Note: Descriptions are shown in the official language in which they were submitted.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
EXCIPIENT COMPOUNDS FOR PROTEIN PROCESSING
RELATED APPLICATION
[0001] This application claims the benefit of U.S. Provisional Application
62/773,018 filed
November 29, 2018. The entire contents of the above application are
incorporated by
reference herein.
FIELD OF APPLICATION
[0002] This application relates generally to formulations for delivering and
processing
1() biopolymers.
BACKGROUND
[0003] Biopolymers may be used for therapeutic or non-therapeutic purposes.
Biopolymer-
based therapeutics, such as antibody or enzyme formulations, are widely used
in treating
disease. Non-therapeutic biopolymers, such as enzymes, peptides, and
structural proteins,
have utility in non-therapeutic applications such as household, nutrition,
commercial, and
industrial uses.
[0004] Biopolymers used in therapeutic applications must be formulated to
permit their
introduction into the body for treatment of disease. For example, it is
advantageous to deliver
antibody and protein/peptide biopolymer formulations by subcutaneous (SC) or
intramuscular
(IM) routes under certain circumstances, instead of administering these
formulations by
intravenous (IV) injections. In order to achieve better patient compliance and
comfort with
SC or IM injection though, the liquid volume in the syringe is typically
limited to 2 to 3 ccs
and the viscosity of the formulation is typically lower than about 20
centipoise (cP) so that
the formulation can be delivered using conventional medical devices and small-
bore needles.
These delivery parameters do not always fit well with the dosage requirements
for the
formulations being delivered.
[0005] Antibodies, for example, may need to be delivered at high dose levels
to exert their
intended therapeutic effect. Using a restricted liquid volume to deliver a
high dose level of an
antibody formulation can require a high concentration of the antibody in the
delivery vehicle,
sometimes exceeding a level of 150 mg/mL. At this dosage level, the viscosity-
versus-
concentration plots of protein solutions lie beyond their linear-nonlinear
transition, such that
the viscosity of the formulation rises dramatically with increasing
concentration. Increased
viscosity, however, is not compatible with standard SC or IM delivery systems.
The
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
solutions of biopolymer-based therapeutics are also prone to stability
problems, such as
precipitation, hazing, opalescence, denaturing, liquid-liquid phase
separation, gel formation,
and reversible or irreversible aggregation. The stability problems limit the
shelf life of the
solutions or require special handling.
.. [0006] One approach to producing protein formulations for injection is to
transform the
therapeutic protein solution into a powder that can be reconstituted to form a
suspension
suitable for SC or IM delivery. Lyophilization is a standard technique to
produce protein
powders. Freeze-drying, spray drying and even precipitation followed by super-
critical-fluid
extraction have been used to generate protein powders for subsequent
reconstitution.
Powdered suspensions are low in viscosity before re-dissolution (compared to
solutions at the
same overall dose) and thus may be suitable for SC or IM injection, provided
the particles are
sufficiently small to fit through the needle. However, protein crystals that
are present in the
powder have the inherent risk of triggering immune response. The uncertain
protein
stability/activity following re-dissolution poses further concerns. There
remains a need in the
art for techniques to produce low viscosity protein formulations for
therapeutic purposes
while avoiding the limitations introduced by protein powder suspensions.
[0007] In addition to the therapeutic applications of proteins described
above, biopolymers
such as enzymes, peptides, and structural proteins can be used in non-
therapeutic
applications. These non-therapeutic biopolymers can be produced from a number
of different
pathways, for example, derived from plant sources, animal sources, or produced
from cell
cultures.
[0008] The non-therapeutic proteins can be produced, transported, stored, and
handled as a
granular or powdered material or as a solution or dispersion, usually in
water. The
biopolymers for non-therapeutic applications can be globular or fibrous
proteins, and certain
forms of these materials can have limited solubility in water or exhibit high
viscosity upon
dissolution. These solution properties can present challenges to the
formulation, handling,
storage, pumping, and performance of the non-therapeutic materials, so there
is a need for
methods to reduce viscosity and improve solubility and stability of non-
therapeutic protein
solutions.
[0009] Proteins are complex biopolymers, each with a uniquely folded 3-D
structure and
surface energy map (hydrophobic/hydrophilic regions and charges). In
concentrated protein
solutions, these macromolecules may strongly interact and even inter-lock in
complicated
ways, depending on their exact shape and surface energy distribution. "Hot-
spots" for strong
specific interactions lead to protein clustering, increasing solution
viscosity. To address these
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
concerns, a number of excipient compounds are used in biotherapeutic
formulations, aiming
to reduce solution viscosity by impeding localized interactions and
clustering. These efforts
are individually tailored, often empirically, sometimes guided by in sit/co
simulations.
Combinations of excipient compounds may be provided, but optimizing such
combinations
again must progress empirically and on a case-by-case basis.
[0010] There remains a need in the art for a truly universal approach to
reducing viscosity
in protein formulations at a given concentration under nonlinear conditions.
There is an
additional need in the art to achieve this viscosity reduction while
preserving the activity of
the protein. It would be further desirable to adapt the viscosity-reduction
system to use with
formulations having tunable and sustained release profiles, and to use with
formulations
adapted for depot injection. In addition, it is desirable to improve processes
for producing
proteins and other biopolymers.
SUMMARY
[0011] Disclosed herein, in embodiments, are liquid formulations comprising a
protein and
an excipient compound selected from the group consisting of hindered amines,
anionic
aromatics, functionalized amino acids, oligopeptides, short-chain organic
acids, low
molecular weight aliphatic polyacids, diones and sulfones, zwitterionic
excipients, and
crowding agents with hydrogen bonding elements, wherein the excipient compound
is added
in a viscosity-reducing amount. In embodiments, the protein is a PEGylated
protein and the
excipient is a low molecular weight aliphatic polyacid. In embodiments, the
formulation is a
pharmaceutical composition, and the therapeutic formulation comprises a
therapeutic protein,
wherein the excipient compound is a pharmaceutically acceptable excipient
compound. In
embodiments, the formulation is a non-therapeutic formulation, and the non-
therapeutic
formulation comprises a non-therapeutic protein. In embodiments, the viscosity-
reducing
amount reduces viscosity of the formulation to a viscosity less than the
viscosity of a control
formulation. In embodiments, the viscosity of the formulation is at least
about 10% less than
the viscosity of the control formulation or is at least about 30% less than
the viscosity of the
control formulation, or is at least about 50% less than the viscosity of the
control formulation,
or is at least about 70% less than the viscosity of the control formulation,
or is at least about
90% less than the viscosity of the control formulation. In embodiments, the
viscosity is less
than about 100 cP, or is less than about 50 cP, or is less than about 20 cP,
or is less than about
10 cP. In embodiments, the excipient compound has a molecular weight of <5000
Da, or
<1500 Da, or <500 Da. In embodiments, the formulation contains at least about
25 mg/mL of
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
the protein, or at least about 100 mg/mL of the protein, or at least about 200
mg/mL of the
protein, or at least about 300 mg/mL of the protein. In embodiments, the
formulation
comprises between about 5 mg/mL to about 300 mg/mL of the excipient compound
or
comprises between about 10 mg/mL to about 200 mg/mL of the excipient compound
or
comprises between about 20 mg/mL to about 100 mg/mL, or comprises between
about 25
mg/mL to about 75 mg/mL of the excipient compound. In embodiments, the
formulation has
an improved stability when compared to the control formulation. In
embodiments, the
excipient compound is a hindered amine. In embodiments, the hindered amine is
selected
from the group consisting of caffeine, theophylline, tyramine, procaine,
lidocaine, imidazole,
.. aspartame, saccharin, and acesulfame potassium. In embodiments, the
hindered amine is
caffeine. In embodiments, the hindered amine is a local injectable anesthetic
compound. The
hindered amine can possess an independent pharmacological property, and the
hindered
amine can be present in the formulation in an amount that has an independent
pharmacological effect. In embodiments the hindered amine can be present in
the formulation
in an amount that is less than a therapeutically effective amount. The
independent
pharmacological activity can be a local anesthetic activity. In embodiments,
the hindered
amine possessing the independent pharmacological activity is combined with a
second
excipient compound that further decreases the viscosity of the formulation.
The second
excipient compound can be selected from the group consisting of caffeine,
theophylline,
tyramine, procaine, lidocaine, imidazole, aspartame, saccharin, and acesulfame
potassium. In
embodiments, the formulation can comprise an additional agent selected from
the group
consisting of preservatives, surfactants, sugars, polysaccharides, arginine,
prolific.;
hyalurorridase, stabilizers, and buffers.
[0012] Further disclosed herein are methods of treating a disease or disorder
in a mammal,
comprising administering to said mammal a liquid therapeutic formulation,
wherein the
therapeutic formulation comprises a therapeutically effective amount of a
therapeutic protein,
and wherein the formulation further comprises an pharmaceutically acceptable
excipient
compound selected from the group consisting of hindered amines, anionic
aromatics,
functionalized amino acids, oligopeptides, short-chain organic acids, low
molecular weight
aliphatic polyacids, diones and sulfones, zwitterionic excipients, and
crowding agents with
hydrogen bonding elements; and wherein the therapeutic formulation is
effective for the
treatment of the disease or disorder. In embodiments, the therapeutic protein
is a PEGylated
protein, and the excipient compound is a low molecular weight aliphatic
polyacid. In
embodiments, the excipient is a hindered amine. In embodiments, the hindered
amine is a
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
local anesthetic compound. In embodiments, the formulation is administered by
subcutaneous injection, or an intramuscular injection, or an intravenous
injection. In
embodiments, the excipient compound is present in the therapeutic formulation
in a viscosity-
reducing amount, and the viscosity-reducing amount reduces viscosity of the
therapeutic
formulation to a viscosity less than the viscosity of a control formulation.
In embodiments,
the therapeutic formulation has an improved stability when compared to the
control
formulation. In embodiments, the excipient compound is essentially pure.
[0013] Further disclosed herein are methods of reducing pain at an injection
site of a
therapeutic protein in a mammal in need thereof, comprising: administering a
liquid
therapeutic formulation by injection, wherein the therapeutic formulation
comprises a
therapeutically effective amount of the therapeutic protein, wherein the
formulation further
comprises an pharmaceutically acceptable excipient compound selected from the
group
consisting of local injectable anesthetic compounds, wherein the
pharmaceutically acceptable
excipient compound is added to the formulation in a viscosity-reducing amount;
and wherein
the mammal experiences less pain with administration of the therapeutic
formulation
comprising the excipient compound than that with administration of a control
therapeutic
formulation, wherein the control therapeutic formulation does not contain the
excipient
compound and is otherwise identical to the therapeutic formulation.
[0014] Disclosed herein, in embodiments, are methods of improving stability of
a liquid
protein formulation, comprising: preparing a liquid protein formulation
comprising a
therapeutic protein and an excipient compound selected from the group selected
from the
group consisting of hindered amines, anionic aromatics, functionalized amino
acids,
oligopeptides, short-chain organic acids, low molecular weight aliphatic
polyacids, diones
and sulfones, zwitterionic excipients, and crowding agents with hydrogen
bonding elements,
wherein the liquid protein formulation demonstrates improved stability
compared to a control
liquid protein formulation, wherein the control liquid protein formulation
does not contain the
excipient compound and is otherwise identical to the liquid protein
formulation. The stability
of the liquid formulation can be a cold storage conditions stability, a room
temperature
stability or an elevated temperature stability.
[0015] Also disclosed herein, in embodiments, are liquid formulations
comprising a protein
and an excipient compound selected from the group consisting of hindered
amines, anionic
aromatics, functionalized amino acids, oligopeptides, short-chain organic
acids, low
molecular weight aliphatic polyacids, diones and sulfones, zwitterionic
excipients, and
crowding agents with hydrogen bonding elements, wherein the presence of the
excipient
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
compound in the formulation results in improved protein-protein interaction
characteristics as
measured by the protein diffusion interaction parameter kr', or the second
virial coefficient
B22. In embodiments, the formulation is a therapeutic formulation, and
comprises a
therapeutic protein. In embodiments, the formulation is a non-therapeutic
formulation, and
comprises a non-therapeutic protein.
[0016] Further disclosed herein, in embodiments, are methods of improving a
protein-
related process comprising providing the liquid formulation described above,
and employing
it in a processing method. In embodiments, the processing method includes
filtration, (e.g.,
tangential flow filtration, sterile filtration, microfiltration,
ultrafiltration, diafiltration,
centrifugal concentration, and in-line filtration), pumping, mixing,
centrifugation, membrane
separation, lyophilization, or chromatography. In embodiments, the processing
method is
selected from the group consisting of cell culture harvest, chromatography,
such as Protein A
chromatography, hydrophobic interaction chromatography, anion exchange
chromatography
and cation exchange chromatography, viral inactivation, and filtration. In
embodiments, the
processing method is a chromatography process or a filtration process. In
embodiments, the
filtration process is a virus filtration process or an
ultrafiltration/diafiltration process.
[0017] Also disclosed herein are methods of improving a parameter of a protein-
related
process, comprising providing a viscosity-reducing excipient additive
comprising at least one
excipient compound selected from the group consisting of hindered amines,
aromatics and
anionic aromatics, functionalized amino acids, oligopeptides, short-chain
organic acids, low
molecular weight aliphatic polyacids, diones and sulfones, zwitterionic
excipients, and
crowding agents with hydrogen bonding elements, and adding a viscosity-
reducing amount of
the at least one excipient compound to a carrier solution for the protein-
related process,
wherein the carrier solution contains a protein of interest, thereby improving
the parameter.
In embodiments, the viscosity-reducing excipient additive in the liquid
formulation comprises
at least one excipient compound selected from the group consisting of
pyrimidines, methyl-
substituted pyrimidines, and phenethylamines. In embodiments, the parameter
can be
selected from the group consisting of cost of protein production, amount of
protein
production, rate of protein production, purity of protein produced, and
efficiency of protein
production. In embodiments, the parameter can be selected from the group
consisting of cost
of protein purification, amount of protein purification, rate of protein
purification, purity of
protein purified, and efficiency of protein purification. The parameter can be
a proxy
parameter, and the proxy parameter can be a reduced protein-protein
interaction The reduced
protein interaction can be determined by a technique selected from the group
consisting of
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
biolayer interferometry, surface plasmon resonance, intrinsic fluorescence
measurement,
extrinsic fluorescence measurement, dynamic light scattering, kD value, static
light
scattering, B22 value, isothermal titration calorimetry, and in sit/co
simulation. In
embodiments, the protein-related process is an upstream processing process.
The carrier
solution for the upstream processing process can be a cell culture medium. In
embodiments,
if the carrier solution is a cell culture medium, the step of adding the
excipient additive to the
carrier solution comprises a first substep of adding the excipient additive to
a supplemental
medium to form an excipient-containing supplemental medium, and a second
substep of
adding the excipient-containing supplemental medium to the cell culture
medium. In other
embodiments, the protein-related process is a downstream processing process.
The
downstream process can be a chromatography process, and the chromatography
process can
be a Protein-A chromatography process. In embodiments, the chromatography
process
recovers the protein of interest, wherein the protein of interest is
characterized by an
improved protein-related parameter selected from the group consisting of
improved purity,
improved yield, fewer particles, less misfolding, improved biological
activity, increased
percentage recovered in a monomeric form, or less aggregation, as compared to
a control
solution. In embodiments, the improved protein-related parameter is improved
yield of the
protein of interest from the chromatography process. In other embodiments, the
protein-
related process is a process selected from the group consisting of filtration,
tangential flow
filtration, sterile filtration, microfiltration, diafiltration, centrifugal
concentration, in-line
filtration, injection, syringing, pumping, mixing, centrifugation, membrane
separation, and
lyophilization, and the selected process can require less force than a process-
specific control
process, wherein the process-specific control process is the protein-related
process performed
in the absence of a viscosity-reducing excipient additive. In embodiments, the
protein-related
process is selected from the group consisting of a cell culture process, a
cell culture
harvesting process, a chromatography process, a viral inactivation process,
and a filtration
process. In embodiments, the protein-related process is the viral inactivation
process, and the
viral inactivation process is conducted at a pH level of about 2.5 to about
5.0, or the viral
inactivation process is conducted at a higher pH than a viral-inactivation-
specific control
process, wherein the viral-inactivation-specific control process is a viral
inactivation process
performed in the absence of a viscosity-reducing excipient additive. In other
embodiments,
the protein-related process is the filtration process. The filtration process
can be a virus
removal filtration process, a sterile filtration process, or an
ultrafiltration/diafiltration process.
The filtration process can be characterized by an improved filtration-related
parameter. The
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
improved filtration-related parameter can be a faster filtration rate than the
filtration rate of
the control solution, wherein the control solution is a solution that does not
contain a
viscosity-reducing excipient additive. The improved filtration-related
parameter can be a
production of a smaller amount of aggregated protein than the amount of
aggregated protein
produced by a control filtration process, wherein the control filtration
process is a filtration
process performed in the absence of a viscosity-reducing excipient additive.
The improved
filtration-related parameter can be a higher mass transfer efficiency than the
mass transfer
efficiency of the control filtration process. The improved filtration-related
parameter can be a
higher concentration or a higher yield of the target protein than a
concentration or yield of the
target protein produced by the control filtration process.
[0018] Further disclosed herein are methods as described above, wherein the
viscosity-
reducing excipient additive comprises two or more excipient compounds. In
embodiments,
the at least one excipient compound is a hindered amine. In embodiments, the
at least one
excipient compound is a pyrimidine, a methyl-substituted pyrimidine, or a
phenethylamine.
.. In embodiments, the hindered amine is a pyrimidine compound. In other
embodiments, the
hindered amine is a phenethylamine compound, which can be a non-psychoactive
phenethylamine. In embodiments, the at least one excipient compound is a
crowding agent
with hydrogen bonding elements, which can be selected from the group
consisting of
raffinose, inulin, pullulan, and sinistrins, or which can be raffinose. In
embodiments, the at
least one excipient compound is selected from the group consisting of
caffeine, saccharin,
acesulfame potassium, aspartame, theophylline, taurine, 1-methyl-2-
pyrrolidone, 2-
pyrrolidinone, niacinamide, and imidazole. In embodiments, the at least one
excipient
compound is selected from the group consisting of caffeine, taurine,
niacinamide, and
imidazole. In embodiments, the at least one excipient compound is selected
from the group
consisting of uracil, 1-methyluracil, 6-methyluracil, 5-methyluracil, 1,3-
dimethyluracil,
cytosine, 5-methylcytosine, 3-methylcytosine, thymine, 1-methylthymine, 0-4-
methylthymine, 1,3-dimethylthymine, and dimethylthymine dimer. In embodiments,
the at
least one excipient compound is selected from the group consisting of
diphenhydramine,
phenethylamine, N-methylphenethylamine, N,N-dimethylphenethylamine, beta-3-
dihydroxyphenethylamine, beta-3-dihydroxy-N-methylphenethylamine, 3-
hydroxyphenethylamine, 4-hydroxyphenethylamine, tyrosinol, tyramine, N-
methyltyramine,
and hordenine. In embodiments, the at least one excipient compound is selected
from the
group consisting of caffeine, nicotinamide, nicotinamide mononucleotide,
diethylnicotinamide, taurine, imidazole, ornithine, iminodiacetic acid,
nicotinic acid, and
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
sulfanilic acid, or is selected from the group consisting of caffeine,
nicotinamide, taurine, and
imidazole, or is caffeine. In embodiments, the at least one excipient compound
is selected
from the group consisting of calcium propionate and potassium sorbate. In
embodiments, the
at least one excipient compound is an aromatic or anionic aromatic excipient,
and, in some
embodiments, the anionic aromatic excipient can be 4-hydroxybenzenesulfonic
acid. In
embodiments, the viscosity-reducing amount is between about 1 mg/mL to about
100 mg/mL
of the at least one excipient compound, or the viscosity-reducing amount is
between about 1
mM to about 400 mM of the at least one excipient compound, or the viscosity-
reducing
amount is between about 1 mM to about 1000 mM of the at least one excipient
compound, or
1() the viscosity-reducing amount is an amount from about 2 mM to about 150
mM. In
embodiments, the carrier solution comprises an additional agent selected from
the group
consisting of preservatives, sugars, polyols, polysaccharides, arginine,
proline, surfactants,
stabilizers, and buffers. In embodiments, the protein of interest is a
therapeutic protein, and
the therapeutic protein can be a recombinant protein, or can be selected from
the group
consisting of a monoclonal antibody, a polyclonal antibody, an antibody
fragment, a fusion
protein, a PEGylated protein, an antibody-drug conjugate, a synthetic
polypeptide, a protein
fragment, a lipoprotein, an enzyme, and a structural peptide. In embodiments,
the methods
further comprise a step of adding a second viscosity-reducing excipient to the
carrier solution,
wherein the step of adding the second viscosity-reducing compound adds an
additional
improvement to the parameter.
[0019] In addition, carrier solutions are disclosed herein, comprising a
liquid medium in
which is dissolved a protein of interest, and a viscosity-reducing additive,
wherein the carrier
solution has a lower viscosity that that of a control solution. The carrier
solution can further
comprise an additional agent selected from the group consisting of
preservatives, sugars,
polyols, polysaccharides, arginine, proline, surfactants, stabilizers, and
buffers.
BRIEF DESCRIPTION OF THE FIGURES
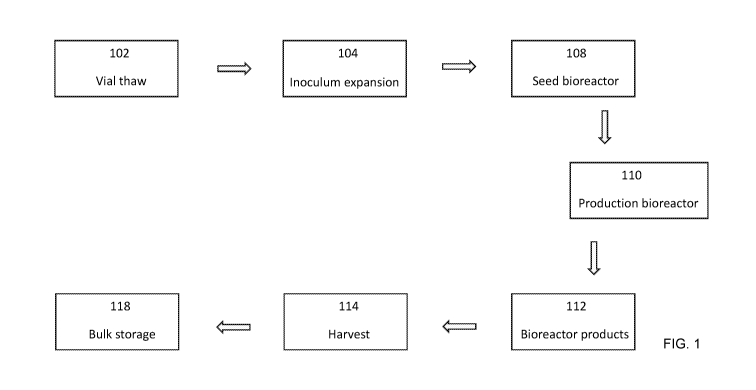
[0020] FIG. 1 presents a block diagram showing the steps in a fermentation
process (an
"upstream processing") for producing therapeutic proteins, for example
monoclonal
.. antibodies.
[0021] FIG. 2 presents a block diagram showing the steps in a purification
process (a
"downstream processing") for producing therapeutic proteins, for example
monoclonal
antibodies.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
[0022] FIGs. 3A and 3B shows graphs of the amount of antibody in the retentate
after
timed intervals of centrifugation.
[0023] FIG. 4 shows a graph of estimated antibody concentration in the
retentate after
timed intervals of centrifugation.
DETAILED DESCRIPTION
[0024] Disclosed herein are formulations and methods for their production that
permit the
delivery of concentrated protein solutions. In embodiments, the approaches
disclosed herein
can yield a lower viscosity liquid formulation or a higher concentration of
therapeutic or
nontherapeutic proteins in the liquid formulation, as compared to traditional
protein solutions.
In embodiments, the approaches disclosed herein can yield a liquid formulation
having
improved stability when compared to a traditional protein solution. A stable
formulation is
one in which the protein contained therein substantially retains its physical
and chemical
stability and its therapeutic or nontherapeutic efficacy upon storage under
storage conditions,
whether cold storage conditions, room temperature conditions, or elevated
temperature
storage conditions. Advantageously, a stable formulation can also offer
protection against
aggregation or precipitation of the proteins dissolved therein. For example,
the cold storage
conditions can entail storage in a refrigerator or freezer. In some examples,
cold storage
conditions can entail storage at a temperature of 100 or less. In additional
examples, the cold
storage conditions entail storage at a temperature from about 2 to about 10
C. In other
examples, the cold storage conditions entail storage at a temperature of about
4 C. In
additional examples, the cold storage conditions entail storage at freezing
temperature such as
about -20 C or lower. In another example, cold storage conditions entail
storage at a
temperature of about -20 C to about 0 C. The room temperature storage
conditions can entail
storage at ambient temperatures, for example, from about 10 C to about 30 C.
Elevated
storage conditions can entail storage at a temperature greater. Elevated
temperature stability,
for example at temperatures from about 30 C to about 50 C, can be used as part
an
accelerated aging study to predict the long-term storage at typical ambient
(10-30 C)
conditions.
[0025] It is well known to those skilled in the art of polymer science and
engineering that
proteins in solution tend to form entanglements, which can limit the
translational mobility of
the entangled chains and interfere with the protein's therapeutic or
nontherapeutic efficacy. In
embodiments, excipient compounds as disclosed herein can suppress protein
clustering due to
specific interactions between the excipient compound and the target protein in
solution.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient compounds as disclosed herein can be natural or synthetic, and
desirably are
substances that the FDA generally recognizes as safe ("GRAS").
1. Definitions
[0026] For the purpose of this disclosure, the term "protein" refers to a
sequence of amino
acids having a chain length long enough to produce a discrete tertiary
structure, typically
having a molecular weight between 1-3000 kDa. In some embodiments, the
molecular weight
of the protein is between about 50-200 kDa; in other embodiments, the
molecular weight of
the protein is between about 20-1000 kDa or between about 20-2000 kDa. In
contrast to the
term "protein," the term "peptide" refers to a sequence of amino acids that
does not have a
discrete tertiary structure. A wide variety of biopolymers are included within
the scope of the
term "protein." For example, the term "protein" can refer to therapeutic or
non-therapeutic
proteins, including antibodies, aptamers, fusion proteins, PEGylated proteins,
synthetic
polypeptides, protein fragments, lipoproteins, enzymes, structural peptides,
and the like.
[0027] As non-limiting examples, therapeutic proteins can include mammalian
proteins
such as hormones and prohormones (e.g., insulin and proinsulin, glucagon,
calcitonin, thyroid
hormones (T3 or T4 or thyroid-stimulating hormone), parathyroid hormone,
follicle-
stimulating hormone, luteinizing hormone, growth hormone, growth hormone
releasing
factor, and the like); clotting and anti-clotting factors (e.g., tissue
factor, von Willebrand's
factor, Factor VIIIC, Factor IX, protein C, plasminogen activators (urokinase,
tissue-type
plasminogen activators), thrombin); cytokines, chemokines, and inflammatory
mediators;
interferons; colony-stimulating factors; interleukins (e.g., IL-1 through IL-
10); growth factors
(e.g., vascular endothelial growth factors, fibroblast growth factor, platelet-
derived growth
factor, transforming growth factor, neurotrophic growth factors, insulin-like
growth factor,
and the like); albumins; collagens and elastins; fibrin sealants;
hematopoietic factors (e.g.,
erythropoietin, thrombopoietin, and the like); osteoinductive factors (e.g.,
bone
morphogenetic protein); receptors (e.g., integrins, cadherins, and the like);
surface membrane
proteins; transport proteins; regulatory proteins; antigenic proteins (e.g., a
viral component
that acts as an antigen); and antibodies. The term "antibody" is used herein
in its broadest
sense, to include as non-limiting examples monoclonal antibodies (including,
for example,
full-length antibodies with an immunoglobulin Fc region), single-chain
molecules, bi-specific
and multi-specific antibodies, diabodies, antibody-drug conjugates, antibody
compositions
having polyepitopic specificity, polyclonal antibodies (such as polyclonal
immunoglobulins
used as therapies for immune-compromised patients), and fragments of
antibodies (including,
for example, Fc, Fab, Fv, and F(ab')2). Antibodies can also be termed
"immunoglobulins."
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
An antibody is understood to be directed against a specific protein or non-
protein "antigen,"
which is a biologically important material; the administration of a
therapeutically effective
amount of an antibody to a patient can complex with the antigen, thereby
altering its
biological properties so that the patient experiences a therapeutic effect.
.. [0028] In embodiments, the proteins are PEGylated, meaning that they
comprise
poly(ethylene glycol) ("PEG") and/or poly(propylene glycol) ("PPG") units.
PEGylated
proteins, or PEG-protein conjugates, have found utility in therapeutic
applications due to their
beneficial properties such as solubility, pharmacokinetics, pharmacodynamics,
immunogenicity, renal clearance, and stability. Non-limiting examples of
PEGylated
.. proteins are PEGylated interferons (PEG-IFN), PEGylated anti-VEGF, PEG
protein
conjugate drugs, Adagen, Pegaspargase, Pegfilgrastim, Pegloticase,
Pegvisomant, PEGylated
epoetin-P, and Certolizumab pegol.
[0029] PEGylated proteins can be synthesized by a variety of methods such as a
reaction of
protein with a PEG reagent having one or more reactive functional groups. The
reactive
functional groups on the PEG reagent can form a linkage with the protein at
targeted protein
sites such as lysine, histidine, cysteine, and the N-terminus. Typical
PEGylation reagents
have reactive functional groups such as aldehyde, maleimide, or succinimide
groups that have
specific reactivity with targeted amino acid residues on proteins. The
PEGylation reagents
can have a PEG chain length from about 1 to about 1000 PEG and/or PPG
repeating units.
.. Other methods of PEGylation include glyco-PEGylation, where the protein is
first
glycosylated and then the glycosylated residues are PEGylated in a second
step. Certain
PEGylation processes are assisted by enzymes like sialyltransferase and
transglutaminase.
[0030] While the PEGylated proteins can offer therapeutic advantages over
native, non-
PEGylated proteins, these materials can have physical or chemical properties
that make them
.. difficult to purify, dissolve, filter, concentrate, and administer. The
PEGylation of a protein
can lead to a higher solution viscosity compared to the native protein, and
this generally
requires the formulation of PEGylated protein solutions at lower
concentrations.
[0031] It is desirable to formulate protein therapeutics in stable, low
viscosity solutions so
they can be administered to patients in a minimal injection volume. For
example, the
.. subcutaneous (SC) or intramuscular (IM) injection of drugs generally
requires a small
injection volume, preferably less than 2 mL. The SC and IM injection routes
are well suited
to self-administered care, and this is a less costly and more accessible form
of treatment
compared with intravenous (IV) injection which is only conducted under direct
medical
supervision. Formulations for SC or IM injection require a low solution
viscosity, generally
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
below 30 cP, and preferably below 20 cP, to allow easy flow of the therapeutic
solution
through a narrow-gauge needle. This combination of small injection volume and
low
viscosity requirements present a challenge to the use of PEGylated protein
therapeutics in SC
or IM injection routes.
[0032] Those proteins having therapeutic effects may be termed "therapeutic
proteins";
formulations containing therapeutic proteins in therapeutically effective
amounts may be
termed "therapeutic formulations." The therapeutic protein contained in a
therapeutic
formulation may also be termed its "protein active ingredient." Typically, a
therapeutic
formulation comprises a therapeutically effective amount of a protein active
ingredient and an
excipient, with or without other optional components. As used herein, the term
"therapeutic"
includes both treatments of existing disorders and preventions of disorders.
Therapeutic
proteins include, for example, proteins such as bevacizumab, trastuzumab,
adalimumab,
infliximab, etanercept, darbepoetin alfa, epoetin alfa, cetuximab, filgrastim,
and rituximab.
Other therapeutic proteins will be familiar to those having ordinary skill in
the art.
[0033] A "treatment" includes any measure intended to cure, heal, alleviate,
improve,
remedy, or otherwise beneficially affect the disorder, including preventing or
delaying the
onset of symptoms and/or alleviating or ameliorating symptoms of the disorder.
Those
patients in need of a treatment include both those who already have a specific
disorder, and
those for whom the prevention of a disorder is desirable. A disorder is any
condition that
alters the homeostatic wellbeing of a mammal, including acute or chronic
diseases, or
pathological conditions that predispose the mammal to an acute or chronic
disease. Non-
limiting examples of disorders include cancers, metabolic disorders (e.g.,
diabetes), allergic
disorders (e.g., asthma), dermatological disorders, cardiovascular disorders,
respiratory
disorders, hematological disorders, musculoskeletal disorders, inflammatory or
rheumatological disorders, autoimmune disorders, gastrointestinal disorders,
urological
disorders, sexual and reproductive disorders, neurological disorders, and the
like. The term
"mammal" for the purposes of treatment can refer to any animal classified as a
mammal,
including humans, domestic animals, pet animals, farm animals, sporting
animals, working
animals, and the like. A "treatment" can therefore include both veterinary and
human
treatments. For convenience, the mammal undergoing such "treatment" can be
referred to as
a "patient." In certain embodiments, the patient can be of any age, including
fetal animals in
utero.
[0034] In embodiments, a treatment involves providing a therapeutically
effective amount
of a therapeutic formulation to a mammal in need thereof. A "therapeutically
effective
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
amount" is at least the minimum concentration of the therapeutic protein
administered to the
mammal in need thereof, to effect a treatment of an existing disorder or a
prevention of an
anticipated disorder (either such treatment or such prevention being a
"therapeutic
intervention"). Therapeutically effective amounts of various therapeutic
proteins that may be
included as active ingredients in the therapeutic formulation may be familiar
in the art; or, for
therapeutic proteins discovered or applied to therapeutic interventions
hereinafter, the
therapeutically effective amount can be determined by standard techniques
carried out by
those having ordinary skill in the art, using no more than routine
experimentation.
[0035] Those proteins used for non-therapeutic purposes (i.e., purposes not
involving
1() treatments), such as household, nutrition, commercial, and industrial
applications, may be
termed "non-therapeutic proteins." Formulations containing non-therapeutic
proteins may be
termed "non-therapeutic formulations". The non-therapeutic proteins can be
derived from
plant sources, animal sources, or produced from cell cultures; they also can
be enzymes or
structural proteins. The non-therapeutic proteins can be used in household,
nutrition,
commercial, and industrial applications such as catalysts, human and animal
nutrition,
processing aids, cleaners, and waste treatment.
[0036] An important category of non-therapeutic biopolymers is enzymes.
Enzymes have a
number of non-therapeutic applications, for example, as catalysts, human and
animal
nutritional ingredients, processing aids, cleaners, and waste treatment
agents. Enzyme
catalysts are used to accelerate a variety of chemical reactions. Examples of
enzyme
catalysts for non-therapeutic uses include catalases, oxidoreductases,
transferases, hydrolases,
lyases, isomerases, and ligases. Human and animal nutritional uses of enzymes
include
nutraceuticals, nutritive sources of protein, chelation or controlled delivery
of micronutrients,
digestion aids, and supplements; these can be derived from amylase, protease,
trypsin,
lactase, and the like. Enzymatic processing aids are used to improve the
production of food
and beverage products in operations like baking, brewing, fermenting, juice
processing, and
winemaking. Examples of these food and beverage processing aids include
amylases,
cellulases, pectinases, glucanases, lipases, and lactases. Enzymes can also be
used in the
production of biofuels. Ethanol for biofuels, for example, can be aided by the
enzymatic
degradation of biomass feedstocks such as cellulosic and lignocellulosic
materials. The
treatment of such feedstock materials with cellulases and ligninases
transforms the biomass
into a substrate that can be fermented into fuels. In other commercial
applications, enzymes
are used as detergents, cleaners, and stain lifting aids for laundry, dish
washing, surface
cleaning, and equipment cleaning applications. Typical enzymes for this
purpose include
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
proteases, cellulases, amylases, and lipases. In addition, non-therapeutic
enzymes are used in
a variety of commercial and industrial processes such as textile softening
with cellulases,
leather processing, waste treatment, contaminated sediment treatment, water
treatment, pulp
bleaching, and pulp softening and debonding. Typical enzymes for these
purposes are
amylases, xylanases, cellulases, and ligninases.
[0037] Other examples of non-therapeutic biopolymers include fibrous or
structural
proteins such as keratins, collagen, gelatin, elastin, fibroin, actin,
tubulin, or the hydrolyzed,
degraded, or derivatized forms thereof These materials are used in the
preparation and
formulation of food ingredients such as gelatin, ice cream, yogurt, and
confections; they are
1() also added to foods as thickeners, rheology modifiers, mouthfeel
improvers, and as a source
of nutritional protein. In the cosmetics and personal care industry, collagen,
elastin, keratin,
and hydrolyzed keratin are widely used as ingredients in skin care and hair
care formulations.
Still other examples of non-therapeutic biopolymers are whey proteins such as
beta-
lactoglobulin, alpha-lactalbumin, and serum albumin. These whey proteins are
produced in
mass scale as a byproduct from dairy operations and have been used for a
variety of non-
therapeutic applications.
2. Measurements
[0038] In embodiments, the protein-containing formulations described herein
are resistant
to monomer loss as measured by size exclusion chromatography (SEC) analysis.
In SEC
analysis as used herein, the main analyte peak is generally associated with
the target protein
contained in the formulation, and this main peak of the protein is referred to
as the monomer
peak. The monomer peak represents the amount of target protein, e.g., a
protein active
ingredient, in the monomeric state, as opposed to aggregated (dimeric,
trimeric, oligomeric,
etc.) or fragmented states. The monomer peak area can be compared with the
total area of the
monomer, aggregate, and fragment peaks associated with the target protein.
Thus, the
stability of a protein-containing formulation can be observed by the relative
amount of
monomer after an elapsed time; an improvement in stability of a protein-
containing
formulation of the invention can therefore be measured as a higher percent
monomer after a
certain elapsed time, as compared to the percent monomer in a control
formulation that does
not contain the excipient.
[0039] In embodiments, an ideal stability result is to have from 98 to 100%
monomer peak
as determined by SEC analysis. In embodiments, an improvement in stability of
a protein-
containing formulation of the invention can be measured as a higher percent
monomer after
exposure to a stress condition, as compared to the percent monomer in a
control formulation
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
that does not contain the excipient when such control formulation is exposed
to the same
stress condition. In embodiments, the stress conditions can be a low
temperature storage,
high temperature storage, exposure to air, exposure to gas bubbles, exposure
to shear
conditions, or exposure to freeze/thaw cycles.
[0040] In embodiments, the protein-containing formulations as described herein
are
resistant to an increase in protein particle size as measured by dynamic light
scattering (DLS)
analysis. In DLS analysis as used herein, the particle size of the protein in
the protein-
containing formulation can be observed as a median particle diameter. Ideally,
the median
particle diameter of the target protein should be relatively unchanged when
subjected to DLS
.. analysis, since the particle diameter represents the active component in
the monomeric state,
as opposed to aggregated (dimeric, trimeric, oligomeric, etc.) or fragmented
states. An
increase of the median particle diameter could represent an aggregated
protein. Thus, the
stability of a protein-containing formulation can be observed by the relative
change in median
particle diameter after an elapsed time.
[0041] In embodiments, the protein-containing formulations as described herein
are
resistant to forming a polydisperse particle size distribution as measured by
dynamic light
scattering (DLS) analysis. In embodiments, a protein-containing formulation
can contain a
monodisperse particle size distribution of colloidal protein particles. In
embodiments, an
ideal stability result is to have less than a 10% change in the median
particle diameter
compared to the initial median particle diameter of the formulation. In
embodiments, an
improvement in stability of a protein-containing formulation of the invention
can be
measured as a lower percent change of the median particle diameter after a
certain elapsed
time, as compared to the median particle diameter in a control formulation
that does not
contain the excipient. In embodiments, an improvement in stability of a
protein-containing
formulation of the invention can be measured as a lower percent change of the
median
particle diameter after exposure to a stress condition, as compared to the
percent change of
the median particle diameter in a control formulation that does not contain
the excipient when
such control formulation is exposed to the same stress condition. In
embodiments, the stress
conditions can be a low temperature storage, high temperature storage,
exposure to air,
exposure to gas bubbles, exposure to shear conditions, or exposure to
freeze/thaw cycles. In
embodiments, an improvement in stability of a protein-containing formulation
therapeutic
formulation of the invention can be measured as a less polydisperse particle
size distribution
as measured by DLS, as compared to the polydispersity of the particle size
distribution in a
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
control formulation that does not contain the excipient when such control
formulation is
exposed to the same stress condition.
[0042] In embodiments, the protein-containing formulations of the invention
are resistant to
precipitation as measured by turbidity, light scattering, and/or particle
counting analysis. In
turbidity, light scattering, or particle counting analysis, a lower value
generally represents a
lower number of suspended particles in a formulation. An increase of
turbidity, light
scattering, or particle counting can indicate that the solution of the target
protein is not stable.
Thus, the stability of a protein-containing formulation can be observed by the
relative amount
of turbidity, light scattering, or particle counting after an elapsed time. In
embodiments, an
ideal stability result is to have a low and relatively constant turbidity,
light scattering, or
particle counting value. In embodiments, an improvement in stability of a
protein-containing
formulation of the invention can be measured as a lower turbidity, lower light
scattering, or
lower particle count after a certain elapsed time, as compared to the
turbidity, light scattering,
or particle count values in a control formulation that does not contain the
excipient. In
embodiments, an improvement in stability of a protein-containing formulation
as described
herein can be measured as a lower turbidity, lower light scattering, or lower
particle count
after exposure to a stress condition, as compared to the turbidity, light
scattering, or particle
count in a control formulation that does not contain the excipient when such
control
formulation is exposed to the same stress condition. In embodiments, the
stress conditions
can be a low temperature storage, high temperature storage, exposure to air,
exposure to gas
bubbles, exposure to shear conditions, or exposure to freeze/thaw cycles.
3. Therapeutic Formulations
[0043] In one aspect, the formulations and methods disclosed herein provide
stable liquid
formulations of improved or reduced viscosity, comprising a therapeutic
protein in a
therapeutically effective amount and an excipient compound. In embodiments,
the
formulation can improve the stability while providing an acceptable
concentration of active
ingredients and an acceptable viscosity. In embodiments, the formulation
provides an
improvement in stability when compared to a control formulation; for the
purposes of this
disclosure, a control formulation is a formulation containing the protein
active ingredient that
is identical on a dry weight basis in every way to the therapeutic formulation
except that it
lacks the excipient compound. In embodiments, improved stability of the
protein containing
formulation is in the form of lower percentage of soluble aggregates,
particulates, subvisible
particles, or gel formation, compared to a control formulation.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
[0044] It is understood that the viscosity of a liquid protein formulation can
be affected by
a variety of factors, including, but not limited to: the nature of the protein
itself (e.g., enzyme,
antibody, receptor, fusion protein, etc.); its size, three-dimensional
structure, chemical
composition, and molecular weight; its concentration in the formulation; the
components of
the formulation besides the protein; the desired pH range; the storage
conditions for the
formulation; and the method of administering the formulation to the patient.
Therapeutic
proteins most suitable for use with the excipient compounds described herein
are preferably
essentially pure, i.e., free from contaminating proteins. In embodiments, an
"essentially
pure" therapeutic protein is a protein composition comprising at least 90% by
weight of the
1() therapeutic protein, or preferably at least 95% by weight, or more
preferably, at least 99% by
weight, all based on the total weight of therapeutic proteins and
contaminating proteins in the
composition. For the purposes of clarity, a protein added as an excipient is
not intended to be
included in this definition. The therapeutic formulations described herein are
intended for
use as pharmaceutical-grade formulations, i.e., formulations intended for use
in treating a
mammal, in such a form that the desired therapeutic efficacy of the protein
active ingredient
can be achieved, and without containing components that are toxic to the
mammal to whom
the formulation is to be administered.
[0045] In embodiments, the therapeutic formulation contains at least 25 mg/mL
of protein
active ingredient. In other embodiments, the therapeutic formulation contains
at least 100
mg/mL of protein active ingredient. In other embodiments, the therapeutic
formulation
contains at least 200 mg/mL of protein active ingredient. In yet other
embodiments, the
therapeutic formulation solution contains at least 300 mg/mL of protein active
ingredient.
Generally, the excipient compounds disclosed herein are added to the
therapeutic formulation
in an amount between about 5 to about 300 mg/mL. In embodiments, the excipient
compound can be added in an amount of about 10 to about 200 mg/mL. In
embodiments, the
excipient compound can be added in an amount of about 20 to about 100 mg/mL.
In
embodiments, the excipient can be added in an amount of about 25 to about 75
mg/mL.
[0046] Excipient compounds of various molecular weights are selected for
specific
advantageous properties when combined with the protein active ingredient in a
formulation.
Examples of therapeutic formulations comprising excipient compounds are
provided below.
In embodiments, the excipient compound has a molecular weight of <5000 Da. In
embodiments, the excipient compound has a molecular weight of <1000 Da. In
embodiments, the excipient compound has a molecular weight of <500 Da.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
[0047] In embodiments, the excipient compounds disclosed herein is added to
the
therapeutic formulation in a viscosity-reducing amount. In embodiments, a
viscosity-
reducing amount is the amount of an excipient compound that reduces the
viscosity of the
formulation at least 10% when compared to a control formulation; for the
purposes of this
disclosure, a control formulation is a formulation containing the protein
active ingredient that
is identical on a dry weight basis in every way to the therapeutic formulation
except that it
lacks the excipient compound. In embodiments, the viscosity-reducing amount is
the amount
of an excipient compound that reduces the viscosity of the formulation at
least 30% when
compared to the control formulation. In embodiments, the viscosity-reducing
amount is the
.. amount of an excipient compound that reduces the viscosity of the
formulation at least 50%
when compared to the control formulation. In embodiments, the viscosity-
reducing amount
is the amount of an excipient compound that reduces the viscosity of the
formulation at least
70% when compared to the control formulation. In embodiments, the viscosity-
reducing
amount is the amount of an excipient compound that reduces the viscosity of
the formulation
at least 90% when compared to the control formulation.
[0048] In embodiments, the viscosity-reducing amount yields a therapeutic
formulation
having a viscosity of less than 100 cP. In other embodiments, the therapeutic
formulation has
a viscosity of less than 50 cP. In other embodiments, the therapeutic
formulation has a
viscosity of less than 20 cP. In yet other embodiments, the therapeutic
formulation has a
viscosity of less than 10 cP. The term "viscosity" as used herein refers to a
dynamic viscosity
value when measured by the methods disclosed herein.
[0049] Therapeutic formulations in accordance with this disclosure have
certain
advantageous properties. In embodiments, the therapeutic formulations are
resistant to shear
degradation, phase separation, clouding out, oxidation, deamidation,
aggregation,
precipitation, and denaturing. In embodiments, the therapeutic formulations
are processed,
purified, stored, syringed, dosed, filtered, and centrifuged more effectively,
compared with a
control formulation. In embodiments, the therapeutic formulations are
administered to a
patient at high concentration of therapeutic protein. In embodiments, the
therapeutic
formulations are administered to patients with less discomfort than would be
experienced
with a similar formulation lacking the therapeutic excipient. In embodiments,
the therapeutic
formulations are administered as a depot injection. In embodiments, the
therapeutic
formulations extend the half-life of the therapeutic protein in the body.
[0050] These features of therapeutic formulations as disclosed herein would
permit the
administration of such formulations by intramuscular or subcutaneous injection
in a clinical
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
situation, i.e., a situation where patient acceptance of an intramuscular
injection would
include the use of small-bore needles typical for IM/SC purposes and the use
of a tolerable
(for example, 2-3 cc) injected volume, and where these conditions result in
the administration
of an effective amount of the formulation in a single injection at a single
injection site. By
contrast, injection of a comparable dosage amount of the therapeutic protein
using
conventional formulation techniques would be limited by the higher viscosity
of the
conventional formulation, so that a SC/IM injection of the conventional
formulation would
not be suitable for a clinical situation. High concentration solutions of
therapeutic proteins
formulated with the excipient compounds described herein can be administered
to patients
1() using syringes or pre-filled syringes.
[0051] In embodiments, the therapeutic excipient has antioxidant properties
that stabilize
the therapeutic protein against oxidative damage. In embodiments, the
therapeutic
formulation is stored at ambient temperatures, or for extended time at
refrigerator conditions
without appreciable loss of potency for the therapeutic protein. In
embodiments, the
therapeutic formulation is dried down for storage until it is needed; then it
is reconstituted
with an appropriate solvent, e.g., water. Advantageously, the formulations
prepared as
described herein can be stable over a prolonged period of time, from months to
years. When
exceptionally long periods of storage are desired, the formulations can be
preserved in a
freezer (and later reactivated) without fear of protein denaturation. In
embodiments,
formulations can be prepared for long-term storage that do not require
refrigeration.
[0052] Methods for preparing therapeutic formulations may be familiar to
skilled artisans.
The therapeutic formulations of the present invention can be prepared, for
example, by
adding the excipient compound to the formulation before or after the
therapeutic protein is
added to the solution. The therapeutic formulation can, for example, be
produced by
combining the therapeutic protein and the excipient at a first (lower)
concentration and then
processed by filtration or centrifugation to produce a second (higher)
concentration of the
therapeutic protein. Therapeutic formulations can be made with one or more of
the excipient
compounds with chaotropes, kosmotropes, hydrotropes, and salts. Therapeutic
formulations
can be made with one or more of the excipient compounds using techniques such
as
encapsulation, dispersion, liposome, vesicle formation, and the like. Methods
for preparing
therapeutic formulations comprising the excipient compounds disclosed herein
can include
combinations of the excipient compounds. In embodiments, combinations of
excipients can
produce benefits in lower viscosity, improved stability, or reduced injection
site pain. Other
additives may be introduced into the therapeutic formulations during their
manufacture,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
including preservatives, surfactants, sugars, sucrose, trehalose,
polysaccharides, arginine,
proline, hyaluronidase, stabilizers, buffers, and the like. As used herein, a
pharmaceutically
acceptable excipient compound is one that is non-toxic and suitable for animal
and/or human
administration.
4. Non-Therapeutic Formulations
[0053] In one aspect, the formulations and methods disclosed herein provide
stable liquid
formulations of improved or reduced viscosity, comprising a non-therapeutic
protein in an
effective amount and an excipient compound. In embodiments, the formulation
improves the
stability while providing an acceptable concentration of active ingredients
and an acceptable
1() viscosity. In embodiments, the formulation provides an improvement in
stability when
compared to a control formulation; for the purposes of this disclosure, a
control formulation
is a formulation containing the protein active ingredient that is identical on
a dry weight basis
in every way to the non-therapeutic formulation except that it lacks the
excipient compound.
[0054] It is understood that the viscosity of a liquid protein formulation can
be affected by
a variety of factors, including but not limited to: the nature of the protein
itself (e.g., enzyme,
structural protein, degree of hydrolysis, etc.); its size, three-dimensional
structure, chemical
composition, and molecular weight; its concentration in the formulation; the
components of
the formulation besides the protein; the desired pH range; and the storage
conditions for the
formulation.
[0055] In embodiments, the non-therapeutic formulation contains at least 25
mg/mL of
protein active ingredient. In other embodiments, the non-therapeutic
formulation contains at
least 100 mg/mL of protein active ingredient. In other embodiments, the non-
therapeutic
formulation contains at least 200 mg/mL of protein active ingredient. In yet
other
embodiments, the non-therapeutic formulation solution contains at least 300
mg/mL of
protein active ingredient. Generally, the excipient compounds disclosed herein
are added to
the non-therapeutic formulation in an amount between about 5 to about 300
mg/mL. In
embodiments, the excipient compound is added in an amount of about 10 to about
200
mg/mL. In embodiments, the excipient compound is added in an amount of about
20 to about
100 mg/mL. In embodiments, the excipient is added in an amount of about 25 to
about 75
mg/mL.
[0056] Excipient compounds of various molecular weights are selected for
specific
advantageous properties when combined with the protein active ingredient in a
formulation.
Examples of non-therapeutic formulations comprising excipient compounds are
provided
below. In embodiments, the excipient compound has a molecular weight of <5000
Da. In
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
embodiments, the excipient compound has a molecular weight of <1000 Da. In
embodiments, the excipient compound has a molecular weight of <500 Da.
[0057] In embodiments, the excipient compounds disclosed herein is added to
the non-
therapeutic formulation in a viscosity-reducing amount. In embodiments, a
viscosity-
reducing amount is the amount of an excipient compound that reduces the
viscosity of the
formulation at least 10% when compared to a control formulation; for the
purposes of this
disclosure, a control formulation is a formulation containing the protein
active ingredient that
is identical on a dry weight basis in every way to the therapeutic formulation
except that it
lacks the excipient compound. In embodiments, the viscosity-reducing amount is
the amount
of an excipient compound that reduces the viscosity of the formulation at
least 30% when
compared to the control formulation. In embodiments, the viscosity-reducing
amount is the
amount of an excipient compound that reduces the viscosity of the formulation
at least 50%
when compared to the control formulation. In embodiments, the viscosity-
reducing amount
is the amount of an excipient compound that reduces the viscosity of the
formulation at least
70% when compared to the control formulation. In embodiments, the viscosity-
reducing
amount is the amount of an excipient compound that reduces the viscosity of
the formulation
at least 90% when compared to the control formulation.
[0058] In embodiments, the viscosity-reducing amount yields a non-therapeutic
formulation having a viscosity of less than 100 cP. In other embodiments, the
non-
therapeutic formulation has a viscosity of less than 50 cP. In other
embodiments, the non-
therapeutic formulation has a viscosity of less than 20 cP. In yet other
embodiments, the non-
therapeutic formulation has a viscosity of less than 10 cP. The term
"viscosity" as used
herein refers to a dynamic viscosity value.
[0059] Non-therapeutic formulations in accordance with this disclosure can
have certain
advantageous properties. In embodiments, the non-therapeutic formulations are
resistant to
shear degradation, phase separation, clouding out, oxidation, deamidation,
aggregation,
precipitation, and denaturing. In embodiments, the therapeutic formulations
can be
processed, purified, stored, pumped, filtered, and centrifuged more
effectively, compared
with a control formulation.
[0060] In embodiments, the non-therapeutic excipient has antioxidant
properties that
stabilize the non-therapeutic protein against oxidative damage. In
embodiments, the non-
therapeutic formulation is stored at ambient temperatures, or for extended
time at refrigerator
conditions without appreciable loss of potency for the non-therapeutic
protein. In
embodiments, the non-therapeutic formulation is dried down for storage until
it is needed;
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
then it can be reconstituted with an appropriate solvent, e.g., water.
Advantageously, the
formulations prepared as described herein is stable over a prolonged period of
time, from
months to years. When exceptionally long periods of storage are desired, the
formulations are
preserved in a freezer (and later reactivated) without fear of protein
denaturation. In
embodiments, formulations are prepared for long-term storage that do not
require
refrigeration.
[0061] Methods for preparing non-therapeutic formulations comprising the
excipient
compounds disclosed herein may be familiar to skilled artisans. For example,
the excipient
compound can be added to the formulation before or after the non-therapeutic
protein is
added to the solution. The non-therapeutic formulation can be produced at a
first (lower)
concentration and then processed by filtration or centrifugation to produce a
second (higher)
concentration. Non-therapeutic formulations can be made with one or more of
the excipient
compounds with chaotropes, kosmotropes, hydrotropes, and salts. Non-
therapeutic
formulations can be made with one or more of the excipient compounds using
techniques
such as encapsulation, dispersion, liposome, vesicle formation, and the like.
Other additives
can be introduced into the non-therapeutic formulations during their
manufacture, including
preservatives, surfactants, stabilizers, and the like.
5. Excipient Compounds
[0062] Several excipient compounds are described herein, each suitable for use
with one or
more therapeutic or non-therapeutic proteins, and each allowing the
formulation to be
composed so that it contains the protein(s) at a high concentration. Some of
the categories of
excipient compounds described below are: (1) hindered amines; (2) anionic
aromatics; (3)
functionalized amino acids; (4) oligopeptides; (5) short-chain organic acids;
(6) low-
molecular-weight aliphatic polyacids; (7) diones and sulfones, (8)
zwitterionic excipients, and
(9) crowding agents with hydrogen bonding elements. Without being bound by
theory, the
excipient compounds described herein are thought to associate with certain
fragments,
sequences, structures, or sections of a therapeutic protein that otherwise
would be involved in
inter-particle (i.e., protein-protein) interactions. The association of these
excipient
compounds with the therapeutic or non-therapeutic protein can mask the inter-
protein
interactions such that the proteins can be formulated in high concentration
without causing
excessive solution viscosity. Excipient compounds advantageously can be water-
soluble,
therefore suitable for use with aqueous vehicles. In embodiments, the
excipient compounds
have a water solubility of >10 mg/mL. In embodiments, the excipient compounds
have a
water solubility of >100 mg/mL. In embodiments, the excipient compounds have a
water
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
solubility of >500 mg/mL. Advantageously for therapeutic proteins, the
excipient
compounds can be derived from materials that are biologically acceptable and
are non-
immunogenic, and are thus suitable for pharmaceutical use. In therapeutic
embodiments, the
excipient compounds can be metabolized in the body to yield biologically
compatible and
non-immunogenic byproducts.
a. Excipient Compound Category 1: Hindered Amines
[0063] High concentration solutions of therapeutic or non-therapeutic proteins
can be
formulated with hindered amine small molecules as excipient compounds. As used
herein,
the term "hindered amine" refers to a small molecule containing at least one
bulky or
sterically hindered group, consistent with the examples below. Hindered amines
can be used
in the free base form, in the protonated form, or a combination of the two. In
protonated
forms, the hindered amines can be associated with an anionic counterion such
as chloride,
hydroxide, bromide, iodide, fluoride, acetate, formate, phosphate, sulfate, or
carboxylate.
Hindered amine compounds useful as excipient compounds can contain secondary
amine,
tertiary amine, quaternary ammonium, pyridinium, pyrrolidone, pyrrolidine,
piperidine,
morpholine, or guanidinium groups, such that the excipient compound has a
cationic charge
in aqueous solution at neutral pH. The hindered amine compounds also contain
at least one
bulky or sterically hindered group, such as cyclic aromatic, cycloaliphatic,
cyclohexyl, or
alkyl groups. In embodiments, the sterically hindered group can itself be an
amine group
such as a dialkylamine, trialkylamine, guanidinium, pyridinium, or quaternary
ammonium
group. Without being bound by theory, the hindered amine compounds are thought
to
associate with aromatic sections of the proteins such as phenylalanine,
tryptophan, and
tyrosine, by a cation pi interaction. In embodiments, the cationic group of
the hindered amine
can have an affinity for the electron rich pi structure of the aromatic amino
acid residues in
the protein, so that they can shield these sections of the protein, thereby
decreasing the
tendency of such shielded proteins to associate and agglomerate.
[0064] In embodiments, the hindered amine excipient compounds has a chemical
structure
comprising imidazole, imidazoline, or imidazolidine groups, or salts thereof,
such as
imidazole, 1-methylimidazole, 4-methylimidazole, 1-hexy1-3-methylimidazolium
chloride, 1-
ethylimidazole, 4-ethylimidazole, 1-hexy1-3-ethylimidazolium chloride,
imidazoline, 2-
imidazoline, imidazolidone, 2-imidazolidone, histamine, 4-methylhistamine,
alpha-
methylhistamine, betahistine, beta-alanine, 2-methyl-2-imidazoline, 1-buty1-3-
methylimidazolium chloride, butyl imidazole, uric acid, potassium urate,
betazole, carnosine,
spermine, spermidine, aspartame, saccharin, acesulfame potassium, xanthine,
theophylline,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
theobromine, caffeine, and anserine. In embodiments, the hindered amine
excipient
compound is a pyrimidine derivative selected from the group consisting of 1,3-
dimethyluracil, 1-methyluracil, 3-methyluracil, 1,3-diethyluracil, 6-
methyluracil, uracil, 1,3-
dimethyl-tetrahydro pyrimidinone, 1-methyl-2-pyrridinone, phenyl serine, DL-3-
phenylserine,
cycloserine, dicyclomine, thymine, 1-methylthymine, 0-4-methylthymine, 1,3-
dimethylthymine, dimethylthymine dimer, cytosine, cysteamine, 5-
methylcytosine, and 3-
methylcytosine. In embodiments, the hindered amine excipient compounds is
selected from
the group consisting of dimethylethanolamine, ethanolamine,
dimethylaminoethanol,
dimethylaminopropylamine, triethanolamine, 1,3-diaminopropane, 1,2-
diaminopropane,
polyetheramines, Jeffamine brand polyetheramines, polyether-monoamines,
polyether-
diamines, polyether-triamines, 1-(1-adamantyl)ethylamine, hordenine,
benzylamine,
dimethylbenzylamine, dimethylcyclohexylamine, diethylcyclohexylamine,
dicyclohexylmethylamine, hexamethylene biguanide, poly(hexamethylene
biguanide),
imidazole, dimethylglycine, meglumine, agmatine, agmatine sulfate,
.. diazabicyclo[2.2.2]octane, tetramethylethylenediamine, N,N-
dimethylethanolamine,
ethanolamine phosphate, glucosamine, choline chloride, phosphocholine,
niacinamide,
isonicotinamide, N,N-diethyl nicotinamide, nicotinic acid, nicotinic acid
sodium salt,
isonicotinic acid, tyramine, N-methyltyramine, 3-aminopyridine, 4-
aminopyridine, 2,4,6-
trimethylpyridine, 3-pyridine methanol, dipyridamole, nicotinamide adenosine
dinucleotide,
biotin, folic acid, folinic acid, folinic acid calcium salt, morpholine, N-
methylpyrrolidone, 2-
pyrrolidinone, procaine, lidocaine, dicyandiamide-taurine adduct, 2-
pyridylethylamine,
dicyandiamide-benzyl amine adduct, dicyandiamide-alkylamine adduct,
dicyandiamide-
cycloalkylamine adduct, and dicyandiamide-aminomethanephosphonic acid adducts.
In
embodiments, a hindered amine compound consistent with this disclosure is
formulated as a
.. protonated ammonium salt. In embodiments, a hindered amine compound
consistent with
this disclosure is formulated as a salt with an inorganic anion or organic
anion as the
counterion. In embodiments, high concentration solutions of therapeutic or non-
therapeutic
proteins are formulated with a combination of caffeine with a benzoic acid, a
hydroxybenzoic
acid, or a benzenesulfonic acid as excipient compounds. In embodiments, the
hindered amine
excipient compounds are metabolized in the body to yield biologically
compatible
byproducts. In some embodiments, the hindered amine excipient compound is
present in the
formulation at a concentration of about 250 mg/mL or less. In additional
embodiments, the
hindered amine excipient compound is present in the formulation at a
concentration of about
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
mg/mL to about 200 mg/mL. In yet additional aspects, the hindered amine
excipient
compound is present in the formulation at a concentration of about 20 to about
120 mg/mL.
[0065] In embodiments, certain hindered amine excipient compounds can possess
other
pharmacological properties. As examples, xanthines are a category of hindered
amines
5 having independent pharmacological properties, including stimulant
properties and
bronchodilator properties when systemically absorbed. Representative xanthines
include
caffeine, aminophylline, 3-isobuty1-1-methylxanthine, paraxanthine,
pentoxifylline,
theobromine, theophylline, and the like. Methylated xanthines are understood
to affect force
of cardiac contraction, heart rate, and bronchodilation. In some embodiments,
the xanthine
10 excipient compound is present in the formulation at a concentration of
about 30 mg/mL or
less.
[0066] Another category of hindered amines having independent pharmacological
properties are the local injectable anesthetic compounds. Local injectable
anesthetic
compounds are hindered amines that have a three-component molecular structure
of (a) a
lipophilic aromatic ring, (b) an intermediate ester or amide linkage, and (c)
a secondary or
tertiary amine. This category of hindered amines is understood to interrupt
neural conduction
by inhibiting the influx of sodium ions, thereby inducing local anesthesia.
The lipophilic
aromatic ring for a local anesthetic compound may be formed of carbon atoms
(e.g., a
benzene ring) or it may comprise heteroatoms (e.g., a thiophene ring).
Representative local
injectable anesthetic compounds include, but are not limited to, amylocaine,
articaine,
bupivicaine, butacaine, butanilicaine, chlorprocaine, cocaine,
cyclomethycaine,
dimethocaine, editocaine, hexylcaine, isobucaine, levobupivacaine, lidocaine,
metabutethamine, metabutoxycaine, mepivacaine, meprylcaine, propoxycaine,
prilocaine,
procaine, piperocaine, tetracaine, trimecaine, and the like. The local
injectable anesthetic
compounds can have multiple benefits in protein therapeutic formulations, such
as reduced
viscosity, improved stability, and reduced pain upon injection. In some
embodiments, the
local anesthetic compound is present in the formulation in a concentration of
about 50 mg/mL
or less.
[0067] In embodiments, a hindered amine having independent pharmacological
properties
is used as an excipient compound in accordance with the formulations and
methods described
herein. In some embodiments, the excipient compounds possessing independent
pharmacological properties are present in an amount that does not have a
pharmacological
effect and/or that is not therapeutically effective. In other embodiments, the
excipient
compounds possessing independent pharmacological properties are present in an
amount that
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
does have a pharmacological effect and/or that is therapeutically effective.
In certain
embodiments, a hindered amine having independent pharmacological properties is
used in
combination with another excipient compound that has been selected to decrease
formulation
viscosity, where the hindered amine having independent pharmacological
properties is used
to impart the benefits of its pharmacological activity. For example, a local
injectable
anesthetic compound can be used to decrease formulation viscosity and also to
reduce pain
upon injection of the formulation. The reduction of injection pain can be
caused by
anesthetic properties; also, a lower injection force can be required when the
viscosity is
reduced by the excipients. Alternatively, a local injectable anesthetic
compound can be used
to impart the desirable pharmacological benefit of decreased local sensation
during
formulation injection, while being combined with another excipient compound
that reduces
the viscosity of the formulation.
b. Excipient Compound Category 2: Anionic Aromatics
[0068] High concentration solutions of therapeutic or non-therapeutic proteins
can be
formulated with anionic aromatic small molecule compounds as excipient
compounds. The
anionic aromatic excipient compounds can contain an aromatic functional group
such as
phenyl, benzyl, aryl, alkylbenzyl, hydroxybenzyl, phenolic, hydroxyaryl,
heteroaromatic
group, or a fused aromatic group. The anionic aromatic excipient compounds
also can
contain an anionic functional group such as carboxylate, oxide, phenoxide,
sulfonate, sulfate,
phosphonate, phosphate, or sulfide. While the anionic aromatic excipients
might be described
as an acid, a sodium salt, or other, it is understood that the excipient can
be used in a variety
of salt forms. Without being bound by theory, an anionic aromatic excipient
compound is
thought to be a bulky, sterically hindered molecule that can associate with
cationic segments
of a protein, so that they can shield these sections of the protein, thereby
decreasing the
interactions between protein molecules that render the protein-containing
formulation
viscous.
[0069] In embodiments, examples of anionic aromatic excipient compounds
include
compounds such as salicylic acid, aminosalicylic acid, hydroxybenzoic acid,
aminobenzoic
acid, para-aminobenzoic acid, benzenesulfonic acid, hydroxybenzenesulfonic
acid,
naphthalenesulfonic acid, naphthalenedisulfonic acid, hydroquinone sulfonic
acid, sulfanilic
acid, vanillic acid, vanillin, vanillin-taurine adduct, aminophenol,
anthranilic acid, cinnamic
acid, coumaric acid, caffeic acid, isonicotinic acid, folic acid, folinic
acid, folinic acid
calcium salt, phenylserine, DL-3-phenylserine, adenosine monophosphate, indole
acetic acid,
potassium urate, furan dicarboxylic acid, furan-2-acrylic acid, 2-
furanpropionic acid, sodium
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
phenylpyruvate, sodium hydroxyphenylpyruvate, dihydroxybenzoic acid,
trihydroxybenzoic
acid, pyrogallol, benzoic acid, and the salts of the foregoing acids. In
embodiments, the
anionic aromatic excipient compounds are formulated in the ionized salt form.
In
embodiments, an anionic aromatic compound is formulated as the salt of a
hindered amine,
such as dimethylcyclohexylammonium hydroxybenzoate. In embodiments, the
anionic
aromatic excipient compounds are formulated with various counterions such as
organic
cations. In embodiments, high concentration solutions of therapeutic or non-
therapeutic
proteins is formulated with anionic aromatic excipient compounds and caffeine.
In
embodiments, the anionic aromatic excipient compounds are metabolized in the
body to yield
biologically compatible byproducts.
c. Excipient Compound Category 3: Functionalized Amino Acids
[0070] High concentration solutions of therapeutic or non-therapeutic proteins
can be
formulated with one or more functionalized amino acids, where a single
functionalized amino
acid or an oligopeptide comprising one or more functionalized amino acids may
be used as
the excipient compound. In embodiments, the functionalized amino acid
compounds
comprise molecules ("amino acid precursors") that can be hydrolyzed or
metabolized to yield
amino acids. In embodiments, the functionalized amino acids can contain an
aromatic
functional group such as phenyl, benzyl, aryl, alkylbenzyl, hydroxybenzyl,
hydroxyaryl,
heteroaromatic group, or a fused aromatic group. In embodiments, the
functionalized amino
acid compounds can contain esterified amino acids, such as methyl, ethyl,
propyl, butyl,
benzyl, cycloalkyl, glyceryl, hydroxyethyl, hydroxypropyl, PEG, and PPG
esters. In
embodiments, the functionalized amino acid compounds are selected from the
group
consisting of arginine ethyl ester, arginine methyl ester, arginine
hydroxyethyl ester, and
arginine hydroxypropyl ester. In embodiments, the functionalized amino acid
compound is a
charged ionic compound in aqueous solution at neutral pH. For example, a
single amino acid
can be derivatized by forming an ester, like an acetate or a benzoate, and the
hydrolysis
products would be acetic acid or benzoic acid, both natural materials, plus
the amino acid. In
embodiments, the functionalized amino acid excipient compounds are metabolized
in the
body to yield biologically compatible byproducts.
d. Excipient Compound Category 4: Oligopeptides
[0071] High concentration solutions of therapeutic or non-therapeutic proteins
can be
formulated with oligopeptides as excipient compounds. In embodiments, the
oligopeptide is
designed such that the structure has a charged section and a bulky section. In
embodiments,
the oligopeptides consist of between 2 and 10 peptide subunits. The
oligopeptide can be bi-
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
functional, for example a cationic amino acid coupled to a non-polar one, or
an anionic one
coupled to a non-polar one. In embodiments, the oligopeptides consist of
between 2 and 5
peptide subunits. In embodiments, the oligopeptides are homopeptides such as
polyglutamic
acid, polyaspartic acid, poly-lysine, poly-arginine, and poly-histidine. In
embodiments, the
oligopeptides have a net cationic charge. In other embodiments, the
oligopeptides are
heteropeptides, such as Trp2Lys3. In embodiments, the oligopeptide can have an
alternating
structure such as an ABA repeating pattern. In embodiments, the oligopeptide
can contain
both anionic and cationic amino acids, for example, Arg-Glu. Without being
bound by
theory, the oligopeptides comprise structures that can associate with proteins
in such a way
that it reduces the intermolecular interactions that lead to high viscosity
solutions; for
example, the oligopeptide-protein association can be a charge-charge
interaction, leaving a
somewhat non-polar amino acid to disrupt hydrogen bonding of the hydration
layer around
the protein, thus lowering viscosity. In some embodiments, the oligopeptide
excipient is
present in the composition in a concentration of about 50 mg/mL or less.
e. Excipient Compound Category 5: Short-Chain Organic Acids
[0072] As used herein, the term "short-chain organic acids" refers to C2-C6
organic acid
compounds and the salts, esters, or lactones thereof. This category includes
saturated and
unsaturated carboxylic acids, hydroxy functionalized carboxylic acids, and
linear, branched,
or cyclic carboxylic acids. In embodiments, the acid group in the short-chain
organic acid is
a carboxylic acid, sulfonic acid, phosphonic acid, or a salt thereof.
[0073] In addition to the four excipient categories above, high concentration
solutions of
therapeutic or non-therapeutic proteins can be formulated with short-chain
organic acids, for
example, the acid or salt forms of sorbic acid, valeric acid, propionic acid,
glucuronic acid,
caproic acid, and ascorbic acid as excipient compounds. Examples of excipient
compounds
in this category include potassium sorbate, taurine, sodium propionate,
calcium propionate,
magnesium propionate, and sodium ascorbate.
f Excipient Compound Category 6: Low Molecular Weight Aliphatic
Polyacids
[0074] High concentration solutions of therapeutic or non-therapeutic
PEGylated proteins
can be formulated with certain excipient compounds that enable lower solution
viscosity,
where such excipient compounds are low molecular weight aliphatic polyacids.
As used
herein, the term "low molecular weight aliphatic polyacids" refers to organic
aliphatic
polyacids having a molecular weight < about 1500, and having at least two
acidic groups,
where an acidic group is understood to be a proton-donating moiety. The acidic
groups can
be in the protonated acid form, the salt form, or a combination thereof. Non-
limiting
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
examples of acidic groups include carboxylate, phosphonate, phosphate,
sulfonate, sulfate,
nitrate, and nitrite groups. Acidic groups on the low molecular weight
aliphatic polyacid can
be in the anionic salt form such as carboxylate, phosphonate, phosphate,
sulfonate, sulfate,
nitrate, and nitrite; their counterions can be sodium, potassium, lithium, and
ammonium.
Specific examples of low molecular weight aliphatic polyacids useful for
interacting with
PEGylated proteins as described herein include maleic acid, tartaric acid,
glutaric acid,
malonic acid, itaconic acid, citric acid, ethylenediaminetetraacetic acid
(EDTA), aspartic
acid, glutamic acid, alendronic acid, etidronic acid and salts thereof.
Further examples of low
molecular weight aliphatic polyacids in their anionic salt form include
phosphate (P043),
1() hydrogen phosphate (HP043), dihydrogen phosphate (H2PO4), sulfate
(5042), bisulfate
(H504), pyrophosphate (P2074), hexametaphosphate, carbonate (C032), and
bicarbonate
(HCO3). The counterion for the anionic salts can be Na, Li, K, or ammonium
ion. These
excipients can also be used in combination with excipients. As used herein,
the low molecular
weight aliphatic polyacid can also be an alpha hydroxy acid, where there is a
hydroxyl group
.. adjacent to a first acidic group, for example glycolic acid, lactic acid,
and gluconic acid and
salts thereof. In embodiments, the low molecular weight aliphatic polyacid is
an oligomeric
form that bears more than two acidic groups, for example polyacrylic acid,
polyphosphates,
polypeptides and salts thereof. In some embodiments, the low molecular weight
aliphatic
polyacid excipient is present in the composition in a concentration of about
50 mg/mL or less.
g. Excipient Compound Category 7: Diones and Sulfones
[0075] An effective viscosity-reducing excipient can be a molecule containing
a sulfone,
sulfonamide, or dione functional group that is soluble in pure water to at
least 1 g/L at 298K
and having a net neutral charge at pH 7. Preferably, the molecule has a
molecular weight of
less than 1000 g/mol and more preferably less than 500 g/mol. The diones and
sulfones
.. effective in reducing viscosity have multiple double bonds, are water
soluble, have no net
charge at pH 7, and are not strong hydrogen bonding donors. Not to be bound by
theory, the
double bond character can allow for weak pi-stacking interactions with
protein. In
embodiments, at high protein concentrations and in proteins that only develop
high viscosity
at high concentration, charged excipients are not effective because
electrostatic interaction is
a longer-range interaction. Solvated protein surfaces are predominantly
hydrophilic, making
them water soluble. The hydrophobic regions of proteins are generally shielded
within the 3-
dimensional structure, but the structure is constantly evolving, unfolding,
and re-folding
(sometimes called "breathing") and the hydrophobic regions of adjacent
proteins can come
into contact with each other, leading to aggregation by hydrophobic
interactions. The pi-
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
stacking feature of dione and sulfone excipients can mask hydrophobic patches
that may be
exposed during such "breathing." Another other important role of the excipient
can be to
disrupt hydrophobic interactions and hydrogen bonding between proteins in
close proximity,
which will effectively reduce solution viscosity. Dione and sulfone compounds
that fit this
description include dimethylsulfone, ethyl methyl sulfone, ethyl methyl
sulfonyl acetate,
ethyl isopropyl sulfone, bis(methylsulfonyl)methane, methane sulfonamide,
methionine
sulfone, sodium bisulfite, menadione sodium bisulfite, 1,2-cyclopentanedione,
1,3-
cyclopentanedione, 1,4-cyclopentanedione, and butane-2,3-dione.
h. Excipient Compound Category 8: Zwitterionic Excipients
[0076] Solutions of therapeutic or non-therapeutic proteins can be formulated
with certain
zwitterionic compounds as excipients to improve stability or reduce viscosity.
As used herein,
the term "zwitterionic" refers to a compound that has a cationic charged
section and an
anionic charged section. In embodiments, the zwitterionic excipient compounds
are amine
oxides. In embodiments, the opposing charges are separated from each other by
2-8 chemical
bonds. In embodiments, the zwitterionic excipient compounds can be small
molecules, such
as those with a molecular weight of about 50 to about 500 g/mol, or can be
medium
molecular weight molecules, such as those with a molecular weight of about 500
to about
2000 g/mol, or can be high molecular weight molecules, such as polymers having
a molecular
weight of about 2000 to about 100,000 g/mol.
[0077] Examples of the zwitterionic excipient compounds include (3-
carboxypropyl)
trimethylammonium chloride, 1-aminocyclohexane carboxylic acid,
homocycloleucine, 1-
methy1-4-imidazoleacetic acid, 3-(1-pyridinio)-1-propanesulfonate, 4-
aminobenzoic acid,
alendronate, aminoethyl sulfonic acid, aminohippuric acid, aspartame,
aminotris
(methylenephosphonic acid) (ATM)), calcobutrol, calteridol, cocamidopropyl
betaine,
cocamidopropyl hydroxysultaine, creatine, cytidine, cytidine monophosphate,
diaminopimelic acid, diethylenetriaminepentaacetic acid, dimethyl
phenylalanine,
methylglycine, sarcosine, dimethylglycine, zwitterionic dipeptides (e.g., Arg-
Glu, Lys-Glu,
His-Glu, Arg-Asp, Lys-Asp, His-Asp, Glu-Arg, Glu-Lys, Glu-His, Asp-Arg, Asp-
Lys, Asp-
His), diethylenetriamine penta(methylene phosphonic acid) (DTPMP), dipalmitoyl
phosphatidylcholine, ectoine, ethylenediamine tetra(methylenephosphonic acid)
(EDTMP),
folate benzoate mixture, folate niacinamide mixture, gelatin, hydroxyproline,
iminodiacetic
acid, isoguvacine, lecithin, myristamine oxide, nicotinamide adenine
dinucleotide (NAD),
aspartic acid, N-methyl aspartic acid, N-methylproline, lysine, N-trimethyl
lysine, ornithine,
oxolinic acid, risendronate, allyl cysteine, S-allyl-L-cysteine, somapacitan,
taurine, theanine,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
trigonelline, vigabatrin, ectoine, 4-(2-hydroxyethyl)-1-
piperazineethanesulfonate, o-
octylphosphoryl choline, nicotinamide mononucleotide, triglycine,
tetraglycine, f3-
guanidinopropionic acid, 5-aminolevulinic acid hydrochloride, picolinic acid,
lidofenin,
phosphocholine, 1-(5-carboxypenty1)-4-methylpyridin-1-ium bromide, L-anserine
nitrate, L-
glutathione reduced, N-ethyl-L-glutamine, N-methyl proline, (Z)-14N-(2-
aminoethyl)-N-(2-
ammonioethyl) amino]diazen-l-ium-1,2-diolate (DETA-NONOate), (Z)-1-[N-(3-
aminopropy1)-N-(3-ammoniopropyl)amino]diazen-l-ium-1,2-diolate (DP TA-NONate),
and
zoledronic acid.
[0078] Not to be bound by theory, the zwitterionic excipient compounds can
exert viscosity
.. reducing or stabilizing effects by interacting with the protein, for
example, by charge
interactions, hydrophobic interactions, and steric interactions, causing the
proteins to be more
resistant to aggregation, or by affecting the bulk properties of the water in
the protein
formulation, such as an electrolyte contribution, a surface tension reduction,
a change in the
amount of unbound water available, or a change in dielectric constant.
i. Excipient Compound Category 9: Crowding Agents with Hydrogen Bonding
Elements
[0079] Solutions of therapeutic or non-therapeutic proteins can be formulated
with
crowding agents with hydrogen bonding elements as excipients to improve
stability or reduce
viscosity. As used herein, the term "crowding agent" refers to a formulation
additive that
.. reduces the amount of water available for dissolving a protein in solution,
increasing the
effective protein concentration. In embodiments, crowding agents can decrease
protein
particle size or reduce the amount of protein unfolding in solution. In
embodiments, the
crowding agents can act as solvent modifiers that cause structuring of the
water by hydrogen
bonding and hydration effects. In embodiments, the crowding agents can reduce
the amount
of intermolecular interactions between proteins in solution. In embodiments,
the crowding
agents have a structure containing at least one hydrogen bond donor element
such as
hydrogen attached to an oxygen, sulfur, or nitrogen atom. In embodiments, the
crowding
agents have a structure containing at least one weakly acidic hydrogen bond
donor element
having a pKa of about 6 to about 11. In embodiments, the crowding agents have
a structure
.. containing between about 2 and about 50 hydrogen bond donor elements. In
embodiments,
the crowding agents have a structure containing at least one hydrogen bond
acceptor element
such as a Lewis base. In embodiments, the crowding agents have a structure
containing
between about 2 and about 50 hydrogen bond acceptor elements. In embodiments,
the
crowding agents have a molecular weight between about 50 and 500 g/mol. In
embodiments,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
the crowding agents have a molecular weight between about 100 and 350 g/mol.
In other
embodiments, the crowding agents can have a molecular weight above 500 g/mol,
such as
raffinose, inulin, pullulan, or sinistrins.
[0080] Examples of the crowding agent excipients with hydrogen bonding
elements include
.. 1,3-Dimethy1-3,4,5,6-tetrahydro-2(1H)-pyrimidione, 15-crown-5, 18-crown-6,
2-butanol, 2-
butanone, 2-phenoxyethanol, acetaminophen, allantoin, arabinose, meglumine,
arabitol,
benzyl acetoacetate, benzyl alcohol, chlorobutanol, cholestanoltetraacetyl-b-
glucoside,
cinnamaldehyde, cyclohexanone, deoxyribose, diethyl carbonate, dimethyl
carbonate,
dimethyl isosorbide, dimethylacetamide, dimethylformamide, dimethylol ethylene
urea,
dimethyluracil, epilactose, erythritol, erythrose, ethyl lactate, ethyl
maltol, ethylene
carbonate, formamide, fucose, galactose, genistein, gentisic acid
ethanolamide,
gluconolactone, glyceraldehyde, glycerol, glycerol carbonate, glycerol formal,
glycerol
urethane, glycyrrhizic acid, gossypin, harpagoside, hederacoside C,
icodextrin, iditol,
imidazolidone, inositol, inulins, isomaltitol, kojic acid, lactitol,
lactobionic acid, lactose,
lactulose, lyxose, madecassoside, maltotriose, mangiferin, mannose, melzitose,
methyl
lactate, methylpyrrolidone, mogroside V, N-acetylgalactosamine, N-
acetylglucosamine, N-
acetylneuraminic acid, N-methyl acetamide, N-methyl formamide, N-methyl
propionamide,
pentaerythritol, pinoresinol diglucoside, glucuronic acid, piracetam, propyl
gallate, propylene
carbonate, psicose, pullulan, pyrogallol, quinic acid, raffinose, rebaudioside
A, rhamnose,
ribitol, ribose, ribulose, saccharin, sedoheptulose, sinistrins, solketal,
stachyose, sucralose,
tagatose, t-butanol, tetraglycol, triacetin, N-acetyl-d-mannosamine, nystose,
kestose,
turanose, acarbose, D-saccharic acid 1,4-lactone, thiodigalactoside, fucoidan,
hydroxysafflor
yellow A, shikimic acid, diosmin, pravastatin sodium salt, D-altrose, L-
gulonic gamma-
lactone, neomycin, rubusoside dihydroartemisinin, phloroglucinol, naringin,
baicalein,
hesperidin, apigenin, pyrogallol, morin, sal salate, kaempferol, myricetin,
3',4',7-
trihydroxyisoflavone, ( )-taxifolin, silybin, perseitol diformal, 4-
hydroxyphenylpyruvic acid,
sulfacetamide, isopropyl 0-D-1-thiogalactopyranoside, ethyl 2,5-
dihydroxybenzoate,
spectinomycin, resveratrol, quercetin, kanamycin sulfate, 1-(2-
Pyrimidyl)piperazine, 2-(2-
pyridyl)ethylamine, 2-imidazolidone, DL-1,2-isopropylideneglycerol, metformin,
m-
xylylenediamine, x-xylylenediamine, demeclocycline, tripropylene glycol,
tubeimoside I,
verbenaloside, xylitol, and xylose.
6. Protein/Excipient Solutions: Properties and Processes
[0081] In certain embodiments, solutions of therapeutic or non-therapeutic
proteins
formulated with above-identified excipient compounds or combinations thereof
(hereinafter,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
"excipient additives"), such as hindered amines, anionic aromatics,
functionalized amino
acids, oligopeptides, or short-chain organic acids, low molecular weight
aliphatic polyacids,
diones and sulfones, zwitterionic excipients, and crowding agents with
hydrogen bonding
elements result in improved protein-protein interaction characteristics or
protein self-
interactions as measured by the protein diffusion interaction parameter, lcD,
by biolayer
interferometry, by surface plasmon resonance, or by determining the second
virial coefficient,
B22, or similar method. As used herein, an "improvement" in one or more
protein-protein
interaction parameters achieved by test formulations using the above-
identified excipient
compounds or combinations thereof can refer to a decrease in attractive
protein-protein
interactions when a test formulation is compared under comparable conditions
with a
comparable formulation that does not contain the excipient compounds or
excipient additives.
Such improvements can be identified by measuring certain parameters that apply
to the
overall process or an aspect thereof, where a parameter is any metric
pertaining to the process
where an alteration can be can be quantified and compared to a previous state
or to a control.
A parameter can pertain to the process itself, such as its efficiency, cost,
yield, or rate.
[0082] A parameter can also be a proxy parameter that pertains to a feature or
an aspect of
the larger process. As an example, parameters such as the lcD or B22
parameters can be
termed proxy parameters. Measurements of lcD and B22 can be made using
standard
techniques in the industry, and can be an indicator of process-related
parameters such as
improved solution properties or stability of the protein in solution. Not to
be bound by
theory, it is understood that a highly negative lcD value can indicate that
the protein has strong
attractive interactions, and this can lead to aggregation, instability, and
rheology problems.
When formulated in the presence of certain of the above-identified excipient
compounds or
combinations thereof, the same protein can have an improved proxy parameter of
a less
negative lcD value, or a lcD value near or above zero, with this improved
proxy parameter
being associated with an improvement in a process-related parameter.
[0083] In embodiments, certain of the above-described excipient compounds or
combinations thereof, such as hindered amines, anionic aromatics,
functionalized amino
acids, oligopeptides, short-chain organic acids, low molecular weight
aliphatic polyacids,
diones and sulfones, zwitterionic excipients, and/or crowding agents with
hydrogen bonding
elements are used to improve a protein-related process, such as the
manufacture, processing,
sterile filling, purification, and analysis of protein-containing solutions,
using processing
methods such as filtration, sterile filtration, depth filtration, syringing,
transferring, pumping,
mixing, heating or cooling by heat transfer, gas transfer, centrifugation,
chromatography,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
membrane separation, centrifugal concentration, tangential flow filtration,
radial flow
filtration, axial flow filtration, lyophilization, and gel electrophoresis. In
these and related
protein-related processes, the protein of interest is dissolved in a solution
that conveys it
through the processing apparatus. Such solutions, referred to herein as
"carrier solutions,"
can include cell culture media (containing, for example, secreted proteins of
interest), lysate
solutions following the lysis of host cells (where the protein of interest
resides in the lysate),
elution solutions (which contain the protein of interest following
chromatographic
separations), electrophoresis solutions, transport solutions for carrying the
protein of interest
through conduits in a processing apparatus, and the like. A carrier solution
containing the
protein of interest may also be termed a protein-containing solution or a
protein solution. As
described in more detail below, one or more viscosity-reducing excipients can
be added to the
protein-containing solution to improve various aspects of processing. As used
herein, the
terms "improve," "improvements," and the like refer to an advantageous change
in a
parameter of interest in a carrier solution when that parameter is compared to
the same
parameter as measured in a control solution. As used herein, a "control
solution" means a
solution that lacks the viscosity-reducing excipient but otherwise
substantially similar to the
carrier solution. As used herein, a "control process," for example a control
filtration process,
a control chromatographic process, and the like, is a protein-related process
that is
substantially similar to the protein-related process of interest and is
performed with a control
solution instead of a carrier solution.
[0084] For example, in processes where a protein-containing solution is pumped
through
conduits (e.g., flow chambers, piping or tubing), adding a viscosity-lowering
excipient to the
protein solution, as described above, before or during the pumping process can
substantially
reduce the force and the power required to pump the solution. It is understood
that fluids
generally exhibit a resistance to flow, i.e., a viscosity, and that a force
must be applied to the
fluid to overcome this viscosity in order to induce and propagate flow. The
power, P,
required for pumping scales with the head, H, and capacity, Q, as shown in the
following
equation:
P HQ
(Eq. 1)
[0085] Viscous fluids tend to increase the power requirements for pumps, to
lower pump
efficiency, to decrease pump head and capacity, and to increase frictional
resistance in piping.
Adding the viscosity-lowering excipients described above to a protein solution
prior to or
during pumping can substantially lower processing costs by decreasing either
the head (H,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Eq. 1) or the capacity (Q, Eq. 1) or both. The benefits of reduced viscosity
can be
manifested, for example, by improved throughput, increased yield, or decreased
processing
time. Moreover, frictional losses from the transmission of a fluid through a
conduit can
account for a significant fraction of the costs associated with conveying such
fluids. Adding
a viscosity-lowering excipient as described above to a protein solution prior
to or during
pumping can substantially lower processing costs by decreasing the friction
accompanying
the pumping process. Measurement of processing costs represents a processing
parameter
that can be improved by using a viscosity-reducing excipient.
[0086] These processes and processing methods for protein solutions can have
improved
efficiency due to the lower viscosity, improved solubility, or improved
stability of the
proteins in the solution during manufacture, processing, purification, and
analysis steps.
Measurement of processing efficiency or measurement of proxy parameters such
as viscosity,
solubility or stability of the proteins in solution represent processing
parameters that can be
improved by using a viscosity-reducing excipient. Several different factors
are understood to
adversely affect protein viscosity, solubility, and stability during
processing. For example,
protein-containing solutions are subject to a variety of physical stressors
during
manufacturing and purification, including significant shear stresses induced
by manipulating
protein solutions through typical processing operations, including, but not
limited to,
pumping, mixing, centrifugation, and filtration. In addition, during these
processing steps, air
bubbles can become entrained within the fluid to which proteins can adsorb.
Such interfacial
tension forces, coupled with typical shear stresses encountered during
processing, can cause
adsorbed protein molecules to unfold and aggregate. Additionally, significant
protein
unfolding can occur during pump cavitation events and during exposure to solid
surfaces
during manufacturing, such as ultrafiltration and diafiltration membranes.
Such events can
impair protein folding and product quality.
[0087] For Newtonian fluids, the stress, T, imposed by a given process scales
with the shear
rate, y, and viscosity of the fluid, r, as shown in the following equation:
T = YTI (Eq.
2)
[0088] By formulating a protein solution with one or more of the above-
described excipient
compounds or combinations thereof, solution viscosity is decreased, thus
decreasing the shear
stress encountered by the protein solution. The decreased shear stress can
improve the
stability of the formulation being processed, as manifested, for example, by a
better or more
desirable measurement of a processing parameter. Such improved processing
parameters can
include metrics such as reduced levels of protein aggregates, particles, or
subvisible particles
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
(manifested macroscopically as turbidity), reduced product losses, or improved
overall yield.
As another example of an improved processing parameter, reducing viscosity of
a protein-
containing solution can decrease the processing time for the solution. The
processing time
for a given unit operation generally scales inversely with the shear rate.
Therefore, for a
.. given characteristic stress, a decrease in protein solution viscosity by
the addition of the
above-described excipient compounds or combinations thereof is associated with
an increase
in shear rate (y, see Eq. 2), and therefore a decrease in the processing time.
[0089] During processing, it is understood that a protein in a solution may be
a desired
protein active ingredient, for example a therapeutic or non-therapeutic
protein. Facilitating
.. the processing of such a protein active ingredient using the excipients
described herein can
increase the yield or the rate of production of the protein active ingredient,
or improve the
efficiency of the particular process, or decrease the energy use, or the like,
any of which
outcomes represent processing parameters that have been improved by the use of
the
viscosity-reducing excipient. It is also understood that protein contaminants
can be formed
during certain processing technologies, for example during the fermentation
and purification
steps of bioprocessing. Removing the contaminants more quickly, more
thoroughly, or more
efficiently can also improve the processing of the desired protein, i.e., the
protein active
ingredient; these outcomes represent processing parameters that have been
improved by the
use of the viscosity-reducing excipient compound or additive. As described
herein, certain
excipients as described herein, by lowering solution viscosity, improving
protein stability,
and/or increasing protein solubility, can improve the transport of desired
protein active
ingredients, and can improve the removal of undesirable protein contaminants;
both effects,
which represent processing parameters that have been improved by the use of
the viscosity-
reducing excipient or additive, show that these excipients or additives
improve the overall
process of protein manufacture. Advantageously, a viscosity-reducing excipient
used in
processing is selected based on its physiological impact or lack thereof on a
potential patient.
For example, while certain substituted phenethylamines are understood to
modulate various
neurotransmitters, such as the monoamine neurotransmitter systems, and these
may have
various psychotropic effects (e.g., stimulant, hallucinogenic, or entactogenic
effects) because
of their impact on the central nervous system, it can be desirable to employ a
viscosity-
reducing phenethylamine excipient in a viscosity-reducing amount that does not
produce
psychotropic effects, or that does not produce clinically problematic
psychotropic effects, or
that may produce psychotropic effects in a dose-related manner, but does not
produce
psychotropic effects at the dosage to be found in a formulation that has been
produced by the
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
processes described herein for improving a processing parameter by adding the
viscosity-
reducing excipient to a step of the process. Similarly, it can be desirable to
employ other
viscosity-reducing excipients that do not produce other physiological effects
(e.g.,
cardiovascular, respiratory, gastrointestinal, genitourinary, and the like),
or that do not
produce clinically problematic physiological effects, or that may produce
physiological
effects in a dose-related manner, but do not produce physiological effects at
the dosage to be
found in a formulation that has been produced by the processes described
herein for
improving a processing parameter by adding the viscosity-reducing excipient to
a step of the
process.
ix) [0090] Specific platform unit operation for therapeutic protein
production and purification
offer further examples of the advantageous uses of viscosity-reducing
excipients as disclosed
herein, and further examples of these excipients' or additives' improving
processing
parameters. For example, introducing one or more of the viscosity-reducing
excipients
described above into these production and purification processes, as described
below, can
provide substantial improvements in molecule stability and recovery, and a
decrease in
operation costs.
[0091] It is understood in the art that the widely practiced technology for
producing and
purifying therapeutic proteins like monoclonal antibodies generally consists
of a fermentation
process followed by a series of steps for purification processing.
Fermentation, or upstream
processing (USP), comprises those steps by which therapeutic proteins are
grown in
bioreactors, typically using bacterial or mammalian cell lines. USP may, in
embodiments,
include steps such as those shown in FIG. 1. Purification, or downstream
processing (DSP)
may, in embodiments, include steps such as those shown in FIG. 2.
[0092] As shown in FIG. 1, USP may commence with the step 102 of thawing of
vials from
a master cell bank (MCB). The MCB can be expanded as shown in step 104, to
form a
working cell bank (not shown) and/or to produce the working stock for further
production.
Cell culture takes place in a series of seed and production bioreactors, as
shown in steps 108
and 110, to yield those bioreactor products 112 from which the desired
therapeutic protein
can be harvested, as shown in step 114. Following harvest 114, the products
can be
.. submitted to further purification (i.e., DSP, as described below in more
detail and as depicted
in FIG. 2), or these products may be stored in bulk, typically by freezing and
storing at a
temperature of approximately -80 C.
[0093] In embodiments, protein production by cell culture techniques can be
improved by
the use of the above-identified excipients, as manifested by improvements in
process-related
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
parameters. In embodiments, the desired excipient can be added during USP in
an amount
effective to reduce the viscosity of the cell culture medium by at least 20%.
In other
embodiments, the desired excipient can be added during USP in an amount
effective to
reduce the viscosity of the cell culture medium by at least 30%. In
embodiments, the desired
excipient can be added to the cell culture medium in an amount of about 1 mM
to about 400
mM. In embodiments, the desired excipient can be added to the cell culture
medium in an
amount of about 20 mM to about 200 mM. In embodiments, the desired excipient
can be
added to the cell culture medium in an amount of about 25 mM to about 100 mM.
The
desired excipient or combination of excipients can be added directly to the
cell culture
medium, or it can be added as a component of a more complex supplemental
medium, for
example a nutrient-containing solution or "feed solution" that is formulated
separately and
added to the cell culture medium. In embodiments, a second viscosity-reducing
compound
can be added to the carrier solution, either directly or via a supplemental
medium, wherein
the second viscosity-reducing compound adds an additional improvement to a
particular
parameter of interest.
[0094] As described below, there are many process-related parameters during
USP that can
be improved by use of one or more viscosity-reducing excipients. For example,
in
embodiments, use of a viscosity-reducing excipient can improve parameters such
as the rate
and/or degree of cell growth during steps such as inoculum expansion 104, and
cell culture
108 and 110, and/or can improve proxy parameters that are correlated with the
improvement
in various process parameters. For example, adding the above-identified
excipients to the
USP process at a step such as the production bioreactor step 110, can decrease
the viscosity
of the cell culture medium, which can subsequently improve heat transfer
efficiency and gas
transfer efficiency. Because the cell culture process requires oxygen infusion
to the cells to
enable protein expression, and the diffusion of oxygen into the cells can
therefore be a rate-
limiting step, improving the rate of oxygen uptake by improving gas transfer
efficiency
through decreasing solution viscosity can improve the rate or amount of
protein expression
and/or its efficiency. In this context, parameters such as the rate of oxygen
uptake and the
rate of gas transfer efficiency can be deemed proxy parameters, whose
improvement is
correlated with an improvement in the process parameter of improved protein
expression or
improved processing efficiency. As another example, the availability of
viscosity-reducing
excipients can improve processing, for example, during the inoculum expansion
step 104 and
during the cell culture steps 108 and 110, by improving a proxy parameter such
as the
solubility of protein growth factors that are required for protein expression;
with improved
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
growth factor solubility, these substances can become more available to the
cells, thereby
facilitating cell growth.
[0095] In embodiments, process parameters such as the amount of protein
recovery or the
rate of protein recovery during USP can be improved by reducing viscosity
during USP by
several mechanisms. For example, the harvest of therapeutic protein at the end
of the lysis
step during harvest 114 from the completed cell culture can be more efficient
or can be
otherwise improved with the use of the above-identified excipients. Not to be
bound by
theory, by reducing viscosity of the expressed protein, these viscosity-
reducing excipients can
increase the efficiency of diffusion of therapeutic protein away from other
lysate components.
In addition, the separation of membranes and other cell debris from the
protein-containing
supernate can be accomplished with a faster separation rate or a higher degree
of supernate
purity, with the use of the viscosity-reducing excipients, thereby improving
the process
parameter of USP efficiency. Furthermore, the protein separation steps that
use
centrifugation or filtration steps can be accomplished faster with the use of
the viscosity-
reducing excipients, since the excipients reduce the viscosity of the medium.
[0096] In embodiments, as an additional benefit, use of the above-described
viscosity-
reducing excipients in cell culture can increase a process parameter such as
protein yield
during USP because protein misfolding and aggregation are reduced. It is
understood that, as
the cell culture is optimized to produce a maximum yield of recombinant
protein, the
resulting protein is expressed in a highly concentrated manner, which can
result in
misfolding; adding a viscosity-reducing excipient can reduce the attractive
protein-protein
interactions that lead to misfolding and aggregation, thereby increasing the
amount of intact
recombinant protein that is available for harvest 114.
[0097] Downstream processing (DSP), depicted in FIG. 2 in an illustrative
embodiment,
involves a sequence of steps that results in the recovery and purification of
therapeutic
proteins, for example monoclonal antibodies, biopharmaceuticals, vaccines, and
other
biologics. At the end of USP, the therapeutic protein of interest can be
dissolved in the cell
culture medium, having been secreted from the host cells. The therapeutic
protein can also
be dissolved in a fluid medium following the lysis of the host cells at the
end of the USP
sequences. DSP is undertaken to retrieve the protein of interest from the
solution in which it
is dissolved (e.g., the culture medium or host cell lysate medium), and to
purify it. During
DSP, (i) various contaminants (such as insoluble cell debris and particulates)
are removed
from the media, (ii) the protein product is isolated through techniques such
as extraction,
precipitation, adsorption or ultrafiltration, (iii) the protein product is
purified through
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
techniques such as affinity chromatography, precipitation, or crystallization,
and (iv) the
product is further polished, and viruses are removed.
[0098] As shown in FIG. 2, a feedstock from cell culture harvest 200 (also as
described in
FIG. 1) is initially subjected to affinity chromatography 204, typically
involving Protein-A
chromatography or other analogous chromatographic steps. The virus
inactivation step 208
typically entails subjecting the feedstock to a low pH hold. One or more
polishing
chromatography steps 210 and 212 are performed to remove impurities, such as
host cell
proteins (HCP), DNA, charge variants, and aggregates. Cation exchange (CEX)
chromatography is commonly used as an initial polishing chromatography step
210, but it
may be accompanied by a second chromatography step 212 that either precedes or
follows it.
The second chromatography step 212 further removes host-cell-related
impurities (e.g., HCP
or DNA), or product related impurities such as aggregates. Anion exchange
(AEX)
chromatography and hydrophobic interaction chromatography (HIC) can be
employed as
second chromatography steps 212. Virus filtration 214 is performed to effect
virus removal.
Final purification steps 218 can include ultrafiltration and diafiltration,
and preparation for
formulation.
[0099] As generally described above, purification processes or DSP following
the
fermentation process can include (1) cell culture harvest, (2) chromatography
(e.g., Protein-A
chromatography and chromatographic polishing steps, including ion exchange and
hydrophobic interaction chromatography), (3) viral inactivation, and (4)
filtration (e.g., viral
filtration, sterile filtration, dialysis, and ultrafiltration and
diafiltration steps to concentrate the
protein and exchange the protein into the formulation buffer). Examples are
provided below
to illustrate the advantages from using a viscosity-reducing excipient as
described herein to
improve process parameters associated with these purification processes. It is
understood
that the viscosity-reducing excipient or combinations thereof can be
introduced at any phase
of DSP by adding it to a carrier solution or in any other way engineering the
contact of the
protein of interest with the excipient, whether in soluble or stabilized form.
In embodiments,
a second viscosity-reducing compound can be added to the carrier solution
during DSP,
wherein the second viscosity-reducing compound adds an additional improvement
to a
particular parameter of interest.
[00100] (1) Cell culture harvest: Cell culture harvest generally
involves
centrifugation and depth filtration operations in which cellular debris is
physically removed
from protein-containing solutions. The centrifugation step can provide a more
complete
separation of soluble protein from cell debris with the benefit of a viscosity-
reducing
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
excipient. Whether done by batch or continuous processing, the centrifuge
separation requires
the dense phase to consolidate as much as possible to maximize recovery of the
target
protein. In embodiments, addition of the above-identified excipients or
combinations thereof
can increase the process parameter of protein yield, for example, by
increasing the yield of
protein-containing centrate that flows away from the dense phase of the
centrifuge separation
process. The depth filtration step is a viscosity-limited step, and thus can
be made more
efficient by using an excipient that reduces solution viscosity. These
processes can also
introduce air bubbles into the protein solution, which can couple with shear-
induced stresses
to destabilize the therapeutic protein molecules being purified. Adding a
viscosity-reducing
excipient to the protein-containing solution, before and/or during cell
culture harvest, as
described above, can protect the protein from these stresses, thereby
improving the process
parameter of quantified product recovery.
[00101] (2) Chromatography: After cell culture harvest by
centrifugation or
filtration, chromatography is typically used to separate the therapeutic
protein from the
fermentation broth. Protein A chromatography is used when the therapeutic
protein is an
antibody: Protein A is selective towards IgG antibodies, which it will bind
dynamically at a
high flow rate and capacity. Cation exchange (CEX) chromatography can be used
as a cost-
effective alternative to Protein A chromatography. If CEX is used, the pH of
the feed must be
adjusted and its conductivity decreased prior to loading onto the column to
optimize the
dynamic binding capacity. Mimetic resins can also be used as an alternative to
Protein A
chromatography. These resins provide ligands to bind immunoglobulins, for
example Ig-
binding proteins like protein G or protein L, synthetic ligands, or protein A-
like porous
polymers.
[00102] Other chromatography processes can be employed during DSP. Ion
exchange
chromatography (IEC) can be used to remove impurities introduced during
previous
processes, for example, leached Protein A, endotoxins or viruses from the cell
line, remaining
host cell proteins or DNA, or media components. IEC, whether CEX or anion
exchange
chromatography, can be applied directly after Protein A chromatography.
Hydrophobic
interaction chromatography (HIC) can complement IEC, generally used as a
polishing step to
.. remove aggregates. In embodiments, the use of the above-identified
excipients can increase
the solubility of, and decrease the viscosity of host cell proteins during
chromatography
column loading steps. In embodiments, the use of the above-identified
excipients can increase
the solubility of, and decrease the viscosity of the therapeutic protein
during chromatography
column loading steps and elution steps.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
[00103] Chromatographic processes during protein purification impose harsh
conditions on
the protein formulation, such as (a) low pH conditions during elution from
Protein-A
chromatography columns, (b) elevated local protein concentration (often on the
order of 300-
400 mg/mL) within the pore-space of chromatographic resin, (c) elevated salt
concentrations
during ion exchange chromatography, and (d) elevated concentrations of salting-
out agents
during elution from HIC columns. Adding a viscosity-reducing excipient to the
protein-
containing solution, before and/or during chromatography, as described above,
can facilitate
the transit of the proteins through the chromatography column so that they are
less exposed to
the potentially damaging conditions imposed by chromatographic processing
steps. In
addition, the elevated local protein concentration within the column pore-
space can result in a
highly viscous material within this space, which places significant back
pressure on the
column. To alleviate this back pressure, media with relatively large pores are
typically used.
However, the resolving power of large-pore media is lower than small-pore
counterparts.
The incorporation of viscosity-modifying excipients as described above can
enable the use of
.. smaller pores in the chromatographic media. In embodiments, the elution
steps from Protein-
A chromatography expose the therapeutic protein to a low pH condition that can
reduce
solubility and increase aggregation of the target protein; addition of the
excipients can
increase the solubility of the target protein such that recovery yield from
the Protein-A
chromatography step is improved. In other embodiments, use of the excipient
can enable
elution of the target protein from Protein-A resin at a higher pH, and this
can reduce chemical
stresses on the target protein, resulting in improving a process parameter of
protein yield by
reducing the amount of protein degradation during processing.
[00104] (3) Viral inactivation: Viral inactivation processes
typically involve holding
the protein solution at a low pH, e.g., pH lower than 4, for an extended
period of time. This
environment, though, can destabilize therapeutic proteins. Formulating the
protein in the
presence of a viscosity-reducing excipient, for example, by adding a viscosity-
reducing
excipient before and/or during a viral inactivation process, can improve
process parameters
such as the stability or solubility of the protein, or its net yield. Also,
formulating the protein
in the presence of a stabilizing excipient, for example, by adding a
stabilizing excipient
before and/or during a viral inactivation process, can improve process
parameters such as the
stability or solubility of the protein, its structural integrity in the
monomeric form, its
resistance to aggregation, or its net yield.
[00105] (4) Filtration: Filtration processes include viral filtration
processes
(nanofiltration) to remove virus particles, microfiltration to remove micron-
scale impurities,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
and ultrafiltration/diafiltration processes to concentrate protein solutions
and to exchange
buffer systems.
[00106] (a) Viral filtration purifies the protein solution by
removing virus
particles, which can be on the order of twice the size of a recombinant human
monoclonal
antibodies. Thus, the filtration membrane for viral filtration can require
nano-sized pores. As
a result of the small pore size through which the proteins must pass, this
filtration step can
introduce stress to the protein, and is accompanied by significant levels of
membrane fouling
from protein aggregate particles. The addition of a viscosity-reducing
excipient, for example,
before and/or during filtration, as described above, can reduce a measurable
parameter such
as back pressure in the filtration system by increasing collective
diffusivity, and can decrease
the tendency for membrane fouling by mitigating the protein-protein
interactions that give
rise to it. The end result is improvement in those parameters indicting
improved performance
of the viral filtration unit during protein purification.
[00107] (b) Ultrafiltration and diafiltration (UF/DF) processes
concentrate
protein solutions and exchange buffer systems by passing the protein-
containing solution
through a filter membrane with a characteristic molecular weight cutoff that
is smaller than
the protein of interest. In this step, the protein solution faces high shear
stresses within the
filter units, elevated protein concentrations, and adsorption of the protein
to the hydrophobic
membranes typically used during UF/DF processes, all of which can increase
protein
aggregation. The addition of a viscosity-reducing excipient, for example,
before and/or
during a UF/DF process, as described above, can reduce back pressure in the
filtration system
by increasing collective diffusivity (measured, for example, by an increase in
kp). This not
only reduces shear stress across the membrane, but also promotes back-
diffusion away from
the filter membrane, thus lowering the effective protein concentration at the
membrane
interface and increasing the permeate flux. As a result, the use of viscosity-
reducing
excipients can improve parameters associated with higher throughput during
these filtration
processes, with reduced product losses and increased net yield. Additionally,
passing viscous
fluids through ultra- and diafilters can produce a large pressure drop across
the filter device,
making the separation inefficient. Formulating the protein solution in the
presence of
viscosity-reducing excipients as described above can substantially reduce the
pressure drop
across the filter device, thereby improving the process parameters of
operation, costs and
processing time by decreasing them both.
[00108] In more detail, UF/DF is an operation of DSP during which the biologic
molecule of
interest is retained while buffer and other analytes pass through the filter
membrane. In
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
UF/DF, the target protein can be retained by the filter membrane itself,
leading to the
formation of a gel layer at the filter membrane surface. This gel layer can
effectively limit
the efficiency of the processing step due to increased protein-protein
interactions (PPIs) in the
region of high local protein concentration at the filter membrane surface,
thereby reducing
filter throughput and/or resulting in aggregation of the protein of interest.
Incorporating a
viscosity-reducing excipient in the protein-containing solution can have the
effect of reducing
PPIs and/or increasing the solubility of the target protein during UF/DF.
Incorporating a
viscosity-reducing excipient can also improve filtration efficiency, decrease
operation time,
and/or increase yield of the target protein. The viscosity reducing excipients
may also
provide benefit during other filtration processes such as viral filtration and
sterile filtration.
[00109] A preferred structure for a viscosity-reducing excipient useful in
UF/DF is a small
molecule having a net charge of 0 at physiological pH, and comprising a
saturated or
unsaturated five-membered or six-membered carbocycle or heterocycle ring. In
embodiments, the ring structure is a heterocycle such as a lactam, a furan, a
tetrahydrofuran,
a pyrrole, a pyrrolidine, a pyran, a pyridine, a piperidine, an imidazole, a
dioxane, a
morpholine, a pyrimidine, a sulfimide, a sulfonamide, or combinations thereof.
In
embodiments, the ring structure is part of a polycyclic ring system in which
the component
rings can be saturated or unsaturated, with optional substitutions that
include short-chain (for
example, Ci-C6) aliphatic or cyclic saturated or unsaturated molecules
containing functional
groups such as hydroxyl, carbonyl, carboxylic acid, amide, and the like.
[00110] Another preferred structure for a viscosity-reducing excipient useful
in UF/DF is a
small molecule having a net positive charge at physiological pH and comprising
an aromatic
ring structure with or without a heteroatom. Other preferred structures
include short-chain
(for example, C3-C6) aliphatic or cyclic saturated or unsaturated molecules
optionally
substituted with functional groups such as hydroxyl, carbonyl, carboxylic
acid, amide, and
the like. Desirable excipients are soluble in the buffer solution used for
processing, and do
not contain a sugar molecule. Examples of viscosity-reducing excipients useful
in UF/DF
include: 1,3-dimethyluracil, 1-methyluracil, 3-methyluracil, 1,3-
diethyluracil, 6-methyluracil,
uracil, thymine, 1-methylthymine, 0-4-methylthymine, 1,3-dimethylthymine,
dimethylthymine dimer, cytosine, 5-methylcytosine, 3-methylcytosine, 2-
pyrrolidinone, N-
methylpyrrolidone, dimethylisosorbide, dimethylphenylalanine, nicotinamide,
isonicotinamide, diethylnicotinamide, 2-butanol, 2-butanone, imidazole,
aspartame,
saccharin, acesulfame potassium, caffeine, theacrine, cyclohexanone,
dimethylsulfone,
piracetam, 1,3-dimethy1-2-oxohexahydropyrimidine, trigonelline, sulfolane,
hordenine,
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
diphenhydramine, phenethylamine, N-methylphenethylamine, N,N-
dimethylphenethylamine,
f3,3-dihydroxyphenethylamine, f3,3-dihydroxy-N-methylphenethylamine, 3-
hydroxyphenethylamine, 4-hydroxyphenethylamine, tyrosinol, tyramine, N-
methyltyramine,
pyridoxine, dicyclomine, 2-pyridylethylamine. Advantageously, the viscosity-
reducing
.. excipients can be used alone or in combinations thereof.
[00111] In embodiments, the viscosity-reducing excipient can be dissolved in
the processing
buffer solution for UF/DF at an effective concentration from about 25 mM to
about 1000
mM, or at an effective concentration from about 50mM to about 500 mM, or at an
effective
concentration from about 75 mM to about 300 mM, or at an effective
concentration from
about 25 mM to about 500 mM, or at an effective concentration from about 50 mM
to about
300 mM. In an exemplary embodiment, the viscosity-reducing excipient is 1,3-
dimethyluracil, added to the processing buffer solution for UF/DF at a
concentration from
about 25 mM to about 1000 mM, or from about 50 mM to about 500 mM, or from
about 75
mM to about 300 mM. In another exemplary embodiment, the viscosity-reducing
excipient is
hordenine HC1, added to the processing buffer solution at a concentration from
about 25 mM
to about 500 mM, or from about 50 mM to about 300 mM, or from about 75 mM to
about
200 mM.
[00112] After the upstream protein processing or downstream purification have
been
completed with the added excipient, the excipient can remain as a part of the
drug substance
.. mixture or it can be separated from the protein active ingredient. Typical
small molecule
separation methods can be used to separate the excipient from the protein
active ingredient,
such as buffer exchange, ion exchange, ultrafiltration, and dialysis. In
addition to the
beneficial effects on the protein purification processes as outlined above,
the use of the
above-identified excipients can protect and preserve equipment used in protein
manufacture,
processing, and purification. For example, equipment-related processes such as
the cleanup,
sterilization, and maintenance of protein processing equipment can be
facilitated by the use of
the above-identified excipients due to decreased fouling, decreased
denaturing, lower
viscosity, and improved solubility of the protein, and parameters associated
with the
improvement of these processes are similarly improved.
[00113] The downstream process can be a chromatography process, and the
chromatography
process can be a Protein-A chromatography process. In embodiments, the
chromatography
process recovers the protein of interest, wherein the protein of interest is
characterized by an
improved protein-related parameter selected from the group consisting of
improved purity,
improved yield, fewer particles, less misfolding, improved biological
activity, or less
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
aggregation, as compared to a control solution. In embodiments, the improved
protein-
related parameter is improved yield of the protein of interest from the
chromatography
process. In embodiments, the chromatography process recovers the protein of
interest,
wherein the protein of interest is characterized by an improved percentage
recovered in the
monomeric form, i.e., with lower level of aggregation compared with the
recovery process in
the absence of the excipient.
[00114] While the use of an excipient compound to improve upstream and/or
downstream
processing has been described extensively herein, it is understood that a
combination of
excipients can be added together in order to achieve a desired effect, such as
an improvement
in a parameter of interest. The term "excipient additive" can refer to either
a single excipient
compound that leads to the desired effect or improved parameter, or to a
combination of
excipient compounds where the combination is responsible for the desired
effect or the
improved parameter.
EXAMPLES
Materials:
= Bovine gamma globulin (BGG), >99% purity, Catalog #G5009, Sigma Aldrich
= Human gamma globulin (HGG), Octagam 10%, Octapharma, Switzerland
= Histidine, Sigma Aldrich
= Other materials described in the examples below were from Sigma Aldrich
unless
otherwise specified.
Example 1: Preparation of formulations containing excipient compounds and test
protein
[00115] Formulations were prepared using an excipient compound and a test
protein, where
the test protein was intended to simulate either a therapeutic protein that
would be used in a
therapeutic formulation, or a non-therapeutic protein that would be used in a
non-therapeutic
formulation. Such formulations were prepared in 50 mM histidine hydrochloride
with
different excipient compounds for viscosity measurement in the following way.
Histidine
hydrochloride was first prepared by dissolving 1.94 g histidine in distilled
water and
adjusting the pH to about 6.0 with 1 M hydrochloric acid (Sigma-Aldrich, St.
Louis, MO) and
then diluting to a final volume of 250 mL with distilled water in a volumetric
flask.
Excipient compounds were then dissolved in 50 mM histidine HC1. Lists of
excipients are
provided below in Examples 4, 5, 6, and 7. In some cases excipient compounds
were adjusted
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
to pH 6 prior to dissolving in 50 mM histidine HC1. In this case the excipient
compounds
were first dissolved in deionized water at about 5 wt% and the pH was adjusted
to about 6.0
with either hydrochloric acid or sodium hydroxide. The prepared salt solution
was then
placed in a convection laboratory oven at about 65 C to evaporate the water
and isolate the
solid excipient. Once excipient solutions in 50 mM histidine HC1 had been
prepared, the test
protein bovine gamma globulin (BGG) was dissolved at a ratio of about 0.336 g
BGG per 1
mL excipient solution. This resulted in a final protein concentration of about
280 mg/mL.
Solutions of BGG in 50 mM histidine HC1 with excipient were formulated in 20
mL vials and
allowed to shake at 100 rpm on an orbital shaker table overnight. BGG
solutions were then
transferred to 2 mL microcentrifuge tubes and centrifuged for ten minutes at
2300 rpm in an
IEC MicroMax microcentrifuge to remove entrained air prior to viscosity
measurement.
Example 2: Viscosity measurement
[00116] Viscosity measurements of formulations prepared as described in
Example 1 were
made with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro,
MA). The viscometer was equipped with a CP-40 cone and was operated at 3 rpm
and 25 C.
The formulation was loaded into the viscometer at a volume of 0.5 mL and
allowed to
incubate at the given shear rate and temperature for 3 minutes, followed by a
measurement
collection period of twenty seconds. This was then followed by 2 additional
steps consisting
of 1 minute of shear incubation and subsequent twenty-second measurement
collection
period. The three data points collected were then averaged and recorded as the
viscosity for
the sample.
Example 3: Protein concentration measurement
[00117] The concentration of the protein in the experimental solutions was
determined by
measuring the optical absorbance of the protein solution at a wavelength of
280 nm in a
UV/VIS Spectrometer (Perkin Elmer Lambda 35). First the instrument was
calibrated to zero
absorbance with a 50 mM histidine buffer at pH 6. Next the protein solutions
were diluted by
a factor of 300 with the same histidine buffer and the absorbance at 280 nm
recorded. The
final concentration of the protein in the solution was calculated by using the
extinction
coefficient value of 1.264 mL/(mg x cm).
Example 4: Formulations with hindered amine excipient compounds
[00118] Formulations containing 280 mg/mL BGG were prepared as described in
Example
1, with some samples containing added excipient compounds. In these tests, the
hydrochloride salts of dimethylcyclohexylamine (DMCHA),
dicyclohexylmethylamine
(DCHMA), dimethylaminopropylamine (DMAPA), triethanolamine (TEA),
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
dimethylethanolamine (DMEA), and niacinamide were tested as examples of the
hindered
amine excipient compounds. Also, a hydroxybenzoic acid salt of DMCHA and a
taurine-
dicyandiamide adduct were tested as examples of the hindered amine excipient
compounds.
The viscosity of each protein solution was measured as described in Example 2,
and the
results are presented in Table 1 below, showing the benefit of the added
excipient compounds
in reducing viscosity.
TABLE 1
Excipient
Test Viscosity Viscosity
Excipient Added Concentration
Number
(cP) Reduction
(mg/mL)
4.1 None 0 79 0%
4.2 DMCHA-HC1 28 50 37%
4.3 DMCHA-HC1 41 43 46%
4.4 DMCHA-HC1 50 45 43%
4.5 DMCHA-HC1 82 36 54%
4.6 DMCHA-HC1 123 35 56%
4.7 DMCHA-HC1 164 40 49%
4.8 DMAPA-HC1 87 57 28%
4.9 DMAPA-HC1 40 54 32%
4.10 DCHMA-HC1 29 51 35%
4.11 DCHMA-HC1 50 51 35%
4.14 TEA-HC1 97 51 35%
4.15 TEA-HC1 38 57 28%
4.16 DMEA-HC1 51 51 35%
4.17 DMEA-HC1 98 47 41%
4.20 DMCHA-hydroxybenzoate 67 46 42%
4.21 DMCHA-hydroxybenzoate 92 42 47%
4.22 Product of Example 8 26 58 27%
4.23 Product of Example 8 58 50 37%
4.24 Product of Example 8 76 49 38%
4.25 Product of Example 8 103 46 42%
4.26 Product of Example 8 129 47 41%
4.27 Product of Example 8 159 42 47%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient
Test Viscosity Viscosity
Excipient Added Concentration
Number
(cP) Reduction
(mg/mL)
4.28 Product of Example 8 163 42
47%
4.29 Niacinamide 48 39 51%
4.30 N-Methyl-2-pyrrolidone 30 45
43%
4.31 N-Methyl-2-pyrrolidone 52 52
34%
Example 5: Formulations with anionic aromatic excipient compounds
[00119] Formulations of 280 mg/mL BGG were prepared as described in Example 1,
with
some samples containing added excipient compounds. The viscosity of each
solution was
measured as described in Example 2, and the results are presented in Table 2
below, showing
the benefit of the added excipient compounds in reducing viscosity.
TABLE 2
Excipient
Test Viscosity Viscosity
Excipient Added Concentration
Number (cP)
Reduction
(mg/mL)
5.1 None 0 79 0%
5.2 Sodium aminobenzoate 43 48
39%
5.3 Sodium hydroxybenzoate 26 50
37%
5.4 Sodium sulfanilate 44 49 38%
5.5 Sodium sulfanilate 96 42 47%
5.6 Sodium indole acetate 52 58
27%
5.7 Sodium indole acetate 27 78
1%
5.8 Vanillic acid, sodium salt 25
56 29%
5.9 Vanillic acid, sodium salt 50
50 37%
5.10 Sodium salicylate 25 57 28%
5.11 Sodium salicylate 50 52 34%
5.12 Adenosine monophosphate 26 47 41%
5.13 Adenosine monophosphate 50 66 16%
5.14 Sodium benzoate 31 61 23%
5.15 Sodium benzoate 56 62 22%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 6: Formulations with oligopeptide excipient compounds
[00120] Oligopeptides (n=5) were synthesized by NeoBioLab Inc. (Woburn, MA) in
>95%
purity with the N terminus as a free amine and the C terminus as a free acid.
Dipeptides
(n=2) were synthesized by LifeTein LLC (Somerset, NJ) in 95% purity.
Formulations of 280
mg/mL BGG were prepared as described in Example 1, with some samples
containing the
synthetic oligopeptides as added excipient compounds. The viscosity of each
solution was
measured as described in Example 2, and the results are presented in Table 3
below, showing
the benefit of the added excipient compounds in reducing viscosity.
TABLE 3
Excipient
Test
Viscosity Viscosity
Excipient Added Concentration
Number (cP)
Reduction
(mg/mL)
6.1 None 0 79 0%
6.2 ArgX5 100 55 30%
6.3 ArgX5 50 54 32%
6.4 HisX5 100 62 22%
6.5 HisX5 50 51 35%
6.6 HisX5 25 60 24%
6.7 Trp2Lys3 100 59 25%
6.8 Trp2Lys3 50 60 24%
6.9 AspX5 100 102 -29%
6.10 AspX5 50 82 -4%
6.11 Dipeptide LE (Leu-Glu) 50 72 9%
6.12 Dipeptide YE (Tyr-Glu) 50 55 30%
6.13 Dipeptide RP (Arg-Pro) 50 51 35%
6.14 Dipeptide RK (Arg-Lys) 50 53 33%
6.15 Dipeptide RH (Arg-His) 50 52 34%
6.16 Dipeptide RR (Arg-Arg) 50 57 28%
6.17 Dipeptide RE (Arg-Glu) 50 50 37%
6.18 Dipeptide LE (Leu-Glu) 100 87 -10%
6.19 Dipeptide YE (Tyr-Glu) 100 68 14%
6.20 Dipeptide RP (Arg-Pro) 100 53 33%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient
Test Viscosity Viscosity
Excipient Added Concentration
Number (cP)
Reduction
(mg/mL)
6.21 Dipeptide RK (Arg-Lys) 100 64 19%
6.22 Dipeptide RH (Arg-His) 100 72 9%
6.23 Dipeptide RR (Arg-Arg) 100 62 22%
6.24 Dipeptide RE (Arg-Glu) 100 66 16%
Example 8: Synthesis of guanyl taurine excipient
[00121] Guanyl taurine was prepared following method described in U.S. Pat.
No.
2,230,965. Taurine (Sigma-Aldrich, St. Louis, MO) 3.53 parts were mixed with
1.42 parts of
dicyandiamide (Sigma-Aldrich, St. Louis, MO) and grinded in a mortar and
pestle until a
homogeneous mixture was obtained. Next the mixture was placed in a flask and
heated at
200 C for 4 hours. The product was used without further purification.
Example 9: Protein formulations containing excipient compounds
[00122] Formulations were prepared using an excipient compound and a test
protein, where
the test protein was intended to simulate either a therapeutic protein that
would be used in a
therapeutic formulation, or a non-therapeutic protein that would be used in a
non-therapeutic
formulation. Such formulations were prepared in 50 mM aqueous histidine
hydrochloride
buffer solution with different excipient compounds for viscosity measurement
in the
following way. Histidine hydrochloride buffer solution was first prepared by
dissolving 1.94
g histidine in distilled water and adjusting the pH to about 6.0 with 1 M
hydrochloric acid
(Sigma-Aldrich, St. Louis, MO) and then diluting to a final volume of 250 mL
with distilled
water in a volumetric flask. Excipient compounds were then dissolved in the 50
mM histidine
HC1 buffer solution. A list of the excipient compounds is provided in Table 4.
In some cases,
excipient compounds were dissolved in 50 mM histidine HC1 buffer solution and
the
resulting solution pH was adjusted with small amounts of sodium hydroxide or
hydrochloric
acid to achieve pH 6 prior to dissolution of the model protein. In some cases,
excipient
compounds were adjusted to pH 6 prior to dissolving in 50 mM histidine HC1. In
this case the
excipient compounds were first dissolved in deionized water at about 5 wt% and
the pH was
adjusted to about 6.0 with either hydrochloric acid or sodium hydroxide. The
prepared salt
solution was then placed in a convection laboratory oven at about 65 C to
evaporate the
water and isolate the solid excipient. Once excipient solutions in 50 mM
histidine HC1 had
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
been prepared, the test protein, bovine gamma globulin (BGG) was dissolved at
a ratio to
achieve a final protein concentration of about 280 mg/mL. Solutions of BGG in
50 mM
histidine HC1 with excipient were formulated in 20 mL vials and allowed to
shake at 100 rpm
on an orbital shaker table overnight. BGG solutions were then transferred to 2
mL
microcentrifuge tubes and centrifuged for ten minutes at 2300 rpm in an IEC
MicroMax
microcentrifuge to remove entrained air prior to viscosity measurement.
[00123] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and subsequent twenty-second measurement collection
period. The three
data points collected were then averaged and recorded as the viscosity for the
sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient. The normalized viscosity is the ratio of the
viscosity of the model
protein solution with excipient to the viscosity of the model protein solution
with no
excipient.
TABLE 4
Test Excipient Added Excipient
Normalized Viscosity
Number
Concentration Viscosity Reduction
(mg/mL) (cP)
9.1 DMCHA-HC1 120 0.44 56%
9.2 Niacinamide 50 0.51 49%
9.3 Isonicotinamide 50 0.48 52%
9.4 Tyramine HC1 70 0.41 59%
9.5 Histamine HC1 50 0.41 59%
9.6 Imidazole HC1 100 0.43 57%
9.7 2-methyl-2-imidazoline HC1 60 0.43 57%
9.8 1-butyl-3-methylimidazolium 100 0.48 52%
chloride
9.9 Procaine HC1 50 0.53 47%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Test Excipient Added Excipient
Normalized Viscosity
Number
Concentration Viscosity Reduction
(mg/mL) (cP)
9.10 3-aminopyridine 50 0.51 49%
9.11 2,4,6-trimethylpyridine 50 0.49 51%
9.12 3-pyridine methanol 50 0.53 47%
Nicotinamide adenine
9.13 20 0.56 44%
dinucleotide
9.15 Sodium phenylpyruvate 55 0.57 43%
9.16 2-Pyrrolidinone 60 0.68 32%
9.17 Morpholine HC1 50 0.60 40%
9.18 Agmatine sulfate 55 0.77 23%
1-butyl-3-methylimidazolium
9.19 60 0.66 34%
iodide
9.21 L-Anserine nitrate 50 0.79 21%
1-hexy1-3-methylimidazolium
9.22 65 0.89 11%
chloride
9.23 N,N-diethyl nicotinamide 50 0.67 33%
9.24 Nicotinic acid, sodium salt 100 0.54 46%
9.25 Biotin 20 0.69 31%
Example 10: Preparation of formulations containing excipient combinations and
test protein
[00124] Formulations were prepared using a primary excipient compound, a
secondary
excipient compound and a test protein, where the test protein was intended to
simulate either
-- a therapeutic protein that would be used in a therapeutic formulation, or a
non-therapeutic
protein that would be used in a non-therapeutic formulation. The primary
excipient
compounds were selected from compounds having both anionic and aromatic
functionality,
as listed below in Table 5. The secondary excipient compounds were selected
from
compounds having either nonionic or cationic charge at pH 6 and either
imidazoline or
benzene rings, as listed below in Table 5. Formulations of these excipients
were prepared in
50 mM histidine hydrochloride buffer solution for viscosity measurement in the
following
way. Histidine hydrochloride was first prepared by dissolving 1.94 g histidine
in distilled
water and adjusting the pH to about 6.0 with 1 M hydrochloric acid (Sigma-
Aldrich, St.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Louis, MO) and then diluting to a final volume of 250 mL with distilled water
in a volumetric
flask. The individual primary or secondary excipient compounds were then
dissolved in 50
mM histidine HC1. Combinations of primary and secondary excipients were
dissolved in 50
mM histidine HC1 and the resulting solution pH adjusted with small amounts of
sodium
hydroxide or hydrochloric acid to achieve pH 6 prior to dissolution of the
model protein.
Once excipient solutions had been prepared as described above, the test
protein bovine
gamma globulin (BGG) was dissolved into each test solution at a ratio to
achieve a final
protein concentration of about 280 mg/mL. Solutions of BGG in 50 mM histidine
HC1 with
excipient were formulated in 20 mL vials and allowed to shake at 100 rpm on an
orbital
shaker table overnight. BGG solutions were then transferred to 2 mL
microcentrifuge tubes
and centrifuged for ten minutes at 2300 rpm in an IEC MicroMax microcentrifuge
to remove
entrained air prior to viscosity measurement.
[00125] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and a subsequent twenty-second measurement collection
period. The
three data points collected were then averaged and recorded as the viscosity
for the sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient, and summarized in Table 5 below. The normalized
viscosity is
the ratio of the viscosity of the model protein solution with excipient to the
viscosity of the
model protein solution with no excipient. The example shows that a combination
of primary
and secondary excipients can give a better result than a single excipient.
TABLE 5
Primary Excipient Secondary Excipient
Test Concentration Concentration Normalized
Name Name
Number (mg/mL) (mg/mL)
Viscosity
10.1 Salicylic Acid 30 None 0 0.79
10.2 Salicylic Acid 25 Imidazole 4 0.59
10.3 4-hydroxybenzoic 30 None 0 0.61
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
10.4 4-hydroxybenzoic 25 Imidazole 5
0.57
10.5 4-hydroxybenzene 31 None 0
0.59
10.6 4-hydroxybenzene 26 Imidazole 5
0.70
10.7 4-hydroxybenzene 25 Caffeine 5
0.69
10.8 None 0 Caffeine 10
0.73
10.9 None 0 Imidazole 5
0.75
Example 11: Preparation of formulations containing excipient combinations and
test protein
[00126] Formulations were prepared using a primary excipient compound, a
secondary
excipient compound and a test protein, where the test protein was intended to
simulate a
therapeutic protein that would be used in a therapeutic formulation, or a non-
therapeutic
protein that would be used in a non-therapeutic formulation. The primary
excipient
compounds were selected from compounds having both anionic and aromatic
functionality,
as listed below in Table 6. The secondary excipient compounds were selected
from
compounds having either nonionic or cationic charge at pH 6 and either
imidazoline or
benzene rings, as listed below in Table 6. Formulations of these excipients
were prepared in
distilled water for viscosity measurement in the following way. Combinations
of primary and
secondary excipients were dissolved in distilled water and the resulting
solution pH adjusted
with small amounts of sodium hydroxide or hydrochloric acid to achieve pH 6
prior to
dissolution of the model protein. Once excipient solutions in distilled water
had been
prepared, the test protein bovine gamma globulin (BGG) was dissolved at a
ratio to achieve a
final protein concentration of about 280 mg/mL. Solutions of BGG in distilled
water with
excipient were formulated in 20 mL vials and allowed to shake at 100 rpm on an
orbital
shaker table overnight. BGG solutions were then transferred to 2 mL
microcentrifuge tubes
and centrifuged for ten minutes at 2300 rpm in an IEC MicroMax microcentrifuge
to remove
entrained air prior to viscosity measurement.
[00127] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and a subsequent twenty-second measurement collection
period. The
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
three data points collected were then averaged and recorded as the viscosity
for the sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient, and summarized in Table 6 below. The normalized
viscosity is
the ratio of the viscosity of the model protein solution with excipient to the
viscosity of the
model protein solution with no excipient. The example shows that a combination
of primary
and secondary excipients can give a better result than a single excipient.
TABLE 6
Primary Excipient Secondary Excipient
Test Concentration
Concentration Normalized
Name Name
Number (mg/mL) (mg/mL)
Viscosity
11.1 Salicylic Acid 20 None 0 0.96
11.2 Salicylic Acid 20 Caffeine 5 0.71
11.3 Salicylic Acid 20 Niacinamide 5 0.76
11.4 Salicylic Acid 20 Imidazole 5 0.73
Example 12: Preparation of formulations containing excipient compounds and PEG
[00128] Materials: All materials were purchased from Sigma-Aldrich, St. Louis,
MO.
Formulations were prepared using an excipient compound and PEG, where the PEG
was
intended to simulate a therapeutic PEGylated protein that would be used in a
therapeutic
formulation. Such formulations were prepared by mixing equal volumes of a
solution of PEG
with a solution of the excipient. Both solutions were prepared in a Tris
buffer consisting of 10
mM Tris, 135 mM NaCl, 1 mM trans-cinnamic acid at pH of 7.3.
[00129] The PEG solution was prepared by mixing 3 g of poly(ethylene oxide)
average Mw
¨1,000,000 (Aldrich Catalog # 372781) with 97 g of the Tris buffer solution.
The mixture
was stirred overnight for complete dissolution.
[00130] An example of the excipient solution preparation is as follows: An
approximately
80 mg/mL solution of citric acid in the Tris buffer was prepared by dissolving
0.4 g of citric
acid (Aldrich cat. # 251275) in 5 mL of the Tris buffer solution and adjusted
the pH to 7.3
with minimum amount of 10 M NaOH solution.
[00131] The PEG excipient solution was prepared by mixing 0.5 mL of the PEG
solution
with 0.5 mL of the excipient solution and mixed by using a vortex for a few
seconds. A
control sample was prepared by mixing 0.5 mL of the PEG solution with 0.5 mL
of the Tris
buffer solution.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 13: Viscosity measurements of formulations containing excipient
compounds and
PEG
[00132] Viscosity measurements of the formulations prepared were made with a
DV-IIT LV
cone and plate viscometer (Brookfield Engineering, Middleboro, MA). The
viscometer was
equipped with a CP-40 cone and was operated at 3 rpm and 25 C. The formulation
was
loaded into the viscometer at a volume of 0.5 mL and allowed to incubate at
the given shear
rate and temperature for 3 minutes, followed by a measurement collection
period of twenty
seconds. This was then followed by 2 additional steps consisting of 1 minute
of shear
incubation and subsequent twenty second measurement collection period. The
three data
1() points collected were then averaged and recorded as the viscosity for
the sample.
[00133] The results presented in Table 7 show the effect of the added
excipient compounds
in reducing viscosity.
TABLE 7
Excipient
Test
Viscosity Viscosity
Excipient Concentration
Number (cP)
Reduction
(mg/mL)
13.1 None 0 104.8 0%
13.2 Citric acid Na salt 40 56.8 44%
13.3 Citric acid Na salt 20 73.3 28%
13.4 glycerol phosphate 40 71.7 30%
13.5 glycerol phosphate 20 83.9 18%
13.6 Ethylene diamine 40 84.7 17%
13.7 Ethylene diamine 20 83.9 15%
13.8 EDTA/K salt 40 67.1 36%
13.9 EDTA/K salt 20 76.9 27%
13.10 EDTA/Na salt 40 68.1 35%
13.11 EDTA/Na salt 20 77.4 26%
13.12 D-Gluconic acid/K salt 40 80.32 23%
13.13 D-Gluconic acid/K salt 20 88.4 16%
13.14 D-Gluconic acid/Na salt 40 81.24 23%
13.15 D-Gluconic acid/Na salt 20 86.6 17%
13.16 lactic acid/K salt 40 80.42 23%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient
Test
Viscosity Viscosity
Excipient Concentration
Number (cP)
Reduction
(mg/mL)
13.17 lactic acid/K salt 20 85.1 19%
13.18 lactic acid/Na salt 40 86.55 17%
13.19 lactic acid/Na salt 20 87.2 17%
13.20 etidronic acid/K salt 24 71.91 31%
13.21 etidronic acid/K salt 12 80.5 23%
13.22 etidronic acid/Na salt 24 71.6 32%
13.23 etidronic acid/Na salt 12 79.4 24%
Example 14: Preparation of PEGylated BSA with 1 PEG chain per BSA molecule
[00134] To a beaker was added 200 mL of a phosphate buffered saline (Aldrich
Cat. #
P4417) and 4 g of BSA (Aldrich Cat. # A7906) and mixed with a magnetic bar.
Next 400 mg
of methoxy polyethylene glycol maleimide, MW=5,000, (Aldrich Cat. #63187) was
added.
The reaction mixture was allowed to react overnight at room temperature. The
following day,
20 drops of HC1 0.1 M were added to stop the reaction. The reaction product
was
characterized by SDS-Page and SEC which clearly showed the PEGylated BSA. The
reaction
mixture was placed in an Amicon centrifuge tube with a molecular weight cutoff
(MWCO) of
30,000 and concentrated to a few milliliters. Next the sample was diluted 20
times with a
histidine buffer, 50 mM at a pH of approximately 6, followed by concentrating
until a high
viscosity fluid was obtained. The final concentration of the protein solution
was obtained by
measuring the absorbance at 280 nm and using a coefficient of extinction for
the BSA of
0.6678. The results indicated that the final concentration of BSA in the
solution was 342
mg/mL.
Example 15: Preparation of PEGylated BSA with multiple PEG chains per BSA
molecule
[00135] A 5 mg/mL solution of BSA (Aldrich A7906) in phosphate buffer, 25 mM
at pH of
7.2, was prepared by mixing 0.5 g of the BSA with 100 mL of the buffer. Next 1
g of a
methoxy PEG propionaldehyde Mw=20,000 (JenKem Technology, Plano, TX 75024) was
added followed by 0.12 g of sodium cyanoborohydride (Aldrich 156159). The
reaction was
allowed to proceed overnight at room temperature. The following day the
reaction mixture
was diluted 13 times with a Tris buffer (10 mM Tris, 135 mM NaCl at pH=7.3)
and
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
concentrated using Amicon centrifuge tubes MWCO of 30,000 until a
concentration of
approximately 150 mg/mL was reached.
Example 16: Preparation of PEGylated lysozyme with multiple PEG chains per
lysozyme
molecule
[00136] A 5 mg/mL solution of lysozyme (Aldrich L6876) in phosphate buffer, 25
mM at
pH of 7.2, was prepared by mixing 0.5 g of the lysozyme with 100 mL of the
buffer. Next 1 g
of a methoxy PEG propionaldehyde Mw=5,000 (JenKem Technology, Plano, TX 75024)
was
added followed by 0.12 g of sodium cyanoborohydride (Aldrich 156159). The
reaction was
allowed to proceed overnight at room temperature. The following day the
reaction mixture
was diluted 49 times with the phosphate buffer, 25 mM at pH of 7.2, and
concentrated using
Amicon centrifuge tubes MWCO of 30,000. The final concentration of the protein
solution
was obtained by measuring the absorbance at 280 nm and using a coefficient of
extinction for
the lysozyme of 2.63. The final concentration of lysozyme in the solution was
140 mg/mL.
Example 17: Effect of excipients on viscosity of PEGylated BSA with 1 PEG
chain per BSA
molecule
[00137] Formulations of PEGylated BSA (from Example 14 above) with excipients
were
prepared by adding 6 or 12 milligrams of the excipient salt to 0.3 mL of the
PEGylated BSA
solution. The solution was mixed by gently shaking and the viscosity was
measured by a
RheoSense microVisc equipped with an A10 channel (100-micron depth) at a shear
rate of
500 sec-1. The viscometer measurements were completed at ambient temperature.
[00138] The results presented in Table 8 shows the effect of the added
excipient compounds
in reducing viscosity.
TABLE 8
Excipient
Test Viscosity
Excipient Concentration Viscosity (cP)
Number
Reduction
(mg/mL)
17.1 None 0 228.6 0%
Alpha-Cyclodextrin
17.2 20 151.5 34%
sulfated Na salt
17.3 K acetate 40 89.5 60%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 18: Effect of excipients on viscosity of PEGylated BSA with multiple
PEG chains
per BSA molecule
[00139] A formulation of PEGylated BSA (from Example 15 above) with citric
acid Na salt
as excipient was prepared by adding 8 milligrams of the excipient salt to 0.2
mL of the
PEGylated BSA solution. The solution was mixed by gently shaking and the
viscosity was
measured by a RheoSense microVisc equipped with an A10 channel (100 micron
depth) at a
shear rate of 500 sec-1. The viscometer measurements were completed at ambient
temperature. The results presented in Table 9 shows the effect of the added
excipient
compounds in reducing viscosity.
TABLE 9
Excipient
Test Viscosity Viscosity
Excipient Added Concentration
Number (cP) Reduction
(mg/mL)
18.1 None 0 56.8 0%
18.2 Citric acid Na salt 40 43.5 23%
Example 19: Effect of excipients on viscosity of PEGylated lysozyme with
multiple PEG
chains per lysozyme molecule
[00140] A formulation of PEGylated lysozyme (from Example 16 above) with
potassium
acetate as excipient was prepared by adding 6 milligrams of the excipient salt
to 0.3 mL of
the PEGylated lysozyme solution. The solution was mixed by gently shaking and
the
viscosity was measured by a RheoSense microVisc equipped with an A10 channel
(100
micron depth) at a shear rate of 500 sec-1. The viscometer measurements were
completed at
ambient temperature. The results presented in the next table shows the benefit
of the added
excipient compounds in reducing viscosity.
TABLE 10
Excipient
Viscosity Viscosity
Test Number Excipient Concentration
(cP) Reduction
(mg/mL)
19.1 None 0 24.6 0%
19.2 K acetate 20 22.6 8%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 20: Protein formulations containing excipient combinations
[00141] Formulations were prepared using an excipient compound or a
combination of two
excipient compounds and a test protein, where the test protein was intended to
simulate a
therapeutic protein that would be used in a therapeutic formulation. These
formulations were
prepared in 20 mM histidine buffer with different excipient compounds for
viscosity
measurement in the following way. Excipient combinations were dissolved in 20
mM
histidine and the resulting solution pH adjusted with small amounts of sodium
hydroxide or
hydrochloric acid to achieve pH 6 prior to dissolution of the model protein.
Excipient
compounds for this Example are listed below in Table 11. Once excipient
solutions had been
1() prepared, the test protein bovine gamma globulin (BGG) was dissolved at
a ratio to achieve a
final protein concentration of about 280 mg/mL. Solutions of BGG in the
excipient solutions
were formulated in 5 mL sterile polypropylene tubes and allowed to shake at 80-
100 rpm on
an orbital shaker table overnight. BGG solutions were then transferred to 2 mL
microcentrifuge tubes and centrifuged for about ten minutes at 2300 rpm in an
IEC
MicroMax microcentrifuge to remove entrained air prior to viscosity
measurement.
[00142] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
.. the given shear rate and temperature for 3 minutes, followed by a
measurement collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and subsequent twenty second measurement collection
period. The three
data points collected were then averaged and recorded as the viscosity for the
sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient, and the results are shown in Table 11 below. The
normalized
viscosity is the ratio of the viscosity of the model protein solution with
excipient to the
viscosity of the model protein solution with no excipient.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 11
Excipient A Excipient B
Normalized
Test #
Conc. Conc. Viscosity
Name Name
(mg/mL) (mg/mL)
20.1 None 0 None 0 1.00
20.2 Aspartame 10 None 0 0.83
20.3 Saccharin 60 None 0 0.51
20.4 Acesulfame K 80 None 0 0.44
20.5 Theophylline 10 None 0 0.84
20.6 Saccharin 30 None 0 0.58
20.7 Acesulfame K 40 None 0 0.61
20.8 Caffeine 15 Taurine 15 0.82
20.9 Caffeine 15 Tyramine 15 0.67
Example 21: Protein formulations containing excipients to reduce viscosity and
injection
pain
[00143] Formulations were prepared using an excipient compound, a second
excipient
compound, and a test protein, where the test protein was intended to simulate
a therapeutic
protein that would be used in a therapeutic formulation. The first excipient
compound,
Excipient A, was selected from a group of compounds having local anesthetic
properties.
The first excipient, Excipient A and the second excipient, Excipient B are
listed in Table 12.
1() These formulations were prepared in 20 mM histidine buffer using
Excipient A and Excipient
B in the following way, so that their viscosities could be measured.
Excipients in the amounts
disclosed in Table 12 were dissolved in 20 mM histidine and the resulting
solutions were pH
adjusted with small amounts of sodium hydroxide or hydrochloric acid to
achieve pH 6 prior
to dissolution of the model protein. Once excipient solutions had been
prepared, the test
protein bovine gamma globulin (BGG) was dissolved in the excipient solution at
a ratio to
achieve a final protein concentration of about 280 mg/mL. Solutions of BGG in
the excipient
solutions were formulated in 5 mL sterile polypropylene tubes and allowed to
shake at 80-
100 rpm on an orbital shaker table overnight. BGG-excipient solutions were
then transferred
to 2 mL microcentrifuge tubes and centrifuged for about ten minutes at 2300
rpm in an IEC
MicroMax microcentrifuge to remove entrained air prior to viscosity
measurement.
[00144] Viscosity measurements of the formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and subsequent twenty second measurement collection
period. The three
data points collected were then averaged and recorded as the viscosity for the
sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient, and the results are shown in Table 12 below. The
normalized
viscosity is the ratio of the viscosity of the model protein solution with
excipient to the
viscosity of the model protein solution with no excipient.
TABLE 12
Excipient A Excipient B
Normalized
Test # Conc. Conc.
Name Name Viscosity
(mg/mL) (mg/mL)
21.1 None 0 None 0 1.00
21.2 Lidocaine 45 None 0 0.73
21.3 Lidocaine 23 None 0 0.74
21.4 Lidocaine 10 Caffeine 15 0.71
21.5 Procaine HC1 40 None 0 0.64
21.6 Procaine HC1 20 Caffeine 15 0.69
Example 22: Formulations containing excipient compounds and PEG
[00145] Formulations were prepared using an excipient compound and PEG, where
the PEG
was intended to simulate a therapeutic PEGylated protein that would be used in
a therapeutic
formulation, and where the excipient compounds were provided in the amounts as
listed in
Table 13. These formulations were prepared by mixing equal volumes of a
solution of PEG
with a solution of the excipient. Both solutions were prepared in deionized
(DI) Water.
[00146] The PEG solution was prepared by mixing 16.5 g of poly(ethylene oxide)
average
Mw ¨100,000 (Aldrich Catalog # 181986) with 83.5 g of DI water. The mixture
was stirred
overnight for complete dissolution.
[00147] The excipient solutions were prepared by this general method and as
detailed in
Table 13 below: An approximately 20 mg/mL solution of potassium phosphate
tribasic
(Aldrich Catalog # P5629) in DI water was prepared by dissolving 0.05 g of
potassium
phosphate in 5 mL of DI water. The PEG excipient solution was prepared by
mixing 0.5 mL
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
of the PEG solution with 0.5 mL of the excipient solution and mixed by using a
vortex for a
few seconds. A control sample was prepared by mixing 0.5 mL of the PEG
solution with 0.5
mL of DI water. Viscosity was measured and results are recorded in Table 13
below.
TABLE 13
Excipient
Test Viscosity
Excipient Concentration Viscosity (cP)
Number
Reduction (%)
(mg/mL)
22.1 None 0 79.7 0
22.2 Citric acid Na salt 10 74.9 6.0
Potassium
22.3 10 72.3 9.3
phosphate
Citric acid Na
22.4 salt/Potassium 10/10 69.1 13.3
phosphate
22.5 Sodium sulfate 10 75.1 5.8
Citric acid Na
22.6 10/10 70.4 11.7
salt/Sodium sulfate
Example 23: Improved processing of protein solutions with excipients
[00148] Two BGG solutions were prepared by mixing 0.25 g of solid BGG with 4
ml of a
buffer solution. For Sample A: Buffer solution was 20 mM histidine buffer
(pH=6.0). For
sample B: Buffer solution was 20 mM histidine buffer containing 15 mg/ml of
caffeine
(pH=6). The dissolution of the solid BGG was carried out by placing the
samples in an orbital
shaker set at 100 rpm. The buffer sample containing caffeine excipient was
observed to
dissolve the protein faster. For the sample with the caffeine excipient
(Sample B) complete
dissolution of the BGG was achieved in 15 minutes. For the sample without the
caffeine
(Sample A) the dissolution needed 35 minutes.
[00149] Next the samples were placed in 2 separate Amicon Ultra 4 Centrifugal
Filter Units
with a 30,000 molecular weight cut off and the samples were centrifuged at
2,500 rpm at 10
minutes intervals. The filtrate volume recovered after each 10 minute
centrifuge run was
recorded. The results in Table 14 show the faster recovery of the filtrate for
Sample B. In
addition, Sample B kept concentrating with every additional run but Sample A
reached a
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
maximum concentration point and further centrifugation did not result in
further sample
concentration.
TABLE 14
Sample A filtrate Sample B filtrate
Centrifuge time (min)
collected (mL) collected (mL)
10 0.28 0.28
20 0.56 0.61
30 0.78 0.88
40 0.99 1.09
50 1.27 1.42
60 1.51 1.71
70 1.64 1.99
80 1.79 2.29
90 1.79 2.39
100 1.79 2.49
Example 24: Protein formulations containing multiple excipients
[00150] This example shows how the combination of caffeine and arginine as
excipients has
a beneficial effect on decreasing viscosity of a BGG solution. Four BGG
solutions were
prepared by mixing 0.18 g of solid BGG with 0.5 mL of a 20 mM Histidine buffer
at pH 6.
Each buffer solution contained different excipient or combination of
excipients as described
in the table below. The viscosity of the solutions was measured as
described in previous
examples. The results show that the hindered amine excipient, caffeine, can be
combined
with known excipients such as arginine, and the combination has better
viscosity reduction
properties than the individual excipients by themselves.
TABLE 15
Viscosity
Sample Excipient added
Viscosity Reduction (%)
(cP)
A None 130.6 0
Caffeine (10mg/m1) 87.9 33
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Viscosity
Sample Excipient added
Viscosity Reduction (%)
(cP)
Caffeine (10mg/m1) / Arginine
66.1 49
(25 mg/ml)
Arginine (25 mg/ml) 76.7 41
[00151] Arginine was added to 280 mg/mL solutions of BGG in histidine buffer
at pH 6. At
levels above 50 mg/mL, adding more arginine did not decrease viscosity
further, as shown in
Table 16.
TABLE 16
Arginine added (mg/mL) Viscosity (cP) Viscosity reduction (%)
0 79.0 0%
53 40.9 48%
79 46.1 42%
105 47.8 40%
132 49.0 38%
158 48.0 39%
174 50.3 36%
211 51.4 35%
[00152] Caffeine was added to 280 mg/mL solutions of BGG in histidine buffer
at pH 6. At
levels above 10 mg/ml, adding more caffeine did not decrease viscosity
further, as shown in
lo Table 17.
TABLE 17
Caffeine added (mg/mL) Viscosity (cP) Viscosity reduction (%)
0 79 0%
60 31%
62 23%
22 50 45%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 25: Caffeine effect during TFF concentration process
[00153] In this Example, bovine gamma globulin (BGG) solutions were
concentrated in the
presence and absence of caffeine using tangential flow filtration (TFF). The
Labscale TFF
System, produced by EMD Millipore (Billerica, MA) was used to perform the
experiments.
The system was fitted with a Pellicon XL TFF cassette that contained an
Ultracel membrane
with 30 kDa molecular weight cutoff (EMD Millipore, Billerica, MA). The
nominal
membrane surface area was 50 cm2. The feed pressure to the cassette was
maintained at 30
psi while the retentate pressure was maintained at 10 psi. The filtrate flux
was monitored
over the course of the experiment by measuring its mass as a function of time.
Approximately 12 grams of BGG were dissolved into 500 mL of buffer containing
15 mg/mL
caffeine, 150 mM NaCl, and 20 mM histidine, adjusted to pH 6. A control sample
was
prepared by dissolving 12 grams of BGG into 500 mL of buffer containing 150 mM
NaCl,
and 20 mM histidine, adjusted to pH 6. The buffer components were purchased
from Sigma-
Aldrich. Both solutions were filtered through a 0.2 [tm PES filter (VWR,
Radnor, PA) prior
to TFF processing. The performance of the test sample and control sample
during TFF were
measured by the mass transfer coefficient. The mass transfer coefficient was
determined for
each sample using the following equation (as described in J. Hung, A. U.
Borwankar, B. J.
Dear, T. M. Truskett, K. P. Johnston, High concentration tangential flow
ultrafiltration of
stable monoclonal antibody solutions with low viscosities. J. Memb. Sci. 508,
113-126
(2016)):
J = Lln(Cw/Cb)
(Eq. 3)
[00154] Eq. 3 describes the filtrate flux J, where L is the mass transfer
coefficient, Cw is the
protein concentration in the vicinity of the membrane, and Cb is the
concentration in the
liquid bulk, and Eq. 3 thereby permits calculation of the mass transfer
coefficient L. A
graph of the calculated flux J against the ln(Cb) yields a linear plot with
slope of -kc. Here the
flux J is calculated by taking the derivative of the filtrate mass with
respect to time and Cb is
calculated using a mass-balance. The best-fit mass transfer coefficients are
listed in Table 18.
The introduction of 15 mg/mL caffeine increased the value of the mass transfer
coefficient by
¨ 13%, from 22.5 to 25.4 Lm-2hr-1 (LMH).
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 18
Sample Mass Transfer Coefficient k (LMH)
Control 22.5 0.1
15 mg/mL caffeine 25.4 0.1
Example 26: Caffeine effect during TFF concentration process
[00155] In this Example, bovine gamma globulin (BGG) solutions were
concentrated in the
presence and absence of caffeine using tangential flow filtration (TFF). The
Labscale TFF
System, produced by EMD Millipore (Billerica, MA) was used to perform the
experiments.
The system was fitted with a Pellicon XL TFF cassette that contained an
Ultracel membrane
with 30 kDa molecular weight cutoff (EMD Millipore, Billerica, MA). The
nominal
membrane surface area was 50 cm2. A control sample was prepared by dissolving
14.6
grams of BGG into 582 mL of buffer containing 150 mM NaCl, and 20 mM
histidine,
adjusted to pH 6, such that the initial BGG concentration was nominally 25.1
mg/mL. The
material was filtered through a 0.2 [tm PES filter (VWR, Radnor, PA) and then
processed in
the TFF device. The pump speed was adjusted such that the feed pressure was
initially 30 psi
and the retentate valve was adjusted such that the retentate pressure was
initially 10 psi. The
material was concentrated without adjusting either the pump speed or retentate
valve for 4.1
hours. The initial and final concentrations were determined to be 25.4 0.6
and 159 6
mg/mL, respectively, by a Bradford assay, as shown in Table 19 below. A
caffeine-
containing sample was prepared by dissolving 14.2 g of BGG into 566 mL of
buffer
containing 15 mg/mL caffeine, 150 mM NaCl, and 20 mM histidine, adjusted to pH
6, such
.. that the initial BGG concentration was nominally 25.1 mg/mL. The material
was filtered
through a 0.2 [tm PES filter (VWR, Radnor, PA) and then processed in the TFF
device. The
pump speed and retentate valve were set to identical levels to those
previously. The feed and
retentate pressures were confirmed to be 30 psi and 10 psi, respectively, as
previously. The
material was concentrated without adjusting either the pump speed or retentate
valve for 4.1
hours. The initial and final concentrations were determined to be 24.4 0.5
and 225 10
mg/mL, respectively, by a Bradford assay, as shown in Table 19 below. The use
of caffeine
during TFF processing increased the final protein concentration by
approximately 42% when
compared to the control, from 159 to 225 mg/mL.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 19
Sample Initial concentration (mg/mL) Final concentration
(mg/mL)
Control 25.4 0.6 159 6
15 mg/mL caffeine 24.4 0.5 225 10
Example 27: Caffeine effect during sterile filtration of BGG solutions
[00156] Bovine gamma globulin (BGG), L-histidine, and caffeine were purchased
from
Sigma-Aldrich (St. Louis, MO, product numbers G5009, H6034, and C7731,
respectively).
Deionized (DI) water was generated from tap water with a Direct-Q 3 UV
purification system
from EMD Millipore (Billerica, MA). 25-mm polyethersulfone (PES) filters with
0.2-[tm
pores were purchased from GE Healthcare (Chicago, IL, catalog number 6780-
2502). 1-mL
Luer-Lok syringes were purchased from Becton, Dickinson and Company (Franklin
Lakes,
NJ, reference number 309628). A 20-mM histidine buffer, pH 6.0 was prepared
using L-
histidine, DI water, and titrated to pH 6.0 with 1 M HC1. A 15 mg/mL solution
of caffeine
was prepared using the histidine buffer. The caffeine-free and caffeine-
containing buffers
were used to reconstitute BGG to a final concentration of about 280 mg/mL. The
protein
concentration, c, was calculated using:
C ______________________________________
b + vm
(Eq. 4)
where mp is the protein mass, b is the volume of buffer added, and v is the
partial specific
volume of BGG, here taken to be 0.74 mL/g. The viscosity of each sample was
measured
using microVisc rheometer (RheoSense, San Ramon, CA) at a temperature of 23 C
and shear
rate of 250 s1. The energies required to pass the BGG solutions through the
sterile filters
were measured using a Tensile Compression Tester (TCT, Instron, Needham, MA,
part
number 3343) fitted with a 100 N load cell (Instron, Needham, MA, part number
2519-103).
The syringe plungers were depressed at a rate of 159 mm/min for a distance of
50 mm. The
energy requirements were calculated by integrating the load-versus-extension
curves
measured by the TCT, and results are summarized in Table 20 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 20
Protein Caffeine
Energy
Sample concentration concentration Viscosity (cP)
requirement (mJ)
(mg/mL) (mg/mL)
1 280 0 106 198
2 280 15.1 68.9 181
Example 28: Excipients to improve Protein-A chromatography elution
[00157] Four purified, research-grade biosimilar antibodies, ipilimumab,
ustekinumab,
omalizumab, and tocilizumab were purchased from Bioceros (Utrecht, The
Netherlands).
They were provided as frozen aliquots at protein concentrations of 20, 26, 15
and 23 mg/mL,
respectively, in an aqueous 40 mM sodium acetate, 50 mM tris-HC1 buffer at pH
5.5. The
protein solutions were thawed at room temperature prior to measurement and
afterwards,
were filtered through a 0.2 [Em polyethersulfone filters. The filtered protein
stock solutions
were mixed in 1:1 ratio of protein stock solution to a binding buffer. The
binding buffer,
used to promote the binding of the antibodies to the Protein-A resin, was
composed of 0.1 M
sodium phosphate and 0.15 sodium chloride at pH 7.2 in deionized (DI) water.
The DI water
was produced by purifying tap water with a Direct-Q 3 UV purification system
from EMD
Millipore (Billerica, MA). These solutions were employed to perform Protein-A
binding and
elution studies using a Pierce Protein-A Spin Plate for IgG Screening
(ThermoFisher
Scientific catalog # 45202). The plate had 96 wells, each containing 50 [EL of
Protein-A resin.
The resin was washed with binding buffer by adding 200 [EL of binding buffer
to each well
and centrifuging the plate at 1000 x g for 1 minute and discarding the flow-
through. All
subsequent centrifugation steps were performed at 1000 x g for 1 minute. This
wash
procedure was repeated once. Following these initial washing steps, the
diluted protein
samples, i.e., samples containing ipilimumab, ustekinumab, omalizumab, and
tocilizumab,
were added to the wells in the plate (200 [EL per well). The plate was then
placed on a
Daigger Scientific (Vernon Hills, IL) Labgenius orbital shaker and agitated at
260 rpm for 30
minutes, following which the plate was centrifuged and the flow-through was
discarded. The
wells were then washed by adding 500 [EL of binding buffer to each well,
centrifuging the
plate and discarding the flow-through. This wash step was repeated twice.
After these
washing steps, the proteins were eluted from the plate using elution buffers
to which different
excipients had been added. For each elution, 50 [EL of a neutralization buffer
consisting of 1
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
M sodium phosphate at pH 7 was added to each well of the collection plate, and
then two
hundred pL of elution buffer was added to each well of the plate. The plate
was agitated at
260 rpm for 1 minute and then centrifuged. The flow-through was recovered for
analysis.
This elution step was repeated once. The control buffer, with no excipients,
contained 20
mM citrate and had a pH of 2.6. Because Protein-A elution buffers often
contain some
amount of salt, an elution buffer of 100 mM NaCl in the citrate buffer was
prepared as a
secondary control.
[00158] Table 21 lists the excipient solutions used in this example, their
concentrations, and
final pH of the elution buffers. All excipients were purchased from Sigma
Aldrich (St. Louis,
MO), with the exception of aspartame, which was purchased from Herb Store USA
(Los
Angeles, CA), trehalose, which was purchased from Cascade Analytical Reagents
and
Biochemicals (Corvallis, OR), and sucrose which was purchased from Research
Products
International (Mt. Prospect, IL, product number S24060). All excipient-
containing elution
buffers were prepared by mixing the appropriate quantity of the excipient with
approximately
10 mL of the salt-free citrate buffer control. The elution buffers were
prepared at
approximately 100 mM excipient. However, not all of the excipients are soluble
at this level;
Table 21 therefore lists all of the excipient concentrations that were used.
The pH of each
elution buffer was adjusted to about 2.6 0.1 using either hydrochloride or
sodium hydroxide
as needed.
[00159] For each protein sample, ASD High performance size-exclusion
chromatography
(SEC) analysis was performed using a TSKgel SuperSW3000 column (30 cm x 4.6 mm
ID,
Tosoh Bioscience, King of Prussia, PA) connected to an HPLC workstation
(Agilent HP
1100 system). The separation was carried out at a flow of 0.35 mL/min at room
temperature.
The mobile phase was an aqueous buffer of 100 mM sodium phosphate, 300 mM
sodium
chloride, pH 7. The protein concentration was monitored by absorbance at 280
nm using an
Agilent 1100 Series G1315B diode array detector. The total amount of protein
eluted from
the Protein-A resin for each protein, i.e., ipilimumab, ustekinumab,
omalizumab, and
tocilizumab, was estimated by integrating the chromatograms. The integrated
peak areas for
each protein, i.e., ipilimumab, ustekinumab, omalizumab, and tocilizumab, are
listed in
Tables 22-25. Tables 22-25 also compare the experimental peak areas to those
of the salt-
free and salt-containing controls. Values greater than 100% indicate that the
elution buffer
recovered more protein from the Protein-A resin than the control whereas
values less than
100% indicate that the elution buffer recovered less protein from the Protein-
A resin than the
control.
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Table 21. Excipients used in Example 28
Sigma-Aldrich product Excipient
Excipient pH
number for excipient concentration (mM)
caffeine C7731 79 2.6
acesulfame potassium 04054 110 2.5
1-methyl-2-pyrrolidone M6762 117 2.6
aspartame N/A 20 2.6
taurine T8691 114 2.5
trehalose N/A 100 2.7
sucrose N/A 101 2.7
niacinamide N5535 99 2.7
sodium chloride control S7653 117 2.6
control N/A N/A 2.5
Table 22. Ipilimumab recovery from Protein-A resin
Peak area Peak area normalized to Peak
area normalized
Excipient
(mAU*min) salt-free control (%) to salt control (%)
citrate 3409 77.9 83.6
Acesulfame
1567 35.8 38.4
potassium
1-methyl-2-
386 8.8 9.5
pyrrolidone
aspartame 4012 91.7 98.3
taurine 3958 90.4 97.0
trehalose 3667 83.8 89.9
sucrose 4585 104.8 112.4
niacinamide 4295 98.2 105.3
sodium chloride
4080 93.2 100.0
control
control 4376 100.0 107.2
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Table 23. Ustekinumab recovery from Protein-A resin
Excipient Integrated peak Peak area normalized Peak area normalized
to
area (mAU*min) to salt-free control (%) salt control (%)
caffeine 2301 86.6 75.2
acesulfame
307 16.2 14.0
potassium
aspartame 417 17.4 15.1
1-methyl-2-
2952 108.8 94.4
pyrrolidone
taurine 3257 118.6 103.0
trehalose 1549 56.6 49.1
sucrose 1274 51.2 44.4
niacinamide 3204 116.1 100.8
sodium chloride
3176 115.2 100.0
control
Table 24. Omalizumab recovery from Protein-A resin
Integrated peak Peak area normalized to Peak area normalized to
Excipient
area (mAU*min) salt-free control (%) salt control (%)
caffeine 4040 105.5 117.5
acesulfame
3620 94.5 105.3
potassium
1-methyl-2-
3334 87.0 97.0
pyrrolidone
aspartame 3605 94.1 104.8
taurine 4337 113.2 126.1
trehalose 3571 93.2 103.8
sucrose 3639 95.0 105.8
niacinamide 4812 125.6 139.9
sodium chloride
3439 89.8 100.0
control
control 3831 100.0 111.4
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Table 25. Tocilizumab recovery from Protein-A resin
Integrated peak Peak area normalized to Peak area normalized to
Excipient
area (mAU*min) salt-free control (%) salt control (%)
caffeine 3120 111.2 100.3
acesulfame
3083 109.9 99.1
potassium
1-methyl -2-
261 9.3 8.4
pyrrolidone
aspartame 556 19.8 17.9
taurine 3054 108.8 98.2
trehalose 2781 99.1 89.4
sucrose 1037 37.0 33.3
niacinamide 2550 90.9 82.0
sodium chloride
3111 110.9 100.0
control
control 2806 100.0 90.2
Example 29: Excipients to improve Protein-A chromatography elution
[00160] The test proteins used in this Example are identical to those in
Example 28, i.e.,
ipilimumab, ustekinumab, omalizumab, and tocilizumab. Protein-A binding and
elution
studies were performed using an identical plate to that in Example 28. The
methods for
loading and eluting the antibodies from the Protein-A plate were identical to
those in
Example 28 with the exception of the elution step. In Example 28, two elution
washes were
performed. However, in this Example, only one wash is performed. As in Example
28,
elution buffers were prepared from a 20 mM citrate, pH 2.6 control buffer. The
elution
buffers are listed in Table 26 below. All of the excipients were purchased
from Sigma-
Aldrich (St. Louis, MO). The recovered protein was analyzed by HPLC in an
identical
fashion to that in Example 28, and results of protein recovery for each
protein, i.e.,
ipilimumab, ustekinumab, omalizumab, and tocilizumab, are documented in Tables
27-30
below.
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Table 26. Excipients used in Example 29
Excipient Sigma-Aldrich Excipient pH
product number concentration (mM)
control N/A N/A 2.5
sodium chloride control S7653 117 2.6
niacinamide N5535 99 2.7
taurine T8691 114 2.5
imidazole 15513 100 2.6
4-hydroxybenzesulfonic acid 171506 107 2.6
Caffeine C7731 79 2.6
Table 27. Ipilimumab recovery from Protein-A resin
Excipient Peak area Peak area normalized to Peak area
normalized
(mAU*min) salt-free control (%) to salt control
(%)
control 4841 100.0 88.3
sodium chloride control 5485 113.3 100.0
niacinamide 6300 130.1 114.8
taurine 7557 156.1 137.8
imidazole 6071 125.4 110.7
4-hydroxybenzesulfonic 5836 120.6 106.4
acid
caffeine 6051 125.0 110.3
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Table 28. Ustekinumab recovery from Protein-A resin
Excipient Integrated
peak Peak area normalized Peak area normalized
area (mAU*min) to salt-free control (%) to salt control
(%)
control 4572 100.0 107.9
sodium chloride control 4238 92.7 100.0
niacinamide 5848 127.9 138.0
taurine 4744 103.8 112.0
imidazole 4617 101.0 108.9
4-hydroxybenzesulfonic 4132 90.4 97.5
acid
caffeine 5084 111.2 120.0
Table 29. Omalizumab recovery from Protein-A resin
Excipient Integrated peak Peak area
normalized Peak area normalized
area (mAU*min) to salt-free control (%) to salt control
(%)
control 4194 100.0 91.7
sodium chloride control 4574 109.1 100.0
niacinamide 5748 137.0 125.7
taurine 4676 111.5 102.2
imidazole 2589 61.7 56.6
4-hydroxybenzesulfonic 3190 76.1 69.7
acid
caffeine 5807 138.5 127.0
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Table 30. Tocilizumab recovery from Protein-A resin
Excipient Integrated peak Peak area normalized Peak area
normalized to
area mAU*min) to salt-free control (%)
salt control (%)
control 4667 100.0 97.5
sodium chloride control 4786 102.6 100.0
niacinamide 5225 111.9 109.2
taurine 5396 115.6 112.7
imidazole 4754 101.9 99.3
4-hydroxybenzesulfonic 4539 97.3 94.8
acid
caffeine 5656 121.2 118.2
Example 30: Excipients that improve omalizumab elution from Protein-A
chromatography
column
[00161] Research-grade omalizumab was purchased from Bioceros (Utrecht, The
Netherlands) and provided frozen at 15 mg/mL in an aqueous 40 mM sodium
acetate, 50 mM
tris-HC1 buffer, pH 5.5. The protein was thawed at room temperature prior to
experiments
and filtered through a 0.2 pm polyethersulfone filter. The filtered material
was mixed in a
1:1 ratio with a binding buffer that consisted of 20 mM sodium phosphate, pH 7
in DI water.
.. Tap water was purified with a Direct-Q 3 UV purification system from EMD
Millipore
(Billerica, MA) to produce the DI water. Protein-A purification was performed
using a
HiTrap Protein-A HP 1 mL column from GE Healthcare (Chicago, IL, product
number
29048576). For each experiment, the column was first equilibrated with 10 mL
of binding
buffer. Following equilibration, 30 mg of protein were loaded onto the Protein-
A column.
The column was then washed with 5 mL of binding buffer. After washing the
column, bound
omalizumab was eluted from the column using fractions of one of the elution
buffers listed in
Table 31 below. The elution buffers were prepared by dissolving the indicated
excipients in a
mM citrate buffer, pH 4Ø All elution buffers were adjusted to pH 4Ø Five 1-
mL
fractions were collected. Finally, Protein-A was regenerated by washing the
column with 5
20 mL of 100 mM citrate, pH 3.0 buffer. The flowrate for each step was 1
mL/min, which was
maintained by a Fusion 100 infusion pump (Chemyx, Stafford, TX). 10-mL
NormJect Luer
Lok syringes were used (Henke Sass Wolf, Tuttlingen, Germany, reference number
4100-
000V0).
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
[00162] Elution fractions, El, E2, E3, E4, and E5, were assayed for total
protein content by
high performance size-exclusion chromatography (SEC) analysis. SEC analysis
was
performed using a TSKgel SuperSW3000 column (30 cm x 4.6 mm ID, Tosoh
Bioscience,
King of Prussia, PA) connected to an HPLC workstation (Agilent HP 1100
system). The
separation was carried out at a flow of 0.35 mL/min at room temperature. The
mobile phase
was an aqueous buffer of 100 mM sodium phosphate, 300 mM sodium chloride, pH
7. The
protein concentration was monitored by absorbance at 280 nm using an Agilent
1100 Series
G1315B diode array detector. The total amount of protein eluted from the
Protein-A resin
was estimated by integrating the chromatograms.
[00163] Citrate is a common excipient used in Protein-A chromatography and was
therefore
used here as a control. The eluate fractions for the control sample exhibited
insoluble
aggregates on storage overnight at 4 C as evidenced by the formation of a
precipitate phase.
Therefore, the peak areas reported in Table 31 below represent the total
soluble protein
amounts in the eluate fractions. We note that insoluble aggregates were only
observed in the
control sample and none of the other samples exhibited such aggregates. Peak
areas greater
than that of the control (using the citrate excipient) indicate that the use
of the test excipient
can enable a more efficient separation of protein from the column.
Table 31. Omalizumab elution from Protein-A column
E4 E5
Total
El peak E2 peak E3 peak peak
peak peak
Elution Elution excipient
area area area area area
area
excipient concentration (mM)
(mAU*min) (mAU*min) (mAU*min) (mAU (mAU* (mAU
*min) min)
*min)
citrate
103 352 9670
4098 4245 2953 21318
(control)
imidazole 100 236 10224
7373 3894 2620 24348
taurine 125 408 17018 7676 3349
2211 30662
niacinamide 102 228 14492
5307 2914 2014 24955
caffeine 81 617 21965 8069 3301 1911 35863
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 31: Formulations of BGG with different amounts of caffeine excipient
[00164] Formulations were prepared with different molar concentrations of
caffeine (at
concentrations listed in Table 32 below) and a test protein, where the test
protein was
intended to simulate a therapeutic protein that would be used in a therapeutic
formulation.
The formulations for this Example were prepared in 20 mM histidine buffer for
viscosity
measurement in the following way. Stock solutions of 0 and 80 mM caffeine were
prepared
in 20 mM histidine and the resulting solution pH adjusted with small amounts
of sodium
hydroxide or hydrochloric acid to achieve pH 6 prior to dissolution of the
model protein.
Additional solutions at various caffeine concentrations were prepared by
blending the two
1() stock solutions at various volume ratios, to provide a series of
caffeine-containing solutions,
at concentrations listed in Table 32 below. Once these excipient solutions had
been prepared,
the test protein bovine gamma globulin (BGG) was dissolved into each test
solution at a ratio
to achieve a final protein concentration of about 280 mg/mL by adding 0.7 mL
of each
excipient solution to 0.25 g lyophilized BGG powder. The BGG-containing
solutions were
formulated in 5 mL sterile polypropylene tubes and allowed to shake at 100 rpm
on an orbital
shaker table overnight. These solutions were then transferred to 2 mL
microcentrifuge tubes
and centrifuged for about five minutes at 2400 rpm in an IEC MicroMax
microcentrifuge to
remove entrained air prior to viscosity measurement.
[00165] Viscosity measurements of formulations prepared as described above
were made
with a microVisc viscometer (RheoSense, San Ramon, CA). The viscometer was
equipped
with an A-10 chip having a channel depth of 100 microns, and was operated at a
shear rate of
250 1/s and 25 C. To measure viscosity, the test formulation was loaded into
the viscometer,
taking care to remove all air bubbles from the pipet. The pipet containing the
loaded sample
formulation was placed in the instrument and allowed to incubate at the
measurement
temperature for about five minutes. The instrument was then run until the
channel was fully
equilibrated with the test fluid, indicated by a stable viscosity reading, and
then the viscosity
recorded in centipoise. Viscosity results that were obtained are presented in
Table 32 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 32
Caffeine conc (mM) Viscosity (cP) Normalized Viscosity
0 83 1.00
67 0.81
70 0.84
77 0.92
63 0.76
65 0.78
65 0.78
57 0.69
50 0.60
50 0.60
Example 32: Preparation of solutions of co-solutes in deionized water
[00166] Compounds used as co-solutes to increase caffeine solubility in water
were obtained
5 from Sigma-Aldrich (St. Louis, MO) and included niacinamide, proline,
procaine HC1,
ascorbic acid, 2,5-dihydroxybenzoic acid, lidocaine, saccharin, acesulfame K,
tyramine, and
aminobenzoic acid. Solutions of each co-solute were prepared by dissolving dry
solid in
deionized water and in some cases adjusting the pH to a value between pH of
about 6 and pH
of about 8 with 5 M hydrochloric acid or 5 M sodium hydroxide as necessary.
Solutions
10 were then diluted to a final volume of either 25 mL or 50 mL using a
Class A volumetric
flask and concentration recorded based on the mass of compound dissolved and
the final
volume of the solution. Prepared solutions were used either neat or diluted
with deionized
water.
15 Example 33: Caffeine solubility testing
[00167] The impact of different co-solutes on the solubility of caffeine at
ambient
temperature (about 23 C) was assessed in the following way. Dry caffeine
powder (Sigma-
Aldrich, St. Louis, MO) was added to 20 mL glass scintillation vials and the
mass of caffeine
recorded. 10 mL of a co-solute solution prepared in accordance with Example 32
was added
20 to the caffeine powder in certain cases; in other cases, a blend of a co-
solute solution and
deionized water was added to the caffeine powder, maintaining a final addition
volume of 10
mL. The volume contribution of the dry caffeine powder was assumed to be
negligible in
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
any of these mixtures. A small magnetic stir bar was added to the vial, and
the solution was
allowed to mix vigorously on a stir plate for about 10 minutes. After about 10
minutes the
vial was observed for dissolution of the dry caffeine powder, and the results
are given in
Table 33 below. These observations indicated that niacinamide, procaine HC1,
2,5-
dihydroxybenzoic acid sodium salt, saccharin sodium salt, and tyramine
chloride salt all
enabled dissolution of caffeine to at least about four times the reported
caffeine solubility
limit (-16 mg/mL at room temperature according to Sigma-Aldrich).
TABLE 33
Co-solute Caffeine
Test No. Observation
Name Conc. (mg/mL)
(mg/mL)
33.1 Proline 100 50 DND
33.2 Niacinamide 100 50 CD
33.3 Niacinamide 100 60 CD
33.4 Niacinamide 100 75 CD
33.5 Niacinamide 100 85 CD
33.6 Niacinamide 100 100 CD
33.7 Niacinamide 80 85 CD
33.8 Niacinamide 50 80 CD
33.9 Procaine HC1 100 85 CD
33.10 Procaine HC1 50 80 CD
33.11 Niacinamide 30 80 DND
33.12 Procaine HC1 30 80 DND
33.13 Niacinamide 40 80 MD
33.14 Procaine HC1 40 80 DND
33.15 Ascorbic acid, Na 50 80 DND
33.16 Ascorbic acid, Na 100 80 DND
33.17 2,5 DHBA, Na 40 80 CD
33.18 2,5 DHBA, Na 20 80 MD
33.19 Lidocaine HC1 40 80 DND
33.20 Saccharin, Na 90 80 CD
33.21 Acesulfame K 80 80 DND
33.22 Tyramine HC1 60 80 CD
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Co-solute Caffeine
Test No. Observation
Name Conc. (mg/mL)
(mg/mL)
33.23 Na Aminobenzoate 46 80 DND
33.24 Saccharin, Na 45 80 DND
33.25 Tyramine HC1 30 80 DND
CD=completely dissolved; MD=mostly dissolved; DND=did not dissolve
Example 34: Profile of HUIMIRA
[00168] HUMIRA (AbbVie Inc., Chicago, IL) is a commercially available
formulation of
the therapeutic monoclonal antibody adalimumab, a TNF-alpha blocker typically
prescribed
to reduce inflammatory responses of autoimmune diseases such as rheumatoid
arthritis,
psoriatic arthritis, ankylosing spondylitis, Crohn's disease, ulcerative
colitis, moderate to
severe chronic psoriasis and juvenile idiopathic arthritis. HUMIRA is sold in
0.8 mL single
use doses containing 40 mg of adalimumab, 4.93 mg sodium chloride, 0.69 mg
sodium
phosphate monobasic dihydrate, 1.22 mg sodium phosphate dibasic dihydrate,
0.24 mg
sodium citrate, 1.04 mg citric acid monohydrate, 9.6 mg mannitol and 0.8 mg
polysorbate 80.
A viscosity vs. concentration profile of this formulation was generated in the
following way.
An Amicon Ultra 15 centrifugal concentrator with a 30 kDa molecular weight cut-
off (EMD-
Millipore, Billerica, MA) was filled with about 15 mL of deionized water and
centrifuged in
a Sorvall Legend RT (ThermoFisher Scientific) at 4000 rpm for 10 minutes to
rinse the
membrane. Afterwards the residual water was removed and 2.4 mL of HUMIRA
liquid
formulation was added to the concentrator tube and was centrifuged at 4000 rpm
for 60
minutes at 25 C. Concentration of the retentate was determined by diluting 10
microliters of
retentate with 1990 microliters of deionized water, measuring absorbance of
the diluted
sample at 280 nm, and calculating the concentration using the dilution factor
and extinction
coefficient of 1.39 mL/mg-cm. Viscosity of the concentrated sample was
measured with a
microVisc viscometer equipped with an A05 chip (RheoSense, San Ramon, CA) at a
shear
rate of 250 5ec1 at 23 C. After viscosity measurement, the sample was diluted
with a small
amount of filtrate and concentration and viscosity measurements were repeated.
This process
was used to generate viscosity values at varying adalimumab concentrations, as
set forth in
Table 34 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 34
Adalimumab concentration (mg/mL) Viscosity (cP)
277 125
253 63
223 34
202 20
182 13
Example 35: Reformulation of HUIMIRA with viscosity-reducing excipient
[00169] The following example describes a general process by which HUMIRA was
.. reformulated in buffer with viscosity-reducing excipient. A solution of the
viscosity-reducing
excipient was prepared in 20 mM histidine by dissolving about 0.15 g histidine
and 0.75 g
caffeine (Sigma-Aldrich, St. Louis, MO) in deionized water. The pH of the
resulting solution
was adjusted to about 5 with 5 M hydrochloric acid. The solution was then
diluted to a final
volume of 50 mL in a volumetric flask with deionized water. The resulting
buffered
viscosity-reducing excipient solution was then used to reformulate HUIIVIIRA
at high mAb
concentrations. Next, about 0.8 mL of HUIMIRA was added to a rinsed Amicon
Ultra 15
centrifugal concentrator tube with a 30 kDa molecular weight cutoff and
centrifuged in a
Sorvall Legend RT at 4000 rpm and 25 C for 8-10 minutes. Afterwards about 14
mL of the
buffered viscosity-reducing excipient solution prepared as described above was
added to the
concentrated HUMIRA in the centrifugal concentrator. After gentle mixing, the
sample was
centrifuged at 4000 rpm and 25 C for about 40-60 minutes. The retentate was a
concentrated
sample of HUIMIRA reformulated in a buffer with viscosity-reducing excipient.
Viscosity
and concentration of the sample were measured, and in some cases then diluted
with a small
amount of filtrate to measure viscosity at a lower concentration. Viscosity
measurements
were completed with a microVisc viscometer in the same way as with the
concentrated
HUIIVIIRA formulation in the previous example. Concentrations were determined
with a
Bradford assay using a standard curve generated from HUIIVIIRA stock solution
diluted in
deionized water. Reformulation of HUIMIRA with the viscosity-reducing
excipient gave
viscosity reductions of 30% to 60% compared to the viscosity values of HUIMIRA
.. concentrated in the commercial buffer without reformulation, as set forth
in Table 35 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 35
Adalimumab concentration (mg/mL) Viscosity
(cP)
290 61
273 48
244 20
205 14
Example 36: Improved stability of adalimumab solutions with caffeine as
excipient
[00170] The stability of adalimumab solutions with and without caffeine
excipient was
evaluated after exposing samples to 2 different stress conditions: agitation
and freeze-thaw.
The adalimumab drug formulation HUMIRA (AbbVie) was used, having properties
described in more detail in Example 34. The HUMIRA sample was concentrated to
200
mg/mL adalimumab concentration in the original buffer solution as described in
Example 39;
this concentrated sample is designated "Sample 1." A second sample was
prepared with
¨200 mg/mL of adalimumab and 15 mg/mL of added caffeine as described in
Example 40;
this concentrated sample with added caffeine is designated "Sample 2." Both
samples were
diluted to a final concentration of 1 mg/mL adalimumab with the diluents as
follows: Sample
1 diluent is the original buffer solution, and Sample 2 diluent is a 20 mM
histidine, 15 mg/mL
caffeine, pH=5. Both HUMIRA dilutions were filtered through a 0.22 p.m
syringe filter.
For every diluted sample, 3 batches of 300 !IL each were prepared in a 2 mL
Eppendorf tube
in a laminar flow hood. The samples were submitted to the following stress
conditions: for
agitation, samples were placed in an orbital shaker at 300 rpm for 91 hours;
for freeze-thaw,
samples were cycled 7 times from -17 to 30 C for an average of 6 hours per
condition. Table
36 describes the samples prepared:
TABLE 36
Sample # Excipient added Stress condition
1-C None None
1-A None Agitation
1-FT None Freeze-Thaw
2-C 15 mg/mL caffeine None
2-A 15 mg/mL caffeine Agitation
2-FT 15 mg/mL caffeine Freeze-Thaw
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 37: Evaluation of stability by Dynamic Light Scattering (DLS)
[00171] A Brookhaven Zeta Plus dynamic light scattering instrument was used to
measure
the hydrodynamic radius of the adalimumab molecules in the samples from
Example 36, and
to look for evidence of the formation of aggregate populations. Table 37 shows
the DLS
results for the 6 samples prepared according to Example 36: some of them (1-A,
1-FT, 2-A,
and 2-FT) had been exposed to stress conditions ("Stressed Samples"), and
others (1-C and 2-
C) had not been stressed. The DLS data in Table 37 show a multimodal particle
size
distribution of the monoclonal antibody in Stressed Samples that do not
contain caffeine. In
the absence of caffeine as an excipient, the Stressed Samples 1-A and 1-FT
showed higher
effective diameter than non-stressed Sample 1-C, and in addition they showed a
second
population of particles of significantly higher diameter; this new grouping of
particles with a
larger diameter is evidence of aggregation into subvisible particles. The
Stressed Samples
containing the caffeine (Samples 2-A and 2-FT) only display one population of
particles, at a
particle diameter similar to the unstressed Sample 2-C. These results
demonstrate that adding
caffeine to these samples reduced the formation of aggregates or subvisible
particles.
TABLE 37
Diameter of % by Intensity Diameter of % by
Effective
Sample # Population of Population Population Intensity of
Diameter (nm)
#1 (nm) #1 #2 (nm)
Population #2
1-C 10.9 10.8 100
1-A 11.5 10.8 87 28.9 13
1-FT 20.4 11.5 66 112.2 44
2-C 10.5 10.5 100
2-A 10.8 10.8 100
2-FT 11.4 11.4 100
[00172] Tables 38A and Table 38B display the DLS raw data of adalimumab
samples from
Example 36 showing the particle size distributions. In these Tables, G(d) is
the intensity-
weighted differential size distribution. C(d) is the cumulative intensity-
weighted differential
size distribution.
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
TABLE 38A
Sample 1-C Sample 1-A Sample 1-FT
Diameter Diameter Diameter
G (d) C(d) G (d) C(d) G (d) C(d)
(nm) (nm) (nm)
10.6 14 4 9.3 13 3 8.2 12 2
10.6 53 20 9.8 47 15 9.2 55 13
10.7 92 46 10.3 87 37 10.3 98 32
10.8 100 76 10.8 100 63 11.5 100 52
10.9 61 93 11.4 67 80 12.9 57 63
10.9 22 100 12 27 87 14.5 14 66
26.1 4 88 89.3 5 67
27.5 10 91 100.1 27 72
28.9 13 94 112.2 52 83
30.5 13 97 125.7 52 93
32.1 7 99 140.8 30 99
33.8 4 100 157.8 7 100
TABLE 38B
Sample 2-C Sample 2-A Sample 2-FT
Diameter Diameter Diameter
G (d) C(d) G (d) C(d) G (d) C(d)
(nm) (nm) (nm)
10.3 14 4 10.6 7 2 11.3 28 9
10.4 52 19 10.6 43 16 11.3 64 29
10.5 91 46 10.7 79 40 11.4 100 60
10.5 100 75 10.8 100 71 11.5 79 85
10.6 62 93 10.8 64 91 11.5 43 98
10.7 23 100 10.9 29 100 11.6 7 100
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Example 38: Evaluation of stability by size-exclusion chromatography (SEC)
[00173] Size exclusion chromatography was used to detect subvisible
particulates of less
than about 0.1 microns in size from the stressed and unstressed adalimumab
samples
described in Example 36. To perform the SEC, a TSKgel SuperSW3000 column
(Tosoh
Biosciences, Montgomeryville, PA) with a guard column was used, and the
elution was
monitored at 280 nm. A total of 10 of each stressed and unstressed sample
from Example
36 was eluted isocratically with a pH 6.2 buffer (100 mM phosphate, 325 mM
NaCl), at a
flow rate of 0.35 mL/min. The retention time of the adalimumab monomer was
approximately 9 minutes. No detectable aggregates were identified in the
samples containing
the caffeine excipient, and the amount of monomer in all 3 samples remained
constant.
Example 39: Viscosity reduction of HERCEPTIN formulation
[00174] The monoclonal antibody trastuzumab (HERCEPTIN from Genentech) was
received as a lyophilized powder and reconstituted to 21 mg/mL in DI water.
The resulting
solution was concentrated as-is in an Amicon Ultra 4 centrifugal concentrator
tube (molecular
weight cut-off, 30 kDa) by centrifuging at 3500 rpm for 1.5 hrs. The
concentration was
measured by diluting the sample 200 times in an appropriate buffer and
measuring
absorbance at 280 nm using the extinction coefficient of 1.48 mL/mg. Viscosity
was
measured using a RheoSense microVisc viscometer.
[00175] Excipient buffers were prepared containing salicylic acid and caffeine
either alone
or in combination by dissolving histidine and excipients in distilled water,
then adjusting pH
to the appropriate level. The conditions of Buffer Systems 1 and 2 are
summarized in Table
39.
TABLE 39
Salicylic Acid Caffeine Osmolality
Buffer System # pH
concentration concentration (mOsm/kg)
1 10 mg/mL 10 mg/mL 145 6
2 0 15 mg/mL 86 6
[00176] HERCEPTIN solutions were diluted in the excipient buffers at a ratio
of ¨1:10 and
concentrated in Amicon Ultra 15 (MWCO 30 kDa) concentrator tubes.
Concentration was
determined using a Bradford assay and compared with a standard calibration
curve made
from the stock HERCEPTIN sample. Viscosity was measured using the RheoSense
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
microVisc viscometer. The concentration and viscosity measurements of the
various
HERCEPTIN solutions are shown in Table 40 below, where Buffer Systems 1 and 2
refer to
those buffers described in Table 39.
TABLE 40
Buffer System 1: Solution
Control solution with no
Buffer System 2: Solution with
with 10 mg/mL Caffeine + 10
added excipients
15 mg/mL Caffeine added
mg/ml Salicylic Acid added
Antibody Antibody
Antibody
Viscosity Viscosity
Concentration Concentration Viscosity (cP)
Concentration
(cP) (cP)
(mg/mL) (mg/mL)
(mg/mL)
37.2 215 9.7 244 23.4 236
9.3 161 7.7 167 12.2 200
3.1 108 2.9 122 5.1 134
1.6 54 2.4 77 2.1 101
[00177] Buffer System 1, containing both salicylic acid and caffeine, had a
maximum
viscosity reduction of 76% at 215 mg/mL compared to the control sample. Buffer
System 2,
containing just caffeine, had viscosity reduction up to 59% at 200 mg/mL.
Example 40: Viscosity reduction of AVASTIN formulation
[00178] AVASTIN (monoclonal antibody bevacizumab formulation marketed by
Genentech) was received as a 25 mg/mL solution in a histidine buffer. The
sample was
concentrated in Amicon Ultra 4 centrifugal concentrator tubes (MWCO 30 kDa) at
3500 rpm.
Viscosity was measured by RheoSense microVisc and concentration was determined
by
absorbance at 280 nm (extinction coefficient, 1.605 mL/mg). The excipient
buffer was
prepared by adding 10 mg/mL caffeine along with 25 mM histidine HC1. AVASTIN
stock
solution was diluted with the excipient buffer then concentrated in Amicon
Ultra 15
centrifugal concentrator tubes (MWCO 30 kDa). The concentration of the
excipient samples
was determined by Bradford assay and the viscosity was measured using the
RheoSense
microVisc. Results are shown in Table 41 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 41
Concentration Viscosity without Viscosity with 10 mg/mL
% Viscosity Reduction
(mg/mL) added excipient (cP) added caffeine excipient (cP)
from Excipient
266 297 113 62%
213 80 22 73%
190 21 13 36%
[00179] AVASTIN showed a maximum viscosity reduction of 73% when concentrated
with 10 mg/mL of caffeine to 213 mg/mL when compared to the control AVASTIN
sample.
Example 41: Preparation of formulations containing caffeine, a secondary
excipient and test
protein
[00180] Formulations were prepared using caffeine as the excipient compound or
a
combination of caffeine and a second excipient compound, and a test protein,
where the test
protein was intended to simulate a therapeutic protein that would be used in a
therapeutic
ix) formulation. Such formulations were prepared in 20 mM histidine buffer
with different
excipient compounds for viscosity measurement in the following way. Excipient
combinations (Excipients A and B, as described in Table 28 below) were
dissolved in 20 mM
histidine and the resulting solution pH adjusted with small amounts of sodium
hydroxide or
hydrochloric acid to achieve pH 6 prior to dissolution of the model protein.
Once excipient
solutions had been prepared, the test protein bovine gamma globulin (BGG) was
dissolved at
a ratio to achieve a final protein concentration of about 280 mg/mL. Solutions
of BGG in the
excipient solutions were formulated in 20 mL glass scintillation vials and
allowed to shake at
80-100 rpm on an orbital shaker table overnight. BGG solutions were then
transferred to 2
mL microcentrifuge tubes and centrifuged for about ten minutes at 2300 rpm in
an IEC
MicroMax microcentrifuge to remove entrained air prior to viscosity
measurement.
[00181] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
The viscometer was equipped with a CP-40 cone and was operated at 3 rpm and 25
C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and subsequent twenty second measurement collection
period. The three
data points collected were then averaged and recorded as the viscosity for the
sample in Table
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
42 below. Viscosities of solutions with excipient were normalized to the
viscosity of the
model protein solution without excipient. The normalized viscosity is the
ratio of the
viscosity of the model protein solution with excipient to the viscosity of the
model protein
solution with no excipient.
TABLE 42
Excipient A Excipient B
Normalized Viscosity
Name Conc. (mg/mL) Name Conc. (mg/mL)
0 0 1.00
Caffeine 15 0 0.77
Sodium
Caffeine 15 12 0.77
acetate
Sodium
Caffeine 15 14 0.78
sulfate
Caffeine 15 Aspartic acid 20 0.73
CaCl2
Caffeine 15 15 0.65
dihydrate
Dimethyl
Caffeine 15 25 0.65
Sulfone
Caffeine 15 Arginine 20 0.63
Caffeine 15 Leucine 20 0.69
Caffeine 15 Phenylalanine 20 0.60
Caffeine 15 Niacinamide 15 0.63
Caffeine 15 Ethanol 22 0.65
Example 42: Preparation of formulations containing dimethyl sulfone and test
protein
[00182] Formulations were prepared using dimethyl sulfone (Jarrow Formulas,
Los Angeles,
1() CA) as the excipient compound and a test protein, where the test
protein was intended to
simulate a therapeutic protein that would be used in a therapeutic
formulation. Such
formulations were prepared in 20 mM histidine buffer for viscosity measurement
in the
following way. Dimethyl sulfone was dissolved in 20 mM histidine and the
resulting solution
pH adjusted with small amounts of sodium hydroxide or hydrochloric acid to
achieve pH 6
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
and then filtered through a 0.22 micron filter prior to dissolution of the
model protein. Once
excipient solutions had been prepared, the test protein bovine gamma globulin
(BGG) was
dissolved at a concentration of about 280 mg/mL. Solutions of BGG in the
excipient
solutions were formulated in 20 mL glass scintillation vials and allowed to
shake at 80-100
rpm on an orbital shaker table overnight. BGG solutions were then transferred
to 2 mL
microcentrifuge tubes and centrifuged for about ten minutes at 2300 rpm in an
IEC
MicroMax microcentrifuge to remove entrained air prior to viscosity
measurement.
[00183] Viscosity measurements of formulations prepared as described above
were made
with a DV-IIT LV cone and plate viscometer (Brookfield Engineering,
Middleboro, MA).
1() The viscometer was equipped with a CP-40 cone and was operated at 3 rpm
and 25 C. The
formulation was loaded into the viscometer at a volume of 0.5 mL and allowed
to incubate at
the given shear rate and temperature for 3 minutes, followed by a measurement
collection
period of twenty seconds. This was then followed by 2 additional steps
consisting of 1 minute
of shear incubation and subsequent twenty second measurement collection
period. The three
data points collected were then averaged and recorded as the viscosity for the
sample.
Viscosities of solutions with excipient were normalized to the viscosity of
the model protein
solution without excipient. The normalized viscosity recorded in Table 43 is
the ratio of the
viscosity of the model protein solution with excipient to the viscosity of the
model protein
solution with no excipient.
TABLE 43
Dimethyl sulfone concentration (mg/mL) Normalized viscosity
0 1.00
15 0.92
0.71
50 0.71
30 0.72
Example 43: Process benefit observed during concentrating HGG by
centrifugation
[00184] Centrifugation was used for quick assessment of the effect of caffeine
in the
25 concentrating process of human-serum-derived gamma globulin (HGG). HGG
stock solution
at 100 mg/mL (10% Octagam) was first exchanged into PBS buffer with or without
50 mM
caffeine using Amicon-15 centrifugal units with 30 kDa-MWCO membrane; 7 mL of
HGG
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
stock was pipetted into an Amicon-15 centrifugal unit, followed by addition of
7 mL of
formulation vehicle into the centrifugal unit. After mixing the solution by
pipetting, the
centrifugal units were centrifuged at 2,844 x g for about 40 min until about 7
mL of filtrate
was collected. The filtrate was discarded. About 7 mL of corresponding
formulation vehicle
was then added to the centrifugal unit and mixed. This process of
centrifugation and dilution
with vehicle was repeated two times, then the buffer-exchanged HGG solution
was collected
from the centrifugal units. The corresponding formulation vehicle was added to
the HGG
solution to a final mass of about 14 g. The HGG concentration in the starting
formulations
was about 50 mg/mL, which was subsequently verified by BCA assay.
[00185] Next, about 13 mL of HGG formulation was added to the outer tubes of
CentriPrep
centrifugal units with a 30 kDa NMWL (nominal molecular weight limit)
membrane. The
formulations were concentrated by centrifugation at 1,300 x g and the mass of
filtrate for
each formulation was recorded every 10 min. HGG concentration in retentate was
estimated
using the mass of filtrate collect using 13 g and 50 mg/mL as the initial
sample weight and
HGG concentration, respectively. This entire process was repeated two times,
to generate the
following data sets: Run #1 was conducted in phosphate buffered saline (PBS)
and this
sample is designated as "PBS-1"; Run #1 was conducted in PBS containing
caffeine and this
sample is designated as "PBS-caffeine-1"; Run #2 was conducted in PBS and this
sample is
designated as "PBS-2"; Run #2 was conducted in PBS containing caffeine and
this sample is
designated as "PBS-caffeine-2". The centrifugation experiments Run #1 and Run
#2 were
conducted separately, so the control data sets (PBS-1 and PBS-2) should be
compared with
their respective caffeine-containing examples (PBS-caffine-1 and PBS-caffeine-
2). The
results of these tests are documented in the Tables 44 (Run #1) and 45 (Run
#2) below,
including mass of filtrate in grams and the concentration of HGG in mg/mL
units. In both
Run #1 and Run #2, the calculated concentration of HGG in the retentate was
higher when
the excipient was added, compared with the control formulation. These results
are also
shown in the graphs of FIGs. 3A and 3B, where an increasing amount of HGG in
the
retentate.
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
TABLE 44
Estimated HGG concentration
Centrifuge Mass of filtrate (g)
in retentate (mg/mL)
Time (min)
PB S -1 PB S - caffeine-1 PB S -1 PB S - caffeine
-1
0 0 0 50 50
1.3428 1.686 56 57
2.8042 3.4284 64 68
4.2015 5.0912 74 82
5.4708 6.5663 86 101
6.5555 7.9021 101 128
7.5734 9.0425 120 164
8.5464 9.9982 146 217
9.3322 10.6264 177 274
9.9302 11.0618 212 335
100 10.3502 11.3234 245 388
110 10.7023 11.5735 283 456
120 10.9384 11.7092 315 504
130 NA NA NA NA
140 NA NA NA NA
TABLE 45
Estimated HGG concentration
Centrifuge Mass of filtrate (g)
in retentate (mg/mL)
Time (min)
PB S-2 PB S-caffeine-2 PB S-2 PB S-caffeine-2
0 0 0 50 50
10 1.3754 1.5748 56 57
20 2.8867 3.1872 64 66
30 4.241 4.623 74 78
40 5.4953 5.9191 87 92
50 6.6583 6.9913 102 108
60 7.3571 7.9329 115 128
70 7.973 8.7046 129 151
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Estimated HGG concentration
Centrifuge Mass of filtrate (g)
in retentate (mg/mL)
Time (min)
PBS-2 PBS-caffeine-2 PBS-2 PBS-caffeine-2
80 8.5071 9.3864 145 180
90 8.9538 9.9148 161 211
100 9.3573 10.3398 178 244
110 9.6367 10.6072 193 272
120 9.8756 10.8152 208 298
130 10.0687 11.0051 222 326
140 10.1515 11.011 228 327
Example 44: DLS viscosity measurements of concentrated human immune globulin
[00186] 10X phosphate buffered saline (PBS) from Fisher Scientific (Hampton,
NH) was
diluted with Milli-Q Type 1 ultra-pure water to 1X concentration prior to use.
Nicotinamide,
acesulfame K, 1,3-dimethyluracil, arginine monohydrochloride, saccharin,
caffeine, tyramine,
and imidazole were purchased from Sigma-Aldrich (St. Louis, MO), sodium
benzoate from
Spectrum Chemical (New Brunswick, NJ) and hordenine HC1 from Bulk Supplements
(Henderson, NV) and all were used as excipients in the following experiment.
[00187] A purified human immune globulin (Octagam 10%) was purchased from NOVA
Biologics, Inc (Oceanside, CA), buffer exchanged into 1X PBS, using a benchtop
EMD
Millipore (Billerica, MA) tangential flow-filtration unit, and concentrated
using an Amicon
Ultra 15 centrifugal concentrator tube with a 30 kDa molecular weight cut-off
(EMD
Millipore, Billerica, MA). Stock excipient solutions were prepared in 1X PBS
at a
concentration of 1 M or the solubility limit of the compound, and pH adjusted
to about 7.4 as
necessary with either concentrated hydrochloric acid or sodium hydroxide. In a
PCR tube, the
concentrated human IgG and excipient solutions were mixed together (9 parts
IgG
concentrate, 1 part excipient solution or buffer). To the mixture was added a
solution of PEG
surface modified gold nanoparticles (nanoComposix, San Diego, CA) in deionized
water.
The resulting mixture of IgG, excipient and particles was loaded in duplicate
into a 384-well
Aurora (Whitefish, MT) microplate. The microplate was then centrifuged at 400
x g in a
Sorvall Legend RT centrifuge, and then placed in a DynaPro II DLS plate reader
(Wyatt
Technology Corp., Goleta, CA) to measure the apparent particle size of the
gold
nanoparticles at 25 degrees Centigrade. The ratio of the apparent particle
size of the gold
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
nanoparticle in a protein formulation to the apparent particle size of the
gold nanoparticle in
buffer (no protein) was used to determine the viscosity of the protein
formulation according
to the stokes-Einstein equation. In this example, the ratio of apparent radius
to the actual
radius of the gold nanoparticle was multiplied by the viscosity of water at 25
degrees
Centigrade to calculate the viscosity of the protein formulation in centipoise
(cP). Viscosity
results in the presence of excipient were compared with results in the
presence of no
excipient to determine magnitude of viscosity reduction achieved.
TABLE 46
DLS viscosity (cP)
Excipient added
replicate 1 replicate 2
1,3-dimethyluracil 42.36 40.44
Hordenine HC1 42.8 43.72
Acesulfame K 45.59 42.28
Nicotinamide 46.4 49.1
Arginine HC1 49.09 52.95
Aspartame 50.64 53.74
Saccharin 51.46 56.22
Caffeine 57.43 47.25
Tyramine 65.76 59.98
Imidazole 61.75 57.41
Sodium benzoate 71.02 87.49
None (control) 67.07 63.39
1() Example 45: DLS viscosity measurements of concentrated human immune
globulin
[00188] To examine the effects of dimethyluracil and hordenine as excipients,
HGG
formulations were prepared by mixing concentrated HGG (215 mg/mL) with
appropriate
amount of PBS and 10x excipient stock to achieve a 50 mg/mL HGG formulations
in PBS
with or without 100 mM excipients. Next, 13 mL of each formulation was added
to a
CentriPrep centrifugal unit and conduct the centrifugal study as described in
Example 43
above. The filtrate volume and retentate concentration were recorded as a
function of
centrifugation time, and the results of these tests with 100 mM concentration
of excipients
dimethyluracil and hordenine vs. control (PBS buffer) are summarized in Table
47 below.
Addition of the excipients hordenine and dimethyluracil resulted in improved
filtration rate
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
compared with the control. The results in Table 47 are also shown in the graph
of FIG. 4,
where an increasing amount of HGG in the retentate correlates with improved
processing
performance.
TABLE 47
Estimated HGG concentration in
Mass of filtrate (g)
Time retentate (mg/mL)
(min) 100 mM 100 mM 100 mM 100 mM
PBS PBS
dimethyluracil hordenine dimethyluracil hordenine
0 0 0 0 50 50 50
0.6804 1.0751 2.2692 53 54 60
1.6566 2.2902 3.2286 57 61 66
2.5385 3.4562 4.1148 62 68 73
3.3715 4.4359 4.9049 66 76 80
4.2098 5.4398 5.6059 71 86 87
4.856 6.2301 6.1031 76 95 93
5.5011 7.0011 6.6934 81 107 102
6.0436 7.6977 7.0824 87 121 108
6.5227 8.2152 7.5251 93 134 117
100 7.0035 8.7504 7.8367 99 151 124
110 7.4411 9.137 8.119 105 165 131
120 7.8312 9.4786 8.3418 111 181 137
130 8.1512 9.7263 8.5544 116 194 143
Example 46: Improving Tangential Flow filtration using caffeine
[00189] 400 mL of human gamma globulin (Octagam, Octapharma, USA) at a
concentration
of 35 mg/mL was prepared by diluting the stock at 100 mg/mL into phosphate
buffered saline
10
(PBS). The buffer was prepared by dissolving 1.8 mM KH2PO4, 10 mM Na2HPO4, 137
mM
NaCl, 2.7 mM KC1 in 1 L of Milli-Q water. A caffeine PBS solution was prepared
by
dissolving 50 mM caffeine, 1.8 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, 2.7 mM
KC1
in 1 L of Milli-Q water. Tap water was purified with a Direct-Q 3 UV
purification system
from EMD Millipore (Billerica, MA) to produce the DI water. The human gamma
globulin
15 (HGG)
solution was transferred to a reservoir of a Labscale tangential flow
filtration (TFF)
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
system (Millipore, Billerica, MA) equipped with 30 KDa MWCO Pellicon XL TFF
cassette
(Millipore, Billerica, MA). Prior to use, the cassette was flushed with Milli-
Q water followed
by PBS and a water permeability test was carried out to ensure membrane
integrity and
efficiency. The HGG solution was pumped using a Quattroflow pump (Cole-Parmer,
IL)
through the cassette with the retentate line going back to the sample
reservoir and the
permeate collected in a graduated measuring cylinder. A stirrer bar ensured
proper mixing of
the feed with the retentate. The feed pump was set to deliver 120 mL/min feed
to the cassette.
The retentate restrictor was used to get the transmembrane pressure (TNIP)
roughly in the 20
to 30 psi range, and it was ensured that the TNIP remained constant throughout
the run by
adjusting the feed pump and retentate restrictor. Data logging of the pressure
and flow rates
was carried out and samples taken every 30 minutes. To calculate the feed
concentration,
samples were analyzed by SE-HPLC where 50 mg was loaded onto an Agilent 1100
HPLC
system fitted with TSKgel SuperSW3000 column (30 cm x 4.6 mm ID, Tosoh
Bioscience,
King of Prussia, PA) and Agilent G1351B Diode array detector. PBS was used as
mobile
phase at a flow rate of 0.35 mL/min. The protein concentration was calculated
by integrating
the area under the peaks. The feed concentration plotted as a function of time
was used to
compare TFF efficiency in presence of caffeine with the TFF using the control
system.
Higher percent concentration change from the initial feed concentration was
observed in a
shorter time with the caffeine as compared to the control, as shown in Table
48 below,
demonstrating increased TFF efficiency.
TABLE 48
Control system, Caffeine
Control system, Caffeine
system,
Time (min) protein conc system, protein
% change % change
(mg/mL) conc (mg/mL)
0 38.62 0 37.07 0
67.42 74.6 82.11 121.5
60 78.14 102.3 120.63 225.4
90 101.94 163.9 141.88 282.7
120 140.81 264.6 198.96 436.7
150 162.39 320.4 222.50 500.2
180 226.06 485.3 286.68 673.3
210 254.76 559.6 305.39 723.8
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
240 281.58 629.0
270 291.99 656.0
Example 47: Using caffeine to improve purification yield from Protein A resin
[00190] Research-grade omalizumab, purchased from Bioceros (Utrecht, The
Netherlands)
at 15 mg/mL in 20 mM sodium phosphate, pH 7 buffer was used as test sample.
This protein
solution was filtered through a 0.2 [tm polyethersulfone (PES) filter. The
filtered material
was mixed in a 1:1 ratio with a binding buffer that consisted of 20 mM sodium
phosphate at
pH 7 in DI water. Tap water was purified with a Direct-Q 3 UV purification
system from
EMD Millipore (Billerica, MA) to produce the DI water. Protein-A purification
was
performed using a HiTrap Protein-A HP 1 mL column from GE Healthcare (Chicago,
IL). 10
mL of binding buffer was used for column equilibration, followed by loading 30
mg of
protein. The column was then washed with 5 mL of binding buffer to remove
unbound
protein. Bound omalizumab was eluted from the column in 1 mL fractions using
either 0.1 M
glycine buffer at pH 3.5 as the control buffer or by using 0.1 M glycine, 50
mM caffeine
buffer at pH 3.5. The control buffer was prepared by dissolving 7.5 g of
glycine into DI
water, adjusting the pH to 3.5 using 6M HC1 and adjusting the volume to 1 L.
Caffeine buffer
was prepared by dissolving 7.5 g of glycine and 10 g of caffeine into DI
water, adjusting the
pH to 3.5 using 6M HC1 and adjusting the volume to 1 L. Five 1 mL fractions
were
collected; these eluted fractions were labeled El, E2, E3, E4, and E5.
Finally, Protein-A was
regenerated by washing the column with 5 mL of 0.1 M glycine pH 3.0 buffer.
The flowrate
for each step was 1 mL/min, which was maintained by a Fusion 100 infusion pump
(Chemyx,
Stafford, TX). 10 mL NormJect Luer Lok syringes were used (Henke Sass Wolf,
Tuttlingen,
Germany, reference number 4100-000V0).
[00191] The 5 eluted fractions, El, E2, E3, E4, and E5, were assayed for total
protein
content by high performance size-exclusion chromatography (SEC) analysis. SEC
analysis
was performed using a TSKgel SuperSW3000 column (30 cm x 4.6 mm ID, Tosoh
Bioscience, King of Prussia, PA) connected to an Agilent HP 1100 HPLC system.
PBS was
used as mobile phase at a flow rate of 0.35 mL/min at room temperature. The
protein
concentration was monitored by absorbance at 280 nm using a G1315B diode array
detector.
The total amount of protein eluted from the Protein-A resin was determined by
integrating the
chromatograms, and about 8% increase in yield was observed in the presence of
caffeine, as
shown in Table 49 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 49
Protein concentration in fractions
Protein concentration in
Sample Fraction eluted without caffeine containing
fractions eluted with caffeine
buffer (mg/mL) containing buffer
(mg/mL)
El 0.277 1.13
E2 2.14 6.72
E3 6.58 4.67
E4 2.24 1.40
E5 1.06 0.708
Total yield 12.29 14.63
% recovery 44.38 52.83
Example 48: Excipients for stabilization during low pH hold
[00192] Research-grade ipilimumab, purchased from Bioceros (Utrecht, The
Netherlands) at
15 mg/mL in 20 mM sodium phosphate, pH 7 buffer was used as test sample. The
protein
solution was filtered through a 0.2 1.tm polyethersulfone (PES) filter.
Raffinose pentahydrate
was obtained from Sigma (St Louis, MO). The excipient stock was prepared by
dissolving the
raffinose pentahydrate at a concentration of 1 M in 0.15 M glycine buffer, pH
2.75. The
buffer was prepared by dissolving 7.5 g of glycine in 0.9 L Milli-Q water,
adjusting the pH to
2.75 using 1 M HC1, and making the volume to 0.1 L. One control formulation
and three
excipient-containing formulations were prepared by adding the excipient to the
ipilimumab
solution, with a final excipient concentration of 0 mM, 100 mM, 200 mM and 400
mM and a
final ipilimumab concentration of 2 mg/mL. The samples were incubated
overnight at the
acidic pH (2.75) for 24 h and the samples were analyzed by SE-HPLC where 50 mg
was
loaded onto an Agilent 1100 HPLC system fitted with TSKgel SuperSW3000 column
(30 cm
x 4.6 mm ID, Tosoh Bioscience, King of Prussia, PA) and Agilent G1351B diode
array
detector. PBS was used as mobile phase at a flow rate of 0.35 mL/min. The
monomer protein
(ipilimumab) concentration was calculated by integrating the areas under the
monomer peak.
The monomer fraction from an untreated sample not exposed to the low pH was
normalized
to 100 % and the monomeric fractions of the treated samples expressed as
percentage change
of this untreated sample. The results in Table 50 below, show that the
presence of raffinose in
the samples resulted in a higher percentage monomeric form of the ipilimumab
after the low
pH hold.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 50
Sample % of ipilimumab in the monomeric form
0 mM raffinose 38.69
100 mM raffinose 52.68
200 mM raffinose 63.07
400 mM raffinose 78.88
Untreated 100
Example 49: Buffer and excipient preparation
[00193] A stock 20 mM histidine hydrochloride (His HC1) buffer was prepared
for use in
formulating excipients and protein buffer exchange. Two liters of His HC1 was
prepared by
dissolving 6.206 grams of histidine (Sigma-Aldrich, St. Louis, MO) in Type 1
ultrapure
water. The solution of dissolved histidine was titrated to pH 6.0 using
concentrated
hydrochloric acid. The His HC1 solution was then brought up to 2 liters using
a volumetric
flask and filtered through a 0.2 p.m membrane bottle-top filter device (Sigma
Aldrich, St.
Louis, MO). Excipients to be tested in Example 51 (listed in Table 51) were
prepared as
excipient solutions for subsequent testing as follows. Each excipient was were
prepared at
10X (1 M) by dissolving it in this His HC1 buffer described above, and
adjusting the pH with
concentrated sodium hydroxide or concentrated hydrochloric acid. Each
excipient solution
was then filtered using 0.2 p.m membrane filter.
Example 50: Protein solution preparation
[00194] Two test proteins, purified omalizumab purchased from Bioceros (The
Netherlands)
and human serum derived polyclonal IgG (Octagam 10%), were buffer exchanged
into His
HC1 (as prepared in Example 49) using 20 kDa molecular weight cut-off dialysis
cassettes
(Fisher Scientific). Each protein solution was transferred into the dialysis
cassette attached to
a buoy and placed in a flask for buffer exchanges. A total of 3 buffer
exchanges were
performed into > 50x the starting protein volume. Upon the final buffer
exchange step, the
protein solution was removed from the dialysis cassette and filtered through
0.2 p.m
membrane filter and protein concentration was measured via A280 by diluting
100-fold into
His HC1 buffer. 100 was then transferred to a UV clear 96 half-well
microplate (Greiner
Bio-One, Austria), and absorbance measured at a wavelength of 280 nm with a
Synergy HT
plate reader (BioTek, Winooski, VT). The blanked, pathlength corrected A280
measurement
was then divided by the respective extinction coefficient and multiplied by
the dilution factor
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
to determine the protein concentration. A subsequent concentration step was
needed to
concentrate the protein in preparation for dynamic light scattering (DLS)
viscosity
measurements in Example 51. Concentration was performed using Amicon-15
centrifugal
devices with a 30 kDa molecular weight cut-off (EMD Millipore, Billerica, MA)
and
concentrated to 175 mg/mL based on retentate mass in the centrifugal device by
centrifuging
at 4000 x g on a benchtop centrifuge (Sorvall Legend RT).
Example 51: DLS measurement of the diffusion interaction parameter
[00195] In this Example, the diffusion interaction parameter (kD) of a dilute
protein solution
was measured by DLS in the presence of 0.1 M excipient solution. The
excipients being
tested are listed in Table 51 For each excipient, a 0.2M solution of the
excipient was prepared
separately from the previously prepared 1 M excipient stock. The kD was
measured by DLS
using 5 different concentrations of omalizumab (prepared as described in
Example 50)
ranging from 10 mg/mL to 0.6 mg/mL in the presence of 0.1M excipient. An
identical set of
control samples was prepared, containing the same concentrations of omalizumab
in the
absence of any excipient. For each test sample, 20 !IL of protein solution was
combined with
tL of 0.2 M excipient solution (1:1 mixture) onto a 384-well plate (Aurora
Microplates,
Whitefish, MT). After loading the samples, the well plate was shaken on a
plate shaker to
mix the contents for 5 minutes. Upon mixing, the well plate was centrifuged at
400 x g in a
Sorvall Legend RT for 1 minute to force out any air pockets. The well plate
was then loaded
20 into a DynaPro II DLS plate reader (Wyatt Technologies Corp., Goleta,
CA) and the
diffusion coefficient of each sample was measured at 25 C. For each excipient,
the measured
diffusion coefficient was plotted as a function of protein concentration, and
the slope of the
linear fit of the data was recorded as the kD. In this example, each
measurement was
normalized to a control average and reported as a percent to the control.
These results are
shown in Table 51 below.
TABLE 51
Excipients Protein
% change in kD value from control
3-(1-Pyridinio)-1-propanesulfonate Omalizumab 22%
Aspartic Acid Omalizumab 72%
Ornithine Omalizumab 49%
beta-alanine Omalizumab 25%
Lysine Omalizumab 47%
CA 03120023 2021-05-13
WO 2020/112855 PCT/US2019/063374
Excipients Protein %
change in kD value from control
Trigonelline Omalizumab 28%
(3-carboxypropyl)
Omalizumab 59%
trimethylammonium chloride
Aminohippuric acid Omalizumab 20%
Arginine Omalizumab 81%
1-Hexy1-3-methylimidazolium
Omalizumab 25%
chloride
NaCl (200 mM) Omalizumab 70%
ethanolamine HCL Omalizumab 25%
spermidine Omalizumab 54%
4-aminopyridine Omalizumab 25%
lysine Omalizumab 66%
cysteamine HC1 Omalizumab 38%
x-xylylenediamine Omalizumab 56%
nicotinic acid Omalizumab 18%
quinic acid Omalizumab 19%
1,3-diaminopropane Omalizumab 62%
lactobionic acid Omalizumab 47%
Glutamic acid Omalizumab 35%
Sodium Ascorbate Omalizumab 35%
sodium propionate Omalizumab 33%
Quinic acid Omalizumab 27%
sodium benzoate Omalizumab 33%
Glucuronic acid Omalizumab 32%
Hydroxybenzoic acid Omalizumab 50%
sodium bisulfite Omalizumab 57%
Salicylic acid Omalizumab 43%
Etidronate Omalizumab 56%
Acesulfame K+ salt Omalizumab 49%
Calcium propionate Omalizumab 83%
Citric acid Omalizumab 47%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipients Protein
% change in kD value from control
hydroquinone sulfonic acid Omalizumab 36%
Menadione sodium bisulfite Omalizumab 25%
2-dimethylaminoethanol Omalizumab 31%
2-methyl-2-imidazoline Omalizumab 17%
cycloserine Omalizumab 9%
3-aminopyridine Omalizumab 6%
4-aminopyridine Omalizumab 23%
agmatine sulfate Omalizumab 69%
cytidine Omalizumab 29%
ethanolamine Omalizumab 29%
meglumine Omalizumab 171%
morpholine Omalizumab 17%
triethanolamine Omalizumab 40%
Example 52: Viscosity measurement by DLS
[00196] Purified omalizumab purchased from Bioceros (The Netherlands) and
human serum
derived polyclonal IgG (Octagam 10%, Pfizer) were used as model protein
systems to
explore viscosity effects of excipients. Concentrated stock solutions of
excipients (listed in
Tables 52 and 53) were prepared at 10X (1M) in His HC1 buffer, following the
protocol
described in Example 49. Omalizumab was buffer exchanged using Amicon-15
centrifugal
(30 kDa MWCO) devices into His HC1 buffer and concentrated to 175 mg/mL based
on
retentate mass in the centrifugal device. Excipient and concentrated protein
were combined in
a 200 !IL PCR tube, adding 1-part 10X excipient and 9 parts protein. An
additional 2 !IL of a
5-fold diluted solution of polyethylene glycol surface modified gold
nanoparticles
(nanoComposix, San Diego, CA) was added to each PCR tube and mixed thoroughly
by
inversion. A control sample was prepared identically, except without adding
any excipient.
Each sample (test samples and the control) was transferred to a 384-well
microplate (Aurora
Microplates, Whitefish, MT) in duplicate (25 per well) and centrifuged at
400 x g for 1
minute before analysis. A DynaPro II DLS plate reader (Wyatt Technology Corp.,
Goleta,
CA) was used to measure apparent particle size of gold nanoparticles at 25 C.
The ratio of
the apparent particle size of the gold nanoparticle to the known particle size
of the gold
nanoparticle in water was used to determine the viscosity of the protein
formulation
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
according to the Stokes-Einstein equation. In this Example, each measurement
was
normalized to a control average and reported as a percent reduction compared
with the
control, and standard deviation is shown, and the results are shown in Tables
52 and 53
below.
TABLE 52
Excipient (100 mM) Protein % Reduction Std. dev.
Dimethyluracil hIgG 36.5% 1.5%
hordenine hIgG 33.7% 0.7%
acesulfame K hIgG 32.6% 2.5%
nicotinamide hIgG 26.8% 2.1%
arginine hIgG 21.8% 3.0%
aspartame hIgG 20.0% 2.4%
saccharin hIgG 17.5% 3.7%
3-(1-Pyridinio)-1-propanesulfonate hIgG 15.0% 1.9%
caffeine hIgG 19.8% 7.8%
imidazole hIgG 8.7% 3.3%
tyramine hIgG 3.6% 4.4%
Control hIgG 0.0% 2.8%
Dimethylglycine hIgG 2.5% 10.0%
4-aminopyridine hIgG 29.7% 2.9%
nicotinamide/caffeine hIgG 30.0% 1.0%
nicotinamide/caffeine hIgG 33.4% 3.5%
hordenine HC1 hIgG 32.9% 5.5%
Dimethyluracil/arginine hIgG 22.2% 0.6%
Jeffamine M600 hIgG 16.5% 2.1%
Dimethyluracil hIgG 18.1% 9.4%
diethylnicotinamide hIgG 11.1% 2.8%
arginine HC1 hIgG 14.6% 6.9%
arginine/glutamic acid hIgG 17.5% 11.2%
nicotinamide hIgG 12.1% 5.8%
serine/theonine hIgG 5.8% 5.1%
isonicotinamide hIgG 8.8% 8.5%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient (100 mM) Protein % Reduction Std. dev.
(3-carboxylpropyl) trimethyl ammonium
hIgG 5.2% 12.1%
chloride
TABLE 53
Excipient (100 mM) Protein %
Reduction Std. dev.
4-(2-hydroxyethyl)-1-
omalizumab 71% 1.3%
piperazineethanesulfonic acid
0-(octylphosphoryl)choline omalizumab 38% 0.1%
Nicotinamide mononucleotide omalizumab 61% 8%
Itaconic acid omalizumab 54% 1%
3-(1-Pyridinio)-1-propanesulfonate omalizumab 4% 0.2%
N-methyl aspartic acid omalizumab 73% 0.6%
L-Ornithine omalizumab 82% 0.0%
beta-alanine omalizumab 25% 5.4%
ethylenediaminetetraacetic acid (EDTA) omalizumab 64%
3.1%
Trigonelline omalizumab 63% 1.1%
(3 -c arb oxypropyl)trim ethyl amm onium
omalizumab 64% 3.6%
chloride
Iminodiacetic acid omalizumab 51% 0.6%
aminohippuric acid omalizumab 66% 2.4%
caffeic acid omalizumab 35% 8.0%
Aspartame (50 mM) omalizumab 32% 2.0%
1-(1-Adamantyl)ethylamine hydrochloride
omalizumab 32% 4.0%
(100 mg/mL)
naphthalenedisulfonic acid (100 mg/mL) omalizumab 9%
8.0%
x-xylylenediamine omalizumab 78% 2.4%
1,3-diaminopropane omalizumab 78% 1.5%
isonicotinic acid omalizumab 45% 17.2%
Lysine omalizumab 54% 3.4%
4-aminopyridine omalizumab 41% 2.8%
Imidazole omalizumab 62% 1.8%
Adenosine monophosphate omalizumab 73% 2.7%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient (100 mM) Protein % Reduction Std. dev.
Dicyclomine (100 mg/mL) omalizumab 43% 2.4%
2-Imidazolidone omalizumab 10% 3.0%
pyridoxine HCl omalizumab 63% 11.0%
2-ethylimidazole omalizumab 51% 1.7%
triethanolamine omalizumab 69% 2.9%
Ethanolamine omalizumab 49% 5.1%
Benzylamine omalizumab 81% 0.6%
1-Butylimidazole omalizumab 60% 4.6%
diphenhydramine HCl omalizumab 77% 0.2%
procaine HCl omalizumab 71% 8.1%
2-dimethylaminoethanol omalizumab 72% 6.7%
Acesulfame K omalizumab 74% 3.3%
sodium ascorbate omalizumab 55% 0.6%
glutamic acid omalizumab 49% 5.0%
Etidronate omalizumab 72% 0.0%
salicylic acid omalizumab 72% 1.1%
quinic acid omalizumab 58% 7.8%
hydroxybenzoic acid omalizumab 68% 5.1%
glucuronic acid omalizumab 60% 6.0%
Lactobionic acid omalizumab 46% 0.1%
sodium hexametaphosphate omalizumab 75% 0.5%
sodium bisulfite omalizumab 71% 6.5%
sodium benzoate omalizumab 60% 12.8%
Calcium propionate omalizumab 75% 3.5%
Sodium propionate omalizumab 56% -
2-dimethylaminoethanol omalizumab 65% 10.6%
2-methyl-2-imidazoline omalizumab 49% 12.6%
cycloserine omalizumab 33% 1.2%
3-aminopyridine omalizumab 59% 4.0%
4-aminopyridine omalizumab 73% 2.3%
agmatine sulfate omalizumab 85% 1.8%
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
Excipient (100 mM) Protein
% Reduction Std. dev.
cytidine omalizumab 68%
1.6%
diphenhydramine omalizumab 81%
0.1%
ethanolamine omalizumab 93%
1.6%
meglumine omalizumab 91%
0.2%
morpholine omalizumab 78%
2.7%
Example 53: Viscosity measurements by viscometer
[00197] Excipient solutions for those excipients listed in Tables 54 and 55
were prepared at
0.1 M or 0.075 M in His HC1 buffer and pH adjusted using concentrated sodium
hydroxide or
concentrated hydrochloric acid. Omalizumab and human IgG were buffer exchanged
into
each excipient formulation using Amicon-15 centrifugal devices (30 kDa MWCO).
After
buffer exchange, the protein solution was concentrated up to 150 mg/mL for
omalizumab and
250 mg/mL for human IgG based on retentate mass in the centrifugal device.
Control
formulations were prepared in identical manner except in the absence of the
excipient.
Viscosity measurements were performed on a RheoSense micro-viscometer using an
A05
chip enclosed in a temperature-controlled enclosure set to 25 C. The shear
rate was set to 250
s1. The viscosity of each formulation was measured 3 times and then diluted by
adding 20 !IL
of the respective buffer and viscosity was measured again. This was repeated 5-
6 times each
to generate viscosity data for 5-6 different protein concentrations. Protein
concentration was
measured by absorbance at 280 nm using an Agilent 1100 series high pressure
liquid
chromatography instrument paired with a size exclusion column (TOSOH TSKgel
SuperSW3000). A scatter plot was generated by plotting viscosity as a function
of
concentration for each excipient formulation. An exponential trendline was
fitted to each
formulation and the viscosity at a concentration was calculated based on the
exponential fit
with the equation y = a*e(b*x), where y is viscosity in cP units, x is
concentration of protein in
mg/mL, a and b are fitting parameters for the equation, and R2 is the
statistical coefficient of
determination. For this example, the viscosity is reported as a function of a
fixed
concentration and results are given in Tables 54 and 55 below.
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
TABLE 54
Exponential Equation Calculations
Excipient Protein Buffer
Calc. Viscosity
a b R2
@ 250 mg/mL
Caffeine human IgG His HC1 pH 5.5 0.469 0.0187 0.9194
50.3
nicotinamide human IgG His HC1 pH 5.5 0.6136 0.0198 0.9329
86.6
hordenine HC1 human IgG His HC1 pH 5.5 0.4116 0.0197 0.9535
56.7
1,3-dimethyluracil human IgG His HC1 pH 5.5 0.0586 0.0282 0.9929
67.6
control human IgG His HC1 pH 5.5 0.2059 0.0248 0.9995
101.5
TABLE 55
Exponential Equation
Excipient Protein Buffer
Calc. Viscosity
a b R2
@120 mg/mL
Sulfanilic Acid omalizumab His HC1 pH 6.0 1.2194 0.0185 0.8218
11.2
Nicotinic acid omalizumab His HC1 pH 6.0 1.0171 0.0244 0.8041
19.0
Ornithine omalizumab His HC1 pH 6.0 0.1068 0.0419 0.6874
16.3
control omalizumab His HC1 pH 6.0 0.8156 0.0319 0.9763
37.5
1,3
omalizumab His HC1 pH 6.0 0.1498 0.0274 0.9835
4.0
diaminopropane
Example 54: BLI measurement of self-interaction
[00198] In this example, biolayer interferometry (BLI) tests were done with a
ForteBio Octet
Red-96 instrument. Amine reactive second-generation (AR2G) biosensors
(Molecular
Devices, CA) were conjugated with omalizumab to detect protein self-
interaction in the
presence of excipients. Excipient solutions for the excipients listed in Table
56 were prepared
1() at 0.1 M in His HC1 buffer. 20 mM sodium phosphate, pH 6.4 buffer was
prepared by
dissolving 1.679 g of dibasic sodium phosphate, heptahydrate (Sigma, St.
Louis) and 1.895 g
of monobasic sodium phosphate, monohydrate (Sigma, St. Louis) in DI water and
adjusting
the volume to 1 L. Research-grade omalizumab, purchased from Bioceros
(Utrecht, The
Netherlands) was buffer exchanged using Amicon-15 centrifugal (30 kDa MWCO)
devices
into phosphate buffer at pH 6.4. This omalizumab stock solution at 15 mg/mL in
20 mM
sodium phosphate, pH 6.4 buffer was further buffer exchanged using Sephadex G-
25 PD-10
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
desalting columns (GE Healthcare Life Sciences) and eluted with the 20 mM
sodium
phosphate, pH 6.4 buffers containing the prepared excipient at 0.1 M. The
control was
similarly prepared by using Sephadex G-25 PD-10 desalting columns (GE
Healthcare Life
Sciences) and eluted with the 20 mM sodium phosphate, pH 6.4 buffer. Protein
concentration
was measured using UV clear 96 half-well microplate (Greiner Bio-One,
Austria), and
absorbance measured at a wavelength of 280 nm with a Synergy HT plate reader
(BioTek,
Winooski, VT). Protein concentration was adjusted to 5 mg/mL by diluting in
the prepared
excipient buffers. In a black bottom 96-well microplate (Greiner Bio-One,
Austria), 250 !IL
of each excipient solution at 0.1 M in 20 mM sodium phosphate, pH 6.4 buffer
was
transferred to column B and 250 !IL of the 5 mg/ml omalizumab solution
containing 0.1 M
excipients was transferred to column C. The 96-well plate was set up so the
columns
represented individual formulations and rows distinguished protein-containing
formulations.
The tray was then transferred into a ForteBio Octet Red-96 for analysis. The
omalizumab
conjugated biosensors were dipped into the formulations containing no protein
for 120
.. seconds to generate a baseline. Biosensors were then removed and dipped
into formulations
containing protein for 300 seconds. In this example, we reported the delta in
a percent of
binding signal at 300 seconds compared to the binding signal of the control,
and the results
are summarized in Table 56 below.
TABLE 56
Excipient Binding (nm) at 300 s % change
control 7 0%
ornithine 2.5 64%
iminodiacetic acid 1.25 82%
nicotinic acid 0.5 93%
sulfanilic acid 0.3 96%
EQUIVALENTS
[00199] While specific embodiments of the subject invention have been
disclosed herein, the
above specification is illustrative and not restrictive. While this invention
has been
particularly shown and described with references to preferred embodiments
thereof, it will be
understood by those skilled in the art that various changes in form and
details may be made
therein without departing from the scope of the invention encompassed by the
appended
claims. Many variations of the invention will become apparent to those of
skilled art upon
CA 03120023 2021-05-13
WO 2020/112855
PCT/US2019/063374
review of this specification. Unless otherwise indicated, all numbers
expressing reaction
conditions, quantities of ingredients, and so forth, as used in this
specification and the claims
are to be understood as being modified in all instances by the term "about."
Accordingly,
unless indicated to the contrary, the numerical parameters set forth herein
are approximations
that can vary depending upon the desired properties sought to be obtained by
the present
invention.