Note: Descriptions are shown in the official language in which they were submitted.
CA 03137198 2021-10-18
FXR SMALL MOLECULE AGONIST AND PREPARATION METHOD THEREFOR
AND USE THEREOF
Technical field
The present invention relates to the field of medicine, and relates to a class
of
compounds as FXR agonist and preparation therefor and use thereof
Specifically, it relates
to a class of non-steroidal compounds that can be used as FXR agonist and the
enantiomer,
diastereomer, tautomer, solvate, prodrug, or pharmaceutically acceptable salt
thereof, the
preparation method therefor and use thereof in the manufacture of a medicament
for the
treatment of FXR-related disease.
Background technique
Nuclear receptors are widely present in organisms and are a type of nuclear
transcription
regulators that rely on specific ligand activation. Metabolic nuclear
receptors are a type of
nuclear receptors that regulate substance metabolism, cell proliferation, and
apoptosis in the
body. Farnesoid X receptor (FXR) is a member of the nuclear receptor
superfamily, which
was first discovered by Foman et al. in 1995 and named because its
transcriptional activity
can be enhanced by farnesoid.
The FXR structure contains ligand-independent transcription activation
function domain
(AF1) at amino-terminal, DNA binding domain (DBD), hinge region, ligand
binding domain
(LBD) and ligand-dependent transcription activation function domain (AF2) at
carbon-terminal, which is a typical nuclear receptor structure. FXR is
activated by bile acids
in the body and participates in the processes of bile acid metabolism, lipid
metabolism, and
sugar metabolism in the living body. The mechanism by which FXR regulates bile
acid
metabolism and transport is mainly accomplished by regulating the
transcription of
cholesterol 7a-hydroxylase (CYP7A1) which is a rate-limiting enzyme of bile
acid synthesis.
Although FXR cannot directly act on the CYP7A1 promoter, it can induce the
expression of
small heterodimer partner (SHP) and combine HNF-4a (hepatocyte nuclear factor
4a) and
LRH-1 (liver receptor homolog) to down-regulate the transcription of CYP7A1.
In the
process of lipid metabolism, FXR in the liver regulates lipid metabolism and
transport to
reduce plasma free fatty acids and triglycerides by directly or indirectly
regulating PPARa,
VLDL receptor (very low density lipoprotein receptor, VLDLR), proprotein
convertase
subtilisin kexin type 9 (PCSK9), scavenger receptor group B type 1 (SRB1),
phosphor lipid
transfer protein (PLTP), liver X receptor (LXR), sterol regulatory element-
binding
protein-1C (SREBP-1C) and fatty acid synthetase (FAS), and activating
lipoprotein lipase
(LPL) and the like. In the process of glucose metabolism, the activation of
FXR can promote
liver glycogen synthesis and increase insulin sensitivity and insulin
secretion to control blood
glucose levels in the body. Since FXR plays an important role in the processes
of bile acid
metabolism, lipid metabolism and glucose metabolism, FXR ligand small molecule
compounds are expected to be used as new medicament for the treatment of
hypertriglyceridemia, type 2 diabetes, metabolic syndrome, NAFLD and other
¨1 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
metabolic-related diseases.
Summary of the invention
The object of the present invention is to provide a FXR small molecule agonist
and
preparation method therefor and use thereof.
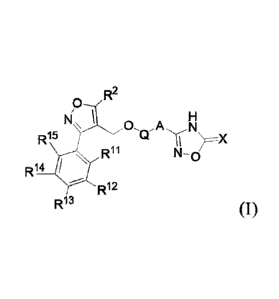
In the first aspect of the present invention, it provides a compound
represented by
general formula I, or enantiomer, diastereomer, tautomer, racemate, solvate,
prodrug or
pharmaceutically acceptable salt thereof,
R2
9
R15 \X
R11 N-0/
R14
R12
R13 (I)
wherein, R", R12, R13, R14 and Ri5
are each independently hydrogen, halogen,
halogenated C1-6 alkyl, halogenated C1-C6 alkoxy, C1-C6 alkyl, C1-6 alkoxy, C3-
C6 cycloalkyl,
C3-C6 cycloalkoxy, cyano or nitro;
R2 is C6-C12 aryl, C1-C6 alkyl or C3-C6 cycloalkyl;
Q is a 4-8 membered heterocyclyl;
A is the following substituted or unsubstituted group: phenyl, pyridyl,
thienyl, furyl,
indazolyl, indolyl, benzothienyl, benzofuranyl, and the "substituted" means
that there is one,
two or three substituents selected from the group consisting of halogen, Ci-C6
alkyl,
halogenated C1-6 alkyl, halogenated C1-C6 alkoxy, C3-C6 cycloalkyl, C1-C6
alkoxy, and C3-C6
cycloalkoxy;
Xis 0 or S.
In another preferred example, R", R12, R13, R14 and K-15
are each independently
hydrogen, fluorine, chlorine, bromine, methyl, ethyl, n-propyl, isopropyl, n-
butyl, isobutyl,
trifluoromethyl, or trifluoromethoxy.
In another preferred example, R12, R13 and R14 are hydrogen.
In another preferred example, R" and R15 are each independently hydrogen,
chlorine,
bromine, trifluoromethyl, or trifluoromethoxy.
In another preferred example, R2 is phenyl, methyl, ethyl, n-propyl,
isopropyl, n-butyl,
isobutyl, cyclopropyl, cyclobutyl or cyclopentyl.
In another preferred example, Q is a 4-8 membered nitrogen-containing
heterocyclyl or
a 4-7 membered nitrogen-containing heterocyclyl.
¨ 2 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
N¨ 140,
In another preferred example, Q is /,
or
In another preferred example, A is the following substituted or unsubstituted
group:
phenyl, pyridyl, thienyl, furyl, indazolyl, indolyl, benzothienyl,
benzofuranyl, and the
"substituted" means that there is one or two substituents selected from the
group consisting
of fluorine, chlorine, bromine, CI-C4 alkyl, C3-C6 cycloalkyl, Ci-C4 alkoxy,
and C3-C6
cycloalkoxy.
In another preferred example, A is the following substituted or unsubstituted
group:
phenyl, pyridyl, thienyl, fury!, indazolyl, indolyl, benzothienyl,
benzofuranyl, and preferably,
A is the following substituted or unsubstituted group: phenyl, pyridyl,
indolyl, and the
"substituted" means that there is one or two substituents selected from the
group consisting
of fluorine, chlorine, bromine, trifluoromethyl, trifluoromethoxy, methyl,
ethyl and propyl.
In the present invention, when there are two or more substituents, each
substituent is the
same or different.
,/sAisL
)55`-N glik Alb A
In another preferred example, A is 'W"-- or F .
In another preferred example, the pharmaceutically acceptable salt in the
present
invention refers to a salt formed from inorganic acid such as phosphoric acid,
sulfuric acid,
hydrochloric acid, etc., or from organic acid such as acetic acid, tartaric
acid, citric acid,
malic acid, etc., or from acidic amino acid such as aspartic acid, glutamic
acid, etc.; or a salt
formed from inorganic base, such as sodium, potassium, calcium, aluminum and
ammonium
salts.
In another preferred example, the compound is:
0_(:)0 HN
14, \ NH 0
' 0\
N
--ON 10
-N
CI CI r CI HN33
0
0
2 3
¨ 3 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 N-o 9 µ
N, ' C:)."-CN * p \ 0.,N
* /1,1,0
= iNo CI CI N,
HN--0
H CI CI
--- N
CI CI HN i
6
6
0 0 ,
14, \ / NC), \ O-CN 0 ,
-ON * NI .0, N N
CI ca HN-- CF3 / '0 OCF3 / '0
0 HN4 FIN4
0 0
7 8 9
0
= * 9
N, \ 0
N --CAN * 9 \ 0--,7-Th
N,,
CI CI , '0
CI "N`,::, V_.../N 1110, ,,,
/-0
HNA CI 0 EN-'I CI EN-'IHN4
0 0
11 12
= N--
_s,,,14,0
CI CI
HN-A) HN 4 HN4
0 0
13 14 15
0 0 \ 0 F
4 = \ CICAN F
--Cis'IN 1 F , \ 00 *
N ,
N
/ '0 N
CI a a 1 a ci
HNA
0
F FIN4
H N4
0
16 17 18
P o
\ NH-0 -.0- N o
CI N
a ICIN .
\ CLç.c1
HN4 HNA
S 0
\
19 20 21
N, --0...0,- .N , I
N, , \
N, F
-0 # N 0 * N
CF3 / '0 / '0
11N4 CF3 CF
0 HN4 FIN4
22 23 0 24 0
0 0
-CN * F N, --C\N N---
CF3 /N '0 ......eN ,0
OCF3
HN4 OCF3 HN-- 27 HN-
F 0 26 0 0
140, \ 0 N 10 N F 140, \ 0 0 to F N rµi , \ N F
N
--O
OCF3 0CF3 OCF3
FIN-4 FIN4 F
28 0
0 29 0
- 4 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 0 \ 0
N ,
' \ C)---'Z"\N N- ,,
F F
FINA HN-A) HN-
31 32 33 0
0 0
N, F
F \ z ,N,0
a CI /
'0
OCF3 F
HN4 0 L.J
HN4 HN4
0
0
34 35 36
0 0
1 \ 0 F 1.1` \ O.-(C\N F
N', \
,N,0 N-
/ '0
CF3 OCF3 HN4 OCF2H
HN4 HN4
o 38 o 39
0
37
o 9 \ F F
N' N
OCF2H, \
\
--CCN--. NIC_ N., --"C\N * N ,
/N -0 / -
0
HN4 HN or 42
A HN4
40 0
41 0
The compound of the present invention has an asymmetric center, a chiral axis,
and a
chiral plane, and can exist in the form of racemate, R-isomer, or S-isomer.
Those skilled in
the art can use conventional technical means to obtain R-isomer and/or S-
isomer from
racemate resolution.
In the second aspect of the present invention, it provides a method for
preparing the
compound according to the first aspect, which includes the following steps:
R2 R2 R2
9 o-
\ 9 ,
N, 0_0-A N, \ 0-n-A N N\ I
H
-CN a ...' -.--.'--/ bH _,,b'
R15 0,Q, A,,N
'
R11 R15 R11 R15 H2N 11 X
Ril N-0
R12 0 R14 R12 R14 R14 1 N
R13 R13 R12
VII VIII R13 (I)
(a') reacting a compound represented by general formula VII with hydroxylamine
hydrochloride to produce a compound represented by general formula VIII;
(b') reacting the compound represented by the general formula VIII under the
action of
phosgene, triphosgene, carbonyldiimidazole or thiocarbonyldiimidazole to
produce the
compound represented by the general formula I,
wherein, the definitions of X, R2, Q, A, R11, R12, R13, R14 and it ¨ is
are described as
above.
In another preferred example, the compound represented by general formula VII
is
- 5 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
prepared by the following steps:
R2
9H a R2
0 0
N ci
N N R I \ Br
N N
R R15 " a RR 15 11 R
RR R COOMe ii R15
Rii R15
12 Ri4
12 14 12 R14
R13 R13 R13 R12 R14
R13
II III IV V
R2
R2 0 \
0 N \ 0 A
rj \ 0-Q CN
Rii R15
R15
Riz R14
Riz R14 R13
R13
VII
VI
a) reacting substituted benzaldehyde compound represented by general formula
II as
starting materials with hydroxylamine hydrochloride to obtain an intermediate
and then
chlorinating the intermediate with N-chlorosuccinimide (NCS) to produce a
compound
represented by general formula III;
b) reacting the compound represented by the general formula III with 3-
oxopropionate
to obtain a compound represented by the general formula IV;
c) reducing the ester in the compound represented by formula IV to produce
alcohol, and
then brominating to produce a compound represented by V;
d) reacting the compound represented by general formula V with Q-OH to produce
a
compound represented by general formula VI;
e) coupling the compound represented by general formula VI with Br-A-CN under
the
catalysis of copper or palladium to obtain the compound represented by general
formula VII,
in each formula, the definitions of R2, Q, A, R", R12, R13, R14 and K-15
are described as
above.
In another preferred example, the compound represented by general formula VII
is
prepared by the following steps:
R2
R2 o
.CN
N \ Br
R11 R15
-OH + F CN HO-Q-A, ___________________________ R15 9
Q CN Riz R14
R12 W4 R13
IX R13
V VII
f) reacting Q-OH with F-A-CN to generate a compound represented by general
formula
IX;
g) reacting a compound represented by the general formula V with the compound
represented by the general formula IX to produce the compound represented by
the general
-6-
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
formula VII,
in each formula, R2, Q, A, R12, R13, R14 and K-15
are defined as above.
In the third aspect of the present invention, it provides a pharmaceutical
composition,
comprising:
the compound represented by the general formula I according to the first
aspect, or the
enantiomer, diastereomer, tautomer, racemate, solvate, prodrug, or
pharmaceutically
acceptable salt thereof; and
a pharmaceutically acceptable carrier.
The compound provided by the present invention can be used alone or mixed with
pharmaceutically acceptable auxiliary material (such as excipient, diluent,
etc.) to prepare
tablet, capsule, granule or syrup for oral administration. The pharmaceutical
composition can
be prepared according to conventional methods in pharmacy.
In the fourth aspect of the present invention, it provides use of the compound
represented by the general formula I according to the first aspect, or the
enantiomer,
diastereomer, tautomer, racemate, solvate, prodrug, or pharmaceutically
acceptable salt
thereof,
(a) as an FXR agonist;
(b) for the manufacture of a medicament for the treatment of FXR-related
diseases;
(c) to reduce the levels of ALP, ALT, AST and TBA in serum;
(d) to reduce the amount of hydroxyproline in liver tissue;
(e) to down-regulate the expression of a-SMA and Collal mRNA in liver tissue;
or
(f) to reduce the content of collagen in the liver.
In another preferred example, the FXR-related disease is a disease related to
bile acid
metabolism, carbohydrate metabolism, lipid metabolism, inflammation, and/or
liver fibrosis.
In another preferred example, the FXR-related disease is non-alcoholic fatty
liver
(NASH), primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC),
gallstone,
non-alcoholic liver cirrhosis, liver fibrosis, cholestatic liver disease,
hyperlipidemia,
.. hypercholesterolemia, or diabetes.
It should be understood that, within the scope of the present invention, each
of the above
technical features of the present invention and each of the technical features
specifically
described below (e.g., examples) can be combined with each other, thereby
forming a new or
.. preferred technical solution. Each feature disclosed in the specification
can be replaced by
any alternative feature that provides the same, equal or similar purpose. Due
to space
limitations, they will not be redundantly described herein.
Brief description of the drawings
Figure 1 shows the effect of compound 1 administered for 4 weeks on ALP in
serum,
hydroxyproline in liver, a-SMA and Coll al mRNA in liver, *P<0.05, "P<0.01,
¨ 7 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
***P<0.001, compared with the model control group (vehicle group).
Figure 2 shows the effect of compound 1 administered for 4 weeks on the
content of
collagen in liver pathological section.
Figure 3 shows the effect of compound 8 administered for 3 and 6 weeks on ALT,
AST,
TBA, and LDH levels in serum, *1"<0.05, "P<0.01, ***P<0.001, compared with the
model
control group (vehicle group).
Figure 4 shows the effect of compound 8 administered for 6 weeks on the
expression of
a-SMA and Coll al mRNA in the liver, *P<0.05, "P<0.01, ***P<0.001, compared
with the
model control group (vehicle group).
Figure 5 shows the effect of compound 8 administered for 4 weeks on the
content of
collagen in liver pathological section, *P<0.05, "P<0.01, ***P<0.001, compared
with the
model control group (vehicle group).
Detailed description
After extensive and intensive researches, the inventors of the present
application
developed a class of non-steroidal compounds that can be used as FXR agonist,
which have
the ability to agonize FXR at the molecular and cellular levels. Studies have
shown that the
compounds of the present application can reduce ALP, ALT, AST, and TBA levels
in serum,
reduce the amount of hydroxyproline in the liver tissue, down-regulate the
expression of
a-SMA and Collal mRNA in the liver tissue, and reduce the content of collagen
in the liver.
The compound of the present invention has the advantages of high FXR agonistic
activity,
simple synthesis, easy availability of raw materials, etc., and can be used
for the manufacture
of a medicament for treating FXR-related diseases. On this basis, the present
invention has
been completed.
Terms
In the present invention, the halogen is F, Cl, Br or I.
In the present invention, unless otherwise specified, the terms used have the
general
meanings known to those skilled in the art.
In the present invention, the term "CI-C6" refers to 1, 2, 3, 4, 5 or 6 carbon
atoms, and
"C-05" refers to 1, 2, 3, 4, 5, 6, 7 or 8 carbon atoms, and so on. "3-10
membered" refers to
3-10 ring atoms, and so on.
In the present invention, the term "alkyl" refers to a saturated linear or
branched
hydrocarbon moiety. For example, the term "Ci-C6 alkyl" refers to a straight
or branched
chain alkyl having 1 to 6 carbon atoms, including but not limited to methyl,
ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl and hexyl, etc.;
preferably ethyl,
n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl and tert-butyl.
In the present invention, the term "alkoxy" means -0-(Ci-C6 alkyl) group. For
example,
the term "Ci-C6 alkoxy" refers to a straight or branched chain alkoxy having 1
to 6 carbon
atoms, including but not limited to methoxy, ethoxy, n-propoxy, isopropoxy,
butoxy and so
on.
¨ 8 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
In the present invention, the term "cycloalkyl" refers to a saturated cyclic
hydrocarbon
moiety, for example, the term "C3-C10 cycloalkyl" refers to a cyclic alkyl
group having 3 to
carbon atoms in the ring, including but not limited to cyclopropyl,
cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl and the like. The terms "C3-C8
cycloalkyl",
5 "C3-C7 cycloalkyl" and "C3-C6 cycloalkyl" have similar meanings.
In the present invention, the term "cycloalkoxy" means cycloalkyl-O-, and
cycloalkyl is
described as above.
In the present invention, the term "4-7 membered nitrogen-containing
heterocyclyl"
refers to a cycloalkyl ring having 3-7 ring atoms and containing 1, 2 or 3 N
atoms, and
10 includes, but not limited to, azacyclopentane ring, azacyclohexane
ring, azacycloheptane ring
and the like.
In the present invention, the term "aryl" means a hydrocarbyl moiety
containing one or
more aromatic rings. For example, the term "C6-C12 aryl" refers to an aromatic
ring group
with 6 to 12 carbon atoms that does not contain heteroatoms in the ring, such
as phenyl,
naphthyl and the like. The term "C6-C aryl" has a similar meaning. Examples of
aryl
include, but are not limited to, phenyl (Ph), naphthyl, pyrenyl, anthracenyl,
and phenanthryl.
In the present invention, the term "heteroaryl" means a moiety containing one
or more
aromatic rings with at least one heteroatom (such as N, 0 or S), for example,
the term "3-12
membered heterocyclyl" means a saturated or unsaturated 3-12 membered ring
group
containing 1 to 3 heteroatoms selected from oxygen, sulfur and nitrogen on the
ring, such as
dioxolyl and the like. The term "3-7 membered heterocyclyl" has a similar
meaning.
Examples of heteroaryl groups include furyl, fluorenyl, pyrrolyl, thienyl,
oxazolyl,
imidazolyl, thiazolyl, pyridyl, pyrimidinyl, quinazolinyl, quinolinyl,
isoquinolinyl, and
indolyl.
In the present invention, the term "heterocyclyl" means a cyclic group
containing at
least one ring heteroatom (such as N, 0 or S), such as furyl, pyrrolyl,
thienyl, oxazolyl,
imidazolyl, thiazolyl, pyridyl, quinolinyl, isoquinolinyl, indolyl,
pyrimidinyl,
tetrahydropyridyl, pyrrolinyl, dihydropyridyl, dihydrofuranyl, dihydrothienyl,
pyranyl.
Unless otherwise specified, the alkyl, alkoxy, cycloalkyl, heterocyclyl, and
aryl
described herein are substituted and unsubstituted groups. Possible
substituents on alkyl,
alkoxy, cycloalkyl, heterocyclyl and aryl include, but are not limited to:
hydroxyl, amino,
nitro, cyano, halogen, C1-C6 alkyl, C2-C10 alkenyl, C2-C10 alkynyl, C3-C20
cycloalkyl, C3-C20
cycloalkenyl, C1-C20 heterocy cloalkyl, C1-C20 heterocycloalkenyl, C1-C6
alkoxy, aryl,
heteroaryl, heteroaryloxy, C1-Cio alkylamino, Ci-C20 dialkylamino, arylamino,
diarylamino,
C1-Cio alkylsulfamoyl, arylsulfamoyl, C1-C10 alkylimino,
alkylsulfoimino,
arylsulfoimino, mercapto, Ci-Cio alkylthio, C1-C10 alkylsulfonyl,
arylsulfonyl, acylamino,
aminoacyl, aminothioacyl, guanidinyl, ureido, cyano, acyl, thioacyl, acyloxy,
carboxyl and
carboxylate group. On the other hand, cycloalkyl, heterocycloalkyl,
heterocycloalkenyl, aryl,
and heteroaryl may also be fused to each other.
In the present invention, the substitution is mono-substitution or poly-
substitution, and
the poly-substitution is di-substitution, tri-substitution, tetra-
substitution, or
- 9 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
penta-substitution. The di-substitution means that there are two substituents,
and so on.
The pharmaceutically acceptable salt of the present invention may be a salt
formed by
an anion and a positively charged group on the compound of formula I. Suitable
anion is
chloride, bromide, iodide, sulfate, nitrate, phosphate, citrate,
methanesulfonate,
trifluoroacetate, acetate, malate, tosyl ate, tartrate, fumarate, glutamate,
glucuronate, lactate,
glutarate or maleate ion. Similarly, a salt can be formed from a cation and a
negatively
charged group on the compound of formula I. Suitable cation includes sodium
ion, potassium
ion, magnesium ion, calcium ion and ammonium ion, such as tetramethylammonium
ion.
In another preferred example, "pharmaceutically acceptable salt" refers to a
salt formed
by a compound of formula I and an acid selected from the group consisting of
hydrofluoric
acid, hydrochloric acid, hydrobromic acid, phosphoric acid, acetic acid,
oxalic acid, sulfuric
acid, nitric acid, methanesulfonic acid, aminosulfonic acid, salicylic acid,
trifluoromethanesulfonic acid, naphthalenesulfonic acid, maleic acid, citric
acid, acetic acid,
lactic acid, tartaric acid, succinic acid, oxalic acid, pyruvic acid, malic
acid, glutamic acid,
p-toluenesulfonic acid, naphthalenesulfonic acid, ethanesulfonic acid,
naphthalenedisulfonic
acid, malonic acid, fumaric acid, propionic acid, oxalic acid, trifluoroacetic
acid, stearic acid,
pamoic acid, hydroxymaleic acid, phenylacetic acid, benzoic acid, glutamic
acid, ascorbic
acid, p-aminobenzenesulfonic acid, 2-acetoxybenzoic acid and isethionic acid,
etc.; or
sodium salt, potassium salt, calcium salt, aluminum salt or ammonium salt
formed by a
compound of formula I and inorganic base; or methylamine salt, ethylamine salt
or
ethanolamine salt formed by a compound of general formula I and organic base.
Preparation method
The preparation method of the compound represented by the general formula I of
the
present invention, the synthetic route is as follows:
R2
0F1 0 R2
0 0
Ns, CI COOMe \
Br
R" R15 R15 R R11 R15
a Rii R"
Riz R14
Riz R14 iz R14
R13 Riz Rizt
R13 R13
R13
II III IV V
¨ 10 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
R2 R2
R2
0 \ 0
\ 0
N N
CN a, 0
/ 'OH
Rii R15 e R1 Ris Rii Ri 5 H2N
Ri2 Ri4 R12 R14 Ri2 Ria
R R13
R13 13
VI VII VIII
R2
b \ 0 A
N
'
R11 R15 HN40
0
R12 R14
R13
The preparation method includes the following steps:
a) reacting substituted benzaldehyde as a starting material with hydroxylamine
hydrochloride under the action of an alkali to obtain an intermediate and then
chlorinating
with N-chlorosuccinimide (NCS) to form a compound represented by the general
formula III;
b) reacting the compound represented by the general formula III with the
corresponding
3-oxopropionate under an alkali condition to form a compound represented by
the general
formula IV;
c) reducing the ester in the compound represented by the general formula IV by
a
reducing agent to generate the corresponding alcohol, and then brominating to
form the
compound represented by V,
d) reacting the compound represented by the general formula V with Q-OH under
the
action of an alkali to form the compound represented by the general formula
VI;
e) coupling the compound represented by the general formula VI with Br-A-CN
under
the catalysis of copper or palladium to obtain a cyano compound represented by
the general
formula VII;
a') reacting the compound represented by the general formula VII with
hydroxylamine
hydrochloride under the action of an alkali to produce a compound represented
by the general
formula VIII;
b') reacting the compound represented by the general formula VIII under the
action of
phosgene, triphosgene, carbonyldiimidazole or thiocarbonyldiimidazole to
produce the
compound represented by the general formula I.
R2
0 ,
IN 0 A
N N
CN
9 RiiI R15
=CN
Q,OH + F.NKCN HO"CrA Ti
V Riz Ria
ix R13
VII
¨ 11 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
The cyano compound represented by the general formula VII can also be prepared
by
the above route, including the following steps:
f) substituting the fluorine in F-A-CN with the amino in Q-OH under the action
of an
alkali to generate compound IX;
g) directly reacting a compound represented by the general formula V with the
prepared
IX under the action of an alkali to form the compound represented by the
general formula VI;
wherein, R2, RH, R12, R13, R14, R15, , ¨
Q A ring and X are defined as above.
The alkali in steps a), b), d), a'), 0 and g) is selected from triethylamine,
diisopropylethylamine, pyridine, 4-dimethylaminopyridine, 1,8-
diazabicycloundec-7-ene,
sodium carbonate, potassium carbonate, lithium hydroxide, sodium hydroxide,
potassium
hydroxide, sodium methoxide, sodium ethoxide, potassium ethoxide, potassium
tert-butoxide,
sodium tert-butoxide, butyl lithium, lithium diisopropylamide.
The alkali in step b) is selected from the group consisting of triethylamine,
diisopropylethylamine, pyridine, DBU, sodium carbonate, potassium carbonate,
lithium
hydroxide, sodium hydroxide, potassium hydroxide, sodium methoxide, sodium
ethoxide,
and potassium ethoxide.
The reducing agent in step c) is selected from the group consisting of sodium
borohydri de, sodium triacetoxyborohydride, sodium cyanoborohydride, lithium
aluminum
hydride, diisopropyl aluminum hydride, and borane.
The copper catalyst in step e) is cuprous iodide, cuprous oxide, and cuprous
sulfate; the
palladium catalyst is palladium acetate, tetrakis(triphenylphosphine)
palladium,
bis(acetonitrile) palladium (II) chloride, dichloride
palladium,
tris(dibenzylideneacetone)dipalladium, bistriphenylphosphorus palladium
dichloride,
tris(dibenzyli deneacetone)dipall adi um-chloroform
adduct, 1,1'-bis(diphenylphosphino)
ferrocene palladium(II) dichloride.
Pharmaceutical composition
The present invention also provides a pharmaceutical composition, which
contains
active ingredient in a safe and effective amount, and a pharmaceutically
acceptable carrier.
The "active ingredient" in the present invention refers to the compound of
formula I in
the present invention.
The "active ingredient" and pharmaceutical composition of the present
invention are
used in the manufacture of a medicament for treating FXR-related diseases. The
"active
ingredient" and pharmaceutical composition of the present invention can be
used as FXR
agonist. In another preferred example, it is used in the manufacture of a
medicament for
preventing and/treating a disease regulated by FXR agonist.
"Safe and effective amount" means that the amount of the active ingredient is
sufficient
to significantly improve the condition without causing serious side effects.
Generally, the
pharmaceutical composition contains 1-2000 mg of active ingredient/dose, more
preferably,
10-200 mg of active ingredient/dose. Preferably, the "one dose" is a tablet.
¨ 12 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
"Pharmaceutically acceptable carrier" refers to one or more compatible solid
or liquid
fillers or gel substances, which are suitable for human use, and must have
sufficient purity
and sufficiently low toxicity. "Compatibility" herein means that each
component in the
composition can be blended with each other and can be blended with the active
ingredient of
the present invention without significantly reducing the efficacy of the
active ingredient.
Examples of pharmaceutically acceptable carriers include cellulose and
derivatives
thereof (such as sodium carboxymethyl cellulose, sodium ethyl cellulose,
cellulose acetate,
etc.), gelatin, talc, solid lubricant (such as stearic acid, magnesium
stearate), calcium sulfate,
vegetable oil (such as soybean oil, sesame oil, peanut oil, olive oil, etc.),
polyol (such as
propylene glycol, glycerin, mannitol, sorbitol, etc.), emulsifier (such as
Tween*), Wetting
agent (such as sodium lauryl sulfate), coloring agent, flavoring agent,
stabilizer, antioxidant,
preservative, pyrogen-free water and the like.
The administration method of the active ingredient or the pharmaceutical
composition of
the present invention is not particularly limited, and representative
administration methods
include (but are not limited to): oral administration, intratumoral
administration, rectal
administration, parenteral (intravenous, intramuscular, or subcutaneous)
administration and
the like.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders and
granules.
Liquid dosage forms for oral administration include pharmaceutically
acceptable
emulsions, solutions, suspensions, syrups or tinctures. In addition to the
active ingredient, the
liquid dosage form may contain inert diluents conventionally used in the art,
such as water or
other solvents, solubilizers and emulsifiers, for example, ethanol,
isopropanol, ethyl
carbonate, ethyl acetate, propylene glycol, 1,3-butanediol, dimethylformamide
and oils,
especially cottonseed oil, peanut oil, corn germ oil, olive oil, castor oil
and sesame oil or
mixtures of these substances. In addition to these inert diluents, the
composition may also
contain adjuvants such as wetting agents, emulsifying agents and suspending
agents,
sweetening agents, flavoring agents and perfumes.
In addition to the active ingredient, the suspension may contain suspending
agent, for
example, ethoxylated isostearyl alcohol, polyoxyethylene sorbitol and
dehydrated sorbitan
ester, microcrystalline cellulose, aluminum methoxide and agar, or mixtures of
these
substances, and the like.
The composition for parenteral injection may contain physiologically
acceptable sterile
aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, and
sterile
powders for reconstitution into sterile injectable solutions or dispersions.
Suitable aqueous
and non-aqueous carriers, diluents, solvents or excipients include water,
ethanol, polyol and
suitable mixtures thereof.
The compound of the present invention can be administered alone or in
combination
with other therapeutic drugs (such as hypolipidemic drugs).
When the pharmaceutical composition is used, a safe and effective amount of
the
compound of the present invention is administered to the mammal (such as a
human) in need
- 13 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
of treatment, wherein the dosage at which the drug is administered is the
pharmaceutically
effective administration dosage. For a person of 60kg body weight, the daily
dose is usually
1-2000 mg, and 20-500 mg is preferred. Centainly, the specific dosage should
be determined
by considering factors such as the route of administration, the patient's
health status, etc.,
which are within the skill range of a skilled physician.
The present invention will be further described below in conjunction with
specific
examples. It should be understood that these examples are only used to
illustrate the present
invention and not to limit the scope of the present invention. The
experimental methods
without specific conditions in the following examples generally follow the
conventional
conditions (eg. the conditions described in Sambrook et al., Molecular
Cloning: Laboratory
Manual (New York: Cold Spring Harbor Laboratory Press, 1989)) or the
conditions
recommended by the manufacturer. Unless stated otherwise, percentages and
parts are
percentages by weight and parts by weight.
Unless otherwise defined, all professional and scientific terms used herein
have the
same meaning as those familiar to the skilled in the art. In addition, any
methods and
materials similar to or equivalent to those described can be applied to the
method of the
present invention. The preferred implementation methods and materials
described herein are
for demonstration purposes only.
The instruments and main experimental materials used are as follows.
The reagents and anhydrous solvents used were purchased from Chinese
commercial
companies. Unless otherwise specified, they were used directly. 11-1 and 13C
NMR were
measured by BrukerAM-400 and Varian Mercury plus-400 nuclear magnetic
resonance
instruments, and mass spectrometry was measured by Agilent 6230 mass
spectrometer and
200- 300 mesh of column chromatography silica gel (Qingdao Ocean Chemical
Factory),
HSGF254 TLC plate (Yantai Chemical Industry Research Institute),
Route 1
R2
9H
o R2
0, N N CI N N COOMe \
Br
Rii R15 Rii R15 RII R15
a Rii R15
Ri2 Ri4
Riz Ria R12 R14
R13 Ri2 R14
R13 RI3
R13
II III IV V
¨14 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
R2 R2 R2
0 0 , 0
1 \ 0'CI
rj , \ 0---Q-A 1 \ 0- -A N
N , 0
\CN a
N,
d R1.1 R15 e _ õ ,i
OH
Rii R15 -...- R" R15
ri2iN
_...
R12R14 R12 R14 R12 Ri4
R R13
R13 13
VII VIII
VI
R2
0 ,
b'
Rii R15 HN,e
R12 1'R14 0
R13
1
Route 2
h
0-0H + F,PrCN H0-0-A
'ON
IX
R2
OH 0 R2
0
... N., CI N `= COOMe
N ,
Ri 1 R15 R11 R15 b R11 R15 c R11
a R15
_,..
R12 R14 R12 R14 R12 R14
R13 R12 R14
R13 R13
R13
V
II III IV
R2 R2
0 , 0 N
Ri R15 0 , R2
0--.Q-A 0-.41-A
tN R
a'
R15 H2Nr OH U
i 11 R11 R15 FIN ,(C)
i
_,...
W2 R14 R12 R14 R12 ^R14 0
R13 R13
R13
VII VIII I
Example 1 LXF-32:
o NI\o0
NH
N \ O Ap
a a
1 (LXF-32)
Synthesis of intermediate VI-1:
¨ 1 5 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OF1
cici
(1)
N CI 0
N' \ 0
CI CI
CI CI
11-1 III-1 11/-1
OH
0
0 N 0
N' \ Br
Boc
____________________________________________________ CI CI
CI CI
VI-1
1/-1
At 0 C, aqueous potassium carbonate solution (3 N, 182 mmol) was added
dropwise to
a stirring solution of hydroxylamine hydrochloride (182 mmol) in ethanol (100
mL),
2,6-dichlorobenzaldehyde (20 g, 114 mmol) was dissolved in 100 ml of ethanol,
and then
added to the hydroxylamine solution. The temperature was raised to 90 C and
the mixture
was reacted for two hours. The mixture was cooled to room temperature and then
concentrated to a solid. A water/ethanol (1000 mL/100 mL) solution was added
and the solid
was stirred to break up, filtered, and dried under vacuum at 50 C overnight
to obtain a
compound intermediate (18.4 g). This intermediate was dissolved in N,N-
dimethylformamide
(50 mL), and added dropwise to N-chlorosuccinimide (97 mmol) solution in
N,N-dimethylformamide (100 mL) at 0 C and stirred overnight. The reaction
solution was
poured into ice water at 0 C, and then extracted with methyl tert-butyl ether
(200 mL each
time, 3 times in total), the organic phase was washed with saturated brine,
and concentrated
to obtain a crude product. N-hexane (600 mL) was added to the flask containing
the crude
product, stirred with a magnetic stir bar, filter, and the solid was dried
under vacuum (30 C.)
to obtain Intermediate III-1 (18.3 g, yield 73%). 1H NMR (400 MHz, CDC13) 8
7.43 - 7.39 (m,
2H), 7.39 - 7.33 (m, 1H).
Triethylamine (8.2 g) was added to methyl 3-cyclopropy1-3-oxopropionate (82
mmol)
and stirred for 30 minutes. Then the mixture was cooled to 10 C, and a
solution of III-1
(18.3 g, 82 mmol) in absolute ethanol (80 mL) was added dropwise (internal
temperature did
not exceed 30 C), and the reaction was kept overnight at room temperature. The
reaction
solution was diluted by adding ethyl acetate (100 mL), washed with water, and
the aqueous
phase was extracted with ethyl acetate (100 mL each time, 3 times in total).
The organic
phases were mixed, washed with saturated brine, and concentrated. 100 mL of
ether was
added to the concentrate and stirred, and the solvent was removed under vacuum
to obtain
solid product IV-1 (21.6 g, yield 84%). 1H NMR (400 MHz, CDC13) 8 7.43 - 7.39
(m, 2H),
7.39 - 7.33 (m, 1H), 3.72 (s, 3H), 2.21 - 2.09 (m, 1H), 1.35 - 1.28 (m, 2H),
1.25 - 1.18 (m,
2H).
¨ 16 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
IV-1 (21.6 g, 69 mmol) was dissolved in tetrahydrofuran (140 mL) and cooled to
0 C.
A solution of diisobutylaluminum hydride (1.5 M, 102mL) in toluene was slowly
added and
the reaction solution is stirred at room temperature for 6h. The reaction
solution was slowly
poured into ice water, and 1M aqueous hydrochloric acid solution was added to
adjust the pH
to about 2. The mixture was extracted with ethyl acetate (100 mL each time,
three times in
total), concentrated, and subjected to column chromatography to obtain the
intermediate
alcohol. This intermediate and triphenyl phosphine (59 mmol) were dissolved in
dichloromethane (60 mL) and cooled to 0 C, and a solution of carbon
tetrabromide (62
mmol) in dichloromethane (60 mL) was added dropwise under the protection of
nitrogen and
reacted at room temperature for 4 h. The solvent was removed from the reaction
solution to
obtain an oily substance, which was subjected to column chromatography to
obtain
intermediate V-1 (15.3 g, yield 96%). 1H NMR (400 MHz, CDC13) 6 7.49 - 7.44
(m, 2H),
7.43 - 7.37 (m, 1H), 4.25 (d, J= 1.3 Hz, 2H), 2.21 - 2.09 (m, 1H), 1.35 - 1.28
(m, 2H), 1.25 -
1.18 (m, 2H).
At 0 C, potassium tert-butoxide (6.5 mmol) was added to a solution of tert-
butyl
4-hydroxypiperidine-1-carboxylate (1.3 g, 6.5 mmol) in anhydrous
tetrahydrofuran (20 mL)
and stirred for 30 minutes, and then a solution of V-1 (4.3 mmol) in anhydrous
tetrahydrofuran (5 mL) was added dropwise, and the reaction was carried out
for 8 h. Water
(20 mL) was added to the reaction solution, extracted with ethyl acetate (15
mL each time, 3
times in total), the organic phase was washed with saturated brine,
concentrated, and
subjected to column chromatography to obtain intermediate tert-butyl
4((5-cy clopropy1)-3 -(2,6-dichl oropheny oxazol-4-yl)methoxy )piperidine-1-
carboxy late
(1.55 g). Intermediate tert-butyl 4-((5-cyclopropy1)-3-(2,6-
dichlorophenyl)isoxazol-
4-yl)methoxy)piperidine-1-carboxylate (1.55 g, 3.3 mmol) was dissolved in
dichloromethane
(8 mL)and cooled to 0 C, and trifluoroacetic acid (8 mL) was added dropwise
and stirred at
room temperature for 3h. The solvent was removed under vacuum, and the residue
was
dissolved in ethyl acetate (20 mL), washed with 2 N sodium hydroxide solution,
saturated
brine, and the solvent was removed to obtain intermediate VI-1 (1.0 g, yield
72%). 1H NMR
(400 MHz, CDC13) 6 7.47 - 7.43 (m, 2H), 7.42 - 7.36 (m, 1H), 4.23 (s, 2H),
3.55 - 3.49 (m,
1H), 3.02 -2.91 (m, 4H), 2.10- 2.02 (m, 1H), 1.93 - 1.76 (m, 2H), 1.75 - 1.62
(m, 2H), 1.26 -
1.06 (m, 4H).
Synthesis of Example Compound 1, namely LXF-32:
-17 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 \
Br CN 0
N \ 0-0
a CI
CN
VI-1
VII-1
0 0
\ CI CI0 0- * rµi \ 0
*--C\N *
CI
-N
VIII-1 H2N
OH HN
e
Intermediate VI-1 (1.0 g, 2.7 mmol), 3-bromobenzonitrile (4.1 mmol), sodium
tert-butoxide (5.4 mmol), palladium acetate (0.14 mmol),
and
1,1'-binaphthalene-2,2'-bisdiphenylphosphine (0.27 mmol) were added to a round
bottom
flask, and toluene (80mL) was added under the protection of nitrogen, heated
to reflux, and
reacted overnight. The reaction solution was cooled to room temperature, and
added with
water, extracted, concentrated, and subjected to column chromatography to
obtain
intermediate VII-1 (0.55 g, yield 43%). 11-1 NMR (400 MHz, CDC13) 6 7.40 (d,
J= 1.2 Hz,
1H), 7.38 (s, 1H), 7.32 - 7.28 (m, 2H), 7.09 - 7.02 (m, 3H), 4.34 (s, 2H),
3.47- 3.41 (m, 1H),
3.31 -3.20 (m, 2H), 2.97 -2.87 (m, 2H), 2.18 - 2.11 (m, 1H), 1.83 - 1.72 (m,
2H), 1.26 (qt, J
= 10.1, 5.1 Hz, 4H), 1.13 (ddd, J= 11.4, 7.0, 4.4 Hz, 2H).
VII-1 (0.4 g, 0.9 mmol), hydroxylamine hydrochloride (2.3 mmol), and absolute
ethanol
(5mL) were added into a round bottom flask and stirred. Triethylamine (2.3
mmol) was
slowly added dropwise, and heated to 80 C to react for 4h. After the mixture
was cooled to
room temperature, the solvent was removed, and the residue was dissolved in
ethyl acetate
(15 mL), and washed with water and saturated brine. The organic phase was
concentrated,
and subjected to silica gel column chromatography to obtain intermediate
3-(4-((5-cyclopropy1-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)piperidin- 1-
y1)-N'-hydro
xybenzamidine VIII-1 (0.41 g, yield 96%).
3-(4-((5-cy clopropy1-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)piperidin-l-
y1)-N'-h
ydroxybenzamidine VIII-1 (0.41 g, 0.83 mmol), N,N'-carbonyldiimidazole (1.0
mmol), and
1,4-dioxane (4 mL) were added to a round bottom flask, and then
1,8-diazabicyclo[5.4.0]undec-7-ene (0.91 mmol) was added, heated to 100 C and
reacted for
3 hours. The reaction solution was cooled to room temperature, diluted with
water (5 mL),
adjusted to pH approximately equal to 2 with a 1M aqueous hydrochloric acid
solution, and
then extracted with ethyl acetate (4 mL each time, 3 times in total). The
organic phases were
combined, washed with saturated brine, and concentrated and the crude product
obtained was
subjected to silica gel column chromatography to obtain the final product 1
(0.28 g, yield
64%). 1-1-1 NMR (400 MHz, CDC13) 6 7.37 (d, J= 7.5 Hz, 2H), 7.31 - 7.26 (m,
2H), 7.17 (d, J
= 10.4 Hz, 2H), 7.07 (d, J= 7.5 Hz, 1H), 6.91 (d, J= 7.6 Hz, 1H), 4.33 (s,
2H), 3.38 (m, 1H),
3.21 (m, 2H), 2.83 (t, Jr 8.6 Hz, 2H), 2.15 (m, 1H), 1.73 (m, 2H), 1.51 (m,
2H), 1.26 (m,
- 18 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
4H), 1.13 (m, 2H). MS(ESI, m/z): 541[M+H].
Example 2:
(LCN
CI CI
0
2
Example 2 was carried out by referring to the operation of example 1. The
compound
was prepared from intermediate VI-1 via route 1. The synthetic route was as
follows.
Synthesis of Example Compound 2:
o CI
0
"-CNH Br =CN =
N
CI
CI CI CN
VI-1
VII-2
0 0
N
--C\N *
CI CI CI a
H2N HN4
0
VIII-2 2
Compound 2 was synthesized from raw material VI-1 according to the synthetic
method
of compound 1, wherein
VIII-2, white solid, yield 42%; 1H NMR (400 MHz, CDC13) 6 7.49 (d, J= 8.7 Hz,
2H),
7.36 (d, J= 7.7 Hz, 2H), 7.30 - 7.25 (m, 1H), 6.81 (d, J= 8.7 Hz, 2H), 5.01
(s, 2H), 4.34 (s,
2H), 3.46 - 3.36 (m, 1H), 3.33 - 3.22 (m, 2H), 2.98 - 2.84 (m, 2H), 2.20 -
2.12 (m, 1H), 1.81 -
1.73 (m, 2H), 1.59 - 1.48 (m, 2H), 1.28 - 1.23 (m, 2H), 1.15 - 1.08 (m, 2H).
MS (EI,m/z):
501[M-fi1.
Compound 2, white solid, yield 71%, 1H NMR (400 MHz, CDC13) 6 7.64 (d, J= 9.0
Hz,
2H), 7.44- 7.36(m, 2H), 7.31 (dd, J= 8.9, 7.1 Hz, 1H), 6.90 (d, J= 9.1 Hz,
2H), 4.37 (s, 2H),
3.50 (tt, Jr 7.2, 3.5 Hz, 1H), 3.43 - 3.32 (m, 2H), 3.13 - 2.99 (m, 2H), 2.16
(d, J= 30.8 Hz,
1H), 1.85 - 1.73 (m, 2H), 1.61 - 1.50 (m, 2H), 1.32 - 1.25 (m, 4H), 1.15 (m,
2H);
MS(ESI,m/z): 527[M+H]+.
Example 3:
0 0-0N -N
CI HNIb
CI
0
3
Example 3 was carried out by referring to the operation of example 1. The
compound
was prepared from intermediate V-1 via route 1. The synthetic route was as
follows.
¨ 19 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OH
JO'
9 \ Br \
ONH Boc
1110 Br eN tk1,ON *
CI CI CI CN
CI CI
V-1 VI-3
VII-3
\ o CI CI "` CI CI _N
HNõO
VIII-3N: CD!
OH
3 11
At 0 C, potassium tert-butoxide (6.5 mmol) was added to a solution of tert-
butyl
4-hydroxyhexahydroazepine-1-carboxylate (6.5 mmol) in anhydrous
tetrahydrofuran (20 mL)
and stirred for 30 minutes, and then a solution of V-1 (4.3 mmol) in anhydrous
tetrahydrofuran (5 mL) was added dropwise, and the reaction was carried out
for 8 h. Water
(20 mL) was added to the reaction solution, which was then extracted with
ethyl acetate (15
mL each time, 3 times in total). The organic phase was washed with saturated
brine,
concentrated, and subjected to column chromatography to obtain an
intermediate. The
intermediate was dissolved in dichloromethane (8 mL) and cooled to 0 C, and
trifluoroacetic
acid (8mL) was added dropwise, and stirred at room temperature for 3h. The
solvent was
removed under vacuum, ethyl acetate (20 mL) was added to dissolve, washed with
2N
sodium hydroxide solution, saturated brine, and the solvent was removed to
obtain
intermediate VI-3 (0.87 g, yield 53%). 1H NMR (400 MHz, CDC13) 8 7.45 - 7.40
(m, 2H),
7.38 - 7.32 (m, 1H), 4.35 -4.23 (m, 2H), 3.49- 3.42 (m, 1H), 3.40 - 3.18 (m,
4H), 2.18 -2.09
(m, 1H), 1.83 - 1.59 (m, 5H), 1.55 - 1.46 (m, 1H), 1.28 - 1.23 (m, 2H), 1.16-
1.10 (m, 2H).
Intermediate VI-3 (0.8 g), 4-bromobenzonitrile (4.1 mmol), sodium tert-
butoxide (5.4
mmol), palladium acetate (0.14 mmol), and 1,1'-binaphthy1-2,2'-
bisdiphenylphosphine (0.27
mmol) were added to a round bottom flask, and toluene (60 mL) was added under
the
protection of nitrogen, heated to reflux, and reacted overnight. The reaction
solution was
cooled to room temperature and water was added. The mixture was extracted,
concentrated,
and subjected to column chromatography to obtain intermediate VII-3 (0.49 g,
yield 48%).
1H NMR (400 MHz, CDC13) 8 7.44 - 7.40 (m, 2H), 7.37 - 7.33 (m, 1H), 7.27 -
7.21 (m, 1H),
6.92- 6.86 (m, 1H), 6.83 - 6.78 (m, 2H), 4.34 - 4.24 (m, 2H), 3.49 - 3.43 (m,
1H), 3.39 - 3.18
(m, 4H), 2.18 - 2.10 (m, 1H), 1.84 - 1.58 (m, 5H), 1.55 - 1.48 (m, 1H), 1.27 -
1.24 (m, 2H),
1.15 - 1.09 (m, 2H).
Compound 3 was synthesized from VII-3 as the raw material according to the
synthesis
method of compound 1, yield 66%, 1H NMR (400 MHz, CDC13) ö 7.42 - 7.38 (m,
2H), 7.36 -
7.31 (m, 1H), 7.30 - 7.25 (m, 1H), 7.03 - 6.99 (m, 2H), 6.82 - 6.75 (m, 1H),
4.35 - 4.23 (m,
2H), 3.48 -3.20 (m, 5H), 2.18 - 2.10 (m, 1H), 1.89 - 1.57 (m, 5H), 1.55 - 1.45
(m, 1H), 1.26 -
1.21 (m, 2H), 1.15 - 1.08 (m, 2H). MS(ESI,m/z): 541[M+H].
Example 4:
-20 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0
cii
01 NO
4
Example 4 was carried out by referring to the operation of example 1. The
compound
was prepared from intermediate VI-3 via route 1. The synthetic route was as
follows.
0 N-
0
rµsi 0, CN
(D¨CN
No
CI CI0H Br SCN CD!
_________________________ - CI _____________ - * CI CI
VI-3
VII-4
4
Compound 4 was synthesized from VI-3 as the raw material according to the
synthesis
method of compound 1, wherein,
VII-4, white solid, yield 59%, NMR (400 MHz, CDC13) 8 7.45 - 7.40 (m, 4H),
7.37 - 7.32
(m, 1H), 6.60 (d, J= 9.1 Hz, 2H), 4.33 - 4.24 (m, 2H), 3.52- 3.19 (m, 5H),
2.17 -2.09 (m, 1H),
1.82 - 1.60 (m, 5H), 1.51 - 1.43 (m, 1H), 1.29 - 1.24 (m, 2H), 1.15 - 1.09 (m,
2H).
Compound 4, white solid, yield 69%, 'H NMR (400 MHz, CDC13) 6 7.60 (d, J= 8.9
Hz, 2H),
7.44 - 7.39 (m, 2H), 7.38 - 7.33 (m, 1H), 6.66 (d, J= 9.1 Hz, 2H), 4.35 -4.23
(m, 2H), 3.51 - 3.19
(m, 5H), 2.18 -2.08 (m, 1H), 1.83 - 1.59 (m, 5H), 1.54 - 1.45 (m, 1H), 1.28 -
1.23 (m, 2H), 1.15 -
1.08 (m, 2H). MS(ESI,m/z): 541[M+H]t
Example 5:
\ 110
CI CI
-N
HN
Example 5 was carried out by referring to the operation of example 1. The
compound was
prepared from intermediate V-1 via route 1. The synthetic route was as
follows.
HO
o ,
Br I----N
Boc Br CN
CI CI CI CI
V-1 VI-5
0
CDI
CN
CI CI CI CI
0
VII-5 5
Compound 5 was synthesized from V-1 as the raw material according to the
synthesis method
of compound 1, wherein
¨21 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
VI-5, colloid, yield 61%, 1H NMR (400 MHz, CDC13) 6 7.45 - 7.40 (m, 2H), 7.39 -
7.32 (m,
1H), 4.36 - 4.22 (m, 2H), 3.98 - 3.92 (m, 1H), 3.40- 3.13 (m, 4H), 2.17 - 2.08
(m, 1H), 1.83 - 1.73
(m, 2H), 1.31 ¨1.24 (m, 2H), 1.17 - 1.11 (m, 2H).
VII-5, white solid, yield 49%, 1H NMR (400 MHz, CDC13) .5 7.33 (dd, J= 7.5,
1.8 Hz, 1H),
.. 7.27 - 7.19 (m, 3H), 6.94 (d, J= 7.5 Hz, 1H), 6.66 - 6.59 (m, 2H), 4.40 -
4.28 (m, 2H), 4.19 - 4.13
(m, 1H), 3.35 - 3.18 (m, 3H), 3.04 (d, J= 10.5 Hz, 1H), 2.18- 2.09(m, 1H),
2.04 - 1.97 (m, 2H),
1.30- 1.25 (m, 2H), 1.16- 1.11 (m, 2H).
5, white solid, yield 63%, 1H NMR (400 MHz, CDC13) 5 7.33 - 7.13 (m, 4H), 7.06
(d, J
= 7.8 Hz, 1H), 6.83 (s, 1H), 6.62 - 6.57 (m, 1H), 4.39 - 4.29 (m, 2H), 3.38 -
3.32 (m,1H), 3.31
.. -3.19 (m, 2H), 3.11 (d, J= 10.3 Hz, 1H),2.18 -2.10 (m, 1H), 2.01 - 1.94 (m,
2H), 1.26- 1.22
(m,2H), 1.16- 1.09 (m, 2H).MS(ESI, m/z): 513[M+H].
Example 6:
r4, \ 0-0 * N-0
FIN-A)CI CI
6
Example 6 was carried out by referring to the operation of example 1. The
compound was
prepared from intermediate VI-5 via route 1. The synthetic route was as
follows.
0 = CN
N-0
0 ,
Br \ 0 = CN HO-N
fri0
---CN I-13C1 _______ /
CI CIr
CI CI CD! cl CI
VI-5
VII-6 6
Compound 6 was synthesized from VI-5 as the raw material according to the
synthesis
method of compound 1, wherein,
VII-6, white solid, yield 38%. 1H NMR (400 MHz, CDC13) 5 7.46 (d, J= 8.9 Hz,
2H),
7.31 (dd, J= 7.9, 1.2 Hz, 1H), 7.25 (dd, J= 8.1, 1.2 Hz, 1H), 7.18 (t, J= 7.9
Hz, 1H), 6.40 (d,
J= 8.9 Hz, 2H), 4.40 - 4.28 (m, 2H), 4.19 - 4.12 (m, 1H), 3.37- 3.22 (m, 3H),
3.11 (d, J=
11.0 Hz, 1H), 2.17- 1.93 (m, 3H), 1.31 - 1.26 (m, 2H), 1.18- 1.11 (m, 2H).
6, white solid, yield 69%. 1H NMR (400 MHz, DMSO-d6) 5 7.59 (d, J= 8.8 Hz,
2H),
7.54- 7.39 (m, 3H), 6.53 (d, J= 8.8 Hz, 2H), 4.32 (q, J= 12.1 Hz, 2H), 4.10
(s, 1H), 3.31 -
3.23 (m, 2H), 3.12 - 3.04 (m, 2H), 2.39 - 2.28 (m, 1H), 2.02 - 1.84 (m, 2H),
1.15 - 1.06 (m,
4H). MS(ESI,m/z): 513[M+H].
Example 7:
P
N OZN ip )4,0
7
¨ 22 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Example 7 was carried out by referring to the operation of example 1. The
compound was
prepared from intermediate V-1 via route 1. The synthetic route was as
follows.
p Br HOZ.., CN
N Boc N
NH Br
CI CI a
v-1 VI-7
\ q/ HO-NH3CI 0 N=CN ION lip /N.0
CI CI ci
HN-
VII-7 7
Compound 7 was synthesized from V-1 as the raw material according to the
synthesis method
of compound 1, wherein,
VI-7, colloid, yield 67%. 1H NMR (400 MHz, CDC13) 6 7.42 - 7.39 (m, 2H), 7.36 -
7.31
(m, 1H), 4.27 - 4.18 (m, 2H), 4.10 - 3.96 (m, 2H), 3.53 (t, J= 4.7 Hz, 1H),
2.16 - 2.07 (m,
1H), 1.91 - 1.69 (m, 6H), 1.64 (d, J= 14.4 Hz, 2H), 1.26 - 1.22 (m, 2H), 1.14 -
1.08 (m, 2H).
VII-7, white solid, yield 54%. 1H NMR (400 MHz, CDC13) 6 7.47 - 7.42 (m, 4H),
7.38 - 7.34
(m, 1H), 6.65 (d, J= 8.9 Hz, 2H), 4.26 (s, 2H), 4.13 - 4.10 (m, 2H), 3.46 -
3.41 (m, 1H), 2.17 -
2.09 (m, 1H), 1.97 - 1.81 (m, 6H), 1.66- 1.61 (m, 2H), 1.28 - 1.25 (m, 2H),
1.17 - 1.11 (m, 2H).
7, white solid, yield 77%. 1H NMR (400 MHz, CDC13) 6 7.60 (d, J= 8.8 Hz, 2H),
7.44 -
7.40 (m, 2H), 7.37 - 7.32 (m, 1H), 6.70 (d, J= 9.0 Hz, 2H), 4.25 (s, 2H), 4.12
- 4.08 (m, 2H),
3.42 (s, 1H), 2.18 - 2.10 (m, 1H), 1.99 - 1.82 (m, 6H), 1.61 (d, J= 14.4 Hz,
2H), 1.26 - 1.22
(m, 2H), 1.16 - 1.10 (m, 2H). MS(ESI,m/z): 553[M+H]
Example 8:
\ (30
CF3
8 (LXF-116)
Example 8, i.e., the preparation of LXF-116 was carried out by referring to
the operation of
example 1. The compound was prepared from intermediate 11-8 via route 1. The
synthetic route
was as follows.
- 23 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OH
O OH
N
\ o 9
N, N,
blOC
CF3 CF3
F30 0¨
F30
11-8 111-8 1V-8 V-8
0 \ 0
N,
CF3 1
F3C (:)0
CN
F3C HN40
VI-8
VII-8 8
Compound 8 was synthesized from 11-8 as the raw material according to the
synthesis method
of compound 1, wherein,
IV-8, white solid, yield 58%. 11-1 NMR (400 MHz, CDC13) 6 7.82 (d, J = 7.5 Hz,
1H),
7.74- 7.59 (m, 2H), 7.56 (d, J= 7.5 Hz, 1H), 3.3.73 (s, 3H), 2.19- 2.09 (m,
1H), 1.33- 1.27
(m, 2H), 1.24 - 1.15 (m, 2H).
V-8, colourless liquid, yield 88%. 1H NMR (400 MHz, CDC13) 8 7.84 (d, J= 7.4
Hz, 1H),
7.73 - 7.61 (m, 2H), 7.57 (d, J= 7.4 Hz, 1H), 4.23 (s, 2H), 2.17 - 2.09 (m,
1H), 1.32 - 1.27 (m, 2H),
1.23- 1.17 (m, 2H).
VI-8, colloid, yield 78%. 1-F1 NMR (400 MHz, CDC13) 6 7.79 (d, J= 7.0 Hz, 1H),
7.66 -
7.56 (m, 2H), 7.41 (d, Jr 7.0 Hz, 1H), 4.23 (s, 2H), 3.55 - 3.49 (m, 1H), 3.03
- 2.91 (m, 4H),
2.10- 2.02 (m, 1H), 1.94 - 1.77 (m, 2H), 1.75 - 1.64 (m, 2H), 1.25 - 1.07 (m,
4H).
VII-8, white solid, yield 48%. 1H NMR (400 MHz, CDC13) 6 7.77 (d, J= 7.2 Hz,
1H),
7.62 - 7.51 (m, 2H), 7.48 - 7.40 (m, 3H), 6.79 (d, J= 9.0 Hz, 2H), 4.27 (s,
2H), 3.49 - 3.35 (m,
3H), 3.12 -2.96 (m, 2H), 2.16- 2.07 (m, 1H), 1.85 - 1.70 (m, 2H), 1.58 - 1.46
(m, 2H), 1.23 -
1.18 (m, 2H), 1.13 - 1.06 (m, 2H).
8, white solid, yield 72%. 1H NMR (400 MHz, DMSO-d6) 6 7.88 (d, J = 7.6 Hz,
1H),
7.80 - 7.67 (m, 2H), 7.64 - 7.56 (m, 3H), 6.99 (d, J= 8.8 Hz, 2H), 4.28 (s,
2H), 3.46 - 3.37 (m,
3H), 3.04 - 2.94 (m, 2H), 2.35 - 2.25 (m, 1H), 1.70 (s, 2H), 1.41 - 1.27 (m,
2H), 1.17 - 1.03
(m, 4H). MS(ESI,m/z): 527[M+H].
Example 9:
N
'0
OCF3
FiN140
9
Example 9 was carried out by referring to the operation of example 1. The
compound was
prepared from intermediate 11-9 via route 1. The synthetic route was as
follows.
¨ 24
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OH
OH
N \ N 0 a \ Br N
00F3- MN Lc
ocF,
F3co 0-- -
F3co
11-9 111-9 IV-9 V-9
9 \
\
N, 'CNN C)-CN N,
F3C0 F3
F3C0 PCN HN-1410
VI-9
VII-9 9
Compound 9 was synthesized from 11-9 as the raw material according to the
synthesis method
of compound 1, wherein,
IV-9, white solid, yield 59%. 1H NMR (400 MHz, CDC13) 6 7.66 - 7.50 (m, 2H),
7.49 -
7.41 (m, 2H), 3.70 (s, 2H), 2.18 - 2.10 (m, 1H), 1.31 - 1.26 (m, 2H), 1.23 -
1.17 (m, 2H).
V-9, colourless liquid, yield 82%. 1H NMR (400 MHz, CDC13) 6 7.65 - 7.52 (m,
2H),
7.49 - 7.40 (m, 2H), 4.36 (s, 2H), 2.18 - 2.10 (m, 1H), 1.31 - 1.26 (m, 2H),
1.23 - 1.17 (m,
2H).
VI-9, colloid, yield 80%. 1H NMR (400 MHz, CDC13) 67.45 - 7.30 (m, 4H), 4.29 -
4.18
(m, 2H), 2H), 3.50 - 3.36 (m, 3H), 3.12 - 3.00 (m, 2H), 2.18 - 2.10 (m, 1H),
1.86 - 1.76 (m,
2H), 1.61 - 1.50 (m, 2H), 1.28 - 1.22 (m, 2H), 1.13 - 1.07 (m, 2H).
VII-9, white solid, yield 55%. 1H NMR (400 MHz, CDC13) 6 7.58 - 7.47 (m, 2H),
7.45 -
7.34 (m, 4H), 6.81 (d, J= 9.0 Hz, 2H), 4.28 (s, 2H), 3.50 - 3.38 (m, 3H), 3.14
- 3.00 (m, 2H),
2.18 -2.10 (m, 1H), 1.85 - 1.76 (m, 2H), 1.61 - 1.50 (m, 2H), 1.27 - 1.22 (m,
2H), 1.13 - 1.07
(m, 2H).
9, white solid, yield 69%. 1H NMR (400 MHz, DMSO) 6 7.69 - 7.48 (m, 6H), 7.00
(d, J
= 8.9 Hz, 2H), 4.37 (s, 2H), 3.55 - 3.40 (m, 3H), 3.06 - 2.96 (m, 2H), 2.37 -
2.28 (m, 1H),
1.80- 1.69 (m, 2H), 1.44 - 1.31 (m, 2H), 1.16 - 1.03 (m, 4H). MS(EsI,m/z):
543[M+H1.
Example 10:
N N
CI CI z
HN40
10
The synthetic route of example 10 was as follows.
- 25 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OH
r'Lci 0 0
\ 0 ' \ Br
N.,
C
IX-10
I CI
CI CIII
CI
III-1 v-10
0
0 \ 0 ---CN
\
/14,0
CI CI
110 CN CICI
HN40
1/11-10 10
CN
HO-CNH + 1101 HO-(N CN
IX-10
At 0 C, sodium methoxide/methanol solution (5.4 M, 4.1 mL) was slowly added
dropwise
into a solution of methyl acetoacetate (22.2 mmol) in anhydrous
tetrahydrofuran (10m1), and then
a solution of III-1 (5 g, 22.2 mmol) ) in anhydrous tetrahydrofuran (10 mL)
was added and stirred
at room temperature for 12 h. Ethyl acetate (40 mL) was added to the reaction
solution, the organic
phase was washed with water and saturated brine, and the solvent was removed
to obtain an oily
substance, which was then subjected to column chromatography to obtain
intermediate IV-10 (3.4
g, yield 54%). 1H NMR (400 MHz, CDC13) 6 7.45 - 7.41 (m, 2H), 7.39 - 7.34 (m,
1H), 3.71 (s, 3H),
2.82 (s, 3H).
4-fluorobenzonitrile (2 g, 16.5 mmol), 4-hydroxypiperidine (18.2 mmol),
anhydrous
potassium carbonate (41.3 mmol) and DMSO (16 mL) were added to a round bottom
flask, heated
to 130 C, and reacted for 12h. The mixture was cooled to room temperature,
added with 30 mL of
water, and filtered. The solid was washed with water to obtain intermediate IX-
10 (3.1 g, yield
93%). 1H NMR (400 MHz, CDC13) 6 7.52 - 7.43 (m, 2H), 6.91 - 6.80 (m, 2H), 4.00
- 3.91 (m, 1H),
3.77 - 3.63 (m, 2H), 3.13 (ddd, J= 13.0, 9.4, 3.3 Hz, 2H), 2.05- 1.95 (m, 2H),
1.70- 1.59 (m, 2H).
The compound intermediate V-10 was synthesized by IV-10 as the raw material
according to
the synthesis method of the compound V-1. At 0 C, potassium tert-butoxide (6.5
mmol) was
added to a solution of IX-10 (1.3 g, 6.5 mmol) in anhydrous tetrahydrofuran
(20 ml) and stirred for
30 minutes, and then a solution of V-10 (4.3 mmol) in anhydrous
tetrahydrofuran (5mL) was
added dropwise and reacted for 8h. Water (20 mL) was added to the reaction
solution, which was
then extracted with ethyl acetate (15 mL x 3). The organic phase was washed
with saturated brine,
concentrated, and subjected to column chromatography to obtain intermediate
VII-10 (1.21 g,
yield 64%). 1H NMR (400 MHz, CDC13) 6 7.47 (d, J= 9.0 Hz, 2H), 7.42 - 7.38 (m,
2H), 7.33 -
7.29 (m, 1H), 6.80 (d, J= 9.0 Hz, 2H), 4.28 (s, 2H), 3.51 - 3.43 (m, 1H), 3.37
- 3.29 (m, 2H), 3.12
- 2.97 (m, 2H), 2.55 (s, 3H), 1.78 - 1.72 (m, 2H), 1.59 - 1.49 (m, 2H).
Compound 10 was synthesized from VII-10 as the raw material according to the
synthesis method of compound 1, white solid, yield 64%. 1H NMR (400 MHz,
CDC13) 6 7.64
(d, Jr 8.9 Hz, 2H), 7.45 - 7.36 (m, 2H), 7.34 - 7.28 (m, 1H), 6.88 (d, J= 8.9
Hz, 2H), 4.29 (s,
- 26
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
2H), 3.51 - 3.27 (m, 3H), 3.14 - 2.99 (m, 2H), 2.55 (s, 3H), 1.85 - 1.70 (m,
2H), 1.62 - 1.47
(m, 2H). MS(ESI,m/z): 501[M+14]+
Example 11:
\
CI a
The synthetic route of example 11 was as follows.
N CI 0 , 0
\ 0 \ Br HO-CN CN
CI CI 0--
C1 CI
III-1 IV-11 V-11
0
\
zrl'O
CI a = CN Ay/
HN40
VII-11 11
Compound intermediate IV-11( yield 61%) was synthesized from III-1 as the raw
material
according to the synthesis method of compound IV-10, wherein, methyl
acetoacetate was replaced
by methyl isobutyryl acetate. Ili NMR (400 MHz, CDC13) 6 7.44 - 7.41 (m, 2H),
7.38 - 7.33 (m,
1H), 3.95 - 3.83 (m, 1H), 3.69(s, 3H), 1.46 (d, J= 7.0 Hz, 6H).
Compound intermediate VH-11 was synthesized from IV-11 as the raw material
according to
the synthesis method of compound VII-10, white solid, yield 71%; 1HNMR (400
MHz, CDC13)
7.43 - 7.38 (m, 2H), 7.37 - 7.32 (m, 2H), 7.28 - 7.23 (m, 1H), 6.79 - 6.71 (m,
2H), 4.23 (s, 2H),
3.46 - 3.36 (m, 1H), 3.34 - 3.23 (m, 2H), 3.07 - 2.98 (m, 2H), 1.73 - 1.63 (m,
2H), 1.50 - 1.45 (m,
2H), 1.38 (d, J= 7.1 Hz, 6H).
Compound 11 was synthesized from VII-11 as the raw material according to the
synthesis
method of compound 1, white solid, yield 66%.11-1NMR (400 MHz, CDC13) 5 7.63
(d, Jr 8.9 Hz,
2H), 7.43 - 7.37 (m, 2H), 7.34 - 7.27 (m, 1H), 6.87 (d, J= 9.0 Hz, 2H), 4.29
(s, 2H), 3.50 - 3.29 (m,
4H), 3.12 - 2.98 (m, 2H), 1.80 - 1.69 (m, 2H), 1.59 - 1.47 (m, 2H), 1.43 (d, J
= 7.0 Hz, 6H).
MS(ESI,m/z): 529[M+H].
Example 12:
0
)r)
HN40
12
The synthetic route of example 12 was as follows.
- 27 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
OH
CI 0 0 \
\ 0 \Br HON =CN
CI CI
o-
IV-12 V-12
0 9 \ * N
z '0
CI CI * CN CI
HN--µ0
VII-12 12
Compound intermediate IV-12 ( yield 67%) was synthesized from III-1 as the raw
material
according to the synthesis method of compound IV-10, wherein, methyl
acetoacetate was replaced
by methyl benzoylacetate. 1H NMR (400 MHz, CDC13) 6 8.10 (d, J= 7.9 Hz, 1H),
7.97 (d, J= 7.9
Hz, 1H), 7.58 - 7.44 (m, 5H), 7.41 - 7.36 (m, 1H), 3.65 (s, 3H).
Compound intermediate VII-12 was synthesized from IV-12 as the raw material
according to
the synthesis method of compound VII-10, white solid, yield 74%; 1H NMR (400
MHz, CDC13)
67.95 (dd, J= 7.8, 1.7 Hz, 2H), 7.64 (d, J= 8.9 Hz, 2H), 7.59 - 7.51 (m, 3H),
7.49 - 7.44 (m, 2H),
7.42 - 7.35 (m, 1H), 6.90 (d, J= 8.9 Hz, 2H), 4.46 (s, 2H), 3.57 - 3.50 (m,
1H), 3.46 - 3.37 (m, 2H),
3.12 - 2.99 (m, 2H), 1.81 - 1.70 (m, 2H), 1.62 - 1.49 (m, 2H).
Compound 12 was synthesized from VII-12 as the raw material according to the
synthesis
method of compound 1, white solid, yield 56%. 1H NMR (400 MHz, CDC13) 6 11.50
(s, 1H), 7.95
(dd, Jr 7.8, 1.7 Hz, 2H), 7.64 (d, Jr 8.9 Hz, 2H), 7.59 - 7.51 (m, 3H), 7.49 -
7.44 (m, 2H), 7.42 -
7.35 (m, 1H), 6.90 (d, J= 8.9 Hz, 2H), 4.46 (s, 2H), 3.57 - 3.50 (m, 1H), 3.46
- 3.37 (m, 2H), 3.12
- 2.99 (m, 2H), 1.81 - 1.70 (m, 2H), 1.62 - 1.49 (m, 2H). MS(ESI, m/z):
562[M+H]+.
Example 13:
, 1 0
N,
HN--\13
13
The synthetic route of example 13 was as follows.
- 28 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
CN
N
HO +
IX-13
O ,
14, Br 0 0
\ CI o \
IX-13 N,
CI CN
CI
V-1 VII-13 13
Compound intermediate IX-13 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 94%; 1H
NMR (400 MHz, CDCI3) 5 8.37 (d, Jr 2.0 Hz, 1H), 7.57 (dd, J= 9.1, 2.0 Hz, 1H),
6.62 (d, J
= 9.1 Hz, 1H), 4.16 - 3.93 (m, 3H), 3.41 - 3.30 (m, 2H), 2.00 - 1.91 (m, 2H),
1.63 - 1.52 (m,
2H).
Compound intermediate VII-13 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 74%; 1H NMR (400
MHz,
CDC13) 6 8.26 (d, J= 2.1 Hz, 1H), 7.47 (dd, J= 9.1, 2.1 Hz, 1H), 7.35 - 7.30
(m, 2H), 7.27 -
7.22 (m, 1H), 6.50 (d, J= 9.1 Hz, 1H), 4.27 (s, 2H), 3.66 - 3.56 (m, 2H), 3.45
- 3.40 (m, 1H),
3.36- 3.24 (m, 2H), 2.13 - 2.04 (m, 1H), 1.65 - 1.58 (m, 3H), 1.45 - 1.35 (m,
2H), 1.16 - 1.12
(m, 2H), 1.07 - 1.00 (m, 2H).
Compound 13 was synthesized from VII-13 as the raw material according to the
synthesis method of compound 1, white solid, yield 71%; 1H NMR (400 MHz,
CDC13) 5 8.52
(d, J= 2.4 Hz, 1H), 7.79 (dd, J= 9.1, 2.5 Hz, 1H), 7.45 -7.31 (m, 3H), 6.67
(d, J= 9.1Hz,
1H), 4.38 (s, 2H), 3.81 - 3.67 (m, 2H), 3.61 - 3.50 (m, 1H), 3.46 - 3.33 (m,
2H), 2.17 (s, 1H),
1.82 - 1.68 (m, 2H), 1.59 - 1.45 (m, 2H), 1.31 - 1.27 (m, 2H), 1.19 - 1.12 (m,
2H).
MS(ESI,m/z): 528[M+1-1]
Example 14:
= \ o
CI CI
HN-Nco
14
The synthetic route of example 14 was as follows.
CN
1,1s1i.,CN
HO
HO IX-14
O 0
\ Br 0
\ 0 \ 0
DC-14 N
CI CI a a -ON_
a a
FiN40
V-1 VII-14 14
¨ 29 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Compound intermediate IX-14 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 91%; 1H
NMR (400 MHz, CDC13) 6 8.26 (d, J = 3.0 Hz, 1H), 7.47 (d, J= 8.8 Hz, 1H), 7.09
(dd, J=
8.8, 3.0 Hz, 1H), 4.03 - 3.95 (m, 1H), 3.76 - 3.65 (m, 2H), 3.25 -3.14 (m,
2H), 2.04 - 1.94 (m,
2H), 1.73 - 1.60 (m, 2H).
Compound intermediate VII-14 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 71%; 11-1 NMR
(400 MHz,
CDC13) 8.27 (d, J = 2.7 Hz, 1H), 7.50 (d, J = 8.9 Hz, 1H), 7.44 - 7.38 (m,
2H), 7.37 -7.29
(m, 1H), 7.23 - 7.17 (m, 1H), 4.27 (s, 2H), 3.58 - 3.35 (m, 3H), 3.24 - 3.14
(m, 2H), 2.08 -
.. 1.96 (m, 1H), 1.84 - 1.77 (m, 2H), 1.67 - 1.59 (m, 2H), 1.30 - 1.25 (m,
2H), 1.18 - 1.11 (m,
2H).
Compound 14 was synthesized from VII-14 as the raw material according to the
synthesis method of compound 1, white solid, yield 55%; 1H NMR (400 MHz,
CDC13) 8.29
(d, J = 2.7 Hz, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.44 - 7.39 (m, 2H), 7.33 (d, J
= 7.1 Hz, 1H),
7.23 - 7.17 (m, 1H), 4.37 (s, 2H), 3.58 - 3.48 (m, 1H), 3.42 - 3.35 (m, 2H),
3.20 - 3.13 (m,
2H), 2.20 - 2.12 (m, 1H), 1.84 - 1.77 (m, 2H), 1.64 - 1.57 (m, 2H), 1.30 -
1.27 (m, 2H), 1.18 -
1.12 (m, 2H). MS(ESI,m/z): 528[M+H]+.
Example 15:
mr
ci CI
20 The synthetic route of example 15 was as follows.
CN
CN
HO
IX-15
0
\ Br 0 0
' \
N,
DC-15 1:3-"CN 40 N
CI CI
CI CI
HN40
V-1 VII-15 15
Compound intermediate IX-15 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 78%; 1H
NMR (400 MHz, CDC13) 6 7.41 - 7.34 (m, 2H), 6.96 (d, J= 8.9 Hz, 1H), 3.90 -
3.80 (m, 1H),
3.18 - 3.10 (m, 2H), 2.78 - 2.70 (m, 2H), 2.26 (s, 3H), 2.03 - 1.97 (m, 2H),
1.77 - 1.67 (m,
2H).
Compound intermediate VII-15 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 62%; 1H NMR (400
MHz,
CDC13) 6 7.38 - 7.33 (m, 4H), 7.30 - 7.25 (m, 1H), 6.87 (d, J= 8.2 Hz, 1H),
4.32 (s, 2H),3.43
- 3.34 (m, 1H), 2.94 - 2.85 (m, 2H), 2.65 - 2.56 (m, 2H), 2.20 (s, 3H), 2.16 -
2.09 (m, 1H),
¨ 30 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
1.82- 1.74 (m, 2H), 1.61 - 1.52 (m, 2H), 1.22- 1.18 (m, 2H), 1.11 - 1.05 (m,
2H).
Compound 15 was synthesized from VII-15 as the raw material according to the
synthesis method of compound 1, white solid, yield 65%; 1H NMR (400 MHz,
CDC13) 6 7.62
(d, J= 1.7 Hz, 1H), 7.59 - 7.54 (m, 1H), 7.46 - 7.42 (m, 2H), 7.38 - 7.32 (m,
1H), 7.00 (d, J=
8.4 Hz, 1H), 4.38 (s, 2H), 3.48 - 3.39 (m, 1H), 3.03 - 2.94 (m, 2H), 2.72 -
2.64 (m, 2H), 2.31
(s, 3H), 2.22 - 2.15 (m, 1H), 1.89 - 1.80 (m, 2H), 1.69 - 1.58 (m, 2H), 1.31 -
1.28 (m, 2H),
1.18- 1.13 (m, 2H).MS(ESI,m/z): 541[M+H]'.
Example 16:
\ 0
CI rh1,0
HN40
16
The synthetic route of example 16 was as follows.
Qi1F1 = CN eji 411
HO F HO
IX-16
0
N', \ Br 0
9 \
IX-16 N, -CN * F
a a
CI CI eN el
HN40
V-1 VII-16 16
Compound intermediate IX-16 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 78%; 1H
NMR (400 MHz, CDC13) 6 7.38 - 7.30 (m, 1H), 6.67 - 6.49 (m, 2H), 4.02 - 3.91
(m, 1H), 3.78
- 3.57 (m, 2H), 3.26 - 3.04 (m, 2H), 2.01 - 1.88 (m, 2H), 1.68 - 1.55 (m, 2H).
Compound intermediate VII-16 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 67%; 1H NMR (400
MHz,
CDC13) 6 7.41 - 7.37 (m, 2H), 7.37 - 7.28 (m, 2H), 6.59 - 6.45 (m, 2H), 4.34
(s, 2H), 3.55 -
3.47 (m, 1H), 3.37 - 3.28 (m, 2H), 3.16 - 3.05 (m, 2H), 2.19 - 2.10 (m, 1H),
1.78 - 1.70 (m,
2H), 1.59 - 1.48 (m, 2H), 1.28 - 1.22 (m, 2H), 1.16 - 1.10 (m, 2H).
Compound 16 was synthesized from VII-16 as the raw material according to the
synthesis method of compound 1, white solid, yield 61%; 1H NMR (400 MHz,
CDC13) 6
10.12 (s, 1H), 7.71 (t, J= 8.8 Hz, 1H), 7.44 - 738 (m, 2H), 7.35 - 7.30 (m,
1H), 6.68 (dd, J-
9.1, 2.2 Hz, 1H), 6.53 (dd, J= 15.5, 2.2 Hz, 1H), 4.36 (s, 2H), 3.56- 3.47 (m,
1H), 3.39 -
3.31 (m, 2H), 3.16 - 3.07 (m, 2H), 2.21 -2.11 (m, 1H), 1.81 - 1.74 (m, 2H),
1.62 - 1.51 (m,
2H), 1.32- 1.27 (m, 2H), 1.18- 1.11 (m, 2H). MS(ESI,m/z): 545[M+Hr
Example 17:
¨ 31 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0
toi F
CI
F HN40
17
The synthetic route of example 17 was as follows.
N
..04H 4. CN
= xii F
HO
F F HO IX-17
0 0
\ Br iak F õ
IX-17 N9, \
ON
wir 2-0
CI F CI CI
HN-A)
V-1 VII-17 17
Compound intermediate IX-17 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 55%; 1H
NMR (400 MHz, CDC13) 8 6.43 - 6.32 (m, 2H), 4.08 - 3.98 (m, 1H), 3.71 - 3.62
(m, 2H), 3.27
- 3.17 (m, 2H), 2.02 - 1.94 (m, 2H), 1.70 - 1.61 (m, 2H).
Compound intermediate VII-17 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 67%; 1H NMR (400
MHz,
CDC13) 8 7.43 - 7.39 (m, 2H), 7.36 - 7.30 (m, 1H), 6.34 - 6.27 (m, 2H), 4.35
(s, 2H), 3.60 -
3.50 (m, 1H), 3.35 - 3.26 (m, 2H), 3.19 - 3.08 (m, 2H), 2.18 -2.10 (m, 1H),
1.74- 1.68 (m,
2H), 1.59 - 1.50 (m, 2H), 1.28 - 1.25 (m, 2H), 1.17 - 1.11 (m, 2H).
Compound 17 was synthesized from VII-16 as the raw material according to the
synthesis method of compound 1, white solid, yield 73%; 1H NMR (400 MHz,
CDC13) 8 7.45
- 7.39 (m, 2H), 7.37 -7.31 (m, 1H), 6.40 (d, J= 13.5 Hz, 2H), 4.36 (s, 2H),
3.58 - 3.48 (m,
1H), 3.36 -3.27 (m, 2H), 3.18 - 3.07 (m, 2H), 2.22 -2.10 (m, 1H), 1.79 - 1.70
(m, 2H), 1.61 -
1.52 (m, 2H), 1.30- 1.26 (m, 2H), 1.18 - 1.12 (m, 2H). MS(ESI,m/z): 563[M+H]
Example 18:
0
\
N.
C
I ,
FIN40
18
The synthetic route of example 18 was as follows.
¨ 32 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
F CN
HOCCN
F
IX-18
0
N', \ Br 0 0
' \ CINFP
IX-17 N, N
CI CI N CI z
CI CI
F HN40
V-1 VII-18 18
Compound intermediate IX-18 was synthesized from 4-hydroxypiperidine as the
raw
material according to the synthesis method of compound IX-10, white solid,
yield 61%; 1H
NMR (400 MHz, DMSO) 5 7.62 - 7.47 (m, 2H), 7.08 (t, J= 8.8 Hz, 1H), 3.71 -
3.62 (m, 1H),
3.43 - 3.37 (m, 2H), 3.01 - 2.79 (m, 2H), 1.92 - 1.74 (m, 2H), 1.57 - 1.42 (m,
2H).
Compound intermediate VII-18 was synthesized from V-1 as the raw material
according
to the synthesis method of compound VII-1, white solid, yield 63%; 1H NMR (400
MHz,
CDC13) 5 7.43 - 7.38 (m, 2H), 7.35 - 7.28 (m, 2H), 7.26 - 7.21 (m, 1H), 6.86
(t, J= 8.6 Hz,
1H), 4.35 (s, 2H), 3.49 - 3.41 (m, 1H), 3.26 - 3.15 (m, 2H), 2.96 - 2.86 (m,
2H), 2.20 - 2.12
(m,1H), 1.86- 1.77 (m, 2H), 1.65 - 1.55 (m, 2H), 1.30 - 1.26 (m, 2H), 1.16-
1.09 (m, 2H).
Compound 18 was synthesized from VII-16 as the raw material according to the
synthesis method of compound 1, white solid, yield 65%; 1-FINMR (400 MHz,
CDC13) 5 7.50
- 7.39 (m, 4H), 7.36 - 7.30 (m, 1H), 6.94 (t, J= 8.5 Hz, 1H), 4.37 (s, 2H),
3.55 - 3.42 (m, 1H),
3.33 - 3.17 (m, 2H), 3.00 -2.84 (m, 2H), 2.22- 2.13 (m, 1H), 1.89 - 1.80 (m,
2H), 1.69 - 1.58
(m, 2H), 1.32- 1.26 (m, 2H), 1.19- 1.12 (m, 2H). MS(ESI,m/z): 545 [M+H]
Example 19:
P
CI
c,
N\
19
The synthetic route of example 19 was as follows.
0 N \ I 0 P
CI 0
NP\
a
CI CI \ CI ci
N
W
VI-1 VII-19 19
Compound intermediate VII-19 was synthesized from VI-1 as the raw material
according to the synthesis method of compound VII-1, white solid, yield 37%;
1H NMR (400
MHz, CDC13) 5 7.67 (s, 1H), 7.56 (s, 1H), 7.37 (d, J= 7.9 Hz, 2H), 7.29 - 7.23
(m, 1H), 7.09
(d, J= 8.9 Hz, 1H), 6.93 (d, J= 8.9 Hz, 1H), 4.31 (s, 2H), 3.52 (s, 3H), 3.37 -
3.27 (m, 1H),
3.24- 3.14 (m, 2H), 2.83 -2.71 (m, 2H), 2.20 - 2.09 (m, 1H), 1.86 - 1.75 (m,
2H), 1.63 - 1.52
(m, 2H), 1.26 - 1.18 (m, 2H), 1.12 - 1.07 (m, 2H).
¨ 33 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Compound 19 was synthesized from VII-19 as the raw material according to the
synthesis
method of compound 1, white solid, yield 29%; 11-1 NMR (400 MHz, DMSO-d6) 6
7.81 (s, 1H),
7.63 (d, J = 7.7 Hz, 2H), 7.55 - 7.50 (m, 1H), 7.43 (d, J= 9.0 Hz, 1H), 7.28
(s, 1H), 7.04 (d, J=
9.0 Hz, 1H), 4.33 (s, 2H), 3.83 (s, 3H), 3.35 (s, 1H), 3.09 (s, 2H), 2.80 -
2.72 (m, 2H), 2.04 - 1.95
(m, 1H), 1.75 (s, 2H), 1.50 - 1.41 (m, 2H), 1.20 - 1.07 (m, 4H). MS(ESI,m/z):
580 [M+H1-F.
Example 20:
0
N
N
CI CI
HNA
The synthetic route of example 20 was as follows.
9
N' N 0 Cs-CN N
CI c NN OH ______________ CI a 'o
H2N HN4s
10 VIII-2 20
VIII-2 (0.41 g, 0.83 mmol), N,N'-thiocarbonyldiimidazole (1.0 mmol) and 1,4-
dioxane (4 mL)
were added into a round bottom flask, and then 1,8-
diazabicyclic[5.4.0]undecarbon-7-ene (0.91
mmol) was heated to 100 C and reacted for 3 hours. The reaction solution was
cooled to room
temperature, diluted with water (5 mL), adjusted to pH approximately equal to
2 with a 1M
15 aqueous hydrochloric acid solution, and then extracted with ethyl
acetate (4 mL each time, 3 times
in total). The organic phases were combined, washed with saturated brine,
concentrated and the
crude product obtained was subjected to silica gel column chromatography to
obtain the final
product 20 (0.22 g, yield 49%). IFINMR (400 MHz, CDC13) 6 7.44 - 7.37 (m, 3H),
7.34 - 7.30 (m,
1H), 6.89 (d, J= 8.9 Hz, 2H), 4.36 (s, 2H), 3.44 - 3.37 (m, 1H), 3.24 - 3.14
(m, 2H), 2.83 - 2.73 (m,
20 2H), 2.21 - 2.14 (m, 1H), 1.81 - 1.76 (m, 2H), 1.61 - 1.54 (m, 2H), 1.32
- 1.26 (m, 2H), 1.17 - 1.09
(m, 2H). MS(ESI,m/z): 543 [M+Hr.
Example 21:
o ,
r4-
CF3
HN40
21
The synthetic route of example 21 was as follows.
-34
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 0
\ Br IX-13 , Nj ,
CF3 CF3
CF3
N
H N40
V-8 VII-21
21
Compound intermediate VII-21 was synthesized from V-8 and IX-13 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 62%; 1H
NMR (400 MHz, CDC13) 6 8.37 (d, J= 1.1 Hz, 1H), 7.79 (d, J= 7.8 Hz, 1H), 7.66-
7.53 (m,
3H), 7.46 (d, J= 6.8 Hz, 1H), 6.57 (d, J= 9.1 Hz, 1H), 4.28 (s, 2H), 3.80 -
3.71 (m, 2H), 3.54
-3.46 (m, 1H), 3.41 -3.33 (m, 2H), 2.16 - 2.08 (m, 1H), 1.79- 1.70 (m, 2H),
1.55- 1.45 (m,
2H), 1.26 - 1.21 (m, 2H), 1.14 - 1.08 (m, 2H).
Compound 21 was synthesized from VII-21 as the raw material according to the
synthesis method of compound 1, white solid, yield 69%; 1H NMR (400 MHz, DMSO)
5 8.46
(d, J= 2.1 Hz, 1H), 7.86 (d, J= 7.8 Hz, 1H), 7.81 - 7.72 (m, 2H), 7.68 (t, J=
7.6 Hz, 1H),
7.58 (d, J= 7.6 Hz, 1H), 6.85 (d, J= 9.2 Hz, 1H), 4.28 (s, 2H), 3.81 -3.70 (m,
2H), 3.45 (s,
1H), 3.25 (t, J= 9.4 Hz, 2H), 2.33 - 2.23 (m, 1H), 1.75 - 1.61 (m, 2H), 1.38 -
1.25 (m,2H),
1.17 - 1.04 (m,4H). MS(ESI,m/z): 528 [M+H].
Example 22:
N
CF3
NO
HN40
22
The synthetic route of example 22 was as follows.
o
\ Br IX-14 NONõ ' 0
CF3 CF3
CF3 "11,0
HNA
V-8 22
Compound intermediate VII-22 was synthesized from V-8 and IX-14 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 71%; 1H
NMR (400 MHz, CDC13) 6 8.23 (d, J= 2.7 Hz, 1H), 7.78 (d, J= 7.2 Hz, 1H), 7.62 -
7.53 (m,
2H), 7.49 - 7.43 (m, 2H), 7.03 (dd, J= 8.8, 2.7 Hz, 1H), 4.28 (s, 2H), 3.53 -
3.46 (m, 1H),
3.43 - 3.36 (m, 2H), 3.18 - 3.10 (m, 2H), 2.15 -2.07 (m, 1H), 1.83 - 1.75 (m,
2H), 1.62 - 1.52
(m, 2H), 1.24- 1.20 (m, 2H), 1.15 - 1.09 (m, 2H).
Compound 22 was synthesized from VII-22 as the raw material according to the
synthesis method of compound 1, white solid, yield 66%; 1H NMR (400 MHz, DMSO)
5 8.33
(d, J= 2.3 Hz, 1H), 7.85 (d, J= 7.7 Hz, 1H), 7.78 - 7.64 (m, 3H), 7.59 (d, J=
7.5 Hz, 1H),
7.32 (dd, J= 9.0, 2.3 Hz, 1H), 4.29 (s, 2H), 3.50 - 3.36 (m, 3H), 3.06 (t, J=
9.1 Hz, 2H), 2.34
- 2.23 (m, 1H), 1.79 - 1.68 (m, 2H), 1.46 - 1.33 (m, 2H), 1.14 - 1.04 (m, 4H).
MS(ESI,m/z):
528 [M+H]
Example 23:
¨35 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 F
14N \ '-eN
CF3 *
HN-0
23
The synthetic route of example 23 was as follows.
NP, \ Br ix-18 N9, \ F \ 0
CF3 CF3 *
CF3 ,N1-0
HN-AD
V-8 VII-23 23
Compound intermediate VII-23 was synthesized from V-8 and IX-18 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 70%; 1H
NMR (400 MHz, CDC13) 6 7.84 - 7.79 (m, 1H), 7.66 - 7.56 (m, 2H), 7.49 (d, J=
6.8 Hz, 1H),
7.35 (dd, Jr 8.4, 1.3 Hz, 1H), 7.28 - 7.24 (m, 1H), 6.87 (t, J= 8.4 Hz, 1H),
4.29 (s, 2H), 3.49
- 3.38 (m, 1H), 3.32 - 3.23 (m, 2H), 2.97 - 2.88 (m, 2H), 2.18 - 2.09 (m, 1H),
1.90 - 1.82 (m,
2H), 1.68 - 1.59 (m, 2H), 1.29 - 1.25 (m, 2H), 1.17 - 1.10 (m, 2H).
Compound 23 was synthesized from VII-23 as the raw material according to the
synthesis method of compound 1, white solid, yield 61%; 1H NMR (400 MHz, DMSO)
6 7.91
(d, J= 7.7 Hz, 1H), 7.83 - 7.70 (m, 2H), 7.60 (d, J= 7.5 Hz, 1H), 7.56- 7.47
(m, 2H), 7.14 -
7.07 (m, 1H), 4.29 (s, 2H), 3.44 - 3.33 (m, 1H), 3.18 - 3.09 (m, 2H), 2.88 -
2.79 (m, 2H), 2.37
- 2.29 (m, 1H), 1.82 - 1.73 (m, 2H), 1.50 - 1.39 (m, 2H), 1.18 - 1.06 (m, 4H).
MS(ESI,m/z):
545 [M+H]+.
Example 24:
*,F
INV '4
N'
CF
40 3 HN0
24
The synthetic route of example 24 was as follows.
\ Br IX-16 NON F N \
CF3 CF3 1110
N CF3
HN--%
V-8
23
Compound intermediate VII-24 was synthesized from V-8 and IX-16 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 70%; 1H
NMR (400 MHz, CDC13) 6 7.82 - 7.75 (m, 1H), 7.64 - 7.54 (m, 2H), 7.47 - 7.43
(m, 1H), 7.38
- 7.31 (m, 1H), 6.57 (dd, J= 8.9, 2.4 Hz, 1H), 6.48 (dd, J= 13.3, 2.4 Hz, 1H),
4.28 (s, 2H),
3.52 - 3.45 (m, 1H), 3.43 -3.34 (m, 2H), 3.15 - 3.04 (m, 2H), 2.16 - 2.07 (m,
1H), 1.83- 1.72
(m, 2H), 1.59 - 1.49 (m, 2H), 1.25 - 1.20 (m, 2H), 1.15 - 1.09 (m, 2H).
Compound 24 was synthesized from VII-24 as the raw material according to the
¨ 36
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
synthesis method of compound 1, white solid, yield 61%; 1H NMR (400 MHz, DMSO)
6 7.89
(d, J = 7.7 Hz, 1H), 7.81¨ 7.68 (m, 2H), 7.59 (d, J = 7.5 Hz, 1H), 7.52 (t, J
= 8.9 Hz, 1H),
6.85 (dd, J= 10.2, 7.5 Hz, 2H), 4.28 (s, 2H), 3.48 - 3.39 (m, 3H), 3.11 - 3.00
(m, 2H), 2.38 -
2.27 (m, 1H), 1.75 - 1.65 (m, 2H), 1.39 - 1.27 (mS, 2H), 1.17 - 1.05 (m, 4H).
MS(ESI,m/z):
545 [M+H]
Example 25:
0
ci¨CN F
F
The synthetic route of example 25 was as follows.
r\i N \ Br ix_17 0--CN F C14 F
CF3 CF3 * CF3
F HN40
V-8 VU-25 25
10 Compound intermediate VII-25 was synthesized from V-8 and IX-17 as the
raw
materials according to the synthesis method of compound VII-10, white solid,
yield 59%; 1H
NMR (400 MHz, CDC13) 6 7.79 - 7.76 (m, 1H), 7.64 - 7.54 (m, 2H), 7.47 - 7.43
(m, 1H), 6.30
(d, J = 11.7 Hz, 2H), 4.27 (s, 2H), 3.54 - 3.46 (m, 1H), 3.40 -3.30 (m, 2H),
3.18- 3.06 (m,
2H), 2.16 -2.05 (m, 1H), 1.82- 1.70 (m, 2H), 1.57 - 1.49 (m, 2H), 1.23 - 1.18
(m, 2H), 1.14 -
15 1.07 (m, 2H).
Compound 25 was synthesized from VII-25 as the raw material according to the
synthesis method of compound 1, white solid, yield 64%; 1H NMR (400 MHz, DMSO)
6 7.89
(d, J = 7.7 Hz, 1H), 7.80 - 7.68 (m, 2H), 7.60 (d, J= 7.4 Hz, 1H), 6.77 (d, J=
13.3 Hz, 2H),
4.29 (s, 2H), 3.50 - 3.38 (m, 3H), 3.18 - 3.03 (m, 2H), 2.37 - 2.25 (m, 1H),
1.78 - 1.64 (m,
20 2H), 1.43 - 1.27 (m, 2H), 1.20 - 1.03 (m, 4H). MS(ESI,m/z): 563
[M+H]
Example 26:
0 ,
0-0,<
ocFa )4,0
HN40
21
The synthetic route of example 26 was as follows.
o
ni, \ IX-13 hi
/r4=-=-1
OCF3 OCF3 OCF3
¨N
HN40
V-9 VII-26 26
25 Compound intermediate VII-26 was synthesized from V-9 and IX-13 as the
raw
materials according to the synthesis method of compound VII-10, white solid,
yield 67%; 1H
NMR (400 MHz, CDC13) 6 8.37 (s, 1H), 7.60 - 7.47 (m, 3H), 7.41 - 7.34 (m, 2H),
6.57 (d, J=
9.1 Hz, 1H), 4.41 (s, 2H), 3.85 - 3.73 (m, 2H), 3.60 - 3.50 (m, 1H), 3.43 -
3.32 (m, 2H), 2.19
¨ 37 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
- 2.09 (m, 1H), 1.81 - 1.73 (m, 2H), 1.57 - 1.47 (m, 2H), 1.25 - 1.20 (m, 2H),
1.14 - 1.07 (m,
2H).
Compound 26 was synthesized from VII-26 as the raw material according to the
synthesis method of compound 1, white solid, yield 68%; 1H NMR (400 MHz, DMSO)
6 8.46
(d, J= 2.3 Hz, 1H), 7.80 (dd, J= 9.1, 13 Hz, 1H), 7.69 -7.61 (m, 2H), 7.57 -
7.49 (m, 2H),
6.92 (d, J= 9.2 Hz, 1H), 4.38 (s, 2H), 3.88 - 3.75 (m, 2H), 3.57 - 3.47 (m,
1H), 3.32 - 3.21 (m,
2H), 2.37 - 2.27 (m, 1H), 1.77 - 1.67 (m, 2H), 1.38 - 1.27 (m, 2H), 1.16 -
1.05 (m, 4H).
MS(ESI,m/z): 544 [M+H1 .
Example 27:
- N
OCF3
HN40
ki 27
The synthetic route of example 27 was as follows.
o
N \ Br IX-14
-N
OCF3 OCF3 z
OCF3
HN4
V-9 VII-27 0
27
Compound intermediate VII-27 was synthesized from V-9 and IX-14 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 74%; 1H
NMR (400 MHz, CDC13) 6 8.23 (s, 1H), 7.58 - 7.43 (m, 3H), 7.37 (d, J= 7.8 Hz,
2H), 7.03
(dd, J= 8.8, 2.7 Hz, 1H), 4.40 (s, 2H), 3.57 - 3.50 (m, 1H), 3.48 - 3.38 (m,
2H), 3.19- 3.09
(m, 2H), 2.17 - 2.08 (m, 1H), 1.87 - 1.75 (m, 2H), 1.65 - 1.54 (m, 2H), 1.24 -
1.18 (m, 2H),
1.13 - 1.07 (m, 2H).
Compound 27 was synthesized from VII-27 as the raw material according to the
synthesis method of compound 1, white solid, yield 61%; 1H NMR (400 MHz, DMSO)
6 8.34
(d, J= 2.4 Hz, 1H), 7.71 (d, J= 8.9 Hz, 1H), 7.67 -7.58 (m, 2H), 7.52 - 7.46
(m, 2H), 7.33
(dd, Jr 8.9, 2.1 Hz, 1H), 4.38 (s, 2H), 3.56 -3.42 (m, 3H), 3.07 (t, J= 9.3
Hz, 2H), 2.34 -
2.23 (m, 1H), 1.83 - 1.72 (m, 2H), 1.49 - 1.37 (m, 2H), 1.14 - 1.03 (m, 4H).
MS(ESI,m/z):
544 [M+H]+.
Example 28:
0
N
-CN
/
OCF3
NW-A)
28
The synthetic route of example 28 was as follows.
NBr0
\ IX-18 NO rµi \
k N
OCF3 OCF3 OCF3
V-9 VII-28
28
¨ 38 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Compound intermediate VII-28 was synthesized from V-9 and IX-18 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 67%; 1H
NMR (400 MHz, CDC13) 6 7.60 - 7.56 (m, 1H), 7.55 - 7.49 (m, 1H), 7.41 - 7.37
(m, 2H), 7.33
(d, J= 8.4 Hz, 1H), 7.25 (d, J= 12.7 Hz, 1H), 6.87 (t, J= 8.6 Hz, 1H), 4.41
(s, 2H), 3.51 -
3.42 (m, 1H), 3.34- 3.23 (m, 2H), 2.97 -2.88 (m, 2H), 2.20 - 2.11 (m, 1H),
1.91 - 1.82 (m,
2H), 1.70 - 1.59 (m, 2H), 1.25 - 1.22 (m, 2H), 1.15 - 1.08 (m, 2H).
Compound 28 was synthesized from VII-28 as the raw material according to the
synthesis method of compound 1, white solid, yield 72%; 1H NMR (400 MHz, DMSO)
87.69
- 7.62 (m, 2H), 7.57 - 7.46 (m, 4H), 7.10 (t, J= 8.7 Hz, 1H), 4.37 (s, 2H),
3.48 - 3.40 (m, 1H),
3.24 - 3.11 (m, 2H), 2.85 (t, J= 9.2 Hz, 2H), 2.40 - 2.25 (m, 1H), 1.87- 1.75
(m, 2H), 1.56 -
1.40 (m, 2H), 1.16- 1.04 (m, 4H). MS(ESI,m/z): 561 [M+H]+.
Example 29:
P
,11,5.0
OCF3
29
The synthetic route of example 29 was as follows.
9 \ 15 B 0 \ 0
N IX-16 NOL-C. F
--C\N
OCF3 OCF3 N ocF3
HN4
V-9 VII-29
29
Compound intermediate VII-29 was synthesized from V-9 and IX-16 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 72%; 1H
NMR (400 MHz, CDC13) 6 7.57 - 7.47 (m, 2H), 7.41 - 7.30 (m, 3H), 6.57 (dd, 1=
8.9, 2.4 Hz,
1H), 6.48 (dd, J= 13.3, 2.4 Hz, 1H), 4.40 (s, 2H), 3.57 -3.49 (m, 1H), 3.46-
3.37 (m, 2H),
3.15 - 3.05 (m, 2H), 2.17 -2.09 (m, 1H), 1.84- 1.74 (m, 2H), 1.60 - 1.50 (m,
2H), 1.23 - 1.18
(m, 2H), 1.13 - 1.06 (m, 2H).
Compound 29 was synthesized from VII-29 as the raw material according to the
synthesis method of compound 1, white solid, yield 60%; 1H NMR (400 MHz, DMSO)
67.69
- 7.61 (m, 2H), 7.58 - 7.48 (m, 3H), 6.91 - 6.80 (m, 2H), 4.38 (s, 2H), 3.55 -
3.43 (m, 3H),
3.14- 2.99 (m, 2H), 2.37 -2.27 (m, 1H), 1.79- 1.66 (m, 2H), 1.43 - 1.30 (m,
2H), 1.18 - 1.03
(m, 4H). MS(ESI,m/z): 561 [M+H].
Example 30:
c)--CN
OCF3
F HN40
The synthetic route of example 30 was as follows.
¨ 39
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0 0 \ 0
\ Br 1X-17
t F \
OCF3 OCF3 'N'O
OCF3
HN4
F 0
V-9 V11-30 30
Compound intermediate VII-30 was synthesized from V-9 and IX-17 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 62%; 1H
NMR (400 MHz, CDC13) 6 7.57 - 7.49 (m, 2H), 7.42 - 7.35 (m, 2H), 6.31 (d, J=
11.6 Hz, 2H),
4.41 (s, 2H), 3.59 - 3.51 (m, 1H), 3.42 - 3.34 (m, 2H), 3.19 - 3.08 (m, 2H),
2.17 - 2.09 (m,
1H), 1.82 - 1.73 (m, 2H), 1.61 - 1.51 (m, 2H), 1.24 - 1.20 (m, 2H), 1.14 -
1.08 (m, 2H).
Compound 30 was synthesized from VII-30 as the raw material according to the
synthesis method of compound 1, white solid, yield 64%; 1H NMR (400 MHz, DMSO)
57.69
- 7.62 (m, 2H), 7.57 - 7.49 (m, 2H), 6.79 (d, J= 13.3 Hz, 2H), 4.38 (s, 2H),
3.57 - 3.45 (m,
3H), 3.17 -3.06 (m, 2H), 2.40- 2.25 (m, 1H), 1.82 - 1.66 (m, 2H), 1.44 - 1.28
(m, 2H), 1.19 -
1.02 (m, 4H). MS(ESI,m/z): 579 [M+11]+.
Example 31:
N' \
0
31
The synthetic route of example 31 was as follows.
HO
X:CNH F HO-ZN_
1X-31
0
F io F
FFF
IX-13
11-31 1V-31 V-31
0 \ 0
/N1,0
HN----µ
0
VII-31
31
Compound intermediate IX-31 was synthesized from nortropine as the raw
material
according to the synthesis method of compound IX-10, white solid, yield 94%;
1H NMR (400
MHz, CDC13) 6 8.41 (d, 1= 1.8 Hz, 1H), 7.58 (dd, J = 9.0, 2.3 Hz, 1H), 6.49
(d, J = 8.9 Hz,
1H), 4.91 -4.26 (m, 2H), 4.12 (t, J= 4.7 Hz, 1H), 2.39 (d, J=-- 7.3 Hz, 2H),
2.18 - 2.04 (m,
4H), 1.81 (d, J= 14.3 Hz, 2H);
Compound V-31 was synthesized from 11-31 as the raw material according to the
synthesis
method of compound 10, wherein,
¨ 40 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
IV-31, white solid, yield 64%. 1H NMR (400 MHz, CDC13) 6 7.43 - 7.34 (m, 1H),
7.00 - 6.91
(m, 2H), 3.69 (s, 3H), 2.92 -2.83 (m, 1H), 1.36 - 1.31 (m, 2H), 1.25 - 1.20
(m, 2H).
V-31, colourless liquid, yield 82%. 1H NMR (400 MHz, CDC13) 6 7.54 - 7.44 (m,
1H), 7.11 -
7.04 (m, 2H), 4.33 (s, 2H), 2.20- 2.08 (m, 1H), 1.34 - 1.17 (m, 4H).
Compound intermediate VII-31 was synthesized from V-31 and IX-13 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 55%. 1H
NMR (400 MHz, CDC13) 6 8.36 (d, J= 2.1 Hz, 1H), 7.53 (dd, J= 9.0, 2.1 Hz, 1H),
7.49 -
6.39 (m, 1H), 7.07 - 6.96 (m, 2H), 6.42 (d, J= 9.0 Hz, 1H), 4.75 -4.18 (m,
4H), 3.48 (t, J=
4.5 Hz, 1H), 2.16- 2.08 (m, 1H), 1.99 - 1.92 (m, 2H), 1.90 - 1.81 (m, 4H),
1.69 (d, J= 14.5
.. Hz, 2H), 1.25 - 1.20 (m, 2H), 1.14- 1.08 (m, 2H).
Compound 31 was synthesized from VII-31 as the raw material according to the
synthesis method of compound 1, white solid, yield 76%. 1H NMR (400 MHz, DMS0)
6 8.46
(d, J= 2.3 Hz, 1H), 7.78 (dd, J= 9.0, 2.3 Hz, 1H), 7.70 - 7.59 (m, 1H), 7.29
(t, J= 8.0 Hz,
2H), 6.76 (d, J= 9.0 Hz, 1H), 4.41 (s, 2H), 4.31 (s, 2H), 3.45 (s, 1H), 2.39 -
2.26 (m, 1H),
1.86 - 1.68 (m, 6H), 1.59 (d, J = 14.4 Hz, 2H), 1.19 - 1.03 (m, 4H).
MS(ESI,m/z):
522[M+1-1]
Example 32:
\
1=1,0
CI CI ,
HN--µ
0
32
The synthetic route of example 32 was as follows.
o o ,
\ Br
IX-31 CL-CN* /4'
CI CI CI cICN CI
0N 3N
HN¨Z
V-1 VII-32 32
Compound intermediate VII-32 was synthesized from V-1 and IX-31 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 69%; 1H
NMR (400 MHz, CDC13) 6 8.34 (d, J= 2.1 Hz, 1H), 7.51 (dd, J= 9.0, 2.1 Hz, 1H),
7.42 -
7.38 (m, 2H), 7.36 - 7.30 (m, 1H), 6.41 (d, J= 9.0 Hz, 1H), 4.63 - 4.16 (m,
4H), 3.46 (t, J=
.. 4.3 Hz, 1H), 2.15 - 2.07 (m, 1H), 1.97 - 1.91 (m, 2H), 1.88 - 1.78 (m, 4H),
1.70 (d, J= 14.5
Hz, 2H), 1.25 - 1.21 (m, 2H), 1.14 - 1.07 (m, 2H).
Compound 32 was synthesized from VII-32 as the raw material according to the
synthesis method of compound 1, white solid, yield 68%; 1H NMR (400 MHz, DMSO)
6 8.45
(d, J= 2.3 Hz, 1H), 7.78 (dd, J= 9.0, 2.3 Hz, 1H), 7.68 -7.60 (m, 2H), 7.59 -
7.53 (m, 1H),
.. 6.77 (d, J= 9.1 Hz, 1H), 4.41 (s, 2H), 4.26 (s, 2H), 3.43 (s, 1H), 2.38 -
2.28 (m, 1H), 1.86 -
1.54 (m, 8H), 1.18 - 1.05 (m, 4H). MS(ESI,m/z): 554 [M+Fl]
Example 33:
¨ 41 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
0
\
CF3
HN--µ
0
33
The synthetic route of example 33 was as follows.
\ Br ,; \ n
IX-31 N
C F3
CF3 U-CN CF3
V-8 VII-33
33
Compound intermediate VII-33 was synthesized from V-8 and IX-31 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 73%; 1H
NMR (400 MHz, CDC13) 6 8.32 (d, J= 2.1 Hz, 1H), 7.76 (d, J= 7.3 Hz, 1H), 7.63 -
7.53 (m,
2H), 7.49 (dd, J= 9.0, 2.1 Hz, 1H), 7.44 - 7.40 (m, 1H), 639 (d, Jr 9.0 Hz,
1H), 4.57 - 4.12
(m, 4H), 3.42 (t, J= 4.3 Hz, 1H), 2.13 -2.04 (m, 1H), 1.98 - 1.91 (m, 2H),
1.88 - 1.78 (m,
4H), 1.69 (d, J= 14.6 Hz, 2H), 1.20- 1.15 (m, 2H), 1.11 - 1.05 (m, 2H).
Compound 33 was synthesized from VII-33 as the raw material according to the
synthesis method of compound 1, white solid, yield 51%; NMR (400 MHz, DMSO)
6 8.45
(d, J= 2.3 Hz, 1H), 7.92 (d, J= 7.4 Hz, 1H), 7.84 - 7.72 (m, 3H), 7.60 (d, J=
7.4 Hz, 1H),
6.79 (d, J= 9.1 Hz, 1H), 4.19 (s, 2H), 4.23 (s, 2H), 3.43 (s, 1H), 2.37 - 2.28
(m, 1H), 1.82 -
1.69 (m, 6H), 1.62 (d, J= 14.4 Hz, 2H), 1.17 - 1.05 (m, 4H). MS(ESI,m/z): 554
[M+H].
Example 34:
P\
/N,0
OCF3
HN4
0
34
The synthetic route of example 34 was as follows.
N' \ Br \ \
ix.31 N 0
OCF3¨' OCF3 OCF3 /
V-9 VII-34
34
Compound intermediate VII-34 was synthesized from V-9 and IX-31 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 76%; Ili
NMR (400 MHz, CDC13) 6 8.35 (d, J= 2.1 Hz, 1H), 7.58 - 7.47 (m, 3H), 7.40 -
7.35 (m, 2H),
6.44 - 6.38 (m, 1H), 4.57 -4.23 (m, 4H), 3.48 (t, J= 4.5 Hz, 1H), 7.16 - 2.06
(m, 1H), 1.98 -
1.92 (m, 2H), 1.90- 1.82 (m, 4H), 1.70 (d, J= 14.5 Hz, 2H), 1.22- 1.18 (m,
2H), 1.13 - 1.07
(m, 2H).
Compound 34 was synthesized from VII-34 as the raw material according to the
¨ 42 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
synthesis method of compound 1, white solid, yield 66%; 1H NMR (400 MHz, DMSO)
6 8.47
(d, J= 2.3 Hz, 1H), 7.79 (dd, J= 9.0, 2.3 Hz, 1H), 7.69 - 7.59 (m, 2H), 7.56 -
7.49 (m, 2H),
6.75 (d, J= 9.1 Hz, 1H), 4.41 (s, 2H), 4.32 (s, 2H), 3.47 (s, 1H), 2.35 -2.26
(m, 1H), 1.86 -
1.68 (m, 6H), 1.63 (d, J= 14.4 Hz, 2H), 1.15 - 1.02 (m, 4H). MS(ESI,m/z): 570
[M+H].
Example 35:
HN4
0
The synthetic route of example 35 was as follows.
HO
-
.
0 tdi F
HO
CN
CN
IX-35
' BrF \ 0
N.\
IX-35 CY¨/"..?CczN N 1110 1,4
CN /
HN40
V-31 VII-35 35
10 Compound intermediate IX-35 was synthesized from nortropine as the raw
material according
to the synthesis method of compound IX-10, white solid, yield 64%; 1H NMR (400
MHz, CDC13)
6 7.42 - 7.34 (m, 1H), 6.48 (dd, J= 8.8, 2.3 Hz, 1H), 6.41 (dd, J= 12.8, 2.3
Hz, 1H), 4.21 (s, 2H),
4.09 (t, J= 4.4 Hz, 1H), 2.44 - 2.36 (m, 2H), 2.18 - 2.05 (m, 4H), 1.74 (d, J=
13.9 Hz, 2H), 1.66 -
1.59 (m, 1H);
15 Compound intermediate VII-35 was synthesized from V-31 and IX-35 as the
raw materials
according to the synthesis method of compound VII-10, white solid, yield 79%;
1H NMR (400
MHz, CDC13) 6 7.49 - 7.41 (m, 1H), 7.37 - 7.30 (m, 1H), 7.07 - 7.00 (m, 2H),
6.44 - 7.31 (m, 2H),
4.31 (s, 2H), 4.06 (s, 2H), 3.46 (t, J= 4.5 Hz, 1H), 2.16 - 2.08 (m, 1H), 1.99
- 1.92 (m, 2H), 1.91 -
1.82 (m, 4H), 1.63 (d, J= 14.5 Hz, 2H), 1.25 - 1.20 (m, 2H), 1.15 - 1.09 (m,
2H).
20 Compound 35 was synthesized from VII-35 as the raw material according to
the
synthesis method of compound 1. White solid, yield 66%; 1H NMR (400 MHz, DMSO)
6
7.70 - 7.59 (m, 1H), 7.50 (t, J= 8.6 Hz, 1H), 7.29 (t, J= 8.0 Hz, 2H), 6.76 -
6.64 (m, 2H),
4.31 (s, 2H), 4.17 (s, 2H), 3.43 (s, 1H), 2.37 -2.26 (m, 1H), 1.83 - 1.67 (m,
6H), 1.52 (d, J=
14.5 Hz, 2H), 1.16 - 1.04 (m, 4H). MS(ESI,m/z): 539 [M+H]
25 Example 36:
CI CI
HN--µ
0
36
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
The synthetic route of example 36 was as follows.
o
\ Br ' \ 0
F N 1111 N --(CN
CI CI ci ci CN
V-1 VII-36
36
Compound intermediate VII-36 was synthesized from V-1 and IX-35 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 73%; 1H
NMR (400 MHz, CDC13) 6 7.46 - 7.41 (m, 2H), 7.39 -7.31 (m, 2H), 6.42 (dd, J=
8.8, 2.1 Hz,
1H), 6.35 (d, J= 12.8 Hz, 1H), 4.26 (s, 2H), 4.07 (s, 2H), 3.46 (t, J= 4.4 Hz,
1H), 2.17 - 2.08
(m, 1H), 2.01 - 1.95 (m, 2H), 1.91 - 1.82 (m, 4H), 1.66 (d, J= 14.7 Hz, 2H),
1.29 - 1.24 (m,
2H), 1.17 - 1.10 (m, 2H).
Compound 36 was synthesized from VII-36 as the raw material according to the
synthesis method of compound 1, white solid, yield 65%; 'H NMR (400 MHz, DMSO)
57.65
- 7.59 (m, 2H), 7.58 - 7.47 (m, 2H), 6.69 (t, J= 12.8 Hz, 2H), 4.25 (s, 2H),
4.17 (s, 2H), 3.41
(s, 1H), 2.38 -2.27 (m, 1H), 1.8. - 1.68 (m, 6H), 1.54 (d, J= 14.5 Hz, 2H),
1.17- 1.05 (m,
4H). MS(ESI,m/z): 571 [M+H]+.
Example 37:
CF3
HN4
37
The synthetic route of example 37 was as follows.
\ \' \
N Br 35 NO* N (:)--(CN N
CF3 CF3 CN CF3 / '0
HN40
V-8 VII-37
37
Compound intermediate VII-37 was synthesized from V-8 and IX-35 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 74%; 1H
NMR (400 MHz, CDC13) 6 7.84 - 7.79 (m, 1H), 7.67 - 7.58 (m, 2H), 7.48 - 7.42
(m, 1H), 7.39
- 7.32 (m, 1H), 6.45 - 6.31 (m, 2H), 4.20 (s, 2H), 4.08 (s, 2H), 3.43 (s, 1H),
2.14 - 2.07 (m,
1H), 2.02 - 1.95 (m, 2H), 1.94 - 1.83 (m, 4H), 1.66 (d, J= 14.7 Hz, 2H), 1.29 -
1.23 (m, 2H),
1.16- 1.09(m, 2H).
Compound 37 was synthesized from VII-37 as the raw material according to the
synthesis method of compound 1, white solid, yield 61%; 1H NMR (400 MHz, DMSO)
57.91
(d, J= 7.6 Hz, 1H), 7.83 - 7.70 (m, 2H), 7.60 (d, J= 7.4 Hz, 1H), 7.50 (t, J=
8.6 Hz, 1H),
6.75 - 6.64 (m, 2H), 4.26 - 4.13 (m, 4H), 3.40 (s, 1H), 2.38 - 2.24 (m, 1H),
1.83 - 1.69 (m,
6H), 1.55 (d, .7= 14.5 Hz, 2H), 1.18- 1.03 (m, 4H). MS(ESI,m/z): 571 [M+H1+.
¨ 44 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Example 38:
\ o¨õC\N
zN
OCF3
HN4
0
38
The synthetic route of example 38 was as follows.
o ,
NiN \ Br \ " 0 N \ 0
1X-35
OCF3 CN OCF3
HN40
V-9 V11-38
38
Compound intermediate VII-38 was synthesized from V-9 and IX-35 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 74%; 1H
NMR (400 MHz, CDC13) 6 7.58 - 7.50 (m, 2H), 7.43 - 7.37 (m, 2H), 7.34 (t, J=
8.2 Hz, 1H),
6.42 (dd, J= 8.8, 2.2 Hz, 1H), 6.35 (dd, J= 12.8, 2.2 Hz, 1H), 4.34 (s, 2H),
4.06 (s, 2H), 3.47
(t, J = 4.5 Hz, 1H), 2.16 - 2.07 (m, 1H), 2.00 - 1.94 (m, 2H), 1.92 - 1.84 (m,
4H), 1.64 (d, J=
14.5 Hz, 2H), 1.26 - 1.20 (m, 2H), 1.15 - 1.09 (m, 2H).
Compound 38 was synthesized from VII-38 as the raw material according to the
synthesis method of compound 1, white solid, yield 67%; 1H NMR (400 MHz, DMSO)
87.71
- 7.60 (m, 2H), 7.58 - 7.46 (m, 3H), 6.77 - 6.64 (m, 2H), 4.32 (s, 2H), 4.19
(s, 2H), 3.45 (s,
1H), 2.37 -2.27 (m, 1H), 1.86 - 1.69 (m, 6H), 1.56 (d, J= 14.5 Hz, 2H), 1.17 -
1.04 (m, 4H).
MS(ESI,m/z): 587 [M+H].
Example 39:
OCF2H
HN--µ
0
39
The synthetic route of example 39 was as follows.
o ,
N \ o \ Br
401 OCF2H 1X-31
H F2C0 HF2C0
11-39 1V-39 V-39
9
Ni \ 0--CN ()TN
OCF2H CN OCF2H /1\1,0
0
VII-39
39
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
Compound V-39 was synthesized from 11-39 as the raw material according to the
synthesis
method of compound 10, wherein,
IV-39, white solid, yield 54%. 1H NMR (400 MHz, CDC13) 6 7.52 - 7.44 (m, 2H),
7.32 - 7.26
(m, 1H), 7.21 (d, J= 8.5 Hz, 1H), 6.46 (t, J= 73.7 Hz, 1H), 3.72 (s, 3H), 2.88
- 2.80 (m, 1H), 1.37
- 1.32 (m, 2H), 1.26 - 1.22 (m, 2H).
V-39, colourless liquid, yield 72%. 1H NMR (400 MHz, CDC13) 6 7.60 - 7.51 (m,
2H), 7.41 -
7.32 (m, 2H), 6.51 (t, J= 73.7 Hz, 1H), 4.38 (s, 2H), 2.18 -2.10 (m, 1H), 1.32
- 1.17 (m, 4H).
Compound intermediate VII-39 was synthesized from V-39 and IX-31 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 61%. 1H
NMR (400 MHz, CDC13) 6 8.37 (d, J= 2.2 Hz, 1H), 7.57 - 7.45 (m, 3H), 7.35 -
7.24 (m, 2H),
6.47 (t, J= 74.1 Hz, 1H), 6.42 (d, J= 8.9 Hz, 1H), 4.34 (s, 4H), 3.49 (t, J =
4.5 Hz, 1H), 2.16
- 2.08 (m, 1H), 2.00 - 1.94 (m, 2H), 1.90 - 1.82 (m, 4H), 1.72 (d, J = 14.4
Hz, 2H), 1.23 -
1.20 (m, 2H), 1.14- 1.07 (m, 2H).
Compound 39 was synthesized from VII-39 as the raw material according to the
synthesis method of compound 1, white solid, yield 78%. 1H NMR (400 MHz, DMSO)
8 8.46
(d, J = 2.3 Hz, 1H), 7.79 (dd, J = 9.0, 2.3 Hz, 1H), 7.64 - 7.55 (m, 1H), 7.54
- 7.49 (m, 1H),
7.40 - 7.33 (m, 2H), 7.23 (d, J = 73.6 Hz, 1H), 6.77 (d, J= 9.0 Hz, 1H), 4.55 -
4.23 (m, 4H),
3.46 (s, 1H), 2.36 - 2.27 (m, 1H), 1.86 - 1.69 (m, 6H), 1.62 (d, J= 14.4 Hz,
2H), 1.15 - 1.05
(m, 4H). MS(ESI,m/z): 552[M+H]
Example 40:
\
HN--µ
ao
The synthetic route of example 40 was as follows.
o ,
\
Br
CY"
=
11-40 1V-40 V-40
0 0
\ 0¨C.1\N N
/
HN---µo
VII-40
25 Compound V-40 was synthesized from 11-40 as the raw material according
to the synthesis
method of compound 10, wherein,
¨ 46
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
IV-40, white solid, yield 54%. 1H NMR (400 MHz, CDC13) 6 7.42 - 7.31 (m, 2H),
7.30 - 7.24
(m, 2H), 3.69 (s, 3H), 2.95 -2.87 (m, 1H), 2.23 (s, 3H), 1.42- 1.36 (m, 2H),
1.30- 1.23 (m, 2H).
V-40, colourless liquid, yield 72%. 1H NMR (400 MHz, CDC13) 6 7.42 - 7.29 (m,
4H), 4.27 (s,
2H), 2.33 (s, 3H), 2.19 - 2.10 (m, 1H), 1.32- 1.27 (m, 2H), 1.23 - 1.16 (m,
2H).
Compound intermediate VII-40 was synthesized from V-40 and IX-31 as the raw
materials
according to the synthesis method of compound V11-10, white solid, yield 61%.
1H NMR (400
MHz, CDC13) 6 8.37 (d, J= 1.8 Hz, 1H), 7.54 (dd, J= 8.8, 2.3 Hz, 1H), 7.37 -
7.22 (m, 4H), 6.43
(d, J= 8.8 Hz, 1H), 4.76 - 4.30 (m, 2H), 4.20 (s, 2H), 3.49 (t, J= 4.4 Hz,
1H), 2.30 (s, 3H), 2.16 -
2.10 (m, 1H), 2.07 - 2.02 (m, 2H), 1.93 - 1.83 (m, 4H), 1.77 (d, J= 14.3 Hz,
2H), 1.26 - 1.21 (m,
2H), 1.15 - 1.08 (m, 2H).
Compound 40 was synthesized from VII-40 as the raw material according to the
synthesis
method of compound 1, white solid, yield 78%. 1H NMR (400 MHz, DMSO) 6 8.47
(d, J= 2.3 Hz,
1H), 7.79 (dd, J= 9.0, 2.3 Hz, 1H), 7.39 - 7.24 (m, 4H), 6.75 (d, J= 9.0 Hz,
1H), 4.43 (s, 2H),
4.19 (s, 2H), 3.45 (s, 1H), 2.33 - 2.24 (m, 1H), 2.21 (s, 3H), 1.92 - 1.64 (m,
8H), 1.13 - 1.04 (m,
4H). MS(ESI,m/z): 500[M+H].
Example 41:
0
OCF2H r '0
HN4
0
41
The synthetic route of example 41 was as follows.
Br Vc Ni, \ / N \ F
IX-35N'0
HF2C0 OCF2H 11, OCF2H
V-39 VII-41 41
Compound intermediate VII-41 was synthesized from V-41 and IX-35 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 64%. 1H
NMR (400 MHz, CDC13) 6 7.55 - 7.48 (m, 2H), 7.38 - 7.29 (m, 3H), 6.47 (t, Jr
74.1 Hz, 1H),
6.45 - 6.40 (m, 1H), 6.38 - 6.32 (m, 1H), 4.34 (s, 2H), 4.10 - 4.03 (m, 2H),
3.52 - 3.44 (m,
1H), 2.16 - 2.09 (m, 1H), 2.00 - 1.95 (m, 2H), 1.92 - 1.85 (m, 4H), 1.65 (d,
J= 15.2 Hz, 2H),
1.25 - 1.22 (m, 2H), 1.15 - 1.09 (m, 2H).
Compound 39 was synthesized from VII-39 as the raw material according to the
synthesis method of compound 1, white solid, yield 71%. 1H NMR (400 MHz, DMSO)
67.60
(td, Jr 8.3, 1.7 Hz, 1H), 7.54- 7.46 (m, 2H), 7.40- 7.34 (m, 2H), 7.23 (t, J=
73.6 Hz, 1H),
6.78 - 6.62 (m, 2H), 4.32 (s, 2H), 4.18 (s, 2H), 3.43 (s, 1H), 2.37 - 2.27 (m,
1H), 1.86 - 1.69
(m, 6H), 1.55 (d, J= 14.5 Hz, 2H), 1.17- 1.02 (m, 4H). MS(ESI,m/z): 569[M+H].
Example 42:
¨ 47 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
NO F
110 /1µ1,0
HN--µ
0
42
The synthetic route of example 42 was as follows.
o ,
\ Br 0
\ 0
NJ NON
N
IX-35
CN
HN---µ
0
V-40
42
Compound intermediate VII-42 was synthesized from V-40 and IX-35 as the raw
materials according to the synthesis method of compound VII-10, white solid,
yield 49%. 1H
NMR (400 MHz, CDC13) 6 7.28 - 7.23 (m, 2H), 7.33 - 7.29 (m, 2H), 7.28 - 7.23
(m, 1H), 6.46
- 6.41 (m, 1H), 6.39 - 6.34 (m, 1H), 4.21 (s, 2H), 4.13 - 4.06 (m, 2H),
3.50 - 3.43 (m, 1H),
2.31 (s, 3H), 2.07 (s, 3H), 1.97 - 1.86 (m, 4H), 1.75 - 1.67 (m, 2H), 1.29 -
1.23 (m, 2H), 1.16
- 1.09 (m, 2H).
Compound 40 was synthesized from VII-40 as the raw material according to the
synthesis method of compound 1, white solid, yield 61%. 1H NMR (400 MHz,
CDC13) 87.66
(t, J= 8.7 Hz, 1H), 7.37 - 7.22 (m, 4H), 6.52 (dd, J= 9.0, 1.9 Hz, 1H), 6.39
(dd, Jr 14.9, 1.9
Hz, 1H), 4.20 (s, 2H), 4.09 (s, 2H), 3.49 - 3.43 (m, 1H), 2.30 (s, 3H), 2.17 -
2.01 (m, 3H),
1.92 (dd, J= 9.6, 3.4 Hz, 4H), 1.69 (d, J= 14.5 Hz, 2H), 1.28 - 1.20 (m, 2H),
1.17 - 1.08 (m,
2H). MS(ESI,m/z): 517[M+H].
Examples of pharmacological experiments
Test method for FXR activity at the molecular level
FXR activity was determined using recombinant GST-FXR fusion protein by Perkin
Elmer's AlphaScreen detection reagent. The reaction in this method was carried
out in a
384-well plate, and the total reaction volume was 15 [IL. The mixture of
protein, agonist,
co-regulatory factor, AlphaScreene acceptor beads and AlphaScreen donor beads
was
reacted in a buffer containing Tris-HC1 50 mM (pH7.4), 50 mM NaC1, BSA 0.1%,
and 1mM
DTT. The FXR activity was reflected by the fluorescence signal intensity at
570nm
wavelength detected by the Envision fluorescence detector. The value of EC50
was
calculated by the software Graphpad Prism 5.
Test method for FXR activity at the cell level
The FXR expression plasmid and FXRE luciferase reporter plasmid at a ratio of
1:9 was
co-transfected into 293T cells, and then the transfected cells were seeded on
a 96-well
flat-bottom microplate (ViewPlate-96, White 96- well Microplate with Clear
Bottom,
PerkinElmer) at 5x105/well. The cells were cultured for 24 hours to ensure
plasmid
expression. Then the FXR receptor agonist to be tested was added and acted for
18 hours.
¨48 ¨
Date Recue/Date Received 2021-10-18
The fluorescence intensity was detected using luciferase kit (steady-Glo
Luciferase Assay
system) to reflect the compound's activation efficiency on the FXR receptor.
OH
Sb
*=
0" CI
C
OCA GW4064
In the preliminary screening, the test compound and the two positive compounds
OCA,
GW4064 acted on the cells at 10 M, and the relative activities of the test
compound to the
two positive compounds were determined respectively (relative activity =
(signal intensity of
test compound -blank)/(signal intensity of positive compound -blank)x100%).
The compound
whose relative activity is higher than 50% of the positive compound enters the
re-screening.
The appropriate concentration interval was selected, and the dose-dependent
relationship,
that is, the EC50 value, was calculated.
Table 1 Activity test results
FXR activity at the FXR
activity at the cell level
molecular level
Test sample EC5() (11M) Activity relative ECso
(11M)
to OCA
(%) 10 M
OCA 0374 100 1.16
GW4064 0.98 106 0.024
Example 1(LXF-32) 0.446 109 0.11
Example 2(LXF-73) 0.072 174 0.001
Example 3(LXF-114) 1.81 82 0.014
Example 4(LXF-111) 0.015 97 0.0006
Example 5(LXF-115) 4.70 83 0.844
Example 6(LXF-112) 3.92 99 0.032
Example 7(LXF-113) 0.0067 88 0.006
Example 8(Compound 8) 0.104 88 0.005
Example 9(LXF-117) 0.055 90 0.004
Example 10(LXF-128) 1.04 202 0.0002
Example 11(LXF-129) 0.439 164 0.0007
Example 12(LXF-130) 2.882 63 0.035
Example 13(LXF-131) 0.178 111 0.0007
Example 14(LXF-132) 0.598 74 0.005
Example 15(LXF-133) 2.653 159 0.003
Example 16(LXF-134) 0.195 168 0.0002
Example 17(LXF-135) 0.257 161 0.0003
Example 18(LXF-143) 0.614 150 0.002
Example 19(LXF-136) 3.138 132 0.285
Example 20(LXF-138) 5.471 146 0.005
Example 21 0.0967 117 0.00132
¨49 ¨
Date Recue/Date Received 2023-02-16
CA 03137198 2021-10-18
Example 22 0.386 108 0.00199
Example 23 0.392 109 0.00222
Example 24 0.0590 105 0.000196
Example 25 0.0847 103 0.00012
Example 26 0.0700 103 0.000254
Example 27 0.0209 106 0.000357
Example 28 0.380 107 0.000617
Example 29 0.108 120 0.000938
Example 30 0.0890 120 0.000255
Example 31 0.0113 116 0.000608
Example 32 0.00433 103 0.0000783
Example 33 0.0188 141 0.000387
Example 34 0.0197 122 0.000388
Example 35 0.0185 117 0.000318
Example 36 0.00422 102 0.0000317
Example 37 0.0506 102 0.000284
Example 38 0.0513 102 0.000304
Example 39 0.0250 108 0.00149
Example 40 0.0944 105 0.00211
Example 41 0.0285 108 0.000718
Example 42 0.0804 106 0.000960
Conclusion: The test results show that the compounds of the present invention
have
good agonistic ability to FXR at the molecular level and the cell level, and
the activities of
several compounds are significantly better than those of the two positive
controls.
In vivo pharmacological activity test of liver fibrosis
1) Pharmacodynamic evaluation of compound 1 (LXF-32) on TAA-induced hepatic
fibrosis model rats
In this experiment, TAA induced hepatic fibrosis model rats were used to
investigate the
effect of long-term oral administration of compound 1 on hepatic fibrosis in
the model rats.
Experimental method: Male SD rats were intraperitoneally injected with
thioacetamide
(TAA, dissolved in normal saline) at a dose of 150 mg/kg three times a week to
induce a liver
fibrosis model. Four weeks after the model was made, the blood was taken from
the
retro-ocular venous plexus of the rats to detect serum ALP indicators.
According to
indicators such as ALP and body weight, the rats were randomly divided into 3
groups, each
with 8 rats, which were respectively the model control group (Vehicle),
Compound 1 group
(20 mg/kg), positive compound OCA group (20 mg/kg), etc., orally administered
by gavage,
once a day. During the administration period, the animal's food intake and
body weight were
monitored. After 2 weeks of administration, the blood was taken from the retro-
ocular venous
plexus of the rats to detect serum ALP indicators. After 4 weeks of
administration, the blood
was taken from the retro-ocular venous plexus and the rats were dislocated and
sacrificed.
The livers were taken out and weighed. Part of the liver was fixed with 4%
paraformaldehyde,
and part of the liver was frozen at -80 C. During the whole experiment,
another 8 rats in the
same cage were injected intraperitoneally with the same volume of normal
saline as the
system normal control group (WT). This experiment detected indicators such as
the level of
- 50 -
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
liver function index ALP in serum, the expression of a-SMA and Collal
(fibrosis-related
genes) gene level in the liver, the content of hydroxyproline (a
characteristic amino acid of
collagen) in the liver, and the pathological changes of the liver (Sirius
scarlet stain), etc.,
thereby reflecting whether the compound has the effect of relieving liver
fibrosis.
The research results showed that the compound 1 of the present invention
significantly
reduced the level of ALP in serum, reduced the content of hydroxyproline in
liver tissue, and
significantly down-regulated the expression of a-SMA and Collal mRNA in liver
tissue
after 4 weeks of administration (Figure 1). In the quantitative analysis of
liver pathological
sections stained with Sirius Scarlet, compound 1 reduced the collagen content
in the liver,
because there were large differences within the model group, and there was no
statistical
difference (Figure 2).
In summary, long-term administration of compound 1 (LXF-32) of the present
invention
could significantly improve the liver function of TAA-induced hepatic fibrosis
rats,
down-regulate the expression of a-SMA and Collal mRNA, reduce the deposition
of
collagen in the liver, and have a certain alleviating effect on liver
fibrosis.
2) Pharmacodynamic evaluation of compound 8 (LXF-116) on CCL4-induced
hepatic fibrosis model mice
In this experiment, CCL4 induced hepatic fibrosis model mice were used to
investigate
the effect of long-term oral administration of compound 8 on hepatic fibrosis
in the model
mice.
Experimental method: Male C57BL/6j mice were injected intraperitoneally with 2
mL/kg, 10% CC14 (dissolved in olive oil) three times a week to induce liver
fibrosis model.
Two weeks after the model was made, the blood was taken from the retro-ocular
venous
plexus of the mice to detect serum ALT, AST, TBA and LDH indicators. According
to ALT,
AST, TBA, LDH, body weight and other indicators, the mice were randomly
divided into 5
groups. There are 10 animals in each group, namely the model control group
(Vehicle),
low-dose compound 8 group (6 mg/kg), high-dose compound 8 group (20 mg/kg),
low-dose
positive compound OCA group (6 mg/kg), high-dose OCA group (20 mg/kg), etc.,
orally
administered by gavage, once a day. During the administration period, the
animals' food
intake and body weight were monitored. After 3 weeks of administration, the
blood was
taken from the retro-ocular venous plexus of the mice to detect serum ALT,
AST, TBA and
LDH indicators. After 6 weeks of administration, the blood was taken from the
retro-ocular
venous plexus and the mice were dislocated and sacrificed. The livers were
taken out and
weighed. Part of the liver was fixed with 4% paraformaldehyde, and part of the
liver was
frozen at -80 C. During the whole experiment, another 10 mice in the same cage
were
intraperitoneally injected with the same volume of olive oil as the system
normal control
group (WT).
This experiment detected indicators such as the levels of liver function
indicators ALT,
AST, TBA, LDH in serum, the expression of a-SMA and Collal gene levels in the
liver, and
the pathological changes of the liver (Sirius scarlet stain), etc., thereby
reflecting whether the
compound has the effect of relieving liver fibrosis.
¨51 ¨
Date Recue/Date Received 2021-10-18
CA 03137198 2021-10-18
The research results showed that the high-dose compound 8 group and low-dose
compound 8 group of the present invention significantly reduced the levels of
ALT, AST and
TBA in serum after 6 weeks of administration, and had little effect on LDH;
the high dose
positive compound OCA group significantly reduced the levels of ALT, AST and
TBA in
serum and the low-dose group only had a lowering effect on TBA; the low-dose
compound 8
group had a slightly better effect than the low-dose OCA group (Figure 3). The
high-dose
compound 8 group significantly down-regulated the expression of a-SMA in the
liver, and
down-regulated the expression of coll al in the liver (Figure 4); in the
quantitative analysis
of liver pathological sections stained with Sirius scarlet, both the high-dose
compound 8
group and low-dose compound 8 group significantly reduced the collagen content
in the liver,
and the effect of low-dose compound 8 group was slightly better than that of
the low-dose
OCA group (Figure 5).
In summary, long-term administration of compound 8 (LXF-116) of the present
invention could significantly improve the liver function of CC14-induced
hepatic fibrosis
mice, down-regulate the expression of a-SMA and Coll al mRNA, reduce the
deposition of
collagen in the liver, and hava a certain alleviating effect on liver fibrosis
All documents mentioned in the present invention are cited as references in
this
application, just as each document is individually cited as a reference. In
addition, it should
be understood that after reading the above teaching content of the present
invention, those
skilled in the art can make various changes or modifications to the present
invention, and
these equivalent forms also fall within the scope defined by the appended
claims of the
present application.
-52 ¨
Date Recue/Date Received 2021-10-18