Note: Descriptions are shown in the official language in which they were submitted.
CA 03151815 2022-02-17
WO 2021/053667 1
PCT/IL2020/051011
COMBINATION CANCER THERAPY AND CYTOKINE CONTROL THERAPY FOR
CANCER TREATMENT
FIELD OF INTEREST
[0001] Disclosed herein are compositions and methods thereof for
inhibiting or reducing the
incidence of cytokine release syndrome (CRS) or a cytokine storm in a subject
undergoing cancer
therapy, for example but not limited to chimeric antigen receptor-expressing T-
cell (CAR T-cell)
cancer therapy. Further, disclosed herein are compositions and methods thereof
for decreasing or
inhibiting cytokine production in a subject experiencing cytokine release
syndrome or a cytokine
storm. Further, compositions disclosed herein may be used for treating,
preventing, inhibiting the
growth of, or reducing the incidence of, a cancer or a tumor in a subject.
Compositions may be used
for increasing survival of a subject suffering from a cancer or a tumor.
Compositions used may be
administered alone or in combination with other cancer therapeutic agents or
chemotherapies.
Methods disclosed herein include those comprising administration of a
combination therapy
comprising an early apoptotic cell population or supernatant thereof and one
or more cancer
.. therapeutic agents, such as CAR T-cells, for slowing, reducing, inhibiting,
or eliminating metastatic
spread of a cancer or a tumor in a subject.
BACKGROUND
[0002] While standard treatments for cancer are surgery, chemotherapy, and
radiation therapy,
improved methods, such as targeted immunological therapies, are currently
being developed and
tested. One promising technique uses adoptive cell transfer (ACT), in which
immune cells are
modified to recognize and attack their tumors. One example of ACT is when a
patient's own cytotoxic
T-cells, or a donor's, are engineered to express a chimeric antigen receptor
(CAR T-cells) targeted to
a tumor specific antigen expressed on the surface of the tumor cells. These
CAR T-cells are then
cytotoxic only to cells expressing the tumor specific antigen. Clinical trials
have shown that CAR T-
cell therapy has great potential in controlling advanced acute lymphoblastic
leukemia (ALL) and
lymphoma, among others.
[0003] However, some patients given CAR T-cell therapy and other immune
therapies experience a
dangerous and sometimes life-threatening side effect called cytokine release
syndrome (CRS), in
which the infused, activated T-cells produce a systemic inflammatory response
in which there is a
rapid and massive release of cytokines into the bloodstream, leading to
dangerously low blood
pressure, high fever and shivering.
[0004] In severe cases of CRS, patients experience a cytokine storm (a.k.a.
cytokine cascade or
hypercytokinemia), in which there is a positive feedback loop between
cytokines and white blood
CA 03151815 2022-02-17
2
WO 2021/053667
PCT/IL2020/051011
cells with highly elevated levels of cytokines. This can lead to potentially
life-threatening
complications including cardiac dysfunction, adult respiratory distress
syndrome, neurologic toxicity,
renal and/or hepatic failure, pulmonary edema and disseminated intravascular
coagulation.
[0005] For example, six patients in a recent phase I trial who were
administered the monoclonal
antibody TGN1412, which binds to the CD28 receptor on T-cells, exhibited
severe cases of cytokine
storm and multi-organ failure. This happened despite the fact that the TGN1412
dose was 500-times
lower than that found to be safe in animals (St. Clair EW: The calm after the
cytokine storm: Lessons
from the TGN1412 trial. J Clin Invest 118: 1344-1347, 2008).
[0006] To date, corticosteroids, biological therapies such as anti-IL6
therapies and anti-inflammatory
drugs are being evaluated to control cytokine release syndrome in patients
administered CAR T-cell
therapy. However, steroids may affect CAR T-cells' activity and/or
proliferation and put the patients
in danger of sepsis and opportunistic infections. Anti-inflammatory drugs may
not be effective in
controlling cytokine release syndromes or cytokine storms, because the
cytokine storm includes a
very large number of cytokines while there is limited ability to infuse
patients with anti-inflammatory
drugs. Novel strategies are needed to control cytokine release syndromes, and
especially cytokine
storms, in order to realize the potential of CAR T-cell therapy.
[0007] Cytokine storms are also a problem after other infectious and non-
infectious stimuli. In a
cytokine storm, numerous proinflammatory cytokines, such as interleukin-1 (IL-
1), IL-6, g-interferon
(g-IFN), and tumor necrosis factor-a (TNFa), are released, resulting in
hypotension, hemorrhage, and,
ultimately, multiorgan failure. The relatively high death rate in young
people, with presumably
healthy immune systems, in the 1918 H1N1 influenza pandemic and the more
recent bird flu H5N1
infection are attributed to cytokine storms. This syndrome has been also known
to occur in advanced
or terminal cases of severe acute respiratory syndrome (SARS), Epstein-Barr
virus-associated
hemophagocytic lymphohistiocytosis, gram-negative sepsis, malaria and numerous
other infectious
diseases, including Ebola infection.
[0008] Cytokine storm may also stem from non-infectious causes, such as acute
pancreatitis, severe
burns or trauma, or acute respiratory distress syndrome. Novel strategies are
therefore needed to
control cytokine release syndrome, and especially cytokine storms.
[0009] Cancer is an abnormal state in which uncontrolled proliferation of one
or more cell
populations interferes with normal biological functioning. The proliferative
changes are usually
accompanied by other changes in cellular properties, including reversion to a
less differentiated, more
developmentally primitive state. The in vitro correlate of cancer is called
cellular transformation.
Transformed cells generally display several or all of the following
properties: spherical morphology,
expression of fetal antigens, growth-factor independence, lack of contact
inhibition, anchorage-
CA 03151815 2022-02-17
3
WO 2021/053667
PCT/IL2020/051011
independence, and growth to high density.
[0010] The primary cause of lethality of malignant diseases such as lung
and skin cancer arises
from metastatic spread. In many cases, it is not possible to prevent the onset
of metastatic disease
since cancers are often metastatic by the time of diagnosis, and even in cases
where cancers are
diagnosed prior to this stage, complete surgical removal or destruction of
primary lesion tissues which
are capable of eventually generating metastases may not be feasible.
Metastatic disease may be
impossible to diagnose at early stages due to the small size of metastatic
lesions, and/or the absence
of reliable markers in primary lesions upon which to reliably predict their
existence. Such lesions may
be difficult or impossible to treat via ablative methods due to their being
inaccessible, disseminated,
and/or poorly localized. Chemotherapy/radiotherapy, the current methods of
choice for treatment of
certain metastatic malignancies are often ineffective or suboptimally
effective, and have the
significant disadvantage of being associated with particularly harmful and/or
potentially lethal side-
effects.
[0011] Immunotherapeutic cancer treatment methods, such as those
involving antigen presenting
cell (APC) vaccinations, have the potential to be optimally effective for
treatment of inaccessible,
disseminated, microscopic, recurrent and/or poorly localized cancer lesions.
One promising
immunotherapy avenue involves the use of professional APCs, such as dendritic
cells (DCs), to elicit
systemic anti-cancer immunity.
[0012] Dendritic cells are antigen-producing and presenting cells of the
mammalian immune
system that process antigen material and present it on the cell surface to the
T-cells of the immune
system and are thereby capable of sensitizing T-cells to both new and recall
antigens. DCs are the
most potent antigen-producing cells, acting as messengers between the innate
and the adaptive
immune systems. DC cells may be used, to prime specific antitumor immunity
through the generation
of effector cells that attack and lyse tumors.
[0013] Apoptotic cells present one pathway of physiological cell death,
most commonly occurring
via apoptosis, which elicits a series of molecular homeostatic mechanisms
comprising recognition, an
immune response and a removal process. Moreover, apoptotic cells are
immunomodulatory cells
capable of directly and indirectly inducing immune tolerance to dendritic
cells and macrophages.
Apoptotic cells have been shown to modulate dendritic cells and macrophages
and to render them
tolerogenic and inhibit proinflammatory activies such as secretion of
proinflammatory cytokiens and
expression of costimulatory molecules.
[0014] There remains an unmet need for compositions and methods for
treating, preventing,
inhibiting the growth of, or reducing the incidence of, a cancer or a tumor in
a subject. The apoptotic
cell preparations, compositions and uses thereof, described herein below,
address this need by
CA 03151815 2022-02-17
4
WO 2021/053667
PCT/IL2020/051011
providing a population of early apoptotic cells that may be used to treat,
prevent, inhibit the growth
or, or reduce the incidence of a cancer or tumor in a subject. Further, the
methods of use described
herein address the need to increasing survival of a subject suffering from a
cancer and tumor,
including increasing remission of the cancer or tumor.
SUMMARY
[0015]
In one aspect, disclosed herein is a method of slowing, reducing,
inhibiting, or eliminating
metastatic spread of a cancer or a tumor, or any combination thereof, in a
subject, said method
comprising a step of administering to a subject a combination therapy
comprising an early apoptotic
cell population and one or more cancer therapeutic agents, wherein said method
slows, reduces,
inhibits, or eliminates metastatic spread of a cancer or a tumor, or any
combination thereof, in said
subject. In a related aspect, a cancer therapeutic agent comprises chimeric
antigen receptor-
expressing T-cells (CAR T-cells).
[0016]
In another related aspect, in a method disclosed herein the survival of
said subject is
increased. In a further related aspect, a subject is a human subject.
[0017] In yet another related aspect, the early apoptotic cell population
comprises a mononuclear
enriched cell population; an apoptotic population stable for greater than 24
hours; an apoptotic
population devoid of cell aggregates; a post apoptotic-cell-induction,
irradiated early apoptotic cell
population; a pooled population of early apoptotic cells; or a mononuclear
apoptotic cell population
comprising a decreased of non-quiescent non-apoptotic cells, a suppressed
cellular activation of any
living non-apoptotic cells, or a reduced proliferation of any living non-
apoptotic cells, or any
combination thereof; or any combination thereof.
[0018]
In a related aspect, the cancer or tumor comprises a solid tumor or non-
solid tumor. In a
further related aspect, the non-solid cancer or tumor comprises a
hematopoietic malignancy, a blood
cell cancer, a leukemia, a myelodysplastic syndrome, a lymphoma, a multiple
myeloma (a plasma
cell myeloma), an acute lymphoblastic leukemia, an acute myelogenous leukemia,
a chronic
myelogenous leukemia, a Hodgkin lymphoma, a non-Hodgkin lymphoma, or plasma
cell leukemia.
In still another further related aspect, the solid tumor comprises a sarcoma
or a carcinoma, a
fibrosarcoma, a myxosarcoma, a liposarcoma, a chondrosarcoma, an osteogenic
sarcoma, a
chordoma, an angio sarcoma, an endothelio sarcoma, a lymphangio sarcoma,
a
lymphangioendotheliosarcoma, a synovioma, a mesothelioma, an Ewing's tumor, a
leiomyosarcoma,
a rhabdomyosarcoma, a colon carcinoma, a pancreatic cancer or tumor, a breast
cancer or tumor, an
ovarian cancer or tumor, a prostate cancer or tumor, a squamous cell
carcinoma, a basal cell
carcinoma, an adenocarcinoma, a sweat gland carcinoma, a sebaceous gland
carcinoma, a papillary
carcinoma, a papillary adenocarcinomas, a cystadenocarcinoma, a medullary
carcinoma, a
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
bronchogenic carcinoma, a renal cell carcinoma, a hepatoma, a bile duct
carcinoma, a
choriocarcinoma, a seminoma, an embryonal carcinoma, a Wilm's tumor, a
cervical cancer or tumor,
a uterine cancer or tumor, a testicular cancer or tumor, a lung carcinoma, a
small cell lung carcinoma,
a bladder carcinoma, an epithelial carcinoma, a glioma, an astrocytoma, a
medulloblastoma, a
5 craniopharyngioma, an ependymoma, a pinealoma, a hemangioblastoma, an
acoustic neuroma, an
oligodenroglioma, a schwannoma, a meningioma, a melanoma, a neuroblastoma, or
a retinoblastoma.
[0019] In a related aspect, a cancer therapeutic agent and said early
apoptotic cells are comprised
in separate compositions. In a related aspect, CAR T-cells and said early
apoptotic cells are comprised
in separate compositions. In yet a further related aspect, the composition
comprising the cancer
therapeutic agent is administered prior to, concurrent with, or following
administration of said early
apoptotic cells. In still a further related aspect, the composition comprising
CAR T-cells is
administered prior to, concurrent with, or following administration of said
early apoptotic cells.
[0020] In one aspect, disclosed herein is a method of slowing, reducing,
inhibiting, or eliminating
metastatic spread of a cancer or a tumor, or any combination thereof, in a
subject undergoing cancer
therapy, the method comprising the step of administering an early apoptotic
cell population to said
subject, wherein said method slows, reduces, inhibits, or eliminates
metastatic spread of a cancer or
a tumor, or any combination thereof, in said subject, compared to a subject
undergoing cancer therapy
and not administered an early apoptotic cell population. In a related aspect,
the cancer therapy
comprises radiation therapy, chemotherapy, transplantation, immunotherapy,
targeted therapy,
hormone therapy, photodynamic therapy, or surgery, or a combination thereof.
In a further related
aspect, cancer therapy comprises chimeric antigen receptor T-cell (CAR T-cell)
therapy.
[0021] In one aspect, disclosed herein is a method of improving cancer
therapy in a subject, the
method comprising the step of administering an early apoptotic cell population
to said subject,
wherein improving cancer therapy comprises increasing the survival time of
said subject, and wherein
said method improves said cancer therapy compared to a subject undergoing
cancer therapy and not
administered an early apoptotic cell population. In a related aspect, a cancer
therapy comprises
radiation therapy, chemotherapy, transplantation, immunotherapy, targeted
therapy, hormone
therapy, photodynamic therapy, or surgery, or a combination thereof. In a
further related aspect, a
cancer therapy comprises chimeric antigen receptor expressing T-cell (CAR T-
cell) therapy.
BRIEF DESCRIPTION OF THE DRAWINGS
[0022] The subject matter disclosed herein is particularly pointed out
and distinctly claimed in the
concluding portion of the specification. The compositions and methods
disclosed herein, however,
both as to organization and method of operation, together with objects,
features, and advantages
thereof, may best be understood by reference to the following detailed
description when read with
CA 03151815 2022-02-17
6
WO 2021/053667
PCT/IL2020/051011
the accompanying drawings.
[0023] The patent application file contains at least one drawing executed
in color. Copies of this
patent or patent application publication with color drawing(s) will be
provided by the office upon
request and payment of the necessary fee
[0024] Figures 1A-1B. Schematic showing standard CAR T-cell therapy (Figure
1A) and
embodiments of a method of safe and efficacious CAR T-cell cancer therapy in a
patient using
patients' own cells (autologous) (Figure 1B) to produce apoptotic cells or an
apoptotic cell
supernatant.
[0025] Figure 2. Schematic showing embodiments of a method of safe and
efficacious CAR T-
1() cell cancer therapy in a patient, using donor cells to produce
apoptotic cells or an apoptotic
supernatant.
[0026] Figure 3. Flow chart presenting the steps during one embodiment of a
manufacturing process
of an early apoptotic cell population, wherein anti-coagulants were included
in the process.
[0027] Figures 4A ¨ 4J.Apoptotic cells prevent cytokine storm in in vitro
model of cytokine
storm induced in LPS-Sterile model of macrophage activation syndrome in a
cancer
environment. Figure 4A shows the reduction of LPS induced IL-10 levels in the
macrophage
activation syndrome model in the presence of cancer following administration
of Apocells at a
macrophage/monocyte:Apocell ratio of 1:8 and 1:16, at two time periods (6
hours and 24 hours).
Figure 4B shows the reduction of LPS induced IL-6 levels in the macrophage
activation syndrome
model following administration of Apocells in the presence of cancer and CAR-
19, at a
macrophage/monocyte:Apocell ratio of 1:8 and1:16, at two time periods (6 hours
and 24 hours).
Figure 4C shows the reduction of LPS induced MIP- 1 a levels in the macrophage
activation
syndrome model in the presence of cancer and CAR-19, following administration
of Apocells at a
macrophage/monocyte:Apocell ratio of 1:8 and1:16, at two time periods (6 hours
and 24 hours).
Figure 4D shows the reduction of LPS induced IL-8 levels in the macrophage
activation syndrome
model in the presence of cancer and CAR-19, following administration of
Apocells at a
macrophage/monocyte:Apocell ratio of 1:8 and1:16, at two time periods (6 hours
and 24 hours).
Figure 4E shows the reduction of LPS induced TNF-a levels in the macrophage
activation syndrome
model in the presence of cancer and CAR-19, following administration of
Apocells at a
macrophage/monocyte:Apocell ratio of 1:8 andl :16, at TWO time periods (6
hours and 24 hours).
Figure 94 shows the reduction of LPS induced MIP- 1 (3 levels in the
macrophage activation syndrome
model in the presence of cancer and CAR-19, following administration of
Apocells at a
macrophage/monocyte:Apocell ratio of 1:4, 1:8, 1:16, 1:32, and 1:64 at 24
hours. Figure 94 shows
the reduction of LPS induced MCP-1 levels in the macrophage activation
syndrome model in the
CA 03151815 2022-02-17
7
WO 2021/053667
PCT/IL2020/051011
presence of cancer and CAR-19, following administration of Apocells at a
macrophage/monocyte:Apocell ratio of 1:4, 1:8, 1:16, 1:32, and 1:64 at 24
hours. Figure 94 shows
the reduction of LPS induced IL-9 levels in the macrophage activation syndrome
model in the
presence of cancer and CAR-19, following administration of Apocells at a
macrophage/monocyte:Apocell ratio of 1:8 and1:16, at two time periods (6 hours
and 24 hours).
Figure 41 shows the increase of LPS induced IL-2R levels in the macrophage
activation syndrome
model in the presence of cancer and CAR-19, following administration of
Apocells at a
macrophage/monocyte:Apocell ratio of 1:4, 1:8, 1:16, 1:32, and 1:64 at 24
hours. Figure 4J shows
that apoptotic cells do not down regulate IL-2 release from cells. Apoptotic
cells were incubated with
macrophages/monocytes in the presence of cancer and CAR-19, over a 24 hour
time period with
increasing doses of apoptotic cells (n=3). Empty bar (outline only) ¨ 2.5 x
106 apoptotic cells per
well; Black ¨ 5 x 106 apoptotic cells per well; Grey ¨ 10 x106 apoptotic cells
per well.
[0028] Figure 5. Verification of Transduction of T-cells showing the flow
cytometry results of
anti-CD124 analysis of transduced T4+ CAR-T cells.
[0029] Figure 6. T4+CAR T-Cells reduced proliferation of SKOV3-luc ovarian
adenocarcinoma cells. The results of the cytotoxicity assay, wherein a
monolayer of SKOV3-luc
cells were cultured either by non-transduced T cells or by T4+ CAR-T cells,
are presented in a bar
graph.
[0030] Figure 7. Apoptotic Cells do not abrogate Tr CAR- T cells anti-tumor
activity. Results
are based on a cytotoxicity assay, wherein a monolayer of SKOV3-luc cells were
cultured either with
non-transduced T cells or with T4 CAR-T cells in the presence of a vehicle
(Hartmann solution), or
apoptotic cells (Apocell), or a supernatant of apoptotic cells (ApoSup), or
supernatant of co-culture
of apoptotic cells and monocytes/macrophages (ApoMon Sup).
[0031] Figure 8.11-6, secreted at high levels during cytotoxicity, is down-
regulated by apoptotic
cells. The results shown here demonstrate the effect of co-culture of SKOV3-
luc and human
monocytes/macrophages were exposed to apoptotic cells (ApoCell), or ApoCell
supernatant
(ApoSup), or apoptotic cells and monocyte/macrophage co-culture (ApoMon Sup).
[0032] Figure 9. Effect of Apoptotic Cells or Apoptotic Cell Supernatant or a
co-culture of
Apoptotic cells and Monocytes following LPS exposure during CAR-T cell
therapy. Extremely
high secretion of IL-6 was documented when lipopolysaccharides LLPS) were
added to the cytotoxic
assay. Results show that exposure to Apoptotic cells (Apocell), or supernatant
of apoptotic cells
(ApoSup) or supernatant of co-culture of apoptotic cells and
monocytes/macrophages (ApoMon Sup),
down regulated IL-6, wherein IL-6 was reduced to acceptable levels.
[0033] Figure 10. Effect of Apoptotic Cells or Apoptotic Cell Supernatant or a
co-culture of
CA 03151815 2022-02-17
8
WO 2021/053667
PCT/IL2020/051011
Apoptotic cells and Monocytes following LPS exposure during CAR T-cell
treatment
mimicking CAR T-cell clinical therapy. Extremely high secretion of IL-6 was
documented when
lipopolysaccharides ILPS) were added to the cytotoxic assay. Results show that
exposure to
Apoptotic cells (Apocell), or supernatant of apoptotic cells (ApoSup) or
supernatant of co-culture of
apoptotic cells and monocytes/macrophages (ApoMon Sup), down regulated IL-6,
wherein IL-6 was
reduced to acceptable levels.
[0034] Figures 11A-11B. Weight and Tumor Size in Mice at time of Culling.
Figure 11A shows
Weight change over the experimental time period. Blue-control no 4.5x106 SKOV3-
luc cells
administrated. Red- 0.5x106 SKOV3-luc cells. Green-1.0x106 SKOV3-luc cells.
Purple-4.5x106
SKOV3-luc cells Figure 11B presents a representative SKOV3-luc tumor for a
mouse receiving
4.5x106 SKOV3-luc cells, 39 days after injection.
[0035] Figure 12. SKOV3-luc Tumor Growth. Mice bearing SKOV3-luc tumors imaged
by
Bioluminescent imaging (BLI) are presented showing the differences between
control (PBS) and
inoculation with 0.5x106, lx106, and 4.5x106 SKOV3-luc cells.
[0036] Figures 13A-13D. SKOV3-luc Tumor Burden. Quantification of
bioluminescence (BLI)
of SKOV3-luc tumors in vivo (See Figure 12). A 600 photon count cut-off was
implemented as
instructed by the manufacturer. Figure 13A, mice inoculated with 0.5x106 SKOV3-
luc. Figure 13B,
mice inoculated with 1x106 SKOV3-luc. Figure 13C, mice inoculated with 4.5x106
SKOV3-luc.
Figure 13D, Average SKOV3-luc tumor growth.
[0037] Figure 14. Cytotoxic Calibration for Raji Burkett Lymphoma Cells. Raji
cells were
plated at various cell densities, with cell lysis occurring immediately prior
to centrifugation. The
results show Raji cell number (x-axis) vs. at absorbance at 492 nm (y-axis).
All cell numbers
exhibited significant readings relative to the unlysed counterpart.
[0038] Figure 15. Addition of early apoptotic cells does not affect CAR T-cell
anti-tumor
activity. E/T ratio shows the CD19+CAR T-cell to HeLa cell ratio. Survival is
of CD19+ Tumor
cells. Filled circle CD19+ Hela; Empty triangle CD19+ Hela + Naive T cells;
Filled triangle CD19+
Hela + CAR T-CD19; Empty circle CD19+ Hela + CAR T-CD19 +ApoCells.
[0039] Figure 16. Cytokine Analysis (GM-CSF) in Raji Burkett Lymphoma Cells in
the
Presence and Absence of Apoptotic cells. The bar graph presents the
concentration measurements
of cytokine GM-CSF (pg/ml) found in culture supernatants of Raji cells
incubated in the presence of
monocytes and LPS, followed by addition of Naive T-cells (Raji + Naive T),
CD19+ CAR T-cells
(Raji + CAR T), CD19+ CAR T-cells and apoptotic cells (ApoCell) at a ratio of
1:8 CAR T-
cells:ApoCells (Raji + CAR T+ApoCell 1:8), CD19+ CAR T-cells and apoptotic
cells (ApoCell) at
a ratio of 1:32 CAR T-cells:ApoCells (Raji + CAR T+ApoCell 1:32), and CD19+
CAR T-cells and
CA 03151815 2022-02-17
9
WO 2021/053667
PCT/IL2020/051011
apoptotic cells (ApoCell) at a ratio of 1:64 CAR T-cells:ApoCells (Raji + CAR
T+ApoCell 1:64).
[0040] Figure 17. Cytokine Analysis (TNF-alpha) in Raji Burkett Lymphoma Cells
in the
Presence and Absence of Apoptotic cells. The bar graph presents the
concentration measurements
of cytokine TNF-alpha (TNF-a) (pg/ml) found in culture supernatants of Raji
cells incubated in the
presence of monocytes and LPS, followed by addition of Naïve T-cells (Raji +
Naïve T), CD19+
CAR T-cells (Raji + CAR T), CD19+ CAR T-cells and apoptotic cells (ApoCell) at
a ratio of 1:8
CAR T-cells:ApoCells (Raji + CAR T+ApoCell 1:8), CD19+ CAR T-cells and
apoptotic cells
(ApoCell) at a ratio of 1:32 CAR T-cells:ApoCells (Raji + CAR T+ApoCell 1:32),
and CD19+ CAR
T-cells and apoptotic cells (ApoCell) at a ratio of 1:64 CAR T-cells:ApoCells
(Raji + CAR
T+ApoCell 1:64).
[0041] Figures 18A and 18B. Experimental Scheme. Figure 18A presents the
experimental
scheme to analyze the influence of apoptotic cells on CAR T-cell therapy. SCID
mice were injected
on day 1 with Raji cancer cells, followed on day 6 by administration of CAR T-
CD19 cells (CART-
cell therapy) and Apoptotic cells. Figure 18B shows that CAR T-cell therapy
was not negatively
influenced by co-administration of ApoCells. Survival Curve: SCID mice were
injected with CD19+
Raji cells with or without addition of early apoptotic cells.
[0042] Figures 19A, 19B, and 19C. Increased release of pro-inflammatory
cytokines from a
tumor, in a solid tumor in vivo model. Figure 19A shows slight increase of IL-
6 released from a
solid tumor present in the peritoneum of BALB/c and SCID mice, wherein the IL-
6 release is
significantly increased in the presence of HeLa CAR-CD-19 CAR T-cells.
Similarly, Figure 19B
shows a slight increase of IP-10 released from a solid tumor present in the
peritoneum of BALB/c
and SCID mice, wherein the IP-10 release is significantly increased in the
presence of HeLa CAR-
CD-19 CAR T-cells, and Figure 19C shows that surprisingly even TNF-a release
is increased by in
the presence of HeLa CAR-CD-19 CAR T-cells.
[0043] Figures 20A and 20B. Testing the efficacy of CD19-CAR-T cells in an IP
model of
HeLaCD19 (Leukemia), in the presence or absence of ApoCell. HeLa-CD19 - Blue;
HeLaCD19+Mock ¨ Green; HeLaCD19 + CAR-T ¨ Purple; and HeLaCD19 + CAR-T +
ApoCell ¨
orange. Figure 20A was with 0.5 x106 CAR-T positive cells. Figure 20B was with
2.2 x 106CAR-T
positive cells.
[0044] Figure 21. Survival curves for in vivo diffuse tumor SCID mouse model.
The curves show
that administration of early apoptotic cells (APO; broad dashed lines ----)
extended survival compared
with mice not administered apoptotic cells (NO APO; dotted line = = = = ),
wherein control SCID mice
showed 100% survival (solid line __ ).
[0045] Figures 22A-22D. Apoptotic cell infusions increased the lifespan of
leukemic mice and
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
increased the number of mice attaining complete remission. Cohorts: No
leukemia (Control-
striped pattern); Leukemia + early apoptotic cells (spotted); Leukemia only
(solid grey). n=51 in total
(p<0.001) Figure 22A. Apoptotic cell infusions increased the percentage of
mice surviving through
the expected life-span post leukemia induction. Figure 22B. Apoptotic cell
infusions increased the
5 percentage of mice surviving up to 12% of the expected life-span post
leukemia induction. Figure
22C. Apoptotic cell infusions increased the percentage of mice surviving up to
30% of the expected
life-span post leukemia induction. Figure 22D. Apoptotic cell infusions
increased the percentage of
mice surviving up to 100% of the expected life-span post leukemia induction
and attaining complete
remission.
10 [0046] Figures 23A-23E. Apoptotic cell infusions increased the life-span
of leukemic mice,
increased the number of mice attaining complete remission, and enhanced the
anti-CD20
monoclonal antibody (mAb) therapeutic effect. Cohorts: Leukemia only (solid
grey); Leukemia +
early apoptotic cells (striped pattern); Leukemia + anti-CD20 mAb (checkered);
Leukemia + anti-
CD20 + early apoptotic cells (spotted). n=28 in total (p<0.002) Figure 23A.
Shows the percent (%)
survival through the expected lifespan of mice following induction of leukemia
with Raji cells. Figure
23B. Apoptotic cell infusions increased the percentage of mice surviving up to
24% longer than the
expected life-span post leukemia induction. Figure 23C. Apoptotic cell
infusions increased the
percentage of mice surviving up to 59% longer than the expected life-span post
leukemia induction
and enhanced the anti-CD20 mAb effect on the life-span of leukemic mice.
Figure 23D. Apoptotic
cell infusions increased the percentage of mice surviving up to 76% longer
than the expected life-
span post leukemia induction and enhanced the anti-CD20 mAb effect on the life-
span of leukemic
mice. Figure 23E. Apoptotic cell infusions increased the percentage of mice
attaining complete
remission.
[0047] Figure 24. Kaplan-Meier survival plot of SCID-Bg mice with Raji
leukemia/lymphoma,
receiving ApoCell. (RPMI group, n = 15; Raji group, n = 23; Raji + ApoCell
group, n = 24) RPMI
(control) ¨ Black; Raji only ¨ Orange; Raji + ApoCell ¨ Blue.
[0048] Figures 25A-25C. Kaplan-Meier survival plots. Figure 25A presents data
from a study
wherein female SCID-Bg mice, 7-weeks-old (ENVIGO, Jerusalem, Israel), were
injected IV with
0.1x106 Raji cells per mouse (n = 10 per group, three groups). Mice received
three IV doses (days 5,
8, 11) of 30x106 ApoCell. (RPMI-light blue; Raji-orange; and Raji + ApoCell ¨
dark blue) Figure
25B presents data from a study wherein female SCID-Bg mice, 7-weeks-old
(ENVIGO, Jerusalem,
Israel), were injected IV with 0.1x106 Raji cells per mouse (n = 10 per group,
three groups). Mice
received three IV doses (days 5, 8, 11) of 30x106 ApoCell. (RPMI-black; Raji-
orange; and Raji +
ApoCell ¨ dark blue) Figure 25C presents data from a study wherein female SCID-
Bg mice, 8-9-
CA 03151815 2022-02-17
11
WO 2021/053667
PCT/IL2020/051011
weeks-old (ENVIGO, Jerusalem, Israel), were injected IV with 0.1x106 Raji
cells per mouse (n=10
per group, 2 groups). Mice received three IV doses (days 5, 8, 12) of 30x106
ApoCell. (Raji-orange;
and Raji + ApoCell ¨ dark blue)
[0049] Figure 26. Kaplan-Meier survival plot of SCID-Bg mice with Raji
leukemia/lymphoma,
receiving RtX and ApoCell. (Raji alone ¨ orange; Raji + ApoCell ¨ blue; Raji +
RtX 2mg ¨ green;
Raji + RtX 2mg + ApoCell ¨ yellow; Raji + RtX 5mg ¨ purple; Raji + RtX 5mg +
ApoCell ¨ grey.)
[0050] Figure 27. Kaplan-Meier survival plot of SCID-Bg mice with Raji
leukemia/lymphoma,
receiving rtx and ApoCell. (Raji alone ¨ orange; Raji + ApoCell ¨ blue; Raji +
RtX 2mg ¨ green; Raji
+ RtX 2mg + ApoCell ¨ yellow.)
[0051] Figure 28. Effect of Pooled ApoCell Preparation. Figure 28 presents a
graph showing
the clear effect (p<0.01) of a single apoptotic cell preparation injection
from multiple individual
donors (blue) on survival. The graph presented is a Kaplan-Meier survival
curve in a GvHD mouse
model that was treated with a single dose irradiated pooled apoptotic cell
preparation from multiple
individual donors.
[0052] Figure 29. Effect of Pooled ApoCell Preparation. Figure 29 presents a
graph showing
the clear effect (p<0.01) of a single apoptotic cell preparation injection
from multiple individual
donors (blue) on percentage of weight loss of the 2 compared groups.
[0053] Figure 30. Comparison of Single Donor versus Pooled ApoCell
Preparation. Figure
30 presents a graph showing comparison between the administration of a single
dose of single-donor
.. and multiple-donor apoptotic cell preparations +1- irradiation on %
survival using a mouse model of
induced GvHD.
[0054] Figures 31A-31B. Potency Test. Figures 31A-31B present the results
of a potency test
that shows the inhibition of maturation of dendritic cells (DCs) following
interaction with apoptotic
cells, measured by expression of HLA-DR. Figure 31A. HLA DR mean fluorescence
of fresh final
product A (t0). Figure 31B. HLA DR mean fluorescence of final product A,
following 24h at 2-8 C.
[0055] Figures 32A-32B. Potency Test. Figures 32A-32B present the results of a
potency test
that shows the inhibition of maturation of dendritic cells (DCs) following
interaction with apoptotic
cells, measured by expression of CD86. Figure 32A. CD86 Mean fluorescence of
fresh final product
A (t0). Figure 32B. CD86 Mean fluorescence of final product A, following 24h
at 2-8 C.
[0056] Figure 33. Schematic of Example 17 experiment scheme showing the
general flow of
treatment of a solid tumor by CAR T-CD19 and ApoCell administration, and the
cell pathology at the
end point.
[0057] Figures 34A-34D. Survival following CAR T-cell therapy alone or with co-
administration
of apoptotic cells. Figure 34A shows survival curves of peritoneal solid tumor
Hela-CD19, treated
CA 03151815 2022-02-17
12
WO 2021/053667
PCT/IL2020/051011
by CAR T-cell therapy alone (SCID-Bg-HeLa-CD19: control no treatment; SCID-Bg-
HeLa-CD19+
CAR-T: includes treatment with CAR T-cells; SCID-Bg-HeLa-CD19+Mock-T: control
addition of
Mock T-cells. Figure 34B is representative of 5 separate experiments, as
described in Example 17
and shows survival curves of peritoneal solid tumor Hela-CD19, treated by CAR
T-cell therapy alone
or CAR T-cell therapy plus co-administration of apoptotic cells. (HeLa-CD19:
control no treatment;
HeLa-CD19+ CAR-T: includes treatment with CAR T-cells; HeLa-CD19+Mock-T:
control addition
of Mock T-cells; Hela-CD19+CAR-T + OTS-ALC-4K: treatment was with CAR T-cells
and "off the
shelf' apoptotic cells irradiated with 4000rad). Figure 34C presents
representative results, as
described in Example 17 and shows survival curves of peritoneal solid tumor
Hela-CD19, treated by
CAR T-cell therapy alone or CAR T-cell therapy plus co-administration of
apoptotic cells. (SCID-
Bg-HeLa-CD19: control no treatment; SCID-Bg-HeLa-CD19+ CAR-T: includes
treatment with
CAR T-cells; SCID-Bg-HeLa-CD19+Mock-T: control addition of Mock T-cells; SCID-
Bg-Hela-
CD19+CAR-T + Allocetra-OTS: treatment was with CAR T-cells and "off the shelf'
apoptotic cells
irradiated with 4000rad). Figure 34D (Luciferase in vivo imaging system (IVIS)
results) shows
progression of tumor as visualized by IVIS representing the development
leading to the survival curve
results presented in Figure 34B. Spread of the tumor can already be visualized
on day 15 (Hela-
CD19-Luc; control no treatment) whereas mice treated with CAR T-cells did not
show any tumor
spread until day 43. When mice receive apoptotic cells in conjunction with CAR
T-cell treatment,
unexpectedly most mice did not show tumor spread until day 50 and tumor size
(see scale for
correlation) was clearly smaller.
[0058] Figures 35A-35B. Survival following CAR T-cell therapy alone or with co-
administration
of apoptotic cells. Figure 35A shows survival curves of peritoneal solid tumor
Hela-CD19, treated
by CAR T-cell therapy alone. Figure 35B shows survival curves of peritoneal
solid tumor Hela-
CD19, treated by CAR T-cell therapy alone or CAR T-cell therapy plus co-
administration of apoptotic
cells. (SCID-Bg-HeLa-CD19: control no treatment; SCID-Bg-HeLa-CD19+ CAR-T:
includes
treatment with CAR T-cells; SCID-Bg-Hela-CD19+CAR-T + Allocetra-OTS: treatment
was with
CAR T-cells and "off the shelf' apoptotic cells irradiated with 4000rad).
[0059] Figures 36A-36C. FACS analysis of macrophages. Figure 36A shows FACS
analysis of
resident peritoneal macrophage markers F4/80, CD1 lb, Tim4, and MerTK, in the
SCID-bg mice
(control, no tumor), in SCID-bg-Hela-CD19 mice (control with tumor, no
treatment), and in SCID-
bg-HeLa-CD19-CART (includes treatment with CAR T-cells). The results seen in
the SCID-bg mice
provide a resident macrophage signature. Comparison of the data shows that
resident peritoneal
macrophages disappear during tumor progression. Figure 36B shows FACS analysis
of tumor
associated macrophage (TAM) markers CCR2, Ly6c, CD206, CD64, CD169, and CD74
during
CA 03151815 2022-02-17
13
WO 2021/053667
PCT/IL2020/051011
tumor progression in the SCID-bg mice (control, no tumor), in SCID-bg-Hela-
CD19 mice (control
with tumor, no treatment), and in SCID-bg-HeLa-CD19-CART (includes treatment
with CAR T-
cells). The results seen in the SCID-bg-CD19 provide a infiltrating macrophage
signature.
Comparison of the data shows that TAM infiltrate during tumor progression.
Figure 36C presents
FACs results showing that resident macrophages in SCID mice are mostly large
peritoneal
macrophages (LPM), and that during tumor progression, most resident
macrophages disappear, and
TAM, monocytes, and dendritic cells appear. Macrophage markers F4/80, CD1 lb,
TIM4, MER-TK,
rIgG2b, and rIgG2a were measured, as was the percent of MHCII positive cells.
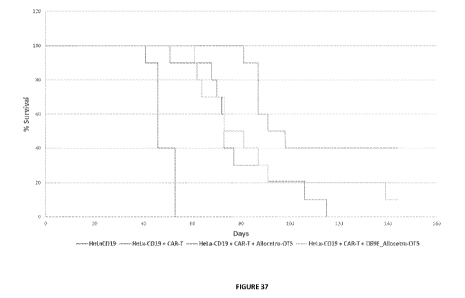
[0060] Figure 37. Percent (%) curves following CAR T-cell therapy alone or
with co-
administration of apoptotic cells or opsonized apoptotic cells. The figure
shows survival curves of
peritoneal solid tumor Hela-CD19, treated by CAR T-cell therapy alone or CAR T-
cell therapy plus
co-administration of apoptotic cells or opsonized apoptotic cells. (SCID-Bg-
HeLa-CD19: control no
treatment; SCID-Bg-HeLa-CD19+ CAR-T: includes treatment with CAR T-cells; SCID-
Bg-Hela-
CD19+CAR-T + Allocetra-OTS: treatment was with CAR T-cells and "off the shelf'
apoptotic cells
irradiated with 4000rad; SCID-Bg-Hela-CD19+CAR-T + D89E Allocetra-OTS:
treatment was with
CAR T-cells and "off the shelf' opsonized apoptotic cells irradiated with
4000rad).
DETAILED DESCRIPTION
[0061] In the following detailed description, numerous specific details
are set forth in order to
provide a thorough understanding of the methods disclosed herein. However, it
will be understood
by those skilled in the art that these methods may be practiced without these
specific details. In other
instances, well-known methods, procedures, and components have not been
described in detail so as
not to obscure the methods disclosed herein.
[0062] Genetic modification of immune cells is well known as a strategy for
immune-cell therapies
against cancer. These immune-cell therapies are based on the manipulation and
administration of
autologous or allogeneic immune cells to a subject in need. Immune-cell based
therapies include
natural killer cells therapies, dendrite cell therapies, and T-cell
immunotherapies including those
utilizing naive T-cells, effector T-cells also known as T-helper cells,
cytotoxic T-cells, and regulatory
T-cells (Tregs).
[0063] In some embodiments, disclosed herein are compositions comprising
genetically modified
immune cells. In another embodiment, the genetically modified immune cell is a
T-cell. In another
embodiment, a T-cell is a naive T-cell. In another embodiment, a T-cell is a
naive CD4+ T-cell. In
another embodiment, a T-cell is a naïve T-cell. In another embodiment, a T-
cell is a naive CD8+ T-
cell. In another embodiment, the genetically modified immune cell is a natural
killer (NK) cell. In
another embodiment, the genetically modified immune cell is a dendritic cell.
In still another
CA 03151815 2022-02-17
14
WO 2021/053667
PCT/IL2020/051011
embodiment, the genetically modified T-cell is a cytotoxic T lymphocyte (CTL
cell). In another
embodiment, the genetically modified T-cell is a regulatory T-cell (Treg). In
another embodiment,
the genetically modified T-cell is a chimeric antigen receptor (CAR) T-cell.
In another embodiment,
the genetically modified T-cell is a genetically modified T-cell receptor
(TCR) cell.
[0064] In some embodiments, disclosed herein are compositions comprising
genetically modified
immune cells and apoptotic cells. In another embodiment, disclosed herein are
compositions
comprising genetically modified immune cells and supernatants from apoptotic
cells. In another
embodiment, the genetically modified immune cell is a T-cell. In another
embodiment, the genetically
modified immune cell is a natural killer (NK) cell. In still another
embodiment, the genetically
modified immune cell is a cytotoxic T lymphocyte (CTL cell). In another
embodiment, the genetically
modified immune cell is a regulatory T lymphocyte (Treg cell).
[0065] In some embodiments, disclosed herein is a method of maintaining or
increasing the
proliferation rate of chimeric antigen receptor-expressing T-cells (CAR T-
cell) during CAR T-cell
cancer therapy, the method comprising the step of administering a composition
comprising apoptotic
cells or an apoptotic cell supernatant to said subject, and wherein said
proliferation rate is maintained
or increased in the subject compared with a subject undergoing CAR T-cell
cancer therapy and not
administered said apoptotic cells or said apoptotic cell supernatant.
[0066] In a related embodiment, the method does not reduce or inhibit the
efficacy of said CAR T-
cell cancer therapy. In a related embodiment, the method improves the efficacy
of said CAR T-cell
cancer therapy. In another related embodiment the incidence of cytokine
release syndrome (CRS) or
a cytokine storm in said subject is inhibited or reduced compared with a
subject not administered said
apoptotic cells or said apoptotic cell supernatant.
[0067] In some embodiments, CRS occurs spontaneously. In another embodiment,
CRS occurs in
response to LPS. In another embodiment, CRS occurs in response to IFN-y.
[0068] In some embodiments, disclosed herein is a method of increasing the
efficacy of chimeric
antigen receptor T-cell (CAR T-cell) cancer therapy, the method comprising the
step of administering
CAR T-cells and an additional agent selected from the group comprising
apoptotic cells, an apoptotic
cell supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or fragment
thereof or analogue
thereof, a tellurium-based compound, or an immune modulating agent, or any
combination thereof,
wherein said efficacy said CAR T-cells is increased in the subject compared
with a subject
undergoing CAR T-cell cancer therapy and not administered said additional
agent. In a related
embodiment, the level of production of at least one pro-inflammatory cytokine
is reduced compared
with the level of said pro-inflammatory cytokine in a subject received CAR T-
cell cancer therapy and
not administered a composition comprising said agent. In another related
embodiment, the pro-
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
inflammatory cytokine comprises IL-6.
[0069] In a related embodiment, when apoptotic cells or an apoptotic cell
supernatant is
administered, said method increases the levels of IL-2 in the subject compared
with a subject
undergoing CAR T-cell cancer therapy and not administered said apoptotic cells
or said apoptotic
5 cell supernatant. In another embodiment, when apoptotic cells or an
apoptotic cell supernatant is
administered, said method maintains the levels of IL-2 in the subject compared
with a subject
undergoing CAR T-cell cancer therapy and not administered said apoptotic cells
or said apoptotic
cell supernatant. In another embodiment, when apoptotic cells or an apoptotic
cell supernatant is
administered, said method maintains or increases the levels of IL-2 in the
subject compared with a
10 subject undergoing CAR T-cell cancer therapy and not administered said
apoptotic cells or said
apoptotic cell supernatant. In another related embodiment, the incidence of
cytokine release
syndrome (CRS) or a cytokine storm in said subject is inhibited or reduced
compared with a subject
not administered said additional agent.
[0070] In a related embodiment, CAR T-cells and said additional agent or any
combination thereof
15 are comprised in a single composition. In another related embodiment,
said CAR T-cell and said
additional cancer therapeutic agent or any combination thereof are comprised
in at least two
compositions.
[0071] In some embodiments, disclosed herein is a method of treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor in
a subject, comprising
the step of administering chimeric antigen receptor-expressing T-cells (CAR T-
cell) and an additional
agent, said additional agent comprising apoptotic cells, apoptotic
supernatants or a CTLA-4 blocking
agent, an alpha-1 anti-trypsin or fragment thereof or analogue thereof, a
tellurium-based compound,
or an immune modulating agent, or any combination thereof, wherein said method
treats, prevents,
inhibits, reduces the incidence of, ameliorates or alleviates a cancer or a
tumor in said subject
compared with a subject administered CAR T-cells and not administered said
additional agent.
[0072] In a related embodiment, said method has increased efficacy treating,
preventing, inhibiting,
reducing the incidence of, ameliorating or alleviating said cancer or said
tumor in said subject
compared with a subject administered CAR T-cells and not administered said
additional agent.
[0073] In another related embodiment, the level of production of at least
one pro-inflammatory
cytokine is reduced compared with the level of said pro-inflammatory cytokine
in a subject
administered said CAR T-cells and not administered a composition comprising
said agent. In another
related embodiment, said pro-inflammatory cytokine comprises IL-6. In another
related embodiment,
said additional agent comprises apoptotic cells or an apoptotic cell
supernatant, said method increases
the levels of IL-2 in the subject compared with a subject administered said
CAR T-cells and not
CA 03151815 2022-02-17
16
WO 2021/053667
PCT/IL2020/051011
administered said apoptotic cells or said apoptotic cell supernatant. In
another related embodiment,
said CAR T-cells and said additional agent or any combination thereof are
comprised in a single
composition. In yet another related embodiment, said CAR T-cells and said
additional agent or any
combination thereof are comprised in at least two compositions.
[0074] In a related embodiment, the administration of said additional agent
occurs prior to,
concurrent with, or following the administration of said CAR T-cells. In
another related embodiment,
said apoptotic cells comprise apoptotic cells in an early-apoptotic state. In
another related
embodiment, said apoptotic cells are autologous to said subject or are pooled
third-party donor cells.
[0075] In a related embodiment, said apoptotic cell supernatant is obtained by
a method comprising
the steps of (a) providing apoptotic cells, (b) culturing the cells of step
(a), and (c) separating the
supernatant from the cells. In another related embodiment, said apoptotic cell
supernatant is an
apoptotic cell-white blood cell supernatant and said method further comprises
the steps of: (d)
providing white blood cells, (e) optionally, washing the apoptotic cells and
the white blood cells, (f)
co-culturing the apoptotic cells and the white blood cells, wherein steps (d)-
(f) are in place of step
(b). In another related embodiment, the provided white blood cells are
selected from the group
consisting of phagocytes, macrophages, dendritic cells, monocytes, B cells, T
cells, and NK cells.
Thus, in some embodiments, apoptotic supernatants comprise a supernatant
produced by culturing
apoptotic cells with macrophages, wherein the macrophage ingests the apoptotic
cells and the
supernatant produced from this co-culturing is used. In some embodiments,
apoptotic supernatants
comprise a supernatant produced by culturing apoptotic cells, wherein the
supernatant is produced
from materials secreted by the apoptotic cells.
[0076] In some embodiments, disclosed herein are compositions comprising
early apoptotic cells.
In some embodiments, disclosed herein are compositions comprising early
apoptotic cells in
combination with an additional agent. In some embodiments, the additional
agent may be a CAR T-
cell. In some embodiments, the additional agent may be an antibody. In some
embodiments, the
antibody comprises rituximab or a functional fragment thereof.
[0077] In some embodiments, compositions of early apoptotic cells
comprise a population of
mononuclear apoptotic cell comprising mononuclear cells in an early-apoptotic
state, wherein said
mononuclear apoptotic cell population comprises: a decreased percent of non-
quiescent non-
apoptotic viable cells; a suppressed cellular activation of any living non-
apoptotic cells; or a reduced
proliferation of any living non-apoptotic cells; or any combination thereof.
[0078] In some embodiments, disclosed herein are compositions comprising
genetically modified T-
cells and apoptotic cells. In another embodiment, disclosed herein are
compositions comprising
genetically modified T-cells and supernatants of apoptotic cells. In another
embodiment, the
CA 03151815 2022-02-17
17
WO 2021/053667
PCT/IL2020/051011
genetically modified T-cell is a chimeric antigen receptor (CAR) T-cell. In
another embodiment, the
genetically modified T-cell is a genetically modified T-cell receptor (TCR)
cell.
[0079] In some embodiments, disclosed herein are compositions comprising CAR T-
cells and
apoptotic cells. In another embodiment, disclosed herein are compositions
comprising genetically
modified T-cell receptor cells (TCRs) and apoptotic cells. In another
embodiment, disclosed herein
are compositions comprising CAR T-cells and supernatants from apoptotic cells.
In another
embodiment, disclosed herein are compositions comprising genetically modified
T-cell receptor cells
(TCRs) and supernatant of apoptotic cells.
[0080] In certain embodiments, genetically modified immune cells and apoptotic
cells or apoptotic
cell supernatants are comprised within a single composition. In other
embodiments, genetically
modified immune cells and apoptotic cells or apoptotic cell supernatants are
comprised in separate
compositions.
[0081] This disclosure provides in some embodiments, a pooled mononuclear
apoptotic cell
preparation comprising mononuclear cells in an early apoptotic state, wherein
said pooled
mononuclear apoptotic cells preparation comprises pooled individual
mononuclear cell populations,
and wherein said pooled mononuclear apoptotic cell preparation comprises a
decreased percent of
living non-apoptotic cells, a suppressed cellular activation of any living non-
apoptotic cells, or a
reduced proliferation of any living non-apoptotic cells, or any combination
thereof. In another
embodiment, the pooled mononuclear apoptotic cells have been irradiated. In
another embodiment,
this disclosure provides a pooled mononuclear apoptotic cell preparation that
in some embodiments,
uses the white blood cell fraction (WBC) obtained from donated blood. Often
this WBC fraction is
discarded at blood banks or is targeted for use in research.
[0082] In some embodiments, a cell population disclosed herein is
inactivated. In another
embodiment, inactivation comprises irradiation. In another embodiment,
inactivation comprises T-
cell receptor inactivation. In another embodiment, inactivation comprises T-
cell receptor editing. In
another embodiment, inactivation comprises suppressing or eliminating an
immune response in said
preparation. In another embodiment, inactivation comprises suppressing or
eliminating cross-
reactivity between multiple individual populations comprised in the
preparation. In other
embodiment, inactivation comprises reducing or eliminating T-cell receptor
activity between multiple
individual populations comprised in the preparation. In another embodiment, an
inactivated cell
preparation comprises a decreased percent of living non-apoptotic cells,
suppressed cellular activation
of any living non-apoptotic cells, or a reduce proliferation of any living non-
apoptotic cells, or any
combination thereof.
[0083] In another embodiment, an inactivated cell population comprises a
reduced number of
CA 03151815 2022-02-17
18
WO 2021/053667
PCT/IL2020/051011
non-quiescent non-apoptotic cells compared with a non-radiated cell
preparation. In some
embodiments, an inactivated cell population comprises 50 percent (%) of living
non-apoptotic cells.
In some embodiments, an inactivated cell population comprises 40% of living
non-apoptotic cells. In
some embodiments, an inactivated cell population comprises 30% of living non-
apoptotic cells. In
some embodiments, an inactivated cell population comprises 20% of living non-
apoptotic cells. In
some embodiments, an inactivated cell population comprises 100% of living non-
apoptotic cells. In
some embodiments, an inactivated cell population comprises 0% of living non-
apoptotic cells.
[0084] In some embodiments, disclosed herein is a method of preparing an
inactivated early
apoptotic cell population. In some embodiments, disclosed herein is a method
for producing a
population of mononuclear apoptotic cell comprising a decreased percent of non-
quiescent non-
apoptotic viable cells; a suppressed cellular activation of any living non-
apoptotic cells; or a reduced
proliferation of any living non-apoptotic cells; or any combination thereof,
said method comprising
the following steps,
obtaining a mononuclear-enriched cell population of peripheral blood;
freezing said mononuclear-enriched cell population in a freezing medium
comprising an
anticoagulant;
thawing said mononuclear-enriched cell population;
incubating said mononuclear-enriched cell population in an apoptosis inducing
incubation
medium comprising methylprednisolone at a final concentration of about 10-100
i.t.g/mL and an
anticoagulant;
resuspending said apoptotic cell population in an administration medium; and
inactivating said mononuclear-enriched population, wherein said inactivation
occurs
following induction,
wherein said method produces a population of mononuclear apoptotic cell
comprising a decreased
percent of non-quiescent non-apoptotic cells; a suppressed cellular activation
of any living non-
apoptotic cells; or a reduced proliferation of any living non-apoptotic cells;
or any combination
thereof.
[0085] In another embodiment, the irradiation comprises gamma irradiation
or UV irradiation. In
yet another embodiment, the irradiated preparation has a reduced number of non-
quiescent non-
apoptotic cells compared with a non-irradiated cell preparation.
[0086] In another embodiment, the pooled mononuclear apoptotic cells have
undergone T-cell
receptor inactivation. In another embodiment, the pooled mononuclear apoptotic
cells have
undergone T-cell receptor editing.
[0087] In some embodiments, pooled blood comprises 3rd party blood from
HLA matched or
CA 03151815 2022-02-17
19
WO 2021/053667
PCT/IL2020/051011
HLA unmatched sources, with respect to a recipient.
[0088] In some embodiments, disclosed herein are compositions comprising
genetically modified
immune cells, for example but not limited to CAR T-cells and an additional
agent selected from the
group comprising apoptotic cells, an apoptotic cell supernatant, a CTLA-4
blocking agent, an alpha-
1 anti-tryp sin or fragment thereof or analogue thereof, a tellurium-based
compound, or an immune
modulating agent, or any combination thereof.
[0089] In some embodiments, this disclosure provides methods of
production of a pharmaceutical
composition comprising a pooled mononuclear apoptotic cell preparation
comprising pooled
individual mononuclear cell populations in an early apoptotic state, wherein
said composition
comprises a decreased percent of living non-apoptotic cells, a preparation
having a suppressed cellular
activation of any living non-apoptotic cells, or a preparation having reduced
proliferation of any living
non-apoptotic cells, or any combination thereof. In another embodiment, the
methods provide a
pharmaceutical composition comprising a pooled mononuclear apoptotic cell
preparation comprising
pooled individual mononuclear cell populations in an early apoptotic state,
wherein said composition
comprises a decreased percent of non-quiescent non-apoptotic cells.
[0090] In some embodiments, disclosed herein is a method of treating,
preventing, inhibiting the
growth of, reducing the incidence of, or any combination thereof, a cancer or
a tumor in a subject,
comprising a step of administering an early apoptotic cell population to said
subject, wherein said
method treats, prevents, inhibits the growth of, reduces the incidence of, or
any combination thereof,
a cancer or a tumor in said subject. In some embodiments, methods herein
comprise treating,
preventing, inhibiting the growth of, delaying disease progression, reducing
the tumor load, or
reducing the incidence of a cancer or a tumor in a subject, or any combination
thereof, comprising a
step of administering a composition comprising an early apoptotic cell
population to said subject. In
some embodiments, the method further comprises administering an additional
immune therapy, a
chemotherapeutic agent, or an immune modulator to said subject, or any
combination thereof. In some
embodiments, the additional immune therapy, a chemotherapeutic agent, or an
immune modulator is
administered prior to, concurrent with, or following administration of said
early apoptotic cells.
[0091] In some embodiments, disclosed herein is a method of increasing
survival of a subject
suffering from a cancer or a tumor, comprising a step of administering an
early apoptotic cell
population to said subject, wherein said method increases survival of said
subject. In some
embodiments, the method further comprises administering an additional cancer
therapy. In some
embodiments, the addition cancer therapy comprises an radiation therapy,
chemotherapy,
transplantation, immunotherapy, targeted therapy, hormone therapy,
photodynamic therapy, an
immune modulator, or surgery, or a combination thereof In some embodiments,
the cancer therapy
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
comprises CAR T-cell therapy. In some embodiments, the additional cancer
therapy or cancer
therapeutic agent, is administered prior to, concurrent with, or following
administration of said early
apoptotic cells.
[0092] In some embodiments, disclosed herein is a method of reducing the
size or reducing the
5 growth rate of a cancer or a tumor, or a combination thereof, in a
subject, comprising a step of
administering an early apoptotic cell population to said subject, wherein said
method reduces the size
or reduces the growth rate. In some embodiments, the method further comprises
administering an
additional immune therapy, a chemotherapeutic agent, or an immune modulator to
said subject, or
any combination thereof. In some embodiments, the additional immune therapy, a
chemotherapeutic
10 agent, or an immune modulator is administered prior to, concurrent with,
or following administration
of said early apoptotic cells.
[0093] In some embodiments, administration of a composition comprising
apoptotic cells does
not affect the efficacy of CAR T-cells to treat, prevent, inhibit, reduce the
incidence of, ameliorating,
reduce the tumor load, or alleviating a cancer or a tumor. In another
embodiment, administration of
15 .. a composition comprising apoptotic cells does not reduce the efficacy of
the CAR T-cells to treat,
prevent, inhibit, reduce the incidence of, ameliorating, reduce the tumor
load, or alleviating a cancer
or a tumor by more than about 5%. In another embodiment, administration of a
composition
comprising apoptotic cells does not reduce the efficacy of the CAR T-cells to
treat, prevent, inhibit,
reduce the incidence of, ameliorating, reduce the tumor load, or alleviating a
cancer or a tumor by
20 more than about 10%. In another embodiment, administration of a
composition comprising apoptotic
cells does not reduce the efficacy of the CAR T-cells to treat, prevent,
inhibit, reduce the incidence
of, ameliorating, reduce the tumor load, or alleviating a cancer or a tumor by
more than about 15%.
In another embodiment, administration of a composition comprising apoptotic
cells does not reduce
the efficacy of the CAR T-cells to treat, prevent, inhibit, reduce the
incidence of, ameliorating, reduce
the tumor load, or alleviating a cancer or a tumor by more than about 20%.
[0094] In some embodiments, administration of apoptotic cells increases
the efficacy of CAR T-
cells. In some embodiments, administration of apoptotic cells increases the
efficacy of CAR T-cells
by at least 5, by at least 10%, by at least 15%, by at least 20%, by at least
25, by at least 30%, by at
least 35%, by at least 40%, by at least 45, or by at least 50%.
[0095] In another embodiment, administration of a composition comprising an
apoptotic cell
supernatant does not reduce the efficacy of the CAR T-cells to treat, prevent,
inhibit, reduce the
incidence of, ameliorating, or alleviating said cancer or said tumor by more
than about 5%. In another
embodiment, administration of a composition comprising an apoptotic cell
supernatant does not
reduce the efficacy of the CAR T-cells to treat, prevent, inhibit, reduce the
incidence of, ameliorating,
CA 03151815 2022-02-17
21
WO 2021/053667
PCT/IL2020/051011
or alleviating said cancer or said tumor by more than about 10%. In another
embodiment,
administration of a composition comprising an apoptotic cell supernatant does
not reduce the efficacy
of the CAR T-cells to treat, prevent, inhibit, reduce the incidence of,
ameliorating, or alleviating said
cancer or said tumor by more than about 15%. In another embodiment,
administration of a
composition comprising an apoptotic cell supernatant does not reduce the
efficacy of the CAR T-cells
to treat, prevent, inhibit, reduce the incidence of, ameliorating, or
alleviating said cancer or said tumor
by more than about 20%. In another embodiment, administration of a composition
comprising the
apoptotic cell supernatant does not affect the efficacy of the CAR T-cells to
treat, prevent, inhibit,
reduce the incidence of, ameliorating, or alleviating said cancer or said
tumor. In another embodiment,
administration of a composition comprising the apoptotic cell supernatant does
not reduce the efficacy
of the CAR T-cells to treat, prevent, inhibit, reduce the incidence of,
ameliorating, or alleviating said
cancer or said tumor.
[0096] In some embodiments, disclosed herein are methods of inhibiting or
reducing the incidence
of cytokine release syndrome (CRS) or cytokine storm in a subject undergoing
CAR T-cell cancer
therapy. In another embodiment, methods disclosed herein decrease or prevent
cytokine production
in a subject undergoing CAR T-cell cancer therapy thereby inhibiting or
reducing the incidence of
cytokine release syndrome (CRS) or cytokine storm in a subject. In another
embodiment, the methods
disclosed herein of inhibiting or reducing the incidence of cytokine release
syndrome (CRS) or
cytokine storm in a subject undergoing CAR T-cell cancer therapy comprise the
step of administering
a composition comprising apoptotic cells to the subject undergoing the cancer
therapy. In yet another
embodiment, methods disclosed herein for decreasing or inhibiting cytokine
production in a subject
undergoing CAR T-cell cancer therapy comprise the step of administering a
composition comprising
apoptotic cells to the subject undergoing the cancer therapy. In another
embodiment, administration
of a composition comprising apoptotic cells does not affect the efficacy of
the CAR T-cell therapy.
In another embodiment, administration of a composition comprising apoptotic
cells or an apoptotic
supernatant does not reduce the efficacy of the CAR T-cell therapy. In another
embodiment,
administration of a composition comprising apoptotic cells or an apoptotic
cell supernatant does not
reduce the efficacy of the CAR T-cells therapy by more than about 5%. In
another embodiment,
administration of a composition comprising apoptotic cells or an apoptotic
cell supernatant does not
reduce the efficacy of the CAR T-cells therapy by more than about 10%. In
another embodiment,
administration of a composition comprising apoptotic cells or an apoptotic
cell supernatant does not
reduce the efficacy of the CAR T-cells therapy by more than about 15%. In
another embodiment,
administration of a composition comprising apoptotic cells or an apoptotic
cell supernatant does not
reduce the efficacy of the CAR T-cells therapy by more than about 20%.
CA 03151815 2022-02-17
22
WO 2021/053667
PCT/IL2020/051011
[0097] In some embodiments, disclosed herein are methods of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm comprising the step of
administering an apoptotic cell
supernatant, as disclosed herein, or a composition comprising said apoptotic
cell supernatant. In
another embodiment, an apoptotic cell supernatant comprises an apoptotic cell-
phagocyte
supernatant.
[0098] In some embodiments, methods disclosed herein for decreasing or
inhibiting cytokine
production in a subject undergoing CAR T-cell cancer therapy comprise the step
of administering a
composition comprising an apoptotic cell supernatant to the subject undergoing
the cancer therapy.
In another embodiment, administration of a composition comprising an apoptotic
cell supernatant
does not affect the efficacy of the CAR T-cell therapy. In another embodiment,
administration of a
composition comprising an apoptotic cell supernatant does not reduce the
efficacy of the CAR T-cell
therapy.
[0099] In some embodiments, a method of inhibiting or reducing the incidence
of a cytokine release
syndrome (CRS) or a cytokine storm in a subject undergoing chimeric antigen
receptor-expressing T-
cell (CAR T-cell) cancer therapy comprises the step of administering a
composition comprising
apoptotic cells or an apoptotic supernatant to said subject. In another
embodiment, a method of
inhibiting or reducing the incidence of a cytokine release syndrome (CRS) or a
cytokine storm in a
subject undergoing chimeric antigen receptor-expressing T-cell (CAR T-cell)
cancer therapy
decreases or inhibits production of at least one pro-inflammatory cytokine in
the subject.
[0100] In another embodiment, this disclosure provides methods of use of
a pooled mononuclear
apoptotic cell preparation comprising mononuclear cells in an early apoptotic
state, as described
herein, for treating, preventing, ameliorating, inhibiting, or reducing the
incidence of an immune
disease, an autoimmune disease, an inflammatory disease, a cytokine release
syndrome (CRS), a
cytokine storm, or infertility in a subject in need thereof. In another
embodiment, disclosed herein is
a pooled mononuclear apoptotic cell preparation, wherein use of such a cell
preparation in certain
embodiments does not require matching donors and recipients, for example by
HLA typing.
[0101] Genetically modified immune cells
[0102] Genetic modification of immune cells is well known as a strategy for
immune-cell therapies
against cancer. These immune-cell therapies are based on the manipulation and
administration of
autologous or allogeneic immune cells to a subject in need. Immune-cell based
therapies include
natural killer cells therapies, dendrite cell therapies, and T-cell
immunotherapies including those
utilizing naive T-cells, effector T-cells also known as T-helper cells,
cytotoxic T-cells, and regulatory
T-cells (Tregs).
CA 03151815 2022-02-17
23
WO 2021/053667
PCT/IL2020/051011
[0103] In one embodiment, disclosed herein are compositions comprising
genetically modified
immune cells. In another embodiment, the genetically modified immune cell is a
T-cell. In another
embodiment, a T-cell is a naive T-cell. In another embodiment, a T-cell is a
naive CD4+ T-cell. In
another embodiment, a T-cell is a naïve T-cell. In another embodiment, a T-
cell is a naive CD8+ T-
.. cell. In another embodiment, the genetically modified immune cell is a
natural killer (NK) cell. In
another embodiment, the genetically modified immune cell is a dendritic cell.
In still another
embodiment, the genetically modified T-cell is a cytotoxic T lymphocyte (CTL
cell). In another
embodiment, the genetically modified T-cell is a regulatory T-cell (Treg). In
another embodiment,
the genetically modified T-cell is a chimeric antigen receptor (CAR) T-cell.
In another embodiment,
the genetically modified T-cell is a genetically modified T-cell receptor
(TCR) cell.
[0104] In one embodiment, disclosed herein are compositions comprising
genetically modified
immune cells and an additional agent selected from the group comprising
apoptotic cells, an apoptotic
cell supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or fragment
thereof or analogue
thereof, a tellurium-based compound, or an immune modulating agent, or any
combination thereof.
In another embodiment, disclosed herein are compositions comprising
genetically modified immune
cells, apoptotic cells, and an additional agent selected from the group
comprising a CTLA-4 blocking
agent, an alpha-1 anti-trypsin or fragment thereof or analogue thereof, a
tellurium-based compound,
or an immune modulating agent, or any combination thereof. In another
embodiment, disclosed
herein are compositions comprising genetically modified immune cells, an
apoptotic cell supernatant,
and an additional agent selected from the group comprising a CTLA-4 blocking
agent, an alpha-1
anti-trypsin or fragment thereof or analogue thereof, a tellurium-based
compound, or an immune
modulating agent, or any combination thereof.
[0105] In one embodiment, the immune cells are cytotoxic. In another
embodiment, cytotoxic cells
for genetic modification can be obtained from bone marrow of the subject
(autologous) or a donor
.. (allogeneic). In other cases, the cells are obtained from a stem cell. For
example, cytotoxic cells can
be derived from human pluripotent stem cells such as human embryonic stem
cells or human induced
pluripotent T-cells. In the case of induced pluripotent stem cells (IPSCs),
such pluripotent T-cells can
be obtained using a somatic cell from the subject to which genetically
modified cytotoxic cells will
be provided. In one embodiment, immune cells may be obtained from a subject or
donor by
harvesting cells by venipuncture, by apheresis methods, by white cell
mobilization followed by
apheresis or venipuncture, or by bone marrow aspiration.
[0106] In one embodiment, immune cells, for example T-cell, are generated and
expanded by the
presence of specific factors in vivo. In another embodiment, T-cell generation
and maintenance is
affected by cytokines in vivo. In another embodiment, cytokines that affect
generation and
CA 03151815 2022-02-17
24
WO 2021/053667
PCT/IL2020/051011
maintenance to T-helper cells in vivo comprise IL-1, IL-2, IL-4, IL-6, IL-12,
IL-21, IL-23, IL-25, IL-
33, and TG93. In another embodiment, Treg cells are generated from naive T-
cells by cytokine
induction in vivo. In still another embodiment, TGF-f3 and/or IL-2 play a role
in differentiating naive
T-cell to become Treg cells.
[0107] In another embodiment, the presence of a cytokine selected from the
group comprising IL-1,
IL-2, IL-4, IL-6, IL-12, IL-21, IL-23, IL-25, IL-33, and TG93, maintains or
increases the proliferation
rate or both, of T-cells in vivo. In another embodiment, the presence of a
cytokine IL-2 and/or TG93,
maintains or increases the proliferation rate or both, of T-cells in vivo. In
another embodiment, the
presence of a cytokine selected from the group comprising IL-1, IL-2, IL-4, IL-
6, IL-12, IL-21, IL-
23, IL-25, IL-33, and TGFP, maintains or increases the proliferation rate or
both, of CAR T-cells in
vivo. In another embodiment, the presence of a cytokine IL-2 and/or TG93,
maintains or increases
the proliferation rate or both, of CAR T-cells in vivo. In another embodiment,
the presence of a
cytokine selected from the group comprising IL-1, IL-2, IL-4, IL-6, IL-12, IL-
21, IL-23, IL-25, IL-
33, and TG93, maintains or increases the proliferation rate or both, of TCR T-
cells in vivo. In another
embodiment, the presence of a cytokine IL-2 and/or TGFP, maintains or
increases the proliferation
rate or both, of TCR T-cells in vivo. In another embodiment, the presence of a
cytokine selected from
the group comprising IL-1, IL-2, IL-4, IL-6, IL-12, IL-21, IL-23, IL-25, IL-
33, and TG93, maintains
or increases the proliferation rate or both, of T-reg cells in vivo. In
another embodiment, the presence
of a cytokine IL-2 and/or TG93, maintains or increases the proliferation rate
or both, of T-reg cells
.. in vivo.
[0108] In one embodiment T-cells having an altered expression or form of
STAT5B encoded protein
or BACH2 encoded protein are maintained for an extended time period or have an
increased
proliferation rate or both. In another embodiment, said altered expression
increases expression
STAT5B polypeptide. In another embodiment, said altered expression increases
expression of
BACH2 polypeptide.
[0109] In another embodiment, T-cells having an altered expression of a STAT5B
encoded protein
are maintained for an extended time period or have an increased proliferation
rate in vivo. In another
embodiment, T-cells having an altered expression of a BACH2 encoded protein
are maintained for
an extended time period or have an increased proliferation rate in vivo. In
another embodiment, T-
.. cells having an altered form of a STAT5B encoded protein are maintained for
an extended time period
or have an increased proliferation rate in vivo. In another embodiment, T-
cells having an altered form
of a BACH2 encoded protein are maintained for an extended time period or have
an increased
proliferation rate in vivo.
[0110] In another embodiment, T-cells having an altered expression of a STAT5B
encoded protein
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
maintain or increase their proliferation rate in vivo for greater than 1 year.
In another embodiment,
T-cells having an altered expression of a STAT5B encoded protein maintain or
increase their
proliferation rate in vivo for greater than 2 years. In another embodiment, T-
cells having an altered
expression of a STAT5B encoded protein maintain or increase their
proliferation rate in vivo for
5 greater than 3 years. In another embodiment, T-cells having an altered
expression of a STAT5B
encoded protein maintain or increase their proliferation rate in vivo for
greater than 4 years. In another
embodiment, T-cells having an altered expression of a STAT5B encoded protein
maintain or increase
their proliferation rate in vivo for greater than 5 years. In another
embodiment, T-cells having an
altered expression of a STAT5B encoded protein maintain or increase their
proliferation rate in vivo
10 for greater than 10 years. In another embodiment, T-cells having an
altered expression of a STAT5B
encoded protein maintain or increase their proliferation rate in vivo for
greater than 20 years.
[0111] In another embodiment, T-cells having an altered expression of a BACH2
encoded protein
maintain or increase their proliferation rate in vivo for greater than 1 year.
In another embodiment,
T-cells having an altered expression of a BACH2 encoded protein maintain or
increase their
15 proliferation rate in vivo for greater than 2 years. In another
embodiment, T-cells having an altered
expression of a BACH2 encoded protein maintain or increase their proliferation
rate in vivo for
greater than 3 years. In another embodiment, T-cells having an altered
expression of a BACH2
encoded protein maintain or increase their proliferation rate in vivo for
greater than 4 years. In another
embodiment, T-cells having an altered expression of a BACH2 encoded protein
maintain or increase
20 their proliferation rate in vivo for greater than 5 years. In another
embodiment, T-cells having an
altered expression of a BACH2 encoded protein maintain or increase their
proliferation rate in vivo
for greater than 10 years. In another embodiment, T-cells having an altered
expression of a BACH2
encoded protein maintain or increase their proliferation rate in vivo for
greater than 20 years.
[0112] In another embodiment, T-cells having an altered form of a STAT5B
encoded protein
25 maintain or increase their proliferation rate in vivo for greater than 1
year. In another embodiment,
T-cells having an altered form of a STAT5B encoded protein maintain or
increase their proliferation
rate in vivo for greater than 2 years. In another embodiment, T-cells having
an altered form of a
STAT5B encoded protein maintain or increase their proliferation rate in vivo
for greater than 3 years.
In another embodiment, T-cells having an altered form of a STAT5B encoded
protein maintain or
increase their proliferation rate in vivo for greater than 4 years. In another
embodiment, T-cells having
an altered form of a STAT5B encoded protein maintain or increase their
proliferation rate in vivo for
greater than 5 years. In another embodiment, T-cells having an altered form of
a STAT5B encoded
protein maintain or increase their proliferation rate in vivo for greater than
10 years. In another
embodiment, T-cells having an altered form of a STAT5B encoded protein
maintain or increase their
CA 03151815 2022-02-17
26
WO 2021/053667
PCT/IL2020/051011
proliferation rate in vivo for greater than 20 years.
[0113] In another embodiment, T-cells having an altered form of a BACH2
encoded protein maintain
or increase their proliferation rate in vivo for greater than 1 year. In
another embodiment, T-cells
having an altered form of a BACH2 encoded protein maintain or increase their
proliferation rate in
vivo for greater than 2 years. In another embodiment, T-cells having an
altered form of a BACH2
encoded protein maintain or increase their proliferation rate in vivo for
greater than 3 years. In another
embodiment, T-cells having an altered form of a BACH2 encoded protein maintain
or increase their
proliferation rate in vivo for greater than 4 years. In another embodiment, T-
cells having an altered
form of a BACH2 encoded protein maintain or increase their proliferation rate
in vivo for greater than
5 years. In another embodiment, T-cells having an altered form of a BACH2
encoded protein
maintain or increase their proliferation rate in vivo for greater than 10
years. In another embodiment,
T-cells having an altered form of a BACH2 encoded protein maintain or increase
their proliferation
rate in vivo for greater than 20 years.
[0114] In another embodiment, CAR T-cells having an altered expression of a
STAT5B encoded
protein are maintained for an extended time period or have an increased
proliferation rate in vivo. In
another embodiment, CAR T-cells having an altered expression of a BACH2
encoded protein are
maintained for an extended time period or have an increased proliferation rate
in vivo. In another
embodiment, CAR T-cells having an altered form of a STAT5B encoded protein are
maintained for
an extended time period or have an increased proliferation rate in vivo. In
another embodiment, CAR
T-cells having an altered form of a BACH2 encoded protein are maintained for
an extended time
period or have an increased proliferation rate in vivo
[0115] In another embodiment, TCR T-cells having an altered expression of a
STAT5B encoded
protein are maintained for an extended time period or have an increased
proliferation rate in vivo. In
another embodiment, TCR T-cells having an altered expression of a BACH2
encoded protein are
maintained for an extended time period or have an increased proliferation rate
in vivo. In another
embodiment, TCR T-cells having an altered form of a STAT5B encoded protein are
maintained for
an extended time period or have an increased proliferation rate in vivo. In
another embodiment, TCR
T-cells having an altered form of a BACH2 encoded protein are maintained for
an extended time
period or have an increased proliferation rate in vivo.
[0116] In another embodiment, Treg-cells having an altered expression of a
STAT5B encoded
protein maintain or increase their proliferation rate in vivo. In another
embodiment, Treg-cells having
an altered expression of a BACH2 encoded protein maintain or increase their
proliferation rate in
vivo. In another embodiment, Treg-cells having an altered form of a STAT5B
encoded protein
maintain or increase their proliferation rate in vivo. In another embodiment,
Treg-cells having an
CA 03151815 2022-02-17
27
WO 2021/053667
PCT/IL2020/051011
altered form of a BACH2 encoded protein maintain or increase their
proliferation rate in vivo.
[0117] In one embodiment, methods for maintaining or increasing the
proliferation rate of a
genetically modified immune cell are disclosed herein, wherein the method
comprises the step of
administering apoptotic cells or an apoptotic supernatant. In another
embodiment, methods for
increasing the efficacy of a genetically modified immune cell are disclosed
herein, wherein the
method comprises the step of administering an additional agent comprising
apoptotic cells, an
apoptotic supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or
fragment thereof or
analogue thereof, a tellurium-based compound, or an immune modulating agent,
or any combination
thereof. In another embodiment, methods for treating, preventing, inhibiting,
reducing the incidence
of, ameliorating, or alleviating a cancer or a tumor disclosed herein
administer a genetically modified
immune cell and an additional agent, wherein said additional agent comprises
apoptotic cells, an
apoptotic supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or
fragment thereof or
analogue thereof, a tellurium-based compound, or an immune modulating agent,
or any combination
thereof.
Chimeric Antigen Receptor-Expressing T-Cells (CAR T-Cells)
[0118] In some embodiments, chimeric antigen receptors (CARs) are a type of
antigen-targeted
receptor composed of intracellular T-cell signaling domains fused to
extracellular tumor-binding
moieties, most commonly single-chain variable fragments (scFvs) from
monoclonal antibodies.
CARs directly recognize cell surface antigens, independent of MHC-mediated
presentation,
permitting the use of a single receptor construct specific for any given
antigen in all patients. Initial
CARs fused antigen-recognition domains to the CD3t activation chain of the T-
cell receptor (TCR)
complex. While these first generation CARs induced T-cell effector function in
vitro, they were
largely limited by poor antitumor efficacy in vivo. Subsequent CAR iterations
have included
secondary costimulatory signals in tandem with CD3c including intracellular
domains from CD28 or
a variety of TNF receptor family molecules such as 4-1BB (CD137) and 0X40
(CD134) .Further,
third generation receptors include two costimulatory signals in addition to
CD3c most commonly
from CD28 and 4-1BB. Second and third generation CARs dramatically improved
antitumor efficacy,
in some cases inducing complete remissions in patients with advanced cancer.
[0119] In some embodiments, a CAR T-cell is an immunoresponsive cell
comprising an antigen
receptor, which is activated when its receptor binds to its antigen.
[0120] In some embodiments, the CAR T-cells used in the compositions and
methods as disclosed
herein are first generation CAR T-cells. In another embodiment, the CAR T-
cells used in the
compositions and methods as disclosed herein are second generation CAR T-
cells. In another
embodiment, the CAR T-cells used in the compositions and methods as disclosed
herein are third
CA 03151815 2022-02-17
28
WO 2021/053667
PCT/IL2020/051011
generation CAR T-cells. In another embodiment, the CAR T-cells used in the
compositions and
methods as disclosed herein are fourth generation CAR T-cells. In some
embodiments, each
generation of CAR T-cells is more potent than the CAR T-cells of earlier
generations.
[0121] In some embodiments, first-generation CARs have one signaling domain,
typically the
cytoplasmic signaling domain of the CD3 TCK chain.
[0122] In another embodiment, the CAR T-cells as disclosed herein are second
generation CAR T-
cells. In another embodiment, CAR T-cells as disclosed herein comprise a
tripartite chimeric receptor
(TPCR). In some embodiments, CAR T-cells as disclosed herein, comprise one or
more signaling
moieties that activate naive T-cells in a co-stimulation independent manner.
In another embodiment,
the CAR T-cells further encode one or more members of the tumor necrosis
factor receptor family,
which in some embodiments, is CD27, 4-1BB (CD137), or 0X40 (CD134), or a
combination thereof.
[0123] Third-generation CAR T-cells attempt to harness the signaling potential
of 2 costimulatory
domains: in some embodiments, the CD28 domain followed by either the 4-1BB or
OX-40 signaling
domains. In another embodiment, the CAR T-cells used in the compositions and
methods as disclosed
herein further encode a co-stimulatory signaling domain, which in one
embodiment is CD28. In
another embodiment, the signaling domain is the CD3-chain, CD97, GDI la-CD18,
CD2, ICOS,
CD27, CD154, CDS, 0X40, 4-1BB, CD28 signaling domain, or combinations thereof.
[0124] In some embodiments, telomere length and replicative capacity correlate
with the engraftment
efficiency and antitumor efficacy of adoptively transferred T-cell lines. In
some embodiments, CD28
stimulation maintains telomere length in T-cells.
[0125] In some embodiments, CAR-modified T-cell potency may be further
enhanced through the
introduction of additional genes, including those encoding proliferative
cytokines (ie, IL-12) or
costimulatory ligands (ie, 4-1BBL), thus producing "armored" fourth-generation
CAR-modified T-
cells. In some embodiments, "armored CAR T-cells," are CAR T-cells which are
protected from the
inhibitory tumor microenvironment. In another embodiment, the "armored" CAR
technology
incorporates the local secretion of soluble signaling proteins to amplify the
immune response within
the tumor microenvironment with the goal of minimizing systemic side effects.
In some
embodiments, the signaling protein signal is IL-12, which can stimulate T-cell
activation and
recruitment. In some embodiments, "armored" CAR technology is especially
useful in solid tumor
indications, in which microenvironment and potent immunosuppressive mechanisms
have the
potential to make the establishment of a robust anti-tumor response more
challenging.
[0126] In some embodiments, CAR T-cells are genetically modified to encode
molecules involved
in the prevention of apoptosis, the remodeling of the tumor microenvironment,
induction of
homeostatic proliferation, and chemokine receptors that promote directed T-
cell homing.
CA 03151815 2022-02-17
29
WO 2021/053667
PCT/IL2020/051011
[0127] In another embodiment, CAR T-cell therapy used in the compositions and
methods as
disclosed herein is enhanced using the expression of cytokine transgenes,
combination therapy with
small molecule inhibitors, or monoclonal antibodies. In another embodiment,
other strategies aimed
at improving CAR T-cell therapy including using dual CARs and chemokine
receptors to more
specifically target tumor cells are to be considered part of the CAR T-cells
and CAR T-cell therapy
as disclosed herein.
[0128] In some embodiments, the CAR T-cells of the compositions and methods as
disclosed herein
comprise a second binding domain that can lead to either an inhibitory or
amplifying signal, in order
to increase specificity of CAR T-cells for cancer cells versus normal cells.
For example, a CAR T-
cell can be engineered such that it would be triggered in the presence of one
target protein, but if a
second protein is present it would be inhibited. Alternatively, it could also
be engineered such that
two target proteins would be required for maximal activation. These approaches
may increase the
specificity of the CAR for tumor relative to normal tissue.
[0129] In some embodiments, the CAR T-cells used in the compositions and
methods as disclosed
herein encode antibody-based external receptor structures and cytosolic
domains that encode signal
transduction modules composed of the immunoreceptor tyrosine-based activation
motif.
[0130] In some embodiments, the CAR T-cell further encodes a single-chain
variable fragment
(scFv) that binds a polypeptide that has immunosuppressive activity. In
another embodiment, the
polypeptide that has immunosuppressive activity is CD47, PD-1, CTLA-4, or a
combination thereof.
[0131] In some embodiments, the CAR T-cell further encodes a single-chain
variable fragment
(scFv) that binds a polypeptide that has immunostimulatory activity. In
another embodiment, the
polypeptide that has immunostimulatory activity is CD28, OX-40, 4-1 BB or a
combination thereof.
In another embodiment, the CAR T-cell further encodes a CD40 ligand (CD4OL),
which, in some
embodiments, enhances the immunostimulatory activity of the antigen.
.. [0132] In some embodiments, the immune cells are cytotoxic. In another
embodiment, cytotoxic cells
for genetic modification can be obtained from bone marrow of the subject or a
donor. In other cases,
the cells are obtained from a stem cell. For example, cytotoxic cells can be
derived from human
pluripotent stem cells such as human embryonic stem cells or human induced
pluripotent T-cells. In
the case of induced pluripotent stem cells (1PSCs), such pluripotent T-cells
can be obtained using a
somatic cell from the subject to which genetically modified cytotoxic cells
will be provided. In some
embodiments, immune cells may be obtained from a subject or donor by
harvesting cells by
venipuncture, by apheresis methods, by white cell mobilization followed by
apheresis or
venipuncture, or by bone marrow aspiration.
[0133] In some embodiments, a method as disclosed herein comprises obtaining
immune cells from
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
a subject, and genetically modifying the immune cells to express a chimeric
antigen receptor. In
another embodiment, a method as disclosed herein comprises obtaining immune
cells from a subject,
genetically modifying the immune cells to express a chimeric antigen receptor
and combining with
apoptotic cell population resulting in reduced cytokine production in a
subject but substantially
5 unaffected cytotoxicity relative to immune cells expressing a CAR not
administered with an apoptotic
cell population (Figures 1A-1B and 2). In another embodiment, a method as
disclosed herein
comprises obtaining immune cells from a subject, genetically modifying the
immune cells to express
a chimeric antigen receptor and combining with an apoptotic cell supernatant
or a composition
comprising the supernatant, resulting in reduced cytokine production in a
subject but substantially
10 unaffected cytotoxicity relative to immune cells expressing a CAR not
administered with an apoptotic
cell supernatant. In another embodiment, administration of an apoptotic cell
population or a
supernatant from apoptotic cells does not reduce the efficacy of the immune
cells expressing the
chimeric antigen receptor.
[0134] In one embodiment, disclosed herein are immune cells, in some
embodiments, CAR T-cells
15 in which the T-cell is autologous to the subject. In another embodiment,
the CAR T-cells are
heterologous to the subject. In some embodiments, the CAR T-cells are
allogeneic. In some
embodiments, the CAR T-cells are universal allogeneic CAR T-cells. In another
embodiment, the T-
cells may be autologous, allogeneic, or derived in vitro from engineered
progenitor or stem cells.
[0135] In another embodiment, the CAR T-cells and apoptotic cells described
herein, are both
20 derived from the same source. In a further embodiment, the CAR T-cells
and apoptotic cells described
herein, are both derived from the subject (Figure 1). In an alternative
embodiment, the CAR T-cells
and apoptotic cells described herein, are derived from different sources. In
yet another embodiment,
the CAR T-cells are autologous, and the apoptotic cells described herein, are
allogeneic (Figure 2).
A skilled artisan would appreciate that similarly, an apoptotic cell
supernatant may be made from
25 cells derived from the same source as the CAR T-cell, which may in one
embodiment be autologous
cells, or an apoptotic cell supernatant may be made from cells derived from a
source different from
the source of CAR T-cells.
[0136] A skilled artisan would appreciate that the term "heterologous" may
encompass a tissue, cell,
nucleic acid molecule or polypeptide that is derived from a different
organism. In some embodiments,
30 a heterologous protein is a protein that was initially cloned from or
derived from a different T-cell
type or a different species from the recipient and that is not normally
present in a cell or sample
obtained from a cell.
[0137] Accordingly, one embodiment as disclosed herein relates to cytotoxic
immune cells (e.g., NK
cells or T-cells) comprising chimeric antigen receptors (CARs) whereby the
cells retain their
CA 03151815 2022-02-17
31
WO 2021/053667
PCT/IL2020/051011
cytotoxic function. In another embodiment, the chimeric antigen receptor is
exogenous to the T-cell.
In another embodiment, the CAR is recombinantly expressed. In another
embodiment, the CAR is
expressed from a vector.
[0138] In some embodiments, the T-cell utilized to generate CAR T-cells is a
naïve CD4+ T-cell. In
another embodiment, the T-cell utilized to generate CAR T-cells is a naïve
CD8+ T-cell. In another
embodiment, the T-cell utilized to generate CAR T-cells is an effector T-cell.
In another embodiment,
the T-cell utilized to generate CAR T-cells is a regulatory T-cell (Treg). In
another embodiment, the
T-cell utilized to generate CAR T-cells is a cytotoxic T-cell.
[0139] Sources for genetically modified immune cells, for example T cells,
have been described
extensively in the literature, see for example Themelli et al. (2015) New Cell
Sources for T Cell
Engineering and Adoptive Immunotherapy. Cell Stem Cell 16: 357-366; Han et al.
(2013) Journal of
Hematology & Oncology 6:47-53; Wilkie et al. (2010) J Bio Chem 285(33):25538-
25544; and van
der Stegen et al. (2013) J. Immunol 191: 4589-4598. CAR T-cells are available
to order from a
commercial source such as Creative Biolabs (NY USA), which provides custom
construction and
production services for Chimeric Antigen Receptors (CAR) and also provides
premade CAR
constructs stock, which can induce protective immunity encode by recombinant
adenovirus
vaccine. Custom made CAR T-cells may also be obtained from Promab
Biotechnologies (CA USA),
which can provide specifically designed CAR T-cells.
T-cell receptors (TCRs) cells
[0140] In one embodiment, compositions and methods as disclosed herein utilize
a designer T-cell
receptor (TCR) cells in addition to or in place of CAR T-cells. The TCR is a
multi-subunit
transmembrane complex that mediates the antigen-specific activation of T-
cells. The TCR is
composed of two different polypeptide chains. The TCR confers antigenic
specificity on the T cell,
by recognizing an antigen epitope on the target cell, for example a tumor or
cancer cell. Following
contact with the antigen present on the tumor or cancer cell, T-cells
proliferate and acquire the
phenotype and function to allow them to eliminate the cancer or tumor cells.
[0141] In one embodiment, TCR T-cell therapy comprises introducing a T-cell
receptor (TCR) that
is specific to an epitope of a protein of interest into a T-cell. In another
embodiment, the protein of
interest is a tumor-associated antigen. In another embodiment, the genetically
engineered TCR
.. recognizes a tumor antigen epitope presented by the major
histocompatibility complex (MHC) on the
tumor cell along with T-cell activating domains. In another embodiment, the T-
cell receptors
recognize antigens irrespectively of their intracellular or membrane
localization. In another
embodiment, TCRs recognize tumor cells that intracellularly express a tumor
associated antigen. In
one embodiment TCRs recognize internal antigens. In another embodiment, TCRs
recognize
CA 03151815 2022-02-17
32
WO 2021/053667
PCT/IL2020/051011
angiogenic factors. In another embodiment, an angiogenic factor is a molecule
involved in the
formation of new blood vessels. Various genetically modified T-cell receptors
and methods of their
production are known in the art.
[0142] In one embodiment, TCR T-cell therapy is used to treat, prevent,
inhibit, ameliorate, reduce
the incidence of, or alleviate a cancer or a tumor. In one embodiment, TCR T-
cell therapy is used to
treat, prevent, inhibit, ameliorate, reduce the incidence of, or alleviate
advanced metastatic disease,
including those with hematological (lymphoma and leukemia) and solid tumors
(refractory
melanoma, sarcoma). In one embodiment, the TCR T-cell therapy used in the
compositions and
methods as disclosed herein treat a malignancy listed in Table 1 of Sadelain
et al., (Cancer Discov.
2013 Apr; 3(4): 388-398).
[0143] In another embodiment, the T-cell receptor is genetically modified to
bind NY-ESO-1
epitopes, and the TCR-engineered T-cell is anti-NY-ESO-1. In another
embodiment, the T-cell
receptor is genetically modified to bind HPV-16 E6 epitopes, and the TCR-
engineered T-cell is anti-
HPV-16 E6. In another embodiment, the T-cell receptor is genetically modified
to bind HPV-16 E7
epitopes, and the TCR-engineered T-cell is anti-HPV-16 E7.In another
embodiment, the T-cell
receptor is genetically modified to bind MAGE A3/A6 epitopes, and the TCR-
engineered T-cell is
anti-MAGE A3/A6. In another embodiment, the T-cell receptor is genetically
modified to bind
MAGE A3 epitopes, and the TCR-engineered T-cell is anti-MAGE A3. In another
embodiment, the
T-cell receptor is genetically modified to bind SSX2 epitopes, and the TCR-
engineered T-cell is anti-
.. SSX2. In another embodiment, the T-cell receptor is genetically modified to
bind a target antigen
disclosed herein. Using the tools well known in the art, a skilled would
appreciate that the T-cell
receptor may be genetically modified to bind a target antigen present on a
cancer or tumor cell,
wherein the TCR-engineer T-cell comprises an anti-tumor or anti-cancer cell.
[0144] In one embodiment, a method as disclosed herein comprises obtaining
immune cells from a
.. subject, and genetically modifying the immune cells to express a
recombinant T-cell receptor (TCR).
In another embodiment, a method as disclosed herein comprises obtaining immune
cells from a
subject, genetically modifying the immune cells to express a recombinant TCR
and combining with
an additional agent, wherein said additional agent comprises an apoptotic cell
population, an
apoptotic cell supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin
or fragment thereof or
analogue thereof, a tellurium-based compound, or an immune modulating agent,
or any combination
thereof.
[0145] In one embodiment, the T-cell utilized to generate TCR T-cells is a
naïve CD4+ T-cell. In
another embodiment, the T-cell utilized to generate TCR T-cells is a naïve
CD8+ T-cell. In another
embodiment, the T-cell utilized to generate TCR T-cells is an effector T-cell.
In another embodiment,
CA 03151815 2022-02-17
33
WO 2021/053667
PCT/IL2020/051011
the T-cell utilized to generate TCR T-cells is a regulatory T-cell (Treg). In
another embodiment, the
T-cell utilized to generate TCR T-cells is a cytotoxic T-cell.
[0146] TCR T-cells have been described extensively in the literature, see for
example Sharpe and
Mount (2015) ibid.; Essand M, Loskog ASI (2013) Genetically engineered T cells
for the treatment
of cancer (Review). J Intern Med 273: 166-181; and Kershaw et al. (2014)
Clinical application of
genetically modified T cells in cancer therapy. Clinical & Translational
Immunology 3:1-7.
Targeting antigens
[0147] In some embodiments, the CAR binds to an epitope of an antigen via an
antibody or an
antibody fragment that is directed to the antigen. In another embodiment, the
antibody is a monoclonal
antibody. In another embodiment, the antibody is a polyclonal antibody. In
another embodiment, the
antibody fragment is a single-chain variable fragment (scFv).
[0148] In one embodiment, the TCR binds to an epitope of an antigen via a
genetically modified T-
cell receptor.
[0149] In another embodiment, the CAR T-cells of the compositions as disclosed
herein bind to a
tumor associated antigen (TAA). In another embodiment, said tumor associated
antigen is: Mucin 1,
cell surface associated (MUC1) or polymorphic epithelial mucin (PEM), Arginine-
rich, mutated in
early stage tumors (Armet), Heat Shock Protein 60 (HSP60), calnexin (CANX),
methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2,
methenyltetrahydrofolate
cyclohydrolase (MTHFD2), fibroblast activation protein (FAP), matrix
metallopeptidase (MMP6), B
Melanoma Antigen-1 (BAGE-1), aberrant transcript of N-acetyl glucosaminyl
transferase V (GnTV),
Q5H943, Carcinoembryonic antigen (CEA), Pmel, Kallikrein-4, Mammaglobin-1,
MART-1,
GPR143-0A1, prostate specific antigen (PSA), TRP1, Tyrosinase, FGP-5, NEU
proto-oncogene, Aft,
MMP-2, prostate specific membrane antigen (PSMA), Telomerase-associated
protein-2, Prostatic
acid phosphatase (PAP), Uroplakin II or Proteinase 3.
[0150] In another embodiment, the CAR binds to CD19 or CD20 to target B cells
in the case where
one would like to destroy B cells as in leukemia. CD19 is a B cell lineage
specific surface receptor
whose broad expression, from pro-B cells to early plasma cells, makes it an
attractive target for the
immunotherapy of B cell malignancies. In another embodiment, the CAR binds to
ROR1, CD22, or
GD2. In another embodiment, the CAR binds to NY-ESO-1. In another embodiment,
the CAR binds
to MAGE family proteins. In another embodiment, the CAR binds to mesothelin.
In another
embodiment, the CAR binds to c-erbB2. In another embodiment, the CAR binds to
mutational
antigens that are tumor specific, such as BRAFV600E mutations and BCR-ABL
translocations. In
another embodiment, the CAR binds to viral antigens which are tumor-specific,
such as EBV in HD,
HPV in cervical cancer, and polyomavirus in Merkel cancer. In another
embodiment, the CAR T-cell
CA 03151815 2022-02-17
34
WO 2021/053667
PCT/IL2020/051011
binds to Her2/neu. In another embodiment, the CAR T-cell binds to EGFRvIII.
[0151] In some embodiments, the chimeric antigen receptor (CAR) T-cell binds
the CD19 antigen.
In another embodiment, the CAR binds the CD22 antigen. In another embodiment,
the CAR binds to
alpha folate receptor. In another embodiment, the CAR binds to CAIX. In
another embodiment, the
CAR binds to CD20. In another embodiment, the CAR binds to CD23. In another
embodiment, the
CAR binds to CD24. In another embodiment, the CAR binds to CD30. In another
embodiment, the
CAR binds to CD33. In another embodiment, the CAR binds to CD38. In another
embodiment, the
CAR binds to CD44v6. In another embodiment, the CAR binds to CD44v7/8. In
another embodiment,
the CAR binds to CD123. In another embodiment, the CAR binds to CD171. In
another embodiment,
the CAR binds to carcinoembryonic antigen (CEA). In another embodiment, the
CAR binds to
EGFRvIII. In another embodiment, the CAR binds to EGP-2. In another
embodiment, the CAR binds
to EGP-40. In another embodiment, the CAR binds to EphA2. In another
embodiment, the CAR binds
to Erb-B2. In another embodiment, the CAR binds to Erb-B 2,3,4. In another
embodiment, the CAR
binds to Erb-B3/4. In another embodiment, the CAR binds to FBP. In another
embodiment, the CAR
binds to fetal acetylcholine receptor. In another embodiment, the CAR binds to
GD2. In another
embodiment, the CAR binds to GD3. In another embodiment, the CAR binds to
HER2. In another
embodiment, the CAR binds to HMW-MAA. In another embodiment, the CAR binds to
IL-11Ralpha.
In another embodiment, the CAR binds toIL-13Ralphal. In another embodiment,
the CAR binds to
KDR. In another embodiment, the CAR binds to kappa-light chain. In another
embodiment, the CAR
binds to Lewis Y. In another embodiment, the CAR binds to Li-cell adhesion
molecule. In another
embodiment, the CAR binds to MAGE-Al. In another embodiment, the CAR binds to
mesothelin. In
another embodiment, the CAR binds to CMV infected cells. In another
embodiment, the CAR binds
to MUCl. In another embodiment, the CAR binds to MUC16. In another embodiment,
the CAR binds
to NKG2D ligands. In another embodiment, the CAR binds to NY-ESO-1 (amino
acids 157-165). In
another embodiment, the CAR binds to oncofetal antigen (h5T4). In another
embodiment, the CAR
binds to PSCA. In another embodiment, the CAR binds to PSMA. In another
embodiment, the CAR
binds to ROR1. In another embodiment, the CAR binds to TAG-72. In another
embodiment, the CAR
binds to VEGF-R2 or other VEGF receptors. In another embodiment, the CAR binds
to B7-H6. In
another embodiment, the CAR binds to CA9. In another embodiment, the CAR binds
to avf36integrin.
In another embodiment, the CAR binds to 8H9. In another embodiment, the CAR
binds to NCAM.
In another embodiment, the CAR binds to fetal acetylcholine receptor.
[0152] In another embodiment, the chimeric antigen receptor (CAR) T-cell
targets the CD19 antigen,
and has a therapeutic effect on subjects with B-cell malignancies, ALL,
Follicular lymphoma, CLL,
and Lymphoma. In another embodiment, the CAR T-cell targets the CD22 antigen,
and has a
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
therapeutic effect on subjects with B-cell malignancies. In another
embodiment, the CAR T-cell
targets alpha folate receptor or folate receptor alpha, and has a therapeutic
effect on subjects with
ovarian cancer or epithelial cancer. In another embodiment, the CAR T-cell
targets CAIX or
G250/CAIX, and has a therapeutic effect on subjects with renal cell carcinoma.
In another
5 embodiment, the CAR T-cell targets CD20, and has a therapeutic effect on
subjects with Lymphomas,
B-cell malignancies, B-cell lymphomas, Mantle cell lymphoma and, indolent B-
cell lymphomas. In
another embodiment, the CAR T-cell targets CD23, and has a therapeutic effect
on subjects with CLL.
In another embodiment, the CAR T-cell targets CD24, and has a therapeutic
effect on subjects with
pancreatic adenocarcinoma. In another embodiment, the CAR T-cell targets CD30,
and has a
10 therapeutic effect on subjects with Lymphomas or Hodgkin lymphoma. In
another embodiment, the
CAR T-cell targets CD33, and has a therapeutic effect on subjects with AML. In
another embodiment,
the CAR T-cell targets CD38, and has a therapeutic effect on subjects with Non-
Hodgkin lymphoma.
In another embodiment, the CAR T-cell targets CD44v6, and has a therapeutic
effect on subjects with
several malignancies. In another embodiment, the CAR T-cell targets CD44v7/8,
and has a
15 therapeutic effect on subjects with cervical carcinoma. In another
embodiment, the CAR T-cell targets
CD123, and has a therapeutic effect on subjects with myeloid malignancies. In
another embodiment,
the CAR T-cell targets CEA, and has a therapeutic effect on subjects with
colorectal cancer. In another
embodiment, the CAR T-cell targets EGFRvIII, and has a therapeutic effect on
subjects with
Glioblastoma. In another embodiment, the CAR T-cell targets EGP-2, and has a
therapeutic effect on
20 subjects with multiple malignancies. In another embodiment, the CAR T-
cell targets EGP-40, and
has a therapeutic effect on subjects with colorectal cancer. In another
embodiment, the CAR T-cell
targets EphA2, and has a therapeutic effect on subjects with Glioblastoma. In
another embodiment,
the CAR T-cell targets Erb-B2 or ErbB3/4, and has a therapeutic effect on
subjects with Breast cancer
and others, prostate cancer, colon cancer, various tumors. In another
embodiment, the CAR T-cell
25 targets Erb-B 2,3,4, and has a therapeutic effect on subjects with
Breast cancer and others. In another
embodiment, the CAR T-cell targets FBP, and has a therapeutic effect on
subjects with Ovarian
cancer. In another embodiment, the CAR T-cell targets fetal acetylcholine
receptor, and has a
therapeutic effect on subjects with Rhabdomyosarcoma. In another embodiment,
the CAR T-cell
targets GD2, and has a therapeutic effect on subjects with Neuroblastoma,
melanoma, or Ewing' s
30 sarcoma. In another embodiment, the CAR T-cell targets GD3, and has a
therapeutic effect on subjects
with Melanoma. In another embodiment, the CAR T-cell targets HER2, and has a
therapeutic effect
on subjects with medulloblastoma, pancreatic adenocarcinoma, Glioblastoma,
Osteosarcoma, or
Ovarian cancer. In another embodiment, the CAR T-cell targets HMW-MAA, and has
a therapeutic
effect on subjects with Melanoma. In another embodiment, the CAR T-cell
targets IL-11Ralpha, and
CA 03151815 2022-02-17
36
WO 2021/053667
PCT/IL2020/051011
has a therapeutic effect on subjects with Osteosarcoma. In another embodiment,
the CAR T-cell
targets IL-13Ralphal, and has a therapeutic effect on subjects with Glioma,
Glioblastoma, or
medulloblastoma. In another embodiment, the CAR T-cell targets IL-13 receptor
alpha2, and has a
therapeutic effect on subjects with several malignancies. In another
embodiment, the CAR T-cell
targets KDR, and has a therapeutic effect on subjects with tumors by targeting
tumor neovasculature.
In another embodiment, the CAR T-cell targets kappa-light chain, and has a
therapeutic effect on
subjects with B-cell malignancies (B-NHL, CLL). In another embodiment, the CAR
T-cell targets
Lewis Y, and has a therapeutic effect on subjects with various carcinomas or
epithelial-derived
tumors. In another embodiment, the CAR T-cell targets Li-cell adhesion
molecule, and has a
therapeutic effect on subjects with Neuroblastoma. In another embodiment, the
CAR T-cell targets
MAGE-A 1 or HLA-Al MAGE Al, and has a therapeutic effect on subjects with
Melanoma. In
another embodiment, the CAR T-cell targets mesothelin, and has a therapeutic
effect on subjects with
Mesothelioma. In another embodiment, the CAR T-cell targets CMV infected
cells, and has a
therapeutic effect on subjects with CMV. In another embodiment, the CAR T-cell
targets MUC1, and
.. has a therapeutic effect on subjects with breast or ovarian cancer. In
another embodiment, the CAR
T-cell targets MUC16, and has a therapeutic effect on subjects with ovarian
cancer. In another
embodiment, the CAR T-cell targets NKG2D ligands, and has a therapeutic effect
on subjects with
myeloma, ovarian, and other tumors. In another embodiment, the CAR T-cell
targets NY-ES0-1
(157-165) or HLA-A2 NY-ES0-1, and has a therapeutic effect on subjects with
multiple myeloma.
.. In another embodiment, the CAR T-cell targets oncofetal antigen (h5T4), and
has a therapeutic effect
on subjects with various tumors. In another embodiment, the CAR T-cell targets
PSCA, and has a
therapeutic effect on subjects with prostate carcinoma. In another embodiment,
the CAR T-cell targets
PSMA, and has a therapeutic effect on subjects with prostate cancer/tumor
vasculature. In another
embodiment, the CAR T-cell targets ROR1, and has a therapeutic effect on
subjects with B-CLL and
mantle cell lymphoma. In another embodiment, the CAR T-cell targets TAG-72,
and has a therapeutic
effect on subjects with adenocarcinomas or gastrointestinal cancers. In
another embodiment, the CAR
T-cell targets VEGF-R2 or other VEGF receptors, and has a therapeutic effect
on subjects with tumors
by targeting tumor neovasculature. In another embodiment, the CAR T-cell
targets CA9, and has a
therapeutic effect on subjects with renal cell carcinoma. In another
embodiment, the CAR T-cell
targets CD171, and has a therapeutic effect on subjects with renal
neuroblastoma. In another
embodiment, the CAR T-cell targets NCAM, and has a therapeutic effect on
subjects with
neuroblastoma. In another embodiment, the CAR T-cell targets fetal
acetylcholine receptor, and has
a therapeutic effect on subjects with rhabdomyosarcoma. In another embodiment,
the CAR binds to
one of the target antigens listed in Table 1 of Sadelain et al. (Cancer
Discov. 2013 Apr; 3(4): 388¨
CA 03151815 2022-02-17
37
WO 2021/053667
PCT/IL2020/051011
398), which is incorporated by reference herein in its entirety. In another
embodiment, CAR T-cells
bind to carbohydrate or glycolipid structures.
[0153] In one embodiment the CAR T-cells binds to an angiogenic factor,
thereby targeting tumor
vasculature. In some embodiments, the angiogenic factor is VEGFR2. in another
embodiment, the
angiogenic factor is endoglin. In another embodiment, an angiogenic factor
disclosed herein is
Angiogenin; Angiopoietin-1; Del-1; Fibroblast growth factors: acidic (aFGF)
and basic (bFGF);
Follistatin; Granulocyte colony-stimulating factor (G-CSF); Hepatocyte growth
factor (HGF) /scatter
factor (SF); Interleukin-8 (IL-8); Leptin; Midkine; Placental growth factor;
Platelet-derived
endothelial cell growth factor (PD-ECGF); Platelet-derived growth factor-BB
(PDGF-BB);
Pleiotrophin (PTN); Progranulin; Proliferin; Transforming growth factor-alpha
(TGF-alpha);
Transforming growth factor-beta (TGF-beta); Tumor necrosis factor-alpha (TNF-
alpha); Vascular
endothelial growth factor (VEGF)/vascular permeability factor (VPF). In
another embodiment, an
angiogenic factor is an angiogenic protein. In some embodiments, a growth
factor is an angiogenic
protein. In some embodiments, an angiogenic protein for use in the
compositions and methods
disclosed herein is Fibroblast growth factors (FGF); VEGF; VEGFR and
Neuropilin 1 (NRP-1);
Angiopoietin 1 (Ang 1) and Tie2; Platelet-derived growth factor (PDGF; BB-
homodimer) and
PDGFR; Transforming growth factor-beta (TGF-f3), endoglin and TGF-f3
receptors; monocyte
chemotactic protein-1 (MCP-1); Integrins aVf33, aVf3.5 and a501; VE-cadherin
and CD31; ephrin;
plasminogen activators; plasminogen activator inhibitor-1; Nitric oxide
synthase (NOS) and COX-2;
.. AC133; or Idl/Id3. In some embodiments, an angiogenic protein for use in
the compositions and
methods disclosed herein is an angiopoietin, which in some embodiments, is
Angiopoietin 1,
Angiopoietin 3, Angiopoietin 4 or Angiopoietin 6. In some embodiments,
endoglin is also known as
CD105; EDG; HHT1; ORW; or ORW1. In some embodiments, endoglin is a TGFbeta co-
receptor.
[0154] In another embodiment, the CAR T-cells bind to an antigen associated
with an infectious
agent. In some embodiments, the infectious agent is Mycobacterium
tuberculosis. In some
embodiments, said Mycobacterium tuberculosis associated antigen is: Antigen
85B, Lipoprotein
IpqH, ATP dependent helicase putative, uncharacterized protein Rv0476/MT04941
precursor or
uncharacterized protein R v1334/M T1376 precursor.
[0155] In another embodiment, the CAR T-cells binds to an antibody. In some
embodiments, the
CAR T-cell is an "antibody-coupled T-cell receptor" (ACTR). According to this
embodiment, the
CAR T-cell is a universal CAR T-cell. In another embodiment, the CAR T-cell
having an antibody
receptor is administered before, after, or at the same time as the antibody is
administered and then
binds to the antibody, bringing the T-cell in close proximity to the tumor or
cancer. In another
embodiment, the antibody is directed against a tumor cell antigen. In another
embodiment, the
CA 03151815 2022-02-17
38
WO 2021/053667
PCT/IL2020/051011
antibody is directed against CD20. In another embodiment, the antibody is
rituximab.
[0156] In another embodiment, the antibody is Trastuzumab (Herceptin;
Genentech): humanized
IgG1 , which is directed against ERBB2. In another embodiment, the antibody is
Bevacizumab
(Avastin; Genentech/Roche): humanized IgG1 , which is directed against VEGF.
In another
embodiment, the antibody is Cetuximab (Erbitux; Bristol-Myers Squibb):
chimeric human¨murine
IgG1 , which is directed against EGFR. In another embodiment, the antibody is
Panitumumab
(Vectibix; Amgen): human IgG2, which is directed against EGFR. In another
embodiment, the
antibody is Ipilimumab (Yervoy; Bristol-Myers Squibb): IgG1 , which is
directed against CTLA4.
[0157] In another embodiment, the antibody is Alemtuzumab (Campath; Genzyme):
humanized
IgGl, which is directed against CD52. In another embodiment, the antibody is
Ofatumumab (Arzerra;
Genmab): human IgG1 , which is directed against CD20. In another embodiment,
the antibody is
Gemtuzumab ozogamicin (Mylotarg; Wyeth): humanized IgG4, which is directed
against CD33. In
another embodiment, the antibody is Brentuximab vedotin (Adcetris; Seattle
Genetics): chimeric
IgG1 , which is directed against CD30. In another embodiment, the antibody is
90Y-labelled
ibritumomab tiuxetan (Zevalin; IDEC Pharmaceuticals): murine IgG 1 , which is
directed against
CD20. In another embodiment, the antibody is 131I-labelled to situmomab
(Bexxar;
GlaxoSmithKline): murine IgG2, which is directed against CD20.
[0158] In another embodiment, the antibody is Ramucirumab, which is directed
against vascular
endothelial growth factor receptor-2 (VEGFR-2). In another embodiment, the
antibody is
ramucirumab (Cyramza Injection, Eli Lilly and Company), blinatumomab
(BLINCYTO, Amgen
Inc.), pembrolizumab (KEYTRUDA, Merck Sharp & Dohme Corp.), obinutuzumab
(GAZYVA,
Genentech, Inc.; previously known as GA101), pertuzumab injection (PERJETA,
Genentech, Inc.),
or denosumab (Xgeva, Amgen Inc.). In another embodiment, the antibody is
Basiliximab (Simulect;
Novartis). In another embodiment, the antibody is Daclizumab (Zenapax; Roche).
[0159] In another embodiment, the antibody to which the CAR T-cell is coupled
is directed to a tumor
or cancer antigen or a fragment thereof, that is described herein and/or that
is known in the art. In
another embodiment, the antibody to which the CAR T-cell is couples is
directed to a tumor-
associated antigen. In another embodiment, the antibody to which the CAR T-
cell is couples is
directed to a tumor-associated antigen or a fragment thereof that is an
angiogenic factor.
[0160] In another embodiment, the antibody to which the CAR T-cell is coupled
is directed to a tumor
or cancer antigen or a fragment thereof, that is described herein and/or that
is known in the art.
[0161] In some embodiments, antibodies described herein may be used in
combination with
compositions described herein, for example but not limited to a composition
comprising CAR-T cells
or early apoptotic cells, or any combination thereof.
CA 03151815 2022-02-17
39
WO 2021/053667
PCT/IL2020/051011
Cytokine Storm and Cytokine Release Syndrome
[0162] In one embodiment, a method as disclosed herein includes providing
immune cells, such as
NK cells, dendritic cells, TCR T-cells, or T-cells comprising engineered
chimeric antigen receptors
(CAR T-cells), with at least an additional agent to decrease toxic cytokine
release or "cytokine release
syndrome" (CRS) or "severe cytokine release syndrome" (sCRS) or "cytokine
storm" that may occur
in the subject. In another embodiment the CRS, sCRS or cytokine storm occurs
as a result of
administration of the immune cells. In another embodiment, the CRS, sCRS or
cytokine storm is the
result of a stimulus, condition, or syndrome separate from the immune cells
(see below). In another
embodiment, a cytokine storm, cytokine cascade, or hypercytokinemia is a more
severe form of
cytokine release syndrome.
[0163] In one embodiment, the additional agent for decreasing harmful cytokine
release comprises
apoptotic cells or a composition comprising said apoptotic cells. In another
embodiment, the
additional agent for decreasing harmful cytokine release comprises an
apoptotic cell supernatant or a
composition comprising said supernatant. In another embodiment, the additional
agent for decreasing
harmful cytokine release comprises a CTLA-4 blocking agent. In another
embodiment, the additional
agent for decreasing harmful cytokine release comprises apoptotic cells or
apoptotic cell supernatants
or compositions thereof, and a CTLA-4 blocking agent. In another embodiment,
the additional agent
for decreasing harmful cytokine release comprises an alpha-1 anti-tryp sin or
fragment thereof or
analogue thereof. In another embodiment, the additional agent for decreasing
harmful cytokine
release comprises apoptotic cells or apoptotic cell supernatants or
compositions thereof, and an alpha-
1 anti-tryp sin or fragment thereof or analogue thereof. In another
embodiment, the additional agent
for decreasing harmful cytokine release comprises a tellurium-based compound.
In another
embodiment, the additional agent for decreasing harmful cytokine release
comprises apoptotic cells
or apoptotic cell supernatants or compositions thereof, and a tellurium-based
compound. In another
embodiment, the additional agent for decreasing harmful cytokine release
comprises an immune
modulating agent. In another embodiment, the additional agent for decreasing
harmful cytokine
release comprises apoptotic cells or apoptotic cell supernatants or
compositions thereof, and an
immune modulating agent. In another embodiment, the additional agent for
decreasing harmful
cytokine release comprises Treg cells. In another embodiment, the additional
agent for decreasing
harmful cytokine release comprises apoptotic cells or apoptotic cell
supernatants or compositions
thereof, and Treg cells.
[0164] A skilled artisan would appreciate that decreasing toxic cytokine
release or toxic cytokine
levels comprises decreasing or inhibiting production of toxic cytokine levels
in a subject, or inhibiting
or reducing the incidence of cytokine release syndrome or a cytokine storm in
a subject. In another
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
embodiment toxic cytokine levels are reduced during CRS or a cytokine storm.
In another
embodiment, decreasing or inhibiting the production of toxic cytokine levels
comprises treating CRS
or a cytokine storm. In another embodiment, decreasing or inhibiting the
production of toxic cytokine
levels comprises preventing CRS or a cytokine storm. In another embodiment,
decreasing or
5 inhibiting the production of toxic cytokine levels comprises alleviating
CRS or a cytokine storm. In
another embodiment, decreasing or inhibiting the production of toxic cytokine
levels comprises
ameliorating CRS or a cytokine storm. In another embodiment, the toxic
cytokines comprise pro-
inflammatory cytokines. In another embodiment, pro-inflammatory cytokines
comprise IL-6. In
another embodiment, pro-inflammatory cytokines comprise IL-1(3. In another
embodiment, pro-
10 inflammatory cytokines comprise TNF-a. In another embodiment, pro-
inflammatory cytokines
comprise IL-6, IL-113, or TNF-a, or any combination thereof.
[0165] In one embodiment, cytokine release syndrome is characterized by
elevated levels of several
inflammatory cytokines and adverse physical reactions in a subject such as low
blood pressure, high
fever and shivering. In another embodiment, inflammatory cytokines comprise IL-
6, IL-113, and TNF-
15 a. In another embodiment, CRS is characterized by elevated levels of IL-
6, IL-1(3, or TNF-a, or any
combination thereof. In another embodiment, CRS is characterized by elevated
levels of IL-8, or IL-
13, or any combination thereof. In another embodiment, a cytokine storm is
characterized by increases
in TNF-alpha, IFN-gamma, IL- lbeta, IL-2, IL-6, IL-8, IL-10, IL-13, GM-CSF, IL-
5, fracktalkine, or
a combination thereof or a subset thereof. In yet another embodiment, IL-6
comprises a marker of
20 CRS or cytokine storm.
[0166] In another embodiment, cytokines increased in CRS or a cytokine storm
in humans and mice
may comprise any combination of cytokines listed in Tables 1 and 2 below.
[0167] Table 1: Panel of Cytokines Increased in CRS or Cytokine Storm in
Humans and/or
Mice
Human Mouse model (pre-clinical)
Cytokine model Cells secreting this
Notes /
CAR-T Mouse Not
(Analyte) (clinical cytokine
other
(H) origin origin specified
trials)
Flt-3L DC (?)
APC, Endothelial cells (?) =
CX3CL1,
Fractalkine
Neurotactin
(Mouse)
M-CSF = CSF1
GM-CSF * (in vitro) T cell, MO
IFN-a T cell, MO, Monocyte
CA 03151815 2022-02-17
41
WO 2021/053667
PCT/IL2020/051011
IFN-13 T cell, MO, Monocyte
cytotoxic T cells, helper T
IFN-y * (in vitro) cells, NK cells, MO,
Monocyte, DC
IL- 1 a Monocyte, MO, Epithel
Macrophages, DCs,
IL- 113 fibroblasts, endothelial
cells, hepatocytes
IL- 1 Ra
IL- 2 * (in vitro) T cells
IL- 2Ra lymphocytes
IL- 4 * (in vitro) Th2 cells
IL-5 T cells
nnonocytes/ macrophages,
dendritic cells, T cells,
fibroblasts, keratinocytes,
IL- 6 endothelial cells,
adipocytes, nnyocytes,
nnesangial cells, and
osteoblasts
IL- 7 In vitro by BM stronnal
cells
IL- 8 Macrophages, nnonocytes
IL -9 T cells, T helper
nnonocytes/nnacrophages,
mast cells, B cells,
IL- 10 * (in vitro)
regulatory T cells, and
helper T cells
MO, Monocyte, DC,
= p70
IL- 12 activated lymphocytes,
(p4O+p35)
neutrophils
IL-13 T cells
[0168] In some embodiments, cytokines F1t-3L, Fractalkine, GM-CSF, IFN-y, IL-
113, IL-2, IL-2Ra,
IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, and IL-13 of Table 1 are
considered to be significant
in CRS or cytokine storm. In another embodiment, IFN-a, IFN-(3, IL-1, and IL-
1Ra of Table 1 appear
to be important in CRS or cytokine storm. In another embodiment, M-CSF has
unknown importance.
In another embodiment, any cytokine listed in Table 1, or combination thereof,
may be used as a
CA 03151815 2022-02-17
42
WO 2021/053667
PCT/IL2020/051011
marker of CRS or cytokine storm.
[0169] Table 2: Panel of Cytokines Increased in CRS or Cytokine Storm in
Humans and/or
Mice
Human model Mouse model (pre-clinical)
Cytokine
(clinical CAR-T (H) Mouse Not Cells secreting this
cytokine Notes / other
(Analyte)
trials) origin origin specified
IL- 15 * * Fibroblasts, monocytes
(7) 22
IL- 17 * * T cells
IL- 18 Macrophages
IL- 21 * T helper cells, NK cells
IL- 22 * activated DC and T cells
IL- 23
IL- 25
Protective?
IL-27 * APC
IP-10 * Monocytes (7)
Endothel, fibroblast, epithel,
MCP-1 * =
CXCL10
monocytes
MCP-3 * PBMCs, MO (7) =
CCL2
MIP-1a * * (in vitro) T
cells = CXCL9
MIP-1f3 * T cells =
CCL3
platelets, endothelial cells,
PAF 7 neutrophils, monocytes,
and = CCL4
macrophages, mesangial cells
Gastrointestinal mucosa and
PGE2 * *
other
RANTES * Monocytes
MO, lymphocytes, endothel,
TGF-f3 * * =
CCL5
platelets ...
TNF-a * * * * (in vitro) Macrophages, NK
cells, T cells
TNF-aR1 *
HGF
T cell chemoattractant, induced
MIG *
by IFN-y
[0170] In one embodiment, IL-15, IL-17, IL-18, IL-21, IL-22, IP-10, MCP-1, MIP-
la, MIP-113, and
TNF-a of Table 2 are considered to be significant in CRS or cytokine storm. In
another embodiment,
IL-27, MCP-3, PGE2, RANTES, TGF-(3, TNF-aRl, and MIG of Table 2 appear to be
important in
CRS or cytokine storm. In another embodiment, IL-23 and IL-25 have unknown
importance. In
another embodiment, any cytokine listed in Table 2, or combination thereof,
may be used as a marker
of CRS or cytokine storm. In another embodiment, mouse cytokines IL-10, IL-
113, IL-2, IP-10, IL-4,
IL-5, IL-6, IFNa, IL-9, IL-13, IFN-y, IL-12p70, GM-CSF, TNF-a, MIP- la, MIP-
113, IL-17A, IL-
CA 03151815 2022-02-17
43
WO 2021/053667
PCT/IL2020/051011
15/IL-15R and IL-7 appear to be important in CRS or cytokine storm.
[0171] A skilled artisan would appreciate that the term "cytokine" may
encompass cytokines (e.g.,
interferon gamma (IFN-y), granulocyte macrophage colony stimulating factor,
tumor necrosis factor
alpha), chemokines (e.g., MIP 1 alpha, MIP 1 beta, RANTES), and other soluble
mediators of
.. inflammation, such as reactive oxygen species and nitric oxide.
[0172] In one embodiment, increased release of a particular cytokine, whether
significant, important
or having unknown importance, does not a priori mean that the particular
cytokine is part of a
cytokine storm. In one embodiment, an increase of at least one cytokine is not
the result of a cytokine
storm or CRS. In another embodiment, CAR T-cells may be the source of
increased levels of a
particular cytokine or group of cytokines.
[0173] In another embodiment, cytokine release syndrome is characterized by
any or all of the
following symptoms: Fever with or without rigors, malaise, fatigue, anorexia,
myalgias, arthalgias,
nausea, vomiting, headache Skin Rash, Nausea, vomiting, diarrhea, Tachypnea,
hypoxemia
Cardiovascular Tachycardia, widened pulse pressure, hypotension, increased
cardiac output (early),
potentially diminished cardiac output (late), Elevated D-dimer,
hypofibrinogenemia with or without
bleeding, Azotemia Hepatic Transaminitis, hyperbilirubinemia, Headache, mental
status changes,
confusion, delirium, word finding difficulty or frank aphasia, hallucinations,
tremor, dymetria, altered
gait, seizures. In another embodiment, a cytokine storm is characterized by IL-
2 release and
lymphoproliferation. In another embodiment, a cytokine storm is characterized
by increases in
cytokines released by CAR T-cells. In another embodiment, a cytokine storm is
characterized by
increases in cytokines released by cells other than CAR T-cells.
[0174] In another embodiment, cytokine storm leads to potentially life-
threatening complications
including cardiac dysfunction, adult respiratory distress syndrome, neurologic
toxicity, renal and/or
hepatic failure, and disseminated intravascular coagulation.
[0175] A skilled artisan would appreciate that the characteristics of a
cytokine release syndrome
(CRS) or cytokine storm are estimated to occur a few days to several weeks
following the trigger for
the CRS or cytokine storm. In one embodiment, CAR T-cells are a trigger for
CRS or a cytokine
storm. In another embodiment, a trigger for CRS or a cytokine storm is not CAR
T-cells.
[0176] In one embodiment, measurement of cytokine levels or concentration, as
an indicator of
cytokine storm, may be expressed as ¨fold increase, per cent (%) increase, net
increase or rate of
change in cytokine levels or concentration. In another embodiment, absolute
cytokine levels or
concentrations above a certain level or concentration may be an indication of
a subject undergoing or
about to experience a cytokine storm. In another embodiment, absolute cytokine
levels or
concentration at a certain level or concentration, for example a level or
concentration normally found
CA 03151815 2022-02-17
44
WO 2021/053667
PCT/IL2020/051011
in a control subject not undergoing CAR-T cell therapy, may be an indication
of a method for
inhibiting or reducing the incidence of a cytokine storm in a subject
undergoing CAR T-cell.
[0177] A skilled artisan would appreciate that the term "cytokine level" may
encompass a measure
of concentration, a measure of fold change, a measure of percent (%) change,
or a measure of rate
change. Further, the methods for measuring cytokines in blood, saliva, serum,
urine, and plasma are
well known in the art.
[0178] In one embodiment, despite the recognition that cytokine storm is
associated with elevation
of several inflammatory cytokines, IL-6 levels may be used as a common measure
of cytokine storm
and/or as a common measure of the effectiveness of a treatment for cytokine
storms. A skilled artisan
would appreciate that other cytokines may be used as markers of a cytokine
storm, for example TNF-
a, IB-la, IL-8, IL-13, or INF-y. Further, that assay methods for measuring
cytokines are well known
in the art. A skilled artisan would appreciate that methods affecting a
cytokine storm may similarly
affect cytokine release syndrome.
[0179] In one embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or a cytokine
storm. In another
embodiment, disclosed herein is a method of decreasing or inhibiting cytokine
production in a subject
vulnerable to experiencing cytokine release syndrome or a cytokine storm. In
another embodiment,
methods disclosed herein decrease or inhibit cytokine production in a subject
experiencing cytokine
release syndrome or a cytokine storm, wherein production of any cytokine or
group of cytokines
listed in Tables 1 and/or 2 is decreased or inhibited. In another embodiment,
cytokine IL-6 production
is decreased or inhibited. In another embodiment, cytokine IL-betal production
is decreased or
inhibited. In another embodiment, cytokine IL-8 production is decreased or
inhibited. In another
embodiment, cytokine IL-13 production is decreased or inhibited. In another
embodiment, cytokine
TNF-alpha production is decreased or inhibited. In another embodiment,
cytokines IL-6 production,
IL- lbeta production, or TNF-alpha production, or any combination thereof is
decreased or inhibited.
[0180] In one embodiment, cytokine release syndrome is graded. In another
embodiment, Grade 1
describes cytokine release syndrome in which symptoms are not life threatening
and require
symptomatic treatment only, e.g., fever, nausea, fatigue, headache, myalgias,
malaise. In another
embodiment, Grade 2 symptoms require and respond to moderate intervention,
such as oxygen, fluids
or vasopressor for hypotension. In another embodiment, Grade 3 symptoms
require and respond to
aggressive intervention. In another embodiment, Grade 4 symptoms are life-
threatening symptoms
and require ventilator and patients display organ toxicity.
[0181] In another embodiment, a cytokine storm is characterized by IL-6 and
interferon gamma
release. In another embodiment, a cytokine storm is characterized by release
of any cytokine or
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
combination thereof, listed in Tables 1 and 2. In another embodiment, a
cytokine storm is
characterized by release of any cytokine or combination thereof, known in the
art.
[0182] In one embodiment, symptoms onset begins minutes to hours after the
infusion begins. In
another embodiment, symptoms coincide with peak cytokine levels.
5 .. [0183] In one embodiment, a method of inhibiting or reducing the
incidence of a cytokine release
syndrome (CRS) or a cytokine storm in a subject undergoing CAR T-cell cancer
therapy comprises
administering an apoptotic cell population or an apoptotic cell supernatant or
compositions thereof.
In another embodiment, the apoptotic cell population or an apoptotic cell
supernatant or compositions
thereof may aid the CAR T-cell therapy. In another embodiment, the apoptotic
cell population or an
10 apoptotic cell supernatant or compositions thereof may aid in the
inhibition or reducing the incidence
of the CRS or cytokine storm. In another embodiment, the apoptotic cell
population or an apoptotic
cell supernatant or compositions thereof may aid in treating the CRS or
cytokine storm. In another
embodiment, the apoptotic cell population or an apoptotic cell supernatant or
compositions thereof
may aid in preventing the CRS or cytokine storm. In another embodiment, the
apoptotic cell
15 .. population or an apoptotic cell supernatant or compositions thereof may
aid in ameliorating the CRS
or cytokine storm. In another embodiment, the apoptotic cell population or an
apoptotic cell
supernatant or compositions thereof may aid in alleviating the CRS or cytokine
storm.
[0184] In one embodiment, a method of inhibiting or reducing the incidence of
a cytokine release
syndrome (CRS) or a cytokine storm in a subject undergoing CAR T-cell cancer
therapy, and being
20 administered an apoptotic cell population or an apoptotic cell
supernatant or compositions thereof,
comprises administering an additional agent. In another embodiment, the
additional agent may aid
the CAR T-cell therapy. In another embodiment, the additional agent may aid in
the inhibition or
reducing the incidence of the CRS or cytokine storm. In another embodiment,
the additional agent
may aid in treating the CRS or cytokine storm. In another embodiment, the
additional agent may aid
25 in preventing the CRS or cytokine storm. In another embodiment, the
additional agent may aid in
ameliorating the CRS or cytokine storm. In another embodiment, the additional
agent may aid in
alleviating the CRS or cytokine storm.
[0185] In one embodiment, a method of inhibiting or reducing the incidence of
a cytokine release
syndrome (CRS) or a cytokine storm in a subject undergoing CAR T-cell cancer
therapy comprises
30 administering an additional agent. In another embodiment, the additional
agent may aid the CAR T-
cell therapy. In one embodiment, a method of inhibiting or reducing the
incidence of a cytokine
release syndrome (CRS) or a cytokine storm in a subject undergoing TCR T-cell
cancer therapy
comprises administering an additional agent. In another embodiment, the
additional agent may aid
the TCR T-cell therapy. In one embodiment, a method of inhibiting or reducing
the incidence of a
CA 03151815 2022-02-17
46
WO 2021/053667
PCT/IL2020/051011
cytokine release syndrome (CRS) or a cytokine storm in a subject undergoing
comprises
administering an additional agent. In another embodiment, the additional agent
may aid the . In one
embodiment, a method of inhibiting or reducing the incidence of a cytokine
release syndrome (CRS)
or a cytokine storm in a subject undergoing NK cell therapy comprises
administering an additional
agent. In another embodiment, the additional agent may aid the NK cell
therapy.
[0186] In another embodiment, the additional agent may aid in the inhibition
or reducing the
incidence of the CRS or cytokine storm. In another embodiment, the additional
agent may aid in
treating the CRS or cytokine storm. In another embodiment, the additional
agent may aid in
preventing the CRS or cytokine storm. In another embodiment, the additional
agent may aid in
ameliorating the CRS or cytokine storm. In another embodiment, the additional
agent may aid in
alleviating the CRS or cytokine storm.
[0187] In one embodiment, the additional agent for decreasing harmful cytokine
release comprises
apoptotic cells or a composition comprising said apoptotic cells. In another
embodiment, the
additional agent for decreasing harmful cytokine release comprises an
apoptotic cell supernatant or a
.. composition comprising said supernatant. In another embodiment, the
additional agent for decreasing
harmful cytokine release comprises a CTLA-4 blocking agent. In another
embodiment, the additional
agent for decreasing harmful cytokine release comprises apoptotic cells or
apoptotic cell supernatants
or compositions thereof, and a CTLA-4 blocking agent. In another embodiment,
the additional agent
for decreasing harmful cytokine release comprises an alpha-1 anti-tryp sin or
fragment thereof or
analogue thereof. In another embodiment, the additional agent for decreasing
harmful cytokine
release comprises apoptotic cells or apoptotic cell supernatants or
compositions thereof, and an alpha-
1 anti-tryp sin or fragment thereof or analogue thereof. In another
embodiment, the additional agent
for decreasing harmful cytokine release comprises a tellurium-based compound.
In another
embodiment, the additional agent for decreasing harmful cytokine release
comprises apoptotic cells
or apoptotic cell supernatants or compositions thereof, and a tellurium-based
compound. In another
embodiment, the additional agent for decreasing harmful cytokine release
comprises an immune
modulating agent. In another embodiment, the additional agent for decreasing
harmful cytokine
release comprises apoptotic cells or apoptotic cell supernatants or
compositions thereof, and an
immune modulating agent.
[0188] In another embodiment, compositions and methods as disclosed herein
utilize combination
therapy of CAR T-cells with one or more CTLA-4-blocking agents such as
Ipilimumab. In another
embodiment, compositions and methods as disclosed herein utilize combined
therapy comprising
apoptotic cells, CAR T-cells, and one or more CTLA-4-blocking agents. In
another embodiment,
compositions and methods as disclosed herein utilize combination therapy of
TCR T-cells with one
CA 03151815 2022-02-17
47
WO 2021/053667
PCT/IL2020/051011
or more CTLA-4-blocking agents such as Ipilimumab. In another embodiment,
compositions and
methods as disclosed herein utilize combined therapy comprising apoptotic
cells, TCR T-cells, and
one or more CTLA-4-blocking agents. In another embodiment, compositions and
methods as
disclosed herein utilize combination therapy of dendritic cells with one or
more CTLA-4-blocking
agents such as Ipilimumab. In another embodiment, compositions and methods as
disclosed herein
utilize combined therapy comprising apoptotic cells, dendritic cells, and one
or more CTLA-4-
blocking agents. In another embodiment, compositions and methods as disclosed
herein utilize
combination therapy of NK cells with one or more CTLA-4-blocking agents such
as Ipilimumab. In
another embodiment, compositions and methods as disclosed herein utilize
combined therapy
comprising apoptotic cells, NK cells, and one or more CTLA-4-blocking agents.
[0189] In another embodiment, CTLA-4 is a potent inhibitor of T-cell
activation that helps to
maintain self-tolerance. In another embodiment, administration of an anti-CTLA-
4 blocking agent,
which in another embodiment, is an antibody, produces a net effect of T-cell
activation.
[0190] In another embodiment, other toxicities resulting from CAR T-cell, TCR
T-cell, dendritic
cell, or NK cell administration that may be treated, prevented, inhibited,
ameliorated, reduced in
incidence or alleviated by the compositions and methods as disclosed herein
comprise B cell aplasia
or tumor lysis syndrome (TLS).
[0191] In one embodiment, a method of inhibiting or reducing the incidence of
a cytokine release
syndrome (CRS) or a cytokine storm in a subject undergoing CAR T-cell cancer
therapy does not
affect the efficacy of the CAR T-cell therapy. In another embodiment, a method
of inhibiting or
reducing the incidence of CRS or a cytokine storm in a subject undergoing CAR
T-cell cancer
therapy, does reduce the efficacy of the CAR T-cells therapy by more than
about 5%. In another
embodiment, a method of inhibiting or reducing the incidence of CRS or a
cytokine storm in a subject
undergoing CAR T-cell cancer therapy, does reduce the efficacy of the CAR T-
cells therapy by more
than about 10%. In another embodiment, a method of inhibiting or reducing the
incidence of CRS or
a cytokine storm in a subject undergoing CAR T-cell cancer therapy, does
reduce the efficacy of the
CAR T-cells therapy by more than about 15%. In another embodiment, a method of
inhibiting or
reducing the incidence of CRS or a cytokine storm in a subject undergoing CAR
T-cell cancer
therapy, does reduce the efficacy of the CAR T-cells therapy by more than
about 20%. In another
embodiment, a method of inhibiting or reducing the incidence of CRS or a
cytokine storm in a subject
undergoing CAR T-cell cancer therapy, increases the efficacy of the CAR T-
cells therapy by more
than about 5%. In another embodiment, a method of inhibiting or reducing the
incidence of CRS or
a cytokine storm in a subject undergoing CAR T-cell cancer therapy, increases
the efficacy of the
CAR T-cells therapy by more than about 10%. In another embodiment, a method of
inhibiting or
CA 03151815 2022-02-17
48
WO 2021/053667
PCT/IL2020/051011
reducing the incidence of CRS or a cytokine storm in a subject undergoing CAR
T-cell cancer
therapy, increases the efficacy of the CAR T-cells therapy by more than about
15%. In another
embodiment, a method of inhibiting or reducing the incidence of CRS or a
cytokine storm in a subject
undergoing CAR T-cell cancer therapy, increases the efficacy of the CAR T-
cells therapy by more
than about 20%.
[0192] Any appropriate method of quantifying cytotoxicity can be used to
determine whether activity
in an immune cell modified to express a CAR remains substantially unchanged.
For example,
cytotoxicity can be quantified using a cell culture-based assay such as the
cytotoxic assays described
in the Examples. Cytotoxicity assays can employ dyes that preferentially stain
the DNA of dead cells.
In other cases, fluorescent and luminescent assays that measure the relative
number of live and dead
cells in a cell population can be used. For such assays, protease activities
serve as markers for cell
viability and cell toxicity, and a labeled cell permeable peptide generates
fluorescent signals that are
proportional to the number of viable cells in the sample. For example, a
cytotoxicity assay may use
7-AAD in a flow cytometry analysis. Kits for various cytotoxicity assays are
commercially available
from manufacturers such as Promega, Abcam, and Life Technologies.
[0193] In another embodiment, a measure of cytotoxicity may be qualitative. In
another embodiment,
a measure of cytotoxicity may be quantitative. In a further embodiment a
measure of cytotoxicity
may be related to the change in expression of a cytotoxic cytokine. In another
embodiment, a measure
of cytotoxicity may be determined by survival curve and tumor load in bone
marrow and liver.
[0194] In one embodiment, the methods as disclosed herein comprise an
additional step that is useful
in overcoming rejection of allogeneic donor cells. In one embodiment, the
methods comprise the step
of full or partial lymphodepletion prior to administration of the CAR T-cells,
which in one
embodiment, are allogeneic CAR T-cells. In another embodiment, the
lymphodepletion is adjusted
so that it delays the host versus graft reaction for a period sufficient to
allow said allogeneic T-cells
to attack the tumor to which they are directed, but to an extent insufficient
to require rescue of the
host immune system by bone marrow transplantation. In another embodiment,
agents that delay
egression of the allogeneic T-cells from lymph nodes, such as 2-amino-2-[2-(4-
octylphenyl)ethyl] prop ane-1,3 -diol (FTY720), 5[4-pheny1-5-
(trifluoromethyl)thiophen-2-yl] -3 43 -
(trifluoromethyl)pheny- 1] 1,2,4-oxadiazole
(SEW2871), 3 -(2-(-hexylphenylamino)-2-
oxoethylamino)propanoic acid (W123), 2-ammonio-4-(2-chloro-4-(3-
phenoxyphenylthio)pheny1)-
2-(hydroxymethyl)but-y1 hydrogen phosphate (KRP-203 phosphate) or other agents
known in the art,
may be used as part of the compositions and methods as disclosed herein to
allow the use of allogeneic
CAR T-cells having efficacy and lacking initiation of graft vs host disease.
In one embodiment, MHC
expression by the allogeneic T-cells is silenced to reduce the rejection of
the allogeneic cells. In
CA 03151815 2022-02-17
49
WO 2021/053667
PCT/IL2020/051011
another embodiment, the apoptotic cells prevent rejection of the allogeneic
cells.
Cytokine Release Associated with CAR T-cell Therapy
[0195] In one embodiment, cytokine release occurs between a few days to 2
weeks after
administration of immune therapy such as CAR T-cell therapy. In one
embodiment, hypotension and
other symptoms follow the cytokine release, i.e. from few days to few weeks.
Therefore, in one
embodiment, apoptotic cells or the apoptotic cell supernatant are administered
to subjects at the same
time as immune therapy as prophylaxis. In another embodiment, apoptotic cells
or supernatant are
administered to subjects 2-3 days after administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 7 days after
administration of immune
therapy. In another embodiment, apoptotic cells or supernatant are
administered to subjects 10 days
after administration of immune therapy. In another embodiment, apoptotic cells
or supernatant are
administered to subjects 14 days after administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 2-14 days after
administration of immune
therapy.
[0196] In another embodiment, apoptotic cells or apoptotic cell supernatant
are administered to
subjects 2-3 hours after administration of immune therapy. In another
embodiment, apoptotic cells
or supernatant are administered to subjects 7 hours after administration of
immune therapy. In another
embodiment, apoptotic cells or supernatant are administered to subjects 10
hours after administration
of immune therapy. In another embodiment, apoptotic cells or supernatant are
administered to
subjects 14 hours after administration of immune therapy. In another
embodiment, apoptotic cells or
supernatant are administered to subjects 2-14 hours after administration of
immune therapy.
[0197] In an alternative embodiment, apoptotic cells or the apoptotic cell
supernatant are
administered to subjects prior to immune therapy as prophylaxis. In another
embodiment, apoptotic
cells or supernatant are administered to subjects 1 day before administration
of immune therapy. In
another embodiment, apoptotic cells or supernatant are administered to
subjects 2-3 days before
administration of immune therapy. In another embodiment, apoptotic cells or
supernatant are
administered to subjects 7 days before administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 10 days before
administration of immune
therapy. In another embodiment, apoptotic cells or supernatant are
administered to subjects 14 days
before administration of immune therapy. In another embodiment, apoptotic
cells or supernatant are
administered to subjects 2-14 days before administration of immune therapy.
[0198] In another embodiment, apoptotic cells or apoptotic cell supernatant
are administered to
subjects 2-3 hours before administration of immune therapy. In another
embodiment, apoptotic cells
or supernatant are administered to subjects 7 hours before administration of
immune therapy. In
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
another embodiment, apoptotic cells or supernatant are administered to
subjects 10 hours before
administration of immune therapy. In another embodiment, apoptotic cells or
supernatant are
administered to subjects 14 hours before administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 2-14 hours before
administration of
5 immune therapy.
[0199] In another embodiment, apoptotic cells or apoptotic cell supernatant
may be administered
therapeutically, once cytokine release syndrome has occurred. In one
embodiment, apoptotic cells or
supernatant may be administered once cytokine release leading up to or
attesting to the beginning of
cytokine release syndrome is detected. In one embodiment, apoptotic cells or
supernatant can
10 terminate the increased cytokine levels, or the cytokine release
syndrome, and avoid its sequelae.
[0200] In another embodiment, apoptotic cells or apoptotic cell supernatant
may be administered
therapeutically, at multiple time points. In another embodiment,
administration of apoptotic cells or
apoptotic cell supernatant is at least at two time points described herein. In
another embodiment,
administration of apoptotic cells or apoptotic cell supernatant is at least at
three time points described
15 herein. In another embodiment, administration of apoptotic cells or
apoptotic cell supernatant is prior
to CRS or a cytokine storm, and once cytokine release syndrome has occurred,
and any combination
thereof.
[0201] In one embodiment, the chimeric antigen receptor-expressing T-cell (CAR
T-cell) therapy
and the apoptotic cell therapy or supernatant are administered together. In
another embodiment, the
20 CAR T-cell therapy is administered after the apoptotic cell therapy or
supernatant. In another
embodiment, the CAR T-cell therapy is administered prior to the apoptotic cell
therapy or
supernatant. According to this aspect and in one embodiment, apoptotic cell
therapy or supernatant
is administered approximately 2-3 weeks after the CAR T-cell therapy. In
another embodiment,
apoptotic cell therapy or supernatant is administered approximately 6-7 weeks
after the CAR T-cell
25 therapy. In another embodiment, apoptotic cell therapy or supernatant is
administered approximately
9 weeks after the CAR T-cell therapy. In another embodiment, apoptotic cell
therapy is administered
up to several months after CAR T-cell therapy.
[0202] Therefore, in one embodiment, apoptotic cells or the apoptotic cell
supernatant are
administered to subjects at the same time as immune therapy as prophylaxis. In
another embodiment,
30 apoptotic cells or supernatant are administered to subjects 2-3 days
after administration of immune
therapy. In another embodiment, apoptotic cells or supernatant are
administered to subjects 7 days
after administration of immune therapy. In another embodiment, apoptotic cells
or supernatant are
administered to subjects 10 days after administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 14 days after
administration of immune
CA 03151815 2022-02-17
51
WO 2021/053667
PCT/IL2020/051011
therapy. In another embodiment, apoptotic cells or supernatant are
administered to subjects 2-14 days
after administration of immune therapy.
[0203] In another embodiment, apoptotic cells or apoptotic cell supernatant
are administered to
subjects 2-3 hours after administration of immune therapy. In another
embodiment, apoptotic cells
or supernatant are administered to subjects 7 hours after administration of
immune therapy. In another
embodiment, apoptotic cells or supernatant are administered to subjects 10
hours after administration
of immune therapy. In another embodiment, apoptotic cells or supernatant are
administered to
subjects 14 hours after administration of immune therapy. In another
embodiment, apoptotic cells or
supernatant are administered to subjects 2-14 hours after administration of
immune therapy.
[0204] In an alternative embodiment, apoptotic cells or the apoptotic cell
supernatant are
administered to subjects prior to immune therapy as prophylaxis. In another
embodiment, apoptotic
cells or supernatant are administered to subjects 1 day before administration
of immune therapy. In
another embodiment, apoptotic cells or supernatant are administered to
subjects 2-3 days before
administration of immune therapy. In another embodiment, apoptotic cells or
supernatant are
administered to subjects 7 days before administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 10 days before
administration of immune
therapy. In another embodiment, apoptotic cells or supernatant are
administered to subjects 14 days
before administration of immune therapy. In another embodiment, apoptotic
cells or supernatant are
administered to subjects 2-14 days before administration of immune therapy.
[0205] In another embodiment, apoptotic cells or apoptotic cell supernatant
are administered to
subjects 2-3 hours before administration of immune therapy. In another
embodiment, apoptotic cells
or supernatant are administered to subjects 7 hours before administration of
immune therapy. In
another embodiment, apoptotic cells or supernatant are administered to
subjects 10 hours before
administration of immune therapy. In another embodiment, apoptotic cells or
supernatant are
administered to subjects 14 hours before administration of immune therapy. In
another embodiment,
apoptotic cells or supernatant are administered to subjects 2-14 hours before
administration of
immune therapy.
[0206] In another embodiment, apoptotic cells or apoptotic cell supernatant
may be administered
therapeutically, once cytokine release syndrome has occurred. In one
embodiment, apoptotic cells or
supernatant may be administered once cytokine release leading up to or
attesting to the beginning of
cytokine release syndrome is detected. In one embodiment, apoptotic cells or
supernatant can
terminate the increased cytokine levels, or the cytokine release syndrome, and
avoid its sequelae.
[0207] In another embodiment, apoptotic cells or apoptotic cell supernatant
may be administered
therapeutically, at multiple time points. In another embodiment,
administration of apoptotic cells or
CA 03151815 2022-02-17
52
WO 2021/053667
PCT/IL2020/051011
apoptotic cell supernatant is at least at two time points described herein. In
another embodiment,
administration of apoptotic cells or apoptotic cell supernatant is at least at
three time points described
herein. In another embodiment, administration of apoptotic cells or apoptotic
cell supernatant is prior
to CRS or a cytokine storm, and once cytokine release syndrome has occurred,
and any combination
thereof.
[0208] In one embodiment, the chimeric antigen receptor-expressing T-cell (CAR
T-cell) therapy
and the apoptotic cell therapy or supernatant are administered together. In
another embodiment, the
CAR T-cell therapy is administered after the apoptotic cell therapy or
supernatant. In another
embodiment, the CAR T-cell therapy is administered prior to the apoptotic cell
therapy or
supernatant. According to this aspect and in one embodiment, apoptotic cell
therapy or supernatant
is administered approximately 2-3 weeks after the CAR T-cell therapy. In
another embodiment,
apoptotic cell therapy or supernatant is administered approximately 6-7 weeks
after the CAR T-cell
therapy. In another embodiment, apoptotic cell therapy or supernatant is
administered approximately
9 weeks after the CAR T-cell therapy. In another embodiment, apoptotic cell
therapy is administered
up to several months after CAR T-cell therapy.
[0209] In other embodiments, an additional agent is administered to subjects
at the same time as
immune therapy as prophylaxis. In one embodiment the additional agent
comprises apoptotic cells,
an apoptotic supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or
fragment thereof or
analogue thereof, of a tellurium-based compound, or an immune-modulating
compounds, or any
combination thereof. In another embodiment, the additional agent is
administered to subjects 2-3 days
after administration of immune therapy. In another embodiment, the additional
agent is administered
to subjects 7 days after administration of immune therapy. In another
embodiment, the additional
agent is administered to subjects 10 days after administration of immune
therapy. In another
embodiment, the additional agent is administered to subjects 14 days after
administration of immune
therapy. In another embodiment, the additional agent is administered to
subjects 2-14 days after
administration of immune therapy.
[0210] In another embodiment, the additional agent is administered to subjects
2-3 hours after
administration of immune therapy. In another embodiment, the additional agent
is administered to
subjects 7 hours after administration of immune therapy. In another embodiment
the additional agent
is administered to subjects 10 hours after administration of immune therapy.
In another embodiment,
the additional agent is administered to subjects 14 hours after administration
of immune therapy. In
another embodiment, the additional agent is administered to subjects 2-14
hours after administration
of immune therapy.
[0211] In an alternative embodiment, the additional agent is administered to
subjects prior to immune
CA 03151815 2022-02-17
53
WO 2021/053667
PCT/IL2020/051011
therapy as prophylaxis. In another embodiment, the additional agent is
administered to subjects 1 day
before administration of immune therapy. In another embodiment, the additional
agent is
administered to subjects 2-3 days before administration of immune therapy. In
another embodiment,
the additional agent is administered to subjects 7 days before administration
of immune therapy. In
another embodiment, the additional agent is administered to subjects 10 days
before administration
of immune therapy. In another embodiment, the additional agent is administered
to subjects 14 days
before administration of immune therapy. In another embodiment, the additional
agent is
administered to subjects 2-14 days before administration of immune therapy.
[0212] In another embodiment, the additional agent is administered to subjects
2-3 hours before
administration of immune therapy. In another embodiment, the additional agent
is administered to
subjects 7 hours before administration of immune therapy. In another
embodiment, the additional
agent is administered to subjects 10 hours before administration of immune
therapy. In another
embodiment, the additional agent is administered to subjects 14 hours before
administration of
immune therapy. In another embodiment, the additional agent is administered to
subjects 2-14 hours
before administration of immune therapy.
[0213] In another embodiment, the additional agent is administered
therapeutically, once cytokine
release syndrome has occurred. In one embodiment, the additional agent is
administered once
cytokine release leading up to or attesting to the beginning of cytokine
release syndrome is detected.
In one embodiment, the additional agent can terminate the increased cytokine
levels, or the cytokine
release syndrome, and avoid its sequelae.
[0214] In another embodiment, the additional agent is administered
therapeutically, at multiple time
points. In another embodiment, administration of the additional agent is at
least at two time points
described herein. In another embodiment, administration of the additional
agent is at least at three
time points described herein. In another embodiment, administration of the
additional agent is prior
to CRS or a cytokine storm, and once cytokine release syndrome has occurred,
and any combination
thereof.
[0215] In one embodiment, the chimeric antigen receptor-expressing T-cell (CAR
T-cell) therapy
and the additional agent is administered together. In another embodiment, the
CAR T-cell therapy is
administered the additional agent. In another embodiment, the CAR T-cell
therapy is administered
prior to the additional agent. According to this aspect and in one embodiment,
the additional agent is
administered approximately 2-3 weeks after the CAR T-cell therapy. In another
embodiment, the
additional agent is administered approximately 6-7 weeks after the CAR T-cell
therapy. In another
embodiment, the additional agent is administered approximately 9 weeks after
the CAR T-cell
therapy. In another embodiment, the additional agent is administered up to
several months after CAR
CA 03151815 2022-02-17
54
WO 2021/053667
PCT/IL2020/051011
T-cell therapy.
[0216] In one embodiment, CAR T-cells are heterologous to the subject. In one
embodiment, CAR
T-cells are derived from one or more donors. In one embodiment, CAR T-cells
are derived from one
or more bone marrow donors. In another embodiment, CAR T-cells are derived
from one or more
blood bank donations. In one embodiment, the donors are matched donors. In one
embodiment, CAR
T-cells are universal allogeneic CAR T-cells. In another embodiment, CAR T-
cells are syngeneic
CAR T-cells. In another embodiment, CAR T-cells are from unmatched third-party
donors. In
another embodiment, CAR T-cells are from pooled third-party donor T-cells. In
one embodiment,
the donor is a bone marrow donor. In another embodiment, the donor is a blood
bank donor. In one
embodiment, CAR T-cells of the compositions and methods as disclosed herein
comprise one or
more MHC unrestricted tumor-directed chimeric receptors. In one embodiment,
non-autologous T-
cells may be engineered or administered according to protocols known in the
art to prevent or
minimize autoimmune reactions, such as described in U.S. Patent Application
No. 20130156794,
which is incorporated herein by references in its entirety.
[0217] In another embodiment, CAR T-cells are autologous to the subject. In
one embodiment, the
patient's own cells are used. In this embodiment, if the patient's own cells
are used, then the CAR T-
cell therapy is administered after the apoptotic cell therapy.
[0218] In one embodiment, apoptotic cells are heterologous to the subject. In
one embodiment,
apoptotic cells are derived from one or more donors. In one embodiment,
apoptotic cells are derived
from one or more bone marrow donors. In another embodiment, apoptotic cells
are derived from one
or more blood bank donations. In one embodiment, the donors are matched
donors. In another
embodiment, apoptotic cells are from unmatched third-party donors. In one
embodiment, apoptotic
cells are universal allogeneic apoptotic cells. In another embodiment,
apoptotic cells are from a
syngeneic donor. In another embodiment, apoptotic cells are from pooled third-
party donor cells. In
one embodiment, the donor is a bone marrow donor. In another embodiment, the
donor is a blood
bank donor. In another embodiment, apoptotic cells are autologous to the
subject. In this embodiment,
the patient's own cells are used.
[0219] According to some embodiments, the therapeutic mononuclear-enriched
cell preparation
disclosed herein, or the apoptotic cell supernatant is administered to the
subject systemically. In
.. another embodiment, administration is via the intravenous route.
Alternately, the therapeutic
mononuclear enriched cell or supernatant may be administered to the subject
according to various
other routes, including, but not limited to, the parenteral, intraperitoneal,
intra-articular, intramuscular
and subcutaneous routes.
[0220] According to some embodiments, the therapeutic mononuclear-enriched
cell preparation
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
disclosed herein, or the additional agent is administered to the subject
systemically. In another
embodiment, administration is via the intravenous route. Alternately, the
therapeutic mononuclear
enriched cell or the additional agent may be administered to the subject
according to various other
routes, including, but not limited to, the parenteral, intraperitoneal, intra-
articular, intramuscular and
5 subcutaneous routes.
[0221] In one embodiment, the preparation is administered in a local rather
than systemic manner,
for example, via injection of the preparation directly into a specific region
of a patient's body. In
another embodiment, a specific region comprises a tumor or cancer.
[0222] In another embodiment, the therapeutic mononuclear enriched cells or
supernatant are
10 administered to the subject suspended in a suitable physiological
buffer, such as, but not limited to,
saline solution, PBS, HBSS, and the like. In addition, the suspension medium
may further comprise
supplements conducive to maintaining the viability of the cells. In another
embodiment, the
additional agent is administered to the subject suspended in a suitable
physiological buffer, such as,
but not limited to, saline solution, PBS, HBSS, and the like.
15 [0223] According to some embodiments the pharmaceutical composition is
administered
intravenously. According to another embodiment, the pharmaceutical composition
is administered in
a single dose. According to alternative embodiments the pharmaceutical
composition is administered
in multiple doses. According to another embodiment, the pharmaceutical
composition is administered
in two doses. According to another embodiment, the pharmaceutical composition
is administered in
20 three doses. According to another embodiment, the pharmaceutical
composition is administered in
four doses. According to another embodiment, the pharmaceutical composition is
administered in
five or more doses. According to some embodiments, the pharmaceutical
composition is formulated
for intravenous injection.
[0224] In one embodiment, any appropriate method of providing modified CAR-
expressing immune
25 cells to a subject can be used for methods described herein. In one
embodiment, methods for
providing cells to a subject comprise hematopoietic cell transplantation
(HCT), infusion of donor-
derived NK cells into cancer patients or a combination thereof.
[0225] In another embodiment, disclosed herein is a method of inhibiting or
reducing the incidence
of cytokine release syndrome or cytokine storm in a subject undergoing
chimeric antigen receptor-
30 expressing T-cell (CAR T-cell) therapy, comprising the step of
administering a composition
comprising apoptotic cells to said subject.
[0226] In another embodiment, disclosed herein is a method of inhibiting or
reducing the incidence
of cytokine release syndrome or cytokine storm in a subject undergoing
chimeric antigen receptor-
expressing T-cell (CAR T-cell) therapy, comprising the step of administering
an apoptotic cell
CA 03151815 2022-02-17
56
WO 2021/053667
PCT/IL2020/051011
supernatant, such as an apoptotic cell-phagocyte supernatant, to said subject.
[0227] In another embodiment, disclosed herein is a method of inhibiting or
reducing the incidence
of cytokine release syndrome or cytokine storm in a subject undergoing
chimeric antigen receptor-
expressing T-cell (CAR T-cell) therapy, comprising the step of administering
an at least one
additional agent to said subject.
[0228] In certain embodiments, a CAR T-cell therapy comprises administering a
composition
disclosed herein comprising CAR T-cells and either apoptotic cells or an
apoptotic cell supernatant,
or another or combination of additional agents as disclosed herein. In
alternative embodiments, a
CAR T-cell therapy comprises administering a composition disclosed herein
comprising CAR T-
cells and a composition comprising either apoptotic cells or an apoptotic cell
supernatant, or an
additional agent or combination thereof as disclosed herein.
Cytokine Release Associated with Non-CAR T-cell Applications
[0229] In one embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm, comprising the step of
administering a composition
comprising apoptotic cells or an apoptotic supernatant to said subject,
wherein said administering
decreases or inhibits cytokine production in said subject. In another
embodiment, decrease or
inhibition of cytokine production is compared with a subject experiencing
cytokine release syndrome
or cytokine storm or vulnerable to cytokine release syndrome or cytokine storm
and not administered
apoptotic cells or an apoptotic supernatant. In another embodiment, methods
for decreasing or
inhibiting cytokine production decrease or inhibit pro-inflammatory cytokine
production. In another
embodiment, methods for decreasing or inhibiting cytokine production decrease
or inhibit production
of at least one pro-inflammatory cytokine. In another embodiment, methods for
decreasing or
inhibiting cytokine production decrease or inhibit production of at least
cytokine IL-6. In another
embodiment, methods for decreasing or inhibiting cytokine production decrease
or inhibit production
of at least cytokine IL- lbeta. In another embodiment, methods for decreasing
or inhibiting cytokine
production decrease or inhibit production of at least cytokine TNF-alpha. In
another embodiment,
methods disclosed herein for decreasing or inhibiting cytokine production,
result in reduction or
inhibition of production of cytokines IL-6, IL-113, or TNF-a, or any
combination in said subject
compared with a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm and not administered apoptotic
cells or an apoptotic
supernatant.
[0230] Cancers or tumors may also affect the absolute level of cytokines
including pro-inflammatory
cytokines. The level of tumor burden in a subject may affect cytokine levels,
particularly
CA 03151815 2022-02-17
57
WO 2021/053667
PCT/IL2020/051011
proOinflammatory cytokines. A skilled artisan would appreciate that the phrase
"decrease or inhibit"
or grammatical variants thereof may encompass fold decrease or inhibition of
cytokine production,
or a net decrease or inhibition of cytokine production, or percent (%)
decrease or inhibition, or may
encompass a rate of change of decrease or inhibition of a cytokine production.
[0231] In another embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm comprising the step of
administering apoptotic cells or
a composition comprising apoptotic cells to said subject.
[0232] In another embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm comprising the step of
administering an apoptotic cell
supernatant, such as an apoptotic cell-phagocyte supernatant, or a composition
comprising said
supernatant to said subject.
[0233] In another embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm comprising the step of
administering an apoptotic cell
supernatant, such as an additional agent selected from the group comprising
apoptotic cells, an
apoptotic supernatant, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or
fragment thereof or
analogue thereof, a tellurium-based compound, or an immune modulating agent,
or any combination
thereof, or a composition comprising said supernatant to said subject.
[0234] In one embodiment, an infection causes the cytokine release syndrome or
cytokine storm in
the subject. In one embodiment, the infection is an influenza infection. In
one embodiment, the
influenza infection is H1N1. In another embodiment, the influenza infection is
an H5N1 bird flu. In
another embodiment, the infection is severe acute respiratory syndrome (SARS).
In another
embodiment, the subject has Epstein-Barr virus-associated hemophagocytic
lymphohistiocytosis
(HLH). In another embodiment, the infection is sepsis. In one embodiment, the
sepsis is gram-
negative. In another embodiment, the infection is malaria. In another
embodiment, the infection is an
Ebola virus infection. In another embodiment, the infection is variola virus.
In another embodiment,
the infection is a systemic Gram-negative bacterial infection. In another
embodiment, the infection is
Jarisch-Herxheimer syndrome.
[0235] In one embodiment, the cause of the cytokine release syndrome or
cytokine storm in a subject
is hemophagocytic lymphohistiocytosis (HLH). In another embodiment, HLH is
sporadic HLH. In
another embodiment, HLH is macrophage activation syndrome (MAS). In another
embodiment, the
cause of the cytokine release syndrome or cytokine storm in a subject is MAS.
CA 03151815 2022-02-17
58
WO 2021/053667
PCT/IL2020/051011
[0236] In one embodiment, the cause of the cytokine release syndrome or
cytokine storm in a subject
is chronic arthritis. In another embodiment, the cause of the cytokine release
syndrome or cytokine
storm in a subject is systemic Juvenile Idiopathic Arthritis (sJIA), also
known as Still's Disease.
[0237] In one embodiment, the cause of the cytokine release syndrome or
cytokine storm in a subject
is Cryopyrin-associated Periodic Syndrome (CAPS). In another embodiment, CAPS
comprises
Familial Cold Auto-inflammatory Syndrome (FCAS), also known as Familial Cold
Urticaria (FCU).
In another embodiment, CAPS comprises Muckle-Well Syndrome (MWS). In another
embodiment,
CAPS comprises Chronic Infantile Neurological Cutaneous and Articular (CINCA)
Syndrome. In
yet another embodiment, CAPS comprises FCAS, FCU, MWS, or CINCA Syndrome, or
any
combination thereof. In another embodiment, the cause of the cytokine release
syndrome or cytokine
storm in a subject is FCAS. In another embodiment, the cause of the cytokine
release syndrome or
cytokine storm in a subject is FCU. In another embodiment, the cause of the
cytokine release
syndrome or cytokine storm in a subject is MWS. In another embodiment, the
cause of the cytokine
release syndrome or cytokine storm in a subject is CINCA Syndrome. In still
another embodiment,
the cause of the cytokine release syndrome or cytokine storm in a subject is
FCAS, FCU, MWS, or
CINCA Syndrome, or any combination thereof.
[0238] In another embodiment, the cause of the cytokine release syndrome or
cytokine storm in a
subject is a cryopyrinopathy comprising inherited or de novo gain of function
mutations in the
NLRP3 gene, also known as the CIASI gene.
[0239] In one embodiment, the cause of the cytokine release syndrome or
cytokine storm in a subject
is a hereditary auto-inflammatory disorder.
[0240] In one embodiment, the trigger for the release of inflammatory
cytokines is a
lipopolysaccharide (LPS), Gram-positive toxins, fungal toxins,
glycosylphosphatidylinositol (GPI)
or modulation of RIG-1 gene expression.
[0241] In another embodiment, the subject experiencing cytokine release
syndrome or cytokine
storm does not have an infectious disease. In one embodiment, the subject has
acute pancreatitis. In
another embodiment, the subject has tissue injury, which in on embodiment, is
severe burns or
trauma. In another embodiment, the subject has acute respiratory distress
syndrome. In another
embodiment, the subject has cytokine release syndrome or cytokine storm
secondary to drug use. In
another embodiment, the subject has cytokine release syndrome or cytokine
storm secondary to toxin
inhalation.
[0242] In another embodiment, the subject has cytokine release syndrome or
cytokine storm
secondary to receipt of immunotherapy, which in one embodiment is
immunotherapy with
superagonistic CD28-specific monoclonal antibodies (CD28SA). In one
embodiment, the CD28SA
CA 03151815 2022-02-17
59
WO 2021/053667
PCT/IL2020/051011
is TGN1412. In another embodiment, the immunotherapy is CAR T-cell therapy.
[0243] In another embodiment, apoptotic cells or supernatant or a CTLA-4
blocking agent, an alpha-
1 anti-trypsin or fragment thereof or analogue thereof, a tellurium-based
compound, or an immune
modulating agent, or any combination thereof, may be used to control cytokine
release syndrome or
cytokine storm that results from administration of a pharmaceutical
composition. In one embodiment,
the pharmaceutical composition is oxaliplatin, cytarabine, lenalidomide, or a
combination thereof.
[0244] In another embodiment, apoptotic cells or the supernatant or a CTLA-4
blocking agent, an
alpha-1 anti-trypsin or fragment thereof or analogue thereof, a tellurium-
based compound, or an
immune modulating agent, or any combination thereof, may be used to control
cytokine release
syndrome or cytokine storm that results from administration of an antibody. In
one embodiment, the
antibody is monoclonal. In another embodiment, the antibody is polyclonal. In
one embodiment, the
antibody is rituximab. In another embodiment, the antibody is Orthoclone OKT3
(muromonab-CD3).
In another embodiment, the antibody is alemtuzumab, tosituzumab, CP-870,893,
LO-CD2a/BTI-322
or TGN1412.
[0245] In another embodiment, examples of diseases for which control of
inflammatory cytokine
production can be beneficial include cancers, allergies, any type of
infection, toxic shock syndrome,
sepsis, any type of autoimmune disease, arthritis, Crohn's disease, lupus,
psoriasis, or any other
disease for which the hallmark feature is toxic cytokine release that causes
deleterious effects in a
subject.
Alpha-1-antitrypsin (AAT)
[0246] Alpha- 1-antitrypsin (AAT) is a circulating 52-kDa glycoprotein that is
produced mainly by
the liver. AAT is primarily known as a serine protease inhibitor and is
encoded by the gene
SERPINAL AAT inhibits neutrophil elastase, and inherited deficiency in
circulating AAT results in
lung-tissue deterioration and liver disease. Serum AAT concentrations in
healthy individuals increase
twofold during inflammation.
[0247] There is a negative association between AAT levels and the severity of
several inflammatory
diseases. For example, reduced levels or activity of AAT have been described
in patients with HIV
infection, diabetes mellitus, hepatitis C infection-induced chronic liver
disease, and several types of
vasculitis.
[0248] Increasing evidence demonstrates that human serum derived alpha- 1-anti-
trypsin (AAT)
reduces production of pro-inflammatory cytokines, induces anti-inflammatory
cytokines, and
interferes with maturation of dendritic cells.
[0249] Indeed, the addition of AAT to human peripheral blood mononuclear cells
(PBMC) inhibits
LPS induced release of TNF-a and IL-10 but increases IL-1 receptor antagonist
(IL-1Ra) and IL-10
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
production.
[0250] AAT reduces in vitro IL-1P¨mediated pancreatic islet toxicity, and AAT
monotherapy
prolongs islet allograft survival, promotes antigen-specific immune tolerance
in mice, and delays the
development of diabetes in non-obese diabetic (NOD) mice. AAT was shown to
inhibit LPS -induced
5 acute lung injury in experimental models. Recently, AAT was shown to
reduce the size of infarct and
the severity of heart failure in a mouse model of acute myocardial ischemia-
reperfusion injury.
[0251] Monotherapy with clinical-grade human AAT (hAAT) reduced circulating
pro-inflammatory
cytokines, diminished Graft vs Host Disease (GvHD) severity, and prolonged
animal survival after
experimental allogeneic bone marrow transfer (Tawara et al., Proc Natl Acad
Sci U S A. 2012 Jan
10 10;109(2):564-9), incorporated herein by reference. AAT treatment
reduced the expansion of
alloreactive T effector cells but enhanced the recovery of T regulatory T-
cells, (Tregs) thus altering
the ratio of donor T effector to T regulatory cells in favor of reducing the
pathological process. In
vitro, AAT suppressed LPS-induced in vitro secretion of proinflammatory
cytokines such as TNF-a
and IL-1(3, enhanced the production of the anti-inflammatory cytokine IL-10,
and impaired NF-KB
15 translocation in the host dendritic cells. Marcondes, Blood. 2014 (Oct
30;124(18):2881-91)
incorporated herein by reference show that treatment with AAT not only
ameliorated GvHD but also
preserved and perhaps even enhanced the graft vs leukemia (GVL) effect.
[0252] In one embodiment, disclosed herein are compositions comprising
chimeric antigen receptor-
expressing T-cells (CAR T-cells) and Alpha- 1 -antitrypsin (AAT). In another
embodiment, CAR T-
20 cells and Alpha- 1 -antitrypsin (AAT) are in separate compositions. In
another embodiment, AAT
comprises a full-length AAT or a functional fragment thereof. In another
embodiment, AA comprises
an analogue of a full-length AAT or a functional fragment thereof. In another
embodiment, a
composition comprising AAT further comprises apoptotic cells or an apoptotic
cell supernatant.
[0253] In another embodiment, disclosed herein is a method of treating,
preventing, inhibiting,
25 reducing the incidence of, ameliorating, or alleviating a cancer or a
tumor in a subject comprising the
step of administering chimeric antigen receptor-expressing T-cells (CAR T-
cells) and a composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject. In another embodiment,
the method further
comprises apoptotic cells or an apoptotic cell supernatant.
[0254] In another embodiment, disclosed herein is a method of inhibiting or
reducing the incidence
30 of cytokine release syndrome or cytokine storm in a subject undergoing
chimeric antigen receptor-
expressing T-cell (CAR T-cell) therapy, comprising the step of administering a
composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject. In another embodiment,
a method of treating
cytokine release syndrome or a cytokine storm in a subject undergoing chimeric
antigen receptor-
expressing T-cell (CAR T-cell) therapy, comprises the step of administering a
composition
CA 03151815 2022-02-17
61
WO 2021/053667
PCT/IL2020/051011
comprising Alpha- 1 -antitrypsin (AAT) to said subject. In another embodiment,
a method of
preventing cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject. In another embodiment,
a method of
ameliorating cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject. In another embodiment,
a method of
alleviating cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject.
[0255] In another embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm, comprising the step of
administering a composition
comprising Alpha- 1 -antitrypsin (AAT) to said subject.
[0256] In one embodiment, AAT is administered alone to control cytokine
release. In another
embodiment, both AAT and apoptotic cells or a composition thereof, or
apoptotic cell supernatants
or a composition thereof, are administered to control cytokine release.
[0257] Irnmuno-Modulatory Agents
[0258] A skilled artisan would appreciate that immune-modulating agents may
encompass
extracellular mediators, receptors, mediators of intracellular signaling
pathways, regulators of
translation and transcription, as well as immune cells. In one embodiment, an
additional agent
disclosed herein is an immune-modulatory agent known in the art. In another
embodiment, use in the
methods disclosed here of an immune-modulatory agent reduces the level of at
least one cytokine. In
another embodiment, use in the methods disclosed here of an immune-modulatory
agent reduces or
inhibits CRS or a cytokine storm. In some embodiments, use in the methods
disclosed herein of an
immune-modulatory agent is for treating, preventing, inhibiting the growth,
delaying disease
progression, reducing the tumor load, or reducing the incidence of a tumor or
a cancer, or any
combination thereof. In some embodiments, use of an immune-modulatory agent is
in combination
with another composition disclosed herein, for example but not limited to a
composition comprising
.. early apoptotic cells or comprising CAR T-cells.
[0259] In one embodiment, an immune-modulatory agent comprises compounds that
block, inhibit
or reduce the release of cytokines or chemokines. In another embodiment, an
immune-modulatory
agent comprises compounds that block, inhibit or reduce the release of IL-21
or IL-23, or a
combination thereof. In another embodiment, an immune-modulatory agent
comprises
CA 03151815 2022-02-17
62
WO 2021/053667
PCT/IL2020/051011
an antiretroviral drug in the chemokine receptor-5 (CCR5) receptor antagonist
class, for example
maraviroc. In another embodiment, an immune-modulatory agent comprises an anti-
DNAM-1
antibody. In another embodiment, an immune-modulatory agent comprises
damage/pathogen-
associated molecules (DAMPs/PAMPs) selected from the group comprising heparin
sulfate, ATP,
and uric acid, or any combination thereof. In another embodiment, an immune-
modulatory agent
comprises a sialic acid binding Ig-like lectin (Siglecs). In another
embodiment, an immune-
modulatory agent comprises a cellular mediator of tolerance, for example
regulatory CD4+ CD25+ T
cells (Tregs) or invariant natural killer T cells (iNK T-cells). In another
embodiment, an immune-
modulatory agent comprises dendritic cells. In another embodiment, an immune-
modulatory agent
comprises monocytes. In another embodiment, an immune-modulatory agent
comprises
macrophages. In another embodiment, an immune-modulatory agent comprises JAK2
or JAK3
inhibitors selected from the group comprising ruxolitinib and tofacitinib. In
another embodiment, an
immune-modulatory agent comprises an inhibitor of spleen tyrosine kinase
(Syk), for example
fostamatinib. In another embodiment, an immune-modulatory agent comprises
histone deacetylase
inhibitor vorinostat acetylated STAT3. In another embodiment, an immune-
modulatory agent
comprises neddylation inhibitors, for example MLN4924. In another embodiment,
an immune-
modulatory agent comprises an miR-142 antagonist. In another embodiment, an
immune-modulatory
agent comprises a chemical analogue of cytidine, for example Azacitidine. In
another embodiment,
an immune-modulatory agent comprises an inhibitor of histone deacetylase, for
example Vorinostat.
In another embodiment, an immune-modulatory agent comprises an inhibitor of
histone methylation.
In another embodiment, an immune-modulatory agent comprises an antibody. In
another
embodiment, the antibody is rituximab (RtX)
Tellurium-based compounds
[0260] Tellurium is a trace element found in the human body. Various tellurium
compounds, have
immune-modulating properties, and have been shown to have beneficial effects
in diverse preclinical
and clinical studies. A particularly effective family of tellurium-containing
compounds is disclosed
for example, in U.S. Patent Nos. 4,752,614; 4,761,490; 4,764,461 and
4,929,739. The immune-
modulating properties of this family of tellurium-containing compounds is
described, for example,
in U.S. Patent Nos. 4,962,207, 5,093,135, 5,102,908 and 5,213,899, which are
all incorporated by
reference as if fully set forth herein.
[0261] One promising compound is ammonium trichloro(dioxyethylene-
0,0')tellurate, which is also
referred to herein and in the art as AS101. AS101, as a representative example
of the family of
tellurium-containing compound discussed hereinabove, exhibits antiviral (Nat.
Immun. Cell Growth
Regul. 7(3):163-8, 1988; AIDS Res Hum Retroviruses. 8(5):613-23, 1992), and
tumoricidal activity
CA 03151815 2022-02-17
63
WO 2021/053667
PCT/IL2020/051011
(Nature 330(6144):173-6, 1987; J. Clin. Oncol. 13(9):2342-53, 1995; J.
Immunol. 161(7):3536-42,
1998). Further, AS101 is characterized by low toxicity.
[0262] In one embodiment, a composition comprising tellurium-containing immune-
modulator
compounds may be used in methods disclosed herein, where the tellurium-based
compound
stimulates the innate and acquired arm of the immune response. For example, it
has been shown that
AS101 is a potent activator of interferon (IFN) in mice (J. Natl. Cancer Inst.
88(18):1276-84, 1996)
and humans (Nat. Immun. Cell Growth Regul. 9(3):182-90, 1990; Immunology
70(4):473-7, 1990;
J. Natl. Cancer Inst. 88(18):1276-84, 1996.)
[0263] In another embodiment, tellurium-based compounds induce the secretion
of a spectrum of
cytokines, such as IL-la, IL-6 and TNF-a.
[0264] In another embodiment, a tellurium-based compound comprises a tellurium-
based compound
known in the art to have immune-modulating properties. In another embodiment,
a tellurium-based
compound comprises ammonium trichloro(dioxyethylene-0,0')tellurate.
[0265] In one embodiment, a tellurium-based compound inhibits the secretion of
at least one
cytokine. In another embodiment, a tellurium-based compound reduces the
secretion of at least one
cytokine. In another embodiment, a tellurium-based compound inhibits or
reduces a cytokine release
syndrome (CRS) of a cytokine storm.
[0266] In one embodiment, disclosed herein are compositions comprising
chimeric antigen receptor-
expressing T-cells (CAR T-cells) and a tellurium-based compound. In another
embodiment, CAR T-
.. cells and Tellurium-based compound are in separate compositions. In another
embodiment, AAT
comprises a full-length AAT or a functional fragment thereof. In another
embodiment, AA comprises
an analogue of a full-length AAT or a functional fragment thereof
[0267] In another embodiment, disclosed herein is a method of treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor in
a subject comprising the
step of administering chimeric antigen receptor-expressing T-cells (CAR T-
cells) and a composition
comprising a Tellurium-based compound to said subject.
[0268] In another embodiment, disclosed herein is a method of inhibiting or
reducing the incidence
of cytokine release syndrome or cytokine storm in a subject undergoing
chimeric antigen receptor-
expressing T-cell (CAR T-cell) therapy, comprising the step of administering a
composition
comprising a Tellurium-based compound to said subject. In another embodiment,
a method of
treating cytokine release syndrome or a cytokine storm in a subject undergoing
chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising a Tellurium-based compound to said subject. In another embodiment,
a method of
preventing cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
CA 03151815 2022-02-17
64
WO 2021/053667
PCT/IL2020/051011
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising a Tellurium-based compound to said subject. In another embodiment,
a method of
ameliorating cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising a Tellurium-based compound to said subject. In another embodiment,
a method of
alleviating cytokine release syndrome or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) therapy, comprises the step of
administering a composition
comprising a Tellurium-based compound to said subject.
[0269] In another embodiment, disclosed herein is a method of decreasing or
inhibiting cytokine
production in a subject experiencing cytokine release syndrome or cytokine
storm or vulnerable to
cytokine release syndrome or cytokine storm, comprising the step of
administering a composition
comprising a Tellurium-based compound to said subject.
[0270] In one embodiment, a tellurium-based compound is administered alone to
control cytokine
release. In another embodiment, both a tellurium-based compound and apoptotic
cells or a
composition thereof, or apoptotic cell supernatants or a composition thereof,
are administered to
control cytokine release.
Dendritic Cells
[0271] In one embodiment, dendritic cells (DCs) are antigen-producing and
presenting cells of the
mammalian immune system that process antigen material and present it on the
cell surface to the T-
cells of the immune system and are thereby capable of sensitizing T-cells to
both new and recall
antigens. In another embodiment, DCs are the most potent antigen-producing
cells, acting as
messengers between the innate and the adaptive immune systems. DC cells may be
used, in one
embodiment, to prime specific antitumor immunity through the generation of
effector cells that attack
and ly se tumors.
[0272] Dendritic cells are present in those tissues that are in contact with
the external environment,
such as the skin (where there is a specialized dendritic cell type called the
Langerhans cell) and the
inner lining of the nose, lungs, stomach and intestines. They can also be
found in an immature state
in the blood. Once activated, they migrate to the lymph nodes where they
interact with T-cells and B
cells to initiate and shape the adaptive immune response. At certain
development stages, they grow
branched projections, the dendrites that give the cell its name. Dendritic
cells may be engineered to
express particular tumor antigens.
[0273] The three signals that are required for T-cell activation are: (i)
presentation of cognate antigen
in self MHC molecules; (ii) costimulation by membrane-bound receptor-ligand
pairs; and (iii) soluble
factors to direct polarization of the ensuing immune response. Dendritic cells
(DCs) are able to
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
provide all of the three signals required for T-cell activation making them an
excellent cancer vaccine
platform.
[0274] Therefore, in one embodiment, disclosed herein are a composition
comprising dendritic cells
and an additional agent, wherein said additional agent comprises apoptotic
cells, apoptotic
5 supernatants, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or
fragment thereof or analogue
thereof, a tellurium-based compound, or an immune modulating agent, or any
combination thereof.
[0275] In another embodiment, disclosed herein is a method of treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor in
a subject comprising the
step of administering dendritic cells and a composition comprising an
additional agent, wherein said
10 agent comprises apoptotic cells, apoptotic supernatants, a CTLA-4
blocking agent, an alpha-1 anti-
trypsin or fragment thereof or analogue thereof, a tellurium-based compound,
or an immune
modulating agent, or any combination thereof, to said subject.
[0276] Genetic modification
[0277] In some embodiments, genetic modification of T-cells, dendritic cells,
and/or apoptotic cells
15 may be accomplished using RNA, DNA, recombinant viruses, or a
combination thereof. In some
embodiments, vectors derived from gamma retroviruses or lentiviruses are used
in the compositions
and methods as disclosed herein. In another embodiment, these vectors can
integrate into the host
genome, with potentially permanent expression of the transgene and have low
intrinsic
immunogenicity. In another embodiment, another vector that integrates into the
host genome and/or
20 has low intrinsic immunogenicity may be used in the compositions and
methods as disclosed herein.
In another embodiment, the non-viral-vector-mediated sleeping beauty
transposon system is used to
insert the CAR and other genes into the T-cell. In another embodiment,
"suicide genes" are integrated
into the T-cells, in which expression of a pro-apoptotic gene is under the
control of an inducible
promoter responsive to a systemically delivered drug.
25 [0278] In some embodiments, genetic modification may be transient. In
another embodiment, genetic
modification may utilize messenger RNA (mRNA). In another embodiment, large
numbers of cells
may be infused on multiple occasions in transiently engineered T-cells, such
as mRNA-transfected
T-cells. In another embodiment, RNA-based electroporation of lymphocytes using
in vitro¨
transcribed mRNA mediates transient expression of proteins for approximately
one week and obviates
30 the risk of integrating viral vectors. In another embodiment, mRNA-
transduced dendritic cells or
mRNA-electroporated T and NK lymphocytes.
[0279] It has been demonstrated that genetically modified T-cells can persist
after adoptive transfer
for more than a decade without adverse effects, indicating that genetically
modifying human T-cells
is fundamentally safe.
CA 03151815 2022-02-17
66
WO 2021/053667
PCT/IL2020/051011
[0280] In another embodiment, the genetic modification of the compositions and
in the methods as
disclosed herein may be any method that is known in the art.
Apoptotic Cells
[0281] Production of apoptotic cells ("ApoCells") for use in compositions
and methods as
disclosed herein, has been described in WO 2014/087408, which is incorporated
by reference herein
in its entirety, and is described in brief in Example 1 below. In another
embodiment, apoptotic cells
for use in compositions and methods as disclosed herein are produced in any
way that is known in
the art. In another embodiment, apoptotic cells for use in compositions and
methods disclosed herein
are autologous with a subject undergoing therapy. In another embodiment,
apoptotic cells for use in
compositions and methods disclosed herein are allogeneic with a subject
undergoing therapy. In
another embodiment, a composition comprising apoptotic cells comprises
apoptotic cells as disclosed
herein or as is known in art.
[0282] A skilled artisan would appreciate that the term "autologous" may
encompass a tissue, cell,
nucleic acid molecule or polypeptide in which the donor and recipient is the
same person.
[0283] A skilled artisan would appreciate that the term "allogeneic" may
encompass a tissue, cell,
nucleic acid molecule or polypeptide that is derived from separate individuals
of the same species. In
some embodiments, allogeneic donor cells are genetically distinct from the
recipient.
[0284] In some embodiments, obtaining a mononuclear-enriched cell composition
according to
the production method disclosed herein is effected by leukapheresis. A skilled
artisan would
appreciate that the term "leukapheresis" may encompass an apheresis procedure
in which leukocytes
are separated from the blood of a donor. In some embodiments, the blood of a
donor undergoes
leukapheresis and thus a mononuclear-enriched cell composition is obtained
according to the
production method disclosed herein. It is to be noted, that the use of at
least one anticoagulant during
leukapheresis is required, as is known in the art, in order to prevent
clotting of the collected cells.
[0285] In some embodiments, the leukapheresis procedure is configured to
allow collection of
mononuclear-enriched cell composition according to the production method
disclosed herein. In some
embodiments, cell collections obtained by leukapheresis comprise at least 65%.
In other
embodiments, at least 70%, or at least 80% mononuclear cells. as disclosed
herein. In some
embodiments, blood plasma from the cell-donor is collected in parallel to
obtaining of the
mononuclear-enriched cell composition in the production method disclosed
herein. In some
embodiments, about 300-600m1 of blood plasma from the cell-donor are collected
in parallel to
obtaining the mononuclear-enriched cell composition according to the
production method disclosed
herein. In some embodiments, blood plasma collected in parallel to obtaining
the mononuclear-
enriched cell composition according to the production method disclosed herein
is used as part of the
CA 03151815 2022-02-17
67
WO 2021/053667
PCT/IL2020/051011
freezing and/or incubation medium. Additional detailed methods of obtaining an
enriched population
of apoptotic cells for use in the compositions and methods as disclosed herein
may be found in WO
2014/087408, which is incorporated herein by reference in its entirety.
[0286] In some embodiments, the early apoptotic cells for use in the
methods disclosed herein
comprise at least 85% mononuclear cells. In further embodiments, the early
apoptotic cells for use in
the methods disclosed herein contains at least 85% mononuclear cells, 90%
mononuclear cells or
alternatively over 90% mononuclear cells. In some embodiments, the early
apoptotic cells for use in
the methods disclosed herein comprise at least 90% mononuclear cells. In some
embodiments, the
early apoptotic cells for use in the methods disclosed herein comprise at
least 95% mononuclear cells.
[0287] It is to be noted that, in some embodiments, while the mononuclear-
enriched cell
preparation at cell collection comprises at least 65%, preferably at least
70%, most preferably at least
80% mononuclear cells, the final pharmaceutical population, following the
production method of the
early apoptotic cells for use in the methods disclosed herein, comprises at
least 85%, preferably at
least 90%, most preferably at least 95% mononuclear cells.
[0288] In certain embodiments, the mononuclear-enriched cell preparation
used for production of
the composition of the early apoptotic cells for use in the methods disclosed
herein comprises at least
50% mononuclear cells at cell collection. In certain embodiments, disclosed
herein is a method for
producing the pharmaceutical population wherein the method comprises obtaining
a mononuclear-
enriched cell preparation from the peripheral blood of a donor, the
mononuclear-enriched cell
preparation comprising at least 50% mononuclear cells. In certain embodiments,
disclosed herein is
a method for producing the pharmaceutical population wherein the method
comprises freezing a
mononuclear-enriched cell preparation comprising at least 50% mononuclear
cells.
[0289] In some embodiments, the cell preparation comprises at least 85%
mononuclear cells,
wherein at least 40% of the cells in the preparation are in an early-apoptotic
state, wherein at least
85% of the cells in the preparation are viable cells. In some embodiments, the
apoptotic cell
preparation comprises no more than 15% 5high expressing cells.
[0290] A skilled artisan would appreciate that the term "early-apoptotic
state" may encompass
cells that show early signs of apoptosis without late signs of apoptosis.
Examples of early signs of
apoptosis in cells include exposure of phosphatidylserine (PS) and the loss of
mitochondrial
membrane potential. Examples of late events include propidium iodide (PI)
admission into the cell
and the final DNA cutting. In order to document that cells are in an "early
apoptotic" state, in some
embodiments, PS exposure detection by Annexin-V and PI staining are used, and
cells that are stained
with Annexin V but not with PI or with only minimal PI staining are considered
to be "early apoptotic
cells" (An+ PI-). In some embodiments, minimal IP staining comprising less
than or equal to (< )15%
CA 03151815 2022-02-17
68
WO 2021/053667
PCT/IL2020/051011
PI+ cells within the population of cells. In some embodiments, minimal IP
staining comprising <
10% PI+ cells within the population of cells. In some embodiments, minimal IP
staining comprising
< 5% PI+ cells within the population of cells. In another embodiment, cells
that are stained by both
Annexin-V FITC and high PI are considered to be "late apoptotic cells". In
some embodiments, high
.. IP staining comprises greater than (>) 15% PI+ cells within the population
of cells. In some
embodiments, high IP staining comprises greater than or equal to (>) 16% PI+
cells within the
population of cells. In another embodiment, cells that do not stain for either
Annexin-V or PI are
considered non-apoptotic viable cells.
[0291] In some embodiments, at least 40% of the cells in a preparation
are in an early apoptotic
state. In some embodiments, at least 45% of the cells in a preparation are in
an early apoptotic state.
In some embodiments, at least 50% of the cells in a preparation are in an
early apoptotic state. In
some embodiments, at least 55% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 60% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 65% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 70% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 75% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 80% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 85% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 90% of the cells in a preparation are in an early
apoptotic state. In some
embodiments, at least 95% of the cells in a preparation are in an early
apoptotic state.
[0292] In some embodiments, an early apoptotic cell preparation comprises
less than or equal to
(<) 15% PI+ cells. In some embodiments, an early apoptotic cell preparation
comprises < 10% PI+
cells. In some embodiments, an early apoptotic cell preparation comprises < 9%
PI+ cells. In some
embodiments, an early apoptotic cell preparation comprises < 8% PI+ cells. In
some embodiments,
an early apoptotic cell preparation comprises <7% PI + cells. In some
embodiments, an early apoptotic
cell preparation comprises < 6% PI+ cells. In some embodiments, an early
apoptotic cell preparation
comprises < 5% PI+ cells. In some embodiments, an early apoptotic cell
preparation comprises < 4%
PI+ cells. In some embodiments, an early apoptotic cell preparation comprises
< 3% PI+ cells. In some
embodiments, an early apoptotic cell preparation comprises < 2% PI+ cells. In
some embodiments,
an early apoptotic cell preparation comprises < 1% PI+ cells.
[0293] In some embodiments, at least 40% of the cells in a preparation
are in an early apoptotic
state (Ant), wherein < 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%,2%, 1%, or 0% of
the cells are PI+.
In some embodiments, at least 45% of the cells in a preparation are in an
early apoptotic state (Ant),
wherein < 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are
PI . In some
CA 03151815 2022-02-17
69
WO 2021/053667
PCT/IL2020/051011
embodiments, at least 50% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI .
In some
embodiments, at least 55% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI .
In some
embodiments, at least 60% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
<15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI . In
some
embodiments, at least 65% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI .
In some
embodiments, at least 70% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI . In
some
embodiments, at least 75% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI .
In some
embodiments, at least 80% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PI .
In some
embodiments, at least 85% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
<15% or <14%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are
PI . In some
embodiments, at least 90% of the cells in a preparation are in an early
apoptotic state (Ant), wherein
< 10%, or < 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or 0% of the cells are PP. In
some embodiments,
at least 95% of the cells in a preparation are in an early apoptotic state
(Ant), wherein < 5%, <4%,
3%,2%, 1%, or 0% of the cells are PP.
[0294] A skilled artisan would appreciate that in some embodiments the terms
"apoptotic cell",
"early apoptotic cell", "Allocetra", Autocetra", "ALC", and "ApoCell", and
grammatical variants
thereof, may be used interchangeably to represent a population of "early
apoptotic cells", wherein
said cell population is enriched for mononuclear cells and has unique
characteristics (See for
example, Example 1). The skilled artisan would appreciate that the
compositions and methods
described herein, in some embodiments comprise early apoptotic cells.
[0295] In some embodiments, Allocetra comprise a population of early
apoptotic cells obtained
from a single allogeneic donor. In some embodiments, Allocetra comprise a
population of early
apoptotic cells obtained from multiple allogeneic donors. In some embodiments,
Allocetra comprise
pooled populations of early apoptotic cells obtained from multiple allogeneic
donors or from cells
obtained from a blood bank. In some embodiments, Allocetra comprise pooled
population of early
apoptotic cells obtained from the same allogeneic donor. In some embodiments,
Allocetra comprise
an irradiated population of early apoptotic cells. In some embodiments, the
term "Allocetra" may be
used interchangeably with the term "Allocetra-OTS". In some embodiments, the
terms "Allocetra"
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
and "Allocetra-OTS" encompass mononuclear early apoptotic cells, prepared as
described in
Example 1, independent of the source of said cells.
[0296] In some embodiments, apoptotic cells comprise cells in an early
apoptotic state. In another
embodiment, apoptotic cells comprise cells wherein at least 90% of said cells
are in an early apoptotic
5 state. In another embodiment, apoptotic cells comprise cells wherein at
least 80% of said cells are in
an early apoptotic state. In another embodiment, apoptotic cells comprise
cells wherein at least 70%
of said cells are in an early apoptotic state. In another embodiment,
apoptotic cells comprise cells
wherein at least 60% of said cells are in an early apoptotic state. In another
embodiment, apoptotic
cells comprise cells wherein at least 50% of said cells are in an early
apoptotic state.
10 [0297] In some embodiments, the composition comprising apoptotic cells
further comprises an anti-
coagulant.
[0298] In some embodiments, early apoptotic cells are stable. A skilled
artisan would appreciate
that in some embodiments, stability encompasses maintaining early apoptotic
cell characteristics over
time, for example, maintaining early apoptotic cell characteristics upon
storage at about 2-8 C. In
15 some embodiments, stability comprises maintaining early apoptotic cell
characteristic upon storage
at freezing temperatures, for example temperatures at or below 0 C.
[0299] In some embodiments, the mononuclear-enriched cell population
obtained according to the
production method of the early apoptotic cells for use in the methods
disclosed herein undergoes
freezing in a freezing medium. In some embodiments, the freezing is gradual.
In some embodiments,
20 following collection the cells are maintained at room temperature until
frozen. In some embodiments,
the cell-preparation undergoes at least one washing step in washing medium
following cell-collection
and prior to freezing.
[0300] As used herein, the terms "obtaining cells" and "cell collection"
may be used
interchangeably. In some embodiments, the cells of the cell preparation are
frozen within 3-6 hours
25 of collection. In some embodiments, the cell preparation is frozen
within up to 6 hours of cell
collection. In some embodiments, the cells of the cell preparations are frozen
within 1, 2, 3, 4, 5, 6, 7,
8 hours of collection. In other embodiments, the cells of the cell
preparations are frozen up to 8, 12,
24, 48, 72 hours of collection. In other embodiments, following collection the
cells are maintained at
2-8 C until frozen.
30 [0301] In some embodiments, freezing according to the production
of an early apoptotic cell
population comprises: freezing the cell preparation at about -18 C to -25 C
followed by freezing the
cell preparation at about -80 C and finally freezing the cell preparation in
liquid nitrogen until
thawing. In some embodiments, the freezing according to the production of an
early apoptotic cell
population comprises: freezing the cell preparation at about -18 C to -25 C
for at least 2 hours,
CA 03151815 2022-02-17
71
WO 2021/053667
PCT/IL2020/051011
freezing the cell preparation at about -80 C for at least 2 hours and finally
freezing the cell preparation
in liquid nitrogen until thawing. In some embodiments, the cells are kept in
liquid nitrogen for at least
8, 10 or 12 hours prior to thawing. In some embodiments, the cells of the cell
preparation are kept in
liquid nitrogen until thawing and incubation with apoptosis-inducing
incubation medium. In some
embodiments, the cells of the cell preparation are kept in liquid nitrogen
until the day of hematopoietic
stem cell transplantation. In non-limiting examples, the time from cell
collection and freezing to
preparation of the final population may be between 1-50 days, alternatively
between 6-30 days. In
alternative embodiments, the cell preparation may be kept in liquid nitrogen
for longer time periods,
such as at least several months.
[0302] In some embodiments, the freezing according to the production of an
early apoptotic cell
population comprises freezing the cell preparation at about -18 C to -25 C for
at least 0.5, 1, 2, 4
hours. In some embodiments, the freezing according to the production of an
early apoptotic cell
population comprises freezing the cell preparation at about -18 C to -25 C for
about 2 hours. In some
embodiments, the freezing In the production of an early apoptotic cell
population comprises freezing
.. the cell preparation at about -80 C for at least 0.5, 1, 2, 4, 12hours.
[0303] In some embodiments, the mononuclear-enriched cell composition may
remain frozen at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 20 months. In some embodiments,
the mononuclear-enriched
cell composition may remain frozen at least 0.5, 1, 2, 3, 4, 5 years. In
certain embodiments, the
mononuclear-enriched cell composition may remain frozen for at least 20
months.
[0304] In some embodiments, the mononuclear-enriched cell composition is
frozen for at least 8,
10, 12, 18, 24 hours. In certain embodiments, freezing the mononuclear-
enriched cell composition is
for a period of at least 8 hours. In some embodiments, the mononuclear-
enriched cell composition is
frozen for at least about 10 hours. In some embodiments, the mononuclear-
enriched cell composition
is frozen for at least about 12 hours. In some embodiments, the mononuclear-
enriched cell
.. composition is frozen for about 12 hours. In some embodiments, the total
freezing time of the
mononuclear-enriched cell composition (at about -18 C to -25 C, at about -80 C
and in liquid
nitrogen) is at least 8, 10, 12, 18, 24 hours.
[0305] In some embodiments, the freezing at least partly induces the
early-apoptotic state in the
cells of the mononuclear-enriched cell composition. In some embodiments, the
freezing medium
comprises RPMI 1640 medium comprising L-glutamine, Hepes, Hes, dimethyl
sulfoxide (DMSO)
and plasma. In some embodiments, the plasma in the freezing medium is an
autologous plasma of the
donor which donated the mononuclear-enriched cells of the population. In some
embodiments, the
freezing medium comprises RPMI 1640 medium comprising 2 mM L-glutamine, 10 mM
Hepes, 5%
Hes, 10% dimethyl sulfoxide and 20% v/v plasma.
CA 03151815 2022-02-17
72
WO 2021/053667
PCT/IL2020/051011
[0306] In some embodiments, the freezing medium comprises an anti-
coagulant. In certain
embodiments, at least some of the media used during the production of an early
apoptotic cell
population, including the freezing medium, the incubation medium and the
washing media comprise
an anti-coagulant. In certain embodiments, all media used during the
production of an early apoptotic
.. cell population which comprise an anti-coagulant comprise the same
concentration of anti-coagulant.
In some embodiments, anti-coagulant is not added to the final suspension
medium of the cell
population.
[0307] In some embodiments, addition of an anti-coagulant at least to the
freezing medium
improves the yield of the cell-preparation. In other embodiments, addition of
an anti-coagulant to the
freezing medium improves the yield of the cell-preparation in the presence of
a high triglyceride level.
As used herein, improvement in the yield of the cell-preparation relates to
improvement in at least
one of: the percentage of viable cells out of cells frozen, the percentage of
early-state apoptotic cells
out of viable cells and a combination thereof.
[0308] In some embodiments, early apoptotic cells are stable for at least
24 hours. In another
embodiment, early apoptotic cells are stable for 24 hours. In another
embodiment, early apoptotic
cells are stable for more than 24 hours. In another embodiment, early
apoptotic cells are stable for at
least 36 hours. In another embodiment, early apoptotic cells are stable for 48
hours. In another
embodiment, early apoptotic cells are stable for at least 36 hours. In another
embodiment, early
apoptotic cells are stable for more than 36 hours. In another embodiment,
early apoptotic cells are
stable for at least 48 hours. In another embodiment, early apoptotic cells are
stable for 48 hours. In
another embodiment, early apoptotic cells are stable for at least 48 hours. In
another embodiment,
early apoptotic cells are stable for more than 48 hours. In another
embodiment, early apoptotic cells
are stable for at least 72 hours. In another embodiment, early apoptotic cells
are stable for 72 hours.
In another embodiment, early apoptotic cells are stable for more than 72
hours.
[0309] A skilled artisan would appreciate that the term "stable"
encompasses apoptotic cells that
remain PS-positive (Phosphatidylserine-positive) with only a very small
percent of PI-positive
(Propidium iodide-positive). PI-positive cells provide an indication of
membrane stability wherein a
PI-positive cells permits admission into the cells, showing that the membrane
is less stable. In some
embodiments, stable early apoptotic cells remain in early apoptosis for at
least 24 hours, for at least
36 hours, for at least 48 hours, or for at least 72 hours. In another
embodiment, stable early apoptotic
cells remain in early apoptosis for 24 hours, for 36 hours, for 48 hours, or
for 72 hours. In another
embodiment, stable early apoptotic cells remain in early apoptosis for more
than 24 hours, for more
than 36 hours, for more than 48 hours, or for more than 72 hours. In another
embodiment, stable early
apoptotic cells maintain their state for an extended time period.
CA 03151815 2022-02-17
73
WO 2021/053667
PCT/IL2020/051011
[0310] In some embodiments, an apoptotic cell population is devoid of
cell aggregates. In some
embodiments, an apoptotic cell population is devoid of large cell aggregates.
In some embodiments,
an apoptotic cell population has a reduced number of cell aggregates compared
to an apoptotic cell
population prepared without adding an anticoagulant in a step other than cell
collection
(leukapheresis) from the donor. In some embodiments, an apoptotic cell
population or a composition
thereof, comprises an anticoagulant.
[0311] In some embodiments, apoptotic cells are devoid of cell
aggregates, wherein said apoptotic
cells were obtained from a subject with high blood triglycerides. In some
embodiments, blood
triglycerides levels of the subject are above 150 mg/dL. In some embodiments,
an apoptotic cell
population is devoid of cell aggregates, wherein said apoptotic cell
population is prepared from cells
obtained from a subject with normal blood triglycerides. In some embodiments,
blood triglycerides
levels of the subject are equal to or below 150 mg/dL. In some embodiments,
cell aggregates produce
cell loss during apoptotic cell production methods.
[0312] A skilled artisan would appreciate that the terms "aggregates" or
"cell aggregates" may
encompass the reversible clumping of blood cells under low shear forces or at
stasis. Cell aggregates
can be visually observed during the incubation steps of the production of the
apoptotic cells. Cell
aggregation can be measured by any method known in the art, for example by
visually imaging
samples under a light microscope or using flow cytometry.
[0313] In some embodiments, the anti-coagulant is selected from the group
comprising: heparin,
acid citrate dextrose (ACD) Formula A and a combination thereof. In some
embodiments, the anti-
coagulant is selected from the group consisting of: heparin, acid citrate
dextrose (ACD) Formula A
and a combination thereof.
[0314] In some embodiments of methods of preparing an early apoptotic cell
population and
compositions thereof, an anticoagulant is added to at least one medium used
during preparation of the
population. In some embodiments, the at least one medium used during
preparation of the population
is selected from the group consisting of: the freezing medium, the washing
medium, the apoptosis
inducing incubation medium, and any combinations thereof.
[0315] In some embodiments, the anti-coagulant is selected from the group
consisting of: Heparin,
ACD Formula A and a combination thereof. It is to be noted that other anti-
coagulants known in the
art may be used, such as, but not limited to Fondaparinaux, Bivalirudin and
Argatroban.
[0316] In some embodiments, at least one medium used during preparation of the
population
contains 5% of ACD formula A solution comprising 10 Wm' heparin. In some
embodiments, anti-
coagulant is not added to the final suspension medium of the cell population.
As used herein, the
terms "final suspension medium" and "administration medium" are used
interchangeably having all
CA 03151815 2022-02-17
74
WO 2021/053667
PCT/IL2020/051011
the same qualities and meanings.
[0317] In some embodiments, at least one medium used during preparation of the
population
comprises heparin at a concentration of between 0.1-2.5 U/ml. In some
embodiments, at least one
medium used during preparation of the population comprises ACD Formula A at a
concentration of
between 1%-15% v/v. In some embodiments, the freezing medium comprises an anti-
coagulant. In
some embodiments, the incubation medium comprises an anti-coagulant. In some
embodiments, both
the freezing medium and incubation medium comprise an anti-coagulant. In some
embodiments the
anti-coagulant is selected from the group consisting of: heparin, ACD Formula
A and a combination
thereof.
[0318] In some embodiments, the heparin in the freezing medium is at a
concentration of between
0.1-2.5 U/ml. In some embodiments, the ACD Formula A in the freezing medium is
at a concentration
of between 1%-15% v/v. In some embodiments, the heparin in the incubation
medium is at a
concentration of between 0.1-2.5 U/ml. In some embodiments, the ACD Formula A
in the incubation
medium is at a concentration of between 1%-15% v/v. In some embodiments, the
anticoagulant is a
solution of acid-citrate-dextrose (ACD) formula A. In some embodiments, the
anticoagulant added to
at least one medium used during preparation of the population is ACD Formula A
containing heparin
at a concentration of 10 U/ml.
[0319] In some embodiments, the apoptosis inducing incubation medium used in
the production
of an early apoptotic cell population comprises an anti-coagulant. In some
embodiments, both the
freezing medium and apoptosis inducing incubation medium used in the
production of an early
apoptotic cell population comprise an anti-coagulant. Without wishing to be
bound by any theory or
mechanism, in order to maintain a high and stable cell yield in different cell
compositions, regardless
of the cell collection protocol, in some embodiments addition of anti-
coagulants comprising adding
the anticoagulant to both the freezing medium and the apoptosis inducing
incubation medium during
production of the apoptotic cell population. In some embodiments, a high and
stable cell yield within
the composition comprises a cell yield of at least 30%, preferably at least
40%, typically at least 50%
cells of the initial population of cells used for induction of apoptosis.
[0320] In some embodiments, both the freezing medium and the incubation medium
comprise an
anti-coagulant. In some embodiments, addition of an anti-coagulant both to the
incubation medium
and freezing medium results in a high and stable cell-yield between different
preparations of the
population regardless of cell-collection conditions, such as, but not limited
to, the timing and/or type
of anti-coagulant added during cell collection. In some embodiments, addition
of an anti-coagulant
both to the incubation medium and freezing medium results in a high and stable
yield of the cell-
preparation regardless of the timing and/or type of anti-coagulant added
during leukapheresis. In some
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
embodiments, production of the cell-preparation in the presence of a high
triglyceride level results in
a low and/or unstable cell-yield between different preparations. In some
embodiments, producing the
cell-preparation from the blood of a donor having high triglyceride level
results in a low and/or
unstable cell-yield of the cell preparation. In some embodiments, the term
"high triglyceride level"
5 refers to a triglyceride level which is above the normal level of a
healthy subject of the same sex and
age. In some embodiments, the term "high triglyceride level" refers to a
triglyceride level above about
1.7 milimole/liter. As used herein, a high and stable yield refers to a cell
yield in the population which
is high enough to enable preparation of a dose which will demonstrate
therapeutic efficiency when
administered to a subject. In some embodiments, therapeutic efficiency refers
to the ability to treat,
10 prevent or ameliorate an immune disease, an autoimmune disease or an
inflammatory disease in a
subject. In some embodiments, a high and stable cell yield is a cell yield of
at least 30%, possibly at
least 40%, typically at least 50% of cells in the population out of cells
initially frozen.
[0321] In some embodiments, in case the cell-preparation is obtained from
a donor having a high
triglyceride level, the donor will take at least one measure selected from the
group consisting of:
15 taking triglyceride-lowering medication prior to donation, such as, but
not limited to: statins and/or
bezafibrate, fasting for a period of at least 8, 10, 12 hours prior to
donation, eating an appropriate diet
to reduce blood triglyceride level at least 24, 48, 72 hours prior to donating
and any combination
thereof..
[0322] In some embodiments, cell yield in the population relates to cell
number in the composition
20 out of the initial number of cells subjected to apoptosis induction. As
used herein, the terms "induction
of early apoptotic state" and "induction of apoptosis" may be used
interchangeably.
[0323] In some embodiments, the mononuclear-enriched cell composition is
incubated in
incubation medium following freezing and thawing. In some embodiments, there
is at least one
washing step between thawing and incubation. As used herein, the terms
"incubation medium" and
25 "apoptosis inducing incubation medium" are used interchangeably. In some
embodiments, the
incubation medium comprises RPMI 1640 medium supplemented with L-glutamine,
Hepes
methylprednisolone and plasma. In some embodiments, the washing medium
comprises 2 mM L-
glutamine, 10 mM Hepes and 10% v/v blood plasma. In some embodiments, the
blood plasma in in
the incubation medium is derived from the same donor from whom the cells of
the cell preparations
30 are derived. In some embodiments, the blood plasma is added to the
incubation medium on the day
of incubation. In some embodiments, incubation is performed at 37 C and 5%
CO2.
[0324] In some embodiments, the incubation medium comprises
methylprednisolone. In some
embodiments, the methylprednisolone within the incubation medium further
induces the cells in the
mononuclear-enriched cell composition to enter an early-apoptotic state. In
some embodiments, the
CA 03151815 2022-02-17
76
WO 2021/053667
PCT/IL2020/051011
cells in the mononuclear-enriched cell composition are induced to enter an
early-apoptotic state both
by freezing and incubating in the presence of methylprednisolone. In some
embodiments, the
production of an early apoptotic cell population advantageously allows
induction of an early-
apoptosis state substantially without induction of necrosis, wherein the cells
remain stable at said
early-apoptotic state for about 24 hours following preparation.
[0325] In some embodiments, the incubation medium comprises methylprednisolone
at a
concentration of about 10-100 .t.g/ml. In some embodiments, the incubation
medium comprises
methylprednisolone at a concentration of about 40-60 jig/ml, alternatively
about 45-55 jig/ml. In some
embodiments, the incubation medium comprises methylprednisolone at a
concentration of 50 jig/ml.
[0326] In some embodiments, the incubation is for about 2-12 hours,
possibly 4-8 hours, typically
for about 5-7 hours. In some embodiments, the incubation is for about 6 hours.
In some embodiments,
the incubation is for at least 6 hours. In a preferred embodiment, the
incubation is for 6 hours.
[0327] In some embodiments, the incubation medium comprises an anti-coagulant.
In some
embodiments, addition of an anti-coagulant to the incubation medium improves
the yield of the cell-
preparation. In some embodiments, the anti-coagulant in the incubation medium
is of the same
concentration as within the freezing medium. In some embodiments, the
incubation medium
comprises an anti-coagulant selected from the group consisting of: heparin,
ACD Formula A and a
combination thereof. In some embodiments, the anti-coagulant used in the
incubation medium is ACD
Formula A containing heparin at a concentration of 10 U/ml.
[0328] In some embodiments, the incubation medium comprises heparin. In some
embodiments,
the heparin in the incubation medium is at a concentration of between 0.1-2.5
U/ml. In some
embodiments, the heparin in the incubation medium is at a concentration of
between 0.1-2.5 U/ml,
possibly between 0.3-0.7 U/ml, typically about 0.5 U/ml. In certain
embodiments, the heparin in the
incubation medium is at a concentration of about 0.5 U/ml.
[0329] In some embodiments, the incubation medium comprises ACD Formula A. In
some
embodiments, the ACD Formula A in the incubation medium is at a concentration
of between 1%-
15% v/v. In some embodiments, the ACD Formula A in the incubation medium is at
a concentration
of between 1%-15% v/v, possibly between 4%-7% v/v, typically about 5% v/v. In
some
embodiments, the ACD Formula A in the incubation medium is at a concentration
of about 5% v/v.
[0330] In some embodiments, improvement in the yield of the cell-
preparation comprises
improvement in the number of the early-apoptotic viable cells of the
preparation out of the number of
frozen cells from which the preparation was produced.
[0331] In some embodiments, addition of an anti-coagulant to the freezing
medium contributes to
a high and stable yield between different preparations of the pharmaceutical
population. In preferable
CA 03151815 2022-02-17
77
WO 2021/053667
PCT/IL2020/051011
embodiments, addition of an anti-coagulant at least to the freezing medium and
incubation medium
results in a high and stable yield between different preparations of the
pharmaceutical composition,
regardless to the cell collection protocol used.
[0332] In some embodiments, the freezing medium comprises an anti-coagulant
selected from the
group consisting of: heparin, ACD Formula A and a combination thereof. In some
embodiments, the
anti-coagulant used in the freezing medium is ACD Formula A containing heparin
at a concentration
of 10 U/ml. In some embodiments, the freezing medium comprises 5% v/v of ACD
Formula A
solution comprising heparin at a concentration of 10 U/ml.
[0333] In some embodiments, the freezing medium comprises heparin. In some
embodiments, the
heparin in the freezing medium is at a concentration of between 0.1-2.5 U/ml.
In some embodiments,
the heparin in the freezing medium is at a concentration of between 0.1-2.5
U/ml, possibly between
0.3-0.7 U/ml, typically about 0.5 U/ml. In certain embodiments, the heparin in
the freezing medium
is at a concentration of about 0.5 U/ml.
[0334] In some embodiments, the freezing medium comprises ACD Formula A. In
some
embodiments, the ACD Formula A in the freezing medium is at a concentration of
between 1%-15%
v/v. In some embodiments, the ACD Formula A in the freezing medium is at a
concentration of
between 1%-15% v/v, possibly between 4%-7% v/v, typically about 5% v/v. In
some embodiments,
the ACD Formula A in the freezing medium is at a concentration of about 5%
v/v.
[0335] In some embodiments, addition of an anti-coagulant to the incubation
medium and/or
freezing medium results in a high and stable cell yield within the population
regardless of the
triglyceride level in the blood of the donor. In some embodiments, addition of
an anti-coagulant to
the incubation medium and/or freezing medium results in a high and stable cell
yield within the
composition when obtained from the blood of a donor having normal or high
triglyceride level. In
some embodiments, addition of an anti-coagulant at least to the incubation
medium, results in a high
and stable cell yield within the composition regardless of the triglyceride
level in the blood of the
donor. In some embodiments, addition of an anti-coagulant to the freezing
medium and incubation
medium results in a high and stable cell yield within the composition
regardless of the triglyceride
level in the blood of the donor.
[0336] In some embodiments, the freezing medium and/or incubation medium
and/or washing
medium comprise heparin at a concentration of at least 0.1 U/ml, possibly at
least 0.3 U/ml, typically
at least 0.5 U/ml. In some embodiments, the freezing medium and/or incubation
medium and/or
washing medium comprise ACD Formula A at a concentration of at least 1% v/v,
possibly at least
3% v/v, typically at least 5% v/v.
[0337] In some embodiments, the mononuclear-enriched cell composition
undergoes at least one
CA 03151815 2022-02-17
78
WO 2021/053667
PCT/IL2020/051011
washing step following cell collection and prior to being re-suspended in the
freezing medium and
frozen. In some embodiments, the mononuclear-enriched cell composition
undergoes at least one
washing step following freezing and thawing. In some embodiments, washing
steps comprise
centrifugation of the mononuclear-enriched cell composition followed by
supernatant extraction and
re-suspension in washing medium.
[0338] In some embodiments, the mononuclear-enriched cell composition
undergoes at least one
washing step between each stage of the production of an early apoptotic cell
population. In some
embodiments, anti-coagulant is added to washing media during washing steps
throughout the
production of an early apoptotic cell population. In some embodiments, the
mononuclear-enriched
cell composition undergoes at least one washing step following incubation. In
some embodiments,
the mononuclear-enriched cell composition undergoes at least one washing step
following incubation
using PBS. In some embodiments, anti-coagulant is not added to the final
washing step prior to re-
suspension of the cell-preparation in the administration medium. In some
embodiments, anti-
coagulant is not added to the PBS used in the final washing step prior to re-
suspension of the cell-
preparation in the administration medium. In certain embodiments, anti-
coagulant is not added to the
administration medium.
[0339] In some embodiments, the cell concentration during incubating is
about 5x106 cells/ml.
[0340] In some embodiments, the mononuclear-enriched cell composition is
suspended in an
administration medium following freezing, thawing and incubating, thereby
resulting in the
pharmaceutical population. In some embodiments, the administration medium
comprises a suitable
physiological buffer. Non-limiting examples of a suitable physiological buffer
are: saline solution,
Phoshpate Buffered Saline (PBS), Hank's Balanced Salt Solution (HBSS), and the
like. In some
embodiments, the administration medium comprises PBS. In some embodiments, the
administration
medium comprises supplements conducive to maintaining the viability of the
cells. In some
embodiments, the mononuclear-enriched cell composition is filtered prior to
administration. In some
embodiments, the mononuclear-enriched cell composition is filtered prior to
administration using a
filter of at least 200i.t.m.
[0341] In some embodiments, the mononuclear-enriched cell population is
re-suspended in an
administration medium such that the final volume of the resulting cell-
preparation is between 100-
1000m1, possibly between 200-800m1, typically between 300-600m1.
[0342] In some embodiments, cell collection refers to obtaining a
mononuclear-enriched cell
composition. In some embodiments, washing steps performed during the
production of an early
apoptotic cell population are performed in a washing medium. In certain
embodiments, washing steps
performed up until the incubation step of the production of an early apoptotic
cell population are
CA 03151815 2022-02-17
79
WO 2021/053667
PCT/IL2020/051011
performed in a washing medium. In some embodiments, the washing medium
comprises RPMI 1640
medium supplemented with L-glutamine and Hepes. In some embodiments, the
washing medium
comprises RPMI 1640 medium supplemented with 2 mM L-glutamine and 10 mM Hepes.
[0343] In some embodiments, the washing medium comprises an anti-coagulant. In
some
embodiments, the washing medium comprises an anti-coagulant selected from the
group consisting
of: heparin, ACD Formula A and a combination thereof. In some embodiments, the
concentration of
the anti-coagulant in the washing medium is the same concentration as in the
freezing medium. In
some embodiments, the concentration of the anti-coagulant in the washing
medium is the same
concentration as in the incubation medium. In some embodiments, the anti-
coagulant used in the
washing medium is ACD Formula A containing heparin at a concentration of 10
U/ml.
[0344] In some embodiments, the washing medium comprises heparin. In some
embodiments, the
heparin in the washing medium is at a concentration of between 0.1-2.5 U/ml.
In some embodiments,
the heparin in the washing medium is at a concentration of between 0.1-2.5
U/ml, possibly between
0.3-0.7 U/ml, typically about 0.5 U/ml. In certain embodiments, the heparin in
the washing medium
is at a concentration of about 0.5 U/ml.
[0345] In some embodiments, the washing medium comprises ACD Formula A. In
some
embodiments, the ACD Formula A in the washing medium is at a concentration of
between 1%-15%
v/v. In some embodiments, the ACD Formula A in the washing medium is at a
concentration of
between 1%-15% v/v, possibly between 4%-7% v/v, typically about 5% v/v. In
some embodiments,
the ACD Formula A in the washing medium is at a concentration of about 5% v/v.
[0346] In some embodiments, the mononuclear-enriched cell composition is
thawed several hours
prior to the intended administration of the population to a subject. In some
embodiments, the
mononuclear-enriched cell composition is thawed at about 33 C-39 C. In some
embodiments, the
mononuclear-enriched cell composition is thawed for about 30-240 seconds,
preferably 40-180
seconds, most preferably 50-120 seconds.
[0347] In some embodiments, the mononuclear-enriched cell composition is
thawed at least 10
hours prior to the intended administration of the population, alternatively at
least 20, 30, 40 or 50
hours prior to the intended administration of the population. In some
embodiments, the mononuclear-
enriched cell composition is thawed at least 15-24 hours prior to the intended
administration of the
population. In some embodiments, the mononuclear-enriched cell composition is
thawed at least
about 24 hours prior to the intended administration of the population. In some
embodiments, the
mononuclear-enriched cell composition is thawed at least 20 hours prior to the
intended
administration of the population. In some embodiments, the mononuclear-
enriched cell composition
is thawed 30 hours prior to the intended administration of the population. In
some embodiments, the
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
mononuclear-enriched cell composition is thawed at least 24 hours prior to the
intended
administration of the population. In some embodiments, the mononuclear-
enriched cell composition
undergoes at least one step of washing in the washing medium before and/or
after thawing.
[0348] In some embodiments, the composition further comprises
methylprednisolone. At some
5 embodiments, the concentration of methylprednisolone does not exceed
30i.tg/ml.
[0349] In some embodiments, the apoptotic cells are used at a high dose.
In some embodiments,
the apoptotic cells are used at a high concentration. In some embodiments,
human apoptotic
polymorphonuclear neutrophils (PMNs) are used. In some embodiments, a group of
cells, of which
50% are apoptotic cells, are used. In some embodiments, apoptotic cells are
verified by May-Giemsa-
10 stained cytopreps. In some embodiments, viability of cells are assessed
by trypan blue exclusion. In
some embodiments, the apoptotic and necrotic status of the cells are confirmed
by annexin
V/propidium iodide staining with detection by FACS.
[0350] In some embodiments, apoptotic cells disclosed herein comprise no
necrotic cells. In some
embodiments, apoptotic cells disclosed herein comprise less than 1% necrotic
cells. In some
15 embodiments, apoptotic cells disclosed herein comprise less than 2%
necrotic cells. In some
embodiments, apoptotic cells disclosed herein comprise less than 3% necrotic
cells. In some
embodiments, apoptotic cells disclosed herein comprise less than 4% necrotic
cells. In some
embodiments, apoptotic cells disclosed herein comprise less than 5% necrotic
cells.
[0351] In some embodiments, a dose of about 140 X 106 - 210 X 106
apoptotic cells are
20 administered. In some embodiments, a dose of about 10-100 x 106
apoptotic cells is administered. In
some embodiments, a dose of about 20 x 106 apoptotic cells is administered. In
some embodiments,
a dose of about 30 x 106 apoptotic cells is administered. In some embodiments,
a dose of about 40 x
106 apoptotic cells is administered. In some embodiments, a dose of about 50 x
106 apoptotic cells is
administered. In some embodiments, 60 x 106 apoptotic cells is administered.
In some embodiments,
25 a dose of about 60 x 106 apoptotic cells is administered. In some
embodiments, a dose of about 70 x
106 apoptotic cells is administered. In some embodiments, a dose of about 80 x
106 apoptotic cells is
administered. In some embodiments, a dose of about 90 x 106 apoptotic cells is
administered. In some
embodiments, a dose of about 1-15 x 107 apoptotic cells is administered. In
some embodiments, a
dose of about 10 x 107 apoptotic cells is administered. In some embodiments, a
dose of about 15 x
30 107 apoptotic cells is administered.
[0352] In some embodiments, a dose of 10x106 apoptotic cells is
administered. In another
embodiment, a dose of 10x107 apoptotic cells is administered. In another
embodiment, a dose of
10x108 apoptotic cells is administered. In another embodiment, a dose of
10x109 apoptotic cells is
administered. In another embodiment, a dose of 10x101 apoptotic cells is
administered. In another
CA 03151815 2022-02-17
81
WO 2021/053667
PCT/IL2020/051011
embodiment, a dose of 10x1011 apoptotic cells is administered. In another
embodiment, a dose of
10x1012 apoptotic cells is administered. In another embodiment, a dose of
10x105 apoptotic cells is
administered. In another embodiment, a dose of 10x104 apoptotic cells is
administered. In another
embodiment, a dose of 10x103 apoptotic cells is administered. In another
embodiment, a dose of
10x102 apoptotic cells is administered.
[0353]
In some embodiments, a high dose of apoptotic cells is administered. In
some
embodiments, a dose of 35x106 apoptotic cells is administered. In another
embodiment, a dose of
210x106 apoptotic cells is administered. In another embodiment, a dose of
70x106 apoptotic cells is
administered. In another embodiment, a dose of 140x106 apoptotic cells is
administered. In another
embodiment, a dose of 35-210x106apoptotic cells is administered.
[0354]
In some embodiments, a single dose of apoptotic cells is administered. In
some
embodiments, multiple doses of apoptotic cells are administered. In some
embodiments, 2 doses of
apoptotic cells are administered. In some embodiments, 3 doses of apoptotic
cells are administered.
In some embodiments, 4 doses of apoptotic cells are administered. In some
embodiments, 5 doses of
apoptotic cells are administered. In some embodiments, 6 doses of apoptotic
cells are administered.
In some embodiments, 7 doses of apoptotic cells are administered. In some
embodiments, 8 doses of
apoptotic cells are administered. In some embodiments, 9 doses of apoptotic
cells are administered.
In some embodiments, more than 9 doses of apoptotic cells are administered. In
some embodiments,
multiple doses of apoptotic cells are administered.
[0355] In some embodiments, the apoptotic cells may be administered by any
method known in
the art including, but not limited to, intravenous, subcutaneous, intranodal,
intratumoral, intrathecal,
intrapleural, intraperitoneal and directly to the thymus.
[0356]
In some embodiments, the apoptotic cells are prepared from cells obtained
from a subject
other than the subject that will receive said apoptotic cells. In some
embodiments, the methods as
disclosed herein comprise an additional step that is useful in overcoming
rejection of allogeneic donor
cells, including one or more steps described in U.S. Patent Application
20130156794, which is
incorporated herein by reference in its entirety. In some embodiments, the
methods comprise the step
of full or partial lymphodepletion prior to administration of the apoptotic
cells, which in some
embodiments, are allogeneic apoptotic cells. In some embodiments, the
lymphodepletion is adjusted
so that it delays the host versus graft reaction for a period sufficient to
allow the allogeneic apoptotic
cells to control cytokine release. In some embodiments, the methods comprise
the step of
administering agents that delay egression of the allogeneic apoptotic T-cells
from lymph nodes, such
as 2- amino-242-(4-octylphenyl)ethyl] prop ane-1,3 -diol
(FTY720), 544-pheny1-5-
(trifluoromethyl)thiophen-2-yl] -3 43 -(trifluoromethyl)pheny - 1] 1,2,4-
oxadiazole (SEW2871), 3 -(2-(-
CA 03151815 2022-02-17
82
WO 2021/053667
PCT/IL2020/051011
hexylphenylamino)-2-oxoethylamino)propanoic acid (W123), 2-ammonio-4-(2-chloro-
4-(3-
phenoxyphenylthio)pheny1)-2-(hydroxymethyl)but-y1 hydrogen phosphate (KRP-203
phosphate) or
other agents known in the art, may be used as part of the compositions and
methods as disclosed
herein to allow the use of allogeneic apoptotic cells having efficacy and
lacking initiation of graft vs
host disease. In another embodiment, MHC expression by the allogeneic
apoptotic T-cells is silenced
to reduce the rejection of the allogeneic cells.
[0357] In some embodiments, methods comprise producing a population of
mononuclear
apoptotic cell comprising a decreased percent of non-quiescent non-apoptotic
viable cells; a
suppressed cellular activation of any living non-apoptotic cells; or a reduced
proliferation of any
living non-apoptotic cells; or any combination thereof, said method comprising
the following steps,
obtaining a mononuclear-enriched cell population of peripheral blood; freezing
said mononuclear-
enriched cell population in a freezing medium comprising an anticoagulant;
thawing said
mononuclear-enriched cell population; incubating said mononuclear-enriched
cell population in an
apoptosis inducing incubation medium comprising methylprednisolone at a final
concentration of
about 10-100 i.t.g/mL and an anticoagulant; resuspending said apoptotic cell
population in an
administration medium; and inactivating said mononuclear-enriched population,
wherein said
inactivation occurs following apoptotic induction, wherein said method
produces a population of
mononuclear apoptotic cell comprising a decreased percent of non-quiescent non-
apoptotic cells; a
suppressed cellular activation of any living non-apoptotic cells; or a reduced
proliferation of any
living non-apoptotic cells; or any combination thereof.
[0358] In some embodiments, the methods comprise the step of irradiating a
population of apoptotic
cells derived from a subject prior to administration of the population of
apoptotic cells to the same
subject (autologous ApoCells). In some embodiments, the methods comprise the
step of irradiating
apoptotic cells derived from a subject prior to administration of the
population of apoptotic cells to a
recipient (allogeneic ApoCells).
[0359] In some embodiments, cells are irradiated in a way that will decrease
proliferation and/or
activation of residual viable cells within the apoptotic cell population. In
some embodiments, cells
are irradiated in a way that reduces the percent of viable non-apoptotic cells
in a population. In some
embodiments, the percent of viable non-apoptotic cells in an inactivated early
apoptotic cell
population is reduced to less than 50% of the population. In some embodiments,
the percent of viable
non-apoptotic cells in an inactivated early apoptotic cell population is
reduced to less than 40% of the
population. In some embodiments, the percent of viable non-apoptotic cells in
an inactivated early
apoptotic cell population is reduced to less than 30% of the population. In
some embodiments, the
percent of viable non-apoptotic cells in an inactivated early apoptotic cell
population is reduced to
CA 03151815 2022-02-17
83
WO 2021/053667
PCT/IL2020/051011
less than 20% of the population. In some embodiments, the percent of viable
non-apoptotic cells in
an inactivated early apoptotic cell population is reduced to less than 10% of
the population. In some
embodiments, the percent of viable non-apoptotic cells in an inactivated early
apoptotic cell
population is reduced to 0% of the population.
[0360] In another embodiment, the irradiated apoptotic cells preserve all
their early apoptotic-,
immune modulation-, stability-properties. In another embodiment, the
irradiation step uses UV
radiation. In another embodiment, the radiation step uses gamma radiation. In
another embodiment,
the apoptotic cells comprise a decreased percent of living non-apoptotic
cells, comprise a preparation
having a suppressed cellular activation of any living non-apoptotic cells
present within the apoptotic
cell preparation, or comprise a preparation having reduced proliferation of
any living non-apoptotic
cells present within the apoptotic cell preparation, or any combination
thereof.
[0361] In some embodiments, irradiation of apoptotic cells does not increase
the population of dead
cells (PI+) compared with apoptotic cells not irradiated. In some embodiments,
irradiation of
apoptotic cells does not increase the population of dead cells (PI+) by more
than about 1% compared
with apoptotic cells not irradiated. In some embodiments, irradiation of
apoptotic cells does not
increase the population of dead cells (PI+) by more than about 2% compared
with apoptotic cells not
irradiated. In some embodiments, irradiation of apoptotic cells does not
increase the population of
dead cells (PI+) by more than about 3% compared with apoptotic cells not
irradiated. In some
embodiments, irradiation of apoptotic cells does not increase the population
of dead cells (PI+) by
more than about 4% compared with apoptotic cells not irradiated. In some
embodiments, irradiation
of apoptotic cells does not increase the population of dead cells (PI+) by
more than about 5%
compared with apoptotic cells not irradiated. In some embodiments, irradiation
of apoptotic cells does
not increase the population of dead cells (PI+) by more than about 6% compared
with apoptotic cells
not irradiated. In some embodiments, irradiation of apoptotic cells does not
increase the population
of dead cells (PI+) by more than about 7% compared with apoptotic cells not
irradiated. In some
embodiments, irradiation of apoptotic cells does not increase the population
of dead cells (PI+) by
more than about 8% compared with apoptotic cells not irradiated. In some
embodiments, irradiation
of apoptotic cells does not increase the population of dead cells (PI+) by
more than about 9%
compared with apoptotic cells not irradiated. In some embodiments, irradiation
of apoptotic cells does
not increase the population of dead cells (PI+) by more than about 10%
compared with apoptotic cells
not irradiated. In some embodiments, irradiation of apoptotic cells does not
increase the population
of dead cells (PI+) by more than about 15% compared with apoptotic cells not
irradiated. In some
embodiments, irradiation of apoptotic cells does not increase the population
of dead cells (PI+) by
more than about 20%, 25%, 30%, 35%, 40%, 45%, or 50% compared with apoptotic
cells not
CA 03151815 2022-02-17
84
WO 2021/053667
PCT/IL2020/051011
irradiated.
[0362] In some embodiments, a cell population comprising a reduced or non-
existent fraction of
living non-apoptotic cells may in one embodiment provide a mononuclear early
apoptotic cell
population that does not have any living / viable cells. In some embodiments,
a cell population
comprising a reduced or non-existent fraction of living non-apoptotic cells
may in one embodiment
provide a mononuclear apoptotic cell population that does not elicit GVHD in a
recipient.
[0363] In some embodiments, use of irradiated ApoCells removes the possible
graft versus leukemia
effect use of an apoptotic population (that includes a minor portion of viable
cells) may cause,
demonstrating that the effects shown here in the Examples (See Example 8)
result from the apoptotic
cells and not from a viable proliferating population of cells with cellular
activity, present within the
apoptotic cell population.
[0364] In another embodiment, the methods comprise the step of irradiating
apoptotic cells derived
from WBCs from a donor prior to administration to a recipient. In some
embodiments, cells are
irradiated in a way that will avoid proliferation and/or activation of
residual viable cells within the
apoptotic cell population. In another embodiment, the irradiated apoptotic
cells preserve all their early
apoptotic-, immune modulation-, stability-properties. In another embodiment,
the irradiation step uses
UV radiation. In another embodiment, the radiation step uses gamma radiation.
In another
embodiment, the apoptotic cells comprise a decreased percent of living non-
apoptotic cells, comprise
a preparation having a suppressed cellular activation of any living non-
apoptotic cells present within
the apoptotic cell preparation, or comprise a preparation having reduced
proliferation of any living
non-apoptotic cells present within the apoptotic cell preparation, or any
combination thereof.
[0365] In some embodiments, apoptotic cells comprise a pooled mononuclear
apoptotic cell
preparation. In some embodiments, a pooled mononuclear apoptotic cell
preparation comprises
mononuclear cells in an early apoptotic state, wherein said pooled mononuclear
apoptotic cells
comprise a decreased percent of living non-apoptotic cells, a preparation
having a suppressed cellular
activation of any living non-apoptotic cells, or a preparation having reduced
proliferation of any living
non-apoptotic cells, or any combination thereof. In another embodiment, the
pooled mononuclear
apoptotic cells have been irradiated. In another embodiment, disclosed herein
is a pooled mononuclear
apoptotic cell preparation that in some embodiments, originates from the white
blood cell fraction
(WBC) obtained from donated blood.
[0366] In some embodiments, the apoptotic cell preparation is irradiated. In
another embodiment,
said irradiation comprises gamma irradiation or UV irradiation. In yet another
embodiment, the
irradiated preparation has a reduced number of non-apoptotic cells compared
with a non-irradiated
apoptotic cell preparation. In another embodiment, the irradiated preparation
has a reduced number
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
of proliferating cells compared with a non-irradiated apoptotic cell
preparation. In another
embodiment, the irradiated preparation has a reduced number of potentially
immunologically active
cells compared with a non-irradiated apoptotic cell population.
[0367] In some embodiments, pooled blood comprises 3rd party blood not matched
between donor
5 and recipient.
[0368] A skilled artisan would appreciate that the term "pooled" may encompass
blood collected
from multiple donors, prepared and possibly stored for later use. This
combined pool of blood may
then be processed to produce a pooled mononuclear apoptotic cell preparation.
In another
embodiment, a pooled mononuclear apoptotic cell preparation ensures that a
readily available supply
10 of mononuclear apoptotic cells is available. In another embodiment,
cells are pooled just prior to the
incubation step wherein apoptosis is induced. In another embodiment, cells are
pooled following the
incubation step at the step of resuspension. In another embodiment, cells are
pooled just prior to an
irradiation step. In another embodiment, cells are pooled following an
irradiation step. In another
embodiment, cells are pooled at any step in the methods of preparation.
15 [0369] In some embodiments, a pooled apoptotic cell preparation is
derived from cells present in
between about 2 and 25 units of blood. In another embodiment, said pooled
apoptotic cell preparation
is comprised of cells present in between about 2-5, 2-10, 2-15, 2-20, 5-10, 5-
15, 5-20, 5-25, 10-15,
10-20, 10-25, 6-13, or 6-25 units of blood. In another embodiment, said pooled
apoptotic cell
preparation is comprised of cells present in about 2, 3, 4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17,
20 18, 19, 20, 21, 22, 23, 24 or 25 units of blood. The number of units of
blood needed is also dependent
upon the efficiency of WBC recovery from blood. For example, low efficiency
WBC recovery would
lead to the need for additional units, while high efficiency WBC recovery
would lead to fewer units
needed. In some embodiments, each unit is a bag of blood. In another
embodiment, a pooled apoptotic
cell preparation is comprised of cells present in at least 25 units of blood,
at least 50 units of blood,
25 or at least 100 units of blood.
[0370] In some embodiments, the units of blood comprise white blood cell (WBC)
fractions from
blood donations. In another embodiment, the donations may be from a blood
center or blood bank. In
another embodiment, the donations may be from donors in a hospital gathered at
the time of
preparation of the pooled apoptotic cell preparation. In another embodiment,
units of blood
30 comprising WBCs from multiple donors are saved and maintained in an
independent blood bank
created for the purpose of compositions and methods thereof as disclosed
herein. In another
embodiment, a blood bank developed for the purpose of compositions and methods
thereof as
disclosed herein, is able to supply units of blood comprising WBC from
multiple donors and
comprises a leukapheresis unit.
CA 03151815 2022-02-17
86
WO 2021/053667
PCT/IL2020/051011
[0371] In some embodiments, the units of pooled WBCs are not restricted by HLA
matching.
Therefore, the resultant pooled apoptotic cell preparation comprises cell
populations not restricted by
HLA matching. Accordingly, in certain embodiments a pooled mononuclear
apoptotic cell
preparation comprises allogeneic cells.
[0372] An advantage of a pooled mononuclear apoptotic cell preparation that is
derived from pooled
WBCs not restricted by HLA matching, is a readily available source of WBCs and
reduced costs of
obtaining WBCs.
[0373] In some embodiments, pooled blood comprises blood from multiple donors
independent of
HLA matching. In another embodiment, pooled blood comprises blood from
multiple donors wherein
HLA matching with the recipient has been taken into consideration. For
example, wherein 1 HLA
allele, 2 HLA alleles, 3 HLA alleles, 4 HLA alleles, 5 HLA alleles, 6 HLA
alleles, or 7 HLA alleles
have been matched between donors and recipient. In another embodiment,
multiple donors are
partially matched, for example some of the donors have been HLA matched
wherein 1 HLA allele, 2
HLA alleles, 3 HLA alleles, 4 HLA alleles, 5 HLA alleles, 6 HLA alleles, or 7
HLA alleles have been
matched between some of the donors and recipient. Each possibility comprises
an embodiment as
disclosed herein.
[0374] In certain embodiments, some viable non-apoptotic cells (apoptosis
resistant) may remain
following the induction of apoptosis step described below (Example 1). The
presence of these viable
non-apoptotic cells is, in some embodiments, is observed prior to an
irradiation step. These viable
non-apoptotic cells may be able to proliferate or be activated. In some
embodiments, the pooled
mononuclear apoptotic cell preparation derived from multiple donors may be
activated against the
host, activated against one another, or both.
[0375] In some embodiments, an irradiated cell preparation as disclosed herein
has suppressed
cellular activation and reduced proliferation compared with a non-irradiated
cell preparation. In
another embodiment, the irradiation comprises gamma irradiation or UV
irradiation. In another
embodiment, an irradiated cell preparation has a reduced number of non-
apoptotic cells compared
with a non-irradiated cell preparation. In another embodiment, the irradiation
comprises about 15
Grey units (Gy). In another embodiment, the irradiation comprises about 20
Grey units (Gy). In
another embodiment, the irradiation comprises about 25 Grey units (Gy). In
another embodiment, the
irradiation comprises about 30 Grey units (Gy). In another embodiment, the
irradiation comprises
about 35 Grey units (Gy). In another embodiment, the irradiation comprises
about 40 Grey units (Gy).
In another embodiment, the irradiation comprises about 45 Grey units (Gy). In
another embodiment,
the irradiation comprises about 50 Grey units (Gy). In another embodiment, the
irradiation comprises
about 55 Grey units (Gy). In another embodiment, the irradiation comprises
about 60 Grey units (Gy).
CA 03151815 2022-02-17
87
WO 2021/053667
PCT/IL2020/051011
In another embodiment, the irradiation comprises about 65 Grey units (Gy). In
another embodiment,
the irradiation comprises up to 2500 Gy. In another embodiment, an irradiated
pooled apoptotic cell
preparation maintains the same or a similar apoptotic profile, stability and
efficacy as a non-irradiated
pooled apoptotic cell preparation.
[0376] In some embodiments, a pooled mononuclear apoptotic cell preparation as
disclosed herein is
stable for up to 24 hours. In another embodiment, a pooled mononuclear
apoptotic cell preparation is
stable for at least 24 hours. In another embodiment, a pooled mononuclear
apoptotic cell preparation
is stable for more than 24 hours. In yet another embodiment, a pooled
mononuclear apoptotic cell
preparation as disclosed herein is stable for up to 36 hours. In still another
embodiment, a pooled
mononuclear apoptotic cell preparation is stable for at least 36 hours. In a
further embodiment, a
pooled mononuclear apoptotic cell preparation is stable for more than 36
hours. In another
embodiment, a pooled mononuclear apoptotic cell preparation as disclosed
herein is stable for up to
48 hours. In another embodiment, a pooled mononuclear apoptotic cell
preparation is stable for at
least 48 hours. In another embodiment, a pooled mononuclear apoptotic cell
preparation is stable for
more than 48 hours.
[0377] In some embodiments, methods of producing the pooled cell preparation
comprising an
irradiation step preserves the early apoptotic, immune modulation, and
stability properties observed
in an apoptotic preparation derived from a single match donor wherein the cell
preparation may not
include an irradiation step. In another embodiment, a pooled mononuclear
apoptotic cell preparation
as disclosed herein does not elicit a graft versus host disease (GVHD)
response.
[0378] Irradiation of the cell preparation is considered safe in the art.
Irradiation procedures are
currently performed on a routine basis to donated blood to prevent reactions
to WBC.
[0379] In another embodiment, the percent of apoptotic cells in a pooled
mononuclear apoptotic cell
preparation as disclosed herein is close to 100%, thereby reducing the
fraction of living non-apoptotic
cells in the cell preparation. In some embodiments, the percent of apoptotic
cells is at least 40%. In
another embodiment, the percent of apoptotic cells is at least 50%. In yet
another embodiment, the
percent of apoptotic cells is at least 60%. In still another embodiment, the
percent of apoptotic cells
is at least 70%. In a further embodiment, the percent of apoptotic cells is at
least 80%. In another
embodiment, the percent of apoptotic cells is at least 90%. In yet another
embodiment, the percent of
apoptotic cells is at least 99%. Accordingly, a cell preparation comprising a
reduced or non-existent
fraction of living non-apoptotic cells may in one embodiment provide a pooled
mononuclear
apoptotic cell preparation that does not elicit GVHD in a recipient. Each
possibility represents an
embodiment as disclosed herein.
[0380] Alternatively, in another embodiment, the percentage of living non-
apoptotic WBC is reduced
CA 03151815 2022-02-17
88
WO 2021/053667
PCT/IL2020/051011
by specifically removing the living cell population, for example by targeted
precipitation. In another
embodiment, the percent of living non-apoptotic cells may be reduced using
magnetic beads that bind
to phosphatidylserine. In another embodiment, the percent of living non-
apoptotic cells may be
reduced using magnetic beads that bind a marker on the cell surface of non-
apoptotic cells but not
.. apoptotic cells. In another embodiment, the apoptotic cells may be selected
for further preparation
using magnetic beads that bind to a marker on the cell surface of apoptotic
cells but not non-apoptotic
cells. In yet another embodiment, the percentage of living non-apoptotic WBC
is reduced by the use
of ultrasound.
[0381] In one embodiment the apoptotic cells are from pooled third-party
donors.
[0382] In some embodiments, a pooled cell preparation comprises at least one
cell type selected from
the group consisting of: lymphocytes, monocytes and natural killer cells. In
another embodiment, a
pooled cell preparation comprises an enriched population of mononuclear cells.
In some
embodiments, a pooled mononuclear is a mononuclear enriched cell preparation
comprises cell types
selected from the group consisting of: lymphocytes, monocytes and natural
killer cells. In another
embodiment, the mononuclear enriched cell preparation comprises no more than
15%, alternatively
no more than 10%, typically no more than 5% polymorphonuclear leukocytes, also
known as
granulocytes (i.e., neutrophils, basophils and eosinophils). In another
embodiment, a pooled
mononuclear cell preparation is devoid of granulocytes.
[0383] In another embodiment, the pooled mononuclear enriched cell preparation
comprises no more
.. than 15%, alternatively no more than 10%, typically no more than 5%
CD15high expressing cells. In
some embodiments, a pooled apoptotic cell preparation comprises less than 15%
CD15 high
expressing cells.
[0384] In some embodiments, the pooled mononuclear enriched cell preparation
disclosed herein
comprises at least 80% mononuclear cells, at least 85% mononuclear cells,
alternatively at least 90%
.. mononuclear cells, or at least 95% mononuclear cells, wherein each
possibility is a separate
embodiment disclosed herein. According to some embodiments, the pooled
mononuclear enriched
cell preparation disclosed herein comprises at least 85% mononuclear cells.
[0385] In another embodiment, any pooled cell preparation that has a final
pooled percent of
mononuclear cells of at least 80% is considered a pooled mononuclear enriched
cell preparation as
disclosed herein. Thus, pooling cell preparations having increased
polymorphonuclear cells (PMN)
with cell preparations having high mononuclear cells with a resultant "pool"
of at least 80%
mononuclear cells comprises a preparation as disclosed herein. According to
some embodiments,
mononuclear cells comprise lymphocytes and monocytes.
[0386] A skilled artisan would appreciate that the term "mononuclear
cells" may encompass
CA 03151815 2022-02-17
89
WO 2021/053667
PCT/IL2020/051011
leukocytes having a one lobed nucleus. In another embodiment, a pooled
apoptotic cell preparation
as disclosed herein comprises less than 5% polymorphonuclear leukocytes.
[0387] In some embodiments, the apoptotic cells are T-cells. In another
embodiment, the apoptotic
cells are derived from the same pooled third-party donor T-cells as the CAR T-
cells. In another
.. embodiment, the apoptotic cells are derived from the CAR T-cell population.
[0388] Surprisingly, the apoptotic cells reduce production of cytokines
associated with the cytokine
storm including but not limited to IL-6, and interferon-gamma (IFN-y), alone
or in combination,
while the effectiveness of CAR T-cell therapy was maintained (Example 2). In
one embodiment, the
apoptotic cells affect cytokine expression levels in macrophages. In another
embodiment, the
apoptotic cells reduce cytokine expression levels in macrophages. In one
embodiment, the apoptotic
cells suppress cytokine expression levels in macrophages. In one embodiment,
the apoptotic cells
inhibit cytokine expression levels in macrophages. In one embodiment, the
apoptotic cells maintain
IFN-y levels to match or nearly match levels present prior to CAR ¨T cell
administration. In another
embodiment, apoptotic cells affect cytokine expression levels in macrophages
but do not affect
cytokine expression levels in the CAR T-cells. In another embodiment, the
apoptotic cells affect
cytokine expression levels in DCs, but do not affect cytokine expression
levels in the CAR T-cells.
It was therefore unexpected that apoptotic cells would be useful in
maintaining the effectiveness CAR
T-cell therapy.
[0389] In another embodiment, the effect of apoptotic cells on cytokine
expression levels in
macrophages, DCs, or a combination thereof, results in reduction of CRS. In
another embodiment,
the effect of apoptotic cells on cytokine expression levels in macrophages,
DCs, or a combination
thereof, results in reduction of severe CRS. In another embodiment, the effect
of apoptotic cells on
cytokine expression levels in macrophages, DCs, or a combination thereof,
results in suppression of
CRS. In another embodiment, the effect of apoptotic cells on cytokine
expression levels in
.. macrophages, DCs, or a combination thereof, results in suppression of
severe CRS. In another
embodiment, the effect of apoptotic cells on cytokine expression levels in
macrophages, DCs, or a
combination thereof, results in inhibition of CRS. In another embodiment, the
effect of apoptotic cells
on cytokine expression levels in macrophages, DCs, or a combination thereof,
results in inhibition of
severe CRS. In another embodiment, the effect of apoptotic cells on cytokine
expression levels in
macrophages, DCs, or a combination thereof, results in prevention of CRS. In
another embodiment,
the effect of apoptotic cells on cytokine expression levels in macrophages,
DCs, or a combination
thereof, results in prevention of severe CRS.
[0390] In another embodiment, the apoptotic cells trigger death of T-cells,
but not via changes in
cytokine expression levels.
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
[0391] In another embodiment, apoptotic cells antagonize the priming of
macrophages and dendritic
cells to secrete cytokines that would otherwise amplify the cytokine storm. In
another embodiment,
apoptotic cells increase Tregs which suppress the inflammatory response and/or
prevent excess
release of cytokines.
5 [0392] In some embodiments, apoptotic cells stabilize the presence
of resident macrophages in the
area of the tumor. In some embodiments, methods of treating a cancer by
combining cancer therapy
with administration of early apoptotic cells, results in a stabilization of
resident macrophages in the
area of the tumor. In some embodiments, methods of treating a cancer by
combining a cancer
therapeutic agent with administration of early apoptotic cells, results in a
stabilization of resident
10 macrophages in the area of the tumor. In some embodiments, methods of
treating a cancer by
combining CAR-T cell therapy with administration of early apoptotic cells,
results in a stabilization
of resident macrophages in the area of the tumor.
[0393] In some embodiments, administration of apoptotic cells inhibits one or
more pro-
inflammatory cytokines. In some embodiments, the pro-inflammatory cytokine
comprises IL- lbeta,
15 IL-6, TNF-alpha, or IFN-gamma, or any combination thereof. In another
embodiment, administration
of apoptotic cells promotes the secretion of one or more anti-inflammatory
cytokines. In some
embodiments, the anti-inflammatory cytokine comprises TGF-beta, IL10, or PGE2,
or any
combination thereof.
[0394] In another embodiment, administration of apoptotic cells inhibits
dendritic cell maturation
20 following exposure to TLR ligands. In another embodiment, administration
of apoptotic cells creates
potentially tolerogenic dendritic cells, which in some embodiments, are
capable of migration, and in
some embodiments, the migration is due to CCR7. In another embodiment,
administration of
apoptotic cells elicits various signaling events which in one embodiment is
TAM receptor signaling
(Tyro3, Axl and Mer) which in some embodiments, inhibits inflammation in
antigen-presenting cells.
25 [0395] In some embodiments, Tyro-3, Axl, and Mer constitute the TAM
family of receptor tyrosine
kinases (RTKs) characterized by a conserved sequence within the kinase domain
and adhesion
molecule-like extracellular domains. In another embodiment, administration of
apoptotic cells
activates signaling through MerTK. In another embodiment, administration of
apoptotic cells
activates the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which in some
embodiments,
30 negatively regulates NF-KB. In another embodiment, administration of
apoptotic cells negatively
regulates the inflammasome which in one embodiment leads to inhibition of pro-
inflammatory
cytokine secretion, DC maturation, or a combination thereof. In another
embodiment, administration
of apoptotic cells upregulates expression of anti-inflammatory genes such as
Nr4a, Thbsl, or a
combination thereof. In another embodiment, administration of apoptotic cells
induces a high level of
CA 03151815 2022-02-17
91
WO 2021/053667
PCT/IL2020/051011
AMP which in some embodiments, is accumulated in a Pannexinl-dependent manner.
In another
embodiment, administration of apoptotic cells suppresses inflammation.
Apoptotic Cell Supernatants (ApoSup and ApoSup Mon)
[0396] In some embodiments, compositions for use in the methods and treatments
as disclosed herein
include an apoptotic cell supernatant as disclosed herein.
[0397] In some embodiments, the apoptotic cell supernatant is obtained by a
method comprising the
steps of a) providing apoptotic cells, b) culturing the apoptotic cells of
step a), and c) separating the
supernatant from the cells.
[0398] In some embodiments, apoptotic cells for use making an apoptotic cell
supernatant as
disclosed herein are autologous with a subject undergoing therapy. In another
embodiment, apoptotic
cells for use in making an apoptotic cell supernatant disclosed herein are
allogeneic with a subject
undergoing therapy.
[0399] The "apoptotic cells" from which the apoptotic cell supernatant is
obtained may be cells
chosen from any cell type of a subject, or any commercially available cell
line, subjected to a method
of inducing apoptosis known to the person skilled in the art. The method of
inducing apoptosis may
be hypoxia, ozone, heat, radiation, chemicals, osmotic pressure, pH shift, X-
ray irradiation, gamma-
ray irradiation, UV irradiation, serum deprivation, corticoids or combinations
thereof, or any other
method described herein or known in the art. In another embodiment, the method
of inducing
apoptosis produces apoptotic cells in an early apoptotic state.
[0400] In some embodiments, the apoptotic cells are leukocytes.
[0401] In an embodiment, said apoptotic leukocytes are derived from peripheral
blood mononuclear
cells (PBMC). In another embodiment, said leukocytes are from pooled third-
party donors. In another
embodiment, said leukocytes are allogeneic.
[0402] According to some embodiments, the apoptotic cells are provided by
selecting non-adherent
leukocytes and submitting them to apoptosis induction, followed by a cell
culture step in culture
medium. "Leukocytes" used to make the apoptotic cell-phagocyte supernatant may
be derived from
any lineage, or sub-lineage, of nucleated cells of the immune system and/or
hematopoietic system,
including but not limited to dendritic cells, macrophages, masT-cells,
basophils, hematopoietic stem
cells, bone marrow cells, natural killer cells, and the like. The leukocytes
may be derived or obtained
in any of various suitable ways, from any of various suitable anatomical
compartments, according to
any of various commonly practiced methods, depending on the application and
purpose, desired
leukocyte lineage, etc. In some embodiments, the source leukocytes are primary
leukocytes. In
another embodiment, the source leukocytes are primary peripheral blood
leukocytes.
[0403] Primary lymphocytes and monocytes may be conveniently derived from
peripheral blood.
CA 03151815 2022-02-17
92
WO 2021/053667
PCT/IL2020/051011
Peripheral blood leukocytes include 70-95 percent lymphocytes, and 5-25
percent monocytes.
[0404] Methods for obtaining specific types of source leukocytes from blood
are routinely practiced.
Obtaining source lymphocytes and/or monocytes can be achieved, for example, by
harvesting blood
in the presence of an anticoagulant, such as heparin or citrate. The harvested
blood is then centrifuged
over a Ficoll cushion to isolate lymphocytes and monocytes at the gradient
interface, and neutrophils
and erythrocytes in the pellet.
[0405] Leukocytes may be separated from each other via standard immunomagnetic
selection or
immunofluorescent flow cytometry techniques according to their specific
surface markers, or via
centrifugal elutriation. For example, monocytes can be selected as the CD14+
fraction, T-
lymphocytes can be selected as CD3+ fraction, B-lymphocytes can be selected as
the CD19+ fraction,
macrophages as the CD206+ fraction.
[0406] Lymphocytes and monocytes may be isolated from each other by subjecting
these cells to
substrate-adherent conditions, such as by static culture in a tissue culture-
treated culturing recipient,
which results in selective adherence of the monocytes, but not of the
lymphocytes, to the cell-adherent
substrate.
[0407] Leukocytes may also be obtained from peripheral blood mononuclear cells
(PBMCs), which
may be isolated as described herein.
[0408] One of ordinary skill in the art will possess the necessary expertise
to suitably culture primary
leukocytes so as to generate desired quantities of cultured source leukocytes
as disclosed herein, and
ample guidance for practicing such culturing methods is available in the
literature of the art.
[0409] One of ordinary skill in the art will further possess the necessary
expertise to establish,
purchase, or otherwise obtain suitable established leukocyte cell lines from
which to derive the
apoptotic leukocytes. Suitable leukocyte cell lines may be obtained from
commercial suppliers, such
as the American Tissue Type Collection (ATCC). It will be evident to the
person skilled in the art that
.. source leukocytes should not be obtained via a technique which will
significantly interfere with their
capacity to produce the apoptotic leukocytes.
[0410] In another embodiment, the apoptotic cells may be apoptotic
lymphocytes. Apoptosis of
lymphocytes, such as primary lymphocytes, may be induced by treating the
primary lymphocytes
with serum deprivation, a corticosteroid, or irradiation. In another
embodiment, inducing apoptosis
of primary lymphocytes via treatment with a corticosteroid is effected by
treating the primary
lymphocytes with dexamethasone. In another embodiment, with dexamethasone at a
concentration of
about 1 micromolar. In another embodiment, inducing apoptosis of primary
lymphocytes via
irradiation is effected by treating the primary lymphocytes with gamma-
irradiation. In another
embodiment, with a dosage of about 66 rad. Such treatment results in the
generation of apoptotic
CA 03151815 2022-02-17
93
WO 2021/053667
PCT/IL2020/051011
lymphocytes suitable for the co-culture step with phagocytes.
[0411] In a further embodiment, apoptotic cells may be apoptotic monocytes,
such as primary
monocytes. To generate apoptotic monocytes the monocytes are subjected to in
vitro conditions of
substrate/surface-adherence under conditions of serum deprivation. Such
treatment results in the
.. generation of non-pro-inflammatory apoptotic monocytes suitable for the co-
culture step with
phagocytes.
[0412] In other embodiments, the apoptotic cells may be any apoptotic cells
described herein,
including allogeneic apoptotic cells, third-party apoptotic cells, and pools
of apoptotic cells.
[0413] In other embodiments, the apoptotic cell supernatant may be obtained
through the co-culture
of apoptotic cells with other cells.
[0414] Thus, in some embodiments, the apoptotic cell supernatant is an
apoptotic cell supernatant
obtained by a method comprising the steps of a) providing apoptotic cells, b)
providing other cells, c)
optionally washing the cells from step a) and b), d) co-culturing the cells of
step a) and b), and
optionally e) separating the supernatant from the cells.
[0415] In some embodiments, the other cells co-cultured with the apoptotic
cells are white blood
cells.
[0416] Thus, in some embodiments, the apoptotic cell supernatant is an
apoptotic cell-white blood
cell supernatant obtained by a method comprising the steps of a) providing
apoptotic cells, b)
providing white blood cells, c) optionally washing the cells from step a) and
b), d) co-culturing the
cells of step a) and b), and optionally e) separating the supernatant from the
cells.
[0417] In some embodiments, the white blood cells may be phagocytes, such as
macrophages,
monocytes or dendritic cells.
[0418] In some embodiments, the white blood cells may be B cells, T-cells, or
natural killer (NK
cells).
[0419] Thus, in some embodiments, compositions for use in the methods and
treatments as disclosed
herein include apoptotic cell-phagocyte supernatants as described in WO
2014/106666, which is
incorporated by reference herein in its entirety. In another embodiment,
apoptotic cell-phagocyte
supernatants for use in compositions and methods as disclosed herein are
produced in any way that is
known in the art.
[0420] In some embodiments, the apoptotic cell-phagocyte supernatant is
obtained from a co-culture
of phagocytes with apoptotic cells,
[0421] In some embodiments, the apoptotic cell-phagocyte supernatant is
obtained by a method
comprising the steps of a) providing phagocytes, b) providing apoptotic cells,
c) optionally washing
the cells from step a) and b), d) co-culturing the cells of step a) and b),
and optionally e) separating
CA 03151815 2022-02-17
94
WO 2021/053667
PCT/IL2020/051011
the supernatant from the cells.
[0422] The term "phagocytes" denotes cells that protect the body by ingesting
(phagocytosing)
harmful foreign particles, bacteria, and dead or dying cells. Phagocytes
include for example cells
called neutrophils, monocytes, macrophages, dendritic cells, and mast T-cells,
preferentially dendritic
cells and monocytes/macrophages. The phagocytes may be dendritic cells (CD4+
HLA-DR+
Lineage- BDCA1 /BDCA3+), macrophages (CD14+ CD206+ HLA-DR+), or derived from
monocytes (CD14+). Techniques to distinguish these different phagocytes are
known to the person
skilled in the art.
[0423] In an embodiment, monocytes are obtained by a plastic adherence step.
Said monocytes can
be distinguished from B and T-cells with the marker CD14+, whereas unwanted B
cells express
CD19+ and T-cells CD3+. After Macrophage Colony Stimulating Factor (M-CSF)
induced
maturation the obtained macrophages are in some embodiments, positive for the
markers CD14+,
CD206+, HLA-DR+.
[0424] In an embodiment, said phagocytes are derived from peripheral blood
mononuclear cells
(PBMC).
[0425] Phagocytes may be provided by any method known in the art for obtaining
phagocytes. In
some embodiments, phagocytes such as macrophages or dendritic cells can be
directly isolated from
a subject or be derived from precursor cells by a maturation step.
[0426] In some embodiments, macrophages may be directly isolated from the
peritoneum cavity of
a subject and cultured in complete RRPMI medium. Macrophages can also be
isolated from the
spleen.
[0427] Phagocytes are also obtainable from peripheral blood monocytes. In said
example, monocytes
when cultured differentiate into monocyte-derived macrophages upon addition
of, without limitation
to, macrophage colony stimulating factor (M-CSF) to the cell culture media.
[0428] For example, phagocytes may be derived from peripheral blood
mononuclear cells (PBMC).
For example, PBMC may be isolated from cytapheresis bag from an individual
through Ficoll
gradient centrifugation, plated in a cell-adherence step for 90 min in
complete RPMI culture medium
(10% FBS, 1 % Penicillin/Streptomycin). Non-adherent T-cells are removed by a
plastic adherence
step, and adherent T-cells cultured in complete RPMI milieu supplemented with
recombinant human
M-CSF. After the culture period, monocyte-derived macrophages are obtained.
[0429] Phagocytes can be selected by a cell-adherence step. Said "cell
adherence step" means that
phagocytes or cells which can mature into phagocytes are selected via
culturing conditions allowing
the adhesion of the cultured cells to a surface, a cell adherent surface (e.g.
a tissue culture dish, a
matrix, a sac or bag with the appropriate type of nylon or plastic). A skilled
artisan would appreciate
CA 03151815 2022-02-17
WO 2021/053667
PCT/IL2020/051011
that the term "Cell adherent surfaces" may encompass hydrophilic and
negatively charged, and may
be obtained in any of various ways known in the art, In another embodiment by
modifying a
polystyrene surface using, for example, corona discharge, or gas-plasma. These
processes generate
highly energetic oxygen ions which graft onto the surface polystyrene chains
so that the surface
5 becomes hydrophilic and negatively charged. Culture recipients designed
for facilitating cell-
adherence thereto are available from various commercial suppliers (e.g.
Corning, Perkin-Elmer,
Fisher Scientific, Evergreen Scientific, Nunc, etc.).
[0430] B cells, T-cells and NK cells may be provided by any method known in
the art for obtaining
such cells. In some embodiments, B cells, T-cells or NK cells can be directly
isolated from a subject
10 or be derived from precursor cells by a maturation step. In another
embodiment, the B, T or NK cells
can be from a B, T or NK cell line. One of ordinary skill in the art will
possess the necessary expertise
to establish, purchase, or otherwise obtain suitable established B cells, T-
cells and NK cell lines.
Suitable cell lines may be obtained from commercial suppliers, such as the
American Tissue Type
Collection (ATCC).
15 [0431] In an embodiment, said apoptotic cells and said white blood
cells, such as the phagocytes, B,
T or NK cells, are cultured individually prior to the co-culture step d).
[0432] The cell maturation of phagocytes takes place during cell culture, for
example due to addition
of maturation factors to the media. In one embodiment said maturation factor
is M-CSF, which may
be used for example to obtain monocyte-derived macrophages.
20 [0433] The culture step used for maturation or selection of phagocytes
might take several hours to
several days. In another embodiment said pre-mature phagocytes are cultured
for 2, 4, 6, 8, 10, 12,
14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50,
52, 54, 56, 58 hours in an
appropriate culture medium.
[0434] The culture medium for phagocytes is known to the person skilled in the
art and can be for
25 example, without limitation, RPMI, DMEM, X-vivo and Ultraculture
milieus.
[0435] In an embodiment, co-culture of apoptotic cells and phagocytes takes
place in a physiological
solution.
[0436] Prior to this "co-culture", the cells may be submitted to a washing
step. In some embodiments,
the white blood cells (e.g. the phagocytes) and the apoptotic cells are washed
before the co-culture
30 step. In another embodiment, the cells are washed with PBS.
[0437] During said co-culture the white blood cells (e.g. the phagocytes such
as macrophages,
monocytes, or phagocytes, or the B, T or NK cells) and the apoptotic cells may
be mixed in a ratio of
10:1, 9:1; 8:1, 7:1, 6:1, 5:1, 4:1, 3:1, 2:1, or 1 :1, or in a ratio of (white
blood cells : apoptotic cells)
1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, or 1:10. In one example, the ratio of
white blood cells to apoptotic
CA 03151815 2022-02-17
96
WO 2021/053667
PCT/IL2020/051011
cells is 1:5.
[0438] The co-culture of the cells might be for several hours to several days.
In some embodiments,
said apoptotic cells are cultured for 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22,
24, 26, 28, 30, 32, 34, 36, 38,
40, 42, 44, 46, 48, 50, 52 hours. A person skilled in the art can evaluate the
optimal time for co-culture
by measuring the presence of anti-inflammatory compounds, the viable amount of
white blood cells
and the amount of apoptotic cells which have not been eliminated so far.
elimination of apoptotic cells
by phagocytes is observable with light microscopy due to the disappearance of
apoptotic cells.
[0439] In some embodiments, the culture of apoptotic cells, such as the co-
culture with culture with
white blood cells (e.g. phagocytes such as macrophages, monocytes, or
phagocytes, or the B, T or
NK cells), takes place in culture medium and/or in a physiological solution
compatible with
administration e.g. injection to a subject.
[0440] A skilled artisan would appreciate that a "physiological solution"
may encompass a
solution which does not lead to the death of white blood cells within the
culture time. In some
embodiments, the physiological solution does not lead to death over 2, 4, 6,
8, 10, 12, 14, 16, 18, 20,
.. 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52 hours. In
other embodiment, 48 hours, or
30 hours.
[0441] In some embodiments, the white blood cells (e.g. phagocytes such as
macrophages,
monocytes, or phagocytes, or the B, T or NK cells) and the apoptotic cells are
incubated in the
physiological solution for at least 30 min. This time of culture allows
phagocytosis initiation and
.. secretion of cytokines and other beneficial substances.
[0442] In an embodiment, such a physiological solution does not inhibit
apoptotic leukocyte
elimination by leukocyte-derived macrophages.
[0443] At the end of the culture or the co-culture step, the supernatant is
optionally separated from
the cultured apoptotic cells or the co-cultured cells. Techniques to separate
the supernatant from the
cells are known in the art. For example, the supernatant can be collected
and/or filtered and/or
centrifuged to eliminate cells and debris. For example, said supernatant may
be centrifuged at 3000
rpm for 15 minutes at room temperature to separate it from the cells.
[0444] The supernatant may be "inactivated" prior to use, for example by
irradiation. Therefore, the
method for preparing the apoptotic cell supernatant may comprise an optional
additional irradiation
step f). Said "irradiation" step can be considered as a disinfection method
that uses X-ray irradiation
(25-45 Gy) at sufficiently rate to kill microorganisms, as routinely performed
to inactivate blood
products.
[0445] Irradiation of the supernatant is considered safe in the art.
Irradiation procedures are currently
performed on a routine basis to donated blood to prevent reactions to WBC.
CA 03151815 2022-02-17
97
WO 2021/053667
PCT/IL2020/051011
[0446] In an embodiment, the apoptotic cell supernatant is formulated into a
pharmaceutical
composition suitable for administration to a subject, as described in detail
herein.
[0447] In some embodiments, the final product is stored at +4 C. In another
embodiment, the final
product is for use in the next 48 hours.
[0448] In some embodiments, the apoptotic cell supernatant, such as an
apoptotic cell-phagocyte
supernatant, or pharmaceutical composition comprising the supernatant, may be
lyophilized, for
example for storage at -80 C.
[0449] In one specific embodiment, as described in Example 1 of WO
2014/106666, an apoptotic
cell-phagocyte supernatant may be made using thymic cells as apoptotic cells.
After isolation, thymic
cells are irradiated (e.g. with a 35 X-Gray irradiation) and cultured in
complete DMEM culture
medium for, for example, 6 hours to allow apoptosis to occur. In parallel,
macrophages are isolated
from the peritoneum cavity, washed and cultured in complete RPMI (10% FBS,
Peni-Strepto, EAA,
Hepes, NaP and 2-MercaptoEthanol). Macrophages and apoptotic cells are then
washed and co-
cultured for another 48 hour period in phenol-free X-vivo medium at a 1/5
macrophage/apoptotic cell
ratio. Then, supernatant is collected, centrifuged to eliminate debris and may
be frozen or lyophilized
for conservation. Macrophage enrichment may be confirmed using positive
staining for F4/80 by
FACS. Apoptosis may be confirmed by FACS using positive staining for Annexin-V
and 7AAD
exclusion.
[0450] In an embodiment, the apoptotic cell supernatant is enriched in TGF-f3
levels both in active
and latent forms of TGF-f3, compared to supernatants obtained from either
macrophages or apoptotic
cells cultured separately. In an embodiment, IL-10 levels are also increased
compared to macrophages
cultured alone and dramatically increased compared to apoptotic cells cultured
alone. In another
embodiment, inflammatory cytokines such as IL-6 are not detectable and IL-1 f3
and TNF are
undetectable or at very low levels.
[0451] In an embodiment, the apoptotic cell supernatant, when compared to
supernatants from
macrophages cultured alone or from apoptotic cells cultured alone, has
increased levels of IL- lra,
TIMP-1, CXCL1/KC and CCL2/JE/MCP1, which might be implicated in a tolerogenic
role of the
supernatant to control inflammation, in addition to TGF-f3 and IL-10.
[0452] In another specific embodiment, as described in Example 3 of WO
2014/106666, human
apoptotic cell-phagocyte supernatant may be made from the co-culture of
macrophages derived from
peripheral blood mononuclear cells (PBMC) cultured with apoptotic PBMC. Thus,
PBMC are
isolated from cytapheresis bag from a healthy volunteer through, for example,
Ficoll gradient
centrifugation. Then PBMC are plated for 90 min in complete RPMI culture
medium (10% FBS, 1
% Penicillin/Streptomycin). Then, non-adherenT-cells are removed and rendered
apoptotic using, for
CA 03151815 2022-02-17
98
WO 2021/053667
PCT/IL2020/051011
example, a 35 Gy dose of X-ray irradiation and cultured in complete RPMI
milieu for 4 days
(including cell wash after the first 48 hrs of culture), in order to allow
apoptosis to occur. In parallel,
adherent T-cells are cultured in complete RPM' milieu supplemented with 50
Ilg/mL of recombinant
human M-CSF for 4 days including cell wash after the first 48 hrs. At the end
of the 4-day culture
period, monocyte-derived macrophages and apoptotic cells are washed and
cultured together in X-
vivo medium for again 48 hours at a one macrophage to 5 apoptotic cell ratio.
Then supernatant from
the latter culture is collected, centrifuged to eliminate cells and debris,
and may be frozen or
lyophilized for conservation and subsequent use.
[0453] In an embodiment, as described in WO 2014/106666, human apoptotic cell-
phagocyte
supernatant may be obtained in 6 days from peripheral blood mononuclear cells
(PBMC). Four days
to obtain PBMC- derived macrophages using M-CSF addition in the culture, and 2
more days for the
co-culture of PBMC-derived macrophages with apoptotic cells, corresponding to
the non-adherent
PBMC isolated at day 0.
[0454] In an embodiment, as described in WO 2014/106666, a standardized human
apoptotic cell-
phagocyte supernatant may be obtained independently of the donor or the source
of PBMC
(cytapheresis or buffy coat). The plastic-adherence step is sufficient to
obtain a significant starting
population of enriched monocytes (20 to 93% of CD14+ cells after adherence on
plastic culture dish).
In addition, such adherent T-cells demonstrate a very low presence of B and T-
cells (1.0% of CD19+
B cells and 12.8% of CD3+ T-cells). After 4 days of culture of adherent T-
cells in the presence of M-
CSF, the proportion of monocytes derived- macrophages is significantly
increased from 0.1 % to
77.7% of CD14+CD206+HLA-DR+ macrophages. At that time, monocyte-derived
macrophages
may be co-cultured with apoptotic non-adherent PBMC (47.6% apoptotic as shown
by annexin V
staining and 7AAD exclusion) to produce the apoptotic cell-phagocyte
supernatant during 48 hours.
[0455] In an embodiment, the collected apoptotic cell-phagocyte supernatant,
contains significantly
more latent TGF than in the culture supernatant of monocyte-derived
macrophages alone or
monocyte-derived macrophages treated in inflammatory conditions (+ LPS), and
only contains trace
or low level of inflammatory cytokines such as IL-10 or TNF.
[0456] In some embodiments, the composition comprising the apoptotic cell
supernatant further
comprises an anti-coagulant. In some embodiments, the anti-coagulant is
selected from the group
consisting of: heparin, acid citrate dextrose (ACD) Formula A and a
combination thereof.
[0457] In another embodiment, an anti-coagulant is added during the process of
manufacturing
apoptotic cells. In another embodiment, the anti-coagulant added is selected
from the group
comprising ACD and heparin, or any combination thereof. In another embodiment,
ACD is at a
concentration of 1%. In another embodiment, ACD is at a concentration of 2%.
In another
CA 03151815 2022-02-17
99
WO 2021/053667
PCT/IL2020/051011
embodiment, ACD is at a concentration of 3%. In another embodiment, ACD is at
a concentration of
4%. In another embodiment, ACD is at a concentration of 5%. In another
embodiment, ACD is at a
concentration of 6%. In another embodiment, ACD is at a concentration of 7%.
In another
embodiment, ACD is at a concentration of 8%. In another embodiment, ACD is at
a concentration of
9%. In another embodiment, ACD is at a concentration of 10%. In another
embodiment, ACD is at a
concentration of between about 1-10%. In another embodiment, ACD is at a
concentration of between
about 2-8 %. In another embodiment, ACD is at a concentration of between about
3-7%. In another
embodiment, ACD is at a concentration of between about 1-5%. In another
embodiment, ACD is at
a concentration of between about 5-10%. In another embodiment, heparin is at a
final concentration
of 0.5 U/ml. In another embodiment, heparin is at a final concentration of
about 0.1 U/m1-1.0 U/ml.
In another embodiment, heparin is at a final concentration of about 0.2 U/m1-
0.9 U/ml. In another
embodiment, heparin is at a final concentration of about 0.3 U/m1-0.7 U/ml. In
another embodiment,
heparin is at a final concentration of about 0. 1 U/m1-0.5 U/ml. In another
embodiment, heparin is at
a final concentration of about 0.5 U/m1-1.0 U/ml. In another embodiment,
heparin is at a final
__ concentration of about 0.01 U/m1-1.0 U/ml. In another embodiment, heparin
is at a final concentration
of 0.1 U/ml. In another embodiment, heparin is at a final concentration of 0.2
U/ml. In another
embodiment, heparin is at a final concentration of 0.3 U/ml. In another
embodiment, heparin is at a
final concentration of 0.4 U/ml. In another embodiment, heparin is at a final
concentration of 0.5
U/ml. In another embodiment, heparin is at a final concentration of 0.6 U/ml.
In another embodiment,
heparin is at a final concentration of 0.7 U/ml. In another embodiment,
heparin is at a final
concentration of 0.8 U/ml. In another embodiment, heparin is at a final
concentration of 0.9 U/ml. In
another embodiment, heparin is at a final concentration of 1.0 U/ml. In
another embodiment, ACD is
at a concentration of 5% and heparin is at a final concentration of 0.5 U/ml.
[0458] In some embodiments, the composition comprising the apoptotic cell
supernatant further
comprises methylprednisolone. At some embodiments, the concentration of
methylprednisolone does
not exceed 30 fig/mi.
[0459] In some embodiments, the composition may be used at a total dose or
aliquot of apoptotic cell
supernatant derived from the co-culture of about 14x109 of CD45+ cells
obtained by cytapheresis
equivalent to about 200 million of cells per kilogram of body weight (for a 70
kg subject). In an
embodiment, such a total dose is administered as unit doses of supernatant
derived from about 100
million cells per kilogram body weight, and/or is administered as unit doses
at weekly intervals, In
another embodiment both of which. Suitable total doses according to this
embodiment include total
doses of supernatant derived from about 10 million to about 4 billion cells
per kilogram body weight.
In another embodiment, the supernatant is derived from about 40 million to
about 1 billion cells per
CA 03151815 2022-02-17
100
WO 2021/053667
PCT/IL2020/051011
kilogram body weight. In yet another embodiment the supernatant is derived
from about 80 million
to about 500 million cells per kilogram body weight. In still another
embodiment, the supernatant is
derived from about 160 million to about 250 million cells per kilogram body
weight. Suitable unit
doses according to this embodiment include unit doses of supernatant derived
from about 4 million
to about 400 million cells per kilogram body weight. In another embodiment,
the supernatant is
derived from about 8 million to about 200 million cells per kilogram body
weight. In another
embodiment, the supernatant is derived from about 16 million to about 100
million cells per kilogram
body weight. In yet another embodiment, the supernatant is derived from about
32 million to about
50 million cells per kilogram body weight.
[0460] In another embodiment, a dose of apoptotic cell supernatant derived
from the co-culture of
about 10x106 apoptotic cells is administered. In another embodiment, a dose
derived from 10x107
apoptotic cells is administered. In another embodiment, a dose derived from
10x108 apoptotic cells is
administered. In another embodiment, a dose derived from 10x109 apoptotic
cells is administered. In
another embodiment, a dose derived from 10x101 apoptotic cells is
administered. In another
embodiment, a dose derived from 10x1011 apoptotic cells is administered. In
another embodiment, a
dose derived from 10x1012 apoptotic cells is administered. In another
embodiment, a dose derived
from 10x105 apoptotic cells is administered. In another embodiment, a dose
derived from 10x104
apoptotic cells is administered. In another embodiment, a dose derived from
10x103 apoptotic cells is
administered. In another embodiment, a dose derived from 10x102 apoptotic
cells is administered.
[0461] In some embodiments, a dose of apoptotic cell supernatant derived from
35x106 apoptotic
cells is administered. In another embodiment, a dose derived from 210x106
apoptotic cells is
administered. In another embodiment, a dose derived from 70x106apoptotic cells
is administered. In
another embodiment, a dose derived from 140x106 apoptotic cells is
administered. In another
embodiment, a dose derived from 35-210x106apoptotic cells is administered.
[0462] In some embodiments, the apoptotic cell supernatant, or composition
comprising said
apoptotic cell supernatant, may be administered by any method known in the art
including, but not
limited to, intravenous, subcutaneous, intranodal, intratumoral, intrathecal,
intrapleural,
intraperitoneal and directly to the thymus, as discussed in detail herein.
[0463] Surprisingly, the apoptotic cell supernatants, such as apoptotic cell-
phagocyte supernatants,
.. reduces production of cytokines associated with the cytokine storm such as
IL-6. Another cytokine,
IL-2, is not involved in cytokine release syndrome although is secreted by DCs
and macrophages in
small quantities. It is, however, required for the survival and proliferation
of CAR-T-cells and is
mostly produced by these T-cells. Unexpectedly, the apoptotic cell
supernatants, such as apoptotic
cell-phagocyte supernatants, do not reduce IL-2 levels sufficiently to
negatively affect the survival of
CA 03151815 2022-02-17
101
WO 2021/053667
PCT/IL2020/051011
CAR T-cells.
[0464] In some embodiments, the apoptotic cell supernatants, such as apoptotic
cell-phagocyte
supernatants, affect cytokine expression levels in macrophages and DCs, but do
not affect cytokine
expression levels in the T-cells themselves. It was therefore unexpected that
apoptotic cell
supernatants would be useful in enhancing CAR T-cell therapy or dendritic cell
therapy.
[0465] In another embodiment, the apoptotic cell supernatants trigger death of
T-cells, but not via
changes in cytokine expression levels.
[0466] In another embodiment, apoptotic cell supernatants, such as apoptotic
cell-phagocyte
supernatants antagonize the priming of macrophages and dendritic cells to
secrete cytokines that
would otherwise amplify the cytokine storm. In another embodiment, apoptotic
cell supernatants
increase Tregs which suppress the inflammatory response and/or prevent excess
release of cytokines.
[0467] In some embodiments, administration of apoptotic cell supernatants,
such as apoptotic cell-
phagocyte supernatants, inhibits one or more pro-inflammatory cytokines. In
some embodiments, the
pro-inflammatory cytokine comprises IL- lbeta, IL-6, TNF-alpha, or IFN-gamma,
or any combination
thereof. In another embodiment, administration of apoptotic cell supernatants
promotes the secretion
of one or more anti-inflammatory cytokines. In some embodiments, the anti-
inflammatory cytokine
comprises TGF-beta, IL10, or PGE2, or any combination thereof.
[0468] In another embodiment, administration of apoptotic cell supernatants,
such as apoptotic cell-
phagocyte supernatants, inhibits dendritic cell maturation following exposure
to TLR ligands. In
another embodiment, administration of apoptotic cell supernatants creates
potentially tolerogenic
dendritic cells, which in some embodiments, are capable of migration, and in
some embodiments, the
migration is due to CCR7. In another embodiment, administration of apoptotic
cell supernatants elicits
various signaling events which in one embodiment is TAM receptor signaling
(Tyro3, Axl and Mer)
which in some embodiments, inhibits inflammation in antigen-presenting cells.
In some
embodiments, Tyro-3, Axl, and Mer constitute the TAM family of receptor
tyrosine kinases (RTKs)
characterized by a conserved sequence within the kinase domain and adhesion
molecule-like
extracellular domains. In another embodiment, administration of apoptotic cell
supernatants activates
signaling through MerTK. In another embodiment, administration of apoptotic
cell supernatants
activates the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which in some
embodiments,
negatively regulates NF-KB. In another embodiment, administration of apoptotic
cell supernatants
negatively regulates the inflammasome which in one embodiment leads to
inhibition of pro-
inflammatory cytokine secretion, DC maturation, or a combination thereof. In
another embodiment,
administration of apoptotic cell supernatants upregulates expression of anti-
inflammatory genes such
as Nr4a, Thbsl, or a combination thereof. In another embodiment,
administration of apoptotic cell
CA 03151815 2022-02-17
102
WO 2021/053667
PCT/IL2020/051011
supernatants induces a high level of AMP which in some embodiments, is
accumulated in a
Pannexinl-dependent manner. In another embodiment, administration of apoptotic
cell supernatants
suppresses inflammation.
Compositions
[0469] As used herein, the terms "composition" and pharmaceutical composition"
may in some
embodiments, be used interchangeably having all the same qualities and
meanings. In some
embodiments, disclosed herein is a pharmaceutical composition for the
treatment of a condition or
disease as described herein.
[0470] In another embodiment, pharmaceutical compositions disclosed here are
for maintaining or
increasing the proliferation rate of a genetically modified immune cells. In a
further embodiment,
methods for maintaining or increasing the proliferation rate of genetically
modified immune cells
further comprise reducing or inhibiting the incidence of cytokine release
syndrome (CRS) or cytokine
storm. In another embodiment, disclosed herein are pharmaceutical compositions
for increasing the
efficacy of a genetically modified immune cell therapy. In another embodiment,
compositions used
in the methods for increasing the efficacy of an immune cell therapy further
comprise reducing or
inhibiting the incidence of CRS or a cytokine storm. In another embodiment,
disclosed herein are
compositions for methods treating, preventing, inhibiting, reducing the
incidence of, ameliorating, or
alleviating a cancer of a tumor in a subject. In another embodiment,
compositions used in the methods
for treating, preventing, reducing the incidence of, ameliorating, or
alleviating a cancer or a tumor in
a subject, further comprise reducing or inhibiting the incidence of CRS or a
cytokine storm.
[0471] In another embodiment, a pharmaceutical composition comprises a
genetically modified
immune cell or a genetically modified receptor thereof. In another embodiment,
a genetically
modified immune cell comprises a T-cell. In another embodiment, a genetically
modified immune
cell comprises a chimeric antigen receptor CAR T-cell. In another embodiment,
a genetically
modified immune cell comprises a chimeric antigen receptor TCR T-cell. In
another embodiment, a
genetically modified immune cell comprises a cytotoxic T lymphocyte. In
another embodiment, a
genetically modified immune cell comprises a dendritic cell. In another
embodiment, a genetically
modified immune cell comprises a natural killer cell. In another embodiment, a
genetically modified
receptor comprises a genetically modified T-cell receptor.
[0472] In another embodiment, a pharmaceutical composition comprises an early
apoptotic cell
population. In another embodiment, a pharmaceutical composition comprises an
apoptotic
supernatant.
[0473] In still another embodiment, a pharmaceutical composition for the
treatment of a condition
or a disease as described herein comprises an effective amount of a
genetically modified immune cell
CA 03151815 2022-02-17
103
WO 2021/053667
PCT/IL2020/051011
or a genetically modified receptor thereof, as described herein in a
pharmaceutically acceptable
excipient. In another embodiment, a pharmaceutical composition for the
treatment of a condition or
a disease as described herein comprises an effective amount of a CAR T-cell as
described herein in,
and a pharmaceutically acceptable excipient. In another embodiment, a
pharmaceutical composition
for the treatment of a condition or a disease as described herein comprises an
effective amount of a
TCR T-cell as described herein in, and a pharmaceutically acceptable
excipient. In another
embodiment, a pharmaceutical composition for the treatment of a condition or a
disease as described
herein comprises an effective amount of a cytotoxic T-cell, as described
herein, and a
pharmaceutically acceptable excipient. In another embodiment, a pharmaceutical
composition for the
treatment of a condition or a disease as described herein comprises an
effective amount of a
genetically modified dendritic cell, as described herein, and a
pharmaceutically acceptable excipient.
In another embodiment, a pharmaceutical composition for the treatment of a
condition or a disease
as described herein comprises an effective amount of a genetically modified
natural killer cell, as
described herein, and a pharmaceutically acceptable excipient. In another
embodiment, a
pharmaceutical composition for the treatment of a condition or a disease as
described herein
comprises an effective amount of a genetically modified T-cell receptor, as
described herein, and a
pharmaceutically acceptable excipient. In still another embodiment, a
pharmaceutical composition
for the treatment of a condition or a disease as described herein comprises an
effective amount of an
early apoptotic cell population, as described herein in a pharmaceutically
acceptable excipient. In still
another embodiment, a pharmaceutical composition for the treatment of a
condition or a disease as
described herein comprises an effective amount of an apoptotic supernatant, as
described herein in a
pharmaceutically acceptable excipient.
[0474] In another embodiment, the condition or disease as described herein is
a tumor or cancer. In
another embodiment, disclosed herein is a composition comprising the
genetically modified immune
cell or receptor thereof, for example a CAR T-cell, that binds to a protein or
peptide of interest as
described herein. In another embodiment, disclosed herein is a composition
comprising the
genetically modified immune cell or receptor thereof, for example a TCR T-
cell, that recognizes and
binds a protein or peptide of interest as described herein. In another
embodiment, the protein or
peptide of interest comprises a tumor antigen or a fragment thereof.
[0475] In another embodiment, a composition disclosed herein and used in
methods disclosed herein
comprises apoptotic cells or an apoptotic cell supernatant, and a
pharmaceutically acceptable
excipient. In some embodiments, a composition comprising apoptotic cells or an
apoptotic cell
supernatant is used in methods disclosed herein for example for treating,
preventing, inhibiting the
growth of, delaying disease progression, reducing the tumor load, or reducing
the incidence of a
CA 03151815 2022-02-17
104
WO 2021/053667
PCT/IL2020/051011
cancer or a tumor in a subject, or any combination thereof.
[0476] In yet another embodiment, a composition comprising an effective amount
of a genetically
modified immune cell or a genetically modified receptor thereof may be the
same composition as
comprises an apoptotic cell population or an apoptotic cell supernatant. In
another embodiment, a
composition comprising an effective amount of a CAR T-cell, or a TCR T-cell,
or a cytotoxic T-cell,
or a genetically modified dendritic cell, or a genetically modified natural
killer cell may be the same
composition as comprises an apoptotic cell population or an apoptotic cell
supernatant. In yet another
embodiment, a composition comprising an effective amount of genetically
modified T-cell receptor
may be the same composition as comprises an apoptotic cell population or an
apoptotic cell
supernatant. In still another embodiment, a composition comprising an
effective amount of a
genetically modified immune cell selected from the group comprising a CAR T-
cell, a TCR T-cell,
a cytotoxic T-cell, a natural killer cell, or a dendritic cell, is not the
same composition as comprises
an apoptotic cell population or an apoptotic cell supernatant. In another
embodiment, a composition
comprises a chimeric antigen receptor-expressing T-cell (CAR T-cell) and
either apoptotic cells or
.. an apoptotic cell supernatant, and a pharmaceutically acceptable excipient.
In another embodiment,
a composition comprises a genetically modified T-cell receptor expressing T-
cell (TCR T-cell) and
either apoptotic cells or an apoptotic cell supernatant, and a
pharmaceutically acceptable excipient.
In another embodiment, a composition comprising an effective amount of a
genetically modified T-
cell receptor is not the same composition as comprises an apoptotic cell
population or an apoptotic
cell supernatant.
[0477] In another embodiment, apoptotic cells comprised in a composition
comprise apoptotic cells
in an early apoptotic state. In another embodiment, apoptotic cells comprised
in a composition are
pooled third-party donor cells. In another embodiment, an apoptotic cell
supernatant comprised in a
composition disclosed herein is collected from early apoptotic cells. In
another embodiment, an
apoptotic cell supernatant comprised in a composition disclosed herein, is
collected pooled third-
party donor cells.
[0478] In one embodiment, a composition comprising a genetically modified
immune cells, for
example a CAR T-cell, further comprises an additional pharmaceutical
composition for preventing,
suppressing, or modulating cytokine release in a patient with cytokine release
syndrome or
experiencing a cytokine storm. In another embodiment, a composition comprising
a genetically
modified immune cells, for example a CAR T-cell, and apoptotic cells further
comprises an additional
pharmaceutical composition for preventing, suppressing, or modulating cytokine
release in a patient
with cytokine release syndrome or experiencing a cytokine storm. In another
embodiment, a
composition comprising a genetically modified immune cells, for example a CAR
T-cell, and an
CA 03151815 2022-02-17
105
WO 2021/053667
PCT/IL2020/051011
apoptotic cell supernatant, further comprises an additional pharmaceutical
composition for
preventing, suppressing, or modulating cytokine release in a patient with
cytokine release syndrome
or experiencing a cytokine storm.
[0479] In one embodiment, a composition comprising a genetically modified
immune cells, for
example a TCR T-cell, further comprises an additional pharmaceutical
composition for preventing,
suppressing, or modulating cytokine release in a patient with cytokine release
syndrome or
experiencing a cytokine storm. In another embodiment, a composition comprising
a genetically
modified immune cells, for example a TCR T-cell, and apoptotic cells further
comprises an additional
pharmaceutical composition for preventing, suppressing, or modulating cytokine
release in a patient
with cytokine release syndrome or experiencing a cytokine storm. In another
embodiment, a
composition comprising a genetically modified immune cells, for example a TCR
T-cell, and an
apoptotic cell supernatant, further comprises an additional pharmaceutical
composition for
preventing, suppressing, or modulating cytokine release in a patient with
cytokine release syndrome
or experiencing a cytokine storm.
[0480] In one embodiment, a composition comprising a genetically modified
immune cells, for
example a dendritic cell, further comprises an additional pharmaceutical
composition for preventing,
suppressing, or modulating cytokine release in a patient with cytokine release
syndrome or
experiencing a cytokine storm. In another embodiment, a composition comprising
a genetically
modified immune cells, for example a dendritic, and apoptotic cells further
comprises an additional
pharmaceutical composition for preventing, suppressing, or modulating cytokine
release in a patient
with cytokine release syndrome or experiencing a cytokine storm. In another
embodiment, a
composition comprising a genetically modified immune cells, for example a
dendritic, and an
apoptotic cell supernatant, further comprises an additional pharmaceutical
composition for
preventing, suppressing, or modulating cytokine release in a patient with
cytokine release syndrome
or experiencing a cytokine storm.
[0481] In one embodiment, a composition comprising a genetically modified
immune cells, for
example a NK cell, further comprises an additional pharmaceutical composition
for preventing,
suppressing, or modulating cytokine release in a patient with cytokine release
syndrome or
experiencing a cytokine storm. In another embodiment, a composition comprising
a genetically
modified immune cells, for example a NK cell, and apoptotic cells further
comprises an additional
pharmaceutical composition for preventing, suppressing, or modulating cytokine
release in a patient
with cytokine release syndrome or experiencing a cytokine storm. In another
embodiment, a
composition comprising a genetically modified immune cells, for example a NK
cell, and an
apoptotic cell supernatant, further comprises an additional pharmaceutical
composition for
CA 03151815 2022-02-17
106
WO 2021/053667
PCT/IL2020/051011
preventing, suppressing, or modulating cytokine release in a patient with
cytokine release syndrome
or experiencing a cytokine storm.
[0482] In one embodiment, the additional pharmaceutical composition comprises
a CTLA-4
blocking agent, which in one embodiment is Ipilimumab. In another embodiment,
the additional
pharmaceutical composition comprises a alpha-1 anti-trypsin, as disclosed
herein, or a fragment
thereof, or an analogue thereof. In another embodiment, the additional
pharmaceutical composition
comprises a tellurium-based compound, a disclosed herein. In another
embodiment, the additional
pharmaceutical composition comprises an immune modulating agent, as disclosed
herein. In another
embodiment, the additional pharmaceutical composition comprises a CTLA-4
blocking agent, an
.. alpha-1 anti-trypsin or fragment thereof or analogue thereof, a tellurium-
based compound, or an
immune modulating compound, or any combination thereof.
[0483] In one embodiment, the composition comprising the genetically modified
immune cell, for
example a CAR T-cell and the pharmaceutical composition comprising any one of
a CTLA-4
blocking agent, an alpha-1 anti-tryp sin or fragment thereof or analogue
thereof, apoptotic cells, or an
apoptotic cell supernatant, a tellurium-based compound, or an immune
modulating agent comprises
a single composition. In another embodiment, the composition comprising the
genetically modified
immune cell, for example CAR T-cells and the pharmaceutical composition
comprising any one of a
CTLA-4 blocking agent, an alpha-1 anti-tryp sin or fragment thereof or
analogue thereof, apoptotic
cells, or an apoptotic cell supernatant, a tellurium-based compound, or an
immune modulating agent,
or any combination thereof, comprises multiple compositions, wherein each of
the genetically
modified immune cell, which in one embodiment is CAR T-cells, the CTLA-4
blocking agent, the
alpha-1 anti-tryp sin or fragment thereof or analogue thereof, the apoptotic
cells, the apoptotic cell
supernatant, the tellurium-based compound, or the immune modulating agent, or
any combination
thereof, are comprised in a separate composition. In yet another embodiment,
the composition
comprising the genetically modified immune cell, which in one embodiment is
CAR T-cells and the
pharmaceutical composition comprising any one of a CTLA-4 blocking agent, an
alpha-1 anti-trypsin
or fragment thereof or analogue thereof, apoptotic cells, an apoptotic cell
supernatant, a tellurium-
based compound, or an immune modulating agent, or any combination thereof,
comprises multiple
compositions, wherein the genetically modified immune cells, which in one
embodiment are CAR
.. T-cells, the CTLA-4 blocking agent, or the alpha-1 anti-trypsin or fragment
thereof or analogue
thereof, the tellurium-based compound, or the immune modulating agent, or any
combination thereof,
or any combination thereof are present in the genetically modified immune
cell, for example a CAR
T-cell, composition, and the apoptotic cells, or the apoptotic cell
supernatant, are comprised in a
separate composition.
CA 03151815 2022-02-17
107
WO 2021/053667
PCT/IL2020/051011
[0484] In one embodiment, the composition comprising the genetically modified
immune cell, for
example a TCR T-cell and the pharmaceutical composition comprising any one of
a CTLA-4
blocking agent, an alpha-1 anti-tryp sin or fragment thereof or analogue
thereof, apoptotic cells, or an
apoptotic cell supernatant, a tellurium-based compound, or an immune
modulating agent comprises
a single composition. In another embodiment, the composition comprising the
genetically modified
immune cell, for example TCR T-cells and the pharmaceutical composition
comprising any one of a
CTLA-4 blocking agent, an alpha-1 anti-tryp sin or fragment thereof or
analogue thereof, apoptotic
cells, or an apoptotic cell supernatant, a tellurium-based compound, or an
immune modulating agent,
or any combination thereof, comprises multiple compositions, wherein each of
the genetically
modified immune cell, which in one embodiment is TCR T-cells, the CTLA-4
blocking agent, the
alpha-1 anti-tryp sin or fragment thereof or analogue thereof, the apoptotic
cells, the apoptotic cell
supernatant, the tellurium-based compound, or the immune modulating agent, or
any combination
thereof, are comprised in a separate composition. In yet another embodiment,
the composition
comprising the genetically modified immune cell, which in one embodiment is
TCR T-cells and the
pharmaceutical composition comprising any one of a CTLA-4 blocking agent, an
alpha-1 anti-trypsin
or fragment thereof or analogue thereof, apoptotic cells, an apoptotic cell
supernatant, a tellurium-
based compound, or an immune modulating agent, or any combination thereof,
comprises multiple
compositions, wherein the genetically modified immune cells, which in one
embodiment are TCR T-
cells, the CTLA-4 blocking agent, or the alpha-1 anti-trypsin or fragment
thereof or analogue thereof,
the tellurium-based compound, or the immune modulating agent, or any
combination thereof, or any
combination thereof are present in the genetically modified immune cell, for
example a TCR T-cell,
composition, and the apoptotic cells, or the apoptotic cell supernatant, are
comprised in a separate
composition.
[0485] In one embodiment, the composition comprising the genetically modified
immune cell, for
example a dendritic cell and the pharmaceutical composition comprising any one
of a CTLA-4
blocking agent, an alpha-1 anti-tryp sin or fragment thereof or analogue
thereof, apoptotic cells, or an
apoptotic cell supernatant, a tellurium-based compound, or an immune
modulating agent comprises
a single composition. In another embodiment, the composition comprising the
genetically modified
immune cell, for example dendritic cells and the pharmaceutical composition
comprising any one of
a CTLA-4 blocking agent, an alpha-1 anti-tryp sin or fragment thereof or
analogue thereof, apoptotic
cells, or an apoptotic cell supernatant, a tellurium-based compound, or an
immune modulating agent,
or any combination thereof, comprises multiple compositions, wherein each of
the genetically
modified immune cell, which in one embodiment is dendritic cells, the CTLA-4
blocking agent, the
alpha-1 anti-tryp sin or fragment thereof or analogue thereof, the apoptotic
cells, the apoptotic cell
CA 03151815 2022-02-17
108
WO 2021/053667
PCT/IL2020/051011
supernatant, the tellurium-based compound, or the immune modulating agent, or
any combination
thereof, are comprised in a separate composition. In yet another embodiment,
the composition
comprising the genetically modified immune cell, which in one embodiment is
dendritic cells and
the pharmaceutical composition comprising any one of a CTLA-4 blocking agent,
an alpha-1 anti-
trypsin or fragment thereof or analogue thereof, apoptotic cells, an apoptotic
cell supernatant, a
tellurium-based compound, or an immune modulating agent, or any combination
thereof, comprises
multiple compositions, wherein the genetically modified immune cells, which in
one embodiment
are dendritic cells, the CTLA-4 blocking agent, or the alpha-1 anti-trypsin or
fragment thereof or
analogue thereof, the tellurium-based compound, or the immune modulating
agent, or any
combination thereof, or any combination thereof are present in the genetically
modified immune cell,
for example a dendritic cell, composition, and the apoptotic cells, or the
apoptotic cell supernatant,
are comprised in a separate composition.
[0486] In one embodiment, the composition comprising the genetically modified
immune cell, for
example a NK cell and the pharmaceutical composition comprising any one of a
CTLA-4 blocking
agent, an alpha-1 anti-tryp sin or fragment thereof or analogue thereof,
apoptotic cells, or an apoptotic
cell supernatant, a tellurium-based compound, or an immune modulating agent
comprises a single
composition. In another embodiment, the composition comprising the genetically
modified immune
cell, for example NK cells and the pharmaceutical composition comprising any
one of a CTLA-4
blocking agent, an alpha-1 anti-tryp sin or fragment thereof or analogue
thereof, apoptotic cells, or an
apoptotic cell supernatant, a tellurium-based compound, or an immune
modulating agent, or any
combination thereof, comprises multiple compositions, wherein each of the
genetically modified
immune cell, which in one embodiment is NK cells, the CTLA-4 blocking agent,
the alpha-1 anti-
tryp sin or fragment thereof or analogue thereof, the apoptotic cells, the
apoptotic cell supernatant, the
tellurium-based compound, or the immune modulating agent, or any combination
thereof, are
comprised in a separate composition. In yet another embodiment, the
composition comprising the
genetically modified immune cell, which in one embodiment is NK cells and the
pharmaceutical
composition comprising any one of a CTLA-4 blocking agent, an alpha-1 anti-
trypsin or fragment
thereof or analogue thereof, apoptotic cells, an apoptotic cell supernatant, a
tellurium-based
compound, or an immune modulating agent, or any combination thereof, comprises
multiple
.. compositions, wherein the genetically modified immune cells, which in one
embodiment are NK
cells, the CTLA-4 blocking agent, or the alpha-1 anti-trypsin or fragment
thereof or analogue thereof,
the tellurium-based compound, or the immune modulating agent, or any
combination thereof, or any
combination thereof are present in the genetically modified immune cell, for
example a NK cell,
composition, and the apoptotic cells, or the apoptotic cell supernatant, are
comprised in a separate
CA 03151815 2022-02-17
109
WO 2021/053667
PCT/IL2020/051011
composition.
[0487] In some embodiments, a composition comprises apoptotic cells and an
additional agent. In
some embodiments, a composition comprises apoptotic cells and an antibody or a
functional fragment
thereof. In some embodiments, a composition comprises apoptotic cells and a
RtX antibody or a
functional fragment thereof. In some embodiments, apoptotic cells and an
antibody or a functional
fragment thereof may be comprised in separate compositions. In some
embodiments, apoptotic cells
and an antibody or a functional fragment thereof may be comprised in the same
composition.
[0488] A skilled artisan would appreciate that a "pharmaceutical composition"
may encompass a
preparation of one or more of the active ingredients described herein with
other chemical components
such as physiologically suitable carriers and excipients. The purpose of a
pharmaceutical composition
is to facilitate administration of a compound to an organism.
[0489] In some embodiments, disclosed herein is a pharmaceutical
composition for treating,
preventing, inhibiting the growth of, or reducing the incidence of a cancer or
a tumor. In some
embodiments, disclosed herein is a pharmaceutical composition for increasing
the survival of a
.. subject suffering from a cancer or a tumor. In some embodiments, disclosed
herein is a pharmaceutical
composition for reducing the size or reducing the growth rate of a tumor or a
cancer. In some
embodiments, disclosed herein is a pharmaceutical comprising for reducing
tumor load in a subject
suffering from a cancer or a tumor. In some embodiments, disclosed herein is a
pharmaceutical
comprising for delaying disease progression in a subject suffering from a
cancer or a tumor. In some
embodiments, disclosed herein is a pharmaceutical comprising for reducing the
incidence of cancer
or a tumor in a subject suffering from a cancer or a tumor. In some
embodiments, disclosed herein is
a pharmaceutical comprising for reducing the size and or growth rate of a
cancer or tumor in a subject
suffering from a cancer or a tumor.
[0490] In some embodiments, a pharmaceutical composition comprises an
early apoptotic cell
.. population as described herein. In some embodiments, a pharmaceutical
composition comprises an
early apoptotic cell population as described herein, and a pharmaceutically
acceptable excipient.
[0491] A skilled artisan would appreciate that the phrases
"physiologically acceptable carrier",
"pharmaceutically acceptable carrier", "physiologically acceptable excipient",
and "pharmaceutically
acceptable excipient", may be used interchangeably may encompass a carrier,
excipient, or a diluent
that does not cause significant irritation to an organism and does not
abrogate the biological activity
and properties of the administered active ingredient.
[0492] A skilled artisan would appreciate that an "excipient" may
encompass an inert substance
added to a pharmaceutical composition to further facilitate administration of
an active ingredient. In
some embodiments, excipients include calcium carbonate, calcium phosphate,
various sugars and
CA 03151815 2022-02-17
110
WO 2021/053667
PCT/IL2020/051011
types of starch, cellulose derivatives, gelatin, vegetable oils and
polyethylene glycols.
[0493] Techniques for formulation and administration of drugs are found
in "Remington' s
Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, latest edition,
which is incorporated
herein by reference.
[0494] In some embodiments, compositions are administered at the same time. In
an alternative
embodiment, compositions are administered at different times. In another
embodiment, compositions
comprising apoptotic cells are administered prior to infusion or genetically
modified immune cells or
receptors thereof. In another embodiment, compositions comprising apoptotic
cells are administered
prior to CAR- T-cell infusion. In another embodiment, compositions comprising
apoptotic cells are
administered prior to cytotoxic T-cell infusion. In another embodiment,
compositions comprising
apoptotic cells are administered prior to natural killer cell infusion. In
another embodiment,
compositions comprising apoptotic cells are administered prior to dendritic
infusion. In another
embodiment, compositions comprising apoptotic cells are administered prior to
infusion of a
genetically modified T-cell receptor.
[0495] In another embodiment, compositions comprising apoptotic cell
supernatants are
administered prior to infusion or genetically modified immune cells or
receptors thereof. In another
embodiment, compositions comprising apoptotic cell supernatants are
administered prior to CAR- T-
cell infusion. In another embodiment, compositions comprising apoptotic cell
supernatants are
administered prior to cytotoxic T-cell infusion. In another embodiment,
compositions comprising
apoptotic cell supernatants are administered prior to natural killer cell
infusion. In another
embodiment, compositions comprising apoptotic cell supernatants are
administered prior to dendritic
infusion. In another embodiment, compositions comprising apoptotic cell
supernatants are
administered prior to infusion of a genetically modified T-cell receptor.
[0496] In another embodiment, compositions comprising apoptotic cell
supernatants are
administered prior to infusion of genetically modified immune cells or
receptors thereof. In another
embodiment, compositions comprising apoptotic cells are administered about 24
hours prior to
genetically modified immune cell or receptor thereof infusion. In another
embodiment, compositions
comprising apoptotic cells are administered about 24 hours prior to CAR T-
cell, or cytotoxic T-cells,
or natural killer cells, or dendritic cell or genetically modified T-cell
receptor infusion. In another
embodiment, compositions comprising apoptotic cell supernatants are
administered about 24 hours
prior to CAR T-cell or cytotoxic T-cells, or natural killer cells, or
dendritic cell or genetically modified
T-cell receptor infusion.
[0497] In some embodiments, compositions are administered at the same time. In
an alternative
embodiment, compositions are administered at different times. In another
embodiment, compositions
CA 03151815 2022-02-17
111
WO 2021/053667
PCT/IL2020/051011
comprising apoptotic cells are administered prior to administration of an
antibody or fragment thereof,
or a composition comprising an antibody or fragment thereof.
[0498] In another embodiment, compositions comprising apoptotic cells are
administered about 2
hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 14 hours, 16 hours, 18
hours 20 hours, 22 hours,
24 hours, 36 hours, 48 hours, 60 hours, or 72 hours prior to an antibody or
fragment thereof, or
composition comprising the antibody or fragment thereof. In another
embodiment, compositions
comprising apoptotic cell supernatants are administered about 2 hours, 4
hours, 6 hours, 8 hours, 10
hours, 12 hours, 14 hours, 16 hours, 18 hours 20 hours, 22 hours, 24 hours, 36
hours, 48 hours, 60
hours, or 72 hours prior to an antibody or fragment thereof, or composition
comprising the antibody
or fragment thereof.
[0499] In another embodiment, compositions comprising apoptotic cells are
administered about 1
day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days,
11 days, 12 days, 13 days
14 days, or 15 days prior to an antibody or fragment thereof, or composition
comprising the antibody
or functional fragment thereof. In another embodiment, compositions comprising
apoptotic cell
supernatants are administered about 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6
weeks, 7 weeks, or 8
weeks prior to an antibody or functional fragment thereof, or composition
comprising the antibody or
functional fragment thereof.
[0500] In another embodiment, compositions comprising apoptotic cells are
administered after
infusion of an antibody or fragment thereof, or composition comprising the
antibody or fragment
thereof. In another embodiment, composition comprising apoptotic cells are
administered after an
antibody or fragment thereof, or composition comprising the antibody or
fragment thereof. In another
embodiment, compositions comprising apoptotic cell supernatants are
administered after
administration of an antibody or fragment thereof, or composition comprising
the antibody or
fragment thereof. In another embodiment, compositions comprising apoptotic
cell supernatants are
administered after administration of an antibody or fragment thereof, or
composition comprising the
antibody or fragment thereof. In another embodiment, compositions comprising
apoptotic cells are
administered about 24 hours after an antibody or fragment thereof, or
composition comprising the
antibody or fragment thereof. In another embodiment, compositions comprising
apoptotic cells are
administered after administration of an antibody or fragment thereof, or
composition comprising the
antibody or fragment thereof. In another embodiment, compositions comprising
apoptotic cells are
administered about 2 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 14
hours, 16 hours, 18
hours 20 hours, 22 hours, 24 hours, 36 hours, 48 hours, 60 hours, or 72 hours
after administration of
an antibody or fragment thereof, or composition comprising the antibody or
fragment thereof. In
another embodiment, compositions comprising apoptotic cell supernatants are
administered about 2
CA 03151815 2022-02-17
112
WO 2021/053667
PCT/IL2020/051011
hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 14 hours, 16 hours, 18
hours 20 hours, 22 hours,
24 hours, 36 hours, 48 hours, 60 hours, or 72 hours after administration of an
antibody or fragment
thereof, or composition comprising the antibody or fragment thereof.
[0501] In another embodiment, compositions comprising apoptotic cells are
administered about 1
day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days,
11 days, 12 days, 13 days
14 days, or 15 days after an antibody or fragment thereof, or composition
comprising the antibody or
functional fragment thereof. In another embodiment, compositions comprising
apoptotic cell
supernatants are administered about 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6
weeks, 7 weeks, or 8
weeks after an antibody or functional fragment thereof, or composition
comprising the antibody or
functional fragment thereof.
[0502] In some embodiments, a composition comprising apoptotic cells is
administered
independent of CAR T-cells. In some embodiments, a composition comprising
apoptotic cells is
administered in combination with an additional agent. In some embodiments, the
additional agent is
an antibody.
[0503] In some embodiments, the composition as disclosed herein comprises a
therapeutic
composition. In some embodiments, the composition as disclosed herein
comprises a therapeutic
efficacy.
[0504] In some embodiments, a composition as disclosed herein is administered
once. In another
embodiment, the composition is administered twice. In another embodiment, the
composition is
administered three times. In another embodiment, the composition is
administered four times. In
another embodiment, the composition is administered at least four times. In
another embodiment, the
composition is administered more than four times.
[0505] In some embodiments, CAR T-cells as disclosed herein are administered
once. In another
embodiment, CAR T-cells are administered twice. In another embodiment, CAR T-
cells are
administered three times. In another embodiment, CAR T-cells are administered
four times. In
another embodiment, CAR T-cells are administered at least four times. In
another embodiment, the
composition is administered more than four times.
[0506] In some embodiments, the composition as disclosed herein is a
therapeutic composition. In
another embodiment, the composition as disclosed herein has therapeutic
efficacy.
.. [0507] In some embodiments, disclosed herein are a composition which
provides reduced
inflammatory cytokine or chemokine release compared to a composition
comprising CAR T-cells
alone, but with comparable cytotoxicity compared to a composition comprising
CAR T-cells alone.
Combined Treatments/Therapies
[0508] In some embodiments, disclosed herein are combination therapies
comprising an early
CA 03151815 2022-02-17
113
WO 2021/053667
PCT/IL2020/051011
apoptotic cell population as described herein and one or more cancer
therapeutic agents. In some
embodiments, the cancer therapeutic agent comprises chimeric antigen receptor-
expressing T-cells
(CAR T-cells) as described herein.
[0509] In some embodiments, disclosed herein is a combination therapy
comprising an early
apoptotic cell population as described herein in detail and one or more
chemotherapeutic agents. In
some embodiments, the term "chemotherapeutic agent" may encompass any chemical
agent that has
therapeutic utility in the treatment of proliferative diseases, such as
cancer.
[0510] In some embodiments, the "cancer therapeutic agent" or
"chemotherapeutic agent"
comprises to a drug selected from one or more of the following: alkylating
agents (including mustard,
nitrogen mustards, methanesulphonate, busulphan, alkyl sulfonates,
nitrosoureas, ethylenimine
derivatives, and triazenes or combinations thereof); anti-angiogenics
(including matrix
metalloproteinase inhibitors); antimetabolites (including adenosine deaminase
inhibitors, folic acid
antagonists, purine analogues, and pyrimidine analogues); antibiotics or
antibodies (including
monoclonal antibodies, CTLA-4 antibodies, anthracyclines); aromatase
inhibitors; cell-cycle
response modifiers; enzymes; farnesyl-protein transferase inhibitors; hormonal
and antihormonal
agents and steroids (including synthetic analogs, glucocorticoids,
estrogens/anti-estrogens [e.g.,
SERMs], androgens/anti-androgens, progestins, progesterone receptor agonists,
and luteinizing
hormone-releasing [LHRH] agonists and antagonists); insulin-like growth factor
(IGF)/insulin-like
growth factor receptor (IGFR) system modulators (including IGFR1 inhibitors);
integrin-signaling
inhibitors; kinase inhibitors (including multi-kinase inhibitors and/or
inhibitors of Src kinase or
Src/ab 1, cyclin dependent kinase [CDK] inhibitors, panHer, Her-1 and Her-2
antibodies, VEGF
inhibitors, including anti-VEGF antibodies, EGFR inhibitors, PARP (poly ADP-
ribose polymerase)
inhibitors, mitogen-activated protein [MAP] inhibitors, MET inhibitors, Aurora
kinase inhibitors,
PDGF inhibitors, and other tyrosine kinase inhibitors or serine/threonine
kinase inhibitors;
microtubule-disruptor agents, such as ecteinascidins or their analogs and
derivatives; microtubule-
stabilizing agents such as taxanes, Platinum-based antineoplastic drugs
(platins) such as cisplatin,
carboplatin, oxaliplatin, nedaplatin, triplatin tetranitrate,
phenanthriplatin, picoplatin and satraplatin
and the naturally-occurring epothilones and their synthetic and semi-synthetic
analogs; microtubule-
binding, destabilizing agents (including vinca alkaloids); topoisomerase
inhibitors; prenyl-protein
transferase inhibitors; platinum coordination complexes; signal transduction
inhibitors; Histone
deacetylase inhibitors (HDIs), inhibitors of Bromodomain and Extra-Terminal
motif (BET),
inhibitors of protein arginine methyltransferase 5 (PRMT5), retinoic acids and
other agents used as
anti-cancer and cytotoxic agents such as biological response modifiers, growth
factors, and immune
modulators.
CA 03151815 2022-02-17
114
WO 2021/053667
PCT/IL2020/051011
[0511] In some embodiments, the "cancer therapeutic agent" or
"chemotherapeutic agent"
comprises an antibody or an antigen binding fragment thereof.
[0512] In some embodiments, methods of use of early apoptotic cells, as
described herein, includes
use of the early apoptotic cells or a composition thereof, in combination with
an antibody. In some
embodiments, the antibody is directed against a tumor cell antigen. In another
embodiment, the
antibody is directed against CD20. In another embodiment, the antibody is
rituximab (Rtx).
[0513] In some embodiments, early apoptotic cells and an antibody are
comprised in the same
composition. In some embodiments, early apoptotic cells and an antibody are
comprised in different
compositions. In some embodiments, administration of a combination of early
apoptotic cells and an
antibody, or composition(s) thereof are concurrent. In some embodiments,
administration of a
combination of early apoptotic cells and an antibody, or composition(s)
thereof comprises
administration of apoptotic cells or a composition thereof, prior to the
antibody. In some
embodiments, administration of a combination of early apoptotic cells and an
antibody, or
composition(s) thereof comprises administration of apoptotic cells or a
composition thereof,
following administration of the antibody.
[0514] In another embodiment, the antibody is Trastuzumab (Herceptin;
Genentech): humanized
IgGl, which is directed against ERBB2. In another embodiment, the antibody is
Bevacizumab
(Avastin; Genentech/Roche): humanized IgGl, which is directed against VEGF. In
another
embodiment, the antibody is Cetuximab (Erbitux; Bristol-Myers Squibb):
chimeric human¨murine
IgGl, which is directed against EGFR. In another embodiment, the antibody is
Panitumumab
(Vectibix; Amgen): human IgG2, which is directed against EGFR. In another
embodiment, the
antibody is Ipilimumab (Yervoy; Bristol-Myers Squibb): IgGl, which is directed
against CTLA4.
[0515] In another embodiment, the antibody is Alemtuzumab (Campath; Genzyme):
humanized
IgGl, which is directed against CD52. In another embodiment, the antibody is
Ofatumumab (Arzerra;
Genmab): human IgGl, which is directed against CD20. In another embodiment,
the antibody is
Gemtuzumab ozogamicin (Mylotarg; Wyeth): humanized IgG4, which is directed
against CD33. In
another embodiment, the antibody is Brentuximab vedotin (Adcetris; Seattle
Genetics): chimeric
IgGl, which is directed against CD30. In another embodiment, the antibody is
90Y-labelled
ibritumomab tiuxetan (Zevalin; IDEC Pharmaceuticals): murine IgG 1, which is
directed against
CD20. In another embodiment, the antibody is 131I-labelled to situmomab
(Bexxar;
GlaxoSmithKline): murine IgG2, which is directed against CD20.
[0516] In another embodiment, the antibody is Ramucirumab, which is directed
against vascular
endothelial growth factor receptor-2 (VEGFR-2). In another embodiment, the
antibody is
ramucirumab (Cyramza Injection, Eli Lilly and Company), blinatumomab
(BLINCYTO, Amgen
CA 03151815 2022-02-17
115
WO 2021/053667
PCT/IL2020/051011
Inc.), pembrolizumab (KEYTRUDA, Merck Sharp & Dohme Corp.), obinutuzumab
(GAZYVA,
Genentech, Inc.; previously known as GA101), pertuzumab injection (PERJETA,
Genentech, Inc.),
or denosumab (Xgeva, Amgen Inc.). In another embodiment, the antibody is
Basiliximab (Simulect;
Novartis). In another embodiment, the antibody is Daclizumab (Zenapax; Roche).
[0517] In another embodiment, the antibody administered in combination with
apoptotic cells is
directed to a tumor or cancer antigen or a fragment thereof, that is described
herein and/or that is
known in the art. In another embodiment, the antibody is directed to a tumor-
associated antigen. In
another embodiment, the antibody is directed to a tumor-associated antigen or
a fragment thereof that
is an angiogenic factor.
.. In some embodiments, antibodies described herein may be used in combination
with compositions
described herein, for example but not limited to a composition comprising
early apoptotic cells.
[0518] In some embodiments, the combination therapy comprises one or more
cancer therapeutic
agents. In some embodiments, the combination therapy comprises at least one
cancer therapeutic
agent. In some embodiments, the combination therapy comprises one cancer
therapeutic agent. In
.. some embodiments, the combination therapy comprises two cancer therapeutic
agents. In some
embodiments, the combination therapy comprises 3 cancer therapeutic agents. In
some embodiments,
the combination therapy comprises 4 cancer therapeutic agents. In some
embodiments, the
combination therapy comprises 5 cancer therapeutic agents. In some
embodiments, the combination
therapy comprises 6 cancer therapeutic agents.
[0519] In some embodiments, disclosed herein are combination therapies
comprising an early
apoptotic cell population as described herein and one or more cancer
therapies. In some embodiments,
a cancer therapy comprises radiation therapy, chemotherapy, transplantation,
immunotherapy,
targeted therapy, hormone therapy, photodynamic therapy, or surgery, or a
combination thereof.
[0520] In some embodiments, a cancer therapy comprises chimeric antigen
receptor-expressing T-
cell (CAR T-cell) therapy, as described herein. In some embodiments, a cancer
therapy comprises an
immunotherapy comprising administration of an antibody or antigen-binding
portion thereof, as
described herein. In some embodiments, a cancer therapy comprises radiation
therapy. In some
embodiments, a cancer therapy comprises chemotherapy. In some embodiments, a
cancer therapy
comprises transplantation. In some embodiments, a cancer therapy comprises an
immunotherapy. In
some embodiments, a cancer therapy comprises a targeted therapy. In some
embodiments, a cancer
therapy comprises a hormone therapy. In some embodiments, a cancer therapy
comprises a
photodynamic therapy. In some embodiments, a cancer therapy comprises surgery.
Formulations
[0521] Pharmaceutical compositions disclosed herein comprising early
apoptotic cell populations,
can be conveniently provided as sterile liquid preparations, e.g., isotonic
aqueous solutions,
CA 03151815 2022-02-17
116
WO 2021/053667
PCT/IL2020/051011
suspensions, emulsions, dispersions, or viscous compositions, which may be
buffered to a selected
pH, Liquid preparations are normally easier to prepare than gels, other
viscous compositions, and
solid compositions. Additionally, liquid compositions are somewhat more
convenient to administer,
especially by injection. Viscous compositions, on the other hand, can be
formulated within the
appropriate viscosity range to provide longer contact periods with specific
tissues. Liquid or viscous
compositions can comprise carriers, which can be a solvent or dispersing
medium containing, for
example, water, saline, phosphate buffered saline, polyol (for example,
glycerol, propylene glycol,
liquid polyethylene glycol, and the like) and suitable mixtures thereof.
[0522] Sterile injectable solutions can be prepared by incorporating the
early apoptotic cell
population described herein and utilized in practicing the methods disclosed
herein, in the required
amount of the appropriate solvent with various amounts of the other
ingredients, as desired. Such
compositions may be in admixture with a suitable carrier, diluent, or
excipient such as sterile water,
physiological saline, glucose, dextrose, or the like. The compositions can
also be lyophilized. The
compositions can contain auxiliary substances such as wetting, dispersing, or
emulsifying agents (e.g.,
methylcellulose), pH buffering agents, gelling or viscosity enhancing
additives, preservatives,
flavoring agents, colors, and the like, depending upon the route of
administration and the preparation
desired. Standard texts, such as "REMINGTON'S PHARMACEUTICAL SCIENCE", 17th
edition,
1985, incorporated herein by reference, may be consulted to prepare suitable
preparations, without
undue experimentation.
[0523] Various additives which enhance the stability and sterility of the
compositions, including
antimicrobial preservatives, antioxidants, chelating agents, and buffers, can
be added. Prevention of
the action of microorganisms can be ensured by various antibacterial and
antifungal agents, for
example, parabens, chlorobutanol, phenol, sorbic acid, and the like. Prolonged
absorption of the
injectable pharmaceutical form can be brought about by the use of agents
delaying absorption, for
example, aluminum monostearate and gelatin. According to the disclosure
herein, however, any
vehicle, diluent, or additive used would have to be compatible with the
genetically modified
immunoresponsive cells or their progenitors.
[0524] The compositions or formulations described herein can be isotonic,
i.e., they can have the
same osmotic pressure as blood and lacrimal fluid. The desired isotonicity of
the compositions as
disclosed herein may be accomplished using sodium chloride, or other
pharmaceutically acceptable
agents such as dextrose, boric acid, sodium tartrate, propylene glycol or
other inorganic or organic
solutes. Sodium chloride may be preferred particularly for buffers containing
sodium ions.
[0525] Viscosity of the compositions, if desired, can be maintained at
the selected level using a
pharmaceutically acceptable thickening agent. Methylcellulose may be preferred
because it is readily
CA 03151815 2022-02-17
117
WO 2021/053667
PCT/IL2020/051011
and economically available and is easy to work with.
[0526] Other suitable thickening agents include, for example, xanthan
gum, carboxymethyl
cellulose, hydroxypropyl cellulose, carbomer, and the like. The preferred
concentration of the
thickener will depend upon the agent selected. The important point is to use
an amount that will
achieve the selected viscosity. Obviously, the choice of suitable carriers and
other additives will
depend on the exact route of administration and the nature of the particular
dosage form, e.g., liquid
dosage form (e.g., whether the composition is to be formulated into a
solution, a suspension, gel or
another liquid form, such as a time release form or liquid-filled form).
[0527] Those skilled in the art will recognize that the components of the
compositions or
.. formulations should be selected to be chemically inert and will not affect
the viability or efficacy of
the early apoptotic cell populations as described herein, for use in the
methods disclosed herein. This
will present no problem to those skilled in chemical and pharmaceutical
principles, or problems can
be readily avoided by reference to standard texts or by simple experiments
(not involving undue
experimentation), from this disclosure and the documents cited herein.
[0528] One consideration concerning the therapeutic use of genetically
modified immunoresponsive
cells disclosed herein is the quantity of cells necessary to achieve an
optimal effect. The quantity of
cells to be administered will vary for the subject being treated. In a some
embodiments, between
104 to 1010, between 105 to 109, or between 106 and 108 genetically modified
immunoresponsive cells
disclosed herein are administered to a human subject. More effective cells may
be administered in
even smaller numbers. In some embodiments, at least about 1 x 108, 2 x 108,
3x108, 4 x 108, and 5 x
108 genetically modified immunoresponsive cells disclosed herein are
administered to a human
subject. The precise determination of what would be considered an effective
dose may be based on
factors individual to each subject, including their size, age, sex, weight,
and condition of the particular
subject. Dosages can be readily ascertained by those skilled in the art from
this disclosure and the
knowledge in the art.
[0529] The skilled artisan can readily determine the amount of cells and
optional additives,
vehicles, and/or carrier in compositions and to be administered in methods
disclosed herein.
Typically, any additives (in addition to the active cell(s) and/or agent(s))
are present in an amount of
0.001 to 50% (weight) solution in phosphate buffered saline, and the active
ingredient is present in
the order of micrograms to milligrams, such as about 0.0001 to about 5 wt %.
In another embodiment
about 0.0001 to about 1 wt %. In still another embodiment, about 0.0001 to
about 0.05 wt% or about
0.001 to about 20 wt %. In a further embodiment, about 0.01 to about 10 wt %.
In another
embodiment, about 0.05 to about 5 wt %. Of course, for any composition to be
administered to an
animal or human, and for any particular method of administration, it is
preferred to determine
CA 03151815 2022-02-17
118
WO 2021/053667
PCT/IL2020/051011
therefore: toxicity, such as by determining the lethal dose (LD) and LD50 in a
suitable animal model
e.g., rodent such as mouse; and, the dosage of the composition(s),
concentration of components
therein and timing of administering the composition(s), which elicit a
suitable response. Such
determinations do not require undue experimentation from the knowledge of the
skilled artisan, this
disclosure and the documents cited herein. And, the time for sequential
administrations can be
ascertained without undue experimentation.
Nucleic acid sequences, vectors, cells
[0530] In some embodiments, disclosed herein are an isolated nucleic acid
sequence encoding a
chimeric antigen receptor (CAR) as described herein for uses in the
compositions and methods as
disclosed herein.
[0531] In another embodiment, disclosed herein are a vector comprising the
nucleic acid sequence
encoding a chimeric antigen receptor (CAR) as described herein.
[0532] In some embodiments, disclosed herein are an isolated nucleic acid
sequence encoding a
genetically modified T-cell receptor (TCR) as described herein for uses in the
compositions and
methods as disclosed herein. In another embodiment, disclosed herein are a
vector comprising the
nucleic acid sequence encoding a genetically modified T-cell receptor (TCR) as
described herein.
[0533] Genetic modification of immunoresponsive cells (e.g., T-cells, CTL
cells, NK cells) can be
accomplished by transducing a substantially homogeneous cell composition with
a recombinant DNA
construct. In some embodiments, a retroviral vector (either gamma- retroviral
or lentiviral) is
employed for the introduction of the DNA construct into the cell. For example,
a polynucleotide
encoding a receptor that binds an antigen (e.g., a tumor antigen, or a
valiant, or a fragment thereof),
can be cloned into a retroviral vector and expression can be driven from its
endogenous promoter,
from the retroviral long terminal repeat, or from a promoter specific for a
targeT-cell type of interest.
Non-viral vectors may be used as well.
[0534] Non-viral approaches can also be employed for the expression of a
protein in cell. For
example, a nucleic acid molecule can be introduced into a cell by
administering the nucleic acid in
the presence of lipofection (Feigner et al., Proc. Natl. Acad. Sci. U.S.A.
84:7413, 1987; Ono et al.,
Neuroscience Letters 17:259, 1990; Brigham et al, Am. J. Med. Sci. 298:278,
1989; Staubinger et al,
Methods in Enzymology 101 :512, 1983), asialoorosomucoid-polyly sine
conjugation (Wu et al.,
Journal of Biological Chemistry 263: 14621, 1988; Wu et al., Journal of
Biological Chemistry
264:16985, 1989), or by micro-injection under surgical conditions (Wolff et
al., Science 247: 1465,
1990). Other non-viral means for gene transfer include transfection in vitro
using calcium phosphate,
DEAE dextran, electroporation, and protoplast fusion. Liposomes can also be
potentially beneficial
for delivery of DNA into a cell. Transplantation of normal genes into the
affected tissues of a subject
CA 03151815 2022-02-17
119
WO 2021/053667
PCT/IL2020/051011
can also be accomplished by transferring a normal nucleic acid into a
cultivatable cell type ex vivo
(e.g., an autologous or heterologous primary cell or progeny thereof), after
which the cell (or its
descendants) are injected into a targeted tissue or are injected systemically.
Recombinant receptors
can also be derived or obtained using transposases or targeted nucleases (e.g.
Zinc finger nucleases,
meganucleases, or TALE nucleases). Transient expression may be obtained by RNA
electroporation.
cDNA expression for use in polynucleotide therapy methods can be directed from
any suitable
promoter (e.g., the human cytomegalovirus (CMV), simian virus 40 (SV40), or
metallothionein
promoters), and regulated by any appropriate mammalian regulatory element or
intron (e.g. the
elongation factor la enhancer/promoter/intron structure). For example, if
desired, enhancers known to
preferentially direct gene expression in specific cell types can be used to
direct the expression of a
nucleic acid. The enhancers used can include, without limitation, those that
are characterized as tissue-
or cell-specific enhancers. Alternatively, if a genomic clone is used as a
therapeutic construct,
regulation can be mediated by the cognate regulatory sequences or, if desired,
by regulatory sequences
derived from a heterologous source, including any of the promoters or
regulatory elements described
above.
[0535] In another embodiment, disclosed herein are a cell comprising the
vector comprising the
nucleic acid sequence encoding a chimeric antigen receptor (CAR) as disclosed
herein.
Methods of Use
[0536] In some embodiments, disclosed herein are methods for slowing,
reducing, inhibiting, or
eliminating metastatic spread of a cancer or a tumor, or any combination
thereof in a subject,
comprising the step of administering to a subject a combination therapy
comprising an early apoptotic
cell population and one or more cancer therapeutic agents as described herein.
In some embodiments,
the cancer therapeutic agent comprises CAR T-cells. In some embodiments, the
cancer therapeutic
agent comprises an antibody or antigen binding portion thereof. In some
embodiments, the method
slows, reduces, inhibits, or eliminates metastatic spread of a cancer or a
tumor, or any combination
thereof, in a subject, compared to a subject not being administered an early
apoptotic cell population.
[0537] A skilled artisan would appreciate that the term "metastasis"
encompasses the spread of
cancer cells from the place where they first formed to another part of the
body. In metastasis, cancer
cells break away from the original (primary) tumor, travel through the blood
or lymph system, and
form a new tumor in other organs or tissues of the body. In some embodiments,
the new, metastatic
tumor is the same type of cancer as the primary tumor. For example, if breast
cancer spreads to the
lung, the cancer cells in the lung are breast cancer cells, not lung cancer
cells.
[0538] A skilled artisan would be familiar with ways for determining the
effectiveness of slowing,
reducing, inhibiting, or eliminating metastatic spread. For example, imaging
methods can be used to
CA 03151815 2022-02-17
120
WO 2021/053667
PCT/IL2020/051011
determine whether a subject's tumor has spread to new or secondary parts or
organs of the body.
[0539] In some embodiments, disclosed herein are methods for slowing,
reducing, inhibiting, or
eliminating metastatic spread of a cancer or a tumor, or any combination
thereof, in a subject
undergoing cancer therapy, the method comprising the step of administering an
early apoptotic cell
population to said subject, wherein said method slows, reduces, inhibits, or
eliminates metastatic
spread of a cancer or a tumor, or any combination thereof, in said subject,
compared to a subject
undergoing cancer therapy and not administered an early apoptotic cell
population. In some
embodiments, the cancer therapy comprises radiation therapy, chemotherapy,
transplantation,
immunotherapy, targeted therapy, hormone therapy, photodynamic therapy, or
surgery, or a
combination thereof. In some embodiments, the cancer therapy comprises a CAR T-
cell therapy. In
some embodiments, the cancer therapy comprises radiation therapy. In some
embodiments, the cancer
therapy comprises a chemotherapy. In some embodiments, the cancer therapy
comprises
transplantation. In some embodiments, the cancer therapy comprises an
immunotherapy. In some
embodiments, the cancer therapy comprises targeted therapy. In some
embodiments, the cancer
therapy comprises a hormone therapy. In some embodiments, the cancer therapy
comprises
photodynamic therapy. In some embodiments, the cancer therapy comprises
surgery.
[0540] In some embodiments, a subject undergoing cancer therapy comprises
a subject being
treated with or who has been previously treated with chimeric antigen receptor
T-cell (CAR T-cell)
therapy, radiation therapy, chemotherapy, transplantation, immunotherapy,
targeted therapy,
hormone therapy, photodynamic therapy, has been surgically treated, or any
combination thereof.
[0541] In some embodiments, the cancer therapy comprises CAR T-cell therapy.
In some
embodiments, the cancer therapy comprises radiation therapy, chemotherapy,
transplantation,
immunotherapy, targeted therapy, hormone therapy, photodynamic therapy, has
been surgically
treated, or any combination thereof.
[0542] In some embodiments, slowing, reducing, inhibiting, or eliminating
metastatic spread of a
cancer or a tumor comprises stabilization of resident macrophages in the area
of the cancer or tumor.
In some embodiments, slowing, reducing, inhibiting, or eliminating metastatic
spread of a cancer or
a tumor comprises increasing the number of resident macrophages in the area of
the cancer or tumor.
In some embodiments, slowing, reducing, inhibiting, or eliminating metastatic
spread of a cancer or
a tumor comprises increasing the anti-tumor macrophage population in the area
of the cancer or
tumor.
[0543] In some embodiments, slowing, reducing, inhibiting, or eliminating
metastatic spread of a
cancer or a tumor comprises reducing the incidence of metastasis to lymph
nodes. In some
embodiments, slowing, reducing, inhibiting, or eliminating metastatic spread
of a cancer or a tumor
CA 03151815 2022-02-17
121
WO 2021/053667
PCT/IL2020/051011
comprises reducing the incidence of angiogenesis. In some embodiments,
slowing, reducing,
inhibiting, or eliminating metastatic spread of a cancer or a tumor comprises
reducing the rate of
metastasis of said tumor or said cancer. In some embodiments, slowing,
reducing, inhibiting, or
eliminating metastatic spread of a cancer or a tumor comprises reducing the
formation of cancer cells
with metastatic potential. In some embodiments, slowing, reducing, inhibiting,
or eliminating
metastatic spread of a cancer or a tumor comprises reducing the migration of
cancer cells.
[0544] In some embodiments, slowing, reducing, inhibiting, or eliminating
metastatic spread of a
cancer or a tumor comprises between 10%-100% less incidence of metastasis. In
some embodiments,
slowing, reducing, inhibiting, or eliminating metastatic spread of a cancer or
a tumor comprises up to
10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% less incidence of
metastasis.
[0545] In some embodiments, the cancer therapeutic agent comprises
chimeric antigen receptor-
expressing T-cells (CAR T-cells). In some embodiments, the CAR T-cells and the
early apoptotic
cells are comprised in separate compositions. In some embodiments, the CAR T-
cells and the early
apoptotic cells are administered together. In some embodiments, the CAR T-
cells and the early
apoptotic cells are administered concurrently. In some embodiments, the CAR T-
cells are
administered prior to the early apoptotic cells. In some embodiments, the CAR
T-cells are
administered subsequent to administration of the early apoptotic cells.
[0546] In some embodiments, the administration of the combination therapy
comprising an early
apoptotic cell population and one or more cancer therapeutic agents occurs at
the same site of
administration. In some embodiments, the early apoptotic cell population and
the one or more cancer
therapeutic agents are administered at different administration sites. In some
embodiments, the early
apoptotic cell population and one or more cancer therapeutic agents are
intravenously administered
to the subject. In some embodiments, the early apoptotic cell population and
one or more cancer
therapeutic agents are orally administered to the subject.
[0547] In some embodiments, the one or more cancer therapeutic agents are
administered several
days before the administration of the early apoptotic cell population. In some
embodiments, one or
more cancer therapeutic agents are administered 1, 2, 3, 4, or 5 days prior to
the administration of the
early apoptotic cell population.
[0548] In some embodiments, the one or more cancer therapeutic agents are
administered several
days after the administration of the early apoptotic cell population. In some
embodiments, one or
more cancer therapeutic agents are administered 1, 2, 3, 4, or 5 days
subsequent to the administration
of the early apoptotic cell population.
[0549] In some embodiments, the early apoptotic cell population and one or
more cancer
therapeutic agents are administered at least once during a treatment cycle. In
some embodiments, the
CA 03151815 2022-02-17
122
WO 2021/053667
PCT/IL2020/051011
early apoptotic cell population and one or more cancer therapeutic agents are
administered to the
subject on the same days. In some embodiments, the early apoptotic cell
population and one or more
cancer therapeutic agents are administered to the subject on different days.
In some embodiments, the
early apoptotic cell population and one or more cancer therapeutic agents are
administered to the
subject on the same days and on different days according to treatment
schedules (or treatment cycles).
[0550] In some embodiments, the treatment cycle comprises at least two,
at least three, at least
four, at least five, at least six, at least seven, at least 14, at least 21,
at least 28, at least 48, or at least
96 days or more. In one embodiment, a treatment cycle is 7 days. In one
embodiment, a treatment
cycle is 14 days. In one embodiment, a treatment cycle is 28 days. In some
embodiments, the early
apoptotic cell population and one or more cancer therapeutic agents are
administered over the same
treatment cycle or concurrently over different treatment cycles assigned to
each. In some
embodiments, the treatment cycle is determined by a health care professional
based on conditions and
needs of the subject.
[0551] In some embodiments, the early apoptotic cell population and one or
more cancer
therapeutic agents are administered on at least one day, at least two days, at
least three days, at least
four days, at least five days, at least six days, at least seven days, at
least eight days, at least nine days,
at least ten days, at least eleven days, at least twelve days, at least 13
days, at least 14 days, at least 21
days, or all 28 days of a 28 day treatment cycle. In some embodiments, the
early apoptotic cell
population and one or more cancer therapeutic agents are administered to a
subject once a day. In
some embodiments, the early apoptotic cell population and one or more cancer
therapeutic agents are
administered twice a day.
[0552] In some embodiments, the early apoptotic cell population and one or
more cancer
therapeutic agents are administered to the subject over one or more treatment
cycles. In some
embodiments, the subject is administered 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10
treatment cycles. In some
embodiments, the early apoptotic cell population and one or more cancer
therapeutic agents as
described herein are administered for several years. In some embodiments, the
early apoptotic cell
population and one or more cancer therapeutic agents as described herein are
administered between
one month to six months.
[0553] The number of times the early apoptotic cell population and one or more
cancer therapeutic
agents are administered to a subject in need thereof depends on the discretion
of a medical
professional, the disorder, the severity of the disorder, and the subject's
response to the combination
therapy. In the case wherein the subject's condition does not improve, upon
the doctor's discretion the
early apoptotic cell population and one or more cancer therapeutic agents are
administered
chronically, that is, for an extended period of time, including throughout the
duration of the subject's
CA 03151815 2022-02-17
123
WO 2021/053667
PCT/IL2020/051011
life in order to slow, reduce, inhibit, or eliminate metastatic spread of a
cancer or a tumor, or otherwise
control or limit the symptoms of the subject's disease or condition.
[0554] In the case where the subject's status does improve, upon the
doctor's discretion the early
apoptotic cell population and one or more cancer therapeutic agents are
administered continuously;
or, the dose being administered may be temporarily reduced or temporarily
suspended for a certain
length of time (i.e., a "drug holiday"). The length of the drug holiday varies
between 2 days and 1
year, including by way of example only, 2 days, 3 days, 4 days, 5 days, 6
days, 7 days, 10 days, 12
days, 15 days, 20 days, 28 days, 35 days, 50 days, 70 days, 100 days, 120
days, 150 days, 180 days,
200 days, 250 days, 280 days, 300 days, 320 days, 350 days, and 365 days. The
dose reduction during
a drug holiday may be from 10%- 100%, including by way of example only 10%,
15%, 20%, 25%,
30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, and
100%.
[0555] In one embodiment, disclosed herein are methods for treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor
comprising the step of
administering a composition as disclosed herein.
[0556] In some embodiments, disclosed herein are methods of treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor in
a subject comprising the
step of administering a composition comprising apoptotic cells. In some
embodiments, disclosed
herein are methods of treating, preventing, inhibiting the growth of, delaying
disease progression,
reducing the tumor load, or reducing the incidence of a cancer or a tumor in a
subject, or any
combination thereof. In some embodiments, methods disclosed herein reduce the
size and or growth
rate of a tumor or cancer. In some embodiments, methods disclosed herein
increase the survival of a
subject suffering from a tumor or cancer. In some embodiments, use of
apoptotic cells or a
composition thereof increases the efficacy of genetically modified immune cell
therapy, for example
but not limited to CAR T-cell therapy.
[0557] In another embodiment, disclosed herein are methods for treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor
comprising the step of
administering genetically modified immune cells and a composition comprising
an additional agent,
wherein said additional agent comprises apoptotic cells, a supernatant from
apoptotic cells, a CTLA-
4 blocking agent, an alpha-1 anti-trypsin or fragment or analog thereof, a
tellurium-based compound,
or an immune modulating agent, or any combination thereof, wherein said method
treats, prevents,
inhibits, reduces the incidence of, ameliorates or alleviates a cancer or a
tumor in said subject
compared with a subject administered said genetically modified immune cells
and not administered
the additional agent. In another embodiment, said genetically modified immune
cells comprise
genetically modified T-cell, cytotoxic T-cells, Treg cells, effector T-cells,
helper T-cells, NK cells,
CA 03151815 2022-02-17
124
WO 2021/053667
PCT/IL2020/051011
or dendritic cells.
[0558] In another embodiment, disclosed herein are methods for treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor
comprising the step of
administering chimeric antigen receptor-expressing T-cells (CAR T-cells) and a
composition
.. comprising an additional agent, wherein said additional agent comprises
apoptotic cells, a supernatant
from apoptotic cells, a CTLA-4 blocking agent, an alpha-1 anti-tryp sin or
fragment or analog thereof,
a tellurium-based compound, or an immune modulating agent, or any combination
thereof, wherein
said method treats, prevents, inhibits, reduces the incidence of, ameliorates
or alleviates a cancer or
a tumor in said subject compared with a subject administered said genetically
modified immune cells
and not administered the additional agent.
[0559] In another embodiment, disclosed herein are methods for treating,
preventing, inhibiting,
reducing the incidence of, ameliorating, or alleviating a cancer or a tumor
comprising the step of
administering genetically modified T-cell receptor cells (TCR T-cells) and a
composition comprising
an additional agent, wherein said additional agent comprises apoptotic cells,
a supernatant from
apoptotic cells, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or fragment
or analog thereof, a
tellurium-based compound, or an immune modulating agent, or any combination
thereof, wherein
said method treats, prevents, inhibits, reduces the incidence of, ameliorates
or alleviates a cancer or
a tumor in said subject compared with a subject administered said genetically
modified immune cells
and not administered the additional agent.
[0560] In another embodiment, administration of apoptotic cells or an
apoptotic supernatant or
compositions thereof does not reduce the efficacy for treating, preventing,
inhibiting, reducing the
incidence of, ameliorating, or alleviating a cancer or a tumor, of said
administering chimeric antigen
receptor-expressing T-cells. In another embodiment, administration of an
additional agent
comprising apoptotic cells, a supernatant from apoptotic cells, a CTLA-4
blocking agent, an alpha-1
anti-trypsin or fragment or analog thereof, a tellurium-based compound, or an
immune modulating
agent, or any combination thereof, or compositions thereof does not reduce the
efficacy for treating,
preventing, inhibiting, reducing the incidence of, ameliorating, or
alleviating a cancer or a tumor, of
said administering chimeric antigen receptor-expressing T-cells.
[0561] In another embodiment, administration of apoptotic cells or an
apoptotic supernatant or
compositions thereof increases the efficacy for treating, preventing,
inhibiting, reducing the incidence
of, ameliorating, or alleviating a cancer or a tumor, of said administering
chimeric antigen receptor-
expressing T-cells. In another embodiment, administration of an additional
agent comprising
apoptotic cells, a supernatant from apoptotic cells, a CTLA-4 blocking agent,
an alpha-1 anti-trypsin
or fragment or analog thereof, a tellurium-based compound, or an immune
modulating agent, or any
CA 03151815 2022-02-17
125
WO 2021/053667
PCT/IL2020/051011
combination thereof, or compositions thereof increases the efficacy for
treating, preventing,
inhibiting, reducing the incidence of, ameliorating, or alleviating a cancer
or a tumor, of said
administering chimeric antigen receptor-expressing T-cells.
[0562] In one embodiment, methods increasing the efficacy of a genetically
modified immune cell
cancer therapy comprise administering said genetically modified immune cells
and an additional
agent comprising apoptotic cells, a supernatant from apoptotic cells, a CTLA-4
blocking agent, an
alpha-1 anti-trypsin or fragment or analog thereof, a tellurium-based
compound, or an immune
modulating agent, or any combination thereof, or compositions thereof, wherein
the efficacy is
increased compared with a subject not administered said additional agent. In
another embodiment
said genetically modified immune cells are T-cells. In another embodiment, a T-
cell is a naive T-cell.
In another embodiment, a T-cell is a naive CD4+ T-cell. In another embodiment,
a T-cell is a naive
T-cell. In another embodiment, a T-cell is a naive CD8+ T-cell. In another
embodiment, the
genetically modified immune cell is a natural killer (NK) cell. In another
embodiment, the genetically
modified immune cell is a dendritic cell. In still another embodiment, the
genetically modified T-cell
is a cytotoxic T lymphocyte (CTL cell). In another embodiment, the genetically
modified T-cell is a
regulatory T-cell (Treg). In another embodiment, the genetically modified T-
cell is a chimeric antigen
receptor (CAR) T-cell. In another embodiment, the genetically modified T-cell
is a genetically
modified T-cell receptor cell (TCR T-cell). In another embodiment, methods
increasing the efficacy
of a CAR T-cell cancer therapy comprise administering said genetically
modified immune cells and
an additional agent comprising apoptotic cells, a supernatant from apoptotic
cells, a CTLA-4 blocking
agent, an alpha-1 anti-trypsin or fragment or analog thereof, a tellurium-
based compound, or an
immune modulating agent, or any combination thereof, or compositions thereof,
wherein the efficacy
is increased compared with a subject not administered said additional agent.
[0563] In another embodiment, methods herein reduce the level of production of
at least one pro-
inflammatory cytokine compared with the level of said pro-inflammatory
cytokine in a subject
receiving an immune cancer therapy and not administered an additional agent.
In another
embodiment, methods herein inhibit or reduce the incidence of cytokine release
syndrome or cytokine
storm in a subject undergoing a genetically modified immune cell cancer
therapy and not
administered an additional agent.
[0564] In another embodiment, methods disclosed herein reduce IL-6.
[0565] In another embodiment, methods herein increase the production of at
least one cytokine
compared with the level of said cytokine in a subject receiving an immune
cancer therapy and not
administered an additional agent. In some embodiments, the additional agent is
apoptotic cells. In
other embodiments, the additional agent is an apoptotic cell supernatant. In
another embodiment,
CA 03151815 2022-02-17
126
WO 2021/053667
PCT/IL2020/051011
methods disclosed herein increase IL-2.
[0566] A skilled artisan would appreciate that the term "production" as used
herein in reference to a
cytokine, may encompass expression of the cytokine as well as secretion of the
cytokine from a cell.
In one embodiment, increased production of a cytokine results in increased
secretion of the cytokine
from the cell. In an alternate embodiment, decreased production of a cytokine
results in decreased
secretion of the cytokine from the cell. In another embodiment, methods
disclosed herein decrease
secretion of at least one cytokine. In another embodiment, methods disclosed
herein decrease
secretion of IL-6. In another embodiment, methods disclosed herein increase
secretion of at least one
cytokine. In another embodiment, methods disclosed herein increase secretion
of IL-2.
[0567] In another embodiment, a cell secreting at least one cytokine is a
tumor cell. In another
embodiment, a cell secreting at least one cytokine is a T-cell. In another
embodiment, a cell secreting
at least one cytokine is an immune cell. In another embodiment, a cell
secreting at least one cytokine
is a macrophage. In another embodiment, a cell secreting at least one cytokine
is a B cell lymphocyte.
In another embodiment, a cell secreting at least one cytokine is a mast cell.
In another embodiment,
a cell secreting at least one cytokine is an endothelial cell. In another
embodiment, a cell secreting at
least one cytokine is a fibroblast. In another embodiment, a cell secreting at
least one cytokine is a
stromal cell. A skilled artisan would recognize that the level of cytokines
may be increased or
decreased in cytokine secreting cells depending on the environment surrounding
the cell.
[0568] In yet another embodiment, an additional agent used in the methods
disclosed herein increases
secretion of at least one cytokine. In yet another embodiment, an additional
agent used in the methods
disclosed herein maintains secretion of at least one cytokine. In still
another embodiment, an
additional agent used in the methods disclosed herein does not decrease
secretion of at least one
cytokine. In another embodiment, an additional agent used in the methods
disclosed herein increases
secretion of IL-2. In another embodiment, an additional agent used in the
methods disclosed herein
increases secretion of IL-2R. In another embodiment, an additional agent used
in the methods
disclosed herein maintains secretion levels of IL-2. In another embodiment, an
additional agent used
in the methods disclosed herein maintains secretion levels of IL-2R. In
another embodiment, an
additional agent used in the methods disclosed herein does not decrease
secretion levels of IL-2R. In
another embodiment, an additional agent used in the methods disclosed herein
maintains or increases
secretion levels of IL-2. In another embodiment, an additional agent used in
the methods disclosed
herein maintains or increases secretion levels of IL-2R. In another
embodiment, an additional agent
used in the methods disclosed herein does not decrease secretion levels of IL-
2R.
[0569] In still a further embodiment, an additional agent used in the methods
disclosed herein
decreases secretion of IL-6. In another embodiment, an additional agent used
in the methods
CA 03151815 2022-02-17
127
WO 2021/053667
PCT/IL2020/051011
disclosed herein maintains, increases, or does not decrease secretion levels
of IL-2 while decreasing
secretion of IL-6. In another embodiment, an additional agent used in the
methods disclosed herein
maintains, increases, or does not decrease secretion levels of IL-2R while
decreasing secretion of IL-
6.
[0570] In one embodiment, methods of increasing the efficacy of a CAR T-cell
cancer therapy
disclosed herein comprises decreasing the level of IL-6 in said subject, said
method comprising
administering CAR T-cells and an additional agent comprising apoptotic cells,
a supernatant from
apoptotic cells, a CTLA-4 blocking agent, an alpha-1 anti-trypsin or fragment
or analog thereof, a
tellurium-based compound, or an immune modulating agent, or any combination
thereof, or
compositions thereof, wherein the efficacy is increased compared with a
subject not administered
said additional agent. In another embodiment, methods of increasing the
efficacy of a CAR T-cell
cancer therapy disclosed herein comprises increasing the level of IL-2 in said
subject, said method
comprising administering CAR T-cells and an additional agent comprising
apoptotic cells, a
supernatant from apoptotic cells, a CTLA-4 blocking agent, an alpha-1 anti-
trypsin or fragment or
analog thereof, a tellurium-based compound, or an immune modulating agent, or
any combination
thereof, or compositions thereof, wherein the efficacy is increased compared
with a subject not
administered said additional agent. In another embodiment, methods of
increasing the efficacy of a
CAR T-cell cancer therapy disclosed herein comprises increasing proliferation
of said CAR T-cells,
said method comprising administering CAR T-cells and an additional agent
comprising apoptotic
cells, a supernatant from apoptotic cells, a CTLA-4 blocking agent, an alpha-1
anti-trypsin or
fragment or analog thereof, a tellurium-based compound, or an immune
modulating agent, or any
combination thereof, or compositions thereof, wherein the efficacy and
proliferation of said CAR T-
cells is increased compared with a subject not administered said additional
agent.
[0571] In one embodiment, methods of increasing the efficacy of CAR T-cell
cancer therapy,
decrease or inhibit cytokine production in the subject, said methods
comprising the step of
administering a composition comprising CAR T-cells and a CTLA-4 blocking
agent, an alpha-1 anti-
trypsin or fragment or analog thereof, a tellurium-based compound, or an
immune modulating agent,
or any combination thereof, or compositions thereof. In another embodiment,
methods of treating,
preventing, inhibiting, reducing the incidence of, ameliorating, or
alleviating a cancer or tumor also
decrease or inhibit cytokine production in the subject, said methods
comprising the step of
administering a composition comprising CAR T-cells and a CTLA-4 blocking
agent, an alpha-1 anti-
trypsin or fragment or analog thereof, a tellurium-based compound, or an
immune modulating agent,
or any combination thereof, or compositions thereof.
[0572] In another embodiment, disclosed herein are methods of treating
cytokine release syndrome
CA 03151815 2022-02-17
128
WO 2021/053667
PCT/IL2020/051011
or cytokine storm in a subject undergoing CAR T-cell cancer therapy.
[0573] In another embodiment, methods of treating, preventing, inhibiting,
reducing the incidence
of, ameliorating, or alleviating a cancer or tumor, decrease or inhibit
cytokine production in a subject,
said methods comprising the step of administering a composition comprising CAR
T-cells and a
CTLA-4 blocking agent, an alpha-1 anti-trypsin or fragment or analog thereof,
a tellurium-based
compound, or an immune modulating agent, or any combination thereof, or
compositions thereof.
[0574] In another embodiment, disclosed herein is a method of treating a
cancer or a tumor in a
subject, said method comprising the step of administering to said subject any
of the compositions as
described herein. In another embodiment, disclosed herein is a method of
preventing a cancer or a
tumor in a subject, said method comprising the step of administering to said
subject any of the
compositions as described herein. In another embodiment, disclosed herein is a
method of inhibiting
a cancer or a tumor in a subject, said method comprising the step of
administering to said subject any
of the compositions as described herein. In another embodiment, disclosed
herein is a method of
reducing a cancer or a tumor in a subject, said method comprising the step of
administering to said
subject any of the compositions as described herein. In another embodiment,
disclosed herein is a
method of ameliorating a cancer or a tumor in a subject, said method
comprising the step of
administering to said subject any of the compositions as described herein. In
another embodiment,
disclosed herein is a method of alleviating a cancer or a tumor in a subject,
said method comprising
the step of administering to said subject any of the compositions as described
herein.
[0575] In one embodiment, disclosed herein are methods of maintaining or
increasing the
proliferation rate of a genetically modified immune cell during an
immunotherapy, the method
comprising the step of administering a composition comprising apoptotic cells
or an apoptotic
supernatant during the immunotherapy. In another embodiment, said genetically
modified immune
cells comprise a T-cell, a naive T-cell, a naive CD4+ T-cell, a naive CD8+ T-
cell, a natural killer
(NK) cell, a dendritic cell, a cytotoxic T lymphocyte (CTL cell), a regulatory
T-cell (Treg), a chimeric
antigen receptor (CAR) T-cell, or a genetically modified T-cell receptor (TCR)
cell. In another
embodiment, disclosed herein are methods of maintaining or increasing the
proliferation rate of a
CAR T-cell during an immunotherapy, the method comprising the step of
administering a
composition comprising apoptotic cells or an apoptotic supernatant during the
immunotherapy.
[0576] In another embodiment, methods of maintaining or increasing the
proliferation rate of the
genetically modified immune cells does not reduce or inhibit the efficacy of
the immunotherapy. For
example, in another embodiment, methods of maintaining or increasing the
proliferation rate of CAR
T-cells does not reduce or inhibit the efficacy of the CAR T-cell cancer
therapy. In another
embodiment, methods of maintaining or increasing the proliferation rate of the
genetically modified
CA 03151815 2022-02-17
129
WO 2021/053667
PCT/IL2020/051011
immune cells, for example CAR T-cells, decrease or inhibit cytokine production
in the subject.
[0577] In another embodiment, compositions and methods as disclosed herein
utilize combination
therapy with apoptotic cells or apoptotic supernatants as disclosed herein,
and one or more CTLA-4-
blocking agents such as Ipilimumab. In one embodiment, CTLA-4 is a potent
inhibitor of T-cell
activation that helps to maintain self-tolerance. In one embodiment,
administration of an anti-CTLA-
4 blocking agent, which in another embodiment, is an antibody, produces a net
effect of T-cell
activation. In another embodiment, compositions and methods as disclosed
herein utilize combined
therapy comprising apoptotic cells, CAR T-cells, and one or more CTLA-4-
blocking agents.
[0578] In some cases, a polypeptide of and for use in the methods as disclosed
herein comprises at
least one conservative amino acid substitution relative to an unmodified amino
acid sequence. In
other cases, the polypeptide comprises a non-conservative amino acid
substitution. In such cases,
polypeptides having such modifications exhibit increased stability or a longer
half-life relative to a
polypeptide lacking such an amino acid substitution.
[0579] In some embodiment, "treating" comprises therapeutic treatment and
"preventing"
comprises prophylactic or preventative measures, wherein the object is to
prevent or lessen the
targeted pathologic condition or disorder as described hereinabove. Thus, in
some embodiments,
treating may include directly affecting or curing, suppressing, inhibiting,
preventing, reducing the
severity of, delaying the onset of, reducing symptoms associated with the
disease, disorder or
condition, or a combination thereof. Thus, in some embodiments, "treating,"
"ameliorating," and
"alleviating" refer inter alia to delaying progression, expediting remission,
inducing remission,
augmenting remission, speeding recovery, increasing efficacy of or decreasing
resistance to
alternative therapeutics, or a combination thereof. In some embodiments,
"preventing" refers, inter
alia, to delaying the onset of symptoms, preventing relapse to a disease,
decreasing the number or
frequency of relapse episodes, increasing latency between symptomatic
episodes, or a combination
thereof. In some embodiments, "suppressing" or "inhibiting", refers inter alia
to reducing the severity
of symptoms, reducing the severity of an acute episode, reducing the number of
symptoms, reducing
the incidence of disease-related symptoms, reducing the latency of symptoms,
ameliorating
symptoms, reducing secondary symptoms, reducing secondary infections,
prolonging patient
survival, or a combination thereof.
[0580] A skilled artisan would appreciate that the term "antigen recognizing
receptor" may
encompass a receptor that is capable of activating an immune cell (e.g., a T-
cell) in response to antigen
binding. Exemplary antigen recognizing receptors may be native or endogenous T-
cell receptors or
chimeric antigen receptors in which a tumor antigen-binding domain is fused to
an intracellular
signaling domain capable of activating an immune cell (e.g., a T-cell).
CA 03151815 2022-02-17
130
WO 2021/053667
PCT/IL2020/051011
[0581] In some embodiments, methods described herein increase the
survival of a subject
suffering from a cancer or a tumor, and comprise administering an early
apoptotic cell population to
said subject, wherein the method increases the survival of the subject.
[0582] A skilled artisan would appreciate that the term "disease" may
encompass any condition or
disorder that damages or interferes with the normal function of a cell,
tissue, or organ. Examples of
diseases include neoplasia or pathogen infection of cell.
[0583] A skilled artisan would appreciate that the term "effective amount" may
encompass an amount
sufficient to have a therapeutic effect. In some embodiments, an "effective
amount" is an amount
sufficient to arrest, ameliorate, or inhibit the continued proliferation,
growth, or metastasis (e.g.,
invasion, or migration) of a neoplasia.
[0584] A skilled artisan would appreciate that the term "neoplasia" may
encompass a disease
characterized by the pathological proliferation of a cell or tissue and its
subsequent migration to or
invasion of other tissues or organs. Neoplasia growth is typically
uncontrolled and progressive, and
occurs under conditions that would not elicit, or would cause cessation of,
multiplication of normal
cells. Neoplasias can affect a variety of cell types, tissues, or organs,
including but not limited to an
organ selected from the group consisting of bladder, bone, brain, breast,
cartilage, glia, esophagus,
fallopian tube, gallbladder, heart, intestines, kidney, liver, lung, lymph
node, nervous tissue, ovaries,
pancreas, prostate, skeletal muscle, skin, spinal cord, spleen, stomach,
testes, thymus, thyroid, trachea,
urogenital tract, ureter, urethra, uterus, and vagina, or a tissue or cell
type thereof. Neoplasias include
cancers, such as sarcomas, carcinomas, or plasmacytomas (malignant tumor of
the plasma cells).
[0585] A skilled artisan would appreciate that the term "pathogen" may
encompass a virus, bacteria,
fungi, parasite or protozoa capable of causing disease.
[0586] A skilled artisan would appreciate that the term "tumor antigen" or
"tumor associated antigen"
may encompass an antigen (e.g., a polypeptide) that is uniquely or
differentially expressed on a tumor
cell compared to a normal or non- IS neoplastic cell. With reference to the
compositions and methods
disclosed herein, a tumor antigen includes any polypeptide expressed by a
tumor that is capable of
activating or inducing an immune response via an antigen recognizing receptor
(e.g., CD 19, MUCI)
or capable of suppressing an immune response via receptor-ligand binding
(e.g., CD47, PD-L1/L2,
B7.1/2).
[0587] A skilled artisan would appreciate that the term "virus antigen" may
encompass a polypeptide
expressed by a virus that is capable of inducing an immune response.
[0588] The terms "comprises", "comprising", and are intended to have the broad
meaning ascribed
to them in U.S. Patent Law and can mean "includes", "including" and the like.
Similarly, the term
"consists of' and "consists essentially of' have the meanings ascribed to them
in U.S. Patent Law.
CA 03151815 2022-02-17
131
WO 2021/053667
PCT/IL2020/051011
The compositions and methods as disclosed herein are envisioned to either
comprise the active
ingredient or specified step, consist of the active ingredient or specified
step, or consist essentially of
the active ingredient or specified step.
[0589] A skilled artisan would appreciate that the term "treatment" may
encompass clinical
.. intervention in an attempt to alter the disease course of the individual or
cell being treated, and can be
performed either for prophylaxis or during the course of clinical pathology.
Therapeutic effects of
treatment include, without limitation, preventing occurrence or recurrence of
disease, alleviation of
symptoms, diminishment of any direct or indirect pathological consequences of
the disease,
preventing metastases, decreasing the rate of disease progression,
amelioration or palliation of the
disease state, and remission or improved prognosis. By preventing progression
of a disease or
disorder, a treatment can prevent deterioration due to a disorder in an
affected or diagnosed subject
or a subject suspected of having the disorder, but also a treatment may
prevent the onset of the disorder
or a symptom of the disorder in a subject at risk for the disorder or
suspected of having the disorder.
[0590] A skilled artisan would appreciate that the term "subject" may
encompass a vertebrate, in
some embodiments, to a mammal, and in some embodiments, to a human. Subject
may also refer, in
some embodiments, to domesticated such as cows, sheep, horses, cats, dogs and
laboratory animals
such as mice, rats, gerbils, hamsters, etc.
[0591] In some embodiments, disclosed herein are CAR T-cells in which the CAR
is directed to a
peptide of interest. In some embodiments, the CAR binds to a peptide of
interest. In another
embodiment, the CAR targets a peptide of interest. In another embodiment, the
CAR activates a
peptide of interest. In another embodiment, the CAR is a ligand of the peptide
of interest. In another
embodiment, the peptide of interest is a ligand of the CAR. Each of these
embodiments is to be
considered part disclosed herein.
[0592] In some embodiments, the immune cell as disclosed herein is not a T-
cell. In another
embodiment, the immune cell as disclosed herein is not an NK cell. In another
embodiment, the
immune cell as disclosed herein is not a CTL. In another embodiment, the
immune cell as disclosed
herein is not a regulatory T-cell. In another embodiment, the immune cell is
not a human embryonic
stem cell. In another embodiment, the immune cell is not a pluripotent stem
cell from which lymphoid
cells may be differentiated.
[0593] One approach to immunotherapy involves engineering a patient's own
immune cells to create
genetically modified immune cells that will recognize and attack their tumor.
Immune cells are
collected and genetically modified, as described herein, for example to
produce chimeric antigen
receptors (CAR) on their cell surface that will allow the immune cell, for
example a T-cell, to
recognize a specific protein antigen on a tumor or cancer cell. An expanded
population of genetically
CA 03151815 2022-02-17
132
WO 2021/053667
PCT/IL2020/051011
modified immune cells, for example CAR T-cells, is then administered to the
patient. In some
embodiments, the administered cells multiply in the patient's body and
recognize and kill cancer and
tumor cells that harbor the antigen on their surface. In another embodiment,
the administered cells
multiply in a patient's body and recognize and kill tumor-associated antigens,
which leads to the death
of cancer and tumor cells.
[0594] In some embodiments, disclosed herein are methods of inhibiting or
reducing the incidence
of cytokine release syndrome or cytokine storm in a subject undergoing CAR T-
cell cancer therapy,
and methods of decreasing or inhibiting cytokine production in a subject
experiencing cytokine
release syndrome or cytokine storm, said methods comprising the step of
administering a composition
comprising apoptotic cells or a supernatant of apoptotic cells. In another
embodiment, disclosed
herein are methods of treating cytokine release syndrome or cytokine storm in
a subject undergoing
CAR T-cell cancer therapy. In another embodiment, disclosed herein are methods
of preventing
cytokine release syndrome or cytokine storm in a subject undergoing CAR T-cell
cancer therapy. In
another embodiment, disclosed herein are methods of alleviating cytokine
release syndrome or
cytokine storm in a subject undergoing CAR T-cell cancer therapy. In another
embodiment, disclosed
herein are methods of ameliorating cytokine release syndrome or cytokine storm
in a subject
undergoing CAR T-cell cancer therapy. In another embodiment, administration of
apoptotic cells or
an apoptotic supernatant or compositions thereof does not reduce the efficacy
of the CAR T-cell
therapy.
[0595] In some embodiments, disclosed herein are methods of inhibiting or
reducing the incidence
of a cytokine release syndrome (CRS) or a cytokine storm in a subject
undergoing chimeric antigen
receptor-expressing T-cell (CAR T-cell) cancer therapy, wherein the method
comprises the step of
administering a composition comprising apoptotic cells or an apoptotic cell
supernatant or
compositions thereof to said subject. In another embodiment, inhibiting or
reducing the incidence of
a cytokine release syndrome (CRS) or a cytokine storm is determined by
measuring cytokine levels
in a subject undergoing chimeric antigen receptor-expressing T-cell cancer
therapy and being
administered apoptotic cells or an apoptotic supernatant. In another
embodiment, measured levels of
cytokines are compared with cytokine levels in a subject not undergoing CAR T-
cell cancer therapy.
In another embodiment, measured cytokine levels are compared with cytokine
levels in a subject not
administered apoptotic cells or an apoptotic supernatant. In yet another
embodiment, measured
cytokine levels are compared with a control subject.
[0596] In another embodiment, the level of pro-inflammatory cytokines are
reduced in the subject
compared with a subject undergoing CAR T-cell cancer therapy and not
administered said apoptotic
cells or said apoptotic cell supernatant or compositions thereof. In another
embodiment, methods
CA 03151815 2022-02-17
133
WO 2021/053667
PCT/IL2020/051011
disclosed herein reduce or inhibit the level of production of at least one pro-
inflammatory cytokines
compared with a subject undergoing CAR T-cell cancer therapy and not
administered said apoptotic
cells or said apoptotic cell supernatant or compositions thereof.
[0597] In another embodiment, a method disclosed herein may further comprise
administration of
additional agents. Alternatively, a method disclosed herein may comprise
administration of additional
agents and not apoptotic cells or an apoptotic cell supernatant. In still a
further embodiment, additional
agents may be those compounds or compositions that enhance or improve, or any
combination
thereof, CAR T-cell cancer therapy. In yet a further embodiment, additional
agents that enhance or
improve CAR T-cell cancer therapy include CTLA-4 blocking agents, an alpha-
lanti-trypsin or
functional fragment thereof, or an analogue thereof, a tellurium-based
compound, or an immune-
modulating agent, or any combination thereof. In another embodiment, an
additional agent includes
apoptotic cells or an apoptotic supernatant. In another embodiment,
administration of an additional
agent, a described herein, is prior to, concurrent with, of following said CAR
T-cell cancer therapy.
[0598] In some embodiments, an IL-6 receptor antagonist, which in one
embodiment is tocilizumab
is used with the compositions and methods as disclosed herein.
[0599] In some embodiments, adoptively transferred T-cells engraft and expand
more efficiently in
a lymphopenic host. Thus, in some embodiments, the subject is subjected to
lymphodepletion prior
to transfer of CAR T-cells or other modified immune cells. In another
embodiment, the subject
receiving the CAR T-cells is given T-cell-supportive cytokines.
[0600] In some embodiments, the T-cells are effector T-cells. In another
embodiment, the T-cells are
naive T-cells. In another embodiment, the T-cells are central memory (Tcm) T-
cells. In another
embodiment, the T-cells are Th17 cells. In another embodiment, the T-cells are
T stem memory cells.
In another embodiment, the T-cells have high replicative capacity. In another
embodiment, T-cell
expansion occurs in the patient. In another embodiment, small numbers of cells
may be transferred to
a patient. In another embodiment, T-cell expansion occurs in vitro. In another
embodiment, large
numbers of cells may be transferred to a patient, cells and/or supernatants
may be transferred to a
patient on multiple occasions, or a combination thereof.
[0601] In some embodiments, an advantage of CAR T-cells is that because they
are specific for cell-
surface molecules, they overcome the constraints of MHC-restricted TCR
recognition and avoid
tumor escape through impairments in antigen presentation or human leukocyte
antigen expression.
[0602] In some embodiments, disclosed herein is a method of reducing a tumor
burden in a subject,
said method comprising the step of administering to said subject any of the
compositions as described
herein.
[0603] In some embodiments, reducing the tumor burden comprises reducing the
number of tumor
CA 03151815 2022-02-17
134
WO 2021/053667
PCT/IL2020/051011
cells in the subject. In another embodiment, reducing the tumor burden
comprises reducing tumor
size in the subject. In another embodiment, reducing the tumor burden
comprises eradicating the
tumor in the subject.
[0604] In another embodiment, disclosed herein is a method of inducing tumor
cell death in a subject,
said method comprising the step of administering to said subject any of the
compositions as described
herein. In another embodiment, a method as disclosed herein for inducing tumor
cell death in a subject
comprises administering immune cells, such as NK cells or T-cells comprising
engineered chimeric
antigen receptors with at least an additional agent to decrease toxic cytokine
release or "cytokine
release syndrome" (CRS) or "severe cytokine release syndrome" (sCRS) or
"cytokine storm" in the
subject.
[0605] In another embodiment, disclosed herein is a method of increasing or
lengthening the survival
of a subject having neoplasia, comprising the step of administering to said
subject any of the
compositions as described herein. In another embodiment, a method of
increasing or lengthening the
survival of a subject comprises administering immune cells, such as NK cells
or T-cells comprising
engineered chimeric antigen receptors with at least an additional agent to
decrease toxic cytokine
release or "cytokine release syndrome" (CRS) or "severe cytokine release
syndrome" (sCRS) or
"cytokine storm" in the subject.
[0606] In another embodiment, disclosed herein is a method of increasing or
lengthening the survival
of a subject having neoplasia, comprising the step of administering to said
subject any of the
compositions as described herein.
[0607] In some embodiments, disclosed herein is a method of delaying cancer
progression in a
subject, comprising a step of administering to the subject any of the
compositions or combinations of
compositions described herein. In some embodiments, disclosed herein is a
method of delaying
progression of a leukemia or lymphoma in a subject, comprising a step of
administering to the subject
any of the compositions or combinations of compositions described herein. In
some embodiments,
disclosed herein is a method of increasing, extending, or prolonging the
survival of a subject suffering
from a cancer or a tumor, comprising a step of administering to the subject
any of the compositions
or combinations of compositions described herein. In some embodiments,
disclosed herein is a
method of increasing, extending, or prolonging the survival of a subject
suffering from a leukemia or
lymphoma, comprising administering to the subject any of the compositions or
combinations of
compositions described herein. In some embodiments, disclosed herein is a
method of reducing the
tumor cell burden in a subject, comprising administering to the subject any of
the compositions or
combinations of compositions described herein. In some embodiments, tumor
burden is reduced the
liver and bone marrow.
CA 03151815 2022-02-17
135
WO 2021/053667
PCT/IL2020/051011
[0608] In another embodiment, disclosed herein is a method of preventing
neoplasia in a subject, said
method comprising the step of administering to the subject any of the
compositions or combinations
of compositions described herein. In some embodiments, the neoplasia is
selected from the group
consisting of blood cancer, B cell leukemia, multiple myeloma, lymphoblastic
leukemia (ALL),
chronic lymphocytic leukemia, non-Hodgkin's lymphoma, ovarian cancer, or a
combination thereof.
[0609] In another embodiment, disclosed herein is a method of treating blood
cancer in a subject in
need thereof, comprising the step of administering to said subject any of the
compositions as described
herein. In some embodiments, the blood cancer is selected from the group
consisting of B cell
leukemia, multiple myeloma, acute lymphoblastic leukemia (ALL), chronic
lymphocytic leukemia,
and non-Hodgkin's lymphoma.
[0610] In some embodiments, administration comprises administering a
composition comprising
CAR T-cells. In some embodiments, administration comprises administering a
composition
comprising early apoptotic cells. In some embodiments, administration
comprises administering a
composition comprising a supernatant obtained from early apoptotic cells. In
some embodiments,
administration comprises administering a combination of compositions described
herein. In some
embodiments, administration comprises administering CAR T-cells and apoptotic
cells in the same
or different compositions. In some embodiments, administration comprises
administering CAR T-
cells in combination with an additional agent as described herein. In some
embodiments,
administration comprises administering apoptotic cells and an antibody or
fragment thereof in the
.. same or different compositions.
[0611] In some embodiments, combination therapy provides a synergistic effect.
In some
embodiments, methods of use an early apoptotic cells in combination with CAR T-
cells increases
CAR T-cell efficacy in comparison to use of CAR T-cells alone. In some
embodiments, methods of
use an early apoptotic cells in combination with CAR T-cells extends the
survival time of a subject
suffering from a cancer or tumor in comparison to use of CAR T-cells alone. In
some embodiments,
methods of use an early apoptotic cells in combination with CAR T-cells
extends the survival time of
a subject suffering from a lymphoma or leukemia in comparison to use of CAR T-
cells alone.
[0612] In some embodiments, methods of use an early apoptotic cells in
combination with an
antibody or fragment thereof delays the onset of cancer or the appearance of a
tumor, in comparison
to use of either apoptotic cells or the antibody alone. In some embodiments,
methods of use an early
apoptotic cells in combination with an antibody or fragment thereof delays the
progression of a cancer,
in comparison to use of either apoptotic cells or the antibody alone. In some
embodiments, methods
of use an early apoptotic cells in combination with an antibody or fragment
thereof delays the growth
of a tumor, in comparison to use of either apoptotic cells or the antibody
alone. In some embodiments,
CA 03151815 2022-02-17
136
WO 2021/053667
PCT/IL2020/051011
methods of use an early apoptotic cells in combination with an antibody or
fragment thereof extends
the survival time of a subject suffering from a cancer or tumor in comparison
to use of either apoptotic
cells or the antibody alone. In some embodiments, methods of use an early
apoptotic cells in
combination with an antibody or fragment thereof extends the survival time of
a subject suffering
from a lymphoma or leukemia in comparison to use of either apoptotic cells or
the antibody alone.
[0613] In some embodiments, methods of use comprising administration of an
early apoptotic cells
in combination with an antibody or fragment thereof comprising RtX, delays the
onset of cancer or
the appearance of a tumor, in comparison to use of either apoptotic cells or
the antibody alone. In
some embodiments, methods of use an early apoptotic cells in combination with
an antibody or
fragment thereof comprising RtX, delays the progression of a cancer, in
comparison to use of either
apoptotic cells or the antibody alone. In some embodiments, methods of use an
early apoptotic cells
in combination with an antibody or fragment thereof comprising RtX , delays
the growth of a tumor,
in comparison to use of either apoptotic cells or the antibody alone. In some
embodiments, methods
of use an early apoptotic cells in combination with an antibody or fragment
thereof comprising RtX,
extends the survival time of a subject suffering from a cancer or tumor in
comparison to use of either
apoptotic cells or the antibody alone. In some embodiments, methods of use an
early apoptotic cells
in combination with an antibody or fragment thereof comprising RtX, extends
the survival time of a
subject suffering from a lymphoma or leukemia in comparison to use of either
apoptotic cells or the
antibody alone.
[0614] In some embodiments, methods of use described herein reduce tumor load.
A skilled artisan
would appreciate that the term "tumor load" may refer to the number of cancer
cells, the size of a
tumor, or the amount of cancer in the body. The term "tumor load" may be used
interchangeably with
the term "tumor burden" having all the same meanings and qualities. In some
embodiments, methods
of use comprising administration of an early apoptotic cells reduces the
number of cancer cells in a
subject, reduces the size of a tumor in a subject, or reduces the amount of
cancer in the body of a
subject, or any combination thereof compared with a subject not administered
apoptotic cells. In some
embodiments, methods of use comprising administration of an early apoptotic
cells in combination
with an antibody or fragment thereof reduces the number of cancer cells in a
subject, reduces the size
of a tumor in a subject, or reduces the amount of cancer in the body of a
subject, or any combination
thereof, compared with a subject not administered apoptotic cells or not
administer the antibody, or
the combination thereof In some embodiments, methods of use comprising
administration of an early
apoptotic cells in combination with a RtX antibody or fragment thereof reduces
the number of cancer
cells in a subject, reduces the size of a tumor in a subject, or reduces the
amount of cancer in the body
of a subject, or any combination thereof, compared with a subject not
administered apoptotic cells,
CA 03151815 2022-02-17
137
WO 2021/053667
PCT/IL2020/051011
the RtX antibody, or the combination thereof.
[0615] In some embodiments, a method of decreasing or inhibiting cytokine
production in a subject
experiencing cytokine release syndrome or cytokine storm or vulnerable to a
cytokine release
syndrome or cytokine storm, as disclosed herein, decreases or inhibits
cytokine production. In another
embodiment, the method decreases or inhibits pro-inflammatory cytokine
production. In a further
embodiment, the method decreases or inhibits at least one pro-inflammatory
cytokine. In another
embodiment, wherein the subject is undergoing CAR T-cell cancer therapy, the
method does not
reduce the efficacy of the CAR T-cell therapy.
[0616] The methods provided herein comprise administering a T-cell, NK cell,
or CTL cell disclosed
herein, in in an amount effective to achieve the desired effect, be it
palliation of an existing condition
or prevention of recurrence. For treatment, the amount administered is an
amount effective in
producing the desired effect. An effective amount can be provided in one or a
series of
administrations. An effective amount can be provided in a bolus or by
continuous perfusion.
[0617] A skilled artisan would recognize that an "effective amount" (or,
"therapeutically effective
amount") may encompass an amount sufficient to effect a beneficial or desired
clinical result upon
treatment. An effective amount can be administered to a subject in one or more
doses. In terms of
treatment, an effective amount is an amount that is sufficient to palliate,
ameliorate, stabilize, reverse
or slow the progression of the disease, or otherwise reduce the pathological
consequences of the
disease. The effective amount is generally determined by the physician on a
case-by-case basis and is
within the skill of one in the art. Several factors are typically taken into
account when determining an
appropriate dosage to achieve an effective amount. These factors include age,
sex and weight of the
subject, the condition being treated, the severity of the condition and the
form and effective
concentration of the antigen-binding fragment administered.
[0618] In one embodiment, methods disclosed herein comprise administering a
composition
comprising a genetically modified cell, and the additional agent or
combination thereof, comprised
in a single composition. In another embodiment, methods comprise administering
a composition
comprising a CAR T-cell, and the additional agent or combination thereof,
comprised in a single
composition. In another embodiment, methods comprise administering a
composition comprising a
TCR T-cell, and the additional agent or combination thereof, comprised in a
single composition.
[0619] In one embodiment, methods disclosed herein comprise administering a
composition
comprising a genetically modified cell, and the additional agent or
combination thereof, comprised
in a at least two compositions. In another embodiment, methods comprise
administering a
composition comprising a CAR T-cell, and the additional agent or combination
thereof, comprised
in at least two compositions. In another embodiment, methods comprise
administering a composition
CA 03151815 2022-02-17
138
WO 2021/053667
PCT/IL2020/051011
comprising a TCR T-cell, and the additional agent or combination thereof,
comprised in at least two
compositions.
[0620] For adoptive immunotherapy using antigen-specific T-cells, for example
CAR T-cells, cell
doses in the range of 106-101 (e.g., 109) are typically infused. Upon
administration of the genetically
modified cells into the host and subsequent differentiation, T-cells are
induced that are specifically
directed against the specific antigen. "Induction" of T-cells may include
inactivation of antigen-
specific T-cells such as by deletion or anergy. Inactivation is particularly
useful to establish or
reestablish tolerance such as in autoimmune disorders. The modified cells can
be administered by any
method known in the art including, but not limited to, intravenous,
subcutaneous, intranodal,
intratumoral, intrathecal, intrapleural, intraperitoneal and directly to the
thymus. In some
embodiments, the T-cells are not administered intraperitoneally. In some
embodiments, the T-cells
are administered intratumorallly.
[0621] Compositions comprising genetically modified immunoresponsive cells as
disclosed herein
(e.g., T-cells, N cells, CTL cells, or their progenitors) can be provided
systemically or directly to a
subject for the treatment of a neoplasia, pathogen infection, or infectious
disease. In some
embodiments, cells disclosed herein are directly injected into an organ of
interest (e.g., an organ
affected by a neoplasia). Alternatively, compositions comprising genetically
modified
immunoresponsive cells are provided indirectly to the organ of interest, for
example, by
administration into the circulatory system (e.g., the tumor vasculature).
Expansion and differentiation
agents can be provided prior to, during or after administration of the cells
to increase production of
T-cells, NK cells, or CTL cells in vitro or in vivo.
[0622] As described above in methods disclosed herein, compositions comprising
additional agents
may be provided prior to, concurrent with, or following administrations of the
genetically modified
immune cells. In one embodiment, in methods disclosed herein genetically
modified immune cells
for example CAR T-cells are administered prior to an additional agent as
disclosed herein. In another
embodiment, in methods disclosed herein genetically modified immune cells for
example CAR T-
cells are administered concurrent with an additional agent, as disclosed
herein. In another
embodiment, in methods disclosed herein genetically modified immune cells for
example CAR T-
cells are administered following administration of an additional agent.
[0623] In one embodiment, methods disclosed herein administer compositions
comprising apoptotic
cells as disclosed herein. In another embodiment, methods disclosed herein
administer compositions
comprising apoptotic cell supernatants as disclosed herein.
[0624] The modified cells can be administered in any physiologically
acceptable vehicle, normally
intravascularly, although they may also be introduced into bone or other
convenient site where the
CA 03151815 2022-02-17
139
WO 2021/053667
PCT/IL2020/051011
cells may find an appropriate site for regeneration and differentiation (e.g.,
thymus). Usually, at least
1x105 cells will be administered, eventually reaching lx101 or more.
Genetically modified
immunoresponsive cells disclosed herein may comprise a purified population of
cells. Those skilled
in the art can readily determine the percentage of genetically modified
immunoresponsive cells in a
population using various well-known methods, such as fluorescence activated
cell sorting (FACS). In
some embodiments, ranges of purity in populations comprising genetically
modified
immunoresponsive cells are about 50 to about 55%, about 55 to about 60%, and
about 65 to about
70%. In other embodiments, the purity is about 70 to about 75%, about 75 to
about 80%, about 80 to
about 85%. In further embodiments, the purity is about 85 to about 90%, about
90 to about 95%, and
about 95 to about 100%. Dosages can be readily adjusted by those skilled in
the art (e.g., a decrease
in purity may require an increase in dosage). The cells can be introduced by
injection, catheter, or the
like. If desired, factors can also be included, including, but not limited to,
interleukins, e.g. IL-2, IL-
3, IL-6, IL-1 1, IL7, IL12, ILIS, IL21, as well as the other interleukins, the
colony stimulating factors,
such as G-, M- and GM-CSF, interferons, e.g. gamma-interferon and
erythropoietin.
[0625] Compositions include pharmaceutical compositions comprising genetically
modified
immunoresponsive cells or their progenitors and a pharmaceutically acceptable
carrier.
Administration can be autologous or heterologous. For example,
immunoresponsive cells, or
progenitors can be obtained from one subject, and administered to the same
subject or a different,
compatible subject. Peripheral blood derived immunoresponsive cells disclosed
herein or their
progeny (e.g., in vivo, ex vivo or in vitro derived) can be administered via
localized injection,
including catheter administration, systemic injection, localized injection,
intravenous injection, or
parenteral administration. When administering a therapeutic composition as
disclosed herein (e.g., a
pharmaceutical composition containing a genetically modified immunoresponsive
cell), it will
generally be formulated in a unit dosage injectable form (solution,
suspension, emulsion).
[0626] In another embodiment, disclosed herein is a method of producing a
composition comprising
CAR T-cells or other immune cells as disclosed herein and apoptotic cells or
an apoptotic cell
supernatant, the method comprising introducing into the T-cell or immune cell
the nucleic acid
sequence encoding the CAR that binds to an antigen of interest. In an
alternative embodiment, the
compositions comprising CAR T-cells or other immune cells as disclosed herein
are separate from
the composition comprising apoptotic cells or an apoptotic supernatant.
[0627] In one embodiment, disclosed herein is a method of treating,
preventing, inhibiting, reducing
the incidence of, ameliorating, or alleviating a malignancy comprising the
step of administering a
composition comprising chimeric antigen receptor-expressing T-cells (CAR T-
cells) and apoptotic
cells or an apoptotic cell supernatant.
CA 03151815 2022-02-17
140
WO 2021/053667
PCT/IL2020/051011
[0628] A skilled artisan would appreciate that an anti-tumor immunity response
elicited by the
genetically modified immune cells, for example CAR-modified T cells, may be an
active or a passive
immune response. In addition, the CAR mediated immune response may be part of
an adoptive
immunotherapy approach in which CAR-modified T-cells induce an immune response
specific to the
antigen binding moiety in the CAR.
[0629] A skilled artisan would appreciate that immunotherapeutics may
encompass the use of
immune effector cells and molecules to target and destroy cancer cells. The
immune effector may be,
for example, an antibody specific for some marker on the surface of a tumor
cell. The antibody alone
may serve as an effector of therapy or it may recruit other cells to actually
effect cell killing. The
antibody also may be conjugated to a drug or toxin (chemotherapeutic,
radionuclide, ricin A chain,
cholera toxin, pertussis toxin, etc.) and serve merely as a targeting agent.
Alternatively, the effector
may be a lymphocyte carrying a surface molecule that interacts, either
directly or indirectly, with a
tumor cell target. Various effector cells include cytotoxic T cells and NK
cells.
[0630] In some embodiments, the early apoptotic cells and compositions
thereof, as disclosed
herein may be used to treat, prevent, inhibit the growth of, or reduce the
incidence of, any
hematological tumor known in the art. In some embodiments, the early apoptotic
cells and
compositions thereof, as disclosed herein may be used to treat, prevent,
inhibit the growth of, or
reduce the incidence of, any diffuse cancer known in the art, for example but
not limited to diffuse
breast cancer, wherein a solid tumor is not formed in the breast. In some
embodiments, the early
apoptotic cells and compositions thereof, as disclosed herein may be used to
extend the survival time
of any hematological tumor known in the art. In some embodiments, the early
apoptotic cells and
compositions thereof, as disclosed herein may be used to extend the survival
time of any diffuse
cancer known in the art, for example but not limited to diffuse breast cancer,
wherein a solid tumor is
not formed in the breast.
[0631] In some embodiments, the early apoptotic cells and compositions
thereof, as disclosed
herein may be used to increase the survival of a subject suffering from any
hematological tumor
known in the art. In some embodiments, the early apoptotic cells and
compositions thereof, as
disclosed herein may be used to increase the survival of a subject suffering
from any diffuse cancer
known in the art, for example but not limited to diffuse breast cancer,
wherein a solid tumor is not
formed in the breast.
[0632] In some embodiments, the early apoptotic cells and compositions
thereof, as disclosed
herein may be used to reduce the growth rate of any hematological tumor known
in the art. In some
embodiments, the early apoptotic cells and compositions thereof, as disclosed
herein may be used to
reduce the growth rate any diffuse cancer known in the art, for example but
not limited to diffuse
CA 03151815 2022-02-17
141
WO 2021/053667
PCT/IL2020/051011
breast cancer, wherein a solid tumor is not formed in the breast.
[0633] In some embodiments, the tumor or cancer being treated comprises a
metastasis of a tumor
or cancer. In some embodiments, methods of use herein prevent or reduce
metastasis of a tumor or
cancer. In some embodiments, methods of use herein inhibit the growth or
reduce the incidence of
metastasis.
[0634] In some embodiments, the subject is a human subject. In some
embodiments, the subject
is a child. In some embodiments, the subject is an adult. In some embodiments,
the subject is animal
mammal.
[0635] In some embodiments, a method disclosed herein comprises administering
an early
apoptotic cell population comprising a mononuclear enriched cell population,
as described in detail
above. In some embodiments, a method disclosed herein comprises administering
an early apoptotic
cell population comprising a stable population cell, wherein said cell
population is stable for greater
than 24 hours. Stable populations of early apoptotic cells have been described
in detail above. In some
embodiments, a method disclosed herein comprises administering an early
apoptotic cell population
comprising a population of cells devoid of cell aggregates. Early apoptotic
cell populations devoid of
aggregates and methods of making them have been described in detail above.
[0636] In some embodiments, a method disclosed herein comprises administering
an autologous
early apoptotic cell population to a subject in need. In some embodiments, a
method disclosed herein
comprises administering an allogeneic early apoptotic cell population to a
subject in need.
[0637] In some embodiments, methods of administration of early apoptotic
cell populations or
compositions thereof comprise administering a single infusion of said
apoptotic cell population or
composition thereof. In some embodiments, a single infusion may be
administered as a prophylactic
to a subject predetermined to be at risk for a cancer or tumor. In some
embodiments, a single infusion
may be administered to a subject having a cancer or tumor on a regular basis
as a part of the subject
therapeutic treatment. In some embodiments, a single infusion may be
administered as a prophylactic
to a subject having a cancer or tumor in order to prevent, reduce the risk of,
or delay the appearance
of metastatic cancer.
[0638] In some embodiments, methods of administration of early apoptotic
cell populations or
compositions thereof comprise administering multiple infusions of said
apoptotic cell population or
composition thereof. In some embodiments, multiple infusions may be
administered as a prophylactic
to a subject predetermined to be at risk for a cancer or tumor. In some
embodiments, multiple infusions
may be administered to a subject having a cancer or tumor on a regular basis
as a part of the subject
therapeutic treatment. In some embodiments, multiple infusions may be
administered as a
prophylactic to a subject having a cancer or tumor in order to prevent, reduce
the risk of, or delay the
CA 03151815 2022-02-17
142
WO 2021/053667
PCT/IL2020/051011
appearance of metastatic cancer.
[0639] In some embodiments, multiple infusions comprise at least two
infusions. In some
embodiments, multiple infusions comprise 2 infusions. In some embodiments,
multiple infusions
comprise more than 2 infusions. In some embodiments, multiple infusions
comprise at least 3
infusions. In some embodiments, multiple infusions comprise 3 infusions. In
some embodiments,
multiple infusions comprise more than 3 infusions. In some embodiments,
multiple infusions
comprise at least 4 infusions. In some embodiments, multiple infusions
comprise 4 infusions. In some
embodiments, multiple infusions comprise more than 4 infusions. In some
embodiments, multiple
infusions comprise at least 5 infusions. In some embodiments, multiple
infusions comprise 5
infusions. In some embodiments, multiple infusions comprise more than 5
infusions. In some
embodiments, multiple infusions comprise at least six infusions. In some
embodiments, multiple
infusions comprise 6 infusions. In some embodiments, multiple infusions
comprise more than 6
infusions. In some embodiments, multiple infusions comprise at least 7
infusions. In some
embodiments, multiple infusions comprise 7 infusions. In some embodiments,
multiple infusions
comprise more than 7 infusions. In some embodiments, multiple infusions
comprise at least 8
infusions. In some embodiments, multiple infusions comprise 8 infusions. In
some embodiments,
multiple infusions comprise more than 8 infusions. In some embodiments,
multiple infusions
comprise at least nine infusions. In some embodiments, multiple infusions
comprise 9 infusions. In
some embodiments, multiple infusions comprise more than 9 infusions. In some
embodiments,
multiple infusions comprise at least 10 infusions. In some embodiments,
multiple infusions comprise
10 infusions. In some embodiments, multiple infusions comprise more than 10
infusions.
[0640] In some embodiments, multiple infusions comprise smaller amounts
of early apoptotic cell,
wherein the total dosage of cells administered is the sum of the infusions.
[0641] In some embodiments, multiple infusions are administered over a
period of hours. In some
embodiments, multiple infusions are administered over a period of days. In
some embodiments,
multiple infusions are administered over a period of hours, wherein there is
at least 12 hours between
infusions. In some embodiments, multiple infusions are administered over a
period of hours, wherein
there is at least 24 hours between infusions. In some embodiments, multiple
infusions are
administered over a period of hours, wherein there is at least a day between
infusions. In some
embodiments, multiple infusions are administered over a period of hours,
wherein there is at least two
days between infusions. In some embodiments, multiple infusions are
administered over a period of
hours, wherein there is at least three days between infusions. In some
embodiments, multiple infusions
are administered over a period of hours, wherein there is at least four days
between infusions. In some
embodiments, multiple infusions are administered over a period of hours,
wherein there is at least five
CA 03151815 2022-02-17
143
WO 2021/053667
PCT/IL2020/051011
days between infusions. In some embodiments, multiple infusions are
administered over a period of
hours, wherein there is at least six days between infusions. In some
embodiments, multiple infusions
are administered over a period of hours, wherein there is at least seven days
between infusions. In
some embodiments, multiple infusions are administered over a period of hours,
wherein there is at
least a week between infusions. In some embodiments, multiple infusions are
administered over a
period of hours, wherein there is at least two weeks between infusions.
[0642] In some embodiments, the amount of cells in multiple infusions is
essentially equivalent
one to the other. In some embodiments, the amount of cells in multiple
infusions is different one to
the other.
[0643] In some embodiments, the methods described herein further comprise
administering an
additional chemotherapeutic agent or an immune modulator to said subject.
[0644] In some embodiments, an additional chemotherapeutic agent or immune
modulator is
administered concurrent or essentially concurrent with the early apoptotic
cells. In some
embodiments, an additional chemotherapeutic agent or immune modulator is
comprised in the same
composition as the early apoptotic cells. In some embodiments, an additional
chemotherapeutic agent
or immune modulator is comprised in a different composition as the early
apoptotic cells.
[0645] In some embodiments, an additional chemotherapeutic agent or immune
modulator is
administered prior to the administration of the early apoptotic cells. In some
embodiments, an
additional chemotherapeutic agent or immune modulator is administered
following the administration
of the early apoptotic cells.
[0646] In some embodiments, the chemotherapeutic agent comprises
alkylating agents, nitrogen
mustards, nitrosoureas, tetrazines, aziridines, cisplatins and derivatives,
non-classical alkylating
agents, mechlorethamine, cyclophosphamide, melphalan, chlorambucil,
ifosfamide, busulfan, N-
Nitro so-N-methylurea (MNU), carmustine (B C NU ) , lomustine (CCNU),
semustine (MeCCNU),
fotemustine, streptozotocin, dacarbazine, mitozolomide, temozolomide,
thiotepa, mitomycin,
diaziquone (AZQ), cisplatin, carboplatin, oxaliplatin, procarbazine,
hexamethylmelamine,
antimetabolites, anti-folates, methotrexate, pemetrexed, fluoropyrimidines,
fluorouracil,
capecitabine, deoxynucleoside analogues, cytarabine, gemcitabine, decitabine,
azacitidine,
fludarabine, nelarabine, cladribine, clofarabine, and pentostatin,
thiopurines, thioguanine,
.. mercaptopurine, anti-microtubule agents, vinca alkaloids, taxanes,
vincristine, vinblastine, semi-
synthetic vinca alkaloids, vinorelbine, vindesine, vinflunine, paclitaxel,
docetaxel, podophyllotoxin,
etoposide, teniposide, topoisomerase inhibitors, irinotecan, topotecan,
camptothecin, etoposide,
doxorubicin, mitoxantrone, teniposide, catalytic inhibitors, novobiocin,
merbarone, aclarubicin,
cytotoxic antibiotics, anthracyclines, bleomycins, mitomycin C, mitoxantrone,
actinomycin,
CA 03151815 2022-02-17
144
WO 2021/053667
PCT/IL2020/051011
doxorubicin, daunorubicin, epirubicin, idarubicin, anthracyclines,
pirarubicin, aclarubicin,
mitoxantrone, bleomycin, mitomycin, targeted therapies, monoclonal antibodies,
naked monoclonal
antibodies, conjugated monoclonal antibodies, chemolabeled antibodies,
bispecific monoclonal
antibodies, or any combination thereof.
[0647] In some embodiments, an immune modulator comprises an antibody or a
functional
fragment thereof. In some embodiments, an antibody or functional fragment
thereof comprises a
monoclonal antibody, a single chain antibody, an Fab fragment, an F(ab')2
fragment, or an Fv
fragment.
[0648] In some embodiments, disclosed herein are active fragments of any one
of the polypeptides
or peptide domains disclosed herein. A skilled artisan would appreciate that
the term "a fragment"
may encompass at least 5, 10, 13, or 15 amino acids. In other embodiments a
fragment is at least 20
contiguous amino acids. Fragments disclosed herein can be generated by methods
known to those
skilled in the art or may result from normal protein processing (e.g., removal
of amino acids from the
nascent polypeptide that are not required for biological activity or removal
of amino acids by
alternative mRNA splicing or alternative protein processing events).
[0649] The terms "antibody" and "immunoglobulin" are used interchangeably in
the broadest
sense and specifically refer to a polyclonal antibody, a monoclonal antibody,
or any fragment thereof,
which retains the binding activity of the antibody. In certain embodiments,
methods disclosed herein
comprise use of a chimeric antibody, a humanized antibody, or a human
antibody.
[0650] In some embodiments, the term "antibody" refers to intact molecules
as well as functional
fragments thereof, such as Fab, F(ab')2, and Fv that are capable of
specifcially interacting with a
desired target as described herein, for example, binding to phagocytic cells.
In some embodiments,
the antibody fragments comprise:
[0651] (1) Fab, the fragment which contains a monovalent antigen-binding
fragment of an
.. antibody molecule, which can be produced by digestion of whole antibody
with the enzyme papain
to yield an intact light chain and a portion of one heavy chain;
[0652] (2) Fab', the fragment of an antibody molecule that can be
obtained by treating whole
antibody with pepsin, followed by reduction, to yield an intact light chain
and a portion of the heavy
chain; two Fab' fragments are obtained per antibody molecule;
[0653] (3) (Fab')2, the fragment of the antibody that can be obtained by
treating whole antibody
with the enzyme pepsin without subsequent reduction; F(ab')2 is a dimer of two
Fab' fragments held
together by two disulfide bonds;
[0654] (4) Fv, a genetically engineered fragment containing the variable
region of the light chain
and the variable region of the heavy chain expressed as two chains; and
CA 03151815 2022-02-17
145
WO 2021/053667
PCT/IL2020/051011
[0655] (5) Single chain antibody ("SCA"), a genetically engineered
molecule containing the
variable region of the light chain and the variable region of the heavy chain,
linked by a suitable
polypeptide linker as a genetically fused single chain molecule.
[0656] Methods of making these fragments are known in the art. (See for
example, Harlow and
Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, New
York, 1988,
incorporated herein by reference).
[0657] In some embodiments, the antibody fragments may be prepared by
proteolytic hydrolysis
of the antibody or by expression in E. coli or mammalian cells (e.g. Chinese
hamster ovary cell culture
or other protein expression systems) of DNA encoding the fragment.
[0658] Antibody fragments can, in some embodiments, be obtained by pepsin
or papain digestion
of whole antibodies by conventional methods. For example, antibody fragments
can be produced by
enzymatic cleavage of antibodies with pepsin to provide a 5S fragment denoted
F(ab')2. This fragment
can be further cleaved using a thiol reducing agent, and optionally a blocking
group for the sulfhydryl
groups resulting from cleavage of disulfide linkages, to produce 3.5S Fab'
monovalent fragments.
Alternatively, an enzymatic cleavage using pepsin produces two monovalent Fab'
fragments and an
Fc fragment directly. These methods are described, for example, by Goldenberg,
U.S. Pat. Nos.
4,036,945 and 4,331,647, and references contained therein, which patents are
hereby incorporated by
reference in their entirety. See also Porter, R. R., Biochem. J., 73: 119-126,
1959. Other methods of
cleaving antibodies, such as separation of heavy chains to form monovalent
light-heavy chain
fragments, further cleavage of fragments, or other enzymatic, chemical, or
genetic techniques may
also be used, so long as the fragments bind to the antigen that is recognized
by the intact antibody.
[0659] Fv fragments comprise an association of VH and VL chains. This
association may be
noncovalent, as described in Inbar et al., Proc. Nat'l Acad. Sci. USA 69:2659-
62, 1972. Alternatively,
the variable chains can be linked by an intermolecular disulfide bond or cross-
linked by chemicals
such as glutaraldehyde. Preferably, the Fv fragments comprise VH and VL chains
connected by a
peptide linker. These single-chain antigen binding proteins (sFv) are prepared
by constructing a
structural gene comprising DNA sequences encoding the VH and VL domains
connected by an
oligonucleotide. The structural gene is inserted into an expression vector,
which is subsequently
introduced into a host cell such as E. coll. The recombinant host cells
synthesize a single polypeptide
chain with a linker peptide bridging the two V domains. Methods for producing
sFvs are described,
for example, by Whitlow and Filpula, Methods, 2: 97-105, 1991; Bird et al.,
Science 242:423-426,
1988; Pack et al., Bio/Technology 11:1271-77, 1993; and Ladner et al., U.S.
Pat. No. 4,946,778,
which is hereby incorporated by reference in its entirety.
[0660] Another form of an antibody fragment is a peptide coding for a single
complementarity-
CA 03151815 2022-02-17
146
WO 2021/053667
PCT/IL2020/051011
determining region (CDR). CDR peptides ("minimal recognition units") can be
obtained by
constructing genes encoding the CDR of an antibody of interest. Such genes are
prepared, for
example, by using the polymerase chain reaction to synthesize the variable
region from RNA of
antibody-producing cells. See, for example, Larrick and Fry, Methods, 2: 106-
10, 1991.
[0661] In some embodiments, the antibodies or fragments as described herein
may comprise
"humanized forms" of antibodies. In some embodiments, the term "humanized
forms of antibodies"
refers to non-human (e.g. murine) antibodies, which are chimeric molecules of
immunoglobulins,
immunoglobulin chains or fragments thereof (such as Fv, Fab, Fab', F(ab').
sub.2 or other antigen-
binding subsequences of antibodies) which contain minimal sequence derived
from non-human
immunoglobulin. Humanized antibodies include human immunoglobulins (recipient
antibody) in
which residues form a complementary determining region (CDR) of the recipient
are replaced by
residues from a CDR of a non-human species (donor antibody) such as mouse, rat
or rabbit having
the desired specificity, affinity and capacity. In some instances, Fv
framework residues of the human
immunoglobulin are replaced by corresponding non-human residues. Humanized
antibodies may also
comprise residues which are found neither in the recipient antibody nor in the
imported CDR or
framework sequences. In general, the humanized antibody will comprise
substantially all of at least
one, and typically two, variable domains, in which all or substantially all of
the CDR regions
correspond to those of a non-human immunoglobulin and all or substantially all
of the FR regions are
those of a human immunoglobulin consensus sequence. The humanized antibody
optimally also will
comprise at least a portion of an immunoglobulin constant region (Fc),
typically that of a human
immunoglobulin [Jones et al., Nature, 321:522-525 (1986); Riechmann et al.,
Nature, 332:323-329
(1988); and Presta, Curr. Op. Struct. Biol., 2:593-596 (1992)].
[0662] Methods for humanizing non-human antibodies are well known in the art.
Generally, a
humanized antibody has one or more amino acid residues introduced into it from
a source which is
non-human. These non-human amino acid residues are often referred to as import
residues, which are
typically taken from an import variable domain. Humanization can be
essentially performed
following the method of Winter and co-workers [Jones et al., Nature, 321:522-
525 (1986);
Riechmann et al., Nature 332:323-327 (1988); Verhoeyen et al., Science,
239:1534-1536 (1988)], by
substituting rodent CDRs or CDR sequences for the corresponding sequences of a
human antibody.
Accordingly, such humanized antibodies are chimeric antibodies (U.S. Pat. No.
4,816,567), wherein
substantially less than an intact human variable domain has been substituted
by the corresponding
sequence from a non-human species. In practice, humanized antibodies are
typically human
antibodies in which some CDR residues and possibly some FR residues are
substituted by residues
from analogous sites in rodent antibodies.
CA 03151815 2022-02-17
147
WO 2021/053667
PCT/IL2020/051011
[0663] Human antibodies can also be produced using various techniques known in
the art, including
phage display libraries [Hoogenboom and Winter, J. Mol. Biol., 227:381 (1991);
Marks et A ,Mol.
Biol., 222:581 (1991)]. The techniques of Cole et al. and Boerner et al. are
also available for the
preparation of human monoclonal antibodies (Cole et al., Monoclonal Antibodies
and Cancer
Therapy, Alan R. Liss, p. 77 (1985) and Boerner et al., J. Immunol., 147(1):86-
95 (1991)]. Similarly,
human can be made by introducing of human immunoglobulin loci into transgenic
animals, e.g. mice
in which the endogenous immunoglobulin genes have been partially or completely
inactivated. Upon
challenge, human antibody production is observed, which closely resembles that
seen in humans in
all respects, including gene rearrangement, assembly, and antibody repertoire.
This approach is
described, for example, in U.S. Pat. Nos. 5,545,807; 5,545,806; 5,569,825;
5,625,126; 5,633,425;
5,661,016, and in the following scientific publications: Marks et al.,
Bio/Technology 10, 779-783
(1992); Lonberg et al., Nature 368 856-859 (1994); Morrison, Nature 368 812-13
(1994); Fishwild et
al., Nature Biotechnology 14, 845-51 (1996); Neuberger, Nature Biotechnology
14, 826 (1996);
Lonberg and Huszar, Intern. Rev. Immunol. 13 65-93 (1995).
[0664] In some embodiments, the immune modulator comprises an anti-CD20
monoclonal
antibody. In some embodiments, the antiCD20 monoclonal antibody is Rituximab.
Rituximab is
commercially available and sold under the name Rituxan , marketed jointly by
Biogen and
Genentech USA, Inc.
[0665] In some embodiments, methods disclosed herein comprise a first-
line therapy.
[0666] A skilled artisan would appreciate that the term "first-line
therapy" may encompass the
first treatment given for a disease. It is often part of a standard set of
treatments, such as surgery
followed by chemotherapy and radiation. When used by itself, first-line
therapy is the one accepted
as the best treatment. If it doesn't cure the disease or it causes severe side
effects, other treatment may
be added or used instead. Also called induction therapy, primary therapy, and
primary treatment.
[0667] In some embodiments, methods disclosed herein comprise an adjuvant
therapy.
[0668] A skilled artisan would appreciate that the term "adjuvant
therapy" may encompass a
treatment that is given in addition to the primary or initial treatment. In
some embodiments, adjuvant
therapy may comprise an additional cancer treatment given prior to the primary
treatment in
preparation of a further treatment. In some embodiments, adjuvant therapy may
comprise an
additional cancer treatment given after the primary treatment to lower the
risk that the cancer will
come back. Adjuvant therapy may include chemotherapy, radiation therapy,
hormone therapy,
targeted therapy, or biological therapy.
[0669] In some embodiments, a method disclosed herein, reduces the
minimal residual disease,
increases remission, increases remission duration, reduces tumor relapse rate,
decreases the size of
CA 03151815 2022-02-17
148
WO 2021/053667
PCT/IL2020/051011
said tumor, decreases growth rate of said tumor or said cancer, prevents
metastasis of said tumor or
said cancer, or reduces the rate of metastasis of said tumor or said cancer,
or any combination thereof.
[0670] A skilled artisan would appreciate that the term "minimal residual
disease" may encompass
small numbers of cancer cells that remain in the patient during treatment or
after treatment when the
patient has no symptoms or signs of disease.
[0671] Additionally, the term "remission" may encompass a decrease or
disappearance of signs
and symptoms of cancer, though cancer may still be in the body. In some
embodiments, remission
may comprise partial remission, wherein some, but not all, signs and symptoms
of cancer have
disappeared. In some embodiments, remission comprises complete remission,
wherein all signs and
symptoms of cancer have disappeared, although cancer still may be in the body.
In some
embodiments, methods disclosed herein may be comprise a remission induction
therapy, wherein the
initial treatment with early apoptotic cells or compositions thereof,
decreases the signs or symptoms
of cancer or make them disappear.
[0672] A skilled artisan would appreciate that the term "relapse" may
encompass the return of a
disease or the signs and symptoms of a disease after a period of improvement.
In some embodiments,
methods used herein lead to a relapse-free survival, wherein the relapse-free
survival encompasses
the length of time after primary treatment for a cancer ends that the patient
survives without any signs
or symptoms of that cancer.
Malignancies
[0673] In some embodiments, CAR T-cells are utilized in methods of
treating, preventing,
inhibiting, reducing the incidence of, ameliorating, or alleviating a cancer
or a tumor wherein the
methods comprise the step of administering chimeric antigen receptor-
expressing T-cells (CAR T-
cells). As disclosed herein, these methods may further comprise administering
an additional agent in
an effort to inhibit or decrease the incidence of CRS or cytokine storm.
[0674] In some embodiments, a method disclosed herein increases the
survival of the subject. In
some embodiments, disclosed herein is a method of increasing or lengthening
the survival of a subject
having a diffuse cancer, comprising the step of administering an early
apoptotic cell population to
said subject, wherein the method increases the survival of the subject.
[0675] In some embodiments, a cancer is a B-cell malignancy. In some
embodiments, the B-cell
malignancy is leukemia. In another embodiment, the B-cell malignancy is acute
lymphoblastic
leukemia (ALL). In another embodiment, the B-cell malignancy is chronic
lymphocytic leukemia.
[0676] In some embodiments, the cancer is leukemia. In some embodiments, the
cancer is lymphoma.
In some embodiments, the lymphoma is large B-cell lymphoma.
[0677] In some embodiments, methods described herein reduce the size or reduce
the growth rate
CA 03151815 2022-02-17
149
WO 2021/053667
PCT/IL2020/051011
of a cancer or a tumor, and comprise administering an early apoptotic cell
population to said subject,
wherein the method reduces the size or the growth rate of a cancer or tumor.
In some embodiments,
disclosed herein is a method of reducing the growth rate of a diffuse cancer,
comprising the step of
administering an early apoptotic cell population to said subject, wherein the
method reduces the
growth rate of the cancer. In some embodiments, disclosed herein is a method
of reducing the size or
reducing the growth rate of a solid cancer or tumor, comprising the step of
administering an early
apoptotic cell population to a subject, wherein the method reduces the size or
reduces the growth rate
of the solid cancer or tumor.
[0678] In some embodiments, a cancer may comprise a solid tumor. In some
embodiments, a solid
tumor comprises an abnormal mass of tissue that usually does not contain cysts
or liquid areas. Solid
tumors may be benign (not cancer), or malignant (cancer). Different types of
solid tumors are named
for the type of cells that form them. Examples of solid tumors are sarcomas,
carcinomas, and
lymphomas. Leukemias (cancers of the blood) generally do not form solid
tumors. In some
embodiments, a solid tumor comprises a sarcoma or a carcinoma.
[0679] In some embodiments, solid tumors are neoplasms (new growth of
cells) or lesions
(damage of anatomic structures or disturbance of physiological functions)
formed by an abnormal
growth of body tissue cells other than blood, bone marrow or lymphatic cells.
In some embodiments,
a solid tumor consists of an abnormal mass of cells which may stem from
different tissue types such
as liver, colon, breast, or lung, and which initially grows in the organ of
its cellular origin. However,
such cancers may spread to other organs through metastatic tumor growth in
advanced stages of the
disease.
[0680] In some embodiments, examples of solid tumors comprise sarcomas,
carcinomas, and
lymphomas. In some embodiments, a solid tumor comprises a sarcoma or a
carcinoma. In some
embodiments, the solid tumor is an intra-peritoneal tumor.
[0681] In some embodiments, a solid tumor comprises, but is not limited to,
lung cancer, breast
cancer, ovarian cancer, stomach cancer, esophageal cancer, cervical cancer,
head and neck cancer,
bladder cancer, liver cancer, and skin cancer. In some embodiments, a solid
tumor comprises a
fibrosarcoma, a myxosarcoma, a liposarcoma, a chondrosarcoma, an osteogenic
sarcoma, a
chordoma, an angio sarcoma, an endothelio sarcoma, a lymphangio
sarcoma, a
lymphangioendotheliosarcoma, a synovioma, a mesothelioma, an Ewing's tumor, a
leiomyosarcoma,
a rhabdomyosarcoma, a colon carcinoma, a pancreatic cancer or tumor, a breast
cancer or tumor, an
ovarian cancer or tumor, a prostate cancer or tumor, a squamous cell
carcinoma, a basal cell
carcinoma, an adenocarcinoma, a sweat gland carcinoma, a sebaceous gland
carcinoma, a papillary
carcinoma, a papillary adenocarcinomas, a cystadenocarcinoma, a medullary
carcinoma, a
CA 03151815 2022-02-17
150
WO 2021/053667
PCT/IL2020/051011
bronchogenic carcinoma, a renal cell carcinoma, a hepatoma, a bile duct
carcinoma, a
choriocarcinoma, a seminoma, an embryonal carcinoma, a Wilm's tumor, a
cervical cancer or tumor,
a uterine cancer or tumor, a testicular cancer or tumor, a lung carcinoma, a
small cell lung carcinoma,
a bladder carcinoma, an epithelial carcinoma, a glioma, an astrocytoma, a
medulloblastoma, a
craniopharyngioma, an ependymoma, a pinealoma, a hemangioblastoma, an acoustic
neuroma, an
oligodenroglioma, a schwannoma, a meningioma, a melanoma, a neuroblastoma, or
a retinoblastoma.
[0682] In some embodiments, the solid tumor comprises an Adrenocortical Tumor
(Adenoma and
Carcinoma), a Carcinoma, a Colorectal Carcinoma, a Desmoid Tumor, a
Desmoplastic Small Round
Cell Tumor, an Endocrine Tumor, an Ewing Sarcoma, a Germ Cell Tumor, a
Hepatoblastoma a
Hepatocellular Carcinoma, a Melanoma, a Neuroblastoma, an Osteosarcoma, a
Retinoblastoma, a
Rhabdomyosarcoma, a Soft Tissue Sarcoma Other Than Rhabdomyosarcoma, and a
Wilms Tumor.
In some embodiments, the solid tumor is a breast tumor. In another embodiment,
the solid tumor is a
prostate cancer. In another embodiment, the solid tumor is a colon cancer. In
some embodiments, the
tumor is a brain tumor. In another embodiment, the tumor is a pancreatic
tumor. In another
.. embodiment, the tumor is a colorectal tumor.
[0683] In some embodiments, early apoptotic cells or compositions thereof
as disclosed herein,
have therapeutic and/or prophylactic efficacy against a cancer or a tumor, for
example sarcomas and
carcinomas (e.g., fibrosarcoma, myxosarcoma, liposarcoma, chondrosarcoma,
osteogenic sarcoma,
chordoma, angiosarcoma, endotheliosarcoma, lymphangiosarcoma,
lymphangioendotheliosarcoma,
synovioma, mesothelioma, Ewing's tumor, leiomyosarcoma, rhabdomyosarcoma,
colon carcinoma,
pancreatic cancer, breast cancer, ovarian cancer, prostate cancer, squamous
cell carcinoma, basal cell
carcinoma, adenocarcinoma, sweat gland carcinoma, sebaceous gland carcinoma,
papillary
carcinoma, papillary adenocarcinomas, cystadenocarcinoma, medullary carcinoma,
bronchogenic
carcinoma, renal cell carcinoma, hepatoma, nile duct carcinoma,
choriocarcinoma, seminoma,
embryonal carcinoma, Wilm's tumor, cervical cancer, uterine cancer, testicular
cancer, lung
carcinoma, small cell lung carcinoma, bladder carcinoma, epithelial carcinoma,
glioma, astrocytoma,
medulloblastoma, craniopharyngioma, ependymoma, pinealoma, hemangioblastoma,
acoustic
neuroma, oligodenroglioma, schwannoma, meningioma, melanoma, neuroblastoma,
and
retinoblastoma).
[0684] In some embodiments, the early apoptotic cells and compositions
thereof as disclosed
herein may be used to treat, prevent, inhibit the growth of, or reduce the
incidence of, any solid tumor
known in the art.
[0685] In some embodiments, the early apoptotic cells and compositions
thereof as disclosed
herein, may be used to increase the survival of a subject suffering from any
solid tumor as disclosed
CA 03151815 2022-02-17
151
WO 2021/053667
PCT/IL2020/051011
herein or known in the art.
[0686] In some embodiments, the early apoptotic cells and compositions
thereof as disclosed
herein, may be used to reduce the size or reduce the growth rate any solid
tumor as disclosed herein
or known in the art.
[0687] In some embodiments, a cancer may be a diffuse cancer, wherein the
cancer is widely
spread; not localized or confined. In some embodiments, a diffuse cancer may
comprise a non-solid
tumor. Examples of diffuse cancers include leukemias. Leukemias comprise a
cancer that starts in
blood-forming tissue, such as the bone marrow, and causes large numbers of
abnormal blood cells to
be produced and enter the bloodstream.
[0688] In some embodiments, a diffuse cancer comprises a B-cell malignancy.
In some
embodiments, the diffuse cancer comprises leukemia. In some embodiments, the
cancer is lymphoma.
In some embodiments, the lymphoma is large B-cell lymphoma.
[0689] In some embodiments, the diffuse cancer or tumor comprises a
hematological tumor. In
some embodiments, hematological tumors are cancer types affecting blood, bone
marrow, and lymph
nodes. Hematological tumors may derive from either of the two major blood cell
lineages: myeloid
and lymphoid cell lines. The myeloid cell line normally produces granulocytes,
erythrocytes,
thrombocytes, macrophages, and masT-cells, whereas the lymphoid cell line
produces B, T, NK and
plasma cells. Lymphomas (e.g. Hodgkin's Lymphoma), lymphocytic leukemias, and
myeloma are
derived from the lymphoid line, while acute and chronic myelogenous leukemia
(AML, CML),
myelodysplastic syndromes and myeloproliferative diseases are myeloid in
origin.
[0690] In some embodiments, a non-solid (diffuse) cancer or tumor
comprises a hematopoietic
malignancy, a blood cell cancer, a leukemia, a myelodysplastic syndrome, a
lymphoma, a multiple
myeloma (a plasma cell myeloma), an acute lymphoblastic leukemia, an acute
myelogenous
leukemia, a chronic myelogenous leukemia, a Hodgkin lymphoma, a non-Hodgkin
lymphoma, or
plasma cell leukemia.
[0691] In another embodiment, early apoptotic cells and compositions
thereof, as disclosed herein
have therapeutic and/or prophylactic efficacy against diffuse cancers, for
example but not limited to
leukemias (e.g., acute leukemia, acute lymphocytic leukemia, acute myelocytic
leukemia, acute
myeloblastic leukemia, acute promyelocyte leukemia, acute myelomonocytic
leukemia, acute
monocytic leukemia, acute erythroleukemia, chronic leukemia, chronic
myelocytic leukemia, chronic
lymphocytic leukemia), polycythemia vera, lymphoma (Hodgkin's disease, non-
Hodgkin's disease),
Waldenstrom's macroglobulinemia, heavy chain disease.
[0692] The compositions and methods as disclosed herein may be used to
treat, prevent, inhibit,
ameliorate, reduce the incidence of, or alleviate any hematological tumor
known in the art.
CA 03151815 2022-02-17
152
WO 2021/053667
PCT/IL2020/051011
[0693] A skilled artisan would appreciate that the use of the term
comprising throughout, may in
certain embodiments be replace by the use of the term consisting essentially
of or consisting of. The
skilled artisan would appreciate that the term "comprising" is intended to
mean that the system
includes the recited elements, but not excluding others which may be optional.
For example a
composition comprising early apoptotic cells but not limited to this
population of cells. Further, the
term "consisting essentially of' may encompass a method that includes the
recited elements, for
example a composition consisting essentially or early apoptotic cells but
exclude other elements that
may have an essential significant effect on the performance of the method.
Thus, such a composition
may still include a pharmaceutically acceptable excipient that does not
comprise an essential activity
in treating cancer. Further, "consisting of' encompasses excluding more than
traces of other elements.
Thus, such a composition consisting of early apoptotic cells would not include
more than traces of
other elements as disclosed herein.
[0694] In some embodiments, methods as disclosed herein may be represented as
uses of the
compositions as described herein for various therapeutic and prophylactic
purposes as described
herein, or alternatively, uses of the compositions as described herein in the
preparation of a
medicament or a therapeutic composition or a composition for various
therapeutic and prophylactic
purposes as described herein.
[0695] In some embodiments, the compositions and methods as disclosed herein
comprise the
various components or steps. However, in another embodiment, the compositions
and methods as
disclosed herein consist essentially of the various components or steps, where
other components or
steps may be included. In another embodiment, the compositions and methods as
disclosed herein
consist of the various components or steps.
[0696] In some embodiments, the term "comprise" may encompass the inclusion of
other
components of the composition which affect the efficacy of the composition
that may be known in
the art. In some embodiments, the term "consisting essentially of' comprises a
composition, which
has chimeric antigen receptor-expressing T-cells (CAR T-cells), and apoptotic
cells or any apoptotic
cell supernatant. However, other components may be included that are not
involved directly in the
utility of the composition. In some embodiments, the term "consisting"
encompasses a composition
having chimeric antigen receptor-expressing T-cells (CAR T-cells), and
apoptotic cells or an
apoptotic cell supernatant as disclosed herein, in any form or embodiment as
described herein.
[0697] A skilled artisan would appreciate that the term "about", may
encompass a deviance of
between 0.0001-5% from the indicated number or range of numbers. Further, it
may encompass a
deviance of between 1 -10% from the indicated number or range of numbers. In
addition, it may
encompass a deviance of up to 25% from the indicated number or range of
numbers.
CA 03151815 2022-02-17
153
WO 2021/053667
PCT/IL2020/051011
[0698] A skilled artisan would appreciate that the singular form "a",
"an" and "the" include plural
references unless the context clearly dictates otherwise. For example, the
term "an agent" or "at least
an agent" may include a plurality of agents, including mixtures thereof.
[0699] Throughout this application, various embodiments disclosed herein
may be presented in a
range format. It should be understood that the description in range format is
merely for convenience
and brevity and should not be construed as an inflexible limitation on the
scope of the disclosure.
Accordingly, the description of a range should be considered to have
specifically disclosed all the
possible sub ranges as well as individual numerical values within that range.
For example, description
of a range such as from 1 to 6 should be considered to have specifically
disclosed sub ranges such as
from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6
etc., as well as individual
numbers within that range, for example, 1, 2, 3, 4, 5, and 6. This applies
regardless of the breadth of
the range.
Whenever a numerical range is indicated herein, it is meant to include any
cited numeral (fractional
or integral) within the indicated range. The phrases "ranging/ranges between"
a first indicated number
and a second indicated number and "ranging/ranges from" a first indicated
number "to" a second
indicated number are used herein interchangeably and are meant to include the
first and second
indicated numbers and all the fractional and integral numerals there between.
[0700] The following examples are presented in order to more fully illustrate
embodiments disclosed
herein. They should in no way be construed, however, as limiting the broad
scope of the disclosure.
EXAMPLES
EXAMPLE 1: Apoptotic Cell Production
[0701] Objective: To produce early-apoptotic cells.
[0702] Methods: Methods of making populations of early-apoptotic cells have
been well
documented in International Publication No. WO 2014/087408 and United States
Application
Publication No. U52015/0275175-A 1, see for example, the Methods section
preceding the Examples
at "Early apoptotic cell population Preparation" and "Generation of apoptotic
cells" (paragraphs
[0223] through [0288]), and Examples 11, 12, 13, and 14, which are
incorporated herein in their
entirety).
[0703] The flow chart presented in Figure 1 provides an overview of one
embodiment of the steps
used during the process of producing a population of early apoptotic cells,
wherein anticoagulants
were included in the thawing and induction of apoptosis steps. As is described
in detailed in Example
14 of International Publication No. WO 2014/087408 and United States
Application Publication No.
US US-2015-0275175-A1, early apoptotic cell populations were prepared wherein
anti-coagulants
CA 03151815 2022-02-17
154
WO 2021/053667
PCT/IL2020/051011
were added at the time of freezing, or at the time of incubation, or at the
time of freezing and at the
time of incubation. The anticoagulant used was acid-citrate dextrose, NIH
Formula A (ACD formula
A) was supplemented with 10 Um' heparin to a final concentration of 5% ACD of
the total volume
and 0.5 Wm' heparin.
[0704] Briefly: The cells were collected and then frozen with addition of
5% anticoagulant citrate
dextrose formula A and 10U/m1 heparin (ACDhep) to the freezing media. Thawing,
incubation in an
apoptosis induction media containing 5% ACDhep, and final product preparation
were performed in
a closed system.
[0705] Apoptosis and viability analysis, potency assay, and cell
population characterization were
performed in each experiment. In order to establish consistence in production
of the early apoptotic
cell product, the final product (FP) of initial batches of apoptotic cells
were stored at 2-8 C and
examined at tO, t24h, t48h and t72h. At each point apoptosis analysis, short
potency assay (Applicants
CD14+ frozen cells), trypan blue measurement and cell population
characterization were performed.
The FP was tested for cell count to assess average cell loss during storage
and apoptosis and viability
analysis.
[0706] The methods sections cited above and Example 11 of International
Publication No. WO
2014/087408 and United States Application Publication No. US US-2015-0275175-
Al provide
details of preparing other embodiment of apoptotic cell populations in the
absence of anti-coagulants,
and are incorporated herein in full.
[0707] Methods of preparing irradiated apoptotic cells: Similar methods were
used to prepare an
inactivated apoptotic cell population, wherein a mononuclear early apoptotic
cell population
comprises a decreased percent of non-quiesnce non-apoptoic cells, or a
population of cells having a
suppressed cellular activation of any living non-apoptotic cells, or a
population of cells having a
reduced proliferation of any living non-apoptotic cells, or any combination
thereof.
[0708] Briefly, an enriched mononuclear cell fraction was collected via
leukapheresis procedure
from healthy, eligible donors. Following apheresis completion, cells were
washed and resuspended
with freezing media comprising 5% Anticoagulant Citrate Dextrose Solution-
Formula A (ACD-A)
and 0.5thml heparin. Cell were then gradually frozen and transferred to liquid
nitrogen for long term
storage.
[0709] For preparation of irradiated ApoCells, cryopreserved cells were
thawed, washed and
resuspended with apoptosis induction media comprising 5% ACD-A, 0.5thml
heparin sodium and
50i.tg/m1 methylprednisolone. Cells were then incubated for 6 hours at 37 C in
5% CO2. At the end
of incubation, cells were collected, washed and resuspended in Hartmann's
solution using a cell
processing system (Fresenius Kabi, Germany). Following manufacturing
completion, ApoCell were
CA 03151815 2022-02-17
155
WO 2021/053667
PCT/IL2020/051011
irradiated at 4000 cGy using g-camera at the radiotherapy unit, Hadassah Ein
Kerem. Apoptosis and
viability of ApoCell determined using AnnexinV and PI (MBL, MA, USA) staining
(> 40% and <
15%, respectively) via Flow cytometer. Results analyzed using FCS express
software.
This irradiated APOcell population is considered to include early apoptotic
cells, wherein any viable
cells present have suppressed cellular activity and reduced or no
proliferation capabilities. In certain
cases, the Apocell population has no viable non-apoptotic cells. Results:
[0710] The stability of the FP produced with inclusion of anticoagulant
at freezing and incubation
(apoptotic induction) and then stored at 2-8 C are shown below in Table 3.
[0711] Table 3: Cell count*- performed using a MICROS 60 hematology analyzer.
FP Time point Cell concentration (x106ce11s\ml) % of cell loss
tO 20.8 NA
t24h 20.0 -3.85
t48h 20.0 -3.85
t72h 19.7 -5.3
* Results Representative of 6 (six) experiments.
[0712] When manufacturing the cells without including an anticoagulant in the
induction medium,
cells were stable for 24 hours and less stable thereafter. Use of
anticoagulants unexpectedly extended
the stability of the apoptotic cell population for at least 72 hours, as shown
in Table 3.
[0713] Table 4: Trypan blue measurement
FP Time point trypan blue positive cells (%)
tO 3.0
t24h 5.9
t48h 5.2
t72h 6.5
[0714] The results of Table 4 show viability of the FP remained high for at
least 72 hours.
[0715] Table 5: Apoptosis analysis- (AnPI staining) performed using Flow
Cytometry
FP Time point 1.5mM Ca
An-PI- (%) An+PI- (%) An+PI+ (%)
tO 44.3 50.9 4.8
t24h 39.0 55.9 5.1
t48h 34.8 60.1 5.1
t72h 33.4 60.5 6.1
[0716] The results of Table 5 show that the percent apoptotic cells
versus necrotic cells was
CA 03151815 2022-02-17
156
WO 2021/053667
PCT/IL2020/051011
maintained over at extended time period of at least 72 hours post preparation
of the cells, as was the
percentage of early apoptotic cells.
[0717] Inclusion of anticoagulants both at the time of freezing and
during induction of apoptosis
resulted in the most consistently high yield of stable early-apoptotic cells
(average yield of early
apoptotic cells 61.3 2.6% % versus 48.4 5.0%, wherein 100% yield is based on
the number of cells
at freezing). This high yield was maintained even after 72 hours storage at 2-
8 C.
[0718] Next a comparison was made between the inclusion of the
anticoagulant at freezing or
thawing or both, wherein percent (%) recovery was measured as well as
stability. Anticoagulant was
included in the apoptotic incubation mix for all populations. Table 6 presents
the results of these
studies.
[0719] Table 6: Yield and stability comparison of final products (FP)
manufactured from cells
collected, with ("+") or without ("-") addition of anticoagulant during
freezing ("F") and thawing
("Tha")
Donor # of % Cell Recovery in Final Product of
Collected Cells
ID Collected FP tO FP t24h*
Cells F-/Tha- F-/Tha+ F+/Tha+ F+/Tha- F-/Tha- F-/Tha+ F+/Tha+ F+/Tha-
(x109,
100%)
13.3 52.1 53.4 62.5 62 52.1 48.9 62.5
62
2 13.6 50.5 36.7 53.5 63.5 47.6 36.7 53.1
59.7
3 15.0 42.7 42 53.6 58.4 42.7 41.7 53.6
57.8
Avg 14.0 48.4 5.0 44.0 8.5 56.5 5.2 61.3 2.6 47.5 4.7
42.4 6.1 56.4 5.3 59.8 2.1
[0720] Additional population analysis comparisons of early apoptotic cell
populations (batches of
cells) prepared with and without anti-coagulant added, show the consistency of
these results.
[0721] Table 7: Cell population analysis comparison between batches prepared
with and
without anticoagulant
Test Specification At ApoCell
ApoCell
Thawing Time 0 h
Time 24 h
Storage
w \o +ACDhep w \o ACDhep
+ACDhep w \o ACDhep +ACDhep
ACDhep
Change in Total >35.0% 85.5 82.8 49.9 66.7
49.0 66.7
Cell Count (79.5-92.5) (67.7-96.4) (46.6-52.3) (62.5-
71.2) 46.6-50.3) (62.5-71.2)
Percent change
(min-max)
CA 03151815 2022-02-17
157
WO 2021/053667
PCT/IL2020/051011
Changes in 90.0 10.0% 100 100 98.2
100
ApoCell (96.2-100)
Percent change
Range (min-
max)
Cell viability PI > 85.0% 98.0 96.0 98.5 94.6
97.7 94.5
exclusion (97.4-98.4) (91.9-98.1) (97.9-99.2)
(93.5-95.5) (96.4-98.6) (93.4-95.1)
Percent viable
Range (min-
max)
Identity/ CD3 (T 75.7 66.5 73.3 62.8 71.6
64.2
Purity cells): (71.6-81.4) (60.1-70.1) (70.3-78.3)
(61.1-65.3) (61.5-79.1) (61.6-68.1)
71.9 (50.0-
Analysis of cell 85.0)
phenotype ApoCell
Average (%) CD3:
(maximal 71.6 (50.0-
calculated 85.0)
range) CD19 (B 7.5 9.8 9.0 9.9 9.5
9.7
cells): (4.0-11.1) (8.6-12.0) (7.6-10.2) (9.3-
10.2) (8.6-10.3) (9.2-10.4)
9.3 (3.0-15.0)
ApoCell
CD19:
9.5 (4-15)
CD14 9.8 14.0 11.6 15.4 9.3
16.1
(monocytes): (6.4-13.0) (8.8-22.1) (10.2-13.3) (8.2-
19.3) (4.8-17.2) (9.0-20.4)
10.1 (2.5-
22.0)
ApoCell
CD14:
10.6 (2.5-
22.0)
CD15 h`gh 0.2 0.46 0.2 0.083 0.1
0.09
(granulocytes): (0-0.3) (0.18-0.69) (0.1-0.4) (0.08-
0.09) (0.1-0.2) (0.07-0.1)
0.4 (0-6.0)
ApoCell
CD15h1gh:
0.2 (0-2.0)
CD 56 (NK): 7.4 10.1 4.7 11.2 4.9
10.0
7.2 (1.5-22.0) (2.4-11.0) (6.6-14.2) (2.7-8.0) (7.2-14.2)
(2.2-9.2) (6.4-13.0)
ApoCell
CD56:
5.2 (1.5-15.0)
[0722] Percentage of final product cells (yield) in the presence or
absence of anticoagulants.
CA 03151815 2022-02-17
158
WO 2021/053667
PCT/IL2020/051011
Similar to the results presented above at Table 3, the data presented in Table
6 demonstrates that early
apoptotic cells manufactured from cells frozen in the presence of
anticoagulant had a beneficial effect
on average yield of fresh final product (FP tO) as compared to cells frozen
without anticoagulant. The
beneficial effect was seen when anticoagulant was used while freezing only
(61.3 2.6% versus
48.4 5.0%), or both freezing and thawing (56.5 5.2% versus 48.4 5.0%). The
beneficial effect was
less significant when anticoagulant was used upon thawing only (44.0 8.5%
versus 48.4 5.0%).
These were non-high triglyceride samples.
[0723] Effect of anticoagulants on aggregation. No cell aggregations were
seen in these 3 non-
high triglyceride samples, or in 21 additional samples (data not shown).
However, in 41 other non-
high triglyceride samples manufactured without anticoagulants (data not
shown), mild aggregates
were seen in 10 (24.4%) and severe aggregates in 5 (12.2%); thus,
anticoagulants avoid completely
cell aggregates.
[0724] Effect of anticoagulants on stability. Fresh FPs manufactured with-
or without
anticoagulants were stored at 2-8 C for 24 hours to determine whether addition
of ACDhep to the
manufacturing procedure impairs the stability of the FP. Cells were sampled
following 24 hours of
storage and yield was calculated In cell count. Similar to the results shown
in Table 3 for extended
time periods (up to 72 hours), Table 6 shows that the beneficial effect was
kept and observed when
anticoagulant was used while freezing only (59.8 2.1% versus 47.5 4.7%), or
both freezing and
thawing (56.4 5.3% versus 47.5 4.7%). The beneficial effect was less
significant when anticoagulant
was added only upon thawing (42.4 6.1% versus 47.5 4.7%). These were all non-
high triglyceride
samples. These results show minimal cell loss following 24 hours of FP storage
in all treatments with
significant advantage to cells treated with anticoagulant during both freezing
and thawing. Average
loss of cells treated with anticoagulant during freezing only was 2.3 3.2%
compared to 1.9 3.3%
without anticoagulants, upon thawing only was 3.0 4.7 compared to 1.9 3.3%
without
anticoagulants, and 0.2 0.4% compared to 1.9 3.3% without anticoagulants when
cells were both
frozen and thawed with ACDhep. In summary, the beneficial effect of
anticoagulants on yield was
kept for at least 24 hours.
[0725] The characteristics of a representative cell population of the FP
are shown below in Table
8.
[0726] Table 8: Characterization of the cell population of fresh (t0) FP
manufactured from cells
collected with ("+") or without ("¨") addition of anticoagulant during
freezing ("F") and thawing
("Tha") procedures.*
Do FP tO
nor F¨/Tha¨ F¨/Tha+ F+/Tha+ F+/Tha¨
ID C CD CD CD CD CD CD CD CD CD C CD CD CD CD C CD CD CD CD
CA 03151815 2022-02-17
159
WO 2021/053667
PCT/IL2020/051011
D3 19+ 56+ 14+ 15+ 3+ 19+ 56+ 14+ 15+ D3 19+ 56+ 14+ 15+ D3 19+ 56+ 14+ 15+
+ (go) (go) (go) (go) (go) (go) (go) (go) (go) + (go) (go) (go) (go) + (go)
(go) (go) (go)
(go (go (go
/- 62. 5.6 9.8 13. 0 61 8.6 8.6 14. 0 63. 7.4 9.4 13. 0 61. 11. 10. 14. 0
3 2 5 0 6.1 1 0 9 3 0 9 5 1 3 0
6.1 o.7 0.9 1.1 0.4 0.9 1.1 5.8 0.6 0.8
1.9 6.0 1.1 1.0 1.3
*Induction of apoptosis was performed using a medium containing anticoagulant
for all batches.
[0727] The results of Table 8 show the cell characteristics of the final
products (FP) manufactured
with or without anticoagulant at freezing and thawing. Batches were sampled,
stained for
mononuclear markers, and analyzed via flow cytometry to determine the cell
distribution in each
sample and to examine whether the addition of anticoagulant affected the cell
population. As
presented in Table 7, there were no significant differences detected in cell
populations manufactured
with or without anticoagulants at freezing or thawing. The average T cell
population (CD3+ cells) in
fresh FP was 62.3 1.2% between treatments compared to 62.9 1.1% before
freezing; the average B
cell population (CD19+ cells) was 8.3 2.5% between treatments compared to 3.1
0.8% before
freezing; the average natural killer cell population (CD56+ cells) was 9.5
0.7% between treatments
compared to 12.9 0.5% before freezing; the average monocyte cell population
(CD14+ cells) was
13.8 0.5% between treatments compared to 17.5 0.3% before freezing; and the
average granulocyte
population (CD15+ cells) was 0.0% in the fresh FP compared to 0.35 0.2% at
freezing.
[0728] The potency of the early apoptotic population was also examined.
[0729] Table 9: Potency analysis of fresh (t0) FP manufactured from cells
with ("+") or without
("-") addition of anticoagulant during freezing ("F") and thawing ("Tha")
procedures.
Donor ID # FP tO
F-/Tha- F-/Tha+ F+/Tha+ F+/Tha-
Treatment
Median DR CD86 DR CD86 DR CD86 DR CD86
fluorescence
DCs 1:2 Early 3% 28% 4% 24% 5% 24% 9% 15%
apoptotic cell up
population from
+LPS LPS
DCs 1:4 Early 4% 38% 6% 35% 6% 34% 6% 24%
apoptotic cell
population
CA 03151815 2022-02-17
160
WO 2021/053667
PCT/IL2020/051011
+LPS
DCs 1:8 Early 13% Not 10% 45% 15 54% 8% 48%
apoptotic cell done %
population
+LPS
[0730] The results presented in Table 9 are from a potency assay
performed to determine the
ability of each final product to enhance a tolerogenic state in immature
dendritic cells (iDCs)
following stimulation with (LPS). The tolerogenic effect was determined by
assessing
downregulation of co-stimulatory molecule HLA-DR and CD86 expression on iDCs
following
interaction with the early apoptotic cell populations and different treatments
leading to LPS
upregulation. The analysis was performed on DCsign+ cells. Results represent
the percent delay in
maturation following interaction with early apoptotic cell population and
following addition of LPS
versus LPS-induced maturation. The experiment tested the potency of fresh FP
(t0) manufactured
with- or without anticoagulant. Results presented in Table 9 show that
apoptotic cells manufactured
with or without anticoagulant enhance the tolerance effect of both co-
stimulatory markers in a dose-
dependent manner.
[0731] The early apoptotic cells produced herein were from non-high
triglyceride samples. This
consistent high yield of stable early apoptotic cells was produced even in the
cases when the donor
plasma is high in triglycerides (See for example, Examples 12 and 13 of
International Publication No.
WO 2014/087408 and United States Application Publication No. US US -2015-
0275175-A1). Note
that anti-coagulants were not added to the PBS media used for formulation of
the final early apoptotic
cell dose for infusion.
[0732] Summary
[0733] The objective of this study was to produce a stable, high yield
early apoptotic cell
population. The rational for use of anticoagulants was that aggregates were
seen first in patients with
high-triglycerides, but later in a significant portion of other patients. A
concern here was the disclosure
in United States Patent No. US 6,489,311 that the use of anticoagulants
prevented cell apoptosis.
[0734] In short, with minimal impact on the composition, viability,
stability, and the apoptotic
nature of the cells, there was a significant improvement of at least 10-20% in
the number of collected
cells in the final product (Yield) when anticoagulant was added. In this study
an up to 13% increase
in yield was shown, which represents 26.8% augmentation in yield in controlled
conditions but in real
GMP conditions it went up to 33% and more augmentations in cell number then
can be produced in
a single collection. This effect is crucial, since it may avoid the need for a
second apheresis from a
CA 03151815 2022-02-17
161
WO 2021/053667
PCT/IL2020/051011
donor.
[0735] This effect was surprising because the anticipated impact was
expected to be dissolution
of mild aggregates. It had been hypothesize that thawing cells with
anticoagulant reduced the amount
of aggregates. When formed, these aggregates eventually lead to massive cell
loss. Cells collected
and frozen without anticoagulant demonstrated aggregate formation at thawing,
immediately after
wash. Furthermore, a high level of aggregates was also detected in cells that
were frozen without
anticoagulant and resuspended with media containing anticoagulant. No
aggregates were seen in cells
that were both frozen and resuspended with media containing anticoagulant.
Taken together, it was
conclude that the addition of anticoagulants during freezing and apoptosis
induction is of high
importance, and did not appear to negatively impact the induction of early
apoptosis on the cell
population.
[0736] Recovery of early apoptotic cells was further tested, for example,
following 24 hours of
storage at 2-8 C, for stability purposes, during which an average cell loss of
3-4.7% was measured,
regardless of manufacturing conditions, with favorable results for cells that
were both frozen and
thawed with media containing anticoagulant (0.2 0.4% cell loss following 24
hours of FP storage),
suggesting that addition of anticoagulant is critical during freezing and
thawing, but once finally
formulated, the early apoptotic cell population is stable. Extended time point
studies showed this
stability to at least 72 hours.
[0737] Apoptosis and viability, as well as cell composition of the FP
product were not significantly
affected by the addition of anticoagulant at the freezing and/or thawing
stage. Values measured from
a wide variety of characteristics were similar, indicating the ACDhep did not
change the early
apoptotic cell characteristics and the final product met the acceptance
criteria of >40% apoptotic cells.
[0738] The assay used to test apoptotic cells potency was based on
immature dendritic cells
(iDCs), DCs that are characterized by functions such as phagocytosis, antigen
presentation, and
cytokine production.
[0739] The HLA-DR (MHC class II) membrane molecule and co-stimulatory molecule
CD86
were selected as markers to detect the tolerogenic effects of antigen-
presenting cells (APCs). Using
flow cytometry, changes in expression of HLA-DR and CD86 on iDCs were measured
following
stimulation with LPS, as well as in the presence of the early apoptotic cell
population manufactured
with- or without anticoagulant and stimulated with LPS. Early apoptotic cell
populations were offered
to DCs in ascending ratios of 1:2, 1:4, and 1:8 iDCs : early apoptotic cell
population. As presented in
Table 6, it was shown that early apoptotic cell population enhanced the
tolerogenic effect over
stimulated DCs in a dose-dependent manner, with slightly better results for
early apoptotic cell
population manufactured with anticoagulant both at freezing and apoptosis
induction.
CA 03151815 2022-02-17
162
WO 2021/053667
PCT/IL2020/051011
[0740] Taken together, it was concluded that addition of anticoagulant to
both freezing and
apoptosis media is of high importance to increase cell recovery and avoid
massive cell loss due to
aggregates, and to avoid in many cases a second round of apheresis from a
donor. It was shown that
all cells met acceptance criteria for the validated FP, indicating that the
addition of anticoagulant does
not impair the FP.
EXAMPLE 2: Effect of Apoptotic Cells on Cytokine Release in an In Vitro
Cytokine Storm
Model
[0741] Objective: Test the effect of apoptotic cells on the level of cytokine
storm markers (cytokines
IL-6, IL-10, MIP- la, IL-8, TNF-a, MIP-10, MCP-1, and IL-9) in a cytokine
storm induced in an
LPS-Sterile model of macrophage activation syndrome.
[0742] Methods:
[0743] Cell lines and culturing reagents
[0744] The human lymphoma cell line Raji (eCACC, UK, access no. 85011429), the
human cervical
adenocarcinoma cell line HeLa (ATCC, USA, number: CCL-2) and HeLa-CD19
(ProMab, USA,
cat. no. PM-Hela-CD19) were cultured in RPMI 1640 (Gibco, ThermoFisher
Scientific, USA, cat.
no. 31870-025) supplemented with 10% FBS (Gibco, ThermoFisher Scietific, South
America, cat.
no. 12657-029), 2 mM GlutaMAX (Gibco, ThermoFisher Scientific, USA, cat. no.
35050-038), and
100 Wm' Penicillin + 100 Um' Streptomycin (Gibco, ThermoFisher Scientific,
USA, cat. no. 15140-
122), henceforth referred to as "Complete Medium". HeLa-CD19 medium was
further supplemented
with 1 1.tg/m1 puromycin (Sigma-Aldrich, USA, cat. no. P9620), as the
selective antibiotics, during
standard culturing.
[0745] All cells were kept in sub-confluent conditions. Raji cells were
maintained in a concentration
range of 0.3x106-2x106 cell/ml. HeLa and HeLa-CD19 cells were passaged when
receptacle was
filled to 90% confluence.
[0746] Primary monocytes were isolated from blood donations buffy coats (Sheba
Medical Center,
Israel). First, peripheral blood mononuclear cells (PBMCs) were isolated on a
Ficoll density gradient
(Ficoll-Paque PLUS, GE Healthcare, UK, cat. no. 17-1440-03). Upon
centrifugation (800x g, 2-8 C,
20 min. with break 0), the interphase containing the PBMCs were transferred to
a fresh test tube and
washed with RPMI-1640 (Lonza, Switzerland, cat. no. BE12-918F) supplemented
with 2 mM L-
glutamine (Lonza, Switzerland, cat. no. BE17-605E) and 10 mM Hepes (Lonza,
Switzerland, cat. no.
BE17-737B), henceforth "Wash Medium", and centrifuged (650x g, 2-8 C, 10
min.). Pelleted cells
were re-suspended in "Wash Medium" to a concentration of 15x106 cell/ml. Cells
were seeded as a
0.9 ml drop at the center of a 35-mm plate (Corning, USA, cat. no. 430165).
Plates were incubated
CA 03151815 2022-02-17
163
WO 2021/053667
PCT/IL2020/051011
for 1.5h in a humidified incubator (37 C, 5% CO2), allowing monocytes to
adhere, and then washed
three times with pre-warmed PBS (Lonza, Switzerland, cat. no. BE17-516F),
removing other cell
types. After washing, cells were cultured in 2 ml RPMI 1640 (Gibco,
ThermoFisher Scientific, USA,
cat. no. 31870-025) supplemented with 10% FBS (Gibco, ThermoFisher Scietific,
South America,
cat. no. 12657-029), 2 mM GlutaMAX (Gibco, ThermoFisher Scientific, USA, cat.
no. 35050-038),
and 100 Um' Penicillin + 100 Um' Streptomycin (Gibco, ThermoFisher Scientific,
USA, cat. no.
15140-122), aka "Complete Medium".
[0747] All cell lines were cultured in a humidified incubator at 37 C and
containing 5% CO2.
[0748] In brief, and following manufacturer's guidelines, target cells (HeLa
or HeLa-CD19) were
cultured alone or in conjunction with monocytes. After target cells adhered to
the plate (6h-
overnight), cultures were exposed to y x106 ApoCells cells for lh, after which
these cells were washed
off by 4-5 washes of RPMI. Removal of ApoCells cells was confirmed visually
under a light
microscope. 10 ng/ml LPS (Sigma-Aldrich, USA, cat. no. L4391) was introduced
to the co-culture
and incubated for lh. After incubation, LPS was removed by 3-5 washing cycles
with RPMI. Viable
CD19-CAR T cells or naïve T cells were added at the designated E/T ratio(s)
and incubated for 4h.
To collect media, plates were centrifuged at 250x g, 2-25 C, 4 min.
(Centrifuge 5810 R, Eppendorf,
Germany) to sediment cells. 50 pl of supernatant medium from each well was
transferred to a fresh
flat-bottom 96-well microplate well (Corning, USA, cat. no. 3596) and 50 1.41
CytoTox 96 Reagent
was added to each well. Plates were incubated in the dark at room temperature
for 30 min., after
which the reaction was terminated by addition of 501.41 Stop Solution per
well. Absorbance was read
at 492 nm using Infinite F50 (Tecan, Switzerland) and captured using Magellan
F50 software. Data
analysis and graph generation was performed using Microsoft Excel 2010.
[0749] Analysis of cytokine release was performed using Liminex technology
following incubation
with apoptotic cells or incubation with supernatant from apoptotic cells.
[0750] Results: Figures 4A through 4H show that there was a significant
reduction in the levels of
cytokine storm markers IL-10, IL-6, MIP-la, IL-8, TNF-a, MIP-10, MCP-1, and IL-
9 which were
induced by LPS in an in vitro model of macrophage activation syndrome. While
administration of
ApoCells to achieve a macrophage:Apocell ratio of 1:8 resulted in
significantly decreased levels of
both IL-10, IL-6, MIP-la, IL-8, TNF-a, MIP-10, MCP-1, and IL-9 released into
the medium
(Figures 4A, 4B, 4C, 4D, 4E, 4F, 4G, 4H), administration of ApoCells to
achieve a macrophage:
ApoCell ratio of 1:16 actually inhibited or nearly inhibited the release of
cytokines IL-10, IL-6, MIP-
la, IL-8, TNF-a, MIP-10, MCP-1, and IL-9 in this model.
[0751] Addition of apoptotic cells resulted in the inhibition of at least 20
pro-inflammatory cytokine
and chemokines induced in macrophage activating, a sample of the results are
shows in Figures 4A-
CA 03151815 2022-02-17
164
WO 2021/053667
PCT/IL2020/051011
4H.The common mechanism for pro-inflammatory cytokine and chemokine release is
NF-KB
inhibition.
[0752] The inhibition of release of pro-inflammatory cytokines and chemokines
appears to be
specific, as examination of cytokine IL-2R (IL-2 receptor) levels under
similar conditions showed
that IL-2R levels released was not influenced in the same manner at the pro-
inflammatory cytokines.
(Figure 41). Addition of apoptotic cells increased the release of IL-2R at 1:4
and 1:8 ratios. Further,
Figure 4J shows that apoptotic cells had no influence on release of IL-2 over
a 24 hour time period.
Activation of the IL-2 receptor is considered to have an essential role in key
functions of the immune
system including tolerance.
[0753] Conclusion: Addition of early apoptotic cells in a cytokine storm model
of macrophage
activation syndrome in the presence of cancer and CAR-19, resulted in
significant reduction and,
surprisingly even prevention of pro-inflammatory cytokines, for example IL-10,
IL-6, MIP- la, IL-8,
TNF-a, MIP-10, MCP-1, and IL-9 , while increasing or not affecting cytokine IL-
2R levels. Thus,
the results here show that while pro-inflammatory cytokines were reduced by
incubation with
apoptotic cells, IL-2 and IL-2R were not influenced in the same manner with
incubation of early
apoptotic cells. Thus, the T-cell associated cytokines are not influenced by
the CAR T-cell therapy +
apoptotic cells, whereas the innate immunity cytokines, for example those
released from monocytes,
macrophages, and dendritic cells are.
EXAMPLE 3: Effect of Apoptotic Cells on Cytokine Storm Without a Negative
Effect on
the CAR-T Cell Efficacy
[0754] Objective: Test the effect of apoptotic cells or supernatants derived
from apoptotic cells on
cytokine storm marker cytokines and CAR T-cell efficacy on tumor cells.
[0755] Methods:
[0756] T4+ CAR T-cells
[0757] A solid tumor model (van der Stegen et al., 2013 ibicl) reported to
induce cytokine storms in
mice was utilized. In this model, T cells were engineered with a chimeric
antigen receptor (CAR)
targeting certain ErbB dimers (Tr CAR-T cells), which are often highly up-
regulated in specific
solid tumors such as head and neck tumors and ovarian cancers. T-cells were
isolated from PBMC
separated from peripheral blood using CD3 micro-beads. Vectors containing the
chimeric T4+
receptor were constructed and transducer into the isolated T-cells, resulting
in T4+ CAR T-cells. For
the experiments performed herein, T4+ CAR T-cells were purchased from Creative
Biolabs (NY
USA) or Promab Biotechnologies (CA USA). Figure 5 presents flow cytometry
curves verifying the
surface expression of 44 chimeric receptor on the T4+ CAR T-cells using an
anti-CD124
monoclonal antibody (Wilkie et al., ibicl). In addition, a PCR procedure was
performed and verified
CA 03151815 2022-02-17
165
WO 2021/053667
PCT/IL2020/051011
the presence of the vector in transduced T cells.
[0758] SKOV3-luc cells
[0759] SKOV3-luc ovarian adenocarcinoma tissue culture cells were purchased
from Cell BioLabs
(cat. #AKR-232). SKOV3-luc highly express ErbB receptors and are a target for
the T4+ CAR-T cells
.. (van der Stegen et al., 2013, ibicl). These cells had been further
manipulated to constitutively express
the firefly luciferase gene, allowing tracking of cell proliferation in vitro
and tumor growth and
recession in vivo.
[0760] Apoptotic cells
[0761] Apoptotic cells were prepared as per Example 1.
[0762] Apoptotic cell supernatants
[0763] Eight (8) million apoptotic cells per seeded per well in a 12-well
plate. After 24 hours the
cells were centrifuge (290g, 4 degrees Celsius, 10 minutes). Supernatant was
collected and frozen in
aliquots at -80 degrees until use. Different numbers of cells are used to make
supernatants. Some
aliquots contain concentrated supernatants.
[0764] Monocyte isolation
[0765] PBMCs were isolated using Ficoll (GE healthcare, United Kingdome) from
peripheral blood\
buffy coat obtained from healthy, eligible donors. Cells were brought to a
concentration of 15x106
cells \ml in RPMI1640 (Gibco, Thermo Fisher Scientific, MA, USA) and seeded in
a 0.9ml drop in
the middle of 35mm plates (Corning, NY, USA). Plates were then incubated at 37
C in 5% CO2 for
.. 1 hour. At the end of incubation, cells were washed three times with PBS
(Biological industries, Beit
Haemek, Israel) and adhesion was determined using a light microscope. Cells
were then incubated
with complete media (RPMI1640+ 10% heat inactivated FBS+ 1% Glutamax+ 1%
PenStrep, all from
Gibco).
[0766] An alternative method of monocyte isolation was also used wherein human
mononuclear
.. cells were isolated from heparinized peripheral blood by density gradient
centrifugation. The isolated
mononuclear cells then were separated into monocyte, B-cell and T-cell
populations by positively
selecting monocytes as the CD14+ fraction by magnetic bead separation
(Miltenyi Biotec., Auburn,
CA, USA), positively selecting B-cells as the CD22+ fraction, and negatively
selecting T-cells as the
CD14-CD22- fraction. Purity was greater than 95 percent for monocytes.
[0767] For macrophage differentiation, at the end of adhesion, cells were
washed three times with
PBS then incubated with RPMI1640+ 1% Glutamax+ 1% PenStrep and 10% heat
inactivated human
AB serum (Sigma, MO, USA). Cells were incubated at 37 C and 5% for 7-9 days,
with media
exchange at day 3 and day 6. Differentiation was determined by morphology via
light microscope.
[0768] Supernatant from apo+ monocytes
CA 03151815 2022-02-17
166
WO 2021/053667
PCT/IL2020/051011
[0769] CD14+ monocytes were cultured with apoptotic cells as prepared above at
a ratio of 1:16, for
24h. The number of monocytes was: 0.5 million cells per well in a 12-well
plate and the number of
apoptotic cells was: 8 million cells per well in a 12-well plate. After
incubation for 24 hours the cells
were centrifuge (290g, 4 degrees Celsius, 10 minutes). Supernatant was
collected and frozen in
aliquots at -80 degrees until use. Similar procedures could be performed at
different ratios of
monocytes:apoptotic cells and/or using other sources of cells, such as
macrophages and dendritic
cells.
[0770] In vitro culturing conditions
[0771] Initial experiments were performed by incubating SKOV3-luc cancer cells
with apoptotic
cells, or apoptotic supernatants, for 1 hour followed by co-culturing with T4+
CAR T-cells (+/-
monocytes-macrophages) for 48 hours.
[0772] In order to simulate in vivo conditions, 1x105 THP-1 cells/ml (HTCC
USA), or monocytes or
macrophages or dendritic cells, will be differentiated with 200 nM (123.4
ng/ml) phorbol myristate
acetate (PMA) for 72 hrs and will then be cultured in complete medium without
PMA for an
additional 24h. Next, cancer or tumor cells ¨ for example SKOV3-luc cells will
be plated in a 24-
well plate at 5x105 SKOV3-luc cells/well on the differentiated THP-1 cells.
Following initial
culturing of the cancer or tumor cells, 4x105-8x105 apoptotic cells (ApoCell)
will be added to the
culture for 1-3h to induce an immunotolerant environment. The ratio of cancer
cell to ApoCell will
be optimized for each cell type. After washing, the co-culture will be treated
with 10 ng/ml LPS after
which 1x106 Tr CAR T cells (or a quantity to be determined by an
effector/target (E/T) ratio graph)
will be added. The ratios of tumor cells and T4+ CAR T-cells will be varied in
order to generate
effector/target (E/T) ratio graphs for each tumor or cancer cell type.
[0773] To assay for SKOV3 cancer cell cytotoxicity, lysates were prepared and
luciferase activity
was determined after the 48 hour incubation period. Additional experiments
will be performed
assaying for cancer or tumor cell cytotoxicity for the other cancer cell types
and at intervals within
the 48h incubation time period. Alternatively, Promega' s CytoTox 96 Non-
Radioactive Cytotoxicity
Assay (Promega, cat #G1780) will be used.
[0774] Lysate Preparation
[0775] SKOV3-luc cell lysates were prepared by washing the SKOV3-luc monolayer
with PBS to
remove any residual serum and adding 70 Ill CCLR lysis buffer x1/well (for 24-
well plates).
Detachment was further enhanced by physical scraping of well bottoms.
Following vortexing for 15
seconds, lysates were centrifuged at 12,000g for 2 minutes at 4 C.
Supernatants were collected and
stored at -80 C.
[0776] In vitro Luciferase Activity
CA 03151815 2022-02-17
167
WO 2021/053667
PCT/IL2020/051011
[0777] To detect luciferase activity in SKOV3-luc cells in culture, Luciferase
Assay System
(Promega, cat. #E1501) was used. Calibration of this kit with the luminometer
reader (Core Facility,
Faculty of Medicine, Ein Kerem, Hebrew University of Jerusalem) was done by
using QuantiLum
recombinant luciferase (Promega, cat. #E170A). 612 ag - 61.2 1.ig (10-20-10-9
moles) was used to
determine detection range and following manufacturer's guidelines. In brief,
each rLuciferase
quantity in 20 Ill volume was placed in a well of black 96-well plates (Nunc).
Each quantity was done
in triplicate. 100 1.11 LAR (luciferin substrate from Luciferase Assay System
kit) was added to each
well and read immediately with a 10 second exposure.
[0778] For luciferase activity reading, lysates were thawed on ice and 20 Ill
samples were placed in
a black 96-well plate (Nunc). Each sample was read in duplicate. 100 Ill LAR
was added and
luminescence was read for 10 second exposure period every 2.5 minutes for 25
minutes and every 40
seconds for the ensuing 10 minutes.
[0779] Cytokine Analysis
[0780] Initial assays for IL-2, IL-2 receptor (IL-2R), IL-6, IL-la, IL-4, IL-
2, TNF-a were performed.
To assay for cytokine release reduction of IL-2, IL-2 receptor (IL-2R), IL-6,
IL- la, IL-4, IL-2, TNF-
a as well as other cytokines, supernatants were be collected and examined for
selected cytokine using
Luminex MagPix reader and ELISA assays.
[0781] Results:
[0782] SKOV3-luc growth
[0783] SKOV3-luc growth was followed using luciferase activity as an
indicator, to determine target
SKOV3-luc cell number in future experiments. 3.8x104-3.8x105 SKOV3-luc
cells/well were plated
in 24-well plates (Corning) and luciferase activity was monitored daily for 3
days. 1.9x105 cells/well
or higher cell number plated reach confluence and present growth saturation
indicated by luciferase
activity 2 days after plating (Figure 6). Note that 3.8x104-1.1x105 SKOV3-luc
cells/well were still
in the linear or exponential growth phase three days after plating (Figure 6,
plots orange, turquoise
and purple). Negative control (3.8x105 SKOV3-luc cells without LAR substrate)
displayed only
background-level reading and demonstrates that bioluminescent readings from
SKOV3-luc cells
result from luciferase activity.
[0784] Verification of T4+ CAR-T cell activity against SKOV3-luc tumor cells
[0785] To corroborate the Tr CAR-T cell activity, monolayers of SKOV3-luc were
exposed to
either 1,000,000 (one million) Tr CAR-T cells or to 1,000,000 (one million)
non-transduced T cells.
After 24h incubation, Tr CAR-T cells reduced SKOV3-luc proliferation by 30%
compared to the
non-transduced T cell control (Figure 7), showing anti-tumor activity of the
Tr CAR-T cells.
[0786] Activity of stand-alone T4+ CAR-T cells against SKOV3-luc tumor cells
was compared to
CA 03151815 2022-02-17
168
WO 2021/053667
PCT/IL2020/051011
activity post exposure to Apoptotic Cells
[0787] Apoptotic cells (ApoCell) and apoptotic cell supernatants (ApoSup and
ApoMon Sup) were
tested to determine if they interfere with T4+ CAR-T cell anti-tumor activity.
The SKOV3-luc tumor
cells were incubate with Apoptotic Cells for one hour, followed by the
addition of T4+ CAR-T cells
(500,000, five hundred thousands) or T4+ non-transduced T cells (500,000, five
hundred thousands)
(ratio of 1:2 Tr CAR-T cells to Apoptotic Cells). The tumor cell/Apoptotic
cell/T4+ CAR T-cells
were then co-cultured for 48h. The control SKOV3-luc tumor cells were co-
cultured with T4+ CAR-
T cells and Hartman solution (the vehicle of Apoptotic Cells), but without
Apoptotic Cells, for 48h.
[0788] The results showed that after 48h incubation, T4+ CAR-T cells anti-
tumor activity was
superior to incubation with non-transduced T cells. Similar incubations were
performed with
apoptotic cells or apoptotic cell supernatants. Surprisingly, T4+ CAR T-cell
anti-tumor activity was
comparable with or without exposure to apoptotic cells or apoptotic cell
supernatants. (Figure 8).
[0789] Effect of Apoptotic Cells on amelioration, reduction or inhibition of
cytokine storms resulting
from CAR-T treatment
[0790] The effect of apoptotic cells to reduce cytokine storms was examined
next. IL-6 is a prototype
pro-inflammatory cytokine that is released in cytokine storms (Lee DW et al.
(2014) Blood 124(2):
188-195) and is often used as a marker of a cytokine storm response.
[0791] Cultures were established to mimic an in vivo CAR T-cell therapy
environment. SKOV3-luc
tumor cells were cultured in the presence of human monocyte-macrophages and
T4+ CAR T-cells.
The concentration of 11-6 measured in the culture media was approximately 500-
600 pg/ml. This
concentration of IL-6 is representative of a cytokine storm.
[0792] Unexpectedly, IL-6 levels measured in the cultured media of SKOV3-luc
tumor cells, human
monocyte-macrophages, T4+ CAR-T cells, wherein the tumor cells had been
previously incubated
with apoptotic cells for one hour (ratio of 1:2 T4+ CAR-T cells to Apoptotic
Cells) were dramatically
reduced. Similarly, IL-6 levels measured in the cultured media of SKOV3-luc
tumor cells, human
monocyte-macrophages, T4+ CAR-T cells, wherein the tumor cells had been
previously incubated
with apoptotic cell supernatants for one hour, were also dramatically reduced.
This reduction in
concentration of IL-6 is representative of a decrease in the cytokine storm
(Figure 9).
[0793] It was concluded that unexpectedly, apoptotic cells and apoptotic
supernatants do not
abrogate the effect of CAR-T cells on tumor cell proliferation while at the
same time they down
regulating pro-inflammatory cytokines such as IL-6, which was been described
as a major cytokine
leading to morbidity.
[0794] Analysis using a wider range of cytokines
[0795] To further evaluate the effect on a possible wider range and levels of
cytokines that are not
CA 03151815 2022-02-17
169
WO 2021/053667
PCT/IL2020/051011
generated during experimental procedures but do appear in clinical settings
during a human
cytokine storm, LPS (10 ng/ml) was added to the SKOV3-luc culture conditions
outlined above.
The addition of LPS is expected to exponentially increase the cytokine storm
level. As expected,
the addition of LPS increased the cytokine storm effect and as a result IL-6
levels increased to
approximately 30,000 pg/ml. Other cytokines known to be expressed in high
levels during a
cytokine storm showed elevated levels, for example: TNF-a (250-300 pg/ml), IL-
10 (200-300
pg/ml), IL1-alpha (40-50 pg/ml) and IL-18 (4-5 pg/ml). As shown in Figure 10,
exposure to
apoptotic cells dramatically reduced the levels of IL-6 even during the
exponential state of the
cytokine storm to almost normal levels that may be seen in clinical settings,
and is not always seen
in experimental procedures with CAR T-cells. This effect was similar across
the other pro-
inflammatory cytokines TNF-alpha, IL-10, ILI¨alpha, IL-113, and IL-18, which
showed a
reduction of between 20-90%. Similar results were found when using apoptotic
cell supernatants in
place of the apoptotic cells.
[0796] Effect of Apoptotic Cells on IL-2 and IL-2R
[0797] The concentration of IL-2 measured in culture supernatants following
incubation of SKOV3-
luc cells with T4+ CAR T-cells was 1084 pg/ml. Surprisingly, when SKOC3-luc
cells were first
incubated with apoptotic cells and then T4+ CAR T-cells the concentration of
IL-2 increased to 1190
pg/ml. Similarly, the concentration of IL-2R measured in culture supernatants
following incubation
of SKOV3-luc cells with T4+ CAR T-cells was 3817 pg/ml. Surprisingly, when
SKOC3-luc cells
were first incubated with apoptotic cells and then T4+ CAR T-cells the
concentration of IL-2R
increased to 4580 pg/ml. In SKOV3-luc alone the concentration of 11-2 was 3.2
pg/ml and with the
addition of apoptotic cells the concentration was 10.6 pg/ml. In SKOV3-luc
alone the concentration
of I1-2R was 26.3 pg/ml and with the addition of apoptotic cells the
concentration was 24.7 pg/ml.
[0798] Conclusion
[0799] CAR-T cell therapy has been documented to cause cytokine storms in a
significant number
of patients. These results demonstrate that apoptotic cells and apoptotic cell
supernatants surprisingly
decreased cytokine storms cytokine markers without affecting CAR-T cell
efficacy against tumor
cells. Moreover, it appears that apoptotic cells increase cytokine IL-2, which
may increase duration
of CAR T-cell therapy by maintaining or increasing CAR T-cell proliferation.
EXAMPLE 4: Apoptotic Cell Therapy Prevents Cytokine Storms in Mice
Administered
CAR T-Cell Therapy
[0800] Objective: Test the in vivo effect of apoptotic cells or apoptotic cell
supernatants in a solid
tumor model (SKOV3 ovarian adenocarcinoma), in order to determine T4+ CAR T-
cell efficacy and
the level of cytokine storm marker cytokines.
CA 03151815 2022-02-17
170
WO 2021/053667
PCT/IL2020/051011
[0801] Materials and Methods
[0802] In vitro studies
[0803] In vitro methods including methods of making, culturing, and analyzing
the results described
above and relevant for use of T4+ CAR T-cells that recognize the ErbB target
antigen (referred to
herein as "T4+ CAR T-cells", SKOV3-luc cells, apoptotic cells, apoptotic
supernatants, monocytes,
macrophages, and the various assays, have all been described above in Example
1. The same methods
were used herein.
[0804] In vivo studies
[0805] Mice
[0806] 7-8 week old SCID-beige mice and NSGS mice were purchased from Harlan
(Israel) and
kept in the SPF animal facility in Sharett Institute.
[0807] SKOV3-luc tumor cells (1 X 106 or 2 X 106) are inoculated into SCID
beige mice or NSGS
mice, by either i.p. in PBS or s.c. in 200 ml Matrigel (BD Biosciences). Tumor
engraftment is
confirmed by bioluminescence imaging (BLI) at about 14-18 days post injection,
and mice are sorted
into groups with similar signal intensity prior to T-cell administration.
[0808] Mice will receive 30 x 106 apoptotic cells either 24 hours prior to
administration of T4+ CAR
T-cells or concurrent with administration of T4+ CAR T-cells (10-30 x 106T4+
CAR T-cells). Tumor
growth will be followed by bioluminescence imaging (BLI) and circulating
cytokine levels will be
determined by Luminex.
[0809] In vivo Luciferase assay
[0810] Tumor growth was monitored weekly through firefly luciferase activity.
In brief, 3 mg D-
luciferin (E1605. Promega, USA)/mouse (100 Ill of 30 mg/ml D-luciferin) was
injected i.p. into
isoflurane-anesthetized mice and ventral images were acquired 10 minutes after
injection using IVIS
Imaging System and Live Image image capture software (both from Perkin Elmer,
USA).
[0811] Image acquisition parameters were chosen for each image session by
imaging mice that
received 0.5x106 5KOV3-luc cells/mouse, 5 minutes post D-luciferin injection
the "auto" option.
Capture parameters were set for binning 4, F/stop 1.2 and exposure of 2-4
minutes using the 24x lens.
Data analysis and quantification was performed with the Live Image software
and graphs were
generated using Microsoft's Excel program.
[0812] In vivo Cytotoxicity
[0813] To assess in vivo toxicity of T-cells, organs are collected from mice,
formalin fixed, and
subjected to histopathologic analysis.
[0814] Cytokine analysis
[0815] Supernatants and sera are analyzed using Luminex MagPix reader and/or
ELISA kits,
CA 03151815 2022-02-17
171
WO 2021/053667
PCT/IL2020/051011
cytometric bead arrays (Th1/Th2/Th17; BD Biosciences) as described by the
manufacturers. For
example, analysis may be for pro-inflammatory cytokine, which in one case
would be IL-6, though,
in some embodiments, any of the cytokines listed in Tables 1 and 2 or known in
the art may be
analyzed herein.
[0816] Results
[0817] Calibrating SKOV3-luc tumors in vivo
[0818] 0.5x106, 1x106 or 4.5x106 SKOV3-luc cells were injected i.p. to S CID
beige mice and
bioluminescence imaging (BLI) was conducted weekly in order to follow tumor
growth, as described
in the Methods (data not shown).
[0819] Clinical score of mice
[0820] Mice displayed no clinical symptoms for the initial 4 weeks. However,
28 days post SKOV3-
luc injection, the mice that received the high dose (4.5x106; purple line)
began to lose weighed
steadily (Figure 11A) and the overall appearance of the mice deteriorated,
manifested in lethargy,
abnormal pacing and general loss of activity. This group was culled at the day
39, and an abdominal
autopsy was performed to expose tumor appearance and size (Figure 11B). SKOV3-
luc tumors were
large, solid, vascularized and displayed a whitish shining complexion. One
large tumor predominated
on the side of the injection (left) either caudal or rostral in the abdominal
cavity. This tumor
encompassed approximately 25-75% of the cavity and clearly pressed and
disturbed the intestines.
Smaller foci were also observed at various locations within the abdominal
cavity. Tumors were
contained within the abdominal cavity and no other tumors were observed in any
other part of the
body in any mice. Mice receiving low (0.5x106) or medium (1x106) dose of SKOV3-
luc cease gaining
weight 40 days after SKOV3-luc injection and began to steadily lose weighed.
Experiment was
terminated 50 days after SKOV3-luc injection.
[0821] SKOV3-luc Tumor Kinetics
[0822] PBS was injected to control for SKOV3-luc cells and these mice did not
exhibit any luciferase
activity throughout the experiment (Figure 12, Left panel). Tumor detection
and growth was dose-
dependent. Lower dose (0.5x106 SKOV3-luc cells) began to display tumors 25
days post-injection
(4/5 animals), medium dose (1x106) injections showed tumors at 18 days post-
injection (4/5 animals),
whereas at higher dose (4.5x106) tumors were detected as early as 11 days post-
injection in 3/5
animals and by day 18 all animals displayed well-established tumors (Figure 12
and Figures 13A-
13D).
[0823] CAR T-cell therapy induces cytokine release syndrome
[0824] Three groups of tumor-free mice as well as mice with tumors are
administered (i.p. or directly
into the tumor) increasing doses of T4+ CAR T-cells (3x106, 10 x106 or
30x106). At the highest dose,
CA 03151815 2022-02-17
172
WO 2021/053667
PCT/IL2020/051011
tumor-free mice and mice with tumors demonstrate subdued behavior,
piloerection, and reduced
mobility within 24 h, accompanied by rapid weight loss followed by death
within 48 hrs. At least
Human interferon-gamma and mouse IL-6 are detectable in blood samples from the
mice given the
highest dose of CAR T-cells. Animals that receive a high dose of CAR T-cells
directed to a different
tumor antigen do not exhibit weight loss or behavioral alterations.
[0825] Administration of apoptotic cells inhibits or reduces the incidence of
cytokine release
syndrome induced by CAR T-cell therapy
[0826] One group of mice given the highest dose of CAR T-cells is
concomitantly administered
2.10x108/kg apoptotic cells, which was previously demonstrated to be a safe
and effective dose. Mice
receiving human CAR T + apoptotic cells have significantly lowered levels of
mouse IL-6, lower
weight loss, and reduced mortality.
EXAMPLE 5: Effect of Combination Immune Therapy on in vitro Diffuse Tumor
Models
[0827] Objective: Test the effect of apoptotic cells or supernatants derived
from apoptotic cells in a
diffuse tumor model where the cancer is widely spread and not localized or
confined, in order to
determine CAR T-cell efficacy on the cancer cells and the level of cytokine
storm marker cytokines.
[0828] Methods:
[0829] CD19+ T4+ CAR T-cells ("CD19+ CAR T-cells")
[0830] CD19-specific CAR-T cells were purchased from ProMab (Lot # 012916).
The T cells
were 30% positive for CAR (according to manufacturer's FACS data ¨ Fab
staining). Briefly, cells
were thawed into AimV + 5% heat-inactivated FBS, centrifuged (300g, 5 minutes,
room-
6
temperature), and resuspended in AimV. On day 6 of the experiment 20x10 cells
were injected
IV per mouse (70% AnnexinPI negative, of which 30% CAR positive).
[0831] Recombinant HeLa cells expressing CD19 will be used as a control cell-
type that also
expresses CD19 on their cell surface.
[0832] CD123+ CAR T-cells
[0833] T4+ CAR T-cells will also be engineered with a CAR targeting CD123
epitopes (referred to
herein as "CD123+ CAR T-cells").
[0834] Raji cells, CD19 expressing HeLa cells, and CD123 expressing leukemic
cells
[0835] Raji cells Raji cells were purchased from ECACC (Cat. #: 85011429), and
routinely cultured
in complete medium (RPMI-1640 supplemented with 10% H.I. FBS, 1% Glutamax, 1%
Penicillin /
5 6
Streptomycin), and maintained at a concentration of 3x10 ¨ 3x10 cells/ml. On
day 1 of the
6
experiment 0.1 x 10 cells were injected IV per mouse.
CA 03151815 2022-02-17
173
WO 2021/053667
PCT/IL2020/051011
[0836] Similarly, CD19 expressing HeLa cells will be generated in the
laboratory and used as a target
for CD19+ CAR T-cells. CD123 expressing leukemic cells will be used as targets
for CD123+ CAR
T-cells. In addition, primary cancer cells will be utilized as a target for
CAR T-cells.
[0837] HeLa cells expressing CD19 were prepared using methods known in the
art. Cells will be
cultured as is well known in the art.
[0838] CD123 is a membrane biomarker and a therapeutic target in hematologic
malignancies.
CD123 expressing leukemic cells, for example leukemic blasts and leukemic stem
cells will be
cultured as is known in the art.
[0839] Apoptotic cells, Apoptotic cell supernatants and rnonocyte isolation,
will be prepared as
described in Example 1. Early apoptotic cells produced were at least 50%
annexin V-positive and
less than 5% PI-positive cells.
[0840] Macrophages. Were generated from CD14positive cells by adherence.
[0841] Dendritic cells. Were CD14 derived grown in the presence of IL4 and
GMCSF.
[0842] Flow-cytornetry. The following antibodies were used :hCD19-PE
(eBiosciences, Cat. # 12-
0198-42); mIgG1 -PE (eBiosciences, Cat. # 12-0198-42); hCD3-FITC
(eBiosciences, Cat. # 11-0037-
42); mIgG2a-FITC (eBiosciences, Cat. # 11-4724-82). Acquisition was performed
using FACS
Calibur, BD.
[0843] Naïve T cells. Naive T cells were isolated from Buffy coat using
magnetic beads (BD), and
cryopreserved in 90% human AB serum and 10% DMSO. Thawing and injection was
identical to
the CAR-T cells.
[0844] In vitro culturing conditions
[0845] Cell lines and culturing reagents
[0846] The human lymphoma cell line Raji (eCACC, UK, access no. 85011429), the
human cervical
adenocarcinoma cell line HeLa (ATCC, USA, number: CCL-2) and HeLa-CD19
(ProMab, USA,
cat. no. PM-Hela-CD19) were cultured in RPMI 1640 (Gibco, ThermoFisher
Scientific, USA, cat.
no. 31870-025) supplemented with 10% FBS (Gibco, ThermoFisher Scietific, South
America, cat.
no. 12657-029), 2 mM GlutaMAX (Gibco, ThermoFisher Scientific, USA, cat. no.
35050-038), and
100 Wm' Penicillin + 100 Um' Streptomycin (Gibco, ThermoFisher Scientific,
USA, cat. no. 15140-
122), henceforth referred to as "Complete Medium". HeLa-CD19 medium was
further supplemented
with 1 1.tg/m1 puromycin (Sigma-Aldrich, USA, cat. no. P9620), as the
selective antibiotics, during
standard culturing.
[0847] All cells were kept in sub-confluent conditions. Raji cells were
maintained in a concentration
range of 0.3x106-2x106 cell/ml. HeLa and HeLa-CD19 cells were passaged when
receptacle was
filled to 90% confluence.
CA 03151815 2022-02-17
174
WO 2021/053667
PCT/IL2020/051011
[0848] Primary monocytes were isolated from blood donations buffy coats (Sheba
Medical Center,
Israel). First, peripheral blood mononuclear cells (PBMCs) were isolated on a
Ficoll density gradient
(Ficoll-Paque PLUS, GE Healthcare, UK, cat. no. 17-1440-03). Upon
centrifugation (800x g, 2-8 C,
20 min. with break 0), the interphase containing the PBMCs were transferred to
a fresh test tube and
washed with RPMI-1640 (Lonza, Switzerland, cat. no. BE12-918F) supplemented
with 2 mM L-
glutamine (Lonza, Switzerland, cat. no. BE17-605E) and 10 mM Hepes (Lonza,
Switzerland, cat. no.
BE17-737B), henceforth "Wash Medium", and centrifuged (650x g, 2-8 C, 10
min.). Pelleted cells
were re-suspended in "Wash Medium" to a concentration of 15x106 cell/ml. Cells
were seeded as a
0.9 ml drop at the center of a 35-mm plate (Corning, USA, cat. no. 430165).
Plates were incubated
for 1.5h in a humidified incubator (37 C, 5% CO2), allowing monocytes to
adhere, and then washed
three times with pre-warmed PBS (Lonza, Switzerland, cat. no. BE17-516F),
removing other cell
types. After washing, cells were cultured in 2 ml RPMI 1640 (Gibco,
ThermoFisher Scientific, USA,
cat. no. 31870-025) supplemented with 10% FBS (Gibco, ThermoFisher Scietific,
South America,
cat. no. 12657-029), 2 mM GlutaMAX (Gibco, ThermoFisher Scientific, USA, cat.
no. 35050-038),
and 100 Um' Penicillin + 100 Um' Streptomycin (Gibco, ThermoFisher Scientific,
USA, cat. no.
15140-122), aka "Complete Medium".
[0849] All cell lines were cultured in a humidified incubator at 37 C and
containing 5% CO2.
[0850] CD19-CAR T cells (ProMab, USA, cat. no. FMC63) were delivered either in
AIM-V medium
or frozen. Cryopreserved CAR T cells for in vitro experiments were thawed on
the day of the
experiment in a 35-38 C bath and immediately immersed in pre-warmed AIM V
medium (Gibco,
ThermoFisher Scientific, USA, cat. no. 12055-091) supplemented with 5% FBS
(Gibco, South
America, cat. no. 12657-029). DMSO was removed by centrifuging the cells (300x
g, room
temperature, 5 min.) and re-suspending in pre-warmed AIM V medium.
Concentration and viability
of CD19-CAR+ cell population was determined by anti-FLAG (BioLegend, USA, cat.
no. 637310)
staining and by Annexin V and PI staining (MEBCYTO Apoptosis kit, MBL, USA,
cat. no. 4700)
read with FACSCalibur flow cytometer (BD, USA).
[0851] For Naïve T cell isolation, PBMCs were extracted either from
leukapheresis fractions
collected from informed consenting eligible donors at Hadassah Medical Center
(Ein Kerem Campus,
Jerusalem, Israel) using a Cobe SpectraTM apheresis apparatus (Gambro BCT,
USA) according to
Leaukapheresis Unit's SOP or from buffy coats (Sheba Medical Center, Israel)
loaded on a Ficoll
density gradient and centrifuged 800x g, 2-8 C, 20 min. T cells were isolated
from the positive
fraction using MagniSort Human CD3 Positive Selection Kit (eBioscience, USA,
cat. no. 8802-6830-
74) following manufacturer's guidelines. T cells were cryopreserved in
"Complete Medium"
(defined above) containing an additional 20% FBS (Gibco, ThermoFisher
Scietific, South America,
CA 03151815 2022-02-17
175
WO 2021/053667
PCT/IL2020/051011
cat. no. 12657-029) and 5% DMSO (CryoSure-DMSO, WAK-Chemie Medical GmbH,
Germany,
cat. no. WAK-DMSO-70) and thawed on the day of experiment parallel to the CD19-
CAR T cells.
[0852] a LDH cytotoxicity assay
[0853] Lactate dehydrogenase (LDH), a stable cytosolic enzyme, is released by
cells undergoing
lysis in a correlative manner. Hence, LDH levels in the medium can be used to
quantify cytotoxic
activity. CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, USA, cat.
no. G1780) is a
colorimetric assay to quantify LDH levels in the medium. A tetrazolium salt
substrate (iodonitro-
tetrazolium violet, TNT) is introduced to the medium in excess and LDH
converts the substrate into
a red formazan product. The amount of red color formed is directly
proportional to the number of
cells lysed.
[0854] In brief, and following manufacturer's guidelines, target cells (HeLa
or HeLa-CD19) were
cultured alone or in conjunction with monocytes. After target cells adhered to
the plate (6h-
overnight), cultures were exposed to y x106 ApoCells cells for lh, after which
these cells were washed
off by 4-5 washes of RPMI. Removal of ApoCells cells was confirmed visually
under a light
microscope. 10 ng/ml LPS (Sigma-Aldrich, USA, cat. no. L4391) was introduced
to the co-culture
and incubated for lh. After incubation, LPS was removed by 3-5 washing cycles
with RPMI. Viable
CD19-CAR T cells or naïve T cells were added at the designated E/T ratio(s)
and incubated for 4h.
To collect media, plates were centrifuged at 250x g, 2-25 C, 4 min.
(Centrifuge 5810 R, Eppendorf,
Germany) to sediment cells. 50111 of supernatant medium from each well was
transferred to a fresh
flat-bottom 96-well microplate well (Corning, USA, cat. no. 3596) and 50 Ill
CytoTox 96 Reagent
was added to each well. Plates were incubated in the dark at room temperature
for 30 min., after
which the reaction was terminated by addition of 50 Ill Stop Solution per
well. Absorbance was read
at 492 nm using Infinite F50 (Tecan, Switzerland) and captured using Magellan
F50 software. Data
analysis and graph generation was performed using Microsoft Excel 2010.
[0855] Flow cytornetry cytotoxicity assay
[0856] HeLa-CD19 (target) and HeLa (control) cells were pre-stained with 51.tM
carboxyfluorescein
succinimidyl ester (CFSE, Life Technologies, USA, cat. no. C1157), mixed
together, and plated on
either fresh plates or on plates populated with isolated primary monocyte.
After target cells adhere to
the plate (6h-overnight), cultures were exposed to y x106 ApoCells cells for
lh. Plates were washed
with RPMI 3-5 times and visually verified that suspended ApoCells cells were
washed off. 10 ng/ml
LPS was introduced to the co-culture and incubated for lh, after which LPS was
removed by 3-5
washing cycles with RPMI. Viable CD19-CAR T cells were then added to the co-
cultures as indicated
by specific E/T ratio(s) and incubated for 4h. After incubation, cells were
harvested by adding trypsin-
EDTA (Biological Industries, Israel, cat. no. 03-052-1B) and incubating for 4
min. at 37 C. To
CA 03151815 2022-02-17
176
WO 2021/053667
PCT/IL2020/051011
terminate the enzymatic activity, two- to four-fold volume of "complete
medium" was added. Cells
were collected, centrifuged at 200x g for 5 min. at room temperature and re-
suspended in 100 Ill
RPMI (Gibco, ThermoFisher Scientific, USA, cat. no. 15140-122). Staining
ensued first against anti-
CD19 (eBioscience, USA, cat. no. 12-0198-42), incubated in dark for 30 min. at
room temperature.
After centrifugation (290x g, 1 min., 2-8 C) and re-suspended in 300 Ill RPMI,
cells were stained
against anti-7AAD (eBioscience, USA, cat. no. 00-6993-50). Analysis was gated
on 7ADD-negative
cells (live cells), where live target cell (HeLa-CD19) and live control cells
(HeLa) was calculated.
Percent survival was calculated by dividing percent live target cells by
percent live control cells. To
correct for variation in starting cell numbers and spontaneous target cell
death, percent survival was
divided by the ratio of percent target cells to percent control cells cultured
without effector cells
(CD19-CAR T cells). Finally, percent cytotoxicity was determined by
subtracting the corrected
survival percentage from 100% 2.
[0857] Initial experiments are performed by incubating Raji cancer cells with
CD19+ CAR T-cells
(+/- monocytes-macrophages) for 48 hours in order to determine optimal ratios
of CD19+ CAR T-
cells to target Raji cancer cells, beginning with 5x104 Raji cells/well in a
96-well plate. An
effector/target (E/T) ratio plate is constructed based on the results.
[0858] Combination immunotherapy experiments are performed by incubating the
Raji cancer cells
with apoptotic cells, or apoptotic supernatants, for 1 hour followed by co-
culturing with CD19+ CAR
T-cells (+/- monocytes-macrophages) for 48 hours.
[0859] In order to simulate in vivo conditions, lx105 THP-1 cells/ml will be
differentiated with 200
nM (123.4 ng/ml) phorbol myristate acetate (PMA) for 72 hrs and will then be
cultured in complete
medium without PMA for an additional 24h. Next, Raji cancer cells will be
plated in a 24-well plate
at 5x105 Raji cells/well on the differentiated THP-1 cells.
[0860] Following initial culturing of the Raji cancer cells, 4x105-8x105
apoptotic cells (ApoCell) will
be added to the culture for 1-3h to induce an immunotolerant environment. The
ratio of cancer cell
to ApoCell will be optimized for each cell type. After washing, the co-culture
will be treated with a
pre-determined number of CD19+ CAR-T cells based on the E/T ratio graph. In
certain experiments,
10 ng/ml LPS will be added to the culture media prior to addition of the CD19+
CAR T-cells. In
other experiments, interferon y (IFN-y) will be added to the culture media
prior to addition of the
CD19+ CAR T-cells. The addition of LPS or IFN-y is expected to exponentially
increase the cytokine
storm level.
[0861] To assay for Raji cancer cell cytotoxicity, lysates are prepared and
viability is determined
after the 48 hour incubation period. Additional experiments will be performed
assaying for Raji cell
cytotoxicity at intervals within the 48h incubation time period.
Alternatively, Promega' s CytoTox 96
CA 03151815 2022-02-17
177
WO 2021/053667
PCT/IL2020/051011
Non-Radioactive Cytotoxicity Assay (Promega, cat #G1780) will be used.
[0862] Similar experiments are run with CD19 expressing HeLa cells and CD19+
CAR T-cells.
[0863] Similar experiments are run with CD123 expressing leukemic cells and
CD123+ CAR T-
cells.
[0864] Cytokine Analysis
[0865] Initial cytokine assays examine the levels of MIP la, IL-4, IL-2, IL-
2R, IL-6, IL8, IL-9, IL-
10, IL-13, IL-15, INF-y, GMCSF, TNF-a, in the culture supernatant.
[0866] Additional cytokine assays examine the level of cytokines IL-10, IL-
113, IL-2, IP-10, IL-4,
IL-5, IL-6, IFNa, IL-9, IL-13, IFN-y, IL-12p70, GM-CSF, TNF-a, MIP- la, MIP-
113, IL-17A, IL-
15/IL-15R, or IL-7, or any combination thereof.
[0867] Cultures were established to mimic an in vivo CAR T-cell therapy
environment. Raji Burkett
Lymphoma cells were cultured in the presence of human monocyte-macrophages,
LPS and CD19+
CAR T-cells without and with the addition of apoptotic cells.
[0868] Raji cells were incubated in the presence of monocytes and LPS,
followed by addition of
Naive T-cells (Raji + Naive T), CD19+ CAR T-cells (Raji + CAR T), CD19+ CAR T-
cells and
apoptotic cells (ApoCell) at a ratio of 1:8 CAR T-cells:ApoCells (Raji + CAR
T+ApoCell 1:8),
CD19+ CAR T-cells and apoptotic cells (ApoCell) at a ratio of 1:32 CAR T-
cells:ApoCells (Raji +
CAR T+ApoCell 1:32), and CD19+ CAR T-cells and apoptotic cells (ApoCell) at a
ratio of 1:64
CAR T-cells:ApoCells (Raji + CAR T+ApoCell 1:64). Concentration measurements
were made
following GM-CSF and TNF-a (TNF-a).
[0869] To assay for cytokine release reduction of IL-6, IL-8, and IL-13, as
well as other cytokines,
supernatants will be collected and examined for selected cytokine using
Luminex MagPix reader and
ELISA assays. Cytokines (mouse or human) may be evaluated by Luminex
technology using MAPIX
system analyzer (Mereck Millipore)) and MILIPLEX analysis software (Merek
Millipore). Mouse
IL-6Ra, MIG (CXCL9) and TGF-(31 were evaluated by Quantikine ELISA (R&D
systems).
[0870] Tissue Analysis
[0871] Bone marrow and liver were evaluated using flow cytometry and
immunohistochemistry.
Upon sacrifice liver and bone marrow were collected for histopathological
analysis. Tissues were
fixed in 4% formalin for 48h at room temperature, and then submitted to the
animal facility at the
Hebrew University for processing. Bones were decalcified prior to processing.
Paraffin sections were
stained for Hematoxylin and Eosin, and CD19.
[0872] IFN-y Effect
[0873] IFN-y effect is evaluated both by STAT1 phosphorylation and biological
products.
[0874] Results:
CA 03151815 2022-02-17
178
WO 2021/053667
PCT/IL2020/051011
[0875] Calibrating cell number for Cytotoxicity assay
[0876] To determine the number of Raji cells to be used in the in vitro model,
sensitivity limits of
the cytotoxicity assay was assessed. 5x104-20x104 Raji cells/well were plated
in a 96-well plate, in
quadruplicate. Lysis was performed on one set of quadruplicate to be compared
with cells that are
still completely viable. Lysis was momentary, adding the lysis solution
immediately prior to
centrifugation to simulate partial cell cytotoxicity. Indeed, all cell
quantities exhibited readings well
above viable cells, with the 5x104 cell number producing the greatest relative
reading (Figure 14;
extrapolation of data). Therefore, subsequent experiments will be using this
cell number as default,
unless otherwise required by experimental deign.
[0877] Verification of CD19+ CAR-T cell activity against Raji Burkett Lymphoma
cells
[0878] To corroborate the CD19+ CAR T-cell activity, monolayers of Raji cancer
cells are exposed
to either 1,000,000 (one million) CD19 + CAR-T cells or to 1,000,000 (one
million) non-transduced
T cells. After 24h incubation, CD19 CAR-T cells reduce Raji cancer cell
proliferation, showing anti-
tumor activity of the CD19+ CAR-T cells.
[0879] Activity of stand-alone CD19+ CAR-T cells against Raji Burkett Lymphoma
cells was
compared to activity post exposure to Apoptotic Cells
[0880] Apoptotic cells (ApoCell) and apoptotic cell supernatants (ApoSup and
ApoMon Sup) are
tested to determine if they interfere with CD19+ CAR-T cell anti-tumor
activity. The Raji Burkett
Lymphoma cells are incubate with Apoptotic Cells for one hour, followed by the
addition of CD19+
CAR-T cells (500,000, five hundred thousands) or CD19+ non-transduced T cells
(500,000, five
hundred thousands) (ratio of 1:2 CD19 + CAR-T cells to Apoptotic Cells). The
tumor cell/Apoptotic
cel1/CD19+ CAR T-cells are then co-cultured for 48h. The control Raji Burkett
Lymphoma cells are
co-cultured with CD19+ CAR-T cells and Hartman solution (the vehicle of
Apoptotic Cells), but
without Apoptotic Cells, for 48h.
[0881] The results are showing that after 48h incubation, CD19+ CAR-T cells
anti-tumor activity
was superior to incubation with non-transduced T cells. Similar incubations
will be performed with
apoptotic cell supernatants. Surprisingly, CD19+ CAR T-cell anti-tumor
activity is comparable with
or without exposure to apoptotic cells or apoptotic cell supernatants.
[0882] No negative effect of apoptotic cells on CAR-modified T cells against
CD19 both in vitro
was seen with comparable E/T ratio results of CAR T in the presence or absence
of apoptotic cells.
[0883] Verification of CD19+ CAR-T cell activity against HeLa Leukemia cells
[0884] HeLa cells are specific CD19 expressing cells, which renders them
susceptible to CAR
CD19+ T-cell activity. In addition, in contrast to Raji cells, which are a non-
adherent cell line, HeLa
cells are adherent.
CA 03151815 2022-02-17
179
WO 2021/053667
PCT/IL2020/051011
[0885] To corroborate the CD19+ CAR T-cell activity, monolayers of HeLa cancer
cells were
exposed to either 1,000,000 (one million) CD19+ CAR-T cells or to 1,000,000
(one million) non-
transduced T cells. After 24h incubation, CD19+ CAR-T cells reduce HeLa cancer
cell proliferation,
showing anti-tumor activity of the CD19+ CAR-T cells (Figure 15 CD19+ + RPMI
and CD19+ +
CAR T-19 cells).
[0886] Activity of stand-alone CD19+ CAR-T cells against CD19 1-1eLa cells was
compared to
activity post exposure to Apoptotic Cells
[0887] Apoptotic cells (ApoCell) were tested to determine if they interfere
with CD19+ CAR-T cell
anti-tumor activity. The HeLa cells were incubated with Apoptotic Cells for
one hour, followed by
the addition of CD19+ CAR-T cells (500,000, five hundred thousand) or CD19+
non-transduced T
cells (Naive T cells; 500,000, five hundred thousand) (ratio of 1:2 CD19+ CAR-
T cells to Apoptotic
Cells). The tumor cell/Apoptotic cel1/CD19+ CAR T-cells were then co-cultured
for 48h. The control
HeLa cells were co-cultured with CD19+ CAR-T cells and RPMI (the vehicle of
Apoptotic Cells),
but without Apoptotic Cells, for 48h. The CD19 CAR-T cell: HeLa cell ratio
(E/T ratio) ranged from
5-20 (Figure 15).
[0888] Figure 15 shows that after 48h incubation, CD19+ CAR-T cells anti-tumor
activity was
superior to incubation with non-transduced T cells (Naive cells) or buffer
alone. Similar incubations
were performed with apoptotic cells. Surprisingly, CD19+ CAR T-cell anti-tumor
activity was
comparable with or without exposure to apoptotic cells. Similar experiments
are performed using
apoptotic cell supernatants. Figure 15 shows the same in vitro cytotoxicity
effect of CAR T-CD19
therapy with or without the addition of ApoCells.
[0889] No negative effect of the apoptotic cells on CAR-modified T cells
against CD19+HeLa cells
was observed at comparable E/T ratios in the presence or absence of apoptotic
cells.
[0890] Thus, the same in vitro cytotoxic effect of the CD19 CAR T-cells was
observed with or
without the addition of early apoptotic cells.
[0891] Effect of Apoptotic Cells on amelioration, reduction or inhibition of
cytokine storms resulting
from CAR-T treatment
[0892] Cytokines IL-8 and IL-13 are measured in the culture media prior to and
following addition
of CD19+ CAR T-cells and are showing a concentration consistent with a
cytokine storm. Addition
of apoptotic cells or apoptotic cell supernatant is showing a reduction of IL-
8 and IL-13
concentrations in the media.
[0893] Analysis using a wider range of cytokines
[0894] To further evaluate the effect on a possible wider range and levels of
cytokines that are not
generated during experimental procedures but do appear in clinical settings
during a human cytokine
CA 03151815 2022-02-17
180
WO 2021/053667
PCT/IL2020/051011
storm, LPS (10 ng/ml) was added to the Raji cell culture conditions outlined
above in the presence
of cancer and CAR-19. The addition of LPS was expected to exponentially
increase the cytokine
storm level. Exposure to apoptotic cells is dramatically reduced the levels of
cytokines. The results
presented in Figure 16 and Figure 17 show that while addition of CD19+ CAR T-
cell greatly
increases cytokine concentration (pg/ml) of GM-CSF and TNF-a in the culture
medium, there is a
significant decrease of both GM-CSF and TNF-a in the presence of apoptotic
cells. The decrease in
the cytokine concentration is dose dependent with respect to apoptotic cell
ratio of CAR T-cells to
apoptotic cells.
[0895] Conclusion:
[0896] Apoptotic cells were able to down regulate cytokine markers of cytokine
storm associated
with CAR T-cell clinical procedures. Significantly, the apoptotic cells did
not show an effect on the
tumor activity of the CAR T-cells. Apoptotic cells decreased pro-inflammatory
cytokines that
originated from innate immunity and inhibit IFN-y effect without harming IFN-y
levels and CAR-T
cytotoxicity.
EXAMPLE 6: Apoptotic Cell Therapy Prevents Cytokine Storms in A Diffuse Cancer
in
vivo Model Administered Car T-Cell Therapy
[0897] Objective: Test the in vivo effect of apoptotic cells or supernatants
derived from apoptotic
cells in a diffuse tumor model, in order to determine CAR T-cell efficacy on
the cancer cells and the
level of cytokine storm marker cytokines.
-- [0898] Materials and Methods
[0899] In vitro studies
[0900] See methods described in Example 5 for in vitro studies.
[0901] Cells and cell culture
[0902] Raji Burkitt lymphoma cells (Sigma-Aldrich cat. # 85011429) were
cultured as per the
-- manufacture's guidelines. CD19+ CAR T-cells, cell cultures, apoptotic
cells, apoptotic cell
supernatants, monocyte isolation, and in vitro measurements are as above for
Examples. Early
apoptotic cells produced were least 50% annexin V-positive and less than 5% PI-
positive cells.
[0903] In vivo studies
[0904] Mice
[0905] 7-8 week old SCID beige mice were purchased from Envigo (formerly known
as Harlan).
Mice were kept in an SPF free animal facility in compliance with institutional
IACUC guidelines.
During the course of the experiments the mice were monitored daily, and
weighted 3 times a week.
Mice showing hind limb paralysis were sacrificed. Upon sacrifice bone marrow
and liver were
collected for FACS analysis and histological processing, and sera were frozen
at -80 C for cytokine
CA 03151815 2022-02-17
181
WO 2021/053667
PCT/IL2020/051011
profiling. In vivo experiments
[0906] SCID beige mice (C.B-17/IcrHsd-Prkdc-SCID-Lyst-bg, Harlan, Israel) were
housed in SPF
conditions at The Authority for Animal Facilities (AAF), The Hebrew University
of Jerusalem (Ein
Kerem Campus, Israel) and following the Association for Assessment and
Accreditation of
Laboratory Animal Care (AAALAC). The studies were approved by The Hebrew
University Ethics
Committee for Animal Experiments, and animal suffering was minimized as
possible.
[0907] (Figure 18A) For the disseminating tumor model, 7-8 week female SCID
beige mice were
injected i.v. with lx105Raji cells suspended in 200 Ill RPMI (Gibcoõ
ThermoFisher Scientific, USA,
cat. no. 15140-122) per mouse (day 1). On day 6, mice of pertinent groups were
inoculated i.v. with
30x106 cells ApoCells in 200 Ill Hartmann' s solution Lactated Ringer's
Injection, Teva Medical,
Israel, cat. no. AWN2324) per mouse. On day 6, mice of relevant groups were
inoculated i.v. with
10x106 viable CD19-CAR T cells or naive T cells in 200111 AIM V per mouse.
Control mice received
equal volume of RPMI for each treatment.
[0908] Mice were examined for clinical indications and weighed twice a week
and were sacrificed
upon development of hind limb paralysis. Pathological samples of bone and
liver were prepared by
the Animal Facility Unit of The Hebrew University of Jerusalem and stained
against human CD20
(Cell Marque, USA, clone L26, cat. no. 120M-84), to detect Raji cells, and
against human CD3 (Cell
Signaling Technology, USA, cat. no. 85061), to detect human T cells.
[0909] In certain experiments, LPS will be administered to the animal subject
prior to addition of the
CD19+ CAR T-cells. In other experiments, interferon-y (IFN-y) will be
administered prior to addition
of the CD19+ CAR T-cells. The addition of LPS or IFN-y is expected to
exponentially increase the
cytokine storm level.
[0910] Cytokine assays examine the level of cytokines including but not
limited to IL-10, IL-113, IL-
2, IP-10, IL-4, IL-5, IL-6, IFNa, IL-9, IL-13, IFN-y, IL-12p70, GM-CSF, TNF-a,
MIP- la, MIP-113,
IL-17A, IL-15/IL-15R, or IL-7, or any combination thereof. Cytokines (mouse or
human) are
evaluated by Luminex technology using MAPIX system analyzer (Mereck
Millipore)) and
MILIPLEX analysis software (Merek Millipore). Mouse IL-6Ra, MIG (CXCL9) and
TGF-(31 are
evaluated by Quantikine ELISA (R&D systems).
[0911] Tissue Analysis
[0912] Bone marrow and liver are evaluated using flow cytometry and
immunohistochemistry. Upon
sacrifice liver and bone marrow were collected for histopathological analysis.
Tissues were fixed in
4% formalin for 48h at room temperature, and then submitted to the animal
facility at the Hebrew
University for processing. Bones were decalcified prior to processing.
Paraffin sections were stained
for Hematoxylin and Eosin, and CD19.
CA 03151815 2022-02-17
182
WO 2021/053667
PCT/IL2020/051011
[0913] IFN-y Effect
[0914] IFN-y effect is evaluated both by STAT1 phosphorylation and biological
products.
[0915] Results
[0916] CAR T-cell therapy induces cytokine release syndrome
[0917] Three groups of tumor-free mice as well as mice with tumors are
administered (i.p. or directly
into the tumor) increasing doses of CD19+ CAR T-cells (3x106, 10 x106 or
30x106). At the highest
dose, tumor-free mice and mice with tumors demonstrate subdued behavior,
piloerection, and
reduced mobility within 24 h, accompanied by rapid weight loss followed by
death within 48 hrs.
Human interferon-gamma, and mouse IL-6 , IL-8, and IL-13 are detectable in
blood samples from
the mice given the highest dose of CD19+ CAR T-cells. Animals that receive a
high dose of CD19+
CAR T-cells directed to a different tumor antigen do not exhibit weight loss
or behavioral alterations.
[0918] Administration of apoptotic cells inhibits or reduces the incidence of
cytokine release
syndrome induced by CAR T-cell therapy
[0919] One group of mice given the highest dose of CD19+ CAR T-cells is
concomitantly
administered 2.10x108/kg apoptotic cells, which was previously demonstrated to
be a safe and
effective dose. Mice receiving human CD19+ CAR T + apoptotic cells have
significantly lowered
levels of at least one mouse pro-inflammatory cytokines, lower weight loss,
and reduced mortality.
[0920] Administration of apoptotic cells in combination with CAR T-cell
administration did not
affect CAR T-cell anti-tumor activity
[0921] Figure 18B shows that the expected death of SCID mice injected with
CD19+ Raji cells
without administration of CD19 CAR T-cells was 18-21 days. Forty percent (40%)
of the mice who
received CD19 CAR T-cells survived to at least day 30 (Figure 18 dash-dot-dash
line and dash-
double dot-dash line). The percentage of survivors was independent of the
addition of apoptotic cells
(Figure 18). The surviving mice were sacrifice on day 30.
[0922] Conclusion: There was comparable survival and no negative effect of
apoptotic cells on CAR-
modified T cells against CD19 in vivo.
[0923] Significant down regulation (p<0.01) of pro-inflammatory cytokines
including, IL-6, IP-10,
TNF-a, MIP-la, MIP-10 was documented. IFN-y was not downregulated but its
effect on
macrophages and dendritic cells was inhibited both at the level of
phosphorylated STAT1 and IFN-
y-induced expression of CXCL10 and CXCL9.
[0924] Conclusion:
[0925] Apoptotic cells decrease pro-inflammatory cytokines that originate from
innate immunity and
inhibit IFN-y effect without harming IFN-y levels and CAR-T cytotoxicity.
CA 03151815 2022-02-17
183
WO 2021/053667
PCT/IL2020/051011
EXAMPLE 7: Apoptotic cell therapy prevents cytokine storms in a solid tumor
cancer in
vivo model administered CAR T-cell therapy
[0926] Objective: Test the in vivo effect of apoptotic cells or supernatants
derived from apoptotic
cells in a solid tumor model, in order to determine CAR T-cell efficacy on the
cancer cells and the
.. level of cytokine storm marker cytokines.
[0927] Materials and Methods
[0928] In vitro studies
[0929] Cells and cell culture
[0930] CD19+ CAR T-cells, Second generation CAR-T-CD19 cells containing TMCD28
were used,
.. cell cultures, apoptotic cells, apoptotic cell supernatants, monocyte
isolation, and in vitro
measurements were as above for Examples 2 & 4 & 6. Early apoptotic cells
produced were least 50%
annexin V-positive and less than 5% PI-positive cells, as described in detail
in Example 1.
[0931] In vivo studies
[0932] Mice
[0933] 7-8 week old SCID-beige mice and NSGS mice were purchased from Harlan
(Israel) and
kept in the SPF animal facility in Sharett Institute.
[0934] SCID beige mice or NSGS mice were inoculated with CD19 expressing Hela
cells, that can
adhere to the peritoneum, in order to form solid intra-peritoneal tumors. Mice
were sorted into groups
prior to T-cell administration.
[0935] Six days post i.v. inoculation, mice were administered 10x106CD19+ CAR
T-cells with and
without apoptotic cell (ApoCell) preconditioning on day 5. Mice receiving pre-
conditioning were
administered 5x106 or 30 x 106 ApoCells. Tumors were surveyed weekly and
circulating cytokine
levels were monitored weekly and determined by the Luminex system. 25 mouse
cytokines and 32
human cytokines were evaluated using the Luminex technology. Upon termination
of the experiment,
mice were culled and organs (bone marrow, liver and spleen) were examined (by
FACS and
immunohistochemistry) for the presence/size of tumors.
[0936] Cytokine assays examined the level of cytokines including but not
limited to GM-CSF, IFNy,
IL-113, IL-10, IL-12p70, IL-13, IL-15, IL-17A, IL-2, IL-4, IL-5, IL-6, IL-7,
IL-9, MIP- la, TNFa,
MIP-113, IFNa, and IP-10. Cytokines (mouse or human) were evaluated by Luminex
technology
using MAPIX system analyzer (Mereck Millipore)) and MILIPLEX analysis software
(Merek
Millipore). Mouse IL-6Ra, MIG (CXCL9) and TGF-(31 were evaluated by Quantikine
ELISA (R&D
systems).
[0937] Tissue Analysis
[0938] Bone marrow and liver are evaluated using flow cytometry and
immunohistochemistry.
CA 03151815 2022-02-17
184
WO 2021/053667
PCT/IL2020/051011
[0939] IFN-y Effect
[0940] IFN-y effect was evaluated both by STAT1 phosphorylation and biological
products.
[0941] Results
[0942] CAR T-cell therapy induces cytokine release syndrome
[0943] Three groups of tumor-free mice as well as mice with tumors were
administered (i.p. or
directly into the tumor) increasing doses of CD19+ CAR T-cells (3x106, 10 x106
or 30x106). At the
highest dose, tumor-free mice and mice with tumors demonstrate subdued
behavior, piloerection, and
reduced mobility within 24 h, accompanied by rapid weight loss followed by
death within 48 hrs.
[0944] Figures 19A-19C graphically show the increased levels of IL-6, IP-10
and surprisingly even
.. TNF-a cytokine release from tumors even before the presence of CAR T-cells.
Figures 19A-19C
show that unexpectedly IL-6, IP-10, and TNF-a were increased by the presence
of cancer cells even
without CAR T-cell therapy. In the presence of CAR T-Cell therapy (Hela-CAR T-
cell CD-19) the
release of cytokines was significantly augmented. These results show that the
tumor itself releases
pro-inflammatory cytokines.
.. [0945] In order to evaluate the benefit of the addition of early apoptotic
cells, cytokines GM-CSF,
IFNy, IL-113, IL-10, IL-12p70, IL-13, IL-15, IL-17A, IL-2, IL-4, IL-5, IL-6,
IL-7, IL-9, MIP- 1 a,
TNFa, MIP-113, IFNa, and IP-10 were measured in three experiments, wherein the
results showed
that macrophage associated cytokines were down-regulated in the presence of
ApoCell
administration, while T-cell associated cytokine levels were not significantly
changed (Table 10).
[0946] Table 10: Cytokine levels from an intra-peritoneum in vivo model that
contained CD19
expressing Hela cells solid tumor, +1- CAR T-cell CD19 therapy, and +1-
ApoCell
Pg,im1 Before Tumor Car or After tumor
+ CAR After tumor, CAR,
Apo +with
ApoCell
GM-CSF 442
88+10 12 4
FNV4 15-18 5 21
14 6 16-1-8
ft-10 764-1 22:74-44 3s 22
1_-.12p7Ct 5- 1 182 -22 12+11
64-2 8 1 844
L-15:4 2
L-2 426 2
26 2 7947
14-2 16. 4 18 .6
8:704-56- 74:1-12
M1P-1C1 8+5 99+13 13 8
TN.Fci 6_1_2 7.60413 17 -15
MtP-i 7 1 1421 -21 21-1-10
4=12 6:84:76 71 14
P40 8 4 182 -33 21 16
[0947]
[0948] Table 10 shows cytokine measurement twenty-four (24) hours after CAR T-
Cell
CA 03151815 2022-02-17
185
WO 2021/053667
PCT/IL2020/051011
administration +/- ApoCells. Resultant cytotoxicity from CAR T-cell therapy
elevated cytokines
including GM-CSF, IL-10, IL-12p70, IL-6, MIP-1 a, TNFa, MIP-10, and IP-10, the
levels of which
were significantly down regulated (p<0.05-0.0001) in the presence of ApoCells.
These cytokines are
mainly associated with macrophages. In contrast, the levels of cytokines
associated with T-cells such
as IL-2, IL-4, IL-13, and IL 15 were not changed significantly.
[0949] The results presented in Figures 19A-C and Table 10, illustrate that
the CRS in the context
of cancer and CAR has several ingredients: a tumor that can secrete cytokines;
an innate immunity
that respond to tumor and to CAR and to other factors; and that CAR T-cells
that secrete cytokines
causes death that influence innate immunity. ApoCells are interacting with
innate immunity, mainly
macrophages, monocytes and dendritic cells, to down regulate the response of
these macrophages,
monocytes and dendritic cells without interacting with T cells or CAR T cells.
[0950] Animals that received a high dose of CD19+ CAR T-cells directed to a
different tumor
antigen do not exhibit weight loss or behavioral alterations.
[0951] Administration of apoptotic cells inhibits or reduces the incidence of
cytokine release
syndrome induced by CAR T-cell therapy
[0952] One group of mice given the highest dose of CD19+ CAR T-cells was
concomitantly
administered 2.10x108/kg apoptotic cells, which was previously demonstrated to
be a safe and
effective dose. Apoptotic cells had no negative effect in vitro or in vivo on
CAR-modified T cells
with specificity against CD19. There were comparable E/T ratios for CAR T-
cells in the
presence/absence of apoptotic cells in vitro, and comparable survival curves
in vivo (Data not shown).
[0953] Mice receiving human CD19+ CAR T + apoptotic cells had significantly
lowered levels of
at least one mouse pro-inflammatory cytokines, lower weight loss, and reduced
mortality.
[0954] No negative effect of apoptotic cells on CAR-modified T cells against
CD19 in vivo was seen
with comparable E/T ratio results of CAR T in the presence or absence of
apoptotic cells, and a
comparable survival curve in vivo.
[0955] Significant down regulation (p<0.01) of pro-inflammatory cytokines
including, IL-6, IP-10,
TNF-a, MIP-la, MIP-10 was documented (Data not shown). IFN-y was not
downregulated but its
effect on macrophages and dendritic cells was inhibited both at the level of
phosphorylated STAT1
and IFN-y-induced expression of CXCL10 and CXCL9 (Data not shown.
[0956] Conclusion:
[0957] . CRS evolves from several factors, including tumor biology,
interaction with
monocytes/macrophages/dendritic cells, and as a response to the CAR T cell
effect and expansion.
Apoptotic cells decrease pro-inflammatory cytokines that originate from innate
immunity and inhibit
the IFN-g effect on monocyte/macrophages/ dendritic cells without harming IFN-
y levels or CAR-T
CA 03151815 2022-02-17
186
WO 2021/053667
PCT/IL2020/051011
cytotoxicity. Thus, apoptotic cells decreased pro-inflammatory cytokines that
originate from innate
immunity and inhibit IFN-y effect without harming IFN-y levels and CAR-T
cytotoxicity. These
results support the safe use of ApoCells for the prevention of CRS in clinical
studies using CAR-T
cell therapy.
EXAMPLE 8: Apoptotic cells downregulate Cytokine Release Syndrome (CRS) and
increase CAR-T-cell efficacy
[0958] Objective: To test the effects of early apoptotic cells on
cytokines and CAR T cell
cytotoxicity over an extended time period. To demonstrate the in vivo efficacy
of CD19-CAR T-cells.
To demonstrate the synergistic effect of early apoptotic cells and CD19-CAR T-
cells.
[0959] Methods: CD19-expressing HeLa cells (ProMab) were used alone or after
co-incubation
with human macrophages for in vitro and intraperitoneal experiments in mice.
Raji was used in vivo
for leukemia induction. LPS and IFN-g were used to trigger additional cytokine
release. Second
generation, CD28-bearing, CD19-specific CAR-modified cells were used (either
ProMab or
produced using a retronectin manufacturing protocol or a polybrene
manufacturing protocol) for anti-
tumor effect against CD19-bearing cells. Cytotoxicity assay was examined in
vivo (7-AAD flow
cytometry) and in vitro (survival curves; tumor load in bone marrow and liver,
flow cytometry and
immunohistochemistry). CRS occurred spontaneously or in response to LPS and
IFN-g. Mouse IL-
10, IL-113, IL-2, IP-10, IL-4, IL-5, IL-6, IFN-a, IL-9, IL-13, IFN-g, IL-
12p70, GM-CSF, TNF-a, MIP-
la, MIP-113, IL-17A, IL-15/IL-15R, IL-7, and 32 human cytokines were evaluated
(Luminex
technology, MAPIX system; MILLIPLEX Analyst, Merck Millipore). Mouse IL-6Ra,
MIG
(CXCL9), and TGF-131 were evaluated (Quantikine ELISA, R&D systems). IFN-y
effect was
evaluated (STAT1 phosphorylation, biological products). Human macrophages and
dendritic cells
were generated from monocytes. Early apoptotic cells were produced generally
as presented in
Example 1 above; >40% of cells were Annexin V-positive; <15% were PI-positive.
[0960] Mice: SCID-Bg mice (female, 7-8 wk) were injected with 2 consecutive
doses of 0.25x106
HeLa-CD19 cells, intra-peritoneally (i.p) on days 1 and 2 of experiment. On
day 9 mice received i.p.
dose of 10x106 4000-rad (cGy) irradiated ApoCell (using a cell processing
system) and an i.p. dose
of a population of 10x106 CAR-T cells comprising either 0.5 x106 CAR-T
positive cells or 2.2x106
CAR-T positive cells, on the following day. As control, mice received 10x106
activated mononuclear
cells or Mock-T cells. Mice were kept in an SPF animal facility in compliance
with institutional
IACUC guidelines. Mice were weighted twice a week and monitored daily for
clinical signs and
peritonitis. End point was defined as severe peritonitis manifested as
enlarged and tense abdomen,
lethargy, reduced mobility or increased respiratory effort. Survival analysis
was performed according
to the Kaplan-Meier method.
CA 03151815 2022-02-17
187
WO 2021/053667
PCT/IL2020/051011
[0961] Results: Significant downregulation (p<0.01) of pro-inflammatory
cytokines, including
IL-6, IP-10, TNF-a, MIP- la, MIP-113, was documented (Data not shown). IFN-g
was not
downregulated, but its effect on macrophages and dendritic cells was inhibited
at the level of
phosphorylated STAT1 (Data not shown). IFN-' induced expression of CXCL10 and
CXCL9 in
macrophages was reduced (Data not shown).
[0962] In the experiment wherein 0.5 x 106 CAR-T positive cells were used, 2
mice per group
were sacrificed on day 17 and 21. HeLa-CD19 treated mice showed peritonitis
manifested as blood
accumulation in the peritoneum, enlarged spleen and tumor loci (Data not
shown). Mice treated with
control MNCs had a little bit less blood in peritoneum and less tumor loci.
Mice treated with CAR-
T or with CAR-T and ApoCell had no signs of peritonitis. This observation
correlated to the survival
curve presented in Figure 20A.
[0963] In the experiment wherein 2.2 x 106 CAR-T positive cells were
used, the same pattern of
effect as seen with four-fold fewer CAR T-cells was observed (Figure 20B). CAR-
T treatment
prolonged survival of mice with peritoneal HeLa-CD19 (p=0.0002). The effect
was more significant
in this experiment, probably due to the higher number of infused CAR T cells
(2.2 versus 0.5 x106
CAR-T positive cells). Even with the more significant and prolonged effect,
the early apoptotic cells
had a synergistic effect and prolonged survival (p= 0.0032). E/T ratios for
CAR T were comparable
in the presence/absence of apoptotic cells in vitro. Surprisingly, CAR T cell
therapy given in the
presence of apoptotic cells ameliorated survival of mice with a significant
and reproducible addition
of at least 12 days (p<0.0032, Figures 20A and 20B) in comparison to CAR
therapy alone.
[0964] Conclusion: CAR-T cell treatment prolonged survival of mice with
peritoneal HeLa-
CD19 cells. Administration of irradiated early apoptotic cells one day before
CAR-T had a synergistic
effect and prolonged mice survival for more than 10 days (p<0.044, log-rank
test), compared to CAR-
T cell treatment alone.
[0965] The irradiated apoptotic cell infusion had a dramatic synergistic
effect to CD19-specific
CAR T cells in treating CD19-bearing Hela cells in SCID mice. In this example,
similar results were
observed to the results presented in Examples 5 and 6. By using irradiated
apoptotic cells in this
Example, compared with Examples 5 and 6, the possibility of a "graft versus
leukemia effect" has
been removed. Thus, this surprising synergistic effect appears to be mediated
via the irradiated
apoptotic cells provided.
[0966] CRS evolves from several factors, including tumor biology, interaction
with
monocytes/macrophages/dendritic cells, and as a response to the CAR T cell
effect and expansion.
Apoptotic cells decrease pro-inflammatory cytokines originating from innate
immunity, and inhibit
the IFN- y effect on monocytes/macrophages/dendritic cells without harming IFN-
y levels or CAR-
CA 03151815 2022-02-17
188
WO 2021/053667
PCT/IL2020/051011
T cytotoxicity, and with significant increase in CAR-T cell efficacy.
Unexpectedly, treatment with
irradiated apoptotic cells complements CAR-T cell therapy, effectively
extending the anti-cancer
effect of the CAR-T cell therapy.
EXAMPLE 9: Effect of Apoptotic Cells Treatment on a non-Solid Tumor Model
[0967] Objective: To test the effect of apoptotic cells on a non-solid tumor
model where the cancer
is widely spread and not localized or confined, in order to determine
apoptotic cells efficacy on the
survival in cancer.
[0968] Methods:
[0969] Raji cells
[0970] Raji cells were purchased from ECACC (Cat. #: 85011429), and routinely
cultured in
complete medium (RPMI-1640 supplemented with 10% H.I. FBS, 1% Glutamax, 1%
Penicillin /
5 6
Streptomycin), and maintained at a concentration of 3x10 ¨ 3x10 cells/ml.
[0971] Apoptotic cells were prepared as described in Example 1. Early
apoptotic cells produced were
at least 50% annexin V-positive and less than 5% PI-positive cells.
[0972] Non-solid (diffuse) tumor model
5
[0973] SCID mice received a single IV injection of 10 Raji cells on day 1 of
the experiment. A
control group of SCID mice received a single IV injection of saline solution.
(3 cohorts were tested;
leukemia was induced in 2 cohorts using Raji cells, and 1 cohort was
maintained as a control.)
[0974] Solid tumor model
5
[0975] SCID mice will receive a single IP injection of 10 Raji cells on day 1
of the experiment,
wherein control groups will receive a single IP injection of saline solution.
[0976] Apoptotic cell treatment
[0977] In a preliminary study, mice received an infusion of early apoptotic
cells 6 days after the
infusion of Raji cells. In later studies, the mice from one of the leukemic
cohorts above received 3
infusions of early apoptotic cells (30 x 106 cells) starting 6 days after the
infusion of Raji cells.
[0978] Results
[0979] SCID mice have no T-cells and therefore no ability to recover from
leukemia without therapy.
[0980] Surprising, in the preliminary study and as shown in Figure 21, the
apoptotic cell infusion
(APO) 6 days after the infusion of Raji, significantly prolonged tumor free
death in SCID injected
with CD19+ Raji, compared with mice that did not receive an apoptotic cell
infusion (NO APO).
[0981] In the leukemic (NO APO) cohort, 70% of mice receiving Raji cells
survived through their
lifespan, compared to 94% of mice receiving both Raji cells and apoptotic
cells (n=51 animals in
CA 03151815 2022-02-17
189
WO 2021/053667
PCT/IL2020/051011
total, p<0.001). As expected, 100% of control mice survived through their
expected lifespan (Figure
22A). In the leukemic cohort, 9% of mice receiving Raji cells and no apoptotic
cells survived through
up to 12% above the expected lifespan, compared to 47% of mice receiving both
Raji cells and
apoptotic cells (Figure 22B). No mice receiving Raji cells and no apoptotic
cells survived through
greater than 30% ofthe expected lifespan, compared to 41% of mice receiving
both Raji cells and
apoptotic cells (Figure 22C). No mice receiving Raji cells and no apoptotic
cells attained complete
remission, compared to 10% of mice receiving both Raji cells and apoptotic
cells (Figure 22D).
[0982] Conclusion:
[0983] Administration of an apoptotic cell infusion maintained and increased
the lifespan of leukemic
mice, wherein in certain instances mice administered early apoptotic cells
attained complete remission
(Figures 2 and 3A-3D).
EXAMPLE 10: Effect of Combined Apoptotic Cell and anti-CD20 mAb Treatment on a
Diffuse Tumor Model
[0984] Objective: To test the effect of administering a combination of early
apoptotic cells and anti-
CD20 mAb on a diffuse (non-solid) tumor model, wherein the cancer is widely
spread and not
localized or confined, in order to determine the efficacy on survival of this
combination therapy.
[0985] Raji cells, apoptotic cells, non-solid (diffuse) tumor model, solid
tumor model, and apoptotic
cell treatment were as described in Examples 1 and 9 above.
[0986] anti-CD20 mAb
[0987] Commercially available anti-CD20 mAb was acquired from Roche.
[0988] anti-CD20 mAb treatment
[0989] Mice received an IV infusion of 5 mg of anti-CD20 mAb.
[0990] Combined Apoptotic cell and anti-CD20 mAb treatment
[0991] Starting at day 6 following Raji cell administration, mice received
three IV infusions of
6
30x10 apoptotic cells each. In addition, mice received an IV infusion of 5 mg
of anti-CD20 mAb.
[0992] Results
[0993] 100% of mice receiving Raji cells, Raji cells + anti-CD20 mAb, and Raji
cells + antiCD20 +
apoptotic cells survived through the expected lifespan of leukemic mice,
compared to 86% of mice
receiving both Raji cells and apoptotic cells (n=28 animals in total,
p<0.0002) (Figure 23A). No mice
receiving Raji cells survived longer than 24% above the expected lifespan,
compared to 29% of mice
receiving both Raji cells + apoptotic cells, and 100% of mice receiving either
Raji cells + anti-CD20
mAb or Raji cells + antiCD20 + apoptotic cells (Figure 23B). No mice receiving
Raji cells survived
longer than 59% above the expected lifespan, compared to 29% of mice receiving
both Raji cells +
CA 03151815 2022-02-17
190
WO 2021/053667
PCT/IL2020/051011
apoptotic cells, 57% of mice receiving Raji cells + anti-CD20 mAb, and 100% of
mice receiving Raji
cells + antiCD20 + apoptotic cells (Figure 23C). No mice receiving Raji cells
survived longer than
76% above the expected lifespan, compared to 29% of mice receiving both Raji
cells + apoptotic
cells, 14% of mice receiving Raji cells + anti-CD20 mAb, and 85% of mice
receiving Raji cells +
antiCD20 + apoptotic cells (Figure 23D). No mice receiving either Raji cells
or Raji cells + anti-
CD20 mAb survived longer than 100% above the expected lifespan of a mouse,
compared to 29% of
mice receiving either Raji cells + apoptotic cells or Raji cells + antiCD20 +
apoptotic cells (Figure
23E).
[0994] Conclusion:
[0995] Apoptotic cell infusions increased the lifespan of leukemic mice,
increased the number of
mice attaining complete remission, and enhanced anti-CD20 mAb therapeutic
effect (Figures 23A-
23E).
EXAMPLE 11: Effect of ApoCell (Early Apoptotic Cells) on Leukemia/Lymphoma
[0996] Objective: The work presented here had three main goals: (1)
Evaluating the effect of
ApoCell in a leukemia-lymphoma mouse model in terms of disease onset,
progression, and ensuing
death; (2) Assessing the distribution of tumor cells in a mouse model of
leukemia-lymphoma after
treatment with ApoCell; and (3) Assessing a possible synergistic effect of
ApoCell and Rituximab
(RtX) in the treatment of leukemia-lymphoma in SCID-Bg mice. As part of the
work to meet these
.. objectives, measurement of the survival of leukemic mice following ApoCell
administration was
measured. As well, the distribution of tumor cells was measured after
treatment with ApoCell.
[0997] Methods:
[0998] Mice. Female SCID-Bg mice, 7 weeks-old (ENVIGO, Jerusalem, Israel),
were injected
intravenously with 0.1x106 Raji cells per mouse. Mice received 3 doses of
30x106 ApoCell
intravenously on days 5, 8, and 11 of the experiment. For combinational
therapy, mice received one
dose (day 8) of RtX (2 or 5 mg/kg; Mabthera, Roche, Basel, Switzerland) 1.5h
after ApoCell
administration.
[0999] Mice were followed daily and weighed twice a week. The endpoint was
defined as death,
or sacrifice due to the development of either of the following symptoms:
paraplegia (lower body
.. paralysis), loss of 20% from mouse start weight, lethargy, reduced
mobility, or increased respiratory
effort.
[1000] Survival analysis was performed according to the Kaplan-Meier method.
Mice were kept
in a specific-pathogen-free (SPF) animal facility in compliance with
institutional Animal Care and
Use Committee (IACUC) guidelines.
CA 03151815 2022-02-17
191
WO 2021/053667
PCT/IL2020/051011
[1001] Raji cell line. This human Burkitt's lymphoma cell line was purchased
from the European
Collection of Authenticated Cell Cultures (ECACC, Cat. #: 85011429), and
routinely cultured in
complete medium (RPMI-1640 supplemented with 10% heat inactivated FBS, 1%
glutamax, 1%
penicillin / streptomycin).
[1002] ApoCell. Essentially, as described in Example 1. Briefly, an
enriched mononuclear cell
fraction was collected via leukapheresis from healthy, eligible donors.
Following apheresis
completion, cells were washed and resuspended with freezing media composed of
PlasmaLyte A pH
7.4, 5% human serum albumin, 10% dimethyl sulfoxide (DMSO), 5% anticoagulant
citrate dextrose
solution formula A (ACD-A) and 0.5thml heparin. Cells were then gradually
frozen and transferred
to liquid nitrogen for long-term storage.
[1003] For preparation of ApoCell, cryopreserved cells were thawed,
washed and resuspended
with apoptosis induction media, composed of RPMI 1640 supplemented with 2 mM L-
glutamine and
10 mM hepes, 10% autologous plasma, 5% ACD-A, 0.5thml heparin sodium and
50i.tg/m1
methylprednisolone. Cells were then incubated for 6 hours at 37 C in 5% CO2.
At the end of
incubation, cells were collected, washed and resuspended in Hartmann's
solution using a cell
processing system. ApoCell was centrifuged at 290g, for 10 min at 2-8 C, and
resuspended in
Hartmann's solution for injection. Apoptosis and viability of ApoCell were
determined using
Annexin V and propidium iodide (PI, Medical & Biological Laboratories, Nagoya,
Japan) using FCS
express software.
[1004] Flow cytomehy. Mouse spleen, liver, and bone marrow were collected from
sacrificed
mice (following deterioration of clinical signs, as defined above) and
analyzed by flow-cytometry
(FACSCalibur, BD, Franklin Lakes, NJ, USA) for the presence of the Raji tumor
(anti-CD20).
[1005] Results:
[1006] Part A: ApoCell delays disease onset and ensuing death in leukemic mice
[1007] Figure 24 is a Kaplan-Meier survival plot presenting 3 individual
experiments (RPMI
group, n = 15; Raji group, n = 23; Raji + ApoCell group, n = 24). In each
experiment, female SCID-
Bg mice (7-8 weeks of age) were injected intravenously with 0.1x106 Raji cells
and a control group
was injected with RPMI. Subsequently, mice were administered with three doses
of 30x106 ApoCell
by intravenous administration (IV), on days 5, 8, and 11. Mice were followed
daily and weighed
twice a week. Endpoint was defined as death, or sacrifice due to the
development of either of the
following symptoms: paraplegia (lower body paralysis), loss of 20% from mouse
start weight,
lethargy, reduced mobility, or increased respiratory effort. Experimental
details are given in Table
11. Significant beneficial effect by ApoCell was seen (p=0.002, Log-rank
(Mantel-Cox) test).
[1008] Table 11: Experimental details of Figure A plot
CA 03151815 2022-02-17
192
WO 2021/053667
PCT/IL2020/051011
Number Survival details
Group of Mean day of Range Notes
mice sacrifice (days)
RPMI 15
DFS* is shown for all
mice
Raji 23 22 21 ¨ 25
2 mice DFS* on day 53
Raji +
24 28 19 ¨ >53
/ 60 (termination of
ApoCell
experiment)
* DFS = disease free survival
[1009] The data for the individual studies is presented in Figures 25A-
25C.
[1010] As depicted above, mice treated with 3 doses of ApoCell after the
administration of Raji
cells had a slower disease progression, and died significantly later
(p=0.0020) than untreated mice.
[1011] Leukemic mice treated with ApoCell had a significant delay in the
onset of symptoms,
demonstrated a slower disease progression, and died later than the control
untreated mice.
Interestingly, about 10 percent of the mice administered with Raji cells and
ApoCell did not develop
any of the expected symptoms characteristic in this leukemia / lymphoma model,
and remained
healthy until termination of the experiments (day 53 or 60).
[1012] ApoCell reduce tumor load in leukemic mice
[1013] Upon sacrifice, following the deterioration of clinical signs as
described above, organs of
interest were collected for analysis, namely the liver, spleen, and bone-
marrow. Cells of these target
organs were analyzed by flow cytometry for the presence of human tumor cells
(Raji cells are positive
for CD20).
[1014] The data below (Table 12) describes the average percent cell
population in target organs
of the sacrificed mice from the 3 experiments described above; values of
individual mice in each
experiment can be found in Tables 13-18, which follow.
[1015] Table 12: Average percent of tumor population in Spleen, Bone Marrow
and Liver
(Flow cytometry)
# of % CD20+ cells (average
Tissue Treatment
mice SD)
RPMI 5 0
Raji 10 0
Spleen
Raji +
8 1.1 3
ApoCell
RPMI 5 0
Bone- Raji 10 16.5 8 . 8
Marrow Raji + 8 6.8 9.2 (P=0.02, t-
ApoCell test)
CA 03151815 2022-02-17
193
WO 2021/053667
PCT/IL2020/051011
RPMI 5 0
Raji 10 11.2 12
Liver
Raji + 8 4.5 7.5 (P=0.06, t-
ApoCell test)
[1016] In vivo experiment 011
[1017] FACS analysis of CD20+ cells in bone marrow, spleen, and liver:
[1018] One mouse receiving three doses of ApoCell was healthy when
sacrificed on day 60.
[1019] Liver, spleen, and bone marrow cells were collected and analyzed by
flow cytometry
(FACSCalibur, BD) for the presence of human CD2O-FITC (Biolegend, Cat. #
302206); mIgGl-
FITC (Biolegend, Cat. # 400110).
[1020] In vivo experiment 019
[1021] FACS analysis of tumor cells in bone marrow, spleen, and liver:
[1022] Mouse spleen, liver, and bone marrow cells were collected from mice
who were sacrificed
following clinical deterioration, as defined in the methods) and analyzed by
flow cytometry
(FACSCalibur, BD) for the presence of human CD20 (FITC).
[1023] The results of analysis of Spleen, Bone marrow, and Liver are
presented in Tables 13-15
below.
[1024] Table 13: Spleen
Spleen
Day of
Mouse Treatment CD20+
sacrifice
Al 0
A2 RPMI 0
A3 0
B2 22 0
B3 22 0
B5 22 0
B6 22 0
B2 Rap
(020) 25 0
B3
0
(020)
Cl 28 0
C2 53 (healthy) 8.7
Raji +
C4 22 0
ApoCell
C6 22 0
C7 28 0
[1025] Table 14: Bone Marrow
Bone marrow
CA 03151815 2022-02-17
194
WO 2021/053667
PCT/IL2020/051011
Day of
Mouse Treatment . . CD20+
sacrifice
Al 0
A2 RPMI 0
A3 0
B2 22 18.7
B3 22 11.3
B5 22 10.5
B6 22 14.5
Rap B2 Ra
25 21
(020)
B3
25 0
(020)
Cl 28 1
53
C2 0.5
Rap + (healthy)
C4 ApoCell 22 0.6
C6 22 17.5
C7 28 1.4
[1026] Table 15: Liver
Liver
Day of
Mouse Treatment CD20+
sacrifice
Al 0
A2 RPMI 0
A3 0
B2 22 4.4
B3 22 6.5
B5 22 1.7
Rap Ra 22 5.4
B2 (020) 25 26.4
B3 (020) 25 5.9
Cl 28 9.8
. 53 0.5
C2 Raji +
A (healthy)
po
C4 22 3
Cell
C6 22 5
C7 28 22.7
[1027] In vivo experiment 023
[1028] Expression (%) of CD20 tumor cells in the spleen, bone marrow, and
liver, as determine
by flow cytometry. Mouse spleen, liver, and bone marrow were collected from
sacrificed mice
(following clinical deterioration, as defined in methods) and analyzed by flow
cytometry
(FACSCalibur, BD) for the presence of human CD20 (FITC). The results for
Spleen, bone marrow,
CA 03151815 2022-02-17
195
WO 2021/053667
PCT/IL2020/051011
and liver for the individual mice are presented in Tables 16-18 below.
[1029] Table 16: Spleen
Spleen
Treatment Day of
Mouse CD20+
sacrifice
B1 22 0
B2 25 0
Raji
B3 22 0
B4 22 0.2
Cl 22 0.2
Raji + C4 22 0.3
ApoCell C5 41 0.1
C6 47 0
El 32 0
Raji + RtX
E2 32 0.7
2mg/kg
E6 32 0
Fl 40 0
Raji + RtX F2 43 0.1
2mg/kg F4 35 0
+ F5 32 0
ApoCell F6 35 0
F7 57 0.4
G1 34 0
G3 34 0.1
Raji + RtX
G4 43 0
5mg/kg
G5 40 0
G7 40 0.1
H1 40 0.4
Raji + RtX H3 36
5mg/kg H4 36
+ H5 40 0
ApoCell H6 40 0
H7 53 0
[1030] Table 17: Bone Marrow
Bone marrow
Treatment Day of
Mouse CD20+
sacrifice
B1 22 18
B2 25 22.5
Raji
B3 22 33.6
B4 22 14.9
Cl 22 24.3
Raji + C4 22 1.8
ApoCell C5 41 7.8
C6 47 0
CA 03151815 2022-02-17
196
WO 2021/053667
PCT/IL2020/051011
El 32 0.8
Raji + RtX
E2 32 3.1
2mg/kg
E6 32 3
Fl 40 4.1
F2 43 0.4
Raji + RtX
F4 35 0.2
2mg/kg +
F5 32 0.8
ApoCell
F6 35 0.2
F7 57 0.4
G1 34 2.1
G3 34 0.5
Raji + RtX
G4 43 4.3
5mg/kg
G5 40 2.2
G7 40 2.6
H1 40 0.9
H3 36 0
Raji + RtX
H4 36 0
5mg/kg +
H5 40 1.6
ApoCell
H6 40 1.3
H7 53 1.8
[1031] Table 18: Liver
Liver
Treatment Day of
Mouse CD20+
sacrifice
B1 22 6.4
B2 25 42.6
Raji
B3 22 5
B4 22 8.1
Cl 22 2.5
Raji + C4 22 2.2
ApoCell C5 41 0.4
C6 47 0
El 32 2.1
Raji + RtX
E2 32 1.2
2mg/kg
E6 32 0.4
Fl 40 0
F2 43
Raji + RtX
F4 35 0
2mg/kg +
F5 32 7.3
ApoCell
F6 35 5
F7 57 0
G1 34 1
G3 34 7.3
Raji + RtX
G4 43 1.4
5mg/kg
G5 40 0.9
G7 40 5.7
CA 03151815 2022-02-17
197
WO 2021/053667
PCT/IL2020/051011
H1 40 0.8
H3 36 0.1
Raji + RtX
H4 36 0
5mg/kg +
H5 40 0.5
ApoCell
H6 40 25.1
H7 53 3.4
[1032] Preliminary Conclusions: In conclusion, tumor distribution in the mouse
organs
correlated the beneficial effect seen in survival plots and was significantly
reduced in bone-marrow
and liver in treated mice.
[1033] Part B: Synergistic effect of ApoCell and Rituximab (RtX) in the
treatment of
leukemia/lymphoma
[1034] Next, it was examined whether the ApoCell treatment was
synergistic with other
conventional treatments of leukemia/lymphoma by evaluating the combined effect
of RtX and
ApoCell on leukemic mice in two experiments.
[1035] Objectives: Measurement of the survival of leukemic mice following RtX
and ApoCell
administration, and detect tumor cells in bone marrow, liver, and spleen in
leukemic mice.
[1036] Methods: The following work is a representative description of the
results obtained in the
combination therapy experiments (ApoCell and rtx). Briefly, female SCID-Beige
mice were injected
intravenously with 0.1x106 Raji cells (n = 7 in all groups). Mice received
three doses of 30x106
ApoCell intravenously on days 5, 8, and 12. On day 8, 1.5 h after ApoCell
injection, the mice received
a single IV dose of 2 or 5mg/kg RtX. Mice were followed daily and weighted
twice a week. The
endpoint was defined as death, or sacrifice due to the development of one or
more of the following
symptoms: paraplegia (lower body paralysis), loss of 20% from starting weight,
lethargy, reduced
mobility, or increased respiratory effort.
[1037] Results:
[1038] As shown in Figure 26, ApoCell had a beneficial effect
corroborating the results presented
in Figure 24. Rituxan (RtX) alone had a superior effect to ApoCell both in 2
mg and 5 mg dosage
but the combination of ApoCell and Rituxan had a synergistic effect at both in
2 mg (p=0.104) and
in 5 mg dosage, although the synergistic effect seen in 5 mg did not reach
statistical significance.
Tumor distribution in the mouse organs correlated the beneficial effect (Table
19).
[1039] Table 19: Statistical analysis of the survival distributions (Log-
rank (Mantel-Cox) test)
Compared grouls Statistical test (P
value)
Log-rank (Mantel-Cox)
Group 1 Group 2
Test
Raji Raji + rtx (2mg/Kg) 0.0002
Raji Raji + rtx (2mg/Kg) + ApoCell 0.0002
Raji Raji + rtx (5mg/Kg) 0.0002
CA 03151815 2022-02-17
198
WO 2021/053667
PCT/IL2020/051011
Raji Raji + rtx (5mg/Kg) + ApoCell 0.0002
Raji + Rituxan Raji + Rituxan (2mg/Kg) +
0.0104
(2mg/Kg) ApoCell
[1040] End of experiment ¨ Day 57
[1041] One mouse (Raji + RtX 2mg/kg + ApoCell) was declared disease free upon
termination
of the experiment.
[1042] The synergistic effect in one 2 mg RtX dose was measured in an
additional experiment.
As was clearly shown in this experiment (Figure 27), the synergistic effect of
ApoCell and RtX was
again verified as significant (p=0.01).
[1043] As shown in Table 20, the spleen was not populated by tumor cells
(0.1-0.3 represents
background staining) and was used as a control. In contrast, bone marrow and
liver were tumor
targets. There was a reduced tumor population (Rajji-cells, as measured using
CD20 marker) in bone
marrow and liver following treatment by ApoCell and RtX separately, and the
benefit increased when
the two were given in combination; p= 0.0034 (**) for Raji + rtx (2mg/Kg) +
ApoCell, and 0.0031
(**) for Raji + rtx (5mg/Kg) + ApoCell (T-test. As expected, RtX significantly
reduces tumor burden
in the target organs of the leukemic mice. Interestingly, treatment with
ApoCell alone reduced tumor
cells in those organs to levels comparable to treatment with conventional RtX
therapy.
[1044] Table 20: Average tumor cell population in the spleen, bone-
marrow, and liver (flow
cytometry)
Statistical
of
Tissue Treatment # test (P
value)
mice
CD20+ T-
Test
Raji 7 0
Raji + ApoCell 4 0.3 0.4
Raji + rtx (2mg/Kg) 3 0.2 0.4
Spleen Raji + rtx (2mg/Kg) +
6 0.1 0.1
ApoCell
Raji + rtx (5 mg/Kg) 7 0
Raji + rtx (5 mg/Kg) +
4 0.1 0.2
ApoCell
Raji 6 18.3 11
Raji + ApoCell 4 8.5 11
Raji + rtx (2mg/Kg) 3 2.3 1.3
Bone Raji + rtx (2mg/Kg) +
0.0034
6 1 1.5
Marrow ApoCell
(**)
Raji + rtx (5 mg/Kg) 7 1.7 1.5
Raji + rtx (5 mg/Kg) +
0.0031
6 0.9 0 . 8
ApoCell
(**)
CA 03151815 2022-02-17
199
WO 2021/053667
PCT/IL2020/051011
15.7
Raji 6
15.4
Raji + ApoCell 4 1.3 1.3
Raji + rtx (2mg/Kg) 3 1.2 0.8
Liver Raji + rtx (2mg/Kg) +
2.5 3.5
ApoCell
Raji + rtx (5 mg/Kg) 7 3 2.5
Raji + rtx (5 mg/Kg) +
7 4.4 9.2
ApoCell
[1045] Conclusion: In summary, the survival plots (Figure 24, Figure 26,
and Figure 27) clearly
demonstrate the beneficial effects of ApoCell and Rtx along as well as the
synergistic effect of
ApoCell and RtX in combination. Of note, when the conventional RtX treatment
was combined with
5 .. ApoCell, survival times increased significantly, regardless of the RtX
dose, indicating a potent
synergistic effect of the two therapies. A supporting clinical observation was
the decrease in the of
tumor cell population in the bone marrow and liver (Table 12).
[1046] Surprisingly, ApoCell preparation had a remarkably beneficial
effect on disease
progression and survival in a leukemic mouse model, independent of any other
treatment. 10-20%
of mice had prolonged survival in Kaplan-Meier analysis (Figure 24, Figure 26,
and Figure 27),
and the tumor cell burden was reduced in the liver and bone marrow.
Furthermore, there was a marked
synergistic effect when ApoCell and RtX were administered in combination,
further delaying disease
onset and progression, and improving survival.
EXAMPLE 12: Use of Pooled Apoptotic Cell Preparation in GVHD Leukemia/Lymphoma
Models
[1047] In the following preliminary work, the effect of the same infusion in
GvHD
leukemia/lymphoma models was examined. The safety and efficacy of an
irradiated multiple donor
single apoptotic cell infusion (a pooled mononuclear irradiated apoptotic cell
preparation) for the
prevention of acute GvHD in mice undergoing bone marrow transplantation (BMT)
was examined.
In this model, BMT rescued irradiated mice (80-100%).
[1048] The question regarding the possible loss of graft versus leukemia
(GvL) effect arises in
every successful treatment that potentially avoids high grade aGVHD, since
this effect was found to
correlate with the severity of GVHD.
[1049] Methods
[1050] Apoptotic cells were prepared as per Example 1 above, except that in
the current
experiments, preparation was done simultaneously from 4 donors. Following
preparation from 4
donors, the cell preparations were combined at the last step (prior to
irradiation), irradiated
CA 03151815 2022-02-17
200
WO 2021/053667
PCT/IL2020/051011
immediately after, and injected immediately after irradiation. Irradiation was
at 25 Gy.
[1051] Results
[1052] The two graphs presented in Figures 28 and 29, show the clear
effect (p<0.01) of a single
injection of apoptotic cell from multiple individual donors (dotted line),
both on survival and weight
loss. Figure 28 is a Kaplan-Meier survival curve in a GvHD mouse model that
was treated with a
single dose irradiated apoptotic cells from multiple individual donors where
survival was significantly
ameliorated. Figure 29 is percentage of weight loss of the 2 compared groups
that follow and correlate
with the findings of Figure 28.
[1053] In summary, the single infusion of multiple-donor irradiated
apoptotic cells successfully
and significantly improved life expectancy in a mouse model of GvHD.
EXAMPLE 13: Stability Criteria for Apoptotic Cells from Multiple Individual
Donors
[1054] The objective of this study is to develop stability criteria for
apoptotic cells from multiple
individual donors with comparability studies to non-irradiated HLA-matched
apoptotic cells
(Mevorach et al. (2014) Biology of Blood and Marrow Transplantation 20(1): 58-
65; Mevorach et al.
(2015) Biology of Blood and Marrow Transplantation 21(2): S339-S340).
[1055] Apoptotic cell final product preparations will be evaluated for
cell number, viability,
apoptotic phenotype and potency after storage at 2 to 8 C for 8, 24, 48, and
60 hours with sampling
at each time point. Apoptotic cell final product lots will be prepared
following standard operating
procedures (SOPs) (Example 1; Example 5) and batch records (BRs; i.e.,
specific manufacturing
procedures). For potency evaluation, samples of early apoptotic cell
preparation final product lots will
be tested for inhibition of lipopolysaccharide (LPS) induced upregulation of
MHC-II expression on
immature dendritic cells (time points 0-24h) or monocytes (time points 0-6)
and will be performed
according to SOPs and recorded on BR. These series of test will be performed
on pooled and non-
pooled products that are in preparations originating from multiple individual
donors and from single
donors, respectively.
[1056] In addition, flow cytometric analysis of CD3 (T cells), CD19 (B
cells), CD14 (monocytes),
CD15high (granulocytes) and CD56 (NK cells) will be documented. The aims of
these studies are to
demonstrate consistency with a narrow range of results. Preliminary results
are consistent with these
goals and no deviations from the SOP are noted and no technical problems are
reported. However,
further studies are needed in order to conclude the range and stability of
effective treatment.
Preliminary results show equivalence in all these parameters. Further, single
donor stability studies
showed stability at least through a 48 hour period (See, Example 1).
CA 03151815 2022-02-17
201
WO 2021/053667
PCT/IL2020/051011
EXAMPLE 14: Safety & Efficacy of Multiple Donor Irradiated Apoptotic Cells as
Prophylaxis For Acute Graft-Versus-Host Disease
[1057] Objective: A phase 1/2a, multicenter, open-label study evaluating
the safety, tolerability
and preliminary efficacy of a single dose administration of irradiated
apoptotic cells, from multi-,
unmatched-donors, for the prevention of graft versus host disease in
hematopoietic malignancies in
human leukocyte antigen-matched, related and unrelated patients undergoing
allogeneic hla-matched
hematopoietic stem cell transplantation
[1058] Primary Objective: To determine safety and tolerability of
multiple donor irradiated
apoptotic cell treatment.
[1059] Secondary Objective: To determine efficacy of irradiated apoptotic
cells from multiple
individual donors as prophylaxis measure for acute GVHD (aGVHD) in patients
with hematopoietic
malignancies scheduled to undergo hematopoietic stem cell transplantation
(HSCT). For the purposes
of this study, HSCT can be either bone marrow transplant (BMT) or peripheral
blood stem cell
transplantation (PB S CT) .
[1060] Therapeutic Indication: Graft vs. Host Disease (GVHD) post-
transplantation in
hematopoietic malignancies in human leukocyte antigen (HLA)-matched, related
and unrelated
patients
[1061] Study Design: This is an open labeled study, multi-center, phase-
1/2a study in patients
diagnosed with hematopoietic malignancies scheduled to undergo HSCT (either
bone marrow
transplantation or peripheral blood stem cell transplantation) from an HLA-
matched related or
unrelated donor, following either full myeloablative or reduced intensity
myeloablative conditioning
regimens.
[1062] After a signing of informed consent by recipient patient, donors
screening period and cell
collection before initiating conditioning regimen, eligible recipient patients
will be assigned (stratified
by prophylactic treatment and related versus non-related transplant donors in
1:1 ratio to receive
intravenous (IV) injection 12-36 hours prior to HSCT transplantation to
either:
[1063] Investigational Arm: single dose of 140x106 20% cell/kg from
multiple individual donors
of irradiated early apoptotic cells/kg body weight in phosphate buffer
solution (PBS).
[1064] All patients will also be treated with the institutional standard
of care (SOC)
immunosuppressive regimen: cyclosporine/methotrexate or
tacrolimus/methotrexate for full
myeloablation and mycofenolate/cyclosporine or mycophenolate/tacrolinus for
reduced intensity.
Patients will be hospitalized as medically indicated.
[1065] Patients will be followed up for 180 days for the secondary
efficacy endpoint and for 1
year for the primary safety and tertiary efficacy endpoints. Number of visits
for patients participating
CA 03151815 2022-02-17
202
WO 2021/053667
PCT/IL2020/051011
in this study will be comparable to those customary for patients in their
condition. For donor, study
specific visit will be for apheresis procedure during the screening period.
[1066] As these patients have many underlying medical conditions and may
experience symptoms
compatible with aGVHD, it may be difficult to absolutely determine if toxicity
is related to apoptotic
cells or not although basic data exist from a former phase 1-2a study using
apoptotic cells for GvHD
prophylaxis (Mevorach et al. (2014) Biology of Blood and Marrow
Transplantation 20(1): 58-65)
Single Infusion of Donor Mononuclear Early Apoptotic Cells as Prophylaxis for
Graft-versus-Host
Disease in Myeloablative HLA-Matched Allogeneic Bone Marrow Transplantation: A
Phase I/IIa
Clinical Trial. BBMT 20(1)58-65).
[1067] Data Safety Monitoring Board (DSMB) will meet as specified in the DSMB
charter,
including at the time of the scheduled interim analysis (180 days) assuming no
safety concerns were
raised beforehand.
[1068] Study Procedures:
[1069] The study will comprise of screening, treatment and follow-up
periods.
[1070] /. Screening Period (Day -60 to Day -2)
[1071] Potential recipient patients will sign informed consent prior to
conduct of any study related
procedures. The standard assessments before approval, will be performed by the
transplantation
center for the donor during the screening period and usually include:
demographic data, medical
history, HLA match status verification (no matching is needed), physical
examination, height and
weight, vital signs, pregnancy test (all women), hematology, blood chemistry,
infectious disease
screen, ECG and urinalysis.
[1072] The recipients (study patients) will undergo the following
assessments during the screening
period: demographic data, medical history, Karnofsky performance status, HLA
match verification,
physical exam, height and weight, vital signs, pregnancy test (all women),
ECG, pulmonary function
test, hematology, blood chemistry, coagulation markers, infectious disease
screen, and urinalysis.
[1073] After the initial screening evaluations, if recipient is eligible
to participate in the study, the
recipient patient will be assigned on the first day of the conditioning
regimen to receive single IV
infusion of 140x106 20% cell/kg of multiple donor apoptotic cells. The
conditioning regimen to be
completed on the day before or day of Apoptotic Cell infusion scheduled for
Study Day -1.
[1074] Apoptotic cell dosage will be calculated for each recipient patient
and presumed apheresis
collection number and number of donors will be decided accordingly.
[1075] For peripheral stern cell transplant donors: Between Days -6 to -
1, the donor will receive
one or more once daily injections of G-CSF to mobilize progenitor cells and on
Day 0 will undergo
apheresis to produce donor hematopoietic blood stem cells for transplantation.
Preparation of the
CA 03151815 2022-02-17
203
WO 2021/053667
PCT/IL2020/051011
hematopoietic blood stem cells for bone marrow transplantation will be
performed in accordance with
the center's standard practice by trained hospital staff. The hematopoietic
blood stem cells for HSCT
will not be manipulated or T cell-depleted prior to administration.
[1076] For bone marrow transplant donors: Bone marrow will harvested and
prepared per center
standard practice and will not be otherwise manipulated.
[1077] 2. Treatment Day (Day -1)
[1078] On Day -1(12-36 hours prior to HSCT), eligible patients will
receive single IV infusion of
either 140x106 20% cell/kg of multiple individual donors irradiated Early
apoptotic Cellsl. Vital
signs will be monitored every hour during infusion and every 4 hours for the
first 24 hours afterwards.
Treatment-related AEs will be assessed immediately following infusion.
[1079] On Day 0, patients will undergo hematopoietic stem cell
transplantation according to local
institution guidelines.
[1080] 3. Short-term Follow-up Period (Day 0 to Day 180)
[1081] Patients will be followed-up to Study Day 180 for assessment of
the primary endpoint
safety and tolerability and secondary and tertiary endpoints: cumulative
incidence of aGVHD grade
II-IV ("modified Glucksberg" consensus based on Przepiorka et al cumulative
incidence of any grade
and high grade aGVHD, i.e., time to development of aGVHD, grades II-IV; any
systemic treatment
of GVHD, and the development of cGVHD.
[1082] The short term follow up visits will be daily while hospitalized
for the transplantation
(usually at least Days -1 to +14 or more) and weekly visits during the first 7
weeks after discharge;
days +7, +14, +21. +28, +35, +42, and then on Days 60, 100, 140, and 180. The
visit window will be
5 days for each weekly visit (first 7 weeks) and 5 days for biweekly or more
visits during the
subsequent follow up period up to 180 days.
[1083] Blood samples will be obtained on days 1, 3, 7, +7, +28, +42, 60,
100, 140 and 180 and
examined for documentation of engraftment, immunological recovery, plasma and
serum biomarkers
("Michigan") and cell subpopulations.
[1084] 4. Long-term Follow up Period (Day 181 to Day 365/1 Year)
[1085] Patients will be followed for one year post-HSCT for the longer
term secondary endpoints:
non-relapse mortality and overall survival (OS), relapse incidence, leukemia
free survival (LFS) and
chronic GVHD. There will be at least two long-term follow-up visits, the last
one being, 12 1 months
following the HSCT.
[1086] Study Duration: For each participating patient, the duration in
the study will be up to 14
months as follows:
Screening Up to 60 days (2 months
CA 03151815 2022-02-17
204
WO 2021/053667
PCT/IL2020/051011
Treatment 1 day
Follow-up 365 days (12 months) consisting of
Short-term: 180 days
Long-term +180 days
[1087] Study Population: A total of 25 patients diagnosed with hematologic
malignancies
scheduled to undergo HSCT (either bone marrow transplantation or peripheral
blood stem cell
transplantation) ,with at least 15 unrelated donors , following either
myeloablative or reduced
intensity conditioning regimens, per center standard practice will be included
in this study and will
be compared to historical controls.
[1088] Inclusion/Exclusion Criteria:
[1089] Recipient Patient Exclusion Criteria
[1090] 1. Patients, Age > 18, who are eligible for allogeneic HSCT for
the following malignancies:
Acute myeloid or undifferentiated or biphenotypic, leukemia, in complete
remission (any
remission) or beyond but with <5% blasts by morphology in bone marrow.
Acute myeloid leukemia (AML) in complete remission if it has evolved from
myelodysplastic syndrome (MDS) (there should be documented diagnosis of MDS at
least 3 months prior to diagnosis of acute myeloid leukemia). Or evolved from
polycythemia vera or essential thrombocytosis.
Acute lymphoblastic leukemia (ALL) in complete remission (any remission) with
<5%
blasts by morphology in bone marrow.
Chronic myeloid leukemia (CML) in chronic or accelerated phase
Myelodysplastic syndromes ¨ refractory cytopenia with multilineage dysplasia
(RCMD),
RA (refractory anemia), RA with ringed sideroblast (RARS; all < 5% blasts), RA
with
excess blasts (RAEB; 5 to 20% blasts).
[1091] The transplant donor and recipient patient must have at least an 8/8
HLA match at the HLA
A, B, C, DQ, and DR loci and no antigen or allele mismatch. However the
donor(s) of leukocytes for
apoptotic cell formation is not restricted to HLA matching.
[1092] Performance status score of at least 70% at time of the screening
visit (Karnofsky for adults
and Lansky for recipient < 16 years old.
[1093] Cardiac left ventricular ejection fraction > 40% in adults within 4
weeks of initiation of
conditioning; MUGA scan or cardiac ECHO required if prior anthracycline
exposure or history of
cardiac disease.
[1094] Pulmonary function test with DLC01, FEV1 (forced expiratory volume) and
FVC (forced
1 Diffusing capacity of the lung for carbon monoxide
CA 03151815 2022-02-17
205
WO 2021/053667
PCT/IL2020/051011
vital capacity) of >60% predicted.
[1095] Oxygen saturation of at least 90% on room air.
[1096] Patients must have adequate organ function as defined below:
AST (SGOT)/ALT (SGPT) <3 x upper limit of normal (ULN).
Serum creatinine <2.0 mg/dL (adults, >16 y) or <0.8 (1-2 y), <1(3-4 y), <1.2(5-
9 y), <1.6(10-
13 y), and 1.8 (14-15 y).
Serum bilirubin <3 mg/dL unless due to Gilbert's disease or hemolysis.
[1097] Signed written informed consent to participate in the study
independently by patient, or
guardian in the case of minors.
[1098] Ability to comply with the requirements of the study.
[1099] For duration of 4 weeks (from day -1), both female and male must
agree to:
Use an acceptable method of birth control or be surgically sterile for the
first month or more if
there are BMT related restrictions.
To have a negative pregnancy test regardless of child-bearing potential.
[1100] Recipient Patient Exclusion Criteria
[1101] All diseases eligible for HSCT not specified in the Inclusion
Criteria.
[1102] Participation in an interventional investigational trial within 30
days of the screening visit.
[1103] Have progressive or poorly controlled malignancies.
[1104] If BMT plan include T-cell depleted allograft
[1105] If BMT plan include anti-thymocyte globulin (ATG) or alemtuzumab as
part of
immunosuppressive regimen or high dose Cyclophosphamide therapy for the
prevention of GVHD
after transplantation
[1106] Uncontrolled infections including sepsis, pneumonia with
hypoxemia, persistent
bacteremia, or meningitis within two weeks of the screening visit.
[1107] Current known active acute or chronic infection with HBV or HCV.
[1108] Known human immunodeficiency virus (HIV) infection.
[1109] Patients with severe or symptomatic restrictive or obstructive
lung disease or respiratory
failure requiring ventilator support.
[1110] Patients with other concurrent severe and/or uncontrolled medical
condition which could
compromise participation in the study (i.e. active infection, uncontrolled
diabetes, uncontrolled
hypertension, congestive cardiac failure, unstable angina, ventricular
arrhythmias, active ischemic
heart disease, myocardial infarction within six months, chronic liver or renal
disease, active upper
gastrointestinal tract ulceration).
[1111] Any chronic or acute condition susceptible of interfering with the
evaluation of
CA 03151815 2022-02-17
206
WO 2021/053667
PCT/IL2020/051011
investigational product effect.
[1112] Any form of substance abuse (including drug or alcohol abuse),
psychiatric disorder or any
chronic condition susceptible, in the opinion of the investigator, of
interfering with the conduct of the
study.
[1113] Organ allograft or previous history of stem cell transplantation
(allogeneic only).
[1114] Breast feeding in women of childbearing potential.
[1115] Patients who are likely to be non-compliant or uncooperative
during the study.
[1116] Investigational Product Route and Dosage Form
[1117] Apoptotic cells will be administered as an IV infusion of 140x106
+ 20% cell/kg of
irradiated multiple donor apoptotic cell product 12-36 hours prior to HSCT.
[1118] Apoptotic cells are a cell-based therapeutic composed of multiple
individual donors
apoptotic cells. The product contains allogeneic donor mononuclear enriched
cells in the form of
liquid suspension with at least 40% early apoptotic cells. The suspension is
prepared from multiple
individual donors with PBS solution in accordance with GMP regulations and
should be stored at 2-
8 C until infusion. The final product will be in a total volume of 300-600 mL
in an opaque transfer
pack and will be irradiated with 25 Gy following preparation. Investigational
product should be
administered to the patient within 48 hours of completing the manufacturing
process.
[1119] Safety Outcomes/Efficacy Endpoints/Outcome Measures
[1120] Primary:
[1121] Safety and tolerability endpoints include time to engraftment and a
physical examination
to determine adverse events, concomitant medications and safety laboratories
on Day 180 and Day
360 (1 year). Further, it is expected that irradiated pooled apoptotic cell
preparations will show a lack
of in vitro and in vivo cell proliferation and lack of in vivo activation.
Such a showing identifies the
pooled apoptotic cell preparation as safe for use.
[1122] Secondary:
Cumulative incidence of aGVHD grade II-IV using "modified Glucksberg"
consensus based on
(Rowlings et al. 1997) IBMTR Severity Index for grading acute graft-versus-
host disease:
retrospective comparison with Glucksberg grade. Br J Haematol. 1997
Jun;97(4):855-64) on Day
180
1-year non-relapse mortality and overall survival (OS)
1-year relapse incidence
1-year leukaemia free survival (LFS)
Maximum grade of aGVHD within the first 180 days
Cumulative incidence of grade III-IV aGVHD
CA 03151815 2022-02-17
207
WO 2021/053667
PCT/IL2020/051011
Incidence of chronic GVHD according to (Jagasia et al., 2015) on Days 180 and
360 (1 year).
Any "systemic treatment" including corticosteroids (both used or not and
cumulative dosage) for
the treatment of aGvHD on Day 20 through Day 180
Immune reconstitution and function on Days +28, 100, 180 and 360 (1 year) in
relation to T, B,
NK, and Monocytes
Major infection rate (including lung infiltrates, CMV reactivation and any
other infections that
require hospitalization) through Day 180 and 1 year.
[1123] Tertiary/Exploratory:
Percent of hospitalization days to total days at risk, or total days alive and
out of the hospital. Or
total hospitalization days till first discharge post transplantation.
Organ specific GVHD
T regs, CD4 Tcon, CD8, NK and B cells levels on Day 180
[1124] Statistical Analysis:
[1125] Study outcome will be compared to historical control with
individuals with comparable
baseline characteristics.
[1126] Descriptive statistics will be used to summarize outcome measures
and baseline
characteristics. In this analysis all available data will be presented with no
imputation for any missing
data. Subjects will contribute the data available up to the point of
withdrawal or study completion or
death. The descriptive statistics such as means, median, standard deviation,
minimum and maximum
will be used to summarize continuous variables. All subjects who receive the
apoptotic cells infusion
will be included in the safety analysis. Subjects who also receive the HSCT
will be included in the
efficacy analysis. As this study is exploratory in nature, ad hoc analyses are
planned.
[1127] Sample size consideration
[1128] A total of 25 patients will be included at least 15 matched
unrelated patients will be
enrolled. Apoptotic cells (active will be given to all, stratifying on GVHD
prophylaxis regimen, and
related versus unrelated transplant donor.
[1129] Population Analysis definition
[1130] All efficacy analyses will be conducted on the Intent-to-Treat
(ITT) population and
compared to adequate historical control. The safety population will be defined
as all patients who
receive a dose of study medication.
[1131] Statistical methods
[1132] Patient, disease, and transplant characteristics will be described
using frequencies and
percentages or median (range) as appropriate.
[1133] Safety analysis
CA 03151815 2022-02-17
208
WO 2021/053667
PCT/IL2020/051011
[1134] Descriptive statistics will be used to summarize safety outcomes
with focus on the AEs
reported between study treatment infusion and HSCT procedure (24-30 hour
window). No alterations
in the conduct of the study will be initiated as a consequence of the DSMB
review, including sample
size adjustment. As such, no penalty adjustment in the overall Type I error as
a consequence of the
interim analysis will be required.
[1135] Secondary Endpoint Analysis
[1136] Grade II-IV aGVHD will be described using the cumulative incidence
estimator with death
prior to aGVHD as a competing event.
[1137] Neutrophil and platelet recovery, Grade III-IV aGVHD, chronic
GVHD, infection, relapse,
and transplant related mortality will be described using cumulative incidence
with relapse as
competing event for TRM and death as the competing event for all others. .
Overall survival and
leukemia free survival will be described using the Kaplan-Meier estimator,
and. The maximum grade
of aGVHD within the first 180 days and the need for steroids at 180 days will
be described using
frequencies and percentages using the Mann-Whitney U-test and chi-square test
respectively.
Immune recovery of each cell subset and TREGs will be described at each time
point using median
and range Mann-Whitney tests.
EXAMPLE 15: Comparison of Pooled Apoptotic Cell Preparation vs. Single Donor
Apoptotic Cell Preparation in GVHD Leukemia/Lymphoma Models
[1138] Objective: Compare the beneficial clinical effect of human early
apoptotic cells obtained
from a single donor on the severity of GvHD in a murine model of GvHD, to the
clinical effect, if
any, of human early apoptotic cells obtained from multiple individual donors
on the severity of GvHD
in the murine model of GvHD, wherein the multiple individual donors
represented HLA-unmatched
heterologous donors.
[1139] Example 12 above shows the beneficial effect of irradiated apoptotic
cells pooled from
multiple individual donors. The results shown in Figure 28 and Figure 29 were
surprising as a skilled
artisan may recognize that the multiple sources of unmatched cells may have
increased the diversity
of antigenicity of the cells, and thus would have expected a dramatic
reduction in the clinical effect.
Unexpectedly, the known, beneficial effect of early apoptotic cells on the
reduction of GvHD severity,
and therefore a prolongation of the number of days till mortality, was also
alleviated by pooled
unmatched early apoptotic cells (Figure 28), which would purportedly have
increased antigenicity
due to the pooled multiple unmatched source cells.
[1140] An additional objective was to understand if there is a difference
between the use of
irradiated early apoptotic cells and non-irradiated apoptotic cells.
CA 03151815 2022-02-17
209
WO 2021/053667
PCT/IL2020/051011
[1141] A skilled artisan would appreciate that unmatched, irradiated
cells keep their antigenic
profile as recognized by the APC mechanism and so by T-Cells of the host into
which they have been
infused. Accordingly, concerns when pooling heterologous unmatched populations
of cells included
cross-reactivity between the individual populations being pooled, mixed-cell
lymphatic reactions of
pooled populations, or T-cell immune reactions between pooled populations that
could reduce or
eliminate cells, or any combination thereof.
[1142] Methods
[1143] Mouse model: Female 7-9 week-old BALB/c mice (H-2d) were used as
recipients and
female 8-9 week-old C57BL/6 mice (H-2b) were used as donors in mismatched GVHD
model.
Recipients were total body irradiated at 850 cGy 24 hours before bone marrow
and splenocyte
transplantation. Donor bone-marrow cells were used for bone-marrow
reconstitution. Bone marrow
cells were extracted from the femoral and tibial bones with RPMI 1640. Red
blood cells were lysed,
then cells were washed and resuspended with PBS. Viability was assessed using
trypan blue dye
exclusion (>90% viability). Donor splenocytes were used for the induction of
GVHD. Spleens were
removed and homogenized and single cell suspension was obtained. Red blood
cells were lysed and
splenocytes were resuspended with PBS. At least 90% viable cells were assessed
using trypan blue
dye.
[1144] Early apoptotic cells: Apoptotic cells were produced from
mononuclear enriched cell
fraction apheresis from healthy donors similar to Example 1. In brief:
[1145] Enriched fractions of mononuclear cells (MNCs) were obtained from
healthy, eligible
donors via leukapheresis procedure. Cells were collected via Spectra OPTIA
apheresis system from
12 liters of blood, in addition to 400-600m1of autologous plasma. The
estimated yield of the enriched
mononuclear cell fraction from a donor was expected to be approximately 1.2-
1.5 x 1010 cells. Prior
to leukapheresis procedure, donors are tested and confirmed negative to the
below viral vectors:
1. Human Immunodeficiency virus (HIV), types 1 and 2;
2. Hepatitis B virus (HB V);
3. Hepatitis C virus (HCV);
4. Cytomegalovirus (CMV);
5. Treponema pallidum (syphilis);
6. Human T-lymphotropic virus (HTLV), types I and II
[1146] Following cell collection, the cells were washed with RPMI and
frozen as follows. The
freezing formulation was composed of PlasmaLyte A for injection pH 7.4, 10%
DMSO, 5% Human
Serum Albumin and 5% Anticoagulant Citrate Dextrose solution inoculated with
101.Aml heparin.
[1147] Freezing media was prepared in bags and the freezing procedure
performed in a closed
CA 03151815 2022-02-17
210
WO 2021/053667
PCT/IL2020/051011
system under cGMP conditions.
[1148] Following leukapheresis procedure completion, enriched MNC fraction was
washed with
PlasmaLyte A and resuspended with ice-cold freezing media to a concentration
of 50-65x106 cells \ml.
Cells were then transferred to freezing bags, bags were transferred to pre-
cooled aluminum cassettes
and cassettes were transferred immediately to -18- (-25) C for two hours.
[1149] Following the two hours, cassettes were transferred to -80 C for
an additional 2 hours and
then to long-term storage in liquid nitrogen (> -135 C).
[1150] Autologous plasma was divided to 50gr aliquots. Plasma aliquots
were transferred to -80 C
for 2 hours and then to a long-term storage in -18- (-25) C.
[1151] For apoptosis induction cells were thawed and washed with pre-warmed
RPMI1640
containing 10mM Hepes buffer, 2mM L-Glutamine and 5% Anticoagulant Citrate
Dextrose solution
inoculated with 101.Aml heparin. After supernatant extraction cells were
resuspended at final
concentration of 5x106/m1 in RPMI 1640 supplemented with 10mM Hepes, 2mM L-
glutamine, with
addition of 10% autologous plasma and. 50i.tg\ml Methylprednisolone and 5%
Anticoagulant Citrate
Dextrose solution inoculated with 101.Am1 heparin. Cells are then transferred
to cell culture bags, and
incubated at humidified incubator 37 C, 5% CO2 for 6 hours to stabilize
apoptosis.
[1152] Following incubation cells were harvested, washed with PBS and
resuspended in PBS.
[1153] Early apoptotic cell product was produced from one single donor or
combined 10 different
individual donors, in which case cells were combined just prior to
irradiation. Since interference may
occur between components in the multiple donor product, for example between
living non-apoptotic
cells, the early apoptotic cell product was subdivided and a sample of early
apoptotic cells to be tested
in vivo was irradiated with 2500 cGy prior to administration (sample F below),
and stored at 2-8 C
until administration. Table 3 of Example 6 below presents details of the
Annexin V
positive/Propidium iodide negative ratio and cell surface markers of the early
apoptotic cell product,
establishing that consistency of apoptotic cells administered to mice is
maintained. The final product
was stable for 48 hours at 2-8 C.
[1154] On the day of transplantation, mice received 5x106 bone-marrow
cells, 3x106 splenocytes
and 30x106 single- or multiple-donor early apoptotic cell product, according
to the following
experimental design:
Irradiation control
Reconstitution control ¨ irradiation + Bone-Marrow transplantation (BM)
GVHD control - irradiation + Bone-Marrow and splenocyte transplantation
Single donor, irradiated - irradiation + Bone-Marrow and splenocyte
transplantation + irradiated
early apoptotic cell product from single donor
CA 03151815 2022-02-17
211
WO 2021/053667
PCT/IL2020/051011
Single donor, non-irradiated - irradiation + Bone-Marrow and splenocyte
transplantation + non-
irradiated early apoptotic cell product from single donor
Multiple donor, irradiated - irradiation + Bone-Marrow and splenocyte
transplantation +
irradiated early apoptotic cell product from multiple donor
Multiple donor, non-irradiated - irradiation + Bone-Marrow and splenocyte
transplantation +
non-irradiated early apoptotic cell product from multiple individual donors.
[1155] Monitoring ¨ Transplanted mice were tagged and survival was monitored.
Body weight
was assessed every two days for the first two weeks of the experiment and then
every day. Loss of
35% from initial body weight was determined as primary end point and mice were
sacrificed and
survival curve was updated accordingly. Body weight results were comparable to
those observed in
Example 12 Figure 29.
[1156] The severity of GVHD was assessed using a known scoring system (Cooke
KR, et al. An
experimental model of idiopathic pneumonia syndrome after bone marrow
transplantation. I. The
roles of minor H antigens and endotoxin. Blood.1996; 8:3230-3239) that
incorporates five clinical
.. parameters: weight loss, posture (hunching), activity, fur texture and skin
integrity. Mice were
evaluated and graded from 0 to 2 for each criterion. By summation of the five
clinical scores a clinical
index value was generated (index number increases with the severity of GVHD).
[1157] Results
[1158] Percent survival of the different population of mice is presented
graphically in Figure 30.
The irradiation only control mice died between day 8 and 12 (n=13), as
expected from mice that did
not receive bone marrow reconstitution. The majority of GVHD control mice
(received bone-marrow
and spleen) died between day 6 and 27. One mouse did not die (n=18). In bone-
marrow reconstitution
control group (BM) 3 out of 7 mice died between day 6 and 8. In the remaining
mice, bone marrow
was reconstituted by donor bone-marrow and mice remained alive (>50 days).
[1159] Single donor, non-irradiated mice died between day 15 and 36. Thus,
as previously shown,
single donor non-irradiated early apoptotic cells had a beneficial effect and
survival was prolonged
(p<0.01).
[1160] Single donor, irradiated mice died between day 7 and 35, one mouse
remained disease free
survival (>50 days). This demonstrated that single donor irradiated apoptotic
cells also provided the
beneficial effect with respect to GVHD. Thus, irradiation did not harm the
immunomodulatory effect
of early apoptotic cells. All had beneficial effect on survival in the GVHD
murine model compared
to GVHD control (p<0.01).
[1161] Non-irradiated multiple donor treatment did not provide a
beneficial effect compared to
GVHD control (n=11). Survival pattern was similar to GVHD control and mice
died between day 6
CA 03151815 2022-02-17
212
WO 2021/053667
PCT/IL2020/051011
and 28 (p=NS -not significant). Surprisingly and in contrast to the non-
irradiated apoptotic cells,
irradiated-multiple individual donor apoptotic cells (treatment F) (n=10) had
a beneficial effect
similar to single donor treatment, as compared with GVHD control. GVHD
symptoms appeared
significantly later and mice died between day 18 and 34 (p<0.01).
[1162] Irradiated-multiple individual donor (n=10), irradiated single donor
(n=10) and non-
irradiated single donor treatment (n=10) had similar survival patterns and no
significant difference in
effect on survival was observed between these three treatment groups.
[1163] The experiments indicated a clear effect of apoptotic cells
infusion in GVHD induced mice.
There was a significant prolonged survival effect for the treatments of
irradiated multiple individual
donors and irradiated- and non-irradiated single donor apoptotic cells.
[1164] Multiple donor treatment did not prolonged survival of mice when
not irradiated but the
irradiation of the apoptotic cell product prior to administration to mice
improved results and treatment
had close survival pattern as single- donor treatments.
[1165] As stated above, Figure 30 shows, non-irradiated apoptotic cells
obtained from multiple
unmatched donors have significantly lower positive clinical effect on
reduction in GvHD and
mortality (% survival), as compared to (1) non-irradiated apoptotic cells
obtained from single
unmatched donors; (2) irradiated apoptotic cells obtained from single
unmatched donors; and (3)
irradiated apoptotic cells obtained from multiple unmatched donors. In
addition, all three (non-
irradiated early apoptotic cells, single donor; irradiated early apoptotic
cells, single donor; and
irradiated early apoptotic cells, multiple individual donors) have similar
effects.
[1166] This data was surprising since the antigenicity of the non-
irradiated apoptotic cells obtained
from multiple individual donors was expected to be similar to that of
irradiated apoptotic cells
obtained from multiple individual donors, why would not both have similar
hostile antigenic reaction
with the implanted bone marrow, and why would both not be able to reduce GvHD
and mortality
rate?
[1167] If antigenicity is the main issue here, it was expected to see
differences between the clinical
effects of non-irradiated apoptotic cells obtained from single donor and
irradiated apoptotic cells
obtained from single donor. However the data does not show this difference.
[1168] One possibility is that the lack of efficacy of non-irradiated
pooled apoptotic cell
preparations prepared from multiple individual donors, resulted from cross-
interaction between the
individual mononuclear populations present in the pooled preparation. These
interactions do not
appear to be directly attributable to antigenicity towards the host, as
irradiated cells maintain their
antigenicity but the efficacy differed significantly from non-irradiated
cells. Therefore, it appears that
the cross-interaction in the pooled early apoptotic cell preparations
receiving irradiation was
CA 03151815 2022-02-17
213
WO 2021/053667
PCT/IL2020/051011
unexpectedly eliminated and the host responded well to administration of the
cells.
[1169] As shown, irradiated pooled donors had essentially the same effect
as a single non
irradiated donor.
EXAMPLE 16: Effect of Irradiation on Final Apoptotic Cell Product
[1170] Apoptotic cells are increasingly used in novel therapeutic
strategies because of their
intrinsic immunomodulatory and anti-inflammatory properties. Early apoptotic
cell preparations may
contain as much as 20-40% viable cells (as measured by lack of PS exposure and
no PI admission;
Annexin V negative and Propidium iodide negative) of which some may be
rendered apoptotic after
use in a transfusion but some will remain viable. In the case of bone marrow
transplantation from a
matched donor, the viable cells do not represent a clinical issue as the
recipient is already receiving
many more viable cells in the actual transplant. However, in the case of a
third-party transfusion, (or
fourth party or more as may be represented in a pooled mononuclear apoptotic
cell preparation) use
of an apoptotic cell population that includes viable cells may introduce a
second GvHD inducer.
Furthermore, the implication of irradiation on the immunomodulatory potential
of early apoptotic
cells has so far been not assessed. A skilled artisan may consider that
additional irradiation of an early
apoptotic cell population may lead cells to progress into later stages of
apoptosis or necrosis. As this
appears a particularly relevant question with regard to clinical applications,
the experiments presented
below were designed to address this issue, with at least one goal being to
improve the biosafety of
functional apoptotic cells.
[1171] Thus, the aim was to facilitate the clinical utilization of
apoptotic cells for many indications
wherein the potency of apoptotic cells may rely on a bystander effect rather
than engraftment of the
transplanted cells.
[1172] Objective: Examine the effect of irradiation on early apoptotic
cells, wherein irradiation
occurs following induction of apoptosis.
[1173] Methods (in brief): The cells were collected according and early
apoptotic cells were
prepared essentially as described in Example 5.
[1174] Three separate early apoptotic cell batches were prepared on
different dates (collections
404-1, 0044-1 and 0043-1).
[1175] Each final product was divided into three groups:
[1176] Untreated
[1177] 2500rad
[1178] 4000rad.
[1179] Following irradiation, early apoptotic cells were tested
immediately (to) for cell count,
CA 03151815 2022-02-17
214
WO 2021/053667
PCT/IL2020/051011
AnnexinV positive-PI negative staining, cell surface markers (% population of
different cell types)
and potency (dendritic cells (DCs)). Following examination at to, early
apoptotic cells were stored at
2-8 C for 24 hours, and examined the next day using the same test panel (t24h)
(cell count, Annexin V
positive-PI negative staining, and cell surface markers and potency).
[1180] Previously, a post-release potency assay was developed, which
assesses the ability of donor
mononuclear early apoptotic cells (Early apoptotic Cells1) to induce tolerance
(Mevorach et al, BBMT
2014 ibid). The assay is based on using flow cytometric evaluation of MHC-
class II molecules (HLA-
DR) and costimulatory molecule (CD86) expression on iDC membranes after
exposure to LPS. As
previously and repeatedly shown, tolerogenic DCs can be generated upon
interaction with apoptotic
cells (Verbovetsky et al., J Exp Med 2002, Krispin et al., Blood 2006), and
inhibition of maturation
of LPS-treated DCs (inhibition of DR and CD86 expression), occurs in a dose
dependent manner.
[1181] During phase 1/2a of the early apoptotic cell clinical study, the
post-release potency assay
was conducted for each early apoptotic cell batch (overall results n=13) in
order to evaluate the ability
of each batch to induce tolerance (Results are shown in Figure 1, Mevorach et
al. (2014) Biology of
Blood and Marrow Transplantation 20(1): 58-65).
[1182] DCs were generated for each early apoptotic cell batch from fresh
buffy coat, collected
from an unknown and unrelated healthy donor, and were combined with early
apoptotic cells at
different ratios (1:2, 1:4 and 1:8 DC:Early apoptotic Cells, respectively).
After incubation with early
apoptotic cells and exposure to LPS, potency was determined based on
downregulation of DC
membrane expression of either HLA DR or CD86 at one or more ratios of DC:
early apoptotic cells.
In all 13 assays, early apoptotic cells demonstrated a tolerogenic effect,
which was seen with
preparations at most DC: early apoptotic cells ratios, and for both markers,
in a dose dependent
manner.
[1183] Monocyte obtained immature DCs (iDCs) were generated from peripheral
blood PBMCs
of healthy donors and cultured in the presence of 1% autologous plasma, G-CSF
and IL-4. iDCs were
then pre-incubated for 2 hours at 1;2, 1;4 and 1;8 ratios with apoptotic cells
either freshly prepared
final product or final product stored at 2-8 C for 24 hours. The two final
products were examined
simultaneously in order to determine whether storage affects potency ability
of apoptotic cells.
Following incubation, LPS was added to designated wells were left for
additional 24 hours. At the
end of incubation, iDCS were collected, washed and stained with both DC-sign
and HLA-DR or
CD86 in order to determine changes in expression. Cells were analyzed using
flow cytometer and
analysis performed using FCS-express software from DC-sign positive cells gate
to assure analysis
on DCs only.
[1184] Figures 31A and 31B and Figures 32A and 32B show potency test of
irradiated pooled
CA 03151815 2022-02-17
215
WO 2021/053667
PCT/IL2020/051011
apoptotic cells compared to non-irradiated single donor cell.
[1185] Results:
[1186] Single Donor preparations
[1187] Table 21 presents the comparative results of non-radiated and
irradiated apoptotic cells;
Average cell loss (%) at 24 hours; Annexin positive() Propidium Iodide (PI)
negative() % at 0 hours
and 24 hrs (% of early apoptotic cells; Annexin positive ( ) Propidium Iodide
(PI) positive ( ) % at 0
hours and 24 hrs (% of late apoptotic cells); presence of cell surface
antigens CD3 (T cells), CD19 (B
cells), CD56 (NK cells), CD14 (monocytes), and
5high (granulocyte), at 0 hours and 24 hours.
[1188] Table 21:
Final product description Apoptotic Cell Apoptotic Cell
Apoptotic Cell
2500rad
4000rad
An+PI- to (%) 59.2 59.6
58.4
Range (min-max) (52.6- 66.1) (51.6- 68.7)
(50.4- 65.1)
An+PI" t24h (%) 62.6 68.1
66.7
Range (min-max) (53.6- 76.3) (52.0- 81.3)
(52.9- 77.1)
An+PI to (%) 4.9 6.0 6.1
Range (min-max) (3.2- 7.0) (5.2- 7.4) (4.0-
9.1)
An+Pr t24h (%) 7.3 8.6
9.0
Range (min-max) (5.0- 11.8) (6.4- 11.8)
(6.0- 14.9)
CD3+ to (%) 56.9 58.3
57.5
Range (min-max) (47.4- 66.3) (48.8- 67.7)
(48.6- 66.4)
CD3+ t24h (%) 56.8 57.1
56.6
Range (min-max) (49.6- 64.0) (48.0- 66.1)
(49.7- 63.4)
CD19+ to (%) 10.6 9.5
9.6
Range (min-max) (10.1- 11.0) (7.7- 11.3)
(8.5- 10.7)
CD19+ ..24h t (%) , - , 11.8 9.2
8.8
Range (min-max) (10.2- 13.4) (6.9- 11.5)
(7.5- 10.1)
CD56+ to (%) 12.2 13.0
14.4
Range (min-max) (7.0- 17.3) (7.6- 18.4)
(21.2- 7.6)
CD56+ .24h I- (%) \ -, 12.9 14.1
17.1
Range (min-max) (8.8- 13.4) (10.4- 17.8)
(10.0- 24.1)
CD14+ to (%) 23.1 25.2
24.6
Range (min-max) (13.1-33.1) (13.8-36.5)
(14.0-35.2)
CD14+ .24h t (%) , - , 21.9 23.7
24.4
Range (min-max) (13.8- 30.0) (13.8- 33.6)
(15.4- 33.4)
CD15 high tO (%) 0.0 0.0
0.01
Range (min-max)
(0.0- 0.02)
CD15 high ..24h t (%) \ -, 0.0 0.0
0.01
Range (min-max)
(0.0- 0.02)
[1189] The results in Table 21 show that both non-irradiated apoptotic
cells and irradiated
CA 03151815 2022-02-17
216
WO 2021/053667
PCT/IL2020/051011
apoptotic cells had comparable percentages of early (rows 2 and 3) and late
(rows 4 and 5) apoptotic
cells. Thus, 25 or 40 Gy irradiation did not accelerate the apoptotic or
necrotic process induced prior
to this high level of gamma-irradiation. Further, there was consistency
between irradiated cell
populations vs. control non-irradiated population with regard to cell type.
[1190] The results of potency assays, presented in Figures 31A-31B (HLA-DR
expression) and
Figures 32A-32B (CD86 expression) show that there was no change in the immune
modulatory
capacity of fresh (Figure 32A, Figure 32A) and 24 hour- stored (Figure 31B and
Figure 32B)
irradiate apoptotic cells when compared with non-irradiated apoptotic cells.
[1191] In both Figures 31A-31B and Figures 32A-32B there is a clear
upregulation in both HLA-
DR and CD86 expression, following exposure to maturation agent LPS.
Significant (p<0.01), dose-
dependent down regulation of both co-stimulatory markers was observed in the
presence of freshly
prepared apoptotic cells both from a single donor or irradiated pooled
donors.. In addition, dose
dependent down regulation was maintained in both markers in the presence of
apoptotic cells stored
at 2-8 C for 24 hours, indicating final product stability and potency
following 24 hours of storage.
[1192] Effect on dendritic cells, In order to test the immunomodulatory
capacity of apoptotic
cells a post release potency assay was used (Mevorach et al., (2014) BBMT,
ibicl). No change in
immune modulatory assay in dendritic cells was observed. (Data not shown)
[1193] Effect on Mixed Lymphocyte Reaction (MLR). In order to further test the
immunomodulatory effect a standardized MLR assay was established. Here, co-
cultivation of
stimulator and responder cells, i.e. a MLR, yielded strong and reliable
proliferation. Upon addition of
non-irradiated apoptotic cells to the MLR, the lymphocyte proliferation was
significantly reduced by
>5- fold, clearly demonstrating cell inhibition of proliferation. Inhibition
of lymphocyte proliferation
in MLRs mediated by irradiated apoptotic cells was completely comparable.
(Data not shown)
[1194] The next step was to evaluate in vivo, irradiated and non-
irradiated apoptotic cells in a
completely mismatched mouse model. As shown in Figures 28 and 29, irradiated
and non-irradiated
early apoptotic cell preparations had comparable in vivo beneficial effect.
[1195] Single Donor Preparations Conclusion:
[1196] In conclusion, irradiation of 25 Gy or 40 Gy did not significantly
accelerate apoptosis or
induced necrosis in populations of apoptotic cells. Significantly, these
populations maintained the
immunomodulatory effect of apoptotic cells both in vitro and in vivo.
[1197] Multiple Donor preparations
[1198] Next, experiments were performed to verify that the phenomenon observed
with single
donor, third-party preparation was also true for multiple third-party donors.
Unexpectedly, when
using pooled individual donor apoptotic cell preparations, the beneficial
effect of a single unmatched
CA 03151815 2022-02-17
217
WO 2021/053667
PCT/IL2020/051011
donor was lost (Figure 30). This was not due to GvHD, as the beneficial effect
of each donor
separately was maintained (test results no shown). One possibility is that the
beneficial effect of the
early apoptotic cell preparation was lost due to the interaction of the
individual donor cells among
themselves.. It was further examined whether this possible interaction of
different donors could be
avoided by gamma irradiation.
[1199] As shown in Figure 30, the beneficial effect of a single donor was
completely restored
following gamma irradiation, wherein the irradiated multiple donor preparation
and the single donor
preparation (irradiated or non-irradiated) had similar survival patterns.
[1200] Conclusion:
[1201] It is shown here for the first time that surprisingly irradiation
(and possibly any method
leading to T-cell Receptor inhibition) not only avoided unwanted proliferation
and activation of T-
cells but also allowed for the beneficial effects of immune modulation when
using a preparation of
multiple donor third-party apoptotic cells.
EXAMPLE 17: Effect of Combined Apoptotic Cell and CAR-T Cell Treatment on a
Solid
Tumor Model
[1202] Objective: To test the effect of CAR-T cell therapy on a human
peritoneal solid tumor
model and evaluate any possible synergistic effect of apoptotic cell infusion
on the CAR T-cell
therapy anti-cancer action.
Methods
[1203] HeLa-CD19 cell line: CD19-expressing HeLa cells were purchased from
ProMab
(ProMab, USA, cat. No. PM-Hela-CD19). In order to follow tumor growth in vivo,
HeLa-CD19 were
stably transduced with pLenti-PGK-V5-Luc-Neo (Addgene, USA, plasmid #21471)
and single clones
were isolated by flow cytometry cell sorter and cultured with 500 iig/m1 G418
(Sigma-Aldrich, USA,
cat. no. A1720-1G) as a selective agent. This cell line stably and
continuously expresses the firefly
/uc gene under the PGK promoter. Genomic integration was verified by PCR and
expression verified
by flow cytometry and bioluminescent imaging (BLI) of the positive clones.
[1204] Cells were cultured in RPMI 1640 (Gibco, ThermoFisher Scientific,
USA, cat. no. 31870-
025) supplemented with 10% FBS (Gibco, ThermoFisher Scientific, South America,
cat. no. 12657-
029), 2 mM GlutaMAX (Gibco, ThermoFisher Scientific, USA, cat. no. 35050-038),
and 100 Wm'
Penicillin + 100 Wm' Streptomycin (Gibco, ThermoFisher Scientific, USA, cat.
no. 15140-122),
referred to as "complete medium". HeLa-CD19 medium was further supplemented
with 1 Ilg/m1
puromycin (Sigma-Aldrich, USA, cat. no. P9620), as the selective antibiotics,
during standard
culturing.
CA 03151815 2022-02-17
218
WO 2021/053667
PCT/IL2020/051011
[1205] For bioluminescent imaging, mice were injected i.p. with 150 mg/kg
XenoLight D-
luciferin (PerkinElmer, USA, cat. no. 122799) and anesthetized with 4%
isoflurane. Ten minutes after
injections, bioluminescence was captured using Caliper IVIS-Kinetics
(PerkinElmer, USA) and
Living Image software, v4.2 (PerkinElmer, USA).
[1206] CD19-CAR-T cells: Fresh mononuclear cells (MNCs) fraction were isolated
from the
peripheral blood of healthy donors using Ficoll gradient. Cells were then
gradually frozen and stored
in liquid nitrogen until use. Following thawing, the cells were washed, and
introduced with
CD28+CD3+ beads (Miltenyi biotech) loaded per manufacturer instructions in a
1:2 bead: cell ratio,
respectively. Cells were then incubated for 3 days in the presence of 300U\ml
recombinant human
IL-2 (rhIL-2, Peprotech). 48 hours prior to MNCs infection, 293 Lenti-X cells
(Clontech-Takara,
632180) were transfected with a 3rd generation CD19-CAR plasmids (Creative
Biolabs) using
JetPrime reagent (Polyplus, 114-01) at 2:1 JetPrime: DNA ratio. 24 hours prior
to infection, non-
treated 24-well plates (SPL, BN30006) were coated with 7.5ug\cm2 of
Retronectin (Takara-Clontech,
T100A) according to manufacturer instructions. On the day of infection, virus-
containing supernatant
collected from the transfected 293 Lenti-X cells was filtered using 0.45um
filter (Millipore),
transferred to the Retronectin-coated plates (0.75m1 of viral supernatant per
well), and centrifuged
according to manufacturer instructions. Activated T-cells were then added to
the plates (0.5x106 cells
per well) and centrifuged as well at 290g. Plates were then incubated for 60
hours in a humidified
incubator. Following incubation, the cells were collected, residual adherent
cells were detached using
trypsin. Cells were washed twice with Hank's balanced salt solution (HBSS,
Biological industries)
supplemented with 2.5% Hepes buffer (Lonza). Cells were re-seeded in the
presence of 300thm1
rhIL-2, and diluted to a concentration of 1.0x106 every 48 hours with fresh
media containing 300U\ml
rhIL-2. On day 12 following infection, cells were again activated with
CD28+CD3+ beads in a 1:2
bead: cell ratio, respectively. Cells were incubated for additional two days,
and then injected (in
RPMI) to animals on day 14 following infection, a total of 10x106 cells per
animal. Mock-T cells
(Mock-T) were prepared as the CAR-T cells with plasmid lacking the anti-CD19
region and injected
as control. PCR and EGFR (Erbitux, Merck) expression were used for
transduction efficacy. An in
vitro cytotoxicity assay was used for functional test of CAR T cells.
[1207] Apoptotic cells: Enriched mononuclear cell fraction was collected
via leukapheresis
procedure from healthy, eligible human donors and prepared by as provided
herein in Example 1 and
gamma irradiated apoptotic cells, as produced herein (2000-6000 rad
irradiation). The irradiated
apoptotic cells used in this example are considered "over the shelf' (OTS),
wherein the donor and
recipient would not necessarily be HLA matched. In some embodiments, for
methods of use herein,
donor and recipient are not HLA matched. In some embodiments, for methods of
use herein, donor
CA 03151815 2022-02-17
219
WO 2021/053667
PCT/IL2020/051011
and recipient are HLA matched. [Apoptosis and viability of apoptotic cells
were determined using
AnnexinV and PI staining (MBL, MA, USA), as described herein. Lack of
proliferation was shown
using Carboxyfluorescein succinimidyl ester (CFSE) staining and bead
stimulation.
[1208] CFSE staining: Cells were brought to final concentration of 10x106
cells/mL with
RPMI1640 and stained with 5i.tM CFSE for 10 minutes at room temperature (RT)
and protected from
light with gentle tilting. Following incubation end time, staining reaction
stopped with complete FBS
for 1 minute. Cells were then washed twice in order to remove excess CFSE dye.
[1209] Cell seeding: Cells were brought to concentration of lx106
cells/mL with culture media
(RPMI1640 and Biotarget, supplemented with 2mM L-Glutamine, 10mM Hepes, 10%
Heat
inactivated FBS and 100U/mL recombinant human IL-2). Cells were then seeded in
24 wells plates,
lmL per well, either with or without CD3+CD28+ loading beads at 1:3 cell:
beads ratio. Plates were
incubated in an incubator (Enlivex Equipment number ENX01051) in the presence
of 5% CO2.
[1210] Sample analysis: Every 48 hours, each preparation was sampled,
counted and analyzed via
flow cytometer in order to access cell number and CFSE staining. Preparations
exceeded
concentration of 1.5x106 cells/mL were diluted to 1x106 cells/mL with culture
media. Preparations
that did not exceeded 1.5x106 cells/mL were treated with culture media
refreshment (0.3mL of media
was discarded and fresh culture media was added). Overall 4 testing points
were performed
throughout 8 days.
[1211] FACS analysis of subpopulations: Peritoneal cells were evaluated
by flow-cytometry
(LSR-II, BD) for the following markers and isotype controls: mMHC-II-PE (12-
5321), mCD19-FITC
(11-0193), mCD1 lc-PE-Cy5.5 (35-0114), mF4/80-eFlour450 (48-4801), mCD1 lb-APC-
eFlour780
(47-0112), rIgG2b-PE (12-4031), rIgG2a-FITC (11-4321), Armenian-hamster IgG-PE-
Cy5.5 (35-
4888), rIgG2a-eFlour450 (48-4801), rIgG2b-APC-eFlour780 (47-4031), hCD19-APC
(17-0198) and
mIgG 1 -APC (17-4714) (eBiosciences).
[1212] Macrophages sub-population characterization was performed according
to Ghosn et al.
(PNAS 2010) February 9, 2010. 107 (6) 2568-2573 and Casado et al. (PlosOne
2011) published July
22, 2011. Briefly, non-CD19, non CD1 lc peritoneal cells were divided by their
CD1 lb and F4/80
expression to Large Peritoneal Macrophages (LPM) ¨ F4/80HighCD1 lbpos, Small
Peritoneal
Macrophages (SPM) ¨ F4/80lowCD1lbpos, and Granulocytes (Gra) ¨
F4/80lowCD11bneg (Ghosn
et al (2010 ibid). Casado et al. (2011 ibid) described the same macrophages
sub-population slightly
differently ¨ non CD19, non CD1 lc, F4/80 positive cells were divided by their
MHC-II and F4/80
expression to LPM ¨ F4/80highMHC-IIneg, SPM- F4/80lowMHC-IIpos, Gra-
F4/80lowMHC-IIneg.
[1213] Single-cell analyses of Tim4,MerTK, F4/80, CD1 lb, CCR2, Ly6c,
CD206, CD64, C169,
and CD74 were confirmed by FACS analysis. Resident peritoneal macrophages were
identified based
CA 03151815 2022-02-17
220
WO 2021/053667
PCT/IL2020/051011
on expression levels of cell surface F4/80, CD1 lb, Tim4, and MerTK.
Infiltrating macrophages were
identified based on expression of cell surface CCR2, Ly6c, CD206, CD64, CD169,
and CD74.
[1214] Luminex Cytokine analysis: Human or mice cytokines were tested by
Luminex cytokine
analysis. Custom Multiplex kits were purchased from eBioscience (Vienna,
Austria) or from R&D
Systems (Minneapolis, USA).
[1215] For intra-peritoneal solid tumor model, SCID-Bg mice were injected
intra-peritoneally
(i.p) with 2 consecutive doses of 0.25x106 human HeLa-CD19-luciferase cells,
on days 1 and 2 of
the experiment. Mice also received 10x106 human apoptotic cells (irradiated as
described above,
2000-6000 rad irradiation) or vehicle, on day 9; and 10x106 CD19-CAR-T (third
generation) cells or
mock T cells on day 10. Mice were weighted twice a week and monitored daily
for clinical signs and
peritoneal fluid accumulations. In some experiments, pre-scheduled sacrifices
were performed to
characterize cell and macrophage sub-population profile. The rest of the mice
were kept for survival
analysis. Survival endpoint was defined by a score based on severe peritoneal
fluid accumulation
manifested as enlarged and tense abdomen, and reduced mobility or increased
respiratory effort.
These clinical findings correlated to large accumulation of HeLa cells in the
peritoneum. Survival
analysis was performed according to the Kaplan-Meier Log rank statistical
test.
[1216] Treatment: Figure 33 presents an experimental scheme.
[1217] Results
[1218] In this model mice survived about 30 5 days (27-37) and died from
the formation of a
solid tumor in the peritoneal cavity followed by accumulation of bloody
peritoneal fluid and clinical
deterioration. Mock treatment showed no significant amelioration of the solid
tumor or change of
mouse survival (34 4 days; range 30-38). CART cell therapy alone significantly
ameliorated mouse
survival to 55 11 (range 34-76, p<0.05) (Figure 34A). However, when mice
received co-
administration of apoptotic cells and CAR T cells, a further significant
increase in survival was seen,
with survival reaching 70 20 days (range 48-90, P<0.05) when compared to CAR T
cell alone.
Furthermore, 2/10 mice were disease free for 150 days (end of experiment).
Figure 34B
(representative of 5 separate experiments) shows survival curves of peritoneal
solid tumor Hela-CD19
under control and treatment conditions. Figure 34C presents survival curves
showing that apoptotic
cells (Allocetra-OTS) dramatically enhanced CAR T anti-cancer function. Figure
34C shows
progression of tumor as visualized by an in vivo imaging system (IVIS),
representative of the results
used for the survival curves of Figure 34B. Spread of the tumor can be
visualized as early as day 15
(Hela-CD19-Luc) whereas mice treated with CAR do not show any tumor spread
until day 43. When
mice received apoptotic cells, most mice do not show tumor spread until day 50
and tumor size (see
scale for correlation) is clearly smaller as reflected in survival curve
(Figure 34B).
CA 03151815 2022-02-17
221
WO 2021/053667
PCT/IL2020/051011
[1219] Figures 35A and 35B provide addition evidence of enhancement of the
effect of CAR T
cells by co-administration with early apoptotic cells (Allocetra-OTS), wherein
survival of mice
bearing a tumor burden was extended with addition of apoptotic cell
administration. SCID-Bg mice
were injected intra-peritoneally with human HeLa-CD19-luciferase cells,
followed by CD19-CAR T
cells, wherein administration of CAR-T cells increased the percent survival
and extended the life
expectancy of the mice (Figure 35A). A similar experiment, wherein SCID-Bg
mice were injected
intra-peritoneally with human HeLa-CD19-luciferase cells, followed by CD19-CAR
T cells, with or
without apoptotic cells (Allocetra-OTS) showed an even greater increased
percentage of survival over
time, wherein some mice (HeLa-CD19 + CAR-T + apoptotic cells) were still alive
at 140 days (Figure
35B).
[1220] Analysis of the macrophage subpopulations over the course of
treatment revealed
surprising results. Figure 36A shows that based on the expression of the
residential peritoneal
macrophage signature of markers F4/80, CD11b, Tim4, and MerTK, resident
macrophages disappear
during tumor progression (compare the top (no tumor) and middle (Hela-CD19
tumor) rows).
Surprisingly, treatment with CAR-T cells resulted in the reappearance of
resident macrophages
(bottom row +CART). Concurrently, measure of the expression of tumor
associated macrophage
(TAM) signature markers CCR2, Ly6c, CD206, CD64, CD169, and CD74 (Figure 36B)
showed that
TAM appeared during tumor progression (compare the top (no tumor) and middle
(Hela-CD19
tumor) rows). Treatment with CART cells resulted in the reduction and
disappearance of these TAM
(bottom row +CART). Resident macrophages in SCID mice were identified as
mostly large peritoneal
macrophages (LPM) (Figure 36C). The results shown in Figures 36A-36C show that
all of the
F4/80high cells are CD1lb positive, and that only 13.5% of the F4/80high cells
were MHCII positive.
Thus, the population of resident macrophages in SCID mice were
F4/80high/TIM4+, MHCIIlow,
MerTK+/CD1 lb+, and during tumor progression this population disappears, and
TAM, monocytes,
and dendritic cells appear. Treatment with CAR T-cells reverses this effect,
wherein resident
macrophages reappear, and TAM disappear.
[1221] To address the role of the resident macrophages present following
CAR T cell, SCID-Bg
mice were injected intra-peritoneally with human HeLa-CD19-luciferase cells,
followed by CD19-
CAR T cells, with or without apoptotic cells (Allocetra-OTS), or opsonized
apoptotic cells
(D89E Allocetra-OTS) that avoids clearance of apoptotic cells by resident
macrophages. The results
unexpectedly showed that in the absence of clearance of administered apoptotic
cells (Hela-CD19
+CAR-T + D89E Allocetra-OTS) the percent survival of mice bearing a tumor
burden was reduced
to almost the level seen with CAR T cell administration alone (Hela-CD19 + CAR-
T) (Figure 37).
This result suggests resident macrophages may, in some embodiments, be an
effective tool during
CA 03151815 2022-02-17
222
WO 2021/053667
PCT/IL2020/051011
cancer therapy, wherein resident macrophage markers could be targeted to
trigger resident
macrophage activation following CAR T-cell administration. Furthermore, one
skilled in the art
would appreciate that the cancer therapy could comprise a cancer therapy other
than CAR T cell
therapy. For example, an immunotherapy, a radiation therapy, a chemotherapy, a
transplantation, a
targeted therapy, a hormone therapy, a photodynamic therapy, or surgery, or a
combination thereof,
wherein stabilization of resident macrophages would be an effective tool for
the cancer therapy.
[1222] Summary: As discussed, Chimeric Antigen Receptor (CAR) T cell therapy
is a novel and
innovative immunotherapy. CAR-T cells are genetically engineered T cells,
carrying MHC
independent specific antigen receptor and co-stimulatory molecule which can
activate an immune
response to a cancer specific antigen. This therapy showed great results in
hematological
malignancies but could not achieve a similar efficacy in solid tumors both
hematological and non-
hematological. Likely reasons for this failure may be lack of antigens, poor
trafficking, and hostile
tumor microenvironment. Here, the effect of CAR T cell therapy on human
peritoneal solid tumor
model in SCID mice was demonstrated, wherein the synergistic effect of
apoptotic cell infusion on
the anti-cancer action of CAR T-cell therapy was significant.
[1223] The results presented here, show that mice survived 30 5 days, and
mock treatment non
significantly ameliorated their survival to 34 4 days. CAR T cell therapy
significantly ameliorated
their survival to 55 11 days. Single-cell analysis confirmed by flow cytometry
revealed that resident
macrophages that were associated with anti-tumor activity, completely
disappeared during tumor
progression, and reappeared during successful CAR T therapy. Apoptotic cells
injected during tumor
progression were able to stabilize the presence of macrophages, as confirmed
by single cell and flow
cytometry analysis, and synergize with the anti-tumor CAR-T cell effect,
resulting in significantly
increased anti-tumor macrophage population and increased survival to 75 10
days (p<0.05).
[1224] While certain features disclosed herein have been illustrated and
described herein, many
modifications, substitutions, changes, and equivalents will now occur to those
of ordinary skill in the
art. It is, therefore, to be understood that the appended claims are intended
to cover all such
modifications and changes as fall within the true spirit disclosed herein.