accordance with any of the herein-described embodiments, at 2.5
hours, preferably at 4 hours, more preferably at 8 hours following the first
dose, the
percentage of proliferating lymphocytes in a whole blood sample from the
subject is
reduced by at least about 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, or
more, relative to the percentage of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the first dose.
In accordance with any of the herein-described embodiments, the subject
is administered the composition for 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, or more,
consecutive days.
In further embodiments, the subject receives a single dose of the
controlled-release composition per day.
In still further embodiments, the administering comprises administering
the single dose for 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or more
days, and
whereupon 24 hours after the dose of each of the second, third, fourth, fifth,
sixth,
seventh, eighth, ninth, tenth, eleventh, twelfth, thirteenth, fourteenth, or
fifteenth days,
and prior to any subsequent dose, the percentage of proliferating lymphocytes
in a whole
blood sample from the subject is reduced by about 35%, about 40%, about 45%,
about
50%, about 55%, about 60%, about 65%, about 70%, about 75%, or more, relative
to the
amount of proliferating lymphocytes in a whole blood sample obtained from the
subject
15 or fewer minutes prior to the dose of the first day. In particular
embodiments, the
administering comprises administering the single dose for 7 or more
consecutive days,
whereupon after the dose of the seventh day, the percentage of proliferating
lymphocytes
in a whole blood sample from the subject is reduced by about 35%, about 40%,
about
45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75%, or
more,
relative to the amount of proliferating lymphocytes in a whole blood sample
obtained
from the subject 15 or fewer minutes prior to the dose of the first day.
In accordance with any of the herein-described embodiments, (i) the
percentage of proliferating lymphocytes in a whole blood sample obtained from
the
41
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
subject at 4 hours, optionally at 8 hours, following administration of a first
dose is lower
than (ii) the percentage of proliferating lymphocytes in a whole blood sample
obtained
from a reference canine subject (i.e., a reference canine subject as described
herein) that
was administered an immediate-release formulation comprising a MPA active
agent.
In accordance with any of the herein-described embodiments, the
administering comprises administering the single dose for 10 or more days,
wherein over
the 10 days, the subject exhibits no adverse gastrointestinal events, wherein
an adverse
gastrointestinal event comprises emesis, diarrhea, soft stool, or any
combination thereof.
In further embodiments, the administering comprises administering the
single dose for 15 or more days, wherein over the 15 days, the subject
exhibits no adverse
gastrointestinal events, wherein an adverse gastrointestinal event comprises
emesis,
diarrhea, soft stool, or any combination thereof.
In further embodiments, the administering comprises administering the
single dose for 10 or more days, wherein over the 10 days, the subject
exhibits a reduced
number, a reduced severity, or both, of an adverse gastrointestinal event as
compared to a
reference canine subject (i.e., a reference canine subject as described
herein) that was
administered an immediate-release formulation comprising a MPA active agent,
wherein
an adverse gastrointestinal event comprises emesis, diarrhea, soft stool, or
any
combination thereof.
In further embodiments, the immediate-release formulation was
administered to the reference canine subject at a twice-daily dose of 7.4
mg/kg MPA,
wherein the immediate-release formulation optionally comprises mycophenolate
mofetil.
In accordance with any of the herein-described embodiments, the MPA
active agent comprises mycophenolate sodium, wherein the mycophenolate sodium
is
.. optionally administered to the canine subject orally.
In further embodiments, the controlled-release composition comprises a
fillable capsule comprising a plurality of particulate subunits that each
comprise: (i) a
core, wherein (a) the MPA active agent is comprised in an active layer
disposed over at
least a portion of the core; or (b) the MPA active agent is disposed within
the core; (ii) a
.. controlled-release layer disposed over at least a portion of (a) the active
layer or (b) the
42
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
core; (iii) an optional seal coat layer disposed between (a) the active layer
and the
controlled-release layer, or (b) the core and the controlled-release layer;
(iv) an optional
protective layer disposed over the controlled-release layer, wherein the MPA
active agent
comprises mycophenolate sodium and is optionally present in the controlled-
release
composition from about 3 mg to about 2.2 g (i.e., about 3, 4, 5, 6, 7, 8, 9,
10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32,
33, 34, 35, 36, 37,
38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56,
57, 58, 59, 60, 61,
62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80,
81, 81, 82, 84, 85,
86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 105, 110, 115,
120, 125, 130,
145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 205, 210, 215,
220, 225, 230,
235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295, 300, 305,
310, 315, 320,
325, 330, 335, 340, 345, 350, 355, 360, 365, 370, 375, 380, 385, 390, 395,
400, 405, 410,
415, 420, 425, 430, 435, 440, 445, 450, 455, 460, 465, 470, 475, 480, 485,
490, 495, 500,
505, 510, 515, 520, 525, 530, 535, 540, 545, 550, 555, 560, 565, 570, 575,
580, 585, 590,
595, 600, 605, 610, 615, 620, 625, 630, 635, 640, 645, 650, 655, 660, 665,
670, 675, 680,
685, 690, 695, 700, 705, 710, 715, 720, 725, 730, 735, 740, 745, 750, 755,
760, 765, 770,
775, 780, 785, 790, 795, 800, 805, 810, 815, 820, 825, 830, 835, 840, 845,
850, 855, 860,
865, 870, 875, 880, 885, 890, 895, 900, 905, 910, 915, 920, 925, 930, 935,
940, 945, 950,
955, 960, 965, 970, 975, 980, 985, 990, 995, 1000, 1100, 1200, 1300, 1400,
1500, 1600,
1700, 1800, 1900, 2000, 2100, or 2200 mg).
In accordance with any of the herein-described embodiments, the canine:
(i) has, or is suspected of having, an autoimmune disease or disorder
associated with
aberrant lymphocyte proliferation and/or activation, wherein the autoimmune
disease or
disorder optionally comprises atopic dermatitis, arthritis, myasthenia gravis,
celiac
.. disease, diabetes mellitus type 1, Grave's disease, inflammatory bowel
disease, multiple
sclerosis, psoriasis, rheumatoid arthritis, systemic lupus erythematosus,
Behcet's disease,
pemphigus vulgaris, refractory incomplete systemic lupus erythematosus, lupus
nephritis, immunoglobulin A nephropathy, small vessel vasculitides,
scleroderma
(systemic sclerosis or SSc), idiopathic thrombocytopenic purpura (ITP),
psoriasis,
apernicious anemia, vitiligo, autoimmune hemolytic disease,
glomerulonephritis,
43
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
immune cytopenias, meningoencephalomyelitis, subepidermal blistering
autoimmune
disease, immunobullous diseases, cutaneous vasculitis, recurrent erythema
multiforme,
erythema nodosum, lichen planus, cutaneous Crohn's disease, sarcoidosis,
hepatitis,
pyoderma gangrenosum, or any combination thereof; (ii) has undergone, is
undergoing,
or will undergo an organ transplant and/or an artificial implant; or (iii)
both of (i) and (ii).
In some embodiments, following the administration, the veterinary
subject exhibits a reduction in the severity of a symptom associated with the
autoimmune
disease or disorder, wherein the symptom is not aberrant lymphocyte
proliferation. Such
symptoms include, for example, rash, elevated levels of autoantibodies and/or
inflammatory cytokine levels, fever, fatigue, stiffness, pain, blisters,
itchiness, discharge,
eczema, swelling, hair loss, loss of appetite, asthma, and foul odor.
Also provided herein are methods of preparing a medicament for
suppressing lymphocyte proliferation and/or activation in a canine, the method
comprising preparing a controlled-release formulation comprising a
mycophenolic acid
(MPA) active agent such that the subject maintains an average plasma [MPA] of
from
about 250 ng/mL to about 3000 ng/mL for about 8 hours following a first dose
of the
controlled-release formulation, such the subject achieves a plasma [MPA] Cmax
of about
3000 ng/mL, optionally about 2500 ng/mL, optionally about 2000 ng/mL,
optionally
about 1500 ng/mL, optionally about 700 ng/mL, optionally about 600 ng/mL,
optionally
about 500 ng/mL, over about 8 hours following the first dose, and a plasma
[MPA] Cmin
of no less than about 250 ng/ml from about 1 to about 8 hours following the
first dose,
and such that at 2.5 hours, 4 hours, and 8 hours following the first dose, the
percentage of
proliferating lymphocytes in a whole blood sample from the subject is reduced
as
compared to the percentage of proliferating lymphocytes in a whole blood
sample
obtained from the subject 15 or fewer minutes prior to the first dose, as
determined using
monoclonal antibody Ki-67.
Dosing and Administration
It will be understood that a variety of dosages of a controlled-release
formulation of the present disclosure (and of a MPA active agent contained
therein) may
be administered to a veterinary subject in accordance with, e.g., the
physiological
44
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
characteristics of the veterinary subject (e.g., size, GI length, digestive
rate, digestive pH,
stomach crushing strength), the state of health of the veterinary subject
(e.g., the urgency
of the need for treatment and of what strength) and other factors. For
example, in certain
embodiments, a dosing regime or schedule comprises a single administration of
a single
dose of a controlled-release formulation as described herein). In certain
embodiments, a
dosage regime or schedule comprises multiple administrations of a single dose
over the
course of, e.g., a day. Alternatively, a dosage regime or schedule may
comprise single or
multiple administrations of multiple doses of a controlled-release
formulation. In
embodiments comprising multiple doses, the doses may be administered
simultaneously,
contemporaneously, or sequentially.
An appropriate dose will be determined according to any one or more of a
variety of factors typically considered when determining an appropriate drug
dose; e.g.,
the size, age, species, gender, and general health of a subject receiving the
dose; the type,
severity, and stage of a disease condition; known PK parameters of the drug,
such as
absorption, in vivo half-life, and the like). Table 1 provides non-limiting
examples of
dosages of a formulation of the present disclosure for canine subjects ranging
in size from
2 to 80 kilograms; it will be understood that for subjects larger than 80
kilograms, or
smaller than 2 kilograms, or in between the shown sizes, and/or of non-canine
species,
dosages may be adjusted accordingly.
Table 1. Exemplary Dosages of a Controlled-Release Formulation
Dosage (mg/kg)
Size (kg) 10 15 20 25 30 35 40
2 20 30 40 50 60 70 80
5 50 75 100 125 150 175 200
10 100 150 200 250 300 350 400
20 200 300 400 500 600 700 800
40 400 600 800 1000 1200 1400 1600
60 600 900 1200 1500 1800 2100 2400
80 800 1200 1600 2000 2400 2800 3200
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
EXAMPLES
EXAMPLE 1:
PREPARATION AND TESTING OF CERTAIN MYCOPHENOLATE CONTROLLED-RELEASE
VETERINARY COMPOSITIONS ACCORDING TO THE PRESENT DISCLOSURE
Preparation of Active Agent-Coated Beads:
Materials
Sugar spheres (#25/30; COLORCON , Harleysville, PA)
OPADRY Clear, hydroxypropyl methylcellulose-based coating
(COLORCON , Harleysville, PA)
Purified Water
Sodium Mycophenolate
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
Table 2: Composition of drug coating solution
Component Batch formula (g)
OPADRY Clear 25
Purified Water 475
Sodium Mycophenolate 240
1. OPADRY Clear was dispersed in purified water and
stirred until
a clear solution was obtained.
2. Mycophenolate sodium was added to the solution and stirred for 1
hour.
3. 500 g of sugar beads as loaded into the fluid bed
chamber.
46
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
4. The bed was fluidized, sugar beads were warmed and the coating
solution prepared as described in Table2 was sprayed onto the fluidized beads.
5. Coating was continued with periodic drying and weighing of the
coated beads.
6. Coating was continued until the beads had gained approximately
40% in weight.
7. Beads were dried overnight (15 hours) at 40 C in a hot
air oven
Exemplary Preparation of Extruded Beads with Active Agent:
Materials
Drug Substance: Sodium mycophenolate: 50 to 90%
Extrusion/Spheronization Aid: Microcrystalline Cellulose: 15 to 20%
Binders: Hydroxypropyl cellulose or hydroxyl propyl methyl cellulose or
Pregelatinized Starch or Ethyl Cellulose or poly(vinyl pyrrolidone) (2 to 5%)
Modified release excipients: HPMC, HPC, acrylic polymers, HEC, EC ¨
these can be incorporated into the beads or applied as coating
Other fillers: lactose, maltodextrin, mannitol, sorbitol, dicalcium
phosphate/ (as needed)
Superdisintegrants: crosslinked PVP, sodium starch glycolate, sodium
croscarmellose (% as needed)
Manufacturing
The drug substance was mixed with microcrystalline cellulose, binder,
and disintegrant in a planetary mixer or a high shear mixer for 10 minutes;
The required amount of water was added to the mixer and mixing
continued for another 5 to 10 minutes;
The resulting wet mass was passed through an extruder to obtain an
extrudate (example equipment: Caleva, LCI, Glatt etc.);
47
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
The extrudate was then spheronized on a spheronizer fitted with a
crosshatch plate to form spheronized beads (example equipment: Caleva, LCI,
Glatt)
The spheronized beads were then dried in a fluid bed dryer till the desired
moisture content (<1%) was reached;
The dried beads were then passed through screens to remove fine beads (<
5001.tm) and coarse beads (>25001.tm);
The dried beads were then loaded into a fluid bed coater and coated with
an appropriate amount of a rate-controlling polymer (15 to 30% range);
Additional enteric coating (optional) was then applied to the coated beads;
The beads were then dried after all coating steps were completed.
Preparation of Seal Coated Beads:
Materials
Drug coated or extruded beads (from above sections)
OPADRY Clear
Talc Purified Water
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
1. OPADRY Clear was dispersed in purified water and stirred to
obtain a clear solution.
2. Talc was dispersed into the OPADRY solution and stirred to
obtain a smooth dispersion.
3. About 300g of the drug coated beads (from above) was loaded into
the fluid bed coater.
4. The bed was fluidized, drug coated beads were warmed,
and the
coating solution prepared as described in Table 3 was sprayed onto the
fluidized beads.
48
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
5. Coating was continued with periodic drying and weighing of the
coated beads.
6. Coating was continued until the beads had gained approximately 8
to 10% in weight.
7. Beads were dried overnight (15 hours) at 40 C in a hot air oven.
Table 3: Composition of seal coat solution
Component Batch formula (g)
OPADRY Clear 10
Talc 20
Purified water 170
Preparation of Controlled-release Layers:
SURELEASE Coating ¨ Option 1
Materials
Seal Coated beads (from above)
SURELEASE E-7 19040, aqueous ethylcellulose rate controlling
polymer (COLORCON , Harleysville, PA) (Other grades of SURELEASE can be used
if desired)
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
Table 4: Composition of SURELEASE Coating solution
Component Batch formula (g)
SURELEASE E7 19040 240
Purified water 170
49
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
1. SURELEASE was dispersed in purified water and stirred to
obtain a smooth dispersion.
2. About 300g of the seal coated beads was loaded into the fluid bed
coater.
3. The bed was fluidized, the seal coated beads were warmed, and the
coating solution prepared as described in Table 4 was sprayed onto the
fluidized beads.
4. Coating was continued with periodic drying and weighing of the
coated beads.
5. Coating was continued until the beads had gained desired coating
weight.
6. Coated bead samples were withdrawn at desired weight gain (e.g.:
15%, 22%, 30%).
7. At the end of the coating, beads were dried overnight in the oven at
60 C/75% relative humidity conditions.
EUDRAGIT Coating ¨ Option 2
Materials
Seal Coated beads (from above)
EUDRAGIT RS (acrylic controlled-release polymer either as powder
or premade dispersion ¨ RS 30D)
EUDRAGIT RL (acrylic controlled-release polymer either as powder or
premade dispersion ¨ RL 30D)
Talc
Triethyl citrate (TEC)
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
Table 5: Sample composition for EUDRAGIT coating
Component Batch formula (g)
EUDRAGIT RL 30D (1 part)* 39.3
EUDRAGIT RS 30D (9 parts)* 352.9
TEC 23.5
Talc 58.8
Purified Water 525.5
*This composition will be referred as EUDRAGIT RS (90): RL (10) or
EUDRAGIT RS/RL: 90/10. This ratio can be altered in any composition,
increasing
amount of RS 100 will reduce membrane permeability with decrease in release
rate from
bead.
1. Talc and TEC were dispersed in purified water and homogenized
until a smooth dispersion is obtained.
2. The dispersion from Step (1) was mixed until a uniform dispersion
was obtained.
3. The dispersion was filtered through 80 mesh sieve to remove any
coarse particles.
4. About 300 g of seal coated beads was loaded into the fluid bed
coater.
5. The bed was
fluidized, seal coated beads were warmed and the
coating solution prepared as described in Table 4 was sprayed onto the
fluidized beads.
6. Coating was continued with periodic drying and weighing of the
coated beads until the desired weight gain was obtained (typically 15% to
30%).
7. Coated beads were dried overnight (15 hours) at 40 C in a hot air
oven.
E.R. Coating ¨ Option 3
Materials
Ethylcellulose 10
51
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Klucel EF (HPC)
Talc
Dibutyl sebacate
DI water
Ethanol
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
Sprayer
Table 6: Sample composition for 30 wt% gain
Component Wt %
Ethylcellulose 10 4.15
Klucel EF (HPC) 0.46
Talc 0.92
Dibutyl Sebacate 0.46
DI water 9.40
Ethanol 190pf 84.61
1. Dibutyl sebacate was dissolved in a mixture of ethanol and
deionized water.
2. The required quantity of HPC was dispersed in the hydroalcoholic
mixture and stirred to obtain a solution.
3. The required quantity of ethylcellulose was dispersed in the above
mixture and stirred until a solution was obtained
4. Talc was dispersed in the above solution and stirred to obtain a
smooth dispersion.
5. The drug coated beads were loaded into the fluid bed
coater
52
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
6. The bed was fluidized, beads were warmed, and the coating
solution prepared as described in step 4 was sprayed onto the fluidized beads.
7. Coating was continued with periodic drying and weighing of the
coated beads
8. Coating was continued until the beads had gained desired coating
weight.
9. Coated bead samples were withdrawn at desired weight
gain (e.g.,
15%, 22%, or 30%)
Preparation of Protective Layer:
Materials
Control release layer coated beads (from above)
EUDRAGIT L30 D 55 (other grades of EUDRAGIT polymers, or
OPADRY polymers that confer enteric protection can also be used)
Triethyl citrate (TEC)
Talc
Equipment
Mechanical stirrer
Fluid bed coater
Hot air oven
Table 7: Composition of Enteric Coating
solution
Component Batch formula (g)
EUDRAGIT L3OD 55 83.3
TEC 2.5
Talc 12.5
Purified Water 101.6
53
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
1. Talc and TEC were dispersed in purified water and homogenized
until a smooth dispersion was obtained.
2. The dispersion from Step (1) was dispersed into EUDRAGIT
L3OD 55 suspension and mix until a uniform dispersion was obtained.
3. The dispersion was filtered through 80 mesh sieve to remove any
coarse particles.
4. About 300 g SURELEASE coated beads was loaded into the
fluid bed coater; alternatively, about 300 g of EUDRAGIT coated beads from
above
may be used.
5. The bed was fluidized, controlled-release coated beads were
warmed and the coating solution prepared as described in Table 7 is sprayed
onto the
fluidized beads.
6. Coating was continued with periodic drying and weighing of the
coated beads until the desired weight gain of EUDRAGIT L 30 D55 was obtained
(typically 8 to 15%).
7. Coated beads were dried overnight (15 hours) at 40 C in a hot air
oven.
Determination of Release Rate
Release rate determinations were performed on beads obtained after
coating with controlled-release layer(s) and optionally a protective layer.
Release rate
determinations were conducted as follows:
Dissolution Apparatus: USP Type 1
Dissolution Media Volume and Speed: 900 mL at 100 rpm
A known quantity of beads were weighed (based on assay of coated
beads) and placed in the USP Type 1 basket apparatus. For a biphasic
dissolution profile
(biphasic media), beads were exposed to pH 1.2 media for 2 hours. After 2
hours, the
basket was moved to buffer media at pH 6.8 and dissolution was continued for
an
additional 10 hours, 12 hours, 14 hours or 24 hours, as desired. Aliquots were
withdrawn
at periodic intervals and analyzed for mycophenolate sodium using a UV
detection
54
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
method. Data from representative experiments are shown in FIGS. 6-9 and Table
8,
below.
FIG. 4 shows the release of sodium mycophenolate from
controlled-release beads in biphasic media as noted above. The bead construct
is sugar
sphere/drug layer/HPMC seal coat/ethyl cellulose (SURELEASE ). The data was
generated for beads with only a controlled-release layer. A protective layer
was not
included. As shown, release from beads coated with a 22% by weight ethyl
cellulose
controlled-release layer was higher than that for beads coated with a 30% by
weight ethyl
cellulose layer. Both compositions maintained their integrity at pH 1.2 (0-2
hours), with
rapid release upon transitioning to pH 6.8 (>2 hours).
FIG. 5 shows the release of sodium mycophenolate from
controlled-release beads in pH 6.8 media. The bead construct is sugar
sphere/drug
layer/HPMC seal coat/acrylic polymer (EUIDRAGIT RS 100). A protective layer
was
not included.
FIG. 6 shows the release of sodium mycophenolate from
controlled-release beads in biphasic media, as described above. The bead
construct is
sugar sphere/drug layer/HMPC seal coat/EUDRAGIT RS 100/EUDRAGIT L3OD 55.
The EUIDRAGIT RS 100 layer provides the controlled-release characteristics,
while the
EUDRAGIT L3OD 55 provides the protective, enteric coating. As noted, little
to no
release occurred at pH 1.2 (0-2 hours), with release occurring once the pH was
raised to
6.8 (>2 hours).
FIG. 7 shows the release of sodium mycophenolate from
controlled-release beads in pH 6.8 media. The bead construct is sugar
sphere/drug
layer/EUDRAGIT RS 100:EUDRAGIT RL 100 (90:10). A seal coat and a protective
layer were not included. Rapid release is noted occurring around hours 2-6.
FIG. 8 shows release of sodium mycophenolate from a particulate
subunits (coated beads) comprising 30 wt% Surelease polymer coat and a
protective
enteric coating. pH was switched from 1.2 to 6.8 following 2h incubation.
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
FIG. 9 shows release of sodium mycophenolate from particulate subunits
(coated beads) comprising a solvent-based coating. pH was switched from 1.2 to
6.8
following 2h incubation.
Table 7 below provides MPA active agent release data from particulate
subunits of two exemplary formulations according to the present disclosure.
Two sets of
extended-release enteric-coating Na.MPA coated beads were prepared as
described
above (22 wt% Surelease coating and 30 wt% coating) and placed in acid (2h, pH
1.2)
followed by a buffer that raised pH to approximately 6.8.
Table 8.
Hours Following % NaMPA released % NaMPA released
Administration (22% Surelease) (30% Surelease)
0.5 0.9 0.0
1 3.2 1.5
2 7.9 4.9
2.5 25.1 13.2
3 35.8 21.4
4 51.3 31.7
5 61.4 40.7
6 67.9 48.0
7 72.8 54.0
8 76.2 59.1
81.5 66.1
14 86.9 76.1
EXAMPLE 2:
PREPARATION OF MYCOPHENOLATE
CONTROLLED-RELEASE MINI-TABLETS
Materials
Mycophenolate Sodium 20-60%, suitably 50%
56
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Microcrystalline cellulose 30-60%, suitably 43%
Poly Vinyl pyrrolidone (PVP) 1-10%, suitably 5%
Magnesium Stearate 0.5-5 %, suitably 2%
The required quantities of mycophenolate sodium and microcrystalline
cellulose were mixed together in a high shear mixer for about 5 minutes.
The required quantity of PVP was dissolved in water to form a 10% w/w
solution of PVP.
The PVP solution was gradually added to the high shear mixer and the
blend was mixed until a wet mass was formed.
The wet mass was transferred to a fluid bed dryer and dried.
The dried granulation was passed through a sieve such that very coarse
and very fine particles were removed.
The sieved granulation was transferred to a V blender and mixed with the
Magnesium stearate for about 3 minutes.
The lubricated granules were compressed into mini-tablets using a 2 mm
round standard concave multi tip tooling on a compression press.
The mini-tablets were coated with a seal coat, a controlled-release layer,
and a protective layer, as described herein.
The appropriate quantity of mini-tablets can be administered to the
veterinary subject, either filled in a capsule, as a slurry, as a sachet, a
dragee, etc.
EXAMPLE 3:
IN VIVO PHARMACOKINETICS OF MPA IN A CANINE MODEL
Two single dose cross-over studies were conducted using a canine model
(male beagle dogs) to evaluate the potential of an enteric coated-extended
release sodium
mycophenolate formulation ("EC-ER-Na.MP"). In a first cross-over study, the
pharmacokinetics of MPA and its metabolites (MPAG and AcMPAG) following oral
dosing of 180 mg of EC-Na.MPA was compared with intracolonic (IC)
administration of
Na.MPA. In a second cross-over study, 270 mg of an EC-ER-Na.MPA formulation
was
57
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
administered in the both fed and fasted states and pharmacokinetics were
compared with
oral administration of 10 mg/kg MMF in the fasted state. Both studies were
conducted by
Absorption Systems (San Diego, CA).
Study 1: For three days prior to IC dosing, dogs (n=5) were offered a soft
diet consisting of canned wet food (Pedigree Choice Cuts). Otherwise, the
dogs were
offered their standard diet (LabDiet 5006 laboratory canine diet). Prior to
each dosing
event, the dogs were fasted for 12 hours prior to dosing until 4 hours post-
dose, when
food was returned. Animals had free access to water throughout the study.
Prior to IC
dosing, each dog was given a non-stimulant enema approximately 1 hour prior to
dosing
to remove feces from the colon. Prior to PO dosing, each dog (n=5) was pre-
treated with
an intramuscular dose of pentagastrin (61.tg/kg) approximately 30 minutes
prior to dosing.
Capsules were administered by placement in the back of the throat followed by
a 10 mL
flush with water.
Each dog received a total dose of 180 mg of MPA for each dose. For IC
dosing, MPA was delivered as a solution via an endoscope, and for PO dosing
each dog
received a single Myfortic 180 mg enteric coated capsule. Following
administration,
blood samples were collected up to 24 hours post-dose. Plasma concentrations
of MPA,
MPAG, and AcMPAG were determined with a qualified LC-MS/MS method, and
pharmacokinetic parameters were determined with WinNonlin v.6.4 software.
Following IC dosing of MPA, maximum plasma concentrations (average
C. of 29460 12587 ng/mL) were observed between 5 and 30 minutes post-dose.
The
average half-life was 5.55 1.77 hours, and the average exposure based on the
dose-normalized AUCIast was 1817 925 hr*kg*ng/mL/mg. MPAG after MPA dosing
had an average Cmax of 4826 1156 ng/mL. The t. for MPAG ranged from 15
minutes
to 1 hour post-dose, and the average AUCIast was 11702 4794 hr*ng/mL. AcMPAG
after MPA dosing had an average Cmax of 303 87.8 ng/mL. The tmax for MPAG
ranged
from 5 to 15 minutes post-dose, and the average AUCIast was 233 160
hr*ng/mL.
Following PO dosing of MPA, maximum plasma concentrations (average
C. of 27320 12037 ng/mL) were observed between 30 minutes and 2 hours post-
dose.
The average half-life, determined in 2 dogs, was 4.49 hours, and the average
exposure
58
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
based on the dose-normalized AUCIast was 2234 799 hr*kg*ng/mL/mg. MPAG after
MPA dosing had an average C. of 14316 5033 ng/mL. The tmax for MPAG ranged
from 1 to 2 hours post-dose, and the average AUClast was 28882 8313
hr*ng/mL.
AcMPAG after MPA dosing had an average C. of 426 113 ng/mL. The tmax for
.. MPAG ranged from 30 minutes to 2 hours post-dose, and the average AUCIast
was 529
217 hr*ng/mL.
Based on average values, systemic exposure to MPA was similar
following IC and PO dosing. The average C. after IC and PO doses were 29460
and
27320 ng/mL, respectively, and the average dose-normalized AUCIast values were
1817
and 2234 hr*kg*ng/mL/mg, respectively. However, MPA did appear to be more
rapidly
absorbed following IC dosing in comparison to the PO dose. MPAG was present at
a
much higher concentration in the plasma than AcMPAG. Systemic exposure to each
of
these glucuronide metabolites after IC dosing was approximately 40% of that
after PO
dosing. The average AUCIast for MPAG was 28882 hr*ng/mL after PO dosing and
11702
hr*ng/mL after IC dosing. The average AUCIast for AcMPAG was 529 hr*ng/mL
after
PO dosing and 233 hr*ng/mL after IC dosing.
The mean pharmacokinetic parameters and the drug to metabolite ratios
are summarized in Table 9. The MPA/MPAG ratio and MPA/AcMPAG ratio were each
observed to be almost 2-fold higher following the IC dosing compared to the
oral
administration.
59
0
Table 9: Mean MPA, MPAG and AcMPAG Pharmacokinetic Parameters following Oral
and t..)
o
t..)
o
Intracolonic Administration of Na.MPA (n=5 male beagle dogs)
O-
u,
o
cio
ORAL MPA MPAG
AcMPAG MPA/MPAG MPA/AcMPAG c,.)
Mean SD Mean SD Mean SD Ratio
Ratio
Animal Weight (kg) 11.1 0.5
Dosed per dog (mg) 180 0
Dose (mg/kg) 16.2 0.7
Cmay, (ng/mL) 27320 12037 14316 5033 426
113 P
,
tmax (hr) 1 0.61 1.4 0.55 1.1
0.55 ,
,
.3
t112 (hr) 4.49 ND 7.12 1.95 ND ND
2
,
MRTIast (hr) 2.86 0.856 3.75 0.371 1.71
0.628 ,
o
,
u2
AUClast (hrng/mL) 36435 13501 28882 8313 529
217 1.42 88.40
AUCõ (hr=ng/mL) 45693 ND 30360 9234 ND ND
1.29 ND
INTRACOLONIC MPA MPAG
AcMPAG MPA/MPAG MPA/AcMPAG
Mean SD Mean SD Mean SD Ratio
Ratio
1-d
n
Animal Weight (kg) 10.7 0.5
Dosed per dog (mg)
180 0 cp
t..)
o
,-,
Dose (mg/kg) 16.8
0.8 cio
O-
u,
o
Cmax (ng/mL) 29460 12587 4826 1156 303
87.8 =
-.1
,.tD
0
INTRACOLONIC MPA MPAG AcMPAG
MPAAVIPAG MPA/AcMPAG
Mean SD Mean SD Mean SD Ratio Ratio
tmay, (hr) 0.23 0.17 0.6 0.38 0.22
0.07 oe
c7,
tin (hr) 5.55 1.77 6.61 2.36 ND ND
MRTIast (hr) 2.76 0.324 4.16 0.688 1.33 0.897
AUCIast (hrng/mL) 30993 17092 11702 4794 233
160 2.86 171.19
AUG, (hr=ng/mL) 31948 17903 13475 4252 ND ND
2.45 ND
C.,: maximum plasma concentration; t.õ: time of maximum plasma concentration;
t112:
half-life; MRT1.: mean residence time, calculated to the last observable time
point; AUCI.:
areaunder the curve, calculated to the last observable time point; AUC.: area
under the curve,
extrapolated to infinity; BLOQ: below the limit of quantitation (1 ng/mL); ND:
not determined.
1-d
oe
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Study 2: In this treatment cross-over study, the pharmacokinetics of MPA
and its metabolites (MPAG and AcMPAG) following 270 mg EC-ER-Na.MP
("OKV-1001") administered in the fed and fasted state were compared with those
following oral administration of immediate release 10 mg/kg MMF in a fasted
state (n=5
per dose group; same group of dogs for each treatment).
For EC-ER-Na.MP and MMF administration in the fasted state, dogs
were fed a certified laboratory diet (5006 laboratory canine diet from
LabDiet) and then
fasted for a minimum of twelve hours prior to dosing. Food was provided
approximately
4 hours post-dose. Water was supplied ad libitum to the animals.
For EC-ER-Na.MP administration in the fed state, dogs were fed a
certified laboratory diet (5006 laboratory canine diet from LabDiet), fasted
for a
minimum of twelve hours, and then fed (Alpo Can food) prior to dosing and then
dosed
no more than 30 minutes post completion of food. The amount of food provided
and
consumed by each animal was recorded. Regular lab diet was provided
approximately 4
hours post-dose. Water was supplied ad libitum to the animals.
For MMF administration, an MMF oral suspension was prepared
according to the instructions for CellCeptg. Leftover dosing solutions were
stored at
room temperature.
For both EC-ER-Na.MP treatments, blood was collected pre-dosing, then
at either: 30 minutes, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 9, 12, and 24 hours
(processed to obtain
plasma); or 1, 2, 3, 4, 6, 8, 12, and 24 hours (processed to PBMC). For the
MMF
treatment, blood was collected at pre-dose, 15 minutes, 30 minutes, 1, 1.5, 2,
2.5, 3õ 4,6,
8, 9, 12, and 18 hours (processed to plasma); or at pre-dose, 1, 2, 3, 4, 6,
8, 12, and 18
hours (processed to obtain PBMCs).
Plasma concentrations of MPA, MPAG, and AcMPAG were determined
with a qualified LC-MS/MS method, and pharmacokinetic parameters were
determined
with WinNonlin v.6.4 software. Plasma concentration curves are shown in FIGS.
10-13.
The mean pharmacokinetic parameters and drug to metabolite ratios are
summarized in
Table 10. Under fasted conditions, the MPA/MPAG ratio was 1.5 to 2.0 fold
higher with
62
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
EC-ER-Na.MP compared to the reference oral dosing (Table 10). The MPA/AcMPAG
ratio also trended to be higher, although to a lesser extent.
63
0
Table 10: Mean Pharmacokinetic Parameters of MPA and Metabolites Following
Oral MMF and t..)
o
t..)
o
EC-ER-Na.MP Administration
u,
o
cio
ORAL MMF MPA MPAG
AcMPAG MPA/MPAG MPA/AcMPAG c,.)
Mean SD Mean SD Mean SD Ratio
Ratio
Animal Weight (kg) 11 1.4
MMF Dose (mg/kg) 10 -
Dose (mg/kg) MPA Equivalent 7.39 -
Cmay, (ng/mL) 1991 1434.5 800.2 300.8
18.25 14.61 P
,
-Lay, (hr) 0.75 0.5 1.1 0.42 0.75
.05 ,
,
.3
t1/2 (hr) I -1 5.78 I 5.68 I 14.30 I 10.91 I 2.42
I 0.65 rõ.
,
,
MRTIast (hr) 5.89 1.19 7.2 0.65 5.83
0.44 .
,
AUCIast (hr?=ng/mL) 5644 2144 3899 1240 44.6 21.7
1.48 139.8
AUC? (hr?=ng/mL) 6543 3277 6615.7 2637.8 50.8 23.7
1.02 133.3
EC-ER-Na.MPA MPA MPAG
AcMPAG MPA/MPAG MPA/AcMPAG
Mean SD Mean SD Mean SD Ratio
Ratio 1-d
n
Animal Weight (kg) 11.1 1.3
cp
Na.MPA Dose (mg)
270 - t..)
o
,-,
cio
Dose (mg/kg) MPA Equivalent 22.94 2.47
u,
o
Cmay, (ng/mL) 2334 823.6 983.8 364.9
20.98 8.63 o
-4
o
0
EC-ER-Na.MPA MPA MPAG AcMPAG
MPAAVIPAG MPA/AcMPAG
Mean SD Mean SD Mean SD Ratio
Ratio
tmax (hr) 2.3 0.27 2.3 0.45 2.1
0.42 oe
c7,
tin (hr)I 7.53 0.45 8.73 1.29 6.62
2.62
MRTIast (hr) 8.44 1.19 9.89 1.77 7.9 2.06
AUCIast (hr?=ng/mL) 15187 2678 7884.5 2771 120.9 50.3
2.18 169.0
AUC? (hr?=ng/mL) 1814 0 3111 10784 5110 145.9 63.6 1.99
163.0
Cf.,: maximum plasma concentration; tinax: time of maximum plasma
concentration; t112: half-life
calculated using 2 points in the terminal phase; MRTIast: mean residence time,
calculated to the
last observable time point; AUCIast: area under the curve, calculated to the
last observable time
point; AUC?: area under the curve, extrapolated to infinity, if ti/2 value was
not available mean
group value was used; BLOQ: below the limit of quantitation (1 ng/mL); ND: not
determined.
1-d
oe
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Study 3: Another cross-over study was performed to determine the
single-dose and steady state pharmacokinetics of MPA and its metabolites (MPAG
and
AcMPAG). The study design is illustrated in FIG. 14. Specifically, three (3)
groups of
healthy male beagle dogs (n= 7 per group for a total of 21 dogs; Marshall
BioResources,
North Rose, NY, USA) participated in a two-treatment, two-period, sequential,
adaptive
cross-over study. Two five-day, repeat-dosing study periods were separated by
a 16-day
washout period. Dogs were at least two years of age at the time of enrollment.
In the first five-day period, three groups of dogs (n=7 per group) were
randomized to receive either OKV-1001 (270 mg) Profile 1 QD (Group A), OKV-
1001
(270 mg) Profile 2 QD (Group B) or MMF (10 mg/kg) oral suspension BID (Group
C).
Profile 1 was formulated for faster release of the MPA active agent as
compared to
Profile 2. MMF oral suspension (CellCept oral suspension, Genentech USA Inc.,
South San Francisco, CA) dosed at 10 mg/kg B.I.D served as the reference
group. A
16-day wash period ensued after the first five-day period, and results from
the first period
were examined. Dogs were then crossed-over in the second period to receive OKV-
1001
(365 mg) Profile 1 QD (Group A) or OKV-1001 (365 mg) Profile 2 QD (Group B) or
either OKV-1001 (270 mg) Profile 1 QD (Group C). The treatments received by
the dogs
in each group and period are summarized in Table 11.
Table 11: Treatments Received by Each Group in Study 3
Period 1 Period 2
Group A
OKV-1001p1 270 mg (QD) OKV-1001p1365 mg (QD)
(n=7)
Group B
OKV-1001p2 270 mg (QD) OKV-1001p2365 mg (QD)
(n=7)
Group
MMF 10 mg/kg (BID) OKV-1001p1 270 mg (QD)
C(n=7)
The two study periods had identical feeding and sampling procedures. On
Day 1, PK samples were collected after a 12-hour fast, while samples were
collected 1
hour after the animals were fed on Day 5. At the time of dosing, any uneaten
food was
removed, and the amount of food provided and consumed by each animal was
recorded.
66
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
On Days 2-5, animals were fed once daily in the morning, 1 hour prior to
administration
of the morning dose.
On Day 1 and Day 5, serial blood samples collected via the jugular vein
were collected prior to dosing (0 min) and at 15 and 30 min, and then 1, 1.5,
2, 2.5, 3, 4, 6,
8, 9 and 12 hours after MMF administration. For OKV-1001, samples were
collected
prior to dosing (0 min), and at 30 min, and then 1, 1.5, 2, 2.5, 3, 3.5, 4, 6,
8, 9, 12 and 24
hours. A qualified and validated GLP ready LC-MS/MS method was used to
quantify
plasma MPA, MPAG, AcMPAG plasma concentrations.
The general health of each animal was assessed at every blood sampling
time point during the course of the study. On study days with no blood
sampling or only
one blood sampling time point, the general health was assessed at least twice
daily (AM
and PM).
Standard non compartmental pharmacokinetic parameters (C., T., t112,
AUCIast and AUCH,ffollowing the Day 1 dosing; C., T.x and AUCtau following the
Day
dosing where tau is 12 h for MMF and 24 h for OKV-1001) were estimated using
Phoenix Winnonlin software 64 (Build 7Ø0.2535) for MPA, MPAG and AcMPAG.
MPA plasma concentration levels for each treatment group on Day 1 (fasted;
single-dose) are shown in FIG. 15. MPA plasma concentration levels for each
treatment
group on Day 5 (fed; steady state) are shown in FIG. 16. Calculated
drug:metabolite
(D:M) ratios for Group C, Group A, and Group B are shown in Tables 12, 13, and
14,
respectively. Reference ratio values from Study 2 are also shown. Non-
compartmental
PK parameter estimates for all treatment groups are provided in Table 15. It
should be
noted that data from Group B/Period 2/Day 5 was not obtained from 5 of the 7
dogs.
67
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
Table 12. Study 3 Group C and Study 2: Mean D:M Ratio Summary
Study 3 (Repeat Dose) Study 2 (Single Dose)
MMF OKV-1001 MMF OKV-1001
(270 mg) (270 mg)
MPAAVIPAG Day 1.6 (0.78-3.07) 1.5 (0.69-2.36) 1.02 1.99
1 (0.70-1.77) (1.10-
3.30)
MPAAVIPAG Day 1.6(0.84-3.35) 0.84
(0.48-1.43)
MPA/AcMPAG 130 (80-219) 90 (65-160) 133 (90-229) 163 (94-380)
Day 1
MPA/AcMPAG 84 (60-168) 76 (53-145)
Day 5
Table 13. Study 3 Group A and Study 2: Mean D:M Ratio Summary
Study 3 (Repeat Dose) Study 2 (Single Dose)
OKV-1001 P1 OKV-1001 P1 MMF OKV-1001
(270 mg) (365 mg) (270 mg)
MPAAVIPAG Day 1.97 1.48 1.02 1.99
1 (1.05-4.73) (0.80-3.18) (0.70-
1.77) (1.10-3.30)
MPAAVIPAG Day 1.27 0.75
5 (0.81-2.71) (0.42-1.52)
MPA/AcMPAG 119 (77-155) 94 (81-130) 133 (90-229) 163 (94-380)
Day 1
MPA/AcMPAG 104 (47-191) 74 (51-128)
Day 5
68
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
Table 14. Study 3 Group B and Study 2: Mean D:M Ratio Summary
Study 3 (Repeat Dose) Study 2 (Single Dose)
OKV-1001 P2 OKV-1001 P2 MMF OKV-1001
(270 mg) (365 mg) (270 mg)
MPAAVIPAG Day 1.95 (1.22-3.5) 1.10(1.0-1.94) 1.02 1.99
1 (0.70-1.77) (1.10-
3.30)
MPAAVIPAG Day 1.6 (0.86-2.96) 0.6 (0.52,
0.70) (n=2)
MPA/AcMPAG 163.5 (91-418) 131 (92-273) 133 (90-229) 163 (94-380)
Day 1
MPA/AcMPAG 145 (63-257) 80 (69, 91)
Day 5 (n=2)
69
0
Table 15. Mean Single-Dose and Steady State I\SPA and MPAG Pharmacokinetic
Parameters - Group A (n=7) t..)
o
t..)
o
Period / Day Tmax Cmax AUCa
Tmax Cmax AUCa -a-,
u,
=
Formulation hr ng/mL hr*ng/mL
hr ng/mL hr*ng/mL oe
c:
GROUP A
MPA
MPAG
1 Mean 3.00 2897.1 18078
3.07 953.6 9873
Period 1/ SD 0.87 1283.8 7615
0.53 146.0 2721
OKV-1001
Profile 1, 270
Mean 2.93 1515.9 14093 3.36 1640.4
12182
mg
SD 0.45 535.8 6429
0.38 704.6 5999 P
.
,
1 Mean 3.00 4895.7 28200"
3.64 2311.4 20426' ,
,
Period 2/ SD 0.58 987.9 11231
0.48 732.9 5572
---A OKV-1001
r.,
r.,
,
Profile 1, 365
'
5 Mean 2.93 1920.3 17464
3.29 3442.9 24234 .
,
mg
.
SD 0.35 924.8 9670
0.49 877.4 10902 u,
GROUP Bd
1 Mean 7.14 835.1 9503
7.57 310.6 4957
Period 1/ SD 1.46 463.8 4215
1.51 65.7 1357
OKV-1001
Profile 2, 270
5 Mean 5.14 854.0 12981
6.57 505.9 6899
mg
Iv
SD 2.78 861.7 15691
7.86 309.2 4649 n
,-i
cp
Period 2/ 1 Mean 4.21 1037.3 11099
7.29 714.4 9747 t..)
o
OKV-1001
oe
-a-,
Profile 2, 365 SD 1.82 654.8 7237
1.98 169.4 2496 vi
o
mg
o
--4
yD
0
GROUP C
t..)
o
t..)
1 Mean 0.32 2950.0 8231 0.43 1032.1 5303
=
C:--,
Period 1/ SD 0.12 1730.4 3412
0.12 368.0 1497 vi
o
Mycophenolate
cio
c:
Mofetil 10
c,.)
5 Mean 0.36 3238.6 8672 0.71 1430.6 5962
mg/kg
SD 0.13 1437.8 2939
0.27 722.7 1687
1 Mean 2.79 3042.9 23324 3.50 1584.3 16749b
Period 2/ SD 0.70 612.9 10366
1.22 333.0 7447
OKV-1001
Profile 1, 270
5 Mean 2.79 1666.1 13023 3.00 2382.9 15814
p
mg
.
SD 0.39 721.3 4738
0.50 547.7 3758
,
,
,
.3
---A a AUCia for Day 1 and AUC0_24 for Day 5
b
2
n=6
,
,
.
c n=5,
5',
d Group B/Period 2/Day 5 is not presented because data are not available from
5 dogs.
,-o
n
,-i
cp
t..)
o
cio
-c=-::.--,
u,
o
o
-.1
o
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
EXAMPLE 4:
MANUFACTURE OF A CONTROLLED-RELEASE MPA COMPOSITION
Controlled-release MPA compositions (two profiles, "Profile 1" and
"Profile 2") were manufactured as follows.
1. Required quantities of Hypromellose 2910 and purified water
were mixed together in a suitable container until the Hypromellose is
completely
dissolved. The required amount of sodium mycophenolate was added to the
hypromellose solution and mixed until all of the drug substance was dissolved.
2. A suitable quantity of microcrystalline cellulose spheres was
loaded into the chamber of an appropriately sized fluid bed coater. The beads
were
pre-warmed to 48 C.
3. The drug¨binder solution prepared in Step 1 was sprayed on to the
microcrystalline spheres in the fluid bed coater while the bed was maintained
at 50 C.
The drug binder solution was sprayed on the spheres until a weight gain of 44%
was
achieved on the dry beads.
4. The drug loaded beads were dried at ambient temperature for
approximately 10 minutes while the air flow was maintained at 75 cubic feet
per minute
(cfm)
5. A subcoating solution was prepared by mixing Opadry Clear and
Purified Water in a suitable mixer until a clear solution was formed.
6. A suitable quantity of drug loaded beads was placed in the
chamber of an appropriately sized fluid bed coater. The beads were pre-warmed
to 50 C
and maintained at 50 C during the subcoating process.
7. The subcoating solution prepared in Step 5 was sprayed on to the
drug-coated beads in the fluid bed coater until a weight gain of 7% was
achieved on the
beads. The beads were dried at ambient temperature for approximately 10
minutes while
the airflow is maintained at 90 cfm.
72
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
8. The rate control membrane solution was prepared as follows:
Required quantities of DI water and ethanol were mixed together in a suitable
mixer until
homogenous. Dibutyl sebacate was added and mixed to form a homogenous
suspension.
Ethyl cellulose and Klucel EF were added and mixed to form a homogenous
suspension.
The required quantity of talc was added and mixed until a uniform suspension
was
formed.
9. A suitable quantity of sub-coated beads was placed in the chamber
of an appropriately sized fluid bed coater. The beads were pre-warmed to 42 C.
10. The rate control membrane prepared in Step 8 was sprayed onto
the drug-coated beads in the fluid bed coater. The membrane coating solution
was
sprayed on the spheres until a weight gain of 30% (Profile 1) or 45% (Profile
2) was
achieved on the beads. After completion of coating, beads were dried under
ambient
temperature for approximately 10 minutes with an air flow volume if 50 cfm.
11. The enteric coat solution was prepared by mixing a required
quantity Plasacryl HTP 20 with required quantity of DI water. Eudragit L3OD 55
was
dispersed and mixed into this solution until a homogenous suspension was
obtained.
12. A suitable quantity of membrane-coated beads was placed in the
chamber of an appropriately sized fluid bed coater. The beads were pre-warmed
to 29 C.
13. The enteric coating solution prepared in Step 11 was sprayed on to
the drug-coated beads in the fluid bed coater. The enteric coating solution
was sprayed
on the spheres until a weight gain of 25% was achieved on the beads for both
Profile 1
and Profile 2 formulations. The beads were dried at ambient temperature for
approximately 10 minutes after coating was complete.
14. The dried beads were manually filled into a Veterinary Size 13
gelatin capsule. The capsule was filled with beads equivalent to either 180 mg
or 270 mg
of mycophenolate sodium. The final MDDS product composition containing 270mg
of
mycophenolate sodium was as shown in Table 16.
73
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
Table 16. Final product composition of OKV-1001 (270mg Na.MPA)
Total
Functional Compendial mg/
Component % w/w weight
layer Grade dosage unit
(mg)
Cellets 700
Drug (Microcrystalline NF Core 635.12
Loaded Cellulose Spheres)
beads Mycophenolate
USP 13.35 270 935.12
(44.0% Sodium
w.g. HPMC 606
theoretical) (Hypromellose 2910) USP 1.65 33.4
Purified Water In-house 85.00 0
Total 100.00 935.12 935.12
Sub Coat Opadry Clear
USP 10.00 65.46 1000.5
(7.0% w.g. 030190001
8
theoretical) Purified Water In-house 90.00 0
100.00 65.46
ECN10
USPNF 4.154 138.54
(Ethylcellulose)
Rate Klucel EF
Control (Hydroxypropyl USPNF 0.462 15.41
membrane* Cellulose) 1200.7
(20.0% 0
Talc USP 0.923 30.78
theoretical _________________________________________________
w.g.) Dibutyl Sebacate USPNF 0.461 15.38
DI Water N/A 9.40 0
Ethanol 190 proof USP 84.60 0
100.00 200.12
Eudragit L30D-55 USP 57.0 205.32
Enteric
Plasacryl HTP20 USP 14.55 34.82 1440.8
coat
4
(20% w.g.) DI Water N/A 28.45 0
100.00 0
74
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
240.14
Finished
Size 13 Gelatin 1440.8
Drug USP 100.00
Capsule 4
Product
EXAMPLE 5:
OPEN-LABEL STUDIES OF MPA PHARMACODYNAMICS IN CANINE
SUBJECTS
The pharmacodynamic effects of MPA in dogs were investigated in
parallel 15-day open-label studies conducted by Absorption Systems (San Diego,
CA).
In one study, male beagle dogs were administered a controlled-release MPA
composition
of the present disclosure ("OKV-1001") under one of three dosing regimens (270
mg
MPA, QD; 180 mg MPA, QD; 180 mg MPA, BID). The study concept is shown
schematically in Figure 17, with the data capture timeline shown at Figure 18.
In a
parallel study, dogs received an immediate-release mycophenolate mofetil ("IR-
MMF")
capsule (120 mg MPA; n=5 dogs, BID) or placebo (n=1). Further details of the
study
designs are provided below.
Dogs
In both studies, healthy male beagle dogs (Marshall Bioresources, North
Rose, NY) with a minimum age of 24 months and an initial body weight of ¨10-14
kg
were used. Dogs were housed one per cage in a single room. Dogs were
identified by ear
tattoo.
Feeding and Water Schedule
For at least 3 days prior to study start, animals were feed-acclimated to a
food schedule (500 g of a 1:1 mixture of a dry certified laboratory diet (5006
laboratory
canine diet from Lab Diet) and canned wet dog food (AlpoTM) lx daily in the
morning).
The feeding schedule was maintained throughout the duration of both studies.
All food
was removed 1 hour prior to administration of the first daily dose.
The quantitative amount of food provided and consumed by each animal
was recorded throughout both studies. Water was supplied ad libitum to the
animals
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
throughout the studies. For dogs that received OKV-1001, body weights for each
animal
were taken prior to dosing on days 1 and 15. For dogs receiving the IR-MMF,
body
weights were taken prior to dosing on days 1, 8, and 15. Laboratory personnel
wore new,
clean, personal protective equipment before entering the room, as well as a
facemask and
hairnet.
Drug Administration
In the OKV-1001 study, animals received oral capsules comprising either
270 mg or 180 mg MPA QD, or 180 mg MPA BID. QD dosing was performed for Days
1-15. BID dosing was performed for Days 1-14, with a final, single dose on Day
15.
In the MMF-IR study, BID dosing was performed for Days 1-14, with a
final, single dose on Day 15.
BID dosing in each study was carried out at approximately 12-hour
intervals.
In both studies, drug was administered at approximately the same time on
all days by placing the capsule in the back of the throat and flushing with 10
mL water.
General Health and Stool Observations
The general health of each animal was assessed at each timepoint during
the course of both studies. If there was not a timepoint, general health
observations were
performed twice daily during the dose administration times.
The stool of all animals was observed and recorded over the active study
duration (Days 1-15). On blood sampling days (Days 1, 8, & 15), the presence
or absence
of stool was noted at each general health observation timepoint. The time(s)
of
defecation (post-dose) were recorded. If defecation occurred between
observation
timepoints, the closest estimation of the time(s) of defecation was recorded.
On
days with no blood sampling, the stool of animals was observed during general
health observations.
The stool of each animal was graded using a modified WALTHAM
feces scoring system, as follows:
Grade 1: Hard, dry
76
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
Grade 2: Well-formed and does not leave a trail when picked up.
Grade 3: Moist and beginning to lose form. Leaves a mark when picked up.
Grade 4: Majority of form is lost, poor consistency; viscous.
Grade 5: Watery diarrhea.
Animal Safety
Removal of animals from the study for any reason was recorded.
PK and PD Time Points
Samples were taken for pharmacokinetic ("PK"; mean plasma
concentration of MPA) and pharmacodynamic ("PD"; amount of lymphocytes
expressing the proliferation marker Ki-67 in a whole blood sample) analysis as
follows:
Table 17. PK and PD Timepoints for 15-Day Study
PK Timepoints PD Timepoints
OKV-1001 Pre-dose; 2.5h(Tmax); 4h; 8h Pre-dose; 2.5h; 4h; 8h
Day 1
OKV-1001 Pre-dose; lh, 2h, 2.5h; 4h; 6h; 8h Pre-dose; 2.5h; 4h; 8h
Day 8
OKV-1001 Pre-dose; 2.5h; 4h; 8h Pre-dose; 2.5h; 4h; 8h
Day 15
IR-MMF Pre-dose; 0.75h (T.); 4h; 8h Pre-dose; 0.75h; 4h; 8h
Days 1, 8,
and 15
Sample Collection, Preparation, and Storage
Blood was collected from the jugular vein or other accessible vessel.
For PK samples, 2 mL whole blood was collected via the jugular vein or other
accessible
vessel directly into 2 mL chilled (purple top) Vacutainers (a needle and
syringe were not used) containing the anticoagulant, K2EDTA, and kept on ice
until
centrifugation. Blood samples were processed to plasma within 60 minutes of
77
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
collection, per SOP. Samples were centrifuged at 4 C, at ¨3,000 x g, for 5
minutes to
obtain plasma. Plasma were collected after centrifugation, and then placed
into tubes
containing a formic acid solution as follows:
1. A 10% formic acid solution in water (1:10 of concentrated, e.g. 88%, formic
acid, final concentration is 8.8%) was prepared;
2. Two aliquots of 300 [IL of plasma was pipetted from each sample supernatant
and added to tubes containing 30 [IL of 10% formic acid in water, capped and
mixed well (to insure compound stabilization of parent molecule) (plasma was
mixed at 10:1 ratio with formic acid, yielding an acidic pH). Note: Two
aliquots were collected.
3. Plasma samples were snap frozen on dry ice and stored frozen at -60 to -
80 C.
For PD samples, 4 mL blood was collected at each timepoint via the
jugular vein or other accessible vessel directly into 4 mL chilled (green top)
Vacutainers
(a needle and syringe were not used) containing the anticoagulant Sodium
Heparin.
Samples were refrigerated and shipped same day to Mann Biologic Laboratories,
Inc., on
ice packs
Sample Processing
For PK studies, plasma concentrations of MPA were determined using the
LC-MS/MS method described in Example 3. For the IR-MMF study, MPA
concentrations were analyzed only for the group receiving MMF.
For PD studies, T lymphocyte proliferation was evaluated using a flow
cytometry assay as previously described (Bishop KA. Pharmacodynamic assessment
of a
panel of immunosuppressant drugs in ex-vivo canine T-lymphocyte proliferation.
(abstract) 2016 Merial NIH National Veterinary Research Scholars Symposium
2016;Ohio State Universtiy, Columbus, OH, USA; Grobman et at., I Vet.
Pharmacol.
Ther. 2017), with modifications as described below:
= Whole blood samples were diluted 1:4 in cRPMI (RPMI 1640 + L-glutamine +
Pen/strep), plated in a 96 well plate, and incubated with either Concanavalin
A
(ConA) at 10[tg/mL or cRPMI for 72 hours at 37 C and 5% CO2.
78
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
= Following incubation, erythrocytes were lysed and leukocytes were stained
with
eBioscienceTm Fixable Viability Dye eFluor 450 (Thermo Fisher Scientific
65-0863-14), Rat a Dog CD5 APC (Bio-Rad MCA1037APC, clone YKIX322.3),
Rat a Dog CD45 RPE (Bio-Rad MCA1037APC, clone YKIX716.13), and Mouse
a Human/Dog Ki-67 FITC (Thermo Fisher Scientific 11-5698-80, clone SolA15).
= Compensation controls used OneComp eBeadsTM Compensation Beads (Thermo
Fisher Scientific 01-1111-41) for the surface and intracellular antibodies,
and
cells for the live/dead marker. Samples were acquired on an Attune NxT flow
cytometer (Thermo Fisher Scientific) at a validated speed of 100 L/min. The
following gating strategy was used: Scatter (FSC / SSC) > Singlets (FSC-A /
FSC-H) > Live (L-D / SSC) > T cells (CD45+ / CD5+) > Proliferating (CD5+ /
Ki-67+). Generated FCS files were analyzed with FlowJo v 10.4.2 (BD
Biosciences).
Results
Lymphocyte Proliferation (Ki-67 Expression)
Lymphocyte PD data are provided in Figures 19-24. Quite surprisingly,
dogs that received OKV-1001 (270 mg) had substantially lower amounts of
proliferating
lymphocytes prior to dosing on Day 8 as compared to the Day 1 pre-dose level,
suggesting that the OKV-1001 formulation had a systemic effect on
proliferation (Figure
19). A similar effect was seen at Day 15. On all days, dogs receiving OKV-1001
had
similar reduced levels of Ki-67-expressing cells at all timepoints after
dosing (decreases
of 50% to 85% relative to baseline), indicating that the OKV-1001 formulations
provide
sustained suppression of lymphocyte proliferation, with little PD variability
over the
course of an 8-hour period following administration. Similar results were
observed in the
dogs that received OKV-1001 (180 mg, QD), as shown in Figure 21.
Day 1 single-dose PD of OKV-1001 (270 mg and 180 mg (the 180 mg
BID group also received only one dose on Day 1)) is shown in Figure 20. The 8-
hour
effect on lymphocyte proliferation is similar to the Day 1 data shown in
Figure 19.
These results were in contrast to the PD data from the IR-MMF group,
shown in Figure 22. In dogs receiving the IR-MMF capsule, an initial sharp
reduction in
79
CA 03111908 2021-03-05
WO 2020/050863
PCT/US2018/050079
Ki-67-expressing cells was observed at 45 minutes after dosing, but the
decrease was not
sustained to the same degree as in the dogs receiving OKV-1001. On Day 1, Ki-
67+ cells
increased above baseline at 4h, and were at 80% of baseline at 8h. On Days 8
and 15,
Ki-67+ cells were reduced at 4h and 8h from the pre-dose baseline, but
increased between
4h and 8h (moreso on Day 8 than Day 15) and were within 60% of baseline by 8h.
Day 1 PD of IR-MMF and OKV-1001 (270mg QD) is directly compared
in Figure 23. Overall, dogs receiving the 0KV-1001 formulation had a greater
sustained
reduction in Ki-67+ cells versus baseline as compared to dogs that received IR-
MMF.
These differences were also seen at Days 8 and 15, as shown in Figure 24.
Safety
Two dogs receiving 180 mg/kg MPA BID were removed on Day 8 due to
severe GI side effects. Interestingly, these dogs had relatively low Cmax MPA
values
(see Figure 26). Dogs that received the OKV-1001 formulations (QD
administration) did
not have any observed severe GI events over the course of the study. This was
in contrast
to the dogs that received IR-MMF; results are shown in Table 18 below.
Table 18. Safety Observations in Dogs Receiving IR-MMF
Timepoint Dog ID
Dog 1 Dog 2 Dog 3 Dog 4 Dog 5
Before Day 10 No diarrhea or Emesis
Diarrhea
emesis observed (Day 6) (Day
8)
Day 10 Diarrhea
Day 11 Soft stool Diarrhea
Day 12 Diarrhea
Day /3 Diarrhea Diarrhea Diarrhea &
Diarrhea
& Emesis Emesis
Day 14 Soft stool Diarrhea
Diarrhea Soft stool
& Emesis
Day 15 Emesis Diarrhea
Diarrhea
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
These results demonstrate surprising advantages of the presently
disclosed MDDS and dosing methods. For example, the daily MPA load in 270 mg
OKV-1001 (252 mg MPA) is 42% higher than that of 120 mg MMF (BID) (177 mg),
but
the OKV-1001 had fewer GI side effects than MMF.
In summary, the data from the parallel 15-day pharmacodynamics studies
show that MDDS of the present disclosure have a faster onset of systemic
action than
IR-MMF, have more consistent pharmacodynamics effects (reduced proliferation
of
lymphocytes) over a 4 and an 8 hour period post-administration, can be
advantageously
administered once daily as compared to IR-MMF (BID), and are safer than IR-
MMF, as
determined by the number and severity of gastrointestinal side effects
experienced by the
dogs.
It will be readily apparent to one of ordinary skill in the relevant arts that
other suitable modifications and adaptations to the methods and applications
described
herein can be made without departing from the scope of any of the embodiments.
.. Further, it is to be understood that while certain embodiments have been
illustrated and
described herein, the claims are not to be limited to the specific forms or
arrangement of
parts described and shown. In the specification, there have been disclosed
illustrative
embodiments and, although specific terms are employed, they are used in a
generic and
descriptive sense only and not for purposes of limitation. Modifications and
variations of
the embodiments are possible in light of the above teachings. It is therefore
to be
understood that the embodiments may be practiced otherwise than as
specifically
described.
The various embodiments described above can be combined to provide
further embodiments. All of the U.S. patents, U.S. patent application
publications, U.S.
patent applications, foreign patents, foreign patent applications and non-
patent
publications referred to in this specification and/or listed in the
Application Data Sheet,
including but not limited to U.S. Provisional Patent Application Serial No.
62/470,806,
and U.S. Provisional Patent Application Serial No. 62/503,270, and PCT
Application No.
PCT/U52018/022266, are incorporated herein by reference, in their entirety.
Aspects of
81
CA 03111908 2021-03-05
WO 2020/050863 PCT/US2018/050079
the embodiments can be modified, if necessary to employ concepts of the
various patents,
applications and publications to provide yet further embodiments.
All publications, patents and patent applications mentioned in this
specification are herein incorporated by reference to the same extent as if
each individual
publication, patent or patent application was specifically and individually
indicated to be
incorporated by reference.
82