Note: Descriptions are shown in the official language in which they were submitted.
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
THERAPEUTIC METHODS AND COMPOSITIONS FOR TREATING CANCER
USING 6,8-BIS-BENZYLTHIO-OCTANOIC ACID AND AN AUTOPHAGY
INHIBITOR
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of and priority to United States
Provisional Patent
Application serial number 62/782,928, filed December 20, 2018; United States
Provisional
Patent Application serial number 62/793,667, filed January 17, 2019; United
States Provisional
Patent Application serial number 62/834,475, filed April 16, 2019; and United
States
Provisional Patent Application serial number 62/854,599, filed May 30, 2019;
the contents of
each of which are hereby incorporated by reference.
FIELD OF THE INVENTION
[0002] The invention provides methods and compositions for treating cancer
by
administration of 6,8-bis-benzylthio-octanoic acid and an autophagy inhibitor.
The invention
further provides methods and compositions for treating a cancer other than
clear cell sarcoma
by administration of 6,8-bis-benzylthio-octanoic acid and an autophagy
inhibitor.
BACKGROUND
[0003] CPI-613 (6,8-bis-benzylthio-octanoic acid) is a first-in-class
investigational small-
molecule (lipoate analog), which targets the altered energy metabolism that is
common to many
cancer cells. CPI-613 has been evaluated in multiple phase I, I/II, and II
clinical studies, and
has been granted orphan drug designation for the treatment of pancreatic
cancer, acute myeloid
leukemia (AML), peripheral T-cell lymphoma (PTCL), Burkitt lymphoma and
myelodysplastic
syndromes (MDS).
[0004] A need exists to improve the safety and efficacy of treating cancer
with CPI-613.
The present invention addresses this need and provides other related
advantages.
SUMMARY
[0005] The invention provides methods and compositions for treating cancer
in a human
patient in need thereof by administering to the patient a therapeutically
effective amount of 6,8-
bis-benzylthio-octanoic acid and an autophagy inhibitor. The cancer may be
relapsed or
refractory. The cancer may be a lymphoma, leukemia, carcinoma, sarcoma,
melanoma,
myeloma, brain or spinal cord cancer, blastoma, germ cell tumor, cancer of the
pancreas,
colorectal cancer, myelodysplastic syndrome, or cancer of the prostate. In
certain embodiments,
1
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
the cancer is a lymphoma, leukemia, carcinoma, sarcoma, melanoma, or myeloma.
In
certain embodiments, the cancer is relapsed or refractory Hodgkin lymphoma,
including
relapsed or refractory Hodgkin lymphoma in a patient who has failed
brentthximab vedotin and
a PD-1 inhibitor, relapsed or refractory T-cell non-Hodgkin lymphoma, relapsed
or refractory
Burkitt's lymphoma, or high-grade B-cell lymphoma with rearrangements ofMYC
and BCL2
and/or BCL6.
[0006] The invention further provides methods and compositions for treating
a cancer other
than clear cell sarcoma in a human patient in need thereof by administering to
the patient a
therapeutically effective amount of 6,8-bis-benzylthio-octanoic acid and an
autophagy
inhibitor. The cancer may be relapsed or refractory. The cancer may be a
lymphoma,
leukemia, carcinoma, non-clear cell sarcoma, melanoma, myeloma, brain or
spinal cord
cancer, blastoma, germ cell tumor, cancer of the pancreas, colorectal cancer,
myelodysplastic
syndrome, or cancer of the prostate. In certain embodiments, the cancer is a
lymphoma,
leukemia, carcinoma, non-clear cell sarcoma, melanoma, or myeloma.
[0007] The foregoing aspects of the invention are described in more detail,
along with
additional embodiments, in the detailed description below.
BRIEF DESCRIPTION OF THE DRAWINGS
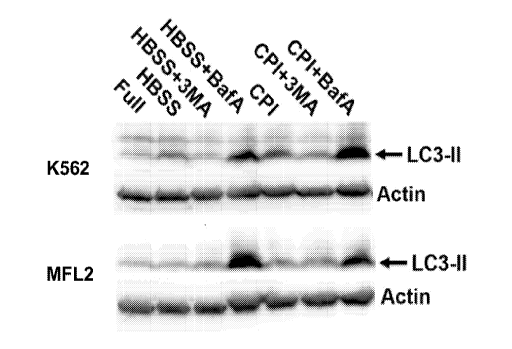
[0008] Fig. 1 depicts induction of autophagy by 6,8-bis-benzylthio-octanoic
acid in K562
and MFL2 AML cells. HBSS=Hanks's balanced salt solution.
[0009] Fig. 2 depicts the in vitro treatment of K562 and OCI-AML3 cells
with a
combination of chloroquine and CPI-613. All concentrations listed are p.M.
[0010] Fig. 3 depicts the treatment of MFL2 syngeneic tumors in C57B1/6
mice with a
combination of CPI-613 and chloroquine.
[0011] Fig. 4 depicts the in vitro treatment of MFL2 cells with a
combination of metformin
and CPI-613. All concentrations listed are p.M. Met=Metformin, CPI=CPI-613
[0012] Fig. 5 depicts the treatment of MFL2 syngeneic tumors in C57B1/6
mice with a
combination of metformin and CPI-613 (Met+CPI).
[0013] Fig. 6 depicts the in vitro treatment of MFL2 cells with a
combination of 2-
deoxyglucose and CPI-613. CPI=CPI-613 (04), 2DG=2-deoxyglucose (mM).
[0014] Fig. 7 depicts the in vitro treatment of OCI-AML3 cells and MFL2
cells with a
combination of chloroquine, 2-deoxyglucose, and CPI-613.
[0015] Fig. 8 depicts the in vitro treatment of PANC-1 cells with
chloroquine,
hydroxychloroquine, or CPI-613 alone or in combination.
2
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0016] Fig. 9 depicts the in vitro treatment of AsPC-1 cells with
chloroquine,
hydroxychloroquine, or CPI-613 alone or in combination.
[0017] Fig. 10 depicts the in vitro treatment of BxPC-3 cells with
chloroquine,
hydroxychloroquine, or CPI-613 alone or in combination.
[0018] Fig. 11 depicts the in vitro treatment of MIA PaCa-2 cells with a
chloroquine or CPI-
613 alone or in combination.
[0019] Fig. 12 depicts the in vitro treatment of CoLo 205 and LoVo cells
with a
combination of chloroquine and CPI-613.
[0020] Fig. 13 depicts the in vitro treatment of SW620 and HT-29 cells with
a combination
of chloroquine and CPI-613.
[0021] Fig. 14 depicts the in vitro treatment of H460 cells with a
combination of
chloroquine and CPI-613.
[0022] Fig. 15 depicts the anti-tumor efficacy of oral 6,8-bis-benzylthio-
octanoic acid in
human non-small cell lung cancer xenografts in mice.
[0023] Fig. 16 depicts the anti-tumor efficacy of oral 6,8-bis-benzylthio-
octanoic acid in
human pancreatic cancer xenografts in mice.
[0024] Fig. 17 presents X-ray powder diffraction patterns of solid
amorphous dispersion
formulations of 6,8-bis-benzylthio-octanoic acid with either Eudragit L100 or
hydroxypropyl
methylcellulose acetate succinate (HPMCAS-M) (top and middle diffraction
patterns,
respectively), and crystalline 6,8-bis-benzylthio-octanoic acid (bottom
diffraction pattern).
DETAILED DESCRIPTION
[0025] The invention provides methods and compositions for treating cancer
in a human
patient in need thereof by administering to the patient a therapeutically
effective amount of 6,8-
bis-benzylthio-octanoic acid and an autophagy inhibitor. The invention further
provides
methods and compositions for treating a cancer other than clear cell sarcoma
in a human patient
in need thereof by administering to the patient a therapeutically effective
amount of 6,8-bis-
benzylthio-octanoic acid and an autophagy inhibitor.
[0026] The practice of the present invention employs, unless otherwise
indicated,
conventional techniques of medicinal chemistry, pharmacology, and
biochemistry. Various
aspects of the invention are set forth below in sections; however, aspects of
the invention
described in one particular section are not to be limited to any particular
section.
3
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
I. DEFINITIONS
[0027] To facilitate an understanding of the present invention, a number of
terms and
phrases are defined below.
[0028] The terms "a," "an" and "the" as used herein mean "one or more" and
include the
plural unless the context is inappropriate
[0029] The term "6,8-bis-benzylthio-octanoic acid" refers to the compound
known as
0
OH
S S
devimistat or CPI-613, having the chemical structure 401 lel .
[0030] Certain compounds contained in compositions of the present invention
may exist in
particular geometric or stereoisomeric forms. The present invention
contemplates all such
compounds, including cis- and trans-isomers, R- and S-enantiomers,
diastereomers, (D)-
isomers, (0-isomers, the racemic mixtures thereof, and other mixtures thereof,
as falling within
the scope of the invention.
[0031] As used herein, the term "patient" refers to a human being in need
of cancer
treatment.
[0032] As used herein, the term "treating" includes any effect, e.g.,
lessening, reducing,
modulating, ameliorating or eliminating, that results in the improvement,
stabilization, or
slowing progression of a condition, disease, disorder, or the like, or a
symptom thereof For
example, treatment can include diminishment of a symptom of a disorder or
complete
eradication of a disorder. As another example, treatment can include slowing
the progression of
a disease, or preventing or delaying its recurrence, such as maintenance
treatment to prevent or
delay relapse.
[0033] "Therapeutically effective amount" refers to an amount of a compound
sufficient to
inhibit, halt, or cause an improvement in a disorder or condition being
treated in a particular
patient or patient population. For example, a therapeutically effective amount
can be an
amount of drug sufficient to slow the progression of a disease, or to prevent
or delay its
recurrence, such as maintenance treatment to prevent or delay relapse. A
therapeutically
effective amount can be determined experimentally in a laboratory or clinical
setting, or may be
the amount required by the guidelines of the United States Food and Drug
Administration, or
equivalent foreign agency, for the particular disease and patient being
treated. It should be
4
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
appreciated that determination of proper dosage forms, dosage amounts, and
routes of
administration is within the level of ordinary skill in the pharmaceutical and
medical arts.
[0034] As used herein, the term "pharmaceutical composition" refers to the
combination of
an active agent with an excipient, inert or active, making the composition
suitable for
administration to a human.
[0035] The phrase "pharmaceutically acceptable" is employed herein to refer
to those
compounds, materials, compositions, and/or dosage forms which are, within the
scope of sound
judgment, suitable for use in contact with the tissues of human beings with
acceptable toxicity,
irritation, allergic response, and other problems or complications
commensurate with a
reasonable benefit/risk ratio.
[0036] As used herein, the term "pharmaceutically acceptable excipient"
refers to any of the
standard pharmaceutical excipients suitable for use in humans. For examples of
excipients, see
e.g., Martin, Remington's Pharmaceutical Sciences, 15th Ed., Mack Publ. Co.,
Easton, PA
[1975].
[0037] As used herein, the term "pharmaceutically acceptable salt" refers
to any salt (e.g.,
acid or base) of a compound of the present invention which is suitable for
administration to a
human. As is known to those of skill in the art, "salts" of the compounds of
the present
invention may be derived from inorganic or organic acids and bases. Examples
of acids
include, but are not limited to, hydrochloric, hydrobromic, sulfuric, nitric,
perchloric, fumaric,
maleic, phosphoric, glycolic, lactic, salicylic, succinic, toluene-p-sulfonic,
tartaric, acetic,
citric, methanesulfonic, ethanesulfonic, formic, benzoic, malonic, naphthalene-
2-sulfonic,
benzenesulfonic acid, and the like. Examples of bases include, but are not
limited to, alkali
metals (e.g., sodium) hydroxides, alkaline earth metals (e.g., magnesium),
hydroxides,
ammonia, and compounds of formula NW3, wherein W is C1-4 alkyl, and the like.
[0038] Further examples of salts include salts made using the ion pairing
agents described in
U.S. Patent No. 8,263,653, the entire disclosure of which is incorporated by
reference herein.
Still further ion pairing agents can be selected with guidance from Handbook
of Pharmaceutical
Salts Properties, Selection and Use, UIPAC, Wiley-VCH, P.H. Stahl, ed., the
entire disclosure
of which is incorporated by reference herein.
[0039] For therapeutic use, salts of the compounds of the present invention
are contemplated
as being pharmaceutically acceptable. However, salts of acids and bases that
are non-
pharmaceutically acceptable may also find use, for example, in the preparation
or purification
of a pharmaceutically acceptable compound.
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
[0040] Throughout the description, where compositions are described as
having, including,
or comprising specific components, or where processes and methods are
described as having,
including, or comprising specific steps, it is contemplated that,
additionally, there are
compositions of the present invention that consist essentially of, or consist
of, the recited
components, and that there are processes and methods according to the present
invention that
consist essentially of, or consist of, the recited steps.
[0041] As a general matter, compositions specifying a percentage are by
weight unless
otherwise specified. Further, if a variable is not accompanied by a
definition, then the previous
definition of the variable controls.
THERAPEUTIC APPLICATIONS
[0042] The invention provides methods and compositions for treating cancer
in a human
patient in need thereof, comprising the step of administering to the patient a
therapeutically
effective amount of 6,8-bis-benzylthio-octanoic acid and an autophagy
inhibitor. The invention
further provides methods and compositions for treating a cancer other than
clear cell sarcoma in
a human patient in need thereof, comprising the step of administering to the
patient a
therapeutically effective amount of 6,8-bis-benzylthio-octanoic acid and an
autophagy
inhibitor.
[0043] The inventors have discovered that when exposed to CPI-613, cancer
cells display
increased autophagy. Without wishing to be bound by theory, the inventors
hypothesize that
when CPI-613 interferes with the altered metabolic pathways of cancer cells,
the cells begin to
starve and respond by using autophagy to help supply their metabolic needs.
Including an
autophagy inhibitor in the treatment regimen inhibits the cancer cells from
using autophagy to
counter the effect of CPI-613, significantly improving efficacy.
Type of Cancer
[0044] In certain embodiments, the cancer is associated with altered energy
metabolism. In
certain embodiments, the cancer displays increased autophagy when contacted
with CPI-613.
As used herein, the term "cancer" is intended to include myelodysplastic
syndromes, and in
certain embodiments of the present invention the cancer is a myelodysplastic
syndrome. In
certain embodiments, the cancer is high risk myelodysplastic syndrome (MDS).
In certain
embodiments, the cancer is high risk MDS in patients who have failed to
respond, progressed,
or relapsed while on hypomethylating therapy.
[0045] The method may be further characterized according to the severity or
type of cancer.
In certain embodiments, the cancer is Stage I or early stage cancer, in which
the cancer is small
6
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
and only in one area. In certain embodiments, the cancer is Stage II or III,
in which the cancer
is larger and has grown into nearby tissues or lymph nodes. In certain
embodiments, the cancer
Stage IV or advanced or metastatic, in which the cancer has spread to other
parts of the body.
[0046] In certain embodiments, the cancer is Stage I lymphoma, in which the
cancer is
found in one lymph node region or the cancer has invaded one extralymphatic
organ or site but
not any lymph node regions. In certain embodiments, the cancer is Stage II
lymphoma, in
which the cancer is found in two or more lymph node regions on the same side
of the
diaphragm or the cancer involves one organ and its regional lymph nodes, with
or without
cancer in other lymph node regions on the same side of the diaphragm. In
certain embodiments,
the cancer is Stage III lymphoma, in which there is cancer in lymph nodes on
both sides of the
diaphragm. In certain embodiments, the cancer is Stage IV lymphoma, in which
the cancer has
spread one or more organs beyond the lymph nodes.
[0047] In certain embodiments, the cancer is progressive or refractory. In
certain
embodiments, the cancer is a metastatic. In certain embodiments, the cancer is
recurrent or
relapsed. In certain embodiments, the cancer is relapsed or refractory. In
certain embodiments,
the cancer is previously untreated. In certain embodiments, the cancer is
previously untreated
with systemic therapies. In certain embodiments, the cancer is previously
untreated with
systemic therapies or local treatment with chemoradiation. In certain
embodiments, the patient
has not received hematopoietic cell transplant. In certain embodiments, the
patient has received
hematopoietic cell transplant.
[0048] In certain embodiments, the cancer is a lymphoma. In certain
embodiments, the
cancer is a T-cell lymphoma. In certain embodiments, the cancer is a B-cell
lymphoma. In
certain embodiments, the cancer is mantle cell lymphoma. In certain
embodiments, the
cancer is a leukemia. In certain embodiments, the cancer is an acute myeloid
leukemia. In
certain embodiments, the cancer is chronic myeloid leukemia. In certain
embodiments, the
cancer is acute lymphoblastic leukemia. In certain embodiments, the cancer is
a carcinoma. In
certain embodiments, the cancer is a sarcoma. In certain embodiments, the
cancer is not a
sarcoma. In certain embodiments, the cancer is a myeloma. In certain
embodiments, the cancer
is a clear cell cancer. In certain embodiments, the cancer is not a clear cell
cancer. In certain
embodiments, the cancer is a clear cell sarcoma. In certain embodiments, the
cancer is not a
clear cell sarcoma. In certain embodiments, the cancer is a non-clear cell
sarcoma. In certain
embodiments, the cancer is a clear cell carcinoma. In certain embodiments, the
cancer is not a
soft tissue cancer. In certain embodiments, the cancer is not a clear cell
carcinoma. In certain
embodiments, the cancer is a brain or spinal cord cancer. In certain
embodiments, the cancer is
7
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
a melanoma. In certain embodiments, the cancer is a blastoma. In certain
embodiments, the
cancer is a germ cell tumor. In certain embodiments, the cancer is a cancer of
the pancreas. In
certain embodiments, the cancer is a metastatic pancreatic cancer. In certain
embodiments, the
cancer is a locally advanced pancreatic cancer. In certain embodiments, the
cancer is a
histologically or cytologically documented and measurable locally advanced
pancreatic
adenocarcinoma. In certain embodiments, the cancer is a histologically or
cytologically
documented and measurable metastatic pancreatic adenocarcinoma. In certain
embodiments,
the cancer is a histologically or cytologically documented and measurable
locally advanced
pancreatic adenocarcinoma that is previously untreated. In certain
embodiments, the cancer is
a histologically or cytologically documented and measurable metastatic
pancreatic
adenocarcinoma that is previously untreated. In certain embodiments, the
cancer is a
histologically or cytologically documented and measurable locally advanced
pancreatic
adenocarcinoma that is previously untreated with systemic therapies. In
certain embodiments,
the cancer is a histologically or cytologically documented and measurable
metastatic pancreatic
adenocarcinoma that is previously untreated with systemic therapies. In
certain embodiments,
the cancer is a histologically or cytologically documented and measurable
locally advanced
pancreatic adenocarcinoma that is previously untreated with systemic therapies
or local
treatment with chemoradiation. In certain embodiments, the cancer is a
histologically or
cytologically documented and measurable metastatic pancreatic adenocarcinoma
that is
previously untreated with systemic therapies or local treatment with
chemoradiation. In certain
embodiments, the cancer is a locally advanced pancreatic adenocarcinoma. In
certain
embodiments, the cancer is a metastatic pancreatic adenocarcinoma. In certain
embodiments,
the cancer is a locally advanced pancreatic adenocarcinoma that is previously
untreated. In
certain embodiments, the cancer is a metastatic pancreatic adenocarcinoma that
is previously
untreated. In certain embodiments, the cancer is a locally advanced pancreatic
adenocarcinoma
that is previously untreated with systemic therapies. In certain embodiments,
the cancer is a
metastatic pancreatic adenocarcinoma that is previously untreated with
systemic therapies. In
certain embodiments, the cancer is a locally advanced pancreatic
adenocarcinoma that is
previously untreated with systemic therapies or local treatment with
chemoradiation. In certain
embodiments, the cancer is a pancreatic adenocarcinoma that is previously
untreated with
systemic therapies or local treatment with chemoradiation.
[0049] In certain embodiments, the cancer is a cancer of the prostate. In
certain
embodiments, the cancer is a castration resistant prostate cancer. In certain
embodiments, the
cancer is a cancer of the lung. In certain embodiment, the cancer is non-small
cell lung cancer.
8
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
In certain embodiments, the cancer is a cancer of the colon. In certain
embodiments, the cancer
is a cancer of the rectum. In certain embodiments, the cancer is a colorectal
cancer. In certain
embodiments, the cancer is a cancer of the cervix. In certain embodiments, the
cancer is a
neuroendocrine tumor. In certain embodiments, the cancer is a
gastroenteropancreatic
neuroendocrine tumor. In certain embodiments, the cancer is a cancer of the
liver. In certain
embodiments, the cancer is a cancer of the uterus. In certain embodiments, the
cancer is a
cancer of the cervix. In certain embodiments, the cancer is a cancer of the
bladder. In certain
embodiments, the cancer is a cancer of the kidney. In certain embodiments, the
cancer is a
cancer of the breast. In certain embodiments, the cancer is a cancer of the
ovary.
[0050] In certain embodiments, the cancer is Burkitt's Lymphoma. In certain
embodiments,
the cancer is relapsed or refractory Burkitt's Lymphoma. In certain
embodiments, the cancer is
relapsed or refractory Burkitt's Lymphoma in which the patient has failed at
least one previous
line of therapy. In certain embodiments, the cancer is relapsed or refractory
Burkitt's
Lymphoma in which the patient has failed prior bone marrow transplant. In
certain
embodiments, the cancer is double hit diffuse large B cell lymphoma. In
certain embodiments,
the cancer is high-grade B cell lymphoma with rearrangements ofMYC and BCL2
and/or BCL6
(DHL/THL). In certain embodiments, the cancer is Hodgkin lymphoma. In certain
embodiments, the cancer is non-Hodgkin lymphoma. In certain embodiments, the
cancer is T-
cell non-Hodgkin lymphoma. In certain embodiments, the cancer is relapsed or
refractory
Hodgkin lymphoma. In certain embodiments, the cancer is relapsed or refractory
non-Hodgkin
lymphoma. In certain embodiments, the cancer is relapsed or refractory T-cell
non-Hodgkin
lymphoma. In certain embodiments, the cancer is Hodgkin lymphoma in which the
patient has
not received hematopoietic cell transplant. In certain embodiments, the cancer
is Hodgkin
lymphoma in which the patient has received hematopoietic cell transplant. In
certain
embodiments, the cancer is non-Hodgkin lymphoma in which the patient has not
received
hematopoietic cell transplant. In certain embodiments, the cancer is non-
Hodgkin lymphoma in
which the patient has received hematopoietic cell transplant. In certain
embodiments, the
cancer is T-cell non-Hodgkin lymphoma in which the patient has not received
hematopoietic
cell transplant. In certain embodiments, the cancer is T-cell non-Hodgkin
lymphoma in which
the patient has received hematopoietic cell transplant. In certain
embodiments, the cancer is
relapsed or refractory Hodgkin lymphoma in which the patient has not received
hematopoietic
cell transplant. In certain embodiments, the cancer is relapsed or refractory
Hodgkin lymphoma
in which the patient has received hematopoietic cell transplant. In certain
embodiments, the
cancer is relapsed or refractory non-Hodgkin lymphoma in which the patient has
not received
9
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
hematopoietic cell transplant. In certain embodiments, the cancer is relapsed
or refractory
Hodgkin lymphoma in which the patient has or has not received hematopoietic
cell transplant.
In certain embodiments, the cancer is relapsed or refractory Hodgkin lymphoma
in which the
patient has failed brentuximab vedotin and a PD-1 inhibitor. In certain
embodiments, the cancer
is relapsed or refractory Hodgkin lymphoma in which the patient has failed
brentuximab
vedotin and a PD-1 inhibitor and has received hematopoietic cell transplant.
In certain
embodiments, the cancer is relapsed or refractory Hodgkin lymphoma in which
the patient has
failed brentuximab vedotin and a PD-1 inhibitor and has not received
hematopoietic cell
transplant. In certain embodiments, the cancer is relapsed or refractory non-
Hodgkin lymphoma
in which the patient has received hematopoietic cell transplant. In certain
embodiments, the
cancer is relapsed or refractory T-cell non-Hodgkin lymphoma in which the
patient has not
received hematopoietic cell transplant. In certain embodiments, the cancer is
relapsed or
refractory T-cell non-Hodgkin lymphoma in which the patient has received
hematopoietic cell
transplant. In certain embodiments, the cancer is relapsed or refractory T-
cell non-Hodgkin
lymphoma in which the patient has or has not received hematopoietic cell
transplant.
General Aspects of Administering a Therapeutic Agent to a Patient
[0051] Generally, the therapeutic agent ¨ i.e., 6,8-bis-benzylthio-octanoic
acid and
autophagy inhibitor ¨ is delivered to the patient in a therapeutically
effective amount, sufficient
to treat cancer. The treatment may involve one or several administrations on
one or more days,
and the dosage may be adjusted by the individual practitioner to achieve a
desired effect.
Preferably, the dosage amount of each agent should be sufficient to interact
primarily with
disease cells, leaving normal cells comparatively unharmed.
[0052] The dosage amount may be administered in a single dose or in the
form of individual
divided doses, such as one, two, three, or four times per day. In certain
embodiments, the daily
dosage amount is administered in a single dose. In the event that the response
in a patient is
insufficient at a certain dose, higher or more frequent doses may be employed
to the extent of
patient tolerance.
[0053] For the present combination therapy, each agent may be administered
in a particular
order and/or on the same or different days according to a treatment cycle. For
example, a dose
of 6,8-bis-benzylthio-octanoic acid may be administered to the patient prior
to administering an
autophagy inhibitor, such as immediately prior, earlier in the day, or on an
earlier day in a
treatment cycle. In certain embodiments, the active agents may be administered
on the same
day of a treatment cycle, for example being co-administered simultaneously or
one right after
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
the other. In certain embodiments, a dose of an autophagy inhibitor is
administered to the
patient prior to administering the 6,8-bis-benzylthio-octanoic acid, such as
immediately prior,
earlier in the day, or on an earlier day in a treatment cycle. In certain
embodiments, treatment
cycles may be repeated one or more times in order to maximize benefit to the
patient.
6,8-Bis-Benzylthio-Octanoic Acid
[0054] The 6,8-bis-benzylthio-octanoic acid may be administered in any
suitable form,
including as a solid or liquid, a free acid or salt. The 6,8-bis-benzylthio-
octanoic acid may be
crystalline, amorphous, or dissolved in solution. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered to the patient as a salt or ion pair.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered to the
patient as a salt or ion
pair with triethanolamine. Exemplary ion pairing agents that may be used
include, for
example, a tertiary amine (such as triethylamine or triethanolamine), other
amines such as
diethylamine, diethanolamine, monoethanolamine, mefenamic acid and
tromethamine, and
combinations thereof In certain embodiments, the ion pairing agent is an
organic Bronsted
base. In certain other embodiments, the ion pairing agent is an amine
compound. In yet other
embodiments, the ion pairing agent is a monoalkylamine, dialkylamine,
trialkylamine, amino-
substituted aliphatic alcohol, hydroxymonoalkylamine, hydroxydialkylamine,
hydroxytrialkylamine, amino-substituted heteroaliphatic alcohol, alkyldiamine,
substituted
alkyldiamine, or optionally substituted heteroaryl group containing at least
one ring nitrogen
atom. In certain embodiments, the therapeutic agent is a salt of 6,8-bis-
benzylthio-octanoic
acid with an ion pairing agent selected with guidance from Berge et al.,
"Pharmaceutical Salts,"
I of Pharmaceutical Science, 1977; 66:1-19 or Handbook of Pharmaceutical Salts
Properties,
Selection and Use, IUPAC, Wiley-VCH, P. H. Stahl, ed., the entire disclosures
of which are
incorporated by reference herein. Ion pairing agents of particular note in the
latter include,
without limitation, those listed in Table 5, p. 342.
[0055] Additional exemplary ion pairing agents include, for example,
polyethyleneimine,
polyglutamic acid, ammonia, L-arginine, benethamine benzathine, betaine,
calcium hydroxide,
choline, deanol, diethanolamine(2,2'-iminobis(ethanol)), diethylamine, 2-
(diethylamino)-
ethanol, ethanolamine, ethylenediamine, N-methyl-glucamine, hydrabamine, 1H-
imidazole,
lysine, magnesium hydroxide, 4-(2-hydroxyethyl)-morpholine, piperazine,
potassium
hydroxide, 1-(2-hydroxyethyl)-pyrrolidine, sodium hydroxide, triethanolamine
(2,2' ,2'
tromethamine, and zinc hydroxide. In certain other embodiments, the ion
pairing agent is diisopropanolamine, 3-amino-1-propanol, meglumine,
morpholine, pyridine,
11
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
niacinamide, tris(hydroxymethyDaminomethane, 2-((2-
dimethylamino)ethoxy)ethanol, 2-
(dimethylamino)ethanol, 1-(2-hydroxyethyl)pyrrolidine, or ammonium hydroxide.
In certain
other embodiments, the ion pairing agent is an alkali metal hydroxide or
alkaline earth metal
hydroxide, such as, for example, cesium hydroxide.
100561 In certain embodiments, the 6,8-bis-benzylthio-octanoic acid has a
purity of at least
about 50% (w/w). In certain embodiments, the 6,8-bis-benzylthio-octanoic acid
has a purity of
at least about 60% (w/w). In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid has a
purity of at least about 70% (w/w). In certain embodiments, the 6,8-bis-
benzylthio-octanoic
acid has a purity of at least about 80% (w/w). In certain embodiments, the 6,8-
bis-benzylthio-
octanoic acid has a purity of at least about 90% (w/w). In certain
embodiments, the 6,8-bis-
benzylthio-octanoic acid has a purity of at least about 95% (w/w). In certain
embodiments, the
6,8-bis-benzylthio-octanoic acid has a purity of at least about 96% (w/w). In
certain
embodiments, the 6,8-bis-benzylthio-octanoic acid has a purity of at least
about 97% (w/w). In
certain embodiments, the 6,8-bis-benzylthio-octanoic acid has a purity of at
least about 98%
(w/w). In certain embodiments, the 6,8-bis-benzylthio-octanoic acid has a
purity of at least
about 99% (w/w).
Autophagy Inhibitor
[0057] The one or more autophagy inhibitors may be administered in any
suitable form,
including as a solid or liquid, a free acid or salt. The autophagy inhibitor
may be crystalline,
amorphous, or dissolved in solution. In certain embodiments, the autophagy
inhibitor is
administered to the patient as a salt or ion pair. When the autophagy
inhibitor is a basic
compound, such as chloroquine or hydroxychloroquine, it may be administered as
an ion pair
with an inorganic or organic acid. Examples of acids include, but are not
limited to,
hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, maleic,
phosphoric, glycolic,
lactic, salicylic, succinic, toluene-p-sulfonic, tartaric, acetic, citric,
methanesulfonic,
ethanesulfonic, formic, benzoic, malonic, naphthalene-2-sulfonic,
benzenesulfonic acid, and the
like. In certain embodiments, the therapeutic agent is a salt of an autophagy
inhibitor with an
ion pairing agent selected with guidance from Berge et al., "Pharmaceutical
Salts," I of
Pharmaceutical Science, 1977; 66:1-19 or Handbook of Pharmaceutical Salts
Properties,
Selection and Use, IUPAC, Wiley-VCH, P. H. Stahl, ed., the entire disclosures
of which are
incorporated by reference herein. Ion pairing agents of particular note in the
latter include,
without limitation, those listed in Table 5, p. 342.
12
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0058] Any suitable autophagy inhibitor may be used. In certain
embodiments, the
autophagy inhibitor is chosen from a 4-aminoquinoline, 3-methyladenine (3-MA,
CAS#5142-
23-4), MHY1485 (CAS#326914-06-1SP600125), 3-methy1-6-(3-methylpiperidin-1-y1)-
3H-
purine, 6-Chloro-N-(1-ethylpiperidin-4-y1)-1,2,3,4-tetrahydroacridin-9-amine,
4-(((1-(2-
Fluorophenyl)cyclopenty1)-amino)methyl)-2-((4-methylpiperazin-1-
y1)methyl)phenol, 6-fluoro-
N44-fluorobenzyllquinazolin-4-amine, N-acetyl-L-cysteine, L-asparagine, N2,N4-
dibenzylquinazoline-2,4-diamine, (2S,3S)-trans-Epoxysuccinyl-L-leucylamido-3-
methylbutane
ethyl ester, N-[6-(4-chlorophenoxy)hexyll-N'-cyano-N"-4-pyridinyl-
guanidine,leupeptin, 2-(4-
Morpholiny1)-8-pheny1-1(4H)-benzopyran-4-one, 4,6-Di-4-morpholinyl-N-(4-
nitropheny1)-
1,3,5-triazin-2-amine, pepstatin A, 2-((5-Bromo-2-((3,4,5-
trimethoxyphenyl)amino)pyrimidin-
4-yl)oxy)-N-methylbenzamide, 6-Fluoro-N-R4-fluorophenyl)methy11-4-
quinazolinamine,
thapsigargin, amodiaquine, artemisinin, mefloquine, primaquine, piperaquine,
quinacrine,
U0126, 3-methyladenine, bafilomycin Al, chloroquine, hydroxychloroquine,
verteporfin,
LY294002, SB202190, SB203580, SC79, and wortmannin. In certain embodiments,
the
autophagy inhibitor is chosen from chloroquine, hydroxychloroquine, and
verteporfin. In
certain embodiments, the autophagy inhibitor is chosen from hydroxychloroquine
and
verteporfin. In certain embodiments, the autophagy inhibitor is a 4-
aminoquinoline. In certain
embodiments, the autophagy inhibitor is chloroquine. In certain embodiments,
the autophagy
inhibitor is chloroquine phosphate. In certain embodiments, the autophagy
inhibitor is
chloroquine sulfate. In certain embodiments, the autophagy inhibitor is
chloroquine
hydrochloride. In certain embodiments, the autophagy inhibitor is
hydroxychloroquine. In
certain embodiments, the autophagy inhibitor is hydroxychloroquine sulfate. In
certain
embodiments, the autophagy inhibitor is verteporfin.
[0059] The autophagy inhibitor may inhibit any suitable type of autophagy
(e.g.,
macroautophagy, microautophagy, chaperone-mediated autophagy, mitophagy, or
lipophagy),
and may do so by any suitable mechanism (e.g., by impacting formation of an
autophagosome
or its cargo). In certain embodiments, the autophagy inhibitor inhibits
macroautophagy or
mitophagy. In certain embodiments, the autophagy inhibitor inhibits
macroautophagy. In
certain embodiments, the autophagy inhibitor inhibits mitophagy. In certain
embodiments, the
mitophagy inhibitor is Mdivi-l. In certain embodiments, the mitophagy
inhibitor is
cyclosporine A. In certain embodiments, the autophagy inhibitor inhibits,
microautophagy. In
certain embodiments, the autophagy inhibitor inhibits chaperone-mediated
autophagy. In
certain embodiments, the autophagy inhibitor inhibits lipophagy.
13
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Route of Administration
[0060] The 6,8-bis-benzylthio-octanoic acid and autophagy inhibitor may be
administered to
the patient by any suitable route. For example, in certain embodiments, the
6,8-bis-benzylthio-
octanoic acid and/or autophagy inhibitor is administered orally to the
patient. In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid and autophagy inhibitor are
administered
orally to the patient. In certain embodiments, the 6,8-bis-benzylthio-octanoic
acid is
administered orally to the patient. In certain embodiments, the autophagy
inhibitor is
administered orally to the patient. In certain embodiments, the 6,8-bis-
benzylthio-octanoic acid
and/or autophagy inhibitor is administered subcutaneously to the patient. In
certain
embodiments, the 6,8-bis-benzylthio-octanoic acid and/or autophagy inhibitor
is administered
intravenously to the patient. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered as an IV infusion over two hours. In certain embodiments, the 6,8-
bis-benzylthio-
octanoic acid is administered as an IV infusion over two hours via a central
venous catheter.
[0061] An advantage of oral dosing of the 6,8-bis-benzylthio-octanoic acid
is that it permits
substantially increased dosing flexibility as compared to IV. In the prior
art, 6,8-bis-benzylthio-
octanoic acid is formulated as a 50 mg/mL solution in 1 M (150 mg/mL) aqueous
triethanolamine, which is diluted from 50 mg/mL to as low as 4 mg/mL (e.g.,
12.5 mg/mL)
with sterile 5% dextrose for injection (D5W) prior to administration as an IV
infusion over 30-
120 minutes via a central venous catheter. Such an infusion is inconvenient
for patients and
effectively precludes regimens involving frequent and/or prolonged dosing.
Since the half-life
of 6,8-bis-benzylthio-octanoic acid after IV dosing is only about 1-2 hours
(Pardee, T.S. et al.,
Clin Cancer Res. 2014, 20, 5255-64), more frequent and/or prolonged dosing
could
advantageously be used to increase the patient's exposure to the drug.
[0062] For example, a possible IV schedule for the treatment of high risk
MDS involves
administering hydroxychloroquine (600 mg to 1,200 mg) orally on days 1-5 of a
28 day cycle,
followed each day by 6,8-bis-benzylthio-octanoic acid (2,000 mg/m2) IV. If
administered
orally, the practitioner would have more flexibility with respect to the 6,8-
bis-benzylthio-
octanoic acid dose and schedule. The 6,8-bis-benzylthio-octanoic acid could be
orally
administered in a single daily dose on days 1-5 of a 28 day cycle as in the IV
schedule.
Alternatively, the 6,8-bis-benzylthio-octanoic acid could be administered in
two or more (e.g.,
three, four, or five) divided doses. The single or divided doses could be
administered on days 1-
of the cycle or on fewer and/or additional days of the cycle, up to and
including every day.
[0063] Another advantage of oral dosing is that it makes maintenance
therapy feasible. For
example, a patient who is treated successfully with first line therapy ¨ with
or without 6,8-bis-
14
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
benzylthio-octanoic acid ¨ and whose cancer is in partial or complete
remission, may be treated
orally with 6,8-bis-benzylthio-octanoic acid and an autophagy inhibitor (e.g.,
hydroxychloroquine) on a chronic basis in order to delay or prevent
recurrence. The
maintenance treatment may involve, for example, one, two, three, four, or five
doses per day of
the 6,8-bis-benzylthio-octanoic acid and autophagy inhibitor on a regular
basis, such as daily or
weekly. In certain embodiments, the maintenance therapy is for the treatment
of pancreatic
cancer.
Pharmaceutical Composition
[0064] Any suitable pharmaceutical composition may be used to administer
the 6,8-bis-
benzylthio-octanoic acid and the autophagy inhibitor to the patient. The
therapeutic agents may
be administered together in the same pharmaceutical composition (e.g., fixed
dose
combination) or separately in different pharmaceutical compositions. There is
a wide variety of
suitable formulations of pharmaceutical compositions of the present invention
(see, e.g.,
Remington: The Science and Practice of Pharmacy, 20th ed., Gennaro et al.
Eds., Lippincott
Williams and Wilkins, 2000). In certain embodiments, one or more of the
therapeutic agents is
administered in a pharmaceutical composition that is a dry oral dosage form.
In certain
embodiments, the pharmaceutical composition is an oral dosage form chosen from
tablet, pill,
capsule, caplet, powder, granule, solution, suspension, and gel. Oral dosage
forms may include
pharmaceutically acceptable excipients, such as carriers, diluents,
stabilizers, plasticizers,
binders, glidants, disintegrants, bulking agents, lubricants, plasticizers,
colorants, film formers,
flavoring agents, preservatives, dosing vehicles, and any combination of any
of the foregoing.
[0065] The pharmaceutical composition will generally include at least one
inert excipient.
Excipients include pharmaceutically compatible binding agents, lubricants,
wetting agents,
disintegrants, and the like. Tablets, pills, capsules, troches and the like
can contain any of the
following excipients, or compounds of a similar nature: a binder such as
microcrystalline
cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose,
a dispersing agent
such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium
stearate; a glidant
such as colloidal silicon dioxide; a sweetening agent such as sucrose or
saccharin; or a
flavoring agent such as peppermint, methyl salicylate, or orange flavoring.
When the dosage
unit form is a capsule, it can contain a liquid excipient such as a fatty oil.
In addition, dosage
unit forms can contain various other materials that modify the physical form
of the dosage unit,
for example, coatings of sugar, shellac, or enteric agents. Further, a syrup
may contain, in
addition to the active compounds, sucrose as a sweetening agent and certain
preservatives,
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
dyes, colorings, and flavorings. In certain embodiments, the pharmaceutical
composition
comprises an excipient in an amount of about 5% to about 99%, such as about
10% to about
85%, by weight of the composition, with the therapeutic agent comprising the
remainder. In
certain embodiments, pharmaceutically acceptable excipients comprise about 20%
to about
80% of the total weight of the composition. In certain embodiments, the
pharmaceutical
composition comprises the therapeutic agent in an amount of at least about 40%
by weight of
the composition, with one or more excipients comprising the remainder. In
certain
embodiments, the pharmaceutical composition comprises the therapeutic agent in
an amount of
at least about 50% by weight of the composition. In certain embodiments, the
pharmaceutical
composition comprises the therapeutic agent in an amount of at least about 60%
by weight of
the composition. In certain embodiments, the pharmaceutical composition
comprises the
therapeutic agent in an amount of at least about 70% by weight of the
composition. In certain
embodiments, the pharmaceutical composition comprises the therapeutic agent in
an amount of
at least about 80% by weight of the composition. In certain embodiments, the
pharmaceutical
composition comprises the therapeutic agent in an amount of at least about 90%
by weight of
the composition.
[0066] Diluents for solid (e.g., oral) compositions include, but are not
limited to,
microcrystalline cellulose (e.g. AVICELO), microfine cellulose, lactose,
starch, pregelatinized
starch, calcium carbonate, calcium sulfate, sugar, dextrates, dextrin,
dextrose, dibasic calcium
phosphate dihydrate, tribasic calcium phosphate, kaolin, magnesium carbonate,
magnesium
oxide, maltodextrin, mannitol, polymethacrylates (e.g. Eudragit), potassium
chloride, powdered
cellulose, sodium chloride, sorbitol and talc.
[0067] Binders for solid (e.g., oral) pharmaceutical compositions include,
but are not limited
to, acacia, tragacanth, sucrose, glucose, alginic acid, carbomer (e.g.
Carbopol),
carboxymethylcellulose sodium, dextrin, ethyl cellulose, gelatin, guar gum,
hydrogenated
vegetable oil, hydroxyethyl cellulose, hydroxypropyl cellulose (e.g. KLUCELO),
hydroxypropyl methyl cellulose (e.g. METHOCELO), liquid glucose, magnesium
aluminum
silicate, maltodextrin, methylcellulose, polymethacrylates, povidone (e.g.
KOLLIDONO,
PLASDONEO), pregelatinized starch, sodium alginate and starch. In certain
embodiments, the
pharmaceutical composition comprises a binder in an amount of about 0.5% to
about 25%, such
as about 0.75% to about 15%, by weight of the composition. In certain
embodiments, the
pharmaceutical composition comprises a binder in an amount of about 1% to
about 10% by
weight of the composition.
16
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0068] The dissolution rate of a compacted solid pharmaceutical composition
in a patient's
stomach may be increased by the addition of a disintegrant to the composition.
Disintegrants
include, but are not limited to, alginic acid, carboxymethylcellulose calcium,
carboxymethylcellulose sodium (e.g. AC-DI-SOLO, PRIMELLOSEO), colloidal
silicon
dioxide, croscarmellose sodium, crospovidone (e.g. KOLLIDONO, POLYPLASDONEO),
guar gum, magnesium aluminum silicate, methyl cellulose, microcrystalline
cellulose,
powdered cellulose, pregelatinized starch, sodium alginate, sodium starch
glycolate (e.g.
EXPLOTABO) and starch. In certain embodiments, the pharmaceutical composition
comprises
a disintegrant in an amount of about 0.2% to about 30%, such as about 0.2% to
about 10%, by
weight of the composition. In certain embodiments, the pharmaceutical
composition comprises
a disintegrant in an amount of about 0.2% to about 5% by weight of the
composition.
[0069] The pharmaceutical composition optionally comprises one or more
pharmaceutically
acceptable wetting agents. Such wetting agents are preferably selected to
maintain the API in
close association with water, a condition that is believed to improve
bioavailability of the
composition. Non-limiting examples of surfactants that can be used as wetting
agents include
quaternary ammonium compounds, for example benzalkonium chloride, benzethonium
chloride
and cetylpyridinium chloride, dioctyl sodium sulfosuccinate, polyoxyethylene
alkylphenyl
ethers, for example nonoxynol 9, nonoxynol 10, and octoxynol 9, poloxamers
(polyoxyethylene
and polyoxypropylene block copolymers), polyoxyethylene fatty acid glycerides
and oils, for
example polyoxyethylene, caprylic/capric mono- and diglycerides (e.g.,
LabrasolTM of
Gattefosse), polyoxyethylene castor oil and polyoxyethylene hydrogenated
castor oil;
polyoxyethylene alkyl ethers, for example polyoxyethylene cetostearyl ether,
polyoxyethylene
fatty acid esters, for example polyoxyethylene stearate, polyoxyethylene
sorbitan esters, for
example polysorbate 20 and polysorbate 80 (e.g., TweenTm 80 of ICI), propylene
glycol fatty
acid esters, for example propylene glycol laurate (e.g., LauroglycolTM of
Gattefosse), sodium
lauryl sulfate, fatty acids and salts thereof, for example oleic acid, sodium
oleate and
triethanolamine oleate, glyceryl fatty acid esters, for example glyceryl
monostearate, sorbitan
esters, for example sorbitan monolaurate, sorbitan monooleate, sorbitan
monopalmitate and
sorbitan monostearate, tyloxapol, and mixtures thereof In certain embodiments,
the
pharmaceutical composition comprises a wetting agent in an amount of about
0.25% to about
15%, such as about 0.4% to about 10%, by weight of the composition. In certain
embodiments,
the pharmaceutical composition comprises a wetting agent in an amount of about
0.5% to about
5% by weight of the composition. In certain embodiments, the pharmaceutical
composition
comprises a wetting agent that is an anionic surfactant. In certain
embodiments, the
17
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
pharmaceutical composition comprises sodium lauryl sulfate as a wetting agent.
In certain
embodiments, the pharmaceutical composition comprises sodium lauryl sulfate in
an amount of
about 0.25% to about 7%, such as about 0.4% to about 4%, by weight of the
composition. In
certain embodiments, the pharmaceutical composition comprises sodium lauryl
sulfate in an
amount of about 0.5% to about 2% by weight of the composition.
[0070] Lubricants (e.g., anti-adherents or glidants) can be added to
improve the flow
properties of solid compositions and/or to reduce friction between the
composition and
equipment during compression of tablet formulations. Excipients that may
function as
lubricants include, but are not limited to, colloidal silicon dioxide,
magnesium trisilicate,
powdered cellulose, starch, talc and tribasic calcium phosphate. Suitable
lubricants further
include glyceryl behapate (e.g., CompritolTM 888 of Gattefosse); stearic acid
and salts thereof,
including magnesium, calcium and sodium stearates; zinc stearate; glyceryl
monostearate;
glyceryl palmitostearate; hydrogenated castor oil; hydrogenated vegetable oils
(e.g., SterotexTM
of Abitec); waxes; boric acid; sodium benzoate; sodium acetate; sodium stearyl
fumarate;
sodium fumarate; sodium chloride; DL-leucine; PEG (e.g., CarbowaxTM 4000 and
CarbowaxTM
6000 of the Dow Chemical Company); sodium oleate; sodium lauryl sulfate; and
magnesium
lauryl sulfate. In certain embodiments, the pharmaceutical compositions
comprises a lubricant
in an amount of about 0.1% to about 10%, such as about 0.2% to about 8%, by
weight of the
composition. In certain embodiments, the pharmaceutical composition comprises
a lubricant in
an amount of about 0.25% to about 5% by weight of the composition. In certain
embodiments,
the pharmaceutical composition comprises magnesium stearate as a lubricant. In
certain
embodiments, the pharmaceutical composition comprises colloidal silicon
dioxide. In certain
embodiments, the pharmaceutical composition comprises talc. In certain
embodiments, the
composition comprises magnesium stearate or talc in an amount of about 0.5% to
about 2% by
weight of the composition.
[0071] Flavoring agents and flavor enhancers make the dosage form more
palatable to the
patient. Common flavoring agents and flavor enhancers for pharmaceutical
products that may
be included in the composition of the present invention include maltol,
vanillin, ethyl vanillin,
menthol, citric acid, fumaric acid ethyl maltol, and tartaric acid.
[0072] Compositions may also be colored using any pharmaceutically
acceptable colorant to
improve their appearance and/or facilitate patient identification of the
product and unit dosage
level. The formulations of the invention may be buffered by the addition of
suitable buffering
agents.
18
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0073] In certain embodiments of the present invention, the therapeutic
agent may be
formulated as a pharmaceutically-acceptable oil; liposome; oil-water or lipid-
oil-water
emulsion or nanoemulsion; or liquid. To facilitate such formulations, the
therapeutic agent
may be combined with a pharmaceutically-acceptable excipient therefor.
[0074] As described in detail below, the pharmaceutical compositions may be
specially
formulated for administration in solid or liquid form, including those adapted
for parenteral
administration, for example, by subcutaneous, intramuscular, intravenous or
epidural injection
as, for example, a sterile solution or suspension, or sustained-release
formulation.
[0075] Further examples of pharmaceutical formulations of 6,8-bis-
benzylthio-octanoic acid
are described in U.S. Patent No. 8,263,653, the entire disclosure of which is
incorporated by
reference herein.
[0076] Methods of preparing pharmaceutical formulations or pharmaceutical
compositions
include the step of bringing into association a compound of the present
invention with the
carrier and, optionally, one or more accessory ingredients. In general, the
formulations are
prepared by uniformly and intimately bringing into association a compound of
the present
invention with liquid carriers, or finely divided solid carriers, or both, and
then, if necessary,
shaping the product.
[0077] In certain embodiments, the pharmaceutical composition comprising
the first
therapeutic agent is a spray-dried dispersion. In certain embodiments, the
pharmaceutical
composition comprising the first therapeutic agent is a spray-dried dispersion
comprising at
least one polymer chosen from polyacrylate, polymethacrylate,
poly(vinylpyrrolidone),
hydroxypropyl methyl cellulose (HPMC), cellulose acetate phthalate (CAP), and
hydroxypropyl methylcellulose acetate succinate (HPMCAS-M). In certain
embodiments, the
pharmaceutical composition comprising the first therapeutic agent is a spray-
dried dispersion
comprising at least one polymer chosen from Eudragit L100,
poly(vinylpyrrolidone),
hydroxypropyl methyl cellulose (HPMC), cellulose acetate phthalate (CAP), and
hydroxypropyl methylcellulose acetate succinate (HPMCAS-M). In certain
embodiments, the
pharmaceutical composition comprising the first therapeutic agent is a spray-
dried dispersion
comprising at least one polymer chosen from Eudragit L100,
poly(vinylpyrrolidone) viscosity
grade K30 (PVP 1(30), hydroxypropyl methyl cellulose (HPMC), cellulose acetate
phthalate
(CAP), and hydroxypropyl methylcellulose acetate succinate (HPMCAS-M). In
certain
embodiments, the pharmaceutical composition comprising the first therapeutic
agent is a spray-
dried dispersion comprising at least one polymer chosen from Eudragit L100 and
19
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
hydroxypropyl methylcellulose acetate succinate (HPMCAS-M). In certain
embodiments, the
pharmaceutical composition comprising the first therapeutic agent is a spray-
dried dispersion
comprising Eudragit L100. In certain embodiments, the pharmaceutical
composition
comprising the first therapeutic agent is a spray-dried dispersion comprising
hydroxypropyl
methylcellulose acetate succinate (HPMCAS-M).
[0078] Pharmaceutical compositions of this invention suitable for
parenteral administration
comprise one or more compounds of the invention in combination with one or
more
pharmaceutically-acceptable sterile isotonic aqueous or nonaqueous solutions,
dispersions,
suspensions or emulsions, or sterile powders which may be reconstituted into
sterile injectable
solutions or dispersions just prior to use, which may contain sugars,
alcohols, antioxidants,
buffers, bacteriostats, solutes which render the formulation isotonic with the
blood of the
intended recipient or suspending or thickening agents.
[0079] In certain embodiments, one or more of the therapeutic agents are
administered by
intraparenteral administration. In certain other embodiments, one or more of
the therapeutic
agents are formulated for inhalational, oral, topical, transdermal, nasal,
ocular, pulmonary,
rectal, transmucosal, intravenous, intramuscular, subcutaneous,
intraperitoneal, intrathoracic,
intrapleural, intrauterine, intratumoral, or infusion methodologies or
administration, or
combinations of any thereof, in the form of aerosols, sprays, powders, gels,
lotions, creams,
suppositories, ointments, and the like. As indicated above, if such a
formulation is desired,
other additives known in the art may be included to impart the desired
consistency and other
properties to the formulation.
[0080] In certain embodiments, the pharmaceutical composition of the
present invention is a
unit dose composition. In certain embodiments, the pharmaceutical composition
contains about
1 mg to about 5000 mg of the therapeutic agent. In certain embodiments, the
pharmaceutical
composition contains about 100 mg to about 3000 mg of the therapeutic agent.
In certain
embodiments, the pharmaceutical composition contains about 200 mg to about
2000 mg of the
therapeutic agent. In certain embodiments, the pharmaceutical composition
contains about 50
mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 700 mg, 800 mg, 900 mg,
1000 mg,
1100 mg, 1200 mg, 1300 mg, 1400 mg, 1500 mg, 1600 mg, 1700 mg, 1800 mg, 1900
mg, 2000
mg, 2500 mg, or 3000 mg of therapeutic agent. In certain embodiments, the
pharmaceutical
composition contains about 300 mg, 500 mg, 700 mg, or 1000 mg of the
therapeutic agent.
[0081] In certain embodiments, the pharmaceutical composition of the
present invention
comprises an emulsion, particle, or gel as described in U.S. Patent No.
7,220,428. In certain
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
embodiments, the pharmaceutical composition is a solid or liquid formulation
having from
about 0.1% to about 75% w/w lipids or fatty acid components. In certain
embodiments, the
formulation contains about 0.1% to about 15% w/v lipids and fatty acid
components. In certain
embodiments, the fatty acid component comprises saturated or unsaturated C4,
C5, C6, C7, C8,
C9, C10, C11, or C12 fatty acids and/or salts of such fatty acids. Lipids may
include cholesterol
and analogs thereof
[0082] In certain embodiments, the pharmaceutical composition of 6,8-bis-
benzylthio-
octanoic acid comprises triethanolamine and 6,8-bis-benzylthio-octanoic acid
in a mole ratio of
triethanolamine to 6,8-bis-benzylthio-octanoic acid of about 10:1 to about
1:10. In certain
embodiments, the mole ratio of triethanolamine to 6,8-bis-benzylthio-octanoic
acid is about
10:1 to about 5:1. In certain embodiments, the mole ratio of triethanolamine
to 6,8-bis-
benzylthio-octanoic acid is about 8:1. In certain embodiments, the
pharmaceutical composition
comprises a 50 mg/mL solution of 6,8-bis-benzylthio-octanoic acid in 1M
aqueous
triethanolamine. In certain embodiments, the pharmaceutical composition
comprises a solution
of 6,8-bis-benzylthio-octanoic acid in 1M aqueous triethanolamine diluted from
50 mg/mL to
as low as 12.5 mg/mL with sterile aqueous 5% dextrose for injection (D5W). In
certain
embodiments, the pharmaceutical composition comprises a solution of 6,8-bis-
benzylthio-
octanoic acid in 1M aqueous triethanolamine diluted from 50 mg/mL to about
12.5 mg/mL
with sterile aqueous 5% dextrose for injection (D5W).
[0083] Several pharmaceutical compositions of autophagy inhibitors are
commercially
available. In certain embodiments, the pharmaceutical composition of the
autophagy inhibitor
is an oral tablet comprising chloroquine phosphate in an amount equivalent to
150 mg of the
free base. In certain embodiments, the pharmaceutical composition of the
autophagy inhibitor is
an oral tablet comprising chloroquine phosphate in an amount equivalent to 300
mg of the free
base. In certain embodiments, the pharmaceutical composition of the autophagy
inhibitor is an
oral tablet comprising 200 mg hydroxychloroquine sulfate, equivalent to 155 mg
of the free
base. In certain embodiments, the pharmaceutical composition of the autophagy
inhibitor is an
injectable liquid comprising chloroquine hydrochloride in an amount equivalent
to 40 mg/mL
of the free base.
Dosing Amounts & Schedules
[0084] The 6,8-bis-benzylthio-octanoic acid and autophagy inhibitor may be
administered to
the patient in any suitable dose according to any suitable schedule. The dose
and schedule will
vary based on the cancer being treated and can be readily determined by those
of ordinary skill
21
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
in the art in view of the 6,8-bis-benzylthio-octanoic acid and autophagy
inhibitor doses and
schedules used in the prior art when administered alone or in combination with
other agents, as
well as the guidance provided herein. In certain embodiments, the dose is the
maximum
tolerated dose.
[0085] In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 150 mg/m2 to about 3000 mg/m2. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 250 mg/m2 to
about 2500
mg/m2. In certain embodiments, the first 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 500 mg/m2 to about 2000 mg/m2. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 150 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 200
mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 250 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 300 mg/m2. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 350 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 400
mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 450 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 500 mg/m2. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 600 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 700
mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 800 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 900 mg/m2. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 1000 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
1100 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 1200 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid
is administered at a daily dose of about 1300 mg/m2. In certain embodiments,
the 6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 1400 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
1500 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 1600 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid
is administered at a daily dose of about 1700 mg/m2. In certain embodiments,
the 6,8-bis-
22
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
benzylthio-octanoic acid is administered at a daily dose of about 1800 mg/m2.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
1900 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 2000 mg/m2. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid
is administered at a daily dose of about 2500 mg/m2. In certain embodiments,
the 6,8-bis-
benzylthio-octanoic acid is administered at a daily dose of about 3000 mg/m2.
[0086] In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 1 mg to about 10,000 mg. In certain embodiments, the 6,8-
bis-benzylthio-
octanoic acid is administered at a daily dose of about 10 mg to about 7,500
mg. In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 100
mg to about 5,000 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic
acid is
administered at a daily dose of about 200 mg to about 4,000 mg. In certain
embodiments, the
6,8-bis-benzylthio-octanoic acid is administered at a daily dose of about 300
mg to about 3,000
mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 400 mg to about 2,500 mg. In certain embodiments, the 6,8-bis-
benzylthio-
octanoic acid is administered at a daily dose of about 500 mg to about 2,000
mg. In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 100
mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 200 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic
acid is
administered at a daily dose of about 300 mg. In certain embodiments, the 6,8-
bis-benzylthio-
octanoic acid is administered at a daily dose of about 400 mg. In certain
embodiments, the 6,8-
bis-benzylthio-octanoic acid is administered at a daily dose of about 500 mg.
In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about 600
mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a daily
dose of about 700 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic
acid is
administered at a daily dose of about 800 mg. In certain embodiments, the 6,8-
bis-benzylthio-
octanoic acid is administered at a daily dose of about 900 mg. In certain
embodiments, the 6,8-
bis-benzylthio-octanoic acid is administered at a daily dose of about 1,000
mg. In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
1,250 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 1,500 mg. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 1,750 mg. In certain embodiments, the
6,8-bis-benzylthio-
octanoic acid is administered at a daily dose of about 2,000 mg. In certain
embodiments, the
6,8-bis-benzylthio-octanoic acid is administered at a daily dose of about
2,500 mg. In certain
23
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
3,000 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 3,500 mg. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 4,000 mg. In certain embodiments, the
6,8-bis-benzylthio-
octanoic acid is administered at a daily dose of about 4,500 mg. In certain
embodiments, the
6,8-bis-benzylthio-octanoic acid is administered at a daily dose of about
5,000 mg. In certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered at a daily
dose of about
6,000 mg. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered at a
daily dose of about 7,000 mg. In certain embodiments, the 6,8-bis-benzylthio-
octanoic acid is
administered at a daily dose of about 8,000 mg. In certain embodiments, the
6,8-bis-benzylthio-
octanoic acid is administered at a daily dose of about 9,000 mg. In certain
embodiments, the
6,8-bis-benzylthio-octanoic acid is administered at a daily dose of about
10,000 mg.
[0087] The daily dose of 6,8-bis-benzylthio-octanoic acid may be
administered as one dose
or divided into two or more doses ¨ e.g., b.i.d. (two times a day), t.i.d.
(three times a day), or
q.i.d. (four times a day). In certain embodiments, the daily dose may be split
into five doses
administered in regular intervals during one day. At higher daily doses and/or
when
administered orally or subcutaneously, it will often be beneficial to
administer the daily dose of
6,8-bis-benzylthio-octanoic acid b.i.d., t.i.d., or q.i.d. Since 6,8-bis-
benzylthio-octanoic acid has
a relatively short half life in the blood, splitting the daily dose may
improve efficacy by
prolonging exposure time and may also improve safety by reducing peak plasma
concentration.
In certain embodiments, each dose of 6,8-bis-benzylthio-octanoic acid or
pharmaceutically
acceptable salt thereof is about 0.5 g to 1.5 g, and is administered once,
twice, three times, four
times, or five times daily. In certain embodiments, each dose of 6,8-bis-
benzylthio-octanoic
acid or pharmaceutically acceptable salt thereof is about 0.5 g to 1.5 g, and
is administered
once daily. In certain embodiments, each dose of 6,8-bis-benzylthio-octanoic
acid or
pharmaceutically acceptable salt thereof is about 0.5 g to 1.5 g, and is
administered twice daily.
In certain embodiments, each dose of 6,8-bis-benzylthio-octanoic acid or
pharmaceutically
acceptable salt thereof is about 0.5 g to 1.5 g, and is administered three
times daily. In certain
embodiments, each dose of 6,8-bis-benzylthio-octanoic acid or pharmaceutically
acceptable
salt thereof is about 0.5 g to 1.5 g, and is administered four times daily. In
certain
embodiments, each dose of 6,8-bis-benzylthio-octanoic acid or pharmaceutically
acceptable
salt thereof is about 0.5 g to 1.5 g, and is administered five times daily. In
certain embodiments,
each dose of 6,8-bis-benzylthio-octanoic acid or pharmaceutically acceptable
salt thereof is
about 1 g, and is administered once, twice, three times, four times, or five
times daily. In
24
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
certain embodiments, each dose of 6,8-bis-benzylthio-octanoic acid or
pharmaceutically
acceptable salt thereof is about 1 g, and is administered once daily. In
certain embodiments,
each dose of 6,8-bis-benzylthio-octanoic acid or pharmaceutically acceptable
salt thereof is
about 1 g, and is administered twice daily. In certain embodiments, each dose
of 6,8-bis-
benzylthio-octanoic acid or pharmaceutically acceptable salt thereof is about
1 g, and is
administered three times daily. In certain embodiments, each dose of 6,8-bis-
benzylthio-
octanoic acid or pharmaceutically acceptable salt thereof is about 1 g, and is
administered four
times daily. In certain embodiments, each dose of 6,8-bis-benzylthio-octanoic
acid or
pharmaceutically acceptable salt thereof is about 1 g, and is administered
five times daily.
[0088] The 6,8-bis-benzylthio-octanoic acid may be administered pursuant to
a treatment
schedule that includes days in which a dose of 6,8-bis-benzylthio-octanoic
acid is administered
and days in which a dose of 6,8-bis-benzylthio-octanoic acid is not
administered. For example,
the 6,8-bis-benzylthio-octanoic acid may be administered pursuant to a
schedule in which 6,8-
bis-benzylthio-octanoic acid is administered during the early days of a cycle
and then not
administered during the latter portion of the cycle. In certain embodiments,
the 6,8-bis-
benzylthio-octanoic acid is administered on days 1-5 of a 28 day cycle. In
certain embodiments,
the 6,8-bis-benzylthio-octanoic acid is administered on days 1, 8, and 15 of a
four week cycle.
In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is administered
on days 1 and 3 of
a two week cycle. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid
is administered
on days 1-5 of a three week cycle. In certain embodiments, the 6,8-bis-
benzylthio-octanoic acid
is administered on days 1-5 of a two week cycle. In certain embodiments, the
6,8-bis-
benzylthio-octanoic acid is administered on days 1-3 of a three week cycle. In
certain
embodiments, the 6,8-bis-benzylthio-octanoic acid is administered on days 1-3
of a two week
cycle. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered every day.
In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is administered
every other day.
In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is administered
three days per
week. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered two days
per week. In certain embodiments, the 6,8-bis-benzylthio-octanoic acid is
administered one day
per week.
[0089] In certain embodiments, hydroxychloroquine sulfate is administered
at a daily dose
of about 50 mg to about 1500 mg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 100 mg to about 1500 mg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 200 mg to
about 1200 mg.
In certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
300 mg to about 1200 mg. In certain embodiments, hydroxychloroquine sulfate is
administered
at a daily dose of about 400 mg to about 1200 mg. In certain embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 600 mg to
about 1200 mg.
In certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about
600 mg to about 1000 mg. In certain embodiments, hydroxychloroquine sulfate is
administered
at a daily dose of about 100 mg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 200 mg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 300 mg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 400 mg. In
certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 500 mg. In
certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 600
mg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 700 mg. In certain embodiments, hydroxychloroquine sulfate is
administered at a daily
dose of about 800 mg. In certain embodiments, hydroxychloroquine sulfate is
administered at a
daily dose of about 900 mg. In certain embodiments, hydroxychloroquine sulfate
is
administered at a daily dose of about 1,000 mg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 1,100 mg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 1,200 mg.
In certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 1,300 mg. In
certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 1400
mg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 1,500 mg.
[0090] In
certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose
of about 2 mg/kg to about 25 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 5 mg/kg to about 20 mg/kg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 6.5 mg/kg
to about 19.5
mg/kg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 2.5 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at a daily
dose of about 3 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at
a daily dose of about 3.5 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 4 mg/kg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 4.5 mg/kg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 5 mg/kg.
In certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 5.5 mg/kg.
26
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
In certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 6
mg/kg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 6.5 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at a daily
dose of about 7 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at
a daily dose of about 7.5 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 8 mg/kg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 8.5 mg/kg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 9 mg/kg.
In certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 9.5 mg/kg.
In certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 10
mg/kg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 10.5 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at a
daily dose of about 11 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 11.5 mg/kg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 12 mg/kg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 12.5
mg/kg. In certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 13 mg/kg. In
certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 13.5
mg/kg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 14 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at a daily
dose of about 14.5 mg/kg. In certain embodiments, hydroxychloroquine sulfate
is administered
at a daily dose of about 15 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 15.5 mg/kg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 16 mg/kg. In certain
embodiments,
hydroxychloroquine sulfate is administered at a daily dose of about 16.5
mg/kg. In certain
embodiments, hydroxychloroquine sulfate is administered at a daily dose of
about 17 mg/kg. In
certain embodiments, hydroxychloroquine sulfate is administered at a daily
dose of about 17.5
mg/kg. In certain embodiments, hydroxychloroquine sulfate is administered at a
daily dose of
about 18 mg/kg. In certain embodiments, hydroxychloroquine sulfate is
administered at a daily
dose of about 18.5 mg/kg. In certain embodiments, hydroxychloroquine sulfate
is administered
at a daily dose of about 19 mg/kg. In certain embodiments, hydroxychloroquine
sulfate is
administered at a daily dose of about 19.5 mg/kg. In certain embodiments,
hydroxychloroquine
sulfate is administered at a daily dose of about 20 mg/kg.
27
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0091] The daily dose of hydroxychloroquine sulfate may be administered as
one dose or
divided into two or more doses ¨ e.g., b.i.d. In certain embodiments, the
daily dose of
hydroxychloroquine sulfate is administered as one dose. In certain
embodiments, the daily dose
of hydroxychloroquine sulfate is divided into two doses and administered
b.i.d.
[0092] In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 50 mg to about 2000 mg, which is equivalent to about 30 mg to about 1200
mg
chloroquine base. In certain embodiments, chloroquine phosphate is
administered at a daily
dose of about 150 mg to about 1800 mg. In certain embodiments, chloroquine
phosphate is
administered at a daily dose of about 250 mg to about 1500 mg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 500 mg to about
1500 mg. In
certain embodiments, chloroquine phosphate is administered at a daily dose of
about 500 mg to
about 1000 mg. In certain embodiments, chloroquine phosphate is administered
at a daily dose
of about 1000 mg to about 1500 mg. In certain embodiments, chloroquine
phosphate is
administered at a daily dose of about 250 mg. In certain embodiments,
chloroquine phosphate
is administered at a daily dose of about 500 mg. In certain embodiments,
chloroquine
phosphate is administered at a daily dose of about 750 mg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 1000 mg. In
certain
embodiments, chloroquine phosphate is administered at a daily dose of about
1,250 mg. In
certain embodiments, chloroquine phosphate is administered at a daily dose of
about 1,500 mg.
In certain embodiments, chloroquine phosphate is administered at a daily dose
of about 1750
mg. In certain embodiments, chloroquine phosphate is administered at a daily
dose of about
2000 mg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 2250 mg. In certain embodiments, chloroquine phosphate is administered
at a daily dose
of about 2500 mg.
[0093] In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 2 mg/kg to about 25 mg/kg. In certain embodiments, chloroquine phosphate
is
administered at a daily dose of about 5 mg/kg to about 20 mg/kg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 6.5 mg/kg to
about 19.5 mg/kg.
In certain embodiments, chloroquine phosphate is administered at a daily dose
of about 2.5
mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of about
3 mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 3.5 mg/kg. In certain embodiments, chloroquine phosphate is administered
at a daily dose
of about 4 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a daily
dose of about 4.5 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a
28
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
daily dose of about 5 mg/kg. In certain embodiments, chloroquine phosphate is
administered at
a daily dose of about 5.5 mg/kg. In certain embodiments, chloroquine phosphate
is
administered at a daily dose of about 6 mg/kg. In certain embodiments,
chloroquine phosphate
is administered at a daily dose of about 6.5 mg/kg. In certain embodiments,
chloroquine
phosphate is administered at a daily dose of about 7 mg/kg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 7.5 mg/kg. In
certain
embodiments, chloroquine phosphate is administered at a daily dose of about 8
mg/kg. In
certain embodiments, chloroquine phosphate is administered at a daily dose of
about 8.5 mg/kg.
In certain embodiments, chloroquine phosphate is administered at a daily dose
of about 9
mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of about
9.5 mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 10 mg/kg. In certain embodiments, chloroquine phosphate is administered
at a daily dose
of about 10.5 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a daily
dose of about 11 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a
daily dose of about 11.5 mg/kg. In certain embodiments, chloroquine phosphate
is administered
at a daily dose of about 12 mg/kg. In certain embodiments, chloroquine
phosphate is
administered at a daily dose of about 12.5 mg/kg. In certain embodiments,
chloroquine
phosphate is administered at a daily dose of about 13 mg/kg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 13.5 mg/kg. In
certain
embodiments, chloroquine phosphate is administered at a daily dose of about 14
mg/kg. In
certain embodiments, chloroquine phosphate is administered at a daily dose of
about 14.5
mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of about
15 mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of
about 15.5 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a daily
dose of about 16 mg/kg. In certain embodiments, chloroquine phosphate is
administered at a
daily dose of about 16.5 mg/kg. In certain embodiments, chloroquine phosphate
is administered
at a daily dose of about 17 mg/kg. In certain embodiments, chloroquine
phosphate is
administered at a daily dose of about 17.5 mg/kg. In certain embodiments,
chloroquine
phosphate is administered at a daily dose of about 18 mg/kg. In certain
embodiments,
chloroquine phosphate is administered at a daily dose of about 18.5 mg/kg. In
certain
embodiments, chloroquine phosphate is administered at a daily dose of about 19
mg/kg. In
certain embodiments, chloroquine phosphate is administered at a daily dose of
about 19.5
mg/kg. In certain embodiments, chloroquine phosphate is administered at a
daily dose of about
20 mg/kg.
29
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
[0094] The daily dose of chloroquine phosphate may be administered as one
dose or
divided into two or more doses ¨ e.g., b.i.d. In certain embodiments, the
daily dose of
chloroquine phosphate is administered as one dose. In certain embodiments, the
daily dose of
chloroquine phosphate is divided into two doses and administered b.i.d.
[0095] For simplicity, the autophagy inhibitor will typically be
administered pursuant to a
treatment cycle that is the same length as each treatment cycle for 6,8-bis-
benzylthio-octanoic
acid (e.g., 2 weeks, three weeks, four weeks, etc.). Like the cycle for 6,8-
bis-benzylthio-
octanoic acid, the autophagy inhibitor cycle may include days in which a dose
of autophagy
inhibitor is administered and days in which a dose of autophagy inhibitor is
not administered.
For example, the autophagy inhibitor may be administered pursuant to a
schedule in which
autophagy inhibitor is administered on the same days that 6,8-bis-benzylthio-
octanoic acid is
administered, and is not administered on the days 6,8-bis-benzylthio-octanoic
acid is not
administered. Alternatively, the autophagy inhibitor may be administered on
some but not all
days in which 6,8-bis-benzylthio-octanoic acid is administered, and/or may be
administered on
some but not all days on which 6,8-bis-benzylthio-octanoic acid is not
administered. In certain
embodiments, the autophagy inhibitor may be administered on each day of the
cycle.
[0096] In certain embodiments, the dosing cycle is repeated at least once.
In certain
embodiments, the method of the present invention comprises treatment with two
cycles or
more. In certain embodiments, the method of the present invention comprises
treatment with
three cycles or more. In certain embodiments, the method of the present
invention comprises
treatment with four cycles or more. In certain embodiments, the method of the
present
invention comprises treatment with five cycles or more. In certain
embodiments, the method of
the present invention comprises treatment with six cycles or more. In certain
embodiments, the
method of the present invention comprises treatment with seven cycles or more.
In certain
embodiments, the method of the present invention comprises treatment with
eight cycles or
more. In certain embodiments, the method of the present invention comprises
treatment with
nine cycles or more. In certain embodiments, the method of the present
invention comprises
treatment with ten cycles or more. In certain embodiments, the method of the
present invention
comprises regular treatment with 6,8-bis-benzylthio-octanoic acid and an
autophagy inhibitor,
including on a daily or weekly basis, for an extended period of time, such as
at least one month,
six months, one year, two years, three years, or longer.
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Patients for Treatment
[0097] The therapeutic methods may be further characterized according to
the patient to be
treated. In the present invention, the patient is a human being. In certain
embodiments, the
patient is an adult. In certain embodiments, the patient is an adult at least
60 years of age. In
certain embodiments, the patient is an adult at least 50 years of age. In
certain embodiments,
the patient is a child.
Treatment Efficacy and Safety
[0098] The therapeutic method of the present invention may be further
characterized by the
efficacy and safety of the treatment. Preferably, the method provides an
acceptable safety
profile, with the benefit of treatment outweighing the risk. When tested in a
phase II or phase
III clinical trial of at least 10 patients with cancer, the method of the
present invention
preferably provides an overall response rate of at least about 10%, a duration
of response of at
least about 1 month, progression-free survival (PFS) of at least about 1
month, and/or overall
survival (OS) of at least about 1 month. Preferably, the phase II or phase III
clinical trial
comprises at least 15 patients. More preferably, the phase II or phase III
clinical trial comprises
at least 20 patients. More preferably, the phase II or phase III clinical
trial comprises at least 25
patients. More preferably, the phase II or phase III clinical trial comprises
at least 50 patients.
More preferably, the phase II or phase III clinical trial comprises at least
100 patients. More
preferably, the phase II or phase III clinical trial comprises at least 200
patients. More
preferably, the phase II or phase III clinical trial comprises at least 300
patients. More
preferably, the phase II or phase III clinical trial comprises at least 400
patients. More
preferably, the phase II or phase III clinical trial comprises at least 500
patients. Preferably, the
method of the present invention provides an overall response rate of at least
about 20% in
patients. More preferably, the method of the present invention provides an
overall response rate
of at least about 30%. More preferably, the method of the present invention
provides an overall
response rate of at least about 40%. More preferably, the method of the
present invention
provides an overall response rate of at least about 50%. More preferably, the
method of the
present invention provides an overall response rate of at least about 60%.
More preferably, the
method of the present invention provides an overall response rate of at least
about 70%. More
preferably, the method of the present invention provides an overall response
rate of at least
about 80%. More preferably, the method of the present invention provides an
overall response
rate of at least about 90%. Preferably, the method of the present invention
provides a duration
of response, PFS, and/or OS of at least about 2 months. Preferably, the method
of the present
31
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
invention provides a duration of response, PFS, and/or OS of at least about 3
months.
Preferably, the method of the present invention provides a duration of
response, PFS, and/or
OS of at least about 4 months. Preferably, the method of the present invention
provides a
duration of response, PFS, and/or OS of at least about 5 months. Preferably,
the method of the
present invention provides a duration of response, PFS, and/or OS of at least
about 6 months.
Preferably, the method of the present invention provides a duration of
response, PFS, and/or
OS of at least about 7 months. Preferably, the method of the present invention
provides a
duration of response, PFS, and/or OS of at least about 8 months. Preferably,
the method of the
present invention provides a duration of response, PFS, and/or OS of at least
about 9 months.
Preferably, the method of the present invention provides a duration of
response, PFS, and/or
OS of at least about 10 months. Preferably, the method of the present
invention provides a
duration of response, PFS, and/or OS of at least about 11 months. Preferably,
the method of the
present invention provides a duration of response, PFS, and/or OS of at least
about 12 months.
Preferably, the method of the present invention provides a duration of
response, PFS, and/or
OS of at least about 14 months. Preferably, the method of the present
invention provides a
duration of response, PFS, and/or OS of at least about 16 months. Preferably,
the method of the
present invention provides a duration of response, PFS, and/or OS of at least
about 18 months.
Preferably, the method of the present invention provides a duration of
response, PFS, and/or
OS of at least about 20 months. Preferably, the method of the present
invention provides a
duration of response, PFS, and/or OS of at least about 24 months. In certain
embodiments, the
overall response rate, duration of response, and progression-free survival
mentioned above are
measured in a phase II clinical trial. In certain embodiments, the overall
response rate, duration
of response, and progression-free survival mentioned above are measured in a
phase III clinical
trial.
Equivalents
[0099] The description above describes multiple aspects and embodiments of
the invention,
including therapeutic applications, treatment methods, and pharmaceutical
compositions. The
patent application specifically contemplates all combinations and permutations
of the aspects
and embodiments.
III. EXAMPLES
[00100] The invention now being generally described, will be more readily
understood by
reference to the following examples, which are included merely for purposes of
illustration of
32
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
certain aspects and embodiments of the present invention, and are not intended
to limit the
invention.
Example 1 ¨ CPI-613 Induces Autophagy in AML Cells In Vitro
[00101] K562 cells in Roswell Park Memorial Institute (RPMI) medium with 10%
fetal
bovine serum (FBS) (100,000 cells/mL) or MFL2 cells (Pardee, T.S. et al.,
Experimental
Hematology, 2011, 39, 473-485) in 45% Iscove's Modified Dulbeccos' Medium
(IMDM)/45%
Dulbecco's Modified Eagle's Medium (DMEM)/10% FBS (50,000 cells/mL) were
incubated
with CPI-613 (200 [tM) at 37 C under 5% CO2 for 4 hours alone or in the
presence of early and
late, respectively, autophagy inhibitors 3-methyladenine (3MA) or Bafilomycin
A (BafA), and
then blotted for LC3-II, which is generated during autophagy (generation
reduced by 3MA) and
consumed upon completion (degradation reduced by BafA). Complete media
("Full") and
Hank's Balanced Salt Solution medium (HBSS) were used as negative and positive
controls of
autophagy, respectively.
[00102] The results are presented in Fig. 1. This example demonstrates that
6,8-bis-
benzylthio-octanoic acid induces autophagy in AML cells.
Example 2¨ Chloroquine Sensitizes AML Cells to CPI-613 In Vitro
[00103] K562 or OCI-AML3 cells in RPMI medium with 10% FBS (100,000 cells/mL)
were
incubated with CPI-613 (100 [tM), chloroquine (25 [tM or 50 [tM), or the
combination of CPI-
613 (100 [tM) and chloroquine (25 [tM or 50 [tM) at 37 C under 5% CO2 for 72
hours and then
viability assessed using the Promega CellTiter-Glo assay.
[00104] The results are presented in Fig. 2. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and chloroquine kills AML cells in vitro
significantly better
than either agent alone.
Example 3¨ Chloroquine Sensitizes AML Cells to CPI-613 In Vivo
[00105] C57B1/6 mice were injected into their tail veins with 1 million MFL2
cells on Day 0
and beginning on Day 7, upon confirmation of engraftment by bioluminescence
imaging, were
treated by gavage with CPI-613 (300 mg/kg of 50 mg/mL solution in 0.05 N NaOH
in 5%
dextrose, adjusted to pH 7.5-8 with 4% glacial acetic acid; 1 animal) daily
except weekends
until death, intraperitoneally (IP) with chloroquine (200 pi (ca. 100 mg/kg)
of 10 mg/mL
solution in PBS; Chlr; 3 animals) daily except weekends until death, or a
combination of both
CPI-613 (300 mg/kg daily gavage as above) and chloroquine (200 [IL daily IP as
above) (4
33
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
animals) and followed for survival. The control animal (1) received both oral
and IP vehicles. P
value was determined by log rank test.
[00106] The results are presented in Fig. 3. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and chloroquine significantly prolongs
survival of AML
tumor-bearing mice in vivo compared to either agent alone at the same
concentration.
Example 4¨ Metformin Sensitizes AML Cells to CPI-613 In Vitro
[00107] MFL2 cells (Pardee, T.S. et al., Experimental Hematology, 2011, 39,
473-485) in
45% IMDM/45% DMEM/10% FBS (50,000 cells/mL) were incubated with CPI-613 (50
[tM),
metformin (1 [tM, 2.5 [tM, or 5 [tM), or the combination of CPI-613 (50 [tM)
and metformin (1
[tM, 2.5 [tM, or 5 [tM) at 37 C under 5% CO2 for 72 hours and then viability
assessed using the
Promega CellTiter-Glo assay.
[00108] The results are presented in Fig. 4. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and metformin kills AML cells in vitro
significantly better
than either agent alone at the same concentration.
Example 5¨ Metformin Sensitizes AML Cells to CPI-613 In Vivo
[00109] C57B1/6 mice were injected into their tail veins with 1 million MFL2
cells on Day 0
and beginning on Day 7, upon confirmation of engraftment by bioluminescence
imaging, were
treated with CPI-613 (300 mg/kg of a 50 mg/mL solution of CPI-613 in 0.05 N
NaOH in 5%
dextrose, adjusted to pH 7.5-8 with 4% glacial acetic acid) administered by
daily gavage except
weekends until death, metformin (1 mg/mL in the drinking water with ad lib
access), or a
combination of metformin (1 mg/mL ad lib in drinking water as above) plus CPI-
613 (300
mg/kg daily gavage as above) (Met+CPI) and followed for survival. P value was
determined by
log rank test.
[00110] The results are presented in Fig. 5. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and metformin significantly prolongs
survival of AML
tumor-bearing mice in vivo.
Example 6¨ 2-Deoxyglucose Sensitizes AML Cells to CPI-613 In Vitro
[00111] MFL2 cells (Pardee, T.S. et al., Experimental Hematology, 2011, 39,
473-485) in
45% IMDM/45% DMEM/10% FBS (50,000 cells/mL) were incubated with CPI-613 (50
[tM),
2-deoxyglucose (0.25 mM or 0.5 mM), or the combination of CPI-613 (50 [tM) and
2-
34
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
deoxyglucose (0.25 mM or 0.5 mM) at 37 C under 5% CO2 for 72 hours and then
viability
assessed using the Promega CellTiter-Glo assay.
[00112] The results are presented in Fig. 6. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and 2-deoxyglucose kills AML cells in
vitro significantly
better than either agent alone at the same concentration.
Example 7¨ Combination of Chloroquine and 2-Deoxyglucose Sensitizes AML Cells
to
CPI-613 In Vitro
[00113] OCI-AML3 cells in RPMI medium with 10% FBS (100,000 cells/mL) or MFL2
cells
(Pardee, T.S. et al., Experimental Hematology, 2011, 39, 473-485) in 45%
IMDM/45%
DMEM/10% FBS (50,000 cells/mL) were incubated with CPI-613 (50 M), chloroquine
(25
1.1.M for OCI; 10 .M for MFL2), 2-deoxyglucose (10 mM for OCI; 0.25 mM for
MFL2) or the
combination of CPI-613 (50 M), chloroquine (25 .M for OCI; 10 .M for MFL2),
and 2-
deoxyglucose (10 mM for OCI; 0.25 mM for MFL2) at 37 C under 5% CO2 for 72
hours and
then viability assessed using the Promega CellTiter-Glo assay.
[00114] The results are presented in Fig. 7. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid, chloroquine, and 2-deoxyglucose kills AML
cells in vitro
significantly better than any of the three agents alone at the same
concentration.
Example 8¨ Chloroquine Sensitizes PDAC Cells to CPI-613 In Vitro
[00115] Pancreatic ductal adenocarcinoma cell (PDAC) lines, AsPC-1, PANC-1,
BxPC-3, or
MIA Paca-2 cells were seeded in complete RPMI medium at 30,000 cells per well
in 96 well
plates and incubated for 18 hrs. Cells were then adapted to nutrient depleted
conditions
resembling tumor nutrient conditions by incubating in RPMI without serum for
20 hrs,
followed by modified Earle's Balanced Salt Solution (EBSS) (CBS2 medium) for 3
hrs. Drugs
were administered in CBS2 medium and incubated for 20 hrs. Drug containing
medium was
replaced with RPMI without serum and incubated overnight. All incubations were
in a
humidified incubator at 37 C and 5% CO2. Cell viability was assessed with
Promega CellTiter-
Glo assay in which luminescence units are proportional to the number of live
cells. Zero (0)
luminescence units indicate that all cells were killed.
[00116] For PANC-1 and ASPC-1 cells the drug treatments were: chloroquine or
hydroxychloroquine at 12.5 1.1.M, 25 1.1.M, 50 M, 100 M, or 200 M, CPI-613 at
10 p.M, 20
[t.M or 40 [t.M, or a combination of chloroquine or hydroxychloroquine with
CPI-613 at these
concentrations. For BxPC-3 cells the drug treatments were: chloroquine or
hydroxychloroquine
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
at 12.5 uM, 25 uM, 50 uM, 100 uM, or 200 uM, CPI-613 7.5 uM, 15 u.M or 30 uM,
or a
combination of chloroquine or hydroxychloroquine with CPI-613 at these
concentrations. For
MIA PaCa-2 cells the drug treatments were: chloroquine at 12.5 uM, 25 uM, 50
uM, 100 uM,
or 200 uM, CPI-613 at 10 uM, 20 u.M or 40 uM, or a combination of chloroquine
and CPI-613
at these concentrations.
[00117] The results are presented in Figs. 8-11. This example demonstrates
that the
combination of 6,8-bis-benzylthio-octanoic acid and chloroquine or
hydroxychloroquine kills
PDAC cells in vitro better than either agent alone.
Example 9 ¨ Chloroquine Sensitizes Colorectal Cancer Cells to CPI-613 In Vitro
[00118] CoLo 205 cells were seeded in complete RPMI medium at 60,000 cells per
well;
LoVo cells were seeded in complete F12-K medium at 60,000 cells per well and
SW620 and
HT29 cells were seeded in complete McCoys medium at 60,000 cells per well in
96 well plates
and incubated for 18 hrs. Cells were then adapted to nutrient depleted
conditions which
resemble tumor nutrient conditions by incubating in their respective media
without serum for
20 hrs followed by modified EBSS (CBS2 medium) for 3 hrs. Drugs were
administered in
CBS2 medium and incubated for 20 hrs. Drug containing medium was replaced with
RPMI
without serum and incubated overnight. All incubations were in a humidified
incubator at 37 C
and 5% CO2. Cell viability was assessed with Promega CellTiter-Glo assay.
[00119] The drug treatments were: chloroquine at 12.5 uM, 25 uM, 50 u.M or 100
uM, CPI-
613 at 12.5 uM, 25 u.M or 50 uM, alone or in combination.
[00120] The results are presented in Figs. 12 and 13. This example
demonstrates that at the
higher CPI-613 concentrations, the combination of 6,8-bis-benzylthio-octanoic
acid and
chloroquine kills colorectal cancer cells in vitro better than either agent
alone.
Example 10 ¨ Chloroquine Sensitizes Non-Small Cell Lung Cancer Cells to CPI-
613 In
Vitro
[00121] H460 cells were seeded in complete RPMI medium at 30,000 cells per
well in 96
well plates and incubated for 18 hrs. Cells were then adapted to nutrient
depleted conditions
which resemble tumor nutrient conditions by incubating in RPMI without serum
for 20 hrs
followed by modified EBSS (CBS2 medium) for 3 hrs. Drugs were administered in
CBS2
medium and incubated for 5 hrs. Drug containing medium was replaced with RPMI
without
serum and incubated overnight. All incubations were in a humidified incubator
at 37 C and 5%
CO2. Cell viability was assessed with Promega CellTiter-Glo assay.
36
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
[00122] The drug treatments were: chloroquine at 12.5 uM, 25 uM, 50 uM, 100
uM, or 200
uM, CPI-613 at 10 uM, 20 u.M or 40 uM, alone or in combination.
[00123] The results are presented in Fig. 14. This example demonstrates that
the combination
of 6,8-bis-benzylthio-octanoic acid and chloroquine kills non-small cell lung
cancer cells in
vitro better than either agent alone.
Example 11 ¨ Treatment of High Risk MDS in Human Patients Who Have Failed
Hypomethylating Therapy Using a Combination of CPI-613 and Hydroxychloroquine
[00124] This is a single-arm open-label study. The investigators and study
subjects are not
blinded to the treatment. Also, the assignment of patients will not be
randomized, since there is
only a single arm in this study.
[00125] The primary objective of this study is to determine the overall
response rate
(complete remission (CR), marrow CR, partial remission (PR), Hematologic
improvement
(HO) of high risk MDS patients who have failed hypomethylating agents, treated
with the
combination of CPI-613 and the maximally tolerated dose of hydroxychloroquine
(HCQ).
Secondary objectives are to evaluate the safety of the combination,
progression-free-survival
(PFS), overall survival (OS) defined as the time from enrolment on study to
death from any
cause, and changes in the frequency of blood transfusions.
Study Design
[00126] The dose of CPI-613 will be 2,000 mg/m2. The maximal tested dose of
HCQ will be
1,200 mg. The sample size will be a total of 17 patients treated at the MTD of
HCQ for this
Phase 1/2 trial. This number is based on Simon's two stage design where 9
patients will be
enrolled in stage 1. If none of the 9 patients have a response the study will
be stopped for lack
of efficacy. If one or more patients has a response the trial will continue
until a total of 17
patients have been treated with the combination at the MTD of HCQ. If 2 or
more of the 17
patients have a response the combination will be considered of sufficient
activity to merit
additional study.
[00127] The initial phase of the study will be a dose escalation of
hydroxychloroquine from
600 mg to 1200 mg PO flat dose given 2 hours before the CPI-613 infusion on
days 1-5 of
every 28. The dose of the CPI-613 will be 2,000 mg/m2 and will not be
escalated. Cohorts of 3
patients each will be treated with 600, then 800 then 1,200 mg of HCQ in a 3+3
dose escalation
design as described below.
37
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
[00128] If no patients in a given cohort develop a dose-limiting toxicity (See
DLT definition
below), dose escalation will continue in cohorts of 3 patients. However, if a
DLT is observed
in a patient (whether it is the first, second or third of the 3 intended
patients) at any dose level,
the cohort of that dose level will be expanded to a maximum of 6 patients. If
no DLT is
observed in another patient out of a maximum of 6 patients, dose escalation
procedure will
continue in 3 patients for each subsequent cohort until a final dose of 1,200
mg is reached.
However, once a DLT is observed in a total of 2 patients at any dose level,
dosing of HCQ in
patients at that dose level will stop immediately, even though the total
number of patients at the
last cohort may be as few as 2. Dose escalation is considered to be complete.
The dose level
that induces a DLT in 2 or more patients is considered to be above MTD, and
the dose level
immediately below the dose level that induced a DLT in >2 patients is
considered the MTD. If
the initial dose of 600 mg proves above the MTD the HCQ dose will drop to dose
level -1(400
mg). Should that dose (400 mg) produce 2 DLTs the study will close and the
toxicity data
reviewed prior to any additional enrollment. Should the 1,200 mg cohort be
completed without
a DLT in the first 3 patients or without a second DLT in the first 6 patients,
dose escalation of
the HCQ will be complete. All subsequent patients will be treated at this dose
level. HCQ dose
levels are summarized in Table 1.
Table 1
Dose level HCQ Dose (flat)
-1 400 mg
1 600 mg
2 800 mg
3 1,200 mg
[00129] Definition of DLT: A dose limiting toxicity is defined as the
occurrence of any
clinically relevant, grade? 3 toxicity attributed as probably or definitely
related to the
combination of HCQ and CPI-613. The following toxicities are excluded from
defining a DLT:
grade 3 nausea and vomiting responsive to anti-emetics, grade 3 diarrhea
responsive to anti-
diarrheal therapy, grade 3 tumor lysis syndrome, grade 3 or 4 metabolic
derangements
attributed to tumor lysis syndrome or antimicrobial medications that correct
with oral or IV
supplementation. Hematologic toxicities can only serve as a DLT if they
represent a significant
(>50%) decline from baseline values. Infectious toxicities can only serve as a
DLT if they are a
consequence of a >20% decline in ANC from baseline in the opinion of the
treating investigator
and not a consequence of the natural history of relapsed or refractory MDS.
[00130]
38
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Inclusion Criteria
[00131] Patients must meet all of the following inclusion criteria before
enrollment:
1) Histologically documented MDS whose disease has failed to respond,
progressed or
relapsed while on a hypomethylating agent.
1) Revised International Prognostic Scoring System (IPSS-R) score of
Intermediate, high
or very high at time of enrollment
2) ECOG Performance Status of < 3.
3) Men and women 18 years of age or older.
4) Expected survival >2 months.
5) Women of child-bearing potential (i.e., women who are pre-menopausal or not
surgically sterile) must use accepted contraceptive methods (abstinence,
intrauterine
device [IUD], oral contraceptive or double barrier device) during the study,
and must
have a negative serum or urine pregnancy test within 1 week prior to treatment
initiation.
6) Fertile men must practice effective contraceptive methods during the study,
unless
documentation of infertility exists.
7) Patients must have fully recovered from the acute, non-hematological, non-
infectious
toxicities of any prior treatment with cytotoxic drugs, radiotherapy or other
anti-cancer
modalities. Patients with persisting, non-hematologic, non-infectious
toxicities from
prior treatment < Grade 2 are eligible, but must be documented as such.
8) Laboratory values obtained < 2 weeks prior to enrollment must demonstrate
adequate
hepatic function, renal function, and coagulation as defined below:
a. aspartate aminotransferase [AST/SGOT] < 3x upper normal limit [UNL]
b. alanine aminotransferase [ALT/SGPT] < 3x UNL
c. bilirubin < 2x UNL
d. serum creatinine < 1.5 mg/dL or 133 umol/L
e. albumin > 2.0 g/dL or > 20 g/L.
9) Mentally competent, ability to understand and willingness to sign an IRB-
approved
written informed consent form.
10)Have access via central line (e.g., portacath).
Exclusion Criteria
[00132] Patients with the following characteristics are excluded:
39
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
1) Serious medical illness, such as significant cardiac disease (e.g.
symptomatic congestive
heart failure, unstable angina pectoris, symptomatic coronary artery disease,
myocardial
infarction within the past 3 months, uncontrolled cardiac arrhythmia,
pericardial disease
or New York Heart Association Class III or IV), or severe debilitating
pulmonary
disease, that would potentially increase patients' risk for toxicity.
2) Patients with active central nervous system (CNS) or epidural tumor.
3) Any active uncontrolled bleeding or bleeding diathesis (e.g., active peptic
ulcer
disease).
4) Any condition or abnormality which may, in the opinion of the investigator,
compromise his or her safety.
5) Pregnant women, or women of child-bearing potential not using reliable
means of
contraception.
6) Fertile men unwilling to practice contraceptive methods during the study
period.
7) Lactating females.
8) Life expectancy less than 2 months.
9) Unwilling or unable to follow protocol requirements.
10) Evidence of ongoing uncontrolled serious infection.
11) Requirement for immediate palliative treatment of any kind including
surgery.
12) Patients with uncontrolled HIV infection.
13) Patients who have received radiotherapy, surgery, treatment with cytotoxic
agents
(except a hypomethylating agent, i.e. azacytidine or decitabine), treatment
with biologic
agents, immunotherapy, or any other anti-cancer therapy of any kind, or any
other
standard or investigational treatment for their cancer, or any other
investigational agent
for any indication, within the past 2 weeks prior to initiation of CPI-613
treatment.
14) Patients that have received a chemotherapy regimen with stem cell support
in the
previous 6 months
Study Procedures
[00133] Table 2 provides an overview of assessments and procedures conducted
during the
pre-study screen and during each treatment cycle.
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
Table 2
Each Treatment Cycle = 4 weeks Follow-Up
Pre-
study8 D1 D2 D3 D4 DS Last day
Assessments of cycle
Medical history
Physical exam, height,
weight, vitals
Pregnancy test
Evaluation of symptoms
and vital signs
ECOG performance
status and survival
Clinical chemistry, Al2 Al2 Al2 Al2 Al2
hematology3,6
CPI-6131
Hydroxychloroquine9
Assessment of -\14
response6'7
Bone marrow exam6 A/4
Optional Blood and
Marrow Aspirate
Samples10
Survival and post-study
follow-up5
D = day, ECOG = Eastern Cooperative Oncology Group; hr = hour; min = minute.
1 CPI-613 is given as a 2-hr IV infusion via a central venous catheter.
2 Creatinine must be performed with results available for review before
administration of the CPI-613.
3 Specific chemistry and hematology are listed in section 5.2.2. Renal
function will be assessed utilizing
the Cockcroft-Gault formula.
4 Cycles 3, 6 and 12 (and every 6 months thereafter and at disease progression
when possible).
Survival and post-study cancer treatment will be monitored by the treating
physician during routine
follow-up visits for 5 years or until death.
6 CR, marrow CR, and PR will be assessed using hematology results from blood
work and bone marrow
exam results, according to the criteria described by Cheson et al. (2006).
7 Frequency of transfusion is defined as the number of transfusions received
during the previous 8 weeks.
The baseline assessment should reflect the number of transfusions received in
the 8 weeks prior to
enrollment. On-treatment assessments should be performed at the end of cycles
4, 8 and 12 (and every 6
months thereafter until disease progression)
Pre-study requirements must be performed within the following time frames:
Within 4 weeks: bone
marrow exam; Within 2 weeks: medical history, physical exam, vital signs,
height, weight, ECOG,
evaluation of symptoms and medications, clinical chemistry, hematology, and
coagulation; Within 1
week: pregnancy test for women of child-bearing potential and frequency of
transfusion.
9 Hydroxychloroquine to be taken 2 hours prior to CPI-613 infusion.
3-5 ml whole blood sample in an EDTA tube taken prior to and at completion of
the CPI-613 infusion
during cycle 1 only. 3-5 ml of marrow aspirate in a heparinized tube (green
top) taken at the time of the
bone marrow exam (see footnote 4). Samples are optional and patients who have
had a baseline marrow
prior to consenting for study do not need it repeated to enroll.
Treatment with CPI-613 and Hydroxychloroquine
[00134] CPI-613 will be administered to patients as shown in Table 3. Briefly,
CPI-613 is
given on Days 1 through 5 of a 28 day cycle with hydroxychloroquine taken
orally 2 hours
41
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
prior to each dose. Patients will receive pre-treatment antiemetics and
supportive measures as
determined by their treating physician. The default premedications will
consist of ondansetron
16 mg either IV infused 15 minutes or oral 30 minutes prior to therapy and
loperamide 2 mg
orally (unless patient has not had a bowel movement in the last 48 hours) 30
min prior to
therapy.
[00135] Baseline transfusion frequency should be recorded as the number of
transfusions
received during the 8 weeks prior to enrollment. Once patients have started
protocol treatment,
transfusion frequency should be assessed at the end of every 2 treatment
cycles.
Table 3
Treatment Cycles
Administration of HCQ Administration of CPI-613
Week Day
Cycle 1 and 1 1-5
Taken orally 2 hours prior to 2-hr IV infusion via a central
beyond (4 CPI-613 infusion venous catheter
weeks) 6-7 Rest Rest
2-4 8-28 Rest Rest
4 Bone
marrow biopsy (week 4
of cycles 3,6 and 12 then
every 6 cycles and at
disease progression)
Safety Assessment
[00136] The safety of CPI-613 will be assessed from the first dose to 1 month
after last dose
of CPI-613. The assessment will be based on: evaluation of symptoms, vital
signs, ECOG
performance status and survival, clinical chemistry (and renal function
utilizing the Cockcroft-
Gault formula), and hematology. All safety assessment tests are performed
during screening
(performed within 2 weeks prior to treatment with CPI-613). Additionally,
evaluation of
symptoms, vital signs, ECOG and survival will be assessed on each treatment
day, with results
available for review within 24 hours before administration CPI-613. Clinical
chemistry (renal
function utilizing the Cockcroft-Gault formula) hematology, and coagulation
will be performed
on Day 1 of each treatment cycle, with results available for review within 24
hours before
administration CPI-613.
[00137] For toxicities attributed as at least possibly related to CPI-613 dose
adjustments will
be as outlined in the following Table 4.
42
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Table 4
Toxicity and Intensity Supportive Care and Dose Adjustment Guidelines
Nausea, Acute (common)
Grade 1 or 2 Maintain dose and schedule. Rule out other causes.
Utilize anti-
(If intolerable or persistent nausea medications including 5-HT3
antagonists.
Grade 2 not responsive to
supportive care, follow
guidelines for Grade 3)
Grade 3-4 Rule out other causes. Utilize anti-nausea
medications including
5-HT3 antagonists.
Interrupt CPI-613 dosing until resolved to Grade 1 or baseline
and reduce CPI-613 dose by 50%.
Diarrhea (common)
Grade 1 Maintain dose and schedule. Rule out other causes
including
drug effects. Treat per institutional guidelines with anti-diarrheals.
Grade 2 Rule out other causes including drug effects. Treat
per
institutional guidelines with anti-diarrheals. Interrupt CPI-613
dosing until resolved to Grade 1 or baseline.
For first occurrence, restart CPI-613 at current dose.
For second occurrence, reduce dose by 25%.
Grade 3 0r4 Interrupt CPI-613 dosing until resolved to Grade 1 or
baseline
and patient is clinically stable. Reduce CPI-613 dose by 50%.
Renal Failure
Grade 1 Maintain dose and schedule.
Grade 2 Hold dose until resolved to grade 1 or baseline and
resume CPI-
613 at a 25% dose reduction.
Grade Hold dose until resolved to grade 1 or baseline and
resume CPI-
613 at a 50% dose reduction.
Non-Hematological Adverse Events
Grade 1 Maintain dose and schedule.
Grade 2 Hold dose until resolved to grade 1 or baseline and
resume CPI-
613 at a 25% dose reduction.
Grade 3 or 4 Hold dose until resolved to grade 1 or baseline and
resume CPI-
613 at a 50% dose reduction.
Hematological Adverse Events
Grade 1-3 Maintain dose and schedule. Utilize growth factor and
transfusion
supportive care asper institutional guidelines.
Grade 4 lasting <7 days Maintain dose and schedule. Utilize growth factor
and transfusion
supportive care asper institutional guidelines.
Grade 4 lasting days Hold dose until resolved to grade 3 or less
and resume CPI-613
at a 50% dose reduction.
[00138] Once toxicities have resolved to less than grade 1 (or returned to
baseline) patients
can have the dose of CPI-613 re-escalated at the discretion of the treating
investigator. Patients
that have recurrence of the original toxicity are not then eligible for dose
re-escalation.
43
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
Assessment of Response
[00139] Tumor response will be assessed based on RR, PFS, and OS (as described
by Cheson
B.D. et al., Clinical application and proposal for modification of the
International Working
Group (IWG) response criteria in myelodysplasia. Blood. 108:419-425, 2006), as
well as
changes in the frequency of transfusion from baseline. RR and PFS, derived
from hematology
and bone marrow exam, will be assessed at baseline, during week 4 of cycles 3,
6, and 12 and
then every 6 treatment cycles thereafter until disease progression.
[00140] Baseline transfusion frequency should be recorded as the number of
transfusions
received during the 8 weeks prior to enrollment. Once patients have started
protocol treatment,
transfusion frequency should be assessed at the end of every 2 treatment
cycles.
[00141] Survival will be assessed during the study and will be monitored by
treating
physician contact after the patients are taken off the trial. The ECOG
Performance Status scales
(Oken M.M. et al., Toxicity And Response Criteria Of The Eastern Cooperative
Oncology
Group. Am J Clin Oncol 5:649-655, 1982) will be used to assess how a patient's
disease is
progressing and assess how the disease affects the daily living abilities of
the patient. ECOG
Grade 0 = Normal activity. Fully active, able to carry on all pre-disease
performance without
restriction. ECOG Grade 1 = Symptoms, but ambulatory; restricted in physically
strenuous
activity, but ambulatory and able to carry out work of a light or sedentary
nature (e.g., light
housework, office work). ECOG Grade 2 = in bed <50% of the time; ambulatory
and capable
of all self-care, but unable to carry out any work activities; up and about
more than 50% of
waking hours. ECOG Grade 3 = In bed >50% of the time; capable of only limited
self-care,
confined to bed or chair more than 50% of waking hours. ECOG Grade 4 = 100%
bedridden;
completely disabled; cannot carry on any self-care; totally confined to bed or
chair. ECOG
Grade 5 = dead.
[00142] The following parameters from bone marrow exam will be recorded:
morphology,
immunophenotype, cellularity, karyotype (cytogenetics and FISH as applicable),
molecular
markers, % of bone marrow myeloblasts, % of dysplasia, WHO classification.
[00143] The Modified International Working Group (IWG)-2006 response criteria
for
altering natural history of MDS are described in the following Table 5.
44
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Table 5
Category Response criteria (responses must last at least 4 wk)
Complete Bone marrow: < 5% myeloblasts with normal maturation of all cell
lines*
remission
Persistent dysplasia will be noted*t
Peripheral blood:
Hgb > 11 g/dL
Platelets? 100 x 109/L
Neutrophils > 1.0 x 109/Lt
Blasts 0%
Partial
Remission All CR criteria if abnormal before treatment except:
Bone marrow blasts decreased by? 50% over pretreatment but still > 5%
Cellularity and morphology not relevant
Marrow CRt Bone marrow: < 5% myeloblasts and decrease by > 50% over
pretreatmentt
Peripheral blood: if HI responses, they will be noted in addition to marrow
CRt
Stable disease Failure to achieve at least PR, but no evidence of progression
for > 8 wks
Failure Death during treatment or disease progression characteized by
worsening of
cytopenias, increase in percentage of bone marrow blasts, or progression to a
more advanced MDS FAB subtype than pretreatment
Relapse after At least 1 of the following:
CR or PR
Return to pretreatment bone marrow blast percentage
Decrement of? 50% from maximum remission/response levels
in granulocytes or platelets
Reduction in Hgb concentration by? 1.5 g/dL or transfusion dependence
Cytogenetic Complete
response
Disappearance of the chromosomal abnormality without appearance of new
ones
Partial
At least 50% reduction of the chromosomal abnormality
For patients with:
Disease
progression
Less than 5% blasts: > 50% increase in blasts to > 5% blasts
5%-10% blasts: > 50% increase to > 10% blasts
10%-20% blasts: > 50% increase to > 20% blasts
20%-30% blasts: > 50% increase to > 30% blasts
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Any of the following:
At least 50% decrement from maximum remission/response in granulocytes or
platelets
Reduction in Hgb by > 2 g/dL
Transfusion dependency
Survival Endpoints:
Overall: death from any cause
Event free: failure or death from any cause
PFS: disease progression or death from MDS
DFS: time to relapse
Cause-specific death: death related to MDS
To convert hemoglobin from grams per deciliter to grams per liter, multiply
grams per deciliter
by O.
MDS indicates myelodysplastic syndromes; figb, hemoglobin; CR, complete
remission; HI,
hematologic improvement; PR, partial remission; FAB, French-American-British.;
AML, acute
myeloid leukemia; PFS, proaression-free survival; DFS, disease-free survival.
* Dysplastic changes should consider the normal range of dysplastic changes
(modification).
t Modification to IWG response criteria.
t In some circumstances, protocol therapy may require the initiation of
further treatment (e.g.,
consolidation, maintenance) before the 4-week period. Such patients can be
included in the
response category into which they fit at the time the therapy is started.
Transient cytopenias
during repeated chemotherapy courses should not be considered as interrupting
durability of
response, as long as they recover to the improved counts of the previous
course.
[00144] The Modified International Working Group (IWG)-2006 response criteria
for
hematologic improvement are described in the following Table 6.
46
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Table 6
Hematologic Response criteria (responses must last at least 8 wk)t
improvement*
Erythroid response Hgb increase by? 1.5 g/dL
(pretreatment, < 11
g/dL)
Relevant reduction of units of RBC transfusions by an absolute number of
at least 4 RBC transfusions/8 wk compared with the pretreatment
transfusion number in the previous 8 wk. Only RBC transfusions given
for a Hgb of < 9.0 g/dL pretreatment will count in the RBC transfusion
response evaluationt
Platelet response Absolute increase of? 30 x 109/L for patients starting
with > 20x 109/L
(pretreatment, platelets
<100 x 109/L)
Increase from < 20 x 109/L to > 20 x 109/L and by at least 100%t
Neutrophil response At least 100% increase and an absolute increase > 0.5 x
109/0
(pretreatment,
< 1.0 x 109/L)
Progression or
relapse after HIT At least 1 of the following:
At least 50% decrement from maximum response levels in granulocytes
or platelets
Reduction in Hgb by? 1.5 g/dL
Transfusion dependence
Deletions to the IWG response criteria are not shown.
Hgb indicates hemoglobin; :RBC: red blood cell; HI: hematologic improvement.
Pretreatment counts averages of at least 2 measurements (not influenced by
transfusions) > I
week apart (modification).
Modification to MG response criteria.
+ In the absence of another explanation, such as acute infection, repeated
courses of
chemotherapy (modification), gastrointestinal bleeding, hernolysis, and so
forth. It is
recommended that the 2 kinds of erythroid and platelet responses be reported
overall as well as
by the individual response pattern.
[00145] Treatment with CPI-613 and HCQ should be continued as long as the
treating
physician believes there is clinical benefit, unless or until: patients
exhibit disease progression;
unacceptable toxicity from CPI-613 and HCQ in spite of dose reduction; patient
withdrawal of
consent; investigator's discretion to withdraw patients from the study because
continued
47
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
participation in the study is not in the patient's best interest; undercurrent
illness (a condition,
injury, or disease unrelated to the intended disease for which the study is
investigating, that
renders continuing the treatment unsafe or regular follow-up impossible);
general or specific
changes in the patient's condition that renders the patient ineligible for
further investigational
treatment; non-compliance with investigational treatment, protocol-required
evaluations or
follow-up visits; or termination of the clinical trial. Upon being taken off
the trial, patient's
survival and post-study cancer treatment will be monitored by follow up
physician visits once
patients are removed from trial. All patients will be followed for 5 years
post treatment, or
until death (unless consent for follow up withdrawn).
CPI-613
[00146] CPI-613 is provided in 10-mL amber glass vials. Each vial contains 10
mL of CPI-
613 at a concentration 50 mg/mL, equivalent to 500 mg of CPI-613. The drug
product of CPI-
613 is a clear and colorless solution that is free of any particulate matter.
[00147] CPI-613 is administered IV by infusion, via an IV catheter with D5W
running at a
rate of about 125-150 mL/hr. To avoid local reactions at and around the site
of administration,
CPI-613 should be administered via a central venous catheter.
[00148] CPI-613 can cause leaching of DEHP from IV infusion sets and IV bags.
Therefore,
DEHP-containing IV infusion sets, IV bags or syringes should not be used in
mixing or
administration of CPI-613. Examples of the IV sets, IV bags and syringes that
do not contains
DEHP and therefore can be used in the administration of CPI-613 are:
= Extension Set for Syringe Pump Use: All extension sets from MED-RX do not
contain
DEHP.
= Syringes: All Monoject syringes are DEHP free.
= IV Infusion Sets: The IV infusion sets that can be used to administer CPI-
613 are:
o PVC material - ADDitIV Primary IV Set with Universal Spike, Backcheck
Valve, 2 Injection Sites, DEHP-Free and Latex-Free, 15 drops/mL, REF
V14453, B Braun Medical Inc.
o Latex material - Interlink System Secondary Medication Set, 10 drops/mL,
2C7451, Baxter Healthcare Corporation
o PVC material - SurshieldTM Safety Winged Infusion Set, 0.19 mL Volume,
Latex-Free, DEHP-Free, SV*S25BLS, Terumo Medical Products Hangzhou Co.
Ltd.
48
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
o Polyethylene material - Interlink System Paclitaxel Set by Baxter
HealthCare,
Non DEHP-free: Polyethylene tubing with a 0.22 microfilter Item # 2C7558 10
drops/mL
= Syringes: CPI-613 drug product (50 mg/mL), and drug product diluted with
D5W to
various concentrations (1.6-25 mg/mL) are compatible with various types of
syringes,
as listed below. Therefore, any of these types of syringes, and syringes that
are made
with the same materials, can be used to administer CPI-613. Also, glass
syringes can
also be used, since glass (such as glass containers) is compatible with CPI-
613 drug
product.
o Norm-Ject, polyethlyene barrel, polyethylene plunger, latex free (Henke
Sass
Wolf GMBH) syringes
o Becton Dickinson syringes
o Terumo syringes
o Monoject syringes
o Glass syringes.
[00149] CPI-613 must be diluted from 50 mg/mL to 12.5 mg/mL with 5% Dextrose
Water
(D5W) (i.e., 1 portion of CPI-613 diluted with 3 portions of D5W) prior to
administration. The
diluted drug product should be visually inspected for clarity. If haziness,
precipitate or
coloration (other than colorless) is observed, do not use the diluted drug
product for dosing.
After dilution with sterile D5W, the solution is clear and has a pH of 8.4-
8.8. The diluted CPI-
613 drug product has been found to be stable for 24 hrs at room temperature
and refrigeration
temperature.
[00150] CPI-613 must be administered IV, via an IV catheter that is free
flowing and free of
air in the dead space of the IV catheter, to minimize vascular irritation,
inflammation and acute
toxicity of CPI-613. Accidental co-administration of extra air in the dead
space of IV catheters
during administration of CPI-613 has demonstrated the potential to induce
acute toxicity of
CPI-613 according to animal studies. Also, accidental leakage of CPI-613 into
the perivascular
space during IV administration, which prolongs exposure of perivascular tissue
to CPI-613, can
induce significant local inflammation according to animal studies. To avoid
local reactions at
and around the site of administration, CPI-613 must be administered via a
central venous
catheter.
[00151] CPI-613 must not be administered as a bolus, but by infusion, at a
rate of ¨0.5
mL/min, via a central venous catheter with D5W running at a rate of about 125-
150 mL/hr.
This is to minimize potential acute toxicity of CPI-613, according to animal
studies.
49
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
[00152] The following precautions must be taken when administering CPI-613:
= Confirmation of the placement of the IV line to ensure a lack of leakage
of CPI-613 into
the perivascular space.
= Confirmation that the IV line is free flowing.
= Confirmation that the IV line is free of dead air space.
= Dilute CPI-613 drug product with D5W, as instructed in the study
protocol.
= Administer CPI-613 by infusion, not as a bolus.
= After administration of CPI-613, flush the IV line with ¨10 mL of D5W to
remove
residual CPI-613.
= To avoid local reactions at and around the site of administration, CPI-
613 should be
administered via a central venous catheter.
[00153] The amount of CPI-613 at each dose level is based on the BSA of the
patient. The
BSA values will be calculated based on the height and body weight taken during
screening and
this BSA value is used throughout the study. This is unless there is a >10%
change in the body
weight from baseline during the study. At that point, BSA should be revised
based on the new
body weight and height. The new BSA values will be used from that point on for
the remainder
of the study, unless there is another >10% change in body weight which will
require another
revision of the BSA.
Other Agents
[00154] Patients cannot receive any standard or investigational treatment
(except CPI-613
and hydroxychloroquine) for their MDS, or any other investigational drugs for
any non-cancer
indications, while on this study. All otherwise permitted concomitant
medications (including
trade and generic names, dosage and dosing schedule) must be recorded.
Treatment of disease-
related symptoms (such as nausea) is permitted. Medications administered in
such instances
will be considered concomitant medications and should be documented
accordingly. Supportive
treatment may include anti-emetic, anti-diarrhea, anti-pyretic, anti-allergic,
anti-hypertensive
medications, analgesics, antibiotics, allopurinol, and others such as blood
products. Patients
may use growth factors as per ASCO guidelines at the discretion of the
treating investigator.
Adverse Events
[00155] The CTEP Active Version of the NCI Common Terminology Criteria for
Adverse
Events (CTCAE 4.0) will be utilized for AE reporting. It is identified and
located on the CTEP
website at http://ctep.cancer.gov/protocolDevelopment/electronic
applications/ctc.htm. All
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
appropriate treatment areas should have access to a copy of the CTEP Active
Version of
CTCAE.
[00156] Attribution of the Adverse Event (AE):
= Definite ¨ The AE is clearly related to the study treatment.
= Probable ¨ The AE is likely related to the study treatment.
= Possible ¨ The AE may be related to the study treatment.
= Unlikely ¨ The AE is doubtfully related to the study treatment.
= Unrelated ¨ The AE is clearly NOT related to the study treatment.
[00157] List of Adverse Events to be Reported: abdominal pain; alkaline
phosphatase; ALT
(SGPT); anorexia; AST (SGOT); bilirubin; (hyperbilirubinemia); calcium
(hypercalcemia,
hypocalcemia); creatinine; diarrhea; flushing; hemoglobin (anemia); injection
site reaction;
leukocytes; lymphopenia; nausea; neutrophils (neutropenia); platelets
(thrombocytopenia);
potassium; sodium; vomiting. All grade 3, 4, 5 adverse events should be
reported on flow
sheets and in ORIS regardless of whether they are on this list. All unexpected
grade 4 and all
grade 5 SAE's on these trials be reported for review.
[00158] Any unanticipated problems involving risks to subjects or others and
adverse events
shall be promptly reported to the IRB. Reporting to the IRB is required
regardless of the
funding source, study sponsor, or whether the event involves an
investigational or marketed
drug, biologic or device. Reportable events are not limited to physical
injury, but include
psychological, economic and social harm. Reportable events may arise as a
result of drugs,
biological agents, devices, procedures or other interventions, or as a result
of questionnaires,
surveys, observations or other interactions with research subjects.
[00159] All members of the research team are responsible for the appropriate
reporting to the
IRB and other applicable parties of unanticipated problems involving risk to
subjects or others.
The Principal Investigator, however, is ultimately responsible for ensuring
the prompt reporting
of unanticipated problems involving risk to subjects or others to the IRB. The
Principal
Investigator is also responsible for ensuring that all reported unanticipated
risks to subjects and
others which they receive are reviewed to determine whether the report
represents a change in
the risks and/or benefits to study participants, and whether any changes in
the informed
consent, protocol or other study-related documents are required.
[00160] Any unanticipated problems involving risks to subjects or others
occurring at a site
where the study has been approved by the IRB (internal events) must be
reported to the IRB.
Any event, incident, experience, or outcome that alters the risk versus
potential benefit of the
research and as a result warrants a substantive change in the research
protocol or informed
51
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
consent process/document in order to insure the safety, rights or welfare of
research subjects
must be reported to the IRB.
Statistical Considerations
[00161] The sample size will be a total of 17 patients treated at the MTD of
HCQ for this
Phase 1/2 trial. This number is based on Simon's two stage design (Simon R.,
Controlled
Clinical Trials, 1989, 10: 1-10) where 9 patients will be enrolled in stage 1.
If none of the 9
patients have a response the study will be stopped for lack of efficacy. If
one or more patients
have a response the trial will continue until a total of 17 patients have been
treated with the
combination at the MTD of HCQ. If 2 or more of the 17 patients have a response
the
combination will be considered of sufficient activity to merit additional
study. These
parameters give an a of 0.0466 and a power of 0.8122 to detect a difference
from no
intervention (set to a response rate of 5%).
Data Management and Reporting Schedule
[00162] Disease response will be assessed based on RR, PFS, and OS (as
described by
Cheson B.D. et al., Clinical application and proposal for modification of the
International
Working Group (IWG) response criteria in myelodysplasia. Blood. 108:419-425,
2006), as
well as changes in the frequency of transfusion from baseline. RR and PFS,
derived from
hematology and bone marrow exam, will be assessed at the following specified
time points:
= Baseline
= Week 4 of every 2 treatment cycles until cycle 6
= Every 3 treatment cycles thereafter (i.e., cycle 9, cycle 12, cycle 15,
etc) until evidence
of disease progression.
Bone marrow exam results should be documented at each specified time point.
Example 12 ¨ Oral Efficacy of 6,8-bis-benzylthio-octanoic acid in Non-Small
Cell Lung
Cancer
[00163] Human H460 NSCLC cells were obtained from American Type Cell Culture
(ATCC) (catalog no. HTB-177, Manassas, VA). These cells tested negative for
viral
contamination using the Mouse Antibody Production (MAP) test, performed by
Charles River
Labs Molecular Division, upon the receipt of the tumor cells from ATCC. The
tumor cells were
maintained at 37 C in a humidified 5% CO2 atmosphere in T225 tissue culture
flasks
containing 50 mL of Roswell Park Memorial Institute (RPMI)-1640 solution with
10% Fetal
Bovine Serum (FBS) and 2 mM L-glutamine. Cells were split at a ratio of 1:10
every 2-3 days
52
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
by trypsinization and resuspended in fresh medium in a new flask. Cells were
harvested for
experiments in the same way at 70-90% confluency.
[00164] CD1-Nu/Nu female mice, ¨4-6 weeks old were obtained from Charles River
Laboratories. Mice were housed 5 to a cage in a micro-isolator room in the
Department of
Animal Laboratory Research of New York State University (SUNY) at Stony Brook.
Light-
dark cycles were 12 h each daily, with light from 7 a.m. to 7 p.m. Food
(Purina Rodent Chow)
and water (distilled sterile-filtered water, pH 7) were provided ad libitum.
Protocols and
procedures were according to the rules of and approved by the SUNY
Institutional Animal Care
and Use Committee (IACUC).
[00165] An acclimation period of 7 days was allowed between the arrival of the
animal at the
study site before tumor inoculation and experimentation. Mice were inoculated
subcutaneously
(SC) in the right flank with 2x106 human H460 NSCLC or BxPC3 pancreatic cancer
cells that
were suspended in 0.1 mL of Dulbeco's Phosphate Buffered Salt (PBS) solution
using a 1 cc
syringe with a 27-5/8 gauge needle. Tumor dimensions (length and width) were
measured daily
before, during and after treatment (using Vernier calipers) and the tumor
volume was calculated
using the prolate ellipsoid formula: (length x width2)/2. Treatment with test
or control articles
began 8 days post tumor cell implantation when the tumor was approximately 300
mm3.
[00166] Oral dosing of 6,8-bis-benzylthio-octanoic acid was at 100mg/kg with
11 animals
per group. 100 mg of 6,8-bis-benzylthio-octanoic acid was suspended in a small
volume 0.01-
0.05N NaOH in 5% dextrose and titrated to pH 7.0 with 4% Glacial Acetic Acid
to 50 mg/mL.
Prior to administration the suspension was diluted with 5% dextrose to 12.5
mg/mL so that the
animals received 100 mg/kg with a dose volume of about 0.2 mL delivered by
gastric gavage.
Post tumor cell implantation, mice were treated on day 8, day 15, day 22, and
day 29.
[00167] A similar study was conducted in CD-1 nude mice (n = 9) inoculated
with 2x106
BxPC-3 cells. The study was initiated when tumors reached an average size of
150 mm3 (day 0)
and CPI-613 was administered at an oral dose of 100 mg/week for 4 weeks. A
comparator arm
(n = 9) was conducted with IP treatment at a weekly dose of 25 mg/kg.
[00168] The results are presented in Figs. 15 and 16. It is evident that the
tumors in the mice
treated with 6,8-bis-benzylthio-octanoic acid grew much more slowly than those
in mice
treated with 5% dextrose or untreated. The effect was especially pronounced in
BxPC3 tumors.
This example demonstrates that 6,8-bis-benzylthio-octanoic acid is effective
to treat cancer
when administered orally.
53
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Example 13¨ Spray Dried Dispersion Oral Formulation of 6,8-bis-benzylthio-
octanoic acid
[00169] Solid amorphous dispersion formulations of 6,8-bis-benzylthio-octanoic
acid (API)
were prepared by mixing the API 1:4 with one of the following polymers:
Eudragit L100,
poly(vinylpyrrolidone) viscosity grade K30 (PVP 1(30), hydroxypropyl methyl
cellulose
(HPMC), cellulose acetate phthalate (CAP), or hydroxypropyl methylcellulose
acetate
succinate (HPMCAS-M), and spray drying from methanol or acetone using a small-
scale Bend
Lab Dryer with 35 kg/hr drying gas flow rate capacity (BLD-35). Conditions,
yields, and
residual solvent levels of two representative spray dried dispersion (SDD)
formulations (75 g
each) are presented in the following table.
Formulation 20% API
: Eudragit L100 20% API : HPMCAS-M
Spray Solution 5% solids in methanol 5% solids in acetone
Outlet Temp 45 C 35 C
Solution Feed Rate 35 g/min
Drying Gas Flow Rate 475-500 g/min
Atomization Pressure 120 psi
Nozzle Schlick 2.0 pressure swirl atomizer
Secondary Drying 20 hr at 30 C
Dry Yield (%) 94 96
Residual Solvent (%) (Wet SDD) 4.21 0.02 (Me0H) 1.01
0.00 (Acetone)
Residual Solvent (%) (Tray-Dried < LOQ < LOQ
Material)
API content by HPLC 201 1.1 mg/g 198 0.2 mg/g
[00170] Scanning electron microscopy (SEM) was used to qualitatively determine
particle
morphology of the two SDD formulations, and to study if any degree of fusion
or crystallinity
was visually present. Particles show collapsed sphere morphology with no
crystallization or
fusion noted.
[00171] X-ray diffraction is typically sensitive to the presence of
crystalline material with an
LOD of about 1% of the sample mass. No crystallinity was detected by PXRD for
either SDD
formulation. Diffractograms in comparison to crystalline 6,8-bis-benzylthio-
octanoic acid API
can be found in Fig. 17, wherein the top diffractogram is the Eudragit L100
formulation, the
middle diffractogram is the HPMCAS-M formulation, and the bottom diffractogram
is
crystalline 6,8-bis-benzylthio-octanoic acid.
54
CA 03121929 2021-06-02
WO 2020/132397
PCT/US2019/067759
Example 14 ¨ Emulsion Oral Formulations of 6,8-bis-benzylthio-octanoic acid
[00172] Monolaurin (131 mg) and 6,8-bis-benzylthio-octanoic acid (93 mg) were
warmed to
50 C in polysorbate-80 (2.5 mL) in a round bottomed flask equipped with a
magnetic stir bar.
After complete dissolution to a clear solution, water (7.5 mL) was added with
vigorous stirring
at 50 C to provide an emulsion.
[00173] 6,8-bis-benzylthio-octanoic acid (312 mg) was combined with
polysorbate 80 (6.25
g), soybean oil (1.25 g), and a lipid mix (100 mg) comprising cholesterol (14
g), cholesteryl
acetate (14 g), cholesteryl benzoate (14 g), monolaurin (25.4 g), and
monopalmitin (32.6 g),
and the mixture heated to 50 C until the solids dissolved (30 min). Dextrose
(11.25g) was
dissolved in 236 mL of water, and the resulting aqueous dextrose solution was
added to the oil
solution above. The resulting two phase mixture was stirred for 30 min at rt,
then vacuum
filtered through a 0.22 um filter.
Example 15 ¨ Liquid Formulations of 6,8-Bis-benzylthio-octanoic Acid
[00174] A 6,8-bis-benzylthio-octanoic acid solution was prepared by the steps
of (a)
providing a 50 mg/mL solution of 6,8-bis-benzylthio-octanoic acid in 1 M
aqueous
triethanolamine, and (b) diluting the 50 mg/mL solution with 5% aqueous
dextrose to a
concentration of 5 mg/mL. The resulting 5 mg/mL solution is identified as
"15A" in Example
16 below.
[00175] A suspension vehicle was prepared by the steps of: (a) combining tris
buffer (48 mg)
and HPMCAS-HF (20 mg) in 14 mL of distilled water, (b) adjusting the pH to 7.4
with dilute
sodium hydroxide to dissolve the HPMCAS-HF, (c) heating the resulting solution
to
approximately 90 C, (d) adding Methocel A4M Premium (100 mg) to the hot
solution, (e)
stirring the mixture vigorously to suspend the undissolved Methocel A4M, (0
cooling and
stirring the mixture with an ice bath until the Methocel A4M dissolves
(approximately 10
minutes), (g) diluting the solution with distilled/deionized water to bring
the total volume to 20
mL, and (h) adjusting the pH to 7.4 with dilute acetic acid or dilute sodium
hydroxide to
provide the suspension vehicle.
[00176] Suspensions of the spray-dried formulations of Example 13 were
prepared by adding
400 mg of the respective SDD formulation to a mortar, slowly adding 4 mL of
the suspension
vehicle (mixing thoroughly with a pestle after each small addition to
uniformly disperse), and
then transferring to a flask and stirring for one minute prior to
administration. The resulting
suspension of the Eudragit L100 SDD formulation (20 mg/mL 6,8-bis-benzylthio-
octanoic
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
acid) is identified as "15B" in Example 16 below. The resulting suspension of
the HPMCAS-M
SDD formulation (20 mg/mL 6,8-bis-benzylthio-octanoic acid) is identified as
"15C" in
Example 16 below.
[00177] In the same way, a 20 mg/mL suspension of 6,8-bis-benzylthio-octanoic
acid was
prepared by adding 80 mg 6,8-bis-benzylthio-octanoic acid to a mortar, slowly
adding 4 mL of
the suspension vehicle (mixing thoroughly with a pestle after each small
addition to uniformly
disperse), and then transferring to a flask and stirring for one minute prior
to administration.
The resulting suspension of 6,8-bis-benzylthio-octanoic acid is identified as
"15D" in Example
16 below.
[00178] A solution of 6,8-bis-benzylthio-octanoic acid was prepared by
dissolving
SOLUTOLO (polyoxyl 15 hydroxystearate; KOLLIPHORO HS 15) (3 grams) in
distilled
water (7 mL) to form a 30% solution, adding 6,8-bis-benzylthio-octanoic acid
(50 mg) to 5 mL
of the 30% solution, vortexing for 1 minute, and then sonicating for 45
minutes to provide a
clear colorless solution (10 mg/mL; pH 7). The resulting solution is
identified as "15E" in
Example 16 below.
Example 16 ¨ Oral Bioavailability of 6,8-Bis-benzylthio-octanoic Acid
[00179] Six groups of 16 BALB/c nude mice (8 males and 8 females) per group
were
administered 6,8-bis-benzylthio-octanoic acid in six different ways: (1) 54/g
IV injection (tail
vein) of the triethanolamine/dextrose aqueous solution of Example 15 (25
mg/kg; 5 mL/kg; Ex.
15A); (2) 5 1,1L/g IP injection of the triethanolamine/dextrose aqueous
solution of Example 15
(25 mg/kg; 5 mL/kg; 15A); (3) 5 1,1L/g oral administration of the Eudragit
L100 SDD
suspension of Example 15 (100 mg/kg; 5 mL/kg; 15B); (4) 54/g oral
administration of the
HPMCAS-M SDD suspension of Example 15 (100 mg/kg; 5 mL/kg; 15C); (5) 54/g oral
administration of the 20 mg/mL 6,8-bis-benzylthio-octanoic acid suspension of
Example 15
(100 mg/kg; 5 mL/kg; 15D); or (6) 104/g oral administration of the 10 mg/mL
SOLUTOL
solution of Example 15 (100 mg/kg; 10 mL/kg; 15E). In each experiment, about
804 of blood
was collected from one subgroup of 4 male and 4 female mice at 0.083, 1, 4,
and 24 hours after
dosing, and from the other subgroup of 4 male and 4 female mice at 0.5, 2, and
8 hours. Plasma
from the collected blood samples was analyzed by LC-MS/MS for the presence of
6,8-bis-
benzylthio-octanoic acid.
56
CA 03121929 2021-06-02
WO 2020/132397 PCT/US2019/067759
Bioavail- AUC
Mice Dose Cmax Tmax
Formulation Route ability Last 1/2
(n) (mg/kg) (uM) (hr)
(%) (uM*hr) (hr)
15A
IV 16 25 36 92
0.08 2.0
(TEA/dextrose)
15A
IP 16 25 83 29 103
0.08 3.9
(TEA/dextrose)
15B (Eudragit
PO 16 100 44 61 94
0.08 2.0
SDD)
15C (HPMCAS-M
FO 16 100 43 60 69
0.08 1.1
SDD)
15D (CPI-613) PO 16 100 57 82 82 0.50 3.7
15E (Solutol) PO 16 100 127 175 229 0.08 4.4
[00180] This example demonstrates that 6,8-bis-benzylthio-octanoic acid is
orally
bioavailable.
INCORPORATION BY REFERENCE
[00181] The entire disclosure of each of the patent documents and scientific
articles referred
to herein is incorporated by reference for all purposes.
EQUIVALENTS
[00182] The invention may be embodied in other specific forms without
departing from the
spirit or essential characteristics thereof The foregoing embodiments are
therefore to be
considered in all respects illustrative rather than limiting the invention
described herein. Scope
of the invention is thus indicated by the appended claims rather than by the
foregoing
description, and all changes that come within the meaning and range of
equivalency of the
claims are intended to be embraced therein.
57