Note: Descriptions are shown in the official language in which they were submitted.
HETEROCYCLIC THR-13 RECEPTOR AGONIST COMPOUND AND PREPARATION
METHOD AND USE THEREFOR
The present application claims the priority rights to the invention patent
application titled
"HETEROCYCLIC THR-f3 RECEPTOR AGONIST COMPOUND AND PREPARATION METHOD
AND USE THEREFOR," having the application number 201910763932.4, and filed
with the State
Intellectual Property Office of the People's Republic of China on August 19,
2019, the disclosure of which is
incorporated herein by reference.
Technical Field
The present invention relates to the field of pharmaceutical synthesis, and,
in particular, to a
compound that may be used as a novel THR-I3 receptor agonist, and a
preparation method and use
therefor.
Background Art
A thyroid hormone (TH) is synthesized in the thyroid in response to thyroid-
stimulating hormone
(TSH) secreted by the pituitary gland. Thyroid hormones play a crucial role in
regulating body growth,
development, and metabolism, and in maintaining a matrix balance. Thyroid
hormones are mainly divided
into two types: 3,3',5-triiodo-L-thyronine (T3) and prohormone thyroxine (T4).
The human body mainly
secretes T4, and in peripheral organs, T4 is converted by deiodinase into T3
that has higher activity. T3
and T4 produced by the thyroid are under the control of negative feedback, and
thyroid-stimulating
hormone (TSH) is responsible for performing normal thyroid functions and
secreting thyroid hormones.
Thyroid-stimulating hormone is synthesized in the anterior lobe of the
pituitary gland, and its secretion is
controlled by thyroid releasing hormone (TRH) synthesized in the hypothalamus.
Thyroid hormones perform functions by binding to thyroid hormone receptors
(THRs). Thyroid
hormone receptors, which are nuclear receptors, regulate the expression of
target genes. Thyroid hormone
receptors are divided into two subtypes: THR-a and THR-13. THR-a, mainly
present in heart tissue, plays
an important role in regulating the function of the heart. The THR-I3 subtype,
mainly expressed in the
liver and the pituitary gland, regulates the metabolism of cholesterol and the
secretion of
thyroid-stimulating hormone.
1
CA 03147789 2022-2-11
At normal levels, the thyroid hormone THs maintains body weight, metabolic
rate, body temperature,
AND emotion, and regulates serum cholesterol. Attempts have been made to
regulate serum cholesterol by
thyroid hormones. However, a natural thyroid hormone, when taken, produces
side effects (such as
tachycardia and arrhythmia, heart failure, and causing the thyroid axis
function, muscle metabolism and
osteoporosis) on the heart, and therefore cannot be used to treat
hypercholesterolemia or obesity. Animal
studies on selective knockout of the THR gene, as well as some studies on
selective THR I igands, showed
that the cardiac side effects caused by these thyroid hormones arc
attributable to THR-a.
The thyroid hormone receptor pathway regulates lipid metabolism, including
metabolism of
cholesterol, triglycerides, and lipoproteins. It has been clinically revealed
that reducing low-density
cholesterol will decrease the incidence of cardiovascular and cerebrovascular
diseases.
Nonalcoholic fatty liver disease (NAFLD) is also a type of metabolic disorder
caused by excessive
accumulation of triglycerides in the liver, which can further cause liver cell
damage and inflammation,
leading to non-alcoholic steatohepatitis (NASH). NASH patients usually also
have type 2 diabetes,
hypercholesterolemia, hyperlipemia, and obesity. There is a high probability
that a NASH patient will
develop liver cirrhosis, liver failure, and ultimately liver cancer. No drug
is available for effective
treatment of NASH. As thyroid hormone performs the function of regulating
lipid metabolism, the thyroid
receptor pathway has become a potential target for the treatment of NASH and
NAFLD. Animal in vivo
studies have confirmed that thyroid hormone analogs can significantly reduce
the degree of liver fat in
an
A selective THR-I3 agonist may be used to avoid cardiac side effects caused by
a conventional THR
receptor agonist, selectively activating only THR-I3 only, which improves
lipid metabolism of the cell, and
performs the function of lowering cholesterol and blood lipids. However, a
selective THR-I3 agonist may
also inhibit the thyroid axis, causing side effects such as depression,
fatigue, and osteoporosis. Therefore,
it is necessary to develop a selective T1-IR-I3 agonist that activates THR-I3
but mitigates the inhibitory
effect on the thyroid axis, thereby avoiding side effects associated with
thyroid axis inhibition.
Patents including W003094845, W02007009913, W02010122980, and W02011038207
disclose
some THR receptor agonists whose structures are almost all designed and
developed on the basis of T3, a
2
CA 03147789 2022-2-11
natural ligand of a THR receptor. Against these backgrounds, a need still
exists to develop a selective
THR-I3 receptor agonist that brings about the beneficial effects of a thyroid
hormone while avoiding
adverse cardiac side effects.
HO I
COOH
NH2
0
T3
Patent W02005051298 also discloses some THR receptor agonists, among which a
good compound
(MB07444) has the following structure:
OH
/
HO
0
HO
MB07444
Patent W02006128058 also discloses some THR receptor agonists, among which
several naphthol
heterocyclic compounds have the following structure. However, the patent does
not disclose any structure
or embodiment similar to a compound of the present invention.
itt
=
PIC),
11 ?
Nõi%
I
u
Ho `1/4õ...r.cs=No,.
a
I
0 P,
3
OH
In the present invention, structural modification was performed on the basis
of T3, a natural ligand of
a THR receptor, and the inventors unexpectedly discovered that after the
structure of the naphthol part
was modified, some compounds unexpectedly improved the agonistic activity of a
THR-I3 receptor
(compared with the code compound 7/MB07444 disclosed by patent W02005051298),
and that almost all
heterocyclic compounds have improved selectivity for THR-ct (compared with
MB07444). in addition,
some compounds of the present invention may be highly enriched in liver target
organs after being
modified by prodrugs, which further reduces distribution thereof in cardiac
organs, thereby potentially
3
CA 03147789 2022-2-11
reducing clinical side effects.
Summary of the Invention
In order to solve the above-described technical problems, the present
invention adopts the following
technical solution:
According to one aspect of the present invention, the present invention
provides a compound shown
in formula (I) below and an isomer thereof or a pharmaceutically acceptable
salt thereof,
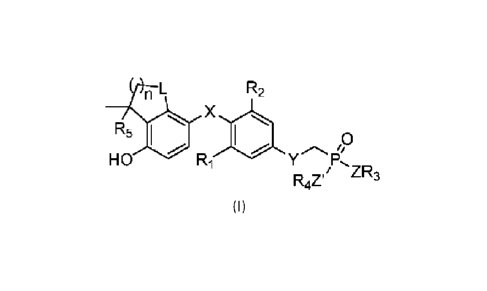
n L R2
R5 X
401 0
.........,, 0
0
HO Ri Y P,
R47/ ZR3
(I)
wherein
R1 and R2 are each independently selected from a halogen atom or a C1-6 alkyl
group;
R3 and R4 are each independently selected from hydrogen, a C1-6 alkyl group,
an unsubstituted
phenyl group, a phenyl group substituted with at least one substituent
selected from a halogen atom, a
trifluoromethyl group, a C1-6 alkyl group, a C1-6 alkoxyl group or a cyano
group, an unsubstituted naphthyl
group, a naphthyl group substituted with at least one substituent selected
from a halogen atom, a
0
trifluoromethyl group, a C1-6 alkyl group, a C1.6 alkoxyl group or a cyano
group, 0,
R7
ekt......ted.a.R8 0
H
0
, or R3, IR4, and adjacent -Z-
P-ZI- jointly form the following six-membered ring
0
6' NO-)
V, wherein V is an unsubstituted five- to ten-membered aryl group, a five- to
ten-membered
aryl group substituted with at least one substituent selected from a halogen
atom, a trifluoromethyl group,
a C2-6 alkyl group, a C2-6 alkoxyl group or a cyano group, an unsubstituted
five- to ten-membered
heteroaryl group containing 1 or 2 heteroatoms selected from N, S and 0, a
five- to ten-membered
4
CA 03147789 2022-2-11
heteroaryl group that is substituted with at least one substituent selected
from a halogen atom, a
trifluoromethyl group, a C1.6 alkyl group, a C1_6 alkoxyl group or a cyano
group and that contains one or
two heteroatoms selected from N, S and 0;
R5 is selected from a H or C16 alkyl group;
R6, R7, and R3 are each independently selected from a C1.6 alkyl group;
X is selected from -0- or -CH2-;
Y is selected from -0- or -CH2-;
Z and Z' are each independently selected from -0- or -NH-;
L is selected from -0-, -S- or -CH2-
n is 1, 2 or 3;
the halogen atom is selected from F, Cl or Br.
According to another aspect of the present invention, preferably, in the
structure shown in formula
(I), R1 and R2 are each independently selected from F, Cl, Br or -CH3.
Further preferably, R1 and R2 are both Cl.
Alternatively, preferably R1 and R2 are both -CH3.
According to another aspect of the present invention, preferably, in the
structure shown in formula
(I), R5 is selected from H or -CH3; according to another aspect of the present
invention, preferably, in the
structure shown in formula (I), n is 1 or 2; further preferably, n is 1;
According to another aspect of the present invention, preferably, in the
structure shown in formula
(I), X is -CH2-;
According to another aspect of the present invention, preferably, in the
structure shown in formula
(I), Y is-O-;
Preferably, in the structure shown in formula (I), V is an unsubstituted
phenyl group, a phenyl group
substituted with at least one substituent selected from a halogen atom, a
trifluoromethyl group, a C1.3
alkyl group and a C1_3 alkoxyl group, an unsubstituted five- to six-membered
monocyclic heteroaryl group
containing 1 or 2 heteroatoms selected from N, S and 0, a five- to six-
membered monocyclic heteroaryl
group that is substituted with at least one substituent selected from a
halogen atom, a trifluoromethyl
CA 03147789 2022-2-11
group, a C1_3 alkyl group and an alkoxyl group and that contains 1 or 2
heteroatoms selected from N, 5,
and 0.
According to another aspect of the present invention, preferably, the compound
shown in formula (I)
and an isomer thereof or a pharmaceutically acceptable salt thereof have a
structure shown in formula (II)
below:
n L R2
so X 0
R5
0
---N. o
HO Ri
Y R.,
' OH
HO
(II)
wherein
R1, R2, R5, X, Y, and n are as defined in the preceding formula (I);
further preferably, in the structure shown in formula (II),
R1 and R2 are both -CH3;
R5 is selected from -CH3;
X is -CH2-;
Y is -0-;
L is -CH2-;
n is 1 or 2.
According to another aspect of the present invention, preferably, the compound
shown in formula (I)
and an isomer thereof or a pharmaceutically acceptable salt thereof have a
structure shown in formula
(Ill):
n L R2
0 x so
R5
0
.....-%, t
HO Ri
Y P
v
(III)
6
CA 03147789 2022-2-11
wherein
R1, R2, R5, X, Y, L, n, and V are as defined in the preceding formula (I);
further preferably, in the structure shown in formula (II),
R1 and R2 are both -CH3;
R5 is selected from -CH3;
X is -CH2-;
Y is -0-;
L is -CH2-;
n is 1 or 2;
preferably, V is an unsubstituted phenyl group, a phenyl group substituted
with at least one
substituent selected from a halogen atom, a trifluoromethyl group, a C1-3
alkyl group and a C1-3 alkoxyl
group, a pyridyl group, and a pyridyl group substituted with at least one
substituent selected from a
halogen atom, a trifluoromethyl group, a C1_3 alkyl group, and a C1_3 alkoxyl
group.
Further preferably, V is an m-chlorophenyl group.
According to another aspect of the present invention, preferably, the compound
shown in formula (I)
and an isomer thereof or a pharmaceutically acceptable salt thereof have a
structure shown in formula
(IV):
( n L R2
101 X
HO
R5 0
0
...........,
Ri
Y P,
R401 R3 !
(IV)
wherein
R1, R2, R5, X, Y, L, and n are as defined in the preceding formula (I);
R3 and R4 are each independently selected from a Ci 6 alkyl group, a phenyl
group, a phenyl group
substituted with at least one substituent selected from a halogen atom, a
trifluoromethyl group, a C1-6
alkyl group, a C1-6 alkoxyl group or a cyano group, a naphthyl group, a
naphthyl group substituted with at
7
CA 03147789 2022-2-11
least one substituent selected from a halogen atom, a trifluoromethyl group, a
C1-6 alkyl group, a C1-6
0
alkoxyl group or a cyano group, Af-M Re,
wherein R6 is selected from a C1_6 alkyl group;
further preferably, in the structure shown in formula (IV),
R1 and R2 are both -CH3;
R6 is selected from -CH;
X is -CH2-;
Y is -0-;
L is -CH2-;
n is 1 or 2;
0
R3 and R4 are both ct Rg , wherein R6 is a
C1_6 alkyl group;
0
more preferably, both R3 and R4 are
According to another aspect of the present invention, preferably, the compound
shown in formula (I)
and an isomer thereof or a pharmaceutically acceptable salt thereof have a
structure shown in formula
(V):
n L R2
ao x
R5
0 R7
,00.,,
HO Y P.
A,0, R8 N
H
0
R4
(V)
wherein
R1, R2, Rs, X, Y, L, and n are as defined in the preceding formula (l);
R4 is selected from a C1-6 alkyl group, a phenyl group, a phenyl group
substituted with at least one
8
CA 03147789 2022-2-11
substituent selected from a halogen atom, a trifluoromethyl group, a Ci_6
alkyl group, a C1_5 a lkoxyl group
or a cyano group, a naphthyl group, a naphthyl group substituted with at least
one substituent selected
from a halogen atom, a trifluoromethyl group, a C1.6 alkyl group, a C1.6 al
koxyl group or a cyano group,
R7 and Rg are each independently selected from a C1.6 alkyl group;
further preferably, in the structure shown in formula (V),
R1 and R2 are both -CH3;
Rg is selected from -CH;
X is -CH2-;
Y is -0-;
L is -CH2-;
n is 1 or 2;
R4 is a phenyl group or a naphthyl group;
R7 is a methyl group;
Rg is an ethyl group or an isopropyl group;
according to another aspect of the present invention, preferably, the compound
and a
pharmaceutically acceptable salt and prodrug thereof are one of the following
compounds:
9
CA 03147789 2022-2-11
,.., ... P
,QH
HO 0 P.õ HO 0 P.,
HO 0..."-.. p....
Hd H HO H
HO -'
OaCIa
CI
/OH
S ir ---. 2
0
Ho 0 1,--0 Ho 0 P.,
......--, 1,
HO Fe OH
H CI 0 P.... "
HO
P
O
HO r He' P
, \ ..er',
0 P
0
HO
00
0
APNNiy y-
õ .
Q0\
0¨\
4. ci
o N'e H 0 I
0
...-41/4 P
Ho a P H0 Orn'P?
If P
d µo d
Cit" HO Or"
* CI
0 0
0 0
* a
P---07-1
HOn..----..c,
,...".. IP
HO 0 _Rs
'-'
L) µ0 0 VI
1
0
1411
...."= P
0
HO 0
6 = 04
111AlicT-
. c
0 0
According to another aspect of the present invention, the present invention
provides a preparation
method for the compound, the preparation method comprising the following
steps:
CA 03147789 2022-2-11
X R' R2
HD, ISO
Ri
crt 't
R,
Y
1-a
'
1-t 1-c
1-9
,9flLriL
R2
H cH X
R5 R5 D
110
1 1- HO * RI .K.-111 1110
VC-2
HO
RI ,i1"
"6:11Pr H R Y
DCG 0 :0)
Hcr OH
V
I 9
II
1) ECG RA CH
3.-171
o
2) SVC:12
-t7
L
H2N fikiin-Rs
31
it:
0
HO R Y
R2
R401 nh3
X
R6 R
CAR, * R7
HO
RI Y R8
Iv
V A, 0
1) Add paraformaldehyde and potassium carbonate to isopropanol, slowly add
diisopropyl phosphite
dropwise when the temperature has been raised to 50 degrees Celsius, and stir
for 2 hours maintaining a
temperature of 50 degrees Celsius. After post-treatment, the compound of
general formula 1-b is obtained;
2) Add compound 1-b and triethylamine to dichloromethane separately, and cool
the system by 4
degrees Celsius with an ice bath. While stirring, slowly add dropwise a p-
toluenesulfonyl chloride
solution into the reaction liquid with a dropping funnel and, after completion
of the dropping, continue
stirring for 2 hours while keeping the ice bath. After completion of the
reaction, the active ester 1-c is
obtained after post-treatment;
3) Add compound 1-c to a mixture of dimethyl sulfoxide, compound 1-d, and
cesium carbonate, raise
the temperature to 55 degrees Celsius in a nitrogen atmosphere, and stir for 6
hours for reaction to obtain
a compound of general formula 1-e;
4) Add compound 1-e to a dichloromethane solution of compound 1-f, cool the
system to 4 degrees
Celsius with an ice bath, and add a trifluoroacetic acid dropwise to catalyze
the reaction. After
post-treatment, a compound of general formula 1-g is obtained;
5) Add trimethylchlorosi lane dropwise to an acetonitri le solution of
compound 1-g and of potassium
iodide, raise the temperature to 50 C, and stir for 2 hours for reaction.
After dealkylation, phosphoric acid
compound II is obtained;
6) Dissolve the phosphoric acid compound II and 1-(3-chlorophenyl)propane-1,3-
diol in pyridine
and DM F, and add condensation reagent DCC at room temperature. Heat to 73 C
and stir for 4 hours, and
11
CA 03147789 2022-2-11
a prodrug compound Ill of II is obtained after post-treatment.
7) Alternatively, add diisopropylethylamine to an acetonitrile solution of
phosphoric acid compound
II at room temperature. Heat to 40 C, stir for half an hour, add iodine, and
continue to stir overnight. A
diesterization reaction occurs to generate phosphate ester prodrug IV.
8) Alternatively, react phosphoric acid compound II with phenol or naphthol R4-
0H under the
promotion by the condensation reagent DCC, and an acyl chloride intermediate
is generated from the
sulfonyl chloride and then is reacted with an amino acid ester to obtain
prodrug compound V of II.
Each of the substituents RI, R2, R3r R4/ R5r R6r R71 RBI X, Y, L, n, and V in
the preceding reaction
formula are as defined in the preceding formula (I).
According to another aspect of the present invention, the present invention
provides a use of the
compound in the preparation of a drug for treating a metabolism-related
disease or fibrosis-related
disease.
According to another aspect of the present invention, the present invention
provides a
pharmaceutical composition comprising a therapeutically effective amount of
the compound according to
the present invention and a pharmaceutically acceptable salt thereof as active
ingredients, and a
pharmaceutically acceptable excipient.
Preferably, the metabolism-related disease is selected from: obesity,
hyperlipidemia,
hypercholesterolemia, diabetes, and non-alcoholic steatohepatitis (NASH),
hepatic steatosis,
atherosclerosis, hypothyroidism and thyroid cancer, liver fibrosis, pulmonary
fibrosis; preferably, the
metabolism-related disease is selected from: non-alcoholic steatohepatitis
(NASH), hypothyroidism and
thyroid cancer, liver fibrosis, pulmonary fibrosis.
According to another aspect of the present invention, the present invention
provides a method for
treating a metabolism-related disease, the method comprising administering to
a subject an effective
amount of the compound according to the present invention or a pharmaceutical
composition comprising
the compound and a pharmaceutically acceptable salt thereof as active
ingredients.
Preferably, according to the method for treating a metabolism-related disease,
the metabolism-related
disease is selected from: obesity, hyperlipidemia, hypercholesterolemia,
diabetes, and non-alcoholic
12
CA 03147789 2022-2-11
steatohepatitis (NASH), hepatic steatosis, atherosclerosis, hypothyroidism and
thyroid cancer; preferably,
the metabolism-related disease is selected from: non-alcoholic steatohepatitis
(NASH), hypothyroidism,
and thyroid cancer.
Preferably, according to another aspect of the present invention, a method for
treating a
metabolism-related disease or fibrosis-related disease is provided, the method
comprising administering
to a subject an effective amount of the compound according to the present
invention or a pharmaceutical
composition containing the compound and an isomer thereof or a
pharmaceutically acceptable salt thereof
as active ingredients.
Specific Embodiments
The present invention will be described in detail below. Before the
description is given, it should be
understood that terms used in the description and the appended claims, instead
of being construed as
being limited to general meanings and dictionary meanings, should be explained
according to meanings
and concepts corresponding to the technical aspects of the present invention
on the basis of the principle
of allowing the inventors to appropriately define terms for optimal
explanation. Therefore, the description
provided herein is only preferred embodiments given for illustrative purposes,
instead of being intended
to limit the scope of the present invention, and, therefore, it should be
understood that other equivalent
embodiments or improved embodiments may be derived therefrom without departing
from the spirit or
scope of the present invention.
According to the present invention, unless otherwise stated, all terms cited
herein have the same
meanings as the terms as commonly understood by those of ordinary skill in the
art.
For example, the term "salt" as used herein refers to a compound containing
cations and anions,
which may be produced by protonation of proton-acceptable sites and/or
deprotonation of
proton-available sites. It is noteworthy that protonation of proton-acceptable
sites leads to the formation
of cationic substances whose charge is balanced by the presence of
physiological anions, while
deprotonation of proton-available sites leads to the formation of anionic
substances whose charge is
balanced by the presence of physiological cations.
The term "pharmaceutically acceptable salt" means that the salt is acceptable
in relation to pharmacy.
13
CA 03147789 2022-2-11
Examples of pharmaceutically acceptable salts include, but are not limited to:
(1) acid addition salts,
formed with inorganic acids, such as hydrochloric acid, hydrobromic acid,
sulfuric acid, nitric acid, and
phosphoric acid; or formed with organic acids, such as glycolic acid, pyruvic
acid, lactic acid, malonic
acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, 3-(4-
hydroxybenzoyl)benzoic acid,
cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-
ethane-disulfonic acid,
2-hydroxyethanesulfonic acid, benzenesulfonic acid, 4-chlorobenzenesulfonic
acid, 2-naphtha lenesulfonic
acid, 4 paratoluenesulfonic acid, camphor acid, lauryl sulfuric acid, gluconic
acid, glutamic acid, salicylic
acid, and cis-adipenedioic acid; or (2) base addition salts, formed with a
conjugate base of any of the
above-mentioned inorganic acids, wherein the conjugate base contains a
cationic component selected
from Nat K+, Mg2+, Ca2+, and NHõR4:, wherein NHõR4.x+ (R is a C1.4 alkyl
group, and the subscript x is
an integer selected from 0, 1, 2, 3, or 4) represents the cation in the
quaternary ammonium salt. It should
be understood that all pharmaceutically acceptable salts involved include the
solvent addition form
(solvate) or crystal form (polymorph) of the same acid addition salt defined
herein.
The term "Ci.m alkyl group" refers to an alkyl group containing 1 - M carbon
atoms, where M is an
integer having the following values: 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19, 20, 21, 22,
23, 24, 25, 26, 27, 28, 29, or 30, for example. For example, the term "C1_6
alkyl group" refers to an alkyl
group containing 1 - 6 carbon atoms. Examples of alkyl groups include, but are
not limited to, lower alkyl
groups, including methyl group, ethyl group, propyl group, isopropyl group, n-
butyl group, isobutyl group,
tert-butyl or pentyl group, isopentyl group, neopentyl group, hexyl group,
heptyl group and octyl group.
The term "aryl group" refers to an aromatic system that can be a single ring
or polyaromatic rings
originally fused or connected together, so that at least a part of the fused
or connected rings form a
conjugated aromatic system. Aryl groups include, but are not limited to:
phenyl group, naphthyl group,
and tetrahydronaphthyl group. An aryl group may be optionally substituted,
such as an aryl or
heterocyclic group which may be substituted with 1 - 4 groups selected from
the group consisting of
halogen, -CN, -OH, -NO2, amino group, alkyl group, cycloalkyl group, alkenyl
group, alkynyl group,
alkoxyl group, aryloxyl group, substituted alkoxyl group, alkylcarbonyl group,
alkylcarboxyl group,
alkylamino group, or arylthio group.
14
CA 03147789 2022-2-11
The term "substituted" means that a reference group is substitutable with one
or more additional
groups, the additional groups being individually and independently selected
from alkyl group, cycloalkyl
group, aryl group, heteroaryl group, heteroalicyclic hydrocarbon, hydroxyl
group, alkoxyl group,
alkylthio group, arylthio group, alkylsulfoxyl group, arylsulfoxyl group,
alkylsulfuryl group, arylsulfuryl
group, cyano group, halogroup, carbonyl group, thiocarbonyl group, nitro
group, haloalkyl group,
fluoroalkyl group and amino group, including mono- and di-substituted amino
groups and protected
derivatives thereof.
A compound shown in formula (I) or a pharmaceutically acceptable salt thereof,
and a
pharmaceutical composition containing the compound provided by the present
invention may be in
various forms, such as tablets, capsules, powders, syrups, solutions, and
suspensions and aerosols, and
may be present in a suitable solid or liquid carrier or diluent and in a
suitable sterilizer for injection or
drip infusion.
Various dosage forms of a pharmaceutical composition of the present invention
may be prepared by
conventional preparation methods in the pharmaceutical field. For example, the
unit dose of the
pharmaceutical formulation contains 0.05 - 200 mg of the compound of formula
(I) or a pharmaceutically
acceptable salt thereof, wherein, preferably, the unit dose of the
pharmaceutical formulation contains
0.1 mg - 100 mg of the compound of formula (I).
The compound and pharmaceutical composition shown in general formula (I) of
the present
invention may be used clinically in mammals, including humans and animals, and
may be administered
orally, nasally, percutaneously, pulmonarily, or through the gastrointestinal
tract. Oral administration is
the most preferred. The most preferred daily dose is 0.01 - 200 mg/kg body
weight, taken at one time, or
0.01 - 100 mg/kg body weight in split doses. Regardless of the adopted method
of administration, the
optimal dose for an individual should be determined on the basis of specific
treatment. Generally, a small
dose should be taken at the beginning, and then the dose may be increased
gradually until the most
suitable dose is found.
In the present invention, the term "effective amount" may refer to an
effective amount for the dosage
and time period required to achieve a desired effect. The effective amount may
vary depending on certain
CA 03147789 2022-2-11
factors, such as type of disease or condition of disease during treatment,
structure of specific target organ
of administration, patient's stature, or severity of disease or of symptom.
Those of ordinary skill in the art
can empirically determine the effective amount of a specific compound without
conducting an excessive
amount of experiments.
A typical formulation is prepared by mixing the compound shown in general
formula (I) of the
present invention, a carrier, a diluent, or an excipient. Suitable carriers,
diluents or excipients are well
known to those of ordinary skill in the art, and include substances such as
carbohydrates, waxes,
water-soluble and/or inflatable polymers, hydrophilic or hydrophobic
substances, gelatin, oils, solvents,
and water.
A specific carrier, diluent or excipient to be used will depend on the method
of and purpose of use of
the compound of the present invention. Solvents are generally selected on the
basis of solvents that those
of ordinary skill in the art believe may be safely and effectively
administered to mammals. Generally, safe
solvents are nontoxic aqueous solvents, such as water, and other nontoxic
solvents that are soluble in or
miscible with water Suitable aqueous solvents include one or more of water,
ethanol, propylene glycol,
and polyethylene glycol (such as PEG400 and PEG300). The formulation may also
comprise one or more
buffers, stabilizers, surfactants, wetting agents, lubricants, emulsifiers,
suspending agents, preservatives,
antioxidants, sunscreens, glidants, processing aids, coloring agents,
sweetening agents, perfuming agents,
flavoring agents or other known additives, so that the drug may be
manufactured or used in an acceptable
form.
When the compound of formula (I) according to the present invention is used in
combination with at
least one other drug, the two or more drugs may be used separately or in
combination, and are preferably
administered in the form of a pharmaceutical composition. The compound or
pharmaceutical composition
of formula (I) of the present invention may be administered separately or
together to a subject in any
known form of medication, for example, oral administration, intravenous
injection, rectal administration,
vaginal administration, cutaneous permeation, or another form of topical or
systemic administration.
These pharmaceutical compositions may also contain one or more buffers,
stabilizers, surfactants,
wetting agents, lubricants, emulsifiers, suspending agents, preservatives,
antioxidants, sunscreens,
16
CA 03147789 2022-2-11
glidants, processing agents, coloring agent, sweetening agent, perfuming
agents, flavoring agents or other
known additives, so that the pharmaceutical composition may be manufactured or
used in an acceptable
form.
A drug of the present invention is preferably administered orally. Solid
dosage forms for oral
administration may include capsules, tablets, powder or granular formulations.
In a solid dosage form, the
compound or pharmaceutical composition of the present invention is mixed with
at least one inert
excipient, diluent or carrier. Suitable excipients, diluents or carriers
include substances such as sodium
citrate or dicalcium phosphate, or starch, lactose, sucrose, mannitol, and
silicic acid; binders such as
carboxymethyl cellulose, alginate, gelatin, polyvinylpyrrolidone, sucrose, and
gum arabic; wetting agents
such as glycerin; disintegrants such as agar, calcium carbonate, potato starch
or tapioca, alginic acid,
specific complex silicate, and sodium carbonate; solution blockers such as
paraffin; absorption enhancers
such as quaternary ammonium compounds; adsorbents such as kaolin and
bentonite; lubricants such as
talc, calcium stearate, magnesium stearate, solid polyethylene glycol, and
sodium lauryl sulfate. In the
case of capsules and tablets, the dosage form may also comprise a buffering
agent. Similar types of solid
compositions may also be used as fillers in soft and hard filled gelatin
capsules, in which lactose,
high-molecular-weight polyethylene glycol, etc. are used as excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions,
solutions, suspensions, syrups and elixirs. In addition to the compound of the
present invention or a
pharmaceutical composition thereof, the liquid dosage form may contain an
inert diluent commonly used
in the art, such as water or another solvent; solubilizers and emulsifiers
such as ethanol, isopropyl alcohol,
ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene
glycol, 1,3-butanediol,
dimethylformamide; oils (such as cottonseed oil, peanut oil, corn germ oil,
olive oil, castor oil, and
sesame oil); glycerin; tetrahydrofurfuryl alcohol; fatty acid esters of
polyethylene glycol and sorbitan; or a
mixture of a plurality of these substances, etc.
In addition to these inert diluents, the composition may also comprise
excipients, such as one or
more of a wetting agent, an emulsifier, a suspending agent, a sweetening
agent, a flavoring agent, and a
perfuming agent.
17
CA 03147789 2022-2-11
With regard to suspension, in addition to the compound shown in general
formula (I) of the present
invention or a pharmaceutically acceptable salt thereof or a pharmaceutical
composition containing the
same, it may further contain a carrier, for example, a suspending agent, such
as ethoxylated isostearyl
alcohol, polyoxyethylene sorbitol, sorbitan ester, microcrystalline cellulose,
aluminum hydroxide,
bentonite, agar and adragant, or a mixture of a plurality of these substances.
The compound shown in general formula (I) of the present invention or a
pharmaceutically
acceptable salt thereof or a pharmaceutical composition containing the same
may be administered in other
dosage forms for topical administration, including ointments, powders, sprays
and inhalants. The drug
may be mixed with a pharmaceutically acceptable excipient, diluent or carrier
and any required
preservatives, buffers or propellants under aseptic conditions. Ophthalmic
formulations, ophthalmic
ointments, powders and solutions are also intended to fall within the scope of
the present invention.
In addition, the present disclosure further covers a kit (such as a
pharmaceutical package). A
provided kit may comprise a pharmaceutical composition or compound described
herein and a container
(for example, a vial, an ampoule, a bottle, a syringe and/or subpackage or
another suitable container). In
some embodiments, a provided kit may optionally further comprise a second
container that contains a
pharmaceutical excipient for diluting or suspending a pharmaceutical
composition or compound described
herein. In some embodiments, a pharmaceutical composition or compound
combination described herein
disposed in the first container and the second container forms a unit dosage
form.
In some embodiments, a kit described herein further comprises instructions on
how to use the
compound or pharmaceutical composition included in the kit. A kit described
herein may further comprise
information required by regulatory agencies, such as the U.S. Food and Drug
Administration (FDA). In
some embodiments, information included in the kit is prescription information.
In certain embodiments, a
kit and instructions provide treatment of proliferative diseases in subjects
in need thereof and/or
prevention of proliferative diseases in subjects in need thereof. A kit
described herein may contain one or
more additional pharmaceutical preparations as separate compositions.
The present invention will be further described in detail below in conjunction
with specific
embodiments, but the present invention is not limited to the embodiments
described below, the
18
CA 03147789 2022-2-11
embodiments being intended to better explain some specific manifestations of
the present invention,
instead of being construed as limiting the scope of the present invention in
any way. Conditions not
specified in the embodiments are conventional conditions. Unless otherwise
specified, the reagents and
instruments used in the following embodiments are all commercially available
products.
The structures of the compounds in the following embodiments were determined
by nuclear
magnetic resonance (NMR) or/and mass spectrometry (MS). The NMR shift (6) was
given in units of
10-6 (ppm). A Bruker AVANCE-400 nuclear magnetic instrument was used for NMR
measurement, the
solvents used were deuterated dimethyl sulfoxide (DMSO-d6), deuterated
chloroform (CDCI3), and
deuterated methanol (CD30D), and the internal standard was tetramethylsilane
(TM 5).
MS measurement was performed with a FINNIGAN LCQAd (ESI) mass spectrometer
(manufacturer:
Thermo, model: Finnigan LCQ advantage MAX).
A Yantai Huanghai HSGF254 or Qingdao GF254 silica gel plate was used as the
thin-layer
chromatography silica gel plate, the specifications of the silica gel plate
used in the thin-layer
chromatography (TLC) was 0.15 mm - 0.2 mm, and the specifications used for
product separation and
purification in the thin-layer chromatography was 0.4 mm - 0.5 mm.
In column chromatography, Yantai Huanghai silica gel, which is 200 - 300 mesh
silica gel, was
generally used as the carrier.
Unless otherwise specified, in an embodiment, the reaction temperature was
room temperature, in
the range of 20 degrees Celsius - 30 degrees Celsius.
In the detection of a reaction process in an embodiment, thin-layer
chromatography (TLC) was
adopted, wherein the developing solvent system used and the elution system of
column chromatography
used to purify the compound included: A: dichloromethane and methanol system,
B: n-hexane and ethyl
acetate system, C: petroleum ether and ethyl acetate system, and D: acetone
and petroleum ether system,
and the volume ratio between the solvents was adjusted according to the
polarity of the compound.
Abbreviations used in the experiments are: DCC, dicyclohexylcarbodiimide;
TMSI, trimethylsilyl
iodide; EA, ethyl acetate; DCM, dichloromethane; h, hour; DM F, N,N-
dimethylamide.
Reference embodiment
A: Preparation of
19
CA 03147789 2022-2-11
((4-((4-hydroxynaphthy1-1-yl)methyl)-3,5-dimethylphenoxy)methyl)phosphoric
acid
fl
OH
HO 0
HO
A
110 OH Step 1
HO ti$
OH
A-1
A-2
HO
Step 2 p._) Step 3
MO\ j.õ..õ Step 4 HO is ¨(to
0 H 0
_____________________________________________________________________________
)10- si 0
d
A-3 A-4
A-6
A-6 )¨
HO
gir A-7
_( Step 6
0
_______________________________________________________________________________
_____ Pm- I OH
Step 5 HO 0
HO 0=e P-,
R b-o
HO '"
A-8
A
Step 1: Synthesis of 4-hydroxymethy1-3,5-dimethylphenol A-2:
Compound A-1 (91.5 g, 750 mmol), water (525 ml), and a NaOH solution (30 ml)
with a
concentration of 50% by mass were mixed and stirred for 1 h until the mixture
was completely clear. The
temperature of the system was cooled to 4 degrees Celsius with an ice water
bath, and a formaldehyde
solution (50 g, 618 mmol) was added all at once. After stirring for 6 hours
with the ice bath kept, the
temperature was naturally raised to room temperature and stirring was
performed for 12 hours. The
reaction solution was poured into a mixed solution of dichloromethane (200 ml)
and ethyl acetate
(200 ml), concentrated HCI (56 ml) was added dropwise to reach pH 5, stirring
was performed for another
6 h, and the precipitated solid was collected by filtration. The filter cake
was washed with water (50 ml)
and dichloromethane (75 ml), and dried to obtain a white solid A-2 (40 g).
1H NMR (400 TNAN7,0130H). 6.47(s, 2H), 460 (s, 2H), 234(s, 6H)
Step 2: Synthesis of hydroxymethyl di isopropyl phosphate A-4:
Paraformaldehyde (9 g, 326 mmol) and potassium carbonate were added to
isopropanol (90 ml),
CA 03147789 2022-2-11
di isopropyl phosphite (45.2 g, 272 mmol) was slowly added dropwise after the
temperature was raised to
50 degrees Celsius, and stirring was performed for 2 hours with the
temperature kept at 50 degrees
Celsius. The temperature of the system was lowered to 35 degrees Celsius,
filtration was performed, the
filter cake was washed twice with isopropanol, and the filtrates were combined
and concentrated under
reduced pressure. Dichloromethane (180 ml) was added to the residue, washed
with 1 N hydrochloric acid
(27 ml) and saturated NaHCO3 (45 ml), dried, and concentrated under reduced
pressure to obtain colorless
liquid A-4 (53.2 g).
111 NMR (400 METz,CDC13): 6.53(m, J=28.0Hz, 2H), 4.60 (m, J=32.0Hz, 2H), 3.64
(m,
J=12.0Hz, 214), 1.24(d, J=8.0Hz, 1214
Step 3: Synthesis of (diisopropoxy phosphate) methyl-4-methylbenzenesulfonate
A-5:
Compound A-4 (49 g, 250 mmol) and triethylamine (69.5 ml, 500 mmol) were added
to
dichloromethane (150 ml), and the system was cooled by 4 degrees Celsius in an
ice bath. While stirring,
a solution of p-toluenesulfonyl chloride (50 g, 263 mmol) in dichloromethane
(350 ml) was slowly added
dropwise with a dropping funnel into the reaction solution (with the
temperature kept below 10 degrees
Celsius), and, after completion of dropping, stirring was continued for 2
hours with the ice bath
maintained. The reaction solution was washed with 1 M hydrochloric acid and
saturated sodium
bicarbonate aqueous solution (300 ml) and dried, and then the organic phase
was concentrated under
reduced pressure and purified by column chromatography to obtain colorless
liquid A-5 (78 g).
NMR (400 MHz,CDC13): 7.80(d, J=8.0Hz, 2H), 7.36 (d, J=8.0Hz, 2H), 4.78 (m,
J-48.0Hz, 2H), 4,17(m, J=46.0Hz, 311), 2,30(s, 1H), 1,32 (m, J=24Hz, 1211),
Step 4: Synthesis of di isopropyl ((4-(hydroxymethyl)-3,5-
dimethylphenoxy)methyl) phosphate A-6:
Compound A-5 (35 g, 100 mmol) was added to a mixture of dimethyl sulf oxide
(85 ml), compound
A-2 (18 g, 120 mmol) and cesium carbonate (52 g, 160 mmol), the temperature
was raised to 55 degrees
Celsius under a nitrogen atmosphere, stirring was performed for 6 hours, and
cooling was performed.
Ethyl acetate (100 ml) and 1% sodium chloride aqueous solution (200 ml) were
added to the system, the
layers were separated, the organic phase was washed with saturated brine, and
the organic phase was
concentrated under reduced pressure to obtain brown oily substance A-6 (45 g).
LH NMR (400 Milz,CDC13). 6.62(s, 2H), 4.85 (m, J=32Hz, 2H), 4.66 (s, 2H),
4.16(d,
J=8.0Hz, 2E1), 2.39(s, 6H), 1.37 (m, J=16.0Hz, 12H).
21
CA 03147789 2022-2-11
Step 5: Synthesis of diisopropyl ((4-((4-hydroxynaphthalene-1-yl)methyl)-3,5-
dimethylphenoxy)methyl)
phosphate A-8:
Compound A-7 (275 mg, 1.91 mmol) was added to compound A-6 (315 mg, 0.95 mmol)
in
dichloromethane (3 ml) solution, the system was cooled to 4 degrees Celsius
with an ice bath,
trifluoroacetic acid (326 mg , 2.86 mmol) was added dropwise, and the TLC dot
plate tracking material
A-6 disappeared. Water (5 ml) was added, the layers were separated, the
organic phase was washed with
water (5 ml), the organic phase was concentrated under reduced pressure to
obtain a brown oily substance,
ether (5 ml) was added, the ambient temperature was minus 18 degrees Celsius,
a switch was made to
room temperature 5 minutes later and stirring was performed for 1 hour, and a
white solid A-8 (100 mg)
was precipitated.
tH NMR (400 11411z,CDC13): 8.32(d, J=8.0Hz, 1H)7 8.18(d, J=8.0Hz, 1H), 7.63(t,
J=12.0Hz, 1H1, 7.56(t, J=16.0Hz, 1H), 6.71 (s, 2H), 6.43(d, J=8,0Hz, 2H), 4.91
(m, J=44.0Hz,
2H), 4.26(m, J=12.0Hz, 4H), 2.15(s, 6H), 1.40(m, J=12.0Hz, 12H).
Step 6: Synthesis of ((44(4-hydroxynaphthy1-1-yl)methyl)-3,5-
dimethylphenoxy)methyl)phosphoric acid
A
Trimethylchlorosilane (76 mg, 0.7 mmol) was added dropwise to a mixture of
compound A-8
(100 mg, 0.22 mmol) and potassium iodide (116 mg, 0.70 mmol) in acetonitrile
(1 ml), the temperature
was raised to 50 C, and the reaction was stirred for 2 hours. Ethyl acetate
(20 ml) and water (20 ml) were
added, the layers were separated, the organic phase was washed with saturated
brine (20 ml) 1 time, and
the solvent was concentrated under reduced pressure to obtain a black solid.
Water (12 ml) was added, the
temperature was raised to 35 - 40 C, and the mixture was stirred for 30
minutes, filtered, and dried to
obtain brown solid reference compound A (35 mg).
11-I NMR (400 1VIIHz,CDC13): 8.26(d, J=8.0Hz, 1H), 8.18(d, J=8.0Hz, 11-0,
7.58(t,
J=16.0Hz, 1H), 7.48(t, J=20.0Hz, 1H), 6.81(m, J=16.0Hz, 2H), 6.57(d, J=8.0H.z,
1H), 6.36(d,
J=8.011z, 11-1), 4.26 (s, 2H), 4.23(d, J=9.0Hz, 211), 2.16(s, 6H).
MS miz (ESI): 371.1 [M-1].
Reference embodiment
B: Preparation of
((44(4-hydroxy-516,718-tetrahydronaphthalene-1-yl)methyl)-3,5-di methyl
phenoxy)methyl)phosphoric
22
CA 03147789 2022-2-11
acid.
OH
1
HO
HO
Using the route of synthesis of reference embodiment A, reference compound B
may be obtained by
replacing the raw material 1-naphthol (A-7) synthesized in Step 5 with
tetrahydronaphthol.
in NMR (400 MI-1750)30H): 6.72(s, 211), 6.35 (d, T=8.0117, 11-0, 6.04(d,
J=8.0Hz, 1 H),
4,18(d, J=12.0Hz, 211), 3,68(s, 211), 2,74(t, J=4,0Hz, 211), 2.67(t, J=12.0Hz,
21-0, 2+11(s, 611),
1.86(m, J=64.0Hz, 411).
MS mlz (ES1): 3711 [M-1].
Embodiment 1:
Preparation of
((4-((7-hydroxy-1,1-dimethy1-2 ,3-di hydro-1H -indene-4-position) methyl )-3,5-
di methyl phenoxy) methyl ) p
hosphoric acid (compound 1).
0
HO
0
OH
1
HO
Using the synthetic route of reference embodiment A, compound 1 may be
obtained by the following
synthetic method.
0 OH
0
CH3MgBr, THF,. io PPA 0 = =
* 13171) ,..r3m HO *
1-1 1-2
1-3 1-4
"
0
e Ho
o
HO
A-5
0, µn_o
`el ),---TMSCI,K1
n
QH
OH
TFA. DCM
01-13011
1-5 1
Step 1: Synthesis of compound 1-2:
Compound 1-1 (1.94 g, 1.0 eq) was dissolved in 20 ml THF, the reaction system
was replaced with
23
CA 03147789 2022-2-11
nitrogen, then the temperature was lowered to around 0 C, 3M methylmagnesium
bromide was slowly
added dropwise, the temperature was kept below 5 C, addition dropwise was
completed, and stirring was
performed for 0.5 hours with the temperature kept. After the reaction was
completed, a saturated
ammonium chloride solution was added dropwise to the reaction solution to
quench the reaction,
extraction was performed with EA (50 mI*3), and the EA phase was washed with
water (50 mI*2),
washed with saturated brine (100 ml), dried with anhydrous sodium sulfate, and
concentrated to obtain
intermediate compound 1-2, 2.0 g.
1-H INNIR (400 MHz, CDC13): 7.26-7.17 (m, 1H), 6.81-6.72 (m, 2H), 3.79 (s,
3H), 2.70-2.66
(m, 2H), 1.81-1.77 (m, 2H)2 1.28 (s, 6H).
Step 2: Synthesis of compound 1-3:
Polyphosphoric acid (PPA) (2.9 g) was added to a reaction flask, then stirring
was started, compound
1-2 (582 mg) was slowly added, and the reaction was stirred at room
temperature for 2 hours. Ice water
(100 ml) was added to quench the reaction, then extraction was performed with
EA (100 mI*3), and the
EA phases were combined, washed with water (50 mI*2) and saturated brine (100
ml), dried with
anhydrous sodium sulfate, concentrated, and column-chromatographed to obtain
compound intermediate
compound 1-3, 50 mg.
1H NMR (400 MHz, CDC13): 7.14-7.10 (m, 1H), 6.80 (d, J=8,0Hz, 1H), 6.69 (d,
J=8,0Hz,
1H), 3.82 (s, 3H), 289-2.85 (m, 211), 1.91-1.88 (m, 2H), 1.36 (s, 6H).
Step 3: Synthesis of compound 1-4:
Compound 1-3 (349 mg, 2 mmol) was dissolved in DCM (20 ml), the temperature
was lowered to
0 C, and BBr3 (2 ml) was added dropwise. After the addition was completed, the
reaction was stirred at
0 C for 1 h. After the reaction, 10m1 of water was added, extraction was
performed with EA to obtain the
organic phase, and the organic phase was washed with 10 ml of brine, dried,
concentrated, and
column-chromatographed to obtain compound intermediate 1-4, 230 mg, which was
directly put into the
next reaction without purification.
Step 4: Synthesis of compound 1-5:
Compound 1-4 (220 mg, 1.4 mmol) and A-6 (2.0 mmol) were dissolved in DCM (5
ml), the
temperature was lowered to -1 C, TFA (307 jil, 4.1 mmol) was added dropwise,
and the reaction was
24
CA 03147789 2022-2-11
stirred for 1 h. After the completion of the reaction, 20 ml of DCM and 10 ml
of water were added,
extraction was performed with EA, the layers were separated to obtain an
organic phase, and the organic
phase was washed with 10 ml of water and 10 ml of brine, respectively. After
drying, concentration and
column chromatography (PE:EA = 1:1), compound 1-5 (110 mg, pale yellow oily
substance) was
obtained.
Step 5: Synthesis of compound 1:
Compound 1-5 (105 mg, 2.2 mmol), KI (118 mg, 0.71 mmol), and TMSC1 (77 mg,
0.71 mmol) were
dissolved in acetonitrile (1 ml), the temperature was raised to 50 C, and the
reaction was stirred for 2 h.
After the reaction, 10 ml of water and 10 ml of EA were added for extraction
to obtain the organic phase,
ml of EA was again added to the aqueous phase for extraction, the organic
phases were combined,
dried, and concentrated, and preparation was performed with a thin-layer
silica gel plate (DCM:Me0H =
8:1) to obtain compound 1(20 mg).
II-1 NMR (400 MHz, DMSO-d6): 6.71 (s, 21-1), 612 (d, J=8.0Hz, 11-1), 6.09 (d,
.I=8.0Hz,
1H), 4.03 (d, J= 12.0Hz, 2H), 3.73 (s, 2H), 2.88-2.85 (m, 2H), 1.94-1.91 (m,
2H), 1.37 (s, 6H).
MS m/z (ESI): 389.1 [M-1].
Embodiment 2: Preparation of
((4-((7-hydroxy-1-methy1-2 ,3-di hydro-1 H -indene-4-position) methyl )-3,5-di
methyl phenoxy)methyl
phosphoric acid (compound 2).
HO
0 ID,
HO/ OH
2
Using the route of synthesis of embodiment 1, compound 2 may be obtained by
replacing the
intermediate 1-4 synthesized in step 4 with 3-methyl-2,3-dihydro-1H-indene-4-
ol.
MS m/z(ESI):375.1[M-1].
Embodiment 3:
Preparation of
((4((4-hydroxy-515-dimethy1-5,6,7,8-tetrahydronaphthalen-1-yl)methyl)-315-
dimethylphenoxy)methyl)ph
osphoric acid (compound 3).
CA 03147789 2022-2-11
õ..,..... pH
HO
0 R.
3 HO
Compound 3 may be obtained using the route of synthesis of embodiment 1,
wherein the synthesis of
intermediate 3-4 is as follows.
, 0 Br
---"->
0 PPA
BBr3
¨1.- 0 ' 3-2 DCIVI OH
3-1
3-3 3-4
HO 0 -(
0
0
..., ,
Ps, 0
,OH
0 0 P
......, 1%.a
HO 0 F
A-8 HO
TMSCI, KI OH
TFA, DCM
CH3CN
3-6
3
Step 1: Synthesis of compound 3-2:
Mg powder (1.56 g, 65.1 mmol) was immersed in anhydrous ether (10 ml), and 1
pellet of 12 was
added. First, 1/3 of the raw material compound 3-1 (10 g, 46.5 mmol) was
slowly added dropwise in an
ether (10 ml) solution, then the remaining raw material compound 3-1 was
slowly added dropwise after
the reaction was induced, and microreflux was continued for 0.5 h. Cul (0.66
g, 3.5 mmol) and THF
(10 ml) were added to another reaction flask, the temperature was lowered to -
20 C, a prepared format
reagent was added dropwise, dimethyl oxirane (5 ml, 55.8 mmol) was added
dropwise after the addition
was completed, and the reaction was stirred for 2 h. After the reaction, 20 ml
of water was added to
quench the reaction, and then 30 ml of EA was added to extract the organic
phase, which was dried,
concentrated, and column-chromatographed (PE:EA = 10:1 - 1:1) to obtain
intermediate compound 3-2
(4.2 g, a light yellow liquid).
Step 2: Synthesis of compound 3-3:
Compound 3-1 (2.2 g, 10.6 mmol) was slowly added dropwise into PPA (10 g), the
temperature was
kept between 15 - 25 C, and, after the dropping was completed, the mixture was
stirred at room
26
CA 03147789 2022-2-11
temperature for 2 h. After the completion of the reaction, 10 ml of water and
10 ml of ether were added
for extraction, and the aqueous phase was extracted 2 times with ether. The
organic phases were
combined, dried, concentrated, and subjected to column chromatography (PE) to
obtain compound 3-3
(230 mg, a pale yellow oily substance).
Step 3: Synthesis of compound 3-3:
Compound 3-2 (230 mg, 1.2 mmol) was dissolved in DCM (10 ml), the temperature
was lowered to
0 C, BBr3 (1 ml) was added dropwise, and the reaction was stirred at 3 C for 1
h. 10 ml of water was
added to quench the reaction, and then 10 ml of DCM was added to extract the
organic phase, which was
dried, concentrated, and column-chromatographed (PE:EA = 10:1) to obtain
compound 3-4 (100 mg, a
light yellow oily substance).
In steps 4 and 5, compound 3 may be prepared by using route of synthesis of
embodiment 1.
NMR (400 MHz, DM50-4): 6.71(s, 21-1), 6.32 (d, J=8.0Hz, 1H), 6.02 (d,
J=12.0Hz,
1H), 4.04 (d, 1=12.0Hz, 2H), 3.67 (s, 2H), 2.73-2.70 (m, 2H), 2.10(s, 6H),
1.85-1.78 (m, 2H),
1,67-1.63 (m, 2W, 1.42(s, 6W.
MS miz (ESI) 403.1 NA]
Embodiment 4:
Preparation of
((4-( (4-hydroxy-5,5-d i methy1-6,7,8,9-tetrahydro-5 H-benzo[7]annun-1-
yl)methyl )-3 ,5-d i methyl phenoxy)
methyl)phosphoric acid (compound 4).
OH
HO
0
HO .C3
4
Using the route of synthesis of embodiment 1, compound 4 may be obtained,
wherein the synthesis
of intermediates 4-7 is as follows.
27
CA 03147789 2022-2-11
0 0
0
0
Br2
LiOH "to
OH
4-1 Br
Br
4-2
4-3
a
cIzIII
0 0
OH
Sodi um
PPA acetate
4-5
TiCIJMA DC M
BBr3
Pc1/0
Br
4-4
4-6 4-7
Step 1: Synthesis of compound 4-2
Compound 4-1 (4.4 g) was put into a reaction flask, chloroform (30 ml) was
added, the temperature
was lowered by stirring to 0 C, then liquid bromine (3.5 g/30 ml chloroform)
was added dropwise, and,
after the addition was completed, the reaction was stirred at room temperature
for 0.5 h. The reaction was
quenched with sodium sulfite solution (20 ml), and extraction with EA (100
mI*3), washing with water
(50 mI*2), washing with saturated brine (50 ml), and drying with anhydrous
sodium sulfate was
performed to obtain a 5.3 g product.
11-1NMR (400 MHz, CDC13)= 7.40 (d, J=80 Hz, 1H), 6,76 (d, J=4.0 Hz, 114), 6.63-
6.60 (m,
111), 3.77 (s, 3H), 3.67 (s, 3H), 2.71-2.67(m, 2H), 2.38-2.34 (m, 214), 1.74-
1.63 (m, 4H).
Step 2: Synthesis of compound 4-3
Compound 4-2 (5.7 g, 1.0 eq) was dissolved in THF (30 ml), water (30 ml) was
added, lithium
hydroxide monohydrate (3.9 g, 5.0 eq) was added with stirring, pH = 2 was
reached by adjustment with
4N hydrochloric acid after stirring overnight, and the mixture was extracted
with EA (100 mI*3), washed
with water (100 mI*2), washed with saturated brine (100 ml), dried with
anhydrous sodium sulfate, and
concentrated to obtain compound 4-3: 5.3 g.
Step 3: Synthesis of compound 4-4
Compound 4-3 (3.6 g) was added dropwise to PPA (240 g), the temperature was
raised to 55 C, and
the reaction was stirred for 4 h. Ice water (400 ml) was added to quench the
reaction, and extraction with
EA (150 mI*3), washing with water (100 mI*2), washing with saturated brine
(100 ml), drying with
anhydrous sodium sulfate, and concentration was performed to obtain oily
compound 4-4: 2.4 g.
1H NIAR (400 MHz, CDC13): 7.54 (d, J=8.0 Hz, 1H), 6.73 (d, J=8.0 Hz, 111),
3.79 (s, 3H),
2,93-2.90 (m, 2H), 2.61-2.60 (in, 2H), 1.76 (m, 4H).
28
CA 03147789 2022-2-11
Step 4: Synthesis of compound 4-5
Compound 4-4 (1.61 g, 1.0 eq) and sodium acetate (0.5 g, 1.0 eq) were put into
a reaction flask,
methanol (8 ml), dioxane (16 ml), and Pd/c (160 mg) were added, replacement
with I-h was performed,
and the reaction was stirred overnight at room temperature under a hydrogen
atmosphere. After filtration
and concentration, the residue EA (200 ml) was dissolved, washed with water
(50 mI*2), washed with
saturated brine (50 ml), dried with anhydrous sodium sulfate, and concentrated
to obtain yellow oily
compound 4-5: 1.2 g.
NMR (400 MHz, CDC13): 7.29-7.25 (m, 111), 6.83 (d, J=8.0 Hz, 111), 6.74 (d,
J=8.0 Hz,
111), 3.81 (s, 3H), 2.75-2/2 (m, 211), 2.65-2.63 (m, 211), 1.82-1.80 (m, 4H).
Step 5: Synthesis of compound 4-6
TiCI4 (7.8 g, 6.8 mmol) was dissolved in DCM (15 ml), the temperature was
lowered to -50 C,
(CH3)2Zn (41 ml, 0.1 M toluene solution) was added, stirring was performed for
0.5 h, 4-5 (1.3 g,
dissolved in 40 ml DCM) was added dropwise, and the temperature was raised
naturally with stirring
overnight. After completion of the reaction, the reaction was quenched by
adding 50 ml of water, the
mixture was extracted 3 times with 50 ml of DCM, and the organic phases were
combined, washed with
50 ml of brine, dried with anhydrous sodium sulfate, and concentrated. Column
chromatography was
performed to yield 4-6 (a 1 g colorless liquid).
Step 6: Synthesis of compound 4-7
Compound 4-6 (1 g, 4.9 mmol) was dissolved in DCM (50 ml), the temperature was
lowered to 0 C,
BBr3 (5 ml) was added, and the mixture was stirred at room temperature for 1
h. After the completion of
the reaction, 50 ml of water was added to quench the reaction, and the DCM
phase was extracted and
washed with 50 ml of brine. It was dried with anhydrous sodium sulfate,
concentrated, and
column-chromatographed (PE:EA = 100:1 - 10:1) to obtain compound 4-7 (335 mg,
a colorless oily
substance).
LH NMR (400 MHz, CD30D): 6.80-6.76 (m, 1H), 6.56 (d, J=12.0 Hz, 1H), 6.48 (d,
J=8.0
Hz, 1H), 2.84 (Ins, 211), 1.86-1.75 (in, 6H), 1.46 (s, 611).
Compound 4 may be prepared by using intermediate 4-7 and following the route
of synthesis of
embodiment 1.
29
CA 03147789 2022-2-11
111 NMR (400 MIL,CD3OD). 9.29 (s, 111), 6.63-6.21 (m, 2H), 6.38 (d, J=12.0Hz,
1H),
6.12 (d, J=12.0 Hz, 1H), 4.42-4.32 (m, 1H), 3.76-3.69 (m, 4H), 2.61-2.55 (m,
2H), 2.05 (s. 6H),
1.65-1.53(m, 6H), 1.11 (d, J=8.0 Hz, 6H).
MS miz (ESI): 417.1 [M-1]
Embodiment 5:
Preparation of
((4-( (7-hydroxy-111-d i methy1-2 ,3-di hydro-1H -indene-4-position) oxy)-3,5 -
d imethylphenoxy)methyl) phos
phoric acid (compound 5).
= 0
/10
HO
OPN
5 HOOH
Using a route of synthesis similar to that in embodiment 1, compound 5 may be
obtained.
MS m/z(ESI): 391.1[M-1].
Embodiment 6:
Preparation of
((315-dichloro-44(7-hydroxy-1,1-dimethy1-2,3-d ihydro-1H-i ndene-4-positi
on)methyl )phenoxy) methyl ) ph
osphoric acid (compound 6).
Cl
,0
ati
HO CI
0 ID,
HO' OH
6
Using a route of synthesis similar to that in embodiment 1, compound 6 may be
obtained.
NMR (400 MHz,CD30D): 7.08 (s, 2H), 6.35 (d, J=8.4Hz, 1H), 6.19 (d, J=8.4 Hz,
1H),
4.66-4.51 (m, 2H), 4.07-4.02 (m, 4H), 2.90 (t, J=14.7 Hz, 2H), 1.93 (t, J =
14.7 Hz, 21-1), 1.37
(m, 6H).
MS mlz (ESI): 429.5 [M-1].
Embodiment 7:
Preparation of
4-(3-chloropheny1)-24 (44 (7-hydroxy-111-d i methy1-2 ,3-di hydro-1H -indene-4-
position)methyl)-3,5-di met
hylphenoxy)methyl)-1,312-dioxaphosphorane 2-oxo (compound 7).
CA 03147789 2022-2-11
0
HO 0 P
it\
0 0
7
fa Cl
The following route of synthesis was used:
OH OH
CI so
HO
01:k
H0 P, _________________________
HO OH
6' 0
DCC
fi CI
1
7
Compound 1 (211 mg, 0.54 mmol) and 1-(3-chlorophenyl) propane-1,3-diol (302
mg, 1.62 mmol)
were dissolved in pyridine (1 ml) and DM F (5 ml), and DCC (334 mg, 1.62 mmol)
was added at room
temperature. The temperature was raised to 70 C and stirring was performed for
4 h. After cooling to
room temperature, the mixture was filtered, concentrated, and separated by
column chromatography to
obtain racemic compound 7 (100 mg).
MS miz(ES1): 541.1[M+1].
Embodiment 8:
Preparation of
(((((4-((7-hydroxy-1,1-dimethyl-2,3-dihydro-1H-indene-4-position)methyl)-3,5-
dimethylphenoxy)methyl
)phosphoryl)bis(oxy))bis(methylene)bispivaloyl (compound 8).
et., at
HO 0 P
0
d o-\
0
8
The following route of synthesis was used:
31
CA 03147789 2022-2-11
I ¨Th,o)Lkis
0 ,,,.... P--%'..071r
HO
R
0 8-1
4 , 0
....----.
0 0 Th
HO 0 13.,
HO OH 8
1
Diisopropylethylamine (140 mg, 1.08 mmol) was added to a solution of compound
1 (211 mg,
0.54 mmol) in acetonitrile (10 ml) at room temperature. The temperature was
raised to 40 C, stirring was
performed for half an hour, then iodide 8-1 (261 mg, 1.08 mmol) was added, and
stirring was continued
overnight. Then, addition of iodide 8-1 (261 mg, 1.08 mmol) and of
diisopropylethylamine (140 mg,
1.08 mmol) was continued, and the reaction was continued at the temperature
for 6 h. The reaction was
quenched with 50 ml of water, and the EA phase was extracted and washed with
50 ml of brine. It was
dried with anhydrous sodium sulfate, concentrated, and separated by column
chromatography to obtain
racemic compound 7 (150 mg).
MS m/z(ESI): 619.2[M+1].
Embodiments 9 and 11: Preparation of compounds 9 and 11.
HO
0N..iy 01/ HO
c
0 H
d 11 I
0
a
9
0 11
SI
The following route of synthesis was used:
0 OH
DCC -79
p
HO 0 K OH HO
0 R_
HO'
1
p hd OH
1 ) 80012
p
Lr HO
0 ==1=k, N 1(0 õ..-i, P
H2N 0,r., 0 H
0 --r-= HO
0 'R. lice0,,v
d [1 2)
0 0 I
49
11
3) Resolution
Step 1: Synthesis of intermediate 9-1:
32
CA 03147789 2022-2-11
DMF (20 ml) and pyridine (4 ml) were added in turn to a mixture of compound
1(780 mg, 2 mmol),
phenol (376 mg, 4 mmol), DCC (1.24 g, 6 mmol), and DMAP (244 mg, 2 mmol), the
temperature was
raised to 80 C after completion of the addition, and the reaction was stirred
for 15 hours. After cooling,
the solvent was directly concentrated under reduced pressure, and intermediate
9-1 (200 mg) was
obtained by column chromatography.
MS miz(ES1): 465.1[M+1].
Step 2: Synthesis of compounds 9 and 11:
Under an ice bath, thionyl chloride (1.72 mmol) was slowly added dropwise to a
solution of compound
9-1 (200 mg, 0.43 mmol) and DMF (32 mg, 0.43 mmol) in dichloromethane (2 m1).
After the dropwise
addition was completed, the reaction system was heated to reflux and stirred
for 3 h. The temperature was
lowered to room temperature, L-alanine isopropyl ester hydrochloride (287 mg,
1.72 mmol) and
di isopropylethylamine (222 mg, 1.72 mmol) were added to the reaction system,
and the mixture was kept
at room temperature and stirred for 15 hours. The reaction was quenched by
adding water (50 ml), and
extraction was performed 1 time with ethyl acetate (50 ml). The organic phase
was concentrated under
reduced pressure and resolved to obtain compounds 9 and 11.
Structure characterization of compound 9:
MS mh (ESI): 580.1 [M+1].
'H NMR (400 MHz,DMSO-d6): 8.79 (s, 11-1), 7.38-7.34 (m, 2H), 7.23-7.15 (m,
3H), 6.70
(m, 2H), 6.35 (d, J=8.0Hz, 1H), 6.01-5.99 (m, 111), 5.90-5.84 (m, 1H), 4.78-
4.70 (m, 1H), 4.32
(d, J=9.6Hz, 2H), 4.00-3.93 (m, 1H), 3.66 (s, 2H), 2.84-2.80 (m. 2H), 2.09 (s,
6H), 1.87-1.83
(m, 211), 1.30 (s, 6H), 1.19 (d, J= 6.8 Hz, 3H)., 1.10-1.08 (m, 611).
Embodiment 10: Preparation of compound 10
Using a route of synthesis similar to that in embodiment 7, compound 10 may be
obtained.
OH OH
CI Ai
DCC HO
P
p 1) e" 10-i
0 P
HO 0 R....nu
_____________________________________________________ 6 \O
Hd 2) Resolution
ci
DMF (20 ml) and pyridine (4 ml) were added in turn to a mixture of compound
1(780 mg, 2 mmol),
compound 10-1 (1.16 g, 6 mmol) and DCC (1.24 g, 6 mmol). After the addition,
the temperature was
33
CA 03147789 2022-2-11
raised to 70 C and stirring was performed for 18 hours. The solvent was
concentrated under reduced
pressure and resolved to obtain compound 10 (150 mg).
MS m/z (ESI): 541.1 [M+1].
NMR (400 MHz,DMSO-d6): 8,81(s, 1H), 7.48(s, 1H), 7.43-7.41(m, 3H), 6,74(s,
2H),
6.35(d, 1=7.6Hz, 1H), 6.02 (d, J=7.6Hz, 1H), 5.82 (m, 1H), 4.62-4.38(m, 411),
3.66(s, 21ft),
2.86-2.77(m, 2H), 2.29-2.18(m, 2H), 2.09(s, 6H), 1.90-1.80(m, 2H), 1.30(s,
6H).
Using compound 2 as the starting material and following the routes of
synthesis of embodiment 7 -
embodiment 11, the following compounds may be prepared.
Embodiment Structure
MS m/z(ESI) [M+1]
12
526.9
0
HO 0 P
\
00
12
CI
13
605.1
0
HO 0 P
\
0
d O\ J/
0
13
14
566.1
0
HO 0-1-41/4Pt
N
0 H
0
14
411:1
34
CA 03147789 2022-2-11
15
526.9
0
HO 0 IR,
00
CI
16
566.1
0
sJyO
HO 0 4
0 H
0
16
Test embodiment 1: Test of the binding ability of the compound to TRa:
1. Main experimental materials and instruments:
Envision 2104 microplate reader
Biotin-SRC2-2 co-activating peptide purchased from Sangon Biotech (Shanghai)
Co., Ltd.
TR-a LBD, GST purchased from Thermo Fisher (product No. PV4762)
Europium-conjugated anti-glutathione antibody purchased from Cisbio (product
No. 61GSTKLB)
Streptavidin-02 purchased from Cisbio (product No. 6105A DAB)
2. Preparation and processing of compounds
2.1 Preparation of compound dimethyl sulfoxide stock solution
All compounds 1 to 6 were dissolved in dimethyl sulfoxide and prepared as a 10
millimole stock
solution.
2.2 Storage of compounds
After being dissolved in dimethyl sulfoxide, compounds 1 to 6 may be stored in
a desiccator at room
temperature for three months. For long-term storage, they may be placed in a
refrigerator at -20 C.
3. Experimental steps
CA 03147789 2022-2-11
3.1 Preparation of a lx reaction buffer
3.2 Screening of compounds:
a) Positive drug triiodothyronine (T3) was diluted from 10 millimoles (100X)
or the test compound
from 1 millimole (100X) in a ratio of 1:3 in geometric proportion with 100%
dimethyl sulfoxide to
achieve a total of 10 concentrations.
b) 4x concentration gradient-diluted compounds were prepared with lx reaction
buffer.
c) 5 microliters of 4x concentration gradient-diluted compounds were added to
a 384-well
experiment plate.
d) 4X TRa LBD and 4X RXRa were prepared with lx reaction buffer.
c) 5 microliters of 4x TRa LBD and 4X RXRa were added to a 384-well experiment
plate.
f) 2X biotin-SRC2-2, 2X europium-conjugated anti-glutathione antibody and 2X
streptavidin-d2
were prepared with lx reaction buffer.
9)10 microliters of 2X mixture (refer to step f) was added to a 384-well
experiment plate.
h) The 384-well experiment plate was centrifuged in a centrifuge at 1000 rpm
for 1 minute.
i) Incubation was carried out in the dark at room temperature for 1 hour.
j) The Envision 2104 microplate reader was used to record the fluorescence
signal values at the
wavelengths of 665 nm and 615 nm in each well of the 384-well experiment
plate, and the fluorescence
ratio of 665 nm/615 nm was calculated.
4. Data analysis
4.1 Calculation of the relative ratio of each hole (Ratio66somm151m-Rationok)
4.2 The activity percentage was calculated as follows:
--T-
Ratioccropound-Ratio Blank
Activity (%) = *100
Ratio positive-Ratio Blank
Ratio Compound: Average of the relative ratios of the compound wells of the
embodiments
Ratio Positive: Average of the relative ratios of all positive control wells.
Ratio Blank: Average of the relative ratios of all negative control wells.
36
CA 03147789 2022-2-11
4.3 Drawing of curves and calculation of EC50:
ECK was calculated by fitting the relationship between activity (%) and
compound logarithmic
concentration with Graphpad 5.0 by a nonlinear regression method.
Y = Bottom + (top - bottom)/(1 + 10"((LogEC50 - X)*slope))
X: Compound logarithmic concentration Y:
Percentage activity
The specific test data are shown in Table 1 below.
Test embodiment 2: Test of the binding ability of the compound to TRJ3
1. Main experimental materials and instruments:
Envision 2104 microplate reader
Biotin-SRC2-2 co-activating peptide purchased from Sangon Biotech (Shanghai)
Co., Ltd.
TRI3 LBD, GST purchased from Thermo Fisher (product No. PV4762)
Europium-conjugated anti-glutathione antibody purchased from Cisbio (product
No. 61GSTKLB)
Streptavidin-D2 purchased from Cisbio (product No. 6105A DAB)
2. Preparation and processing of compounds
2.1 Preparation of compound dimethyl sulfoxide stock solution
All compounds 1 to 6 were dissolved in dimethyl sulfoxide and prepared as a 10
millimole stock
solution.
2.2 Storage of compounds
After being dissolved in dimethyl sulfoxide, the compounds may be stored in a
desiccator at room
temperature for three months. For long-term storage, they may be placed in a
refrigerator at -20 C.
3. Experimental steps
3.1 Preparation of a lx reaction buffer
3.2 Screening of compounds:
a) Positive drug triiodothyronine (T3) was diluted from 10 micromoles (100X)
or the test compound
from 1 millimole (100X) in a ratio of 1:3 in geometric proportion with 100%
dimethyl sulfoxide to
achieve a total of 10 concentrations.
b) 4x concentration gradient-diluted compounds were prepared with lx reaction
buffer.
37
CA 03147789 2022-2-11
c) 5 microliters of 4x concentration gradient-diluted compounds were added to
a 384-well
experiment plate.
d) 4X TRI3 LBD and 4X RXRI3 were prepared with lx reaction buffer.
c) Add 5 microliters of 4x TRI3 LBD and 4X RXRI3 to a 384-well experiment
plate.
f) 2X biotin-SRC2-2, 2X europium-conjugated anti-glutathione antibody and 2X
streptavidin-d2
were prepared with lx reaction buffer.
9)10 microliters of 2X mixture (refer to step f) was added to a 384-well
experiment plate.
h) The 384-well experiment plate was centrifuged in a centrifuge at 1000 rpm
for 1 minute.
i) Incubation was carried out in the dark at room temperature for 1 hour
j) The Envision 2104 microplate reader was used to record the fluorescence
signal values at 665 nm
and 615 nm in each well of the 384-well experiment plate, and the ratio 665
nm/615 nm was calculated.
4. Data analysis
4.1 Calculation of the relative ratio of each hole (Ratio665nrnisisirn-
Rationnk)
4.2 The activity percentage was calculated as follows:
--T-
Ratiocornpound-Ratio Blank
Activity (%) =
_________________________________________________________________________ *100
1.711.7 ¨
NMI() Positive-nal-10 Blank
Ratio compound: Average of the relative ratios of the compound wells of the
embodiments
Ratio Positive: Average of the relative ratios of all positive control wells.
Ratio Blank= = Average of the relative ratios of all negative control wells.
4.3 Drawing of curves and calculation of EC50:
EC50 was calculated by fitting the relationship between activity (%) and
compound logarithmic
concentration with Graphpad 5.0 by a nonlinear regression method.
Y = Bottom + (top - bottom)/(1 + 10"((LogEC50 - X)*slope))
X: Compound logarithmic concentration Y:
Percentage activity
The specific test data are shown in Table 1 below. The selectivity algorithm
was calculated after
being standardized with T3 on the basis of the literature (A Pharmacology
Primer Techniques for More
38
CA 03147789 2022-2-11
Effective and Strategic Drug Discovery, 4th Edition, Page 220).
Table 1 Binding activities of the compounds to thyroxine receptor 13:
Compound THR-13 THR-a
EC.50(uM) Emax(%) EC50(UM)
EMU(%) a/13 selectivity (multiple)
Reference
0.109 100.00 0.280 106.50
4.7
compound A
Reference
0.041 110.70 0.23 105.70 11.4
compound B
1 0.012 104.10
0.041 96.42 7.0
2 <0.100 103.60 <0.200
98.30
3 0.104 100.00
0.270 104.80 4.8
4 >1 >10
<0.100 109.50 <0.200 102.10
6 0.108 79. 04 0.05
99.15 0.7
MB07444 0.033 113.00 0.051 112.60
2.9
T3 0.0004 98.31
0.0002 106.70 1.0
Conclusion: Compared with the published comparative compound MB07444, most
compounds of
the present invention unexpectedly showed very high selectivity; in addition,
the activity of compound 1
for THR-I3 was much higher than that of the contrast compound MB07444. Even
compared with the
naphthol reference compound A, a compound of the present invention still had
higher activity and
selectivity.
Test 3: Drug metabolism experiment on prodrug in SD rats
In the experiment, two groups of 12 male SD rats having similar body weights
were selected and
orally administered compound 9, compound 10, and the control drug VK2809 (the
prodrug of MB07444,
structured as shown below) at a dose of 3 mg/kg at a single time,
respectively, and blood and liver
samples were collected at different time points.
39
CA 03147789 2022-2-11
Metabolism by
liver
0 ----,.... ----) I I OH
HO ,..... 0
0 P, ¨
HO
OP
do
60H
. CI
VK2809
MI307444
Preparation of test products
Solutions of compound 9/compound 10 and VK2809 with a final concentration of
0.6 mg/mL were
respectively prepared, and the ratio between the solvents used for the
preparation was PEG400:pure water
= 50:50 (v/v).
Group and dose
No randomization was performed. The weights of the animals were measured
before administration,
and healthy animals with similar body weights were selected for inclusion in
the experiment. The oral
dose was 3 mg/kg.
Sample collection
At least 0.2 mL of blood was collected from the tail vein or jugular vein, and
heparin sodium was
used as the anticoagulant.
Acquisition time
15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h after administration.
Sample treatment
After being collected, a blood sample was placed in a marked ice-water bath
centrifuge tube, plasma
was quickly separated by centrifugation under centrifugation conditions: 3500
rpm, 10 minutes, and 4 C,
and the plasma was stored under -40 C for testing.
After the liver sample was collected, its surface was washed with normal
saline, dried with medical
gauze, placed in a labeled small ziplock bag, and stored under -40 C for
testing.
Sample analysis
Liquid chromatography-mass spectrography conditions
Liquid chromatography conditions:
HPLC: LC-20ADxR, SHIMADZU
CA 03147789 2022-2-11
Liquid phase pump: LC-20ADxR
Column thermostat: CTO-20A
Autosampler: SI L-20ACxR
Controller: CBM-20A
Degasser: DUG-20A3R chromatographic column: ZORBAX Eclipse Plus C18 2.1*50 mm,
3.5 gm,
Agi lent
Precolumn: Guard column C18 4 *2.0 mm, Phenomenex
Mobile phase: A: 2mM ammonium acetate aqueous solution;
B: Acetonitri le;
Autosampler needle washing liquid: 80% acetonitrile aqueous solution
Autosampler needle washing program: washing mode: before and after suction
Flushing volume: 200 L
Flushing speed: 35 pL/sec
Mobile phase gradient:
Time (min) A%
B%
0.01 75
25
0.50 10
90
1.85 10
90
1.90 75
25
2.50 Stop
Flow rate: 0.75 mL/min
Autosampler temperature: 42C
Injection volume: 2 RL
Running time: 2.50 min
Mass spectrometry analysis conditions:
Q TRAP6500 mass spectrometer with ESI source was used to perform negative ion
MRM scanning.
An LC-MS/MS analysis method was used to detect the contents of compound 9,
compound 10 and its
41
CA 03147789 2022-2-11
active metabolite 1, VK2809 and its active metabolite MB07444 in the plasma
and liver. The metabolic
kinetics data analysis software WinNonlin 7.0 was used to calculate the
concentration data of plasma, and
the non-compartmental model (NCA) method was used to calculate the
pharmacokinetic parameters.
Results and analysis:
The main pharmacokinetic parameters of the plasma of the SD rats are listed in
Table 2 below:
Table 2
Kel TMEIX
Cmax AUCO-t AUC0,0 t1/2
Administration Compound
(11h) (h)
(nglmL) (ng=h/mL) (ng-h/mL) (h)
Compound 9 - -
BLQ BLQ BLQ -
Compound 9
Compound 1 0.147 4.67
134 1287 1347 4.82
Compound 10 0.428 4.67
19.9 69.2 89.3 1.73
Compound 10
Compound 1 0.154 6.67
45.1 586 613 4.59
VK2809 0.273 6.00
9.9 43.6 83.3 4.81
VK2809
MB07444 0.131 7.33
38.8 420 448 5.29
Conclusion: After being orally administered to the rats, the prodrug compounds
9 and 10, like the
control drug VK2809, were quickly transformed into the active parent drug
compound 1. In addition, the
concentrations of both in plasma were not high.
The parameters of concentration in the livers of the SD rats and a comparison
with the concentration
in plasma are shown in Table 3 below:
Table 3
Administration Ct(ng/g)
Test drug
3 mg/kg 1 h
4h 8h 24h
Blood BLQ
BLQ BLQ BLQ
Compound 9 Liver BLQ
BLQ BLQ BLQ
Compound 9 (prodrug) Liver-to-blood
_ ratio
Compound 1 Blood 15.0
96.3 29.9 8.4
42
CA 03147789 2022-2-11
(parent drug) Liver
312 1563 762 396
Liver-to-blood
20.8 16.2 25.5 47.1
ratio
Blood
8.8 7.9 10.3 BLQ
Compound 10 Liver
141 223 46.5 BLQ
(prodrug) Liver-to-blood
16.2 7.8 4.7 -
ratio
Compound 10
_______________________________________________________________________________
___________________________________
Blood
5.4 28.4 43.6 3.83
Compound 1 Liver
693 2578 1710 431
(parent drug) Liver-to-blood
29.1 20.6 17.7 121
ratio
Blood
2.9 4.8 5.6 BLQ
VK-2809 Liver 39
30 17 BLQ
(prodrug) Liver-to-blood
8.6 5.2 4.0 -
ratio
VK-2809
Blood
5.9 16.9 29.6 3.6
MB07444 Liver
214 952 542 37
(parent drug) Liver-to-blood
15.9 14.0 13.6 9.9
ratio
Conclusion: The ability of the prodrug compound 10 prepared according to the
present invention to
transform into active metabolite drug 1 in the liver was much better than that
of VK2809. Under the same
prodrug dose, the absolute concentration of active metabolite compound 1 in
the liver was at least three
times that of the active metabolite drug MB07444 of the control drug VK2809,
and its liver-to-blood ratio
was also significantly higher than that of the control drug. Similarly,
prodrug compound 9 may also be
rapidly metabolized in the liver to produce active metabolite compound 1, and
its absolute concentration
is also higher than that of the control drug VK2809. The above data show that
compounds of the present
invention and prodrugs thereof are drugs with better liver-targeting
properties, and have unparalleled
43
CA 03147789 2022-2-11
pharmaceutical quality.
The above-described embodiments are intended only to explain the technical
ideas and
characteristics of the present invention, serving the purpose of enabling
those of ordinary skill in the art to
understand the contents of the present invention and implement the present
invention, instead of limiting
the scope of the present invention. Modifications or alterations made to the
above-mentioned
embodiments without departing from the spirit of the present invention shall
fall within the scope of the
present invention.
44
CA 03147789 2022-2-11