Note: Descriptions are shown in the official language in which they were submitted.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
MODIFIED CELL EXPANSION AND USES THEREOF
CROSS REFERENCE TO RELATED PATENT APPLICATIONS
[0001] This application is a continuation-in-part of U.S. Application
16/445,965, filed June 19,
2019; and U.S. Application 16/387,166, filed April 17, 2019. This application
also claims the
benefit of U.S. Provisional Application 62/932,587, filed November 8, 2019;
U.S. Provisional
Application 62/902,766, filed September 19, 2019; U.S. Provisional Application
62/891,131, filed
August 23, 2019; U.S. Provisional Application 62/889,926, filed August 21,
2019; U.S. Provisional
Application 62/848,961, filed May 16, 2019; U.S. Provisional Application
62/846,563, filed May
10, 2019; U.S. Provisional Application 62/817,322, filed March 12, 2019; U.S.
Provisional
Application 62/816,497, filed March 11, 2019; U.S. Provisional Application
62/799,462, filed
January 31, 2019; and U.S. Provisional Application 62/790,783, filed January
10, 2019; which are
hereby incorporated by reference in their entirety.
SEQUENCE LISTING INFORMATION
[0002] A computer readable textfile, entitled "Sequence Listing_5T25.bd,"
created on or
about January 6, 2020 with a file size of about 1.20 MB, contains the sequence
listing for this
application and is hereby incorporated by reference in its entirety.
TECHNICAL FIELD
[0003] The present disclosure relates to compositions and methods for
expanding and
maintaining modified cells including genetically modified cells, and uses
thereof in the treatment
of diseases, including cancer.
BACKGROUND
[0004] Chimeric Antigen Receptor (CAR) T cell therapy has achieved good
clinical efficacy
in cancer such as B-cell acute lymphoblastic leukemia (B-ALL), chronic
lymphocytic leukemia
(CLL), and lymphoma. However, progress is relatively slow for treatment of
solid tumors. For
CAR T cell therapy to be effective, long-term maintenance of CAR T cells in a
patient is
important for the prognosis of the patient in the treatment of tumors. For
example, if the long-
term presence of CAR T cells can be maintained, this technology may
effectively reduce tumor
recurrence.
[0005] Cancer is known as malignant tumors involving abnormal cell growth
with the
potential to invade or spread to other parts of the body. In humans, there are
more than one
hundred types of cancer. One example is breast cancer occurring in the
epithelial tissue of the
breast. Since breast cancer cells lose the characteristics of normal cells,
the connection
between breast cancer cells is lost. Once cancer cells are exfoliated, they
spread over the entire
body via the blood and/or lymph systems and therefore become life-threatening.
Currently,
breast cancer has become one of the common threats to women's physical and
mental health.
Although immunotherapy (e.g., CAR T) has been proven to be effective for
treating some
1
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cancers, there is still a need to improve immunotherapy so that it is
effective in treating more
cancers including those involving solid tumors.
SUMMARY
[0006] Since a patient can survive the depletion of B cells, B cells of the
patient may be
used to expand the CAR T cells in the patient using a first antigen binding
domain of the CAR T
cell. Accordingly, more CAR T cells may be timely expanded in the patient,
increasing the
potency of CAR T cells. The timely expanded CAR T cells in the patient may
increase the
chances for the CAR T cells to come in contact with tumor cells, especially
solid tumor cells
having the antigen that a second CAR binds.
[0007] The present disclosure describes genetically modified cells that
include one or more
different antigen binding domains. The genetically modified cells can include
at least two
different antigen binding domains: a first antigen binding domain for
expanding and/or
maintaining the genetically modified cells, and a second antigen binding
domain for killing a
target cell, such as a tumor cell. For example, the first antigen binding
domain binds a surface
marker, such as a cell surface molecule of a white blood cell (WBC), and the
second antigen
binding domain binds a target antigen of tumor cells. In embodiments, the cell
surface molecule
is a surface antigen of a WBC. A CAR can comprise the first or second antigen
binding domain.
The modified cells comprise the first and second antigen binding domains. In
embodiments, the
modified cells comprise modified cells comprising (1) a first group of
modified cells comprising
the first antigen binding domain and (2) a second group of modified cells
comprising the second
binding domain. In embodiments, the modified cells are a mixed population
comprising two
different groups of modified cells
[0008] The CAR can be a bispecific CAR. For example, the two antigen
binding domains
are on the same CAR (a bispecific CAR or tandem CAR (tanCAR)), on different
CAR molecules,
or on a CAR and T cell receptor (TCR). A single CAR can include at least two
different antigen
binding domains, or the two different antigen binding domains are each on a
separate CAR.
[0009] The present disclosure also describes one or more nucleic acids
encoding a first
CAR molecule and a second CAR molecule or a TCR. The first CAR includes the
first antigen
binding domain and the second CAR or TCR includes the second antigen binding
domain. In
embodiments, the first CAR and the second CAR or TCR are expressed as separate
polypeptides and encoded by at least two separate nucleic acids. In
embodiments, a single
CAR contains at least the first and second antigen binding domains described
herein and is
encoded by a single nucleic acid. In embodiments, the two different antigen
binding domains
can be encoded by more than one nucleic acids. Moreover, the present
disclosure describes
vectors containing the nucleic acids described herein and cells comprising the
nucleic acids
described herein. In embodiments, the cells include genetically modified
cells, for example
genetically modified T cells, such as CAR T cells.
2
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[0010] The present disclosure also describes a population of modified
cells, such as a
mixed population of modified T cells, effective for expanding and/or
maintaining the genetically
modified cells in a patient. In embodiments, the mixed population of
genetically modified cells
includes at least two different genetically modified cells, a first
genetically modified cell
expressing an antigen binding domain for expanding and/or maintaining the
modified cells and a
second genetically modified cell expressing an antigen binding domain for
killing a target cell,
such as a tumor cell. The two antigen binding domains are different molecules
and bind
different antigens. In embodiments, the mixed population of genetically
modified cells further
includes a third genetically modified cell expressing at least two different
antigen binding
domains, a first antigen binding domain for expanding and/or maintaining the
genetically
modified cell and a second antigen binding domain for killing a target cell
(wherein the two
different antigen binding domains are expressed on the same cell).
[0011] In embodiments, the mixed population of modified cells includes
genetically modified
cells expressing at least two different antigen binding domains, a first
antigen binding domain
for expanding and/or maintaining the modified cells and a second antigen
binding domain for
killing a target cell (wherein the two different antigen binding domains are
expressed on the
same cell).
[0012] In embodiments, the mixed population of modified cells includes a
modified cell
expressing an antigen binding domain for killing a target cell and a modified
cell expressing at
least two antigen binding domains, a first antigen binding domain for
expanding and/or
maintaining the modified T cells and a second antigen binding domain for
killing a target cell
(wherein the two different antigen binding domains are expressed on the same
modified cell).
[0013] In embodiments, the mixed population of modified cells includes a
modified cell
expressing an antigen binding domain for expanding and/or maintaining the
modified T cells
and a modified cell expressing at least two antigen binding domains, a first
antigen binding
domain for expanding and/or maintaining the modified cell and a second antigen
binding
domain for killing a target cell (wherein the two different antigen binding
domains are expressed
on the same modified cell).
[0014] The present disclosure describes compositions comprising the mixed
population of
modified cells described herein.
[0015] In embodiments, the modified cell is a modified T cell, a modified
NK cell, a modified
macrophage, or a modified dendritic cell. In embodiments, the modified T cell
is a CAR T cell. In
embodiments, the modified cell expressing two different antigen binding
domains can be a
single CAR T cell. In embodiments, the single CAR T cell can be a bispecific
CAR T cell.
[0016] In embodiments, the antigen binding domain for expanding and/or
maintaining the
modified cell binds the surface antigen of a WBC, and the antigen binding
domain for killing a
target cell binds a tumor antigen. In embodiments, the WBC is a B cell. In
embodiments, the
3
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
surface antigen of a B cell is CD19, and the tumor antigen is tMUC1, TSHR,
GUCY2C, ACPP,
CLDN18.2 (18.2), PSMA, UPK2, or other tumor antigens.
[0017] Furthermore, the present disclosure describes the use of the
composition or the
mixed population of modified cells described herein for enhancing expansion
and/or
maintenance of CAR T cells in patients in need thereof. The enhanced expansion
and
maintenance of CAR T cells improves the efficacy of the CAR T cell therapy.
The present
disclosure describes a method of treating a patient having tumor with a mixed
population of
modified cells described herein. In embodiments, the mixed population of
genetically modified
cells expands and/or maintains the modified cells in the patient and
effectively inhibits the
growth of the tumor. In embodiments, the tumor is a solid tumor.
[0018] Additionally, the present disclosure describes the release of
cytokines in response to
the introduction of the mixed population of modified cells.
[0019] This Summary is not intended to identify key features or essential
features of the
claimed subject matter, nor is it intended to be used to limit the scope of
the claimed subject
matter.
BRIEF DESCRIPTION OF THE DRAWINGS
[0020] The Detailed Description is described with reference to the
accompanying figures.
The use of the same reference numbers in different figures indicates similar
or identical items.
[0021] FIG. 1 is a schematic diagram of an exemplary portion of a cell
membrane of a
modified cell including two CAR molecules.
[0022] FIG. 2 is a schematic diagram showing a mixed population of modified
cells including
two modified cells having different CAR molecules.
[0023] FIG. 3 is a schematic diagram showing an exemplary portion of a cell
membrane
comprising a CAR and a TCR molecules.
[0024] FIG. 4 is a schematic diagram showing a mixed population of modified
cells including
a modified cell comprising a CAR molecule and a modified cell comprising a T
cell receptor
(TCR).
[0025] FIG. 5 is a schematic diagram showing an exemplary portion of a cell
membrane
comprising a bispecific CAR molecule.
[0026] FIG. 6 shows cytokine data of peripheral blood samples from mice.
[0027] FIG. 7 shows a design of the bispecific CAR and results of
expression assays.
[0028] FIG. 8 shows cytokine release of T cells expressing a bispecific
CAR.
[0029] FIG. 9 shows results of co-culturing assay of T cells expressing a
bispecific CAR and
corresponding target cells.
[0030] FIG. 10 shows another design of bispecific CAR and results of
expression assays.
[0031] FIG. 11 shows results of an expression assay of the bispecific CAR
used in the
assay of FIG. 10.
[0032] FIG. 12 shows schematic diagrams of nucleic acid constructs of CAR
molecules.
4
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[0033] FIG. 13 shows expression of the CAR molecules shown in FIG. 12.
[0034] FIG. 14 shows results of IFNy (IFNg) release of co-culturing CAR T
cells with tumor
cells.
[0035] FIG. 15 shows flow cytometry results depicting 0D137 expression for
co-culturing of
CAR T cells and tumor cells.
[0036] FIG. 16 shows changes in CAR copy number of patients with respect to
days after
infusion of T cells expressing a single CAR (tMUC1 CAR or TSHR CAR).
[0037] FIG. 17 shows changes in CAR copy number of patients with respect to
days after
infusion of T cells expressing tMUC1 CAR and CD19 CAR.
[0038] FIG. 18 shows changes in CAR T cell number of a patient with respect
to days after
infusion of T cells expressing tMUC1 CAR.
[0039] FIG. 19 shows changes in CAR T cell number of a patient with respect
to days after
infusion of mixed population of CAR T cells expressing tMUC1 CAR and CD19 CAR.
[0040] FIGS. 20 and 21 show changes in CAR T cell number of several
patients with
respect to days after infusion of mixed CAR T cells expressing MUC1 CAR and
CD19 CAR.
[0041] FIGS. 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, and 32 show results of
various assays
for patients in response to infusion of mixed CAR T cells.
[0042] FIGS. 33, 34, and 35 show CT and/or PET CT scanning images of
patients before
and after the infusion of mixed CAR T cells.
[0043] FIG. 36 shows results of flow cytometry analysis of CD19 CAR T cells
co-cultured
with tMUC1 CAR T cells in the presence or the absence of K19 cells.
[0044] FIG. 37 shows the activation of PBMC and monocytes in the cell
cultures used in the
assay of FIG. 36.
[0045] FIG. 38 shows IFNy release by tMUC1 CAR T cells and CD19 CAR T
cells.
[0046] FIG. 39 shows GZMB release by tMUC1CAR T cells and CD19 CAR T cells.
[0047] FIGS. 40 and 41 show proliferation of MUC1CAR T cells in various
embodiments.
[0048] FIG. 42 shows proliferation of CD19 CAR T cells in various
embodiments.
[0049] FIG. 43 shows cytokine releases in embodiments.
[0050] FIG. 44 shows CD137 expression in various cell cultures.
[0051] FIG. 45 shows results of flow cytometry analysis of cell activation.
[0052] FIG. 46 shows the activation of PBMC and monocyte in the cell
cultures described in
FIG. 44.
[0053] FIG. 47 shows that activation of CD19 CAR T cells causes ACPP CAR T
cells to
release intracellular IFNy.
[0054] FIGS. 48 and 49 show cytokine releases after cells are co-cultured
for 24h0ur5 (hrs)
in cell cultures.
[0055] FIG. 50 shows CD137 expression in various cell cultures.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[0056] FIG. 51 shows results of flow cytometry analysis of various CAR T
cells co-cultured
with KAT03+ cells for 48 hours.
[0057] FIG. 52 shows the activation of PBMC and monocyte in the systems
described in
FIG. 50.
[0058] FIGS. 53 and 54 show activation of CLDN18.2 CAR T cells causes CD19
CAR T
cells to release intracellular IFNy.
[0059] FIG. 55 shows results of killing assays of various cell cultures.
[0060] FIG. 56 shows proliferation of CLDN18.2 CAR T cells.
[0061] FIG. 57 shows proliferation of CD19 CAR T cells in CLDN18.2 CAR and
CD19 CAR
systems.
[0062] FIGS. 58, 59, and 60 show cytokine release in various cell cultures.
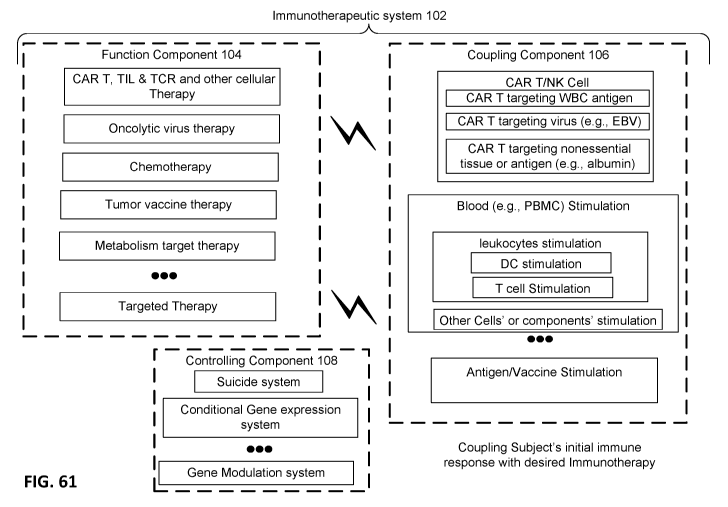
[0063] FIG. 61 shows a schematic overview of an immunotherapeutic system.
[0064] FIG. 62 shows a schematic overview of an implementation of the
immunotherapeutic
system in FIG. 61.
[0065] FIG. 63 shows a schematic overview of another implementation of the
immunotherapeutic system in FIG. 61.
[0066] FIG. 64 is a schematic diagram of exemplary conditional gene
expression systems.
[0067] FIG. 65 is a schematic diagram of exemplary implementations of
dendritic cell
activation.
[0068] FIG. 66 shows expression of several markers on CAR T cells and
TanCAR T cells
using flow cytometry analysis.
[0069] FIG. 67 shows cytokine release of CAR T cells and TanCAR T cells.
[0070] FIG. 68 shows the expansion of cells in each group after 5 days of
stimulation with
the corresponding substrate cells.
[0071] FIG. 69 shows results of killing assay indicating that 6917
inhibited MCF-7 and 6921
inhibited PC3-ACPP.
[0072] FIG. 70 shows expression of several markers on CAR T cells and
TanCAR T cells
and the cytokine release of CAR T cells and TanCAR T cells measured using flow
cytometry
analysis.
[0073] FIG. 71 shows cytokine release of various CAR T cells and TanCAR T
cells in
response to substrate cells.
[0074] FIG. 72 shows PDL1 expression of monocytes in Patient 009.
[0075] FIGS. 73, 74, and 75 show expansion of CAR T cell in Patient 011 in
response to
infusion of modified T cells.
[0076] FIG. 76 shows cytokine release in Patient 011 in response to
infusion of modified T
cells.
[0077] FIGs. 77A and B illustrate exemplary structures of binding
molecules.
6
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[0078] FIG. 78 illustrates the determination of phenotype and expression of
a gene of
interest using flow cytometry.
[0079] FIG. 79 shows the identification of co-cultured cells using flow
cytometry.
[0080] FIG. 80 shows results of flow cytometry analysis on activation of co-
cultured cells
including CD19 CAR T cells and NYESO-1 TORTS. Arrows 114 and 116 as well as
boxes 102,
104, 106, and 108 refer to comparison groups.
[0081] FIG. 81 show results of flow cytometry analysis on the proliferation
of co-cultured
cells including CD19 CAR T cells and NYESO-1 TORTS. Arrow 208 as well as boxes
202, 204,
and 206 refer to comparison groups.
[0082] FIG. 82 show results of flow cytometry analysis on activation of co-
cultured cells
including CD19 CAR T cells and AFP TORTS. Arrows 314 and 316 as well as boxes
302, 304,
306, and 308 refer to comparison groups.
[0083] FIG. 83 show results of flow cytometry analysis on the proliferation
of co-cultured
cells including 0D19 CAR T cells and AFP TORTS. Arrow 408 as well as boxes
402, 404, and
406 refer to comparison groups.
[0084] FIG. 84 shows other histograms of 0D137 expression in various cell
cultures.
[0085] FIG. 85 shows the proliferation of GUCY2C CAR T cells.
[0086] FIG. 86 shows cytokine release after cells were co-cultured for 24
hrs in cell cultures.
[0087] FIGs. 87A-87D illustrate exemplary constructs of polynucleotides
encoding
recombinant proteins and exemplary structure of antibodies.
DETAILED DESCRIPTION
[0088] Unless defined otherwise, all technical and scientific terms used
herein have the
same meaning as commonly understood by those of ordinary skill in the art to
which the
disclosure belongs. Although any method and material similar or equivalent to
those described
herein can be used in the practice or testing of the present disclosure,
preferred methods and
materials are described. For the purposes of the present disclosure, the
following terms are
defined below.
[0089] The articles "a" and "an" are used herein to refer to one or to more
than one (i.e., to
at least one) of the grammatical object of the article. By way of example, "an
element" means
one element or more than one element.
[0090] By "about" is meant a quantity, level, value, number, frequency,
percentage,
dimension, size, amount, weight or length that varies by as much as 20, 15,
10, 9, 8, 7, 6, 5, 4,
3, 2 or 1% to a reference quantity, level, value, number, frequency,
percentage, dimension,
size, amount, weight or length.
[0091] The term "activation," as used herein, refers to the state of a cell
that has been
sufficiently stimulated to induce detectable cellular proliferation.
Activation can also be
7
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
associated with induced cytokine production and detectable effector functions.
The term
"activated T cells" refers to, among other things, T cells that are undergoing
cell division.
[0092] The term "antibody" is used in the broadest sense and refers to
monoclonal
antibodies (including full length monoclonal antibodies), polyclonal
antibodies, multi-specific
antibodies (e.g., bispecific antibodies), and antibody fragments so long as
they exhibit the
desired biological activity or function. The antibodies in the present
disclosure may exist in a
variety of forms including, for example, polyclonal antibodies; monoclonal
antibodies; Fv, Fab,
Fab', and F(ab')2 fragments; as well as single chain antibodies and humanized
antibodies
(Harlow et al., 1999, In: Using Antibodies: A Laboratory Manual, Cold Spring
Harbor Laboratory
Press, NY; Harlow et al., 1989, In: Antibodies: A Laboratory Manual, Cold
Spring Harbor, New
York; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et
al., 1988, Science
242:423-426).
[0093] The term "antibody fragments" refers to a portion of a full-length
antibody, for
example, the antigen binding or variable region of the antibody. Other
examples of antibody
fragments include Fab, Fab', F(ab')2, and Fv fragments; diabodies; linear
antibodies; single-
chain antibody molecules; and multi-specific antibodies formed from antibody
fragments.
[0094] The term "Fv" refers to the minimum antibody fragment which contains
a complete
antigen-recognition and -binding site. This fragment consists of a dimer of
one heavy- and one
light-chain variable region domain in tight, non-covalent association. From
the folding of these
two domains emanates six hypervariable loops (3 loops each from the H and L
chain) that
contribute amino acid residues for antigen binding and confer antigen binding
specificity to the
antibody. However, even a single variable domain (or half of an Fv including
only three
complementarity determining regions (CDRs) specific for an antigen) has the
ability to recognize
and bind antigen, although at a lower affinity than the entire binding site
(the dimer).
[0095] An "antibody heavy chain," as used herein, refers to the larger of
the two types of
polypeptide chains present in all antibody molecules in their naturally
occurring conformations.
An "antibody light chain," as used herein, refers to the smaller of the two
types of polypeptide
chains present in all antibody molecules in their naturally occurring
conformations. K and A light
chains refer to the two major antibody light chain isotypes.
[0096] The term "synthetic antibody" refers to an antibody which is
generated using
recombinant DNA technology, such as, for example, an antibody expressed by a
bacteriophage.
The term also includes an antibody which has been generated by the synthesis
of a DNA
molecule encoding the antibody and the expression of the DNA molecule to
obtain the antibody
or to obtain an amino acid encoding the antibody. The synthetic DNA is
obtained using
technology that is available and well known in the art.
[0097] The term "antigen" refers to a molecule that provokes an immune
response, which
may involve either antibody production, or the activation of specific
immunologically-competent
cells, or both. Antigens include any macromolecule, including all proteins or
peptides, or
8
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
molecules derived from recombinant or genomic DNA. For example, DNA including
a nucleotide
sequence or a partial nucleotide sequence encoding a protein or peptide that
elicits an immune
response, and therefore, encodes an "antigen" as the term is used herein. An
antigen need not
be encoded solely by a full-length nucleotide sequence of a gene. An antigen
can be generated,
synthesized or derived from a biological sample including a tissue sample, a
tumor sample, a
cell, or a biological fluid.
[0098] The term "anti-tumor effect" as used herein, refers to a biological
effect associated
with a decrease in tumor volume, a decrease in the number of tumor cells, a
decrease in the
number of metastases, decrease in tumor cell proliferation, decrease in tumor
cell survival, an
increase in life expectancy of a subject having tumor cells, or amelioration
of various
physiological symptoms associated with the cancerous condition. An "anti-tumor
effect" can also
be manifested by the ability of the peptides, polynucleotides, cells, and
antibodies in the
prevention of the occurrence of tumor in the first place.
[0099] The term "auto-antigen" refers to an endogenous antigen mistakenly
recognized by
the immune system as being foreign. Auto-antigens include cellular proteins,
phosphoproteins,
cellular surface proteins, cellular lipids, nucleic acids, glycoproteins,
including cell surface
receptors.
[00100] The term "autologous" is used to describe a material derived from a
subject which is
subsequently re-introduced into the same subject.
[00101] The term "allogeneic" is used to describe a graft derived from a
different subject of
the same species. As an example, a donor subject may be a related or unrelated
to the
recipient subject, but the donor subject has immune system markers which are
similar to the
recipient subject.
[00102] The term "xenogeneic" is used to describe a graft derived from a
subject of a
different species. As an example, the donor subject is from a different
species than a recipient
subject, and the donor subject and the recipient subject can be genetically
and immunologically
incompatible.
[00103] The term "cancer" is used to refer to a disease characterized by
the rapid and
uncontrolled growth of aberrant cells. Cancer cells can spread locally or
through the
bloodstream and lymphatic system to other parts of the body. Examples of
various cancers
include breast cancer, prostate cancer, ovarian cancer, cervical cancer, skin
cancer, pancreatic
cancer, colorectal cancer, renal cancer, liver cancer, brain cancer, lymphoma,
leukemia, lung
cancer, and the like.
[00104] Throughout this specification, unless the context requires
otherwise, the words
"comprise," "includes" and "including" will be understood to imply the
inclusion of a stated step
or element or group of steps or elements but not the exclusion of any other
step or element or
group of steps or elements.
9
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00105] The phrase "consisting of" is meant to include, and is limited to,
whatever follows the
phrase "consisting of." Thus, the phrase "consisting of" indicates that the
listed elements are
required or mandatory and that no other elements may be present.
[00106] The phrase "consisting essentially of" is meant to include any
element listed after the
phrase and can include other elements that do not interfere with or contribute
to the activity or
action specified in the disclosure for the listed elements. Thus, the phrase
"consisting
essentially of' indicates that the listed elements are required or mandatory,
but that other
elements are optional and may or may not be present depending upon whether or
not they
affect the activity or action of the listed elements.
[00107] The terms "complementary" and "complementarity" refer to
polynucleotides (i.e., a
sequence of nucleotides) related by the base-pairing rules. For example, the
sequence "A-G-T,"
is complementary to the sequence "T-C-A." Complementarity may be "partial," in
which only
some of the nucleic acids' bases are matched according to the base pairing
rules, or there may
be "complete" or "total" complementarity between the nucleic acids. The degree
of
complementarity between nucleic acid strands has significant effects on the
efficiency and
strength of hybridization between nucleic acid strands.
[00108] The term "corresponds to" or "corresponding to" refers to (a) a
polynucleotide having
a nucleotide sequence that is substantially identical or complementary to all
or a portion of a
reference polynucleotide sequence or encoding an amino acid sequence identical
to an amino
acid sequence in a peptide or protein; or (b) a peptide or polypeptide having
an amino acid
sequence that is substantially identical to a sequence of amino acids in a
reference peptide or
protein.
[00109] The term "co-stimulatory ligand," refers to a molecule on an
antigen presenting cell
(e.g., an APC, dendritic cell, B cell, and the like) that specifically binds a
cognate co-stimulatory
molecule on a T cell, thereby providing a signal which, in addition to the
primary signal provided
by, for instance, binding of a TCR/CD3 complex with an MHC molecule loaded
with peptide,
mediates a T cell response, including at least one of proliferation,
activation, differentiation, and
other cellular responses. A co-stimulatory ligand can include B7-1 (CD80), B7-
2 (0D86), PD-L1,
PD-L2, 4-1BBL, OX4OL, inducible co-stimulatory ligand (ICOS-L), intercellular
adhesion
molecule (ICAM), CD3OL, CD40, CD70, 0D83, HLA-G, MICA, MICB, HVEM, lymphotoxin
beta
receptor, 3/TR6, ILT3, ILT4, HVEM, a ligand for CD7, an agonist or antibody
that binds the Toll
ligand receptor, and a ligand that specifically binds with B7-H3. A co-
stimulatory ligand also
includes, inter alia, an agonist or an antibody that specifically binds with a
co-stimulatory
molecule present on a T cell, such as CD27, CD28, 4-i BB, 0X40, CD30, CD40, PD-
1, ICOS,
lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-
H3, and a
ligand that specifically binds CD83.
[00110] The term "co-stimulatory molecule" refers to the cognate binding
partner on a T cell
that specifically binds with a co-stimulatory ligand, thereby mediating a co-
stimulatory response
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
by the T cell, such as proliferation. Co-stimulatory molecules include an MHC
class I molecule,
BTLA, and a Toll-like receptor.
[00111] The term "co-stimulatory signal" refers to a signal, which in
combination with a
primary signal, such as TCR/CD3 ligation, leads to T cell proliferation and/or
upregulation or
downregulation of key molecules.
[00112] The terms "disease" and "condition" may be used interchangeably or
may be
different in that the particular malady or condition may not have a known
causative agent (so
that etiology has not yet been worked out), and it is therefore not yet
recognized as a disease
but only as an undesirable condition or syndrome, wherein a more or less
specific set of
symptoms have been identified by clinicians. The term "disease" is a state of
health of a subject
wherein the subject cannot maintain homeostasis, and wherein if the disease is
not ameliorated
then the subject's health continues to deteriorate. In contrast, a "disorder"
in a subject is a state
of health in which the animal is able to maintain homeostasis, but in which
the animal's state of
health is less favorable than it would be in the absence of the disorder. Left
untreated, a
disorder does not necessarily cause a further decrease in the animal's state
of health.
[00113] The term "effective" refers to adequate to accomplish a desired,
expected, or
intended result. For example, an "effective amount" in the context of
treatment may be an
amount of a compound sufficient to produce a therapeutic or prophylactic
benefit.
[00114] The term "encoding" refers to the inherent property of specific
sequences of
nucleotides in a polynucleotide, such as a gene, a cDNA, or an mRNA, to serve
as a template
for synthesis of other polymers and macromolecules in biological processes
having either a
defined sequence of nucleotides (i.e., rRNA, tRNA and mRNA) or a defined
sequence of amino
acids and the biological properties resulting therefrom. Thus, a gene encodes
a protein if
transcription and translation of mRNA corresponding to that gene produces the
protein in a cell
or other biological system. Both the coding strand, the nucleotide sequence of
which is identical
to the mRNA sequence (except that a "T" is replaced by a "U") and is usually
provided in
sequence listings, and the non-coding strand, used as the template for
transcription of a gene or
cDNA, can be referred to as encoding the protein or other product of that gene
or cDNA.
[00115] The term "exogenous" refers to a molecule that does not naturally
occur in a wild-
type cell or organism but is typically introduced into the cell by molecular
biological techniques.
Examples of exogenous polynucleotides include vectors, plasmids, and/or man-
made nucleic
acid constructs encoding the desired protein. VVith regard to polynucleotides
and proteins, the
term "endogenous" or "native" refers to naturally-occurring polynucleotide or
amino acid
sequences that may be found in a given wild-type cell or organism. Also, a
particular
polynucleotide sequence that is isolated from a first organism and transferred
to a second
organism by molecular biological techniques is typically considered an
"exogenous"
polynucleotide or amino acid sequence with respect to the second organism. In
specific
embodiments, polynucleotide sequences can be "introduced" by molecular
biological techniques
11
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
into a microorganism that already contains such a polynucleotide sequence, for
instance, to
create one or more additional copies of an otherwise naturally-occurring
polynucleotide
sequence, and thereby facilitate overexpression of the encoded polypeptide.
[00116] The term "expression or overexpression" refers to the transcription
and/or translation
of a particular nucleotide sequence into a precursor or mature protein, for
example, driven by its
promoter. "Overexpression" refers to the production of a gene product in
transgenic organisms
or cells that exceeds levels of production in normal or non-transformed
organisms or cells. As
defined herein, the term "expression" refers to expression or overexpression.
[00117] The term "expression vector" refers to a vector including a
recombinant
polynucleotide including expression control (regulatory) sequences operably
linked to a
nucleotide sequence to be expressed. An expression vector includes sufficient
cis-acting
elements for expression; other elements for expression can be supplied by the
host cell or in an
in vitro expression system. Expression vectors include all those known in the
art, such as
cosmids, plasmids (e.g., naked or contained in liposomes) and viruses (e.g.,
lentiviruses,
retroviruses, adenoviruses, and adeno-associated viruses) that incorporate the
recombinant
polynucleotide.
[00118] Viruses can be used to deliver nucleic acids into a cell in vitro
and in vivo (in a
subject). Examples of viruses useful for delivery of nucleic acids into cells
include retrovirus,
adenovirus, herpes simplex virus, vaccinia virus, and adeno-associated virus.
[00119] There also exist non-viral methods for delivering nucleic acids
into a cell, for
example, electroporation, gene gun, sonoporation, magnetofection, and the use
of
oligonucleotides, lipoplexes, dendrimers, and inorganic nanoparticles.
[00120] The term "homologous" refers to sequence similarity or sequence
identity between
two polypeptides or between two polynucleotides when a position in both of the
two compared
sequences is occupied by the same base or amino acid monomer subunit, e.g., if
a position in
each of two DNA molecules is occupied by adenine, then the molecules are
homologous at that
position. The percent of homology between two sequences is a function of the
number of
matching or homologous positions shared by the two sequences divided by the
number of
positions compared x100. For example, if 6 of 10 of the positions in two
sequences are
matched or homologous, then the two sequences are 60% homologous. By way of
example, the
DNA sequences ATTGCC and TATGGC share 50% homology. A comparison is made when
two sequences are aligned to give maximum homology.
[00121] The term "immunoglobulin" or "Ig," refers to a class of proteins,
which function as
antibodies. The five members included in this class of proteins are IgA, IgG,
IgM, IgD, and IgE.
IgA is the primary antibody that is present in body secretions, such as
saliva, tears, breast milk,
gastrointestinal secretions and mucus secretions of the respiratory and
genitourinary tracts. IgG
is the most common circulating antibody. IgM is the main immunoglobulin
produced in the
primary immune response in most subjects. It is the most efficient
immunoglobulin in
12
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
agglutination, complement fixation, and other antibody responses, and is
important in defense
against bacteria and viruses. IgD is the immunoglobulin that has no known
antibody function but
may serve as an antigen receptor. IgE is the immunoglobulin that mediates
immediate
hypersensitivity by causing the release of mediators from mast cells and
basophils upon
exposure to the allergen.
[00122] The term "isolated" refers to a material that is substantially or
essentially free from
components that normally accompany it in its native state. The material can be
a cell or a
macromolecule such as a protein or nucleic acid. For example, an "isolated
polynucleotide," as
used herein, refers to a polynucleotide, which has been purified from the
sequences which flank
it in a naturally-occurring state, e.g., a DNA fragment which has been removed
from the
sequences that are normally adjacent to the fragment. Alternatively, an
"isolated peptide" or an
"isolated polypeptide" and the like, as used herein, refer to in vitro
isolation and/or purification of
a peptide or polypeptide molecule from its natural cellular environment, and
from association
with other components of the cell.
[00123] The term "substantially purified" refers to a material that is
substantially free from
components that are normally associated with it in its native state. For
example, a substantially
purified cell refers to a cell that has been separated from other cell types
with which it is
normally associated in its naturally occurring or native state. In some
instances, a population of
substantially purified cells refers to a homogenous population of cells. In
other instances, this
term refers simply to a cell that has been separated from the cells with which
they are naturally
associated in their natural state. In embodiments, the cells are cultured in
vitro. In embodiments,
the cells are not cultured in vitro.
[00124] In the context of the present disclosure, the following
abbreviations for the commonly
occurring nucleic acid bases are used. "A" refers to adenosine, "C" refers to
cytosine, "G" refers
to guanosine, "T" refers to thymidine, and "U" refers to uridine.
[00125] Unless otherwise specified, a "nucleotide sequence encoding an
amino acid
sequence" includes all nucleotide sequences that are degenerate versions of
each other and
that encode the same amino acid sequence. The phrase nucleotide sequence that
encodes a
protein or an RNA may also include introns to the extent that the nucleotide
sequence encoding
the protein may in some version contain an intron(s).
[00126] The term "lentivirus" refers to a genus of the Retroviridae family.
Lentiviruses are
unique among the retroviruses in being able to infect non-dividing cells; they
can deliver a
significant amount of genetic information into the DNA of the host cell, so
they are one of the
most efficient methods of a gene delivery vector. Moreover, the use of
lentiviruses enables
integration of the genetic information into the host chromosome resulting in
stably transduced
genetic information. HIV, Sly, and FIV are all examples of lentiviruses.
Vectors derived from
lentiviruses offer the means to achieve significant levels of gene transfer in
vivo.
13
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00127] The term "modulating," refers to mediating a detectable increase or
decrease in the
level of a response in a subject compared with the level of a response in the
subject in the
absence of a treatment or compound, and/or compared with the level of a
response in an
otherwise identical but untreated subject. The term encompasses perturbing
and/or affecting a
native signal or response thereby mediating a beneficial therapeutic response
in a subject,
preferably, a human.
[00128] Nucleic acid is "operably linked" when it is placed into a
functional relationship with
another nucleic acid sequence. For example, DNA for a presequence or secretory
leader is
operably linked to DNA for a polypeptide if it is expressed as a preprotein
that participates in the
secretion of the polypeptide; a promoter or enhancer is operably linked to a
coding sequence if
it affects the transcription of the sequence; or a ribosome binding site is
operably linked to a
coding sequence if it is positioned so as to facilitate translation.
[00129] The term "under transcriptional control" refers to a promoter being
operably linked to
and in the correct location and orientation in relation to a polynucleotide to
control (regulate) the
initiation of transcription by RNA polymerase and expression of the
polynucleotide.
[00130] The term "overexpressed" tumor antigen or "overexpression" of the
tumor antigen is
intended to indicate an abnormal level of expression of the tumor antigen in a
cell from a
disease area such as a solid tumor within a specific tissue or organ of the
patient relative to the
level of expression in a normal cell from that tissue or organ. Patients
having solid tumor or a
hematological malignancy characterized by overexpression of the tumor antigen
can be
determined by standard assays known in the art.
[00131] Solid tumors are abnormal masses of tissue that usually do not
contain cysts or
liquid areas. Solid tumors can be benign or malignant. Different types of
solid tumors are named
for the type of cells that form them (such as sarcomas, carcinomas, and
lymphomas). Examples
of solid tumors, such as sarcomas and carcinomas, include fibrosarcoma,
myxosarcoma,
liposarcoma, chondrosarcoma, osteosarcoma, synovioma, mesothelioma, Ewing's
tumor,
leiomyosarcoma, rhabdomyosarcoma, colon carcinoma, lymphoid malignancy,
pancreatic
cancer, breast cancer, lung cancers, ovarian cancer, prostate cancer,
hepatocellular carcinoma,
squamous cell carcinoma, basal cell carcinoma, adenocarcinoma, sweat gland
carcinoma,
medullary thyroid carcinoma, papillary thyroid carcinoma, pheochromocytomas
sebaceous
gland carcinoma, papillary carcinoma, papillary adenocarcinomas, medullary
carcinoma,
bronchogenic carcinoma, renal cell carcinoma, hepatoma, bile duct carcinoma,
choriocarcinoma, VVilms' tumor, cervical cancer, testicular tumor, seminoma,
bladder carcinoma,
melanoma, and CNS tumors (such as a glioma (such as brainstem glioma and mixed
gliomas),
glioblastoma (also known as glioblastoma multiforme), astrocytoma, CNS
lymphoma,
germinoma, medulloblastoma, Schwannoma craniopharyogioma, ependymoma,
pinealoma,
hemangioblastoma, acoustic neuroma, oligodendroglioma, menangioma,
neuroblastoma,
retinoblastoma, and brain metastases).
14
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00132] A solid tumor antigen is an antigen expressed on a solid tumor. In
embodiments,
solid tumor antigens are also expressed at low levels on healthy tissue.
Examples of solid tumor
antigens and their related disease tumors are provided in Table 1.
Table 1
Solid Tumor antigen Disease tumor
PRLR Breast Cancer
CLCA1 colorectal Cancer
MUC12 colorectal Cancer
GUCY2C colorectal Cancer
GPR35 colorectal Cancer
CR1L Gastric Cancer
MUC 17 Gastric Cancer
TMPRSS11B esophageal Cancer
MUC21 esophageal Cancer
TMPRSS11E esophageal Cancer
CD207 bladder Cancer
SLC30A8 pancreatic Cancer
CFC1 pancreatic Cancer
SLC12A3 Cervical Cancer
SSTR1 Cervical tumor
GPR27 Ovary tumor
FZD10 Ovary tumor
TSHR Thyroid Tumor
SIGLEC15 Urothelial cancer
SLC6A3 Renal cancer
KISS1R Renal cancer
QRFPR Renal cancer:
GPR119 Pancreatic cancer
CLDN6 Endometrial cancer/ Urothelial cancer
UPK2 Urothelial cancer (including bladder cancer)
ADAM12 Breast cancer, pancreatic cancer and the like
SLC45A3 Prostate cancer
ACPP Prostate cancer
MUC21 Esophageal cancer
MUC16 Ovarian cancer
MS4Al2 Colorectal cancer
ALPP Endometrial cancer
CEA Colorectal carcinoma
EphA2 Glioma
FAP Mesotelioma
GPC3 Lung squamous cell carcinoma
1L13-Ra2 Glioma
Mesothelin Metastatic cancer
PSMA Prostate cancer
ROR1 Breast lung carcinoma
VEGFR-II Metastatic cancer
GD2 Neuroblastoma
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
FR-a Ovarian carcinoma
ErbB2 Carcinomasb
EpCAM Carcinomasa
EGFRvIll Glioma¨Glioblastoma
EGFR Glioma¨NSCL cancer
tMUC1 Cholangiocarcinoma, Pancreatic cancer, Breast
PSCA pancreas, stomach, or prostate cancer
FCER2, GPR18, FCRLA, breast cancer
CXCR5, FCRL3, FCRL2,
HTR3A, and CLEC17A
TRPMI, SLC45A2, and lymphoma
SLC24A5
DPEP3 melanoma
KCNK16 ovarian, testis
LIM2 or KCNV2 pancreatic
SLC26A4 thyroid cancer
CD171 Neuroblastoma
Glypican-3 Sarcoma
IL-13 Glioma
CD79a/b Lymphoma
[00133] The term "parenteral administration" of a composition includes,
e.g., subcutaneous
(s.c.), intravenous (i.v.), intramuscular (i.m.), intrasternal injection, or
infusion techniques.
[00134] The terms "patient," "subject," and "individual," and the like are
used interchangeably
herein and refer to any human, or animal, amenable to the methods described
herein. In certain
non-limiting embodiments, the patient, subject, or individual is a human or
animal. In
embodiments, the term "subject" is intended to include living organisms in
which an immune
response can be elicited (e.g., mammals). Examples of subjects include humans,
and animals,
such as dogs, cats, mice, rats, and transgenic species thereof.
[00135] A subject in need of treatment or in need thereof includes a
subject having a
disease, condition, or disorder that needs to be treated. A subject in need
thereof also includes
a subject that needs treatment for prevention of a disease, condition, or
disorder.
[00136] The term "polynucleotide" or "nucleic acid" refers to mRNA, RNA,
cRNA, rRNA,
cDNA or DNA. The term typically refers to a polymeric form of nucleotides of
at least 10 bases
in length, either ribonucleotides or deoxynucleotides or a modified form of
either type of
nucleotide. The term includes all forms of nucleic acids including single and
double-stranded
forms of nucleic acids.
[00137] The terms "polynucleotide variant" and "variant" and the like refer
to polynucleotides
displaying substantial sequence identity with a reference polynucleotide
sequence or
polynucleotides that hybridize with a reference sequence under stringent
conditions that are
defined hereinafter. These terms also encompass polynucleotides that are
distinguished from a
reference polynucleotide by the addition, deletion or substitution of at least
one nucleotide.
Accordingly, the terms "polynucleotide variant" and "variant" include
polynucleotides in which
16
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
one or more nucleotides have been added or deleted or replaced with different
nucleotides. In
this regard, it is well understood in the art that certain alterations
inclusive of mutations,
additions, deletions, and substitutions can be made to a reference
polynucleotide whereby the
altered polynucleotide retains the biological function or activity of the
reference polynucleotide
or has increased activity in relation to the reference polynucleotide (i.e.,
optimized).
Polynucleotide variants include, for example, polynucleotides having at least
50% (and at least
51% to at least 99% and all integer percentages in between, e.g., 90%, 95%, or
98%) sequence
identity with a reference polynucleotide sequence described herein. The terms
"polynucleotide
variant" and "variant" also include naturally-occurring allelic variants and
orthologs.
[00138] The terms "polypeptide," "polypeptide fragment," "peptide," and
"protein" are used
interchangeably herein to refer to a polymer of amino acid residues and to
variants and
synthetic analogues of the same. Thus, these terms apply to amino acid
polymers in which one
or more amino acid residues are synthetic non-naturally occurring amino acids,
such as a
chemical analogue of a corresponding naturally occurring amino acid, as well
as to naturally-
occurring amino acid polymers. In certain aspects, polypeptides may include
enzymatic
polypeptides, or "enzymes," which typically catalyze (i.e., increase the rate
of) various chemical
reactions.
[00139] The term "polypeptide variant" refers to polypeptides that are
distinguished from a
reference polypeptide sequence by the addition, deletion, or substitution of
at least one amino
acid residue. In certain embodiments, a polypeptide variant is distinguished
from a reference
polypeptide by one or more substitutions, which may be conservative or non-
conservative. In
certain embodiments, the polypeptide variant comprises conservative
substitutions and, in this
regard, it is well understood in the art that some amino acids may be changed
to others with
broadly similar properties without changing the nature of the activity of the
polypeptide.
Polypeptide variants also encompass polypeptides in which one or more amino
acids have
been added or deleted or replaced with different amino acid residues.
[00140] The term "promoter" refers to a DNA sequence recognized by the
synthetic
machinery of the cell or introduced synthetic machinery, required to initiate
the specific
transcription of a polynucleotide sequence. The term "expression control
(regulatory)
sequences" refers to DNA sequences necessary for the expression of an operably
linked coding
sequence in a particular host organism. The control sequences that are
suitable for prokaryotes,
for example, include a promoter, optionally an operator sequence, and a
ribosome binding site.
Eukaryotic cells are known to utilize promoters, polyadenylation signals, and
enhancers.
[00141] The term "bind," "binds," or "interacts with" refers to a molecule
recognizing and
adhering to a second molecule in a sample or organism but does not
substantially recognize or
adhere to other structurally unrelated molecules in the sample. The term
"specifically binds," as
used herein with respect to an antibody, refers to an antibody which
recognizes a specific
antigen, but does not substantially recognize or bind other molecules in a
sample. For example,
17
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
an antibody that specifically binds an antigen from one species may also bind
that antigen from
one or more species. But, such cross-species reactivity does not itself alter
the classification of
an antibody as specific. In another example, an antibody that specifically
binds an antigen may
also bind different allelic forms of the antigen. However, such cross
reactivity does not itself
alter the classification of an antibody as specific. In some instances, the
terms "specific binding"
or "specifically binding," can be used in reference to the interaction of an
antibody, a protein, or
a peptide with a second chemical species, to mean that the interaction is
dependent upon the
presence of a particular structure (e.g., an antigenic determinant or epitope)
on the chemical
species; for example, an antibody recognizes and binds a specific protein
structure rather than
to any protein. If an antibody is specific for epitope "A," the presence of a
molecule containing
epitope A (or free, unlabeled A), in a reaction containing labeled "A" and the
antibody, will
reduce the amount of labeled A bound to the antibody.
[00142] By "statistically significant," it is meant that the result was
unlikely to have occurred
by chance. Statistical significance can be determined by any method known in
the art.
Commonly used measures of significance include the p-value, which is the
frequency or
probability with which the observed event would occur if the null hypothesis
were true. If the
obtained p-value is smaller than the significance level, then the null
hypothesis is rejected. In
simple cases, the significance level is defined at a p-value of 0.05 or less.
A "decreased" or
"reduced" or "lesser" amount is typically a "statistically significant" or a
physiologically significant
amount, and may include a decrease that is about 1.1, 1.2, 1.3, 1.4, 1.5, 1.6
1.7, 1.8, 1.9, 2, 2.5,
3, 3.5, 4, 4.5, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, or 50 or more times (e.g.,
100, 500, 1000 times)
(including all integers and decimal points in between and above 1, e.g., 1.5,
1.6, 1.7. 1.8, etc.)
an amount or level described herein.
[00143] The term "stimulation," refers to a primary response induced by
binding of a
stimulatory molecule (e.g., a TCR/CD3 complex) with its cognate ligand thereby
mediating a
signal transduction event, such as signal transduction via the TCR/CD3
complex. Stimulation
can mediate altered expression of certain molecules, such as downregulation of
TGF-13, and/or
reorganization of cytoskeletal structures.
[00144] The term "stimulatory molecule" refers to a molecule on a T cell
that specifically
binds a cognate stimulatory ligand present on an antigen presenting cell. For
example, a
functional signaling domain derived from a stimulatory molecule is the zeta
chain associated
with the T cell receptor complex. The stimulatory molecule includes a domain
responsible for
signal transduction.
[00145] The term "stimulatory ligand" refers to a ligand that when present
on an antigen
presenting cell (e.g., an APC, a dendritic cell, a B-cell, and the like.) can
specifically bind with a
cognate binding partner (referred to herein as a "stimulatory molecule") on a
cell, for example a
T cell, thereby mediating a primary response by the T cell, including
activation, initiation of an
immune response, proliferation, and similar processes. Stimulatory ligands are
well-known in
18
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
the art and encompass, inter alia, an MHC Class I molecule loaded with a
peptide, an anti-CD3
antibody, a superagonist anti-CD28 antibody, and a superagonist anti-CD2
antibody.
[00146] The term "therapeutic" refers to a treatment and/or prophylaxis. A
therapeutic effect
is obtained by suppression, remission, or eradication of a disease state or
alleviating the
symptoms of a disease state.
[00147] The term "therapeutically effective amount" refers to the amount of
the subject
compound that will elicit the biological or medical response of a tissue,
system, or subject that is
being sought by the researcher, veterinarian, medical doctor or another
clinician. The term
"therapeutically effective amount" includes that amount of a compound that,
when administered,
is sufficient to prevent the development of, or alleviate to some extent, one
or more of the signs
or symptoms of the disorder or disease being treated. The therapeutically
effective amount will
vary depending on the compound, the disease and its severity and the age,
weight, etc., of the
subject to be treated.
[00148] The term "treat a disease" refers to the reduction of the frequency
or severity of at
least one sign or symptom of a disease or disorder experienced by a subject.
[00149] The term "transfected" or "transformed" or "transduced" refers to a
process by which
an exogenous nucleic acid is transferred or introduced into the host cell. A
"transfected" or
"transformed" or "transduced" cell is one which has been transfected,
transformed, or
transduced with exogenous nucleic acid. The cell includes the primary subject
cell and its
progeny.
[00150] The term "vector" refers to a polynucleotide that comprises an
isolated nucleic acid
and which can be used to deliver the isolated nucleic acid to the interior of
a cell. Numerous
vectors are known in the art including linear polynucleotides, polynucleotides
associated with
ionic or amphiphilic compounds, plasmids, and viruses. Thus, the term "vector"
includes an
autonomously replicating plasmid or a virus. The term also includes non-
plasmid and non-viral
compounds which facilitate the transfer of nucleic acid into cells, such as,
for example,
polylysine compounds, liposomes, and the like. Examples of viral vectors
include adenoviral
vectors, adeno-associated virus vectors, retroviral vectors, and others. For
example, lentiviruses
are complex retroviruses, which, in addition to the common retroviral genes
gag, pol, and env,
contain other genes with regulatory or structural function. Lentiviral vectors
are well known in
the art. Some examples of lentivirus include the Human Immunodeficiency
Viruses: HIV-1, HIV-
2, and the Simian Immunodeficiency Virus: SIV. Lentiviral vectors have been
generated by
multiply attenuating the HIV virulence genes, for example, the genes env, vif,
vpr, vpu, and nef
are deleted making the vector biologically safe.
[00151] Ranges: throughout this disclosure, various aspects of the
disclosure can be
presented in a range format. It should be understood that the description in
range format is
merely for convenience and brevity and should not be construed as an
inflexible limitation on
the scope of the disclosure. Accordingly, the description of a range should be
considered to
19
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
have specifically disclosed all the possible subranges as well as individual
numerical values
within that range. For example, description of a range such as from 1 to 6
should be considered
to have specifically disclosed subranges such as from 1 to 3, from 1 to 4,
from 1 to 5, from 2 to
4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that
range, for example, 1,
2, 2.7, 3, 4, 5, 5.3, and 6. This applies regardless of the breadth of the
range.
[00152] A "chimeric antigen receptor" (CAR) molecule is a recombinant
polypeptide including
at least an extracellular domain, a transmembrane domain and a cytoplasmic
domain or
intracellular domain. In embodiments, the domains of the CAR are on the same
polypeptide
chain, for example a chimeric fusion protein. In embodiments, the domains are
on different
polypeptide chains, for example the domains are not contiguous.
[00153] The extracellular domain of a CAR molecule includes an antigen
binding domain.
The antigen binding domain is for expanding and/or maintaining the modified
cells, such as a
CAR T cell or for killing a tumor cell, such as a solid tumor. In embodiments,
the antigen binding
domain for expanding and/or maintaining modified cells binds an antigen, for
example, a cell
surface molecule or marker, on the surface of a WBC. In embodiments, the WBC
is at least one
of GMP (granulocyte macrophage precursor), MDP (monocyte-macrophage/dendritic
cell
precursors), cMoP (common monocyte precursor), basophil, eosinophil,
neutrophil, SatM
(Segerate-nucleus-containing atypical monocyte), macrophage, monocyte, CDP
(common
dendritic cell precursor), cDC (conventional DC), pDC (plasmacytoid DC), CLP
(common
lymphocyte precursor), B cell, ILC (Innate Lymphocyte), NK cell,
megakaryocyte, myeloblast,
pro - myelocyte, myelocyte, meta - myelocyte, band cells, lymphoblast,
prolymphocyte,
monoblast, megakaryoblast, promegakaryocyte, megakaryocyte, platelets, or MSDC
(Myeloid-
derived suppressor cell). In embodiments, the WBC is a granulocyte, monocyte
and or
lymphocyte. In embodiments, the WBC is a lymphocyte, for example, a B cell. In
embodiments,
the WBC is a B cell. In embodiments, the cell surface molecule of a B cell
includes CD19,
0D22, CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, CD30, CD14, 0D68, CD11b, CD18,
0D169, CD1c, 0D33, 0D38, 0D138, or CD13. In embodiments, the cell surface
molecule of the
B cell is CD19, CD20, 0D22, or BCMA. In embodiments, the cell surface molecule
of the B cell
is CD19.
[00154] The cells described herein, including modified cells such as CAR
cells and modified
T cells can be derived from stem cells. Stem cells may be adult stem cells,
embryonic stem
cells, more particularly non-human stem cells, cord blood stem cells,
progenitor cells, bone
marrow stem cells, induced pluripotent stem cells, totipotent stem cells or
hematopoietic stem
cells. A modified cell may also be a dendritic cell, a NK-cell, a B-cell or a
T cell selected from the
group consisting of inflammatory T-lymphocytes, cytotoxic T-lymphocytes,
regulatory T
lymphocytes or helper T-lymphocytes. In embodiments, Modified cells may be
derived from the
group consisting of CD4+ T lymphocytes and CD8+ T lymphocytes. Prior to
expansion and
genetic modification of the cells of the invention, a source of cells may be
obtained from a
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
subject through a variety of non-limiting methods. T cells may be obtained
from a number of
non-limiting sources, including peripheral blood mononuclear cells, bone
marrow, lymph node
tissue, cord blood, thymus tissue, tissue from a site of infection, ascites,
pleural effusion, spleen
tissue, and tumors. In certain embodiments of the present invention, any
number of T cell lines
available and known to those skilled in the art, may be used. In embodiments,
modified cells
may be derived from a healthy donor, from a patient diagnosed with cancer or
from a patient
diagnosed with an infection. In embodiments, a modified cell is part of a
mixed population of
cells which present different phenotypic characteristics.
[00155] A population of cells refers to a group of two or more cells. The
cells of the
population could be the same, such that the population is a homogenous
population of cells.
The cells of the population could be different, such that the population is a
mixed population or a
heterogeneous population of cells. For example, a mixed population of cells
could include
modified cells comprising a first CAR and cells comprising a second CAR,
wherein the first CAR
and the second CAR bind different antigens.
[00156] The term "stem cell" refers to any of certain types of cell which
have the capacity for
self-renewal and the ability to differentiate into other kind(s) of cell. For
example, a stem cell
gives rise either to two daughter stem cells (as occurs in vitro with
embryonic stem cells in
culture) or to one stem cell and a cell that undergoes differentiation (as
occurs e.g. in
hematopoietic stem cells, which give rise to blood cells). Different
categories of stem cells may
be distinguished on the basis of their origin and/or on the extent of their
capacity for
differentiation into other types of cell. For example, stem cells may include
embryonic stem (ES)
cells (i.e., pluripotent stem cells), somatic stem cells, induced pluripotent
stem cells, and any
other types of stem cells.
[00157] The pluripotent embryonic stem cells are found in the inner cell
mass of a blastocyst
and have an innate capacity for differentiation. For example, pluripotent
embryonic stem cells
have the potential to form any type of cell in the body. When grown in vitro
for long periods of
time, ES cells maintain pluripotency as progeny cells retain the potential for
multilineage
differentiation.
[00158] Somatic stem cells can include fetal stem cells (from the fetus)
and adult stem cells
(found in various tissues, such as bone marrow). These cells have been
regarded as having a
capacity for differentiation that is lower than that of the pluripotent ES
cells ¨ with the capacity of
fetal stem cells being greater than that of adult stem cells. Somatic stem
cells apparently
differentiate into only a limited number of types of cells and have been
described as multipotent.
The "tissue-specific" stem cells normally give rise to only one type of cell.
For example,
embryonic stem cells may be differentiated into blood stem cells (e.g.,
Hematopoietic stem cells
(HSCs)), which may be further differentiated into various blood cells (e.g.,
red blood cells,
platelets, white blood cells, etc.).
21
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00159] Induced pluripotent stem cells (i.e., iPS cells or iPSCs) may
include a type of
pluripotent stem cell artificially derived from a non-pluripotent cell (e.g.,
an adult somatic cell) by
inducing an expression of specific genes. Induced pluripotent stem cells are
similar to natural
pluripotent stem cells, such as embryonic stem (ES) cells, in many aspects,
such as the
expression of certain stem cell genes and proteins, chromatin methylation
patterns, doubling
time, embryoid body formation, teratoma formation, viable chimera formation,
and potency and
differentiability. Induced pluripotent cells can be obtained from adult
stomach, liver, skin, and
blood cells.
[00160] In embodiments, the antigen binding domain for killing a tumor,
binds an antigen on
the surface of a tumor, for example a tumor antigen or tumor marker. Tumor
antigens are
proteins that are produced by tumor cells that elicit an immune response,
particularly T cell
mediated immune responses. Tumor antigens are well known in the art and
include, for
example, tumor associated MUC1 (tMUC1), a glioma-associated antigen,
carcinoembryonic
antigen (CEA), 13-human chorionic gonadotropin, alphafetoprotein (AFP), lectin-
reactive AFP,
thyroglobulin, RAGE-1, MN-CA IX, human telomerase reverse transcriptase, RU1,
RU2 (AS),
intestinal carboxyl esterase, mut h5p70-2, M-CSF, prostase, prostate-specific
antigen (PSA),
PAP, NY-ESO-1, LAGE-1 a, p53, prostein, PSMA, Her2/neu, surviving, telomerase,
prostate-
carcinoma tumor antigen-1 (PCTA-1), MAGE, ELF2M, neutrophil elastase,
ephrinB2, CD22,
insulin growth factor (IGF)-I, IGF-II, IGF-I receptor, CD19, and mesothelin.
For example, when
the tumor antigen is CD19, the CAR thereof can be referred to as CD19 CAR or
19CAR, which
is a CAR molecule that includes an antigen binding domain that binds CD19.
[00161] In embodiments, the extracellular antigen binding domain of a CAR
includes at least
one scFv or at least a single domain antibody. As an example, there can be two
scFvs on a
CAR. The scFv includes a light chain variable (VL) region and a heavy chain
variable (VH)
region of a target antigen-specific monoclonal antibody joined by a flexible
linker. Single chain
variable region fragments can be made by linking light and/or heavy chain
variable regions by
using a short linking peptide (Bird et al., Science 242:423-426, 1988). An
example of a linking
peptide is the GS linker having the amino acid sequence (GGGGS)3 (SEQ ID NO:
278), which
bridges approximately 3.5 nm between the carboxy terminus of one variable
region and the
amino terminus of the other variable region. Linkers of other sequences have
been designed
and used (Bird et al., 1988, supra). In general, linkers can be short,
flexible polypeptides and
preferably comprised of about 20 or fewer amino acid residues. The single
chain variants can
be produced either recombinantly or synthetically. For synthetic production of
scFv, an
automated synthesizer can be used. For recombinant production of scFv, a
suitable plasmid
containing polynucleotide that encodes the scFv can be introduced into a
suitable host cell,
either eukaryotic, such as yeast, plant, insect or mammalian cells, or
prokaryotic, such as E.
coli. Polynucleotides encoding the scFv of interest can be made by routine
manipulations such
22
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
as ligation of polynucleotides. The resultant scFv can be isolated using
standard protein
purification techniques known in the art.
[00162] The cytoplasmic domain of the CAR molecules described herein
includes one or
more co-stimulatory domains and one or more signaling domains. The co-
stimulatory and
signaling domains function to transmit the signal and activate molecules, such
as T cells, in
response to antigen binding. The one or more co-stimulatory domains are
derived from
stimulatory molecules and/or co-stimulatory molecules, and the signaling
domain is derived
from a primary signaling domain, such as the CD3 zeta domain. In embodiments,
the signaling
domain further includes one or more functional signaling domains derived from
a co-stimulatory
molecule. In embodiments, the co-stimulatory molecules are cell surface
molecules (other than
antigens receptors or their ligands) that are required for activating a
cellular response to an
antigen.
[00163] In embodiments, the co-stimulatory domain includes the
intracellular domain of
0D27, 0D28, 4-1BB, 0X40, CD30, CD40, PD-1, ICOS, lymphocyte function-
associated
antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, a ligand that specifically
binds with
0D83, or any combination thereof. In embodiments, the signaling domain
includes a CD3 zeta
domain derived from a T cell receptor.
[00164] The CAR molecules described herein also include a transmembrane
domain. The
incorporation of a transmembrane domain in the CAR molecules stabilizes the
molecule. In
embodiments, the transmembrane domain of the CAR molecules is the
transmembrane domain
of a 0D28 or 4-1BB molecule.
[00165] Between the extracellular domain and the transmembrane domain of
the CAR, there
may be incorporated a spacer domain. As used herein, the term "spacer domain"
generally
means any oligo- or polypeptide that functions to link the transmembrane
domain to the
extracellular domain and/or the cytoplasmic domain on the polypeptide chain. A
spacer domain
may include up to 300 amino acids, preferably 10 to 100 amino acids, and most
preferably 25 to
50 amino acids.
[00166] The present disclosure describes a method for in vitro cell
preparation, the method
comprising: preparing cells; contacting the cells with (1) a first vector
comprising a
polynucleotide encoding a first antigen binding molecule that binds a first
antigen and (2) a
second vector comprising a polynucleotide encoding a second antigen binding
molecule that
binds a second antigen to obtain a population of modified cells, wherein the
first antigen is
different from the second antigen.
[00167] The present disclosure also describes a method for enhancing cell
expansion in a
subject having cancer, the method comprising: obtaining cells from the subject
or a healthy
donor; contacting the cells with (1) a first vector comprising a
polynucleotide encoding a first
antigen binding molecule that binds a first antigen and (2) a second vector
comprising a
polynucleotide encoding a second antigen binding molecule that binds a second
antigen to
23
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
obtain a population of modified cells; and administering an effective amount
of modified cells to
the subject, wherein: the first antigen is different from the second antigen;
and the level of cell
expansion in the subject is higher than the level of cell expansion in a
subject administered with
an effective amount of cells that have been contacted with the first vector
but not the second
vector.
[00168] The present disclosure also describes a method for treating a
subject having cancer,
the method comprising: obtaining cells from the subject or a healthy donor;
contacting the cells
with (1) a first vector comprising a polynucleotide encoding a first antigen
binding molecule that
binds a first antigen and (2) a second vector comprising a polynucleotide
encoding a second
antigen binding molecule that binds a second antigen to obtain a population of
modified cells;
and administering an effective amount of modified cells to the subject,
wherein: the first antigen
is different form the second antigen.
[00169] The present disclosure also describes a method for enhancing
treatment of a subject
having cancer, the method comprising: obtaining cells from the subject or a
healthy donor;
contacting the cells with (1) a first vector comprising a polynucleotide
encoding a first antigen
binding molecule that binds a first antigen and (2) a second vector comprising
a polynucleotide
encoding a second antigen binding molecule that binds a second antigen to
obtain a population
of modified cells; and administering an effective amount of modified cells to
the subject,
wherein: the first antigen is different from the second antigen; and the level
of inhibition of tumor
growth by the effective amount of modified cells is higher than the level of
inhibition of tumor
growth by the effective amount of cells that have been contacted with the
second vector but not
the first vector.
[00170] The present disclosure also describes a method for in vitro cell
preparation, the
method comprising: introducing a first vector comprising a polynucleotide
encoding a first
antigen binding molecule that binds a first antigen into a first population of
cells; introducing a
second vector comprising a polynucleotide encoding a second antigen binding
molecule that
binds a second antigen into a second population of cells; and culturing the
first and second
population of cells, wherein the first antigen is different from the second
antigen.
[00171] The present disclosure also describes a method for enhancing cell
expansion in a
subject having cancer, the method comprising: introducing a first vector
comprising a
polynucleotide encoding a first antigen binding molecule that binds a first
antigen into a first
population of cells to obtain a first population of modified cells;
introducing a second vector
comprising a polynucleotide encoding a second antigen binding molecule that
binds a second
antigen into a second population of cells to obtain a second population of
modified cells; and
administering an effective amount of the first and second population of
modified cells to the
subject, wherein: the first antigen is different from the second antigen; and
the level of cell
expansion in the subject is higher than the level of cell expansion in a
subject administered an
24
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
effective amount of the second population of modified cells but not the first
population of
modified cells.
[00172] The present disclosure also describes a method for treating a
subject having cancer,
the method comprising: introducing a first vector comprising a polynucleotide
encoding a first
antigen binding molecule that binds a first antigen into a first population of
cells to obtain a first
population of modified cells; introducing a second vector comprising a
polynucleotide encoding
a second antigen binding molecule that binds a second antigen into a second
population of cells
to obtain a second population of modified cells; and administering an
effective amount of the
first and second population of modified cells to the subject, wherein: the
first antigen is different
from the second antigen.
[00173] The present disclosure also describes a method for enhancing
treatment of a subject
having cancer, the method comprising: introducing a first vector comprising a
polynucleotide
encoding a first antigen binding molecule that binds a first antigen into a
first population of cells
to obtain a first population of modified cells; introducing a second vector
comprising a
polynucleotide encoding a second antigen binding molecule that binds a second
antigen into a
second population of cells to obtain a second population of modified cells;
and administering an
effective amount of the first and second population of modified cells to the
subject, wherein: the
first antigen is different from the second antigen; and the level of
inhibition of tumor growth in
the subject by the effective amount of first population of modified cells is
higher than the level of
inhibition of tumor growth in the subject by the effective amount of the
second population of
modified cells that is not administered the first population of modified
cells.
[00174] The present disclosure also describes a method for enhancing T cell
response, the
method comprising: introducing a first vector comprising a polynucleotide
encoding a first
antigen binding molecule that binds a first antigen into a first population of
cells; introducing a
second vector comprising a polynucleotide encoding a second antigen binding
molecule that
binds a second antigen into a second population of cells; contacting cells
expressing the second
antigen with the first population of cells and the second population of cells;
and measuring a
level of the T cell response, wherein the level is higher than a level of the
T cell response in
response to the cells contacted with the second population of cells without
the first population.
[00175] The present disclosure also describes a method for enhancing T cell
response, the
method comprising: contacting a population of cells with a first vector
comprising a
polynucleotide encoding a first antigen binding molecule that binds a first
antigen and a second
vector comprising a polynucleotide encoding a second antigen binding molecule
that binds a
second antigen to obtain a population of modified cells; contacting cells
expressing the second
antigen with a population of modified cells; and measuring the level of the T
cell response,
wherein the level of T cell response is higher than the level of T cell
response in cells contacted
with the population of cells that have been contacted with the second vector
but not the first
vector.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00176] The cells include macrophages, dendritic cells, or lymphocytes such
as T cells or NK
cells. In embodiments, the cells are T cells. In embodiments, the first
antigen binding molecule
binds a cell surface molecule of a WBC. In embodiments, the WBC is a
granulocyte, a
monocyte, or lymphocyte. In embodiments, the WBC is a B cell. In embodiments,
the cell
surface molecule of the WBC is CD19, 0D22, CD20, BCMA, CD5, CD7, CD2, CD16,
0D56,
CD30, CD14, 0D68, CD11b, CD18, 0D169, CD1c, 0D33, 0D38, 0D138, or CD13. In
embodiments, the cell surface molecule of the WBC is CD19, CD20, 0D22, or
BCMA. In
embodiments, the cell surface molecule of the WBC is CD19.
[00177] In embodiments, the second antigen binding molecule binds a solid
tumor antigen. In
embodiments, the solid tumor antigen is tumor associated MUC1 (tMUC1), PRLR,
CLCA1,
MUC12, GUCY2C, GPR35, CR1L, MUC 17, TMPRSS11B, MUC21, TMPRSS11E, 0D207,
SLC30A8, CFC1, SLC12A3, SSTR1, GPR27, FZD10, TSHR, SIGLE015, SLC6A3, CLDN
18.2,
KISS1R, QRFPR, GPR119, CLDN6, UPK2, ADAM12, SLC45A3, ACPP, MUC21, MUC16,
MS4Al2, ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2, Mesothelin, PSMA, ROR1, VEGFR-
II,
GD2, FR-a, ErbB2, EpCAM, EGFRvIll, or EGFR.
[00178] In embodiments, the first and second binding molecules are CARs. In
embodiments,
the CAR comprises an extracellular domain, a transmembrane domain, and an
intracellular
domain, and the extracellular domain binds a tumor antigen. In embodiments,
the intracellular
domain comprising a co-stimulatory domain comprises an intracellular domain of
a co-
stimulatory molecule selected from the group consisting of 0D27, 0D28, 4-1BB,
0X40, CD30,
CD40, PD-1, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7,
LIGHT,
NKG2C, B7-H3, and any combination thereof. In embodiments, the intracellular
domain
comprises a CD3 zeta signaling domain.
[00179] In embodiments, the first binding molecule is a CAR, and the second
binding
molecule is TCR. In embodiments, the T cell comprises a modified T Cell
Receptor (TCR). In
embodiments, the TCR is derived from spontaneously occurring tumor-specific T
cells in
patients. In embodiments, the TCR binds a tumor antigen. In embodiments, the
tumor antigen
comprises CEA, gp100, MART-1, p53, MAGE-A3, or NY-ESO-1. In embodiments, the
TCR
comprises TCRy and TORO chains, or TCRa and TCR[3 chains, or a combination
thereof.
[00180] In embodiments, the second population of cells are derived from
tumor-infiltrating
lymphocytes (TILs). In embodiments, a T cell clone that expresses a TCR with a
high affinity for
the target antigen may be isolated. TILs or peripheral blood mononuclear cells
(PBMCs) can be
cultured in the presence of antigen-presenting cells (APCs) pulsed with a
peptide representing
an epitope known to elicit a dominant T cell response when presented in the
context of a
defined HLA allele. High-affinity clones may be then selected on the basis of
MHC-peptide
tetramer staining and/or the ability to recognize and lyse target cells pulsed
with low titrated
concentrations of cognate peptide antigen. After the clone has been selected,
the TCRa and
TCR[3 chains or TCRy and TORO chains are identified and isolated by molecular
cloning. For
26
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
example, for TCRa and TCR8 chains, the TCRa and TCR8 gene sequences are then
used to
generate an expression construct that ideally promotes stable, high-level
expression of both
TCR chains in human T cells. The transduction vehicle, for example, a
gammaretrovirus or
lentivirus, can then be generated and tested for functionality (antigen
specificity and functional
avidity) and used to produce a clinical lot of the vector. An aliquot of the
final product can then
be used to transduce the target T cell population (generally purified from
patient PBMCs), which
is expanded before infusion into the patient.
[00181] Various methods may be implemented to obtain genes encoding tumor-
reactive
TCR. More information is provided in Kershaw et al., Olin Trans! Immunology.
2014 May; 3(5):
e16. In embodiments, specific TCR can be derived from spontaneously occurring
tumor-specific
T cells in patients. Antigens included in this category include the melanocyte
differentiation
antigens MART-1 and gp100, as well as the MAGE antigens and NY-ESO-1, with
expression in
a broader range of cancers. TCRs specific for viral-associated malignancies
can also be
isolated, as long as viral proteins are expressed by transformed cells.
Malignancies in this
category include liver and cervical cancer, those associated with hepatitis
and papilloma
viruses, and Epstein-Barr virus-associated malignancies. In embodiments,
target antigens of the
TCR include CEA (e.g., for colorectal cancer), gp100, MART-1, p53 (e.g., for
melanoma),
MAGE-A3 (e.g., melanoma, esophageal and synovial sarcoma), and NY-ESO-1 (e.g.,
for
nelanoma and sarcoma as well as multiple myelomas).
[00182] In embodiments, preparation and transfusion of tumor infiltrating
lymphocytes (TIL)
may be implemented in the following manner. For example, tumor tissue coming
from surgical
or biopsy specimens, can be obtained under aseptic conditions and transported
to the cell
culture chamber in ice box. Necrotic tissue and adipose tissue can be removed.
The tumor
tissue can be cut into small pieces of about 1-3 cubic millimeter.
Collagenase, hyaluronidase
and DNA enzyme can be added, and digested overnight at 4 C. Filtering with
0.2 um filter, cells
can be separated and collected by lymphocyte separation fluid, under 1500 rpm
for 5 min.
Expanding the cells in a culture medium comprising PHA, 2-mercaptoethanol, and
CD3
monoclonal antibody, and a small dose of IL-2 (10-20 IU / ml) may be added to
induce
activation and proliferation. The cell density may be carefully measured and
maintained within
the range of 0.5-2x106/ml for 7-14 days at a temperature of 37 C with 5% 002.
TIL positive
cells having the ability to kill homologous cancer cell can be screened out by
co-culture. The TIL
positive cells can be amplified in a serum-free medium containing a high dose
of IL-2 (5000-
6000 IU/m1) until greater than 1x1011 TILs can be obtained. To administer
TILs, they are first
collected in saline using continuous-flow centrifugation and then filtered
through a platelet-
administration set into a volume of 200-300 mL containing 5% albumin and
450000 IU of IL-2.
The TILs can be infused into patients through a central venous catheter over a
period of 30-60
minutes. In embodiments, TILs can be infused in two to four separate bags, and
the individual
infusions can be separated by several hours.
27
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00183] In embodiments, the population of modified cells comprise cells
comprising the first
binding molecule and cells comprising the second binding molecules. In
embodiments, the
population of modified cells comprise cells comprising the first binding
molecule, cells
comprising the second binding molecules, and cells comprising both the first
binding molecule
and the second binding molecule.
[00184] In embodiments, the increase in T cell response is based on the
increase in the
number of copies of CAR(s) and/or the amount of cytokine released (e.g., IL-6
and IFN-y. In
embodiments, the T cell response comprises cytokine releases, cell expansion,
and/or
activation levels. In embodiments, the first vector further comprises a
polynucleotide encoding
IL-6 or IFNy, or a combination thereof. In embodiments, the first vector
further comprises a
polynucleotide encoding IL-12. In embodiments, the polynucleotide comprises a
polynucleotide
encoding NFAT and/or VHL. In embodiments, the population of modified cells
comprises cells
expressing the first binding molecule and IL-6 or IFNy, or a combination
thereof, cells
expressing the second binding molecules, cells expressing the first and second
molecules,
and/or cells expressing the first binding molecule and IL-12. In embodiments,
the population of
modified cells comprises cells expressing the second binding molecule and IL-6
or IFNy, or a
combination thereof, cells expressing the second binding molecules, cells
expressing the first
and second molecules, and/or cells expressing the first binding molecule and
IL-12. In
embodiments, the population of modified cells comprises cells expressing the
second binding
molecule and IL-6 or IFNy, or a combination thereof, cells expressing the
second binding
molecules, cells expressing the first and second molecules, and/or cells
expressing the second
binding molecule and IL-12. In embodiments, the population of modified cells
comprises cells
expressing a dominant negative form of PD-1.
[00185] The present disclosure describes nucleic acids encoding at least
two different
antigen binding domains. In embodiments, there is a first antigen binding
domain that binds an
antigen on the surface of a WBC, and there is a second antigen binding domain
that binds an
antigen on a tumor that is different from the antigen on the surface of a WBC.
The first antigen
binding domain functions to expand the cells that it is introduced into, while
the second antigen
binding domain functions to inhibit the growth of or kill tumor cells
containing the target tumor
antigen upon binding to the target antigen. In embodiments, a nucleic acid
described herein
encodes both the first and second antigen binding domains on the same nucleic
acid molecule.
In embodiments, the two antigen binding domains are encoded by two separate
nucleic acid
molecules. For example, a first nucleic acid encodes a first antigen binding
domain and a
second nucleic acid encodes a second antigen binding domain.
[00186] In embodiments, the present disclosure describes nucleic acids
encoding a first
antigen binding domain of a binding molecule and a second antigen binding
domain of a binding
molecule, wherein the first antigen binding domain binds a cell surface
molecule of a WBC, and
the second antigen binding domain binds an antigen different from the cell
surface molecule of
28
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
the WBC. In embodiments, the first antigen binding domain binds a cell surface
antigen of a B
cell or a B cell marker. In embodiments, the second binding domain does not
bind a B cell
marker. In embodiments, the second binding domain includes a scFv comprising
an amino acid
sequence of SEQ ID No: 264 or 265. For example, the second antigen binding
domain is on a
CAR having one of the amino acid sequences of SEQ ID NOs: 271-277.
[00187] In embodiments, the first and second antigen binding domains are on
two different
binding molecules (first and second binding molecules) such as a first CAR and
a second CAR.
As an example, a first CAR includes an extracellular binding domain that binds
a marker on the
surface of a B cell, and a second CAR includes an extracellular binding domain
that binds a
target antigen of a tumor cell. In embodiments, the first CAR and second CAR
are encoded by
different nucleic acids. In embodiments, the first CAR and second CAR are two
different binding
molecules but are encoded by a single nucleic acid.
[00188] In embodiments, the two different antigen binding domains can be on
the same
binding molecule, for example on a bispecific CAR, and encoded by a single
nucleic acid. In
embodiments, the bispecific CAR can have two different scFv molecules joined
together by
linkers. Examples of the bispecific CAR are provided in Table 2.
[00189] An example of a bispecific CAR is shown in FIG. 5. As shown in FIG.
5, a bispecific
CAR (or tandem CAR (tanCAR)) may include two binding domains: scFv1 and scFv2.
In
embodiments, scFv1 binds an antigen of a white blood cell (e.g., CD19), and
scFv2 binds a
solid tumor antigen (e.g., tMUC1). In embodiments, scFv1 binds a solid tumor
antigen, and
scFv2 binds another solid tumor antigen (e.g., tMUC1 and CLDN 18.2).
Claudin18.2 (CLDN
18.2) is a stomach-specific isoform of Claudin-18. CLDN 18.2 is highly
expressed in gastric and
pancreatic adenocarcinoma. In embodiments, scFv1 binds an antigen expressed on
tumor cells
but not on normal tissues (e.g., tMUC1); scFv2 binds an antigen expressed on
nonessential
tissues associated with solid tumor; and the killing of normal cells of the
tissue does not cause a
life-threatening event (e.g., complications) to the subject (e.g., TSHR,
GUCY2C). Examples of
the nonessential tissues include organs such as prostate, breast, or
melanocyte. In
embodiments, scFv1 and scFv2 bind to different antigens that expressed on the
same
nonessential tissue (e.g., ACPP and SLC45A3 for Prostate cancer, and 5IGLE015
and UPK2
for Urothelial cancer). The sequences of the bispecific CARs and their
components may be
found in Table 5.
Table 2
Variable Linker 1 Variable Linker 2 Variable Linker3
Variable
domain 1 domain 3 domain 5 domain 7
Anti-TSHR- 3*GGGGS Anti-TSHR- 4*GGGGS humanized- 3*GGGGS humanized-
VL linker VH bispecific anti CD19- linker anti CD19-
VL
CAR linker VH
Anti-TSHR- 3*GGGGS Anti-TSHR- 4*GGGGS humanized- 3*GGGGS humanized-
VH linker VL bispecific anti CD19- linker anti CD19-
VH
CAR linker VL
29
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Tumor 3*GGGGS Tumor 4*GGGGS anti CD19- 3*GGGGS anti CD19-VH
associated linker associated bispecific VL linker
MUC1 MUC1 CAR linker
scFv-1 0r2 scFv-1 0r2
VL VH
Tumor 3*GGGGS Tumor 4*GGGGS anti CD19- 3*GGGGS anti CD19-VL
associated linker associated bispecific VH linker
MUC1 MUC1 CAR linker
scFv-1 0r2 scFv-1 0r2
VH VL
humanized- 3*GGGGS humanized- 4*GGGGS Tumor 3*GGGGS Tumor
anti CD19- linker anti CD19- bispecific associated linker associated
VH VL CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
Tumor 3*GGGGS Tumor 4*GGGGS Anti-TSHR- 3*GGGGS Anti-TSHR-VH
associated linker associated bispecific VL linker
MUC1 MUC1 CAR linker
scFv-1 0r2 scFv-1 0r2
VL VH
Anti-TSHR- 3*GGGGS Anti-TSHR- 4*GGGGS Tumor 3*GGGGS Tumor
VL linker VH bispecific associated linker associated
CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
Tumor 3*GGGGS Tumor 4*GGGGS Anti- 3*GGGGS Anti-GUCY2C-
associated linker associated bispecific GUCY2C- linker VL or VH
MUC1 MUC1 CAR linker VH or VL
scFv-1 0r2 scFv-1 0r2
VL VH
Anti- 3*GGGGS Anti- 4*GGGGS Tumor 3*GGGGS Tumor
GUCY2C- linker GUCY2C- bispecific associated linker associated
VH or VL VL or VH CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
Tumor 3*GGGGS Tumor 4*GGGGS Anti-ACPP- 3*GGGGS Anti- ACPP -
associated linker associated bispecific VH or VL linker VL or
VH
MUC1 MUC1 CAR linker
scFv-1 0r2 scFv-1 0r2
VL VH
Anti-ACPP- 3*GGGGS Anti- ACPP 4*GGGGS Tumor 3*GGGGS Tumor
VH or VL linker -VL or VH bispecific associated
linker associated
CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
Tumor 3*GGGGS Tumor 4*GGGGS Anti- 3*GGGGS Anti-
associated linker associated bispecific CLDN18.2- linker CLDN18.2 -VL
MUC1 MUC1 CAR linker VH or VL or VH
scFv-1 0r2 scFv-1 0r2
VL VH
Anti- 3*GGGGS Anti- 4*GGGGS Tumor 3*GGGGS Tumor
CLDN18.2- linker CLDN18.2 - bispecific associated linker associated
VH or VL VL or VH CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Tumor 3*GGGGS Tumor 4*GGGGS Anti-UPK2- 3*GGGGS Anti- UPK2 -
associated linker associated bispecific VH or VL linker VL
or VH
MUC1 MUC1 CAR linker
scFv-1 0r2 scFv-1 0r2
VL VH
Anti-UPK2- 3*GGGGS Anti- UPK2 4*GGGGS Tumor 3*GGGGS Tumor
VH or VL linker -VL or VH bispecific associated
linker associated
CAR linker MUC1 MUC1 scFv-1
scFv-1 0r2 or 2 VH
VL
Tumor 3*GGGGS Tumor 4*GGGGS Anti- 3*GGGGS Anti-
associated linker associated bispecific SIGLE015- linker SIGLE015-VL
MUC1 MUC1 CAR linker VH or VL or VH
scFv-1 0r2 scFv-1 0r2
VL VH
Anti- 3*GGGGS Anti- 4*GGGGS Tumor 3*GGGGS Tumor
SIGLE015- linker SIGLE015- bispecific associated linker associated
VH or VL VL or VH CAR linker MUC1 MUC1 scFv-1
scFv-1 or 2 or 2 VH
VL
3*(GGGGS) is (GGGGS)3and 4*(GGGGS) is (GGGGS)4.
[00190] In embodiments, the two different antigen binding domains can be on
a CAR and a T
cell receptor (TCR) and are encoded by separate nucleic acids. The binding
domain of a TCR
can target a specific tumor antigen or tumor marker on the cell of a tumor. In
embodiments the
TCR binding domain is a TCR alpha binding domain or TCR beta binding domain
that targets a
specific tumor antigen. In embodiments, the TCR comprises the TCRy and TORO
chains or the
TCRa and TCR[3 chains.
[00191] The present disclosure also describes vectors including the nucleic
acids described
herein. In embodiments, a single vector contains the nucleic acid encoding the
first CAR and
second CAR or TCR (containing the second antigen binding domain). In
embodiments, a first
vector contains the first nucleic acid encoding a first CAR, and a second
vector contains the
nucleic acid encoding the second CAR or TCR. In embodiments, the vector
includes the nucleic
acid encoding a bispecific CAR including at least the two different antigen
binding domains. In
embodiments, the vectors including the nucleic acids described herein are
lentiviral vectors.
[00192] Moreover, the present disclosure describes modified cells
comprising the nucleic
acids or vectors described herein. The cells have been introduced with the
nucleic acids or
vectors described herein and express at least one or more different antigen
binding domains. In
embodiments, the cells express one antigen binding domain. In embodiments, the
cells include
a first antigen binding domain and a second antigen binding domain, wherein
the first antigen
binding domain binds a cell surface molecule of a WBC, and the second antigen
binding domain
binds an antigen different from the cell surface molecule of a WBC. In
embodiments, the second
antigen binding domain binds a tumor antigen. In embodiments, the cells are
modified T cells. In
embodiments, the modified T cells are CAR T cells including one or more
nucleic acids
encoding a first antigen binding domain and/or a second antigen binding
domain. In
31
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
embodiments, the modified cells include T cells containing a TCR including the
second antigen
binding domain.
[00193] Further, the present disclosure describes compositions including a
mixed population
of the modified cells described herein. In embodiments, the modified cells
include modified
lymphocytes, modified dendritic cells, and modified macrophages. In
embodiments, the
modified lymphocytes are modified T cells or modified NK cell. In embodiments,
the modified T
cells are CAR T cells.
[00194] The present disclosure describes a mixed population of modified
cells effective for
expanding and/or maintaining the modified cells in a patient. In embodiments,
examples of a
mixed population of modified cells include the following: (1) a first modified
cell expressing an
antigen binding domain for expanding and/or maintaining the modified cells and
a second
modified cell expressing an antigen binding domain for killing a target cell,
such as a tumor cell;
(2) the modified cells of (1) and a further modified cell expressing at least
two different antigen
binding domains, a first antigen binding domain for expanding and/or
maintaining the modified
cells and a second antigen binding domain for killing a target cell (wherein
the two different
antigen binding domains are expressed on the same cell); (3) a modified cell
expressing at least
two different antigen binding domains, a first antigen binding domain for
expanding and/or
maintaining the modified cells and a second antigen binding domain for killing
a target cell
(wherein the two different antigen binding domains are expressed on the same
cell); (4) a
modified cell expressing an antigen binding domain for killing a target cell
and a modified cell
expressing at least two antigen binding domains, a first antigen binding
domain for expanding
and/or maintaining the modified cells and a second antigen binding domain for
killing a target
cell (wherein the two different antigen binding domains are expressed on the
same modified
cell); or (5) a modified cell expressing an antigen binding domain for
expanding and/or
maintaining the modified cells and a modified cell expressing at least two
antigen binding
domains, a first antigen binding domain for expanding and/or maintaining the
modified cells and
a second antigen binding domain for killing a target cell (wherein the two
different antigen
binding domains are expressed on the same modified cell). In embodiments, the
two antigen
binding domains are different molecules. In embodiments, the antigen binding
domain for
expanding the modified cells (the first antigen binding domain) is an antigen
binding domain that
binds a WBC, such as a B cell, and the antigen binding domain for killing a
target cell, such as
tumor cell, (the second antigen binding domain) is an antigen binding domain
that binds a
tumor. In embodiments, the antigen binding domain binding a B cell binds the
surface antigen of
the B cell, for example, CD19, and the antigen binding domain binding a tumor
binds an antigen
of a tumor, for example tMUC1. In embodiments, the tumor cell is a solid tumor
cell.
[00195] In embodiments, the mixed population of modified cells may include
at least one of
the following modified cells: a first modified cell expressing an antigen
binding domain for
expanding and/or maintaining the modified cells, a second modified cell
expressing an antigen
32
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
binding domain for killing a target cell, such as a tumor cell, and a third
modified cell expressing
both the antigen binding domain for expanding and/or maintaining the modified
cells and the
antigen binding domain for killing a target cell. For example, the mixed
population of modified
cells includes the first and second modified cells, the first and third
modified cells, or the second
and third modified cells. In embodiments, the first modified cell expresses a
CAR binding an
antigen of WBC (e.g., CD19); the second modified cell expresses a CAR or TCR
binding a solid
tumor antigen; and the third modified cell expresses the CAR binding the
antigen of WBC and
the CAR/TCR binding the solid tumor antigen. It has been reported that
persistent antigen
exposure can cause T cell exhaustion. Thus, a population of modified cells
including the third
modified cell can exhaust at a higher rate than the mixed population of
modified cells. For
example, the population of modified cells including the third modified cell
alone can exhaust at a
higher rate than the mixed population of modified cells including the first
and the second
modified cells in the presence of the antigen of WBC. Examples of the solid
tumor antigens of
TCR comprise TPO, TGM3, TDGF1, TROP2, LY6K, TNFSF13B, HEG1, LY75, HLA-G,
CEACAM8, CEACAM6, EPHA2, GPRC5D, PLXDC2, HAVCR1, CLEC12A, CD79B, 0R51E2,
CDH17, IFITM1, MELTF, DR5, SLC6A3, ITGAM, SLC44A1, RHOC, CD109, ABCG2, ABCA10,
ABCG8, 5t4, HHLA2, PRAME, CDH6, ESR1, SLC2A1, GJA5, ALPP, FGD2, PMEL, CYP19A1,
MLANA, STEAP1, SSX2, PLAC1, ANKRD30A, CPA2, TTN, ZDHHC23, ARPP21, RBPMS,
PAX5, MIA, CIZ1, AMACR, BAP31, ID01, PGR, RAD51, USP17L2, OLAH, IGF2BP3, STS,
IGF2, ACTA1, or CTAG1.
[00196] The mixed population of modified cells described herein includes
about 1% to 10%
modified cells expressing the first antigen binding domain, 50% to 60%
modified cells
expressing a second antigen binding domain, and about 10% modified cells
expressing both the
first antigen binding domain and the second antigen binding domain (wherein
the first and
second antigen binding domains are expressed in a single cell).
[00197] The present disclosure also describes methods of culturing cells
described herein.
The methods described herein include obtaining a cell comprising a first
antigen binding domain
and/or a second antigen binding domain, wherein the first antigen binding
domain binds a cell
surface molecule of a WBC, and the second antigen binding domain binds an
antigen different
from the cell surface molecule of the WBC; and culturing the cell in the
presence of an agent
derived from a cell surface molecule of the WBC or from an antigen to which
the second antigen
binding domain binds. In embodiments, the agent is an extracellular domain of
a cell surface
molecule of a WBC.
[00198] The present disclosure also describes methods of culturing mixed
population of cells
described herein. The methods described herein include obtaining a mixed
population of cells
comprising a first antigen binding domain and/or a second antigen binding
domain, wherein the
first antigen binding domain binds a cell surface molecule of a WBC, and the
second antigen
binding domain binds an antigen different from the cell surface molecule of
the WBC; and
33
CA 03125646 2021-06-30
WO 2020/146743
PCT/US2020/013099
culturing the cells in the presence of an agent derived from a cell surface
molecule of the WBC
or from an antigen to which the second antigen binding domain binds. In
embodiments, the
agent is an extracellular domain of a cell surface molecule of a WBC.
[00199] The
present disclose describes methods for in vitro cell preparation, wherein the
method includes providing cells; introducing one or more nucleic acids
described herein
encoding a first antigen binding domain and/or a second antigen binding domain
into the cells,
wherein the first antigen binding domain binds a cell surface molecule of a
WBC, and the
second antigen binding domain binds an antigen different from the cell surface
molecule of the
WBC; and culturing the cells in the presence of an agent derived from the cell
surface molecule
of the WBC or from an antigen to which the second antigen binding domain
binds. The methods
provide genetically modified cells including a first antigen binding domain,
cells including a
second binding domain, and cells including both the first and second antigen
binding domain.
The methods provide cells with single binding domains and cells expressing
both antigen
binding domains. The methods also provide a mixed population of cells
including cells including
a single binding domain and cells expressing both antigen binding domains.
Additionally, the
methods provide compositions including a mixed population of cells described
herein.
[00200] The
present disclosure describes using the prepared cell preparation, the mixed
population of cells, or the compositions of mixed population of cells to
enhance and maintain the
T cell expansion in a subject having cancer, in order to be effective in
killing the tumorigenic
cells in the subject. In embodiments, the method comprises introducing a
plurality of nucleic
acids described herein into T cells to obtain a mixed population of modified T
cells, the plurality
of nucleic acids encoding a chimeric antigen receptor (CAR) or TCR binding a
solid tumor
antigen and/or encoding a CAR binding an antigen of a WBC; and administering
an effective
amount of a mixed population of modified cells to the subject, wherein
examples of a mixed
population of modified cells include the following: (1) T cells containing a
CAR or TCR binding a
solid tumor antigen and T cells containing a CAR binding an antigen of a WBC;
(2) the T cells of
(1) and further T cells containing both (i) a CAR or TCR binding a solid tumor
antigen, and (ii) a
CAR binding an antigen of a WBC (both (i) and (ii) are in a single modified T
cell); (3) T cells
containing both (i) the CAR or TCR binding a solid tumor antigen, and (ii) a
CAR binding an
antigen of a WBC (both (i) and (ii) are in a single modified T cell); (4) T
cells containing a CAR
or TCR binding a solid tumor antigen and T cells containing both (i) a CAR or
TCR binding a
solid tumor antigen and (ii) a CAR binding an antigen of a WBC (both (i) and
(ii) are in a single
modified T cell); or (5) T cells containing a CAR binding an antigen of a WBC
and T cells
containing both (i) a CAR or TCR binding a solid tumor antigen and (ii) a CAR
binding an
antigen of a WBC (both (i) and (ii) are in a single modified T cell). In
embodiments, the WBC is a
B cell. Additionally, the present disclosure describes methods for introducing
and/or enhancing
lymphocyte (T cell) response in a subject wherein the response is to a
therapeutic agent (e.g.,
cytokines) or a therapy for treating the subject. Embodiments described herein
involve a
34
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
mechanism that expands and/or maintains the lymphocytes and a mechanism that
relates to
binding of a CAR to a tumor cell. In embodiments, the first mechanism involves
a molecule
involved in expanding and/or maintaining the lymphocytes in a subject, and an
additional
mechanism involves a molecule directed to inhibiting the growth of, or the
killing of a tumor cell
in the subject. In embodiments, the mechanisms involve signal transduction and
molecules or
domains of a molecules responsible for signal transduction are involved in the
mechanisms
described herein. For example, the first mechanism includes a CAR binding an
antigen
associated with blood, such as blood cells and blood plasma, or non-essential
tissues, and the
additional mechanism includes a CAR or TCR targeting an antigen associated
with the tumor
cell. Examples of non-essential tissues include the mammary gland, colon,
gastric gland, ovary,
blood components (such as WBC), and thyroid. In embodiments, the first
mechanism involves a
first antigen binding domain of a molecule, and the additional mechanism
involves a second
antigen binding domain of a molecule. In embodiments, the first mechanism and
the additional
mechanism are performed by a mixed population of modified cells. In
embodiments, the
mechanism involves a cell expressing an antigen associated with a tumor cell,
and the
additional mechanism involves a lymphocyte, such as a B cell, expressing a
cell surface
antigen. In embodiments, the CAR binding a solid tumor antigen is a bispecific
CAR. In
embodiments, the CAR binding an antigen of WBC is a bispecific CAR.
[00201] The methods described herein involves lymphocytes expressing an
expansion
molecule and a function molecule. In embodiments, the expansion molecule
expands and/or
maintains the lymphocytes in a subject, and the function molecule inhibits the
growth of or kills a
tumor cell in the subject. In embodiments, the expansion molecule and the
function molecule
are on a single CAR molecule, for example a bispecific CAR molecule. In
embodiments, the
expansion molecule and the function molecule are on separate molecules, for
example, CAR
and TCR or two different CARs. The expansion molecule can include a CAR
binding to an
antigen associated with blood (e.g., blood cells and blood plasma) or non-
essential tissues, and
the function molecule can include a CAR or TCR targeting an antigen associated
with a tumor
cell.
[00202] Lymphocyte or T cell response in a subject refers to cell-mediated
immunity
associated with a helper, killer, regulatory, and other types of T cells. For
example, T cell
response may include activities such as assisting other WBCs in immunologic
processes and
identifying and destroying virus-infected cells and tumor cells. T cell
response in the subject can
be measured via various indicators such as a number of virus-infected cells
and /or tumor cells
that T cells kill, the amount of cytokines (e.g., IL-6 and IFN-y) that T cells
release in vivo and/or
in co-culturing with virus-infected cells and/or tumor cells, indicates a
level of proliferation of T
cells in the subject, a phenotype change of T cells, for example, changes to
memory T cells,
and a level longevity or lifetime of T cells in the subject.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00203] In embodiments, the method of enhancing T cell response described
herein can
effectively treat a subject in need thereof, for example, a subject diagnosed
with a tumor. The
term tumor refers to a mass, which can be a collection of fluid, such as
blood, or a solid mass. A
tumor can be malignant (cancerous) or benign. Examples of blood cancers
include chronic
lymphocytic leukemia, acute myeloid leukemia, acute lymphoblastic leukemia,
and multiple
myeloma.
[00204] Solid tumors usually do not contain cysts or liquid areas. The
major types of
malignant solid tumors include sarcomas and carcinomas. Sarcomas are tumors
that develop in
soft tissue cells called mesenchymal cells, which can be found in blood
vessels, bone, fat
tissues, ligament lymph vessels, nerves, cartilage, muscle, ligaments, or
tendon, while
carcinomas are tumors that form in epithelial cells, which are found in the
skin and mucous
membranes. The most common types of sarcomas include undifferentiated
pleomorphic
sarcoma which involves soft tissue and bone cells; leiomyosarcoma which
involves smooth
muscle cells that line blood vessels, gastrointestinal tract, and uterus;
osteosarcoma which
involves bone cells, and liposarcoma which involves fat cells. Some examples
of sarcomas
include Ewing sarcoma, Rhabdomyosarcoma, chondosarcoma, mesothelioma,
fibrosarcoma,
fibrosarcoma, and glioma.
[00205] The five most common carcinomas include adrenocarcinoma which involves
organs
that produce fluids or mucous, such as the breasts and prostate; basal cell
carcinoma which
involves cells of the outer-most layer of the skin, for example, skin cancer;
squamous cell
carcinoma which involves the basal cells of the skin; and transitional cell
carcinoma which
affects transitional cells in the urinary tract which includes the bladder,
kidneys, and ureter.
Examples of carcinomas include cancers of the thyroid, breast, prostate, lung,
intestine, skin,
pancreas, liver, kidneys, and bladder, and cholangiocarcinoma.
[00206] The methods described herein can be used to treat a subject
diagnosed with cancer.
The cancer can be a blood cancer or can be a solid tumor, such as a sarcoma or
carcinoma.
The method of treating includes administering an effective amount of a mixed
population of T
cells described herein comprising a first antigen binding domain and/or a
second antigen
binding domain to the subject to provide a T-cell response, wherein the first
antigen binding
domain binds a cell surface molecule of a WBC, and the second antigen binding
domain binds
an antigen different from the cell surface molecule of the WBC. In
embodiments, enhancing the
T cell response in the subject includes selectively enhancing proliferation of
T cell expressing
the first antigen binding domain and the second antigen binding domain in
vivo.
[00207] The methods for enhancing T cell response in a subject include
administering to the
subject T cells comprising a CAR or a bispecific CAR including two different
antigen binding
domains and T cells comprising a first CAR and a second CAR, wherein the first
CAR and the
second CAR, each includes a different antigen binding domain.
36
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00208] In embodiments, methods for enhancing T cell response in a subject
described
herein include administering to the subject T cells including a CAR molecule
and a TCR
molecule. The CAR molecule targets or binds a surface marker of a white blood
cell, and the
TCR molecule binds a marker or an antigen of the tumor that is expressed on
the surface or
inside the tumor cell.
[00209] In embodiments, the methods for enhancing T cell response in a
subject in need
thereof include administering to the subject, a mixed population of modified
cells or a
composition comprising a mixed population of modified cells. Examples of a
mixed population of
modified T cells include the following: (1) T cells containing a CAR binding
an antigen of a WBC
and T cells containing a CAR or TCR binding a tumor antigen; (2) the T cells
of (1) and further T
cells containing both (i) the CAR or TCR binding a tumor antigen, and (ii) a
CAR binding an
antigen of a WBC (both (i) and (ii) are in a single modified T cell); (3) T
cells containing both (i) a
CAR or TCR binding a tumor antigen, and (ii) a CAR binding an antigen of a WBC
(both (i) and
(ii) are in a single modified T cell); (4) T cells containing a CAR or TCR
binding a tumor antigen
and T cells containing both (i) a CAR or TCR binding a solid tumor antigen and
(ii) a CAR
binding an antigen of a WBC; or (5) T cells containing a CAR binding an
antigen of a WBC and
T cells containing both (i) a CAR or TCR binding a solid tumor antigen and
(ii) the CAR binding
the antigen of a WBC (both (i) and (ii) are in a single modified T cell). In
embodiments, the
subject is diagnosed with a solid tumor. In embodiments, the tumor antigen is
a solid tumor
antigen, for example tM UC1. In embodiments, the WBC is a B cell, and the
antigen is a B cell
antigen. In embodiments, the B cell antigen is CD19. In embodiments, the tumor
antigen is
tMUC1 and the antigen of a WBC is CD19.
[00210] The present disclosure describes methods of expanding and/or
maintaining cells
expressing an antigen binding domain in vivo. The method includes
administering an effective
amount of a mixed population of modified cells or a composition including a
mixed population of
modified cells described herein to a subject These methods described herein
are useful for
expanding T cells, NK cells, macrophages and/or dendritic cells.
[00211] The mixed population of modified T cells described herein include a
first CAR and/or
a second CAR or TCR. In embodiments, the first CAR contains a first antigen
binding domain
and the second CAR or TCR contains a second antigen binding domain. For
example, the first
CAR and the second CAR or TCR include an extracellular antigen binding domain,
a
transmembrane domain, and a cytoplasmic domain. The cytoplasmic domain of the
first CAR
and second CAR include a co-stimulatory domain and a CD3 zeta domain for
transmitting
signals for activation of cellular responses. In embodiments, the first CAR
and second CAR or
TCR are expressed on different modified T cells. In embodiments, the first CAR
and second
CAR or TCR are expressed on the same modified T cell.
[00212] In embodiments, in the mixed population of modified T cells
described herein, the
cytoplasmic domain of the first CAR, which contains an antigen binding domain
for expanding
37
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
and/or maintaining modified T cells, includes one or more co-stimulatory
domains in the
absence of a CD3 zeta domain such that activation or stimulation of the first
CAR expands
WBCs, such as lymphocytes, without introducing and/or activating the killing
function of the
modified T cells targeting the WBCs. In embodiments, the lymphocytes are T
cells. In
embodiments, when the cytoplasmic domain of the first CAR includes one or more
co-
stimulatory domains in the absence of a CD3 zeta domain, the second CAR
includes a CD3
zeta domain.
[00213] In embodiments, the first and second antigen binding domains are on
the same CAR
(the first CAR), for example, a bispecific CAR with an extracellular antigen
binding domain, a
transmembrane domain, and a cytoplasmic domain. The extracellular antigen
binding domain
includes at least two scFvs and at least one of the scFvs function as a first
antigen binding
domain for binding a cell surface molecule of a WBC. In embodiments, the
bispecific CAR is
expressed on a modified T cell.
[00214] In embodiments, the antigen different from the cell surface
molecule of the WBC is
0D19, 0D22, CD20, BCMA, CD5, CD7, CD2, 0D16, 0D56, CD30, 0D14, 0D68, CD11b,
0D18,
0D169, CD1c, 0D33, 0D38, 0D138, CD13, B7-H3, CAIX, 0D123, 0D133, CD171,
CD171/L1-
CAM, CEA, Claudin 18.2, cMet, CS1, CSPG4, Dectin1, EGFR, EGFR vIII, EphA2,
ERBB
receptors, ErbB T4, ERBB2, FAP, Folate receptor 1, FITC, Folate receptor 1,
FSH, GD2, GPC3,
HA-1 H/HLA- A2, HER2, IL-11Ra, IL13 receptor a2, IL13R, IL13Ra2 (zetakine),
Kappa,
Leukemia, LewisY, Mesothelin, MUC1, NKG2D, NY-ESO-1, PSMA, ROR-1, TRAIL-
receptor1,
or VEGFR2.
[00215] In embodiments, the MUC1 is a tumor-exclusive epitope of a human
MUC1, and the
first CAR and the second CAR or the TCR are expressed as separate
polypeptides. In
embodiments, the MUC1 is a tumor form of human MUC1 (tMUC1).
[00216] In embodiments, in the mixed population of modified cells described
herein, the first
CAR, which includes an antigen binding domain for expanding and/or maintaining
modified
cells, may include a co-stimulatory domain without a signaling domain of CD3
zeta domain, and
the CAR (second CAR) may comprise the MUC1 binding domain, a transmembrane
domain, a
co-stimulatory, and a CD3 zeta domain.
[00217] As used herein, the term "MUC1" refers to a molecule defined as
follows. MUC1 is
one of the epithelial mucin family of molecules. MUC1 is a transmembrane mucin
glycoprotein
that is normally expressed on all glandular epithelial cells of the major
organs. In normal cells,
MUC1 is only expressed on the apical surface and is heavily glycosylated with
its core proteins
sequestered by the carbohydrates. As cells transform to a malignant phenotype,
expression of
MUC1 increases several folds, and the expression is no longer restricted to
the apical surface,
but it is found all around the cell surface and in the cytoplasm. In addition,
the glycosylation of
tumor associated MUC1 (tMUC1) is aberrant, with greater exposure of the
peptide core than is
found on MUC1 expressed in normal tissues.
38
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00218] MUC1 is widely expressed on a large number of epithelial cancers
and is aberrantly
glycosylated making it structurally and antigenically distinct from that
expressed by non-
malignant cells (see, e.g., Barratt-Boyes, 1996; Price et al., 1998; Peterson
et al., 1991). The
dominant form of MUC1 is a high molecular weight molecule comprising a large
highly
immunogenic extracellular mucin-like domain with a large number of twenty
amino acid tandem
repeats, a transmembrane region, and a cytoplasmic tail (Quin et al., 2000;
McGucken et al.,
1995; Dong et al., 1997).
[00219] In most epithelial adenocarcinomas including breast and pancreas,
MUC1 is
overexpressed and aberrantly glycosylated. Adenocarcinoma of the breast and
pancreas not
only overexpress MUC1 but also shed MUC1 into the circulation. High MUC1 serum
levels are
associated with progressive disease. MUC1 has been exploited as a prospective
biomarker
because of the complex and heterogeneous nature of the epitopes expressed
within the
antigen. MUC1 synthesized by cancerous tissues (e.g., tumor associated MUC1)
usually
displays an aberrant oligosaccharide profile, which gives rise to the
expression of neomarkers
such as sialyl-Lea (assayed in the CA19-9 test), sialyl-Lex, and sialyl-Tn
(TAG-72), as well as
the cryptic epitopes such as Tn.
[00220] Several antibodies are being developed against MUC1 for therapeutic
use.
Pemtumomab (also known as HMFG1) is in Phase III clinical trials as a carrier
to deliver the
radioisotope Yttrium-90 into tumors in ovarian cancer (reviewed in Scott et
al., 2012). CA15-3
(also the HMFG1 antibody), 0A27-29, and CA19-9 are all antibodies to MUC1 that
are used to
assess levels of circulating MUC1 in patients with cancer. However, these
antibodies have
shown limited utility as therapeutic agents or as biomarkers because they
cannot distinguish
effectively between MUC1 expressed on normal versus transformed tumor
epithelia. In other
words, none of these antibodies appear to be targeted to a tumor associated
MUC1 (tMUC1)
epitope.
[00221] A new antibody that is highly specific for a tumor associated form
of MUC1 (tMUC1)
is designated TAB-004 and is described in U.S. Pat. No. 8,518,405 (see also
Curry et al., 2013).
While Pemtumomab (HMFG1) was developed using human milk fat globules as the
antigen
(Parham et al., 1988), TAB-004 was developed using tumors expressing an
altered form of
MUC1 (Tinder et al., 2008). TAB-004 recognizes the altered glycosylated
epitope within the
MUC1 tandem repeat sequence. This area is accessible for antigenic detection
in tMUC but is
blocked from antigenic detection in normal MUC1 by large branches of
glycosylation (Gendler,
2001; Mukherjee et al., 2003b; Hollingsworth & Swanson, 2004; Kufe, 2009).
Importantly, TAB-
004 is different from the epitopes recognized by other MUC1 antibody and has
unique
complementary determinant regions (CDRs) of the heavy and light chains. The
antibody binds
the target antigen with a high binding affinity at 3 ng/ml (20 pM) and does
not bind unrelated
antigens (Curry et al., 2013). Thus, TAB-004 distinguishes between normal and
tumor form of
MUC1 while HMFG1 (Pemtumomab) does not (see U.S. Pat. No. 8,518,405).
39
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00222] In embodiments, the first CAR comprises the first antigen binding
domain, a
transmembrane domain, a co-stimulatory domain, and a CD3 zeta domain, and/or
the second
CAR comprises the second antigen binding domain, a transmembrane domain, a co-
stimulatory
domain, and a CD3 zeta domain.
[00223] In embodiments, the antigen binding domain is a Fab or a scFv. In
embodiments, the
first CAR comprises the amino acid sequence of one of SEQ ID NO: 5, 6, and 53-
58; and the
second CAR comprises the amino acid sequence of one of SEQ ID NOs: 5-17, 29,
33, 37, 71,
and 72, or the amino acid sequence encoded by the nucleic acid sequence of one
of SEQ ID
NOs: 41, 45, 63, 67, and 68. In embodiments, a nucleic acid sequence encoding
the first CAR
comprises the nucleic acid sequence of SEQ ID NO: 59 or 60, and a nucleic acid
sequence
encoding the second CAR comprises the nucleic acid sequence of SEQ ID NO: 61.
In
embodiments, the nucleic acid comprises one of the nucleic acid sequence of
SEQ ID NO: 62-
69. In embodiments, the first CAR and the second CAR are expressed as separate
polypeptides.
[00224] In embodiments, the first antigen binding domain is on a CAR and
the second
antigen binding domain is on a T Cell Receptor (TCR). In embodiments, the TCR
is a modified
TCR. In embodiments, the TCR is derived from spontaneously occurring tumor-
specific T cells
in patients. In embodiments, the TCR binds a tumor antigen. In embodiments,
the tumor antigen
comprises CEA, gp100, tMUC1, MART-1, p53, MAGE-A3, or NY-ESO-1.
[00225] As used herein, "a thyroid antigen" refers to an antigen expressed
on or by a thyroid
cell. Examples of thyroid cells include follicular cells and parafollicular
cells. A human TSHR is a
receptor for thyroid-stimulating hormone (TSH) which is present on the thyroid
membrane (SEQ
ID NO: 20). When TSH secreted from the pituitary gland binds to TSHR on the
thyroid follicle
cell membrane, the thyroid gland secretes T3 and T4 having metabolic
functions. TSHR is a
seven-transmembrane receptor having a molecular weight of about 95,000 to
100,000 Da!tons.
It was reported that the human thyrotropin receptor (TSHR) includes three
domains: a leucine-
rich domain (LRD; amino acids 36-281), a cleavage domain (CD; amino acids 282-
409), and a
transmembrane domain (TMD; amino acids 410-699). Human thyrotropin (hTSH) a
chains were
found to bind many amino acids on the LRD surface and CD surface. As used
herein, "TSHR"
refers to human thyroid stimulating hormone receptor. The term should be
construed to include
not only human thyroid stimulating hormone receptor, but variants, homologs,
fragments and
portions thereof to the extent that such variants, homologs, fragments and
portions thereof
retain the ability of human thyroid stimulating hormone receptor to bind to
antibodies or ligands
of human thyroid stimulating hormone receptor as disclosed herein.
[00226] In certain embodiments, the antigen is a stomach or colon antigen.
For example, the
colon antigen is Guanylate cyclase 2C (GUCY2C) having SEQ ID NO: 23. As used
herein, "a
colon antigen" refers to an antigen expressed on or by a colon cell. Examples
of colon cells
include goblet cells and enterocytes. Guanylyl cyclase 2C (GUCY2C) is
principally expressed in
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
intestinal epithelial cells. GUCY2C is the receptor for diarrheagenic
bacterial enterotoxins (STs)
and the gut paracrine hormones, guanylin, and uroguanylin. These ligands
regulate water and
electrolyte transport in the intestinal and renal epithelia and are ultimately
responsible for acute
secretory diarrhea. As used herein, "GUCY2C" refers to human Guanylyl cyclase
20. The term
should be construed to include not only human Guanylyl cyclase 20, but also
variants,
homologs, fragments and portions thereof to the extent that such variants,
homologs, fragments
and portions thereof retain the ability of Guanylyl cyclase 20 to bind
antibodies or ligands of
human Guanylyl cyclase 20 as disclosed herein. In embodiments, the amino acid
sequence of
at least a portion of GUCY2C comprises SEQ ID NO: 23. Claudin18.2 (CLDN 18.2)
is a
stomach-specific isoform of Claudin-18 and is highly expressed in gastric and
pancreatic
adenocarcinoma.
[00227] In embodiments, a T cell clone that expresses a TCR with high
affinity for the target
antigen may be isolated. Tumor-infiltrating lymphocytes (TI Ls) or peripheral
blood mononuclear
cells (PBMCs) can be cultured in the presence of antigen-presenting cells
(APCs) pulsed with a
peptide representing an epitope known to elicit a dominant T cell response
when presented in
the context of a defined HLA allele. High-affinity clones may then be selected
on the basis of
MHC¨peptide tetramer staining and/or the ability to recognize and lyse target
cells pulsed with
low titrated concentrations of cognate peptide antigen. After the clone has
been selected, the
TCRa and TCR[3 chains or TCRy and TORO chains are identified and isolated by
molecular
cloning. For example, for TCRa and TCR[3 chains, the TCRa and TCR[3 gene
sequences are
then used to generate an expression construct that ideally promotes stable,
high-level
expression of both TCR chains in human T cells. The transduction vehicle, for
example, a
gammaretrovirus or lentivirus, can then be generated and tested for
functionality (antigen
specificity and functional avidity) and used to produce a clinical lot of the
vector. An aliquot of
the final product can then be used to transduce the target T cell population
(generally purified
from patient PBMCs), which is expanded before infusion into the patient.
[00228] Various methods may be implemented to obtain genes encoding tumor-
reactive
TCR. More information is provided in Kershaw et al., Olin Trans! Immunology.
2014 May; 3(5):
e16. In embodiments, specific TCR can be derived from spontaneously occurring
tumor-specific
T cells in patients. Antigens included in this category include the melanocyte
differentiation
antigens MART-1 and gp100, as well as the MAGE antigens and NY-ESO-1, with
expression in
a broader range of cancers. TCRs specific for viral-associated malignancies
can also be
isolated, as long as viral proteins are expressed by transformed cells.
Malignancies in this
category include liver and cervical cancer, associated with hepatitis and
papilloma viruses, and
Epstein-Barr virus-associated malignancies. In embodiments, target antigens of
the TCR may
include CEA (e.g., for colorectal cancer), gp100, MART-1, p53 (e.g., for
Melanoma), MAGE-A3
(e.g., Melanoma, esophageal and synovial sarcoma), NY-ESO-1 (e.g., for
Melanoma and
sarcoma as well as Multiple myelomas).
41
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00229] In embodiments, a binding domain of the first CAR binds CD19, and a
binding
domain of the second CAR binds tumor associated MUC1 (tMUC1). In embodiments,
the
binding domain of the second CAR comprises: (i) a heavy chain complementary
determining
region 1 comprising the amino acid sequence of SEQ ID: 76 or 85, a heavy chain
complementary determining region 2 comprising the amino acid sequence of SEQ
ID: 77 or 86,
and a heavy chain complementary determining region 3 comprising the amino acid
sequence of
SEQ ID: 78 or 87; and (ii) a light chain complementary determining region 1
comprising the
amino acid sequence of SEQ ID: 73 or 82, a light chain complementary
determining region 2
comprising the amino acid sequence of TRP-ALA-SER (WAS) or SEQ ID: 83, and a
light chain
complementary determining region 3 comprising the amino acid sequence of SEQ
ID: 75 or 84.
[00230] In embodiments, the binding domain of the second CAR comprises: (i)
a heavy chain
complementary determining region 1 comprising the amino acid sequence of SEQ
ID: 76, a
heavy chain complementary determining region 2 comprising the amino acid
sequence of SEQ
ID: 77, and a heavy chain complementary determining region 3 comprising the
amino acid
sequence of SEQ ID: 78; and (ii) a light chain complementary determining
region 1 comprising
the amino acid sequence of SEQ ID: 73, a light chain complementary determining
region 2
comprising the amino acid sequence of TRP-ALA-SER (WAS), and a light chain
complementary
determining region 3 comprising the amino acid sequence of SEQ ID: 75.
[00231] In embodiments, the binding domain of the second CAR comprises: (i)
a heavy
chain complementary determining region 1 comprising the amino acid sequence of
SEQ ID: 85,
a heavy chain complementary determining region 2 comprising the amino acid
sequence of
SEQ ID: 86, and a heavy chain complementary determining region 3 comprising
the amino acid
sequence of SEQ ID: 87; and (ii) a light chain complementary determining
region 1 comprising
the amino acid sequence of SEQ ID: 82, a light chain complementary determining
region 2
comprising the amino acid sequence of SEQ ID: 83, and a light chain
complementary
determining region 3 comprising the amino acid sequence of SEQ ID: 84. In
embodiments, the
binding domain of the first CAR comprises the amino acid sequence of SEQ ID: 5
or 6. In
embodiments, the binding domain of the second CAR comprises one of the amino
acid
sequences of SEQ ID: 70 -72 and 79-81.
[00232] In embodiments, the first CAR comprises the first antigen binding
domain, a
transmembrane domain, a co-stimulatory domain, and a CD3 zeta domain and/or
the second
CAR comprises the second antigen binding domain, a transmembrane domain, a co-
stimulatory
domain, and a CD3 zeta domain.
[00233] In embodiments, the first CAR and the second CAR are expressed as
separate
polypeptides.
[00234] In embodiments, the cytoplasmic domain or the transmembrane domain
of the
second CAR is modified such that the second CAR is capable of activating the
modified T cell
via cells expressing CD19 without damaging the cells expressing CD19.
42
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00235] Embodiments described herein relate to a bispecific chimeric
antigen receptor,
comprising: a first antigen binding domain, a second antigen binding domain, a
cytoplasmic
domain, and transmembrane domain, wherein the first antigen binding domain
recognizes a first
antigen, and the second antigen binding domain recognizes a second antigen,
the first antigen
is different from the second antigen.
[00236] In embodiments, the first antigen and the second antigen do not
express on the
same cell. In embodiments, the first antigen is an antigen of a blood
component, and the
second antigen is an antigen of a solid tumor.
[00237] Blood cells refer to red blood cells (RBCs), white blood cells
(WBCs), platelets, or
other blood cells. For example, RBCs are blood cells of delivering oxygen (02)
to the body
tissues via the blood flow through the circulatory system. Platelets are cells
that are involved in
hemostasis, leading to the formation of blood clots. WBCs are cells of the
immune system
involved in defending the body against both infectious disease and foreign
materials. There are
a number of different types and sub-types of WBCs and each has a different
role to play. For
example, granulocytes, monocytes, and lymphocytes are 3 major types of white
blood cell.
There are three different forms of granulocytes: Neutrophils, Eosinophils,
Basophils.
[00238] A cell surface molecule of a WBC refers to a molecule expressed on the
surface of
the WBC. For example, the cell surface molecule of a lymphocyte may include
CD19, 0D22,
CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, and CD30. The cell surface molecule of
a B cell
may include CD19, CD20, 0D22, BCMA. The cell surface molecule of a monocyte
may include
CD14, 0D68, CD11 b, CD18, 0D169, and CD1c. The cell surface molecule of
granulocyte may
include 0D33, 0D38, 0D138, and CD13.
[00239] In embodiments, the first antigen is CD19, and the second antigen
is a tumor
associated MUC1 (tMUC1). In embodiments, the first antigen binding domain
comprises one of
the amino acid sequences of SEQ ID: 5 and 6. In embodiments, the second
antigen binding
domain comprises one of the amino acid sequences of SEQ ID: 70 -72 and 79-81.
[00240] In embodiments, the present disclosure describes a method of
enhancing T cell
response in a subject in need thereof or treating a tumor of a subject, the
method comprising:
administering an effective amount of a mixed population of modified T cells or
a composition of
a mixed population of modified T cells, described herein, to the subject to
provide a T cell
response such that the CAR T cell is expanded in the blood of the subject via
cells expressing
CD19. In embodiments, the method may further comprise infusing B cells into
the subject to
continue to activate and/or expand the CAR T cells. For example, the B cells
of the subject or
genetically modified B cells from healthy donor may be obtained and stored
before CAR T cell
infusion. In embodiments, the method may further comprise administering a cell
expressing
CD19 or a polypeptide comprising at least an extracellular domain of CD19 or
the antigen that
the CAR T cells recognize. For example, the cell expressing CD19 may include
cell lines such
as K562 and NK92 that are transduced with nucleic acid sequences encoding
CD19. In
43
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
embodiments, the method may further comprise identifying CAR T cells
expressing both first
and second CAR, as well as administering the identifier CAR T cells to the
subject. For
example, MUC1 may be associated as a sorting marker such that CAR T cells
expressing
MUC1 may be identified timely.
[00241] In embodiments, the tumor associated MUC1 (tMUC1) is expressed on
tumor cells,
but not on corresponding non-malignant cells. In embodiments, a scFv against
the tumor
associated MUC1 directly interacts with an o-glycosylated GSTA motif (SEQ ID
NO. 88).
[00242] In embodiments, the present disclosure describes a method of in
vivo cell expansion
and maintenance. In embodiments, the method may include administering an
effective amount
of a mixed population of modified T cells described herein to the subject in
need thereof to
provide a T cell response; and administering an effective amount of presenting
cells (e.g., T
cells) expressing a soluble agent that an extracellular domain of the CAR
recognizes. In
embodiments, the method may be implemented to enhance T cell response in a
subject in need
thereof. The method may include administering an effective amount of a mixed
population of
modified T cells comprising a CAR to the subject to provide a T cell response
and administering
an effective amount of presenting cells expressing a soluble agent that an
extracellular domain
of the CAR recognizes to enhance the T cell response in the subject. In
certain embodiments,
the presenting cells are T cells, dendritic cells, and/or antigen presenting
cells. In certain
embodiments, the enhancing T cell response in the subject may include
selectively enhancing
proliferation of T cell comprising the CAR. In embodiments, the method may be
used to
enhance treatment of a condition of a subject using modified T cells. The
method may include
administering a population of cells that express an agent or administering an
agent that is
formulated as a vaccine. In these instances, the modified T cells include a
nucleic acid that
encodes a CAR, and an extracellular domain of the CAR recognize the agent. In
embodiments,
the method may be implemented to enhance proliferation of the modified T cells
in a subject
having a disease. The method may include preparing the modified T cells
comprising a CAR;
administering an effective amount of the modified T cells to the subject;
introducing, into cells, a
nucleic acid encoding an agent that an extracellular domain of the CAR
recognizes; and
administering an effective amount of the cells (introduced with the nucleic
acid encoding the
agent) to the subject. In embodiments, the T cell expansion may be measured
based on an
increase in copy number of CAR molecules in genomic DNA of the T cells. In
embodiments, the
T cell expansion may be measured based on flow cytometry analysis on molecules
expressed
on the T cells.
[00243] Embodiments described herein relate to mixed population of modified
T cells
comprising a first CAR and a second CAR or TCR in separate T cells and/or in
the same T cells,
wherein an antigen binding domain of the first CAR binds an antigen such as
CD19, 0D33,
CD14, and BCMA, and an antigen binding domain of the second CAR binds a tumor
associated
MUC. In embodiments, the tumor associated MUC is MUC1 (for example tMUC1) or
MUC2.
44
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Embodiments described herein relate to a composition comprising a mixed
population of the
modified T cells and to a method of enhancing T cell response in a subject in
need thereof or
treating a tumor of a subject, the method comprising: administering an
effective amount of the
mixed population of modified T cells.
[00244] In embodiments, the first CAR comprises the amino acid sequence of
SEQ ID NO:
207, and the second CAR comprises the amino acid sequence of SEQ ID: 202. In
embodiments, the first CAR comprises the amino acid sequence of SEQ ID NO:
203, 207, 216,
or 219, and the second CAR comprises the amino acid sequence of SEQ ID: 202 or
205. In
embodiments, the antigen binding domain of the second CAR comprises the amino
acid
sequence of SEQ ID NO: 70. In embodiments, the antigen binding domain of the
second CAR
comprises the amino acid sequence of SEQ ID NO: 5 or 6. In embodiments, the a
modified T
cell described herein comprises a nucleic acid sequences of SEQ ID NO: 201,
204, 206, 208,
215, 217, 218, or 220. In embodiments, each of the first CAR and the second
CAR comprises
an antigen binding domain, a transmembrane domain, and a cytoplasmic domain.
[00245] In embodiments, the cytoplasmic domain of the CAR molecules
described herein
comprise a co-stimulatory domain and a CD3 zeta domain. In embodiments, the
CAR
molecules described herein may include a co-stimulatory domain without a
corresponding
component of CD3 zeta domain. In embodiments, the CAR molecules described
herein may
include a CD3 zeta domain without a co-stimulatory domain.
[00246] In embodiments, the modified cell comprises a dominant negative
variant of a
receptor of programmed death 1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-
4), B- and T-
lymphocyte attenuator (BTLA), T cell immunoglobulin mucin-3 (TIM-3),
lymphocyte-activation
protein 3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT),
leukocyte-
associated immunoglobulin-like receptor 1 (LAIR!), natural killer cell
receptor 2B4 (2B4), or CD
160. In embodiments, the modified cell further comprises a nucleic acid
sequence encoding a
suicide gene, and/or the suicide gene comprises a HSV-TK suicide gene system.
In
embodiments, the isolated T cell comprises a reduced amount of TCR, as
compared to the
corresponding wide-type T cell.
[00247] Dominant negative mutations have an altered gene product that acts
antagonistically
to the wild-type allele. These mutations usually result in an altered
molecular function (often
inactive) and are characterized by a dominant or semi-dominant phenotype. In
embodiments,
the modified cells described herein comprise the dominant negative (DN) form
of the PD-1
receptor. In embodiments, the expression of the DN PD-1 receptor in the
modified cells
described herein is regulated by an inducible gene expression system. In
embodiments, the
inducible gene expression system is a lac system, a tetracycline system, or a
galactose system.
[00248] The present disclosure describes pharmaceutical compositions. The
pharmaceutical
compositions include one or more of the following: CAR molecules, TCR
molecules, modified
CAR T cells, modified cells comprising CAR or TCR, mix population of modified
cells, nucleic
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
acids, and vectors described herein. Pharmaceutical compositions are
administered in a
manner appropriate to the disease to be treated (or prevented). The quantity
and frequency of
administration will be determined by such factors as the condition of the
patient, and the type
and severity of the patient's disease, although appropriate dosages may be
determined by
clinical trials.
[00249] The term "pharmaceutically acceptable" means approved by a
regulatory agency of
the US. Federal or a state government or the EMA (European Medicines Agency)
or listed in
the U.S. Pharmacopeia Pharmacopeia (United States Pharmacopeia- 33/National
Formulary-28
Reissue, published by the United States Pharmacopeia Convention; lnc,,
Rockville Md.,
publication date: April 2010) or other generally recognized phari-nacopeia for
use in animals,
and more particularly in humans.
[00250] The term "carrier" refers to a diluent, adjuvant {e,g,, Freund's
adjuvant (complete and
incomplete)), excipient, or vehicle with which the therapeutic is
administered. Pharmaceutical
carriers can be sterile liquids, such as water and oils, including those of
petroleum, animal,
vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oii,
sesame oil and the
like. Water is a preferred carrier when the pharmaceutical composition is
administered
intravenously. Saline solutions and aqueous dextrose and glycerol solutions
can also be
employed as liquid carriers, particularly for injectable solutions,
[00251] The present disclosure also describes a pharmaceutical composition
comprising the
first and the second population of cells, described herein. The pharmaceutical
composition
described herein, comprising a first population of cells comprising a first
antigen binding
molecule and a second population of cells comprising a second antigen binding
domain, are
suitable for cancer therapy. For example, the binding of first antigen binding
molecule with an
antigen enhances expansion of the cells suitable for cancer therapy.
[00252] The present disclosure also describes a method for enhancing cancer
therapy using
the cells described herein that are suitable for cancer therapy. The method
comprises
administering an effective amount of a first composition to the subject having
a form of cancer
expressing a tumor antigen, the first composition comprising a first
population of cells (e.g., T
cells) comprising a first antigen binding molecule (e.g., CAR) binding a first
antigen; and
administering an effective amount of a second composition to the subject, the
second
composition comprising a population of the cells comprising a second antigen
binding molecule.
Administration of the first and second compositions can be performed
simultaneously or
separately, for example sequentially. More information about the cells
suitable for cancer
therapy can be found at Eyileten et al., Immune Cells in Cancer Therapy and
Drug Delivery,
Mediators Inflamm. 2016; 2016: 5230219, which is incorporated herein for
reference.
[00253] In embodiments, the method comprises administering an effective
amount of a
population of CAR T cells binding a WBC antigen; and administering an
effective amount of a
population of CAR T cells binding a solid tumor antigen. In embodiments, the
method comprises
46
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
administering an effective amount of a population of CAR T cells binding a WBC
antigen; and
administering an effective amount of a population of T cells binding a solid
tumor antigen (T
cells used in TCR and TIL therapies). In embodiments, the method comprises
administering an
effective amount of a population of CAR T cells binding a WBC antigen; and
administering an
effective amount of a population of NK cells or NK cells expressing CAR
binding a solid tumor
antigen. In embodiments, the method comprises administering an effective
amount of a
population of CAR T cells binding a WBC antigen; and administering an
effective amount of a
population of NK cells or NK cells expressing CAR binding a solid tumor
antigen. In
embodiments, the method comprises administering an effective amount of a
population of CAR
T cells binding a WBC antigen; and administering an effective amount of a
population of DCs or
DCs expressing CAR binding a solid tumor antigen. In embodiments, the method
comprises
administering an effective amount of a population of CAR T cells binding a WBC
antigen; and
administering an effective amount of a population of macrophages or
macrophages expressing
CAR binding a solid tumor antigen. In embodiments, the method comprises
administering an
effective amount of a population of CAR T cells binding a WBC antigen; and
administering an
effective amount of a population of neutrophils or neutrophils expressing CAR
binding a solid
tumor antigen. In embodiments, the method comprises administering an effective
amount of a
population of CAR T cells binding a WBC antigen; and administering an
effective amount of a
population of lymphocytes binding or targeting a solid tumor antigen. In
embodiments, the solid
tumor antigen can be located on the cell surface (e.g., TSHR), on the
extracellular matrix of
tumor microenvironment (e.g., av135 integrin), and/or inside of tumor cells
(e.g., gp100).
[00254] When "an immunologically effective amount", "an anti-tumor
effective amount", "a
tumor-inhibiting effective amount", or "a therapeutically effective amount" is
indicated, the
precise amount of the compositions of the present disclosure to be
administered can be
determined by a physician with consideration of individual differences in age,
weight, tumor
size, extent of infection or metastasis, and condition of the patient
(subject). It can be stated that
a pharmaceutical composition comprising the modified cells described herein
may be
administered at a dosage of 10 to 109cells/kg body weight, preferably
105to106cells/kg body
weight, including all integer values within those ranges. Modified cell
compositions may also be
administered multiple times at these dosages. The cells can be administered by
using infusion
techniques that are commonly known in immunotherapy (see, e.g., Rosenberg et
al., New Eng.
J. of Med. 319:1676, 1988). The optimal dosage and treatment regime for a
particular patient
can readily be determined by one skilled in the art of medicine by monitoring
the patient for
signs of disease and adjusting the treatment accordingly. In certain
embodiments, it may be
desired to administer activated T cells to a subject and then subsequently
redraw the blood (or
have apheresis performed), collect the activated and expanded T cells, and
reinfuse the patient
with these activated and expanded T cells. This process can be carried out
multiple times every
few weeks. In certain embodiments, T cells can be activated from blood draws
of from 10 cc to
47
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
400 cc. In certain embodiments, T cells are activated from blood draws of 20
cc, 30 cc, 40 cc,
50 cc, 60 cc, 70 cc, 80 cc, 90 cc, or 100 cc. Not to be bound by theory, using
this multiple blood
draw/multiple reinfusion protocols, may select out certain populations of T
cells.
[00255] In embodiments, a mixed population of therapeutically effective
amount of modified
cells can be administered to the subject in need thereof sequentially or
simultaneously. As an
example, for a mixed population of two different modified cells, a
therapeutically effective
amount of the modified cells containing the antigen binding domain for
expanding and/or
maintaining the modified cells can be administered before, after, or at the
same time a
therapeutically effective amount of the modified cells containing the antigen
binding domain for
killing a target cell. As another example of a mixed population of two
different modified cells, a
therapeutically effective amount of the modified cells containing the antigen
binding domain for
killing a target cell can be administered before, after, or at the same time a
therapeutically
effective amount of the modified cells containing both the antigen binding
domains of expanding
and/or maintaining the modified cells and of killing a target cell (in a
single modified cell). As an
example, for a mixed population of three different modified cells including
(1) modified cells
containing an antigen binding domain for expanding and/or maintaining the
modified cells, (2)
modified cells containing an antigen binding domain for killing a target cell,
and (3) modified
cells containing both the antigen binding domains of expanding and/or
maintaining the modified
cells and of killing a target cell (in a single modified cell), a
therapeutically effective amount of
(1), (2), and (3) can be administered sequentially in any order (1,2, 3; 2, 3,
1; 3, 1,2; 1, 3, 2; 2,
1,3; or 3, 2, 1) or simultaneously (1+2+3 at the same time). Moreover, two of
the three modified
cells can be combined and administered together with the third one being
administered before
or after the combination. For example, the combination of (1) and (2) can be
administered
before or after (3); or the combination of (1) and (3) can be administered
before or after (2); or
the combination of (2) and (3) can be administered before or after (1).
[00256] The administration of the pharmaceutical compositions described
herein may be
carried out in any convenient manner, including by aerosol inhalation,
injection, ingestion,
transfusion, implantation, or transplantation. The compositions described
herein may be
administered to a patient subcutaneously, intradermally, intratumorally,
intranodally,
intramedullary, intramuscularly, by intravenous (i. v.) injection, or
intraperitoneally. In
embodiments, the modified cell compositions described herein are administered
to subjects by
intradermal or subcutaneous injection. In embodiments, the T cell compositions
of the present
disclosure are administered by i.v. injection. The compositions of modified
cells may be injected
directly into a tumor, lymph node, or site of infection. In embodiments, cells
activated and
expanded using the methods described herein, or other methods known in the art
where T cells
are expanded to therapeutic levels, are administered to patients in
conjunction with (e.g.,
before, simultaneously or following) any number of relevant treatment
modalities, for example
as a combination therapy, including but not limited to treatment with agents
for antiviral therapy,
48
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cidofovir and interleukin-2, Cytarabine (also known as ARA-C); or natalizumab
treatment for MS
patients; or efalizumab treatment for psoriasis patients or other treatments
for PML patients. In
further embodiments, the T cells described herein can be used in combination
with
chemotherapy, radiation, immunosuppressive agents, such as cyclosporin,
azathioprine,
methotrexate, mycophenolate, and FK506, antibodies, or other immunoablative
agents such as
CAM PATH, anti-CD3 antibodies or other antibody therapies, cytoxin,
fludaribine, cyclosporin,
FK506, rapamycin, mycophenolic acid, steroids, FR901228, cytokines, and
irradiation. These
drugs inhibit either the calcium dependent phosphatase calcineurin
(cyclosporine and FK506) or
inhibit the p70S6 kinase that is important for growth factor induced signaling
(rapamycin). (Liu et
al., Cell 66:807-815, 1991; Henderson et al., lmmun 73:316-321, 1991; Bierer
et al., Curr. Opin.
lmmun 5:763-773, 1993; lsoniemi (supra)). In embodiments, the cell
compositions described
herein are administered to a subject in conjunction with (e.g., before,
simultaneously or
following) bone marrow transplantation, T cell ablative therapy using either
chemotherapy
agents such as, fludarabine, external-beam radiation therapy (XRT),
cyclophosphamide, or
antibodies such as OKT3 or CAM PATH. In embodiments, the cell compositions
described
herein are administered following B-cell ablative therapy. For example, agents
that react with
CD20, e.g., Rituxan may be administered to patients. In embodiments, subjects
may undergo
standard treatment with high dose chemotherapy followed by peripheral blood
stem cell
transplantation. In certain embodiments, following the transplant, subjects
receive an infusion of
the expanded immune cells of the present disclosure. In embodiments, expanded
cells are
administered before or following surgery. The dosage of the above treatments
to be
administered to a subject in need thereof will vary with the precise nature of
the condition being
treated and the recipient of the treatment. The scaling of dosages for human
administration can
be performed according to art-accepted practices by a physician depending on
various factors.
Additional information on the methods of cancer treatment using modified cells
is provided in
U.S. Pat. No. U58,906,682, incorporated by reference in its entirety.
[00257] Embodiments described herein relate to an in vitro method for
preparing modified
cells. The method may include obtaining a sample of cells from a subject. For
example, the
sample may include T cells or T cell progenitors. The method may further
include transfecting
the sample of cells with a DNA encoding at least a CAR and culturing the
sample of cells ex
vivo in a medium that selectively enhances proliferation of CAR-expressing T
cells. The sample
of cells can be a mixed population of modified cells described herein.
[00258] In embodiments, the sample is a cryopreserved sample. In
embodiments, the
sample of cells is from umbilical cord blood or a peripheral blood sample from
the subject. In
embodiments, the sample of cells is obtained by apheresis or venipuncture. In
embodiments,
the sample of cells is a subpopulation of T cells.
[00259] Embodiments of the present disclosure relate to a Zinc Finger
Nuclease (ZFN)
comprising a DNA-binding domain comprising zinc finger DNA binding proteins
and a DNA-
49
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cleaving domain comprising a cleavage domain and/or a cleavage half-domain.
The zinc finger
DNA binding proteins may include 1, 2, 3, 4, 5, 6 or more zinc fingers, each
zinc finger having a
recognition helix that binds a target subsite in the target gene. In
embodiments, the zinc finger
proteins comprise 3, 4, 5, 6 fingers (where the fingers are designated F1, F2,
F3, F4, F5 and F6
and are ordered F1 to F3, F4 or F5 or F6 from the N-terminus to the C-
terminus), and the
fingers comprise the amino acid sequence of the recognition regions shown in
Table 5.
Examples of cleavage domains and/or cleavage half-domains include wild-type or
engineered
Fokl cleavage half-domain. In embodiments, the DNA cleaving domain comprises a
wild-type
cleavage domain or cleavage half-domain (e.g., a Fokl cleavage half-domain).
In embodiments,
the cleavage domain and/or cleavage half-domain comprise engineered (non-
naturally
occurring) cleavage domains or cleavage half-domains, for example, engineered
Fokl cleavage
half-domains that form obligate heterodimers. In embodiments, the gene is a
human gene. In
embodiments, the cleavage domain comprises a wild-type or engineered Fokl
cleavage domain.
Embodiments relate to a polynucleotide encoding the isolated ZFN as described
herein.
Embodiments relate to a vector comprising the polynucleotide. In embodiments,
the vector is an
adenoviral or lentiviral vector. Embodiments relate to an isolated cell or a
cell line comprising
the isolated ZFN described herein. In embodiments, the isolated cell is a stem
cell, a T cell, or a
Natural Killer (NK) cell. In embodiments, the cell is a T cell derived from a
primary human T cell
isolated from a human donor. In embodiments, the cell has reduced expression
of an
endogenous gene of CTLA4, LAG3, BTLA, TIM3, FOXP3, SIVA1, or LGALS9. In
embodiments,
various gene editing techniques or overexpression techniques (e.g., Cas9,
TALEN, and ZFN)
can be used to regulate T/NK cell functions by knocking out, knocking down,
overexpressing, or
inserting one or more genes. For example, the modified cell has reduced or
increased
expression of one or more genes of a biosynthesis or transportation pathway of
a peptide in List
1 and List 2 (see Paragraph 268), as compared to the corresponding wild-type
cell. In
embodiments, the target gene is Runx3. For example, the modified T/NK cell has
increased
expression of Runx3 as compared to the corresponding wild-type cell. As an
example, the
increased expression of Runx3 helps the infiltration of T cells or their long-
term residence within
tumor cells, therefore increasing T cell killing effects. In embodiments, the
modified cell is a
modified stem cell, a modified T cell, or a modified Natural Killer (NK) cell.
In embodiments, the
modified cell is a T cell derived from a primary human T cell isolated from a
human donor. In
embodiments, the cell has a reduced expression of an endogenous gene of CTLA4,
LAG3,
BTLA, TIM3, FOXP3, SIVA1, and LGALS9.
[00260] CTLA4 is an inhibitory receptor acting as a major negative
regulator of T-cell
responses. T lymphocyte receptor CTLA-4 binds co-stimulatory molecules CD80
(B7-1)
andCD86 (B7-2) with higher avidity than stimulatory co-receptor CD28 and
negatively regulates
T cell activation. LAG3 is a member of the immunoglobulin superfamily and is
expressed on the
surface of activated T and NK cells. LAG3 has also been detected on the
surface of B cells,
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
dendritic cells, TILs and Tregs. Blockage of LAG3 significantly increases T
cell proliferation and
function. TIM3 is an immune checkpoint receptor constitutively expressed by
CD4+ T helper 1
(Th1), CD8+ T cytotoxic 1 cells (Tc1) and Th17 cells. The interaction between
TIM3 and its
ligand galectin-9 LGALS9 is believed to result in suppression of T-cell
responses. FOXP3 is a
member of the forkhead/winged-helix family of transcriptional regulators,
which is crucial for the
development and inhibitory function of regulatory T-cells (Treg). SIVA1
induces 0D27-mediated
apoptosis, inhibits BCL2L1 isoform BcI-x(L) anti-apoptotic activity, inhibits
activation of NF-
kappa-B, and promotes T-cell receptor-mediated apoptosis.
[00261] Embodiments relate to modified cells comprising isolated nucleic
acid sequence
encoding a chimeric antigen receptor (CAR), wherein an endogenous gene is
inactivated using
the ZFN.
[00262] In embodiments, the CAR comprises an antigen binding domain, a
transmembrane
domain, a co-stimulatory domain, and a CD3 zeta signaling domain.
[00263] In embodiments, the modified T cell has a reduced graft-versus-host
disease
(GVHD) response in a bioincompatible human recipient as compared to the GVHD
response of
the primary human T cell.
[00264] In embodiments, the antigen binding domain of the CAR binds FZD10,
TSHR,
PRLR, Muc17, GUCY2C, 0D207, CD19, or CD20.
[00265] In embodiments, the antigen binding domain of the CAR binds at
least one of B7,
BCMA, CAIX, 0D123, 0D133, 0D138, CD171, CD171/L1-CAM, CD19, CD2, 0D22, CD30,
0D33, CEA, cMet, CS1, CSPG4, Dectin1, EGFR, EGFR vIII, EphA2, ERBB receptors,
ErbB T4,
ERBB2, FAP, Folate receptor 1, FITC, Folate receptor 1, GD2, GPC3, HA-1 H/HLA-
A2, HER2,
IL-11Ra, IL13 receptor a2, IL13R, IL13Ra2 (zetakine), Kappa, LewisY,
Mesothelin, MUC1,
NKG2D, NY-ESO-1, PSMA, ROR-1, TRAIL-receptor1, or VEGFR2.
[00266] In embodiments, the co-stimulatory domain of the CAR comprises the
intracellular
domain of a co-stimulatory molecule selected from the group consisting of
0D27, 0D28, 4-i BB,
0X40, CD30, CD40, PD-1, ICOS, lymphocyte function-associated antigen-1 (LFA-
1), CD2,
CD7, LIGHT, NKG2C, B7-H3, and any combination thereof.
[00267] In embodiments, the modified cells include a nucleic acid sequence
encoding hTERT
or a nucleic acid encoding SV4OLT, or a combination thereof. In embodiments,
the modified
cells include a nucleic acid sequence encoding hTERT and a nucleic acid
encoding SV4OLT. In
embodiments, the expression of hTERT is regulated by an inducible expression
system. In
embodiments, the expression of SV4OLT gene is regulated by an inducible
expression system.
In embodiments, the inducible expression system is rTTA-TRE, which increases
or activates the
expression of SV4OLT gene or hTERT gene, or a combination thereof. In
embodiments, the
modified cells include a nucleic acid sequence encoding a suicide gene. In
embodiments, the
suicide gene includes an HSV-TK suicide gene system. In these instances, the
modified cell
can be induced to undergo apoptosis.
Si
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00268] The present disclosure describes methods of treating cancer in a
subject, the
methods comprising administering a mixed population of modified cells
described herein to the
subject, wherein the cancer is selected from the group consisting of a lung
carcinoma,
pancreatic cancer, liver cancer, bone cancer, breast cancer, colorectal
cancer, leukemia,
ovarian cancer, lymphoma, and brain cancer.
[00269] The methods described herein include a modified T cell and/or
modified NK cell
comprising a reduced amount of one or more peptides including PD1, PDL1, PDL2,
CTLA4,
LRBA, LAG3, Tim3, BILA, CD160, 2B4, SOCS1, SOCS3, Foxp3, CCR4, PVRIG, CD16B,
SIVA1, 0D33, LAGLS9, 0D122, ID01, 0D45, Cvp1b1, TNFAIP8L2, ID02, TD02, DNMT3A,
and/or Ceacam-1 (List 1), as compared to a corresponding wild-type cell. In
embodiments, the
methods of treating cancer in a subject including enhancing the modified T
cell and/or NK cell
response of these T cells and/or NK cells (having a reduced amount of one or
more peptides
listed immediately above) when the mixed population of genetically modified T
cells is
administrated into a subject The methods include a modified T cell and/or
modified NK cell
comprising an increased amount of one or more peptides including Runx3, lexm,
PILRA,
Ptnns1L3, Fcgr3a, Nat8, CcI9, Hck, Trem2, CcI6, Cd36, Igf1, Ctss, Gzmc, Batf,
Cxcl2,
TNFAIP8L3, II1b, TRPV1, TRPV2, TRPV3, TRPV4, Rgs1, PLSCR1, ITGB2, C3AR1,
ITGA3,
ITGA5, ITGAL, batf, batf3, Cxcl2, CARD11, and/or 0D83 (List 2), as compared to
a
corresponding wild-type cell. In embodiments, the methods of treating cancer
in a subject
include enhancing the T cell and/or NK cell response of these T cells and/or
NK cells (having an
increased amount of the one or more peptides listed immediately above) when
the modified T
cells and/or modified NK cells are administrated to a subject. In embodiments,
various gene
editing techniques or overexpression techniques (e.g., Cas9, TALEN, and ZFN)
may be used to
regulate the functions of T cell and/or NK cell by knocking out/knocking
down/overexpressing/inserting one or more genes encoding one or more peptides
in list 1 or 2.
For example, the genetically modified T cell has reduced or increased
expression of one or
more genes of a biosynthesis or transportation pathway of a peptide in list 1
and list 2 (see
above), as compared to the corresponding wild-type cell.
[00270] In embodiments, the target gene is Runx3. For example, the modified
T cells have
increased expression of Runx3 as compared to the corresponding wild-type cell.
In these
instances, the increased expression of Runx3 may help, for example, the
infiltration or long-term
residence of the modified T cells within the tumor cells, therefore increasing
T cell killing effects.
[00271] For example, T cell response in a subject refers to cell-mediated
immunity
associated with helper, killer, regulatory, and other types T cells. For
example, T cell response
may include activities such as assistance to other white blood cells in
immunologic processes
and identifying and destroying virus-infected cells and tumor cells. T cell
response in the subject
may be measured via various indicators such as a number of virus-infected
cells and/or tumor
cells that the T cells kill, an amount of cytokines that the T cells release
in co-culturing with
52
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
virus-infected cells and/or tumor cells, a level of proliferation of the T
cells in the subject, a
phenotype change of the T cells (e.g., changes to memory T cells), and the
longevity or the
length of the lifetime of the T cells in the subject.
[00272] T cell response also includes the release of cytokines. Although
cytokine release is
often associated with systemic inflammation and complication of disease, the
release of
cytokines appears to be also associated with the efficacy of a CAR T cell
therapy. The release
of cytokines may correlate with expansion and progressive immune activation of
adoptively
transferred cells, such as in CAR T cell therapy. The present disclosure
describes the release of
effector cytokines, such as IFN-y, and pro- and anti-inflammatory cytokines,
such as IL-6, in
response to mixed population of modified T cells described herein, especially
in response to the
presence of a first CAR including an antigen binding domain for expanding
cells and a second
CAR or TCR including an antigen binding domain for killing a target cell. In
embodiments, the
present disclosure describes the release of IL-6 and IFN-y in a subject
introduced with the first
CAR and second CAR or TCR described herein. In embodiments, the subject is in
need of
cancer treatment, and the cancer treatment is pancreatic cancer treatment. In
embodiments, the
present disclosure describes determining the efficacy or monitoring the
efficacy of a CAR T cell
therapy by measuring the level of cytokine release. In embodiments, the
release of cytokines
(e.g., IL-6 and/or IFN-y) in the subject in response to CAR T cell therapy
using mixed population
of modified T cells described herein is more than that using T cells
comprising the second CAR
without the first CAR.
[00273] In embodiments, the modified cells described herein may further
comprise a
dominant negative variant of a receptor of programmed death 1 (PD-1),
cytotoxic T lymphocyte
antigen-4 (CTLA- 4), B- and T-lymphocyte attenuator (BTLA), T cell
immunoglobulin mucin-3
(TIM-3), lymphocyte-activation protein 3 (LAG-3), T cell immunoreceptor with
Ig and ITIM
domains (TIGIT), leukocyte-associated immunoglobulin-like receptor 1 (LAIR!),
natural killer cell
receptor 2B4 (2B4), or CD 160 such that the T cell response induced by the
mixed population of
modified cells may be enhanced. In embodiments, the modified cells described
herein may
further comprise a nucleic acid sequence encoding a suicide gene, and/or a
suicide gene
comprising an HSV-TK suicide gene system such that the fate of the modified
cell may be
controlled. For example, the T cell can be induced to undergo apoptosis if the
therapy imposes
risks to the subject, and/or the subject encounters adverse effects, or if the
therapy has been
completed, a certain required condition has been met, and/or a predetermined
time has passed.
[00274] The present disclosure describes a composition comprising a mixed
population of
modified cells described herein. In embodiments, there is a first population
of modified cells
comprising a first CAR binding a first antigen, and a second population of
modified cells
comprising a second CAR or TCR binding a second antigen that is different from
the first
antigen. The first antigen can be an antigen of a WBC, such as a B cell, while
the second
antigen is a tumor antigen. The present disclosure describes a method of
enhancing expansion
53
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
and maintenance of the second population of modified cells for killing tumor
cells. The method
includes administering an effective amount of the composition comprising a
mixed population of
modified cells to a subject having a form of cancer associated with the tumor
antigen which the
second CAR recognizes and binds. Embodiments also include a method of
enhancing T cell
response in a subject in need thereof or treating a subject having cancer. The
method includes
administering an effective amount of the composition described herein to the
subject having a
form of cancer associated with the tumor antigen which the second CAR
recognizes and binds.
Further the embodiments include a method of enhancing expansion and/or
maintenance of
modified cells in a subject, the method comprising: contacting T cells with a
first vector
comprising a first nucleic acid sequence encoding the first CAR and a second
vector comprising
a second nucleic acid sequence encoding the second CAR to obtain the
composition described
herein of a mixed population of modified cells; and administering an effective
amount of the
composition to the subject having a form of cancer associated with the tumor
antigen which the
second CAR recognizes and binds. Additional embodiments include a method of
enhancing T
cell response in a subject in need thereof or treating a subject having
cancer, the method
comprising: contacting T cells with a first vector comprising a first nucleic
acid sequence
encoding the first CAR and a second vector comprising a second nucleic acid
sequence
encoding the second CAR to obtain the composition described herein of a mixed
population of
modified cells; and administering an effective amount of the composition to
the subject having a
form of cancer associated with the tumor antigen, which the second CAR
recognizes and binds.
Embodiments include a method of enhancing expansion and maintenance of the
modified cells
in a subject, the method comprising: administering an effective amount of the
composition
described herein of a mixed population of modified cells.
[00275] In embodiments, the composition comprises at least the first
population and second
population of modified cells. The first population of modified cells comprises
a polynucleotide
encoding the first CAR (e.g., CD19, 0D22, and BCMA CARs) and a polynucleotide
encoding
one or more cytokines (e.g., IL-6, 1L12, and IFNy). The second population of
modified cells
comprises a polynucleotide encoding the second CAR binding a solid tumor
antigen. For
example, the composition comprises the first population, the second, the
third, and the fourth
populations of modified cells. The first population of modified cells
comprises a polynucleotide
encoding CAR binding a WBC antigen and IL-6 (e.g., FIG. 87B). The second
population of
modified cells comprises a polynucleotide encoding CAR binding a solid tumor
antigen (e.g.,
FIG. 87A). The third population of modified cells comprises a polynucleotide
encoding CAR
binding a WBC antigen and IL-12 (e.g., FIG. 87B). The fourth population of
modified cells
comprises a polynucleotide encoding CAR binding a WBC antigen and IFNy (e.g.,
FIG. 87B).
These WBC antigens can be the same (e.g., CD19) or different (e.g., CD19 and
BCMA). The
first, the third, and the fourth populations of modified cells can be mixed
based on a first
predetermined ratio to obtain a group of modified cells, which can be then
mixed based on a
54
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
second predetermined ratio with the second population of modified cells to
obtain a composition
comprising a mixed population of modified cells. The predetermined ratio is
used to control the
amount of expression of the one or more cytokines in the subject to achieve
controllable,
lasting, and efficient cytokine effects in the subject while having less
cytotoxicity. In
embodiments, the first predetermined ratio ratio the first, the third, and the
fourth populations of
modified cells is set such that there are more of modified cells comprising
the polynucleotide
encoding IFNy than the modified cells comprising the polynucleotide encoding
IL-12 or IL-6. For
example, the first predetermined ratio is 1:1:10. In embodiments, the second
predetermined
ratio is determined such that there are more of the modified cells comprising
the polynucleotide
encoding the second CAR (e.g., the second population of modified cells) than
the modified cells
comprising the polynucleotide encoding the first CAR (e.g., the first, the
second, and/or the third
populations of modified cells). For example, the second predetermined ratio of
the first
population of modified cells and the second population of modified cells is
less than 1:1 but
more than 1:10,000. In embodiments, the second predetermined ratio is 1:1,
1:10, 1:100,
1:1000, and 1:104, as well as individual numbers within that range, for
example, 1:10, 1:100, or
1:1000. In embodiments, the second predetermined ratio is between 1:10 and
1:1000. In
embodiments, the second predetermined ratio is between 1:10 and 1:100. In
embodiments, the
second predetermined ratio is between 1:1 and 1:100. In embodiments, the cells
(e.g., NK cells,
T cells, B cells, myeloid-derived cells, etc.) are obtained from a subject or
a healthy donor and
divided into at least two groups. These groups of cells may be transferred
with two or more
vectors, respectively. These cells can be further modified if obtained from a
healthy donor. In
embodiments, the second population of modified cells does not express the one
or more
cytokines.
[00276] In embodiments, a polynucleotide encoding the first CAR is present
in the modified
cell in a recombinant DNA construct, in an mRNA, or in a viral vector. In
embodiments, the
polynucleotide is an mRNA, which is not integrated into the genome of the
modified cell, such
that the modified cell expresses the first CAR (e.g., CD19 CAR) for a finite
period of time.
[00277] In embodiments, the mixed population of modified cells further
includes a third
population of modified cells expressing a third CAR and/or a fourth population
of modified cells
expressing a fourth CAR such that immune responses caused by the various
population of
modified cells can be coupled to boost CAR T treatment. In embodiments, CARs
may be
replaced by TCRs or a combination of CAR and TCR.
[00278] Embodiments relate to a method of enhancing CAR T therapy by
implementing
multiple infusion of CAR T cells timely. The method includes obtaining PBMC
from a subject or
a healthy donor, preparing CAR T cells using the obtained PBMC, culturing the
CAR T cells, for
example, for a predetermined amount of time, administering a portion of the
cultured CAR T
cells to the subject, observing and/or measuring the CAR T cells in the blood
of the subject,
administering a second portion of the cultured CAR T cells when the level of
the CAR T cells in
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
the blood reaches a predetermined value or when the CART cells home to an
organ (e.g.,
lymph node). For example, the first infused CAR T cells can be selectively
activated and
expanded in the organ and cause an immune response by the subject. Thus,
infusion of the
second portion of CAR T cells can be coupled with the immune response to
enhance the
activation and/or expansion of the second population of CAR T cells, thus
enhancing the CAR T
therapy.
[00279] The present disclosure describes a composition including a
population of modified
cells including a first population of modified cells that comprises a first
CAR without a second
CAR, and/or a second population of modified cells that comprises a second CAR
without a first
CAR. The present disclosure also describes a composition including a
population of modified
cells comprising the first CAR and second CAR (in a single modified cell). In
embodiments, the
composition includes a first and a second population of modified cells and a
third population of
modified cells comprising one or more nucleic acid sequences encoding the
first CAR and the
second CAR in the same modified cell. In embodiments, the composition
comprises a second
population of modified cells, in the absence of a first population of
genetically modified cells,
and a third population of modified cells comprising one or more nucleic acid
sequences
encoding the first CAR and the second CAR in the same modified cells.
[00280] Embodiments relate to a method of using or the use of
polynucleotide encoding the
antigen binding molecule and/or therapeutic agent(s) to enhance the expansion
of the modified
cells or to enhance the T cell response in a subject. The method or use
includes: providing a
viral particle (e.g., AAV, lentivirus or their variants) comprising a vector
genome, the vector
genome comprising the polynucleotide, wherein the polynucleotide is operably
linked to an
expression control element conferring transcription of the polynucleotide; and
administering an
amount of the viral particle to the subject such that the polynucleotide is
expressed in the
subject. In embodiments, the AAV preparation may include AAV vector particles,
empty capsids
and host cell impurities, thereby providing an AAV product substantially free
of AAV empty
capsids. More information of the administration and preparation of the viral
particle may be
found at the US Patent NO: 9840719 and Milani et al., Sci. Trans!. Med. 11,
eaav7325 (2019)
22 May 2019, which are incorporated herein by reference.
[00281] In embodiments, the polynucleotide may integrate into the genome of
the modified
cell and the progeny of the modified cell will also express the
polynucleotide, resulting in a
stably transfected modified cell. In embodiments, the modified cell expresses
the polynucleotide
encoding the CAR but the polynucleotide does not integrate into the genome of
the modified cell
such that the modified cell expresses the transiently transfected
polynucleotide for a finite
period of time (e.g., several days), after which the polynucleotide is lost
through cell division or
other factors. For example, the polynucleotide is present in the modified cell
in a recombinant
DNA construct, in an mRNA, or in a viral vector, and/or the polynucleotide is
an mRNA, which is
not integrated into the genome of the modified cell.
56
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00282] In embodiments, the first population of cells comprises the first
CAR and the second
CAR, and the second population of cells comprises the first CAR but does not
comprise the
second CAR. In embodiments, the first population of cells comprises the first
CAR and the
second CAR, and the second population of cells comprises the first CAR and the
second CAR.
In embodiments, first population of cells comprises the first CAR but does not
comprise the
second CAR, the second population of cells comprises the first CAR and the
second CAR. In
embodiments, the first population of cells comprises the first CAR but does
not contain the
second CAR, and the second population of cells comprise the second CAR but
does comprise
first CAR. In embodiments, first population of cells comprises the second CAR
but does not
comprise the first CAR and the second population of cells comprises the first
CAR and the
second CAR. In embodiments, the first population of cells comprises the first
CAR but does not
comprise the second CAR; the second population comprises a second CAR but does
not
comprise the first CAR; and a third population comprises the first CAR and the
second CAR. As
described herein, the first CAR includes an antigen binding domain for
expanding and/or
maintaining the modified cells, and the second CAR includes an antigen binding
domain for
killing target cells, such as tumors.
[00283] In embodiments, the antigen binding domain binds an antigen that is
or that
comprises a cell surface molecule of a white blood cell (WBC), a tumor
antigen, or a solid tumor
antigen. In embodiments, the WBCs are T cells, NK cells, or dendritic cells.
[00284] In embodiments, the WBC is a granulocyte, a monocyte, or
lymphocyte. In
embodiments, the WBC is a B cell. In embodiments, the cell surface molecule or
antigen of the
B cell is CD19, 0D22, CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, CD30, CD14, 0D68,
CD11b, CD18, 0D169, CD1c, 0D33, 0D38, 0D138, or CD13. In embodiments, the cell
surface
molecule or antigen of the B cell is CD19, CD20, 0D22, or BCMA. In
embodiments, the cell
surface molecule or antigen of the B cell is CD19.
[00285] In embodiments, the tumor antigen is a solid tumor antigen. In
embodiments, the
solid tumor antigen is tMUC1, PRLR, CLCA1, MUC12, GUCY2C, GPR35, CR1L, MUC 17,
TMPRSS11B, MUC21, TMPRSS11E, 0D207, SLC30A8, CFC1, SLC12A3, SSTR1, GPR27,
FZD10, TSHR, SIGLE015, SLC6A3, KISS1R, QRFPR, GPR119, CLDN6, UPK2, ADAM12,
SLC45A3, ACPP, MUC21, MUC16, MS4Al2, ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2,
Mesothelin, PSMA, ROR1, VEGFR-II, GD2, FR-a, ErbB2, EpCAM, EGFRvIll, B7-H3, or
EGFR.
In embodiments, the solid tumor antigen is or comprises tumor associated MUC1
(tMUC1),
TSHR, GUCY2C, ACPP, CLDN18.2 (18.2), PSMA, or UPK2.
[00286] In embodiments, the CAR comprises the antigen binding domain, a
transmembrane
domain, a co-stimulatory domain, and a CD3 zeta domain. In embodiments, the co-
stimulatory
domain comprises the intracellular domain of 0D27, 0D28, 4-1BB, 0X40, CD30,
CD40, PD-1,
ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT,
NKG2C, B7-H3, a
ligand that specifically binds with 0D83, or a combination thereof. In
embodiments, the second
57
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CAR includes a binding domain that binds tMUC1 and a co-stimulatory domain
that includes an
intracellular domain of 0D28; and/or the first CAR includes a binding domain
that binds 0D19
and a co-stimulatory domain that includes an intracellular domain of 4-i BB.
[00287] In embodiments, the first population of cells and/or the second
population of cells
further comprise a dominant negative form of a checkpoint protein or of the
checkpoint protein's
receptor present on T cells (e.g., PD-1). In embodiments, the first population
of cells comprise a
vector comprising a nucleic acid encoding the first CAR and the dominant
negative form of PD-
1.
[00288] In embodiments, the second CAR comprises a scFv binding tMUC1, an
intracellular
domain of 4-i BB or 0D28, CD3 zeta domain, and the second CAR comprises a scFv
binding
CD19, an intracellular domain of 4-i BB or 0D28, CD3 zeta domain. In
embodiments, the first
CAR comprises a scFv, which is SEQ ID NO: 5, and the second CAR comprise a
scFv, which is
the SEQ ID NO: 70. Corresponding sequences are listed in Table 5.
[00289] Embodiments relate to a method comprising administering an
effective amount of the
second population of T cells comprising a second CAR comprising a scFv binding
tMUC1 to a
patient having cancer. The second CAR may further comprise an intracellular
domain of 4-i BB
or CD28, CD3 zeta domain. In embodiments, the method further comprises
administering an
effective amount of the first population of T cells comprising a first CAR
comprising a scFv
binding CD19 to the patient, thereby enhancing expansion of the second
population of T cells in
the patient. The CAR may further comprise an intracellular domain of 4-i BB or
CD28, and CD3
zeta domain.
[00290] In embodiments, the second CAR comprises the intracellular domain
of CD28, and
the first CAR comprises the intracellular domain of 4-i BB. In this instance,
the first population of
T cells comprising CD19 may cause less adverse effect on the patient (e.g.,
CRS), and/or the
second population of T cells comprising tMUC1 may cause enhanced T cell
response (e.g.,
killing) as compared to those of the second CAR comprising the intracellular
domain of 4-i BB
and/or the first CAR comprising the intracellular domain of CD28. In
embodiments, the second
CAR comprises the intracellular domain of CD28 such that the second population
of T cells may
cause enhanced T cell response (e.g., killing) as compared to that of the
second CAR
comprising the intracellular domain of 4-i BB. In embodiments, the first CAR
comprises the
intracellular domain of 4-i BB such that the first population of T cells may
cause less adverse
effect on the patient (e.g., CRS) as compared to that of the first CAR
comprising the intracellular
domain of CD28.
[00291] In embodiments, the second population of cells comprises the scFv
binding a solid
tumor antigen but do not comprise the scFv binding a B cell antigen, and the
first population of
cells comprises the scFV binding an antigen different from the solid tumor
antigen (e.g., a WBC
antigen or a B cell antigen) but do not comprise the scFV binding the tumor
antigen. In these
instances, the T cell response of the patient induced by binding between the
first population of T
58
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cells and the antigen (e.g., CD19) may cause both the first and second
populations of T cells to
expand. Accordingly, the patient may be administered with a mixed population
of genetically
engineered T cells consisting essentially of the first population of cells and
the second
population of cells. In embodiments, the patient may be administered with the
second
population of genetically engineered T cells and one or more recombinant
proteins (e.g.,
cytokine such as IL6 and/or INFy) or cells expressing and secretion of the one
or more
recombinant proteins, which may induce similar or enhanced T cell response
caused by the first
population of T cells. In embodiments, the patient may be administered with
the second
population of T cells and a hormone drug (e.g., fulvestrant), which may induce
similar or
enhanced T cell response caused by the first population of T cells.
[00292] In embodiments, the first population of modified cells can further
comprise a third
CAR comprising the scFy binding tMUC1, the intracellular domain of 4-1BB or
0D28, and the
CD3 zeta domain. In embodiments, the second population of cells does not
comprise the scFy
binding CD19. In embodiments, the first population of cells does not comprise
the scFy binding
tMUC1.
[00293] In embodiments, the methods described herein of enhancing cell
expansion and/or
cell response in a subject are compared to methods in which the subject is
administered with
only one CAR (for example, only the first CAR or only the second CAR) and/or
the subject is not
administered with a mixed population of cells described herein. In
embodiments, the mixed
population of cells described herein enhances the expansion of the cells
and/or the cell
response.
[00294] Embodiments relate to a composition and a method for treating a
subject having
cancer or enhancing T cell response of the subject. The method includes
administering to the
subject an effective amount of a population of modified cells having a first
CAR. The first CAR
includes an antigen binding domain, a transmembrane domain, a co-stimulatory
domain of
0D28, and/or a CD3 zeta domain. The method can further include monitoring
and/or measuring
one or more parameters of T cell response induced by the modified cells. For
example, the one
or more parameters include cytokine release, lymphocyte numbers, and a level
of CAR T cell
expansion and exhaustion. The method can further include administering an
effective amount of
a population of modified cells including a second CAR to the subject in
response to a
predetermined time (e.g., one or two weeks after the infusion) and/or
condition, which may be
associated with the measured parameters (e.g., a copy number of CAR and
numbers of CAR T
cells). The second CAR includes an antigen binding domain, a transmembrane
domain, a co-
stimulatory domain of 4-1BB, and/or a CD3 zeta domain. It has been reported
that 0D28 CAR T
cells and 4-1BB CAR T cells behave differently in the lab and in the clinic.
Accordingly, the
method combines the advantages of the two co-stimulatory domains by coupling
the strong
initial immune response with the long and persistent immune response. For
example, the first
CAR including 0D28 elicits a robust T cell activation and is associated with
effector-like
59
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
differentiation. While the first CAR can cause T cell exhaustion, it is
designed to induce a strong
initial response of the subject's immune system. The second CAR including the
4-1BB reduces
T cell exhaustion, enhance persistence, and increases central memory
differentiation and
mitochondrial biogenesis, which are designed for persistent CAR T therapy. In
embodiments,
the initial response induced by the first CAR can enhance the persistent CAR T
therapy. In
embodiments, the population of modified cells including the first CAR and the
population of
modified cells including the second CAR may be administered to the subject at
the same time.
For example, the composition may include the population of modified cells
including the first
CAR and the population of modified cells including the second CAR. In
embodiments, the first
CAR binds an antigen of WBC, and the second CAR binds a solid tumor antigen.
In
embodiments, the first CAR and the second CAR bind the same or different solid
tumor
antigens. For example, a population of modified cells including a CAR that
binds a solid tumor
antigen (e.g., TSHR) and includes 4-1BB co-stimulatory domain and a population
of modified
cells including a CAR that binds the solid tumor antigen (e.g., TSHR) or
another solid tumor
antigen (e.g., tMuc1) and includes 0D28 co-stimulatory domain were mixed
together to
obtained a mixed modified cells. In embodiments, the modified cells may be
further
administered to the subject. In embodiments, the modified cells may be further
administered to
the subject along with a population of modified cells including a CAR binding
a WBC antigen
(e.g., CD19).
[00295] In embodiments, the CAR molecules described herein comprise one or
more
complementarity-determining regions (CDRs) for binding an antigen of interest.
CDRs are part
of the variable domains in immunoglobulins and T cell receptors for binding a
specific antigen.
There are three CDRs for each variable domain. Since there is a variable heavy
domain and a
variable light domain, there are six CDRs for binding an antigen. Further
since an antibody has
two heavy chains and two light chains, an antibody has twelve CDRs altogether
for binding
antigens. In embodiments, the CAR molecules described herein comprise one or
more CDRs
for binding antigens. In embodiments, the one or more CDRs bind the antigen of
a WBC, such
as a B cell. As an example, the one or more CDRs bind CD19, the cell surface
antigen of a B
cell. In embodiments, the one or more CDRs bind a tumor antigen, for example,
tMUC1. TSHR,
GUCY2C, ACPP, CLDN18.2 (18.2), PSMA, or UPK2.
[00296] Embodiments relate to an immunotherapeutic system and its use for
treating cancer
of a subject. As shown in FIG. 61, the immunotherapeutic system 102 includes
function
component 104 configured to inhibit growth of tumor cells, coupling component
106 configured
to couple the subject's immune response with the inhibition of the growth of
tumor cells, and
controlling component 108 configured to control the inhibition and/or
coupling. In embodiments,
the immunotherapeutic system 102 is a composition comprising one or more
pharmaceutical
compositions (e.g. antibodies and cells) suitable for treating cancer.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00297] Examples of function component 104 include CAR T, TIL, and TCR and
other
cellular therapies, an oncolytic virus therapy, a chemotherapy, a tumor
vaccine therapy, a
metabolism target therapy, and targeted therapy. In embodiments, function
component 104
includes at least one of the inhibitors that regulate immune metabolism (e.g.,
IDO inhibitors and
adenosine inhibitors); the immunomodulators (e.g., IMiDs); the agonists
against monocytes or
dendritic cells (e.g., TLRs/STING); an oncolytic virus therapy; the tumor
vaccines (e.g., DC
vaccines); the tumor infiltrating T cells (e.g., Tils); the macrophage-
reprogramming agents (e.g.,
CCR2-CCL2 inhibitor, CSF-1Rs inhibitor, PPAR-gamma agonist/inhibitor and CD-40
agonist);
the chemotherapy drugs (e.g., cyclophosphamide, fludarabine and ibrutinib);
the monoclonal
antibody targeting drugs (e.g., anti-her2); or the targeted drugs for non-
monoclonal antibodies
(e.g., ALK inhibitors, EGF/VEGF inhibitors). Example targets of TCR therapy
are listed in Table
6. In embodiments, function component 104 can be implemented by a Bite
molecule (e.g.,
TSHR-CD3). In embodiment, a Bite molecule comprises a first and a second
binding domain,
wherein the first binding domain binds to a solid tumor antigen, and the
second binding domain
binds, for example, the T cell CD3 receptor complex or 0D28, as illustrated in
FIG. 77A. The
second binding domain can also bind other T cell molecules such as 4-1BB,
0X40, GTTR,
ICOS, NKG20, etc.
[00298] Examples of coupling component 106 include immune response elicited
by CAR
T/NK cells, DC stimulation, T cell stimulation, and antigen/vaccine
stimulation. The CAR T/NK
cells include the modified cells described in the present disclosure. For
example, the modified
cell includes a CAR binding an antigen of WBC (e.g., CD19), an antigen of EBV,
and/or
albumin. T cell stimulation may be implemented by a Bite molecule (e.g., CD19-
CD3). DC cell
stimulation may be implemented by administering CAR T/NK cells to the subject,
or
administering a small molecule, small peptide, vaccine, or antigen to lymphoid
organs (e.g.,
lymph node) of the subject. In embodiment, a Bite molecule may comprise a
first and a second
binding domain, wherein the first binding domain binds to an antigen, and the
second binding
domain binds, for example, the T cell CD3 receptor complex or 0D28, as
illustrated in FIG. 77A.
The second binding domain can bind other T cell molecules such as 4-1BB, 0X40,
GITR, ICOS,
NKG20, etc. The antigen may bind a WBC antigen (e.g., CD19 and BCMA). In
embodiments,
CAR T cells may express the Bite molecule. In embodiments, CAR T cells and the
Bite
molecule may be administered to the subject at same time or separately.
[00299] In embodiments, the immunotherapeutic system 102 can comprise
various Bite
antibodies to treat cancer. In embodiments, the immunotherapeutic system 102
comprises a
first Bite molecule and a second Bite molecule. The first Bite molecule can
comprise a first and
a second binding domain, wherein the first binding domain binds to a solid
tumor antigen, and
the second binding domain binds, for example, the T cell CD3 receptor complex
or 0D28. The
second Bite molecule can comprise a third and a fourth binding domain, wherein
the third
binding domain binds to an antigen, and the fourth binding domain binds, for
example, the T cell
61
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CD3 receptor complex or 0D28. In embodiments, the immunotherapeutic system 102
comprises modified bispecific antibodies or trispecific antibody (e.g., FIGs.
870 and 87D) as
well as the first Bite and/or the second Bite antibodies. In these instances,
antibody techniques
can be used to stimulate cells to secrete one or more cytokines (e.g., IL-6,
IL-12, IL-15, IL-7,
and IFNy) in or close to tumor microenvironment. Component 8702 can be
implemented to
function as a stimulator that stimulate various cells to enhance cytokine
releases. For example,
the stimulator can comprise agonists or ligands directly or indirectly cause a
subject to secrete
one or more cytokines (e.g., IL-6, IL-12, IL-7, IL-15, and IFNy). In
embodiments, uses of the first
and/or the second Bite molecules can be combined with the administration of
human
recombinant forms of the one or more cytokines. In embodiments, the
therapeutic agent can be
isolated, synthetic, native, or recombinant human cytokines. In embodiments,
administering an
effective amount of the human recombinant cytokine comprises intravenous
delivery of an
amount of IL-6 in the range of about 0.5-50 ug per kilogram of body weight. In
embodiments,
the human recombinant cytokine comprises IL-6 or IL-7. Recombinant IL-15 can
be
administered as a daily bolus infusion for a predetermined time or days at 3
mcg/kg/day and 1
mcg/kg/ day. Recombinant IFNy can be administered at a dose of 2 million units
daily for 5 days
per week over a predetermined time. In embodiments, administering the
effective amount of the
human recombinant cytokine comprises administering an effective amount of the
human
recombinant cytokine such that concentrations of the cytokines, such as IL-6
and/or IFN-y, in
the blood of the subject can increase 5-1000 times (e.g., 50 times). Methods
of administering IL-
6, IL-15, and/or IFNy can be found in U.S. Patent Application U55178856A and
Cytokines in the
Treatment of Cancer, Volume 00, Number 00, 2018 of Journal of Interferon &
Cytokine
Research, which are incorporated herein by reference in their entirety. In
embodiments,
recombinant IL-12 can be administered at 30 ng/kg as a starting dose and
escalated to 500
ng/kg twice weekly after the infusion of CAR T cells. Methods of administering
of IL-12 can be
found in Leuk Res. 2009 November; 33(11): 1485-1489, which is incorporated
here by
reference. In embodiments, the human recombinant cytokine can be administered
to the subject
starting from day 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20, 21, 22, 23,
24, 25, 26, 27, 28, 29, or 30 days after administration.
[00300] In embodiments, the coupling component 106 and the function
component 104 may
combined and implemented using lentiviral vectors encoding the CAR binding a
solid tumor
antigen and a superantigen that result in excessive activation of the immune
system of the
subject. For example, the population of modified cells comprise a lentiviral
vector encoding the
CAR and a superantigen, the superantigen is Aravan virus Nucleoprotein,
Australian bat
lyssavirus Nucleoprotein, Duvenhage virus Nucleoprotein, European bat
lyssavirus 1
Nucleoprotein, lrkut virus Nucleoprotein, Khujand virus Nucleoprotein, Maize
mosaic virus
Nucleoprotein, Mokola virus Nucleoprotein, Mouse mammary tumor virus Protein
PR73, Rabies
virus Nucleoprotein, Rice yellow stunt virus Nucleoprotein, Staphylococcus
aureus Enterotoxin,
62
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Taro vein chlorosis virus Nucleoprotein or West Caucasian bat virus
Nucleoprotein. The
nucleoproteins may be modified with addition of an extracellular signal
peptide. In
embodiments, CAR T cells may be combined with bispecific or trispecific
antibodies to treat
tumors. The CAR T cells may bind a solid tumor antigen. In embodiments, CAR T
cells and the
antibodies may be administered to the subject at same time or separately. In
embodiments,
CAR T cells may express the antibodies. The bispecific antibody may comprise a
first antibody
fragment targeting CD3, CD28, 41-BB, GITR, 0X40, etc. and a second antibody
fragment
targeting a solid tumor antigen or a WBC antigen. The trispecific antibody may
comprise a first
antibody fragment targeting, for example, CD3, TLR, FcR or NKG2D, a second
antibody
fragment targeting, for example, CD28, 41-BB, GITR, or 0X40, and a third
antibody fragment
targeting, for example, a WBC antigen or a solid tumor antigen, as illustrated
in FIG. 77B.
[00301] The present disclosure also describes a population of modified
cells comprising a
polynucleotide encoding a CAR and the bispecific antibody or the trispecific
antibody described
above. The present disclosure also describes a population of modified cell
expressing a CAR
and the bispecific antibody or the trispecific antibody described above.
[00302] As shown in FIG. 65, there are three ways to activate dendritic
cells (DCs). The fist
way is to deliver the antigen (e.g., CEA, PSA or TERT) to the DCs. For
example, cancer vaccine
or Nanoparticles comprising the antigen can activate DCs which in turn can
activate the immune
system. The second way is by delivering an agonist (e.g., cytokines) to
accelerate the DCs'
maturation and release related cytokines directly or indirectly. The third way
is to deliver
cytokines or proteins that helps the activation of DCs. Other methods can also
be implemented
to activate DCs. For example, DC may be stimulated by various methods such as
LPS, various
viruses, Plasmodium antigen, cytokines, and vaccine. In embodiments, a small
molecule (e.g.,
CpG oligonucleotides and imiquimod, prototypic drugs) can be associated with
an albumin to be
delivered to a lymph node to stimulate DCs, which can then selectively cause
expansion of CAR
T cells homing to the lymph node. The Examples of the present disclosure show
that some T
cells (e.g., central memory T cells) do not stably remain in the blood after
infusions but enter
lymphoid organs such as lymph nodes due to molecules such as CCR7 and CD62L on
the T
cells. Thus, direct and/or indirect stimulation of DCs can selectively expand
and/or activate CAR
T cells showing more memory-like phenotypes, thus, enhancing efficacy of T
therapy. More
information about the implementation can be found in Ma et al., Science 365,
162-168 (2019),
which is incorporated by reference.
[00303] Antigen/vaccine stimulation may be implemented by the following
embodiments. As
an example, the method comprises: administering an effective amount of T cells
(e.g., TILs,
CAR T, TCR cells) to a subject in need thereof to treat tumor (e.g., solid
tumor), and
administering an effective amount of an agent that directly or indirectly
activates the T cells. In
embodiments, the agent includes an antigen that the T cells recognize. In
embodiments, the
agent includes presenting cells expressing a soluble agent that the
extracellular domain of the
63
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CAR recognizes. In embodiments, the agent includes vaccines derived from the
antigen. For
example, the agent includes the antigen associated with albumin such that the
agent activates
the T cells in, for example, the lymph nodes and then activate DCs, eliciting
expansion of the T
cells.
[00304] Examples of controlling component 108 include a suicide system
(e.g., suicide
gene), conditional gene expression system (e.g., lac, tetracycline, or
galactose systems), and
gene modulation system (e.g., Hif1a, NFAT, FOXP3, and/or NFkB).
[00305] FIG. 62 shows an immunotherapeutic system, for example
immunotherapeutic
system 102. In embodiments, the population of modified cells comprises two
types of cells:
function component cells and coupling component cells. The function component
cells are
capable of inhibiting tumor cells. In embodiments, the function components
cells include a
binding molecule binding a tumor antigen (e.g., a solid tumor antigen). For
example, the binding
molecule is or includes a CAR or a TCR that binds a solid tumor. In
embodiments, the coupling
component cells include a CAR targeting a white blood cell antigen. In
embodiments, the
coupling component cells include modified cells including a nucleic acid
sequence encoding
IL12 linked to a HIF VHL binding domain, and/or modified cells including a
nucleic acid
sequence encoding IL6 and IFNy linked by a 2A peptide.
[00306] FIG. 62 shows a schematic overview of an example process for the
combination of
CAR T cells and tumor-infiltrating lymphocytes (TIL). PBMCs of a subject can
be obtained and
CAR T targeting an antigen of WBC (e.g., CD19) can be prepared using various
methods
described in the present disclosure. In embodiments, the CAR T cells can be
Coupling
Component cells described in FIG. 61. The subject can then be lymphodepleted.
TILs can be
prepared using various methods. An example of the methods is the preparation
of TIL 102. For
example, after excision, the tumor metastasis is digested into a single cell
suspension in 24 well
plates. These suspensions/fragments are then cultured in the presence of IL-2.
In
embodiments, the cultures are tested for recognition of autologous melanoma
cells (for
example, melanoma cell lines or freshly frozen tumor digest, and if not
available a panel of HLA-
matched allogeneic tumor cell lines), by measuring IFNy secreted in the medium
using an IFNy
ELISA. In embodiments, the selection step for tumor reactivity can be omitted.
TIL cultures are
then expanded to treatment levels by stimulation with soluble anti-CD3
monoclonal antibody
and high concentration of IL-2, and irradiated allogeneic feeder cells. After
the TIL cultures are
purified to obtain the product cells, the product cells are ready to be
infused with CAR T cells
that enhance TIL expansion in the subject. Information on TILs preparations
may be found in
International Application NOs: W02018/081473 and W0201S/094167 and Molecular
Oncology,
Volume 9, Issue 10, December 2015, Pages 1918-1935, which are incorporated
herein by
reference.
[00307] There are three theoretical problems that need to be resolved for T
cells to overcome
solid tumors. The first problem is the identification of the T cells that
recognize the tumor.
64
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Instead of identifying only one target, it is necessary to identify as many
heterogeneous cancer
cells as possible. In this regard, TIL (Tumor Infiltrating T Lymphocyte)
Therapy seems
promising. The second challenge is to allow these screened T cells that
recognize tumors to
overcome the suppression of the tumor microenvironment. The third challenge is
to allow these
screened population of T cells that recognize tumors and overcome the
microenvironmental
inhibition and expand sufficiently to fight advanced tumors and reverse the
course of the
disease. Ordinary TIL technology is amplified in large quantities in vitro,
but at a high cost and
long cycles. Excessive costs can lead to high drug prices in the future, and
too long a cycle can
make advanced cancer patients unable to afford, which will challenge future
applications of the
product to treatment. Accordingly, lmmunotherapeutic system 102 can be helpful
for the latter
two challenges. Coupling component 106 can couple a subject's immune response
with TIL
therapy, for example, to expand TILs in the subject, reducing the cost and
shortening the cycle
associated with the TIL therapy and/or overcoming the suppression of the tumor
microenvironment by maintaining the population of TILs in the subject.
[00308] The present disclosure describes a composition for treating blood
cancer (e.g.,
leukemia, melanoma, and lymphoma). Example of blood cancers include Chronic
lymphocytic
leukemia (CLL) and Non-Hodgkin lymphoma (NHL). The composition comprises mixed
population of modified cells comprising at least two groups of modified cells,
each having a
polynucleotide encoding a CAR binding a blood cancer antigen (e.g., CD19,
CD20, and BCMA).
One group of the mixed population of modified cells further comprises a
polynucleotide
encoding one or more recombinant proteins (e.g., IL-6, IL-12, IL-7, IL-15, and
IFNy). For
example, the mixed population of modified cells comprises a first group of
modified cells
comprising a polynucleotide encoding CD19 CAR (e.g., FIG. 87A) and at least
one of a second
group of modified cells comprising a polynucleotide encoding CD19 CAR and IL-
6, a third group
of modified cells comprising a polynucleotide encoding CD19 CAR and IL-12, and
a fourth
group of modified cells comprising a polynucleotide encoding CD19 CAR and IFNy
(e.g., FIG.
87B). These groups of modified cells can be mixed to obtain the mixed
population of modified
cells, which are administered to a subject having B cell leukemia and
lymphoma. In
embodiments, the mixed population of modified cells can be mixed based on a
predetermined
ratio to obtain the mixed population of modified cells. The predetermined
ratio is used to control
the amount of expression of the one or more cytokines in the subject to
achieve controllable,
lasting, and efficient cytokine effects in the subject while experiencing less
cytotoxic effects. In
embodiments, the predetermined ratio for the first, the second, the third, and
the fourth groups
of modified cells is set such that there are more of the first group of
modified cells than the
second, third, or fourth group of modified cells in the mixed population of
modified cells. For
example, the predetermined ratio of the first group of modified cells and the
second, the third, or
the fourth group of modified cells is 10:1. In embodiments, the predetermined
ratio is 1:1, 10:1,
100:1, 1000:1, and 104:1, as well as individual numbers within that range, for
example, 10:1,
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
100:1, or 1000:1. In embodiments, the second predetermined ratio is between
10:1 and 1000:1.
In embodiments, the second predetermined ratio is between 10:1 and 1:100. In
embodiments,
the second predetermined ratio is between 1:1 and 100:1.
[00309] The present disclosure describes a composition for treating solid
tumor. The
composition comprises two populations of modified cells. The first population
of modified cells
comprises two or more groups of modified cells. One group of modified cells
comprises a
polynucleotide encoding the first CAR (e.g., CD19, 0D22, BCMA CARs) and at
least one other
group of modified cells comprises a polynucleotide encoding one or more
cytokines (e.g., IL-6,
IL12, and IFN) or encoding the one or more cytokines and the first CAR. In
embodiments, the
first CAR binds a WBC antigen. For example, the first population of modified
cells comprises a
first group of modified cells comprising a polynucleotide encoding CD19 CAR
(e.g., FIG. 87A)
and a second group of modified cells comprising a polynucleotide encoding CD19
CAR and a
cytokine (e.g., Embodiment 2 of FIG. 87). The first and second groups of
modified cells are
mixed to obtain the first population of modified cells. In embodiments, the
first and second
groups of modified cells are mixed based on a third predetermined ratio such
that there are
more of the first group of modified cells than the second group of modified
cells in the first
population of modified cells. For example, the third predetermined ratio of
the first group of cells
to the second group of modified cells is 10:1. In embodiments, the second
population of
modified cells comprises a CAR binding a solid tumor antigen. In embodiments,
the second
population of modified cell does not express the one or more cytokines. The
first population and
the second population of modified cells can be mixed to obtain the mixed
population of modified
cells, which are infused in the subject. In embodiments, the first population
and the second
population of modified cells can be mixed based on a fourth predetermined
ratio such that there
are more of the second population of modified cells than the first population
of modified cells.
For example, the second predetermined ratio of the first population and the
second population
of modified cells is less than 1:1 but more than 1:10,000. In embodiments, the
fourth
predetermined ratio is 1:1, 1:10, 1:100, 1:1000, and 1:104, as well as
individual numbers within
that range, preferably 1: 10, 1: 100, or 1:1000. In embodiments, the fourth
predetermined ratio
is between 1:10 and 1:1000. In embodiments, the second predetermined ratio is
between 1:10
and 1:100. In embodiments, the second predetermined ratio is between 1:1 and
1:100. The
predetermined ratio is used to control the amount of expression of the one or
more cytokines in
the subject to achieve controllable, lasting, and efficient cytokine effects
in the subject while
having less cytotoxicity.
[00310] The present disclosure is further described by reference to the
following exemplary
embodiments and examples. These exemplary embodiments and examples are
provided for
purposes of illustration only and are not intended to be limiting unless
otherwise specified. Thus,
the present disclosure should in no way be construed as being limited to the
following
66
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
exemplary embodiments and examples, but rather, should be construed to
encompass any and
all variations which become evident as a result of the teaching provided
herein.
EXEMPLARY EMBODIMENTS
[00311] The following are exemplary embodiments:
1. A population of modified cells effective for expanding and/or maintaining
the modified cells in
a patient, wherein the population of modified cells comprise at least two
different modified cells:
a first modified cell comprising an antigen binding domain for expanding
and/or maintaining the
modified cells; and a second modified cell comprising an antigen binding
domain for killing a
target cell, such as a tumor cell. In embodiments, the modified cells are
modified T cells. In
embodiments, the at least two different modified cells include two different
modified T cells, two
different modified immune cells, or a combination thereof. In embodiments, the
modified
immune cells include modified T cells, DC cells, and/or macrophages.
2. The population of modified cells of embodiment 1, wherein the antigen
binding domains bind
different antigens.
3. The population of modified cells of embodiment 1, wherein the population of
modified cells
further comprises a third modified cell expressing at least two different
antigen binding domains,
a first antigen binding domain for expanding and/or maintaining the modified
cells and a second
antigen binding domain for killing a target cell, and wherein the two
different antigen binding
domains are expressed on the same cell.
4. The population of modified cells of embodiment 1, wherein the population of
modified cells
comprises a modified cell expressing an antigen binding domain for killing a
target cell and a
modified cell expressing at least two antigen binding domains, a first antigen
binding domain for
expanding and/or maintaining the modified cells and a second antigen binding
domain for killing
a target cell, and wherein the two different antigen binding domains are
expressed on the same
modified cell.
5. The population of modified cells of embodiment 1, wherein the population of
modified cells
includes a modified cell expressing an antigen binding domain for expanding
and/or maintaining
the modified cells and a modified cell expressing at least two antigen binding
domains, a first
antigen binding domain for expanding and/or maintaining the modified cells and
a second
antigen binding domain for killing a target cell, and wherein the two
different antigen binding
domains are expressed on the same modified cell.
6. The population of modified cells of any one of embodiments 1-5, wherein the
modified cell is
a modified T cell, a modified NK cell, a modified macrophage, or a modified
dendritic cell.
7. The population of modified cells of any one of embodiments 1-6, wherein the
antigen binding
domain for expanding/or and maintaining the modified cells bind the surface
antigen of a WBC,
and the antigen binding domain for killing a target cell binds a tumor
antigen.
8. The population of modified cells of embodiment 7, wherein the WBC is a B
cell.
67
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
9. The population of modified cells of embodiment 7, wherein the cell surface
antigen of the
WBC is CD19, 0D22, CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, CD30, CD14, 0D68,
CD11b, CD18, 0D169, CD1c, 0D33, 0D38, 0D138, or CD13.
10. The population of modified cells of any one of embodiments 1-9, wherein
the solid tumor
antigen is tMUC1, PRLR, CLCA1, MUC12, GUCY2C, GPR35, CR1L, MUC 17, TMPRSS11B,
MUC21, TMPRSS11E, 0D207, SLC30A8, CFC1, SLC12A3, SSTR1, GPR27, FZD10, TSHR,
SIGLE015, SLC6A3, KISS1R, QRFPR, GPR119, CLDN6, UPK2, ADAM12, SLC45A3, ACPP,
MUC21, MUC16, MS4Al2, ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2, Mesothelin, PSMA,
ROR1, VEGFR-II, GD2, FR-a, ErbB2, EpCAM, EGFRvIll, B7-H3, EGFR, or one of
those listed
in Table 1.
11. The population of modified cells of embodiment 7, wherein the cell surface
antigen of the
WBC is CD19, CD20, 0D22, or BCMA.
12. The population of modified cells of embodiment 7, wherein the cell surface
antigen of a B
cell is CD19, and the tumor antigen is tMUC1, TSHR, GUCY2C, ACPP, CLDN18.2
(18.2),
PSMA, or UPK2.
13. A composition comprising a first population of cells comprising a first
CAR binding a first
antigen and a second population of cells comprising a second CAR binding a
second antigen,
wherein the second antigen is a tumor antigen and the first antigen and second
antigen are
different antigens.
14. The composition of embodiment 13, wherein the first population of cells
does not comprise
the second CAR, and/or the second population of cells does not comprise the
first CAR.
15. The composition of embodiment 14, wherein the composition further
comprises a third
population of cells comprising the first CAR and the second CAR.
16. The composition of embodiment 13, wherein the second population of cells
further
comprises the first CAR, and the first population of cells do not comprise the
second CAR; or
the first population of cells further comprises the second CAR.
17. The composition of embodiment 13, wherein second population of cells does
not comprise
the first CAR, and the first population of cells comprise the second CAR.
18. A method of enhancing expansion of the second population of cells, wherein
the second
population of cells are cells targeting a solid tumor, the method comprising
administering an
effective amount of the composition of any one of embodiments 13-17 to a
subject having a
form of cancer associated with or expressing the tumor antigen.
19. A method of enhancing T cell response in a subject or treating a subject
having cancer, the
method comprising administering an effective amount of the composition of any
one of
embodiments 13-17 to the subject having a form of cancer associated with or
expressing the
tumor antigen.
20. A method of enhancing expansion of cells in a subject, the method
comprising: contacting
cells with a first vector comprising a first nucleic acid sequence encoding a
first CAR and a
68
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
second vector comprising a second nucleic acid sequence encoding a second CAR
to obtain
the composition of any one of embodiments 13-17; and administering an
effective amount of
the composition to the subject having a form of cancer associated with or
expresses the tumor
antigen.
21. A method of enhancing T cell response in a subject in need thereof or
treating a subject
having cancer, the method comprising: contacting cells with a first vector
comprising a first
nucleic acid sequence encoding a first CAR and a second vector comprising a
second nucleic
acid sequence encoding a second CAR to obtain the composition of any one of
embodiments
13-17; and administering an effective amount of the composition to the subject
having a form of
cancer associated with or expressing the tumor antigen.
22. A method of enhancing expansion of cells in a subject, the method
comprising:
administering an effective amount of the first population of cells of the
composition of any one of
embodiments 13-17; and administering an effective amount of the second
population of cells.
23. The method of any one of embodiments 20-22, wherein the first vector and
the second
vector comprise lentiviral vectors.
24. The composition or the method of any one of embodiments 13-23, wherein the
first or
second antigen is or comprises a surface molecule of a white blood cell (WBC),
a tumor
antigen, or a solid tumor antigen.
25. The composition or the method of any one of embodiments 13-24, wherein the
cells are
modified T cells, modified NK cells, modified macrophages, or modified
dendritic cells.
26. The composition or the method of embodiment 24, wherein the WBC is a
granulocyte, a
monocyte, or a lymphocyte.
27. The composition or the method of embodiment 26, wherein the WBC is a B
cell.
28. The composition or the method of embodiment 27, wherein the cell surface
molecule of the
WBC is CD19, 0D22, CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, CD30, CD14, 0D68,
CD11b, CD18, 0D169, CD1c, 0D33, 0D38, 0D138, or CD13.
29. The composition or the method of embodiment 26, wherein the cell surface
molecule of the
WBC is CD19, CD20, 0D22, or BCMA.
30. The composition or the method of embodiment 26, wherein the cell surface
molecule of the
WBC is CD19.
31. The composition or the method of embodiment 26, wherein the tumor antigen
is a solid
tumor antigen.
32. The composition or the method of embodiment 26, wherein the solid tumor
antigen is
tMUC1, PRLR, CLCA1, MUC12, GUCY2C, GPR35, CR1L, MUC 17, TMPRSS11B, MUC21,
TMPRSS11E, 0D207, SLC30A8, CFC1, SLC12A3, SSTR1, GPR27, FZD10, TSHR,
SIGLEC15, SLC6A3, KISS1R, QRFPR, GPR119, CLDN6, UPK2, ADAM12, SLC45A3, ACPP,
MUC21, MUC16, MS4Al2, ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2, Mesothelin, PSMA,
ROR1, VEGFR-II, GD2, FR-a, ErbB2, EpCAM, EGFRvIll, B7-H3, CLDN18.2, or EGFR.
69
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
33. The composition or the method of embodiment 26, wherein the solid tumor
antigen is or
comprises tMUC1.
34. The composition or the method of any one of embodiments 13-33, wherein the
CAR
comprises the antigen binding domain, a transmembrane domain, a co-stimulatory
domain, and
a CD3 zeta domain.
35. The composition or the method of embodiment 34, wherein the co-stimulatory
domain
comprises the intracellular domain of 0D27, 0D28, 4-1BB, 0X40, CD30, CD40, PD-
1, ICOS,
lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-
H3, a ligand
that specifically binds with 0D83, or a combination thereof.
36. The composition or the method of embodiment 34, wherein the co-stimulatory
domain of the
second CAR comprises or is an intracellular domain of 4-i BB, and the antigen
binding domain
of the second CAR binds tMUC1; and/or the antigen binding domain of the first
CAR binds
CD19 and the co-stimulatory domain of the second CAR comprises or is an
intracellular domain
of CD28.
37. The composition or the method of any one of embodiments 13-36, wherein the
first
population of cells and/or the second population of cells further comprise a
dominant negative
form of PD-1.
38. The composition or the method of embodiment 37, wherein the first
population of cells
comprise a vector encoding the first CAR and the dominant negative form of PD-
1.
39. The composition or the method of any one of embodiments 13-38, wherein the
first CAR
comprises a scFy binding tMUC1, an intracellular domain of 4-i BB or 0D28, and
a CD3 zeta
domain, and the second CAR comprises a scFy binding CD19, an intracellular
domain of 4-i BB
or 0D28, and a CD3 zeta domain.
40. The composition or the method of any one of embodiments 13-39, wherein the
first CAR
comprises SEQ ID NO: 5, and the second CAR comprise SEQ ID NO: 70.
41. The composition or the method of any one of embodiments 13-40, wherein the
second
population of cells comprises a lentiviral vector encoding the first CAR and a
therapeutic agent
and the first population of cells comprises a lentiviral vector encoding the
second CAR and a
dominant negative form of PD-1.
42. The composition or the method of any one of embodiments 13-41, wherein the
first
population of cells comprise the first CAR and a therapeutic agent and the
second population of
cells comprise the second CAR and a dominant negative form of PD-1.
43. The composition or the method of embodiment 41 or 42, wherein the
therapeutic agent
comprises or is a cytokine.
44. The composition or the method of embodiment 43, wherein the cytokine is
IL6 and/or INFy.
45. A method comprising administering an effective amount of a first
population of T cells
comprising a CAR comprising a scFy binding CD19, an intracellular domain of 4-
i BB or 0D28,
and a CD3 zeta domain to a subject, thereby enhancing expansion of the first
population of T
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cells in the subject; and administering an effective amount of a second
population of T cells
comprising a CAR comprising a scFv binding tMUC1, an intracellular domain of 4-
1BB or 0D28,
and a CD3 zeta domain to the patient.
46. The method of embodiment 45, wherein first population of cells further
comprises an
additional CAR comprising the scFv binding tMUC1, the intracellular domain of
4-1BB or 0D28,
and the CD3 zeta domain.
47. The method of embodiment 45, wherein the second population of cells does
not comprise
the scFv binding CD19.
48. The method of embodiment 45, wherein the first population of cells does
not comprise the
scFv binding tMUC1.
49. A method for enhancing treatment of a subject with cancer, the method
comprising:
administering to the subject with CAR T cells targeting an antigen of WBC; and
administering to the subject tumor infiltrating lymphocytes (TILs).
50. A method for expanding TILs in a subject with cancer, the method
comprising:
administering to the subject with CAR T cells targeting an antigen of WBC; and
administering to the subject tumor infiltrating lymphocytes (TILs).
51. The method of embodiment 49 or 50, wherein the TI Ls are prepared by:
(i) obtaining a first population of TILs from a tumor resected from the
subject;
(ii) performing a first expansion by culturing the first population of TI Ls
in a cell culture medium
comprising IL-2 to produce a second population of TILs;
(iii) performing a second expansion by supplementing the cell culture medium
of the second
population of TILs with additional IL-2, OKT-3, and antigen presenting cells
(APCs), to produce
a third population of TILs, wherein the third population of TI Ls is at least
100-fold greater in
number than the second population of TILs, and wherein the second expansion is
performed for
at least 14 days in order to obtain the third population of TILs, wherein the
third population of
TILs is a therapeutic population of TI Ls which comprises an increased
subpopulation of effector
T cells and/or central memory T cells relative to the second population of
TILs; and
(iv) administering a therapeutically effective dosage of the third population
of TI Ls to the subject.
52. The method of embodiment 51, wherein the method further comprises prior to
step (iv) a
step of performing an additional second expansion by supplementing the cell
culture medium of
the third population of TILs with additional IL-2, additional OKT-3, and
additional APCs, wherein
the additional second expansion is performed for at least 14 days to obtain a
larger therapeutic
population of TILs than obtained in step (iii), wherein the larger therapeutic
population of TI Ls
comprises an increased subpopulation of effector T cells and/or central memory
T cells relative
to the third population of TILs.
53. The method of embodiment 51, wherein after step (ii) the cells are removed
from the cell
culture medium and cryopreserved in a storage medium prior to the second
expansion of
embodiment 51.
71
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
54. The method of embodiment 53, wherein the cells are thawed prior to the
second expansion
of embodiment 51.
55. The method of embodiment 51, wherein step (iii) is repeated one to four
times in order to
obtain sufficient TI Ls in the therapeutic population of TI Ls for a
therapeutically effective dosage
of the TI Ls.
56. The method of any one of embodiments 49 to 55, wherein the APCs are
peripheral blood
mononuclear cells (PBMCs).
57. The method of any one of embodiments 49 to 55, wherein the effector T
cells and/or central
memory T cells exhibit one or more characteristics selected from the group
consisting of
expression of 0D27, expression of 0D28, longer telomeres, increased 0D57
expression, and
decreased 0D56 expression, relative to effector T cells and/or central memory
T cells in the
third population of cells.
58. The method of any one of embodiments 49 to 55, wherein the effector T
cells and/or central
memory T cells exhibit increased 0D57 expression and decreased 0D56
expression, relative to
effector T cells and/or central memory T cells in the third population of
cells.
59. The method of any one of embodiments 49 to 55, wherein the cancer is
selected from the
group consisting of melanoma, cervical cancer, head and neck cancer,
glioblastoma, ovarian
cancer, sarcoma, pancreatic cancer, bladder cancer, breast cancer, triple
negative breast
cancer, and non-small cell lung carcinoma.
60. The method of any one of embodiments 49-59, wherein the CAR binds CD19,
CD20, 0D22,
or BCMA.
61. The method of any one of embodiments 49-60, wherein number of TI Ls in a
subject infused
with both CAR T cells and TI Ls is more than number of TI Ls in a subject
infused with TILs.
62. The method of any one of embodiments 49-60, wherein the CAR T cells
comprise the
modified cell 2 and modified cell 1 of FIG. 63.
63. A method of enhancing expansion of cells in a subject in need thereof or
treating a subject
having cancer, the method comprising:
administering an effective amount of a composition to the subject having a
form of cancer
expressing a tumor antigen, the composition comprising a first population of
cells comprising a
first CAR binding a first antigen, and a second population of cells comprising
a second CAR
binding a second antigen, wherein the second antigen is a tumor antigen and is
different from
the first antigen.
64. The method of embodiment 63, wherein the cells are T cells, NK cells, or
dendritic cells.
65. The method of embodiment 63, wherein the first antigen comprises a cell
surface molecule
of a white blood cell (WBC), a tumor antigen, or a solid tumor antigen.
66. The method of embodiment 65, wherein the WBC is a granulocyte, a monocyte,
or
lymphocyte.
67. The method of embodiment 66, wherein the lymphocyte is a B cell.
72
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
68. The method of embodiment 65, wherein the cell surface molecule of the WBC
is 0D19,
0D22, CD20, BCMA, CD5, CD7, CD2, 0D16, 0D56, CD30, 0D14, 0D68, CD11b, 0D18,
0D169, CD1c, 0D33, 0D38, 0D138, or CD13.
69. The method of embodiment 65, wherein the cell surface molecule of the WBC
is 0D19,
CD20, 0D22, or BCMA.
70. The method of embodiment 65, wherein the cell surface molecule of the WBC
is 0D19.
71. The method of embodiment 63, wherein the tumor antigen is a solid tumor
antigen.
72. The method of embodiment 71, wherein the solid tumor antigen is tMUC1,
PRLR, CLCA1,
MUC12, GUCY2C, GPR35, CR1L, MUC 17, TMPRSS11B, MU021, TMPRSS11E, 0D207,
SLC30A8, CFC1, SLC12A3, SSTR1, GPR27, FZD10, TSHR, SIGLE015, SLC6A3, KISS1R,
QRFPR, GPR119, CLDN6, UPK2, ADAM12, SLC45A3, ACPP, MU021, MU016, MS4Al2,
ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2, Mesothelin, PSMA, ROR1, VEGFR-II, GD2,
FR-a,
ErbB2, EpCAM, EGFRvIll, B7-H3, CLDN18.2, or EGFR.
73. The method of embodiment 71, wherein the solid tumor antigen comprises
tMUC1.
74. The method of embodiment 63, wherein the CAR comprises an antigen binding
domain, a
transmembrane domain, a co-stimulatory domain, and a CD3 zeta domain.
75. The method of embodiment 74, wherein the co-stimulatory domain comprises
the
intracellular domain of 0D27, 0D28, 4-1BB, 0X40, CD30, CD40, PD-1, ICOS,
lymphocyte
function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, a ligand
that binds
0D83, or a combination thereof.
76. The method of embodiment 63, wherein the first CAR comprises a scFy
binding CD19, an
intracellular domain of 4-i BB or 0D28, and a CD3 zeta domain, and the second
CAR
comprises a scFy binding tMUC1, an intracellular domain of 4-i BB or 0D28, and
a CD3 zeta
domain.
77. The method of embodiment 63, wherein an antigen binding domain of the
first CAR
comprises SEQ ID NO: 5 and an antigen binding domain of the second CAR
comprises SEQ ID
NO: 70.
78. The method of embodiment 63, wherein the second population of cells
comprises a lentiviral
vector encoding the second CAR and a dominant negative form of PD-1.
79. The method of embodiment 63, wherein the first population of cells
comprises a lentiviral
vector encoding the first CAR and a therapeutic agent.
80. The method of embodiment 79, wherein the therapeutic agent comprises a
cytokine.
81. The method of embodiment 80, wherein the cytokine is IL6 and/or INFy.
82. The method of embodiment 80, wherein the cytokine is at least one of IL6,
IL12, IL7, IL15,
TNF-a, or IFNy.
83. A method for in vitro cell preparation, the method comprising: contacting
cells with (1) a first
vector comprising a polynucleotide encoding a first antigen binding molecule
that binds a first
antigen and (2) a second vector comprising a polynucleotide encoding a second
antigen binding
73
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
molecule that binds a second antigen to obtain a population of modified cells,
to obtain a mixed
population of modified cells, wherein the first antigen is different from the
second antigen.
84. A method for enhancing cell expansion in a subject having cancer, the
method comprising:
obtaining cells from the subject or a healthy donor; contacting the cells with
(1) a first vector
comprising a polynucleotide encoding a first antigen binding molecule that
binds a first antigen
and (2) a second vector comprising a polynucleotide encoding a second antigen
binding
molecule that binds a second antigen to obtain a mixed population of modified
cells; and
administering an effective amount of the mixed population of modified cells to
the subject;
wherein: the first antigen is different from the second antigen; and a level
of the cell expansion
in the subject is higher than a level of the cell expansion in a subject
administered an effective
amount of a population of modified cells that have been contacted with the
first vector but not
the first vector.
85. A method for treating a subject having cancer, the method comprising:
obtaining cells from
the subject or a healthy donor; contacting the cells with (1) a first vector
comprising a
polynucleotide encoding a first antigen binding molecule that binds a first
antigen and (2) a
second vector comprising a polynucleotide encoding a second antigen binding
molecule that
binds a second antigen to obtain a mixed population of modified cells; and
administering an
effective amount of the mixed population of modified cells to the subject;
wherein: the first
antigen is different from the second antigen.
86. A method for enhancing treatment of a subject having cancer, the method
comprising:
obtaining cells from the subject or a healthy donor; contacting the cells with
(1) a first vector
comprising a polynucleotide encoding a first antigen binding molecule that
binds a first antigen
and (2) a second vector comprising a polynucleotide encoding a second antigen
binding
molecule that binds a second antigen to obtain a mixed population of modified
cells; and
administering an effective amount of the mixed population of modified cells to
the subject;
wherein: the first antigen is different from the second antigen; and a level
of inhibition of tumor
growth in the subject is higher than a level of inhibition of tumor growth in
a subject
administered with an effective amount of a population of modified cells that
have been
contacted with the second vector but not the first vector.
87. A method for in vitro cell preparation, the method comprising: introducing
a first vector
comprising a polynucleotide encoding a first antigen binding molecule that
binds a first antigen
into a first population of cells; and introducing a second vector comprising a
polynucleotide
encoding a second antigen binding molecule that binds a second antigen into a
second
population of cells; and culturing the first and second population of cells
separately; wherein the
first antigen is different form the second antigen.
88. A method for enhancing cell expansion in a subject having cancer, the
method comprising:
introducing a first vector comprising a polynucleotide encoding a first
antigen binding molecule
that binds a first antigen into a first population of cells to obtain a first
population of modified
74
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cells; introducing a second vector comprising a polynucleotide encoding a
second antigen
binding molecule that binds a second antigen into a second population of cells
to obtain a
second population of modified cells; and administering an effective amount of
the first and
second population of modified cells to the subject; wherein: the first antigen
is different from the
second antigen; and a level of the cell expansion in the subject is higher
than a level of the cell
expansion in a subject administered an effective amount of the second
population of modified
cells but not the first population of modified cells. In embodiments, the
first population of
modified cells and the second population of modified cells are administered
simultaneously or
sequentially.
89. A method for treating a subject having cancer, the method comprising:
introducing a first
vector comprising a polynucleotide encoding a first antigen binding molecule
that binds a first
antigen into a first population of cells to obtain a first population of
modified cells; introducing a
second vector comprising a polynucleotide encoding a second antigen binding
molecule that
binds a second antigen into a second population of cells to obtain a second
population of
modified cells; and administering an effective amount of the first and second
population of
modified cells to the subject; wherein the first antigen is different from the
second antigen. In
embodiments, the first population of modified cells and the second population
of modified cells
are administered simultaneously or sequentially.
90. A method for enhancing treatment of a subject having cancer, the method
comprising:
introducing a first vector comprising a polynucleotide encoding a first
antigen binding molecule
that binds a first antigen into a first population of cells to obtain a first
population of modified
cells; introducing a second vector comprising a polynucleotide encoding a
second antigen
binding molecule that binds a second antigen into a second population of cells
to obtain a
second population of modified cells; and administering an effective amount of
the first and
second population of modified cells to the subject, wherein: the first antigen
is different from the
second antigen; and a level of inhibition of tumor growth in the subject is
higher than a level of
inhibition of tumor growth in a subject administered with an effective amount
of the second
population of modified cells in the absence of the first population of
modified cells. In
embodiments, the first population of modified cells and the second population
of modified cells
are administered simultaneously or sequentially.
91. A method for enhancing T cell response, the method comprising: introducing
a first vector
comprising a polynucleotide encoding a first antigen binding molecule that
binds a first antigen
into a first population of cells; introducing a second vector comprising a
polynucleotide encoding
a second antigen binding molecule that binds a second antigen into a second
population of
cells; contacting cells expressing the second antigen with the first
population of cells and the
second population of cells; and measuring a level of the T cell response,
wherein the level of T
cell response is higher in the contacted cells than a level of the T cell
response in cells
contacted with the second population of cells without the first population of
cells.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
92. A method for enhancing T cell response, the method comprising: contacting
a population of
cells with a first vector comprising a polynucleotide encoding a first antigen
binding molecule
that binds a first antigen and a second vector comprising a polynucleotide
encoding a second
antigen binding molecule that binds a second antigen to obtain a mixed
population of modified
cells; contacting cells expressing the second antigen with the mixed
population of modified
cells; and measuring a level of the T cell response, wherein the level of T
cell response is higher
in the contacted cells than a level of the T cell response in cells contacted
with the a population
of cells contacted with the second vector without the first vector.
93. The method of any one of embodiments 83-92, wherein the cells are T cells,
NK cells, or
dendritic cells. In embodiments, the cells T cells.
94. The method of any one of embodiments 83-93, wherein the first antigen
binding molecule
binds a cell surface molecule of a WBC.
95. The method of embodiment 94, wherein the WBC is a granulocyte, a monocyte,
or
lymphocyte.
96. The method of embodiment 94, wherein the WBC is a B cell.
97. The method of embodiment 94, wherein the cell surface molecule of the WBC
is CD19,
0D22, CD20, BCMA, CD5, CD7, CD2, CD16, 0D56, CD30, CD14, 0D68, CD11b, CD18,
0D169, CD1c, 0D33, 0D38, 0D138, or CD13.
98. The method of embodiment 94, wherein the cell surface molecule of the WBC
is CD19,
CD20, 0D22, or BCMA.
99. The method of embodiment 94, wherein the cell surface molecule of the WBC
is CD19.
100. The method of any one of embodiments 83-99, wherein the second antigen
binding
molecule binds to a solid tumor antigen.
101. The method of embodiment 100, wherein the solid tumor antigen is tMUC1,
PRLR,
CLCA1, MUC12, GUCY2C, GPR35, CR1L, MUC 17, TMPRSS11B, MUC21, TMPRSS11E,
0D207, SLC30A8, CFC1, SLC12A3, SSTR1, GPR27, FZD10, TSHR, SIGLEC15, SLC6A3,
KISS1R, QRFPR, GPR119, CLDN6, UPK2, ADAM12, SLC45A3, ACPP, MUC21, MUC16,
MS4Al2, ALPP, CEA, EphA2, FAP, GPC3, 1L13-Ra2, Mesothelin, PSMA, ROR1, VEGFR-
II,
GD2, FR-a, ErbB2, EpCAM, EGFRvIll, CLDN18.2, or EGFR.
102. The method of any one of embodiments 83-101, wherein the first and second
binding
molecules are CARs.
103. The method of embodiment 102, wherein the CAR comprises an extracellular
domain, a
transmembrane domain, and an intracellular domain, and the extracellular
domain binds a
tumor antigen.
104. The method of embodiment 103, wherein the intracellular domain comprising
a co-
stimulatory domain that comprises an intracellular domain of a co-stimulatory
molecule selected
from the group consisting of 0D27, 0D28, 4-1BB, 0X40, CD30, CD40, PD-1, ICOS,
lymphocyte
76
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, or a
combination
thereof.
105. The method of embodiment 105, wherein the intracellular domain comprises
a CD3 zeta
signaling domain.
106. The method of any one of embodiments 83-101, wherein the first binding
molecule is a
CAR, and the second binding molecule is a TCR.
107. The method of embodiment 106, wherein the T cell comprises a modified T
Cell Receptor
(TCR).
108. The method of embodiment 106, wherein the TCR is derived from
spontaneously occurring
tumor-specific T cells in patients.
109. The method of embodiment 106, wherein the TCR binds a tumor antigen.
110. The method of embodiment 109, wherein the tumor antigen comprises CEA,
gp100,
MART-1, p53, MAGE-A3, or NY-ESO-1.
111. The method of embodiment 106, wherein the TCR comprises TCRy and TORO
chains,
TCRa and TCRf3 chains, or a combination thereof.
112. The method of embodiment 106, wherein the second population of cells are
derived from
TILs.
113. The method of any one of embodiments 83-112, wherein the population of
modified cells
comprise cells comprising the first binding molecule and cells comprising the
second binding
molecules.
114. The method of any one of embodiments 83-112, wherein the population of
modified cells
comprise cells comprising the first binding molecule, cells comprising the
second binding
molecules, and cells comprising both the first binding molecule and the second
binding
molecule.
115. The method of any one of embodiments 83-112, wherein the T cell response
is measured
by the number of copies of CAR(s) and/or the amount of cytokine released. In
embodiments,
the cytokine released are IL-6 and/or IFNy.
116. The method of any one of embodiments 83-112, wherein the T cell response
comprises
cytokine release, cell expansion, and/or activation levels.
117. The method of any one of embodiments 83-112, wherein the first vector
further comprises
a polynucleotide encoding IL-6, IFNy, or a combination thereof.
118. The method of any one of embodiments 83-112, wherein the first vector
further comprises
a polynucleotide encoding IL-12.
119. The method of any one of embodiments 116 and 117, wherein the
polynucleotide
comprises a polynucleotide encoding NFAT and/or VHL.
120. The method of any one of embodiments 83-119, wherein the population of
modified cells
comprise cells expressing the first binding molecule and IL-6, IFNy, or a
combination thereof,
77
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cells expressing the second binding molecules, cells expressing the first and
second molecules,
and/or cells expressing the first binding molecule and IL-12.
121. The method of any one of embodiments 83-120, wherein the population of
modified cells
comprise cells expressing the second binding molecule and IL-6, IFNy, or a
combination
thereof, cells expressing the second binding molecules, cells expressing the
first and second
molecules, and/or cells expressing the first binding molecule and IL-12.
122. The method of any one of embodiments 83-121, wherein the population of
modified cells
comprise cells expressing the second binding molecule and IL-6, IFNy, or a
combination
thereof, cells expressing the second binding molecule, cells expressing the
first and second
molecules, and/or cells expressing the second binding molecule and IL-12.
123. The method of any one of embodiments 83-122, wherein the population of
modified cells
comprise cells expressing a dominant negative form of PD-1.
124. A bispecific chimeric antigen receptor, comprising: a first antigen
binding domain, a second
antigen binding domain, a cytoplasmic domain, and transmembrane domain,
wherein the first
antigen binding domain recognizes a first antigen, and the second antigen
binding domain
recognize a second antigen, and the first antigen is different from the second
antigen.
125. The bispecific chimeric antigen receptor of embodiment 124, wherein the
first antigen and
the second antigen are not expressed on the same cell.
126. The bispecific chimeric antigen receptor of embodiment 124 or 125,
wherein the first
antigen is an antigen of a blood component, and the second antigen is an
antigen of a solid
tumor.
127. The bispecific chimeric antigen receptor of any one of embodiments 124-
126, wherein the
first antigen is 0D19, and the second antigen is a tumor associated MUC1.
128. The bispecific chimeric antigen receptor of any one of embodiments 124-
128, wherein the
first antigen binding domain comprises amino acid sequence SEQ ID: 5 or 6.
129. The bispecific chimeric antigen receptor of any one of embodiments 124-
128, wherein the
second antigen binding domain comprises one of amino acid sequence SEQ ID: 70,
71, 72, 79,
80, or 81.
130. The bispecific chimeric antigen receptor of embodiment 124, wherein the
CAR comprises
amino acid sequence of any one of tanCARs listed in Table 5.
131. The bispecific chimeric antigen receptor of embodiment 124, wherein the
first binding
domain binds an antigen of nonessential tissues, and the second binding domain
binds an
antigen of tumor tissue. In embodiments, the first binding domain binds TSHR
or GUCY2C. In
embodiments, the second binding domain binds tMUC1, MAGE-E1, or Epithelial
tumor antigen
(ETA).
132. The bispecific chimeric antigen receptor of embodiment 124, wherein the
first binding
domain binds a tissue specific antigen, and the second binding domain binds an
antigen
78
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
expressed on more than one tissue. In embodiments, the first binding domain
binds TSHR or
PRLR. In embodiments, the second binding domain binds tMUC1, MAG-E1, or ETA.
133. The bispecific chimeric antigen receptor of embodiment 124, wherein the
first binding
domain binds an antigen of normal tissue, and the second binding domain binds
an antigen
expressed on tumor tissue. In embodiments, the first binding domain binds
ACPP, TSHR,
GUCY2C, UPK2, CLDN18.2, PSMA, DPEP3, CXCR5, B7-H3, MUC16, SIGLEC-15, CLDN6,
Muc17, PRLR, or FZD10. In embodiments, the second binding domain binds tMUC1,
MAG-E1,
or ETA.
134. The bispecific chimeric antigen receptor of any one of embodiments 123,
wherein the first
binding domain binds to an antigen that is expressed on non-malignant cells,
and the second
binding domain binds an antigen that is expressed on tumor cells and not on
corresponding
non-malignant cells.
135. A cell comprising the bispecific CAR of any one of embodiments 123-134.
136. A nucleic acid encoding the bispecific CAR of any one of embodiments 123-
134.
137. A method of enhancing T cell response, enhancing treatment of cancer,
treating cancer in
a subject, treating a subject having a tumor, or inhibiting the growth of a
tumor, the method
comprising: administering an effective amount of cell of embodiment 135.
136. The use of the cell, the bispecific CAR, population of modified cells,
the composition, or the
method of any one of embodiments 1-135 for the treatment of a subject in need
thereof.
137. The use of the cell, the bispecific CAR, population of modified cells,
the composition, or the
method of embodiment 136, wherein the subject has cancer.
EXAMPLES
Example 1. Bispecific CARs
[00312] Lentiviral vectors that encode individual CAR molecules were
generated and
transfected with T cells, which are elaborated below. Techniques related to
cell cultures,
construction of cytotoxic T lymphocyte assay may be found in "Control of
large, established
tumor xenografts with genetically retargeted human T cells containing CD28 and
CD137
domains," PNAS, March 3, 2009, vol. 106 no. 9, 3360-3365 and "Chimeric
Receptors
Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T
Cells and
Increased Antileukemic Efficacy In Vivo," Molecular Therapy, Aug. 2009, vol.
17 no. 8, 1453-
1464, which are incorporated herein by reference in its entirety.
[00313] On Day 0, peripheral blood was drawn from healthy volunteers and
sorted to collect
CD3+ T cells. CD3/CD28 Dynabeads was added to the collected CD3+ T cells in a
1:1 ratio. On
Day 1, the activated CD3+ T cells were transfected with vectors including CD19
CAR (M01=15;
the binding domain of the CAR being SEQ ID NO: 5) and vectors including TSHR
CAR (M01=92;
the binding domain of the CAR being SEQ ID NO: 8), and vectors including TSHR-
CD19 bispecific
CAR (M01=92; the binding domain of the CAR being SEQ ID NO: 435). More
structure and
sequence information are provided in FIG. 7 and Table 5. On Day 2, culture
media were changed.,
79
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
The lentivirus was removed, and the cells were resuspended in fresh media. On
Day 5, flow
detection of CAR expression was performed. Various expression rates were
observed (CD19
CAR 17.45%, TSHR CAR 76.84%, and TSHR-CD19 bispecific CAR 20.59%). Also, on
Day 0,
peripheral blood was drawn from healthy volunteers and sorted to collect CD3+
T cells.
CD3/0D28 Dynabeads were added to the collected CD3+ T cells in a 1:1 ratio. On
Day 1, the
activated CD3+ T cells were transfected with vectors including CD19 CAR
(M01=2; the binding
domain of the CAR being SEQ ID NO: 5) and vectors including tMUC1 CAR (M01=30;
the binding
domain of the CAR being SEQ ID NO: 70), and vectors including tMUC1-CD19
bispecific CAR
(M01=95; the binding domain of the CAR being SEQ ID NO: 437), and vectors
including
CLDN18.2-CD19 (18.2-CD19) bispecific CAR (M01=180, the binding domain of the
CAR being
SEQ ID No: 439). More sequence information is provided in FIGS. 10, 12, and 13
and Table 5.
On Day 2, culture media were changed. The lentivirus was removed, and the
cells were
resuspended in fresh media. On Day 5, flow detection of CAR expression was
performed. Various
expression rates were observed (CD19CAR 68.28%, tMUC1CAR 31.58%, and tMUC1CD19
bispecific CAR 28.11% and 35.11%).
[00314] As shown in FIG. 8, 0.2 or 1 x 104 CAR T cells and 1x104 Nalm6 or B-
CPAB-B tumor
cells were co-cultured for 24 hours (hrs), and the supernatant was collected.
IFNy release was
detected. Nalm6 was a CD19-positive tumor cell, and B-CPAB-B was a TSHR
positive tumor cell.
As shown in the left panel of FIG. 8, CD19 CAR T cells released more IFNy in
response to Nalm6
as compared to that released in response to B-CPAB-B. As shown in the middle
panel, TSHR
CAR T cells released more IFNy in response to B-CPAB-B as compared to that
released in
response to Nalm6. As shown in the right panel, bispecific CAR T cells
released significant
amount of IFNy in response to each of Nalm6 and B-CPAB-B. These results
indicated that
bispecific CAR T cells can be stimulated by both CD19-positive or TSHR-
positive cells. 105 CAR
T cells and 105 Nalm6 or B-CPAB-B tumor cells were co-cultured for 24 hours,
and 0D137
expression of CAR T CD8 positive cells was then detected by flow cytometry.
The left panel of
FIG. 9 showed CD137 expression of CAR T cells not co-cultured with tumor
cells, while the middle
and right showed CD137 expression of CAR T cells co-cultured with Nalm6 or B-
CPAB-B. The
results demonstrated that bispecific CAR T (TSHR-CD19 bispecific CAR) were
activated by both
Nalm6 and B-CPAB-B. Similar cytokine release assays were performed and showed
that
bispecific CART (CLDN18.2-CD19 bispecific CAR or CLDN18.2-19tan CAR) cells
were activated
by both Nalm6 and cells expressing CLDN18.2 (FIGS. 12-15).
[00315] FIG. 12 shows schematic structure of constructs of vectors encoding
CAR molecules.
FIG. 13 shows expression of the CAR molecules shown in FIG. 12. Since CD19 CAR
included a
humanized antibody, 18.2 CAR is a murine antibody. Therefore, human and murine
CAR
antibodies were used for detection. The ratio of expression of the two
antibodies was detected
with bispecific CAR, which was close to 1:1, indicating that the expression of
bispecific CAR was
as expected. FIG. 14 shows results of IFNy release of co-culturing CAR T cells
and tumor cells.
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
The experiment was carried out by co-culturing with 0.2 or 1 x 104 CAR T cells
and 1 x 104 293T
or KATO 111-18.2+ or Nalm-6 cells. The supernatant was collected after 24 hrs
to detect IFN-y.
Nalm-6 is a CD19 T cell; KATO 111-18.2+ is a cell that overexpresses CLDN18.2;
and 293T is a
double-negative cell that does not express CD19 and CLDN18.2. As shown, 18.2
CAR T showed
significant IFN-y release when co-cultured with KAT0111-18.2+ cells,
indicating that KAT0111-18.2+
can be recognized by 18.2 CAR T cells and released IFN-y to kill target cells;
Nalm-6 was also
recognized by CD19 CART cells and released IFN-y to kill target cells; 18.2-
CD19 bispecific CAR
(18.2-19tan CAR) had significant IFN-y release when co-cultured with KAT0111-
18.2+ and Nalm-
6. In addition, Nalm-6 could not stimulate the release of IFN-y from 18.2 CAR
T cells, and CD19
CAR T cells could not stimulate the release of IFN-y by KATO 111-18.2+,
indicating that both CAR
T cells are specific. In conclusion, 18.2-CD19 bispecific CAR T cells can
specifically recognize
18.2 and CD19-positive target cells, and release IFN-y to kill target cells.
[00316] FIG. 15 shows flow cytometry results depicting CD137 expression for
co-culturing of
CAR T cells and tumor cells. 1 X 104 CAR T cells were co-cultured with 1 x 104
293T-VVT or
KAT0111-18.2+ or Nalm-6 cells. CD137 expression of CAR T CD8+ cells was
measured by flow
cytometry after 48 Hours. The left column shows CD137 expression of CAR T
cells co-cultured
with 293T. CD19 CAR expression is absent in the CD19 CAR group, the 18.2 CAR
group, and
the 18.2-19 tan CAR group. It can be seen that the 293T has no specific
antigen expression and
cannot activate CAR T cells. In the middle column, CAR T cells were co-
cultured with KATO III-
18.2+ cells with high expression of 18.2 protein. The expression of CD137 in
the 18.2 CAR group
was 8.77%, and the expression of CD137 in the 18.2-19 bispecific CAR group was
6.36%. The
expression of CD137 was not observed in the CD19 CAR group. 18.2-CAR T and
18.2-CD19
bispecific CAR T recognized and activated the 18.2 protein in KAT0111-18.2+;
CD19 CAR T did
not. The right column is a co-culture of CAR T cells with Nalm-6 cells, which
are CD19+ cells that
are specifically recognized and activated by CD19 CAR T cells. The results
showed that the
expression of CD137 in the CD19 CAR group was 11.14%, the expression of CD137
in the 18.2-
19 bispecific CAR group was 10.55%, and the expression in the 18.2 CAR group
was not
detectable. CD19 CAR and 18.2-CD19 bispecific CAR can be activated by Nalm-6,
while 18.2
CAR failed to activate Nalm-6. In conclusion, it was demonstrated that 18.2-
CD19 bispecific CAR
T cells can specifically recognize the 18.2 antigen and the CD19 antigen.
Since CD137 is a
marker protein for the activation of T cells, the level of CD137 up-regulation
of CAR T cells, after
co-culturing with CAR T cells and substrate target cells, can be used to
determine whether CAR
T cells are activated.
[00317] Abnormal glycosylation is known to be common in many tumors, such as
the abnormal
glycosylation of MUC1 (tMUC1). A CAR binding tMUC1 may include a scFv based on
the 5E5
antibody. Many tumors specifically express certain characteristic targets.
More information of
tumor markers and their corresponding cancer types are listed in Table 3. The
examples include
two scFvs joined by linker to form a tandem CAR (tanCAR) comprising the two
scFvs.
81
CA 03125646 2021-06-30
WO 2020/146743
PCT/US2020/013099
Table 3: CAR T cells and substrate cells
Numbering Remarks
6917 CAR-5e528z
6921 CAR-ACPP-28z
2529 TanCAR 28z ACPP+G4S+5e5LH
2530 TanCAR 28z ACPP+G4S+5e5HL
2533 TanCAR 28z 5e5 +G4S+ACPP LH
2534 TanCAR 28z 5e5 +G4S+ACPP HL
MCF-7 Breast cancer cell lines with high expression of tnMUC1
PC3-ACPP Human prostate cancer cells that overexpress ACPP
2407 tMUC1CAR-41bb
163 CLDN 18.2CAR
2517 TanCAR 5E5 LH+G4S+163LH
1604 TSHR - 4-1BB
2407 t MUC1 - 4-1BB
2519 TSHR - G4S - tMUC1 - 4-1BB
2521 tMUC1 - G4S - TSHR - 4-1BB
[00318] On Day 0, peripheral blood was drawn from healthy volunteers. CD3+
T cells were
sorted with pan T kit and activated by CD3/0D28 Dynabeads at a ratio of 3:1.
On Day 1, the
activated CD3+ T cells were infected. Several groups of cells (each 1.00E+06 T
cells) were
infected with Vectors based on the Table 4, and remaining cells were used as
NT (non-
transfected). On Day 2, the lentivirus and the Dynabeads were removed, and the
culture media
were replaced. On Day 6, the CAR ratio and cell phenotype of CAR T cells were
measured in
each group using flow cytometry assay. Since anti-ACPP antibodies are a
humanized antibody
and anti-MUC1 antibodies are a murine antibody, a rabbit anti-human CAR
antibody and rabbit
anti-mouse CAR antibody were used to detect expression of these two scFvs,
respectively. On
Day 7, the experiment was carried out according to Table 4. The samples were
flow-stained after
24 hours of full activation. The supernatant was collected for detection of
Cytometric Bead Array
(CBA), and carboxyfluorescein succinimidyl ester (CFSE) staining was performed
to observe the
proliferation. The cells were co-cultured with fluorescent substrate cells,
and the survival of cells
with fluorescent substrates was observed to determine the killing effect.
Table 4: cells for co-culturing assay
ID Vectors SEQ MOI Number
of hCAR+ mCAR+ Co-culture
ID cells (ACPP) (tMUC1)
6917 CAR-5e528z 13.15 1.00E+06
62.2 Co-cultured
6921 CAR-ACPP-28z 23.05 1.00E+06 61.18 with
2529 TanCAR 28z 51.65 1.00E+06 55.21 49.54
substrate
ACPP+G45+5e5LH
cells (MCF-7,
2530 TanCAR 28z 52.49 1.00E+06 70.51 60.49 PC3-
ACPP,
ACPP+G45+5e5HL 293T)
2533 TanCAR 28z 5e5 51.04 1.00E+06 55.94 63.1
+G45+ACPP LH
82
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
2534 TanCAR 28z 5e5 52.28 1.00E+06 34.21 31.47
+G4S+ACPP HL
2407 tMUC1CAR-41bb 8.44 1.00E+06 25.68 Co-cultured
163 CLDN 18.2CAR 64.8 1.00E+06 41.06 with
2517 TanCAR 5E5 59.7 1.00E+06 44.42 substrate
LH+G4S+163LH cells (MCF-
7, KAT03+,
293T)
[00319] FIG. 66 provides histograms showing expression of several markers
on CAR T cells
and tanCAR T cells using flow cytometry assay. NT, 6917, 6921, 2529, 2530,
2533, as well as
2534 and substrate cells (MCF-7, PC3-acpp, 293T cells) were co-cultured for 24
hours and flow
cytometry assay was performed on Day 8. CAR T cells and three substrate cells
(293T, MCF-7,
PC3-acpp) were co-cultured for 24 hrs. Flow cytometry assay was performed
after the activation
of CAR T cells. In FIG. 66, the vertical coordinates are CAR+0D137+ cells (the
total CAR+ cells)
and CAR+0D25+ (total CAR+ cells), respectively. From the expression of 0D137
and 0D25, four
types of tanCAR cells were effectively activated by corresponding substrate
cells. The statistical
analysis of the expression of CD4OL by flow cytometry was performed after CAR
T cells was co-
cultured with substrate cells (293T, MCF-7 and PC3-acpp) for 24h. Four types
of tanCAR cells
expressed CD4OL, which can activate CD40+ and other immune cells of the immune
system,
such as B cells, activated monocytes, DCs, etc.
[00320] FIG. 67 provides histograms showing cytokine release of CAR T cells
and tanCAR T
cells. NT, 6917, 6921, 6921, 2529, 2530, 2533, as well as 2534 and substrate
cells (MCF-7, P03-
acpp, 293T cells), were co-cultured for 24 hours and cytokine release was
measured on Day 8.
[00321] FIG. 68 shows the expansion of cells in each group after 5 days of
stimulation with the
corresponding substrate cells. As compared to control groups, tanCAR groups
showed apparent
expansion in response to both substrate cells. Proliferation of 6917, 6921,
2529, 2530, 2533,
2534, and NT was measured on Day 12 after co-culturing with substrate cells
(MCF-7, PC3-acpp,
293T cells) for 5 days.
[00322] FIG. 69 shows killing assay results. The results indicate that 6917
inhibited MCF-7
and 6921 inhibited PC3-ACPP. The four groups of tanCAR T cells killed both
substrate cells. NT
was negative for the experiment. The control contained only tumor cells.
Killing assay was
performed for 6917, 6921, 2529, 2530, 2533, 2534 and NT cells after co-
culturing with substrate
cells for five days.
[00323] FIG. 70 provides histograms showing expression of several markers
on other CAR T
cells and tanCAR T cells and cytokine release using flow cytometry assay.
2407, 163, and 2517
were co-cultured with MCF-7, KAT03+, and 293T cells for 24 hours on Day 8, and
cytokine
release assay were performed. TanCAR 2517 were activated by both MCF-7 and
KAT03+
substrate cells, and the intensity and proportion were close to single CAR.
The corresponding
83
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CAR T cells were co-cultured with substrate cells (293T, MCF-7, and KAT03+)
for 24 hrs, and
the expression of CD4OL was detected by flow cytometry.
[00324] FIG. 71 cytokine release of various CAR T cells and tanCAR T cells
in response to
substrate cells. The experimental process and experimental design were similar
to those of
experiments above.
Table 5: Sequence IDs and corresponding identifiers
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
SP 1 UPK2 101 Construct of 201
MUC1-5E5-A-
IRES-CD19-A
Hinge & 2 ADAM12 102 CAR 1 of MUC1- 202
transmembrane 5E5-A-IRES-
domain CD19-A
Co-stimulatory 3 SLC45A3 103 CAR 2 of MUC1- 203
domain 5E5-A-IRES-
CD19-A
CD3-zeta 4 ACPP 104 Construct of 204
MUC1-5E5-B-
IRES-CD19-A
scFv Humanized 5 MUC21 105 CAR 1 of MUC1- 205
CD19 5E5-B-IRES-
CD19-A
scFv CD19 6 MUC16 106 CAR 2 of MUC1- 203
5E5-B-IRES-
CD19-A
scFv FZD10 7 MS4Al2 107 Construct of 206
MUC1-5E5-A-
IRES-CD19-B
scFv TSHR 8 ALPP 108 CAR 1 of MUC1- 202
5E5-A-IRES-
CD19-B
scFv PRLR 9 SLC2A14 109 CAR 2 of MUC1- 207
5E5-A-IRES-
CD19-B
scFv Muc 17 10 GS1-259H13.2 110 Construct of 208
MUC1-5E5-B-
IRES-CD19-B
scFv GUCY2C 11 ERVFRD-1 111 CAR 1 of MUC1- 205
5E5-B-IRES-
CD19-B
scFv CD207 12 ADGRG2 112 CAR 2 of MUC1- 207
5E5-B-IRES-
CD19-B
Prolactin (ligand) 13 ECEL1 113 Construct of 209
MUC1-2-A-IRES-
CD19-A
scFv CD3 14 CHRNA2 114 CAR 1 of MUC1-2- 210
A-IRES-CD19-A
scFv CD4 15 GP2 115 CAR 2 of MUC1-2- 203
A-IRES-CD19-A
84
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ
ID
NO: ID No:
NO:
scFv CD4-2 16 PSG9 116 Construct of 211
MUC1-2-B-IRES-
CD19-A
scFv CD5 17 SIGLEC15 117 CAR 1 of MUC1-2- 212
B-IRES-CD19-A
CD19 antigen 18 SLC6A3 118 CAR 2 of MUC1-2- 203
B-IRES-CD19-A
FZD10 antigen 19 KISS1R 119 Construct of 213
MUC1-2-A-IRES-
CD19-B
TSHR antigen 20 QRFPR 120 CAR 1 of MUC1-2- 210
A-IRES-CD19-B
PRLR antigen 21 GPR119 121 CAR 2 of MUC1-2- 207
A-IRES-CD19-B
Muc 17 antigen 22 CLDN6 122 Construct of 214
MUC1-2-B-IRES-
CD19-B
GUCY2C antigen 23 SP-2 123 CAR 1 of MUC1-2- 212
B-IRES-CD19-B
CD207 antigen 24 Linker-2 124 CAR 2 of MUC1-2- 207
B-IRES-CD19-B
CD3 antigen 25 Hinge-2 125 Construct of 215
MUC1-5E5-A-
IRES-hCD19-A
CD4 antigen 26 TM-2 126 CAR 1 of MUC1- 202
5E5-A-IRES-
hCD19-A
CD5 antigen 27 4-1BB-2 127 CAR 2 of MUC1- 216
5E5-A-IRES-
hCD19-A
CAR CD19 nucleic 28 CD3 zeta-2 128 Construct of 217
acid MUC1-5E5-B-
IRES-hCD19-A
Hinge & TM 29 CLDN6-CAR-1 129 CAR 1 of MUC1- 205
domain B 5E5-B-IRES-
hCD19-A
Hinge & TM 30 ScFv CLDN6-CAR- 130 CAR 2 of MUC1- 216
domain A 1 5E5-B-IRES-
hCD19-A
Hinge & TM 31 ScFv VL CLDN6- 131 Construct of 218
domain D CAR-1 MUC1-5E5-A-
IRES-hCD19-B
Hinge & TM 32 ScFv VH CLDN6- 132 CAR 1 of MUC1- 202
domain C CAR-1 5E5-A-IRES-
hCD19-B
Hinge domain D 33 CLDN6-CAR-2 133 CAR 2 of MUC1- 219
5E5-A-IRES-
hCD19-B
Hinge domain C 34 ScFv CLDN6-CAR- 134 Construct of 220
2 MUC1-5E5-B-
IRES-hCD19-B
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
Hinge domain B 35 ScFv VL CLDN6- 135 CAR 1 of MUC1- 205
CAR-2 5E5-B-IRES-
hCD19-B
Hinge domain A 36 ScFv VH CLDN6- 136 CAR 2 of MUC1- 219
CAR-2 5E5-B-IRES-
hCD19-B
TM domain D 37 CLDN6-CAR-3 137 Construct of 221
MUC1-2-A-IRES-
hCD19-A
TM domain A 38 scFv CLDN6-CAR- 138 CAR 1 of MUC1-2- 210
3 A-IRES-hCD19-A
CD19 extracellular 39 scFv VL CLDN6- 139 CAR 2 of MUC1-2- 216
domain CAR-3 A-IRES-hCD19-A
TM domain C or B 40 scFv VH CLDN6- 140 Construct of 222
CAR-3 MUC1-2-B-IRES-
hCD19-A
VVTCD3zeta 41 CLDN6-CAR-4 141 CAR 2CAR 1 of 212
MUC1-2-B-IRES-
hCD19-A
VVTCD3zeta- 42 scFv CLDN6-CAR- 142 Construct of 216
BCMACAR full 4 MUC1-2-B-IRES-
length hCD19-A
BCMA 43 scFv VL CLDN6- 143 Construct of 223
CAR-4 MUC1-2-A-IRES-
hCD19-B
BCMA CAR vector 44 scFv VH CLDN6- 144 CAR 1 of MUC1-2- 210
CAR-4 A-IRES-hCD19-B
BCMA CAR vector 45 SIGLEC-15-CAR-1 145 CAR 2 of MUC1-2- 219
A-IRES-hCD19-B
VL anti-CD5 46 scFv SIGLEC-15- 146 Construct of 224
CAR-1 MUC1-2-B-IRES-
hCD19-B
VH anti-CD5 47 scFv VL SIGLEC- 147 CAR 1 of MUC1-2- 212
15-CAR-1 B-IRES-hCD19-B
VL anti-CD4 48 scFv VH SIGLEC- 148 CAR 2 of MUC1-2- 219
15-CAR-1 B-IRES-hCD19-B
VH anti-CD4 49 VL1 VH1 SIGLEC- 149 Construct of 225
15-CAR-2 MUC1-5E5-A-
IRES-CD22-A
VL anti-CD3 50 VL1 VH2 SIGLEC- 150 CAR 1 of MUC1- 202
15-CAR-3 5E5-A-IRES-
CD22-A
VH anti-CD3 Si VL1 VH3 SIGLEC- 151 CAR 2 of MUC1- 226
15-CAR-4 5E5-A-IRES-
CD22-A
TSHR extracellular 52 VL1 VH 4 SIGLEC- 52 Construct of 227
domain 15-CAR-5 MUC1-5E5-B-
IRES-CD22-A
VH region of 53 VL2 VH 1 SIGLEC- 153 CAR 1 of MUC1- 205
BCMA scFv 15-CAR-6 5E5-A-IRES-
CD22-A
86
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ
ID
NO: ID No:
NO:
VL region of BCMA 54 VL2 VH2 SIGLEC- 154 CAR 2 of MUC1- 226
scFv 15-CAR-7 5E5-A-IRES-
0D22-A
VH region of CD14 55 VL2 VH3 SIGLEC- 155 Construct of 228
scFv 15-CAR-8 MUC1-5E5-A-
IRES-CD22-B
VL region of CD14 56 VL2 VH4 SIGLEC- 156 MUC1-5E5-A- 202
scFv 15-CAR-9 IRES-CD22-B CAR
1
VH region of CD33 57 VL1 SIGLEC-15- 157 MUC1-5E5-
A- 229
scFv CAR IRES-CD22-B CAR
2
VL region of CD33 58 VL2 SIGLEC-15- 158 MUC1-5E5-B- 230
scFv CAR IRES-CD22-B
CD22CAR 59 VH1 SIGLEC-15- 159 CAR 1 of MUC1- 205
CAR 5E5-B-IRES-
CD22-B
BCMACAR 60 VH2 SIGLEC-15- 160 CAR 2 of MUC1- 229
CAR 5E5-B-IRES-
CD22-B
MUC1CAR 61 VH3 SIGLEC-15- 161 Construct of 231
CAR MUC1-2-A-IRES-
CD22-A
m19CAR-IRES- 62 VH4 SIGLEC-15- 162 CAR 1 of MUC1-2- 210
MUC1CAR CAR A-IRES-CD22-A
hCD19CAR-IRES- 63 MUC16-CAR-1 163 CAR 2 of MUC1-2- 226
MUC1CAR A-IRES-CD22-A
hCD22CAR-IRES- 64 scFv MUC16-CAR- 164 MUC1-2-B-IRES- 232
MUC1CAR 1 CD22-A
BCMACAR-IRES- 65 scFv VL MUC16- 165 MUC1-2-B-IRES- 212
MUC1CAR CAR-1 CD22-A CAR 1
mCD19CAR-2A- 66 scFv VH MUC16- 166 MUC1-2-B-IRES- 226
MUC1CAR CAR-1 CD22-A CAR 2
hCD19CAR-2A- 67 MUC16-CAR-2 167 MUC1-2-A-IRES- 233
MUC1CAR CD22-B
hCD22CAR-2A- 68 scFv MUC16-CAR- 168 MUC1-2-A-IRES- 210
MUC1CAR 2 CD22-B CAR 1
BCMA-2A- 69 scFv VL MUC16- 169 MUC1-2-A-IRES- 229
MUC1CAR CAR-2 CD22-B CAR 2
Tumor associated 70 scFv VH MUC16- 170 Construct of 234
MUC1 scFv 1 CAR-2 MUC1-2-B-IRES-
CD22-B
Tumor associated 71 KISS1R-CAR 171 CAR 1 of MUC1-2- 212
MUC1 scFv-1 VH B-IRES-CD22-B
Tumor associated 72 Ligent peptide 172 CAR 2 of MUC1-2-
229
MUC1 scFv-1 VL KISS1R-CAR B-IRES-CD22-B
Tumor associated 73 ZFLm1 ( left ) RS 173 Construct of 235
MUC1 scFv-1 VL MUC1-5E5-A-
CDR 1 aa IRES-CD14-A
L2D8-2 (hCAR VL) 74 ZFLm1 ( left ) F1 174 CAR 1 of MUC1- 202
5E5-A-IRES-
CD14-A
87
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
Tumor associated 75 ZFLm1 ( left ) F2 174 CAR 2 of MU01- 236
MUC1 scFv-1 VL 5E5-A-IRES-
CDR 3 CD14-A
Tumor associated 76 ZFLm1 ( left ) F3 176 Construct of 237
MUC1 scFv-1 VH MUC1-5E5-B-
CDR 1 IRES-CD14-A
Tumor associated 77 ZFLm1 ( left ) F4 177 CAR 1 of MUC1- 205
MUC1 scFv-1 VH 5E5-B-IRES-
CDR 2 CD14-A
Tumor associated 78 ZFLm1 ( left ) F5 178 CAR 2 of MUC1- 236
MUC1 scFv-1 VH 5E5-B-IRES-
CDR 3 CD14-A
Tumor associated 79 ZFLm1 ( left ) F6 179 Construct of 238
MUC1 scFv2 MUC1-5E5-A-
IRES-CD14-B
Tumor associated 80 ZFRm1-4 ( right ) 180 CAR 1 of MUC1- 202
MUC1 scFv2 VH R 5E5-A-IRES-
S aa
CD14-B
Tumor associated 81 ZFRm1-4 ( right ) 181 CAR 2 of MUC1- 239
MUC1 scFv2 VL
F1 5E5-A-IRES-
CD14-B
Tumor associated 82 ZFRm1-4 ( right ) 182 Construct of 240
MUC1 scFv-2 VL MUC1-2-A-IRES-
CDR 1 F2
CD14-A
Tumor associated 83 ZFRm1-4 ( right ) 184 CAR 1 of MUC1-2- 210
MUC1 scFv-2 VL F3 A-IRES-CD14-A
CDR 2
Tumor associated 84 ZFRm1-4 ( right ) 184 CAR 2 of MUC1-2- 236
MUC1 scFv-2 VL F4 A-IRES-CD14-A
CDR 3
'Tumor associated 85 6 chain-1 of 185 Construct of 241
MUC1 scFv-2VH Vy9V62 MUC1-2-B-IRES-
CDR 1 CD14-A
Tumor associated 86 y chain-2 of Vy9V62 186 CAR 1 of MUC1-2- 212
MUC1 scFv-2 VH B-IRES-CD14-A
CDR 2
Tumor associated 87 6 chain-2 of Vy9V62 187 CAR 2 of MUC1-2- 236
MUC1 scFv-2 VH B-IRES-CD14-A
CDR 3
GSTA motif 88 Vy9V62 TCR-1 : 188 Construct of 242
DG. SF13 MUC1-2-A-IRES-
CD14-B
y chain
Modified PD-1 89 Vy9V62 TCR-1 : 189 CAR 1
of MUC1-2- 210
intracellular A-IRES-CD14-B
domain -1 DG. SF13 6 chain
Modified PD-1 90 Vy9V62 TCR-2: 190 CAR 2 of
MUC1-2- 239
intracellular DG. SF68: A-IRES-CD14-B
domain -2 y chain
Modified PD-1 91 Vy9V62 TCR-2 : 191
Construct of 243
intracellular MUC1-2-B-IRES-
domain -3 DG. SF68 : CD14-B
6 chain
88
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ
ID
NO: ID No:
NO:
Modified PD-1 92 Vy9V62 TCR-3: 192 CAR 1 of
MUC1-2- 212
intracellular 12G12: B-IRES-CD14-B
domain -4 y chain
Modified PD-1 93 Vy9V62 TCR-3: 193 CAR 2 of
MUC1-2- 239
intracellular 12G12: B-IRES-CD14-B
domain -5 6 chain
Removed PD-1 94 Vy9V62 TCR-4: 194
Construct of 244
intracellular CP.1.15 MUC1-5E5-A-
domain -1 y chain IRES-BCMA-A
Removed PD-1 95 TCR-4: CP.1.156 195
CAR 1 of MUC1- 202
intracellular chain 5E5-A-IRES-
domain -2 BCMA-A
Fokl WC 96 VVT CD3-zeta 196 CAR 2 of MUC1- 245
5E5-A-IRES-
BCMA-A
M Fokl 97 Invariant sequence 197 Construct of 246
for iNKT a chain MUC1-5E5-B-
(hVa24-JaQ-TRAC) IRES-BCMA-A
M Fokl 98 An example for 198 CAR 1 of MUC1- 205
iNKT 13 chain 5E5-B-IRES-
sequence BCMA-A
(containing V[311):
y chain-1 of 99 Invariant sequence 199 CAR 2 of MUC1- 245
Vy9V62 for MAIT a chain 5E5-B-IRES-
(hAV7S2-AJ33 a BCMA-A
chain) ( version1 )
VL anti-CD4-2 100 VH anti- CD4-2 200
Construct of 247
MUC1-5E5-A-
IRES-BCMA-B
CAR 1 of MUC1-2- 210 CAR 1 of MUC1- 205 CAR 1 of MUC1- 202
A-IRES-CD33-A 5E5-B-IRES-CD33- 5E5-A-IRES-
A BCMA-B
CAR 2 of MUC1-2- 255 CAR 2 of MUC1- 255 CAR 2 of MUC1- 248
A-IRES-CD33-A 5E5-B-IRES-CD33- 5E5-A-IRES-
A BCMA-B
Construct 261 Construct ofMUC1- 257 Construct of 249
ofMUC1-2-B- 5E5-A-lRES-CD33- MUC1-5E5-B-
IRES-CD33-A B IRES-BCMA-B
CAR 1 of MUC1-2- 212 CAR 1 of MUC1- 202 CAR 1 of MUC1- 205
B-IRES-CD33-A 5E5-A-IRES-CD33- 5E5-B-IRES-
BCMA-B
CAR 2 of MUC1-2- 255 CAR 2 of MUC1- 258 CAR 2 of MUC1- 245
B-IRES-CD33-A 5E5-A-IRES-CD33- 5E5-B-IRES-
BCMA-B
Construct 262 Construct ofMUC1- 259 Construct of 250
ofMUC1-2-A- 5E5-B-lRES-CD33- MUC1-2-A-IRES-
IRES-CD33-B B BCMA-A
CAR 1 of MUC1-2- 210 CAR 1 of MUC1- 205 CAR
1 of MUC1-2- 210
A-IRES-CD33-B 5E5-B-lRES-CD33- A-IRES-BCMA-A
89
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
CAR 2 of MUC1-2- 258 CAR 2 of MUC1- 258 CAR 2 of MUC1-2- 245
A-IRES-CD33-B 5E5-B-IRES-0D33- A-IRES-BCMA-A
B
Construct 263 Construct ofMUC1- 260 Construct of 251
ofMUC1-2-B- 2-A-IRES-CD33-A MUC1-2-B-IRES-
IRES-CD33-B BCMA-A
CAR 1 of MUC1-2- 212 Construct ofMUC1- 253 CAR 1 of MUC1-2- 212
B-IRES-CD33-B 2-B-IRES-BCMA-B B-IRES-BCMA-A
CAR 2 of MUC1-2- 258 CAR 1 of MUC1-2- 212 CAR 2 of MUC1-2- 245
B-IRES-CD33-B B-IRES-BCMA-B B-IRES-BCMA-A
Construct 254 MUC1-2-B-IRES- 248 Construct of 252
ofMUC1-5E5-A- BCMA-B CAR 2 MUC1-2-A-IRES-
IRES-CD33-A BCMA-B
CAR 1 of MUC1- 202 MUC1-5E5-B- 256 CAR 1 of MUC1-2- 210
5E5-A-lRES- IRES-CD33-A A-IRES-BCMA-B
CD33-A
CAR 2 of MUC1- 255 CAR 2 of MUC1-2- 248
Mcu1-5e5Panko- 264
5E5-A-lRES- A-IRES-BCMA-B enhanced
CD33-A scFc
Mcu1-Panko5e5 ¨ 265 hinge and/or 266 hinge and/or 267
enhanced scFc transmembrane transmembrane
domain A domain B
hinge and/or 268 hinge and/or 269 Mcu1-5e5Panko- 270
transmembrane transmembrane enhanced
domain C domain D scFc A 41BB CD2
zeta
Mcu1-5e5Panko- 271 Mcu1-5e5Panko- 272 Mcu1-5e5Panko- 273
enhanced enhanced enhanced
scFc B 41BB CD2 scFc C 41BB CD2 scFc D 41BB CD2
zeta zeta zeta
Mcu1-Panko5e5- 274 Mcu1-Panko5e5- 275 Mcu1-Panko5e5- 276
enhanced enhanced enhanced
scFc A 41BB CD2 scFc B 41BB CD2 scFc C 41BB CD2
zeta zeta zeta
Mcu1-Panko5e5- 277 GS linker 278 Construct of TSHR 279
enhanced CAR
scFc D 41BB CD2
zeta
M Fokl-1 280 M Fokl-2 281 Fokl WC 282
PSCA-CAR ScFv 356 CD8sp 428 Anti-TSHR-VL 429
3*GGGGS linker 278 Anti-TSHR-VH 430 4*GGGGS 431
bispecific CAR
linker
humanized-anti 432 humanized-anti 433 B7-H3 scFv 1
434
CD19-VH CD19-VL
B7-H3 scFv 2 435 B7-H3 scFv 3 436 Anti- CLDN 18.2 437
(175) -VL
Anti- CLDN 18.2 438 CLDN 18.2 ( 175 ) 439
(175) -VH CAR Binding
domain
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
tMUC1-CLDN 18.2 440 tMUC1-CLDN 18.2 452 scfv TSHR LH 466
tanCAR binding tanCAR
domain 5e5/175LH-1
175/5e5LH
tMUC1-CLDN 18.2 441 tMUC1-CLDN 18.2 453 scfv TSHR HL 467
tanCAR binding tanCAR
domain 5e5/175HL-1
175/5e5HL
tMUC1-CLDN 18.2 442 tMUC1-CLDN 18.2 454 scfv GUCY2C LH 468
tanCAR binding tanCAR
domain 5e5/163LH-1
163/5e5LH
tMUC1-CLDN 18.2 443 tMUC1-CLDN 18.2 455 scfv GUCY2C HL 469
tanCAR binding tanCAR
domain 5e5/163HL-1
163/5e5HL
tMUC1-CLDN 18.2 444 tMUC1-CLDN 18.2 456 scfv ACPP LH 470
tanCAR binding tanCAR
domain 175/5e5LH-2
5e5/175LH
tMUC1-CLDN 18.2 445 tMUC1-CLDN 18.2 457 scfv ACPP HL 471
tanCAR binding tanCAR
domain 175/5e5HL-2
5e5/175HL
tMUC1-CLDN 18.2 446 tMUC1-CLDN 18.2 458 scfv UPK2 LH (1) 472
tanCAR binding tanCAR
domain 163/5e5LH-2
5e5/163LH
tMUC1-CLDN 18.2 447 tMUC1-CLDN 18.2 459 scfv UPK2 HL (1) 473
tanCAR binding tanCAR
domain 163/5e5HL-2
5e5/163HL
tMUC1-CLDN 18.2 448 tMUC1-CLDN 18.2 460 scfv UPK2 LH (2) 474
tanCAR tanCAR
175/5e5LH-1 5e5/175LH-2
tMUC1-CLDN 18.2 449 tMUC1-CLDN 18.2 461 scfv UPK2 HL (2) 475
tanCAR tanCAR
175/5e5HL-1 5e5/175HL-2
tMUC1-CLDN 18.2 450 tMUC1-CLDN 18.2 462 scfv PSMA LH 476
tanCAR tanCAR
163/5e5LH-1 5e5/163LH-2
91
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Name SEQ ID Name SEQ Name SEQ ID
NO: ID No:
NO:
tMUC1-CLDN 18.2 451 tMUC1-CLDN 18.2 463 scfv PSMA HL 477
tanCAR tanCAR
163/5e5HL-1 5e5/163HL-2
scfv CD19 HL 465 scfv CD19 LH 464 anti CXCR5 Scfy 478
Anti DPEP3 Scfv 479 hCD19-CAR (4- 480 NFAT6x + minimal 481
1BB+ CD3 zeta) - IL12 promoter
NATF-1L6-2A-IFNy
IL-6 482 2A 483 IFN-y aa 484
aa Sequence
hCD19-CAR (4- 485 IL12 aa 486 Hif VHL-interaction 487
1BB+ CD3 zeta)- domain : Hif
NATF-1L12-VHL
amino acid 344-
417
GUCY2C-CAR 488 scFy 6503 55D1 489 163: cldn18.2 490
scfv : CD8-signal
peptide+cldn18.2V
L+GS
linker+cldn18.2VH
6921: ACPP scFv: 491 2517: tMUC1, 492 2519: tMUC1, 493
CD8-signal cldn18.2 tanCAR cldn18.2 tanCAR
peptide+acpp-
VL+GS
linker+acpp-VH
2521: TSHR, 494 2529: ACPP, 495 2530: ACPP, 496
tMUC1 tanCAR tMUC1 tanCAR tMUC1 tanCAR
2533: ACPP, 497 2534: ACPP, 498 scFy target PSMA 499
tMUC1 tanCAR tMUC1 tanCAR
scFy target 500 scFy target 501 scFy target CEA 502
Mesothelin EGFRvIll
scFy target 503 scFy target IL-13 504
Glypican-3
= 3*GGGGS is (GGGGS)3and 4*GGGGS is (GGGGS)4
= CD8sp-- tMUC1-VL--3*GGGGS linker-- tMUC1-VH--4*GGGGS bispecific CAR
linker--
humanized-CD19-VH--3*GGGGS linker--humanized-CD19-VL (2501)
= CD8sp-- tMUC1-VL--3*GGGGS linker-- tMUC1-VH--4*GGGGS bispecific CAR
linker--
humanized-CD19-VL--3*GGGGS linker-- humanized- CD19-VH (2504)
= CD8sp-- humanized- CD19-VL--3*GGGGS linker-- humanized- CD19-VH --4*GGGGS
bispecific CAR linker-- tMUC1-VL--3*GGGGS linker-- tMUC1-VH
= CD8sp-- humanized-CD19-VL--3*GGGGS linker-- humanized- CD19-VH --4*GGGGS
bispecific CAR linker-- tMUC1-VH--3*GGGGS linker-- tMUC1-VL
Table 6: Example targets of TCR therapy
TCL1 B cell lymphoma
NY-ESO-1 Urinary squamous cell carcinoma/melanoma
MAGA1/2/3 Lung cancer / pancreatic cancer / gastric cancer / breast
cancer
MAGE Lung cancer / pancreatic cancer / gastric cancer / breast
cancer
A3/A6/A10/Al2
HPV-16 E6/E7 Cervical cancer/ head and neck cancer/ anal cancer
WT-1 MDS & AML
92
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
SSX2 Hepatocellular carcinoma / melanoma / prostate cancer
KRAS Multiple malignant tumors
Neoantigen Multiple malignant tumors
LM P7 Brain cancer/HIV infection/cervical cancer in situ, cutaneous
basal cell
carcinoma or squamous cell carcinoma, localized prostate cancer or ductal
carcinoma in situ
AFP In theory, CART's maker (film surface) is also possible (as
long as TCR
can recognize)
HA1 Multiple malignant tumors
P53 Multiple leukemia + lymphoma
GP100 Multiple malignant tumors
LMP1, LMP2 Melanoma
and EBNA1
MCPyV EBV
CEA Merkel cell cancer
LAGE-1A Multiple malignant tumors
MART-1 Urinary squamous cell carcinoma/melanoma
Example 2. CART Cell Expansion and Anti-tumor Activity in Patients
[00325] Clinical studies were designed to assess the safety and efficacy of
infusing
autologous T cells modified to express several solid tumor markers specific
CAR/4-1BB/CD3-
into patients. On the first arm of the studies, patients received solid tumor
marker-specific CAR
T cells only. The solid tumor marker included TSHR and tM UC1. On the second
arm, patients
received CAR T cells directed to CD19 and a solid tumor antigen (e.g., TSHR,
tMUC1, or
GUCY2C). T cells of the patients were obtained, modified, and infused to the
patients. T cell
responses of patients from the first and second arms were measured and
compared using the
following protocols, which were approved by the hospitals where the trials
were conducted. All
patients were provided with written informed consent. Information regarding
these patients are
provide below in Table 9 (SD: stable disease; PD: progressive disease; PR:
partial remission;
CR: complete remission; NR: no response).
[00326] PBMCs were obtained from patients. Various lentiviral vectors were
generated and
then transfected to the T cells, which were further cultured for several days
before the co-
cultivation assay. More information can be found in Tables 7, 9, and 10 below.
Techniques related
to cell cultures, construction of cytotoxic T-lymphocyte assay can be found in
"Control of large,
established tumor xenografts with genetically retargeted human T cells
containing CD28 and
CD137 domains," PNAS, March 3, 2009, vol. 106 no. 9, 3360-3365, which is
incorporated herein
by reference in its entirety.
[00327] Several methods were used to generate CAR T cells. For Patients 001-
003, CD3+
cells were obtained from PBMCs and were cultured using X-vivo 15 media
containing IL-2. For
example, CD3+ T cells can be collected using antibody kits including CD14,
CD15, CD16, CD19,
CD34, CD36, CD56, CD123, and CD235a to remove undesired cells. The CD3+ T
cells were
activated using CD3/CD28 Dynabeads and then sampled as well counted before
infection. The
number of cells to be infected was obtained. The cell number of Group 1 was 6
x 107, and the cell
93
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
number of Group 2 was 7 x 107. The number of corresponding carriers and the
volume of the
carrier were calculated according to the required carrier MOI (See Table 10).
For Patients 004-
010, PBMCs were cultured using TEXMACS culture media containing IL-2. CD4 and
CD8
magnetic beads were used to sort and select T cells in the PBMCs. The
appropriate starting
culture amount was selected and Transact activator was used to activate T
cells. MACS GM P
T Cell TransActTm includes a colloidal polymeric nanomatrix covalently
attached to humanized
recombinant agonists against human CD3 and CD28. Due to the nanomatrix MACS
GMP T Cell
TransAct can be sterile filtered and excess reagent can be removed by
centrifugation and
following conventional supernatant replacement or simply by media wash. This
reagent is suitable
for use in automated culture systems, such as the CliniMACS Prodigy
Instrument. The number
of corresponding carriers and the volume of the carrier were calculated
according to the required
carrier MOI (See Table 10). Specifically, for Patients 004-008 and 010,
lentiviral vectors
containing multiple vectors were mixed with the T cells for 24 hours. The T
cells were further
washed and cultured for 8 days before being transported to the hospital. For
Patient 009, the T
cells were divided into four groups, and each group of T cells was mixed with
lentiviral vectors,
for 24 hours, which contain one or more vectors (See Table 7), and these T
cells were washed
and cultured for 8 days. These four groups of transfected T cells were mixed
and then transported
to the hospital.
Table 7:
Group Vector MOI Day6 Total CAR copy Ratio to Total
number per ugDNA
hCAR+/CD3+ CD19 scFv + IL- IL12
TSHR
/CD3+ 6/IFN-y
1 1 50 42.64% 12.37% 112238.97 8.27% 0.21% 91.52%
2 10
3 1
2 1 50 59.28% 33.32% 118925.1 17.77% 0.34% 81.89%
2 10
3 1
3 3 20 19.52% 18.16% 19408.75 0.00% 100.00%0.00%
4 3 20 26.70% 26.59% 17874.89 0.00% 100.00%0.00%
[00328] For fresh cells, after removing the magnetic beads, the transduced
cells were
centrifuged or replaced with a solution of 95% compound electrolyte and 5%
human albumin,
loaded into a return bag, and transported at 15-25 C after sealing. Fresh
preparations are
94
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
returned directly. For cryopreserved cells, the media including 33.75%
compound electrolyte
solution, 33.75% dextran 40 glucose solution, 25% human blood albumin, and
7.5% dimethyl
sulfoxide was used for cryopreservation. The cell suspension was loaded into a
cryopreservation bag and then the bag was cooled to -90 C and transferred to
a gas phase
liquid nitrogen tank for storage. The reconstitution of the frozen
preparations was completed
within 30 minutes after resuscitation of the frozen preparations. Peripheral
blood mononuclear
cells (PBMCs) were obtained from patients by leukapheresis for CAR T cell
preparation, and the
first day of CAR T infusion was set as study day 0.
[00329] Several patients were given a conditioning treatment for
lymphodepletion for CAR T
cell infusion. Fludarabine- and cyclophosphamide-based conditioning treatment
varied
according to the tumor burden in the bone marrow (BM) and peripheral blood
(PB). Some
patients were administered a long-acting G-CSF for 1-3 days after the
conditioning treatment at
a dose of about 6 mg each or 100pg/kg of body weight to boost the patient's
neutrophils which
are critical to fight off infections. CAR T cells were transfused to the
patients. Each day CAR T
cells were transported to hospital, washed, counted, checked for viability and
then prepared for
administration to patients, who were then observed closely for at least 2
hours. Cytokine
Release Syndrome (CRS) was graded according to a revised grading system (See
Lee DW. et
al, Blood 2014;124:188-95). Other toxicities during and after therapy were
assessed according
to the National Institutes of Health Common Terminology Criteria for Adverse
Events Version
4.0 (http://ctep.cancer.gov/). Therapy responses were assessed by flow
cytometry and
morphological analysis. When possible, patients were assessed by chimeric gene
expression
levels.
[00330] Bone Marrow (BM) and peripheral blood (PB) samples after CAR T cell
infusion were
collected in K2EDTA BD vacutainer tubes. The persistence of CD19 CAR T cells
in PB and BM
of patients was determined by FACS. Circulating CAR T cell numbers per pl were
calculated on
the basis of measured absolute CD3+ T lymphocyte counts. Simultaneously, CAR
DNA copies
were evaluated as another method of determining CAR T cell expansion and
persistence.
Genomic DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen) from
cryopreserved
PB and BM. CAR DNA copies were assessed by quantitative real-time PCR as
described in the
supplementary materials. The levels of cytokines I FN-y, TNF-a, IL-4, IL-6, IL-
10, IL-17, etc. in
serum and CSF were measured in a multiplex format according to the
manufacturer's
instructions.
[00331] Genomic DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen)
from
cryopreserved peripheral blood and bone marrow. Quantitative PCR (qPCR) was
performed in
real-time in triplicates using the ABI 2xTaqMan Universal Master Mix with
AmpErase UNG
(Applied Biosystems) in a 7500 real-time PCR system (Applied Biosystems). Copy
numbers per
microgram of genomic DNA were calculated from a standard curve of 10-fold
serial dilutions of
purified CAR plasmid containing 102-108 copies/pL. Amplification of an
internal control gene
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
was used for normalization of DNA quantities. Primers/probes specific for the
CAR transgene
and an internal control gene were as previously described (see GOkbuget N. et
al., Blood
2012;120:2032-41 and O'Brien S. et al, J Olin Oncol 2013;31:676-83).
[00332] CAR T cell expansion was observed based on CAR copy numbers of
individual
CARs and shown in FIGS. 16 and 17. As shown in these figures, CAR T cells
expansion in
Patients 004 and 005 were significantly higher than those in Patients 002,
003, and 001,
indicating that T cells expressing CD19 CAR and/or CD19 CAR and tMUC1 CAR
enhanced
CAR T cell expansion (Also see Table 10). T cells expressing CD19 CAR, solid
tumor CAR
(e.g., tMUC1, TSHR, GUCY2C CARs), and double CARs (CD19 CAR and solid tumor
CAR)
were calculated. For example, T cells expressing CD19 CAR, tMUC1 CAR, and
double CARs
(CD19 CAR and tMUC1 CAR) were calculated using the following equations:
WBC X CD3% X ((tMUC1CAR+ CD19CAR-)/CD3),
WBC X CD3% X ((tMUC1CAR- CD19CAR+)/CD3), and
WBC X CD3% X ((tMUC1CAR+ CD19CAR+)/CD3),
wherein WBC is the number of WBC; CD3% is the percentage of CD3 positive cells
in WBC;
(tMUC1CAR+ CD19CAR-)/CD3 is the percentage of T cells expressing tMUC1 CAR,
with no
CD19 CAR in CD3 positive cells; (tMUC1CAR- CD19CAR+)/CD3 is the percentage of
T cells
expressing CD19 CAR, with no tMUC1 CAR in CD3 positive cells; and (tMUC1CAR +
CD19CAR+)/CD3 is the percentage of T cells expressing CD19 CAR and tMUC1 CAR
in CD3
positive cells. The results are shown in FIGS. 18 and 19. As shown in these
figures, CD19 CAR
cells significantly increased expansion of tMUC1 CAR T cells, indicating that
presence of CD19
CAR enhances increased expansion of tMUC1 CAR T cells. Similar results were
observed in
Patients 006-010 (See FIG. 20 and 21). Combination of in vitro results above
and in vivo results
in the Example below shows that activation of CAR T cells targeting WBC
antigens can
enhance expansion of CAR T cells targeting solid tumor antigen.
[00333] Patient 008 had undergone thyroidectomy. 28 days after the
infusion, the right tumor
disappeared, and the size of the left tumor reduced. Examples of PET CT
scanning images are
shown in FIG. 33. Three months after the infusion, the right tumor did not
recur, and the left
tumor disappeared. The PET CT images (not shown) showed that there was no
tumor
recurrence or recurrence in the surgical area. After the scanning signal is
enhanced, no
abnormal enhancement signal is observed in the above areas. The double neck II
and III areas
showed multiple small lymph nodes with a maximum short diameter of no more
than 10 mm.
There were no abnormalities in the bilateral submandibular gland morphology
and signal. At the
same time, the cervical spinal cord morphology and CT signal were normal. It
appeared that the
patient had achieved at least partial remission (PR). During the treatment, no
severe CRS (e.g.,
no greater than level 2 CRS) was observed in Patient 008. The patient was
evaluated to
achieve PR.
96
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00334] Patient 009 was diagnosed with poorly differentiated follicular
papillary carcinoma
with neuroendocrine carcinoma in the thyroid gland. Patient 009 underwent
thyroid double lobe
resection and was later examined and confirmed to have multiple lung
metastases. Multiple
enlarged lymph nodes were found in the mediastinum. 30 days after the infusion
of CAR T cells,
CT scanning showed that small tumors disappeared, and the size of the two
major tumors was
reduced by more than 70% (see Table 8). FIG. 34 shows that the major tumor
shrunk, and the
small tumor disappeared (see lines as well circles in FIG. 34). The patient
was evaluated to
have achieved PR.
Table 8: Reduction of tumor sizes on Patient 009
Major tumor Before Estimated Day 4 Estimated Day
Estimate Reduce
Infusion( Volume (mm) Volume 30 d Volume d
mm) (mm3) (mm3)
(mm) (mm3) Volume
Right lower 68x60 128112.0 70x6
136312.6 42x3 33431.58 73.90%
lobe 0 1 3 9
Mediastinum 25 65416.67 25 65416.67 15
14130.00 78.40%
and double
hilar
[00335] Patient 010 was diagnosed with colorectal cancer and went through 8
cycles
chemotherapy as well as other treatments such as surgery before CAR T cell
infusion. One
month after infusion, PET-CT scanning results show most of the target lesions
were significantly
reduced (more than 50%), and the comprehensive calculation of tumor reduction
was 44.7%.
The patient was evaluated to have achieved PR (See arrows in FIG. 35).
[00336]
Patient 011 was diagnosed with thyroid cancer. The patient's PBMC was
collected
and sorted using Prodigy to obtain CD3+ cells, which were then divided into
six groups. Each of
the six groups of cells was mixed with media containing a corresponding
vector, as shown in
Table 19. In these six groups of cells, there were no cells expressing both
CD19 CAR and
TSHR CAR. Subsequently, the six groups of cells were cultured with media
without vectors to
Day 7 under appropriate conditions, and cell numbers were calculated. A
certain number of
cells were then obtained from each group and mixed together as shown in Table
19 to obtain a
mixed population of cells, which were transported to the hospital for
infusion. FIG. 73 shows
increases of lymphocytes including CAR T cell, natural killer cells (NK
cells), natural killer T cells
(NKT cells), and monocytes of Patient 011 in response to the infusion. FIGS.
74 and 75 show
the increases of the number of individual CAR T cells and the total number of
CAR T cells of
Patient 011 in response to the cell infusion. Copy numbers of individual CAR T
cell were
measured to calculate the number of each type of CAR T cells and the total
number of CAR T
cells in the blood of Patient 011. The copy numbers and flow cytometry data
were used to
perform linear regression analysis and to calculate the numbers of individual
CAR T cells. The
linear regression analysis and expansion of the individual CAR T cells are
shown in FIG. 75.
These data as well as data from previous patients show that (1) CD19 CAR T
cells enhanced
the expansion of solid tumor CAR T cells (e.g., TSHR CAR) and (2) CD19 CAR T
cells
97
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
enhanced the expansion of non-CAR T cells (see individual lymphocyte number
increases in
FIG. 73). Further, these data indicate that this enhancement is triggered by
activation of CD19
CAR and mediated by the immune cells in the patients' body, for example, the
DCs. Thus, CAR
T cells binding a WBC antigen (e.g., CD19 and BCMA) can also be used to
enhance other T
cell-based therapies (e.g., NK, TCR and TIL). For example, CD19 CAR T cells
can be
administered to patients combining with NK and/or T cells expressing
manipulated TCR or TILs,
and activation of CD19 CAR T cells can enhance expansion of these lymphocytes
in the
patients. FIG. 76 shows cytokine release of Patient 011 in response to cell
infusion.
Table 19 CAR T cells and vectors used for Patient 011.
Cell types Group Vectors MOI Infusion
% in
CAR% CAR- infused
T/KG
cells
1 TSHR scfv-41BB-CD3Z 36.64% 36.64% 36.64% 36.64%
TSHR2 TSHR scfv-41BB-CD3Z 39.04% 39.04% 39.04% 39.04%
CAR-T
3 TSHR scfv-41BB-CD3Z 15.52% 15.52% 15.52% 15.52%
4 CD19 scfv-41BB-CD3Z- 7.22% 7.22% 7.22% 7.22%
NFAT-IFNg
CD19 5 CD19 scfv-41BB-CD3Z- 0.82% 0.82% 0.82%
0.82%
CAR-T NFAT-1L6
6 CD19 scfv-41BB-NFAT- 0.82% 0.82% 0.82% 0.82%
1L12-VHL
Total for infusion 40.26%
1.25x107 100%
[00337] The
combination of in vitro results above and in vivo results in this Example
shows
that activation of CAR T cells targeting WBC antigens can enhance anti-tumor
activity of CAR T
cells targeting solid tumor antigen.
[00338]
FIG. 72 shows PDL1 expression of monocytes in Patient 009 on Day 0, Day 1, and
Day 4. Monocytes were obtained from several patients, before and after
infusion of the mixed
CAR T cells (CD19CAR + tMuc1 CAR, CD19CAR + GUCY2C CAR, and CD19CAR + TSHR
CAR) into the patients. The monocytes were analyzed using flow cytometry to
measure the
expression of markers such as PDL1. The flow cytometry results showed that the
PDL1
expression was up-regulated in monocytes of patients after infusion of mixed
CAR T cells. An
example is shown in FIG. 72. The upregulation of PDL1 in monocytes showed the
activation of
monocytes, further proving the immune system of the patients was activated.
Table 9: clinical trial data
Patient's Cancer Infusion CAR T cell types Percentage CSR Efficacy
ID CART/kg before infusion >2
001 Breast 1x105 N/A No PD (One
cancer 1.24 x106 Month)
0.966 x107
002 Pancre 1.28x106 N/A No NR (One
atic 1.29 x107 Month)
cancer
003 Thyroid 1.1X106 N/A No NR (One
cancer Month)
98
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
004 Pancre 1.18x106 = MUC1 CART cells No PD (One
atic /CD3+= 50% Month)
cancer = CD19 CAR T cells
/CD3+=15%
= MUC1&CD19 CART
cells/CD3+=6.8%
005 Breast 1.03x106 N/A No SD (One
cancer Month)
006 Breast 0.11 x 106 N/A No SD (One
cancer Month)
007 Breast 1.52 x 106 N/A No SD (One
cancer Month)
008 Thyroid 2.08x109 = TSHR+CD19 CAR T No PR (See
cancer cells/CD3+=33.04% FIG. 33)
= CD19 CAR T
cells/CD3+=21.96%
009 Thyroid 1.36x109 = TSHR+CD19 CART No PR (See
cancer cells/CD3+=42.64% FIG. 34)
= CD19 CAR T
cells/CD3+=12.37%
010 Colorec 3.78x108 = GUCY2C+CD19 CAR T No PR (See
tal cells/CD3+=15.55% FIG. 35)
cancer
011 Thyroid 1.25x107 = CAR/CD3+=40.26% No N/A
cancer = CD19 CAR/CD3+=2.25%
= TSHR
CAR/CD3+=38.1%
Table 10: cell manufacture for clinical trials
Patient's Vectors and MOI Infusion Pre-treatment
ID Methods
001 Vector 1: MUC1-CAR (scFv of the CAR is Fresh (first
cyclophosphamide
SEQ ID NO: 70): 30:1 infusion) and 1.5 grams/m2
in -3
Cryopreserved days
(second
infusion) cells
002 Vector 1: MUC1-CAR (scFv of the CAR is Fresh cells No
SEQ ID NO: 70): 30:1
003 Vector 2: TSHR-CAR (scFv of the CAR is Fresh cells FC regimen
at -5 to
SEQ ID NO: 8): 100:1 -3 days
(cyclophosphamide
500 mg/m2,
fludarabine 30
mg/m2)
004 Vector 1: MUC1-CAR (scFv of the CAR is Fresh cells FC regimen
at -5 to
SEQ ID NO: 70): 19:1 -3 days
(cyclophosphamide
99
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
Vector 2: hCD19-CAR (scFv of the CAR is 500 mg/m2,
SEQ ID 5): 5:1 fludarabine 30
mg/m2)
005 Vector 1: MUC1-CAR /Dominant Negative Fresh cells No
PD-1(scFv of the CAR is SEQ ID NO: 70
and 89): 18:1
Vector 2: hCD19-CAR (scFv of the CAR is
SEQ ID 5): 5:1
006 Vector 1: MUC1-CAR (scFv of the CAR is Fresh cells
cyclophosphamide
SEQ ID NO: 70): 19:1 1.5 grams/m2 in -3
Vector 2: hCD19-CAR (scFv of the CAR is days
SEQ ID 5): 5:1
007 Vector 1: MUC1-CAR (scFv of the CAR is Cryopreserved
cyclophosphamide
SEQ ID NO: 70): 19:1 cells 1.5 grams/m2 in -3
Vector 2: hCD19-CAR (scFv of the CAR is days
SEQ ID 5): 5:1
008 Vector 1: TSHR-CAR (CAR: SEQ ID NO: Fresh cells FC regimen at -
5 to
279, scFv of the CAR: SEQ ID NO: 8): -3 days
19:1(M01); and Vector 2: hCD19-CAR- (cyclophosphamide
NATF-1L6-2A-IFNy (Vector SEQ ID NO: 500 mg/m2,
480, scFv of CD19 CAR: SEQ ID 5, fludarabine 30
6xNFAT: SEQ ID: 481, aa of 1L6: SEQ ID mg/m2)
NO: 482, 2A is SEQ ID NO: 483, and aa of
IFN-y: SEQ ID NO: 484 (See the construct
of Embodiment 1 of FIG. 63)): 5:1(M01)
009 Vector 1: TSHR-CAR (CAR: SEQ ID NO: Fresh cells FC regimen at -
5 to
279, scFv of the CAR: SEQ ID NO: 8): (See -3 days
MOI in Table 7); Vector 2: hCD19-CAR- (cyclophosphamide
NATF-1L6-2A-IFNy (Vector SEQ ID NO: 600 mg,
480, scFv of CD19 CAR: SEQ ID 5, fludarabine 50 mg)
6xNFAT: SEQ ID: 481, aa of 1L6: SEQ ID
NO: 482, 2A is SEQ ID NO: 483, and aa of
IFN-y: SEQ ID NO: 484 (See the construct
in Embodiment 1 of FIG. 63)) (See MOI in
Table 7); and Vector 3: hCD19-CAR-NATF-
IL12-VHL (Vector SEQ ID NO: 485, scFv of
CD19 CAR: SEQ ID 5, 6xNFAT: SEQ ID:
100
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
481, aa of 1L12: SEQ ID NO: 486, VHL:
SEQ ID NO: 487 (See the construct of
Embodiment 3 of FIG. 63) (See MOI in
Table 7).
010 Vector 4: GUCY2C-CAR (CAR: SEQ ID Fresh cells FC regimen at -2
NO: 488, scFv of the CAR: SEQ ID NO: days
11): 50:1(M01); and Vector 2: hCD19-CAR- (cyclophosphamide
NATF-1L6-2A-IFNy (Vector SEQ ID NO: 500 mg/m2,
480, scFv of CD19 CAR: SEQ ID 5, fludarabine 30
6xNFAT: SEQ ID: 481, aa of 1L6: SEQ ID mg/m2)
NO: 482, 2A is SEQ ID NO: 483, and aa of
IFN-y: SEQ ID NO: 484 (See the construct
of Embodiment 1 of FIG. 63)): 10:1(M01)
011 Vector 1: TSHR-CAR (CAR: SEQ ID NO: Fresh cells FC regimen at -
2
279, scFv of the CAR: SEQ ID NO: 8) (See days
MOI in Table 19); Vector 3: hCD19-CAR- (cyclophosphamide
NATF-1L12-VHL (Vector SEQ ID NO: 485, 500 mg/m2,
scFv of CD19 CAR: SEQ ID 5, 6xNFAT: fludarabine 30
SEQ ID: 481, aa of 1L12: SEQ ID NO: 486, mg/m2)
VHL: SEQ ID NO: 487 (See the construct of
Embodiment 3 of FIG. 63) (See MOI in
Table 19); and Vector 4: hCD19-CAR-
NATF-IFNy (Vector SEQ ID NO: 480, scFv
of CD19 CAR: SEQ ID 5, 6xNFAT: SEQ ID:
481, and aa of IFN-y: SEQ ID NO: 484 (See
the construct in Embodiment 5 of FIG. 63))
(See MOI in Table 19); and Vector 5:
hCD19-CAR-NATF-1L6 (Vector SEQ ID NO:
480, scFv of CD19 CAR: SEQ ID 5,
6xNFAT: SEQ ID: 481, and aa of 1L6: SEQ
ID NO: 482 (See the construct in
Embodiment 4 of FIG. 63)) (See MOI in
Table 19);
Example 3. Activation of Coupled/mixed T Cells
[00339] Mixed CAR T cells (Coupled CAR T cells) were divided into three
Groups for assays
related to activation: CD19 CAR and tMUC1 CAR (Group 1), anti-CD19 CAR and
ACPP CAR
(Group 2), and CD19 and CLDN18.2 CAR (Group 3). Peripheral blood of healthy
volunteers was
101
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
collected. CD3+ T cells were sorted using Pan T kits, and CD3/0D28 Dynabeads
were added at
a 1:1 ratio. CD3+ T cells were then transfected with lentivirus. The
lentivirus and the Dynabeads
were removed, and fresh media were added. CAR ratios and cell phenotype were
determined.
Expression of CAR in these three groups of cells was measured. CD19 CART
cells, tMUC1 CAR
T cells, and target cells were selected and mixed for 24 hours or 48 hours.
Expression of various
markers in corresponding cells was measured. 20 x 104 CAR T cells and 20 x 104
substrate cells
were co-cultured for 24 hrs. The expression of molecules such as hCAR
(humanized scFv),
mCAR (murine scFv), CD25, and CD137 in T cells was measured by flow cytometry.
For example,
CD25 and CD137 positive staining indicated that T cells were activated.
Amounts of cytokines
released from various T cells were measured in response to the antigen
activation, and
background of the corresponding T cells was subtracted.
[00340] Tables 11, 12, and 13 provide information for CAR T cells and
corresponding substrate
cells of Group 1, Group 2, and Group 3, respectively. For example, CAR 1204 is
a human-derived
CAR, which can be labeled with human CAR antibody and CD137 antibody. CAR 2407
(tMUC1
CAR) is a murine CAR that can be labeled for activation with a murine CAR
antibody and a CD137
antibody. Cells expressing CAR 1204 (CD19 CAR T cells) can be activated by
K562 cells
expressing CD19, resulting in up-regulated CD137 expression. CAR 1204 cells,
CAR 2407 cells,
and K562 cells expressing CD19 were co-cultured to induce CD19 CAR T cell
activation. The
binding domains of CD19 CAR and tMUC1 CAR include SEQ ID NOs: 5 and 70,
respectively.
The activation of 2407 CAR T cells was detected and measured based on the
expression of
CD137, which evidence the indirect activation of CD19 CAR T cells.
Table 11: CAR T cells and substrate cells used in Group 1
Coupled CAR T CD19 CAR T Cells & tMUC1 CAR T Cells
Cells
CAR T cell ID CAR Substrate cell ID Notes
1234 CD19 CAR K19 CD19 positive Cell
2407 tMUC1 CAR MCF-17 5E5 (tMUC1) positive cell
[00341] FIG. 36 shows results of flow cytometry analysis of CD19 CAR T
cells co-cultured with
tMUC1 CAR T cells in the presence or absence of K19 cells.
[00342] Peripheral blood of healthy volunteers was collected on Day 0. CD3+
T cells were
sorted using Pan T kits, and CD3/CD28 Dynabeads were added to the collected
CD3+ T cells at
a 1:1 ratio. On Day 1, the activated CD3+ T cells were divided into two
subgroups, each
transfected with lentivirus encoding a single CAR (CD19 CAR or tMUC1 CAR).
Thus, two
subgroups of CAR T cells were obtained: a subgroup of CAR T cells expressing
CD19 CAR, and
another subgroup of CAR T cells expressing tMUC1 CAR. The binding domains of
CD19 CAR
and tMUC1 CAR include SEQ ID NOs: 5 and 70, respectively. On Day 2, the
lentivirus and the
Dynabeads were removed, and fresh media were added. On Day 7, CAR T cells and
target cells
102
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
were co-cultured for 24 hours, and various assays were performed on Day 8.
Subgroups of cells
can be mixed and co-cultured with corresponding substrate cells (see FIGS. 36-
60).
[00343] FIG. 36 provides histograms showing 0D137 expression in various
cell cultures. In
each cell culture, CAR T cells were cultured with the corresponding substrate
cells, and 0D137
expression was measured using flow cytometry (Gate mCAR+: tMUC1CAR). The cell
cultures
include (1) tMUC1CAR T cells and K19, (2) tMUC1CAR T cells, K19, and PBMC, (3)
tMUC1CAR
T cells, CD19CAR T cells and K19, (4) tMUC1CAR CAR T cells, CD19 CAR T cells,
K19, and
PBMC. The CD8+ T cells were also counted. As shown in FIG. 36, the activation
of tMUC1CAR
T cells (i.e., expression of 0D137) was observed in the presence of K19, and
the activation level
of MUC1 CAR T cells was higher than that of the single group. Further, the
level of activation was
higher after adding PBMC (e.g., MFI of CD137). These results indicate that the
activation of CD19
CART cells by K19 activates tMUC1 CART cells in the absence of the antigen
that tMUC1 CAR
binds (tMUC1), and this activation is enhanced by the presence of PBMC. The
experimental
results are based on the expression ratio as the main basis for measuring the
difference (left).
When the proportional difference is not significant, the expression intensity
(MFI) is used as a
measure of the difference (right).
[00344] FIG. 37 shows the activation of PBMC and monocytes in the cell
cultures described in
FIG. 36. Flow cytometry assays of monocyte (CD14+) and activated monocyte
(CD14+CD80+)
were performed in PBMC, and FIG. 37 shows a histogram of statistical analysis
of the assays.
h19CAR is a humanized CD19CAR, and the cell cultures include (1) PBMC alone,
(2)
PBMC+K19, (3) PBMC and CD19CAR T cells, (4) PBMC, K19, and CD19CAR T cells. As
shown
in FIG. 37, the last group of PBMCs showed activation (CD80 expression). These
results show
that activation of the CAR T cells is capable of activating PBMC including
monocytes.
Combination of the results shown in FIGS. 36 and 37 indicates that activation
of CD19 CAR T
cells by K19 activates tMUC1CAR T cells in the absence of the antigen that
tMUC1 CAR binds,
and this activation can be mediated at least partially through PBMC.
[00345] FIG. 38 provides a histogram showing IFNy release by tMUC1 CAR T
cells and CD19
CAR T cells. Various cells were cultured on Day 7, and flow cytometry assays
were performed
on Day 8. The graph is a statistical analysis of the convective graph. In
these assays, NT (non-
transfected T cells) was used as a control. Compared with the control, cell
cultures including
CD19 CAR T cells and tMUC1 CAR T cells showed an increase of intracellular
IFNy in CD19
CAR T and MUC1 CAR T cells, indicating that CD19CAR T cells activated by K19
released IFNy
and activated tMUC1CAR T cells to release IFNy. The PBMC group up-regulated
the ratio of IFNy
released by CD19CAR T cells and by tMUC1CAR T cells. IFNy cumulated in the
coupled CAR
group was more than that in the cells expressing a single CAR (CD19 CAR or
tMUC1 CAR), and
the addition of PBMC upregulated this effect. The mCAR-group is not all CD19
CAR positive cells,
and the statistical value is relative. The results show that activation of
CD19 CAR T cells induced
103
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
tMUC1 CAR T cells to express more IFNy and thus release IFNy in the absence of
the antigen
that tMUC1 CAR binds (tMUC1).
[00346] FIG. 39 provides a histogram showing GZMB release by tMUC1 CAR T
cells and
CD19 CAR T cells. Various cells were cultured on Day 7, and flow cytometry
assays were
performed on Day 8. Flow cytometry assays showed GZMB release by the activated
CD19 CAR
T cells and MUC1 CAR T cells. The statistical analysis of the convective
graphs (proportional
comparison MFI) indicates that the activation of CD19 CAR T cells can cause
MUC1 CAR T cells
to release GZMB, and such release was enhanced in the presence of PBMC. The
mCAR-group
is not all CD19 CAR positive cells, and the statistical value is relative.
These results show that
activation of CD19 CAR T cells induced MUC1 CAR T cells to release
intracellular GZMB.
[00347] FIG. 40 and 41 show proliferation of MUC1 CAR T cells in various
embodiments.
CFSE reactions were performed and used to indicate levels of cell
proliferation. Various cells
were cultured on Day 7, and flow cytometry assays were performed on Day 8. As
shown on FIG.
40, the first row is the experimental group of coupled CAR T cells co-cultured
with two substrate
cells, and the second row is the control group of MUC1 CAR T cells co-cultured
with two substrate
cells. As shown in the third and fourth columns of the first and second rows,
activation of CD19
CAR T cells with K19 induces the proliferation of MUC1 CAR T cells. The fifth
and sixth columns
show that MCF-7 activates and incudes the proliferation of MUC1CAR T cells.
FIG. 41 shows
counting results from the flow cytometry shown in FIG. 40. The volume
calibration was performed,
tMUC1 CAR cell population was gated, and the number of cells of each group of
tMUC1 CAR
was statistically analyzed. As shown in FIG. 41, the number of cells in the
group including CD19
CART cells and tMUC1CAR T cells was higher than that in the control group, and
the proliferation
of the group including CD19 CAR T cells and tMUC1 CAR T cells in the presence
of PBMC was
the highest. The results show that activation of CD19 CAR T cells can enhance
the proliferation
of MUC1 CAR T cells, which can be enhanced and/or medicated through PBMC.
[00348] FIG. 12 shows the proliferation CD19 CART cells in various
embodiments. CFSE
reactions were performed and used to indicate levels of cell proliferation.
Various cells were
cultured on Day 7, and flow cytometry assays were performed on Day 8. The
groups of cells
comprising CD19 CAR T cells, tMUC1 CAR T cells, MCF-7 in the presence or
absence of PBMC
showed the proliferation of CD19 CAR T cells. These results show that
activation of tMUC1 CAR
T cells can enhance the proliferation of CD19 CAR T cells, which may be
enhanced and/or
medicated through PBMC. The combination of the results shown in FIGS. 40-42
indicates that
the mixture of CD19 CAR T cells and tMUC1 CAR T cells may form a positive
circle through
PBMC such that activation of CD19 CAR T cells or tMUC1 CAR T cells may further
activate each
other to enhance the proliferation of CD19 CART cells and tMUC1 CART cells
and/or the release
of cytokines by CD19 CAR T cells and tMUC1 CAR T cells, which may be mediated
and/or
enhanced by PBMC (See FIG. 62). These results can also explain the reason that
tMUC1 CAR
T cells expanded more in subjects infused with a population of cells
comprising coupled CAR T
104
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
cells (e.g., Patients 001-003) than subjects infused with a population of
cells including a single
type of CAR T cells (e.g., Patient 004-010). Coupled CAR T cells (e.g., CD19
CAR T cells and
tMUC1 CAR T cells) can contribute to this enhanced cell expansion.
[00349] FIG. 43 shows cytokine release in embodiments. Various cells were
cultured on Day
7, and flow cytometry assays were performed on Day 8. As shown in FIG. 43, IFN-
y release in
the control group is limited. The coupled CAR group and single CAR group are
labeled using the
solid line and the dotted line, respectively. The levels of IFN-y released
were similar in the
absence of PBMC. When PBMC was added, the levels of IFN-y released increased.
IL6 was
mainly secreted by PBMC, and the released amount in the activated system is
increased. Here,
the amount of tMUC1CAR cytokine released was relatively low.
Table 12: CAR T cells and substrate cells used in Group 2
Coupled CAR T cells CD19 CAR T cells & ACPP CAR cells
CAR T Cell ID CAR Substrate Cell Notes
1234 CD19 CAR Nalm CD19 positive Cell
6503 ACPP CAR PC3-ACPP ACPP positive cell
[00350] FIG. 44 shows other histograms of CD137 expression in various cell
cultures.
Peripheral blood of healthy volunteers was collected on Day 0. CD3+ T cells
were sorted and
collected using Pan T kits, and CD3/CD28 Dynabeads were added at a 1:1 ratio
to the collected
CD3+ T cells. Day 1, CD3+ T cells were transfected with lentivirus encoding
CD19 CAR and
ACPP CAR, respectively. The binding domains of CD19 CAR and ACPP CAR include
SEQ ID
NOs: 5 and 489, respectively. On Day 2, the lentivirus and the Dynabeads were
removed, and
fresh media were added. On Day 7, CAR T cells and target cells were co-
cultured for 24 hours
and various assays were performed on Day 8. Flow cytometry assays were
performed, and the
results show expression of CD19 CAR and ACPP CAR T cells. As shown in FIG. 44,
the activation
of the ACPP CAR T cells was higher, and the activation was increased in
presence of PBMC.
These results show activation of CD19 CAR T cells by na1m6 can activate ACPP
CAR T cells,
and this effect is enhanced by PBMC.
[00351] FIG. 45 shows flow cytometry assays of activation analysis. CD45R0
and CD62L can
be used to divide CART cells into four states. Nalm6 activated expression of
CD45R0 and CD62L
on CD19 CAR T cells, and the proportion of effector cells in ACPP CAR T cells
increased. These
results show that the activation of CD19 CAR T cells induced ACPP CAR T cells
to a functional
state, which acted as the pre-activation of ACPP CAR T cells.
[00352] FIG. 46 shows the activation of PBMC and monocyte in the cell
cultures described in
FIG. 44. Flow cytometry assays showed monocyte (CD14+) and activated monocyte
(CD14+&CD80+) in PBMC. h19CAR is a humanized CD19CAR, and the groups include
(1)
PBMC alone, (2) PBMC and K19, (3) PBMC andCD19CAR T cells, (4) PBMC and K19
and
105
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CD19CAR T cells. These results indicate that activation the CAR T cells is
capable of activating
PBMC.
[00353] FIG. 47 shows that activation of CD19 CAR T cells induces ACPP CAR
T cells to
release intracellular IFNy. Similar to above, various cells were cultured on
Day 7, and flow
cytometry assays were performed on Day 8. When both CAR T cells were present
and there was
PBMC in the system, ACPP CAR T cells also showed enhanced IFNy release.
[00354] FIG. 48 and 49 show cytokine release after cells were co-cultured
for 24 hrs in cell
cultures. There are limited amounts of TNF-a, IFN-y, GZMB released in the
control group. The
coupled CAR group (CD19 CAR T cells and ACPP CAR T cells) and single CAR group
(CD19
CAR T cells or ACPP CAR T cells) were labeled with solid line and dotted line,
respectively. The
levels of TNF-a, IFN-y, GZMB released are similar in the absence of PBMC. When
PBMC was
added, the amount of TNF-a, IFN-y, GZMB released increased. IL6 was mainly
secreted by
PBMC, and the amount of released cytokines was enhanced in the coupled CAR
group in the
presence of PBMC.
Table 13: CAR T cells and substrate cells used in Group 3
Coupled CAR T cells CD19 CAR T cells & CLDN 18.2 CAR T cells
CAR T Cell ID CAR Substrate Cell Note
1234 CD19 CAR K19 CD19 positive Cell
6503 CLDN 18.2 CAR KATO3 CLDN 18.2 positive cell
[00355] FIG. 50 provides additional histograms showing CD137 expression in
various cell
cultures. Peripheral blood of healthy volunteers was collected on Day 0. CD3+
T cells were sorted
using Pan T kits, and CD3/CD28 Dynabeads were added at a 1:1 ratio. On Day 1,
CD3+ T cells
were transfected with lentivirus encoding CD19 CAR and CLDN 18.2 CAR,
respectively. The
binding domains of CD19 CAR and CLDN 18.2 CAR include SEQ ID NOs: 5 and 437,
respectively. On Day 2, the lentivirus and the Dynabeads were removed, and
fresh media were
added. On Day 7, CAR T cells and target cells were cocultured for 24 or 48
hours and various
assays were performed on Day 8. As shown in FIG. 50, the activation of the
CLDN18.2 CAR T
cells was higher, and the activation was enhanced in the presence of PBMC.
These results show
that activation of CD19 CAR T cells by K19 can indirectly activate CLDN18.2
CAR T cells, and
this effect is enhanced by PBMC.
[00356] FIG. 51 shows results of flow cytometry analysis of various CAR T
cells cocultured
with KAT03+ cells for 48 hours. It can be seen from the histograms that the
level of activation of
CD19 CAR T cells in the coupled CAR T group (CD19 CAR T cells and CLDN 18.2
CAR) was
higher than in the single CAR T group (CD19 CAR T cells or CLDN 18.2 CAR) in
the presence of
KAT03+ cells. The level of activation of CD19 CAR T cells was higher after
being activated in the
presence of PBMC (e.g., the ratio of CD25 and CD137), indicating that CD19 CAR
T cells can be
activated by activation of CLDN18.2 CAR T cells by KAT03+ cells, which was
enhanced by
106
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
PBMC. CD4OL is mainly expressed by CD4 T cells (interacting with CD40L+ cells
in PBMC, such
as B cells, activated monocytes, DC). The results show that activation of
CLDN18.2 CAR T cells
by KAT03+ cells can up-regulate the expression of CD4OL of CD19 CAR T cells,
which can
activate B cells and mononuclear cells. This effect was enhanced by PBMC.
[00357] FIG. 52 shows the activation of PBMC and monocyte in the systems
described in FIG.
50. h19CAR is a humanized CD19 CAR, and the groups include (1) PBMC alone, (2)
PBMC and
K19, (3) PBMC and CD19 CART cells, (4) PBMC, K19, and CD19 CART cells. As
shown in FIG.
52, last column of PBMCs shows activation, indicating that activation of the
CART cells is capable
of activating PBMC.
[00358] FIG. 53 and 54 show that activation of CLDN18.2 CAR T cells induces
CD19 CAR T
cells to release intracellular IFNy. Similar to those in FIGS. 39 and 39, the
amount of IFNy
released in the coupled CAR T cell group (CD19 CAR T cells and CLDN 18.2 CAR)
was more
than that of the single type CAR T cell group (CD19 CAR T cells or CLDN 18.2
CAR), and the
addition of PBMC can upregulate this effect.
[00359] FIG. 55 shows killing assays for various cell cultures. The
starting amount of both
substrate cells is 2.0x105/600u1 or 3.33x105/ml. FIG. 55 shows the cell
density of the substrate
cells after three days of killing. PBMC helped the killing of the substrate
cells, and the coupled
CAR T cell group (CD19 CAR T cells and CLDN 18.2 CAR) enhanced the killing
effect of CD19
CAR T cells alone or CLDN18.2 CAR T cells alone. In the presence of PBMC, the
coupled CAR
T cells had better killing effects, demonstrating that the activated CAR T
cells can activate PBMC
and further activate another type of CAR T cells in the coupled CAR T cell
group to release
cytokines and enhance the efficacy when a type of CAR T cells in a coupled CAR
T system is
activated.
[00360] FIG. 56 shows the proliferation of CLDN18.2 CAR T cells. Various
cells were cultured
on Day 7, and flow cytometry assays were performed on Day 8. Further, CFSE
reaction was
measured to evaluate levels of proliferation. As shown in FIG. 56, the first
row is the experimental
group comprising coupled CAR co-cultured with two substrate cells, and the
second row is the
control group comprising CLDN18.2CAR co-cultured with two substrate cells.
FIG. 56 shows that
the activation of CD19 CAR T cells with K19 can induce the proliferation of
CLDN18.2CAR T
cells. KATO3 cells can be effectively activated by CLDN18.2 CAR T cells and
then were
proliferated. The presence of PBMC can further enhance proliferation. The
results demonstrate
that CD19 CAR is efficiently activated by K19 in the coupled CAR group and
activated CD19
CART can activate CLDN18.2 CAR T cells to promote proliferation CLDN18.2
cells, which can
be further enhanced by the PBMC.
[00361] FIG. 57 shows proliferation of CD19 CAR T cells. Various cells were
cultured on Day
7, and flow cytometry assays were performed on Day 8. Further, CFSE reaction
was measured
to evaluate the levels of proliferation. Further, CFSE reaction was measured
to evaluate the levels
of proliferation. As shown on FIG. 57, the first row is the experimental group
comprising couple
107
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
CAR T cells co-cultured with two substrate cells, and the second row
comprising the control group
CD19 CAR T cells co-cultured with two substrate cells. FIG. 57 shows that
activation of CLDN18.2
CAR T cells with KAT03+ cells can induce the proliferation of CD19 CAR T
cells. The fifth and
sixth columns show that PBMC can further enhance proliferation of CD19 CART
cells. The results
demonstrate that CLDN18.2 CAR T cells were activated by KAT03+ cells in the
coupled CAR
group and activated CLDN18.2 CAR T cells can activate CD19 CAR T cells to
promote the
proliferation of CD19 CAR T cells, which can be further enhanced by PBMC.
[00362] FIGS. 58-60 show cytokine release in various cell cultures. Various
cells were cultured
on Day 7, and flow cytometry assays were performed on Day 8. As shown, limited
amounts of
IL12, IFNy and GZMB were released in the control group. The coupled CAR T cell
group and
single CAR T cell group were labeled with solid line and dotted line,
respectively. The amount of
IL12, I FNy and GZMB released is similar in the absence of PBMC. When PBMC was
added, the
amount of 1L12, IFNy and GZMB released increased.
Table 20: CAR T cells and substrate cells used in Group 4
Coupled CAR T cells CD19 CAR T cells & ACPP CAR cells
CAR T Cell ID CAR Substrate Cell Notes
6404 BCMA CAR 8226 BCMA positive Cell
6701 GUCY2C CAR T84 GUCY2C positive cell
[00363] FIG. 84 shows other histograms of CD137 expression in various cell
cultures.
Peripheral blood of healthy volunteers was collected on Day 0. CD3+ T cells
were sorted and
collected using Pan T kits, and CD3/CD28 Dynabeads were added at a 1:1 ratio
to the collected
CD3+ T cells. On Day 1, CD3+ T cells were transfected with lentivirus encoding
BCMA CAR and
GUCY2C CAR, respectively. The binding domains of CD19 CAR and ACPP CAR include
SEQ
ID NOs: 60 and 488, respectively. On Day 2, the lentivirus and the Dynabeads
were removed,
and fresh media were added. On Day 7, CAR T cells and target cells (e.g.,
8226) were co-cultured
for 24 hours and various assays were performed on Day 8. Flow cytometry assays
were
performed, and the results show expression of CD19 CAR and ACPP CAR T cells.
As shown in
FIG. 84, the activation of the GUCY2C CAR T cells was higher, and the
activation was increased
in the presence of PBMC. These results show activation of BCMA CAR T cells by
8226 can
activate GUCY2C CAR T cells, and this effect is enhanced by PBMC. Since PBMC
includes B
cells and plasma cells that include BCMA, PBMC can activate BCMA CAR T cells.
The activation
of BCMA CAR T cells by PMBC is enhanced by GUCY2C CAR T cells.
[00364] FIG. 85 shows the proliferation of GUCY2C CAR T cells. Various
cells were cultured
on Day 7, and flow cytometry assays were performed on Day 8. Further, CFSE
reaction was
measured to evaluate levels of proliferation. PMBC includes B cells and plasma
cells, which
include BCMA. As shown in FIG. 85, the activation of BCMA CAR T cells with
PMBC can induce
the proliferation of GUCY2C CAR T cells.
108
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
[00365] FIG. 86 shows cytokine release after cells were co-cultured for 24
hrs in cell cultures.
There are limited amounts of IL-6, I FN-y, GZMB released in the control group.
The levels of IL-6
and GZMB released are similar in the absence of PBMC. When PBMC was added, the
amount
of IL-6 and GZMB released increased. The amount of released cytokines was
enhanced in the
coupled CAR group in the presence of PBMC.
[00366] NY-ESO-1 transduced T cells (NYESO-1 TORTS or 8302) and AFP transduced
T cells
(AFP TORTS or DW105) were mixed with 0D19 CAR T cells (1234), respectively,
and co-cultured
with various corresponding target cells (e.g., K19: K562-0D19). FIG. 78
illustrates the
determination of phenotype and expression of a gene of interest using flow
cytometry. After mixed
cells were co-cultured for 7 days, flow cytometry was used to detect the
phenotype of the cells
and the expression of the gene of interest. For example, an approximate range
of live cells was
delineated (A), the adhesion cells were removed (B), DAPI staining was
performed to delineate
the living cell population (C), and the 0D3-positive cell population (i.e., T
cells) was delineated
(D). Flow cytometry was used to determine the cell phenotype and CAR
expression. For NT (T
cells not expressing CAR) and 0D19 CAR T Groups, 0D8 percentages of NYESO-1
TORTS and
AFP TORTS were 70.32%, 56.44%, 73.85% and 72.74% respectively. 0D19 CAR
expression
was 63.71%, the expression of NYESO-1 TOR was 88.80%, and the expression of
AFP TOR was
71.61%. The expression phenotypes of the cells were normal; the expression of
CD137 was low;
and the cells were already in a resting state, which could be used for
subsequent experiments.
[00367] FIG. 79 shows the identification of co-cultured cells using flow
cytometry. In order to
distinguish the two kinds of T cells after co-culturing, 0D19 CAR cells were
stained with VIOLET
to be labeled with purple fluorescence. Cells were divided into two groups by
flow cytometry V450-
PB channel: the positive group was 0D19 CAR cells, and the negative group was
NYES0-1/AFP
TORTS (C). The 0D3 positive population was the T cell.
[00368] FIG. 80 shows results of flow cytometry analysis on activation of
co-cultured cells
including 0D19 CAR T cells and NYESO-1 TORTS. Various groups of cells were co-
cultured for
24 hours, and activation of these cells was measured using flow cytometry. The
activation of
NYESO-1 TORTS was very low in the control group NO (1.43% MFI = 5559). The
activation of
NYESO-1 TORTS in the PC group was normally (15.02%, MFI = 23301). The
activation of
NYESO-1 TORTS in group A (2.56%, MFI = 6087) was higher than that in NO group
(See 102
and 104). The activation of NYESO-1 TORTS in group B (5.28%, MFI = 12352) was
higher than
that of group A (2.56%, MFI = 6087) (See 106 and 108). The activation of NYESO-
1 TORTS in
group C (6.80%, MFI = 12352) was higher than that in group B (5.28%, MFI =
12352) (See 110
and 112). The activation of NYESO-1 TORTS in group C was higher than in group
A (See 114
and 116).
[00369] FIG. 81 show results of flow cytometry analysis on the
proliferation of co-cultured cells
including 0D19 CAR T cells and NYESO-1 TORTS. Various groups of cells were co-
cultured for
96 hours, and the proliferation of these cells was measured using flow
cytometry. A comparison
109
CA 03125646 2021-06-30
WO 2020/146743
PCT/US2020/013099
of cell proliferation was performed. The proliferation of NYESO-1 TORTS cells
in the NC control
group was 2.46%. The proliferation of NYESO-1 TORTS cells in group A was
28.17%, which was
increased compared to the NO group (See 202). The proliferation of NYESO-1
TORTS cells of
group B was 41.60% higher than group A (See 204). The proliferation of NYESO-1
TORTS cells
of group C was 47.79%, which was higher than that of group B 41.60% (206) and
higher than that
of group A (See 208).
[00370] FIG. 82 show results of flow cytometry analysis on activation of co-
cultured cells
including 0D19 CAR T cells and AFP TORTS. Various groups of cells were co-
cultured for 24
hours, and activation of these cells was measured using flow cytometry. The
AFP TORTS of the
control group NO was not activated (0.70% MFI = 4568). The activation of AFP
TORTS of PC
group was normally (38.58%, MFI = 23327). The activation of AFP TORTS of group
A (1.24%,
MFI = 4884) was higher than that of NO group (See 302 and 304). The activation
of AFP TORTS
of group B (4.17%, MFI = 13112) was higher than that of group A (1.24%, MFI =
4884) (see 306
and 308). The activation of AFP TORTS of group C (6.47%, MFI = 14218) was
higher than that
in group B (4.17%, MFI = 13112) (see 310 and 312) and was higher than that in
group A (see 314
and 316). In addition, TOR negative T cells were also partially activated (NC
= 0.51%; A = 1.46%;
B = 2.84%; C = 5.12%). The relationship among the groups was the same as that
of the positive
part.
[00371] FIG. 83 shows results of flow cytometry analysis on the
proliferation of co-cultured
cells including 0D19 CAR T cells and AFP TORTS. Various groups of cells were
co-cultured for
96 hours, and activation of these cells was measured using flow cytometry. A
comparison of cell
proliferation was performed. The proliferation rate of AFP TORTS of the NO
control group was
3.11%. The proliferation rate of AFP TORTS of group A was 36.44%, which was
increased
compared to NO group (402). The proliferation rate of AFP TORTS of group B was
39.59%, which
was higher than 36.44% of group A (404). The proliferation rate of AFP TORTS
of group C was
51.97%, which was higher than 39.59% (406) in group B and higher than group A
(408). Thus,
0D19 CAR T cells enhance TORT cells' expansion by increasing their
proliferation rates.
[00372] These
data show that the activated first type of CAR T cells can activate the
second type of CAR T cells in coupled CAR T cells (e.g., 0D19 CAR T cells and
CLDN18.2
CAR T cells). For example, the activated first type of CAR T cells enhanced
the activation,
cytokine releases, and cell proliferation of the second type of CAR T cells.
This effect was
enhanced when PBMC was present. Given that PBMC and monocytes were activated,
the first
type of CAR T cells can activate monocytes (e.g., DCs), which can then
activate the second
type of CAR T cells. The combination of the data presented here and the data
shown in the in
vivo Examples of the present disclosure shows that subjects' dendritic cells
(DCs) work as a
medium to associate the activation of the first type of CAR T cells with the
activation of the
second type of CAR T cells and form a positive circle of activations, which
may contribute to the
expansion of CAR T cells observed in the subjects (Patients 004-011) due to
amplification of
110
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
immune homeostasis. These data and the clinical data above indicate that
coupled or mixed T
cells achieve enhanced T cell response including the expansion of T cells
and/or cytokine
release. Examples of the coupled or mixed T cells include BCMA and GUCY2C CAR
T cells as
well as CD19 CART cells and NYESO-1 TORTS. After infusion of the mixed T cells
to patients,
a first group of T cells (e.g., CD19 and BCMA CAR T cells) bind an antigen of
B cells and are
activated. After their activation, the first group of CAR T cells up-regulates
certain membrane
molecules (e.g., CD28, 0X40, 4-1BB, CD4OL, etc.) and release certain cytokines
(e.g., IFNy
and GM-CSF). These surface molecules and cytokines activate and/or recruit
cells such as
monocytes (e.g. DCs) and neutrophils. The recruited and/or activated cells
release cytokines
(e.g., TNFa, IL6, IL12) to form an inflammatory-like environment. In light of
the inflammatory-like
environment, these activated immune cells up-regulate some proteins (e.g.,
CD80, CD80, and
CD40), which activate a second group of T cells (e.g., NT, CAR T cells
targeting solid tumors,
and NYESO-1 TORTS). Also, cytokines (e.g., IFNy) secreted by the first group
of T cells also
activate the second group of T cells.
Example 4. Modified Cells using ZFNs. TALENs, and/or Cas9
[00373] Multiple gene-specific ZFNs were constructed to enable the site-
specific introduction
of mutations. Various ZFNs were designed and incorporated into plasmid vectors
essentially as
described in Mala et al. (2005) Biochem Biophys Res Commun 335(2):447-57, Liu
et al. (2002) J
Bio Chem 277(6):3850-6, Sander et al. (2011) Nat Methods. 8(1):67-9, Urnov et
al. (2005) Nature
435(7042):646-651, and U.S. Patent Publication 2008/0131962. The ZFNs included
various
combinations of Zinc finger binding domains (e.g., ZFN-left and ZFN-right
binding domains),
which are listed in Table 14 and Table 15. The cleavage domain of the ZFNs
comprised an
engineered Fokl cleavage domain (SEQ ID NOs: 280, 281, or 282).
Table 14: Exemplary ZFN pairs as well as the target sequences (Target site:
the target sequence
for ZFN include two 9-bp recognition sites (i.e., upper case letters) are
separated by a 6-bp
spacer)
SEQ SEQ SEQ
Gene ICT ZFN Target site.
SEQ
ZFN Finger 1 ID Finger 2 ID Finger 3 ID
ID
Name Number
NO: NO: NO: NO:
ICTZF1L ZFN-L QSGSLTR 301 DRSDLTR 302 QSSALTR 303 TGCGGCAA 304
CT LA4 CctacatGATG
ICTZF1R ZFN-R QRSNLVR 305 RSDHLTR 306 TSANLSR 307 GGGAA
ICTZF2L ZFN-L QSSDLTR 308 RSDNLAR 309 QSGHLQR 310 AGCCTCTCC 311
LAG3 agccaGGGG
ICTZF2R ZFN-R RSDNLAR 309 QSSDLTR 308 RSDHLSR 312 CTGAG
ICTZF3L ZFN-L RSDHLAR 313 QSGDLTR 314 RSDDLTR 315 CCCTGCCG 319
LAG3 CctgcctGCTG
ICTZF3R ZFN-R RSDALTR 316 TSGHLVR 317 QSSDLQR 318 GTGTG
ICTZF4L ZFN-L GGTALRM 320 QRSSLVR 321 QRNNLGR 322 GTTTACTTC 326
BTLA ctttggGGGGA
ICTZF4R ZFN-R RKRNLIM 323 VRHNLTR 324 RGDKLGP 325 TTGC
ICTZF5L ZFN-L SRFTLGR 327 RREHLVR 328 QTATLKR 329 CACCCCTG 333
TIM3 õ, CaccgacTCG
ICTZF5R ZFN-R KHSNLAR 330 QSTTLKR 331 RADGLQL =-)=-)4 GCAGAG
111
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
ICTZF6L ZFN-L TRQKLET 334 RQDNLGR 335 QQHGLRH 336 ACCCTCACA 340
TIM3 accttTGGGTT
ICTZF6R ZFN-R TKKILTV 337 HKSSLTR 338 RSDHLSL 339 GTC
ICTZF7L ZFN-L RKHHLGR 341 RREHLVR 28 VSNSLAR 342 CCCCCCAG 346
FOXP3 CacccttTCGG
ICTZF7R ZFN-R RTSSLKR 343 QRSDLTR 344 RSDGLRG 345 CTGTG
ICTZF8L ZFN-L DSPTLRR 347 QSAHLKR 348 QDVSLVR 349 GGCTCCTG 352
FOXP3 CtgcatcGTAG
ICTZF8R ZFN-R MKNTLTR 350 QRSDLTR 344 QSGTLTR 351 CTGCT
ICTZF9L ZFN-L TKQILGR 353 QSTTLKR 331 RTEHLAR 354 AGCTGCCC 296
SIVA1 ,õ CttcgcgGACG
ICTZF9R ZFN-R KRRDLDR 355 RREVLEN 300 DPSNLRR L" TGGCC
ICTZF10 298 324 297
ZFN-L MKHHLDA VRHNLTR HHNSLTR ACCATCAAC
293
LGALS L
agactgGAAG
9 ICTZF10 295 300 294
ZFN-R KKDHLHR RREVLEN QTVNLDR TGGGG
R
ICTZF11 301 292 312
289
ZFN-L QSGSLTR RSDDLQR RSDHLSR TGCCGCCC
L
CD33 CtactgtGGAG
ICTZF11 291 290 310
ZFN-R QSGHLAR TSGNLVR QSGHLQR ATGGA
R
1CTZF12 308 314 312
285
ZFN-L QSSDLTR QSGDLTR RSDHLSR AGCTGCCC
L
SIVA1 CttcgcgGACG
ICTZF12 288 287 286
ZFN-R ERGTLAR RSDALSR DRSNLTR TGGCC
R
ICTZF13 288 310 314 GGCTCCTG 283
ZFN-L ERGTLAR QSGHLQR QSGDLTR
L CtgcatcGTAG
FOXP3 308 308 284 CTGCT
ICTZF13
ZFN-R QSSDLTR QSSDLTR QRASLTR
R
[00374] ZFN-left arm plasmid vectors and ZFN-right arm plasmid vectors were
transfected into
Hela cells using fugene transfection reagent, respectively. 24 hours after
transfection, Hela cells
were treated with 1 pg/ml puromycin for 48 hours to obtain cells rich in ZFNs.
Hela cells were
then collected. Lysed DNA fragments containing ZFNs were amplified by PCR
using primers
specific to various genes (i.e., CTLA4, LAG3, BTLA, TIM3, FOXP3, SIVA1, or
LGALS9) and the
genome of Hela cells as templates. The DNA fragments were sequenced using
forward primers.
The DNA fragments were cloned into vectors. The DNA fragments of about 30
monoclonal cells
were sequenced to determine whether the DNA fragments include mutations. The
results of the
sequencing are shown in Table 15.
Table 15: Monoclonal sequencing results for ZFNs that target gene fragments,
which are
amplified by PCR.
ZFN Number of clones
Number of clones
Mutation frequency
Left Right analyzed mutated
ICTZF1L ICTZF1R 31 6 19%
ICTZF2L ICTZF2R 26 3 12%
ICTZF3L ICTZF3R 30 9 30%
ICTZF4L ICTZF4R 30 4 13%
112
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
ICTZF5L ICTZF5R 29 1 3%
ICTZF6L ICTZF6R 30 4 13%
ICTZF7L ICTZF7R 29 5 17%
ICTZF8L ICTZF8R 33 7 21%
ICTZF9L ICTZF9R 34 10 29%
ICTZF1OL ICTZF1OR 27 2 7%
ICTZF11L ICTZF11R 34 14 41%
I0TZF12L I0TZF12R 24 3 13%
I0TZF13L I0TZF13R 21 5 24%
[00375] Multiple gene-specific ZFNs were constructed to enable site-
specific introduction of
mutations. Various ZFNs were designed and incorporated into plasmid vectors
essentially as
described (Mala et al. (2005) Biochem Biophys Res Commun 335(2):447-57; Liu et
al. (2002) J
Bio Chem 277(6):3850-6; Sander et al. (2011) Nat Methods. 8(1):67-9; Handel et
al. (2009) Mol
Ther. Jan;17(1):104-11; Urnov et al. (2005) Nature 435(7042):646-651; and U.S.
Patent
Publication 2008/0131962; which are incorporated by reference in their
entireties). The ZFNs
included various combinations of Zinc finger binding domains (e.g., ZFN-left
and ZFN-right
binding domains), which are listed in Table 16. The cleavage domain of the
ZFNs comprises an
engineered Fokl cleavage domain (SEQ ID NOS.: 96, 97, or 98).
Table 16: Exemplary ZFN pairs as well as the target sequences (Target site:
the target sequence
for ZFN include two 9-bp recognition sites (i.e., uppercase letters) separated
by a 6-bp spacer).
Gene ICT Linker between
ZFN Finger 1 Finger 2 Finger 3 Target site
Name ZFN # ZFP and Fok1
QRSDLT TGPGAAARA
RTSSLKR RSDHLSL
ICTZF R (SEQ ID NO: 417)
ZFN-L (SEQ ID (SEQ ID CACAGCCC
14L
NO: 357) NO: 359) AagatagtTAA
(NS0E:Q358)ID
B2M GTGGGG
KKDHLH QRGNLN TGPGAAARA
RREVLEN (SEQ ID NO:
ICTZF R M (SEQ ID NO: 417)
ZFN-R (SEQ ID 360)
14R (SEQ ID No: 362\ (SEQ ID
NO:361) I NO: 363)
QDGNLT LRGSQ (SEQ ID
VPSKLKR EAHHLSR
ICTZF R
ZFN-L (SEQ ID (SEQ ID GCCACCTT NO.= 418)
15L (SEQ ID
NO: 364) NO: 265) NO: 366) CccccaGCTG
AAGTC
QQTNLT LRGSQ (SEQ ID
TSTLLNR VGNSLTR (SEQ ID NO:
ICTZF R NO: 418)
ZFN-R (SEQ ID (SEQ ID 400)
15R (SEQ ID
NO: 367) NO: 369)
CIITA NO: 368)
QSGSLT QSGDLT LRGSQ (SEQ ID
ZFN -L
TSGNLVR
ICTZF R R TGCATCTG NO: 418) 16L
(SEQ ID (SEQ ID NO: 371) (SEQ ID CgacgtgGGA
NO: 370) NO: 372) GCCGAG
DRSDLTR (SEQ ID NO: LRGSQ (SEQ ID
ICTZF RSDNLA QSGHLQ
ZFN-R (SEQ ID 0 401) NO: 418)
16R R
NO: 374) "
113
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
(SEQ ID (SEQ ID
NO: 373) NO: 375)
QSSDLQ LRGSQ (SEQ ID
DRSHLTR RSDALSR
ICTZF
ZFN-L (SEQ ID (SEQ ID GCCCACAG NO: 418)
17L (SEQ ID
NO: 376) NO: 377) NO: 378) CcactcGTGG
CGGCC
RSDDLQ LRGSQ (SEQ ID
ERGTLAR RSDALTR (SEQ ID NO:
ICTZF NO: 418)
ZFN-R (SEQ ID (SEQ ID 402)
17R (SEQ ID
NO: 379) NO: 381)
NO: 380)
RSDHLA QSGDLT QSGHLQ LRGSQ (SEQ ID
ICTZF
ZFN-L CCCTGCTC NO: 418)
18L (SEQ ID (SEQ ID (SEQ ID
NO: 382) NO: 383) NO: 384) CctccgGGGG
CTGCT
RSDHLS LRGSQ (SEQ ID
QSSDLTR QSSDLTR (SEQ ID NO:
ICTZF RNO: 418)
ZFN-R (SEQ ID (SEQ ID 403)
18R (SEQ ID
NO: 385) NO: 385)
NO: 387)
QSGHLA QSG N LA LRGSQ (SEQ ID
DRSDLTR
ICTZF
ZFN-L (SEQ ID TCCGGCTT NO: 407)
19L (SEQ ID (SEQ ID
NO: 389) CtccatGGAG
NO: 388) NO: 390)
CAGGC
QSGDLT QSGHLQ LRGSQ (SEQ ID
DRSHLTR (SEQ ID
ICTZF R RNO: 418)
ZFN-R (SEQ ID NO:404)
19R (SEQ ID (SEQ ID
NO: 391)
NO: 392) NO: 393)
DGG H LT LRGSQ (SEQ ID
RSH I LTN QSTTLKR
ICTZF
ZFN-L (SEQ ID (SEQ ID CACTGCGC NO: 418)
20L (SEQ ID
NO: 394) NO: 395) NO: 396) CcacgagGCC
GAGGAG
RQMNLD RQDNLG LRGSQ (SEQ ID
DKSVLAR (SEQ ID NO:
ICTZF R R NO: 418)
ZFN-R (SEQ ID 405)
20R (SEQ ID (SEQ ID
NO: 399)
NO: 397) NO: 398)
[00376] ZFN-left arm plasmid vectors and ZFN-right arm plasmid vectors were
transfected into
Hela cells using fugene transfection reagent, respectively. 24 hours after
transfection, Hela cells
were treated with 1 pg/ml puromycin for 48 hours to obtain cells rich in ZFNs.
Hela cells were
then collected. Lysed DNA fragments containing ZFNs were amplified by PCR
using primers
specific to the various genes (i.e., B2M and CIITA) and the genome of Hela
cells as templates.
The DNA fragments were sequenced using forward primers. The DNA fragments were
cloned
into vectors. The DNA fragments of about 30 monoclonal cells were sequenced to
determine
whether the DNA fragments include mutations. The results of the sequencing are
shown in Table
18. T cells were introduced with TRAC-specific ZFNs constructed to enable the
site-specific
introduction of mutations at TRAC gene. Various ZFNs were designed and
incorporated into
plasmids vectors essentially as described (Urnov et al. (2005) Nature
435(7042):646-651;
Lombardo et al. (2007) Nat Biotechnol. November, 25(11):1298-306; and U.S.
Patent Publication
2008/0131962; which are incorporated by reference in their entireties). The
ZFNs included
various combinations of Zinc finger binding domains (e.g., ZFN-left and ZFN-
right binding
domains), which are listed in Table 17. The cleavage domain of the ZFNs
comprises a Fokl
114
CA 03125646 2021-06-30
WO 2020/146743
PCT/US2020/013099
cleavage domain (SEQ ID NOs: 96, 97, or 98). mRNA encoding a pair of ZFNs (see
Table 17)
was introduced into the transduced cells to modify a target genomic locus
associated with the a
chain of TCR.
Table 17
Recognition
ZFN F1 F2 F3 F4 F5 F6
Sequence
GTTGCTCCAGG QSGDLT QWGTRY ERGTLA RSDNLR QSGDLT TSGALT
ZFLm1
CCACAGCA R (SEQ R (SEQ R (SEQ E (SEQ R (SEQ R (SEQ
(left)
(SEQ ID: 408) ID: 409) ID: 410) ID: 411) ID: 386) ID:
412) ID: 413)
ZFRm WRSSLA QSGSLTR HKVVVLR DRSNLT
GACTTTGCATG
1-4 S (SEQ (SEQ ID: Q (SEQ R (SEQ
(right) T (SEQ ID: 414)
ID: 415) 416) ID: 406) ID: 407)
[00377] TALENs for CIITA were designed to target exon 2 (2L1:
gctgaccccctgtgcct (SEQ ID
NO: 426); 2L2: gaccccctgtgcctct (SEQ ID NO: 427); 2R1: ctccagccaggtccatct (SEQ
ID NO: 419);
2R2: tctccagccaggtccat (SEQ ID NO: 420)) and exon 3 (3L1: tcagcaggctgttgt (SEQ
ID NO: 421);
3L2: tcagcaggctgttgtgt (SEQ ID NO: 422); 3R1: ccctggtctcttcat (SEQ ID NO:
423); 3R2:
aagcctccctggtctt (SEQ ID NO: 424); 3R3: aagcctccctggtct (SEQ ID NO: 425)). The
TALENs were
constructed using the FastTALE TALEN Assembly Kit (Sidansai), and their
activities were
confirmed in 293T cells as previously described. The constructed TALENs were
transfected into
293T cells and selected with 2 pg/ml puromycin (Sigma). The genomic DNA of
293T cells was
harvested after selection. Subsequently, PCR and sequencing were performed to
examine the
efficiency of the TALENs. The plasmids expressing Cas9 and gRNA were co-
transfected into
293T cells with fugene transfection reagent. After 72 hours, 293T cells were
collected and the
expression of B2m and HLA proteins was detected by flow cytometry.
Table 18: Monoclonal sequencing results for ZFNs that target various gene
fragments, which are
amplified by PCR.
ZFN Number of clones Number of clones Mutation
Left Right analyzed mutated
frequency
I0TZF14L I0TZF14R 33 10 30%
I0TZF15L I0TZF15R 26 3 12%
I0TZF16L I0TZF16R 31 7 23%
I0TZF17L I0TZF17R 35 3 9%
I0TZF18L I0TZF18R 27 6 22%
I0TZF19L I0TZF19R 32 9 28%
ICTZF20L ICTZF20R 30 1 3%
[00378] All publications, patents and patent applications cited in this
specification are
incorporated herein by reference in their entireties as if each individual
publication, patent or
patent application were specifically and individually indicated to be
incorporated by reference.
115
CA 03125646 2021-06-30
WO 2020/146743 PCT/US2020/013099
While the foregoing has been described in terms of various embodiments, the
skilled artisan will
appreciate that various modifications, substitutions, omissions, and changes
may be made
without departing from the spirit thereof.
116