Note: Descriptions are shown in the official language in which they were submitted.
WO 2021/165370
PCT/EP2021/053973
- 1 -
MACROCYCLIC INDOLE DERIVATIVES AS INHIBITORS OF MCL-1
FIELD OF THE INVENTION
The present invention relates to pharmaceutical agents useful for therapy
and/or
prophylaxis in a subject, pharmaceutical composition comprising such
compounds, and
their use as MCL-1 inhibitors, useful for treating or preventing diseases such
as cancer.
BACKGROUND OF THE INVENTION
Cellular apoptosis or programmed cell death is critical to the development and
homeostasis of many organs including the hematopoietic system. Apoptosis can
be
initiated via the extrinsic pathway, which is mediated by death receptors, or
by the
intrinsic pathway using the B cell lymphoma (BCL-2) family of proteins.
Myeloid cell
leukemia-1 (MC1,-1) is a member of the BCT,-2 family of cell survival
regulators and is
a critical mediator of the intrinsic apoptosis pathway. MCL-1 is one of five
principal
anti-apoptotic BCL-2 proteins (MCL-1, BCL-2, BCL-XL, BCL-w, and BFL 1/A1)
responsible for maintaining cell survival. MCL-1 continuously and directly
represses the
activity of the pro-apoptotic BCL-2 family proteins Bak and Bax and indirectly
blocks
apoptosis by sequestering BH3 only apoptotic sensitizer proteins such as Bim
and Noxa.
The activation of BaldBax following various types of cellular stress leads to
aggregation
on the mitochondrial outer membrane and this aggregation facilitates pore
formation,
loss of mitochondrial outer membrane potential, and subsequent release of
cytochrome
C into the cytosol. Cytosolic cytochrome C binds Apaf-1 and initiates
recruitment of
procaspase 9 to form apoptosome structures (Cheng etal. eLife 2016; 5:
e17755). The
assembly of apoptosomes activates the executioner cysteine proteases 3/7 and
these
effector caspases then cleave a variety of cytoplasmic and nuclear proteins to
induce cell
death (Julian etal. Cell Death and Differentiation 2017; 24, 1380-1389).
Avoiding apoptosis is an established hallmark of cancer development and
facilitates the survival of tumor cells that would otherwise be eliminated due
to
oncogenic stresses, growth factor deprivation, or DNA damage (Hanahan and
Weinberg.
Cell 2011;1-44). Thus, unsurprisingly, MCL-1 is highly upregulated in many
solid and
hematologic cancers relative to normal non-transformed tissue counterparts.
The
overexpression of MCL-1 has been implicated in the pathogenesis of several
cancers
where it correlated with poor outcome, relapse, and aggressive disease.
Additionally,
overexpression of MCL-1 has been implicated in the pathogenesis of the
following
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 2 -
cancers: prostate, lung, pancreatic, breast, ovarian, cervical, melanoma, B-
cell chronic
lymphocytic leukemia (CLL), acute myeloid leukemia (AML), and acute
lymphoblastic
leukemia (ALL). The human MCL-1 genetic locus (1q21) is frequently amplified
in
tumors and quantitatively increases total MCL-1 protein levels (Beroukhim
etal. Nature
2010;463 (7283) 899-905). MCL-1 also mediates resistance to conventional
cancer
therapeutics and is transcriptionally upregulated in response to inhibition of
BCL-2
function (Yecies etal. Blood 2010;115 (16)3304-3313).
A small molecule BH3 inhibitor of BCL-2 has demonstrated clinical efficacy in
patients with chronic lymphocytic leukemia and is FDA approved for patients
with CLL
or AML (Roberts et al. NE,JM 2016;374:311-322). The clinical success of BCL-2
antagonism led to the development of several MCL-1 BH3 mimetics that show
efficacy
in preclinical models of both hematologic malignancies and solid tumors
(Kotschy etal.
Nature 2016;538 477-486, Merino et Sci Transl. Med;2017 (9)).
MCL-1 regulates several cellular processes in addition to its canonical role
in
mediating cell survival including mitochondrial integrity and non-homologous
end
joining following DNA damage (Chen etal. JCI 2018;128(1):500-516). The genetic
loss
of MCL-1 shows a range of phenotypes depending on the developmental timing and
tissue deletion. MCL-1 knockout models reveal there are multiple roles for MCL-
1 and
loss of function impacts a wide range of phenotypes. Global MCL-1-deficient
mice
display embryonic lethality and studies using conditional genetic deletion
have reported
mitochondrial dysfunction, impaired activation of autophagy, reductions in B
and T
lymphocytes, increased B and T cell apoptosis, and the development of heart
failure/
cardiomyopathy (Wang etal. Genes and Dev 2013;27 1351-1364, Steimer et aL
Blood
2009;(113) 2805-2815).
W02018178226 discloses MCL-1 inhibitors and methods of use thereof.
W02017182625 discloses macrocyclic MCL-1 inhibitors for treating cancer.
W02018178227 discloses the synthesis of MCL-1 inhibitors.
W02020063792 discloses indole macrocyclic derivatives.
CN110845520 discloses macrocyclic indoles as MCL-1 inhibitors.
W02020103864 discloses macrocyclic indoles as MCL-1 inhibitors.
There remains a need for MCL-1 inhibitors, useful for the treatment or
prevention
of cancers such as prostate, lung, pancreatic, breast, ovarian, cervical,
melanoma, B-cell
chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML), and acute
lymphoblastic leukemia (ALL).
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 3 -
SUMMARY OF THE INVENTION
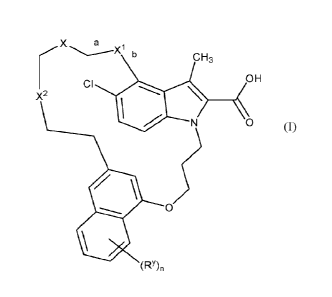
The present invention concerns novel compounds of Formula (I)
X a
b
CH3
OH
CI
X2
0 (I)
05
(RY),
and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
R1
R1
\õ.
N¨N 1=41\1
Or
b b
wherein 'a' and b' indicate how variable X1 is attached to the remainder of
the
molecule;
and R2 each independently represent hydrogen; methyl, or C2_6alkyl optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -0R3, and -NR43R4b;
Het' represents morpholinyl or tetrahydropyranyl;
12.1 represents hydrogen, Ci_4alkyl, -C2_4alkyl-O-C1_4a1ky1, -C2_4alkyl-OH, or
-C
R4a and R4b are each independently selected from the group consisting of
hydrogen and
Ci_4alkyl;
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 4 -
X2 represents
Nni
R2/ .%
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)-, -S(=0)2-, or -N(R)-;
IV represents hydrogen, methyl, C2.6allcyl, -S(=0)2-C1-6allcyl,
C3.6cycloalkyl, -C(=0)-C3.6cyc1oallcy1, or -S(-0)2-C3_6cyc1oalkyl; wherein
C2.6alkyl,
-C(=0)-C14allcyl, -S(=0)2-C1-6a1lcy1, Cmcycloallcyl, -C(=0)-C34cycloallcyl,
and
-S(=0)2-C3.6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci_aallcyl and C _Al icy]
substituted with one,
two or three halo atoms;
RY represents halo;
n represents 0, 1 or 2;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention also relates to a pharmaceutical composition comprising
a
therapeutically effective amount of a compound of Formula (1), a
pharmaceutically
acceptable salt, or a solvate thereof, and a pharmaceutically acceptable
carrier or
excipient.
Additionally, the invention relates to a compound of Formula (I), a
pharmaceutically acceptable salt, or a solvate thereof, for use as a
medicament, and to a
compound of Formula (I), a pharmaceutically acceptable salt, or a solvate
thereof, for
use in the treatment or in the prevention of cancer.
In a particular embodiment, the invention relates to a compound of Formula
(I),
a pharmaceutically acceptable salt, or a solvate thereof, for use in the
treatment or in the
prevention of cancer.
The invention also relates to the use of a compound of Formula (I), a
pharmaceutically acceptable salt, or a solvate thereof, in combination with an
additional
pharmaceutical agent for use in the treatment or prevention of cancer.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 5 -
Furthermore, the invention relates to a process for preparing a pharmaceutical
composition according to the invention, characterized in that a
pharmaceutically
acceptable carrier is intimately mixed with a therapeutically effective amount
of a
compound of Formula (I), a pharmaceutically acceptable salt, or a solvate
thereof.
The invention al so relates to a product comprising a compound of Formula (I),
a
pharmaceutically acceptable salt, or a solvate thereof, and an additional
pharmaceutical
agent, as a combined preparation for simultaneous, separate or sequential use
in the
treatment or prevention of cancer.
Additionally, the invention relates to a method of treating or preventing a
cell
proliferative disease in a subject which comprises administering to the said
subject an
effective amount of a compound of Formula (I), a pharmaceutically acceptable
salt, or a
solvate thereof, as defined herein, or a pharmaceutical composition or
combination as
defined herein.
DETAILED DESCRIPTION OF THE INVENTION
The term 'halo' or 'halogen' as used herein represents fluor , chloro, bromo
and
iodo.
The prefix
(where x and y are integers) as used herein refers to the number
of carbon atoms in a given group. Thus, a C1-6a1kyl group contains from 1 to 6
carbon
atoms, and so on.
The term `Ci_iallcyr as used herein as a group or part of a group represents a
straight or branched chain fully saturated hydrocarbon radical having from 1
to 4 carbon
atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl
and the like.
The term 'CI-6alkyr as used herein as a group or part of a group represents a
straight or branched chain fully saturated hydrocarbon radical having from 1
to 6 carbon
atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl,
n-pentyl, 11-
hexyl and the like.
The term `C24allcyr as used herein as a group or part of a group represents a
straight or branched chain fully saturated hydrocarbon radical having from 2
to 4 carbon
atoms, such as ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl and the
like.
The term `C2-6alkyr as used herein as a group or part of a group represents a
straight or branched chain fully saturated hydrocarbon radical having from 2
to 6 carbon
atoms, such as ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl, n-
pentyl, n-hexyl and
the like.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 6 -
The term `C3_6cycloalkyl' as used herein as a group or part of a group defines
a
fully saturated, cyclic hydrocarbon radical having from 3 to 6 carbon atoms,
such as
cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
It will be clear for the skilled person that S(=0)2 or SO2 represents a
sulfonyl
moiety.
It will be clear for the skilled person that CO or C(=0) represents a carbonyl
moiety.
In general, whenever the term 'substituted' is used in the present invention,
it is
meant, unless otherwise indicated or clear from the context, to indicate that
one or more
hydrogens, in particular from 1 to 4 hydrogens, more in particular from 1 to 3
hydrogens,
preferably 1 or 2 hydrogens, more preferably 1 hydrogen, on the atom or
radical indicated
in the expression using 'substituted' are replaced with a selection from the
indicated
group, provided that the normal valency is not exceeded, and that the
substitution results
in a chemically stable compound, i.e. a compound that is sufficiently robust
to survive
isolation to a useful degree of purity from a reaction mixture.
Combinations of substituents and/or variables are permissible only if such
combinations result in chemically stable compounds. 'Stable compound' is meant
to
indicate a compound that is sufficiently robust to survive isolation to a
useful degree of
purity from a reaction mixture.
The skilled person will understand that the term 'optionally substituted'
means
that the atom or radical indicated in the expression using 'optionally
substituted' may or
may not be substituted (this means substituted or unsubstituted respectively).
When two or more substituents are present on a moiety they may, where possible
and unless otherwise indicated or clear from the context, replace hydrogens on
the same
atom or they may replace hydrogen atoms on different atoms in the moiety.
It will be clear for the skilled person that
R1
R1
N-N N-N
is an alternative representation for a j/
b b
CA 03168355 2022 8 17
WO 2021/165370
PCT/EP2021/053973
- 7 -
It will be clear for the skilled person that
R R _NI
_N
is an alternative representation for H 3
b ib
It will be clear that a Compound of Formula (I) includes Compounds of
/
2/ %=.
Formula (I-x) and (I-y) (both directions of X2 being R
_a yi
a _Ari
CH3
CH3
N / CI OH
OH
\ CI
0
R2
0
(I-x)
(1-y)
0
4110 0
11111
(R Y), (RY),,
When any variable occurs more than one time in any constituent or in any
formula (e.g.
Formula (1)), each definition is independent.
The term -subject" as used herein, refers to an animal, preferably a mammal
(e.g. cat,
dog, primate or human), more preferably a human, who is or has been the object
of
treatment, observation or experiment.
The tei ____________ in "therapeutically effective amount" as used herein,
means that amount of active
compound or pharmaceutical agent that elicits the biological or medicinal
response in a
tissue system, or subject (e.g., human) that is being sought by a researcher,
veterinarian,
medicinal doctor or other clinician, which includes alleviation or reversal of
the
symptoms of the disease or disorder being treated.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 8 -
The term "composition" is intended to encompass a product comprising the
specified
ingredients in the specified amounts, as well as any product which results,
directly or
indirectly, from combinations of the specified ingredients in the specified
amounts.
The term "treatment", as used herein, is intended to refer to all processes
wherein there
may be a slowing, interrupting, arresting or stopping of the progression of a
disease, but
does not necessarily indicate a total elimination of all symptoms.
The term "compound(s) of the (present) invention" or "compound(s) according to
the
(present) invention" as used herein, is meant to include the compounds of
Formula (I)
and the pharmaceutically acceptable salts, and the solvates thereof.
As used herein, any chemical formula with bonds shown only as solid lines and
not as
solid wedged or hashed wedged bonds, or otherwise indicated as having a
particular
configuration (e.g. R, ,S) around one or more atoms, contemplates each
possible
stereoisomer, or mixture of two or more stereoisomers.
Hereinbefore and hereinafter, the term "compound(s) of Formula (1)" is meant
to include
the tautomers thereof and the stereoisomeric forms thereof.
The terms "stereoisomers", "stereoisomeric forms" or "stereochemically
isomeric
forms" hereinbefore or hereinafter are used interchangeably.
The invention includes all stereoisomers of the compounds of the invention
either as a
pure stereoisomer or as a mixture of two or more stereoisomers.
Enantiomers are stereoisomers that are non-superimposable mirror images of
each other.
A 1:1 mixture of a pair of enantiomers is a racemate or racemic mixture.
Atropisomers (or atropoisomers) are stereoisomers which have a particular
spatial
configuration, resulting from a restricted rotation about a single bond, due
to large steric
hindrance. All atropisomeric forms of the compounds of Formula (I) are
intended to be
included within the scope of the present invention.
In particular, the compounds disclosed herein possess axial chirality, by
virtue of
restricted rotation around a biaryl bond and as such may exist as mixtures of
atropisomers. When a compound is a pure atropisomer, the stereochemistry at
each chiral
center may be specified by either Ra or S.. Such designations may also be used
for
mixtures that are enriched in one atropisomer. Further description of
atropisomerism and
axial chirality and rules for assignment of configuration can be found in
Eliel, E.L. &
Wilen, S. H. 'Stereochemistry of Organic Compounds' John Wiley and Sons, Inc.
1994.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 9 -
Di astereomers (or di astereoi somers) are stereoisomers that are not
enantiomers, i .e. they
are not related as mirror images. If a compound contains a double bond, the
substituents
may be in the E or the Z configuration.
Substituents on bivalent cyclic saturated or partially saturated radicals may
have either
the cis- or trans-configuration; for example if a compound contains a di sub
stituted
cycloalkyl group, the substituents may be in the cis or trans configuration.
Therefore, the invention includes enantiomers, atropisomers, diastereomers,
racemates,
E isomers, Z isomers, cis isomers, trans isomers and mixtures thereof,
whenever
chemically possible
The meaning of all those terms, i.e. enantiomers, atropisomers, diastereomers,
racemates,
E isomers, Z isomers, cis isomers, trans isomers and mixtures thereof are
known to the
skilled person.
The absolute configuration is specified according to the Cahn-Ingold-Prelog
system. The
configuration at an asymmetric atom is specified by either R or S. Resolved
stereoisomers whose absolute configuration is not known can be designated by
(+) or
(-) depending on the direction in which they rotate plane polarized light. For
instance,
resolved enantiomers whose absolute configuration is not known can be
designated by
(+) or (-) depending on the direction in which they rotate plane polarized
light. Optically
active (Ra)- and (Sa)-atropisomers may be prepared using chiral synthons,
chiral reagents
or chiral catalysts, or resolved using conventional techniques well known in
the art, such
as chiral HPLC.
When a specific stereoisomer is identified, this means that said stereoisomer
is
substantially free, i.e. associated with less than 50%, preferably less than
20%, more
preferably less than 10%, even more preferably less than 5%, in particular
less than 2%
and most preferably less than 1%, of the other stereoisomers. Thus, when a
compound
of Formula (I) is for instance specified as (R), this means that the compound
is
substantially free of the (S) isomer; when a compound of Formula (I) is for
instance
specified as E, this means that the compound is substantially free of the Z
isomer; when
a compound of Formula (I) is for instance specified as cis, this means that
the compound
is substantially free of the trans isomer; when a compound of Formula (I) is
for instance
specified as Ra, this means that the compound is substantially free of the Sa
atropisomer.
Pharmaceutically acceptable salts, in particular pharmaceutically acceptable
additions
salts, include acid addition salts and base addition salts. Such salts may be
formed by
conventional means, for example by reaction of a free acid or a free base form
with one
or more equivalents of an appropriate base or acid, optionally in a solvent,
or in a medium
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 10 -
in which the salt is insoluble, followed by removal of said solvent, or said
medium, using
standard techniques (e.g. in vacuo, by freeze-drying or by filtration). Salts
may also be
prepared by exchanging a counter-ion of a compound of the invention in the
form of a
salt with another counter-ion, for example using a suitable ion exchange
resin.
The pharmaceutically acceptable salts as mentioned hereinabove or hereinafter
are meant
to comprise the therapeutically active non-toxic acid and base salt forms
which the
compounds of Formula (I), and solvates thereof, are able to form.
Appropriate acids comprise, for example, inorganic acids such as hydrohalic
acids, e.g.
hydrochloric or hydrobrom ic acid, sulfuric, nitric, phosphoric and the like
acids; or
organic acids such as, for example, acetic, propanoic, hydroxyacetic, lactic,
pyruvic,
oxalic (i.e. ethanedioic), malonic, succinic (i.e. butanedioic acid), maleic,
fumaric, malic,
tartaric, citric, methanesulfonic, ethanesulfonic, benzenesulfonic, p-
toluenesulfonic,
cyclamic, salicylic, p-aminosalicylic, pamoic and the like acids. Conversely
said salt
forms can be converted by treatment with an appropriate base into the free
base form.
I 5 The compounds of Formula (I) and solvates thereof containing an acidic
proton may also
be converted into their non-toxic metal or amine salt forms by treatment with
appropriate
organic and inorganic bases.
Appropriate base salt forms comprise, for example, the ammonium salts, the
alkali and
earth alkaline metal salts, e.g. the lithium, sodium, potassium, cesium,
magnesium,
calcium salts and the like, salts with organic bases, e.g. primary, secondary
and tertiary
aliphatic and aromatic amines such as methylamine, ethylamine, propylamine,
isopropylamine, the four butylamine isomers, dimethylamine, diethylamine,
diethanolamine, dipropylamine, diisopropylamine, di-n-butylamine, pyrrolidine,
piperidine, morpholine, trimethylamine, triethylamine, tripropylamine,
quinuclidine,
pyridine, quinoline and isoquinoline; the benzathine, N-methyl-D-glucamine,
hydrabamine salts, and salts with amino acids such as, for example, arginine,
lysine and
the like. Conversely the salt form can be converted by treatment with acid
into the free
acid form.
The term solvate comprises the solvent addition forms as well as the salts
thereof, which
the compounds of Formula (I) are able to form. Examples of such solvent
addition forms
are e.g. hydrates, alcoholates and the like.
The compounds of the invention as prepared in the processes described below
may be
synthesized in the form of mixtures of enantiomers, in particular racemic
mixtures of
enantiomers, that can be separated from one another following art-known
resolution
procedures. A manner of separating the enantiomeric forms of the compounds of
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 11 -
Formula (I), and pharmaceutically acceptable salts, and solvates thereof,
involves liquid
chromatography using a chiral stationary phase. Said pure stereochemically
isomeric
forms may also be derived from the corresponding pure stereochemically
isomeric forms
of the appropriate starting materials, provided that the reaction occurs
stereospecifically.
Preferably if a specific stereoisomer is desired, said compound would be
synthesized by
stereospecific methods of preparation. These methods will advantageously
employ
enantiomerically pure starting materials.
The term "enantiomerically pure" as used herein means that the product
contains at least
80% by weight of one enantiomer and 20% by weight or less of the other
enantiomer.
Preferably the product contains at least 90% by weight of one enantiomer and
10% by
weight or less of the other enantiomer. In the most preferred embodiment the
term
"enantiomerically pure" means that the composition contains at least 99% by
weight of
one enantiomer and 1% or less of the other enantiomer.
The present invention also embraces isotopically-labeled compounds of the
present
invention which are identical to those recited herein, but for the fact that
one or more
atoms are replaced by an atom having an atomic mass or mass number different
from the
atomic mass or mass number usually found in nature (or the most abundant one
found in
nature).
All isotopes and isotopic mixtures of any particular atom or element as
specified herein
are contemplated within the scope of the compounds of the invention, either
naturally
occurring or synthetically produced, either with natural abundance or in an
isotopically
enriched form. Exemplary isotopes that can be incorporated into compounds of
the
invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus,
sulfur,
fluorine, chlorine and iodine, such as 2H, 3H, "C, 13c, '4C, 13N, 150, 170,
180, 32p, 33F,
35s, 18F, 36c1, 1221, 1231, 1251, 131-,
1 75Br, 76Br, 77Br and 'Br. Preferably, the isotope is
selected from the group of 2H, 3H, tic and 18F. More preferably, the isotope
is 2H. In
particular, deuterated compounds are intended to be included within the scope
of the
present invention.
Certain isotopically-labeled compounds of the present invention (e.g., those
labeled with
3H and I4C) may be useful for example in substrate tissue distribution assays.
Tritiated
(3H) and carbon-14 ('4C) isotopes are useful for their ease of preparation and
detectability. Further, substitution with heavier isotopes such as deuterium
(i.e., 2H) may
afford certain therapeutic advantages resulting from greater metabolic
stability (e.g.,
increased in vivo half-life or reduced dosage requirements) and hence may be
preferred
in some circumstances. Positron emitting isotopes such as 150 "N, "C and '8F
are useful
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 12 -
for positron emission tomography (PET) studies. PET imaging in cancer finds
utility in
helping locate and identify tumours, stage the disease and determine suitable
treatment.
Human cancer cells overexpress many receptors or proteins that are potential
disease-
specific molecular targets. Radiolabelled tracers that bind with high affinity
and
specificity to such receptors or proteins on tumour cells have great potential
for
diagnostic imaging and targeted radionuclide therapy (Charron, Carlie L. et
al.
Tetrahedron Lett. 2016, 57(37), 4119-4127).
Additionally, target-specific PET
radiotracers may be used as biomarkers to examine and evaluate pathology, by
for
example, measuring target expression and treatment response (Austin R. et al.
Cancer
Letters (2016), doi: 10.1016/j.canlet.2016.05.008).
The present invention relates in particular to compounds of Formula (I) as
defined
herein, and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
R1
R1
,s.
N¨N 1,11=4
or
b b
wherein 'a' and 'b' indicate how variable X1 is attached to the remainder of
the
molecule;
RI and R2 each independently represent hydrogen; methyl; or C2_6alkyl
optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -0R3, and -NR4aR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, Ci_4alkyl, -C2_4a1ky1-O-C14alkyl, or
kyl -0 -C1_4alkyl ;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
CI _4alkyl;
X2 represents
NN
2/ s==,
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 13 -
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(-0)2-, or -N(R)c)-;
Rx represents hydrogen, methyl, C2_6alkyl, -C(=0)-C1_6alky1, -S(-0)2-
C1_6a1ky1,
C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl, or -S(=0)2-C3_6cyc10a1ky1; wherein
C2_6a1kyl,
-C(=0)-Ci_6a1ky1, -S(=0)2-Ci_6a1ky1, C3-6cycloalkyl, -C(=0)-C3_6cycloalkyl,
and
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, C3_4alkyl and Ci_4alkyl
substituted with one,
two or three halo atoms;
RY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (1) as
defined
herein, and the tautomers and the stereoisomeric foi __ ins thereof, wherein
XI represents
R1 1
RN
N¨N N¨N
or
b b
wherein 'a' and 'b' indicate how variable X1 is attached to the remainder of
the
molecule;
RI and R2 each independently represent hydrogen; methyl; or C2_6alkyl
optionally
substituted with one or two sub stituents each independently selected from the
group
consisting of Het', -0R3, and -NR4aR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, Ci_4alkyl, -C2_4alkyl-O-C1_4alkyl, or
-C2_4alkyl-O-C2_4alky1-0-C1_4alkyl;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
Ci ialkyl;
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 14 -
X2 represents
\N /
2/ s=
which can be attached to the remainder of the molecule in both directions;
X represents 0 , S , S(-0)2-, or
Rµ represents hydrogen, methyl, C2_6alky1, -C(=0)-Ci_6a1ky1, -S(=0)2-
Ct_6alkyl,
C3_6cycloalkyl, -C(-0)-C3_6cycloa1kyl, or -S(=0)2-C3_6cyc1oalkyl; wherein
C2_6alkyl,
-C(=0)-C1_6a1kyl, -S(=0)2-C1_6a1kyl, C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl,
and
-S(=0)-C3_6cycloalky1 are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci_4alkyl and C1_4alkyl
substituted with one,
two or three halo atoms;
BY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof;
provided that
z
OH
CI
N
\N
0
o
and the tautomers and the stereoisomeric forms thereof are excluded
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 15 -
XI represents
R1
R1
N-N
N-N
or
ib
wherein 'a' and `b' indicate how variable X1 is attached to the remainder of
the
molecule;
and R2 each independently represent hydrogen; methyl, or C2_6alkyl optionally
substituted with one substituent selected from the group consisting of [let', -
OR3, and
_NR4aR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, C1_4a1kyl, or -C2_4alky1-O-C1_4alkyl;
R4a and RTh are each independently selected from the group consisting of
hydrogen and
Ci_4alkyl;
X2 represents
NN _________________
2/ N,
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
ft' represents hydrogen, methyl, C2_6alkyl, -C(=0)-C,_6alkyl, -S(-0)2-
C1_6alkyl,
C3-6cyc1oa1ky1, -C(=0)-C3-6cycloalkyl, or -S(=0)2-C3-6cycloalkyl; wherein
C2_6a1ky1,
-S(=0)2-C1_6a1ky1, C3-6cyc10a1ky1, -C(=0)-C3-6cyc10a1ky1, and
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, C1_4alkyl and Ci_4a1ky1
substituted with one,
two or three halo atoms;
R) represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 16 -
The present invention relates in particular to compounds of Formula (1) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X' represents
R\N-N
LL
b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the
molecule;
It' and R2 each independently represent hydrogen; methyl; or C2-6a1lcy1
optionally
substituted with one substituent selected from the group consisting of Het', -
OW, and
_NR4aR4b;
Het" represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, CI-alkyl, or -C2_4a1lcy1-0-CI-4a1ky1;
It4a and R41' are each independently selected from the group consisting of
hydrogen and
X2 represents
NN /
2/
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or -N(Rx)-;
Itx represents hydrogen, methyl, C2.6a1lcy1, -C(=0)-C1-6allcyl, -S(=0)2-C1-
6allcyl,
C3-6cyc10a1lcy1, -C(=0)-C3-6cycloallcyl, or -S(=0)2-C3-6cycloallcyl; wherein
C2-6allcyl,
-C(=0)-Ci_6a1ky1, -S(=0)2-C1.6a1ky1, Cmcycloalkyl, -C(=0)-C3cycloalkyl, and
-S(=0)2-C3.6cyc10a1ky1 are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci-aalkyl and C14allcyl
substituted with one,
two or three halo atoms;
W represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 17 -
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
R1
R1
\N¨N
N¨N
or
ib
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
Rl and R2 each independently represent hydrogen; methyl; or C2.6alkyl
optionally
substituted with one substituent selected from the group consisting of Het', -
OW, and -
NleaR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, Ci_4a1kyl, or -C24alky1-0-Ci_4alkyl;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
C1_4alkyl;
X2 represents
NN
2/ ss,
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
represents hydrogen, methyl, C2_6alky1, -C(=0)-C1_6alkyl, -S(=0)2-Ci_6a1kyl,
C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl, or -S(=0)2-C3_6cycloalky1; wherein
C2_6alkyl,
-S(=0)2-Ci_6a1ky1, C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl, and
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci_4alkyl and Ci_4alkyl
substituted with one,
two or three halo atoms;
RY represents halo;
n represents 1;
and the pharmaceutically acceptable salts and the solvates thereof.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 18 -
The present invention relates in particular to compounds of Formula (1) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X' represents
RI
\N¨N
b
wherein 'a' and 'b' indicate how variable X3 is attached to the remainder of
the molecule;
and R2 each independently represent hydrogen; methyl; or C2.6a1lcy1 optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -Ole, and -MeaR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, C14alkyl, -C24a1lcyl-O-Ci4a1lcy1, or
-C24alky1-0-C2-4a1ky1-0-C14allcyl;
R4a and leb are each independently selected from the group consisting of
hydrogen and
Ci_aallcyl;
X2 represents
N
N
2/
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or -N(W);
Rx represents hydrogen, methyl, C2.6allcyl, -C(=0)-C1-6a1lcy1, -S(0)2-C1-
alkyl,
C3-6cyc10a1lcy1, -C(=0)-C3-6cycloallcyl, or -S(=0)2-C3-6cyc1oallcyl; wherein
C2-6allcyl,
-C(=0)-Ci_6a1ky1, -S(=0)2-C1.6a1ky1, Cmcycloalkyl, -C(=0)-C3cycloalkyl, and
-S(=0)2-C3.6cyc1oa1ky1 are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci-aallcyl and C14allcyl
substituted with one,
two or three halo atoms;
W represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 19 -
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X1 represents
R1
R1
\N¨N
N¨N
or
b b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
R1 and R2 each independently represent hydrogen; methyl; or C2_6alkyl
optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -0R3, and -NleaR4b;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, Ci_4alkyl, -C2_4alky1-O-Ci_4a1ky1, or
-C2_4a1kyl-O-C2_4alky1-0-C1_4a1ky1;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
Ci_4alkyl;
X2 represents
N
NN _________________
2/
which can be attached to the remainder of the molecule in both directions;
X represents 0 , S , S(-0)2-, or
IV represents hydrogen, methyl, C2_6alky1, -C(=0)-Ci_6a1ky1, -S(=0)2-
C1_6alkyl,
C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl, or -S(=0)2-C3_6cycloalky1; wherein
C2_6a1kyl,
-C(=0)-Ci_6a1kyl, -S(=0)2-Ci_6alkyl, C3_6cycloalkyl, -C(=0)-C3_6cycloalkyl,
and
-S(=0)2-C3_6cycloalky1 are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci_zialkyl and CiAalkyl
substituted with one,
two or three halo atoms;
RY represents halo,
n represents 1;
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-20 -
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X1 represents
R1
R1
N-N
N-N
or
b b
wherein 'a' and 'b' indicate how variable Xl is attached to the remainder of
the molecule;
RI represents C2_6alkyl substituted with two substituents each independently
selected
b
from the group consisting of Het', -0R3, and _NR4aR4; wherein R3 represents
hydrogen, Ci_4alkyl, -C2_4alky1-O-C1_4a1ky1, or
or
R1 represents C2_6alkyl substituted with one or two -Ole substituents; wherein
R3
represents -C2_4alkyl-O-C2_4alky1-0-C1_4a1ky1;
R2 represents methyl;
Het' represents morpholinyl or tetrahydropyranyl;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
Ci_4alkyl;
X2 represents
2/
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
It,' represents hydrogen, methyl, C2_6alkyl, -C(=0)-Ci_6a1ky1, -S(=0)2-
C1_6alkyl,
C3_6cycloalkyl, -C(=0)-C3-6cycloalkyl, or -S(=0)2-C3-6cycloalkyl; wherein
C6a1ky1,
-C(=0)-Ci_6alkyl, -S(=0)2-C1_6alkyl, C3-6cycloalkyl, -C(=0)-C3-6cycloalkyl,
and
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-21 -
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, Ci_4alky1 and Ci_4alkyl
substituted with one,
two or three halo atoms;
RY represents halo,
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X' represents
R1
R1
Nk.
N¨N 1,11
or
b b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
Rl represents C2.6alkyl substituted with two substituents each independently
selected
from the group consisting of Het', -0R3, and -NR4aR4b,
R2 represents methyl;
Het' represents morpholinyl or tetrahydropyranyl;
R3 represents hydrogen, C1_4alkyl, -C2_4alkyl-O-C1_4a1kyl, or
-C2_4a1kyl-O-C2-4alky1-0-Ci_4a1ky1;
R4a and R4b are each independently selected from the group consisting of
hydrogen and
C14alkyl;
X2 represents
N
NN /
2/ ss,
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-22 -
Rx represents hydrogen, methyl, C2_6alkyl, -C(=0)-C1_6alkyl, -S(=0)2-
C1_6,alkyl,
C3_6cycloalkyl, -C(=0)-C3-6cycloalkyl, or -S(=0)2-C3_6cycloalkyl; wherein
C2_6alky1,
-C(=0)-C1_6a1ky1, -S(=0)2-C1_6alkyl, C3_6cycloalkyl, -C(=0)-C3-6cycloalkyl,
and
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, C1_4alkyl and C1_4alkyl
substituted with one,
two or three halo atoms;
BY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
X1 represents
R1
R1
NN¨N
N¨N
or
b b
wherein 'a' and 'b' indicate how variable X1 is attached to the remainder of
the molecule;
R1 represents C2_6alkyl substituted with one or two -Ole substituents;
R2 represents methyl;
R3 represents -C2_4alkyl-O-C2_4alky1-0-Ch4alkyl;
X2 represents
N
NN /
2/ ==
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
Rx represents hydrogen, methyl, C2_6alkyl, -C(=0)-C1_6alkyl, -S(=0)2-
C1_6alkyl,
C3_6cycloalkyl, -C(=0)-C3-6cycloalky1, or -S(=0)2-C3-6cycloalkyl; wherein
C2_6alkyl,
-C(=0)-C1_6alkyl, -S(=0)2-Ci_6alkyl, C3-6cycloa1kyl, -C(=0)-C3_6cycloalkyl,
and
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-23 -
-S(=0)2-C3_6cycloalkyl are optionally substituted with one, two or three
substituents
selected from the group consisting of halo, C1_4alkyl and C1_4alkyl
substituted with one,
two or three halo atoms;
RY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
R1
R1
N-N 1,11
or
b b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
Rl and R2 each independently represent methyl, or C2.6alkyl optionally
substituted with
one or two substituents each independently selected from the group consisting
of Heti, -
OR3, and -NR4914b;
Het' represents tetrahydropyranyl;
R3 represents Ci_4alkyl, -C2_4alkyl-O-C1_4alkyl, or -C2_4alkyl-O-C2_4alky1-0-
C1_4alky1;
R4a and R4b represent hydrogen;
X2 represents
N
NN
2
/
which can be attached to the remainder of the molecule in both directions;
X represents -S-, -S(=0)2-, or -N(Rx)-;
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
-24 -
Rx represents methyl;
RY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
1 1
R R\rs. rs.
N¨N
or
b b
wherein 'a' and 'b' indicate how variable X3 is attached to the remainder of
the molecule;
R' and R2 each independently represent hydrogen; methyl; or C2_6a1ky1
optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -OW, and -NW'R4b,
Het' represents tetrahydropyranyl;
R3 represents Ci4alkyl, -C24alkyl-O-C1_4a1ky1, or -C2_4a1ky1-O-C2_4a1ky1-0-
C14alky1;
R4a and R4b represent hydrogen;
X' represents
N
2/
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(=0)2-, or
Rx represents methyl;
R3" represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
-25 -
The present invention relates in particular to compounds of Formula (I) as
defined herein,
and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
1 1
R R\N¨N
N¨N
or
b b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
Rl and R2 each independently represent hydrogen; methyl; or C2_6alkyl
optionally
substituted with one or two substituents each independently selected from the
group
consisting of Het', -0R3, and -NR4aR4b;
Het' represents tetrahydropyranyl;
R3 represents hydrogen, Ci_4a1ky1, -C2_4alky1-O-Ci_4a1ky1, -C2_4alkyl-OH, or
-C2_4a1kyl-O-C2_4alky1-0-C1_4a1ky1;
R4a and R4b represent hydrogen or Ci_4a1ky1;
X2 represents
N
NN
2/ ss,
which can be attached to the remainder of the molecule in both directions;
X represents -0-, -S-, -S(-0)-, -S(-0)2-, or
Rx represents methyl;
RY represents halo;
n represents 0, 1 or 2;
and the pharmaceutically acceptable salts and the solvates thereof.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-26 -
The present invention relates in particular to compounds of Formula (I) as
defined
herein, and the tautomers and the stereoisomeric foi __ ins thereof, wherein
Xl represents
R1
N -N
b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
Rl and R2 represent methyl;
X2 represents
which can be attached to the remainder of the molecule in both directions;
X represents -S-, -S(=0)2-, or
Rx represents methyl;
RY represents halo;
n represents 0 or 1;
and the phaimaceutically acceptable salts and the solvates thereof
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -N(Rx)-.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -S-.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 27 -
as mentioned in any of the other embodiments, wherein
X represents -0-.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -N(Rx)-; and It' represents hydrogen.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -N(Rx)-; and It' represents methyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -0-, -S-, -S(=0)2-, or -N(11.x)-
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein W represents fluoro.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
n represents 1; and
RY represents fluoro.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
n represents 2; and
W represents fluoro or chloro.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
n represents 2; and
RY represents fluoro.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
-28 -
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
XI represents
R1
N-N
b
=
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the phainiaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
XI represents
R
N-N
b
=
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n represents 1; and
XI represents
RixN-N
b
=
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n represents 2; and
XI represents
R
b
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 29 -
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein 12' represents hydrogen.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein RI represents C2-
6allcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein 12' represents methyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein R2 represents hydrogen.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein R2 represents
C2_6allcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein R2 represents methyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n represents 0.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n represents 2.
In an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
123 represents -C2-4a1ky1-O-C2-4alkyl-O-C14alkyl .
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 30 -
In an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
R3 represents hydrogen, Ci4a1lcyl, -C24a1lcy1-0-Ci4a1lcy1, or
-C24a1ky1-O-C2.4a1kyl-0-C 4alkyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
RN N¨N
b
;and
R3 represents -C2-4allcy1-0-C24alkyl-O-C14allcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
R3 represents -C2-4a1ky1-0-C24alkyl-0-C14alkyl; and n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
R N¨N
! b
R3 represents -C2-4alkyl-O-C2-4alkyl -0-CI4 al kyl ; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
It' represents C2-6alkyl substituted with one or two -0R3 substituents;
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 31 -
R2 represents methyl; and
R3 represents -C2_4alkyl-O-C2_4alky1-0-Ci_4a1ky1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
XI represents
R
NN-N
b
RI represents C2_6alkyl substituted with one or two -OR' substituents,
R2 represents methyl; and
R3 represents -C2_4alkyl-O-C2_4alkyl-0-C1_4alkyl
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof', or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
RI represents C2_6alkyl substituted with one or two -0R3 substituents;
R2 represents methyl;
R3 represents -C2-4alkyl-O-C2_4alky1-0-Ci_4a1ky1; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
Xl represents
R
NN-N
b
RI represents C/_6alkyl substituted with one or two -OW substituents;
R2 represents methyl;
R3 represents -C2_4alkyl-O-C2_4alkyl-O-C1_4a1ky1;
and n represents 1.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 32 -
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
It' represents C2_6allcyl substituted with two substituents each independently
selected
from the group consisting of Het', -01e, and -NR4aR4b; wherein R3 represents
hydrogen, C i_aallcyl, -C2_4a1kyl-O-Ci4allcyl, or -C2_4allcyl-O-C24allcyl-0-
Ci4a1lcyl;
or
It' represents C2-6allcyl substituted with one or two -OW substituents;
wherein R3
represents -C24a1lcyl-O-C24a1lcyl-0-C1.4allcyl;
R2 represents hydrogen; methyl; or C2-6allcyl optionally substituted with one
substituent
selected from the group consisting of Het', -0R3, and -NR' R4b; wherein R3
represents
hydrogen, Ci4allcyl, or -C24a1lcyl-O-Ci_aallcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
111 represents C2_6allcyl substituted with two substituents each independently
selected
-
from the group consisting of Het', _oR3, and _NR4aR4b ; wherein R3 represents
hydrogen, C1-4alkyl, -C24a1lcy1-0-C14allcyl, or
-C24a1kyl-O-C2_4alky1-0-C14alkyl;
or
RI represents C2.6a1lcy1 substituted with one or two -0113 substituents;
wherein R3
represents -C24alkyl-O-C24alkyl-O-C1-4allcyl;
R2 represents methyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
RNN ¨N
b
IV represents C2-6alkyl substituted with two substituents each independently
selected
from the group consisting of Heti, _0R3, and _NR4aR4b, wherein R3 represents
hydrogen, Ci-aallcyl, -C2-4alkyl-O-C1-4allcyl, or -C24allcyl-O-C24alkyl-0-
Ci4a1kyl;
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 33 -
or
RI represents C2-6allcyl substituted with one or two -0R3substituents; wherein
R3
represents -C2-alkyl-O-C2-4alky1-0-C 4alkyl ;
R2 represents methyl.
in an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
IV represents C2_6allcyl substituted with two substituents each independently
selected
from the group consisting of Het', _0R3, and _NRita¨ab
; wherein R3 represents
hydrogen, C14allcyl, -C24a1kyl-O-CI4alicyl, or -C24alkyl-O-C24alkyl-0-
C14a1ky1;
or
RI represents C2-6allcyl substituted with one or two -0R3 substituents;
wherein R3
represents -C24a1kyl-O-C24allcyl-0-C14allcyl;
R2 represents methyl;
and n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
R
b
=
IV represents C2.6allcyl substituted with two substituents each independently
selected
from the group consisting of Het _0R3, and _NR4aR4b ; wherein R3 represents
hydrogen, Ci4allcyl, -C24alkyl-0-C14allcyl, or -C2_4a1lcy1-0-C2_4a1lcy1-0-
C14allcyl;
or
RI represents C2.6allcyl substituted with one or two -0R3 substituents;
wherein R3
represents -C24alkyl-O-C24alky1-0-C14alkyl;
R2 represents methyl;
and n represents 1
In an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
R.' represents C24allcyl substituted with two -0R3subsfituents;
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 34 -
R2 represents methyl;
R3 represents CI-alkyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
R N -N
b
IV represents C2.6alkyl substituted with two -0R3substituents;
R2 represents methyl;
10 represents Ci-talky!.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
R.' represents C2.6allcyl substituted with two -0R3substituents;
R2 represents methyl;
R3 represents C1-alkyl; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X1 represents
1
RNN-N
b
11' represents C24alkyl substituted with two -OW substituents;
R2 represents methyl;
R3 represents CI-alkyl; and
n represents 1.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 35 -
In an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
It' represents C2_6allcyl substituted with one -0R3substituent;
R2 represents methyl; and
R3 represents -C2.4allcyl-0-C2_4alkyl-0-C14allcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
R N-N
b
RI represents Cmalkyl substituted with one -0R3substituent;
R2 represents methyl;
R3 represents -C2-4a1lcy1-0-C24alkyl-0-C14allcyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
11' represents C2-6a1lcy1 substituted with one -0R3substituent;
R2 represents methyl;
R3 represents -C2-4a1ky1-O-C24allcyl-0-C14allcyl; and
n represents =1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
RN N-N
b
IV represents C2.6alkyl substituted with one -0R3substituent;
R2 represents methyl;
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 36 -
R3 represents -C2.4alkyl-O-C24alky1-0-C14a1kyl and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
represents C2_6alkyl substituted with one substituent selected from the group
consisting of Het' or -0R3;
R2 represents methyl;
10 R3 represents -C2-4a1ky1-O-Ci4alkyl or -C2-4a1kyl-O-C2-4a1kyl-0-C1-
4a1kyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
RNN-N
b
= 15
11' represents C2.6alkyl substituted with one substituent selected from the
group
consisting of Het' or -0R3;
R2 represents methyl;
R3 represents -C2-4allcy1-0-Ci4alkyl or -C2-4a1lcy1-0-C24a1kyl-0-C1-aa1kyl.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
IV represents C2-6a1ky1 substituted with one substituent selected from the
group
consisting of Het' or -0R3;
R2 represents methyl;
R3 represents -C2-4a1ky1-O-C14alkyl or -C2-4alkyl-O-C24alkyl-O-C1-4a1kyl; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 37 -
as mentioned in any of the other embodiments, wherein
Xl represents
R
N-N
b
RI represents C2_6alky1 substituted with one substituent selected from the
group
consisting of Het' or -0R3;
R2 represents methyl;
R3 represents -C,Aalkyl-O-Ci-talkyl or -C2_4a1kyl-O-C2.4alky1-0-Ci_4alkyl; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
Rl represents C2_6alkyl substituted with one -0R3substituent;
R2 represents methyl;
R3 represents -C2_4alky1-0-Ci-4alkyl or -C2_4a1kyl-O-C2_4alky1-0-Ci_4a1ky1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
Xl represents
R
IN-IN
b
RI represents C2_6alkyl substituted with one -0R3substituent;
R2 represents methyl;
R3 represents -C2-4alkyl-O-Ci_4a1ky1 or -C2_4alkyl-O-C2_4alky1-0-C14alkyl.
In an embodiment, the present invention relates to those compounds of Formula
(1) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
RI represents Cmalkyl substituted with one -0R3substituent;
R2 represents methyl;
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 38 -
R3 represents -C2-4a1 ky I -0-C14a1 kyl or -C2-4a1 kyl-O-C24a1 ky1-047 -4a1 ky
I ; and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
0) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X' represents
R N -N
b
11' represents C2.6alkyl substituted with one -OW substituent;
R2 represents methyl;
R3 represents -C.2.4alky1-0-Ci4alkyl or -C.2.4alkyl-O-C2...ialkyl-O-C14allcyl;
and
n represents 1.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -N(Rx)-; and BY represents halo.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -N(Rx)-; and BY represents fluoro.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -S-; and RY represents halo.
In an embodiment, the present invention relates to those compounds of Formula
0) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein
X represents -S-; and BY represents fluoro.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 39 -
as mentioned in any of the other embodiments, wherein n is 1 and wherein RY is
in
position 3 as indicated below:
4 11110
1
R Y 3 2
=
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein n is 1 and wherein R) is
in
position 3 as indicated below; and wherein RY represents fluoro:
4
R Y 3 2
=
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein the compounds of Formula
(1)
are restricted to compounds of Formula (I-x):
X
3
N / CI CH OH
/
0
R2
=
1111
(RY),
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-40 -
It will be clear that all variables in the structure of Formula (I-x), are
defined as defined
for the compounds of Formula (1) or any subgroup thereof as mentioned in any
of the
other embodiments.
The present invention relates in particular to compounds of Formula (I-x) as
defined
herein, and the tautomers and the stereoisomeric forms thereof, wherein
X' represents
1
N-N
a.
b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the molecule;
R' and R2 represent methyl;
X represents -S-, -S(=0)2-, or
It,' represents methyl;
RY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I-x) as
defined
herein, and the tautomers and the stereoisomeric forms thereof, wherein
X' represents
R
IN-IN
b
wherein 'a' and 'b' indicate how variable X1 is attached to the remainder of
the molecule;
R1 represents C2_6a1ky1 substituted with one -OW substituent;
R2 represent methyl;
R3 represents -C2-4alkyl-O-Ci-4alkyl;
X represents -S-, -S(=0)2-, or
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 41 -
It' represents methyl;
RY represents halo;
n represents 0 or 1;
and the pharmaceutically acceptable salts and the solvates thereof.
The present invention relates in particular to compounds of Formula (I-x) as
defined
herein, and the tautomers and the stereoisomeric forms thereof, wherein
XI represents
RNN-N
b
wherein 'a' and 'b' indicate how variable X' is attached to the remainder of
the
molecule;
11' represents C2_6a1lcy1 substituted with one -010 substituent;
R2 represent methyl;
R3 represents -C2.4allcyl-0-C14allcyl;
X represents -S-;
RY represents halo; in particular F;
n represents 1;
and the pharmaceutically acceptable salts and the solvates thereof.
in an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein the compounds are Ra
atropisomers.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein the compounds are Sa
atropisomers.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
-42 -
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, wherein the compounds of Formula
(I)
are restricted to compounds of Formula (I-y)-
X
CH3
R2 ci OH
N-
0
4110 (I-Y)
(RY),
It will be clear that all variables in the structure of Formula (I-y), are
defined as defined
for the compounds of Formula (I) or any subgroup thereof as mentioned in any
of the
other embodiments.
In an embodiment, the present invention relates to those compounds of Formula
(I) and
the pharmaceutically acceptable salts, and the solvates thereof, or any
subgroup thereof
as mentioned in any of the other embodiments, provided that
N-N
CI OH
N V
\N
0
and the tautomers and the stereoisomeric forms thereof are excluded. In an
embodiment,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-43 -
the scope of the present invention does not include said excluded compound,
and the
pharmaceutically acceptable salts thereof. In an embodiment, the scope of the
present
invention does not include said excluded compounds, and the pharmaceutically
acceptable salts and the solvates thereof
In an embodiment, the present invention relates to a subgroup of Formula (I)
as defined
in the general reaction schemes.
In an embodiment the compound of Foimula (I) is selected from the group
consisting of
any of the exemplified compounds,
tautomers and stereoisomeric forms thereof,
any pharmaceutically acceptable salts, and the solvates thereof
All possible combinations of the above indicated embodiments are considered to
be
embraced within the scope of the invention.
METHODS FOR THE PREPARATION OF COMPOUNDS
In this section, as in all other sections unless the context indicates
otherwise, references
to Formula (I) also include all other sub-groups and examples thereof as
defined herein.
The general preparation of some typical examples of the compounds of Formula
(I) is
described hereunder and in the specific examples, and are generally prepared
from
starting materials which are either commercially available or can be prepared
by standard
synthetic processes commonly used by those skilled in the art of organic
chemistry. The
following schemes are only meant to represent examples of the invention and
are in no
way meant to be a limit of the invention.
Alternatively, compounds of the present invention may also be prepared by
analogous
reaction protocols as described in the general schemes below, combined with
standard
synthetic processes commonly used by those skilled in the art.
The skilled person will realize that in the reactions described in the
Schemes, although
this is not always explicitly shown, it may be necessary to protect reactive
functional
groups (for example hydroxy, amino, or carboxy groups) where these are desired
in the
final product, to avoid their unwanted participation in the reactions. In
general,
conventional protecting groups can be used in accordance with standard
practice. The
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-44 -
protecting groups may be removed at a convenient subsequent stage using
methods
known from the art.
The skilled person will realize that in the reactions described in the
Schemes, it may be
advisable or necessary to perform the reaction under an inert atmosphere, such
as for
example under N2-gas atmosphere.
It will be apparent for the skilled person that it may be necessary to cool
the reaction
mixture before reaction work-up (refers to the series of manipulations
required to isolate
and purify the product(s) of a chemical reaction such as for example
quenching, column
chromatography, extraction).
The skilled person will realize that heating the reaction mixture under
stirring may
enhance the reaction outcome. In some reactions microwave heating may be used
instead
of conventional heating to shorten the overall reaction time.
The skilled person will realize that another sequence of the chemical
reactions shown in
the Schemes below, may also result in the desired compound of Formula (I).
The skilled person will realize that intermediates and final compounds shown
in the
Schemes below may be further functionalized according to methods well-known by
the
person skilled in the art. The intermediates and compounds described herein
can be
isolated in free form or as a salt, or a solvate thereof. The intermediates
and compounds
described herein may be synthesized in the form of mixtures of tautomers and
stereoisomeric forms that can be separated from one another following art-
known
resolution procedures
R1
N-N
X
CI OH
\N
0
0
(I-a)
Compounds of Formula (I-a) can be prepared according to Scheme 1,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-45 -
R1 R1
N¨N N¨N
X / X
CI 0 N CI
0
N / V
0
0
e
e---OP1 OH
))o i2 OH
(Rnn
(IV) (III)
Ri
R1
NN
X / N¨N
X /
CI OH
N
0
CI
N N
0 1\1 /
R2
5)0
0 /C
0
(R)r,
(Ron
(I-a)
(I I-a)
Scheme 1
¨ by reacting an intermediate of Formula (II-a) where X,
It', R2, and (RY). are
defined as in Formula (I), with a suitable base such as, for example, LiOH or
NaOH, in a suitable solvent such as water or a mixture of water and a suitable
organic solvent such as dioxane or tetrahydrofuran (THF), or a mixture of
methanol (Me0H) and THF, at a suitable temperature such as room temperature
or 60 C.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-46 -
¨ Intermediates of Formula (II-a) can be prepared by reacting an intermediate
of
Formula (III) where X, R1, and (RY)n are defined as in Formula (I), and R2 is
a
suitable protecting group such as, for example, paramethoxybenzyl (PMB),
dimethoxylbenzyl (DMB), or tetrahydropyranyl (THP), or can also be a suitable
alkyl substituent such as, for example, methyl, with a suitable reagent, such
as,
for example, diethyl azodicarboxylate (DEAD) or di-tert-butyl azodicarboxylate
(DTBAD), in the presence of a suitable phosphine such as, for example, PPh3,
in
a suitable solvent such as, for example, THF, toluene, or a mixture thereof,
at a
suitable temperature such as, for example, room temperature or 70 C.
¨ Intermediates of Formula (III) can be prepared by reacting an intermediate
of
Formula (IV) where X, R1, R2, and (RY)n are as defined in Formula (III), and
P1
as well as P2 are suitable protecting groups, such as, for example, tert-
butyldimethylsily1 (TBDMS) or tert-butyldiphenylsilyl (TBDPS), with a suitable
deprotecting reagent such as, for example, tetrabutylammonium fluoride (TB
AF),
in a suitable solvent such as, for example, THF, at a suitable temperature
such as,
for example, room temperature or 60 C.
¨ Alternatively, when P2 in intermediates of Formula (IV) is a PMB group, an
additional deprotection step might be necessary, using a suitable deprotection
reagent such as, for example, TFA or 2,3-Dichloro-5,6-dicyano-1,4-
benzoquinone (DDQ), in a suitable solvent such as, for example,
dichloromethane (DCM), at a suitable temperature such as, for example, room
temperature.
An intermediate of Formula (II-a) might have a protecting group in the R1
position such
as, for example, tetrahydropyranyl. In such a case, the intermediate of
Formula (II) is
reacted with a suitable deprotection reagent, such as, for example, pTs0H (p-
toluenesulfonic acid) or HC1, in a suitable solvent such as, for example,
iPrOH (2-
propanol), at a suitable temperature such as, for example, room temperature.
In a next
step the obtained unprotected intermediate can be reacted with a suitable
alkylating agent
R1L (where L is as suitable leaving group) such as, for example, an alkyl
halide, in the
presence of a suitable base such as, for example, Cs/CO3, in a suitable
solvent such as,
for example, DMF (N,N-dimethylformamide), at a suitable temperature such as,
for
example, room temperature or 60 'C.
It will be clear for someone skilled in the art, that orthogonality of
protective groups will
have to be considered in this case, for instance when R1 is a
tetrahydropyranyl, 131 and P2
should be preferably TBDMS or TBDPS groups.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-47 -
R1
X
C I N OH
0
05)
(R nn
(I-b)
Similarly, compounds of Formula (I-b) can be prepared as described for
compounds of
Formula (I-a), but starting from the regioisomer of intermediates of Formula
(XXI)
(where le is on the other pyrazole nitrogen). It will be clear for a skilled
person that in
the final synthesis step, an intermediate of Formula (II-b) (where R2 is on
the other
pyrazole nitrogen) is reacted to a compound of Formula (I-b) in that case.
Alternatively, both intermediates of Formula (II-a) and (II-b), where le is
defined as in
compounds of Formula (I-a) and (I-b), respectively, can be prepared in two
steps.
¨ First, by reacting an intermediate of Formula (II-a) or (II-b),
respectively, where
R2 is then defined as a suitable protecting group such as, for example, THP,
with
a suitable deprotection reagent such as, for example, HC1, in a suitable
solvent
such as, for example, dioxane or isopropanol, at a suitable temperature such
as,
for example, room temperature.
R1
X
H N
0
(R
0
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-48 -
¨ Then, by reacting the obtained intermediate of Formula (The) with a suitable
alkylating agent R2L, such as, for example, an alkyl halide, in a suitable
solvent,
such as, for example, DMF, or acetonitrile, in the presence of a suitable
base,
such as, for example, trietylamine (Et3N), N,N-Diisopropylethylamine
(iPr2EtN),
Cs2CO3, or 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), at a suitable
temperature, such as, for example, room temperature or 60 C, followed by a
suitable separation of the isomers (II-a) and (II-b), such as, for example, a
chromatographic separation.
Alternatively, compounds of Formula (I) where Rl, R2, and (RY). are as defined
in
Formula (I-a), and X is defined as N(CH3), can be prepared according to Scheme
2,
r_, r 1
H /
\ \
0 CI 0
, CI
N / \
\ \ / -,.... \ /
N N 0 N N 0
0 0
,R1 ,.,,,--'..--''''
N--1\1 R1
/
i
X / \ K(RY),
\
\
OH
N ,
\
0
NN
1 /
0
CI
0 / \
\ ,
RZN N 0
(II)
(I-a)
0
\ /
(VII)
Scheme 2
¨ by reacting an intermediate of Formula (V) with a suitable base such as, for
example, LiOH or NaOH, in a suitable solvent such as water or a mixture of
water
and a suitable organic solvent such as dioxane or THF, or a mixture of Me0H
and THF, at a suitable temperature such as room temperature or 60 C.
¨ Intermediates of Formula (V) can be prepared by reacting an intermediate of
Formula (VI) with a suitable aldehyde such as, for example, formaldehyde, and
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-49 -
a suitable reducing agent such as, for example, NaBH(OAc)3 or NaBH3CN, in
the presence of a suitable acid such as, for example, AcOH, in a suitable
solvent
such as, for example, CH2C12, at a suitable temperature such as, for example,
room temperature.
¨ Intermediates of Formula (VI) can be be prepared by reacting an
intermediate of
Formula (II) where X is defined as nitrogen protected by a protecting group
such
as, for example, 2-nitrophenylsulfonyl, with a suitable deprotecting agent
such
as, for example, thiophenol, in the presence of a suitable base such as, for
example, K2CO3, in a suitable solvent such as, for example, acetonitrile, at a
suitable temperature such as, for example, room temperature.
Alternatively, compounds of Formula (I) where R1, R2, and (RY). are as defined
in
Formula (I), and X is defined as S(0)2, can be prepared according to Scheme 2,
¨ by reacting an intermediate of Formula (VII) with a suitable base such as,
for
example, LiOH or NaOH, in a suitable solvent such as water or a mixture of
water
and a suitable organic solvent such as dioxane or THE, or a mixture of Me0H
and THF, at a suitable temperature such as room temperature or 60 C.
¨ Intermediates of Formula (VII) can be prepared by reacting an intermediate
of
Formula (II) where le, R2, (RY). are as defined in Formula (I), and X is
defined
as S (sulfur), with a suitable oxidizing agent such as, for example, mCPBA, in
a
suitable solvent such as, for example, CH2C12, at a suitable temperature such
as,
for example 0 C or room temperature.
When X is defined as S (sulfur), intermediates of Formula (IV) can be prepared
according
to Scheme 3,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 50 -
R1
NO2
N¨N
H
S,N
0411
0 a OMe
N 0
(XII) e--0P1
OH
N¨N
HO NN
OP2
CI OMe
\o
\ (XIII)
CI
N
N 0
N 0
L2
e-
OP \1/4
(XI) N./
OP2
/R1
NN
\ X (v)
Ll
(RY)n I
CI OMe
OP2
N 0 (IX)
(x)
e_0,1 (RY)(R),IR1
N¨N
AcS
\
CI o
N 0
(VIII) ""OP1
Scheme 3
¨ by reacting an intermediate of Formula (VIII), where Pl
is a suitable protecting
groups such as, for example, tert-butyldimethylsilyl (TBDMS), with an
intermediate of Formula (IX) where L2 is a suitable leaving group such as, for
example, chloride or mesylate, and P2 is a suitable protecting group such as,
for
example, TBDPS, in the presence of a suitable base such as, for example,
K2CO3,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-51 -
in a suitable solvent, such as for example, Me0H, at a suitable temperature
such
as, for example, room temperature.
¨ Intermediates of Formula (VIII) can be prepared by
reacting an intermediate of
Formula (X), where Ll is a suitable leaving group such as, for example, iodide
or
mesylate, with KSAc, in a suitable solvent such as, for example, acetonitrile,
at a
suitable temperature such as, for example, room temperature.
¨ Intermediates of Formula (IX) can be prepared by reacting an intermediate
of
Formula (XIII), with a suitable reagent such as for example, mesyl chloride or
thionyl chloride, if necessary in the presence of a suitable base such as, for
example, triethylamine, in a suitable solvent such as, for example, CH2C12, at
a
suitable temperature such as, for example, 0 C or room temperature.
¨ Intermediates of Formula (X) can be prepared by reacting an intermediate
of
Formula (XI), with a suitable alkylsulfonyl chloride such as, for example,
mesyl
chloride, in the presence of a suitable base such as, for example,
triethylamine, in
a suitable solvent such as, for example, CH2C12, at a suitable temperature,
such
as for example, room temperature.
¨ Alternatively, intermediates of Formula (X) can be prepared in two steps,
by
reacting an intermediate of Formula (XI), with a suitable alkyl sulfonyl
chloride,
such as for example, mesyl chloride, in the presence of a suitable base such
as,
for example, triethylamine, in a suitable solvent such as, for example,
CH2C12, at
a suitable temperature such as, for example, room temperature; followed by
reaction with a suitable metal halide such as, for example, potassium iodide,
in a
suitable solvent such as, for example, acetonitrile, at a suitable
temperature, such
as for example, room temperature or 60 C.
Alternatively, when X is defined as nitrogen protected by a protecting group
such as for
example 2-nitrophenylsulfonyl, intermediates of Formula (IV) can be prepared
according
to Scheme 3,
¨ by reacting an intermediate of Formula (XII) with an intermediate of Formula
(XIII) in the presence of a suitable reagent such as, for example, DEAD or
DBAD, in the presence of a suitable phosphine such as, for example,
triphenylphosphine (PPh3), in a suitable solvent such as, for example, THF,
toluene, or a mixture thereof, at a suitable temperature such as, for example,
room
temperature or 60 C.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 52 -
¨ Intermediates of Formula (XII) can be prepared by reacting an
intermediate of
Formula (XI) with a suitable protected nitrogen such as, for example, 2-
nitrophenylsulfonamide, in the presence of a suitable reagent such as, for
example, DEAD or DBAD, in the presence of a suitable phosphine such as, for
example, PPh3, in a suitable solvent such as, for example, THF, toluene, or a
mixture thereof, at a suitable temperature such as, for example, room
temperature
or 60 C.
Intermediates of Formula (XI), where Pl is a suitable protecting group, such
as, for
example, TBDMS, can be prepared according to Scheme 4,
R1
Br Me0
CI 401 Br
0 Me =
CI O
N 0 -B.. CI
Me
NH 0
NH 0
mixture of (E) and (Z) isomers
(XVII) (XVI)
(XV)
R1
N¨N
HO
HO
CI 0Me
Me
CI
N
NH 0
(XIV)
(XI)
Scheme 4
¨ by reacting an intermediate of Formula (XIV), with a suitable 0-protected
propyl
halide or alkylsulfonate such as, for example, (3-bromopropoxy)(tert-
butyl)dimethylsilane, in the presence of a suitable base such as, for example,
Cs2CO3, in a suitable solvent such as, for example, DMF, at a suitable
temperature such as, for example, room temperature.
¨ Intermediates of Formula (XIV) can be prepared by reacting an
intermediate of
Formula (XV), with a suitable deprotecting agent such as, for example,
trifluoromethanesulfonic acid, TFA, or DDQ, in a suitable solvent such as, for
example, CH2C12, at a suitable temperature such as, for example, room
temperature.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 53 -
¨ Intermediates of Formula (XV) can be prepared by reacting an intermediate of
Formula (XVI), with a suitable substituted pyrazole derivative such as, for
example, 3-(((4-methoxybenzypoxy)methyl)-1,5-
dimethyl-4-(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-y1)-1H-pyrazole , in the presence of a
suitable
catalyst such as, for example, Pd2(dba)3, in the presence of a suitable
phosphine
ligand such as, for example, S-Phos, in the presence of a suitable base such
as,
for example, sodium bicarbonate, in a suitable solvent such as, for example,
dioxane, water, or a mixture thereof, at a suitable temperature such as, for
example, 100 C. A skilled person will realize that other suitable substituted
pyrazole derivatives, can be for example derivatives wherein the p-
methoxybenzyl moiety is replaced by hydrogen or TBDMS.
¨ Intermediates of Formula (XVI) can be prepared by
reacting an intermediate of
Formula (XVII), with a suitable acid, such as, for example, sulfuric acid, in
a
suitable solvent, such as, for example, acetic acid, at a suitable
temperature, such
as, for example, 70 C.
¨ An intermediate of Formula (XVII) can be prepared by reacting (3-bromo-4-
chlorophenyl)hydrazine with methyl 2-oxobutanoate, in the presence of a
suitable
acid, such as, for example, hydrochloric acid, in a suitable solvent, such as,
for
example, methanol, at a suitable temperature, such as, for example, 65 C.
Intermediates of Formula (XIII), wherein R2 and (R)), are defined as in
Formula (I), P2
is a suitable protecting group, such as, for example, TBDPS, can be prepared
according
to Scheme 5,
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 54 -
Hal * =
Hal
OP2
OP2
(RY)r,
(XXXVII)
(RY),
(XXXII)
OH 0
op2 OP2 0
(Rnn (Rnn N
µN undetermined
mixture of E/Z
(XXII) (XX)
O(I)
RS
o 0
N
(X µNi
OP2
(XIX)
R2
Cl-
0 Ph3P
OH OH
OP2
NJ,Ns"..
undetermined
(Rnn X I
mixture of E/Z
(O(III) R2 R2
OP2
OP2
0 (Rnn
(Rnn
(XIII)
(XVIII)
OH
(RY)n
(XXIV)
Scheme 5
¨ by reacting an intermediate of Formula (XVIII) with a suitable hydrogenating
reagent such as, for example, hydrogen gas, in the presence of a suitable
catalyst
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 55 -
such as, for example, Pd/C, in a suitable solvent such as, for example, Me0H,
at
a suitable temperature such as, for example, room temperature.
¨ Intermediates of Formula (XVIII) can be prepared by reacting an
intermediate of
Formula (XIX) with a suitable reducing agent such as, for example, LiA1H4, in
a
suitable solvent such as, for example, THF, at a suitable temperature such as,
for
example, 0 C.
¨ Intermediates of Formula (XIX) can be prepared by reacting an
intermediate of
Formula (X() with an intermediate of Formula (XXI), in the presence of a
suitable base such as, for example, Nail-I, in a suitable solvent such as, for
example, THF, at a suitable temperature such as, for example, -10 C.
¨ Intermediates of Formula (XX) can be prepared by reacting an intermediate
of
Formula (XXII) with a suitable oxidizing agent such as, for example, Mn02, in
a
suitable solvent such as, for example, acetonitrile, at a suitable temperature
such
as, for example, 60 C
¨ Intermediates of Formula (XXII) can be prepared by reacting an intermediate
of
Formula (XXIII) with a suitable reducing agent such as, for example, Li AlH4,
in
a suitable solvent such as, for example, THF, at a suitable temperature such
as,
for example, 0 C.
¨ Intermediates of Formula (XXIII) can be prepared by reacting an
intermediate of
Formula (XXIV) with a suitable protecting reagent such as, for example, tert-
butyl(chloro)diphenylsilane (TBDP SC1) or 4-methoxybenzyl chloride (PMBC1),
in the presence of a suitable base such as, for example, imidazole or NaH, in
a
suitable solvent such as, for example, DMF, at a suitable temperature such as,
for
example, room temperature.
¨ Intermediates of Formula (XXI) and Intermediates of Formula (XXIV) are
commercially available or can be prepared according to procedures described in
literature.
Alternatively, intermediates of Formula (III), wherein Rl, R2 and (RY)n are
defined as in
Formula (I), and X is 0 (oxygen), can be prepared according to Scheme 6,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 56 -
191 t
/
N¨N /RI /R1
1
N¨N
HO N
\='_ NN
1 X
/
N¨(
Me R2 x /
/
CI OW
\ \
\
...õ CI
(XXXI)
N 0 \ N
N
0
OP1 R2 OP3 R2
OH
...OH
(XI) (XXX)
(XXIX)
R1
/
N¨N
1 R
X / 11 ,
R1
\ NN /
N¨N
0 / /
N(
\
R2 ---
...-,,D * . N( /
,N : IR
R2 ¨0 0
\:\I N
._ -
---0 N 0
* OP2 ¨ I P. *
IP y,
(R /n Hal
(XXVII) OP1
....OH
(XXVIII)
OP2
(XXV i)
(Rnn
(XXXi I)
R1
R1 /
/ N¨N
N¨N /
I x /
x / \
\ ci
\
1 / N N 0
Pi N 0 R5 OH
..
R2
di 2 OP1 4 OH
OP
IP 111111(RY)n
(RY)õ
(III)
(X(V)
Scheme 6
¨ by reacting an intermediate of Formula (XXV) with a suitable deprotection
agent
such as, for example, p-toluenesulfonic acid (PTSA), in a suitable solvent
such
as, for example, Me0H, at a suitable temperature such as, for example, room
temperature.
¨ Intermediates of Formula (XXV) can be prepared by reacting an
intermediate of
Formula (XXVI) with a suitable hydrogenating reagent such as, for example,
hydrogen gas, in the presence of a suitable catalyst such as Pd/C, in a
suitable
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 57 -
solvent such as, for example, ethyl acetate (Et0Ac), at a suitable temperature
such as, for example, room temperature.
¨ Intermediates of Formula (XXVI) can be prepared by
reacting an intermediate of
Formula (XXVII) with an intermediate of Formula (XXXII), in the presence of a
suitable base such as, for example, NaH, in a suitable solvent such as, for
example, THF, at a suitable temperature such as, for example, 0 C or room
temperature.
¨ Intermediates of Formula (XXVII) can be prepared by
reacting an intermediate
of Formula (XXVIII) with a suitable protecting group precursor, such as, for
example, tert-butyldimethylsilyl chloride (TBDMSC1), in the presence of a
suitable base such as, for example, imidazole, in a suitable solvent such as,
for
example, DCM, at a suitable temperature such as, for example, room
temperature.
¨ Intermediates of Formula (XXVIII) can be prepared by reacting an
intermediate
of Formula (XXIX) with a suitable oxidizing agent such as, for example, Mn07,
in a suitable solvent such as, for example, DCM, at a suitable temperature
such
as, for example, room temperature.
¨ Intermediates of Formula (XXIX) can be prepared by reacting an
intermediate of
Formula (X)0() with with a suitable deprotection agent such as, for example,
PTSA, in a suitable in a suitable solvent such as, for example, Me0H, at a
suitable
temperature such as, for example, 0 C or room temperature.
¨ Intermediates of Formula (XXX) can be prepared by
reacting an intermediate of
Formula (XI) with an intermediate of Formula (XXXI), in the presence of a
suitable base such as, for example, NaH, in a suitable solvent such as, for
example, at a suitable temperature such as, for
example, 0 C or room
temperature.
¨ It will be clear for someone skilled in the art, that orthogonality of
protective
groups will have to be considered, for instance when R1 is a
tetrahydropyranyl,
P1, 132 and P3 should be preferably TBDMS or TBDPS groups.
Intermediates of Formula ()(XXII) can be prepared according to Scheme 5
¨ by reacting an intermediate of Formula (X)0(VII) with a suitable phosphine
such
as, for example, triphenylphosphine (PPh3), in a suitable solvent such as, for
example, DCM, at a suitable temperature such as, for example, room temperature
¨ Intermediates of Formula (XXXVII) can be prepared by reacting an
intermediate
of Formula (XXII) with a suitable activating agent such as, for example,
thionyl
chloride, in a suitable solvent such as, for example, DCM, at a suitable
temperature such as, for example, room temperature.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 58 -
Intermediates of Formula (XXXI), wherein P3 is a suitable protecting group
such as, for
example, TBDMS, and L is a suitable leaving group such as, for example, I
(iodide), can
be prepared according to Scheme 7,
0 O 0O 0O OH
/
,N /N /N
R2 R2 R2
R2 0 R2
0 HO P30 P30 P30
(XXXVI) (XXXV) (XXXIV) (XXXIII) (XXXI)
Scheme 7
- by reacting an intermediate of Formula (XXXIII) with a suitable
activating agent
such as, for example, mesyl chloride (MsC1), in the presence of a suitable
base
such as, for example, triethylamine (Et3N), followed by addition of a suitable
leaving group precursor such as, for example, NaI, in a suitable solvent such
as,
for example, THF, at a suitable temperature such as, for example, room
temperature.
- Intermediates of Formula (XXXIII) can be prepared by reacting an
intermediate
of Formula (XXXIV) with a suitable reducing agent such as, for example,
LiA1H4, in a suitable solvent such as, for example, THF, at a suitable
temperature
such as, for example, 0 C.
- Intermediates of Formula (XXXIV) can be prepared by reacting an
intermediate
of Formula (XXXV) with a suitable protecting group precursor such as, for
example, TBDMSC1, in the presence of a suitable base such as, for example,
imidazole, in a suitable solvent such as, for example, DCM, at a suitable
temperature such as, for example, 0 C or room temperature.
- Intermediates of Formula (XXXV) can be prepared by reacting an
intermediate
of Formula (XXXVI) with a suitable reducing agent such as, for example,
NaBH4, in a suitable solvent such as, for example, Me0H, 2-
methyltetrahydrofuran (2-Me-THF), or a mixture thereof, at a suitable
temperature such as, for example, 0 C or room temperature.
- Intermediates of Formula (XXXVI) are commercially available or can be
prepared according to reaction protocols known by a skilled person.
It will be clear for a skilled person that, in case le is a protective group,
the
protective groups P3 will have to be an orthogonal protective group.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 59 -
A skilled person will understand that analogous reaction protocols can also be
used to
prepare Compounds of the invention wherein Xl represents
R
NN-N
b
To obtain such compounds, an alternative pyrazole-boronate of Formula
(XXXVIII)
should be used, which can be prepared according to Scheme 8.
tBr N
/
/
/
0 0 R1 R1
0 0 0 H 0
(XLIV) (XLIII) (XLII) (XLI)
N B(OR)2 N 111(0R)2
Br
R1 R1 R1
HO P40 P40
(XXXVIII) (XXXIX) (XL)
Scheme 8
¨ by reacting intermediates of Formula (XXXIX) in which P4 is a suitable
protective group such as, for example, TBDMS, with a suitable deprotecting
reagent such as, for example, tetrabutylammonium fluoride (TBAF), in a
suitable
solvent such as, for example, THF, at a suitable temperature such as, for
example,
room temperature or 60 C.
- Intermediates of Formula (XXXIX) can be prepared by reacting an
intermediate
of Formula (XL) with a suitable borylating reagent such as, for example, 2-
isopropoxy-4,4,5,5-tetramethy1-1,3,2-dioxaborolane, in the presence of a
suitable
metalation reagent such as, for example, n-butyllithium, in a suitable solvent
such
as, for example, THF, at a suitable temperature such as, for example, -78 C.
- Intermediates of Formula (XL) can be prepared by reacting an intermediate
of
Formula (XLI) with a suitable protecting group precursor such as, for example,
TBDMSC1, in the presence of a suitable base such as, for example, imidazole,
in
a suitable solvent such as, for example, DCM, at a suitable temperature such
as,
for example, 0 C or room temperature.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 60 -
- Intermediates of Formula (XLI) can be prepared by
reacting an intermediate of
Formula (XLII) with a suitable reducing agent such as, for example, NaBH4, in
a
suitable solvent such as, for example, Me0H or THE, or a mixture thereof, at a
suitable temperature such as, for example, 0 C or room temperature.
- Intermediates of Formula (XLII) can be prepared by reacting an
intermediate of
Formula (XLIII) with a suitable alcohol such as, for example 2-(2-
methoxyethoxy)ethanol in the presence of a suitable phosphorane such as, for
example, cyanomethylenetributylphosphorane, in a suitable solvent such as, for
example THE, at a suitable temperature such as, for example, 0 C to room
temperature.
- Intermediates of Formula (XLIII) can be prepared by
reacting an intermediate of
Formula (XLIV) with a suitable brominating reagent such as, for example, N-
bromosuccinimide (NBS), in a suitable solvent such as, for example DCM, at a
suitable temperature such as, for example, room temperature
- Intermediates of Formula (X,Tv) are commercially available or can be
prepared
according to procedures described in literature.
Using a similar procedure as described in Scheme 4 for intermediates of
Formula (XI),
the alternative pyrazole-boronate of Formula (XXXVIII) or its precursor of
Formula
(XXXIX) can provide intermediates of Formula (XLV) carrying RI at the other
pyrazole
nitrogen position compared to intermediates of Formula (XI).
Alternatively intermediates of Formula (XLV), wherein
is defined as in Formula (I),
and Pl is a suitable protecting group, such as, for example, TBDMS, can be
prepared
according to Scheme 9,
RI
N¨N
Rµl
N¨N
B(OR) 2 HO
Br Br
CI
CI Me Me (X.XXVIII) O
CI Me
NH 0 N 0 N 0
(XVI) (XLVI)\ \ ¨op (XLV)\--
\-0Pi
Scheme 9
¨ by reacting an intermediate of Formula (XLVI) with an
intermediate of Formula
(XXXVIII), in the presence of a suitable catalyst such as, for example,
dichlorobis[di-tert-butyl(p-dimethylaminophenyl)phosphino]palladium(II)
(Pd(amphos)2C12), in the presence of a suitable base such as, for example,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 61 -
potassium carbonate, in a suitable solvent such as, for example, dioxane,
water,
or a mixture thereof, at a suitable temperature such as, for example, 65 'C.
¨
Intermediates of Formula (XLVI) can be prepared by reacting an
intermediate of
Formula (XVI) with a suitable 0-protected propyl halide or alkylsulfonate such
as, for example, (3-bromopropoxy)(tert-butyl)dimethylsilane, in the presence
of
a suitable base such as, for example, Cs2CO3, in a suitable solvent such as,
for
example, DMF, at a suitable temperature such as, for example, room
temperature.
¨ It will be clear to a skilled person that this alternative sequence can also
be used
to synthesize intermediates of Formula (XI).
It will be clear for a skilled person that Intermediates of Formula (XLV) can
be converted
to compounds of Formula (I) in a similar way as described for intermediates of
Formula
(XI).
It will be appreciated that where appropriate functional groups exist,
compounds of
various formulae or any intermediates used in their preparation may be further
derivatized by one or more standard synthetic methods employing condensation,
substitution, oxidation, reduction, or cleavage reactions. Particular
substitution
approaches include conventional alkylation, arylation, heteroarylation,
acylation,
sulfonylation, halogenation, nitration, formylation and coupling procedures.
The compounds of Formula (I) may be synthesized in the form of racemic
mixtures of
enantiomers which can be separated from one another following art-known
resolution
procedures. The racemic compounds of Formula (I) containing a basic nitrogen
atom
may be converted into the corresponding diastereomeric salt forms by reaction
with a
suitable chiral acid. Said diastereomeric salt forms are subsequently
separated, for
example, by selective or fractional crystallization and the enantiomers are
liberated
therefrom by alkali. An alternative manner of separating the enantiomeric
forms of the
compounds of Formula (I) involves liquid chromatography using a chiral
stationary
phase. Said pure stereochemically isomeric forms may also be derived from the
corresponding pure stereochemically isomeric forms of the appropriate starting
materials, provided that the reaction occurs stereospecifically.
In the preparation of compounds of the present invention, protection of remote
functionality (e.g., primary or secondary amine) of intei
__________________________ mediates may be necessary. The
need for such protection will vary depending on the nature of the remote
functionality
and the conditions of the preparation methods. Suitable amino-protecting
groups (NH-
Pg) include acetyl, trifluoroacetyl, t-butoxycarbonyl (Boc), benzyloxycarbonyl
(CBz)
and 9-fluorenylmethyleneoxycarbonyl (Fmoc). The need for such protection is
readily
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 62 -
determined by one skilled in the art. For a general description of protecting
groups and
their use, see T. W. Greene and P. G. M. Wuts, Protective Groups in Organic
Synthesis,
4th ed., Wiley, Hoboken, New Jersey, 2007.
PHARMACOLOGY OF COMPOUNDS
It has been found that the compounds of the present invention inhibit one of
more
MCL-1 activities, such as MCL-1 antiapoptotic activity.
An MCL-1 inhibitor is a compound that blocks one or more MCL-1 functions,
such as the ability to bind and repress proapoptotic effectors Bak and Bax or
BH3 only
sensitizers such as Bim, Noxa or Puma.
The compounds of the present invention can inhibit the MCL-1 pro-survival
functions. Therefore, the compounds of the present invention may be useful in
treating
and / or preventing, in particular treating, diseases that are susceptible to
the effects of
the immune system such as cancer.
In another embodiment of the present invention, the compounds of the present
invention exhibit anti-tumoral properties, for example, through immune
modulation.
In an embodiment, the present invention is directed to methods for treating
and /
or preventing a cancer, wherein the cancer is selected from those described
herein,
comprising administering to a subject in need thereof (preferably a human), a
therapeutically effective amount of a compound of Formula (I), or
pharmaceutically
acceptable salt, or a solvate thereof.
In an embodiment, the present invention is directed to a method for treating
and
/ or preventing cancer comprising administering to a subject in need thereof,
preferably
a human, a therapeutically effective amount of a compound of Formula (I), or a
pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer is
selected from
the group consisting of acute lymphoblastic leukemia (ALL), acute myeloid
leukemia
(AML), B cells acute lymphoblastic leukemia, B-cell chronic lymphocytic
leukemia
(CLL), bladder cancer, breast cancer, chronic lymphocytic leukemia, chronic
myeloid
leukemia, colon adenocarcinoma, diffuse large B cell lymphoma, esophageal
cancer,
follicular lymphoma, gastric cancer, head and neck cancer (including, but not
limited to
head and neck squamous cell carcinoma), hematopoietic cancer, hepatocellular
carcinoma, Hodgkin lymphoma, liver cancer, lung cancer (including but not
limited to
lung adenocarcinoma), lymphoma, medulloblastoma, melanoma, monoclonal
gammopathy of undetermined significance, multiple myeloma, myelodysplastic
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
-63 -
syndromes, myel ofi brosi s, myeloprol iferative neoplasms, ovarian cancer,
ovarian clear
cell carcinoma, ovarian serous cystadenoma, pancreatic cancer, polycythemia
vera,
prostate cancer, rectum adenocarcinoma, renal cell carcinoma, smoldering
multiple
myeloma, T cell acute lymphoblastic leukemia, T cell lymphoma, and
Waldenstroms
macroglobulinemia.
In another embodiment, the present invention is directed to a method for
treating
and / or preventing cancer comprising administering to a subject in need
thereof,
preferably a human, a therapeutically effective amount of a compound of
Formula (I), or
a pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer
is preferably
selected from the group consisting of acute lymphoblastic leukemia (ALL),
acute
myeloid leukemia (AML), B cells acute lymphoblastic leukemia, B-cell chronic
lymphocytic leukemia (CLL), breast cancer, chronic lymphocytic leukemia,
chronic
myeloid leukemia, diffuse large B cell lymphoma, follicular lymphoma,
hematopoietic
cancer, Hodgkin lymphoma, lung cancer (including, but not limited to lung
adenocarcinoma) lymphoma, monoclonal gammopathy of undetermined significance,
multiple myeloma, myelodysplastic syndromes, myelofibrosis, myeloproliferative
neoplasms, smoldering multiple myeloma, T cell acute lymphoblastic leukemia, T
cell
lymphoma and Waldenstroms macroglobulinemia.
In another embodiment, the present invention is directed to a method for
treating
and / or preventing cancer comprising administering to a subject in need
thereof,
preferably a human, a therapeutically effective amount of a compound of
Formula (I), or
a pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer
is selected
from the group consisting of adenocarcinoma, benign monoclonal gammopathy,
biliary
cancer (including, but not limited to, cholangiocarcinoma), bladder cancer,
breast cancer
(including, but not limited to, adenocarcinoma of the breast, papillary
carcinoma of the
breast, mammary cancer, medullary carcinoma of the breast), brain cancer
(including,
but not limited to, meningioma), glioma (including, but not limited to,
astrocytoma,
oligodendroglioma; medulloblastoma), bronchus cancer, cervical cancer
(including, but
not limited to, cervical adenocarcinoma), chordoma, choriocarcinoma,
colorectal cancer
(including, but not limited to, colon cancer, rectal cancer, colorectal
adenocarcinoma),
epithelial carcinoma, endothelial sarcoma (including, but not limited to,
Kaposi's
sarcoma, multiple idiopathic hemorrhagic sarcoma), endometrial cancer
(including, but
not limited to, uterine cancer, uterine sarcoma), esophageal cancer
(including, but not
limited to, adenocarcinoma of the esophagus, Barrett' s adenocarinoma), Ewing
sarcoma,
gastric cancer (including, but not limited to, stomach adenocarcinoma),
gastrointestinal
stromal tumor (GIST), head and neck cancer (including, but not limited to,
head and neck
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 64 -
squamous cell carcinoma), hematopoietic cancers (including, but not limited
to, leukemia
such as acute lymphocytic leukemia (ALL) (including, but not limited to, B-
cell ALL,
T-cell ALL), acute myelocytic leukemia (AML) (e.g. B-cell AML, T-cell AML),
chronic
myelocytic leukemia (CML) (e.g. B-cell CML, T-cell CML), and chronic
lymphocytic
leukemia (CLL) (e.g. B-cell CLL, T- cell CLL), lymphoma such as Hodgkin
lymphoma
(HL) (including, but not limited to, B-cell HL, T-cell HL) and non-Hodgkin
lymphoma
(NHL) (e.g. B-cell NHL such as diffuse large cell lymphoma (DLCL) (e.g.
diffuse large
B-cell lymphoma (DLBCL)), follicular lymphoma, chronic lymphocytic
leukemia/small
lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL), marginal zone B-
cell lymphomas (including, but not limited to, mucosa-associated lymphoid
tissue
(MALT) lymphomas, nodal marginal zone B-cell lymphoma. splenic marginal zone B-
cell lymphoma), primary mediastinal B-cell lymphoma, Burkitt lymphoma,
lymphoplasmacytic lymphoma (including, but not limited to, Waldenstrom's macro
gl obul nemi a), i mmunobl asti c large cell lymphoma, hairy cell leukemia
(HCL),
precursor B -Iymphoblastic lymphoma and primary central nervous system (CNS)
lymphoma, T-cell NHL such as precursor T-I ym ph obl asti c lymphoma/1 eukemi
a,
peripheral T-cell lymphoma (PTCL) (e.g. cutaneous T-cell lymphoma (CTCL)
(including, but not limited to, mycosis fungiodes, Sezary syndrome),
angioimmunoblastic T-cell lymphoma, extranodal natural killer T-cell lymphoma,
enteropathy type T-cell lymphoma, subcutaneous panniculitis-like T-cell
lymphoma,
anaplastic large cell lymphoma, a mixture of one or more leukemia/lymphoma as
described above, multiple myeloma (MM), heavy chain disease (including, but
not
limited to, alpha chain disease, gamma chain disease, mu chain disease),
immunocytic
amyloidosis, kidney cancer (including, but not limited to, nephroblastoma
a.k.a. Wilms'
tumor, renal cell carcinoma), liver cancer (including, but not limited to,
hepatocellular
cancer (HCC), malignant hepatoma), lung cancer (including, but not limited to,
bronchogenic carcinoma, non-small cell lung cancer (NSCLC), squamous lung
cancer
(SLC), adenocarcinoma of the lung, Lewis lung carcinoma, lung neuroendocrine
tumors,
typical carcinoid, atypical carcinoid, small cell lung cancer (SCLC), and
large cell
neuroendocrine carcinoma), myelodysplastic syndromes (MD S),
myeloproliferative
disorder (MPD), polycythemia vera (PV), essential thrombocytosis (ET),
agnogenic
myeloid metaplasia (AMM) a.k.a. myelofibrosis (MF), chronic idiopathic
myelofibrosis,
chronic myelocytic leukemia (CML), chronic neutrophilic leukemia (CNL),
hypereosinophilic syndrome (HES), ovarian cancer (including, but not limited
to,
cystadenocarcinoma, ovarian embryonal carcinoma, ovarian adenocarcinoma),
pancreatic cancer (including, but not limited to, pancreatic andenocarcinoma,
intraductal
papillary mucinous neoplasm (IPMN), Islet cell tumors), prostate cancer
(including, but
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
-65 -
not limited to, prostate adenocarcinoma), skin cancer (including, but not
limited to,
squamous cell carcinoma (SCC), keratoacanthoma (KA), melanoma, basal cell
carcinoma (BCC)) and soft tissue sarcoma (e.g. malignant fibrous histiocytoma
(MFH),
liposarcoma, malignant peripheral nerve sheath tumor (MPNST), chondrosarcoma,
fibrosarcoma, myxosarcoma).
In another embodiment, the present invention is directed to a method for
treating
and / or preventing cancer comprising administering to a subject in need
thereof,
preferably a human, a therapeutically effective amount of a compound of
Formula (1), or
a pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer
is selected
from the group consisting of benign monoclonal gammopathy, breast cancer
(including,
but not limited to, adenocarcinoma of the breast, papillary carcinoma of the
breast,
mammary cancer, medullary carcinoma of the breast), hematopoietic cancers
(including,
but not limited to, leukemia such as acute lymphocytic leukemia (ALL)
(including, but
not limited to, B-cell ALL, T-cell ALL), acute myelocytic leukemia (AML) (e.g.
B-cell
AML, T-cell AML), chronic myelocytic leukemia (CML) (e.g. B-cell CML, T-cell
CML), and chronic lymphocytic leukemia (CLL) (e.g. B-cell CLL, T- cell CLL),
lymphoma such as Hodgkin lymphoma (HL) (including, but not limited to, B-cell
HL,
T-cell HL) and non-Hodgkin lymphoma (NHL) (e.g. B-cell NHL such as diffuse
large
cell lymphoma (DLCL) (e.g. diffuse large B-cell lymphoma (DLBCL)), follicular
lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL),
mantle cell lymphoma (MCL), marginal zone B-cell lymphomas (including, but not
limited to, mucosa-associated lymphoid tissue (MALT) lymphomas, nodal marginal
zone B-cell lymphoma. splenic marginal zone B-cell lymphoma), primary
mediastinal
B-cell lymphoma, Burkitt lymphoma, lymphoplasmacytic lymphoma (including, but
not
limited to, Waldenstrom's macro globulinemia), immunoblastic large cell
lymphoma,
hairy cell leukemia (HCL), precursor B -Iymphoblastic lymphoma and primary
central
nervous system (CNS) lymphoma, T-cell NHL such as precursor T-Iymphoblastic
lymphoma/leukemia, peripheral T-cell lymphoma (PTCL) (e.g. cutaneous T-cell
lymphoma (CTCL) (including, but not limited to, mycosis fungiodes, Sezary
syndrome),
angioimmunoblastic T-cell lymphoma, extranodal natural killer T-cell lymphoma,
enteropathy type T-cell lymphoma, subcutaneous panniculitis-like T-cell
lymphoma,
anaplastic large cell lymphoma, a mixture of one or more leukemia/lymphoma as
described above, multiple myeloma (MM), heavy chain disease (including, but
not
limited to, alpha chain disease, gamma chain disease, mu chain disease),
immunocytic
amyloidosis, liver cancer (including, but not limited to, hepatocellular
cancer (HCC),
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 66 -
malignant hepatoma), lung cancer (including, but not limited to, bronchogenic
carcinoma, non-small cell lung cancer (NSCLC), squamous lung cancer (SLC),
adenocarcinoma of the lung, Lewis lung carcinoma, lung neuroendocrine tumors,
typical
carcinoid, atypical carcinoid, small cell lung cancer (SCLC), and large cell
neuroendocrine carcinoma), myelodysplastic syndromes (MDS), myeloproliferative
disorder (MPD), and prostate cancer (including, but not limited to, prostate
adenocarcinoma).
In another embodiment, the present invention is directed to a method for
treating
and / or preventing cancer comprising administering to a subject in need
thereof,
preferably a human, a therapeutically effective amount of a compound of
Formula (I), or
a pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer
is selected
from the group consisting of prostate, lung, pancreatic, breast, ovarian,
cervical,
melanoma, B-cell chronic lymphocytic leukemia (CLL), acute myeloid leukemia
(AML), and acute lymphoblastic leukemia (ALL).
In another embodiment, the present invention is directed to a method for
treating
and / or preventing cancer comprising administering to a subject in need
thereof,
preferably a human, a therapeutically effective amount of a compound of
Formula (I), or
a pharmaceutically acceptable salt, or a solvate thereof, wherein the cancer
is multiple
myeloma.
The compounds according to the present invention or pharmaceutical
compositions comprising said compounds, may also have therapeutic applications
in
combination with immune modulatory agents, such as inhibitors of the PD1/PDL1
immune checkpoint axis, for example antibodies (or peptides) that bind to
and/or inhibit
the activity of PD-1 or the activity of PD-Li and or CTLA-4 or engineered
chimeric
antigen receptor T cells (CART) targeting tumor associated antigens.
The compounds according to the present invention or pharmaceutical
compositions comprising said compounds, may also be combined with radiotherapy
or
chemotherapeutic agents (including, but not limited to, anti-cancer agents) or
any other
pharmaceutical agent which is administered to a subject having cancer for the
treatment
of said subject's cancer or for the treatment or prevention of side effects
associated with
the treatment of said subject's cancer.
The compounds according to the present invention or pharmaceutical
compositions comprising said compounds, may also be combined with other agents
that
stimulate or enhance the immune response, such as vaccines.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 67 -
In an embodiment, the present invention is directed to methods for treating
and /
or preventing a cancer (wherein the cancer is selected from those described
herein)
comprising administering to a subject in need thereof (preferably a human), a
therapeutically effective amount of co-therapy or combination therapy; wherein
the co-
therapy or combination therapy comprises a compound of Formula (I) of the
present
invention and one or more anti-cancer agent(s) selected from the group
consisting of (a)
immune modulatory agent (such as inhibitors of the PD1/PDL1 immune checkpoint
axis,
for example antibodies (or peptides) that bind to and/or inhibit the activity
of PD-1 or the
activity of PD-Li and or CTLA-4); (b) engineered chimeric antigen receptor T
cells
(CART) targeting tumor associated antigens; (c) radiotherapy; (d)
chemotherapy; and (e)
agents that stimulate or enhance the immune response, such as vaccines.
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for use as a
medicament.
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for use in the
inhibition of MCL-
1 activity.
As used herein, unless otherwise noted, the term "anti-cancer agents" shall
encompass "anti -tumor cell growth agents" and " anti -n eopl astic agents".
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for use in treating
and / or
preventing diseases (preferably cancers) mentioned above.
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for treating and / or
preventing
diseases (preferably cancers) mentioned above.
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for treating and / or
preventing,
in particular for treating, a disease, preferably a cancer, as described
herein (for example,
multiple myeloma).
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for use in treating
and / or
preventing, in particular for treating, a disease, preferably a cancer, as
described herein
(for example, multiple myeloma).
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for treating and / or
preventing,
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
-68 -
in particular for treating, MCL-1 mediated diseases or conditions, preferably
cancer,
more preferably a cancer as herein described (for example, multiple myeloma).
The present invention is directed to compounds of Formula (I) and
pharmaceutically acceptable salts, and solvates thereof, for use in treating
and / or
preventing, in particular for use in treating, MCL-1 mediated diseases or
conditions,
preferably cancer, more preferably a cancer as herein described (for example,
multiple
myeloma).
The present invention relates to compounds of Formula (I) and pharmaceutically
acceptable salts, and solvates thereof, for the manufacture of a medicament.
The present invention relates to compounds of Formula (1) and pharmaceutically
acceptable salts, and solvates thereof, for the manufacture of a medicament
for the
inhibition of MCL-1.
The present invention relates to compounds of Formula (I) and pharmaceutically
acceptable salts, and solvates thereof, for the manufacture of a medicament
for treating
and / or preventing, in particular for treating, a cancer, preferably a cancer
as herein
described. More particularly, the cancer is a cancer which responds to
inhibition of
MCL-1 (for example, multiple myeloma).
The present invention is directed to compounds of Formula (1) and
pharmaceutically acceptable salts, and solvates thereof, for the manufacture
of a
medicament for treating and / or preventing, in particular for treating, any
one of the
disease conditions mentioned hereinbefore.
The present invention is directed to compounds of Formula (1) and
pharmaceutically acceptable salts, and solvates thereof, for the manufacture
of a
medicament for treating and / or preventing any one of the disease conditions
mentioned
hereinbefore.
The compounds of Formula (1) and pharmaceutically acceptable salts, and
solvates thereof, can be administered to subjects, preferably humans, for
treating and / or
preventing of any one of the diseases mentioned hereinbefore.
In view of the utility of the compounds of Formula (I) and pharmaceutically
acceptable salts, and solvates thereof, there is provided a method of treating
subjects,
preferably mammals such as humans, suffering from any of the diseases
mentioned
hereinbefore; or a method of slowing the progression of any of the diseases
mentioned
hereinbefore in subject, humans; or a method of preventing subjects,
preferably
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 69 -
mammals such as humans, from suffering from any one of the diseases mentioned
hereinbefore.
Said methods comprise the administration, i.e. the systemic or topical
administration, preferably oral or intravenous administration, more preferably
oral
administration, of an effective amount of a compound of Formula (I) or a
pharmaceutically acceptable salt, or a solvate thereof, to subjects such as
humans.
One skilled in the art will recognize that a therapeutically effective amount
of the
compounds of the present invention is the amount sufficient to have
therapeutic activity
and that this amount varies inter alias, depending on the type of disease, the
concentration of the compound in the therapeutic formulation, and the
condition of the
patient. In an embodiment, a therapeutically effective daily amount may be
from about
0.005 mg/kg to 100 mg/kg.
The amount of a compound according to the present invention, also referred to
herein as the active ingredient, which is required to achieve a therapeutic
effect may vary
on case-by-case basis, for example with the specific compound, the route of
administration, the age and condition of the recipient, and the particular
disorder or
disease being treated. The methods of the present invention may also include
administering the active ingredient on a regimen of between one and four
intakes per
day. In these methods of the present invention, the compounds according to the
invention
are preferably formulated prior to administration.
The present invention also provides compositions for treating and / or
preventing
the disorders (preferably a cancer as described herein) referred to herein.
Said
compositions comprise a therapeutically effective amount of a compound of
Formula (I),
or a pharmaceutically acceptable salt, or a solvate thereof, and a
pharmaceutically
acceptable carrier or diluent.
While it is possible for the active ingredient (e.g. a compound of the present
invention) to be administered alone, it is preferable to administer it as a
pharmaceutical
composition. Accordingly, the present invention further provides a
pharmaceutical
composition comprising a compound according to the present invention, together
with a
pharmaceutically acceptable carrier or diluent. The carrier or diluent must be
"acceptable" in the sense of being compatible with the other ingredients of
the
composition and not deleterious to the recipients thereof.
The pharmaceutical compositions of the present invention may be prepared by
any methods well known in the art of pharmacy, for example, using methods such
as
those described in, for example, Gennaro et al. Remington's Pharmaceutical
Sciences
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 70 -
(18th ed., Mack Publishing Company, 1990, see especially Part 8 :
Pharmaceutical
preparations and their Manufacture).
The compounds of the present invention may be administered alone or in
combination
with one or more additional therapeutic agents. Combination therapy includes
administration of a single pharmaceutical dosage formulation which contains a
compound according to the present invention and one or more additional
therapeutic
agents, as well as administration of the compound according to the present
invention and
each additional therapeutic agent in its own separate pharmaceutical dosage
formulation.
Therefore, in an embodiment, the present invention is directed to a product
comprising, as a first active ingredient a compound according to the invention
and as
further, as an additional active ingredient one or more anti-cancer agent(s),
as a combined
preparation for simultaneous, separate or sequential use in the treatment of
patients
suffering from cancer.
The one or more other anti-cancer agents and the compound according to the
I 5 present invention may be administered simultaneously (e.g. in separate
or unitary
compositions) or sequentially, in either order. in an embodiment, the two or
more
compounds are administered within a period and / or in an amount and / or a
manner that
is sufficient to ensure that an advantageous or synergistic effect is
achieved. It will be
appreciated that the preferred method and order of administration and the
respective
dosage amounts and regimes for each component of the combination will depend
on the
particular other anti-cancer agent and the compound of the present invention
being
administered, their route of administration, the particular condition, in
particular tumor,
being treated and the particular host being treated.
The following examples further illustrate the present invention.
EXAMPLES
Several methods for preparing the Compounds of this invention are illustrated
in the
following examples. Unless otherwise noted, all starting materials were
obtained from
commercial suppliers and used without further purification, or alternatively
can be
synthesized by a skilled person by using well-known methods.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 71 -
Abbreviation Meaning
ACN acetonitrile
AcOH acetic acid
Celite diatomaceous earth
Co Compound
Co. No. Compound Number
DCM dichloromethane
DDQ 2,3-Dichloro-5,6-dicyano- 1,4-benzoqui none
Dess-Martin 3 -oxo- 1,3 -dihydro- 1k5,2-benziodoxole- 1,
1, 1-triy1 triacetate
periodinane
DIBAL diisobutylaluminium hydride
DicaliteCD diatomaceous earth
DIPE diisopropyl ether
DIPEA N,N-diisopropylethylamine
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
DTBAD Di-tert-butyl Azodicarboxylate
eq. equivalent(s)
Et3N or TEA trietylamine
Et3N.(1IF)3 triethylamine trihydrofluoride
Et0Ac or AcOEt ethyl acetate
Et0H ethanol
Et20 diethyl ether
hour(s)
HPLC high peiformance liquid chromatography
iPrNH2 isopropylamine
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 72 -
Abbreviation Meaning
IPA or iPrOH isopropanol
mCPBA meta-chloroperoxybenzoic acid
KSAc potassium thioacetate
Me methyl
Met methyl iodide
Me0H methanol
2-Me-THF 2-methyltetrahydrofuran
MP melting point
MsCI methanesulfonyl chloride
NH3-Me0H ammonia solution in methanol
nBuI,i n-butyllithium
NaBH(OAc)3 sodium triacetoxyborohydride
Na0Ac sodium acetate
Pd/C palladium on carbon
Pd(amphos)2C12 dichlorobis[di-tert-butyl(p-
dimethylaminophenyl)phosphino]palladium(II)
Pd2(dba)3 tris(dibenzylideneacetone)dipalladium(0)
Pd(dtbpf)C12 1,1'-Bis(di-t-butylphosphino)ferrocene palladium
dichloride
PPh3 triphenylphosphine
pTs0H p-toluenesulfonic acid
pTs0H.H20 p-toluenesulfonic acid monohydrate
rac racemic
Rochelle salt potassium sodium tartrate tetrahydrate
RP reversed phase
SFC superciitical fluid chromatography
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 73 -
Abbreviation Meaning
S-Phos 2-dicyclohexylphosphino-2',6'-
dimethoxybiphenyl
TBAF tetrabutylammonium fluoride
TBDMSC1 tert-butyldimethylsilyl chloride
TB DPSCI tert-butyl(chl oro)di phenyl si
lane
TIIF tetrahydrofuran
TLC thin layer chromatography
As understood by a person skilled in the art, Compounds synthesized using the
protocols
as indicated may contain residual solvent or minor impurities.
A skilled person will realize that, even where not mentioned explicitly in the
experimental protocols below, typically after a column chromatography
purification, the
desired fractions were collected and the solvent was evaporated.
In case no stereochemistry is indicated, this means it is a mixture of
stereoisomers, unless
otherwise is indicated or is clear from the context.
Compounds or intermediates indicated as -Sa or Ra atropisomer" or ¨Ra or Sa
atropisomer" are compounds or intermediates which are one atropisomeric form
but for
which the absolute stereochemistry is undetermined.
Preparation of intermediates
For intermediates that were used in a next reaction step as a crude or as a
partially purified
intermediate, in some cases no mol amounts are mentioned for such intermediate
in the
next reaction step or alternatively estimated mol amounts or theoretical mol
amounts for
such intermediate in the next reaction step are indicated in the reaction
protocols
described below.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 74 -
Intermediate 1
Br
CI
40,
0
A solution of (3-bromo-4-chlorophenyl)hydrazine (4.655 g, 18.047 mmol) and
methyl
2-oxobutanoate (1.02 eq) in HC1 (93 mL, 1.25 M in Me0H) was refluxed for 90
min.
The reaction mixture was cooled to room temperature and volatiles were removed
under reduced pressure to give 5.768 g of Intermediate 1 as a brown oily
residue that
solidified within minutes, used without further purification in the next step.
Intermediate 2
Br
CI 0
O¨
H
A suspension of Intermediate 1 (5.768 g, 18 mmol) in acetic acid (37 mL) was
heated
to 70 C. Sulfuric acid (4.81 mL, 5 eq.) was added dropwise over 10 min
(exotherm
developed and a precipitate formed). After 15 additional min, the reaction
mixture was
cooled to room temperature and then to 0 C by adding ice. The solid
precipitate was
filtered and washed with water until the filtrate was of neutral pH. The solid
was
triturated with cold heptane/diisopropylether (8/2, 50 mL) to give an off-
white solid.
This solid was purified by preparative SFC (Stationary phase: Chiralpak Daicel
IG 20
x 250 mm, Mobile phase: CO2, Et0H + 0.4 % iPrINTI2) to give Intermediate 2
(1.745 g,
32 %).
Intermediate 3
Me0
N¨N
0
\o
CI
NH 0
Intermediate 2 (500 mg, 1.65 mmol), 3-0(4-methoxybenzyl)oxy)rnethyl)-1,5-
dimethyl-
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-y1)-1H-pyrazole [2143010-90-4]
(800 mg,
1.3 eq.), Pd2(dba)3 (76 mg, 0.05 eq.), and S-Phos (68 mg, 0.1 eq.) were
weighed in a
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 75 -
pressure tube under nitrogen atmosphere. Dioxane (10.5 mL) and a saturated
aqueous
NaHCO3 solution (4.5 mL) were added and the mixture was heated at 100 C for 2
h.
The reaction mixture was cooled to room temperature, diluted with Et0Ac (40
mL) and
water (40 mL). The organic layer was separated and the aqueous one was
extracted
with Et0Ac (40 mL). The combined organic layer was dried over MgSO4, filtered
and
evaporated. The crude mixture was purified by flash chromatography on silica
gel (40
g, gradient: from heptane 100 % up to heptane/Et0Ac 4/6). Intermediate 3 (790
mg, 89
%) was obtained as a yellowish oil that solidified on standing . Intermediate
3 was used
without further purification in the next reaction step.
Intermediate 4
N¨N
HO / r
CI 0
NH 0
Tfifluoromethanesulfonic acid (0.888 mL, 5 eq.) was added to a solution of
Intermediate 3 (1080 mg, 2 mmol) in DCM (25 mL). The reaction mixture was
stirred
at room temperature for 1 h. The reaction mixture was diluted with DCM (100
mL) and
treated with saturated aqueous NaHCO3 (30 mL). The organic layer was separated
and
the aqueous one was extracted with DCM (50 mL x 3). The combined organic layer
was dried over MgSO4, filtered, and evaporated. Intermediate 4 (625 mg, 89 %)
was
obtained as a yellowish solid, used without further purification in the next
step.
Intermediate 5
N¨N
HO /
CI 0
0
"-OTBDMS
Cesium carbonate (732 mg, 1.25 eq.) was added to a solution of Intermediate 4
(625
mg, 1.79 mmol) in DMF (10 mL) under nitrogen atmosphere. (3-Bromopropoxy)(tert-
butyl)dimethylsilane (0.458 mL, 1.1 eq.) was added dropwise and the reaction
mixture
was stirred at room temperature overnight. The reaction mixture was diluted
with
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 76 -
Et0Ac (100 mL) and water (50 mL). The organic layer was separated and washed
with
brine (2 x 30 mL). The combined aqueous layers were extracted with Et0Ac (50
mL).
The combined organic layer was then dried over MgSO4, filtered and evaporated.
The
crude mixture was purified by flash chromatography on silica gel (40 g,
gradient: from
heptane 100 % up to Et0Ac 100 %) to afford Intermediate 5 (360 mg, 38 %) as a
white
solid.
Intermediate 6
N¨N
CI 0
0
""--OTBDMS
Mesyl chloride (0.12 mL, 2.5 eq.) was added dropwi se to a solution of
Intermediate 5
(320 mg, 0.61 mmol) and triethylamine (0.256 mL, 3 eq.) in DCM (10 mL)
stirring at 0
C under nitrogen. The reaction mixture was then allowed to warm up to room
temperature and was stirred at room temperature for 1 h. Additional
triethylamine (3
eq.) and mesyl chloride (2.5 eq.) were added and stirring was continued at
room
temperature for 1 h. The reaction mixture was diluted with DCM (10 mL) and
treated
with saturated aqueous NaHCO3 (5 mL). The organic layer was separated and the
aqueous one was extracted with DCM (10 mL). The combined organic layer was
dried
over MgSO4, filtered, and evaporated to give Intermediate 6 (368 mg,
quantitative),
used without further purification in the next step.
Intermediate 7
N¨N
CI 0
0
OTBDMS
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 77 -
Potassium iodide (1.021 g, 10 eq.) was added to a solution of Intermediate 6
(368 mg,
0.61 mmol) in acetonitrile (5 mL). The reaction was stirred at room
temperature
overnight. The reaction mixture was diluted with Et0Ac (50 mL) and filtered
over
Dicalitee. Water (25 mL) was added to the filtrate and, after some stirring,
the organic
layer was separated. The aqueous layer was back-extracted with Et0Ac (25 mL).
The
combined organic layer was dried over MgSO4, filtered, and evaporated to give
Intermediate 7, used without further purification in the next step.
Intermediate 8
N¨N
0 ci 0
0
.--OTBDMS
KSAc (400 mg, 1.5 eq.) was added to a degassed solution of Intermediate 7
(1.55 g,
2.337 mmol) in ACN (25 mL) at room temperature. The resulting reaction mixture
was
stirred at room temperature for 16 h. The reaction mixture was filtered
through a pad of
Celite and concentrated. The crude product was purified by flash column
chromatography on silica gel (heptane:Et0Ac - 1:0 to 6:4) to give Intermediate
8 (1.15
g, yield: 80 %) as a yellow oil.
Intermediate 9
* o
TBDPSC1 (6.41 mL, 1.25 eq.) was added dropwise to a solution of methyl 4-
hydroxy-
2-naphthoate ([34205-71-5], 4g, 19.78 mmol) and imidazole (2.35 g, 1.75 eq.)
in DMF
(70 mL), cooled to 0 C. Once the addition was complete, the reaction mixture
was
stirred at room temperature for 14 h. The reaction mixture was diluted with
Et0Ac (40
mL) and washed subsequently with water, dilute aqueous HCI (0.1 M), saturated
aqueous NaHCO3, and brine (each 30 mL). The organic layer was dried over
MgSO4,
filtered, and concentrated. The residue was purified by column chromatography
on
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 78 -
silica gel (heptane:Et0Ac ¨ 1:0 to 9:1) to afford Intermediate 9(8.81 g,
yield: 91 %) as
a yellow oil.
Intermediate 10
OH
=
Li AlE14 (2 M solution in TI-117, 9.44 mL, 1.05 eq.) was slowly added to a
solution of
Intermediate 9 (8.8 g, 17.97 mmol) in TI-1F (70 mL) cooled to 0 C. Once the
addition
was complete, the reaction mixture was stirred at 0 C for 30 min. The
reaction was
quenched by slow addition of Et0Ac (20 mL) followed by a saturated solution of
Rochelle salt. The heterogeneous mixture was stirred at room temperature for 2
h. The
aqueous layer was extracted with Et0Ac (2 x 65 mL), and the combined organic
extracts were washed with brine (20 mL), dried over MgSO4, filtered, and
concentrated.The residue was purified by flash column chromatography on silica
gel
(heptane:Et0Ac - 1:0 to 3:1) to give Intermediate 10 (5_81 g, yield: 74 %) as
a white
solid.
Intermediate 11
= ¨o
=
Mn02 (5.81 g, 5 eq.) was added to a solution of Intermediate 10 (5.81 g, 13.38
mmol)
in ACN (60 mL) at room temperature. The resulting solution was stirred at 60
C for 2
h. The reaction mixture was filtered over a pad of Dicaliteg and concentrated
to give
Intermediate 11(5.47 g, yield: 94 %) as a white solid, used without further
purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 79 -
Intermediate 12
c!) 0
N 7
\N
111101
undetermined mixture of E/Z
41101 0 Si (
NaH (653 mg, 1.1 eq.) was added to a suspension of intermediate 105 (8.094 g,
1.1 eq.)
in TI-if (90 mL) at 0 C. The resulting solution (solution A) was stirred at 0
C for 45
min before it was cooled to -25 'C. A solution of Intermediate 11(6.7 g, 15.5
mmol) in
11-IF (16 mL) was added slowly to solution A while maintaining the temperature
between -20 C and -30 'C. Once the addition was complete, the reaction
mixture was
stirred at -10 C for 1 h. The reaction was quenched by slow addition of
saturated
aqueous NI-IdC1 (10 mL) at -10 C and was diluted with Et0Ac (100 mL). The
layers
were separated and the aqueous layer was extracted with Et0Ac (2 x 100 mL).
The
combined organic layer was dried over MgSO4, filtered, and concentrated. The
residue
was purified by column chromatography on silica gel (heptane:Et0Ac - 1:0 to
7:3) to
afford Intermediate 12 (6.75 g, yield: 75 A) as a white foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 80 -
Intermediate 13
OH
N
undetermined mixture of E/Z
140
LiA1H4 (2 M solution in THF, 6.1 mL, 1.05 eq.) was slowly added to a solution
of
Intermediate 12 (6.7 g, 11.64 mmol) in THF (45 mL) cooled to 0 C. Once the
addition
was complete, the reaction mixture was stirred at 0 C for 30 min. The
reaction was
quenched by slow addition of Et0Ac (20 mL) followed by a saturated solution of
Rochelle salt. The heterogeneous mixture was stirred at room temperature for 2
h. The
aqueous layer was extracted with Et0Ac (2 x 65 mL), and the combined organic
extracts were washed with brine (20 mL), dried over MgSO4, filtered, and
concentrated
to afford Intermediate 13 (6.01 g, yield: 94 %) as a white foam, used without
further
purification.
Intermediate 14
OH
N
0¨Si (
Intermediate 13 (5.95 g, 10.89 mmol) was dissolved in Me0H (280 mL). Pd/C (10
%,
1159 mg, 0.1 eq.) was added under nitrogen atmosphere. The reaction mixture
was then
flushed with hydrogen gas and vacuum (3 times), then hydrogen (atmospheric
pressure,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 81 -
244 mL, 1 eq.) was taken up while stirring at room temperature. The reaction
mixture
was filtered over a pad of Dicaliteg and concentrated to give Intermediate 14
(5.9 g,
yield: 98 %) as a glassy yellow solid, used without further purification.
Intermediate 15
CI
N
0-Si (
411111
Thionyl chloride (459 tiIõ 1.15 eq.) was added to a solution of Intermediate
14 (3 g,
5.47 mmol) in DCM (23 mL) cooled to 0 C. Once the addition was complete, the
reaction was allowed to warm to room temperature and was stirred for 1 h. The
reaction
mixture was diluted with DCM (35 mL), washed with saturated aqueous NaHCO3 (2
x
50 mL) and brine (50 mL). The organic layer was dried over MgSO4, filtered,
and
concentrated. The residue was purified by flash column chromatography on
silica gel
(heptane:Et0Ac - 1:0 to 8:2) to give Intermediate 15 (2.65 g, yield: 85 %) as
a colorless
oil that crystallized on standing to a white amorphous solid.
Intermediate 16
N-N
N--
/ CI
N
0
OH OH
Intermediate 8 (500 mg, 0.821 mmol) and Intermediate 15 (559 mg, 1.2 eq.) were
dissolved in Me0H (10 mL). The reaction mixture was degassed and re-filled
with
nitrogen three times. The reaction mixture was then cooled to 0 C before
addition of
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 82 -
K2CO3 (227 mg, 2 eq.). After that addition, the reaction mixture was again
degassed
and re-filled with nitrogen twice. The reaction mixture was allowed to warm to
room
temperature and was stirred for 3 h. The reaction mixture was concentrated and
the
residue was partitioned between water (10 mL) and Et0Ac (15 mL). The layers
were
separated and the aqueous layer was extracted with Et0Ac (2 x 20 mL). The
combined
organic layer was washed with brine (30 mL), dried over MgSO4, filtered, and
concentrated to give a pale yellow foam.
This crude foam was dissolved in Me0H (10 mL) and pTs0H (469 mg, 3 eq.) was
added. The resulting reaction mixture was stirred at room temperature for 30
min. The
reaction mixture was concentrated. The residue was dissolved in Et0Ac (20 mL)
and
washed with saturated aqueous NaHCO3 (15 mL). The aqueous layer was extracted
with Et0Ac (2 x 20 mL), and the combined organic layer was washed with brine
(30
mL), dried over MgSO4, filtered, and concentrated. The crude product was
purified by
flash column chromatography on silica gel (heptane:Et0Ac - 6:4 to 0:1) to give
Intermediate 16 (695 mg, yield: 92 %) as a yellow oil.
Intermediate 17 and Intermediate 18
N¨N
CI
N j
/
0
or
Intermediate 17: Sa or Ra atropisomer; Intermediate 18: Ra or Sa atropisomer
A solution of Intermediate 16 (690 mg, 0.754 mmol) and DTBAD (694 mg, 4 eq.)
dissolved in a mixture of toluene (22 mL) and THE (4.5 mL) was added dropwise
with
a syringe pump (0.1 inlimin) to a solution of PP113 (791 mg, 4 eq.) in toluene
(22 inL)
at 70 C. At the end of the addition, the reaction mixture was concentrated.
The residue
was purified by flash column chromatography on silica gel (heptane:Et0Ac - 6:4
to
0:1) to give the racemic mixture (320 mg, yield: 60 %) of Intermediate 17 and
Intermediate 18 as a white foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 83 -
200 mg of the isolated mixture were purified by preparative SFC (Stationary
phase:
Chiralpak Daicel ID 20 x 250 mm, Mobile phase: CO2, Et0H-iPrOH (50-50) + 0.4 %
iPrNH2) to give Intermediate 17 (56 mg, yield: 10 %) and Intermediate 18 (68
mg,
yield: 13 %) as colourless oils.
Intermediate 19a and Intermediate 19b (mixture of regioisomers)
= o /¨
=
o 0/¨
* ,o
F
Intermediate 19a Intermediate 19b
TBDPSC1 (3.726 mL, 3 eq.) was added dropwise to a 5:1 mixture of ethyl 7-
fluoro-4-
hydroxy-2-naphthoate [1093083-28-3], ethyl 5-fluoro-4-hydroxy-2-naphthoate
[1093083-27-2] (2244 mg, 9.58 mmol), and imidazole (1141 mg, 3.5 eq.) in DMF
(25
mL), cooled to 0 C. Once the addition was complete, the reaction was stirred
at room
temperature for 12 h. The reaction mixture was diluted with Et0Ac (40 mL) and
washed successively with water, dilute aqueous HC1 (0.1 M), saturated aqueous
NaFIC03, and brine (each 30 mL). The organic layer was dried over IsvIgSO4,
filtered,
and concentrated to afford a pale yellow oil. This oil was purified by column
chromatography on silica gel (heptane:Et0Ac - 1:0 to 9:1) to afford the
mixture of
Intemiediate 19a and Intermediate 19b (2:1 ratio, 5.65 g, still impure, yield
considered
quantitative) as a yellow oil, used without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 84 -
Intermediate 20a and Intermediate 20b (mixture of regioisomers)
= = OH OH
i
F
Intermediate 20a Intermediate 20b
LiA1H4 (2 Mmn THF, 4.888 mL, 1.05 eq.) was added slowly to the mixture of
Intermediate 19a and Intermediate 19b (4.4 g, 9.31 mmol) in THE (35 mL),
cooled to 0
'C. Once the addition was complete, the reaction mixture was stirred at 0 C
for 2 h.
The reaction was quenched by slow addition of Et0Ac (20 ml-) followed by a
saturated
aqueous solution of Rochelle salt. The heterogeneous mixture was stirred at
room
temperature for 2 h. The aqueous layer was extracted with Et0Ac (2 x 65 mL),
the
combined organic extract was washed with brine (20 mL), dried over MgSO4,
filtered,
and concentrated to afford an orange oil. The crude product was purified by
flash
column chromatography on silica gel (heptane:Et0Ac - 1:0 to 3:1) to give the
mixture
of Intermediate 20a and Intermediate 20b (4.2 g, yield: 94 %) as a white
solid.
Intermediate 21a and Intermediate 21b (mixture of regioisomers)
¨o
= ¨o
Y-siõo
= F
Intermediate 21a Intermediate 21b
Mn02 (5.907 g, 5 eq.) was added to the mixture of Intermediate 20a and
Intermediate
20b (6.501 g, 13.589 mmol) in ACN (60 mL) at room temperature. The resulting
solution was stirred at 60 'V for 2 h. The reaction mixture was filtered over
a pad of
Dicalite and concentrated to give the mixture of Intermediate 21a and
Intermediate
21b (4.45 g, 80 % pure, yield: 61 %) as a white solid, used without further
purification.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 85 -
Intermediate 22a and Intermediate 22b (mixture of regioisomers)
0 0
0 0
N
\N undetermined E/Z mixture
N
/ undetermined E/Z mixture
0-Si (
Si
411111 40 0
411 F
Intermediate 22a Intermediate 22b
NaII (60% in mineral oil, 354 mg, 1.1 eq.) was added to a suspension of
intermediate
105 (4.386 g, 1.1 eq.) in THE' (50 mL) at 0 C. The resulting solution was
stirred at this
temperature for 45 min before being cooled to -25 C. A solution of the
mixture of
Intermediate 21a and Intermediate 21b (4.5 g, 8.4 mmol) in THY (9 mL) was
added
slowly to the solution while maintaining the temperature between -20 C and -
30 C.
Once the addition was complete, the reaction was stirred at -10 C for 1.5 h.
The
reaction was quenched by slow addition of saturated aqueous NI-I4C1 (10 mL) at
- 10
'C. The mixture was diluted with Et0Ac (50 mL). The layers were separated and
the
aqueous layer was extracted with Et0Ac (2 x 50 mL). The combined organic layer
was
dried over MgSO4, filtered, and concentrated under reduced pressure. The crude
product was purified by column chromatography on silica gel (heptane:Et0Ac -
1:0 to
7:3) to afford the mixture of Intennediate 22a and Intermediate 22b (3.98 g,
yield: 79
%) as a white foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 86 -
Intermediate 23a and Intermediate 23b (mixture of regioisomers)
OH
OH
\N
undetermined E/Z mixture
\N / undetermined E/Z mixture
ID 0¨Si (
101
F
Intermediate 23a Intermediate 23b
LiA1H4 (2 M in THF, 4.1 mL, 1.25 eq.) was slowly added to the mixture of
Intermediate 22a and Intermediate 22b (3.9 g, 6.561 mmol) in THF (50 mL),
cooled to
0 C. Once the addition was complete, the reaction mixture was stirred at 0 C
for 30
min. The reaction was quenched by slow addition of Et0Ac (10 mL) followed by a
saturated aqueous Rochelle salt solution (50 mL). The aqueous layer was
extracted with
Et0Ac (2 x 45 mL). The combined organic extract was washed with brine (20 mL),
dried over MgSO4, filtered, and concentrated to afford the mixture of
Intermediate 23a
and Intermediate 23b (3.51 g, yield: 94%) as a pale yellow foam, used without
further
purification.
Intermediate 24a and Intermediate 24b (mixture of regioisomers)
OH
OH
N
\N
N
\N
F
Intermediate 24a Intermediate 24b
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 87 -
The mixture of Intermediate 23a and Intermediate 23b (3.5 g, 6.195 mmol) was
dissolved in Me01-1 (160 mL). Pd/C (10 %, 659 mg, 0.1 eq.) was added under
nitrogen
atmosphere. The reaction mixture was then flushed with hydrogen gas and vacuum
(3
times). The reaction mixture was then stirred at room temperature under an
hydrogen
atmosphere (1 atm) until 1 equivalent of hydrogen was taken up. The reaction
mixture
was filtered over a pad of Dicaliteg and concentrated to give the mixture of
Inteimediate 24a and Intermediate 24b (3.16 g, yield: 90%) as an off-white
solid, used
without further purification.
Intermediate 25a and Intermediate 25b (mixture of regioisomers)
ci
N
/\N
F
Intermediate 25a Intermediate 25b
SOC12 (0.274 mL, 1.5 eq.) was added to the mixture of Intermediate 24a and
Intermediate 24b (1.85 g, 3.262 mmol) in DCM (20 mL), cooled to 0 C. Once the
addition was complete, the reaction mixture was allowed to warm to room
temperature
and was stirred for 1 h. The reaction mixture was diluted with DCM (35 mL),
washed
with saturated aqueous NaHC0.3 (2 x 50 mL) and brine (50 mL). The organic
layer was
dried over MgSO4, filtered, and concentrated to give an orange oil. This oil
was
purified by flash column chromatography on silica gel (heptane: Et0Ac - 1:0 to
8:2) to
give the mixture of Intermediate 25a and Intermediate 25b (1.75g. yield: 91 %)
as a
colorless oil which crystallized on standing to a white solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 88 -
Intermediate 26a and Intermediate 26b (mixture of regioisomers)
N-N
N-N
N CI 0-
I
0
0
OH
OH
OH
OH
Intermediate 26a Intermediate 26b
Intermediate 8 (600 mg, 0.986 mmol) and the mixture of Intermediate 25a and
Intermediate 25b (659 mg, 1.2 eq.) were dissolved in Me0H (12 mL). The
reaction
mixture was degassed and re-filled with nitrogen three times. The reaction
mixture was
then cooled to 0 C before addition of1(?CO3 (272 mg, 2 eq.). After that
addition, the
reaction mixture was again degassed and re-filled with nitrogen twice. The
reaction
mixture was allowed to warm to room temperature and was stirred for 60 h. The
reaction mixture was concentrated and the residue was partitioned between
water (10
mL) and Et0Ac (15 mL). The layers were separated and the aqueous layer was
extracted with Et0Ac (2 x 20 mL). The combined organic layer was washed with
brine
(30 mL), dried over MgSO4, filtered, and concentrated.
The crude foam was dissolved in Me0H (10 mL) and pTs0H (469 mg, 3 eq.) was
added. The resulting reaction mixture was stirred at room temperature for 30
min. The
reaction mixture was concentrated. The residue was dissolved in Et0Ac (20 mL)
and
washed with saturated aqueous NaHCO3 (15 mL). The aqueous layer was extracted
with Et0Ac (2 x 20 mL). The combined organic layer was washed with brine (30
mL),
dried over MgSO4, filtered, and concentrated. The crude product was purified
by flash
column chromatography on silica gel (heptane:Et0Ac - 6:4 to 0:1) to give the
mixture
of Intermediate 26a and Intermediate 26b (620 mg, yield: 85 %) as a white
foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 89 -
Intermediate 27, Intermediate 28, and Intermediate 29
N¨N
rN-
/
0
0
Intermediate 27: Sa or IV, atropisomer
Intermediate 28: Ra or Sa atropisomer
N-N
N
/
0
Intermediate 29: Mixture of atropisomers
A solution of the mixture of Intermediate 26a and Intermediate 26b (600 mg,
0.809
mmol) and DTBAD (745 mg, 4 eq.) in a mixture of toluene (23 mL) and THF (4.8
mL)
was added dropwise with a syringe pump (0.1 mL/min) to a solution of PPh3 (849
mg,
4 eq.) in toluene (23 mL) at 70 C. After completion of the addition, the
reaction
mixture was concentrated and the crude product was purified by flash column
chromatography on silica gel (heptane:Et0Ac - 6:4 to 0:1) to give the mixture
of
Intermediate 27, Intermediate 28, and Intermediate 29 (450 mg, yield: 81 %) as
a white
foam. 225 mg of the isolated product were purified by preparative SFC
(Stationary
phase: Chiralpak Daicel ID 20 x 250 mm, Mobile phase: CO2, Et0H + 0.4 %
iPrNH2)
to give Intermediate 27 (71 mg, yield: 12 %) and a mixture of Intermediate 28
and
Intermediate 29. This mixture was purified again by preparative SFC
(Stationary phase:
Chiralpak Daicel AS 20 x 250 mm, Mobile phase: CO2, Et0H 0.4 % iPrNH2), to
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 90 -
afford Intermediate 28 (61 mg, yield: 10 %) and Intermediate 29 (29 mg, yield:
4 %) as
pale yellow oils that crystallized on standing.
Intermediate 30 and Intermediate 31
o, N¨N
CI
0
o/C
Intermediate 30: Sa or Ra atropisomer
Intermediate 31: Ra or Sa atropisomer
mCPBA (124 mg, 2.1 eq.) was added to the racemic mixture of Intermediate 17
and
Intermediate 18 (180 mg, 0.256 mmol) in DCM (10 mL), cooled in an ice bath.
After
15 min at 0 C, the reaction mixture was allowed to warm to room temperature
and was
stirred overnight. The reaction mixture was concentrated. The crude product
was
purified by flash column chromatography (heptane:Et0Ac - 3:1 to 2:8) to give
the
racemic mixture of Intermediate 30 and Intermediate 31(160 mg, yield: 80%) as
a
yellow solid. The atropisomers were then separated by preparative SFC
(Stationary
phase: Chiralpak Diacel AS 20 x 250 mm, Mobile phase: CO2, Et0H + 0.4 %
iPrNH2),
affording Intermediate 30 (34 mg, yield: 18 %) and Intermediate 31 (27 mg,
yield: 14
%) as white solids.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 91 -
Intermediate 32
NO2
0
N¨N
HN
\o
CI
0
OTBDMS
A solution of di-tert-butyl azodicarboxylate (78 mg, 2 eq.) in DCM (1 mL) was
added
dropwise to a suspension of Intermediate 5 (88 mg, 0.17 mmol), 2-
nitrobenzenesulfonamide (38 mg, 1.1 eq.), and triphenylphospine (89 mg, 2 eq.)
in
DCM (2.5 mL) stirring at room temperature under nitrogen atmosphere. After 15
min,
the reaction mixture was directly loaded onto a silica gel column (12 g) and
the product
was purified eluting with a gradient from heptane 100 % up to heptane/Et0Ac
1/1.
Intermediate 32 (120 mg, quantitative) was obtained as a yellow solid.
Intermediate 33a and Intermediate 33b (mixture of regioisomers)
*0 u 0 u
N-N N-N
N / N /
/ CI 0- / CI
N 0 N 0
4111 4110
= *
0, 0_Si
F
Intermediate 33a Intermediate 33b
To a suspension of Intermediate 32 (1450 mg, 1.133 mmol), the mixture of
Inteimediate 24a and Intermediate 24b (642 mg, 1 eq.) and PPh3 (594, 2 eq.) in
DCM
(17 mL) was added a solution of DTBAD (521 mg, 2 eq.) in DCM (5 mL). The
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 92 -
resulting reaction mixture was stirred at room temperature for 16 h. The
reaction
mixture was concentrated and the crude product was purified by flash column
chromatography on silica gel (heptane:Et0Ac - 1:0 to 1:1) to give the mixture
of
Intermediate 33a and Intermediate 33b (1.21 g, yield: 61%) as a yellow foam.
Intermediate 34a and Intermediate 34b (mixture of regioisomers)
40, O. =
1\1µ.
0-
0
0
N-N SN
%(:) N-N
N
CI 0-
CI 0-
/N
/N
0
0
H
41111 ik OH
Or OH
Intermediate 34a Intermediate 34b
The mixture of Intermediate 33a and Intermediate 33b (2156 mg, 1.232 mmol) was
dissolved in Me0H (15 mL) and pTs0H (782 mg, 6 eq.) was added. The resulting
reaction mixture was stirred at room temperature for 30 min. The reaction
mixture was
concentrated to give a yellow oil. The oil was dissolved in Et0Ac (20 mL) and
was
washed with saturated aqueous NaHCO3 (15 mL). The aqueous layer was extracted
with Et0Ac (2 x 20 mL). The combined organic layer was washed with brine (30
mL),
dried over MgSO4, filtered, and concentrated to give a yellow oil. This yellow
oil was
dissolved in Me01-I (15 mL) and K2CO3 (284 mg, 3 eq.) was added. The reaction
mixture was stirred at room temperature for 14 h. The reaction mixture was
concentrated and the residue was partitioned between DCM (20 mL) and saturated
aqueous NELIC1 (20 mL). The aqueous layer was extracted with DCM (20 mL), the
combined organic layer was dried over MgSO4, filtered, and evaporated. The
crude
product was purified by flash column chromatography on silica gel
(heptane:Et0Ac -
6:4 to 0:1) to give the mixture of Intermediate 34a and Intermediate 34b (630
mg,
yield: 73 %) as a yellow foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 93 -
Intermediate 35a and Intermediate 35b (mixture of regioisomers)
o
II
N'
140 ,)0 ¨N/ .--0-
s N¨N
N /
CI 0¨
\ ¨ N "
\
/
* 0
110 41 i
F 10 F
Inteiiiiedi ate 35a Intermediate 35b
A solution of the mixture of Intermediate 34a and Intermediate 34b (625 mg,
0.502
mmol) and DTBAD (462 mg, 4 eq.) in a mixture of toluene (15 mL) and THY (3 mL)
was added dropwise with a syringe pump (0.1 mL/min) to a solution of PPh3 (526
mg,
4 eq.) in toluene (15 mL) at 70 C. The reaction mixture was concentrated. The
residue
was purified by flash column chromatography on silica gel (heptane:Et0Ac - 6:4
to
2:8) to yield a mixture of Intermediate 35a and Intermediate 35b (507 mg,
yield: 83 %)
as a white foam.
Intermediate 36a and Intermediate 36b (mixture of regioisomers)
/
/
N¨N 0)) /
H / N¨N
N / H i
N /
_
N\ AP
/ CI \ 0¨
CI
N N 0 \ /
N N 0
/
411 At 0)
F 111./ F
Intermediate 36a
Intermediate 36b
To a suspension of a mixture of Intermediate 35a and Intermediate 35b (500 mg,
0.41
mmol) and K2CO3 (566 mg, 10 eq.) in anhydrous ACN (10 mL) was added dropwise
thiophenol (0.421 mL, 10 eq.). The reaction mixture was stirred overnight at
room
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 94 -
temperature. The reaction mixture was filtered over a pad of Dicalite and the
filtrate
was evaporated. The crude product was purified by column chromatography on
silica
gel (DCM:Me0H - 1:0 to 9:1) to give a mixture of Intermediate 36a and
Intermediate
36b (185 mg, yield: 64 %) as a white foam.
Intermediate 37, Intermediate 38, Intermediate 39, and Intermediate 40
N¨N
CI O¨
N
0
=
41110
Intermediate 37: Sa or Ra atropisomer
Intermediate 38: Ra or Sa atropisomer
CI O¨
N "r
0
41111P
F
Intermediate 39: Sa or Ra atropisomer
Intermediate 40: Ita or Sa atropisomer
Foi ____________ inaldehyde (37% aqueous solution, 57 tL, 3 eq.) was added to
a solution of a
mixture of Intermediate 36a and Intermediate 36b (180 mg, 0.256 mmol) and AcOH
(44 tifõ 3 eq.) in DCM (3 mL) at room temperature. Then, NaBH(OAc)3 (162 mg, 3
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-95 -
eq.) was added and the reaction mixture was stirred at room temperature for 1
h. The
reaction was quenched by addition of saturated aqueous NaHCO3 (2.5 mL) and was
diluted with water (2.5 mL) and DCM (10 mL). The organic layer was separated
and
the aqueous layer was extracted with DCM (2 x 10 mL). The combined organic
layer
was dried over MgSO4, filtered, and evaporated. The residue was purified by
preparative SFC (Stationary phase: Chiralpak Daicel ID 20 x 250 mm, Mobile
phase:
CO2, iPrOH + 0.4 % iPrNH2) to give a mixture of Intermediate 37 and
Intermediate 38
and a mixture of Intermediate 39 and Intermediate 40. The first mixture was
purified by
preparative SFC (Stationary phase: Chiralcel Diacel OD 20 x 250 mm, Mobile
phase:
CO2, Et0H + 0.4 % iPrNH2) to give Intermediate 37(40 mg, yield: 22 %) and
Intermediate 38 (41 mg, yield: 22 %). The second mixture was purified by
preparative
SFC (Stationary phase: Chiralcel Diacel OD 20 x 250 mm, Mobile phase: CO2,
Et0H +
0.4 % iPrNH2) to give Intermediate 39(14 mg, yield: 7 %) and Intermediate 40
(13 mg,
yield: 7%).
Intermediate 41
0 OH
Sodium ethoxide (12.918 g, 2 eq.) was slowly added to anhydrous Et0H (175 mL)
at
room temperature, under nitrogen atmosphere. Once the addition was complete,
the
reaction mixture was warmed to 50 C and was stirred for 1 h. A solution of 2-
fluorobenzaldehyde (10 mL, 94.912 mmol) and diethyl succinate (16.581 mL, 1.05
eq.)
dissolved in Et0H (30 mL) were then added dropwise at 50 C via syringe pump
(0.5
mL/min). Once the addition was complete, the reaction mixture was refluxed for
3 h.
The reaction mixture was concentrated under reduced pressure and the residue
was
partitioned between 1 M aqueous HC1 (150 mL) and Et0Ac (200 mL). The layers
were
separated and the aqueous layer was extracted with Et0Ac (2 x 200 mL). The
combined organic layer was washed with brine, dried over MgSO4, filtered, and
concentrated to afford Intermediate 41(26.5 g, yield: 50 %) as an orange oil,
used
without further purification.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 96 -
Intermediate 42
0L
Liyo
-`
0
Sodium acetate (8.456 g, 1 eq.) was added to Intermediate 41(26 g, 103.07
mmol) in
acetic anhydride (80 mL). The resulting solution was refluxed for 1.5 h. After
cooling,
the reaction mixture was concentrated under reduced pressure. The residue was
partitioned between Et0Ac and water (200 mL each). The layers were separated
and
the aqueous layer was extracted with Et0Ac (3 x 350 mL). The combined organic
layer
was carefully quenched with saturated aqueous NaHCO3 and then solid NaHCO3
until
the pH reached 8. The organic layer was washed one more time with saturated
aqueous
NaHCO3 (400 mL) and then with brine (400 mL). The organic layer was dried on
MgSO4, filtered, and evaporated. The crude product was purified by flash
column
chromatography on silica gel (heptane:Et0Ac - 1:0 to 8:2) to give Intermediate
42
(4.45 g, yield: 12 %) as a yellow solid.
Intermediate 43
OH
O
K2CO3 (2.852g. 2 eq.) was added to Intermediate 42 (3800 mg, 10.316 mmol) in a
mixture of Et0H (40 mL), Me0H (5 mL) and THE (10 mL) and the reaction mixture
was stirred for 16 h at room temperature. The reaction mixture was filtered to
remove
the residual potassium carbonate and concentrated under reduced pressure. The
dark
brown oil was dissolved in Et0A_c (70 mL) and washed with saturated aqueous
NH4C1
(50 mL). The aqueous layer was extracted with Et0Ac (2 x 60 mL). The combined
organic layer was washed with brine (100 mL), dried over MgSO4, filtered, and
concentrated under reduced pressure. The crude product was purified by column
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 97 -
chromatography on silica gel (heptane:Et0Ac - 1:0 to 7:3) to give Intermediate
43
(2.42 g, yield: 90 %) as an orange solid.
Intermediate 44 and Intermediate 45
N ¨N
N
I C I 0
N /
0
Intermediate 44: Ra or Sa atropisomer
Intermediate 45: Sa or Ra atropisomer
Intemiediate 44 and Intermediate 45 were prepared following the same synthetic
pathway as for Intermediate 27 and Intermediate 28, respectively, starting
initially from
Intermediate 43 instead of the mixture of ethyl 7-fluoro-4-hydroxy-2-
naphthoate and
ethyl 5-fluoro-4-hydroxy-2-naphthoate.
Intermediate 46 and Intermediate 47
0 NTN
CI O¨
N V
0
05)
Intermediate 46: Sa or Ra atropisomer
Intermediate 47: Ita or Sa atropisomer
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 98 -
Intermediate 46 and Intermediate 47 were prepared using an analogous procedure
as for
Intermediate 30 and Intermediate 31, starting from the pure atropisomers
Intermediate
27 and Intermediate 28, respectively, instead of the racemic mixture of
Intermediate 17
and Intermediate 18.
Intermediate 48
4100. o
Intermediate 48
TBDPSC1 (14.66 g, 1.5 eq.) was added to a solution of methyl 7-fluoro-4-
hydroxy-2-
naphthoate (CAS [2092726-85-5]) (8 g, 35.555 mmol) and imidazole (7.26, 3 eq.)
in
DCM (500 nit), cooled to 0 C under nitrogen atmosphere. Once the addition was
complete, the reaction was stirred at room temperature overnight. The reaction
was
quenched by addition of water (100 mL). The mixture was extracted with Et0Ac
(3 x
200 mL). The combined organic layer was dried over Na2SO4, filtered, and
concentrated to afford a yellow oil. This oil was purified by flash column
chromatography on silica gel (petroleum ether:Et0Ac - 1:0 to 1:1) to afford
Inteimediate 48 (14 g, yield: 86 %) as a yellow oil.
Intermediate 20a
OH
=
Intermediate 20a
LiA1H4 (1.39 g , 1.2 eq.) was added slowly to a solution of Intermediate 48
(14 g,
30.528 mmol) in TI-1F (200 mL), cooled to 0 'V under nitrogen atmosphere. Once
the
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 99 -
addition was complete the reaction mixture was stirred at 0 C for 2 h. The
reaction was
quenched by slow addition of water (2 mL) followed by a 10% aqueous NaOH
solution (2 mL) at 0 'C. The heterogeneous mixture was filtered, and the
filter cake was
washed with DCM (200 mL). The filtrate was evaporated and the residue was
purified
by flash column chromatography on silica gel (petroleum ether:Et0Ac - 1:0 to
1:1) to
give Intermediate 20a (12 g, yield: 90%) as a yellow solid.
Intermediate 21a
¨0
O
=
Intermediate 21a
Mn02 (29.074 g, 12 eq.) was added to a solution of Intermediate 20a (12g. 27
869
mmol) in DCM (200 mL) at room temperature. The resulting solution was stirred
at
room temperature overnight. The reaction mixture was filtered and the filtrate
was
concentrated. The residue was purified by flash column chromatography over
silica gel
(eluent: petroleum ether/Et0Ac, 100/0 to 50/50) to afford Intermediate 21a (12
g, yield:
99%) as a yellow oil.
Intermediate 22a
0
N
\N
undetermined E/Z mixture
0¨Si (
Intermediate 22a
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 100 -
NaH (60 % in mineral oil, 1.448 g, 1.3 eq.) was added to a suspension of
intermediate
105 (13.812 g, 1.1 eq.) in THF (200 mL) at 0 C. The resulting solution was
stirred at
this temperature for 1 h before being cooled to -30 'C. Intermediate 21a (12
g, 27.847
mmol) was added slowly to the solution while maintaining the temperature
between -20
C and -30 C. Once the addition was complete, the reaction was stirred at -30
C for 2
h. The reaction was quenched by slow addition of water (100 mL). The mixture
was
extracted with DCM (3 x 300 mL). The combined organic layer was dried over
Na2SO4, filtered, and concentrated under reduced pressure. The crude product
was
purified by column chromatography on silica gel (petroleum ether:Et0Ac - 1:0
to 1:1)
to afford Intermediate 22a (13 g, yield: 82%) as a white solid.
Intermediate 49
o
/
01 I
0 S i (
Intermediate 49
A solution of Intermediate 22a (13 g, 23.02 mmol) in Me0H (75 mL) and THF (75
mL) was hydrogenated at 25 'V (15 psi H2) in the presence of Pd/C (2 g; 10 %).
The
reaction mixture was stirred for 16 It After uptake of H2 (1 eq.), the
catalyst was
filtered off and the filtrate was evaporated to afford Intermediate 49 (13 g,
yield: 100
%) as a colorless oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 101 -
Intermediate 24a
0 H
N
/
0 - S (
Intermediate 24a
LiA1H4 (1.045 g, 1.2 eq.) was added portionwise to a solution of Intermediate
49 (13 g,
22.94 mmol) in TT-IF (200 mL) at 0 C, under nitrogen atmosphere. The reaction
mixture was stirred at 0 C for 2 h. Water (1 mL) was then added dropwise,
followed
by a 10 % aqueous NaOH solution (1 mL), at 0 C. The reaction mixture was
filtered,
the filter cake was washed with DCM (200 mL), and the filtrate was evaporated.
The
crude product was purified by flash column chromatography over silica gel
(eluent:
petroleum ether/Et0Ac, 100/0 to 0/100) to afford Intermediate 24a (10.4 g,
yield: 84
%) as a white solid.
Intermediate 25a
C I
N V
/
111101
0 -S ______________________________________________________
Intermediate 25a
S0C12 (0.78 mL, 1.15 eq.) was added dropwise to a solution of Intermediate
24a(5 g,
9.28 mmol) in anhydrous DCM (57 mL) under nitrogen atmosphere at 0 C. Once
the
addition was complete, the reaction mixture was allowed to warm to room
temperature
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 102 -
and was stirred for 1.5 h. The reaction was diluted with DCM, washed with a
saturated
aqueous NaHCO3 solution (x 2) and brine. The combined aqueous extracts were
extracted with DCM (x 3). The combined organic extract was dried over MgSO4,
filtered, and concentrated under reduced pressure to give a pale yellow solid.
This solid
was purified by flash column chromatography (SiO2, 40 g RediSep,
heptane/Et0Ac,
100/0 to 0/100) to afford intermediate 25a (4.55g, yield: 87%) as a white
solid.
Intermediate 50
QRS
N-N
Br
Intermediate 50
pTs0H (5.4 g, 0.1 eq.) was added to 1H-pyrazole-3-carboxylic acid, 4-bromo-5-
methyl-, methyl ester (CAS [1232838-31-1]) (76 g, 315 mmol) and 3,4-dihydro-2H-
pyran (53 g, 2 eq.) in DCM (600 mL). The reaction mixture was stirred at room
temperature for 2 h. The reaction was quenched by addition of water (300 mL)
and the
mixture was extracted with DCM (500 mL x 2). The combined organic layer was
washed with brine (200 mL), dried with Na2SO4, and filtered. The filtrate was
evaporated and the residue was purified by column chromatography over silica
gel
(eluent: petroleum ether/ Et0Ac 100:0 to 80:20) to give Intermediate 50 (130 g
crude,
78 % pure, quantitative) as a yellow solid.
Intermediate 51
RS
N-N
OH
Br
Intermediate 51
LiA1H4 (14.4 g, 2 eq.) was added portionwise to THJF (1 L) at 0 C. The
mixture was
stirred at 0 C for 5 min. Then, Intermediate 50 (64 g, 190 mmol) was added
portionwise. The reaction mixture was stirred at 0 C for 1 h. Water (14 mL)
was added
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 103 -
dropwi se, followed by aqueous NaOH (2 M, 14 mL), and finally MgSO4 (10 g).
The
mixture was filtered over a pad of Celite and the filter cake was washed with
DC1\4 (1 L
x 2). The combined organic layer was evaporated to give a yellow oil. This oil
was
purified by flash column chromatography over silica gel (petroleum ether/Et0Ac
from
100/0 to 40/60) to give Intermediate 51 (two fractions: 20 g (98% pure, yield:
37 %)
and 26 g (68% pure, yield: 47 %)) as a white solid.
Intermediate 52
QRS
N-N
Br
Intermediate 52
DMAP (814 mg, 0.4 eq.) and Et3N (4.6 mL, 2 eq.) were added to a solution of
Intermediate 51 (5 g, 16.66 mmol) in THE (50 mL) at room temperature. TBDMSC1
(3.77 g, 1.5 eq.) was added and the reaction mixture was stirred at room
temperature for
4 h. The reaction was quenched by addition of saturated aqueous NaHCO3 (50 mL)
and
the mixture was extracted with Et0Ac (50 mL x 2). The combined organic layer
was
washed with brine (50 mL), dried with Na2SO4, filtered, and evaporated. The
residue
was purified by column chromatography over silica gel (eluent: petroleum
ether/
Et0Ac 100:0 to 20:80) to give Intermediate 52 (6.31 g, yield: 96 %) as a
colorless oil.
Intermediate 53
QRS
N-N
Intermediate 53
nBuLi (59 mL, 1.2 eq.) was slowly added to a solution of Intermediate 52 (48
g, 123
mmol) in TI-IF (1 L) at -78 'V under nitrogen atmosphere. The reaction mixture
was
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 104 -
stirred at -78 C for 1 h. 2-Isopropoxy-4,4,5,5-tetramethy1-1,3,2-
dioxaborolane [61676-
62-8] (34.4g. 1.5 eq.) was slowly added to the reaction mixture. The reaction
mixture
was stirred at room temperature for 2 h. The reaction mixture was slowly added
to
saturated aqueous NH4C1 (200 mL). The resulting mixture was extracted with
Et0Ac
(500 mL x 2), and the combined organic layer was washed with brine (100 mL),
dried
on Na2SO4, filtered, and evaporated. The residue was purified by flash column
chromatography over silica gel (eluent: petroleum ether/ethyl acetate from
100/0 to
90/10) to give Intermediate 53 (50 g, yield: 69 %) as a yellow oil.
Intermediate 54
N¨N
HoJL
Intermediate 54
TBAF (1 M in THF, 54.99 mL, 1.2 eq.) was added to an ice-cooled solution of
Intermediate 53 (20 g, 46 mmol) in anhydrous 2-Me-TFIF (287 mL) under nitrogen
atmosphere. The ice bath was removed and the resulting reaction mixture was
stirred at
room temperature for 19 h. The reaction mixture was diluted with Et0Ac and the
layers
were separated. The organic layer was washed with an aqueous saturated
solution of
NaHCO3 and brine. The combined aqueous layer was extracted with Et0Ac (x 3)
and
the combined organic extract was dried over MgSO4, filtered, and concentrated
under
reduced pressure. The residue was purified by flash column chromatography
(SiO2, 120
g RediSep column, heptane/Et0Ac, gradient 100/0 to 0/100) to afford
Intermediate 54
(12 g, yield: 80 %) as a colorless oil that solidified to a white solid upon
standing.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 105 -
Intermediate 55
Br
CI 0-
\
0
0
Intermediate 55
A mixture of Intermediate 2 (37.4 g, 123.6 mmol), (3-bromopropoxy)-tert-
butyldimethylsilane (CAS [89031-84-51) (37.567 g, 1.2 eq.), and K2CO3 (51.25g.
3 eq.)
in ACN (300 mL) was stirred at 80 C overnight. The reaction mixture was
cooled to
room temperature and was filtered. The filter cake was washed with Et0Ac (100
mL).
The filtrate was concentrated and the residue was purified by column
chromatography
over silica gel (eluent: petroleum etlier/Et0Ac from 100/0 to 10/90) to afford
Intermediate 55 (42 g, 71 %) as a red gum.
Intermediate 56
RSQ)
N¨N
HO
CI 0
0¨
o
Intermediate 56
A pressure tube was charged with Intermediate 55(5 g, 10.17 mmol),
Intermediate 54
(4.18 g, 1.19 eq.), Pd(amphos)2C12 (CAS [887919-35-9]) (364 mg, 0.05 eq.), and
K2CO3 (2.84 g, 2 eq.) under nitrogen atmosphere. A mixture of 1,4-di oxane (49
mL)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 106 -
and water (12.5 mL), previously nitrogen-purged for 35 min, was added under
nitrogen
atmosphere to the reaction tube. The tube was sealed and the reaction mixture
was
heated for 3.5 h at 70 C. After cooling to room temperature, the reaction
mixture was
diluted with water and Et0Ac. The layers were separated and the aqueous layer
was
extracted with Et0Ac (x 3). The combined organic layer was dried over MgSO4,
filtered, and concentrated. The residue was purified by flash column
chromatography
(SiO2, 120 g Redi Sep column, heptane/Et0Ac, gradient 100/0 to 0/100) to give
Intermediate 56 (4.74 g, yield: 76 %) as a pale yellow foam.
Intermediate 57
RsQ
OrsN¨N
CI 0
0 ¨
i
Intermediate 57
Et3N (1.21 inL,1.5 eq.), followed by MsC1 (0.56 InL, 1.25 eq.) were added
dropwise to
a solution of Intermediate 56(3.57 g, 5.81 mmol) in anhydrous THE (71 mL,
degassed
by bubbling nitrogen for 15 min) under nitrogen atmosphere at 0 C The
reaction
mixture was stirred at 0 "V for 5 min, and then at room temperature for 1 h.
The
reaction mixture containing the intermediate mesylate was degassed by bubbling
nitrogen for 10 min. Then, a nitrogen purged solution of KSAc (6.63 g, 10 eq.)
in
anhydrous DMF (112 mL, nitrogen-purged for 30 min) was added in one portion to
the
reaction mixture at room temperature. The resulting mixture was nitrogen-
purged for 5
min and then stirred at room temperature for 30 min. The reaction mixture was
diluted
with Et0Ac and water. The aqueous layer was separated and extracted with Et0Ac
(x
3). The combined organic layer was washed with brine (x 3), dried over MgSO4,
filtered, and evaporated. The residue was purified by flash column
chromatography
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 107 -
(Si02, 220 g Redi Sep column, heptanefEt0Ac, gradient 100/0 to 0/100) to
afford
Intermediate 57 (3.9 g, yield: 93 %) as an orange oil.
Intermediate 58
RS
N-N
OH
N
CI O-
N /
0
OH
Intermediate 58
Intermediate 57 (3.9 g, 5.41 mmol), Intermediate 25a (3.69 g, 1.2 eq.), and
PPh3 (142
mg, 0.1 eq.) were charged in a 500 mL round bottom flask. The mixture was
degassed
and re-filled with nitrogen three times. Dry Me0H (200 mL, degassed by
bubbling
nitrogen for 20 min) was added. The mixture was degassed and re-filled with
nitrogen
three times, then degassed by bubbling nitrogen for 15 min. The resulting
suspension
was cooled to 0 C before addition of K2CO3 (2.24 g, 3 eq.). The reaction
mixture was
degassed and re-filled again with nitrogen three times, then degassed by
bubbling
nitrogen for 5 min. 'the reaction mixture was allowed to warm to room
temperature and
was stirred for 1.5 h. The reaction mixture was concentrated under reduced
pressure.
The residue was partitioned between water and Et0Ac. The layers were separated
and
the aqueous layer was extracted with Et0Ac (x 3). The combined organic layer
was
washed with brine, dried over MgSO4, filtered, and evaporated. The residue was
dissolved in TUT' (110 mL) and the solution was cooled to 0 C. TBAF (1M in
THF,
32.48 mL, 6 eq.) was added and the reaction mixture was allowed to warm to
room
temperature and was stirred for 30 min. Additional TBAF (1M in THF, 10.83 mL,
2
eq.) was added and the reaction mixture was stirred at room temperature for 40
min
The reaction was quenched by addition of saturated aqueous NH4C1. The layers
were
separated. The organic layer was washed with brine (x 2), the combined aqueous
layer
was extracted with Et0Ac (x 3) and DCM and the combined organic extract was
dried
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 108 -
over MgSO4, filtered, and concentrated under reduced pressure. The residue was
purified by flash column chromatography (SiO2, 40 g RediSep, heptane/Et0Ac,
100/0
to 0/100) to give impure Intermediate 58. This impure product was purified
again by
flash column chromatography (SiO2, 120 g RediSep, DCM/Me0H, 100/0 to 90/10) to
afford Intermediate 58(4.13 g, yield: 98%) as a brownish foam.
Intermediate 59
RSQ
N-N
N
1 C I
N
0
411 0/
/PI
Intermediate 59
A solution of PP113 (1.07 g, 4 eq.) in toluene (31 mL) was degassed and re-
filled with
nitrogen three times (Solution A). A solution of Intermediate 58 (789 mg, 1.02
mmol)
and DTBAD (938 mg, 4 eq.) in a mixture of toluene (31 mL) and Tiff (6 mL) was
degassed and re-filled with nitrogen three times (Solution B). Solution B was
added via
syringe pump (0.1 ml/min) to Solution A, stirred at 70 C under nitrogen
atmosphere.
Once the addition was complete, the reaction mixture was stirred for 15 min at
70 C.
The reaction mixture was cooled to room temperature, and the solvents were
evaporated. The residue was purified by flash column chromatography (SiO2, 80
g
RediSep, DCM/Me01-I, 100/0 to 90/10) to afford Intermediate 59 (2 g, impure,
yield
considered quantitative) as a yellow oil, used without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 109 -
Intermediate 60 and Intermediate 61
N¨NH N¨NH
N N
CI 0¨ CI
O¨
N / N
0
0
o/
o/
Intermediate 60 Intermediate 61
Ra or Sa atropisomer Sa or Ra atropisomer
HC1 (1.25 M in Me0H, 192 mL, 50 eq.) was added dropwise to a solution of
Intermediate 59 (3.62 g, 4.79 mmol) in anhydrous THF (190 mL) at 0 C. The
reaction
mixture was stirred at room temperature for 3 h. The reaction mixture was
concentrated
under reduced pressure. The residue was purified by preparative HPLC
(Stationary
phase: RP )(Bridge Prep C18 OBD-10 hrn,50 x 150 mm, Mobile phase: 0.25 %
NI-14HCO3 solution in water, CH3CN) to give the racemic mixture of
Intermediate 60
and Intermediate 61. This mixture was separated into its atropisomers by
preparative
SFC (Stationary phase: Chiralcel Diacel OJ 20 x 250 mm, Mobile phase: CO2,
Et0H +
0.4 % iPrNH2) to afford Intermediate 60 (892 mg, yield: 28 %) and Intermediate
61
(932 mg, yield: 29 %).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 110 -
Intermediate 62 and Intermediate 63
\o
o¨\\_
c)\
N¨N N¨N
µ'N
N
NI j CI I j CI
0
0
o/
o/
Intermediate 62 Intermediate 63
Ra or Sa atropisomer Ra or Sa atropisomer
Diethylene glycol 2-bromoethyl methyl ether (CAS 1172593-77-2D (63 mg, 2.5
eq.) was
added to a solution of Intermediate 60 (75 mg, 0.11 mmol) and Cs2CO3 (182 mg,
5 eq.)
in anhydrous DMF (2 mL), stirred at room temperature, under nitrogen
atmosphere.
The vial was sealed and the reaction mixture was stirred at 60 C. for 4 h.
The solvent
was evaporated and the residue was diluted with Et0Ac and water. The aqueous
layer
was extracted with Et0Ac (3 x). The combined organic layer was washed with
brine,
dried over MgSO4, filtered, and evaporated to give as a colorless oil. This
oil was
purified by preparative SFC (Stationary phase: Chiralpak Diacel AD 20 x 250
mm,
Mobile phase: CO2, iPrOH + 0.4 % iPrNH2) to afford Intermediate 62 (21 mg,
yield: 23
%) and Intermediate 63 (12 mg, yield: 13 %).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 1 1 1 -
Intermediate 64 and Intermediate 65
N¨N N¨N
N
O¨
N / N
0 0
o/
0
Intermediate 64 Intermediate 65
Ra or Sa atropisomer Ra or Sa atropisomer
2-Bromoethyl methyl ether (CAS [6482-24-2]) (45 L, 2.6 eq.) was added to a
solution
of Intermediate 60(121 mg, 0.181 mmol) and Cs7CO3 (178 mg, 3 eq.) in anhydrous
DMF (3 mL) at room temperature under nitrogen atmosphere. The reaction mixture
was stirred at room temperature for 6 h. The reaction mixture was diluted with
Et0Ac
and water. The layers were separated. The organic layer was washed with brine
(x 3),
and the combined aqueous extract was extracted with Et0Ac (x 2) and with DCM
(x
3). The combined organic layer was dried over MgSO4, filtered, and evaporated.
The
residue was purified by preparative SEC (Stationary phase: Chiralpak Daicel IC
20 x
250 mm, Mobile phase: CO2. Et0H + 0.4 % iPrNtb) to afford Intermediate 64 (54
mg,
yield: 41 %) and Intermediate 65 (54 mg, yield: 41 %), both as white solids.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 112 -
Intermediate 66 and Intermediate 67
¨o\ cp¨
N¨N N¨N
CI 0¨ CI O¨
N / N
0 0
o 0
Intermediate 66 Intermediate 67
Ra or Sa atropisomer Ra or Sa atropisomer
1,3-dimethoxypropan-2-y1 methanesulfonate (CAS [215453-88-6]) (142 mg, 5 eq.)
and
Intermediate 60 (96 mg, 0.144 mmol) were dissolved in anhydrous DME (2 mL)
under
nitrogen atmosphere. Cs2CO3 (141 mg, 3 eq.) was added at room temperature. The
vial
was sealed and the reaction mixture was stirred at 70 C for 16 h. To push the
reaction
to completion, additional 1,3-dimethoxypropan-2-y1 methanesulfonate [215453-88-
6]
(142 mg, 5 eq.) was added under nitrogen atmosphere and the reaction mixture
was
stirred at 100 C for 6 h. Again, additional 1,3-dimethoxypropan-2-y1
methanesulfonate
[215453-88-6] (142 mg, 5 eq.) was added and the reaction mixture was stirred
at 100
"V for 3 h. The reaction mixture was cooled to room temperature and was
stirred for 17
h. The solvent was evaporated, and the resulting crude mixture was diluted
with Et0Ac
and water. The layers were separated. The organic layer was washed with brine
(x 3),
and the combined aqueous extract was extracted with Et0Ac (x 3) and DCM. The
combined organic layer was dried over MgSO4, filtered, and evaporated. The
residue
was purified by flash column chromatography (SiO2, 40 g RediSep, DCM/Me0H,
100/0 to 90/10) to give a yellow oil. This oil was further purified by
preparative 1-IPLC
(Stationary phase: RP X13ridge Prep C18 013D- 5 p.m, 50x250 mm, Mobile phase:
0.25 % N1-141-1CO3 solution in water, CH3CN), followed by preparative SFC
(Stationary
phase: Chiralpak Diacel AD 20 x 250 mm, Mobile phase: CO2, Et0H + 0.4 %
iPrNH2)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 113 -
to afford Intermediate 66 (5 mg, yield: 5 %) and Intermediate 67 (7 mg, yield:
7 %),
both as white solids.
Intermediate 68 and Intermediate 69
0,
N¨N N¨N
N
/ CI
N CI 0-
0
o/
Intermediate 68 Intermediate 69
Ra or Sa atropisomer Ra or Sa atropisomer
4-(2-Bromoethyl)tetrahydropyran (CAS [4677-20-7]) (62 mg, 2.7 eq.) was added
to a
solution of Intermediate 60 (80 mg, 0.12 mmol) and Cs2CO3 (117 mg, 3 eq.) in
anhydrous DMF (2 mL) at room temperature, under nitrogen atmosphere. The
reaction
mixture was stirred at room temperature for 4.5 h. The solvent was evaporated
and the
residue was diluted with DCM and water. The layers were separated and the
organic
layer was washed with brine (x 3). The combined aqueous layer was extracted
with
Et0Ac (x 2) and with DCM (x 3). The combined organic layer was dried over
MgSO4,
filtered, and evaporated to give a colorless oil. This oil was purified by
preparative SFC
(Stationary phase: Chiralpak Diacel AD 20 x 250 mm, Mobile phase: CO2, Et0I-1
+ 0.4
"Yo iPrNH2) to afford Intermediate 68 (39 mg, yield: 42 %) and Intermediate 69
(30 mg,
yield: 32 %), both as white solids.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 114 -
Intermediate 74 and Intermediate 75
N¨N N¨N
N---- N
N CI 0¨
N CI 0-
0 0
o/
0
Intermediate 74 Intermediate 75
Ra or S. atropisomer R. or S. atropisomer
Mel (21 pL, 2.5 eq.) was added to a mixture of Intermediate 60 (90 mg, 0.134
mmol)
and Cs2CO3 (132 mg, 3 eq.) in anhydrous DMF (2 mL) at room temperature under
nitrogen atmosphere. The reaction mixture was stirred at room temperature for
4 h. The
solvent was evaporated. The residue was diluted with DCM and water and the
layers
were separated. The organic layer was washed with brine (x 3). The combined
aqueous
layer was extracted with DCM (x 4) and Et0Ac. The combined organic layer was
dried
over MgSO4, filtered, and evaporated. The residue was purified by flash column
chromatography (SiO2, 24 g RediSep, DCM/Me0H, 100/0 to 90/10) followed by
preparative HPLC (Stationary phase: RP XBridge Prep C18 OBD- 5 ttm, 50 x 250
mm, Mobile phase: 0.25 % NH4HCO3 solution in water, CH3CN), and finally by
preparative SFC (Stationary phase: Chiralpak Daicel ID 20 x 250 mm, Mobile
phase:
CO2, Et0H + 0.4 % iPrNH2) to give Intermediate 74 (7 mg, yield: 7 %) and
Intetinediate 75 (1 mg, yield: 1 %), both as white solids.
Intermediate 76
Br
N.,
H
0,õ1
Intermediate 76
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 115 -
Cyanomethylenetributylphosphorane (CAS [157141-27-0]) (45.02 mL, 1 eq.) was
added
dropwise to a solution of 1H-pyrazole-3-carboxylic acid, 4-bromo-5-methyl-,
ethyl ester
[6076-14-8] (20 g, 85.81 mmol) and 2-(2-methoxyethoxy)ethanol [111-77-3]
(14.15 mL,
1.4 eq.) in TI-IF (1.9 L) at room temperature. The reaction mixture was
stirred at room
temperature overnight. The reaction mixture was poured into water (100 mL) and
the
mixture was extracted with Et0Ac (3 x 100 mL). The combined organic layer was
washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The
crude
product was purified via flash column chromatography on silica gel
(heptane/Et0Ac,
100/0 to 50/50) to give Intermediate 76 (24 g, yield: 83 %).
Intermediate 77
Br
Intermediate 77
Sodium borohydride (4.26 g, 5 eq.) was added to a solution of Intermediate 76
(7.45 g,
22.23 mmol) in a mixture of THF (130 mL) and Me0H (34 mL) at 0 C. After 5
min, the
resulting mixture was allowed to reach room temperature and was stirred for 3
h. The
reaction mixture was diluted by very slow addition of acetone (80 mL) and
water (80
mL), followed by Et0Ac (100 mL). The layers were separated and the aqueous
layer was
extracted with Et0Ac (2 x 50 mL) followed by a 1:1 mixture of Et0Ac/THE (2 x
50
mL). The combined organic layer was dried over MgSO4, filtered, and evaporated
to
afford Intermediate 77 (7.24 g, quantitative) as a tan oil, used without
further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 116 -
Intermediate 78
Br
0
Intermediate 78
TBDMSC1 (617 mg, 1.2 eq.) was added portionwise at 0 C to a stirred and
previously
degassed (nitrogen) solution of Intermediate 77 (1 g, 3.41 mmol) and imidazole
(325 mg,
1.4 eq.) in dry DCM (10 mL). The reaction mixture was stirred at room
temperature
under nitrogen for 2 h. To push the reaction to completion, additional TBDMSC1
(150
mg, 0.3 eq.) was added and the reaction mixture was stirred at room
temperature for
another 1.5 h. Saturated aqueous NH4C1 was added and the layers were
separated. The
organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The
resulting
tan oil residue was purified by flash chromatography on silica gel
(Et0Ac/heptane 0/100
to 30/70) to give Intermediate 78 (1.09 g, yield: 78 %) as a light tan clear
oil.
Intermediate 79
(3-h<
B-0
0
Intermediate 79
A solution of Intermediate 78 (1.06 g, 2.60 mmol) in dry THF (11 mL) was
cooled to -
78 C under nitrogen atmosphere. nBuLi (2.5 M in hexanes; 1.3 mL, 1.25 eq.)
was added
dropwise. The reaction mixture was stirred at -78 C for 1 h. 2-Isopropoxy-
4,4,5,5-
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 117 -
tetramethy1-1,3,2-dioxaborolane (CAS [61676-62-8]) (0.64 mL, 1.2 eq.) was
added
dropwise. After the addition, the reaction mixture was allowed to warm to room
temperature and was stirred for 1 h. The reaction was quenched by slow
addition of
Et0Ac (25 mL), followed by saturated aqueous NH4C1 (20 mL). The layers were
separated and the aqueous layer was extracted with Et0Ac (2 x 20 mL). The
combined
organic layer was washed with brine (20 mL), dried over MgSO4, filtered, and
concentrated in vacuo. The residue was purified by flash column chromatography
on
silica gel (Et0Ac/heptane 0/100 to 50/50) to afford Intermediate 79 (934 mg,
yield: 79
%) as a yellow clear oil.
Intermediate 80
ZL'i<
B-0
Co
Intermediate 80
TBAF (1.0 M in THF, 1.2 eq.) was added to a solution of Intermediate 79 (930
mg, 2.05
mmol) in anhydrous 2-Me-THF (12 mL) under nitrogen atmosphere, at 0 C. The
ice
bath was removed and the resulting mixture was stirred at room temperature for
16 h.
The reaction mixture was diluted with Et0Ac and saturated aqueous NH4C1 was
added.
The layers were separated and the aqueous layer was extracted twice with
Et0Ac. The
combined organic layer was washed with brine, dried over MgSO4, filtered, and
concentrated in vacuo. The residue was purified by flash column chromatography
on
silica gel (Me0H in DCM 0/100 to 5/95) to afford Intermediate 80 (580 mg,
yield: 83
%) as a light yellow clear oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 118 -
intermediate 81
o¨\\_
N-N
HON
CI 0
0 -
S
Intermediate 81
Pd(amphos)2C12 (CAS [887919-35-9]) (51 mg, 0.05 eq.) was added to a stirred
and
previously nitrogen-degassed mixture of Intermediate 55 (706 mg, 1.44 mmol),
Intermediate 80 (580 mg, 1.2 eq.) and K2CO3 (400 mg, 2 eq.) in water (2 mL)
and 1,4-
dioxane (8 mL) in a microwave tube at room temperature and under nitrogen. The
reaction mixture was degassed by bubbling nitrogen through. The vial was
sealed and
the reaction mixture was stirred at 65 C for 2 h. The reaction mixture was
diluted with
Et0Ac and water and the layers were separated. The aqueous layer was extracted
twice
with Et0Ac. The combined organic layer was dried over MgSO4, filtered, and
evaporated. The residue was purified by flash column chromatography (silica;
Et0Ac in
n-heptane 0/100 to 100/0) to yield Intermediate 81 (700 mg, yield: 80%) as a
yellow oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 119 -
Intermediate 82
o¨\\_
N-N
0
CI 0
0-
Intermediate 82
MsC1 (0.11 mL 1.25 eq.) was added dropwise to a previously nitrogen-degassed
solution of Intermediate 81 (700 mg, 1.15 mmol) and Et3N (0.24 mL, 1.5 eq.) in
THF
(10 mL), under nitrogen at 0 'C. The resulting mixture was allowed to warm up
to
room temperature and was stirred for 1 h. A previously nitrogen-degassed
solution of
KSAc (657 mg, 5 eq.) in DMF (20 mL) was added and stirring was continued at
room
temperature for 2 h. To push the reaction to completion, a nitrogen-degassed
solution of
KSAc (394 mg, 3 eq.) in DMF (10 mL) was added. The reaction mixture was
further
stirred for 1 h. the reaction mixture was diluted with Et0Ac and water. The
layers were
separated and the aqueous layer was extracted twice with Et0Ac. The combined
organic layer was washed with brine, dried over MgSO4, filtered, and
evaporated. The
residue was purified by flash column chromatography (silica, 120 g, Et0Ac in n-
heptane 30/70 to 70130) to afford Intermediate 82 (287 mg, yield: 37 %).
Impure
fractions were purified again by flash column chromatography (silica, 80 g,
Et0Ac in
n-heptane 0/100 to 70/30) to yield another batch of Intermediate 82 (172 mg,
yield: 22
%)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 120 -
Intermediate 83
o¨\\_
N-N
N---
CI
N
F OH
0
OH
Intermediate 83
Intermediate 82 (460 mg, 0.69 mmol), Intermediate 25a (466 mg, 1.2 eq.), and
PPh3 (18
mg, 0.1 eq.) were charged in a 100 mL round bottom flask. The mixture was
degassed
and re-filled with nitrogen three times. Anhydrous Me0H (25 mL; degassed by
bubbling nitrogen for 30 min) was added. The suspension was degassed and re-
filled
with nitrogen three times. The reaction mixture was cooled to 0 C before
addition of
K2CO3 (286 mg, 3 eq.). The reaction mixture was degassed and re-filled with
nitrogen
three times. The reaction mixture was allowed to warm to room temperature and
was
stirred for 1.5 h. The reaction mixture was concentrated under vacuum and the
resulting
slurry was partitioned between water and Et0Ac. The layers were separated and
the
aqueous layer was extracted twice with Et0Ac. The combined organic layer was
washed with brine, dried over MgSO4, filtered, and concentrated under reduced
pressure. This residue was dissolved in Me0H (25 mL), and pTs0H.H20 (394 mg, 3
eq.) was added at room temperature. The solution was stirred for 40 min at
room
temperature. The solvent was evaporated and the residue was dissolved in Et0Ac
and
water, and saturated aqueous NaHCO3 was added. The layers were separated and
the
aqueous layer was extracted twice with Et0Ac. The combined organic layer was
washed with brine, dried over MgSO4, filtered, and concentrated under reduced
pressure. The residue was purified by flash column chromatography (silica, 120
g;
Me0H in DCM 0/100 to 5/95) to afford Intermediate 83 (511 mg, yield: 93 %).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 121 -
Intermediate 84 and Intermediate 85
o¨\\_
N¨N N¨N
N N---
O¨
N / N
0 0
o/
o/
Intermediate 84
Intermediate 85
Ra or Sa atropisomer Sa or Ra atropisomer
PPlet3 (650 mg, 4 eq.) was dissolved in dry toluene (19 mL, previously vacuum-
degassed and re-filled with nitrogen three times) to give Solution A. DTBAD
(571 mg,
4 eq.) was added to a solution of Intermediate 83 (491 mg, 0.62 mmol) in dry
THF (4
mL, previously vacuum-degassed and re-filled with nitrogen three times) and
dry
toluene (19 mL, previously vacuum-degassed and re-filled with nitrogen three
times) to
give Solution B. Solution B was added to solution A via syringe pump (0.1
mL/min) at
70 C. Once the addition was complete, the reaction mixture was stirred for 20
min at
70 C. After cooling to room temperature, the solvents were evaporated and the
residue
was purified by flash column chromatography (silica, 120 g; Et0Ac in n-heptane
0/100
to 20/80, then 100 % Et0Ac, and finally, Me0II in DCM 5/95) to yield a light
yellow
solid. This solid was purified by preparative SFC (Stationary phase: Chiralpak
Diacel
AD 20 x 250 mm, Mobile phase: CO2, Et0H + 0.4 % iPrNH2) to afford Intermediate
84 (135 mg, yield: 28 %) and Intermediate 85 (148 mg, yield: 31 %), both as
off-white
solids.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 122 -
Intermediate 86
o¨\\_
N-N
N
I CI O-
N
0
OH OH
Intermediate 86
Intermediate 82 (1.96 g, 2.94 mmol), Intermediate 15 (2 g, 1.2 eq.), and PPh3
(77 mg,
0.1 eq.) were charged in a 500 mL round bottom flask. The mixture was degassed
and
re-filled with nitrogen three times. Dry Me0H (200 mL, degassed by bubbling
nitrogen
for 30 min) was added. The suspension was degassed and re-filled with nitrogen
three
times. The reaction mixture was cooled to 0 C before addition of K2CO3 (1.22
g, 3
eq.). After this addition, the reaction mixture was degassed and re-filled
with nitrogen
three times. The reaction mixture was allowed to warm to room temperature and
was
stirred for 1.5 h, then heated up to 40 C and stirred for 1.5 h. The reaction
mixture was
concentrated under reduced pressure and the resulting slurry was partitioned
between
water and Et0Ac. The layers were separated and the aqueous layer was extracted
twice
with Et0Ac. The combined organic layer was washed with brine, dried over
MgS0.4,
filtered, and concentrated under reduced pressure. The residue was dissolved
in Me0H
(200 mL) and pTs0H.H20 (1.68 g, 3 eq.) was added at room temperature. The
reaction
mixture was stirred at room temperature for 30 min. The solvent was evaporated
and
the residue was partitioned between Et0Ac and water. Saturated aqueous NaHCO3
was
added. The aqueous layer was extracted twice with Et0Ac. The combined organic
layer
was washed with brine, dried over MgSO4, filtered, and concentrated under
reduced
pressure. The residue was purified by flash column chromatography (silica, 220
g,
Et0Ac in n-heptane 0/100 to 100/0 followed by Me0H in DCM 0/100 to 5/95) to
yield
Intermediate 86 (1.89 g, yield: 76%) as a tan clear oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 123 -
Intermediate 87 and Intermediate 88
\__\\
N¨N N¨N
N N
I /
0 0
o/
o/
Intermediate 87
Intermediate 88
Ra or Sa atropisomer Sa or Ra atropisomer
Intermediate 87 and Intermediate 88 were prepared according to an analogous
procedure as for Intermediate 84 and Intermediate 85, respectively, starting
from
Intermediate 86 instead of Intermediate 83.
Intermediate 89
=
,Si
0 110
CI
Intermediate 89
TBDPSC1 (4.93 g, 1.5 eq) was added dropwi se to a solution of ethyl 7-chloro-4-
hydroxy-2-naphthoate (CAS [2122548-70-11) (3 g, 11.97 mmol) and imidazole
(1.22g.
1.5 eq) in thy DWIF (60 mL) at 0 'C. The resulting mixture was stirred
overnight at
room temperature under nitrogen atmosphere. The reaction mixture was diluted
with
Et0Ac (100 mL) and washed with water. The organic layer was dried with Na2SO4,
filtered, and concentrated under reduced pressure. The residue was purified by
silica
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 124 -
gel column chromatography to give Intermediate 89 (5.8 g, yield: 90 % yield)
as a
yellow oil.
Intermediate 90
= H
,,Si
10. c
CI
Intermediate 90
DIBAL (1 M in hexane, 5.11 mL, 2.5 eq) was added dropwise to a solution of
Intermediate 89 (1 g, 2.045 mmol) in dry toluene (40 mL) at -78 C. The
reaction
mixture was stirred at -78 C for 10 min under nitrogen atmosphere, then
warmed to 0
C and kept at this temperature for 1 h. The reaction was quenched by addition
of
saturated aqueous NI-14C1 and the reaction mixture was extracted with Et0Ac.
The
organic layer was dried over Na2SO4, filtered, and evaporated. The residue was
purified
by silica gel chromatography (Et0Acipetroleum ether 0/100 to 15/85) to afford
Intermediate 90 (463 mg, yield: 51 %) as a pale yellow solid.
Intermediate 91
,Si
sip 0
CI
Intermediate 91
Dess-Martin periodinane (440 mg, 1 eq) was added to a solution of Intermediate
90
(463 mg, 1.037 mmol) in DCM (40 mL) at room temperature. The reaction mixture
was
stirred at room temperature for 2 h.The reaction was quenched by addition of
saturated
aqueous Na2S03, and the mixture was extracted with Et0Ac. The organic layer
was
dried over Na2SO4, filtered, and evaporated. The residue was purified by
silica gel
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 125 -
chromatography (Et0Ac/petroleum ether 0/100 to 10/90) to afford Intermediate
91
(439 mg, yield: 95 %) as a pale yellow solid.
Intermediate 92
/N 0
¨N
CIOH
undetermined E/Z mixture
Intermediate 92
NaH (60 % in mineral oil, 20 mg, 1.1 eq) was added to a suspension of
intermediate
105 (223 mg, 1.1 eq) in dry THY (5 mL) at 0 C, under nitrogen atmosphere.
After
stirring at 0 C for 40 min, the reaction mixture was cooled to -20 C and
Intermediate
91(200 mg, 0.449 mmol) in THE (1 mL) was added slowly at -20 'C. After the
addition, the reaction mixture was stirred at -10 C for 2 h. Water was added
to quench
the reaction at 0 C. The resulting mixture was extracted with Et0Ac. The
separated
organic layer was dried over Na2SO4, filtered, and evaporated. The residue was
purified
by silica gel column chromatography (petroleum ether/Et0Ac 100/0 to 30/70) to
afford
Intermediate 92 (150 mg, yield: 97 %) as a white solid.
Intermediate 93
0
¨N
CI Cr¨Si
Intermediate 93
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 126 -
TBDPSC1 (307 mg, 1.5 eq) was added dropwise to a mixture of Intermediate 92
(255
mg, 0.744 mmol) and imidazole (76 mg, 1.5 eq) in dry DMF (10 mL) at 0 C. The
reaction mixture was stirred overnight at room temperature under nitrogen
atmosphere.
The reaction mixture was diluted with Et0Ac (30 mL) and washed with water. The
organic layer was dried with Na2SO4, filtered, and concentrated under reduced
pressure. The residue was purified by silica gel column chromatography to give
Intermediate 93 (400 mg, yield: 92 %) as a white solid.
Intermediate 94
0
-N
Si
CI 10
Intermediate 94
Pd/C (10%, 37 mg, 0.8 eq.) was added to a solution of Intermediate 93 (250 mg,
0.43
mmol) in dry Et0Ac (5 mL). The reaction mixture was degassed, filled with H2
three
times, and stirred under an atmosphere of H2 at room temperature for 16 h. The
reaction
mixture was filtered through a Celite pad and the solid cake was washed with
Et0Ac.
The filtrate was evaporated and the residue was purified by silica gel column
chromatography to afford Intermediate 94 (245 mg, yield: 97 %) as a colorless
oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 127 -
Intermediate 95
OH
-N
CI 0--Si
=
Intermediate 95
DIBAL (1.5 M in toluene, 1.05 mL, 3.5 eq) was added dropwise to a solution of
Intermediate 94 (263 mg, 0.451 mmol) in dry toluene (5 mL) at -78 C. The
reaction
mixture was stirred at -78 C for 10 min then warmed to 0 C and kept at this
temperature for 1 h. The reaction was quenched by addition of saturated
aqueous
NI-I4C1 and the mixture was extracted with Et0Ac. The organic layer was dried
over
Na2SO4, filtered, and evaporated. The residue was purified by silica gel
chromatography (DCM/Me0H 100/0 to 90/10) to afford Intermediate 95 (214 mg,
yield: 85 % yield) as a white solid.
Intermediate 96
/N CI
-N
CI 0--Si
Intermediate 96
Thionyl chloride (32 [IL, L15 eq) was added dropwise to a solution of
Intermediate 95
(214 mg, 0.385 mmol) in dry DCM (5 mL) at 0 'C. The reaction mixture was
stirred at
0 'C. under nitrogen atmosphere for 10 min then warmed to room temperature and
kept
at this temperature for 1 h. The reaction was quenched by addition of
saturated aqueous
NH4C1 and the mixture was extracted with Et0Ac. the organic layer was dried
over
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 128 -
Na2SO4, filtered, and evaporated to afford Intermediate 96 (223 mg, considered
quantitative), used without purification.
Intermediate 97
N-N
.//
0--
/N
0
OH
\Si (
I
ci
Intermediate 97
Intermediate 8(1.243 g, 2.15 mmol) and Intermediate 96 (1.418 g, 1.15 eq) were
dissolved in Me0H (15 mL). The reaction mixture was degassed and re-filled
with
nitrogen five times. K2CO3 (594 mg, 2 eq) was then added and the reaction
mixture was
stirred at room temperature overnight. The solvent was evaporated and the
residue was
partitioned between water and Et0Ac. The layers were separated and the organic
layer
was washed with brine, dried over Na2SO4, filtered, and evaporated. The
residue was
purified by silica gel chromatography (hexane/Et0Ac 100/0 to 20/80) to afford
Intermediate 97 (1.294 g, yield: 71 %) as an off-white foamy solid.
Intermediate 98
N-N
/N
0
OH
OH
CI
Intermediate 98
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 129 -
pTs0H.H20 (324 mg, 1.1 eq) was added to a solution of Intermediate 97 (1.294
g,
1.549 mmol) in Me0H (30 mL). The reaction mixture was stirred at room
temperature
for 1.5 h. The solvent was evaporated and the residue was partitioned between
water
and Et0Ac. The layers were separated and the organic layer was washed with
brine,
dried over Na2SO4, filtered, and evaporated. The residue was purified by
silica gel
chromatography (DCM/Me0H 100/0 to 95/5) to afford Intermediate 98 (930 mg,
yield:
83 %) as a pale yellow foamy solid.
Intermediate 99 and Intermediate 100
N¨N N¨N
N CI 0¨ N CI 0_
\N \N
0 0
o?
CI CI
Intermediate 99 Intermediate 100
Sa or Ra atropisomer Ra or Sa atropisomer
A solution of Intermediate 98 (1.506 g, 2.165 mmol) and DTBAD (1.994g. 4 eq.)
in
toluene (55 niL) arid THE' (8 mL) was added dropwise over 60 min, at 70 "V
under
nitrogen to a solution of PPh3 (2.271 g, 4 eq) in toluene (55 mL). After the
addition, the
reaction mixture was further stirred at the same temperature for 1 h. The
solvents were
evaporated and the residue was partitioned between water and DCM. The layers
were
separated and the aqueous layer was extracted with DCM (50 mL x 3). The
combined
organic layer was washed with brine, dried over Na2SO4, filtered, and
evaporated. The
residue was purified by silica gel chromatography (hexane/Et0Ac 100/0 to
20/80) to
give the racemic mixture of Intermediate 99 and Intermediate 100. This racemic
mixture was separated by preparative chiral-HPLC (Column: CHIRAL ART Cellulose-
SB, 30*250 mm,5 urn; Mobile Phase A: CO2, Mobile Phase B: IPA (0.5 % 2 M NI-I3-
Me0I I); Flow rate:50 mL/min; Gradient: 40 % B) to afford Intermediate 99 (490
mg,
yield: 32 %) and Intermediate 100 (420 mg, yield: 27 %), both as a pale yellow
foamy
solids.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 130 -
Intermediate 101 and Intermediate 102
o
) _________________________________ 0
HN
o
_________________________________________________________ HN¨\
\N¨N
N¨N
N---
I /
N CI
0
0
0 0
Intermediate 101 Intermediate 102
Ra or Sa atropisomer Ra or Sa atropisomer
3-(Boc-amino)propyl bromide (CAS [83948-53-2]) (191 mg, 3 eq.) was added to a
stirred mixture of Intermediate 60 (180 mg, 0.268 mmol) and Cs2CO3 (264 mg, 3
eq.)
in anhydrous MIT' (4 mL) at room temperature, under nitrogen atmosphere. The
reaction mixture was stirred at room temperature under nitrogen atmosphere for
18 h.
The solvent was removed under reduced pressure. The residue was diluted with
DCM
and brine. The layers were separated and the organic layer was washed with
brine (x 3).
The combined aqueous layer was extracted with DCM (x 4). The combined organic
layer was dried over MgSO4, filtered, and evaporated to give a colorless oil.
This oil
was further purified by preparative SFC (Stationary phase: Chiralpak Daicel ID
20 x
250 mm, Mobile phase: CO2, iPrOH + 0.4 % iPrNH2) to afford Intermediate 101
(90
mg, yield. 40 %) and Intermediate 102 (93 mg, yield: 42 %), both as pale
yellow oils.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 131 -
Intermediate 103
H2N
N¨N
N---
/
0
0 HCl salt
Intermediate 103
Ra or Sa atropisomer (HCl salt)
HCl (6 M in iPrOH, 1.81 mL, 100 eq.) was added to a solution of Intermediate
101 (90
mg, 0.109 mmol) in Me0H (2 mL) at room temperature under nitrogen atmosphere.
The reaction mixture was stirred at room temperature for 5 h. The solvent was
evaporated to give Intermediate 103 (96 mg, considered quantitative) as a pale
yellow
solid, used without further purification
Intermediate 104
H2N
\N¨N
N
N CI
0
o/
HCI salt
Intermediate 104
Ita or Sa atropisomer (HC1 salt)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 132 -
Intermediate 104 was prepared according to an analogous procedure as for
Intermediate
103, starting from Intermediate 102 instead of Intermediate 101.
Intermediate 105
=
= = o
Cl-
Intermediate 105
A solution of 5-(chloromethyl)-1-methy1-1H-pyrazole-3-carboxylic acid, methyl
ester
(CAS [2245938-86-5]) (24 g, 0.127 mol) and PPly3 (37 g, 1 eq.) in ACN (250 mL)
was
stirred under reflux for 16 h. The white suspension was concentrated in vacuo
and
triturated with Et0Ac (100 mL). The resulting solid was collected by
filtration and
dried to afford Intermediate 105 (54.8 g, yield: 96 %) as a white solid.
Intermediate 106
=
o
Intermediate 106
Thionyl chloride (13 g, 1.5 eq.) was added to a solution of Intermediate
20a(31 g, 72
mmol) in DCM (300 mL) at room temperature and the reaction mixture was stirred
at
room temperature for 3 h. The reaction mixture was concentrated under reduced
pressure to give Intermediate 106 (32 g, yield: 99 %) as a yellow oil, used
without
further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 133 -
Intermediate 107
C I -
P 4101
110
Intermediate 107
PP11.3 (37.68 g, 2 eq.) was added to a solution of Intermediate 106 (32 g,
71.26 mmol) in
DCM (300 mL) at room temperature. The solvent was evaporated and the residue
was
stirred at 140 C for 16 h (neat reaction). The resulting residue was
triturated with
Et0Ac (150 mL) and filtered to give Intermediate 107 (27 g, yield: 46 %) as a
white
solid.
Intermediate 108
11
0
Si-
/ __________________________________________________
Intermediate 108
TBDMSC1 (77 g, 1.1 eq.), followed by imidazole (35 g, 1.1 eq.) were added to a
solution of methyl 5-hydroxymethyl-1-methyl-lh-pyrazole-3-carboxylate (CAS
[1208081-63-3], 79 g, 464 mmol) in DCM (800 mL) and the resulting solution was
stirred at room temperature for 16 h. The solvent was evaporated and the
residue was
purified by silica gel column chromatography (Et0Ac/petroleum ether, 3/1) to
give
Intermediate 108 (126 g, yield: 78 %) as a light yellow oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 134 -
Intermediate 109
OH
/
0
Si-
/X
Intermediate 109
DIBAL (1 Mmn hexane, 1.33 L, 3 eq.) was added dropwise at 0 C to a solution
of
Intermediate 108 (126 g, 443 mmol) in THF (1 L). The reaction mixture was
stirred for
2 h at 0 C, then allowed to warm to room temperature. The reaction mixture
was
carefully poured into a Rochelle salt solution (1.5 L). Et0Ac (1.5 L) was
added and the
resulting biphasic mixture was stirred for 1.5 h. The aqueous layer was
separated and
then extracted with Et0Ac (2 x 1.5 L). The combined organic layer was dried
over
MgSO4, filtered, and evaporated to give Intermediate 109 (108 g, yield: 87 %)
as a
white solid, used without further purification.
Intermediate 110
1\1===
I /
0
Si
Intermediate 110
Intermediate 109 (81 g, 315.8 mmol), followed by methanesulfonic anhydride
(71.5 g,
1.4 eq.), were added to a solution of DIPEA (61.2 g, 1.5 eq.) in THF (900 mL)
at 0 C.
The resulting mixture was stirred at 0 C for 5 min, then at room temperature
for 30
min. NaI (213 g, 4.5 eq.) was then added to the reaction mixture and it was
stirred at
50 C for 2 h. After cooling, the solvent was evaporated. The residue was
partitioned
between Et0Ac and water. The organic layer was separated and the aqueous layer
was
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 135 -
extracted with Et0Ac. The combined organic layer was washed with brine, dried
over
Na2SO4, filtered, and evaporated. The residue was purified by flash column
chromatography on silica gel (petroleum ether/Et0Ac 4/1) to afford
Intermediate 110
(30 g, yield: 26 %) as a yellow oil.
Intermediate 111
N¨N
0
CI
O¨
N /
0
0
Si¨
/XSi
Intermediate 111
NaH (60 % in mineral oil, 415 mg, 1.2 eq.) was added at 0 C to a solution of
Intermediate 5 (4.5 g, 8.65 mmol) in anhydrous THF (90 mL) under nitrogen
atmosphere. The reaction mixture was stirred at 0 C for 30 min before
addition of a
solution of Intermediate 110 (3.80 g, 1.2 eq.) in THF (10 mL). After stirring
at 0 C for
10 min, the mixture was warmed to room temperature and stirred for 4 h. The
reaction
was quenched by addition of a solution of saturated aqueous NH4C1 and Et0Ac
was
added. The organic layer was separated, dried over Na2SO4, filtered, and
concentrated
under reduced pressure. The residue was purified by silica gel chromatography
(hexane/Et0Ac 100/0 to 20/80) to afford Intermediate 111 (5 g, yield: 76 %) as
yellow
oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 136 -
Intermediate 112
N-N
0
CI
0-
k /
0
H 0
Intermediate 112
pTs0H.H20 (2.89 g, 2.4 eq.) was added at 0 C to a solution of Intermediate 111
(4.8
g, 6.33 mmol) in Me0H (100 mL) under nitrogen atmosphere. The reaction mixture
was stirred at 0 C for 10 min. Then the reaction mixture was warmed to room
temperature and stirred for 3 h before being quenched with water (50 mL). The
volatiles were removed under reduced pressure and the aqueous residue was
extracted
with DCM (3 x 50 mL). The combined organic layer was dried over Na2SO4,
filtered,
and evaporated. The residue was purified by column chromatography on silica
gel
(Me0H/DCM 0/100 to 10/90) to afford Intermediate 112 (3.2 g, yield: 94 %) as a
white
solid.
Intermediate 113
N-N
0
/
0
0
Intermediate 113
Activated Mn02 (7.8 g, 15 eq.) was added at 0 C to a solution of Intermediate
112 (3.2
g, 6.04 mmol) in DCM (100 mL) under nitrogen atmosphere. The reaction mixture
was
stirred at room temperature overnight. It was then filtered and the filter pad
was washed
with DCM (200 mL). The combined filtrate was concentrated in vacuo to afford
Intermediate 113 (3.3 g, yield: 90 %) as a yellow oil, used without further
purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 137 -
Intermediate 114
N-N
)NCI O-
7
0
0
Intermediate 114
TBDMSC1 (548 mg, 1.2 eq.), followed by imidazole (248 mg, 1.2 eq.) were added
to a
solution of Intermediate 113(1.6 g, 3.03 mmol) in DCM (15 mL) and the reaction
mixture was stirred at room temperature for 4 h under nitrogen atmosphere. The
reaction mixture was filtered through a Celite pad, and the filtrate was
concentrated
under reduced pressure. The residue was combined with a residue coming from
the
same reaction performed with another batch of Intermediate 113. The combined
residue
was purified by flash column chromatography on silica gel (petroleum
ether/Et0Ac
2/1) to afford Intermediate 114 (2.7g) as a yellow oil.
Intermediate 115
N-N
0
CI
0-
/
0
OH
Intermediate 115
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 138 -
NaH (60 % in mineral oil, 146 mg, 1.5 eq.) was added to a solution of
Intermediate 114
(2.6 g, 4.048 mmol) and Intermediate 107 (3.2 g, 4.45 mmol) in TI-IF (30 mL)
cooled to
0 'V under nitrogen atmosphere. The reaction mixture was stirred at room
temperature
for 48 h, then it was quenched with aqueous NH4C1 (100 mL) and extracted with
Et20
(3 x 100 mL). The organic layer was dried over Na2SO4, filtered, and
evaporated. The
residue was purified by flash column chromatography on silica gel (petroleum
ether/Et0Ac, 1/1) to afford Intermediate 115 (2 g, yield: 62%) as a yellow
oil.
Intermediate 116
N-N
0
CI
/
0
Si
OH
Intermediate 116
Pd/C (2 g, 1 eq.) was added to a solution of Intermediate 115 (2 g, 2.5 mmol)
in Et0Ac
(100 mL). The reaction mixture was stirred at 35 C for 16 h under an hydrogen
atmosphere, then it was filtered through a Celite pad. The filtrate was
concentrated
under reduced pressure to afford Intermediate 116 (1.9 g, yield: 95 %) as a
yellow oil,
used without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 139 -
Intermediate 117
N¨N
0
CI
OH
1 /
0
lip OH
110
Intermediate 117
pTs0H.H20 (1 g, 1.1 eq.) was added to a solution of Intermediate 116 (4.9 g,
4.71
mmol) in Me0H (100 mL) at room temperature. The reaction mixture was stirred
at
room temperature for 1 h. The reaction was quenched by addition of water. The
mixture was extracted with Et20. The organic layer was washed with brine (100
mL)
followed by aqueous NaHC,03 (100 mL). The organic layer was dried over Na2SO4
and
concentrated under reduced pressure. The residue was purified by flash column
chromatography on silica gel (DCMNIe0H 20/1) to afford Intermediate 117 (1.2
g,
yield: 37 %) as a yellow oil.
Intermediate 118 and Intermediate 119
N¨N
0 N¨N
0
CI
N 0¨ CI
1
0¨
/
N/.
I /
0 /N
0
o?
* OS)
Intermediate 118 Intermediate 119
Sa or Ra atropisomer Ra or Sa
atropisomer
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 140 -
DTBAD (502 mg, 1.5 eq.) was added to a solution of Intermediate 117 (1 g,
1.453
mmol) in THF (2 mL) and toluene (15 mL). The resulting mixture was filled with
nitrogen, stirred for 15 min at room temperature, and then added dropwise to a
solution
of PPh3 (572 mg, 1.5 eq.) in toluene (5 mL) at 70 "V under nitrogen
atmosphere. The
reaction mixture was stirred for 15 min at 70 C under nitrogen atmosphere.
After
cooling, the reaction mixture was concentrated under reduced pressure. The
residue
was purified by reverse phase chromatography (Column: C18 spherical, 20-35 pm,
100A, 330 g; Mobile Phase A: ACN, Mobile Phase B: H20 (0.05% 0.5 M NH4HCO3-
H20); Gradient: A/B 40/60 to 100/0) to afford the racemic mixture of
Intermediate 118
and Intermediate 119. The atropisomers were separated by preparative chiral
SFC
(Column: CHIRAL ART Cellulose-SB, 3 x 25 cm, 5 um; Mobile Phase A:CO2, Mobile
Phase B: Me0H (0. 5 % 2 M NH3 in Me0H) to afford Intermediate 118 (90 mg,
yield:
9 %) and Intermediate 119 (110 mg, yield: 11 %), both as white solids.
intermediate 120
N¨N
Br
Lithium borohydride (32.2 g, 4 eq.) was added slowly to a solution of 1H-
pyrazole-3-
carboxylic acid, 4-bromo-5-methyl-1-(tetrahydro-2H-pyran-2-y1)-, ethyl ester
(CAS
[2246368-58-9D (130 g, 369.7 mmol) in 2-Me-THF (1 L) at 0 C. The reaction
mixture
was allowed to warm to room temperature and was left stirring at room
temperature
overnight. The reaction was quenched by addition of water (800 mL). The
mixture was
extracted with Et0Ac (800 mL x 2). The combined organic layer was washed with
brine
(500 mL), dried with Na2SO4, filtered, and evaporated to afford Intermediate
120 (105
g, yield: 94 %) as a white solid.
Intermediate 121
/
si___
N¨N
0 /
Br
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 141 -
DMAP (16.28 g, 0.4 eq.) and Et3N (92.38 mL, 2 eq.) were added to a solution of
Intermediate 120 (100 g, 333.2 mmol) in TI-IF (1 L). TBDMSC1 (75.3 g, 1.5 eq.)
was
added at room temperature and the reaction mixture was stirred for 16 h. The
reaction
was quenched by addition of saturated aqueous NaHCO3 (800 mL) and the mixture
was
extracted with Et0Ac (1 L x 2). The combined organic layer was washed with
brine (800
mL), dried with Na2SO4, filtered, and evaporated. The residue was purified by
column
chromatography over silica gel (petroleum ether/ Et0Ac 100/0 to 30/70) to
afford
Intermediate 121 (130 g, yield: 94%) as a colorless oil.
Intermediate 122
/
Si_
N-N
nBuLi (104.55 mL, 1 eq.) was slowly added to a solution of Intermediate 121
(108 g,
261.4 mmol) in TI-1F (1 L) at -78 'V, under nitrogen atmosphere, and the
reaction mixture
was stirred at -78 C for 1 h. Then, 2-isopropoxy-4,4,5,5-tetramethy1-1,3,2-
dioxaborolane (97.2 g, 2 eq.) was added slowly and the reaction mixture was
stirred at
room temperature for 2 h. Saturated aqueous NRIC1 (800 mL) was added slowly to
quench the reaction. The mixture was extracted with Et0Ac (1 L x 2). The
combined
organic layer was washed with brine (800 mL), dried with Na2SO4, filtered, and
evaporated to afford Intermediate 122 (140 g, assumed quantitative) as a
yellow oil.
Intermediate 123
N-N
0
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 142 -
TBAF (1 M in TT-IF, 192.4 mL, 1.2 eq.) was added dropwise to a solution of
Intermediate
122 (70 g, 160 mmol) in DCM (700 mL) at room temperature under nitrogen
atmosphere.
The reaction mixture was stirred overnight at room temperature. The reaction
mixture
was added to a stirring solution of saturated aqueous NaHCO3 (500 mL) and this
mixture
was extracted with Et0Ac (700 mL x 2). The combined organic layer was washed
with
brine (500 mL), dried with Na7SO4, Filtered, and evaporated. The residue was
purified
by column chromatography over silica gel (petroleum ether/ Et0Ac 100/0 to
50/50) to
afford Intermediate 123 (35 g, yield: 62 %) as a white solid.
Intermediate 124
c¨O)
N-N
HO
CI 0¨
\
0
K2CO3 (6.9 g, 2 eq.) was added to a solution of Intermediate 55 (12 g, 24.9
mmol) and
Intermediate 123 (9.6 g, 1.2 eq.) in water (40 mL) and dioxane (200 mL).
Pd(amphos)2C12 (CAS [887919-35-9]) (0.8 g, 0.05 eq.) was added under nitrogen
atmosphere and the reaction mixture was stirred at 60 'V, for 2 h. Water (40
mL) was
added to the mixture and it was extracted with Et0Ac (60 mi., x2). The
combined organic
layer was washed with brine, dried with Na2SO4, filtered, and evaporated. The
residue
was purified by flash column chromatography over silica gel (petroleum
ether/Et0Ac
100/0 to 60/40) to afford Intermediate 124 (15 g, yield: 99 %) as a yellow
solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 143 -
Intermediate 125
N¨N
Nr-S
0 \o
CI
0
Si---n
Et3N (5.1 mL, 1.5 eq.) followed by MsC1 (2.4 mL, 1.25 eq.) were added dropwise
to a
solution of Intermediate 124 (14.5 g, 24.567 mmol) in dry TI-1F (180 mL)
(degassed by
bubbling nitrogen for 15 min) at 0 C under nitrogen atmosphere. The reaction
mixture
was stirred for 10 min at room temperature. Then, a degassed solution
(degassed by
bubbling nitrogen for 30 min) of potassium thioacetate (28.1 g, 10 eq.) in DMF
(400
mL) (previously degassed by bubbling nitrogen for 30 min) was added at room
temperature. The resulting mixture was degassed by bubbling nitrogen for 5 min
and
was then stirred at room temperature for 30 min. The reaction mixture was
diluted with
Et0Ac (500 mL) and water (300 mL). The layers were separated and the aqueous
layer
was extracted with Et0Ac (2 x 500 mL). The combined organic layer was washed
with
brine (3 x 300 mL), dried over Na2SO4, filtered, and concentrated. The residue
was
purified by silica gel chromatography (Et0Ac/petroleum ether 0/100 to 30/70)
to afford
Intermediate 125 (15.3 g, yield: 96 %) as a brown oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 144 -
Intermediate 126
N¨N
/
CI
0
0
OH
(
CI
Intermediate 96 (8.005 g, 1.2 eq.) was added to a solution of Intermediate 125
(7.54 g,
11.63 mmol) in Me0H (100 mL). The reaction mixture was degassed and re-filled
with
nitrogen five times. Then, K2CO3 (3.215 g, 2 eq.) was added. The resulting
mixture was
stirred at room temperature overnight. The reaction mixture was concentrated
under
reduced pressure. The residue was diluted with Et0Ac (300 mL) and water (300
mL).
The layers were separated and the aqueous layer was extracted with Et0Ac (2 x
300
mL). The combined organic layer was washed with brine (3 x 300 mL), dried over
Na2SO4, filtered, and concentrated. The residue was purified by silica gel
chromatography (Et0Aclpetroleum ether 25/75 to 50/50) to afford Intermediate
126 (7
g, yield: 66 %) as a yellow solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 145 -
Intermediate 127
N-N
/ CI
N
0
OH
OH
CI
Et3N.(HF)3 (1.857 g, 1.5 eq.) was added to a solution of Intermediate 126
(6.95 g, 7.679
mmol) in TI-IF (70 mL) at room temperature under nitrogen atmosphere. The
reaction
mixture was stirred at room temperature for 18 h. The reaction mixture was
diluted
with Et0Ac (200 mL) and water (200 mL). The layers were separated and the
aqueous
layer was extracted with Et0Ac (2 x 200 mL). The combined organic layer was
washed
with brine (2 x 100 mL), dried over Na2SO4, filtered, and concentrated to
afford
Intermediate 127 (6 g, yield: 99%) as a light yellow solid, used without
further
purification.
Intermediate 128
N-N
1 /
0
0
CI
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 146 -
DTBAD (6.988 g, 4 eq.) was added to a solution of Intermediate 127(6 g, 7.587
mnnol)
in TI-IF (40 mL) and toluene (80 mL) (both degassed and re-filled with
nitrogen five
times). The reaction mixture was stirred for 15 min at room temperature. Then,
this
solution was added dropwise to a solution of PPh3 (7.960 mg, 4 eq.) in toluene
(80 mL)
at 70 '12 under nitrogen atmosphere. The reaction mixture was stirred for 10
min at 70
'V under nitrogen atmosphere. After cooling, the reaction mixture was diluted
with
water (150 mL) and Et0Ac (3 x200 mL). The layers were separated and the
organic
layer was washed with brine (3 x 200 mL), dried over Na2SO4, filtered, and
concentrated. The residue was purified by silica gel chromatography
(Et0Ac/petroleum
ether 25/75 to 70/30) to afford Intermediate 128 (5 g, yield: 85 %) as a
yellow solid.
Intermediate 129 and Intermediate 130
N¨NH
/
0
CI
Intermediate 129 Ra or Sa atropisomer
Intermediate 130: Sa or Ra atropisomer
Intermediate 128 (5 g, 6.470 mmol) was dissolved in a 4 M solution of I-ICI in
1,4-
dioxane (30 mL). The reaction mixture was stirred at room temperature for 2 h.
The
reaction mixture was then concentrated under reduced pressure. The residue was
purified by reverse phase chromatography (ACN/H20 - 5 mmol NH4HCO3, 50/50 to
90/10) to afford the racemic mixture of Intermediate 129 and Intermediate 130
as a
light yellow solid. This solid was separated into its atropisomers by
preparative chiral
SFC (Column: CHIRAL ART Cellulose-SB, 3 x 25cm, 5 urn; Mobile Phase A:CO2,
Mobile Phase B:IPA(0.5 % 2 M NH3-Me0H); A/B 50/50) to afford Intermediate 129
(800 mg, yield: 18 %) and Intermediate 130 (800 mg, yield: 18 %)
Intermediate 129: OR: [a]= +18.6 (589 nm, 28.7 C, 5.0 mg in 10 mI_, Me0H).
Intermediate 130: OR: [a]= -23.9 (589 nm, 28.7 C, 5.0 mg in 10 mL Me0H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 147 -
Intermediate 131 and Intermediate 132
0
0 (0
N¨N) N¨N
N CI 0 N CI
0
0
0
0 0
CI CI
Intermediate 131: Ra or Sa atropisomer
Intermediate 132: Ra or Sa atropisomer
Cs2CO3 (397 mg, 3 eq.) was added to a solution of Intermediate 129 (280 mg,
0.407
mmol) in DMF (5 mL) under nitrogen atmosphere. 1-Bromo-2-(2-
methoxyethoxy)ethane (223 mg, 3 eq.) was added and the resulting mixture was
stirred
at room temperature under nitrogen for 16 h. The reaction mixture was diluted
with
H20 (20 mL) and Et0Ac (20 mL). The layers were separated and the aqueous layer
was extracted again with Et0Ac (2 x 20 mL). The combined organic layer was
washed
with brine (3 x 20 mL), dried over Na2SO4, filtered, and concentrated to
afford the
mixture of Intermediate 131 and Intermediate 132 (300 mg) as a light yellow
solid,
used without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 148 -
Intermediate 133 and Intermediate 134
0'
¨0
ri
N¨N N¨N
\o \
N N
0 0
CI CI
Intermediate 133: Ra or Sa atropisomer
Intermediate 134: Ra or Sa atropisomer
The mixture of Intermediate 133 and Intermediate 134 was prepared according to
the
same procedure as for the mixture of Intermediate 131 and Inteiniediate 132,
using 1-
bromo-2-(2-methoxy)ethane instead of 1-bromo-2-(2-methoxyethoxy)ethane.
Intermediate 135 and Intermediate 136
/
0
\
0 (0
N¨N) N¨N
N. 7
\ \
N N
5)
0 0
CI CI
Intermediate 135: Sa or Ra atropisomer
Intermediate 136: Sa or Ra atropisomer
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 149 -
The mixture of Intermediate 135 and Intermediate 136 was prepared according to
the
same procedure as for the mixture of Intermediate 131 and Intermediate 132,
starting
from Intermediate 130 instead of Intermediate 129.
The mixture of Intermediate 135 and Intermediate 136 was then separated by
preparative
chiral HPLC (Column: CHIRAL ART Cellulose-SC, 2 x 25 cm, 5 urn; Mobile Phase
A:
hexane:DCM 3:1(0.5 % 2 M N13-Me0H), Mobile Phase B: Et0H; 95 % Al 5 % B) to
afford pure Intermediate 135 and Intermediate 136.
Intermediate 137 and Intermediate 138
¨0\
T-1
N¨N N¨N
N CI N CI
\o
0
0
0 0
CI CI
Intermediate 137: Sa or Ra atropisomer
Intermediate 138: Sa or Ra atropisomer
The mixture of Intermediate 137 and Intermediate 138 was prepared according to
the
same procedure as for the mixture of Intermediate 133 and Inteimediate 134,
starting
from Intermediate 130 instead of Intermediate 129.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 150 -
Intermediate 139
Br
H
0 0
Intermediate 139
Cyanomethylenetributylphosphorane (CAS [157141-27-0], 37.15 mL, 141.6 mmol,
1.5
eq.) was added dropwise to a solution of 1H-pyrazole-3-carboxylic acid, 4-
bromo-5-
methyl-, ethyl ester (CAS [6076-14-8], 22 g, 94.4 mmol) and 2-(tetrahydro-2H-
pyran-
2-yloxy)ethanol (CAS [2162-31-4], 15.7 mL, 113.3 mmol, 1.2 eq.) in THF (100
mL) at
0 C and the mixture was stirred overnight at room temperature. The solvent
was
evaporated and the residue was taken up in Et0Ac/water. The organic layer was
separated, dried over MgSO4, filtered, and evaporated. The residue was
purified by
column chromatography on silica gel (heptane/Et0Ac, 100/0 to 80/20) to afford
Intettnediate 139 (17.2 g, yield: 50 A).
Intermediate 140
Br
0 0
**"..
Intermediate 140
NaBH4 (226 mg, 5.979 mmol, 2 eq.) was added to a solution of Intermediate 139
(1.08
g, 2.99 mmol) in THE (18 mL) and Me0H (4 mL) at 0 C. The reaction mixture was
then stirred at room temperature for 24 h. To push the reaction to completion,
more
NaBH4 (679 mg, 17.94 mmol, 6 eq.) was added and the reaction mixture was
stirred at
room temperature overnight The reaction mixture was cooled to 0 C, treated
with
NT-14C1 and AcOEt, stirred for 15 min at room temperature, and extracted with
more
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 151 -
AcOEt. The combined organic layer was dried on 1V1gSO4, filtered, and
evaporated to
give Intermediate 140 (917 mg, yield: 96 %), used without further
purification.
Intermediate 141
B--0
N
0 0
Intermediate 141
Et3N (7.708 mL, 55.451 mmol, 3 eq.) followed by pinacolborane (CAS [25015-63-
8],
5.9 mL, 39.441 mmol, 2.1 eq.) were added dropwise to a nitrogen-degassed
solution of
Intermediate 140 (5.9 g, 18.484 mmol), bis(acetonitrile)dichloropalladium (II)
(CAS
[14592-56-4], 240 mg, 0.924 mmol, 0.05 eq.). and 2-dicyclohexylphosphino-2',6'-
dimethoxybiphenyl (CAS [657408-07-6], 1.518 g, 3.697 mmol, 0.2 eq.) in 1,4-
dioxane
(65 mL). The reaction mixture was stirred at 80 C for 1 h. The mixture was
diluted
with of water (20 mL) and was extracted with Et0Ac (3 x). The combined organic
layer was washed with water and brine, dried (MgSO4), filtered, and
evaporated. The
residue was purified by column chromatography on silica gel (heptane/Et0Ac,
100/0 to
50/50) to afford Intermediate 141 (4.7 g, yield: 69 %).
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 152 -
intermediate 142
N¨N
HO
CI 0
0¨
Si-,
Intermediate 142
A 20 mL vial was charged with a solution of Intermediate 55 (1.23 g, 2.5 mmol)
and
Intermediate 141 (1.1 g, 3 mmol, 1.2 eq.) in 1,4-dioxane (15 mL) and this was
purged
with nitrogen for 15 min. Bis(di-tert-buty1(4-
dimethylaminophenyl)phosphine)dichloropalladium(11) (CAS [887919-35-9], 88 mg,
0.12 mmol, 0.05 eq.) and a solution of K2CO3 (0.69 g, 5 mmol, 2 eq.) in water
(3 mL)
were added. The vial was capped and heated at 65 C for 2h. The reaction
mixture was
diluted with water and Et0Ac. The layers were separated and the aqueous layer
was
extracted with Et0Ac. The combined organic layer was dried with MgSO4 and a
little
Norit, filtered, and concentrated in vacuo. The residue was purified by flash
column
chromatographyy (40 g Redisep Flash column eluting with heptane/Et0Ac 100/0 to
50/50) to afford Intermediate 142 (1.13 g, yield: 71%) as a colorless oil.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 153 -
Intermediate 143
N-N
\
0
CI 0
0-
Intermediate 143
Methanesulfonyl chloride (175 p.Iõ 2.24 mmol, 1.25 eq.) was added dropwise to
an ice-
cooled solution of Intermediate 142 (1.13 g, 1.78 mmol) and Et3N (375 uL, 2.71
mmol,
1.5 eq.) in dry TI-IF (15 mL). The ice bath was removed and stirring was
continued for
30 min. A solution of potassium thioacetate (2.03 g, 17.82 mmol, 10 eq.) in
dry DME
(30 mL) was added and the mixture was diluted with THE (15 mL). After 30 min
at
room temperature, the orange viscous solution was partitioned between
saturated
aqueous NaHCO3 and Et0Ac, and the layers were separated. The organic layer was
washed with brine, dried on MgSO4, filtered, and concentrated in vacuo. The
residue
was purified by flash column chromatography (40 g Redisep column eluting with
heptane/Et0Ac 100/0 to 50/50) to afford Intermediate 143 (1.22 g, yield: 100
%) as a
tan oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 154 -
intermediate 144
N-N
N.
CI O-
N /
0
OH
Intermediate 144
A solution of Intermediate 143 (1.23 g, 1.78 mmol), Intermediate 25a(1.19 g,
2.13
mmol, 1.2 eq.), and triphenylphosphine (49 mg, 0.19 mmol, 0.1 eq.) in Me0H
(110
mL) was degassed and re-filled with nitrogen three times. The suspension was
cooled
to 0 C before addition of K2CO3 (0.75 g, 5.43 mmol, 3 eq.). The reaction
mixture was
degassed with nitrogen again and was stirred at room temperature for 3.5 h.
The
reaction mixture was concentrated under reduced pressure and the resulting
slurry was
partitioned between water and Et0Ac. The layers were separated and aqueous
layer
was extracted with Et0Ac (3 x). The combined organic layer was washed with
brine,
dried over MgSO4, filtered, and concentrated under reduced pressure to afford
Intermediate 144 (833 mg, yield: 50 %), used without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 155 -
intermediate 145
N¨N
N.
CI O¨
N /
0
OH
OH
Intermediate 145
TBAF (1M in THE, 1.33 mL, 1.33 mmol, 1.5 eq.) was added to a solution of
Intermediate 144 (0.83 g, 0.88 mmol) in THE (20 mL) at 0 C. The reaction
mixture
was stirred at room temperature for 4.5 h. After cooling to 0 C, the reaction
mixture
was treated with saturated aqueous NI-14C1 and was stirred for 15 min. The
mixture was
extracted with Et0Ac (3 x). The combined organic layer was washed with brine,
dried
(MgSO4), filtered, and evaporatyed. The residue was purified by flash column
chromatography on silica gel (DCM/Me0H, 100/0 to 95/5) to afford Intermediate
145
(410 mg, yield: 57 %) as a off-white foam.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 156 -
Intermediate 146
N¨N
CI
N
0
o/
Intermediate 146
A solution of Intermediate 145 (3.89 g, 0.0048 mol) and di-tert-hutyl
azodicarhoxylate
(4.5 g, 0.02 mol, 4.1 eq.) in previously nitrogen-degassed THF/toluene (10
mL/50 mL)
was added dropwise via a syringe pump (0.3 mL/min) to a previously thoroughly
nitrogen-degassed solution of triphenylphosphine (5 1 g, 0.019 mol, 4.1 eq.)
in toluene
(600 mL), stirring at 70 C. When the addition was complete, the solution was
cooled to
room temperature and concentrated in vacuo. The residue was purified by flash
column
chromatography (220 g Redisep flash column, DCM/Me0H 100/0 to 98/2) to afford
Intermediate 146 (1.56g. yield: 41 %) as a tan oil.
Intermediate 147 and Intermediate 148
HO
N¨N
N
CI O¨
N /
0
o/
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 157 -
intermediate 147: Ra or Sa; pure atropisomer but absolute stereochemistry
undetermined
Intermediate 148: Sa or Ra; pure atropisomer but absolute stereochemistry
undetermined
p-Toluenesulfonic acid monohydrate (0.56 g, 2.92 mmol, 1.5 eq.) was added to a
solution of Intermediate 146 (1.56 g, 1.95 mmol) in Me0H (50 mL) and the
reaction
mixture was stirred at room temperature for 16 h. The solvent was evaporated
and the
residual oil was partitioned between DCM and saturated aqueous NaHCO3. The
layers
were separated and the organic layer was dried over MgSO4, filtered, and
concentrated
in vacuo. The residue was purified by preparative SFC (Stationary phase:
Chiralpak
Daicel IG 20 x 250 mm, Mobile phase: CO2, Et0H + 0.4 % iPrNH2) to afford
Intermediate 147 (502 mg, yield: 36 %) and Intermediate 148 (476 mg, yield: 34
%),
both as white solids.
Intermediate 149
o =
F
Intermediate 149
NaH (60% in mineral oil, 61.9 g, 1548.2 mmol, 1.1 eq.) was added to a solution
of 4-
(tert-butyl) 1-ethyl 2-(diethoxyphosphoryl)succinate (CAS [77924-28-8], 523.8
g,
1548.2 mmol, 1.1 eq.) in THF (3500 mL) at 0 C. The resulting solution was
stirred at
0 'V for 1 h. Then, 2,3-difluorobenzaldehyde (200 g, 1407.4 mmol), dissolved
in THF
(1500 mL), was added to the solution and the reaction mixture was stirred at
room
temperature for 3 h. The reaction was quenched by addition of cold water (2000
mL).
The resulting mixture was extracted with Et0Ac (3 x 3000 mL). The combined
organic
layer was dried over Na2SO4, filtered, and concentrated to afford Intermediate
149 (538
g, assumed quantitative) as a yellow oil, used without further purification.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 158 -
Intermediate 150
OO
OH
F
Intermediate 150
Inteimediate 149 (538 g, 1648.6 mmol) was dissolved in TFA (2000 mL) and the
reaction mixture was stirred at room temperature for 1 h. The reaction mixture
was
concentrated under reduced pressure. Toluene was added and evaporated under
reduced
pressure to afford Intermediate 150 (533 g, assumed quantitative) as a yellow
solid,
used without further purification.
Intermediate 151
Intermediate 151
Na0Ac (161.8 g, 1972.4 mmol, 1 eq.) was added to a solution of Intermediate
150 (533
g, 1972.4 mmol) in acetic anhydride (3600 mL). The resulting solution was
stirred at
130 C for 1 h. After cooling down to room temperature, the reaction mixture
was
concentrated under reduced pressure. The residue was diluted with water (1000
mL)
and extracted with Et0Ac (3 x 3000 mL). The combined organic layer was dried
over
Na2SO4, filtered, and concentrated. The residue was purified by silica gel
chromatography (Et0Ac/petrol eum ether 0/100 to 30/70) to afford Intermediate
151
(190 g, yield: 33 %) as a yellow solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 159 -
Intermediate 152
OH
Intermediate 152
K2CO3 (75.86 g, 548.85 mmol, 1.7 eq.) was added to a solution of Intermediate
151(95
g, 322.85 mmol) in Et0H (1500 mL). The resulting solution was stirred at room
temperature for 1 h. The solution was filtered and concentrated under reduced
pressure.
Aqueous HCl (0.5 M, 500 mL) was added to the residue and the mixture was
extracted
with Et0Ac (3 x 2000 mL). The combined organic layer was dried over Na2SO4,
filtered, and concentrated to afford Intei
_______________________________________ mediate 152 (70.4 g, yield: 86 %) as a
yellow
solid, used without further purification.
Intermediate 153
= 01
si
Intermediate 153
Tert-butylchlorodiphenylsilane (92.066 g, 334.955 mmol, 1.2 eq.) and DMAP
(6.820 g,
55.826 mmol, 0.2 eq.) were added to a solution of Intermediate 152 (70.4g.
279.129
mmol) in THE (1500 mL) under nitrogen atmosphere. Imidazole (28.471 g, 418.694
mmol, 1.5 eq.) was then added. The resulting solution was stirred at 50 C for
16 h.
After cooling down to room temperature, the reaction was quenched with water
(500
mL). The resulting mixture was extracted with Et0Ac (3 x 1000 mL). The
combined
organic layer was combined, dried over Na2SO4, filtered, and concentrated. The
residue
was purified by silica gel chromatography (Et0Acipetroleum ether 0/100 to
20/80) to
afford Intermediate 153 (114g. yield: 83%) as a yellow solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 160 -
Intermediate 154
= SI
si
OH
Intermediate 154
LiA1H4 (10.596 g, 278.835 mmol, 1.2 eq.) dissolved in THY (200 mL) was added
to a
solution of Intermediate 153 (114 g, 232.362 mmol) in THF (1500 mL) at 0 C.
The
resulting solution was stirred at room temperature for 1 h. The reaction was
quenched
by addition of sodium sulfate decahydrate. The resulting mixture was filtered
and the
filter cake was washed with Et0Ac (3 x 1000 mL). The combined organic layer
was
concentrated to afford Intel ______ mediate 154 (94.6 g, yield: 91 %) as a
white solid, used
without further purification.
Intermediate 155
101
si
Intermediate 155
Dess-Martin periodinane (CAS [87413-09-0], 267.773 g,631.331 mmol, 3 eq.) was
added to a solution of Intermediate 154 (94.4 g, 210.444 mmol) in DCM (1500
mL).
The resulting mixture was stirred at room temperature for 1 h. The reaction
was
quenched by addition of saturated aqueous sodium thiosulfate (1000 mL). The
resulting
mixture was extracted with DCM (3 x 2000 mL). The combined organic layer was
dried over Na2SO4, filtered, and concentrated. The residue was purified by
silica gel
chromatography (petroleum ether/Et0Ac 100/0 to 50/50) to afford Intermediate
155
(70 g, yield: 74 %) as a white solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 161 -
Intermediate 156
0 0
N
/
=
0 4*
Intermediate 156
Intermediate 105 (61.794 g, 137.047 mmol, 1.2 eq.) was added to a mixture of
Intermediate 155 (51 g, 114.206 mmol) in THF (2 L). NaH (60 % in mineral oil,
6.8 g,
171.309 mmol, 1.5 eq.) was added to the reaction mixture at 0 C and the
mixture was
stirred at room temperature for 40 min. The reaction was quenched by addition
of
saturated aqueous NFI4C1 (2 L). The mixture was extracted with Et0Ac (3 x 1
L). The
combined organic layer was dried over Na2SO4, filtered, and concentrated under
reduced pressure. The residue was purified by column chromatography on silica
gel
(petroleum ether/Et0Ac 8/1) to afford Intermediate 156 (59 g, yield: 88 %) as
a white
solid.
Intermediate 157
0 0
N
/
=
0 IO
Intermediate 157
Pd/C (10%, 10 g, 0.17 eq.) was added to a solution of Intermediate 156 (58 g,
99.535
mmol) in Et0Ac (1 L) and THE (200 mL). The mixture was stirred at 40 C for 16
h
under hydrogen atmosphere. The reaction mixture was filtered through a celite
pad and
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 162 -
the filtrate was concentrated under reduced pressure. The residue was purified
by
column chromatography (petroleum ether/Et0Ac 5/1) to afford Intermediate 157
(38 g,
yield: 65 %) as a colorless oil, used without further purification.
Intermediate 158
OH
N
k
\< =
Si
Intermediate 158
LiA1H4 (2.885 g, 75.933 mmol, 1.2 eq.) dissolved in TI-1F (20 mL) was added to
a
solution of Intermediate 157 (37 g, 63.277 mmol) in TI-IF (240 mL) at 0 C.
The
resulting solution was stirred at room temperature for 1 h. The reaction was
quenched
by addition of sodium sulfate decahydrate. The resulting mixture was filtered
and the
filter cake was washed with Et0Ac (3 x 200 mL). The combined organic layer was
concentrated and the residue was triturated with petroleum ether and diethyl
ether to
afford Intermediate 158 as a white solid (15.5 g, yield: 41 %), used without
further
purification.
Intermediate 159
ci
N
k
=
0 410
Intermediate 159
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 163 -
A solution of Intermediate 158 (1.0 g, 1.696 mmol) in dry DCM (15 mL) was
cooled to
0 'V under nitrogen atmosphere. SOC12 (0.141 mL, 1.950 mmol, 1.15 eq.) was
added
dropwise and the reaction mixture was stirred at room temperature for 1 h. The
reaction
mixture was diluted with DCM (35 mL) and saturated aqueous NaHCO3 (15 mL). The
layers were separated and the organic one was washed with saturated aqueous
NaHCO3
(15 mL) and brine (15 mL). The organic layer was dried over MgSO4, filtered,
and
concentrated under reduced pressure to give Intermediate 159 (1030 mg, yield:
98 %)
as a colorless paste, used without further purification.
Intermediate 160
N-N
0--
N CI
0
OH
Intermediate 160
K2CO3 (620 mg, 4.496 mmol, 2 eq.) was added to a solution of Intermediate 8
(1.3 g,
2.248 mmol) and Intermediate 159 (1.4 g, 2.473 mmol, 1.1 equiv) in Me0H (30
mL)
under nitrogen atmosphere. The reaction mixture was stirred at room
temperature for
16 h. The reaction was quenched by adding water (50 mL). The resulting mixture
was
extracted with Et0Ac (3 x 50 mL). The combined organic layer was dried over
Na2SO4, filtered, and concentrated. The residue was purified by silica gel
chromatography (petroleum ether/Et0Ac 100/0 to 20/80) to afford Intermediate
160 as
a yellow oil (1.7 g, yield: 90 %).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 164 -
intermediate 161
N-N
0--
N CI
/N
0
OH
OH
Intermediate 161
pTs0H.H20 (375 mg, 1.972 mmol, 1.1 eq.) was added to a solution of
Intermediate
160 (1.5 g, 1.793 mmol) in Me0H (30 mL). The reaction mixture was stirred at
room
temperature for 1.5 h. The solvent was evaporated and the residue was diluted
with
water and DCM. The layers were separated and the aqueous layer was extracted
with
DCM (40 mL x 3). The combined organic layer was washed with aqueous NaIIC03
(30
mL), brine (30 mL), dried over Na2SO4, filtered, and evaporated to afford
Intermediate
161 (1.2g, yield: 93 %) as a white solid.
Intermediate 162 and intermediate 163
N-N
N CI 0-
1 /
0
Intermediate 162: Ra or Sa; pure atropisomer but absolute stereochemistry
undetermined
Intermediate 163: Sa or Ra; pure atropisomer but absolute stereochemistry
undetermined
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 165 -
Intermediate 161 (1.05 g, 1.454 mmol) and DTBAD (502 mg, 2.181 mmol, 1.5 eq.)
in
toluene (10 mL) and TI-IF (1 mL) was added dropwise over 10 min to a solution
of
triphenylphosphine (571 mg, 2.181 mmol, 1.5 eq.) in toluene (10 mL) at 70 C
under
nitrogen atmosphere. After the addition was complete, the reaction mixture was
further
stirred at the same temperature for 10 min. The solvents were evaporated and
the
residue was extracted with DCM (10 mL x 3). The combined organic layer was
washed
with brine (10 mL), dried over Na2SO4, filtered, and evaporated. The residue
was
purified by reverse phase flash chromatography (40 ¨ 100 % 0.05 % NII4FIC03
H20/CH3CN) followed by preparative SFC (CHIRALPAK IG, 3 * 25 cm, 5 um;
Mobile Phase A: CO2, Mobile Phase B: IPA:ACN = 1:1(0.1 % 2 M NH3-Me0H);
Gradient: 50 % B) to afford Intermediate 162 (300 mg, yield: 29 A) and
Intermediate
163 (300 mg, yield: 29 %), both as pale yellow foamy solids.
Intermediate 164
L.
=
o ,
Intermediate 164
A suspension of sodium hydride (27.7 g, 693.76 mmol, 1 eq.) in THF was added
dropwise to a stirred solution of 4-(tert-butyl) 1-ethyl 2-
(diethoxyphosphoryl)succinate
(CAS [77924-28-8], 258.2 g, 763.13 mmol. 1.1eq) in TI-IF (1.5 L) at 0 C . The
reaction mixture was stirred for 1 h at room temperature before 3-chloro-2-
fluorobenzaldehyde (110 g, 693.8 mmol) was added at room temperature. The
reaction
was further stirred at room temperature for 3 h. The reaction was quenched by
adding
ice/water (500 mL) and the mixture was extracted by Et0Ac (300 mL x 3). The
organic
layer was dried over Na2SO4, filtered, and concentrated under reduced pressure
to
afford Intermediate 164 (237 g, assumed quantitative), used without further
purification.
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 166 -
intermediate 165
Lo
I
OH
CI
Intermediate 165
A solution of Intermediate 164 (543 g, 1584 mmol) in TFA (1.5 L) was stirred
at 25 C
for 1 h. The mixture was concentrated under reduced pressure to afford
Intermediate
165 (454 g, assumed quantitative), used without further purification.
Intermediate 166
OH
CI
Intermediate 166
Sodium acetate (0.486 g, 5.93 mmol, 1.7 eq.) was added to a solution of
Intermediate
165 (1 g, 3.49 mmol) in TFA (10 mL) and the reaction mixture was stirred at
130 C
for 2 h. The mixture was concentrated under reduced pressure. The residue was
dissolved in Et0H (10 mL) and K2CO3 (0.756 g, 5.471 mmol, 1.7 eq.) was added.
The
reaction mixture was stirred at room temperature for 2 h. The solvent was
evaporated to
give Intermediate 166, used in the next step without further purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 167 -
Intermediate 167
Si
ci
Intermediate 167
Imidazole (24.7 g, 362.9 mmol, 1.5 eq.), tert-butylchlorodiphenylsilane (79.8
g, 290.3
mmol, 1.2 eq.) and DMAP (5.9 g, 48.4 mmol, 0.2 eq.) were added to a solution
of
Intermediate 166 (65 g, 241.9 mmol) in TI-IF (1 L). The reaction mixture was
stirred at
room temperature overnight. The reaction was quenched by addition of water (1
L).
The resulting mixture was extracted with Et0Ac (3 x 500 mL). The organic layer
was
washed with brine (1 L), dried over Na7SO4, filtered through a celite pad, and
concentrated under reduced pressure. The residue was purified by flash
chromatography on silica gel (petroleum etheilEt0Ac = 4/1) to afford
Intermediate 167
(105 g, yield: 85 % yield) as a yellow oil.
Intermediate 168
osi
OH
CI
Intermediate 168
LiA1H4 (9.43g, 248.5 mmol, 1.2 eq.) was added portionwise to a solution of
Intermediate 167 (105 g, 207.1 mmol) in THE (1 L) at 0 C. The reaction
mixture was
stirred at room temperature for 1 h. The reaction was quenched by addition of
sodium
sulfate decahydrate (10 g). The resulting mixture was filtered and the
filtrate was
combined and concentrated. The crude product was triturated with petroleum
ether and
diethyl ether to afford Intermediate 168 (95 g, yield; 93 %) as a white solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 168 -
Intermediate 169
Si
10/
c,
Intermediate 169
Dess-Martin periodinane (CAS [87413-09-0], 150.5 g, 354.8 mmol, 3 eq.) was
added to
a mixture of Intermediate 168 (55 g, 118.3 mmol) in DCM (1 L). The reaction
mixture
was stirred at room temperature for 1 h. The resulting mixture was filtered
through a
celite pad. The filtrate was diluted with water (1 L) and was extracted with
DCM (500
mL x 3). The combined organic layer was washed with brine (2 L), dried over
MgSO4,
filtered through a celite pad, and concentrated under reduced pressure. The
crude
product was triturated with petroleum ether (100 mL) and diethyl ether (100
mL) to
afford Intermediate 169 (45 g, yield: 82 %) as a white solid.
Intermediate 170
11,
N
CI
Intermediate 170
Sodium hydride (60% in mineral oil, 7.1 g, 178.2 mmol, 1.5 eq.) was added to a
solution of Intermediate 169 (55 g, 118.8 mmol) and Intermediate 105 (53.5 g,
118.8
mmol, 1.5 eq.) in THF (600 mL) at 0 'V and the resulting solution was stirred
at room
temperature for 1 h. The reaction was quenched by adding saturated aqueous
NH4C1
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 169 -
(100 mL) and the resulting mixture was extracted with Et0Ac (3 x 500 mL). The
organic layer was washed with brine (1 L), dried over Na2SO4, filtered through
a celite
pad, and concentrated under reduced pressure. The residue was purified by
flash
chromatography on silica gel (petroleum ether/Et0Ac 4/1) to afford
Intermediate 170
(38 g, yield: 53 %) as a white solid.
Intermediate 171
0
o/
N_
________________________________ 411
Si
0
CI
Intermediate 171
Pd/C (10 %, 15 g, 140.9 mmol, 0.225 eq.) was added to a solution of
Inteimediate 170
(37.5 g, 62.6 mmol) in Et0Ac (500 mL) under nitrogen atmosphere and the
resulting
solution was stirred under hydrogen atmosphere at room temperature for 16 h.
The
reaction mixture was filtered through a celite pad and the filtrate was
concentrated
under reduced pressure. The residue was purified by flash chromatography on
silica gel
(petroleum ether/Et0Ac 4/1) to afford Intermediate 171 (25 g, yield: 66 %
yield) as a
white solid
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 170 -
Intermediate 172
OH
N_
,-N
\\<
Si
0
CI
=
Intermediate 172
Diisobutylaluminium hydride (1 M in toluene, 83.2 mL, 124.7 mmol, 3 eq.) was
added
dropwise to a mixture of Intermediate 171 (25 g, 41.6 mmol) in DCM (500 mL)
under
nitrogen atmosphere at -78 'C. The reaction mixture was stirred at room
temperature
for 1 h. The reaction was quenched by adding saturated aqueoud potassium
sodium
tartrate (200 mL). The resulting mixture was filtered and the filtrate was
extracted with
DCM (3 x 200 mL). The combined organic layer was evaporated and the crude
product
was triturated with petroleum ether (100 mL) and diethyl ether (100 mL) to
afford
Intermediate 172 (19 g, yield: 78%) as a white solid.
Intermediate 173
ci
N_
41111
Si
0
CI
=
Intermediate 173
SOC12 (1.08 g, 9.07 mmol, 1.3 eq.) was added to a solution of Intermediate 172
(4 g,
6.98 mmol, 1 eq.) in DCM (100 mL) at 0 C. The reaction mixture was stirred at
room
temperature for 1 h. The reaction was quenched by adding saturated aqueous
NaHCO3
(100 mL) The mixture was extracted with DCM (100 ml, x 3).The combined organic
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 171 -
layer was washed with brine (100 mL), dried over Na2SO4, filtered, and
concentrated
under reduced pressure to afford Intermediate 173 (4.1 g, yield: 99 %) as a
white solid.
Intermediate 174
z
N CI
\ ________________________________________ 0
OH
Intermediate 174
Intermediate 173 (2.25 g, 3.80 mmol, 1.1 eq.) was added to a solution of
Intermediate 8
(2 g, 3.46 mmol) in Me0H (50 mL) at room temperature. The reaction mixture was
stirred at room temperature for 10 min under nitrogen atmosphere. K2CO3 (0.96
g, 6.92
mmol, 2 eq.) was added and the mixture was stirred at room temperature for 16
h under
nitrogen atmosphere. Water (30 mL) was added and the mixture was extracted
with
Et0Ac (30 mL x 3). The organic layer was washed with brine (30 mL), dried over
Na2SO4, and concentrated under reduced pressure. The residue was purified by
flash
chromatography (petroleum ether/Et0Ac /2) to afford Intermediate 174 (2.2 g,
yield:
51 %) as a red solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 172 -
intermediate 175
N-N
z
0--
ci
N
0
OH
HO
CI
Intermediate 175
p-Toluenesulfonic acid (806 mg, 2.95 mmol, 1.2 eq.) was added to a solution of
Intermediate 174 (2.1 g, 2.46 mmol) in Me0H (30 mL) and the reaction mixture
was
stirred at room temperature for 1 h. Water (30 mL) was added and the mixture
was
extracted with Et0Ac (30 mL x 3). The organic layer was washed with saturated
aqueous NaHCO3 (30 mL x 2) and brine (50 mL). The organic layer was
concentrated
under reduced pressure to afford Intermediate 175 (1.6 g, yield: 73 %), used
without
further purification.
Intermediate 176 and intermediate 177
N-N
CI CY"-
0
CI
Intermediate 176: Ita or Sa, pure atropisomer but absolute stereochemistry
undetermined
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 173 -
Intermediate 177: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
A solution of Intermediate 175 (1.5 g, 2.03 mmol, 1 eq.) and di-tert-Butyl
azodicarboxylate (0.9 g, 4.06 mmol, 2 eq.) in toluene (30 mL) and TI-IF (5 mL)
was
added dropwise over 10 min to a solution of triphenylphosphine (1.1 g, 4.06
mmol, 2
eq.) in toluene (30 mL) at 70 C under nitrogen atmosphere. After the addition
was
complete, the reaction mixture was further stirred at the same temperature for
10 min.
The mixture was concentrated under reduced pressure and the residue was
purified by
reverse-phase flash chromatography (50 ¨ 99 % ACN/Water - 5 mmol NH4HCO3)
followed by prepative chiral HPLC (Column: CHIRAL ART Amylose-SA S, 3 * 25
cm, 5 [tm; Mobile Phase A: CO2, Mobile Phase B: IPA:ACN = 1:1(0.1 % 2 M NH3-
Me0H), Gradient:50 % B) to afford Intermediate 176 (250 mg, yield: 17 %) and
Inteimediate 177 (350 mg, yield: 24 %), both as a white solids.
Intermediate 178
z
N CI
0
OH
CI
Intermediate 178
Intermediate 173 (3.7g, 6.28 mmol, 1.1 eq.) was added to a solution of
Intermediate 125
(3.7 g, 5.71 mmol) in Me0H (100 mL) under nitrogen atmosphere. K2CO3 (1.57 g,
11.41
mmol, 2 eq.) was added and the reaction mixture was stirred at room
temperature for 16
h under nitrogen atmosphere. The reaction was quenched by adding water (100
mL). The
resulting mixture was extracted with Et0Ac (3 x 100 mL). The combined organic
layer
was dried over Na2SO4, filtered, and evaporated. The residue was purified by
silica gel
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 174 -
chromatography (petroleum etherfEt0Ac 100/0 to 80/20) to afford Intermediate
178 (4.2
g, yield: 80 %) as a yellow oil.
Intermediate 179
N_N
0--
N CI
0
\--O
OH H
ci
Intermediate 179
Triethylamine trihydrofluoride (1.1 g, 6.82 mmol, 1.5 eq.) was added to a
solution of
Intermediate 178 (4.2 g, 4.55 mmol) in TI-IF (100 mL) and the reaction mixture
was
stirred at room temperature for 16 h. The reaction was quenched by adding
water (100
mL). The resulting mixture was extracted with Et0Ac (3 x 50 mL). The combined
organic layer was washed with brine, dried over Na2SO4, filtered, and
concentrated to
afford Intermediate 179 (3.6 g, yield: 98 %) as a yellow solid, used without
further
purification.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 175 -
intermediate 180
N-N
N
/
0
CI
Intermediate 180
A solution of Intermediate 179 (3.6 g, 4.45 mmol) and DTBAD (3.0 g, 13.35
mmol, 3
eq.) in toluene (50 mL) and THT (5 mL) was added dropwise over 5 min to a
solution
of PPh3 (3.5 g, 13.31 mmol, 3 eq.) in toluene (50 mL), stirring at 70 C under
nitrogen
atmosphere. After the addition, the reaction mixture was further stirred at
the same
temperature for 20 min.The solvents were evaporated and the residue was
partitioned
between water and DCM. The layers were separated and the aqueous layer was
extracted with DCM (50 mL x 3). The organic layer was washed with brine (50
mL),
dried over Na2SO4, filtered, and evaporated. The residue was purified by
reverse phase
flash chromatography (40 ¨ 100 % 0.05 % Na4HCO3 H20/C1-13CN) to afford
Intermediate 180 (1.9 g, yield. 54%) as a yellow solid.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 176 -
Intermediate 181 and Intermediate 182
N¨NH
N === CI 0¨
k /
/N
0
o
CI
Intermediate 181: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 182: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
A solution of Intermediate 180 (1.9 g, 2.40 mmol) in HC1 (4 M in dioxane, 30
mL) was
stirred at room temperature for 16 h. The solid that appeared was collected by
filtration.
The residue was purified by preparative chiral SFC (Column: CHIRALPAK IF, 30 *
250 mm, S lam; Mobile Phase A: CO2, Mobile Phase B: iPrOH:ACN = 1:1 (0.1 % 2 M
NH3-Me0H), Gradient: 50 B) to afford Intermediate 181 (370 mg, yield: 22 %)
and
Intermediate 182 (410 mg, yield: 23 %), both as off-white solids.
Intermediate 181: OR: +44 (589 nm, 22.4 C, 5 mg in 10 mL Me0H)
Intermediate 182: OR: -42 (589 nm, 22.4 "V, 5 mg in 10 mL Me0H)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 177 -
Intermediate 183 and Intermediate 184
0/
0¨\_0
N¨N>
N¨N
o
N CI N CI 0¨
\ / \
/N
0 /N
0
0 0
01 a
Intermediate 183 Intermediate 184
Intermediate 183: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 184: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
1-Bromo-2-(2-methoxyethoxy)ethane (145 mg, 0.79 mmol, 2 eq.) and Cs2CO3 (386
mg, 1.19 mmol, 3 eq.) were added to a solution of Intermediate 181 (280 mg,
0.40
mmol) in DMF (15 mL). The reaction mixture was stirred at 35 C for 48 h. The
reaction was quenched by adding water (20 mL). The mixture was extracted with
Et0Ac (3 x 20 mL). The combined organic layer was dried over Na2SO4, filtered,
and
evaporated. The residue was purified by reverse phase flash chromatography (40
¨ 100
% 0.05 % NH4HCO3 H20/CH3CN) followed by preparative chiral HPLC (Column:
CHIRALPAK IC, 3 * 25 cm, 5 um; Mobile Phase A: hexane (0.5 % 2 M NI-13-Me0F1),
Mobile Phase B: EtOR Gradient:40 % B to 40 c1/0 B in 15 min) to afford
Intermediate
183 (130 mg, yield: 41 %) and Intermediate 184 (130 mg, yield: 41 %), both as
yellow
oils.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 178 -
intermediate 185 and intermediate 186
o/
O
N-N)\N¨N
\
0¨
\ / \
0 0
o.?
CI CI
Intermediate 185 Intermediate 186
Intermediate 185. Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 186: Sa or Ri, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 185 and Intermediate 186 were prepared according to an analogous
procedure as for Intermediate 183 and Intermediate 184, respectively, starting
from
Intermediate 182 instead of Intermediate 181.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 179 -
Intermediate 187
CI
0
0
OH
Intermediate 187
Intermediate 187 was prepared according to an analogous procedure as for
Intermediate
178, starting from Intermediate 159 instead of Intermediate 173.
Intermediate 188
0--
N CI
/N 0
\--O
OH H
Intermediate 188
Intermediate 188 was prepared according to an analogous procedure as for
Intermediate
179, starting from Intermediate 187 instead of Intermediate 178.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 180 -
Intermediate 189
N CI
0
Intermediate 189
A solution of Intermediate 188 (3 g, 3.786 mmol) and di-tert-butyl
azodicarboxylate
(2.615 g, 11.359 mmol, 3 eq.) in toluene (40 mL) and THE (10 mL) was added
dropwise
over 10 min to a solution of triphenylphosphine (2.979g, 11.359 mmol, 3 eq.)
in toluene
(40 mL) while stirring at 70 C under nitrogen atmosphere. After the addition
was
complete, the reaction mixture was further stirred at the same temperature for
10 min.
The mixture was concentrated under reduced pressure and the residue was
purified by
reverse-phase flash chromatography (50-99 % ACN / Water - 5 mmol NFI4HCO3) to
afford Intermediate 189 (1.8g, yield: 55%) as a white solid.
Intermediate 190 and Intermediate 191
N¨NH
N CI
/
0
o
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 181 -
Intermediate 190: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 191: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
A solution of Intermediate 189 (1.7 g, 2.19 mmol) in HCl (4 M in dioxane, 50
mL) was
stirred at room temperature for 2 h. The reaction mixture was concentrated
under
reduced pressure and the residue was purified by reverse-phase flash
chromatography
(50-99 % ACN/Water - 5 mmol NH4HCO3) followed by preparative chiral 1-113LC
(Column: CHIRALPAK IG, 3 * 25 cm, 5 um, Mobile Phase A:CO2, Mobile Phase B:
iPrOH (0.5 % 2 M NH3-Me0H); Gradient:50 c/o B) to afford Intermediate 190 (350
mg,
yield: 22 %) and Intermediate 191 (330 mg, yield: 21 %), both as white solids.
Intermediate 190: OR: ¨ +67.5 ( 589 nm, 22.5 C, 5.0 mg in 10 mL Me0H).
Intermediate 191: OR: -47.5 ( 589 nm, 22.5 C, 5.0 mg in 10 mL Me0H).
Intermediate 192 and Intermediate 193
0/
N¨N N¨N
\o
N CI N CI 0¨
\ /
0 0
o/
Intermediate 192: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 193: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 182 -
Intermediate 192 and Intennediate 193 were prepared according to an analogous
procedure as for Intermediate 183 and Intermediate 184, respectively, starting
from
Intelmediate 190 instead of Intermediate 181.
Intermediate 194 and Intermediate 195
o/
o¨\\_
(o
N¨N N¨N
\o
N CI N CI 0¨
\ /
0 0
Intermediate 194: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 195: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
Intermediate 194 and Intermediate 195 were prepared according to an analogous
procedure as for Intermediate 183 and Intermediate 184, respectively, starting
from
Intermediate 191 instead of Intermediate 181.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 183 -
Intermediate 196
N
0 S i __
Intermediate 196
DIPEA (0.64 mL, 2 eq.) followed by methanesulfonic anhydride (0.65 g, 2 eq.)
was
added to a solution of Intermediate 24a (1.0 g, 1.86 mmol) in TI-if (45 mL),
cooled to 0
'C. The reaction mixture was stirred at room temperature for 0.5 h. Sodium
iodide (1.39
g, 5 eq.) was then added to the mixture and it was further stirred at room
temperature for
1 h. The reaction mixture was diluted with DCM (100 mL) and washed with water
(20
mL). The aqueous layer was extracted with DCM/iPrOH 3:1 (2 x 30 mL), the
combined
organic layer was dried over MgSO4, and concentrated under reduced pressure to
give a
dark yellow oil. This oil was purified by flash column chromatography on
silica gel
(SiO2, 24 g column, 0-3 % Me0II in DCM) to give Intermediate 196 (1.1 g,
yield: 91 %)
Intermediate 197
oo
N-N
0
N
0-
\
0
OH
Intermediate 197
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 184 -
A solution of Intermediate 81(540 mg, 0.888 mmol) and Intermediate 196 (691mg,
1.065
mmol, 1.2 eq.) in THF (18 mL) was added dropvvise over 20 min to a suspension
of NaH
(60 % in mineral oil, 43 mg, 1.776 mmol, 2 eq.) in THF (18 mL) at 0 C. The
reaction
mixture was stirred at 0 C for 1 h. The reaction was quenched by adding Me01-
1 (5 mL).
The solvents were evaporated and the residue was purified by preparative TLC
(Et0Ac)
to afford Intermediate 197 (410 mg, yield: 52%) as a yellow oil.
Intermediate 198
NN
0
/ CI
0
OH
OH
Intermediate 198
p-Toluenesulfonic acid (95 mg, 0.55 mmol, 1.2 eq.) was added to a solution of
Intermediate 197 (410 mg, 0.46 mmol) in Me0H (5 mL). The reaction mixture was
stirred at room temperature for 1 h. Water (5 mL) was added and the mixture
was
extracted with Et0Ac (5 mL x 3). The combined organic layer was washed with
saturated
aqueous NaHCO3 (10 mL), brine (10 mL), dried over Naz SO4, filtered, and
concentrated
under reduced pressure. The residue was purified by preparative TLC (Et0Ac) to
afford
Intermediate 198 (250 mg, yield: 70%) as a yellow oil.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 185 -
Intermediate 199
o¨\\_
N¨N
0
Nfr..- CI
/
/N
0
o$)
Intermediate 199
Intermediate 199 was prepared according to an analogous procedure as for
Intermediate
180, starting from Intermediate 198 instead of Intermediate 179.
Intermediate 200 and Intermediate 201
\ 0
) 0 /
0 \¨\
N¨N N¨N
1 S /
Ra or Sa Ra or
Sa
N-- N--
NI / CI 0¨ CI 0¨
N 0 N 0
)
Alt 0 0/
Intermediate 200 Intermediate 201
Pure stereoisomers but absolute stereochemistry undetermined
A solution of Intermediate 60 (200 mg, 0.3 mmol), tert-butyl (2-
chloroethyl)(methyl)carbamate (CAS [220074-38-4], 202 mg, 1.04 mmol, 3.5 eq.),
and
Cs2CO3 (291 mg, 0.89 mmol, 3 eq.) in dry DMF (4.6 mL) was stirred at 60 C
under
nitrogen atmosphere for 6.5 h. Additional tert-butyl (2-
chloroethyl)(methyl)carbamate
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 186 -
(202 mg, 1.04 mmol, 3.5 eq.) was added and the mixture was stirred at 60 C
for 16 h.
Again, additional tert-butyl (2-chloroethyl)(methyl)carbamate (202 mg, 1.04
mmol, 3.5
eq.) was added and the mixture was stirred at 60 C for 3.5 h. The solvent was
removed
under reduced pressure and the residue was taken up with DCM and brine. The
layers
were separated and the organic layer was washed with brine (x 3). The combined
aqueous
layer was extracted with DCM (x 5) and the combined organic layer was dried
over
MgSO4, filtered, and concentrated under reduced pressure. The residue was
purified by
flash column chromatography (SiO2, 12 g RediSep, DCM/Me0H 100/0 to 90/10)
followed by preparative SFC (stationary phase: Chi ralpak Daicel IG 20 x 250
mm,
Mobile phase: CO2, Et0H + 0.4 % iPrNH2) to give Intermediate 200 (86 mg,
yield: 35
%) and Intermediate 201 (95 mg, yield: 38 %), both as pale yellow solids.
Intermediate 202
HN
N¨N
N--
Ra or So
NI / CI 0-
0
=
z
HC1 salt
Pure stereoisomer but absolute stereochemistry undetermined
HC1 (6 M in iPrOH, 2.6 mL, 15.59 mmol, 150 eq.) was added to a solution of
Intermediate 200 (86 mg, 0.104 mmol) in Me0H (2 mL) under nitrogen atmosphere.
The
reaction mixture was stirred at room temperature for 4 h. More HC1 (6 M in
iPrOH, 0.52
mL, 3.12 mmol, 30 eq.) was added again and the mixture was stirred at room
temperature
for 1 h. The solvent was removed under reduced pressure and the solid was
rinsed twice
with Me0I-I to give Intermediate 202 (1-Id 1 salt, 82.5 mg, yield:
quantitative) as a pale
yellow solid.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 187 -
Intermediate 203
NH
N¨N
N¨
Ra or Sa
I CI 0-
0
0
Pure stereoisomer but absolute stereochemistry undetermined
Intermediate 203 was prepared according to the same procedure as for
Intermediate 202,
starting from Intermediate 201 instead of Intermediate 200.
Preparation of Compounds
Compound 1
N¨N
z
CI OH
N
0
/
o/
)N
Sa or Ra atropisomer
LiOH (28 mg, 15 eq.) was added to a solution of Intermediate 17 (55 mg, 0.078
mmol)
in a mixture of THF (1.25 mL), Me0H (1.25 mL) and water (0.625 mL) at room
temperature. The resulting reaction mixture was stirred for 2 h at 60 C. The
reaction
mixture was concentrated to give a white solid. The solid was dissolved in
water (5 mL)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 188 -
and acidified with aqueous HC1 (1 M) to pH 3, a white precipitate forming upon
acidification. The aqueous layer was extracted with Et0Ac (20 mL) and then DCM
(3 x
20 mL), the combined organic layer was dried over MgSO4, filtered, and
concentrated.
The crude product was purified by flash column chromatography on silica gel
(DCM:Me0H - 1:0 to 95:5) to give a white solid that was triturated in DIPE and
filtered
to afford Compound 1 (32 mg, yield: 59 %) as a white solid.
11-1 NAIR (400 MHz, DMSO-d6) 6 ppm 1.91 (s, 3 H), 2.04 (s, 3 H), 2.20 -2.39
(m, 2 H),
2.74- 285 (m, 3 H), 2.96 -3.06 (m, 3 H), 3.29 - 3.30 (m, 2 H), 3.40 (s, 3 I-
I), 3.55 (q,
J=8.1 Hz, 1 H), 3.70 -3.79 (m, 4 H), 4.61 (ddd, J=14.2, 9.7, 4.0 Hz, 1 H),
4.95 (s, 1 H),
5.00 (dt, J=14.6, 4.8 Hz, 1 H), 6.12 (d, J=1.4 Hz, 1 H), 7.04 (d, J=9.0 Hz, 1
H), 7.22 (s,
1 H), 7.41 -7.51 (m, 2 H), 7.53 (d, J=9.1 Hz, 1 H), 7.71 -7.78 (m, 1 H), 8.15 -
8.23 (m,
1 H), 12.85- 13.63 (m, 1 H).
Compound 2
CI
N
N 0
o
Ra or Sa atropisomer
Compound 2 was prepared according to the same procedure as for Compound 1,
starting from Intermediate 18 instead of Intermediate 17.
lEINMIR (400 MHz, DMSO-d6) 6 ppm 1.91 (s, 3 H), 2.04 (s, 3 H), 2.21 -2.38 (m,
2 H),
2.74 - 2.86 (m, 3 H), 2.96 - 3.06 (m, 3 H), 3.29 - 3.30 (m, 2 H), 3.40 (s, 3
H), 3.55 (q,
J=8.3 Hz, 1 H), 3.69 - 3.78 (m, 4 H), 4.61 (ddd, J=14.1, 9.7, 4.1 Hz, 1 H),
4.95 (s, 1 H),
5.00 (dt, J=14.6, 4.8 Hz, 1 H), 6.12 (s, 1 H), 7.04 (d, .1=9.0 Hz, 1 H), 7.22
(s, 1 H), 7.41
- 7.50 (m, 2 H), 7.53 (d, J=9.0 Hz, 1 H), 7.70 - 7.79 (m, 1 H), 8.19 (d, J=7.9
Hz, 1 H),
12.65 - 13.84 (m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 189 -
Compound 3
N-N/
S
N.---
I CI OH
f
0
orj
Sa or Ita atropisomer
LiOH (32 mg, 15 eq.) was added to a solution of Intermediate 27 (65 mg, 0.09
mmol) in
a mixture of TI-IF (2 mL), Me0H (2 mL), and water (1 mL). The resulting
reaction
mixture was stirred for 4 h at 60 'C. The reaction mixture was concentrated to
give a
white solid. The solid was dissolved in water (5 mL) and acidified with
aqueous HC1 (1
M) to pH 4-5, a white precipitate forming upon acidification. The aqueous
layer was
extracted with DCM (3 x 20 mL), the combined organic layer was dried over
MgSO4,
and concentrated to give a white solid. This crude product was purified by
flash column
chromatography on silica gel (DCM:Me0H - 1:0 to 97:3). The purest fractions
were
combined to give a yellow solid that was triturated in Et20 and filtered to
afford
Compound 3 (18 mg, yield: 28 %) as a pale yellow solid. A second fraction of
Compound
3 (14 mg, yield: 22 %) with slightly lower purity was also isolated as a pale
yellow solid.
1H NMR (400 MHz, DMSO-do) a ppm 1.91 (s, 3 H), 2_04 (s, 3 H), 219- 2.39 (m, 2
H),
2.75 - 2.86 (m, 3 H), 3.00 (br d, J=13.8 Hz, 3 H), 3.28 - 3.29 (m, 2 H), 3.40
(s, 3 H),
3.55 (br d, J=9.2 Hz, 1 H), 3.69 - 3.79 (m, 4 H), 4.61 (br s, 1 H), 4.95 (s, 1
H), 4.97 -
5.06 (m, 1 H), 6.10 (s, 1 H), 7.08 (d, J=9.0 Hz, 1 H), 7.20 (s, 1 H), 7.32
(td, J=8.9, 2.6
Hz, 1 H), 7.47 - 7.56 (m, 2 H), 8.24 (dd, J=9.2, 5.9 Hz, 1 H), 12.88 - 13.64
(m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 190 -
Compound 4
N-N
S
N---
.1. CI OH
../1
0
of)
Ra or Sa atropisomer
Compound 4 was prepared according to the same procedure as for Compound 3,
starting
from Intermediate 28 instead of Intermediate 27.
11-INMIR (400 MHz, DMSO-d6) 5 ppm 1.91 (s, 3 H), 2.04 (s, 3 H), 2.20- 2.38 (m,
2 H),
2.76 - 2.86 (m, 3 H), 2.96 - 3.06 (m, 3 H), 3.28 - 3.29 (m, 2 H), 3.40 (s, 3
H), 3.55 (q,
J=8.0 Hz, 1 H), 3.69 - 3.79 (m, 4 H), 4.54 - 4.67 (m, 1 H), 4.95 (s, 1 H),
4.97 - 5.06 (m,
1 H), 6.10 (s, 1 H), 7.08 (d, J=9.0 Hz, 1 H), 7.20 (s, 1 H), 7.32 (td, J=8.9,
2.6 Hz, 1 H),
7.47 - 7.57 (m, 2 H), 8.24 (dd, J=9.2, 5.8 Hz, 1 H), 12.86 - 13.61 (m, 1 H).
Compound 5
N-N/
S
N--- / CI OH
/IN
0
ori
Mixture of atropisomers
Compound 5 was prepared according to the same procedure as for Compound 3,
starting
from Intermediate 29 instead of Intermediate 27.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 191 -
1F1 NIVIR (400 MHz, DMSO-d6) 6 ppm 1.92 (s, 3 H) 2.06 (s, 3 H) 2.16 -2.33 (m,
2 H)
2.72- 2.86 (m, 3 H) 2.94 - 3.09 (m, 3 H) 3.37 (br d, J=5.5 Hz, 10 H) 3.35 -
3.38 (m, 4
H) 3.39 (s, 5 H) 3.40 - 3.42 (m, 2 H) 3.69 - 3.74 (m, 1 H) 3.75 (s, 4 H) 4.50 -
4.61 (m, 1
H) 4.91 - 4.99 (m, 1 II) 5.00 (s, 1 H) 6.16 (s, 1 11) 7.00 (d, J=9.0 Hz, 1 H)
7.18 (dd,
J=13.3, 7.6 Hz, 1 H) 7.33 (s, 1 H) 7.43 (td, J=8.0, 4.8 Hz, 1 H) 7.54 (d, J-
9.1 Hz, 1 H)
7.59 (d, J=8.3 Hz, 1 H).
Compound 6
o N-N
OH
CI
N\
N 0
o./(\
Sa or Ra atropisomer
Compound 6 was prepared according to the same procedure as for Compound 3,
starting from Intermediate 30 instead of Intermediate 27.
1II NMR (400 MHz, DMSO-do) 6 ppm 1.93 (s, 3 II), 2.08 (s, 3 II), 2.16 - 2.36
(m, 2 II),
2.68 -2.86 (m, 2 H), 2.97 (s, 3 H), 2.99 - 3.11 (m, 3 H), 3.25 (br d, J=14.5
Hz, 1 H),
3.49 - 3.57 (m, 1 H), 3.66 - 3.72 (m, 1 H), 3.77 (dd, J=14.8, 2.1 Hz, 1 H),
3.82 (s, 3 H),
4.64 (br s, 1 H), 4.82 (d, J=14.8 Hz, 1 H), 4.93 (br d, J=14.3 Hz, 1 H), 5.62
(s, 2 H),
7.03 (d, J=9.0 Hz, 1 H), 7.35 (s, 1 H), 7.44 (d, J=9.1 Hz, 1 H), 7.47 - 7.57
(m, 2 H),
7.77 - 7.84 (m, 1 H), 8.25 - 8.32 (m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 192 -
Compound 7
o N-N
r
CI OH
/
/
N 0
Ra or Sa atropisomer
Compound 7 was prepared according to the same procedure as for Compound 3,
starting from Intermediate 31 instead of Intermediate 27.
1H NMR. (400 MHz, DMSO-d6) ppm 1.93 (s, 3 H), 2.08 (s, 3 H), 2.16- 2.35 (m, 2
H),
267- 286 (m, 2 H), 2_97 (s, 3 H), 2.99 - 3.11 (m, 3 H), 3.25 (hr d, J=14,3 Hz,
1 H),
3.53 (dt, J=9.7, 4.9 Hz, 1 H), 3.65 -3.73 (m, 1 H), 3.74 - 3.81 (m, 1 H), 3.83
(s, 3 H),
4.58 - 4.69 (m, 1 H), 4.82 (d, J=14.8 Hz, 1 H), 4.89 - 4.97 (m, 1 H), 5.62 (s,
2 H), 7.03
(d, J=9.0 Hz, 1 H), 7.35 (s, 1 H), 7.44 (d, J-9.1 Hz, 1 H), 7.46 - 7.56 (m, 2
H), 7.77 -
7.84 (m, 1 H), 8.26 - 8.32 (m, 1 H).
Compound 8
N-N
CI OH
N V /
\ /
N 0
Sa or Ra atropisomer
To a solution of Intermediate 37 (40 mg, 0.055 mmol) in a mixture of THF (2
mL),
Me0H (2 mL) and water (1 mL) was added LiOH (20 mg, 15 eq.). The resulting
reaction
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 193 -
mixture was stirred for 2 h at 60 C. The reaction mixture was concentrated to
give a
white solid. The solid was purified by preparative 1-[PLC (Stationary phase:
RP )(Bridge
Prep C18 OBD- 5 p.m, 50 x 250 mm, Mobile phase: 0.25 A.NI-1.4HCO3 solution in
water,
CH3CN) to give a yellow solid that was triturated in Et20 and filtered to
afford
Compound 8 (24 mg, yield: 61 %) as a pale yellow solid.
IFINMR (400 MHz, DMSO-d6) 6 ppm 1.75 (s, 3 H), 1.88- 1.93 (m, 3 H), 1.98 (s, 3
H),
2.24 - 2.42 (m, 2 H), 2.80 (dd, J=28.9, 12.9 Hz, 2 H), 2.87 - 2.98 (m, 3 H),
3.06 - 3.12
(m, 2 I-1), 3.62 (s, 3 II), 3.72- 3.82 (m, 5 IT), 4.03 -4.13 (m, 1 H), 4.50
(ddd, J=14.1,
9.5, 4.1 Hz, 1 H), 4.63 (s, 1 H), 5.06 (dt, J=14.5, 4.8 Hz, 1 H), 6.57 (s, 1
H), 7.09 (s, 1
H), 7.21 -7.31 (m, 2 H), 7.43 (dd, J=10.5, 2.6 Hz, 1 H), 7.69 (d, J-=9.0 Hz, 1
H), 8.09
(dd, J-9.2, 5.9 Hz, 1 H).
Compound 9
N-N
OH
CI
N 0
o
Ra or Sa atropisomer
Compound 9 was prepared according to the same procedure as for Compound 8,
starting from Intermediate 38 instead of Intermediate 37.
11-I NMR (400 MHz, DMSO-d6) 6 ppm 1.75 (s, 3 H), 1.90 (s, 3 H), 1.98 (s, 3 1-
1), 2.24 -
2.41 (m, 211), 2.79 (dd, J=28.8, 12.9 Hz, 2 I-1), 2,86 - 2.98 (m, 3 H), 3.04 -
3.13 (m, 3
H), 3.62 (s, 3 H), 3.73 - 3.82 (m, 4 H), 4.08 (br d, J=8.6 Hz, 1 H), 4.43 -
4.55 (m, 1 H),
4.62(s, 1 H), 5.01 -5.10 (m, 1 H), 6.57 (s, 1 H), 7.09 (s, 1 H), 7.20 - 7.29
(m, 2 H),
7.43 (dd, J=10.5, 2.6 Hz, 1 H), 7.68 (d, J=9.0 Hz, 1 H), 8.09 (dd, J=9.2, 5.9
Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 194 -
Compound 10
N-N
S
N---
CI
N
0
orj
Ra or Sa atropisomer
A solution of LiOH (68 mg, 15 eq.) in water (2 mL) was added to a solution of
Intermediate 44 (130 mg, 0.19 mmol) in a mixture of THF (4 mL) and Me0H (4
mL).
The reaction mixture was heated at 60 C for 3 h. After cooling to room
temperature, the
reaction mixture was diluted with Me0H and directly injected on preparative
HPLC
(Stationary phase: RP )(Bridge Prep C18 OBD-10 nm, 30x 150 mm, Mobile phase:
0.25
% Na4HCO3 solution in water, CH3CN) to give Compound 10 (104 mg, yield: 81 %)
as
a white solid.
NMR: 1H NMIR (400 MHz, DMSO-d6) 6 ppm 1.91 (s, 3 H), 2.04 (s, 3 H), 2.21 -2.37
(m, 2 H), 2.77 (d, J=13.4 Hz, 1 H), 2.80 - 2.90 (m, 2 H), 2.99 (d, J=13.4 Hz,
1 H), 3.01
-3.09 (m, 2 H), 3.25 (d, J=14.1 Hz, 1 H), 3.30 (d, J=14.1 Hz, 1 H), 3.41 (s, 3
H), 3.51 -
3.60 (m, 1 H), 3.72 - 3.80 (m, 4 H), 4.56 -4.65 (m, 1 H), 4.95 (s, 1 H), 5.01
(dt, J=14.5,
4.7 Hz, 1 H), 6.21 (s, 1 H), 7.07 (d, J=9.0 Hz, 1 H), 7.27 - 7.33 (m, 1 H),
7.35 (s, 1 H),
7.42 (td, J=8.1, 5.5 Hz, 1 H), 7.53 (d, J=9.0 Hz, 1 H), 8.02 (d, J=8.4 Hz, 1
H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 195 -
Compound 11
N-N
S
---
N
I / CI
or)
Sa or Ra atropisomer
Compound 11 was obtained using an analogous method as for Compound 10,
starting
from Intermediate 45 instead of Intermediate 44.
NMR: 'HNMR (400 MHz, DMSO-d6) 6 ppm 1.91 (s, 3 H), 2.03 (s, 3 H), 2.21 - 2.35
(m, 2 H), 2.78 (d, J=13.6 Hz, 1 H), 2.80 - 2.91 (m, 2 H), 2.99 (d, J=13.6 Hz,
1 H), 3.01
- 3.09 (m, 1 H), 3.26 (d, J=14.1 Hz, 2 H), 3.30 (d, J=14.1 Hz, 1 H), 3.41 (s,
3 H), 3.51 -
3.60 (m, 1 H), 3.72 - 3.79 (m, 4 H), 4.55 -4.65 (m, 1 H), 4.95 (s, 1 H), 5.01
(dt, J=14.5,
4.7 Hz, 1 H), 6.21 (s, 1 H), 7.06 (d, J=9.0 Hz, 1 H), 7.27 - 7.33 (m, 1 H),
7.35 (s, 1 H),
7.42 (td, J=8.1, 5.5 Hz, 1 H), 7.52 (d, J=9.0 Hz, 1 H), 8.02 (d, J=8.4 Hz, 1
H).
Compound 12
N-N
N
N---
CI
N
orj
Sa or Ra atropisomer
Compound 12 was obtained using an analogous method method as for Compound 10,
starting from Intermediate 39 instead of Intermediate 44.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 196 -
NMR: 11-INMR (400 MHz, CDCb, 27 C) 6 ppm 1.93 -2.12 (m, 7 H) 2.16 -2.33 (m, 4
H) 2.59 -2.80 (m, 3 H) 2.93 -3.19 (m, 4 H) 3.35 -3.45 (m, 2 H) 3.49 (s, 2 H)
3.55 -
3.89 (m, 5 H) 4.41 -4.61 (m, 1 H) 4.99 -5.26 (m, 1 H) 5.30 (s, 1 H) 5.79 -
6.00 (m, 1
H) 6.78 -6.93 (m, 1 H) 6.99 - 7.13 (m, 1 H) 7.14 - 7.24 (m, 1 H) 7.36 (br s, 2
H) 7.47 -
7.53 (m, 1 H).
Compound 13
N-N
N
N---
0
or)
Ra or Sa atropisomer
Compound 13 was obtained using an analogous method as for Compound 10,
starting
from Intermediate 40 instead of Intermediate 44.
NMR: 1H NMR (600 MHz, DMSO-d6, 77 C) 6 ppm 1.87 (br s, 3 H) 1.95 (s, 3 H) 2.02
(s, 3 H) 2.20 -2.34 (m, 2 H) 2.86 - 2.93 (m, 1 H) 2.93 -2.98 (m, 1 H) 2.98 -
3.04 (m, 2
H) 3.01 -3.08 (m, 2 H) 3.15 - 3.16 (m, 1 1-1)3.41 - 3.46 (m, 1 H) 3.54 (s, 3
H) 3.71 -
3.77 (m, 2 H) 3.78 (s, 3 H) 4.48 -4.57 (m, 1 H) 4.96 (br s, 1 H) 5.01
(dt,J=14.6, 4.9
Hz, 1 H) 6.48 (br s, 1 H) 7.08 (dd, J=13.1, 7.5 Hz, 1 H) 7.18 (d, J=8.9 Hz, 1
H) 7.23 (s,
1 H) 7.36 (td, J=7.9, 4.8 Hz, 1 H) 7.51 (d, J=8.1 Hz, 1 H) 7.62 (d, 1=9.1 Hz,
1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 197 -
Compound 14
N-N
S /
N---
I / CI OH
N
0
or)
Itc, or Sa atropisomer
Compound 14 was prepared according to an analogous procedure as for Compound
7,
starting from Intermediate 47 instead of Intermediate 31.
ifINMR (400 MHz, DMSO-d6) ppm 1.93 (s, 3 H); 2.08 (s, 3 H); 2.16 - 2.31 (m, 2
H); 2.67 - 2.86 (m, 2 H); 2.97 - 3.10 (m, 4 H); 2.99 (s, 3 H); 3.23 - 3.28 (m,
1 El), 3.50
- 3.57 (m, 1 H); 3_69 (br d, J=14.31 Hz, 1 H); 3.76- 3.81 (m, 1 H); 3.83 (s, 3
H); 4.64
(br t, J=10.78 Hz, 1 H); 4.81 (d, J=14.75 Hz, 1 H); 4.87 - 4.97 (m, 1 H); 5.60
(s, 1 H);
5.62 (s, 1 H); 7.06 (d, J=9.02 Hz, 1 H); 7.31 - 7.45 (m, 3 H); 7.58 (dd,
J=10.45, 2.53
Hz, 1 H); 8.35 (dd, J=9.13, 5.83 Hz, 1 H).
Compound 15
N-N/
0 0
\=
S /
N---
I / CI OH
0
Sa orRa atropisomer
Compound 15 was prepared according to an analogous procedure as for Compound
7,
starting from Intermediate 46 instead of Intermediate 31.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 198 -
1H NIMR (400 MHz, DMSO-d6) 6 ppm 1.93 (s, 3 H); 2.08 (s, 3 H), 2.16 -2.34 (m,
2 H),
2.66 - 2.85 (m, 2 H), 2.94 -3.10 (m, 4 H), 2.98 (s, 3 H), 3.27 (br s, 3 H),
3.49 -3.56
(m, 1 H), 3.69 (br d, J=14.32 Hz, 1 H), 3.78 (br d, J=14.95 Hz, 1 H), 3.83 (s,
3 H), 4.64
(br t, J=10.97 Hz, 1 H), 4.81 (d, J=14.63 Hz, 1 H), 4.88 - 4.97 (m, 1 H),
5.60(s, 1 H),
5.62 (s, 1 H), 7.07 (d, J=8.99 Hz, 1 H), 7.31 - 7.45 (m, 3 H), 7.58 (dd,
J=10.45, 2.61
Hz, 1 H), 8.35 (dd, J=9.25, 5.90 Hz, 1 H).
Compound 16
-o\
o-\_0
N---
N 0
Ra or Sa atropisomer
LiOH (19 mg, 30 eq.) was added to a solution of Intermediate 62 (21 mg, 0.026
mmol)
in a mixture of TI-IF (1 mL), MeOH (1 mL) and water (0.5 mL) at room
temperature.
The resulting reaction mixture was stirred overnight at 45 'C. The reaction
mixture was
concentrated, the residue was dissolved in water (5 mL), and was acidified
with
aqueous HC1 (1 M). The aqueous layer was extracted with CHC13 (3 x). The
combined
organic layer was washed with brine, dried over MgSO4, filtered, and
evaporated to
afford Compound 16 (20 mg, yield: 97 A).
1FINMR (400 MHz, CDC13) 5 ppm 2.03 (s, 3 H); 2.18 (s, 3 H); 2.34 (br d, J=3.66
Hz, 2
H); 2.84 - 2.93 (m, 5 H); 3.08 (s, 3 H); 3.21 (d, J=12.54 Hz, 1 H); 3.34 (br
t, J=5.33 Hz,
2 H); 3.36 (s, 3 H); 3.39 (d, J=15.57 Hz, 1 H); 3.50 -3.67 (m, 9 H); 3.78 (d,
115.57
Hz, 1 H); 3.85 - 3.97 (m, 2 H); 4.24 -4.34 (m, 2 H); 4.54 (ddd, J-14.63, 6.58,
3.66 Hz,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 199 -
1 H); 5.21 (ddd, J=14.76, 7.92, 3.87 Hz, 1 H); 5.45 (s, 1 H); 5.48 (s, 1 H);
7.14 - 7.18
(m, 2 H); 7.23 - 7.36 (m, 3 H); 8.33 (dd, J=9.20, 5.75 Hz, 1 H).
Compound 17
\\c)
o
so
N-N
S
/ CI OH
N 0
Ra or Sa atropisomer
Compound 17 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 63 instead of Intermediate 62.
1H NMIR (400 MHz, CDC13) 5 ppm 2.07 (s, 3 H); 2.20 (s, 3 H); 2.29 (br d,
J=7.42 Hz, 2
H); 2.78 -2.97 (m, 5 H); 3.08 (s, 3 H); 3.16 (d, J=12.23 Hz, 1 H); 3.24 - 3.39
(m, 6 H);
3.47 - 3.63 (m, 8 H); 3.66 - 3.73 (m, 1 H); 3.83 - 3.93 (m, 2 H); 4.28 (t,
J=5.43 Hz, 2
H); 4.46 -4.60 (m, 1 H); 5.15 - 5.27 (m, 1 H); 5.46 (s, 1 H); 5.49 (s, 1 H);
7.13 - 7.20
(m, 2 H); 7.22 - 7.37 (m, 3 H); 8.33 (dd, J=9.25, 5.80 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 200 -
Compound 18
-q\\
N z CI
N 0
Ra or Sa atropisomer
Compound 18 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 64 instead of Intermediate 62.
lEINM_R (400 MHz, CDC13) 6 ppm 2.04 (s, 3 H), 2.16 (s, 3 H), 2.31 (br s, 2 H),
2.79
(d, J=10.2 Hz, 2 H), 2.93 (s, 2 H), 2.95 (s, 3 H), 3.18 (hr d, J=4.0 Hz, 1 H),
3.21 -3.30
(m, 2 H), 3.31 (s, 3 H), 3.38 - 3.44 (m, 1 H), 3.73 -3.82 (m, 3 H), 4.21 -4.29
(m, 2 H),
4.52 (s, 1 H), 5.16- 5.29 (m, 1 H), 5.36 (s, 1 H), 5.59 (s, 1 H), 7.15 (s, 1
H), 7.22 -7.25
(m, 2 H), 7.27 - 7.33 (m, 3 H), 8.29 (dd, J=9.2, 5.9 Hz, 1 H).
Compound 19
\o
N-N
S
N
CI OH
N
N 0
Ra or Sa atropisomer
Compound 19 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 65 instead of Intermediate 62.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 201 -
1H NMR (400 MHz, CDC13) 6 ppm 2.07 (s, 3 H), 2.20 (s, 3 H), 2.23 -2.42 (m, 2
H),
2.73 -2.82 (m, 2 H), 2.94 (s, 3 H), 2.94 -2.99 (m, 2 H), 3.15 -3.28 (m, 3 H),
3.29 (s, 3
H), 3.37 - 3.44 (m, 1 H), 3.72 - 3.79 (m, 3 H), 4.23 -4.28 (m, 2 H), 4.46 -
4.54 (m, 1
H), 5.20 - 5.28 (m, 1 H), 5.37 (s, 1 H), 5.64 (s, 1 H), 7.18 (s, 1 H), 7.22 -
7.25 (m, 2 H),
7.27 - 7.34 (m, 3 H), 8.31 (dd, J=9.1, 5.8 Hz, 1 H).
Compound 20
o/
-o
N-N
N---
/ CI
/IN
N 0
Ra or Sa atropisomer
LiOH (2.5 mg, 15 eq.) was added to a solution of Intermediate 66 (5.4 mg,
0.007 mmol)
in a mixture of Me0H (200 !AL), THF (200 !AL), and water (90 L). The
resulting reaction
mixture was stirred for 4 h at 50 C. The reaction mixture was concentrated
under
reduced pressure to give a pale yellow solid. This solid was dissolved in
water and DCM
and acidified with 1 M aqueous HC1 to pH 4-5, a pale yellow precipitate
forming upon
acidification. The aqueous layer was extracted with DCM (x 4). The combined
organic
layer was dried over MgSO4, filtered, and evaporated to give Compound 20 (4
mg, yield:
79 %) as a pale yellow solid.
11-1NMIR (400 MHz, CDC13) 6 ppm 2.03 (s, 3 H), 2.14 (s, 3 H), 2.15 -2.42 (m, 3
H),
2.79 (br d, J=9.7 Hz, 2 H), 2.92 (br s, 2 H), 2.97 (br s, 3 H), 3.15 - 3.21
(m, 1 H), 3.22 -
3.26 (m, 2 H), 3.27 (s, 3 H), 3.28 - 3.30 (m, 1 H), 3.34 (s, 3 H), 3.37 - 3.45
(m, 1 H),
3.72 - 3.79 (m, 2 H), 3.79 - 3.84 (m, 1 H), 3.84 - 3.93 (m, 2 H), 4.43 -4.55
(m, 2 H),
5.23 (br d, J=4.5 Hz, 1 H), 5.38 (br s, 1 H), 5.59 (br s, 1 H), 7.14 (s, 1 H),
7.27 -7.33
(m, 3 H), 8.25 - 8.35 (m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 202 -
Compound 21
\o
xo-
N-N
S
N---
I / CI OH
N 0
Ra or Sa atropisomer
Compound 21 was prepared according to an analogous procedure as for Compound
20,
starting from Intermediate 67 instead of Intermediate 66.
IHNMR (400 MHz, CDC13) 5 ppm 1.85 - 2.01 (m, 1 H), 2.05 (s, 3 H), 2.17 - 2.22
(m, 3
H), 2.22 -2.40 (m, 2 H), 2.70 -2.97 (m, 5 H), 3.00 (s, 3 H), 3.16 (d, J=11.8
Hz, 1 H),
3.23 (br d, J=8.3 Hz, 1 H), 3.29 (s, 3 H), 3.32 (s, 3 H), 3.34 - 3.43 (m, 1
H), 3.70 - 3.91
(m, 5 H), 4.45 -4.56 (m, 2 H), 5.18 -5.27 (m, 1 H), 5.40 (s, 1 H), 5.59 (s, 1
H), 7.18 (s,
1 H), 7.21 (s, 1 H), 7.25 (br s, 1 H), 7.27 - 7.30 (m, 1 H), 7.30 - 7.34 (m, 1
H), 8.32 (dd,
J=9.2, 5.7 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 203 -
Compound 22
\N-N
N---
I / CI OH
z
N 0
07
Ra or Sa atropisomer
LiOH (18 mg, 15 eq.) was added to a solution of Intermediate 68 (39 mg, 0.05
mmol) in
a mixture of Me0H (1.2 mL), THF (1.2 mL), and water (0.6 mL). The resulting
reaction
mixture was stirred for 4 h at 50 C. The reaction mixture was concentrated
under
reduced pressure to give a pale yellow solid. This solid was dissolved in
water and DCM
and acidified with 1 M aqueous HC1 to pH 4-5, a pale yellow precipitate
forming upon
acidification. The aqueous layer was extracted with DCM (x 4). The combined
organic
layer was dried over MgSO4, filtered, and evaporated to give Compound 22 (33
mg,
yield: 86 %) as a pale yellow solid.
1H NMR (400 MHz, CDC13) 6 ppm 1.38 (br d, J=31.5 Hz, 3 H), 1.60 - 1.67 (m, 3
H),
1.85 - 1.95 (m, 2 H), 2.04 (s, 3 H), 2.17 (s, 3 H), 2.32 (br d, J=8.4 Hz, 2
H), 2.81 (d,
J=10.3 Hz, 2 H), 2.92 (s, 2 H), 2.95 (s, 3 H), 3.18 (br d, J=4.4 Hz, 1 H),
3.23 (d, J=11.7
Hz, 1 H), 3.30 - 3.35 (m, 2 H), 3.35 - 3.40 (m, 2 H), 3.55 (d, J=15.4 Hz, 1
H), 3.89 -
3.97 (m, 2 H), 4.07 (t, J=7.0 Hz, 2 H), 4.51 (br d, J=14.7 Hz, 1 H), 5.17 -
5.30 (m, 1 H),
5.35 (s, 1 H), 5.59 (s, 1 H), 7.16 (s, 1 H), 7.23 -7.25 (m, 1 H), 7.27 - 7.28
(m, 1 H),
7.31 (s, 1 H), 7.32 - 7.34 (m, 1 H), 8.30 (dd, J=9.1, 5.8 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 204 -
Compound 23
N-N
S
N---
I CI OH
N 0
Ra or Sa atropisomer
Compound 23 was prepared according to an analogous procedure as for Compound
22,
starting from Intermediate 69 instead of Intermediate 68.
1HNMIR (400 MHz, CDC13) 6 ppm 1.35 (br d, J=7.9 Hz, 1 H), 1.57- 1.70 (m, 4 H),
1.80 (t, J=6.5 Hz, 2 H), 2.05 (s, 3 H), 2.20 (s, 3 H), 2.25 - 2.37 (m, 2 H),
2.77 (d, J=9.2
Hz, 2 H), 2.90 (s, 3 H), 2.93 -3.01 (m, 3 H), 3.14 (br d, J=3.7 Hz, 1 H), 3.18
(d, J=11.4
Hz, 1 H), 3.22 (d, J=15.0 Hz, 1 H), 3.31 - 3.44 (m, 3 H), 3.74 (s, 1 H), 3.94
(br d,
J=11.9 Hz, 2 H), 4.14 (t, J=7.4 Hz, 2 H), 4.44 - 4.55 (m, 1 H), 5.30 (s, 1 H),
5.35 (s, 1
H), 5.67 (s, 1 H), 7.18 (s, 1 H), 7.27 (br d, J=1.3 Hz, 1 H), 7.29 - 7.31 (m,
1 H), 7.32 (s,
1 H), 7.32 - 7.34 (m, 1 H), 8.30 (dd, J=9.0, 5.9 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 205 -
Compound 28
N-N
N----
/ CI OH
N 0
o
R. or S. atropisomer
Compound 28 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 74 instead of Intermediate 62.
1H NMR (400 MHz, CDC13) 6 ppm: 2.03 (s, 3 H), 2.18 (s, 3 H), 2.32 (br s, 2 H),
2.77 -
2.85 (m, 1 H), 2.87 (br s, 3 H), 2.92 (br d, J=12.2 Hz, 1 H), 3.04 (br s, 3
H), 3.23 (d,
J=12.5 Hz, 1 H), 3.26 - 3.36 (m, 211), 3.37 - 3.47 (m, 1 H), 3.47- 3.55 (m, 1
H), 3.85
(s, 3 H), 4.54 (br d, J=15.4 Hz, 1 H), 5.20 (br d, J=9.0 Hz, 1 H), 5.46 (br s,
2 H), 7.16
(s, 2 H), 7.24 (br d, J=2.5 Hz, I H), 7.27 - 7.34 (m, 2 H), 8.32 (dd, J=9.0,
5.7 Hz, 1 H).
Compound 29
N-NH
S
N---
/ CI OH
N 0
o
R. or S. atropisomer
Compound 29 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 60 instead of Intermediate 62.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 206 -11-1NMR (400 MHz, CDC13) 6 ppm 2.16 (s, 2 H), 2.19 (s, 3 H), 2.34 (br
d, J=5.3 Hz, 2
H), 2.86 (s, 3 H), 2.95 (d, J=12.6 Hz, 1 H), 3.10 (s, 3 H), 3.19 (d, J=12.5
Hz, 1 H), 3.35
(br d, J=4.5 Hz, 2 H), 3.41 (br d, J=14.8 Hz, 1 H), 3.63 (br d, J=15.0 Hz, 1
H), 4.56 (br
d, J=15.4 Hz, 1 H), 5.19- 5.28 (m, 1 H), 5.43 (s, 1 H), 5.50 (s, 1 H), 7.13
(s, 1 H), 7.17
(d, J=8.9 Hz, 1 H), 7.19 - 7.25 (m, 2 H), 7.30 (dd, J=10.0, 2.4 Hz, 1 H), 7.34
(d, J=9.1
Hz, 1 H), 8.31 (dd, J=9.1, 5.7 Hz, 1 H).
Compound 30
N-NH
S
z CI OH
/IN
N 0
)
Sa or 11,, atropisomer
Compound 30 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 61 instead of Intermediate 62.
1H NMR (400 MHz, CDC13) 6 ppm 2.12 (s, 2 H), 2.19 (s, 3 H), 2.34 (br d, J=4.6
Hz, 2
H), 2.86 (br d, J=6.1 Hz, 3 H), 2.97 (d, J=12.4 Hz, 1 H), 3.09 (s, 3 H), 3.20
(d, J=12.4
Hz, 1 H), 3 28 - 3.36 (m, 2 H), 3.36 - 3.40 (m, 1 H), 3.58 (d, J=15.3 Hz, 1
H), 4.49 -
4.59 (m, 1 H), 5.19 - 5.27 (m, 1 H), 5.46 (s, 1 H), 5.49 (s, 1 H), 7.12 (s, 1
H), 7.19 (d,
J=8.9 Hz, 1 H), 7.21 - 7.25 (m, 2 H), 7.28 - 7.32 (m, 1 H), 7.32 - 7.35 (m, 1
H), 8.29
(dd, J=9.2, 5.7 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 207 -
Compound 31
oo
N-N
N---
I / CI OH
./N
N 0
o/
Ra or Sa atropisomer
LiOH (52 mg, 15 eq.) was added to a stirred solution of Intermediate 84 (112
mg, 0.144
mmol) in water (1.7 mL), THF (3.4 mL), and Me0H (3.4 mL) at room temperature.
The
reaction mixture was stirred at 50 'DC overnight. The reaction mixture was
concentrated
under reduced pressure and the residue was diluted with water (15 mL) and
acidified
with 1 M aqueous HCl until acidic pH. This aqueous solution was extracted
twice with
DCM (10 mL), then with a 1:1 mixture of Et0Ac:THF (10 mL). The combined
organic
layer was dried over MgSO4, filtered, and evaporated. The residue was co-
evaporated
with DCM and tBuOMe to yield Compound 31(109 mg, yield: 99 %) as an off-white
solid.
1F1 NMIt (400 MHz, CDC13) 6 ppm 2.04 (s, 3 H) 2.18 (s, 3 H) 2.24 -2.42 (m, 2
H) 2.78
- 2.96 (m, 5 H) 3.01 (s, 3 H) 3 20 - 3.25 (m, 2 H) 3.32 - 3.39 (m, 5 H) 3.46 -
3.50 (m, 2
H) 3.51 -3.65 (m, 2 H) 3.82 (d, J=15.57 Hz, 1 H) 3.86 -3.98 (m, 2 H) 4.21 -
4.35 (m, 2
H) 4.47 - 4.59 (m, 1 H) 5.23 (ddd, J=14.84, 8.94, 3.61 Hz, 1 H) 5.41 (s, 1 H)
5.56 (s, 1
H) 7.17 (s, 1 H) 7.20 - 7.26 (m, 2 H) 7.28 - 7.35 (m, 2 H) 8.32 (dd, J=9.14,
5.80 Hz, 1
H).
OR = + 102.2 (c = 0.21 w/v%, DMF, 20 C).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 208 -
Compound 32
o-\_0
N-N
N
z CI OH
N 0
Sa or Ra atropisomer
Compound 32 was prepared according to an analogous procedure as for Compound
16,
starting from Intermediate 85 instead of Intermediate 62.
11-1NMIR (400 MHz, CDC13) 6 ppm 2.03 (s, 3 H) 2.19 (s, 3 H) 2.25 - 2.41 (m, 2
H) 2.81
-2.95 (m, 5 H) 3.10 (s, 3 H) 3.22 (d, J=12.76 Hz, 1 H) 3.29 -3.38 (m, 5 H)
3.42 (d,
J=15.63 Hz, 1 H) 3.46 - 3.56 (m, 3 H) 3.57 - 3.65 (m, 1 H) 3.76 (d, J=15.63
Hz, 1 H)
3.86 - 3.98 (m, 2 H) 4.25 - 4.36 (m, 2 H) 4.54 (ddd, J=14.52, 6.82, 3.74 Hz, 1
H) 5.21
(ddd, J=14.69, 7.65, 3.85 Hz, 1 H) 5.44 (s, 1 H) 5.50(s, 1 H) 7.11 -7.26 (m,
2H) 7.27 -
7.37 (m, 2 H) 8.33 (dd, J=9.24, 5.72 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 209 -
Compound 33
oo
N-N
N---
/ CI OH
N 0
o/
Ra or Sa atropisomer
LiOH (183 mg, 15 eq.) was added to a stirred solution of Intermediate 87 (389
mg, 0.514
mmol) in water (6 mL), THF (12 mL), and Me0H (12 mL) at room temperature. The
reaction mixture was stirred at 50 C for 18 h. The reaction mixture was
concentrated
under reduced pressure and then diluted with water (30 mL) and acidified with
1 M
aqueous HC1 until acidic pH. This aqueous phase was extracted twice with DCM
(25
mL), then with a 1:1 mixture of Et0Ac:THF (25 mL). The combined organic layer
was
dried over MgSO4, filtered, and evaporated. The residue was coevaporated a
couple of
times with n-heptane. The obtained solid was purified by flash column
chromatography
(silica; Me0H in DCM 0/100 to 5/95) to yield Compound 33 (332 mg, yield: 87 %)
as
an off-white solid.
11-INMR (400 MHz, CDC13) 6 ppm 2.03 (s, 3 H) 2.19 (s, 3 H) 2.26 -2.43 (m, 2 H)
2.83
-2.95 (m, 5 H) 3.12 (s, 3 H) 3.22 (d, J=12.75 Hz, 1 H) 3.27 -3.37 (m, 4 H)
3.43 (br d,
J=15.68 Hz, 2 H) 3.46 - 3.51 (m, 2 H) 3.51 -3.56 (m, 1 H) 3.56 - 3.63 (m, 1 H)
3.75 (d,
J=15.57 Hz, 1 H) 3.86 - 3.98 (m, 2 H) 4.25 - 4.35 (m, 2 H) 4.57 (ddd, J=14.79,
7.11,
3.71 Hz, 1 H) 5.20 (ddd, J=14.68, 7.47, 3.76 Hz, 1 H) 5.40 (s, 1 H) 5.55 (s, 1
H) 7.11
(d, J=8.99 Hz, 1 H) 7.23 (s, 1 H) 7.31 (d, J=8.99 Hz, 1 H) 7.46 - 7.54 (m, 2
H) 7.70 -
7.76(m, 1 H) 8.31- 8.37(m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 210 -
Compound 34
o-\_0
N-N
N---
/ CI OH
N 0
0/
Sa or ita atropisomer
Compound 34 was prepared according to an analogous procedure as for Compound
33,
starting from Intermediate 88 instead of Intermediate 87.
111NMR (400 MHz, CDC13) 6 ppm 2.03 (s, 3 H) 2.19 (s, 3 H) 2.25 -2.44 (m, 2 H)
2.83
- 2.94 (m, 5 H) 3.09 (s, 3 H) 3.22 (d, J=12.65 Hz, 1 H) 3.29 - 3.45 (m, 6 H)
3.46 - 3.51
(m, 2 El) 3.51 -3.56 (m, 1 H) 3.56 - 3.64 (m, 1 H) 3.76 (d, J=15.47 Hz, 1 H)
3.86 - 3.98
(m, 211) 4.24 - 4.36 (m, 2 II) 4.57 (ddd, J=14.47, 6.95, 3.87 Hz, 111) 5.21
(ddd,
J=14.84, 7.79, 3.61 Hz, 1 H) 5.43 (s, 1 H) 5.53 (d, J=0.84 Hz, 1 H) 7.13 (d,
J=8.99 Hz,
1 H) 7.23 (s, 1 H) 7.31 (d, J=8.99 Hz, 1 H) 7.46 - 7.54 (m, 2 H) 7.70 - 7.76
(m, 1 H)
8.31 -8.37 (m, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 211 -
Compound 35
N-N
S
Nk/ cl OH
N 0
CI
Ra or Sa atropisomer
LiOH (2 M in water, 4.5 mL, 15 eq.) was added to a solution of Intermediate
100 (420
mg, 0.598 mmol) in Me0H (10 mL) and THF (10 mL). The reaction mixture was
stirred
at 60 C for 4 h. After cooling, the reaction mixture was concentrated under
vacuum and
then diluted with water (5 mL). The pH of the solution was adjusted to 1-2
with 2 M
aqueous HC1. The resulting mixture was extracted with Et0Ac (3 x 50 mL). The
combined organic layer was combined, dried over Na2SO4, filtered, and
evaporated. The
residue was purified by reverse-phase flash chromatography (Column. Sunfire
Prep C18
OBD Column, 30*100 mm 5 um 10 nm; Mobile Phase A: Water (10 mM NH4HCO3),
Mobile Phase B: ACN; Flow rate: 60 mL/min) to afford Compound 35 (209 mg,
yield:
51 %), as an off-white solid.
MP: 220 C (Tianjin RY-2 type melting point apparatus)
OR: +32.9 (c = 0.1 w/v; DMSO; 589 nm; 26.5 C); +71.8 (c = 0.1 w/v; Me0H;
589
nm; 21.6 C)
1H NMR (300 MHz, DMSO-d6) 6 ppm 8.19 (d, J = 9.0 Hz, 1H), 7.84 (d, J = 2.1 Hz,
1H), 7.48 ¨7.41 (m, 2H), 7.19 (s, 1H), 7.01 (d, J = 8.9 Hz, 1H), 6.21 (s, 1H),
5.06 (d, J
= 14.2 Hz, 1H), 4.94 (s, 1H), 4.56¨ 4.51(m, 1H), 3.75 (s, 4H), 3.60 ¨ 3.51 (m,
1H),
3.43 ¨3.28 (m, 5H), 3.17 (s, 1H), 3.07 ¨2.92 (m, 3H), 2.87 ¨ 2.73 (m, 3H),
2.29 (s,
2H), 2.00 (s, 3H), 1.91 (s, 3H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 212 -
Compound 36
N-N
S
N/ CI OH
N 0
CI
Sa or Ra atropisomer
Compound 36 was prepared according to an analogous procedure as for Compound
35,
starting from Intermediate 99 instead of Intermediate 100.
MP: 211 C (Tianjin RY-2 type melting point apparatus)
OR: 49.20-
(c = 0.1 w/v; DMSO; 589 nm; 27.1 C); -76.9 (c = 0.1 w/v; Me0H; 589
nm; 22.1 C)
lEINMIR (300 MHz, DMSO-d6) 6 ppm 8.18 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 2.2
Hz,
1H), 7.47 -7.40 (m, 2H), 7.18 (s, 1H), 7.00 (d, J = 8.8 Hz, 1H), 6.21 (s, 1H),
5.09 (d, J
= 13.8 Hz, 1H), 4.93 (s, 1H), 4.55 - 4.50 (m, 1H), 3.75 (s, 4 H), 3.55 (d, J =
7.4 Hz,
1H), 3.43 (s, 3H), 3.23 (d, J = 32.7 Hz, 2H), 3.18 (s, 1H), 3.07 -2.99 (m,
3H), 2.83 -
2.78 (m, 3H), 2.42 - 2.29 (m, 2H), 2.02 (s, 3H), 1.91 (s, 3H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 213 -
Compound 37
H2N
N¨N
I / CI OH
/
0
o/
Ra or Sa atropisomer
Compound 37 was prepared according to an analogous procedure as for Compound
8,
starting from Intermediate 103 instead of Intermediate 37.
Compound 38
H2
\N¨N
I / CI OH
0
o/
Ra or Sa atropisomer
Compound 38 was prepared according to an analogous procedure as for Compound
8,
starting from Intermediate 104 instead of Intermediate 37.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 214 -
Compound 39 and Compound 40
\o o/
o
S
NN--
N CI
.1\1
N 0 N 0
o/ o/
CI ci
Ra or Sa atropisomer Ra or Sa atropisomer
Compound 39 Compound 40
LiOH (55 mg, 12 eq.) was added to the mixture of Inteimediate 131 and
Inteimediate
132 (300 mg, 0.379 mmol) in THE (4 mL) and H20 (4 mL) under nitrogen
atmosphere.
The resulting mixture was stirred at room temperature under nitrogen
atmosphere for 48
h. The reaction mixture was concentrated under vacuum and then diluted with
water (5
mL). The pH of the solution was adjusted to 1-2 with 3 M aqueous HC1. The
resulting
mixture was extracted with Et0Ac (3 x 10 mL). The combined organic layer was
dried
over Na2SO4, filtered, and concentrated. The residue was purified by
preparative chiral
SFC (Column: Phenomenex Lux 5u Cellulose-3, 5 x 25 cm, 5 pm; Mobile Phase A:
CO2,
Mobile Phase B: Me0H/ACN 1/1 (0.1 % 2 M NH3-Me0H); Gradient: 40% B) to afford
Compound 39 (28 mg, yield: 9 %) and Compound 40 (25 mg, yield: 16 %), both as
a
light yellow solids.
Compound 39
11-1 NAIR (300 MHz, CDC13) 5 ppm 8.25 (d, J = 6.0 Hz, 1H), 7.70 (s, 1H), 7.50 -
7.36
(m, 2H), 7.04 (s, 2H), 5.75 (s, 1H), 5.19 (d, J = 9 Hz, 1H), 5.12 (s, 1H),
4.93 (s, 1H),
4.63 (s, 2H), 4.10 (s, 2H), 3.63 (s, 4H), 3.51 (s, 6H), 3.34 (s, 5H), 3.34 ¨
2.91 (m, 3H),
2.81 (s, 2H), 2.48 (s, 211), 2.26 (s, 314), 2.14 (s, 311).
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
-215 -
Compound 40
IFINMR (300 MHz, CDC13) 6 ppm 8.32 (d, J = 9.0 Hz, 1H), 7.77 (s, 1H), 7.56 -
7.31
(m, 2H), 7.20 (s, 1H), 6.93 (s, 1H), 5.89 (s, 1H), 5.18 (s, 2H), 4.64 (s, 2H),
4.07 (s, 2H),
3.90 - 3.40 (m, 9H), 3.34 (s, 3H), 3.26 - 2.60 (m, 9H), 2.43 (s, 2H), 2.20 (s,
6H).
Compound 41 and Compound 42
<
N¨N N¨N
r
I CI I / CI
0 0
0/
o/
CI CI
Ra or Sa atropisomer Ra or Sa atropisomer
Compound 41 Compound 42
LiOH (77 mg, 12 eq.) was added to the mixture of Intermediate 133 and
Intermediate
134 (400 mg, 0.536 mmol) in THF (4 mL) and 1120 (4 mL) under nitrogen
atmosphere.
The resulting mixture was stirred at 40 C under nitrogen atmosphere for 48 h.
The
reaction mixture was concentrated under vacuum and then diluted with water (10
mL).
The pH of the solution was adjusted to 1-2 with 3 M aqueous HCl. The resulting
mixture
was extracted with Et0Ac (3 x 10 mL). The combined organic layer was dried
over
Na2SO4, filtered, and concentrated. The residue was purified by preparative
HPLC
(Column: XSelect CSH Prep C18 OBD, 5 um, 19 x 150 mm; Mobile Phase A: Water
(0.05 % HCl), Mobile Phase B: ACN, Gradient:63 % B to 78 % B in 7 min) to
afford
Compound 41(89 mg, yield: 43 %) and Compound 42 (89 mg, yield: 43 %), both as
light
yell ow solids.
A sample of Compound 41(52 mg, 0.068 mmol) was dissolved in Me0H (2 mL) and
NaOH (1 M in H20, 68 I-, 1 eq.) was added. The mixture was stirred for a few
min,
then volatiles were removed under reduced pressure. The residue was suspended
in D1PE
(2 mL) and evaporated to dryness. The residue was then triturated with DIPE,
filtered,
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 216 -
and dried under vacum at 55 C for 2 h to afford the sodium salt of Compound
41(40
mg, yield: 73 %) as an off-white solid.
Compound 41
lEINMR (300 MHz, CDC13) 5 ppm 8.15 (d, J = 9.0 Hz, 1H), 7.65(s, 1H), 7.50 -
7.39
(m, 2H), 7.08 (s, 1H), 6.88 (s, 1H), 5.82 (s, 111), 5.22 (d, J = 14.1 Hz, 1H),
4.90 (s, 2H),
4.64 (s, 2H), 3.97 (s, 2H), 3.85 (s, 1H), 3.54 (s, 3H), 3.52 ¨ 3.43 (m, 2H),
3.37 (s, 3H),
3.33 ¨2.89 (m, 5H), 2.85 ¨ 2.63 (m, 2H), 2.63 ¨ 2.31 (m, 2H), 2.22 (s, 3H),
2.09 (s,
3H).
Compound 42
1HNMR (300 MHz, CDC13) 6 ppm 8.35 (d, J = 9.0 Hz, 1H), 7.78 (s, 1H), 7.48 (d,
J =
9.0 Hz, 1H), 7.34 (d, J ¨9.0 Hz, 1H), 7.20 (s, 1H), 6.87 - 6.84 (m, 1H), 5.98
(s, 1H),
5.17 (d, J = 14.1 Hz, 1H), 5.04 (s, 1H), 4.78 -4.47 (m, 311), 4.11 (s, 1H),
4.02 - 3.58
(m, 6H), 3.35 (s, 5H), 3.07(s, 3H), 2.83 (s, 211), 2.64 ¨ 2.31 (m, 3H), 2.24
(s, 3H), 2.18
(s, 311).
Compound 43
ck)
<
N¨N
N
ci
/IN
N 0
CI
Sa or Ra atropisomer
LiOH (13 mg, 6 eq.) was added to a solution of Intermediate 135 (70 mg, 0.089
namol)
in THF (2 mL) and 1-120 (2 mL) under nitrogen atmosphere. The resulting
mixture was
stirred under nitrogen atmosphere at room temperature for 48 h. The mixture
was
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 217 -
concentrated under vacuum and then diluted with water (5 mL). The pH of the
solution
was adjusted to 1-2 with 3 M HC1. The resulting mixture was extracted with
Et0Ac (3 x
mL). The combined organic layer was combined, dried over Na2SO4, filtered, and
concentrated under reduced pressure. The residue was purified by preparative
HPLC
5 (Column: XBridge Prep OBD C18 Column, 19 x 250 mm, 5 um; Mobile Phase A:
Water
(0.05 % HC1), Mobile Phase B: ACN; Gradient:73 % B to 83 % B in 7 min) to
afford
Compound 43 (25 mg, yield: 37 %) as a light yellow solid.
1H NMR (300 MHz, CDC13) 6 ppm 8.28 (d, J = 9.0 Hz, 1H), 7.71 (s, 1H), 7.51 -
7.37
(m, 2H), 7.11 (s, 2H), 5.61 (s, 1H), 5.32 (s, 111), 5.22 (d, J = 15.0 Hz, 1H),
4.90 (s, 1H),
10 4.60 (s, 2H), 4.08 (s, 21-1), 3.62 (s, 5H), 3.38 (s, 2H), 3.33 (s, 31-
1), 3.21 (s, 5H), 2.99 (s,
3H), 2.81 (s, 2H), 2.43 (s, 2H), 2.28 (s, 3H), 2.18 (s, 3H).
Compound 44
o/
(0
N¨N
S
I /
N
N 0
o
ci
Sa or Ra atropisomer
Compound 44 was prepared according to an analogous procedure as for Compound
43,
starting from Intermediate 136 instead of Intermediate 135.
1H NMR (300 MHz, CDC13) 6 ppm 8.29 (d, J = 9.0 Hz, III), 7.74 (s, HI), 7.50 -
7.31
(m, 2H), 7.21 - 7.04 (m, 2H), 5.63 (s, 111), 5.37 (s, 1H), 5.22 (d, J = 9.0
Hz, 1H), 4.56
(s, 3H), 4.00 (s, 2H), 3.75 (d, J = 15.0 Hz, 1H), 3.65 - 3.12 (m, 14H), 3.92
(s, 511), 2.38
(s, 2H), 2.18 (d, J = 12.0 Hz, 6H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 218 -
Compound 45
N¨N
0 /
N
A / CI OH
N 0
o
Ra or Sa atropisomer
LiOH (18 mg, 6 eq.) was added to a solution of Intermediate 119 (85 mg, 0.127
mmol)
in Me0H (0.5 mL), THF (3 mL), and water (3 mL). The reaction mixture was
stirred at
40 C for 16 h under a nitrogen atmosphere. After cooling, the reaction
mixture was
concentrated under vacuum and then diluted with water (5 mL) and diethyl ether
(5 mL).
The layers were separated and the aqueous layer was extracted with diethyl
ether (3 x 10
mL). The pH of the aqueous layer was then adjusted to 3-4 with 2 M aqueous
HC1. The
resulting precipitate was filtered to afford Compound 45 (53 mg, yield: 63 %)
as an off-
white solid.
IHNMR (400 MHz, CD30D) 6 ppm 8.24 (m, 1H), d 7.48 (m, 1H), d 7.34 (m, 1H), d
7.19(m, 1H), d 7.10 (m, 2H), d6.21 (s, 1H), d5.21 (s, 2H), d 4.62 (m, 1H), d
4.16 (m,
2H), d 3.90 (m, 5H), d 3.76 (s, 1H), d 3.64 (s, 4H), d 3.06 (m, 2H), d 2.93
(m, 2H), d
2.37 (s, 2H), d 2.06 (m, 6H).
"F NMR (376 MHz, CD30D) 6 -117.2.
OR: +5.12 (c = 0.5 w/v. Me0H. 28.8 C).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 219 -
Compound 46
N-N/
0 /
N--
CI OH
N
N 0
01
Sa or Ra atropisomer
Compound 46 was prepared according to an analogous procedure as for Compound
45,
starting from Intermediate 118 instead of Intermediate 119.
IIINMIt (400 MHz, CD30D) 6 ppm 8.24 (m, 1H), d 7.48 (d, J = 8.0 Hz, 1H), d
7.34
(m, 1H), d 7.19 (m, 1H), d 7.10 (m, 2H), d 6.21 (s, 1H), d 5.21 (s, 2H), d
4.61 (m, 1H),
d 4.17 (m, 2H), d 3.99 (d, J = 12.0 Hz, 1H), d 3.90 (d, J= 12.0 Hz, 1H), d
3.85 (s, 3H),
d 3.77 (m, 1II), d 3.63 (m, 111), d 3.55 (m, 311), d 3.06 (m, 2H), d 2.94 (m,
2H), d 2.37
(s, 2H), d 2.06 (m, 6H).
19F NIVIR (376 MHz, CD30D) 6 -117.2
OR: -9.06 (c = 0.5 w/v. Me0H. 28.8 C).
Compound 47 and Compound 48
o )(c)
N-N N-N
S
N
N N
0/ 0/
CI CI
Sa or Ra atropisomer Sa orRa atropisomer
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 220 -
Compound 47 Compound 48
Compound 47 and Compound 48 were prepared prepared according to an analogous
procedure as for Compound 41 and Compound 42, starting from the mixture of
Intermediate 137 and Intermediate 138 instead of the mixture of Intermediate
133 and
Intermediate 134.
Compound 47
1H NMR (300 MHz, CDC13) 6 ppm 8.17 (d, J = 9.0 Hz, 1H), 7.65 (s, 1H), 7.43 -
7.35
(m, 2H), 7.08 (d, J= 8.1 Hz, 1H), 6.88 (s, 1H), 5.82 (s, 1H), 5.22 (d, J= 14.1
Hz, 1H),
4.91 (s, 2H), 4.64 (s, 2H), 3.97 (s, 2H), 3.84 (s, 1H), 3.53 (s, 3H), 3.52 ¨
3.43 (m, 2H),
3.37(s, 3H), 3.30 ¨3.05 (m, 5H), 2.83 ¨2.61 (m, 2H), 2.51 (s, 2H), 2.22 (s,
3H), 2.10
(s, 3H).
Compound 48
1H NMR (300 MHz, CDC13) 6 ppm 8.35 (d, J = 9.0 Hz, 1H), 7.78 (s, 1H), 7.48 (d,
J =
9.0 Hz, 1H), 7.34 (d, J = 9.0 Hz, 1H), 7.19 (s, 1H), 6.87 (d, J = 9.0 Hz, 1H),
5.94 (s,
1H), 5.17 (d, J ¨ 14.1 Hz, 1H), 5.06 (s, 1H), 4.82 - 4.55 (m, 3H), 4.09 (s,
1H), 3.99 -
3.82 (m, 3H), 3.67 (s, 3H), 3.34 (s, 5H), 3.18 - 2.92 (m, 3H), 2.83 (s, 2H),
2.59 (s, 1H),
2.43 (s, 2H), 2.23 (s, 3H), 2.17 (s, 3H).
Compound 49 and Compound 50
o¨\_0 0¨\_o
S or R
0 \--\\ R or S
N¨N -V=== N¨N
os"S
Ra or Sa Ra or Sa
N--
N
N 0 N 0
IIII
Compound 49 Compound 50
Both compounds are pure stereoisomers but absolute stereochemistry
undetermined
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 221 -
A cooled (0 C) solution of Compound 31 (150 mg, 0.2 mmol) in Me0H (2 mL) was
added to a cold (0 C) solution of sodium periodate (55 mg, 0.26 mmol, 1.3
eq.) in Me0H
(4 mL). The reaction mixture was stirred at room temperature overnight. The
solvent was
evaporated and the residue was dissolved in DCM and washed with water and
brine. The
organic layer was dried over MgSO4, filtered, and evaporated. The residue was
purified
by preparative HPLC (Stationary phase: RP )(Bridge Prep C18 OBD- 5 pm, 50 x
250
mm, Mobile phase: 0.25 % NH4HCO3 solution in water, CH3CN) to afford Compound
49 (41 mg, yield: 27 %) and Compound 50 (19 mg, yield: 13 %).
Compound 49
11-INMR (400 MHz, CDC13) 5 ppm 2.01 (s, 3 H); 2.03 (s, 1 H); 2.09 (s, 3 H);
2.33 (br s,
2 H); 2.83 (br d, J=12.75 Hz, 2 H); 2.86 (s, 3 H); 2.94 (br d, J=11.29 Hz, 2
H); 3.10 -
3.26 (m, 2 H); 3.31 (s, 3 H), 3.30 - 3.37 (m, 1 H); 3.42 - 3.48 (m, 2 H); 3.48
- 3.55 (m,
1 H); 3.55 - 3.62 (m, 1 H); 3.75 (d, J=13.69 Hz, 1 H); 3.86 -4.00 (m, 2 H);
4.06 (br d,
J=14.00 Hz, 1 H); 4.39 (dt, J=14.47, 4.00 Hz, 1 H); 4.46 - 4.63 (m, 2 H); 4.49
- 4.57
(m, 1 H); 5.12 - 5.25 (m, 1 H); 5.47 (s, 1 H); 5.83 (s, 1 H); 7.13 (d, J=8.99
Hz, 1 H);
7.20 (s, 1 H); 7.23 - 7.29 (m, 2 H); 7.30 (d, J-9.09 Hz, 1 H); 7.34 (dd, J-
9.98, 2.46 Hz,
1 H); 8.34 (dd, J=9.20, 5.75 Hz, 1 H).
Compound 50
11-INMR (400 MHz, CDC13, 51 'V) 6 ppm 2.00 (s, 3 H); 2.25 (s, 3 H); 2.32 (br
s, 2
H); 2.58 -2.84 (m, 4 H); 2.86 - 3.04 (m, 7 H); 3.12 (br d, J=5.33 Hz, 2 H);
3.31 (s, 4
H); 3.39 - 3.60 (m, 6 H); 3.86 - 3.97 (m, 2 H); 4.12 (d, J=14.74 Hz, 1 H);
4.27 - 4.38
(m, 1 H); 4.41 - 4.56 (m, 3 H); 5.14 (br d, J=14.63 Hz, 1 H); 5.53 (s, 2 H);
7.01 (d,
J=8.91 Hz, 1 H); 7.15 - 7.25 (m, 3 H); 7.32 (d, J=9.88 Hz, 1 H); 8.31 (dd,
J=9.14, 5.80
Hz, 1 H)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 222 -
Compound 51
HO-\
0\
N-N
)2 CI
N 0
Compound 51
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
Trimethylsilyl iodide (CAS [16029-98-4], 1 M in DCM, 025 mL, 0.25 mmol, 3 eq.)
was
added to a slurry of Compound 31(62 mg, 0.082 mmol) in ACN (4 mL) at 10 C.
The
resulting dark yellow solution was stirred at reflux for 1 h. The reaction
mixture was
cooled to 10 C, then treated with aqueous NaOH (1 M, 1 mL), and stirred at
room
temperature for 20 min. The solvents were evaporated and the residue was
dissolved in
water, cooled to 0 C, then treated with aqueous HC1 (1M, 1 mL). The aqueous
layer was
extracted with CHC13 (3 x). The combined organic layer was dried over MgSO4,
filtered,
and evaporated. The residue was purified by preparative HPLC (Stationary
phase: RP
)(Bridge Prep C18 OBD- 104m, 30x 150 mm, Mobile phase: 0.25 % NH4HCO3 solution
in water, CH3CN) to afford Compound 51(32 mg, yield: 52 %).
1E1 NMR (400 MHz, CDC13) ö ppm 1.96 (s, 3 H); 2.14 (s, 3 H); 2.28 -2.38 (m, 2
H);
2.72 - 2.85 (m, 2 H); 2.85 -2.96 (m, 3 H); 3.18 (d, J=13.38 Hz, 1 H); 3.22 (s,
3 H); 3.22
-3.30 (m, 1 H); 3.46 - 3.62 (m, 4 H); 3.49 -3.54 (m, 1 H); 3.66 -3.71 (m, 2
H); 3.92
(br t, J=4.96 Hz, 2 H); 4.19 (br s, 1 H); 4.28 (br t, J=4.86 Hz, 2 H); 4.43 -
4.52 (m, 1
H); 5.04 - 5.16 (m, 2 H); 5.07 - 5.09 (m, 1 H); 5.19 (s, 1 H); 5.61 (s, 1 H);
6.98 (d,
J=8.97 Hz, 1 H); 7.12 (s, 1 H); 7.20 (d, J=9.30 Hz, 1 H); 7.21 - 7.25 (m, 1
H); 7.32 (d,
J=9.84 Hz, 1 H); 8.30 (dd, J=9.14, 5.80 Hz, 1 H).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 223 -
Compound 52
HC\
N-N
N---
CI
N
0
o/
Compound 52
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
A solution of lithium hydroxide (0.71 mL, 1 M in water, 0.7 mmol, 10 eq.) was
added to
a suspension of Intermediate 147 (50 mg, 0.07 mmol) in Me0H/THF (2 mL/2 mL)
and
the resulting solution was heated at 50 C for 16 h. The solvents were
evaporated and the
residue was diluted with DCM (5 mL), treated with water (1 mL) and aqueous HC1
(1
M) until pH ¨ 1, and the layers were separated. The aqueous layer was
extracted with
DCM (3 x) and the combined organic layer was dried over MgSO4, filtered, and
evaporated. The residual oil was dissolved in DCM/Me0H (5 mL/5 mL) and then
slowly
evaporated to afford Compound 52 (45 mg, yield: 92 %) as a white solid.
111NMIR (400 MHz, CDC13) 5 ppm 2.00 - 2.20 (m, 6 H) 2.35 (br s, 2 H) 2.80 -
2.97 (m,
5 H) 3.11 (s, 3 H) 3.22 (d, J=12.75 Hz, 1 H) 3.29 - 3.55 (m, 4 H) 3.73 (q,
J=7.00 Hz, 1
H) 4.02 - 4.26 (m, 4 H) 4.48 - 4.65 (m, 1 I-1) 5.14 - 5.27 (m, 1 IT) 5.45 (d,
J=39.92 Hz, 2
H) 7.08 - 7.26 (m, 3 H) 7.28 - 7.48 (m, 3 H) 8.33 (dd, J=9.14, 5.80 Hz, 1 H)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 224 -
Compound 53
HO\
N¨N
I / CI OH
N 0
o/
Compound 53
Sa or It<i; pure atropisomer but absolute stereochemistry undetermined
Compound 53 was prepared according to an analogous procedure as for Compound
52,
starting from Intermediate 148 instead of Intermediate 147.
111NMR (400 MHz, CDC13) 5 ppm 2.00 -2.10 (m, 4 H) 2.16 (s, 3 H) 2.25 -2.42 (m,
3
H) 2.83 -2.94 (m, 5 H) 3.06 (s, 3 H) 3.18 - 3.26 (m, 1 H) 3.32 (br t, J=5.38
Hz, 2 H)
3.37 - 3.45 (m, 1 H) 3.47 - 3.59 (m, 1 H) 4.02 - 4.24 (m, 4 H) 4.50 - 4.59 (m,
1 H) 5.21
(ddd, J=14.79, 7.73, 4.13 Hz, 1 H) 5.45 (s, 2 H) 7.12 - 7.25 (m, 3 H) 7.27 -
7.34 (m, 2
H) 8.32 (dd, J=9.14, 5.80 Hz, 1 H)
Compound 54
N¨N/
S
/ CI OH
/N
)N 0
o
Compound 54
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 225 -
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
A solution of LiOH (61 mg, 2.556 mmol, 6 eq.) in water (5 mL) was added to a
solution
of Intermediate 162 (300 mg, 0.426 mmol) in THF (5 mL) and the mixture was
stirred at
40 C for 48 h. Most of the THF was removed under reduced pressure and the
mixture
was extracted with Et20 (5 mL x 3). The aqueous layer was acidified with
aqueous HC1
(2 M) to pH = 3. The solid that appeared was collected by filtration and was
triturated
with DCM/petroleum ether (lm L/10 mL). The solid was filtered to afford
Compound
54 (115 mg, yield: 36 %) as a white solid.
OR: +46 (589 nm, 24.7 C, 5 mg in 10 mL Me0H)
1H NMR (300 MHz, Methanol-d4) 6 (ppm) 8.06 - 8.09 (m, 1H), 7.01 - 7.45 (M,
3H),
7.00 (d, J = 9.0 Hz, 1H), 6.05 (s, 1H), 5.13 -5.19 (m, 1H), 4.88 (s, 1H), 4.60
- 4.67 (m,
1H), 3.81 -3.85 (m, 4H), 3.49 - 3.54 (m, 4H), 3.01 -3.01 (m, 5H), 2.70 -2.87
(m, 3H),
2 34 - 2 40 (m, 2H), 2.12 (s, 3H), 1.98 (s, 31-I)
19F NMR (300 MHz, Methanol-d4) 6 (ppm) -144.0, -152.0
Compound 55
N¨N/
S
N CI OH
/N
)N 0
o
Compound 55
Sa or Ra; pure atropisomer but absolute stereochemistry undetermined
Compound 55 was prepared according to an analogous procedure as for Compound
54,
starting from Intermediate 163 instead of Intermediate 162.
OR: -32 (589 nm, 24.7 C, 5 mg in 10 mL Me0H)
1H NEVER (300 MHz, Methanol-d4) 6 (ppm) 8.04 - 8.06 (m, 1H), 7.44 (d, J = 9.0
Hz,
1H), 7.29 - 7.36 (m, 1H), 7.27 (s, 1H), 7.00 (d, J = 9.0 Hz, 1H), 6.05 (s,
1H), 5.13 -
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 226 -
5.19(m, 1H), 4.88 (s, 1H), 4.60 - 4.67 (m, 1H), 3.81 - 3.85 (m, 4H), 3.49 -
3.54 (m,
4H), 3.01 -3.01 (m, 5H), 2.70 - 2.87 (m, 3H), 2.34 - 2.40 (m, 2H), 2.12(s,
3H), 1.98 (s,
3H).
19F NMR (300 MHz, Methanol-d4) 6 (ppm) -144.0, -151.9
Compound 56
NN/
S
CI
N
k
N 0
05)
CI
Compound 56
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
A solution of LiOH (50 mg, 2.08 mmol, 6 eq.) in water (5 mL) was added to a
solution
of Intermediate 176 (250 mg, 0.35 mmol) in THF (5 mL). The reaction mixture
was
stirred at 40 C for 16 h. Most of the solvent was removed under reduced
pressure. The
mixture was extracted with Et20 (5 mL x 3). The aqueous layer was acidified
with
aqueous HC1 (2 M) to pH = 3. The solid that appeared was collected by
filtration. The
crude product was triturated with DCM/petroleum ether (1 mL/10 mL) and
filtered to
afford Compound 56 (115 mg, yield: 36%. as a white solid.
OR: +32 (589 nm, 22.5 C, 5 mg in 10 mL Me0H)
lfl NMR (300 MHz, CDC13) 6 (ppm) 8.09 (d, J = 9.1 Hz, 1H), 7.30 - 7.45 (m,
2H),7.25
-7.29 (in, 1H), 7.12 (d, J- 9.3 Hz, 1H), 5.69 (s, 1H), 5.39 (s, 1H), 5.20 -
5.25 (iii, 1H),
4.52 - 4.56 (m, 1H), 3.91 (s, 3H), 3.70 (d, J = 14.8 Hz, 1H), 3.16-
3.44(m,7H), 2.88 -
2.91 (m, 5H), 2.22 - 2.33 (m, 5H), 2.07 (s, 3H)
19F NMR (300 MHz, CDC13) 6 (ppm) -124.39
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 227 -
Compound 57
NN/
S /
N CI
/N
)N o
CI
Compound 57
Sa or Ra; pure atropisomer but absolute stereochemistry undetermined
Compound 57 was prepared according to an analogous procedure as for Compound
56,
starting from Intermediate 177 instead of Intermediate 176.
OR: -38 (589 nm, 22.5 C, 5 mg in 10 mL Me0H)
1H NMR (300 MHz, CDC13) 6 ppm 8.09 (d, J = 9.0 Hz, 1H), 7.31 - 7.47 (m, 2H),
7.28 -
7.29 (s, 1II), 7.16 (d, J = 8.9 Hz, 1II), 5.65 (s, 1II), 5.45 (s, 111), 5.18 -
5.23 (m,
4.51 -4.59 (m, 1H), 3.90 (s, 3H), 3.71 (d, J = 14.8 Hz, 1H), 3.31 -3.45 (m,
3H), 3.19 -
3.20 (m, 4H), 2.91 - 2.94 (m, 5H), 2.32 (s, 2H), 2.23 (s, 3H), 2.07 (s, 3H)
19F NMR (300 MHz, CDC13) 6 ppm -124.42
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 228 -
Compound 58
o-\\_
N-N
CI OH
1 /
N 0
o?
CI
Compound 58
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
A solution of LiOH (23 mg, 0.96 mmol, 6 eq.) in water (3 mL) was added to a
solution
of Intermediate 183 (130 mg, 0.16 mmol) in THF (3 mL). The reaction mixture
was
stirred at 40 C for 48 h. Most of the THE was removed under reduced pressure.
The
mixture was extracted with Et20 (5 mL x 3). The aqueous layer was acidified
with
aqueous HC1 (2 M) to pH = 3. The solid formed was collected by filtration and
this crude
product was triturated with DCM/petroleum ether (1 mL/10 mL) and filtered to
afford
Compound 58 (56 mg, yield: 43 %) as a white solid.
NMR (300 MHz, CDC13) 6 ppm 8.07 (d, J = 9.0 Hz, 1H), 7.44 - 7.47 (m, 2H), 7.28
-
7.31 (m, 1H), 7.19 - 7.21 (m, 1H), 5.55 (d, J= 12.7 Hz, 2H), 5.20 - 5.28 (m,
1H), 4.55
(d, J = 14.9 Hz, 1H), 4.33 (t, J = 5.7 Hz, 2H), 3.78 - 4.01 (m, 3H), 3.47 -
3.63 (m, 5H),
3.19 - 3.41 (m, 6H), 3.09 (s, 3H), 2.91 -2.98 (m, 5H), 2.34 (s, 2H), 2.20 (s,
3H), 2.05
(s, 3H)
19F NMR (300 MHz, CDC13) 6 ppm -124.42
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 229 -
Compound 59
o/
N-5
s /
N CI
\
)N 0
0)
CI
Compound 59
Ra or Sa; pure atropisomer but absolute stereochemistry undetermined
Compound 59 was prepared according to an analogous procedure as for Compound
58,
starting from Intermediate 184 instead of Intermediate 183.
1H NM_R (300 MHz, CDC13) 6 ppm 8.08 (d, J = 9.1 Hz, 1H), 7.38 - 7.47 (m, 2H),
7.30
(s, 1H), 7.17 (d, J = 8.9 Hz, 1H), 5.49- 5.63(m, 2H), 5.19 - 5.26 (m, 1H),
4.51 -4.54
(m, 1H), 4.33 (t, J = 5.5 Hz, 2H), 3.90 - 3.92 (m, 2H), 3.70 - 3.75 (m, 1H),
3.46 -3.57
(m, 4H), 3.36 - 3.41 (m, 6H), 3.16 - 3.20 (m, 4H), 2.87 - 3.01 (m, 5H),2.31
(s, 2H), 2.23
(s, 3H), 210 (s, 3H).
19F NMR (300 MHz, CDC13) 6 ppm -124.42
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 230 -
Compound 60
o¨\\_
N¨N
CI OH
1 /
N 0
o?
CI
Compound 60
Sa or Ra; pure atropisomer but absolute stereochemistry undetermined
Compound 60 was prepared according to an analogous procedure as for Compound
58,
starting from Intermediate 185 instead of Intermediate 183.
1H NM_R (300 MHz, CDC13) 6 ppm 8.07 (d, J = 9.0 Hz, 1H), 7.44 - 7.47 (m, 2H),
7.28 -
7.31(m, 1H), 7.19 - 7.21 (m, 1H), 5.55 (d, J = 12.7 Hz, 2H), 5.20 - 5.28 (m,
1H),4.55
(d, J = 14.9 Hz, 1H), 4.33 (t, J = 5.7 Hz, 2H), 3.78 - 4.01 (m, 3H), 3.47 -
3.63 (m, 5H),
3.19 - 3.41 (m, 6H), 3.09 (s, 3H), 2.91 -2.98 (m, 5H), 2.34 (s, 2H), 2.20 (s,
3H), 2.05
(s, 3H).
19F NMR (300 MHz, CDC13) 6 ppm -124.38
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 231 -
Compound 61
o/
N-5
s /
N CI
\
)N 0
0)
CI
Compound 61
Sa or Ra; pure atropisomer but absolute stereochemistry undetermined
Compound 61 was prepared according to an analogous procedure as for Compound
58,
starting from Intermediate 186 instead of Intermediate 183.
1H NM_R (300 MHz, CDC13) 6 ppm 8.08 (d, J = 9.1 Hz, 1H), 7.38 - 7.47 (m, 2H),
7.30
(s, 1H), 7.17 (d, J = 8.9 Hz, 1H), 5.49- 5.63(m, 2H), 5.19 - 5.26 (m, 1H),
4.51 -4.54
(m, 1H), 4.33 (t, J = 5.5 Hz, 2H), 3.90 - 3.92 (m, 2H), 3.70 - 3.75 (m, 1H),
3.49 -3.54
(m, 2H), 3.47 - 3.49 (m, 2H), 3.36 - 3.41 (m, 6H), 3.16 - 3.20 (m, 4H), 2.87 -
3.01 (m,
5H),2.3 I (s, 2H), 2.23 (s, 3H), 2.10 (s, 3H).
19F NMR (300 MHz, CDC13) 6 ppm -124.42
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 232 -
Compound 62
o¨\\_
N-N
N CI OH
/N
N 0
Compound 62: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
A solution of Li OH (18 mg, 0.76 mmol, 6 eq.) in water (4 mL) was added to a
solution
of Intermediate 192 (100 mg, 0.13 mmol) in THF (4 mL). The reaction mixture
was
stirred at 40 C for 16 h. Most of the THF was removed under reduced pressure.
The
mixture was extracted with Et20 (5 mL x 3). The aqueous layer was acidified
with
aqueous HC1 (2 M) to pH = 3. The solid formed was collected by filtration and
this
crude product was triturated with Et0Ac/petroleum ether (1 mL/10 mL) and
filtered to
afford Compound 62 (47 mg, yield: 47 %) as an off-white solid.
1LINMR (300 MHz, CDC13) 6 ppm 8.10 (d, J = 6 Hz, 1H), 7.48 (s, 1H), 7.35 (d, J
= 15
Hz, 2H), 7.23 (d, J = 9 Hz, 1H), 5.57 ¨ 5.49 (m, 2H), 5.26¨ 5.18 (m, 1H), 4.54
(d, J =
Hz, 1H), 4.33 (s, 2H), 3.95 ¨ 3.83 (m, 3H), 3.68 ¨ 3.42 (m, 5H), 3.36 ¨ 3.17
(m,
6H), 3.05 (s, 3H), 2.92 (d, J = 12 Hz, 5H), 2.34 (s, 2H), 2.20 (s, 3H), 2.08
(s, 3H)
15 19F NMR (282 MHz, CDC13) 6 ppm -140.708--140.775,-149.626--149.693.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 233 -
Compound 63
o/
(o
N¨N
S
CI OH
zN
N 0
o)
Compound 63: Ita or Sa, pure atropisomer but absolute stereochemistry
undetermined
Compound 63 was prepared according to an analogous procedure as for Compound
62,
starting from Intermediate 193 instead of Intermediate 192.
1H NM_R (300 MHz, CDC13) 6 ppm 8.11 (s, 1H), 7.49 (s, 1H), 7.33 (d, J = 9 Hz,
2H),
7.21 (d, J = 9 Hz, 1H), 5.55 (s, 2H), 5.24 (s, 1H), 4.57 ¨4.37 (s, 3H), 3.93
(s, 2H), 3.77
(d, J = 12 Hz, 1H), 3.57 ¨ 3.49 (d, J = 15, 4H), 3.36 (s, 6H), 3.15 ¨2.93 (m,
9H), 2.34 ¨
2.12 (m, 8H)
19F NMR. (282 MHz, CDC13) 6 ppm -140.739 ¨ -140.806, -149.580 ¨ -149.648.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 234 -
Compound 64
o¨\\_
o\
N¨N
CI OH
/N
)N 0
o
Compound 64: S. or R., pure atropisomer but absolute stereochemistry
undetermined
Compound 64 was prepared according to an analogous procedure as for Compound
62,
starting from Intermediate 194 instead of Intermediate 192.
NMR (300 MHz, CDC13) 6 ppm 8.10 (d, J = 9 Hz, 1H), 7.49 (s, 1H), 7.43 ¨ 7.28
(m,
2H), 7.28 (d, J = 6, 1H), 5.60 ¨ 5.47 (d, J = 12, 2H), 5.22 (m, 1H), 4.54 (d,
J = 15 Hz,
1H), 4.35 (s, 2H), 3.96 ¨3.87 (m, 3H), 3.61 ¨3.23 (m, 11H), 3.02 ¨2.93 (m,
8H), 2.34
(s, 2H), 2.20 ¨209 (d, J = 18, 6H)
19F NMR (282 MHz, CDC13) 6 ppm -140.711 --140.779,-149.618--149.686.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 235 -
Compound 65
(o
N¨N
S
CI OH
zN
N 0
o)
Compound 65: Sa or Ra, pure atropisomer but absolute stereochemistry
undetermined
Compound 65 was prepared according to an analogous procedure as for Compound
62,
starting from Intermediate 195 instead of Intermediate 192.
1H NMR (300 MHz, CDC13) 6 ppm 8.10 (m, 1H), 7.49 (s, 1H), 7.33 (d, J = 9 Hz,
2H),
7.24 (d, J = 9 Hz, 1H), 5.61 ¨ 5.50 (d, J = 15, 2H), 5.25 (m, 1H), 4.53 ¨4.35
(m, 3H),
3.91 ¨3.78 (m, 3H), 3.56 ¨ 3.49 (m, 4H), 3.36 ¨3.20 (m, 6H), 3.19 ¨2.88 (m,
9H), 2.32
(s, 2H), 2.22 (s, 3H), 2.12 (s, 3H)
19F NMR (282 MHz, CDC13) 6 ppm -140.797 ¨ -140.863, -149.672 ¨ -149.739.
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 236 -
Compound 66
o¨\\_
N¨N
0
N CI OH
N 0
o
Compound 66: mixture of atropisomers
A solution of LiOH (4 mg, 0.13 mmol, 10 eq.) in water (0.5 mL) was added to a
solution of Intermediate 199 (10 mg, 0.013 mmol) in THE (0.5 mL). The reaction
mixture was stirred at 40 'V for 3 days. Most of the THF was removed under
reduced
pressure. The aqueous layer was acidified with aqueous HC1 (2 M) to pH = 3.
The solid
that appeared was collected by filtration to afford Compound 66 (3 mg, yield:
29 %) as
a white solid.
IHNMR (300 MHz, Methanol-d4)6 ppm 8.17 (m, 1H), 7.58 (d, J = 6 Hz, 1H), 7.28 ¨
7.10 (m, 3H), 6.82 (s, 1H), 6.27 (s, 1H), 5.22 (m, 1H), 4.67 (m, 1H), 4.35 (m,
1H), 4.27
(m, 1H), 4.05 (m, 3H), 3.99 (m, 3H), 3.77 (m, 1H), 3.69 (m, 4H), 3.65 (s, 2H),
3.66 ¨
3.55 (m, 2H), 3.35 (s, 3H), 3.14 ¨ 2.94 (m, 2H), 2.86 (d, J = 9 Hz, 2H), 2.44
(s, 2H),
2.04 (m, 1H), 1.96 (d, J = 6 Hz, 6H)
19F NMR (282 MHz, Methanol-d4) 6 ppm -117.23.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 237 -
Compound 67
oo
N¨N
N
I /
N o
o/
Compound 67: Ra or Sa, pure atropisomer but absolute stereochemistry
undetermined
mCPBA (31 mg, 0.177 mmol, 2.2 eq.) was added in one portion to a solution of
Compound 31(61 mg, 0.080 mmol) in DCM (10 mL) at room temperature. The
reaction mixture was stirred for 5 h at room temperature. Water was added to
the
reaction mixture and the layers were separated. The combined organic layer was
dried
by filtration on Extrelut NT3, and evaporated. The residue was purified by
column
chromatography (Biotage Sfar 10 g; eluent: DCM/Me0H 100:0 -> 90:10) to give
Compound 67 (35 mg, yield: 55 %) as a white solid.
NMR (400 MHz, CDC13) 6 ppm 2.01 (s, 3 H) 2.25 (s, 3 H) 2.35 (br s, 2 H) 2.67 -
2.77(m, 5 H) 2.86 - 3.06 (m, 3 H) 3.30 (s, 3 H) 3.41 - 3.51 (m, 5 H) 3.56(t,
1=4.8 Hz, 1
H) 3.57 - 3.64 (m, 1 H) 3.88 - 3.96 (m, 2 H) 4.35 - 4.46 (m, 2 H) 4.55 - 4.68
(m, 2 H)
5.01 - 5.16(m, 2 H) 5.34 (s, 1 H) 5.89 (s, 1 H) 7.12 (d, J=9.0 Hz, 1 H)
7.22(s, 1 H)
7.27 - 7.33 (m, 2 H) 7.36 (dd, J=9.9, 2.4 Hz, 1 H) 8.37 (dd, J=9.1, 5.6 Hz, 1
H)
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 238 -
Compound 68
NH
N-N
S /
Ra or Sa
CI OH
N 0
0
Pure stereoisomer but absolute stereochemistry undetermined
LiOH (13 mg, 0.54 mmol, 20 eq.) was added to a solution of Intermediate 203
(20.6 mg,
0.027 mmol) in a mixture of Me0H (0.7 mL), TT-IF (0.7 mL), and water (0.4 mL).
The
reaction mixture was stirred for 4 h at 50 C. The solvents were evaporated
and the
residue was purified by preparative HPLC (stationary phase: RP )(Bridge Prep
C18
OBD- 5 pm, 50 x 250 mm, Mobile phase: 0.25 % NH4HCO3 solution in water, CH3CN)
to give Compound 68 (14 mg, yield: 73 %) as a pale yellow solid.
1HNMIR (400 MHz, DMSO-d6) 6 ppm 1.86 (br s, 3 H), 1.95 (s, 3 H), 2.23 -2.31
(m, 2
H), 2.42 - 2.46 (m, 3 H), 2.75 - 2.93 (m, 4 H), 3.03 (br d, J=13.7 Hz, 6 H),
3.45 (s, 3 H),
3.54 (br d, J=8.8 Hz, 2 H), 3.74 (br s, I H), 4.17 - 4.49 (m, 3 H), 4.99 (s, 1
H), 5.10 (br
s, 1 H), 6.20 (s, 1 H), 6.93 (d, J=8.6 Hz, 1 H), 7.20 (s, 1 H), 7.31 (td,
J=8.9, 2.8 Hz, 1
H), 7.39 (d, J=8.9 Hz, 1 H), 7.51 (dd, J=10.5, 2.6 Hz, 1 H), 8.22 (dd, J=9.2,
6.1 Hz, 1
H).
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 239 -
Compound 69
HN
N-N
Ra or Sa
N
/ CI OH
N 0
tdif
Pure stereoisomer but absolute stereochemistry undetermined
Compound 69 was prepared according to the same procedure as for Compound 68,
starting from Intermediate 202 instead of Intermediate 203.
11-1NMR (400 MHz, DMSO-d6) 6 ppm 1.82 (s, 3 H), 1.86 - 1.93 (m, 3 H), 2.28 (br
s, 2
H), 2.43 - 2.47 (m, 3 H), 2.69 - 2.86 (m, 3 H), 2.89 (d, J=13.9 Hz, 1 H), 2.96
- 3.02 (m,
2 H), 3.03 -3.13 (m, 4 H), 3.44 - 3.47 (m, 3 H), 3.49 - 3.54 (m, 2 H), 3.76 -
3.83 (m, 1
H), 4.13 -4.29 (m, 2 H), 4.41 -4.53 (in, 1 H), 4.87 (s, 1 H), 5.06 (br d,
J=14.6 Hz, 1 H),
6.13 (s, 1 H), 6.83 (d, J=8.8 Hz, 1 H), 7.16 (s, 1 H), 7.29 -7.37 (m, 2 H),
7.50 (dd,
J=10.4, 2.6 Hz, 1 H), 8.29 (dd, J=9.1, 5.8 Hz, 1 H).
LCMS results (RT means retention time)
Compound LCMS results
number
1 confirms the MW (RT: 1.75, [M+H]+ 654, LCMS Method
4)
2 confirms the MW (RT: 1.75, [M+H]+ 654, LCMS Method
4)
3 confirms the MW (RT: 1.82, [M+H]+ 672, LCMS Method
2)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 240 -
Compound LCMS results
number
4 confirms the MW (RT: 1.82, [M+H]+ 672, LCMS Method
2)
confirms the MW (RT: 0.90, [M+H]+ 672, LCMS Method 3)
6 confirms the MW (RT: 1.73, [M+H]+ 686, LCMS Method
4)
7 confirms the MW (RT: 1.73, [M+H]+ 686, LCMS Method
4)
8 confirms the MW (RT: 1.70, [M+H]+ 669, LCMS Method
1)
9 confirms the MW (RT: 1.70, [M+H]+ 669, LCMS Method
1)
confirms the MW (RT: 0.93, [M+H]+ 672, LCMS Method 3)
11 confirms the MW (RT: 0.93, [M+H]+ 672, LCMS Method
3)
12 confirms the MW (RT. 1.69, [M+H]+ 669, LCMS Method
5)
13 confirms the MW (RT: 1.70, [M+H]+ 629, LCMS Method
1)
14 confirms the MW (RT: 1.88, [M+H]+ 704, LCMS Method:
2)
confirms the MW (RT: 1.87, [M+H]+ 704, LCMS Method: 2)
16 confirms the MW (RT: 1.03, [M+H]+ 804, LCMS Method
6)
17 confirms the MW (RT: 0.98, [M+H]+ 804, LCMS Method
6)
18 confirms the MW (RT: 0_93, [M+H]+ 716, LCMS Method
3)
19 confirms the MW (RT: 0.93, [M+H]+ 716, LCMS Method
3)
confirms the MW (RT: 0.95, [MI I 760, LCMS Method 3)
21 confirms the MW (RT: 0.96, [M+H]+ 760, LCMS Method
3)
22 confirms the MW (RT: 0.97, [M+H]+ 770, LCMS Method
3)
23 confirms the MW (RT: 0.97, [M+H]+ 770, LCMS Method
3)
28 confirms the MW (RT: 0.92, [M+H]+ 672, LCMS Method
3)
29 confirms the MW (RT: 0.88, [M+H]+ 658, LCMS Method
3)
confirms the MW (RT: 0.88, [M+H]+ 658, LCMS Method 3)
31 confirms the MW (RT: 0.93, [M+H]+ 760, LCMS Method
3)
32 confirms thc MW (RT: 0.93, [M+H]+ 760, LCMS Method
3)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 241 -
Compound LCMS results
number
33 confirms the MW (RT: 1.75, [M+H]+ 742, LCMS Method
8)
34 confirms the MW (RT: 1.75, [M+H]+ 742, LCMS Method
8)
35 confirms the MW (RT: 1.35, [M+H]+ 688, LCMS Method
9)
36 confirms the MW (RT: 1.37, [M+H]+ 688, LCMS Method
9)
37 confirms the MW (RT: 0.96, [M+H]+ 715, LCMS Method
7)
38 confirms the MW (RT: 0.96, [M+H]+ 715, LCMS Method
7)
39 confirms the MW (RT: 1.56, [M+H]+ 776, LCMS Method
10
40 confirms the MW (RT: 2.90, [M+H]+ 776, LCMS Method
11
41 confirms the MW (RT. 2.96, [M+H]+ 732, LCMS Method
12
42 confirms the MW (RT: 3.13, [M+H]+ 732, LCMS Method
12
43 confirms the MW (RT: 2.84, [M+H]+ 776, LCMS Method
11
44 confirms the MW (RT: 1.59, [M+H]+ 776, LCMS Method
10
45 confirms the MW (RT: 1.62, [M+H]+ 656, LCMS Method
13
46 confirms the MW (RT: 1.62, [M+H]+ 656, LCMS Method
13
47 confirms the MW (RT: 2_79, [M+H]+ 732, LCMS Method
11
48 confirms the MW (RT: 2.93, [M+H]+ 732, LCMS Method
11
49 confirms the MW (RT: 1.70, [MI I 776, LCMS Method 4
50 confirms the MW (RT: 1.71, [M+H]+ 776, LCMS Method 4
51 confirms the MW (RT: 1.68, [M+H]+ 746, LCMS Method 4
52 confirms the MW (RT: 2.34, [M+H]+ 702, LCMS Method
14
53 confirms the MW (RT: 2.33, [M+H]+ 702, LCMS Method
14
54 confirms the MW (RT: 2.74, [M+H]+ 690, LCMS Method
15
55 confirms the MW (RT: 1.51, [M+H]+ 690, LCMS Method
10
56 confirms the MW (RT: 2.92, [M+H]+ 706, LCMS Method
16
57 confirms the MW (RT: 1.59, [M+H]+ 706, LCMS Method
10
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 242 -
Compound LCMS results
number
58 confirms the MW (RT: 2.64, [M+H]+ 706, LCMS Method
17
59 confirms the MW (RT: 2.97, [M+H]+ 794, LCMS Method
11
60 confirms the MW (RT: 2.89, [M+H]+ 794, LCMS Method
11
61 confirms the MW (RT: 1.61, [M+H]+ 794, LCMS Method
10
62 confirms the MW (RT: 1.51, [M+H]+ 778, LCMS Method
10
63 confirms the MW (RT: 1.58, [M+H]+ 778, LCMS Method
10
64 confirms the MW (RT: 1.51, [M+H]+ 778, LCMS Method
10
65 confirms the MW (RT: 1.53, [M+H]+ 778, LCMS Method
10
66 confirms the MW (RT. 1.53, [M+H]+ 778, LCMS Method
18
67 confirms the MW (RT: 0.99, [M+H]+ 792, LCMS Method 3
68 confirms the MW (RT: 1.69, [M+H]+ 715, LCMS Method
19)
69 confirms the MW (RT: 0.90, [M+H]+ 715, LCMS Method
3)
Table: Analytical SFC data ¨ Rt means retention time (in minutes), [M+H]+
means the
protonated mass of the compound, method refers to the method used for (SFC)MS
analysis of enantiomerically pure compounds. No. means number.
Compound
SFC Method Rt 1M+111+
No.
1 1 4.32 654
2 1 3.93 654
3 2 4.48 672
4 2 4.87 672
6 3 7.94 686
7 3 7.26 686
8 4 6.59 669
9 4 7.26 669
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 243 -
Cornpound
SFC Method Rt 1M+H1+
No.
5 4.35 672
11 6 3.57 672
14 7 7.88 704
7 6.94 704
18 6 3.49 716
19 2 4.47 716
9 3.94 760
21 9 3.96 760
22 10 4.39 770
23 10 4.90 770
28 5 4.07 672
29 6 3.63 658
6 4.06 658
31 10 3.99 760
32 10 4.46 760
12 1.45 688
36 12 1.36 688
39 13 1.70 776
13 1.73 776
41 13 1.73 732
42 13 1.76 732
43 14 1.97 776
44 14 2.00 776
15 1.93 656
46 16 2.11 656
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 244 -
Cornpound
SFC Method Rt 1M+1-11+
No.
47 13 1.82 732
48 13 1.78 732
52 17 4.94 702
53 17 4.44 702
54 15 1.91 690
55 18 1.84 690
56 18 2.08 706
57 15 2.00 706
58 14 1.80 794
59 20 4.15 794
60 14 1.86 794
61 20 5.51 794
62 19 0.93 778
63 21 1.61 778
64 19 1.29 778
65 21 1.61 778
68 7 6.51 715
69 7 6.50 715
Analytical Analysis
The High Performance Liquid Chromatography (HPLC) measurement was performed
using a LC pump, a diode-array (DAD) or a UV detector and a column as
specified in
the respective methods. If necessary, additional detectors were included (see
table of
methods below).
Flow from the column was brought to the Mass Spectrometer (MS) which was
configured with an atmospheric pressure ion source. It is within the knowledge
of the
skilled person to set the tune parameters (e.g. scanning range, dwell time...)
in order to
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 245 -
obtain ions allowing the identification of the compound's nominal monoisotopic
molecular weight (MW). Data acquisition was performed with appropriate
software.
Compounds are described by their experimental retention times (Re) and ions.
If not
specified differently in the table of data, the reported molecular ion
corresponds to the
[M+H] (protonated molecule) and/or [M-H] (deprotonated molecule). in case the
compound was not directly ionizable the type of adduct is specified (i.e.
[M+NHa],
[M+HCOO], etc...). For molecules with multiple isotopic patterns (Br, Cl), the
reported
value is the one obtained for the lowest isotope mass. All results were
obtained with
experimental uncertainties that are commonly associated with the method used.
Hereinafter, "SQD" means Single Quadrupole Detector, "MSD" Mass Selective
Detector, "RT" room temperature, "BEH" bridged ethylsiloxane/silica hybrid,
"DAD"
Diode Array Detector, "HSS" High Strength silica.
LCMS Method Codes (Flow expressed in mL/min; column temperature (T) in C; Run
time in minutes)
LC-MS methods:
CA 03168355 2022-8- 17
WO 2021/165370
PCT/EP2021/053973
- 246 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
From
100 %
A to 5
% A in
Waters:
Waters: A: 10 mM 2.1
Acquity 0.6
EB H (1.8 CH3COONH4 in 95 % min, to
1 UPLC -
3.5
am, 2.1*100 H20 + 5 % CH3CN 0 % A
DAD and 55
mm) B: CH3CN in 0.9
SQD
min, to
%A
in 0.5
min
From
100 %
A to
5 %A
Waters:
Waters A: 10 mM in 2.1
Acquity 0.7
:BEH CH3COONH4 in 95 % min,
2 UPLC -
3.5
(1.8am, H20 + 5 % CH3CN to 0 %
DAD and 55
2. 1*100mm) B: CH3CN A in 0.9
SQD
min,
to 5 %
A in 0.5
min
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 247 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
From
95 % A
Waters:
,) Waters : A: 10 mM to 5 %
Acquity 0.8
BEH C18 CH3COONH4 in 95 % A in 1.3
3 UPLC -
2
(1.7um, H20 + 5% CH3CN min,
DAD and 55
2.1*50mm) B: CH3CN held for
SQD
0.7
min.
From
100 %
A to
5% A
Waters:
Waters A: 10 mM in 2.10
Acquity- 0.7
:BEH CH3COONH4 in 95 % min, to
4 UPLC -
3.5
(1.8um, H20 + 5 % CH3CN 0 % A
DAD and 55
2.1*100mm) B: CH3CN in 0.9
SQD
min, to
%A
in 0.5
min
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 248 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
From
100 %
A to
%A
Waters:
Waters A: 0.1 % NH4HCO3 in 2.10
Acquity 0.6
:BEH in 95 % H20 + 5 % min,
5 UPLC -
4.5
(1.81.Lm, CH3CN to 0 %
DAD and 55
2.1*100mm) B: CH3CN A in 0.9
SQD
min,
to 5 %
A in 0.5
min
From
100 %
Waters:
Waters A: 0.1% NH4HCO3 hi A to 5
Acquity 0.8
:BEH (1.8 95 % H20 + 5% % A in
6 UPLC -
2.0
2.1*50 CH3CN 1.3
DAD and 55
mm) B: CH3CN min,
SQD
hold 0.7
min
From
100 %
Waters:
Waters A: 0.1 % NH4HCO3 in A to 5
Acquity 0.8
:BEH (1.8 95 % H20 + 5 % % A in
7 UPLC -
2.0
im,2.1*50 CH3CN 1.3
DAD and 55
mm) B: CH3CN min,
SQD2
hold
0.7min
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 249 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
From
100 %
Waters: A to 5
, Waters A: 0.1 % NH4HCO3 in
Acquity % A in 0.6
:BEH (1.8 95% 1120 + 5%
8 UPLC - 2.10
3.5
m, 2.1*100 CH3CN
DAD and min, to 55
mm) B: CH3CN
SQD2 0 % A
in 1.4
min
10% A
to 95 %
A in 2.0
min,
hold 0.7
Kinetex min at
Shimadzu EVO C18 A. Water - 5 mM 95 % 1.2
9 LCMS- 100A, 3.0 * NI-14HCO3 A, 95
2.85
2020 50 mm, 2.6 B: CH3CN % A to 40
urn 10 % A
in 0.05
min,
hold 0.1
min at
10% A
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 250 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
% A
to 100
HALO 90 A % A in
Shimadzu 1.5
C18, 3.0 x A: Water/0.05 %TFA 2 min,
LCMS-
3
30 mm, 2.0 B: ACN/0.05 %TFA hold 0.7
2020 40
urn min at
100 %
A
5 % A
to 70 %
A in 3
min, 70
HALO 90 A % A to
Shimadzu 1.5
C18, 3.0 x A: Water/0.05 %TFA 95 % A
11 LCMS-
4
30 mm, 2.0 B. ACN/0.05 %TFA in 0.3
2020 40
urn min,
hold
0.45
min at
95 % A
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
-251 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
% A
to 65 %
A in 3
min, 65
HALO 90 A % A to
Shimadzu 1.5
C18, 3.0 x A: Water/0.05 %TFA 95 % A
12 LCMS-
4
30 mm, 2.0 B: ACN/0.05 %TFA in 0.3
2020 40
um min,
hold
0.45
min at
95 % A
5 % A
to 100
Ascentis % A in
Shimadzu Express 2 min,
1.5
A: Water/0.05 %TFA
13 LCMS- C18, 3.0x hold
3
B: ACN/0.05 %TFA
2020 50 mm, 2.7 0.70 40
urn min at
100 %
A
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 252 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
From
100 %
A to 5
% A in
Waters:
Waters A: 10mM NFLEIC03 2.10
Acquity 0.6
:BEH 1.8 in 95 % H20 + 5 % min,
14 UPLC -
3.5
pm, 2.1 x CH3CN to 0 %
DAD and 55
100 mm) B: Me0H A in 0.9
SQD
min,
to 5 %
A in 0.5
min
10% to
50% in
3.0
min,
HALO 90 A A: Water/6.5 mM 50 % to
Shimadzu 1.2
C18, 3.0x NH4HCO3 -F NH4OH 95 % in
15 LCMS-
4
30 mm, 2.0 (pH = 10) 0.3
2020 40
urn B: ACN min,
hold
0.45
min at
95 %
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 253 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
% A
to 70 %
A in 3
min, 70
% A to
HALO 90 A
Shimadzu 95 % A 1.5
C18, 3.0 x A: Water/0.05 %TFA
16 LCMS- in 0.3
4
30 mm, 2.0 B: ACN/0.05 %TFA .
2020 min, 40
Urn
hold
0.45
min at
100 %
A
30% A
to 70 %
A in 3
min, 70
% A to
HALO 90 A
Shimadzu 100% 1.5
C18, 3.0 x A: Water/0.05 %TFA
17 LCMS- A in 0.3
4
30 mm, 2.0 B: ACN/0.05 %TFA
2020 min, 40
urn
hold
0.45
min at
100 %
A
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 254 -
Flow
Method
Run
Instrument column mobile phase gradient
Code
Col time
30% A
to 70%
Amn 1.7
min, 70
HALO 90 A
Shimadzu % A to 1.5
C18, 3.0 x A: Water/0.05 %TFA
18 LCMS- 95 % A
3
30 mm, 2.0 B: ACN/0.05 %TFA
2020 in 0.6 40
urn
min,
hold 0.5
min at
95 % A
From
100 %
A to 5
% A in
Waters: A: 10 mM
Waters 2.10
Acquity - CH3COONH4 0.6
:BEH mm n
19 UPLC - in , 95 %
H20 + 5 % 3.5
(1.8 um, 2.1 to 0 %
DAD and CH3CN 55
* 100 mm) A in 0.9
SQD B: CH3CN
min,
to 5 %
A in 0.5
min
SFC-MS methods:
The SFC measurement was performed using an Analytical Supercritical fluid
chromatography (SFC) system composed by a binary pump for delivering carbon
dioxide
(CO2) and modifier, an autosampler, a column oven, a diode array detector
equipped with
a high-pressure flow cell standing up to 400 bars. If configured with a Mass
Spectrometer
(MS) the flow from the column was brought to the (MS). It is within the
knowledge of
the skilled person to set the tune parameters (e.g. scanning range, dwell
time...) in order
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 255 -
to obtain ions allowing the identification of the compound's nominal
monoisotopic
molecular weight (MW). Data acquisition was performed with appropriate
software.Analytical SFC-MS Methods (Flow expressed in mL/min; column
temperature
(Col I) in C; Run time in minutes, Backpressure (BPR) in bars.
"iPrN1-L2- means isopropylamine, "iPrOH" means 2-propanol, "Et0H" means
ethanol,
"min" mean minutes, "DEA" means diethylamine.
SFC methods:
Run
Flow
SFC
time
Column mobile phase gradient
Method
Col T
BPR
Daicel
5% B hold 6
Chiralpak AS3 A:CO2 2.5
9.5
min, to 50% in 1
1 column (3.0 B: Et0H + 0.2 %
min hold 2.5
urn, 150 x4.6 iPrNH2 40
130
min
mm)
Daicel
5 B hold 6
Chiralpak A:CO2 2.5
9.5
min, to 50 % in
2 AD3 column B: Et0H + 0.2 %
1 min hold 2.5
(3.0 um, 150 x iPrNH2 40
130
min
4.6 mm)
3 Daicel
5 c1/0 B hold 6
Chiralpak ID3 A:CO2 2.5
9.5
min, to 50 % in
column (3.0 B: Et0H + 0.2 %
1 min hold 2.5
um, 150 x4.6 iPrNH2 40
130
min
mm)
4 Daicel
5 B hold 6
Chiralpak IG3 A:CO2 2.5
9.5
min, to 50 % in
column (3.0 B: iPrOH + 0.2 %
1 min hold 2.5
um, 150 x4.6 iPrNH2 40
130
min
mm)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 256 -
Run
Flow
SFC
time
Column mobile phase gradient
Method
Col T
BPR
Daicel
Chiralpak A:CO2 10 % - 50 % B 2.5 9.5
AD3 column B: Et0H + 0.2 % in 6 min, hold
(3.0 tun, 150 x iPrNH2 3.5 min 40 130
4.6 mm)
6 Daicel
Chiralpak 0J3 A:CO2 10 % - 50 % B 2.5 9.5
column (3.0 B: Et0H + 0.2% in 6 min, hold
nm, 150 x 4.6 iPrNH2 3.5 min 40 130
mm)
7 Daicel
Chiralpak IG3 A:CO2 10%-50% B in 6 2.5 9.5
column (3.0 B: Et0H+0.2% min, hold 3.5
um, 150 x 4.6 iPrNH2 min 40 130
mm)
Daicel
Chiralpak AS3 A:CO2 10 % - 50 % B 2.5
9.5
9 column (3.0 B: Et0H + 0.2% in
6 min, hold
nin, 150 x 4.6 iPrN1-T2 3.5 min 40 130
mm)
Daicel
Chiralpak A:CO2 10 % - 50 % B 2.5 9.5
AD3 column B: Et0H + 0.2% in 6 min, hold
(3.0 tun, 150 x iPrNH2 3.5 min 40 130
4.6 mm)
Daicel
Chiralpak IC3 A:CO2 10 % - 50 % B 2.5 9.5
11 column (3.0 B: Et0H + 0.2% in
6 min, hold
um, 150 x 4.6 iPrNH2 3.5 min 40 130
mm)
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 257 -
Run
Flow
SFC
time
Column mobile phase gradient
Method
Col T
BPR
CHIRALPAK A:CO2 10 % to 50 % in 2
3
12 AS, 3.0 x 50 B: Me0H+ 0.1% 2 min, hold 1
mm, 3 um Et2NH min at 50 % 35
100
CHIRALPAK A: CO2 10 % to 50 % in 2
3
13 0J-3, 4.6 x 50 B: Me0H (0.1 % 2.0 min, hold 1
mm, 3 um DEA) min at 50 % 35
103
CHIRALPAK A: CO2 10 % to 50 % in 2
3
14 IA-3 3.0 x 50 B: Me0H (1 % 2 2.0 min, hold
mm 3 um M NIL in Me0II) 1.0 min at 50% 35
100
CHIRALPAK A: CO2 10 % to 50 % in 4
3
15 IB N-3, 4.6x B: Me0H (0.1 % 2.0 min, hold
100 mm, 3 um DEA) 1.0 min at 50% 35
103
CHIRALPAK A: CO2 10 % to 50 % in 2
3
16 OD, 3.0 x 100 B: Me0H (0.1 % 2.0 min, hold
mm, 3 urn DEA) 1.0 min at 50 % 35
103
Daicel
ArCO2 10 % - 50 % B 2.5
9.5
Chiralpak IH3
17 B: Et0H + 0.2 % in 6 min, hold
column (3.0 p.m,
iPrNH2 3.5 min 40
130
150 x 4 6 mm)
(S,S) Whelk-01, A:CO2 10 % - 50 % B 4
3
18 4.6 * 100 mm, 5 B: Me0H + 0.1% in 2 min, hold 1
pm Et2NH min at 50% 35
103
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 258 -
Run
Flow
SFC
time
Column mobile phase gradient
Method
Col T
BPR
CHIRALPAK 4
3
Me0H (1 % 2 M 30 % to 30 % in
19 IG-3 4.6 * 50
NH3 in Me0H) 3.0 min
mm 3 um 35
100
CHIRALPAK MeOH:ACN:DCM 2
7
30% to 30% in
20 IC 3.0 x 100 = 1:1:1 (0.1 %
7.0 min
mm, 3 um DEA) 35
103
Lux 3u 10 % to 50 % B 4
3
Me0H (1 % 2M
21 Cellulose-3 4.6 in 2.0 min, hold
N113 in Me0H)
* 100 mm 3 um 1.0 min at 50 % 35
100
NMR
1H NMR and 19F NMR spectra were recorded on Bruker Avance III 400M1-1z and
Avance
NEO 400MHz spectrometers. CDC13 was used as solvent, unless otherwise
mentioned.
The chemical shifts are expressed in ppm relative to tetramethylsilane.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 259 -
Pharmacological Analysis
Biological Example 1
Terbium labeled Myeloid Cell Leukemia l(Mc1-1) homogeneous time-resolved
fluorescence (HTRF) binding assay utilizing the BIM BH3 peptide (H2N-
(C/Cy5Mal)
WIAQELRRIGDEFN-OH) as the binding partner for Mc1-1.
Apoptosis, or programmed cell death, ensures normal tissue homeostasis, and
its
dysregulation can lead to several human pathologies, including cancer. Whilst
the
extrinsic apoptosis pathway is initiated through the activation of cell-
surface receptors,
the intrinsic apoptosis pathway occurs at the mitochondrial outer membrane and
is
governed by the binding interactions between pro- and anti-apoptotic Bc1-2
family
proteins, including Mc1-1. In many cancers, the anti-apoptotic Bc1-2
protein(s), such as
the Mc1-1, are upregulated, and in this way the cancer cells can evade
apoptosis. Thus,
inhibition of the Bc1-2 protein(s), such as Mc1-1, may lead to apoptosis in
cancer cells,
providing a method for the treatment of said cancers.
This assay evaluated inhibition of the BH3 domain: Mc1-1 interaction by
measuring the
displacement of Cy5-labeled BIM BH3 peptide (H2N-(C/Cy5Mal)
WIAQELRRIGDEFN-OH) in the HTRF assay format.
Assay Procedure
The following assay and stock buffers were prepared for use in the assay: (a)
Stock
buffer: 10 mM Tris-HC1, pH = 7.5 + 150 mM NaCl, filtered, sterilized, and
stored at 4 C;
and (b) IX assay buffer, where the following ingredients were added fresh to
stock
buffer: 2 mM dithiothreitol (DTT), 0.0025% Tween-20, 0.1 mg/mL bovine serum
albumin (BSA). The 1X Tb-Mcl-1 + Cy5 Bim peptide solution was prepared by
diluting
the protein stock solution using the lx assay buffer (b) to 25 pM Tb-Mcl-1 and
8 nM
Cy5 Bim peptide.
Using the Acoustic ECHO, 100 nL of 100x test compound(s) were dispensed into
individual wells of a white 384-well Perkin Elmer Proxiplate, for a final
compound
concentration of lx and final DMSO concentration of 1%. Inhibitor control and
neutral
control (NC, 100 nL of 100% DMSO) were stamped into columns 23 and 24 of assay
plate, respectively. Into each well of the plate was then dispensed 10 I, of
the 1X Tb-
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 260 -
Mc1-1 + Cy5 Bim peptide solution. The plate was centrifuged with a cover plate
at 1000
rpm for 1 minute, then incubated for 60 minutes at room temperature with
plates covered.
The TR-FRET signal was read on an BMG PHERAStar FSX MicroPlate Reader at room
temperature using the HTRF optic module (HTRF: excitation: 337nm, light
source: laser,
emission A: 665 nm, emission B: 620 nm, integration start: 60 ius, integration
time: 400
s).
Data Analysis
The BMG PHERAStar FSX MicroPlate Reader was used to measure fluorescence
intensity at two emission wavelengths ¨ 665 nm and 620 nm - and report
relative
fluorescence units (RFU) for both emissions, as well as a ratio of the
emissions (665
nm/620 nm)*10,000. The RFU values were normalized to percent inhibition as
follows:
% inhibition = (((NC - IC) - (compound - IC)) / (MC - IC)) *100
where IC (inhibitor control, low signal) = mean signal of 1 X Tb-MC1-I + Cy5
Bim
peptide+ inhibitor control or 100% inhibition of Mc1-1; NC (neutral control,
high
signal) = mean signal 1X Tb-MC1-1 + Cy5 Bim peptide with DMSO only or 0%
inhibition
An 11-point dose response curve was generated to determine IC50 values (using
GenData) based on the following equation:
Y ¨Bottom -h (Top-Bottom)/(1-h_10^((logIC50-X)*HillSlope))
where Y = % inhibition in the presence of X inhibitor concentration; Top =
100%
inhibition derived from the IC (mean signal of Mc1-1 + inhibitor control);
Bottom = 0%
inhibition derived from the NC (mean signal of Mcl-1 + DMS0); Hillslope = Hill
coefficient; and /C50 = concentration of compound with 50% inhibition in
relation to
top/neutral control (NC).
Ki = IC50 / (1 + [L]/Kd)
In this assay [L] = 8 nM and Kd = 10 nM
Representative compounds of the present invention were tested according to the
procedure as described above, with results as listed in the Table below (n.d.
means not
determined).
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 261 -
Compound TB-MCL1 Ki (nM) Compound TB-MCL1 Ki
(nM)
1 NT 38 0.019
2 0.483 39 0.032
3 0.023 40 0.053
4 0.713 41 0.66
5 0.411 42 0.030
6 0.059 43 2.50
7 1.71 44 3.45
8 0.027 45 1.17
9 0.509 46 0.018
10 0.042 47 2.95
11 7.48 48 4.35
12 0.141 49 0.010
13 15.34 50 0.013
14 0.021 51 0.013
15 28.02 52 0.013
16 0.024 53 0.306
17 0.018 54 0.020
18 0.025 55 3.96
19 0.035 56 0.038
20 0.026 57 2.97
21 0.066 58 0.081
22 0.029 59 0.104
23 0.016 60 4.73
28 0.028 61 114.90
29 0.033 62 0.027
30 6.95 63 0.026
31 0.015 64 2.33
32 1.51 65 2.98
33 0.027 66 0.015
34 2.36 67 0.017
35 0.026 68 0.008
36 3.07 69 0.014
Biological Example 2
MCL-1 is a regulator of apoptosis and is highly over-expressed in tumor cells
that
escape cell death. The assay evaluates the cellular potency of small-molecule
compounds targeting regulators of the apoptosis pathway, primarily MCL-1, Bfl-
1, Bel-
2, and other proteins of the Bc1-2 family. Protein-protein inhibitors
disrupting the
interaction of anti-apoptotic regulators with BH3-domain proteins initiate
apoptosis.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 262 -
The Caspase-Glo 3/7 Assay is a luminescent assay that measures caspase-3 and
-7 activities in purified enzyme preparations or cultures of adherent or
suspension cells.
The assay provides a proluminescent caspase-3/7 substrate, which contains the
tetrapeptide sequence DEVD. This substrate is cleaved to release
aminoluciferin, a
substrate of luciferase used in the production of light. Addition of the
single Caspase-
Glo 3/7 Reagent in an "add-mix-measure" format results in cell lysis,
followed by
caspase cleavage of the substrate and generation of a "glow-type- luminescent
signal.
This assay uses the MOLP-8 human multiple myeloma cell line, which is
sensitive to
MCL-1 inhibition.
Materials:
= Perkin Elmer Envision
= Multidrop 384 and small volume dispensing cassettes
= Centrifuge
= Countess automated cell counter
= Countess counting chamber slides
= Assay plate: ProxiPlate-384 Plus, White 384-shallow well Microplate
= Sealing tape: Topseal A plus
= T175 culture flask
Product Units Storage
RPMI1640 (no L-Glutamine, no
500 mL 4 C
phenol red)
Foetal Bovine Serum (FBS) (Heat
500 mL 4 C
inactivated)
L-Glutamine (200 mM) 100 ml -20 C
Gentamicin (50 mg/mL) 100 mL 4 C
100 mL
Caspase 3/7 Detection kit -20 C
10 x 10 mL
CA 03168355 2022- 8- 17
WO 2021/165370 PCT/EP2021/053973
- 263 -
Cell culture media:
MOLP8
RPMI-1640 medium 500 mL
20 % FBS (heat inactivated) 120 mL
2 mM L-Glutamine 6.2 mL
50 iig/mL Gentamicin 620 iaL
Assay media
RPMI-1640 medium 500 mL
% FBS (Heat inactivated) 57 mL
2 mM L-Glutamine 5.7 mL
50 iig/mL Gentamicin 570 1.iL
5 Cell culture:
Cell cultures were maintained between 0.2 and 2.0 x106 cells/mL. The cells
were
harvested by collection in 50 mL conical tubes. The cells were then pelleted
at 500 g for
5 mins before removing supernatant and resuspension in fresh pre-warmed
culture
medium. The cells were counted and diluted as needed.
Caspase-Glo reagent:
The assay reagent was prepared by transferring the buffer solution to the
substrate vial
and mixing. The solution may be stored for up to 1 week at 4 C with
negligible loss of
signal.
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 264 -
Assay procedure:
Compounds were delivered in assay-ready plates (Proxiplate) and stored at -20
C.
Assays always include 1 reference compound plate containing reference
compounds.
The plates were spotted with 40 nL of compounds (0.5 % DMSO final in cells;
serial
dilution; 30 tM highest conc. 1/3 dilution, 10 doses, duplicates). The
compounds were
used at room temperature and 4 pL of pre-warmed media was added to all wells
except
column 2 and 23. The negative control was prepared by adding 1 % DMSO in
media.
The positive control was prepared by adding the appropriate positive control
compound
in final concentration of 60 pM in media. The plate was prepared by adding 4
tit negative
control to column 23, 4 pL positive control to column 2 and 4 pL cell
suspension to all
wells in the plate. The plate with cells was then incubated at 37 C for 2
hours. The assay
signal reagent is the Caspase-Glo solution described above, and 8 pL was added
to all
wells. The plates were then sealed and measured after 30 minutes.
The activity of a test compound was calculated as percent change in apoptosis
induction as follows:
LC = median of the Low Control values
= Central Reference in Screener
= DMSO
= 0 %
HC = Median of the High Control values
= Scale Reference in Screener
= 30 [iM of positive control
= 100 % apoptosis induction
%Effect (AC50) = 100 ¨ ((sample-LC) / (1-1C-LC)) *100
%Control = (sample /HC)*100
%Control min = ((sample-LC) / (HC-LC)) *100
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 265 -
Table: Measured AC50 for Representative Compounds of Formula (I). Averaged
values
are reported over all runs on all batches of a particular compound.
MOLP8
Caspase-Glo
AC50 (nM)
Compound LD value
1 NT
2 1265.0
3 21.0
4 247.5
5 1287.9
6 930.7
7 11888.0
8 68.8
9 1169.5
10 37.6
11 8928.9
12 3416.6
13 24998.0
14 343.9
15 >30000
16 19.7
17 46.8
18 17.0
19 20.7
20 8.8
21 49.5
22 20.1
23 30.6
28 16.7
29 126.4
30 20578
31 11.5
32 743.4
33 26.1
34 2087.8
35 54.1
36 2900
38 539.9
39 28.5
40 57.6
41 568.5
CA 03168355 2022- 8- 17
WO 2021/165370
PCT/EP2021/053973
- 266 -
MOLP8
Caspase-Glo
AC50 (nM)
Compound LD value
42 59.8
43 2007.5
44 4263
45 1597
46 14.6
47 2714.6
48 3693.8
49 141.2
50 83.6
51 87.3
52 39.4
53 1249.7
54 10.7
55 1642.9
56 43.6
57 4812.8
58 29.7
59 82.4
60 2152.3
61 >30000
62 11.3
63 22.0
64 2102.3
65 1597.7
66 58.2
67 104.1
68 439.8
69 401.9
CA 03168355 2022- 8- 17