Note: Descriptions are shown in the official language in which they were submitted.
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
ANTI-INTEGRIN BETA7 ANTIBODY FORMULATIONS AND DEVICES
CROSS-REFERENCE TO RELAQTED APPLICATIONS
This application claims priority to U.S. Provisional Patent Application Serial
No.
63/059,427, filed July 31, 2020, the content of which is incorporated by
reference in its
entirety, and to which priority is claimed.
FIELD
Formulations comprising an anti-integrin beta7 antibody or an antigen-binding
fragment thereof are provided, including pharmaceutical formulations and
devices
comprising such formulations and methods of using such formulations and
devices.
SEQUENCE LISTING
The instant application contains a Sequence Listing which has been submitted
in
ASCII format via EFS-Web and is hereby incorporated by reference in its
entirety. Said
ASCII copy, created on July 27, 2021, is named 00B2061132SL.txt and is 13,697
bytes
in size.
BACKGROUND
The integrins are 43 heterodimeric cell surface receptors involved in numerous
cellular processes from cell adhesion to gene regulation (Hynes Cell
(1992);69:11-25;
and Hemler, Annu. Rev. Immunol. (1990), 8:365-368). Several integrins have
been
implicated in disease processes and have generated widespread interest as
potential
targets for drug discovery (Sharar et al., Springer Semin. Immunopathol.
(1995);16:359-
378). In the immune system, integrins are involved in leukocyte trafficking,
adhesion and
infiltration during inflammatory processes (Nakajima et al., I Exp. Med.
(1994);179:1145-1154). Differential expression of integrins regulates the
adhesive
properties of cells and different integrins are involved in different
inflammatory
responses (Butcher et al., Science (1996);272:60-66. The beta7 integrins
(i.e., a4(37 and
alphaEf37) are expressed primarily on monocytes, lymphocytes, eosinophils,
basophils,
and macrophages but not on neutrophils (Elices et al., Cell (1990);60:577-
584). The
primary ligands for a4(37 integrin are the endothelial surface proteins
mucosal address in
cell adhesion molecule (MAdCAM) and vascular cell adhesion molecule (VCAM-1)
(Makarem et al., I Biol. Chem. (1994);269:4005-4011). The binding of the a4(37
to
MAdCAM and/or VCAM expressed on high endothelial venules (HEVs) at sites of
inflammation results in firm adhesion of the leukocyte to the endothelium
followed by
extravasation into the inflamed tissue (Chuluyan et al., Springer Semin.
Immunopathol.,
1
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
1995, 16:391404). Monoclonal antibodies directed against a407, MAdCAM or VCAM
have been shown to be effective modulators in animal models of chronic
inflammatory
diseases such as asthma (Laberge et al., Am. I Respir. Crit. Care Med.
(1995);151:822-
829.), rheumatoid arthritis (Barbadillo et at., Springer Semin. Immunopathol.
(1995);16:375-379), colitis (Viney, I Immunol. (1996);157: 2488-2497) and
inflammatory bowel diseases (Podalski, N. Eng. I Med. (1991);325:928-937;
Powrie et
at., Ther. Immunol. (1995);2:115-123).
Humanized anti-integrin beta7 antibodies and antigen-binding fragments thereof
have been described. See, e.g., Intn'l Patent Publication No. W02006/026759.
One
particular anti-integrin beta7 antibody, etrolizumab, has been clinically
investigated for
treating gastrointestinal inflammatory disorders, e.g., inflammatory bowel
disease, e.g.,
ulcerative colitis and Crohn's disease. The results of a Phase 1 study of
etrolizumab in
moderate to severe ulcerative colitis were described in Rutgeerts PJ, et al.
Gut
2013;62:1122-1130 reporting no clinically significant safety signals observed.
The
results of a global multicenter Phase 2 study of etrolizumab in moderate to
severe
ulcerative colitis showed evidence of clinical efficacy of etrolizumab
treatment as
measured by induction of clinical remission (Vermeire, S. et al., Lancet 2014;
384: 309-
18). In addition, clinically meaningful remission was observed following
treatment of
moderate to severe Crohn's Disease patients with etrolizumab (Sandborn et al.,
presentation entitled "Etrolizumab as Induction Therapy in Moderate to Severe
Crohn's
Disease: Results from Bergamot Cohort 1," presented at United European
Gastroenterology Week Congress, Oct. 28-Nov. 2, 2017). Accordingly,
etrolizumab has
demonstrated promise as a therapeutic treatment option in inflammatory bowel
diseases
and further studies are ongoing to refine the safety and efficacy profile of
etrolizumab.
In each of the reported studies to date, etrolizumab was administered by a
health
care provider in a clinical setting either intravenously or subcutaneously.
For
subcutaneous administration, a vial and syringe with a vial concentration of
150 mg/ml
was used. Because inflammatory bowel diseases such as ulcerative colitis and
Crohn's
Disease are chronic diseases, long-term therapeutic treatment with etrolizumab
may be
needed. For optimal patient convenience and compliance among other advantages,
self-
administration of etrolizumab or administration in the home by a caregiver or
healthcare
professional is desirable. Accordingly, development of self-administration
devices and
formulations of etrolizumab compatible with such devices would be
advantageous.
2
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Because proteins, including antibodies such as etrolizumab, are larger and
more
complex than traditional organic and inorganic drugs (e.g., possessing
multiple
functional groups in addition to complex three-dimensional structures), the
formulation
of such proteins poses special problems. For a protein to remain biologically
active, a
formulation must preserve intact the conformational integrity of at least a
core sequence
of the protein's amino acids while at the same time protecting the protein's
multiple
functional groups from degradation. Degradation pathways for proteins can
involve
chemical instability (e.g., any process which involves modification of the
protein by
bond formation or cleavage resulting in a new chemical entity) or physical
instability
.. (e.g., changes in the higher order structure of the protein). Chemical
instability can
result from deamidation, racemization, hydrolysis, oxidation, beta elimination
or
disulfide exchange. Physical instability can result from denaturation,
aggregation,
precipitation or adsorption, for example. The three most common protein
degradation
pathways are protein aggregation, deamidation and oxidation. Cleland et al
Critical
Reviews in Therapeutic Drug Carrier Systems 10(4): 307-377 (1993).
High concentration (e.g., > 100 mg/mL) liquid antibody formulations are
desirable, for example, for routes of therapeutic administration or for
therapeutic
applications where small volumes of drug product are advisable, for example,
for
subcutaneous injection including, for example, using a prefilled syringe or
self-
administration device. High concentration antibody formulations, however, pose
numerous challenges and problems including challenges and problems associated
with
use of prefilled syringes or self-administration devices. One problem is
instability due to
the formation of particulates. With reconstituted liquid formulations, this
problem has
been addressed through the use of surfactants (e.g., a polysorbate), but
surfactants are
sometimes thought unsuitable for liquid formulations, because they render
further
processing difficult. Moreover, surfactants further do not reduce the
increased viscosity
caused as a result of numerous intermolecular interactions from the
macromolecular
nature of antibodies.
Selection of pH and optimal excipients is important for preventing particle
formation resulting from polysorbate-induced degradation, preventing
isomerization of
certain amino acids and formation of undersirable intermediates and for
extending shelf-
life in addition to providing advantages for manufacturing. Selection of pH
and optimal
excipients is also important for development of formulations, e.g., for
compatibility with
storage conditions, for administration by prefilled syringe, including
prefilled syringe
3
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
containing devices such as a prefilled syringe with a needle safety device or
an
autoinjector or self-administration devices, for example, to ensure
compatibility with
device components and to provide low injection forces.
It would be highly advantageous to have formulations comprising an anti-beta7
antibody, including etrolizumab, having extended stability and low viscosity
at high
antibody concentrations. High antibody concentration formulations having such
properties would be highly advantageous for certain routes of administration,
e.g., for
subcutaneous administration, including use with prefilled syringes and self-
administration devices. The formulations provided herein address these needs
and
provide other useful benefits.
It would be highly advantageous to have formulations comprising an anti-beta7
antibody having extended stability and low viscosity at high antibody
concentrations.
High antibody concentration formulations having such properties would be
highly
advantageous for certain routes of administration, e.g., for subcutaneous
administration.
The formulations provided herein address these needs and provide other useful
benefits.
All references cited herein, including patent applications and publications,
are
incorporated by reference in their entirety for any purpose.
SUMMARY
The formulations of the present disclosure are based, at least in part, on the
discovery that an anti-integrin beta7 antibody described herein, etrolizumab,
can be
formulated at a high concentration (about > 100 mg/mL) in a histidine buffer,
and
arginine succinate, and a surfactant and that such high antibody concentration
formulation is of low viscosity, has extended physical and chemical stability
and
maintains potency. The presently disclosed formulations are optimally
compatible for
self-administration devices such as a prefilled syringe with a needle safety
device (PFS-
NSD). In certain embodiments, the prefilled syringe is assembled into an
autoinjector.
The presently disclosed formulation can be packaged into subcutaneous
administration
devices as described herein with maintenance of, for example, product
stability and other
desirable attributes. Formulations of the present disclosure are useful for,
e.g., the
treatment of a gastrointestinal inflammatory disorder, e.g., an inflammatory
bowel
disease, e.g., ulcerative colitis and Crohn's disease.
Accordingly, in one aspect, a formulation comprising an anti-integrin beta7
antibody or an antigen-binding fragment thereof is provided. In certain
embodiments,
the concentration of the antibody or antigen-binding fragment thereof in the
formulation
4
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
is at least about 100 mg/mL and the viscosity of the formulation is less than
about 20
centipoise (cP) at 25 C. In certain embodiments, the viscosity of the
formulation is less
than about 7 cP at 25 C.
In certain embodiments, the anti-integrin beta7 antibody is a monoclonal
antibody. In certain embodiments, the anti-integrin beta7 antibody is a
humanized
antibody. In certain embodiments, the anti-integrin beta7 antibody or antigen-
binding
fragment thereof comprises three light chain hypervariable regions (HVRs), HVR-
L1,
HVR-L2, and HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3,
wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID NO:6 or
SEQ ID NO:7. In certain embodiments, the anti-integrin beta7 antibody or
antigen-
binding fragment thereof comprises a light chain variable region comprising
the amino
acid sequence set forth in SEQ ID NO: 8, and a heavy chain variable region
comprising
the amino acid sequence set forth in SEQ ID NO: 9. In certain embodiments, the
anti-
integrin beta7 antibody or antigen-binding fragment thereof comprises a light
chain
comprising the amino acid sequence set forth in SEQ ID NO:10 and a heavy chain
comprising the amino acid sequence set forth in SEQ ID NO: 11. In certain
embodiments, the anti-integrin beta7 antibody or antigen-binding fragment
thereof
comprises a light chain comprising the amino acid sequence set forth in SEQ ID
NO:10
and a heavy chain comprising the amino acid sequence set forth in SEQ ID NO:
12. In
certain embodiments, the anti-integrin beta7 antibody is etrolizumab. In
certain
embodiments, the concentration of the anti-integrin beta7 antibody or antigen-
binding
fragment thereof in the formulation is between about 100 mg/ml and about 220
mg/ml.
In certain embodiments, the concentration of the anti-integrin beta7 antibody
or antigen-
binding fragment thereof in the formulation is about 150 mg/ml.
In certain embodiments, the pH of the formulation is greater than 5.0 and up
to
7Ø In certain embodiments, the pH of the formulation is greater than 5.5. In
certain
embodiments, the pH of the formulation is between 5.6 and 6.1. In certain
embodiments,
the pH of the formulation is 5.8, between 5.7 and 5.9, or between 5.75 and
5.85.
5
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
In certain embodiments, the formulation comprises arginine-succinate. In
certain
embodiments, the concentration of the arginine succinate in the formulation is
between
about 100 mM and about 300 mM. In certain embodiments, the concentration of
the
arginine succinate in the formulation is between about 150 mM and about 300
mM. In
certain embodiments, the concentration of the arginine succinate in the
formulation is
between about 150 mM and about 250 mM. In certain embodiments, the
concentration
of the arginine succinate in the formulation is about 200 mM.
In certain embodiments, the formulation further comprises a surfactant, and
the
concentration of the surfactant in the formulation is greater than 0.01%
weight/volume
(w/v) and up to about 1% w/v. In certain embodiments, the concentration of the
surfactant in the formulation is between 0.03% w/v and 0.06% w/v. In certain
embodiments, the concentration of the surfactant in the formulation is 0.04%
w/v or
about 0.04% w/v. In certain embodiments, the surfactant is polysorbate 20.
In certain embodiments, the formulation further comprises histidine. In
certain
embodiments, the concentration of the histidine in the formulation is between
about 5
mM and about 40 mM. In certain embodiments, the concentration of the histidine
in the
formulation is 20 mM or about 20 mM.
In certain embodiments, the formulation has extended stability. In certain
embodiments, the anti-integrin beta7 antibody is stable for at least about
seven years at -
20 C. In certain embodiments, the anti-integrin beta7 antibody or antigen-
binding
fragment thereof is stable for at least about one year at 5 C. In certain
embodiments, the
anti-integrin beta7 antibody or antigen-binding fragment thereof is stable for
at least
about five years at 5 C. In certain embodiments, the anti-integrin beta7
antibody or
antigen-binding fragment thereof is stable for about six years at 5 C.
In certain embodiments, the anti-integrin beta7 antibody or antigen-binding
fragment thereof is stable for at least about 1 day at room temperature. In
certain
embodiments, the anti-integrin beta7 antibody or antigen-binding fragment
thereof is
stable for up to about one month at room temperature.
In another aspect, the present disclosure provides a formulation comprising an
anti-integrin beta7 antibody or an antigen-binding fragment thereof in 20 mM
or about
20 mM histidine buffer, pH 5.8, 0.04% polysorbate 20, and 200 mM or about 200
mM
arginine succinate, wherein the concentration of the anti-integrin beta7
antibody is about
150 mg/ml, and wherein the anti-integrin beta7 antibody comprises three light
chain
hypervariable regions (HVRs), HVR-L1, HVR-L2, and HVR-L3, and three heavy
chain
6
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
HVRs, HVR-H1, HVR-H2, and HVR-H3, wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID NO:6 or
SEQ ID NO:7.
In certain embodiments, the formulation has a pH of between 5.7 and 5.9 or
between 5.75 and 5.85. In certain embodiments, the formulation comprises 0.04%
polysorbate 20 or about 0.04% polysorbate 20.
The presently disclosed subject matter provides a formulation comprising an
anti-
integrin beta7 antibody, in 20 mM histidine buffer or about 20 mM histidine
buffer, pH
5.8 or pH between 5.7 and 5.9 or pH between 5.75 and 5.85, 0.04% polysorbate
20 or
about 0.04% polysorbate 20, and 200 mM arginine succinate or about 200 mM
arginine
succinate, and wherein the anti-integrin beta7 antibody comprises three light
chain
hypervariable regions (HVRs), HVR-L1, HVR-L2, and HVR-L3, and three heavy
chain
HVRs, HVR-H1, HVR-H2, and HVR-H3, wherein: (i) the HVR-L1 comprises the amino
acid sequence set forth in SEQ ID NO:1; (ii) the HVR-L2 comprises the amino
acid
sequence set forth in SEQ ID NO:2; (iii) the HVR-L3 comprises the amino acid
sequence set forth in SEQ ID NO:3; (iv) the HVR-H1 comprises the amino acid
sequence set forth in SEQ ID NO:4; (v) the HVR-H2 comprises the amino acid
sequence
SEQ ID NO:5; and (vi) the HVR-H3 comprises the amino acid sequence set forth
in SEQ
ID NO:6 or SEQ ID NO:7. In certain embodiments, the anti-integrin beta7
antibody
comprises a light chain comprising the amino acid sequence set forth in SEQ ID
NO:10
and a heavy chain comprising the amino acid sequence set forth in SEQ ID NO:
11. In
certain embodiments, the anti-integrin beta7 antibody comprises a light chain
comprising
the amino acid sequence set forth in SEQ ID NO:10 and a heavy chain comprising
the
amino acid sequence set forth in SEQ ID NO: 12. In certain embodiments, the
anti-
integrin beta7 antibody is etrolizumab.
In certain embodiments, the anti-integrin beta7 antibody or antigen-binding
fragment thereof comprises a light chain variable region comprising the amino
acid
sequence set forth in SEQ ID NO: 8, and a heavy chain variable region
comprising the
amino acid sequence set forth in SEQ ID NO: 9. In certain embodiments, the
anti-
7
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
integrin beta7 antibody or antigen-binding fragment thereof comprises a light
chain
comprising the amino acid sequence set forth in SEQ ID NO:10 and a heavy chain
comprising the amino acid sequence set forth in SEQ ID NO: 11. In certain
embodiments, the anti-integrin beta7 antibody or antigen-binding fragment
thereof
comprises a light chain comprising the amino acid sequence set forth in SEQ ID
NO:10
and a heavy chain comprising the amino acid sequence set forth in SEQ ID NO:
12.
In still a further aspect, an article of manufacture comprising a subcutaneous
administration device is provided. In certain embodiments, the subcutaneous
administration device delivers to a subject a flat dose of an anti-integrin
beta7 antibody
or an antigen-binding fragment thereof. In certain embodiments, the flat dose
is about
100 mg. In certain embodiments, the flat dose is 105 mg. In certain
embodiments, the
flat dose is about 200 mg. In certain embodiments, the flat dose is 210 mg. In
certain
embodiments, the anti-integrin beta7 antibody is etrolizumab. The anti-
integrin beta7
antibody or antigen-binding fragment thereof in the subcutaneous
administration device
is formulated as described above such that it is provided in a stable
pharmaceutical
formulation.
In certain embodiments, the subcutaneous administration device is a needle
safety
device. In certain embodiments, the needle safety device comprises a prefilled
syringe.
In certain embodiments, the anti-integrin beta7 antibody is stable at the
subcutaneous
administration device for at least about 60 months at 5 C, or at least about 3
months at
C. In certain embodiments, the prefilled syringe comprises a glass barrel, a
plunger
stopper, a needle, and needle shield or a tip cap. In certain embodiments, the
needle
shield is a rigid needle shield. In certain embodiments, the rigid needle
shield comprises
a rubber formulation having low zinc content. In certain embodiments, the
rigid needle
25 shield comprises an elastomeric component, and a rigid shield. In
certain embodiments,
the prefilled syringe is assembled into an autoinjector.
In certain embodiments, the volume of the formulation contained in the
prefilled
syringe is between about 0.5 mL and about 2.0 mL. In certain embodiments, the
volume
of the formulation contained in the prefilled syringe is between about 0.5 mL
and about
1.0 mL. In certain embodiments, the volume of the formulation contained in the
prefilled syringe is about 0.7 mL. In certain embodiments, the volume of the
formulation contained in the prefilled syringe is about 0.75 mL. In certain
embodiments,
the volume of the formulation contained in the prefilled syringe is about 1.0
mL. In
certain embodiments, the volume of the formulation contained in the prefilled
syringe is
8
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
between about 1.0 mL and about 1.5 mL. In certain embodiments, the volume of
the
formulation contained in the prefilled syringe is about 1.4 mL. In certain
embodiments,
the volume of the formulation contained in the prefilled syringe is about 1.5
mL. In
certain embodiments, the volume of the formulation contained in the prefilled
syringe is
about 1.45 mL. In certain embodiments, the prefilled syringe has a syringe
capacity of 1
mL. In certain embodiments, the prefilled syringe has a syringe capacity of
2.25 mL.
In certain embodiments, the prefilled syringe comprises silicone oil. In
certain
embodiments, the amount of silicone oil in the prefilled syringe is not
greater than about
1 mg. In certain embodiments, the amount of silicone oil in the prefilled
syringe is
between about 0.1 mg and about 1 mg. In certain embodiments, wherein the
amount of
silicone oil in the prefilled syringe is between about 0.2 mg and about 0.6
mg. In certain
embodiments, the amount of silicone oil in the prefilled syringe is between
about 0.5 mg
and 0.9 mg.
In certain embodiments, the needle safety device has an injection forces that
is
not greater than about 50 Newton (N). In certain embodiments, the needle
safety device
has an injection force that is not greater than about 35 Newton (N). In
certain
embodiments, the needle safety device has an injection force that is not
greater than
about 33 Newton (N).
The presently disclosed subject matter provides an article of manufacture
comprising about 0.7 mL of a formulation and a subcutaneous administration
device,
wherein
(a) the formulation comprises an anti-integrin beta7 antibody, in 20 mM
histidine
buffer or about 20 mM histidine buffer, pH 5.8 or pH between 5.7 and 5.9 or pH
between
5.75 and 5.85, 0.04% polysorbate 20 or about 0.04% polysorbate 20, and 200 mM
arginine succinate or about 200 mM arginine succinate, and wherein the anti-
integrin
beta7 antibody comprises three light chain hypervariable regions (HVRs), HVR-
L1,
HVR-L2, and HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3,
wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID NO:6 or
9
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
SEQ ID NO:7, and
(b) the subcutaneous administration device is a needle safety device that
comprises 1 mL of a prefilled syringe with a syringe capacity of 1 mL.
In certain embodiments, the anti-integrin beta7 antibody is present in the
formation at a concentration of 150 mg/mL or about 150 mg/ml.
The presently disclosed subject matter provides an article of manufacture
comprising about 1.4 mL of a formulation and a subcutaneous administration
device,
wherein
(a) the formulation comprises an anti-integrin beta7 antibody, in 20 mM
histidine
buffer or about 20 mM histidine buffer, pH 5.8 or pH between 5.7 and 5.9 or pH
between
5.75 and 5.85, 0.04% polysorbate 20 or about 0.04% polysorbate 20, and 200 mM
arginine succinate or about 200 mM arginine succinate, and wherein the anti-
integrin
beta7 antibody comprises three light chain hypervariable regions (HVRs), HVR-
L1,
HVR-L2, and HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3,
wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID
NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID
NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID
NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID
NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID
NO:6 or SEQ ID NO:7, and
(b) the subcutaneous administration device is a needle safety device that
comprises 1 mL of a prefilled syringe with a syringe capacity of 2.25 mL.
In certain embodiments, the anti-integrin beta7 antibody is present in the
formation at a concentration of 150 mg/mL or about 150 mg/ml.
The presently disclosed subject matter further provides autoinjectors
comprising
the article of manufacture disclosed herein.
The presently disclosed subject matter provides an autoinjector comprising an
article of manufacture comprising about 0.7 mL of a formulation and a
subcutaneous
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
administration device, wherein
(a) the formulation comprises an anti-integrin beta7 antibody, in 20 mM
histidine
buffer or about 20 mM histidine buffer, pH 5.8 or pH between 5.7 and 5.9 or pH
between
5.75 and 5.85, 0.04% polysorbate 20 or about 0.04% polysorbate 20, and 200 mM
arginine succinate or about 200 mM arginine succinate, and wherein the anti-
integrin
beta7 antibody comprises three light chain hypervariable regions (HVRs), HVR-
L1,
HVR-L2, and HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3,
wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID
NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID
NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID
NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID
NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID
NO:6 or SEQ ID NO:7, and
(b) the subcutaneous administration device is a needle safety device that
comprises 1 mL of a prefilled syringe with a syringe capacity of 1 mL.
In certain embodiments, the anti-integrin beta7 antibody is present in the
formation at a concentration of 150 mg/mL or about 150 mg/ml.
The presently disclosed subject matter provides an autoinjector comprising
article
of manufacture comprising about 1.4 mL of a formulation and a subcutaneous
administration device, wherein
(a) the formulation comprises an anti-integrin beta7 antibody, in 20 mM
histidine
buffer or about 20 mM histidine buffer, pH 5.8 or pH between 5.7 and 5.9 or pH
between
5.75 and 5.85, 0.04% polysorbate 20 or about 0.04% polysorbate 20, and 200 mM
arginine succinate or about 200 mM arginine succinate, and wherein the anti-
integrin
beta7 antibody comprises three light chain hypervariable regions (HVRs), HVR-
L1,
HVR-L2, and HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3,
wherein:
(i) the HVR-L1 comprises the amino acid sequence set forth in SEQ ID
11
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
NO:1;
(ii) the HVR-L2 comprises the amino acid sequence set forth in SEQ ID
NO:2;
(iii) the HVR-L3 comprises the amino acid sequence set forth in SEQ ID
NO:3;
(iv) the HVR-H1 comprises the amino acid sequence set forth in SEQ ID
NO:4;
(v) the HVR-H2 comprises the amino acid sequence SEQ ID NO:5; and
(vi) the HVR-H3 comprises the amino acid sequence set forth in SEQ ID
NO:6 or SEQ ID NO:7, and
(b) the subcutaneous administration device is a needle safety device that
comprises 1 mL of a prefilled syringe with a syringe capacity of 2.25 mL.
In certain embodiments, the anti-integrin beta7 antibody is present in the
formation at a concentration of 150 mg/mL or about 150 mg/ml.
In yet another aspect, a method of treating a gastrointestinal inflammatory
disorder in a subject is provided. In certain embodiments, the method
comprises
administering to the subject an effective amount of any of the above
formulations.
In still yet another aspect, methods of administering subcutaneously a
formulation comprising an anti-integrin beta7 antibody or an antigen-binding
fragment
thereof are provided. Such methods comprise administering subcutaneously any
of the
formulations described above. In certain embodiments, the methods comprise a
subcutaneous administration device according to any of the devices described
above. In
certain embodiments, the method comprises the autoinjector disclosed herein.
In certain
embodiments, the administering results in mild pain or no pain. In certain
embodiments,
the administering results in a transient and mild injection site reaction. In
certain
embodiments, the full dose is administered or at least 90% of the full dose is
administered. In certain embodiments, the administering provides an equivalent
exposure to etrolizumab compared to a prefilled syringe with needle safety
device.
The presently disclosed subject matter also provides uses of the formulation,
the
article of manufacture, or the autoinjector disclosed herein in a therapy.
Furthermore, the presently disclosed subject matter provides uses of the
formulation, the article of manufacture, or the autoinjector disclosed herein
in treating a
gastrointestinal inflammatory disorder in a subject. In certain embodiments,
the
12
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
gastrointestinal inflammatory disorder is an inflammatory bowel disease. In
certain
embodiments, the inflammatory bowel disease is ulcerative colitis or Crohn's
disease.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts solubility curves for lauric acid, myristic acid, and
palmitic acid
.. as a function of pH and polysorbate 20 (PS20) concentration as described in
Example 1.
Figure 2 depicts impact of arginine on solution viscosity as described in
Example
1.
Figure 3 depicts impact of protein concentration on viscosity of formulations
comprising 200mM arginine succinate as described in Example 1.
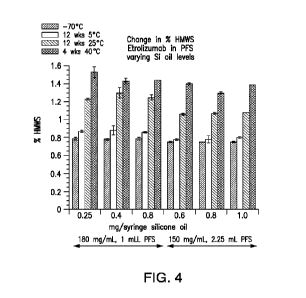
Figure 4 depicts the change in the percentage of high molecular weight species
(HMWS) of etrolizumab in pre-filled syringes with varying quantities of
silicone oil as
described in Example 1. The term "high molecular weight species (HMWS)" and
the
term "high molecular weight forms (HMWF)" are used interchangeability herein.
Figure 5 depicts the percentage of HMWS of etrolizumab in varying
concentrations of tungsten over time as described in Example 1.
Figure 6 depicts the impact of zinc on the viscosity of etrolizumab at varying
protein concentrations in formulations comprising 20 mM histidine, 200 mM
arginine
succinate, pH 5.8 at 25 C as described in Example 1.
Figure 7 shows protein aggregate formation by 50 mM zinc and 150 mg/mL
.. etrolizumab at room temperature as described in Example 1.
Figure 8 depicts the HMWS formation by 10 mM zinc and 10 mg/mL or 50
mg/mL etrolizumab at 40 C as described in Example 1.
Figure 9 depicts the impact of histidine on HMWS formation in formulations
comprising 10 mM zinc and 10 mg/mL etrolizumab at 40 C as described in Example
1.
Figure 10 depicts the impact of succinate on HMWS formation in formulations
comprising10 mM zinc and 10 mg/mL etrolizumab at 40 C as described in Example
1.
Figure 11 shows varying concentrations of histidine and succinate and the
combined impact on HMWS formation in formulations comprising 10 mM zinc and 50
mg/mL etrolizumab at 40 C as described in Example 1.
Figure 12 shows exemplary prefilled syringes (top two) and autoinjector (last)
as
described in Example 2.
Figure 13 shows prefilled autoinjector of etrolizumab as described in Example
2.
13
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Figure 14 shows autoinjector (AI) tolerability and human factors study design
as
described in Example 2.
Figure 15 shows graph indicating pain over time by intensity (7-point Visual
Descriptive Scale) as described in Example 2.
Figure 16 depicts graphs indicating pain over time by injection site (7-point
Visual Descriptive Scale) as described in Example 2.
Figure 17 shows the two-part pharmacokinetic bridging study design as
described
in Example 2. *Justification of geometric mean ratio (GMR) of 1.15 as decision
point
was based on the assumption that at 15% difference between prefilled syringe
with
needle safety device (PFS-NSD) was likely to be real and, therefore, the study
would be
unable to demonstrate bioequivalence. tAdjustment to N driven by GMR from
pilot
study. AT autoinjector; AUC area under the curve; C. maximum serum
concentration
of etrolizumab.
Figure 18 shows participant disposition as described in Example 3. Al
autoinjector, AUC area under the curve, PFS-NSD prefilled syringe with needle
safety
device, PK pharmacokinetic, SC subcutaneous. 'Excluded because of eligibility
criteria
(weight restriction). Participants were excluded from specific PK analyses
because of
insufficient PK data for calculations.
Figure 19 shows the impact of body weight on etrolizumab C. (top) or AUC0-Inf
(Bottom) as described in Example 3. Al autoinjector, AUC area under the curve,
AUCo_inf
AUC extrapolated to infinity, Cm maximum concentration, PFS-NSD prefilled
syringe
with needle safety device, SC subcutaneous.
Figure 20 shows etrolizumab serum concentrations over time with AT and PFS-
NSD on a linear scale (A) and semi-logarithmic scale (B) in the pivotal study
as
described in Example 3. Al autoinjector, PFS-NSD prefilled syringe with needle
safety
device.
Figure 21 shows etrolizumab serum concentrations over time by ADA status with
the AT (N=73 total, with 20 subjects being ADA postive, top) and PFS-NSD
(N=75, with
24 subjects being ADA positive, bottom) as described in Example 3. ADA
antidrug
antibody, Al autoinjector, PFS-NSD prefilled syringe with needle safety
device.
Figure 22 shows the different force definitions during injection as described
in
Example 4.
14
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
DETAILED DESCRIPTION
Unless defined otherwise, technical and scientific terms used herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Singleton et al., Dictionary of Microbiology and Molecular
Biology
2nd ed., J. Wiley & Sons (New York, N.Y. 1994), and March, Advanced Organic
Chemistry Reactions, Mechanisms and Structure 4th ed., John Wiley & Sons (New
York, N.Y. 1992), provide one skilled in the art with a general guide to many
of the
terms used in the present application.
CERTAIN DEFINITIONS
For purposes of interpreting this specification, the following definitions
will
apply and whenever appropriate, terms used in the singular will also include
the plural
and vice versa. In the event that any definition set forth below conflicts
with any
document incorporated herein by reference, the definition set forth below
shall control.
As used in this specification and the appended claims, the singular forms "a,"
"an" and "the" include plural referents unless the context clearly dictates
otherwise.
Thus, for example, reference to "a protein" or an "antibody" includes a
plurality of
proteins or antibodies, respectively; reference to "a cell" includes mixtures
of cells, and
the like.
As used herein, the term "about" or "approximately" means within an acceptable
error range for the particular value as determined by one of ordinary skill in
the art,
which will depend in part on how the value is measured or determined, i.e.,
the
limitations of the measurement system. For example, "about" can mean within 3
or
more than 3 standard deviations, per the practice in the art. Alternatively,
"about" can
mean a range of up to 20%, preferably up to 10%, more preferably up to 5%, and
more
preferably still up to 1% of a given value. Alternatively, particularly with
respect to
biological systems or processes, the term can mean within an order of
magnitude,
preferably within 5-fold, and more preferably within 2-fold, of a value.
Reference to
"about" a value or parameter herein includes (and describes) embodiments that
are
directed to that value or parameter per se. For example, description referring
to "about
X" includes description of "X."
The term "pharmaceutical formulation" refers to a preparation which is in such
form as to permit the biological activity of the active ingredient to be
effective, and
which contains no additional components which are unacceptably toxic to a
subject to
which the formulation would be administered. Such formulations are sterile.
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
"Pharmaceutically acceptable" excipients (vehicles, additives) are those which
can
reasonably be administered to a subject mammal to provide an effective dose of
the
active ingredient employed.
A "sterile" formulation is aseptic or free or essentially free from all living
microorganisms and their spores.
A "frozen" formulation is one at a temperature below 0 C. Generally, the
frozen
formulation is not freeze-dried, nor is it subjected to prior, or subsequent,
lyophilization.
In certain embodiments, the frozen formulation comprises frozen drug substance
for
storage (in stainless steel tank) or frozen drug product (in final vial
configuration).
A "stable" formulation is one in which the protein therein essentially retains
its
physical stability and/or chemical stability and/or biological activity upon
storage. In
certain embodiments, the formulation essentially retains its physical and
chemical
stability, as well as its biological activity upon storage. The storage period
is generally
selected based on the intended shelf-life of the formulation.
As used herein, a formulation having "extended stability" means one in which
the
protein therein essentially retains its physical stability, chemical
stability, and biological
activity upon storage at 5 C for one year or more. In certain embodiments, the
storage is
at 5 C for one year or more. In certain embodiments, the storage is at 5 C
for up to five
years or six years. In certain embodiments, the anti-integrin beta7 antibody
is stable for
at least about 1 day at room temperature. In certain embodiments, the anti-
integrin beta7
antibody is stable for up to about one month at room temperature. As used
herein, the
room temperature is between about 20 and about 22 C. In certain embodiments,
the
room temperature is about 20 C.
A protein "retains its physical stability" in a pharmaceutical formulation if
it
shows no signs or very little of aggregation, precipitation and/or
denaturation upon visual
examination of color and/or clarity, or as measured by UV light scattering or
by size
exclusion chromatography.
A protein "retains its chemical stability" in a pharmaceutical formulation, if
the
chemical stability at a given time is such that the protein is considered to
still retain its
biological activity as defined below. Chemical stability can be assessed by
detecting and
quantifying chemically altered forms of the protein. Chemical alteration may
involve
size modification (e.g. clipping) which can be evaluated using size exclusion
chromatography, SDS-PAGE and/or matrix-assisted laser desorption
ionization/time-of-
flight mass spectrometry (MALDI/TOF MS), for example. Other types of chemical
16
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
alteration include charge alteration (e.g. occurring as a result of
deamidation) which can
be evaluated by ion-exchange chromatography or imaged capillary isoelectric
focusing
(icIEF), for example.
An antibody "retains its biological activity" in a pharmaceutical formulation,
if
the biological activity of the antibody at a given time is within about 20%
(within the
errors of the assay) of the biological activity exhibited at the time the
pharmaceutical
formulation was prepared as determined in an antigen binding assay or a
potency assay,
for example.
Herein, "biological activity" of a monoclonal antibody refers to the ability
of the
antibody to bind to antigen. It can further include antibody binding to
antigen and
resulting in a measurable biological response which can be measured in vitro
or in vivo.
Such activity may be antagonistic or agonistic.
The antibody which is formulated is essentially pure and desirably essentially
homogeneous (e.g., free from contaminating proteins etc.). "Essentially pure"
antibody
means a composition comprising at least about 90% by weight of the antibody,
based on
total weight of the composition, or at least about 95% by weight. "Essentially
homogeneous" antibody means a composition comprising at least about 99% by
weight
of antibody, based on total weight of the composition.
By "isotonic" is meant that the formulation of interest has essentially the
same
osmotic pressure as human blood. Isotonic formulations will generally have an
osmotic
pressure from about 250 to 350 mOsm. Isotonicity can be measured using a vapor
pressure or ice-freezing type osmometer, for example.
As used herein, "buffer" refers to a buffered solution that resists changes in
pH
by the action of its acid-base conjugate components.
Herein, a "surfactant" refers to a surface-active agent, typically a nonionic
surfactant. Examples of surfactants herein include polysorbate (for example,
polysorbate
20 and, polysorbate 80); poloxamer (e.g. poloxamer 188); Triton; sodium
dodecyl sulfate
(SDS); sodium laurel sulfate; sodium octyl glycoside; lauryl-, myristyl-,
linoleyl-, or
stearyl-sulfobetaine; lauryl-, myristyl-, linoleyl- or stearyl-sarcosine;
linoleyl-, myristyl-,
or cetyl-betaine; lauroamidopropyl-, cocamidopropyl-, linoleamidopropyl-,
myristamidopropyl-, palmidopropyl-, or isostearamidopropyl-betaine (e.g.
lauroamidopropyl); myristamidopropyl-, palmidopropyl-, or isostearamidopropyl-
dimethylamine; sodium methyl cocoyl-, or disodium methyl oleyl-taurate; and
the
MONAQUATTM series (Mona Industries, Inc., Paterson, N.J.); polyethyl glycol,
17
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
polypropyl glycol, and copolymers of ethylene and propylene glycol (e.g.
Pluronics,
PF68 etc); etc. In certain embodiments, the surfactant is polysorbate 20.
As used herein, "treatment" refers to clinical intervention in an attempt to
alter
the natural course of the individual or cell being treated, and can be
performed before or
during the course of clinical pathology. Desirable effects of treatment
include preventing
the occurrence or recurrence of a disease or a condition or symptom thereof,
alleviating a
condition or symptom of the disease, diminishing any direct or indirect
pathological
consequences of the disease, decreasing the rate of disease progression,
ameliorating or
palliating the disease state, and achieving remission or improved prognosis.
An "effective amount" refers to an amount effective, at dosages and for
periods
of time necessary, to achieve the desired therapeutic or prophylactic result.
A
"therapeutically effective amount" of a therapeutic agent may vary according
to factors
such as the disease state, age, sex, and weight of the individual, and the
ability of the
antibody to elicit a desired response in the individual. A therapeutically
effective
amount is also one in which any toxic or detrimental effects of the
therapeutic agent are
outweighed by the therapeutically beneficial effects.
An "individual," "subject" or "patient" is a vertebrate. In certain
embodiments,
the vertebrate is a mammal. Mammals include, but are not limited to, primates
(including human and non-human primates) and rodents (e.g., mice and rats). In
certain
embodiments, a mammal is a human.
A "medicament" is an active drug to treat a disease, disorder, and/or
condition.
"Antibodies" (Abs) and "immunoglobulins" (Igs) refer to glycoproteins having
similar structural characteristics. While antibodies exhibit binding
specificity to a
specific antigen, immunoglobulins include both antibodies and other antibody-
like
molecules which generally lack antigen specificity. Polypeptides of the latter
kind are,
for example, produced at low levels by the lymph system and at increased
levels by
myelomas.
The terms "antibody" and "immunoglobulin" are used interchangeably in the
broadest sense and include monoclonal antibodies (e.g., full length or intact
monoclonal
antibodies), polyclonal antibodies, monovalent antibodies, multivalent
antibodies,
multispecific antibodies (e.g., bispecific antibodies so long as they exhibit
the desired
biological activity) and may also include certain antibody fragments (as
described in
greater detail herein). An antibody can be chimeric, human, humanized and/or
affinity
matured.
18
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
The terms "full length antibody," "intact antibody" and "whole antibody" are
used herein interchangeably to refer to an antibody in its substantially
intact form, not
antibody fragments as defined below. The terms particularly refer to an
antibody with
heavy chains that contain the Fc region.
"Antibody fragments" comprise a portion of an intact antibody, preferably
comprising the antigen binding region thereof Examples of antibody fragments
include
Fab, Fab', F(ab')2, and Fv fragments; diabodies; linear antibodies; single-
chain antibody
molecules; and multispecific antibodies formed from antibody fragments.
Papain digestion of antibodies produces two identical antigen-binding
fragments,
called "Fab" fragments, each with a single antigen-binding site, and a
residual "Fc"
fragment, whose name reflects its ability to crystallize readily. Pepsin
treatment yields
an F(ab')2 fragment that has two antigen-combining sites and is still capable
of cross-
linking antigen.
"Fv" is a minimum antibody fragment which contains a complete antigen-binding
site. In one embodiment, a two-chain Fv species consists of a dimer of one
heavy- and
one light-chain variable domain in tight, non-covalent association.
Collectively, the six
CDRs of an Fv confer antigen-binding specificity to the antibody. However,
even a
single variable domain (or half of an Fv comprising only three CDRs specific
for an
antigen) has the ability to recognize and bind antigen, although at a lower
affinity than
the entire binding site.
The Fab fragment contains the heavy- and light-chain variable domains and also
contains the constant domain of the light chain and the first constant domain
(CH1) of
the heavy chain. Fab' fragments differ from Fab fragments by the addition of a
few
residues at the carboxy terminus of the heavy chain CH1 domain including one
or more
cysteines from the antibody hinge region. Fab'-SH is the designation herein
for Fab' in
which the cysteine residue(s) of the constant domains bear a free thiol group.
F(ab')2
antibody fragments originally were produced as pairs of Fab' fragments which
have
hinge cysteines between them. Other chemical couplings of antibody fragments
are also
known.
The term "monoclonal antibody" as used herein refers to an antibody obtained
from a population of substantially homogeneous antibodies, i.e., the
individual
antibodies comprising the population are identical except for possible
mutations, e.g.,
naturally occurring mutations, that may be present in minor amounts. Thus, the
modifier
"monoclonal" indicates the character of the antibody as not being a mixture of
discrete
19
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
antibodies. In certain embodiments, such a monoclonal antibody typically
includes an
antibody comprising a polypeptide sequence that binds a target, wherein the
target-
binding polypeptide sequence was obtained by a process that includes the
selection of a
single target binding polypeptide sequence from a plurality of polypeptide
sequences.
For example, the selection process can be the selection of a unique clone from
a plurality
of clones, such as a pool of hybridoma clones, phage clones, or recombinant
DNA
clones. It should be understood that a selected target binding sequence can be
further
altered, for example, to improve affinity for the target, to humanize the
target binding
sequence, to improve its production in cell culture, to reduce its
immunogenicity in vivo,
to create a multispecific antibody, etc., and that an antibody comprising the
altered target
binding sequence is also a monoclonal antibody of this invention. In contrast
to
polyclonal antibody preparations which typically include different antibodies
directed
against different determinants (epitopes), each monoclonal antibody of a
monoclonal
antibody preparation is directed against a single determinant on an antigen.
In addition
to their specificity, monoclonal antibody preparations are advantageous in
that they are
typically uncontaminated by other immunoglobulins.
The modifier "monoclonal" indicates the character of the antibody as being
obtained from a substantially homogeneous population of antibodies, and is not
to be
construed as requiring production of the antibody by any particular method.
For
example, the monoclonal antibodies to be used in accordance with the present
invention
may be made by a variety of techniques, including, for example, the hybridoma
method
(e.g., Kohler et al., Nature, 256: 495 (1975); Harlow et al., Antibodies: A
Laboratory
Manual (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling et al.,
in:
Monoclonal Antibodies and T-Cell Hybridomas 563-681 (Elsevier, N.Y., 1981)),
recombinant DNA methods (see, e.g., U.S. Patent No. 4,816,567), phage display
technologies (see, e.g., Clackson et al., Nature, 352: 624-628 (1991); Marks
et al.,
Mol. Biol. 222: 581-597 (1992); Sidhu et al., I Mol. Biol. 338(2): 299-310
(2004); Lee et
al., J. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci.
USA
101(34): 12467-12472 (2004); and Lee et al., I Immunol. Methods 284(1-2): 119-
132(2004), and technologies for producing human or human-like antibodies in
animals
that have parts or all of the human immunoglobulin loci or genes encoding
human
immunoglobulin sequences (see, e.g., W098/24893; W096/34096; W096/33735;
W091/10741; Jakobovits et al., Proc. Natl. Acad. Sci. USA 90: 2551 (1993);
Jakobovits
et al., Nature 362: 255-258 (1993); Bruggemann et al., Year in Immunol. 7:33
(1993);
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
U.S. Patent Nos. 5,545,807; 5,545,806; 5,569,825; 5,625,126; 5,633,425;
5,661,016;
Marks et al., Bio.Technology 10: 779-783 (1992); Lonberg et al., Nature 368:
856-859
(1994); Morrison, Nature 368: 812-813 (1994); Fishwild et al., Nature
Biotechnol. 14:
845-851 (1996); Neuberger, Nature Biotechnol. 14: 826 (1996) and Lonberg and
Huszar,
Intern. Rev. Immunol. 13: 65-93 (1995).
The monoclonal antibodies herein specifically include "chimeric" antibodies in
which a portion of the heavy and/or light chain is identical with or
homologous to
corresponding sequences in antibodies derived from a particular species or
belonging to
a particular antibody class or subclass, while the remainder of the chain(s)
is identical
with or homologous to corresponding sequences in antibodies derived from
another
species or belonging to another antibody class or subclass, as well as
fragments of such
antibodies, so long as they exhibit the desired biological activity (U.S.
Patent No.
4,816,567; and Morrison etal., Proc. Natl. Acad. Sci. USA 81:6855-9855
(1984)).
"Native antibodies" refer to naturally occurring immunoglobulin molecules with
varying structures. For example, native IgG antibodies are heterotetrameric
glycoproteins of about 150,000 daltons, composed of two identical light chains
and two
identical heavy chains that are disulfide-bonded. From N- to C-terminus, each
heavy
chain has a variable region (VH), also called a variable heavy domain or a
heavy chain
variable domain, followed by three constant domains (CHL CH2, and CH3).
Similarly,
.. from N- to C-terminus, each light chain has a variable region (VL), also
called a variable
light domain or a light chain variable domain, followed by a constant light
(CL) domain.
The light chain of an antibody may be assigned to one of two types, called
kappa and
lambda, based on the amino acid sequence of its constant domain.
The term "variable region" or "variable domain" refers to the domain of an
antibody heavy or light chain that is involved in binding the antibody to
antigen. The
variable domains of the heavy chain and light chain (VH and VL, respectively)
of a native
antibody generally have similar structures, with each domain comprising four
conserved
framework regions (FRs) and three hypervariable regions (HVRs). (See, e.g.,
Kindt et al.
Kuby Immunology, 6th ed., W.H. Freeman and Co., page 91 (2007).) A single VH
or VL
domain may be sufficient to confer antigen-binding specificity. Furthermore,
antibodies
that bind a particular antigen may be isolated using a VH or VL domain from an
antibody
that binds the antigen to screen a library of complementary VL or VH domains,
respectively. See e.g., Portolano etal., I Immunol. 150:880-887 (1993);
Clarkson et al.,
Nature 352:624-628 (1991).
21
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
A "humanized" antibody refers to a chimeric antibody comprising amino acid
residues from non-human HVRs and amino acid residues from human FRs. In
certain
embodiments, a humanized antibody will comprise substantially all of at least
one, and
typically two, variable domains, in which all or substantially all of the HVRs
(e.g.,
CDRs) correspond to those of a non-human antibody, and all or substantially
all of the
FRs correspond to those of a human antibody. A humanized antibody optionally
may
comprise at least a portion of an antibody constant region derived from a
human
antibody. A "humanized form" of an antibody, e.g., a non-human antibody,
refers to an
antibody that has undergone humanization.
The term "hypervariable region," "HVR," or "HV," when used herein refers to
the regions of an antibody variable domain which are hypervariable in sequence
and/or
form structurally defined loops. Generally, antibodies comprise six
hypervariable
regions; three in the VH (H1, H2, H3), and three in the VL (L1, L2, L3). A
number of
hypervariable region delineations are in use and are encompassed herein. The
Kabat
Complementarity Determining Regions (CDRs) are based on sequence variability
and
are the most commonly used (Kabat et al., Sequences of Proteins of
Immunological
Interest, 5th Ed. Public Health Service, National Institutes of Health,
Bethesda, Md.
(1991)). Chothia refers instead to the location of the structural loops
(Chothia and Lesk J.
Mol. Biol. 196:901-917 (1987)). The AbM hypervariable regions represent a
compromise between the Kabat CDRs and Chothia structural loops, and are used
by
Oxford Molecular's AbM antibody modeling software. The "contact" hypervariable
regions are based on an analysis of the available complex crystal structures.
The
residues from each of these HVRs are noted below.
Loop Kabat AbM Chothia Contact
Li L24-L34 L24-L34 L26-L32 L30-L36
L2 L50-L56 L50-L56 L50-L52 L46-L55
L3 L89-L97 L89-L97 L91-L96 L89-L96
H1 H31-H35B H26-H35B H26-H32 H30-H35B (Kabat Numbering)
H1 H31-H35 H26-H35 H26-H32 H30-H35 (Chothia Numbering)
H2 H50-H65 H50-H58 H53-H55 H47-H58
H3 H95-H102 H95-H102 H96-H101 H93-H101
22
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Hypervariable regions may comprise "extended hypervariable regions" as
follows: 24-36 or 24-34 (L1), 46-56 or 49-56 or 50-56 or 52-56 (L2) and 89-97
(L3) in
the VL and 26-35 (H1), 50-65 or 49-65 (H2) and 93-102, 94-102 or 95-102 (H3)
in the
VH. The variable domain residues are numbered according to Kabat et al., supra
for
each of these definitions.
Depending on the amino acid sequences of the constant domains of their heavy
chains, antibodies (immunoglobulins) can be assigned to different classes.
There are five
major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of
these
may be further divided into subclasses (isotypes), e.g., IgGi, IgG2, IgG3,
IgG4, IgAi, and
IgA2. The heavy-chain constant domains that correspond to the different
classes of
immunoglobulins are called a, 6, 6, y, and , respectively. The subunit
structures and
three-dimensional configurations of different classes of immunoglobulins are
well
known and described generally in, for example, Abbas et al. Cellular and Mol.
Immunology, 4th ed. (W. B. Saunders, Co., 2000). An antibody may be part of a
larger
fusion molecule, formed by covalent or non-covalent association of the
antibody with
one or more other proteins or peptides.
The "class" of an antibody refers to the type of constant domain or constant
region possessed by its heavy chain. There are five major classes of
antibodies: IgA,
IgD, IgE, IgG, and IgM, and several of these may be further divided into
subclasses
(isotypes), e.g., IgGl, IgG2, IgG3, IgG4, IgAl , and IgA2. The heavy chain
constant
domains that correspond to the different classes of immunoglobulins are called
a, 6, 8,y,
and II., respectively.
An "isolated" biological molecule, such as a nucleic acid, polypeptide, or
antibody, is one which has been identified and separated and/or recovered from
at least
one component of its natural environment.
A "subcutaneous administration device" refers to a device which is adapted or
designed to administer a drug, for example a therapeutic antibody, or
pharmaceutical
formulation by the subcutaneous route. Exemplary subcutaneous administration
devices
include, but are not limited to, a needle safety device (e.g., one comprising
a pre-filled
syringe), an injection device, including an autoinjector (e.g., one comprising
a pre-filled
syringe), infusion pump, injector pen, needleless device, and patch delivery
system. A
subcutaneous administration device administers a certain volume of the
pharmaceutical
23
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
formulation, for example about 0.5 mL, about 0.7 mL, about 1.0 mL, about 1.25
mL,
about 1.4 mL, about 1.5 mL, about 1.75 mL, about 2.0 mL or about 5.0 mL.
A "package insert" or "label" is used to refer to instructions customarily
included
in commercial packages of therapeutic products or medicaments, that contain
information about the indications, usage, dosage, administration,
contraindications, other
therapeutic products to be combined with the packaged product, and/or warnings
concerning the use of such therapeutic products or medicaments and the like.
As used herein, the term "gastrointestinal inflammatory disorders" refer to a
group of chronic disorders that cause inflammation and/or ulceration in the
mucous
membrane. These disorders include, for example, inflammatory bowel disease
(e.g.,
Crohn's disease, ulcerative colitis, indeterminate colitis and infectious
colitis), mucositis
(e.g., oral mucositis, gastrointestinal mucositis, nasal mucositis and
proctitis), necrotizing
enterocolitis, and esophagitis.
"Inflammatory Bowel Disease" or "MD" is used interchangeably herein to refer
to diseases of the bowel that cause inflammation and/or ulceration and
includes without
limitation Crohn's disease and ulcerative colitis.
"Crohn's disease (CD)" or "ulcerative colitis (UC)" are chronic inflammatory
bowel diseases of unknown etiology. Crohn's disease, unlike ulcerative
colitis, can affect
any part of the bowel. The most prominent feature Crohn's disease is the
granular,
reddish-purple edmatous thickening of the bowel wall. With the development of
inflammation, these granulomas often lose their circumscribed borders and
integrate with
the surrounding tissue. Diarrhea and obstruction of the bowel are the
predominant
clinical features. As with ulcerative colitis, the course of Crohn's disease
may be
continuous or relapsing, mild or severe, but unlike ulcerative colitis,
Crohn's disease is
not curable by resection of the involved segment of bowel. Most patients with
Crohn's
disease require surgery at some point, but subsequent relapse is common and
continuous
medical treatment is usual.
Crohn's disease may involve any part of the alimentary tract from the mouth to
the anus, although typically it appears in the ileocolic, small-intestinal or
colonic-
anorectal regions. Histopathologically, the disease manifests by discontinuous
granulomatomas, crypt abscesses, fissures and aphthous ulcers. The
inflammatory
infiltrate is mixed, consisting of lymphocytes (both T and B cells), plasma
cells,
macrophages, and neutrophils. There is a disproportionate increase in IgM- and
IgG-
secreting plasma cells, macrophages and neutrophils. Anti-inflammatory drugs
24
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
sulfasalazine and 5-aminosalisylic acid (5-ASA) are useful for treating mildly
active
colonic Crohn's disease and is commonly prescribed to maintain remission of
the
disease. Metroidazole and ciprofloxacin are similar in efficacy to
sulfasalazine and
appear to be particularly useful for treating perianal disease. In more severe
cases,
corticosteroids are effective in treating active exacerbations and can even
maintain
remission. Azathioprine and 6-mercaptopurine have also shown success in
patients who
require chronic administration of cortico steroids. It is also possible that
these drugs may
play a role in the long-term prophylaxis. Unfortunately, there can be a very
long delay
(up to six months) before onset of action in some patients.
Antidiarrheal drugs can also provide symptomatic relief in some patients.
Nutritional therapy or elemental diet can improve the nutritional status of
patients and
induce symtomatic improvement of acute disease, but it does not induce
sustained
clinical remissions. Antibiotics are used in treating secondary small bowel
bacterial
overgrowth and in treatment of pyogenic complications.
"Ulcerative colitis (UC)" afflicts the large intestine. The course of the
disease
may be continuous or relapsing, mild or severe. The earliest lesion is an
inflammatory
infiltration with abscess formation at the base of the crypts of Lieberkan.
Coalescence
of these distended and raptured crypts tends to separate the overlying mucosa
from its
blood supply, leading to ulceration. Symptoms of the disease include cramping,
lower
.. abdominal pain, rectal bleeding, and frequent, loose discharges consisting
mainly of
blood, pus and mucus with scanty fecal particles. A total colectomy may be
required for
acute, severe or chronic, unremitting ulcerative colitis. The clinical
features of UC are
highly variable, and the onset may be insidious or abrupt, and may include
diarrhea,
tenesmus and relapsing rectal bleeding. With fulminant involvement of the
entire colon,
toxic megacolon, a life-threatening emergency, may occur. Extraintestinal
manifestations
include arthritis, pyoderma gangrenoum, uveitis, and erythema nodosum.
The term "serum sample" refers to any serum sample obtained from an
individual. Methods for obtaining sera from mammals are well known in the art.
The term "whole blood" refers to any whole blood sample obtained from an
.. individual. Typically, whole blood contains all of the blood components,
e.g., cellular
components and plasma. Methods for obtaining whole blood from mammals are well
known in the art.
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
THERAPEUTIC AGENTS
A therapeutic agent for treating gastrointestinal inflammatory disorders
(e.g.,
Inflammatory bowel disease) is provided herein. Inflammatory bowel disease
(IBD) is a
chronic gastrointestinal disease that severely affects patient quality of life
and often
results in the need for surgical intervention (Casellas et al., Dig Dis.
1999;17(4):208-18;
Carter et al., Gut. 2004;53(Suppl 5):V1-16; Borren et al., Nat Rev
Gastroenterol
Hepatol. 2019;16(4):247-59). The predominant forms of IBD are ulcerative
colitis (UC)
and Crohn's disease, 2 distinct conditions that share some common symptoms and
exhibit a partially overlapping etiology (Zhang and Li, World J Gastroenterol.
2014;20(1):91-9; Abraham and Cho, N Engl J Med. 2009;361(21):2066-78). Current
pharmacologic therapies for IBD are not curative. In addition, many
pharmacologic
therapies for IBD lose efficacy over the duration of the disease and can
result in systemic
side effects (Abraham and Cho, N Engl J Med. 2009;361(21):2066-78; Rogler,
Best
Pract Res Clin Gastroenterol. 2010;24(2):157-65). Etrolizumab is an anti-07
integrin
monoclonal antibody in development for patients with UC and Crohn's disease.
Etrolizumab selectively inhibits a407 and aFf37 to reduce trafficking of
immune cells
into the gut and subsequent inflammatory effects on the gut lining (Zundler et
al., Gut.
2019;68(9):1688-700). The efficacy and safety of etrolizumab in patients with
UC was
demonstrated in the phase 2 EUCALYPTUS study (Vermeire et al., Lancet.
2014;384(9940):309-18).
In certain embodiments, the therapeutic agent is an anti-integrin beta7
antibody
or an antigen-binding fragment thereof. In certain embodiments, the anti-
integrin beta7
antibody is a humanized monoclonal anti-integrin beta7 antibody. In certain
such
embodiments, the anti-integrin beta7 antibody or antigen-binding fragment
thereof
comprises three light chain hypervariable regions (HVRs), HVR-L1, HVR-L2, and
HVR-L3, and three heavy chain HVRs, HVR-H1, HVR-H2, and HVR-H3, wherein: (i)
the HVR-L1 comprises the amino acid sequence set forth SEQ ID NO: 1; (ii) the
HVR-
L2 comprises the amino acid sequence set forth in SEQ ID NO: 2; (iii) the HVR-
L3
comprises the amino acid sequence set forth in SEQ ID NO: 3; (iv) the HVR-H1
comprises the amino acid sequence set forth in SEQ ID NO: 4; (v) the HVR-H2
comprises the amino acid sequence set forth in SEQ ID NO: 5; and (vi) the HVR-
H3
comprises the amino acid sequence set forth in SEQ ID NO: 6 or SEQ ID NO: 7.
In certain such embodiments, the anti-integrin beta7 antibody or antigen-
binding
fragment thereof further comprises a light chain variable region domain
comprising the
26
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
amino acid sequence set forth in SEQ ID NO:8 and a heavy chain variable region
domain
comprising the amino acid sequence set forth in SEQ ID NO:9. In certain
embodiments,
the anti-integrin beta7 antibody or antigen-binding fragment thereof comprises
a light
chain variable region domain comprising the amino acid sequence set forth in
SEQ ID
NO: 8 and a heavy chain comprising the amino acid sequence set forth in SEQ ID
NO:
11 or a heavy chain comprising the amino acid sequence set forth in SEQ ID NO:
12. In
certain embodiments, the anti-integrin beta7 antibody or antigen-binding
fragment
thereof comprises a light chain comprising the amino acid sequence set forth
in SEQ ID
NO: 10 and a heavy chain variable region domain comprising the amino acid
sequence
set forth in SEQ ID NO: 9.
In certain such embodiments, the anti-integrin beta7 antibody or antigen-
binding
fragment thereof further comprises a light chain comprising the amino acid
sequence set
forth in SEQ ID NO:10 and a heavy chain comprising the amino acid sequence set
forth
in SEQ ID NO: 11 or a heavy chain comprising the amino acid sequence set forth
in SEQ
ID NO: 12. In certain such embodiments, the anti-integrin beta7 antibody is
etrolizumab. SEQ ID NOS: 1-12 are provided below. Anti-integrin beta7
antibodies or
antigen-binding fragments thereof are further described in Intn'l Pub. No.
W02006/026759, which is incorporated herein by reference.
SEQ ID Description Sequence
NO
1 anti-integrin beta7 antibody HVR-L1 RASESVDDLLH
2 anti-integrin beta7 antibody HVR-L2 KYASQSIS
3 anti-integrin beta7 antibody HVR-L3 QQGNSLPNT
4 anti-integrin beta7 antibody HVR-Hl GFFITNNYWG
5 anti-integrin beta7 antibody HVR-H2 GYISYSGSTSYNPSLKS
6 anti-integrin beta7 antibody HVR- RTGSSGYFDF
H3 .v1
7 anti-integrin beta7 antibody HVR- ARTGSSGYFDF
H3.v2
8 anti-integrin beta7 antibody VL DIQMTQSPSSLSASVGDRVTITCRASESVD
DLLHWYQQKPGKAPKLLIKYASQSISGVPS
RFSGSGSGTDFTLTISSLQPEDFATYYCQQ
GNSLPNTFGQGTKVEIKR
9 anti-integrin beta7 antibody VH EVQLVESGGGLVQPGGSLRLSCAASGFFIT
NNYWGWVRQAPGKGLEWVGYISYSGSTSYN
PSLKSRFTIS RDTSKNTFYLQMNSLRAEDT
AVYYCARTGSSGYFDFWGQGTLVTVSS
27
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
anti-integrin beta7 antibody LC DIQMTQSPSS LSASVGDRVT ITCRASESVD
DLLHWYQQKPGKAPKLLIKYASQSISGVPS
RFSGSGSGTD FTLTISSLQPEDFATYYCQQ
GNSLPNTFGQGTKVEIKRTVAAPSVFIFPP
SDEQLKSGTASVVCLLNNFYPREAKVQWKV
DNALQSGNSQESV1EQDSKDSTYSLSSTLT
LSKADYEKHKVYACEVTHQGLSSPVTKSFN RGEC
11 anti-integrin beta7 antibody HC.v 1 EVQLVESGGG LVQPGGSLRLSCAASGFFIT
NNYWGWVRQAPGKGLEWVGYISYSGSTSYN
PSLKSRFTIS RDTSKNTFYLQMNSLRAEDT
AVYYCARTGSSGYFDFWGQG TLVTVS SA ST
KGPSVFPLAPSSKSTSGGTAALGCLVKDYF
PEPVTVS WNS GALT SGVHTFPAVLQS SGLY
SLSSVVTVPSSSLGTQTYICNVNHKPSNTK
VDKKVEPKSCDKTHTCPPCPAPELLGGPSV
FLFPPKPKDTLMISRTPEVTCVVVDVSHED
PEVKFNWYVDGVEVHNAKTKPREEQYNSTY
RVVSVLTVLHQDWLNGKEYKCKVSNKALPA
PIEKTISKAKGQPREPQVYTLPPSREEMTK
NQVSLTCLVKGFYPSDIAVEWESNGQPENN
YKTTPPVLDSDGSFFLYSKLTVDKSRWQQG
NVFSCSVMHEALHNHYTQKSLSL SP G
12 anti-integrin beta7 antibody HC.v2 EVQLVESGGG LVQPGGSLRLSCAASGFFIT
NNYWGWVRQAPGKGLEWVGYISYSGSTSYN
PSLKSRFTIS RDTSKNTFYLQMNSLRAEDT
AVYYCARTGSSGYFDFWGQG TLVTVS SA ST
KGPSVFPLAPSSKSTSGGTAALGCLVKDYF
PEPVTVS WNS GALT SGVHTFPAVLQS SGLY
SLSSVVTVPSSSLGTQTYICNVNHKPSNTK
VDKKVEPKSCDKTHTCPPCPAPELLGGPSV
FLFPPKPKDTLMISRTPEVTCVVVDVSHED
PEVKFNWYVDGVEVHNAKTKPREEQYNSTY
RVVSVLTVLHQDWLNGKEYKCKVSNKALPA
PIEKTISKAKGQPREPQVYTLPPSREEMTK
NQVSLTCLVKGFYPSDIAVEWESNGQPENN
YKTTPPVLDSDGSFFLYSKLTVDKSRWQQG
NVFSCSVMHEALHNHYTQKSLSL SP GK
CERTAIN MOLECULAR BIOMARKERS
[0001] In certain instances, biomarkers are quantitated in a biological
sample
obtained from a subject as a means of selecting subjects for treatment with a
given
therapeutic agent. International Pat. Nos. W02014160753, W02015148809, and
5 W02009140684 describe methods of predicting the responsiveness of
subjects having a
gastrointestinal inflammatory disorder to anti-integrin beta7 antibody
formulations
described herein, and methods of selecting subjects having a gastrointestinal
inflammatory disorder for treatment with anti-integrin beta7 antibody
formulations
described herein.
28
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
GENERAL TECHNIQUES FOR FORMULATIONS
Formulations comprising anti-integrin beta7 antibodies or antigen-binding
fragments thereof may be prepared and analyzed using certain excipients and
techniques
known in the art and as further described herein. In certain embodiments, the
antibody to
be formulated has not been subjected to prior lyophilization and the
formulation of
interest herein is an aqueous formulation. In certain embodiments, the
antibody is a full-
length antibody. In certain embodiments, the antibody in the formulation is an
antibody
fragment, such as an F(ab')2, in which case problems that may not occur for
the full-
length antibody (such as clipping of the antibody to Fab) may need to be
addressed. The
therapeutically effective amount of antibody present in the formulation is
determined by
taking into account the desired dose volumes and mode(s) of administration. In
certain
embodiments, from about 0.1 mg/mL to about 250 mg/mL, or from about 10 mg/mL
to
about 220 mg/mL, or from about 50 mg/mL to about 220 mg/mL, or from about 100
mg/mL to about 220 mg/mL, or from about 100 mg/mL to about 150 mg/mL, or from
about 150 mg/mL to about 200 mg/mL is an exemplary antibody concentration in
the
formulation. In certain embodiments, the anti-integrin beta7 antibody is
formulated at a
concentration of 150 mg/mL.
An aqueous formulation is prepared comprising the anti-integrin beta7 antibody
or an antigen-binding fragment thereof in a pH-buffered solution. The buffer
can have a
pH in the range from about 4.5 to about 6.5. In certain embodiments, the pH is
greater
than 5.0 and up to 7Ø In certain embodiments, the pH is greater than 5.5. In
certain
embodiments, the pH is between 5.5 and 6.1. In certain embodiments, the pH is
between
5.6 and 6.1. In certain embodiments, the pH is 5.8 or about 5.8. In certain
embodiments,
the pH is 5.8. In certain embodiments, the pH is between 5.7 and 5.9. In
certain
embodiments, the pH is between 5.75 and 5.85. The pH of the presently
disclosed
formulation is higher than standard for an antibody formulation with similar
excipient
composition. Typical antibody formulations have a pH of 5.5, whereas the
presented
disclosed formulation has a pH of greater than 5.5, e.g., a pH of 5.8, between
5.7 and 5.9
or between 5.75 and 5.85. The higher formulation pH lowers the risk of
particle
formation as a result of polysorbate degradation during long term storage in a
pre-filled
syringe at high protein concentration. The risk of particle formation is
lowered due to the
increased solubility of free fatty acids at the higher pH which can result
from polysorbate
degradation. A pH of greater than 5.5., e.g., a pH of 5.8, balances the risk
of particle
formation with the chemical and physical stability of the antibody. A pH of
greater than
29
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
5.5., e.g., a pH of 5.8, minimizes the rate of Asp isomerization and
succinimide
intermediate formation, which allows for patient convenience in combination
with the
device by allowing storage at ambient temperatures without substantially
impacting the
chemical stability of the antibody and thereby the product quality.
Examples of buffers that will control the pH within this range include acetate
(e.g. histidine acetate, arginine acetate, sodium acetate), succinate (such as
histidine
succinate, arginine succinate, sodium succinate), gluconate, citrate and other
organic acid
buffers and combinations thereof. The buffer concentration can be from about 1
mM to
about 600 mM, depending, for example, on the buffer and the desired
isotonicity of the
formulation. In certain embodiments, the buffer comprises histidine. The
presence of
histidine in the formulation can greatly reduce the rate of high molecular
weight species
(HMWS) formation in the presence of zinc. The concentration of the histidine
in the
formulation can be between about 5 mM and about 40 mM, between about 5 mM and
about 30 mM, between about 10 mM and about 40 mM, between about 10 mM and
about 30 mM, between about 15 mM and about 25 mM, between about 10 mM and
about 20 mM, or between about 15 mM and about 20 mM. In certain embodiments,
the
concentration of the histidine in the formulation is about 20 mM.
In certain embodiments, the buffer is 20 mM histidine, pH 5.8.
In certain embodiments, the formulation comprises arginine succinate. In
certain
embodiments, the concentration of the arginine succinate in the formulation is
from
about 20 mM to 300 mM. In certain embodiments, the concentration of the
arginine
succinate in the formulation is from about 100 mM to 300 mM, from about 100 mM
to
about 200 mM, from about 150 mM to about 300 mM, from about 200 mM to about
300
mM, from about 100 mM to about250 mM, from about 150 mM to about 250 mM, or
from about 150 mM to about 200 mM. In certain embodiments, the concentration
of the
arginine succinate in the formulation is about 200 mM. The high arginine
concentration,
and high conductivity formulation shields charge on the antibody and prevents
shifts in
pH. In addition, arginine can impact the viscosity of the formulation, e.g.,
formulation
viscosity is decreased by the addition of arginine to the formulation.
The formulation has a viscosity of less than about 20 centipoise (cP) at 25 C.
In
certain embodiments, the viscosity of the formulation is between about 1 cP
and about 20
cP at 25 C, between about 5 cP and about 20 cP at 25 C, between about 5 cP and
about
15 cP at 25 C, between about 1 cP and about 10 cP at 25 C, or between about 5
cP and
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
about 10 cP at 25 C. In certain embodiments, the viscosity of the formulation
is about 7
cP at 25 C.
In certain embodiments, the antibody formulation comprises a surfactant.
Exemplary surfactants include nonionic surfactants. Suitable non-ionic
surfactants
include polysorbates (20, 40, 60, 65, 80, etc.), poloxamers (184, 188, etc.),
Pluronic
polyols, Triton , polyoxyethylene sorbitan monoethers (Tween -20, Tween -80,
etc.),
lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor
oil 10, 50
and 60, glycerol monostearate, sucrose fatty acid ester, methyl celluose and
carboxymethyl cellulose. Anionic detergents that can be used include sodium
lauryl
sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate. Cationic
detergents
include benzalkonium chloride or benzethonium chloride. In certain
embodiments, the
surfactant is polysorbate 20.
Non-ionic surfactants can help solubilize the therapeutic agent as well as to
protect the therapeutic protein against agitation-induced aggregation, which
also permits
the formulation to be exposed to shear surface stress without causing
denaturation of the
active therapeutic protein or antibody.
The amount of surfactant added is such that it reduces aggregation of the
formulated antibody and/or minimizes the formation of particulates in the
formulation
and/or reduces adsorption. For example, the surfactant may be present in the
formulation
in an amount of greater than 0.005% weight/volume (w/v). In certain
embodiments, the
concentration of the surfactant in the formulation is greater than 0.005% w/v
and up to
about 1% w/v. The concentration of the surfactant in the formulation can be
between
about 0.005% and about 0.5% w/v, between about 0.02% w/v and about 0.5% w/v,
between about 0.03% w/v and about 0.5% w/v, between 0.03% w/v and 0.1% w/v. In
certain embodiments, concentration of the surfactant in the formulation is
0.04% w/v. In
certain embodiments, the surfactant is polysorbate 20 present in the
formulation in an
amount of 0.04% w/v. The typical concentration of polysorbate 20 for an
antibody
formulation is 0.02% (w/v). The presently disclosed formulation comprises
0.04% w/v
Polysorbate 20. The higher concentration of polysorbate 20 helps solubilize
free fatty
acids, which can be generated as a result of polysorbate degradation, thereby
lowering
the risk of forming particles.
In certain embodiment, the formulation contains the above-identified agents
(e.g.,
antibody, buffer, and surfactant) and is essentially free of one or more
preservatives,
31
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
such as benzyl alcohol, phenol, m-cresol, chlorobutanol and benzethonium Cl.
In certain
embodiments, the formulation does not comprise a preservative. In certain
embodiments, a preservative may be included in the formulation, particularly
where the
formulation is a multidose formulation. The concentration of preservative may
be in the
range from about 0.1% to about 2%, or from about 0.5% to about 1%. One or more
other pharmaceutically acceptable carriers, excipients or stabilizers such as
those
described in Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed.
(1980)
may be included in the formulation provided that they do not adversely affect
the desired
characteristics of the formulation. Acceptable carriers, excipients or
stabilizers are
nontoxic to recipients at the dosages and concentrations employed and include;
additional buffering agents; co-solvents; anti-oxidants including ascorbic
acid and
methionine; chelating agents such as EDTA; metal complexes (e.g. Zn-protein
complexes); biodegradable polymers such as polyesters; and/or salt-forming
counterions.
While the various descriptions of chelators herein often focus on EDTA, it
will
be appreciated that other metal ion chelators are also encompassed within the
invention.
Metal ion chelators are well known by those of skill in the art and include,
but are not
necessarily limited to aminopolycarboxylates, EDTA (ethylenediaminetetraacetic
acid),
EGTA (ethylene glycol-bis(beta-aminoethyl ether)-N,N,N',N'-tetraacetic acid),
NTA
(nitrilotriacetic acid), EDDS (ethylene diamine disuccinate), PDTA (1,3-
propylenediaminetetraacetic acid), DTPA (diethylenetriaminepentaacetic acid),
ADA
(beta-alaninediacetic acid), MGCA (methylglycinediacetic acid), etc.
Additionally,
some embodiments herein comprise phosphonates/phosphonic acid chelators.
Tonicity agents, sometimes known as "stabilizers" are present to adjust or
maintain the tonicity of a liquid composition. When used with large, charged
biomolecules such as proteins and antibodies, they are often termed
"stabilizers" because
the can interact with the charged groups of the amino acid side chains,
thereby lessening
the potential for inter and intra-molecular interactions. Tonicity agents can
be present in
any amount between 0.1% to 25% by weight, or 1 to 5%, taking into account the
relative
amounts of the other ingredients. Tonicity agents include polyhydric sugar
alcohols,
thrihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol,
xylitol, sorbitol
and mannitol.
Additional stabilizers include a broad range of excipients which range in
function
from bulking agents to solubility enhancers, to agents preventing denaturation
or
adherence to the container wall. Stabilizers can be present in the range from
0.1 to
32
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
10,000 parts per weight active protein or antibody. Typical stabilizers
include:
polyhydric sugar alcohols (enumerated above); amino acids such as alanine,
glycine,
glutamine, asparagine, histidine, arginine, lysine, methionine, ornithine,
leucine, 2-
phenylalanine, glutamic acid, threonine, etc.; organic sugars or sugar
alcohols such as
.. sucrose, lactose, lactitol, trehalose, stachyose, mannose, sorbose, xylose,
ribose, ribitol,
myoinisitose, myoinisitol, galactose, gal actitol, glycerol, cyclitols (e.g.,
inositol),
polyethylene glycol; sulfur containing reducing agents, such as urea,
glutathione, thioctic
acid, sodium thioglycolate, thioglycerol, a-monothioglycerol and sodium thio
sulfate;
low molecular weight proteins such as human serum albumin, bovine serum
albumin,
gelatin or other immunoglobulins; hydrophilic polymers such as
polyvinylpyrrolidone;
monosaccharides (e.g., xylose, mannose, fructose, glucose; disaccharides
(e.g., lactose,
maltose, sucrose); trisaccharides such as raffinose; and polysaccharides such
as dextrin
or dextran.
Various analytical techniques for measuring protein stability are available in
the
art and are reviewed in Peptide and Protein Drug Delivery, 247-301, Vincent
Lee Ed.,
Marcel Dekker, Inc., New York, N.Y., Pubs. (1991) and Jones, A. Adv. Drug
Delivery
Rev. 10: 2990 (1993), for example. Stability can be measured at a selected
temperature
for a selected time period. In certain embodiments, the formulation is stable
at about
40 C for at least about 1 week. In certain embodiments, the formulation is
stable at
about 5 C for at least about 12 months, and/or or stable at about 5 C for at
least about 18
months, and/or or stable at about 5 C for at least about 2 years, and/or or
stable at about
5 C for at least about 3 years, and/or or stable at about 5 C for at least
about 4 years,
and/or or stable at about 5 C for at least about 5 years. In certain
embodiments, the
formulation is stable at about -20 C for at least 2 years, and/or stable at
about -20 C for
at least 4 years, and/or stable at about -20 C for at least about 5 years,
and/or stable at
about -20 C for at least about 6 years, and/or stable at about -20 C for at
least about 7
years. In certain embodiments, the formulation is stable at about 25 C for at
least about
1 week, and/or stable at about 25 C for at least about 2 weeks, or stable at
about 25 C for
at least about 4 weeks. In certain embodiments, the formulation is stable
following
freezing (to, e.g., -70 C) and thawing of the formulation, for example
following 1, 2, 3,
4, or 5 cycles of freezing and thawing. Stability can be evaluated
qualitatively and/or
quantitatively in a variety of different ways, including evaluation of
aggregate formation
(for example using size exclusion chromatography, by measuring turbidity,
and/or by
visual inspection); by assessing charge heterogeneity using cation exchange
33
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
chromatography, image capillary isoelectric focusing (icIEF) or capillary zone
electrophoresis; amino-terminal or carboxy-terminal sequence analysis; mass
spectrometric analysis; SDS-PAGE analysis to compare reduced and intact
antibody;
peptide map (for example tryptic or LYS-C) analysis; evaluating biological
activity or
antigen binding function of the antibody; etc. Instability may involve any one
or more
of: aggregation, deamidation (e.g. Asn deamidation), oxidation (e.g. Met
oxidation),
isomerization (e.g. Asp isomerization), clipping/hydrolysis/fragmentation
(e.g. hinge
region fragmentation), succinimide formation, unpaired cysteine(s), N-terminal
extension, C-terminal processing, glycosylation differences, etc.
The formulations to be used for in vivo administration should be sterile. This
is
readily accomplished by filtration through sterile filtration membranes, prior
to, or
following, preparation of the formulation.
In certain embodiments, the anti-integrin beta7 antibody or antigen-binding
fragment thereof is administered using, for example, a self-inject device,
autoinjector
device, or other device designed for self-administration. In certain
embodiments, the
anti-integrin beta7 antibody or antigen-binding fragment thereof is
administered using a
subcutaneous administration device. Various self-inject devices and
subcutaneous
administration devices, including autoinjector devices, are known in the art
and are
commercially available. Exemplary devices include, but are not limited to,
prefilled
syringes (such as BD HYPAK SCF , BD NEOPAKTM, READYFILLTm, and
STERIFILL SCFTM from Becton Dickinson; CLEARSHOTTm copolymer prefilled
syringes from Baxter; and Daikyo Seiko CRYSTAL ZENITH prefilled syringes
available from West Pharmaceutical Services); disposable pen injection devices
such as
BD Pen from Becton Dickinson; ultra-sharp and microneedle devices (such as
INJECT-
EASE' and microinfuser devices from Becton Dickinson; and H-PATCHTm available
from Valeritas) as well as needle-free injection devices (such as BIOJECTOR
and
IJECT available from Bioject; and SOF-SERTER and patch devices available
from
Medtronic). Certain embodiments of subcutaneous administration devices are
described
further herein. Co-formulations or co-administrations with such self-inject
devices or
subcutaneous administration devices of an anti-integrin beta7 antibody or an
antigen-
binding fragment thereof with at least a second therapeutic compound are
envisioned.
RECOMBINANT METHODS
Antibodies may be produced using recombinant methods and compositions, e.g.,
as described in U.S. Patent No. 4,816,567. In certain embodiments, a nucleic
acid
34
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
molecule encoding an anti-integrin beta7 antibody or an antigen-binding
fragment
thereof described herein is provided. Such nucleic acid molecule may encode an
amino
acid sequence comprising the VL and/or an amino acid sequence comprising the
VH of
the antibody (e.g., the light and/or heavy chains of the antibody). In certain
embodiments, one or more vectors (e.g., expression vectors) comprising such
nucleic
acid molecules are provided. In certain embodiments, a host cell comprising
such
nucleic acid molecule is provided. In certain embodiments, a host cell
comprises (e.g.,
has been transformed with): (1) a vector comprising a nucleic acid molecule
that encodes
an amino acid sequence comprising the VL of the antibody and an amino acid
sequence
comprising the VH of the antibody, or (2) a first vector comprising a nucleic
acid
molecule that encodes an amino acid sequence comprising the VL of the antibody
and a
second vector comprising a nucleic acid molecule that encodes an amino acid
sequence
comprising the VH of the antibody. In certain embodiments, the host cell is
eukaryotic,
e.g. a Chinese Hamster Ovary (CHO) cell or lymphoid cell (e.g., YO, NSO, Sp20
cell).
In certain embodiments, a method of making an anti-integrin beta7 antibody is
provided,
wherein the method comprises culturing a host cell comprising a nucleic acid
molecule
encoding the antibody, as provided above, under conditions suitable for
expression of the
antibody, and optionally recovering the antibody from the host cell (or host
cell culture
medium).
For recombinant production of an anti-integrin beta7 antibody, nucleic acid
molecules encoding an antibody, e.g., as described above, is isolated and
inserted into
one or more vectors for further cloning and/or expression in a host cell. Such
nucleic
acid molecules may be readily isolated and sequenced using conventional
procedures
(e.g., by using oligonucleotide probes that are capable of binding
specifically to genes
encoding the heavy and light chains of the antibody).
Suitable host cells for cloning or expression of antibody-encoding vectors
include
prokaryotic or eukaryotic cells described herein. For example, antibodies may
be
produced in bacteria, in particular when glycosylation and Fc effector
function are not
needed. For expression of antibody fragments and polypeptides in bacteria,
see, e.g.,
U.S. Patent Nos. 5,648,237, 5,789,199, and 5,840,523. (See also Charlton,
Methods in
Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003),
pp.
245-254, describing expression of antibody fragments in E. colt) After
expression, the
antibody may be isolated from the bacterial cell paste in a soluble fraction
and can be
further purified.
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or
yeast are suitable cloning or expression hosts for antibody-encoding vectors,
including
fungi and yeast strains whose glycosylation pathways have been "humanized,"
resulting
in the production of an antibody with a partially or fully human glycosylation
pattern.
See Gerngross, Nat. Biotech. 22:1409-1414 (2004), and Li et al., Nat. Biotech.
24:210-
215 (2006).
Suitable host cells for the expression of glycosylated antibody are also
derived
from multicellular organisms (invertebrates and vertebrates). Examples of
invertebrate
cells include plant and insect cells. Numerous baculoviral strains have been
identified
which may be used in conjunction with insect cells, particularly for
transfection of
Spodoptera frugiperda cells.
Plant cell cultures can also be utilized as hosts. See, e.g., US Patent Nos.
5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429 (describing
PLANTIBODIESTM technology for producing antibodies in transgenic plants).
Vertebrate cells may also be used as hosts. For example, mammalian cell lines
that are adapted to grow in suspension may be useful. Other examples of useful
mammalian host cell lines are monkey kidney CV1 line transformed by 5V40 (COS-
7);
human embryonic kidney line (293 or 293 cells as described, e.g., in Graham et
al., J.
Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK); mouse sertoli cells
(TM4
cells as described, e.g., in Mather, Biol. Reprod. 23:243-251 (1980)); monkey
kidney
cells (CV1); African green monkey kidney cells (VERO-76); human cervical
carcinoma
cells (HELA); canine kidney cells (MDCK; buffalo rat liver cells (BRL 3A);
human lung
cells (W138); human liver cells (Hep G2); mouse mammary tumor (MMT 060562);
TRI
cells, as described, e.g., in Mather et al., Annals N.Y. Acad. Sci. 383:44-68
(1982); MRC
5 cells; and F54 cells. Other useful mammalian host cell lines include Chinese
hamster
ovary (CHO) cells, including DHFR-CHO cells (Urlaub et al., Proc. Natl. Acad.
Sci.
USA 77:4216 (1980)); and myeloma cell lines such as YO, NSO and Sp2/0. For a
review of certain mammalian host cell lines suitable for antibody production,
see, e.g.,
Yazaki and Wu, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana
Press, Totowa, NJ), pp. 255-268 (2003).
36
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
ASSAYS
Anti-integrin beta7 antibodies provided herein may be identified, screened
for, or
characterized for their physical/chemical properties and/or biological
activities by
various assays known in the art.
The etrolizumab potency assay measures the ability of etrolizumab to inhibit
RPMI8866 B cell binding to MAdCAM. In this assay, MAdCAM was coated onto a
96-well microtiter plate. Following overnight incubation, etrolizumab
standards,
controls, and samples were added to the plate, along with a fixed amount of
cells. The
plate was incubated at 37 C in a humidified incubator to allow binding of the
cells to the
MAdCAM. A wash step was performed to remove non-adherent cells, and the
remaining live cells were quantified by adding the redox dye alamar Blue,
which is blue
and non-fluorescent in its oxidized state but is reduced by the intracellular
environment
into a pink form that is highly fluorescent. Thus, changes in color and
fluorescence were
proportional to the number of bound viable cells. The results were expressed
in RFU
and plotted against etrolizumab concentration. Parallel curve analysis was
used to
estimate the activity of the etrolizumab sample(s) relative to the Reference
Material.
Potency Assay
ARTICLES OF MANUFACTURE AND KITS
An article of manufacture is provided which comprises the formulation and
provides instructions for its use. The article of manufacture comprises a
container.
In certain embodiments, an article of manufacture comprising a subcutaneous
administration device is provided which delivers to a subject a flat dose of
an anti-
integrin beta7 antibody or an antigen-binding fragment thereof, wherein the
flat dose is
for example, but not limited to, 105 mg, or 210 mg. In certain embodiments,
the anti-
integrin beta7 antibody is etrolizumab. The anti-integrin beta7 antibody or
antigen-
binding fragment thereof in the subcutaneous administration device is
formulated in a
buffer, for example, histidine pH 5.8, and other excipients, for example,
polysorbate and
arginine succinate, such that it is provided in a stable pharmaceutical
formulation.
In certain embodiments, the subcutaneous administration device is a prefilled
syringe comprising a glass barrel with needle and optionally, a needle shield
and also
optionally, a needle shield device. In certain embodiments, the volume of the
formulation contained in the syringe is between about 0.1 mL and about 2 mL,
between
about 0.1 mL and about 2 mL, between about 0.5 mL and about 2 mL, or between
about
37
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
1 mL and about 2 mL. In certain embodiments, the volume of the formulation
contained
in the syringe is between about 0.5 mL and about 2 mL. In certain embodiments,
the
volume of the formulation contained in the syringe is about 0.5 mL, about 0.7
mL, about
1 mL, about 1.4 mL, about 1.5 mL, or about 2.0 mL. In certain embodiments, the
volume of the formulation contained in the syringe is about 0.7 mL. In certain
embodiments, the volume of the formulation contained in the syringe is about
0.75 mL.
In certain embodiments, the volume of the formulation contained in the syringe
is about
1 mL. In certain embodiments, the volume of the formulation contained in the
prefilled
syringe is between about 0.5 mL and about 1.0 mL. In certain embodiments, the
volume
of the formulation contained in the prefilled syringe is between about 1.0 mL
and about
1.5 mL. In certain embodiments, the volume of the formulation contained in the
prefilled syringe is about 1.4 mL. In certain embodiments, the volume of the
formulation
contained in the prefilled syringe is about 1.5 mL. In certain embodiments,
the volume
of the formulation contained in the prefilled syringe is about 1.45 mL.
In certain embodiments, the needle is a staked-in needle comprising a 3-bevel
tip
or a 5-bevel tip. In certain embodiments, the needle is between 25 gauge (G)
and 30G.
In certain embodiments, the needle is between 1/2 inch long and 5/8 inch long.
In
certain embodiments, the subcutaneous administration device comprises a
prefilled 1.0
mL low tungsten borosilicate glass (type I) syringe and a stainless steel 5-
bevel 27G 1/2
inch long thin-wall staked-in needle. In certain embodiments, the subcutaneous
administration device comprises a rigid needle shield. In certain embodiments,
the rigid
needle shield comprises a rubber formulation having low zinc content and
comprises a
rigid polypropylene shield. In certain embodiments, the rubber plunger stopper
comprises Daikyo 777-7 rubber and FluroTec ethylene-tetrafluoroethylene
(ETFE)
coating (West Pharmaceutical Services, Inc.). In certain embodiments, the
subcutaneous
administration device comprises a needle safety device. Exemplary needle
safety
devices include, but are not limited to, Ultrasafe Passive Needle Guard X100L
(Becton
Dickinson and Company) and Rexam Safe n Sound Tm (Rexam).
In certain embodiments, the injection device is a prefilled syringe. Non-
limiting
.. examples of prefilled syringes include BD HYPAK SCF , READYFILLTm, and
STERIFILL SCFTM from Becton Dickinson; CLEARSHOTTm copolymer prefilled
syringes from Baxter; and Daikyo Seiko CRYSTAL ZENITH prefilled syringes
available from West Pharmaceutical Services. In certain embodiments, the
prefilled
syringe comprises silicone oil. The etrolizumab prefilled syringe was
developed with
38
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
optimal levels of silicone oil in order to ensure low injection forces while
maintaining a
low risk of particle formation. Free fatty acids from degraded polysorbate
partition to
silicone oil to a greater or lesser extent depending on the pH and the
Polysorbate
concentration. Hence, the specific combination of the formulation excipients
at the
target pH and the target levels of silicone oil in combination are uniquely
well suited to
long term storage of a high concentration antibody formulation stored in a
prefilled
syringe. In certain embodiments, the amount of silicone oil in the prefilled
syringe is not
greater than about 1 mg. In certain embodiments, the amount of silicone oil in
the
prefilled syringe is between about 0.1 mg and about 1 mg. In certain
embodiments, the
.. amount of silicone oil in the prefilled syringe is between about 0.2 mg and
about 0.6 mg.
In certain embodiments, the amount of silicone oil in the prefilled syringe is
between
about 0.5 mg and 0.9 mg.
The prefilled syringe may have any suitable syringe capacity. In certain
embodiments, the prefilled syringe has a syringe capacity of between about 0.5
mL and
about 10 mL, between about 0.5 mL and about 5 mL, between about 0.5 mL and
about
2.5 mL, between about 1 mL and about 5 mL, or between about 1 mL and about 2.5
mL.
In certain embodiments, the prefilled syringe has a syringe capacity of 1 mL.
In certain
embodiments, the prefilled syringe has a syringe capacity of 2.25 mL.
In certain embodiments, the injection device is an autoinjector, e.g.,
autoinjectors
disclosed in U.S. Pat. Nos. 2014/0148763 and 2014/0114247, which are
incorporated by
reference herein. In certain embodiments, the autoinjector is a single-use
autoinjector.
In certain embodiments, the autoinjector is based on Rotaject technology
(e.g.,
accessible at https://www.shl.group/products-and-services/rotaject-technology-
auto-
injector/).
Additional devices suitable for subcutaneous delivery include for example, but
not limited to, an injection device such as INJECT-EASE Tm and GENJECTIm
devices;
an infusion pump such as ACCU-CHECK; an injector pen such as BD VystraTM from
Becton Dickinson, a needleless device such as MEDDCTORTm and BIOJECTORTm and
IJECTTm, and a subcutaneous patch delivery system such as BD Libertas TM, H-
PATCH TM available from Valeritas, and SOF-SERTERTM.
Kits will typically comprise the container described above and one or more
other
containers comprising materials desirable from a commercial and user
standpoint,
including buffers, diluents, filters, needles, syringes, and package inserts
with
39
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
instructions for use. A label may be present on the container to indicate that
the
composition is used for a specific therapy.
EXAMPLES
The following are examples of the formulations and methods of the invention.
It
is understood that various other embodiments may be practiced, given the
general
description provided above.
Example 1
Important aspects of the optimization of the etrolizumab drug product
.. formulation and configuration are minimizing viscosity to minimize
injection force,
maximizing the accuracy of the injection volume and optimizing compatibility
with
device components. Balancing the optimal formulation excipients to
sufficiently protect
the antibody from physical and chemical degradation, while maintaining
biological
activity (potency) and achieving high concentration of antibody while
maintaining low
viscosity is important. These aspects of the formulation must be balanced in
concert
with the prefilled syringe configuration and specifications such as silicone
oil
concentration, syringe materials of construction, barrel width, and needle
wall thickness,
which allow for lower injection force, shorter injection time and less pain as
the result of
injection.
In the present example, an etrolizumab formulation comprising 150 mg/ml
etrolizumab, 20 mM histidine, 200 mM arginine succinate, and 0.04% w/v of
polysorbate 20 (PS20) at pH 5.8 was prepared. It was shown that this
etrolizumab
formulation was unexpectedly well suited for long-term storage at high
concentration in
a pre-filled syringe. Furthermore, when paired with prefilled syringes, this
formulation
minimized the risk of subvisible and visible particle formation upon long-term
storage,
which allow for extended shelf-life and patient-convenience. Impacts of the
formulation
pH, arginine, succinate, polysorbate 20, antibody concentration, and histidine
on the
viscosity and stability (e.g., aggregate formulation, charge variants, etc.)
of an
etrolizumab formulation were investigated.
pH and polysorbate 20
The impacts of the polysorbate 20 and the pH of the solution (formulation
buffer)
on the solubility of lauric acid, myristic acid, and palmitic acid were
investigated. As
shown in Figure 1, increasing levels of polysorbate 20 increases the
solubility of lauric
acid, myristic acid, and palmitic acid (which can be generated as a result of
polysorbate
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
degradation) thereby lowering the risk of forming particles. The results shown
in Figure
1 also suggest that increasing pH increases the solubility of free fatty
acids. pH 5.8 is an
optimal formulation pH which balances the risk of particle formation with the
chemical
and physical stability of the antibody. A formulation of this pH minimizes the
rate of
aspartic acid (Asp) isomerization and succinimide intermediate formation,
which allows
for patient convenience in combination with the device by allowing storage at
ambient
temperatures without substantially impacting the chemical stability of the
antibody and
thereby the product quality. Furthermore, the risk of particle formation is
lowered due to
the increased solubility of free fatty acids at the higher pH which can result
from
polysorbate degradation.
Arginine
Low viscosity is desired, as low viscosity can reduce injection force and
ensure
accuracy of injection volume. Use of arginine hydrochloride or arginine
succinate in
antibody formulations has been described previously. See, e.g., U.S. Patent
No.
8,142,776, and International Patent Application Publication Nos. W02006065746
and
W02010102241. The impact of the arginine succinate on the viscosity of the
formulation
was investigated. First, the viscosity of an etrolizumab formulation with
arginine
succinate was compared to the viscosity of etrolizumab formulation absent
arginine
succinate, and the results are shown in Figure 2. As shown in Figure 2,
solution viscosity
was decreased significantly by the addition of arginine succinate to the
formulation.
Next, the inventors investigated whether arginine, in this case, 200 mM
arginine
succinate, can impact the viscosity of an etrolizumab formulation of different
antibody
concentration. The viscosities of an etrolizumab formulation comprising 100
mg/mL,
150 mg/mL, 180 mg/mL, 200 mg/mL, and 220 mg/mL were measured, and the results
are shown in Figure 3. As shown in Figure 3, with arginine succinate (e.g.,
200 mM
arginine succinate), formulations with antibody concentrations as high as 200
mg/mL
have viscosities suitable for pre-filled syringe (PFS) administration.
Silicone oil
Because prefilled syringe drug products typically contain silicone oil and
because it is known that silicone oil may cause protein aggregation and/or
particle
formation over time, the inventors investigated the effect of silicone oil on
etrolizumab
formulations.
To test the effect of silicone oil, the interior of pre-filled syringe glass
barrels was
sprayed with silicone oil before filling with the etrolizumab formulation. To
assess
41
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
protein aggregation, high molecular weight species (HMWS) of etrolizumab were
determined using size exclusion chromatography (SEC) for the determination of
the
aggregation and oligomeric state of antibodies including HMWS in the
etrolizumab
formulation. SEC was conducted on a Agilent high performance liquid
chromatography
(HPLC) system with a Tosoh TSKgel column and a mobile phase consisting of
potassium phosphate and potassium chloride at a flowrate of 0.5 mL/min and
quantified
by UV absorbance and peak area integration. Briefly, the effect of different
amounts of
silicone oil on the formation of HMWS was determined in etrolizumab
formulations
comprising 150 mg/mL etrolizumab or 180 mg/mL etrolizumab. As shown in Figure
4,
the increased levels of silicone oil did not impact the physical stability of
all tested
etrolizumab formulations under various conditions.
Hence, the specific combination of the formulation excipients at the target pH
and in the presence of silicone oil are uniquely well suited to long term
storage of a high
concentration antibody formulation stored in a prefilled syringe. This finding
is
.. supported by about 6 years (e.g., 74 months) of long-term stability of
etrolizumab in
prefilled syringes without the observation of visible particles.
Succinate and Histidine
The presently disclosed formulations have the advantage to protect the
antibody
from additional stresses which can results from the storage in a prefilled
syringe.
Leachables from the prefilled syringe can lead to chemical and physical
degradation of
the antibody. For example, zinc and tungsten can contribute to metal-catalyzed
degradation. During the syringe manufacturing process, a hot tungsten pin is
inserted
into the glass barrel to make the hole for needle insertion. This process can
leave residual
tungsten particles in the syringe barrel, which can interact with the drug
solution causing
the formation of aggregates. Tungsten can induce protein aggregation and
formation of
proteinaceous particles. Protein oxidation can be induced by tungsten as well
leading to
protein aggregation. Succinic acid protected from zinc-based metal-catalyzed
degradation. Further, the presently disclosed formulation was not susceptible
to
tungsten-spiking mediated aggregation.
To determine the impact of tungsten exposure on the physical stability an
etrolizumab formulation, the percentages of HMWS were determined after
exposure to
different quantities of tungsten over time. As shown in Figure 5, tungsten did
not impact
the product quality of etrolizumab.
42
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Both the plunger stopper and the needle shield may be composed of rubber
material which may leach zinc into the formulation. It is known that zinc may
complex
with protein leading to protein aggregates (e.g., HMWS) and increased
viscosity of
antibody formulations. The effect of zinc on the viscosity of etrolizumab
formulations at
.. varying protein concentrations was investigated. As shown in Figure 6,
there was no
difference in viscosity between formulations lacking zinc and formulations
containing 10
mM zinc when the concentration of etrolizumab was 150 mg/mL or less in
formulations
comprising 20 mM histidine, 200 mM arginine succinate at pH 5.8. Zinc
increased the
viscosity at etrolizumab concentrations higher than 150 mg/mL. In addition,
Figure 7
shows the formation of protein aggregates in formulations comprising 150 mg/mL
etrolizumab and 50 mM zinc Figure 8 shows that the addition of zinc increases
the
percentage of HMWS and that the increase in percentage of HMWS is higher at 50
mg/mL etrolizumab compared to 10 mg/mL etrolizumab.
The inventors then tested whether histidine can reduce the formation of HMWS.
As shown in Figure 9, the presence of histidine in the formulation greatly
reduced the
rate of HMWS formation in presence of zinc. Furthermore, as shown in Figure
10,
succinate also suppressed the interaction between etrolizumab and zinc to form
HMWS.
Next, the impact of varying concentrations of histidine and succinate on the
HMWS
formation was studied. As shown in Figure 11, the combination of histidine and
succinate minimized the HMWS formation in the presence of zinc.
Drug Substance and Drug Substance Stability
The stability of etrolizumab drug substance (DS) formulated at 150 mg/mL in
20mM histidine, pH 5.8, 200mM arginine succinate, 0.04% polysorbate 20 was
evaluated in at 30 C, 5 C, and -20 C. The stability was also evaluated up to
five
.. freeze/thaw cycles to support at-scale storage and handling. No changes in
the chemical,
physical, or bioactivity properties were observed after five freeze/thaw
cycles or after
storage of the DS at -20 C for seven years (84 months). After storage at 5 C
for six
months, there was no change observed by all assays except ion exchange
chromatography. After storage at 30 C for 14 days, degradation was measured by
.. nonreduced capillary electrophoresis, ion exchange, and size exclusion
chromatography.
Deamidation and isomerization are degradation routes that can occur on
stability.
Ion exchange chromatography (IEC) was used to monitor chemical degradation and
quantitates acidic variants, basic variants, and main peak. No change was
observed in
main peak, acidic variants, or basic variants after five freeze/thaw cycles or
after 84
43
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
months of storage at -20 C. After six months of storage at 5 C, a 1.7% loss of
main peak
with concomitant 1.0% and 0.8% increases in acidic and basic variants,
respectively, was
observed. After 14 days of storage at 30 C, there was a 4.5% loss of main peak
with
concomitant 2.1% and 2.4% increases in acidic and basic variants,
respectively.
Qualitatively, no change in ion exchange profile or new peaks were observed.
After 14 days of storage at 30 C, the following changes were observed: by non-
reduced capillary electrophoresis-SDS, 1.0% loss of main peak observed with
concomitant 0.82% and 0.14% increases in pre-peaks and post-peaks
respectively; by
IEC, there was a 4.5% loss of main peak with concomitant 2.1% and 2.4%
increases in
acidic and basic variants, respectively; by size exclusion chromatography
(SEC),
increases of .28% and .09% are observed in HMWS and LMWS, respectively.
The stability of etrolizumab drug product formulated at 150 mg/mL in 20mM
histidine, pH 5.8, 200mM arginine succinate, 0.04% polysorbate 20 in a 105mg
(0.7 ml)
prefilled syringe configuration was investigated. Etrolizumab showed no change
in
product quality after 60 months at 5 C, as assessed by clarity, opalescence,
and
coloration (COC), pH, protein concentration, size exclusion chromatography
(SEC), and
potency. There was a 7.7% loss of main peak by ion exchange chromatography
(IEC)
with concomitant increases of 4.2% and 3.4%acidic and basic variants,
respectively, and
a 1.0% loss of main peak by CE-SDS non-reduced. After storage at 25 C for 3
months
there was: no change in clarity, opalescence, and coloration (COC), pH,
protein
concentration, or potency; a loss of 0.4% main peak by size exclusion
chromatography
with 0.2% increases in both high molecular weight and low molecular weight
species; a
1.3% loss of main peak by CE-SDS; a 9.8% loss of main peak by ion exchange
chromatography with a 5.1% increase in acidic variants and a 4.6% increase in
basic
variants. Etrolizumab drug product stored at 5 C for 60 months was practically
free of
visible particles.
Conclusion
Unique aspects of the specific excipient combination of the etrolizumab
formulation can protect the antibody from additional stresses which can result
from the
storage of an antibody in a prefilled syringe, e.g., histidine and succinate
can reduce
HMWS formation by zinc and the antibody. Typical antibody formulations
comprise
polysorbate 20 at 0.02% w/v and typical antibody formulations are at pH 5.5.
The higher
than typical polysorbate 20 concentration (e.g., 0.04% w/v) and higher than
typical pH
(e.g., pH 5.8) can solubilize free fatty acids, thereby lowering the risk of
particle
44
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
formation as a result of polysorbate degradation, e.g., during long term
storage in a pre-
filled syringe. The excipient combination of the etrolizumab formulation is
especially
effective at controlling pH during Tangential Flow Filtration (TFF) at large
scale
manufacturing. The high arginine concentration and high conductivity
formulation
shields charge on the antibody and prevents shifts in pH. The histidine
concentration
effectively buffers the formulation at the target pH. This is important to
ensure robust
control of the pH during manufacturing and allow for a narrow pH range of the
formulation to enable this optimal configuration. The formulation also
maintained
chemical and physical stability and maintained potency over various time
periods and at
various temperatures as described above.
Example 2 - Clinical Strategies and Initial Outcomes in Support of an
Etrolizumab
Autoinjector in Healthy Volunteers
Summary
Etrolizumab is a novel, dual-action, anti-07 integrin antibody in development
for
patients with moderate to severe ulcerative colitis or Crohn's disease. Phase
3 studies
utilize a prefilled syringe (PFS) for etrolizumab administration. In parallel,
an
autoinjector (AI) is being developed in order to increase delivery options for
patients.
This Example describe the overall development strategy and detail the first-in-
human
study of this developmental AT.
This open-label study of healthy volunteers (HVs) evaluated the tolerability
and
usability of the etrolizumab AT under development. The primary end point was
the
proportion of participants with greater than mild pain following injection.
Adverse
events (AEs) and usage errors were also assessed. Results were reported by
injection site
(thigh vs abdomen) and needle training (experienced vs naive). Pharmacokinetic
(PK)
variability between participants was included as an exploratory end point.
Thirty participants completed the study. A total of 97% of participants never
experienced greater than mild pain during the study; 50% did not experience
any pain.
Three usage errors were observed, 1 of which resulted in a partial dose
delivery of
etrolizumab. No patterns of usage errors were observed. Mild injection-site
reactions
(ISRs) were reported; all resolved by the end of the study. Participants
injecting into the
abdomen reported more ISRs than those injecting into the thigh; needle
training did not
appear to influence incidence or severity of AEs.
Results from this first-in-human study demonstrate that single injections of
etrolizumab 105 mg using an AT are well tolerated in HVs, with transient, mild
pain and
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
minimal usage errors. Results from this study also informed the design of a
subsequent
PK comparability study comparing the PFS and AT. Overall, the availability of
an AT
may provide an attractive option for patients desiring a convenient, easy to
use delivery
mechanism for etrolizumab.
Introduction
Etrolizumab is being evaluated in an extensive clinical program of phase 3
studies in patients with moderate to severe UC and Crohn's disease (Etro
Studies. The
Etro Studies: Explore Innovation: Contribute to Science. Genentech; 2019.
Accessed
July 26, 2019), where etrolizumab is administered once per month via
subcutaneous (SC)
injection using a prefilled syringe (PFS) with a needle safety device (NSD).
Single-use, prefilled autoinjectors (AIs) have many potential advantages over
PFS-NSDs; most notably, their ability to keep the needle out of sight of the
user at all
times during injection. Exemplary PFS and AT are shown in Figure 12. AIs also
offer
increased convenience, ease of use, reduced risk of dosage error, and improved
patient
comfort. Studies have consistently shown that many patients who self-
administer prefer
an AT over a syringe-based device (Kivitz et al., Clin Ther. .
2006;28(10):1619-29; Kivitz
and Segurado, Expert Rev Med Devices. 2007;4(2):109-16; Borras-Blasco et al.,
Expert
Opin Biol Ther. . 2010;10(3):301-7; Vermeire et al., Patient Prefer Adherence.
2018;12:1193-202). For example, a recent study of golimumab in patients with
UC
demonstrated that a majority of patients preferred administration with an AT
versus a
PFS, citing increased ease of use and reduced discomfort with injection
(Vermeire et al.,
Patient Prefer Adherence. 2018;12:1193-202).
The AT currently under development consists of an automated delivery system
encasing the same PFS used in the phase 3 studies (see Figure 13). The drug
product
contained in both the AT and the PFS-NSD consists of a liquid formulation of
105 mg of
etrolizumab solution (0.7 mL, nominal volume of 150 mg/mL) for single-dose
administration. The entire dose is typically administered in about 2 seconds.
The AT includes many features aimed to improve the patient experience and
increase patient comfort with self-administration. The automated drug delivery
system is
activated by lightly pressing the device onto skin perpendicularly. Once
activated, the AT
automatically inserts the needle and dispenses the syringe contents upon
activation. Once
injection is complete, a needle cover extends and locks over the needle,
keeping the
needle out of view at all times during injection and protecting the user and
others from
accidental contact with the used needle. The AT also incorporates both visual
and
46
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
auditory mechanisms designed to assist users with self-injection; a visible
spinning top
and an audible clicking sound both indicate whether drug administration is
ongoing or
completed. In addition, a visible plunger rod moves across the viewing window
while the
injection is in progress.
Here, the overall development strategy for a novel AT for etrolizumab, and
findings from a first-in-human study of this device are shown. The primary
objectives of
this study (NCT02629744) were to evaluate the safety and tolerability of
etrolizumab
administered by the AT and primarily injection site pain following self-
injection with the
AT and to document critical use errors. In addition, the inventors evaluated
this relatively
unique study design, which combined usage error assessments (traditionally
conducted
as simulated studies) with a tolerability, safety, and exploratory PK study.
Methods
Study Design and Procedures. This first-in-human AT tolerability study was an
open-label, single-arm study in healthy volunteers evaluating pain, safety,
and usability
of an AT when self-administered subcutaneously. Participants were assigned
(1:1) into 2
groups. In order to simulate prior experience of self-injection, 1 group
("needle-
experienced") received training before self-injection with the AT; the other
group
("needle-naïve") did not. Training involved simulated needle experience by
self-injection
with placebo. Before etrolizumab administration, all participants
(irrespective of needle
experience group) received an instructions for use (IFU) leaflet regarding the
AT for
review before self-injection. Subjects were randomly assigned to administer
study drug
to their abdomen or anterior thigh. All participants self-administered a
single SC dose of
etrolizumab on day 1 of the study in a simulated home setting (see Figure 14).
Participants were monitored during self-injection, discharged at study day 3,
and
returned for follow-up visits on study days 8, 29, 43, 57, and 85 (study
completion).
Participants. Eligible participants were to be between the ages of 18 and 65
years, have a body mass index (BMI) of between 18.0 and 32.0 kg/m2
(inclusive), and be
in good health with no significant medical history or laboratory test
abnormalities. Both
men and women were enrolled, with the target of 55% to 60% male participants
to
mimic the sex distribution of patients with MD. Participants with any prior
use of anti-
integrin therapies (including etrolizumab) or immunosuppressive drugs were
excluded,
as were participants with a recent history of corticosteroid use. Participants
with a history
of tuberculosis were also excluded.
47
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Procedures. Participants assigned to the needle-experienced group received
training (simulated self-injection experience) 5 and 7 days before etrolizumab
injection.
During training, participants were instructed by a healthcare professional on
the use of a
needle and syringe. Following this instruction, participants practiced self-
injection with a
placebo solution 3 times using a needle and syringe. Needle-experienced
participants
deemed to be suitable by the healthcare professional (based on their
interactions with
syringes) progressed through the study. Both needle-experienced and needle-
naïve
participants were randomized, stratified by sex and needle experience, to
inject into
either the abdomen or anterior thigh. On study day 1, participants self-
administered a
single SC dose of etrolizumab 105 mg into their abdomen or anterior thigh
using the AT.
Participants were assessed for operational difficulties and usage errors and
reported pain
during and immediately following the injection. One serum sample was taken for
exploratory pharmacokinetic assessment 7 days following the SC injection
(study day 8).
Approaches for Assessing Pain. Pain was assessed via 2 independent methods,
both of which were administered by study site personnel. The 7-point
categorical Verbal
Descriptive Scale (VDS-7) was the primary measure of pain for this study.
During VDS-
7 administration, patients were asked to choose the number from 1 to 7 that
best
represents the pain associated with the injection (scale as follows: 1 = no
pain, 2 = very
mild pain, 3 = mild pain, 4 = not very severe pain, 5 = quite severe pain, 6 =
very severe
pain, 7 = almost unbearable pain). As a confirmatory assessment, pain was also
assessed
via a 100-point continuous visual analog scale (VAS). For the VAS, patients
were asked
to mark a line on a horizontal 100 mm scale that best represents their pain
(scale as
follows: 0 mm = no pain, 100 mm = worst possible pain). Study personnel then
measured the distance between the 0 mm point and the patient's mark to
determine their
VAS score.
Outcomes. The primary end point was the proportion of participants with
greater
than mild pain (VDS-7 > 3) immediately following injection. To meet the
primary end
point, the upper bound of the 2-sided 95% confidence interval (CI) around the
proportion
of participants experiencing greater than mild pain immediately following
injection must
not exceed 30%. Secondary end points included the proportion of participants
experiencing greater than mild pain at 5, 10, 20, 60, and 240 minutes (4
hours) following
injection, and the proportion of participants in each VDS-7 category over
time.
Tolerability was assessed intensively in this study via active monitoring for
injection site
reactions (ISRs) on study day 1 at 5, 60, and 240 minutes after injection, and
on study
48
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
days 2, 8, 43, and 85. To identify ISRs, a local injection site symptom
assessment
(LISSA) was performed to assess for burning, itching, bruising, redness,
and/or
formation of hives, and the size of the reaction if present. All ISRs were
categorized and
reported as an adverse event (AE) or serious AE as appropriate. Usage errors
and
operational difficulties during use of the AT were documented. In addition,
participants'
knowledge of the IFU and their overall opinions of the AT experience were
collected.
Safety was assessed via AE monitoring, laboratory assessment, vital signs,
physical
examinations, electrocardiograms (ECGs), and immunogenicity. For this study,
no
formal statistical testing was planned. Determination of PK variability on a
single time
point of study day 8 following self-injection was assessed as an exploratory
end point.
Results
Thirty healthy participants were enrolled and randomized (stratified by sex
and
needle experience) to inject etrolizumab into the abdomen or anterior thigh.
All
participants completed the study; however, 1 volunteer (needle-experienced,
thigh
injection) did not receive a full dose of etrolizumab because of a usage error
(will be
discussed further). The enrolled population was broadly representative of the
IBD
population. The median age of enrolled participants was 36 years, the mean BMI
was
26.1 kg/m2, and the majority of the participants were white (60%) and not
Hispanic or
Latino (83%). Approximately half (47%) of the participants were male.
Pain and Tolerability. Half of the participants did not report pain at any
timepoint following injection (see Figure 15). For those participants who
reported pain,
all but one reported "very mild" or "mild" pain, the majority of which
subsided within 60
minutes after drug administration. A single volunteer reported greater than
mild pain
(VDS-7 = 5) immediately following the injection, which subsided to mild pain
at 5
minutes following injection. Similar data were reported when using the VAS
(data not
shown). Reported pain differed between injection sites. Injection into the
thigh led to a
greater proportion of participants reporting pain compared with participants
injecting
into the abdomen (60% vs 40%, respectively) (see Figure 16). The single
volunteer
reporting greater than mild pain was assigned to the thigh administration
group.
Participants injecting into the thigh also reported a longer duration of pain
than those
injecting into the abdomen; all pain experienced following abdominal injection
subsided
within 5 minutes, and the majority of pain following thigh injection subsided
within 60
minutes. Needle-experience training did not appear to impact reported pain
following
etrolizumab injection. Using the intensive, LISSA-based monitoring scheme
described
49
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
above, 40% of participants (12/30) experienced an ISR during the study, all of
which
occurred within 1 hour following etrolizumab injection (Table 1).
Table 1. Summary of local injection site symptom assessments over time by
injection
site and needle-experience groups
ISR Timepoi Reactio Abdomen Thigh Overa
nt, mm n size, Need! Needle- Tot Need! Needle- Tot
11 (n =
mm e- experienc al (n e- experienc al (n 30)
naïve ed (n = 9) = naïve ed (n = 9) =
(n = 6) 15) (n = 6) 15)
Hive 60 18 1(11.1) 1 1(3.3)
formati (6.7)
on
Redness 5 18 1 1 1(3.3)
(16.7) (6.7)
21 1(11.1) 1 2 2 3
(6.7) (33.3) (13.3
(10.0)
24 1 1 1(3.3)
(16.7) (6.7)
60 <18 1(11.1) 1 1(3.3)
(6.7)
18 1(11.1) 1 1(3.3)
(6.7)
24 2 1(11.1) 3 3
(23.3) (20.0 (10.0)
31 1 1 1(3.3)
(16.7) (6.7)
ISR injection site reaction
All reported ISRs were mild (grade 1) and transient; all resolved by study
completion. The most frequent ISR was redness, which ranged from < 18 to 31 mm
in
diameter. Most ISRs resolved within 60 minutes following injection. One
volunteer
reported formation of hives (18 mm in diameter) at the abdominal injection
site 60
minutes postdose; the hives resolved within 3 hours without treatment.
Injection site did
not appear to affect either the frequency or severity of ISRs.
Usage Errors. Twenty-seven of the 30 participants (90%) were able to
successfully self-administer etrolizumab using the AT without significant
usage errors
regardless of needle experience training. No complaints about the AT were
registered,
and no pattern of usage errors was observed. A total of 3 usage errors were
observed
during the study, only 1 of which occurred during the injection. One volunteer
began to
remove the AT prematurely during the injection, resulting in a droplet of
liquid remaining
on the volunteer's skin. Of the 2 usage errors not occurring during injection,
1 volunteer
was unsure when to remove the cap from the AT, and another incorrectly
reported the
simulated expiration date. These 2 usage errors were associated with
misunderstanding
of the labeling and IFU of the AT; neither of these errors impacted the dose
of
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
etrolizumab being administered. Most participants noted that they found the AT
easy to
use. Participants reported that the audible and visual feedback mechanisms
were helpful
for determining when the injection had started and stopped, and to verify that
a complete
dose had been administered. However, some participants stated that it was
difficult to
view the visual spinning top while injecting into their abdomen. During IFU
comprehension probes, some confusion was noted related to acceptable injection
sites,
medication warm-up time, and product storage.
Pharmacokinetics (Exploratory). On study day 8 (7 days following injection),
the mean ( SD) serum concentration of etrolizumab across all participants was
13.6 (
3.66) g/mL (median 13.8). Serum concentrations ranged from 5.8 ps/mL to 20.0
pg/mL, a roughly 31% between-participant variability. Neither injection site
nor needle
training appeared to affect serum etrolizumab concentration at day 8 based on
the limited
data set.
Safety. Twenty-nine participants (97%) received the full 105 mg dose of
etrolizumab; 1 volunteer received approximately 90% of the 105 mg dose.
Overall,
single 105 mg SC doses of etrolizumab were safe and well-tolerated when self-
administered using the AT. Around half of participants experienced a treatment-
emergent
adverse event (TEAE), most of which were related to the injection site. All
TEAEs were
mild (grade 1) in severity and had resolved by study completion. No
significant changes
were noted in clinical laboratory evaluations, vital sign measurements, body
weight
measurements, or 12-lead ECGs during this study. Interestingly, more TEAEs
were
reported by participants injecting into the abdomen compared with the thigh
(19 vs 9
TEAEs, respectively). Fewer TEAEs were reported by needle-experienced
participants
than needle-naïve participants. A summary of TEAEs can be found in Table 2.
Table 2. Summary of treatment-emergent adverse events
Abdomen Thigh Overall
Needle- Needle- Total (n Needle- Needle- Total
(n (n = 30)
naive (n = experienced = 15) naive = experienced = 15)
6) (n = 9) 6) (n = 9)
Participants 3 (50.0) 5 (55.6) 8 (53.3) 5 (83.3) 3
(33.3) 8 (53.3) 16 (53.3)
with any
TEAE
Number of 13 6 19 6 3 9 28
TEAEs
Suspected to 2(33.3) 2 (22.2) 4(26.7) 3 (50.0) 1(11.1)
4(26.7) 8 (26.7)
be caused by
study dmg
Other causes 2(33.3) 3 (33.3) 5(33.3) 2(33.3) 2(22.2) 4
(26.7) 9(30.0)
Severity
Mild 3 (50.0) 5 (55.6) 8 (53.3) 5 (83.3) 3 (33.3)
8 (53.3) 16 (53.3)
(grade 1)
51
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Abdomen Thigh Overall
Needle- Needle- Total (n Needle- Needle- Total
(n = 30)
naive (n = experienced = 15) naive = experienced = 15)
6) (n = 9) 6) (n = 9)
Total 3 (50.0) 5 (55.6) 8 (53.3) 5 (83.3) 3 (33.3)
8 (53.3) 16 (53.3)
Values are n (%). TEAE treatment-emergent adverse event
Discussion
In healthy participants, a single, self-administered, SC dose of etrolizumab
using
the AT was well-tolerated and resulted in mild pain for the majority of
participants. This
study met its primary end point with only a single volunteer experiencing
greater than
mild pain following injection. Overall, the data presented here are consistent
with data of
AIs used in the treatment of other chronic diseases, including rheumatoid
arthritis (RA)
and chronic kidney disease. These studies suggest many patients prefer the
convenience
of an AT compared with a PFS. Patients commonly report that AIs are associated
with
less pain than PFS devices and perceive AIs as more portable and easier to use
(Kivitz et
al., Clin Ther. . 2006;28(10):1619-29; Kivitz and Segurado, Expert Rev Med
Devices.
2007;4(2):109-16; Borras-Blasco et al., Expert Opin Biol Ther. .
2010;10(3):301-7; Lim
et al., Clin Ther. . 2012;34(9):1948-53). A recent study in patients with UC
reported
similar findings, noting that around three-quarters of patients in the study
preferred
injection with an AT compared with a PFS (Vermeire et al., Patient Prefer
Adherence.
2018;12:1193-202). In a recent multinational survey, 200 patients with RA and
100
nurses were asked to rate the relative importance of various components of AIs
(Tischer
and Mehl, Patient Prefer Adherence. 2018;12:1413-24). Both patients and nurses
rated
"easy to perform the self-injection with the pen (ie, autoinjector)" as the
most important
attribute. Other key attributes, as reported by both patients and nurses,
included
"injection needle is safely concealed in the injector body," "audible feedback
after
completion of the injection," and "visual feedback after completion of the
injection"; all
of these features are built into the etrolizumab AT. Similar results were
reported in a
European study of 220 patients with RA (Thakur et al., Rheumatol Ther. .
2016;3(2):245-
56). It is worth noting that the proportion of patients experiencing mild ISRs
in this study
is higher than was observed in the phase 2 EUCALYPTUS study, in which
etrolizumab
was administered by vial and syringe (Vermeire et al., Lancet.
2014;384(9940):309-18).
It was believed that this likely reflects differences in the study design, as
this study
assessed tolerability intensively, using LISSA to actively monitor for ISRs at
scheduled
intervals and possibly resulting in overreporting of ISRs. This first-in-human
study is
relatively unique in that it combined tolerability assessments, actual-use
human factor
52
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
assessments (such as usage errors), and an initial PK assessment into a single
trial. This
novel approach, in part, aimed to assess overall risk associated with AT in a
way that
would minimize the number of clinical studies necessary, hence reducing the
overall
time and cost of AT development. PK assessment was incorporated into the study
protocol with a single blood sample taken on study day 8, around the time of
maximum
serum concentration following a single SC dose. The intent of this exploratory
PK
assessment was to understand the intersubject exposure variability following
etrolizumab
SC delivery by AT. In addition, these preliminary PK data helped to evaluate
potential
differences in exposure following AT injection compared with the predicted
exposure
using a model generated based on PK data from administration with a vial and
syringe in
patients with UC. Of note, the day 8 etrolizumab exposure observed in this
analysis was
approximately 75% higher than the predicted value (predicted day 8 median
etrolizumab
serum concentration ,=,'7.9 pg/mL [90% CI 4.15-16.3], data not shown). The PK
variability and unexpected higher day 8 exposure from this analysis informed
the
decision to conduct a 2-part study comparing the pharmacokinetics of
etrolizumab
delivered by the AT and PFS-NSD in healthy volunteers (see Figure 17 and
Example 3).
Results from this study effectively eliminated the requirement for additional
AT ease of
use and/or clinical studies. In addition, these results influenced the design
of a
subsequent study to compare PK properties between administrations by the PFS-
NSD
and AI, mitigating the risk of failing the comparability study and minimizing
unnecessary exposure of healthy volunteers to biological treatment. As a
result of the PK
findings reported here, a pilot cohort was added to the originally proposed
single-part
device PK comparability study design. Results from this pilot cohort served to
optimize
the design of the pivotal cohort by informing the proper sample size, sample
collection
duration, and body weight range. Information gained from the human factors
component
of this study resulted in small amendments to the IFU, implemented before the
PK
comparability study. Results from the device PK comparability are reported in
Example
3.
Conclusion
Results from this study demonstrate that single Sc injections of etrolizumab
with
an AT are well-tolerated in healthy volunteers, with tolerable levels of pain
following
injection. Most participants found the AT easy to use and experienced only
minimal
usage errors. The AT may be an appropriate delivery mechanism for certain
patients with
MD who desire the safety and convenience of self-injection with an invisible
needle.
53
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
The positive results from this first-in-human tolerability study, in
combination with data
gathered during the subsequent 2-part PK comparability study, comprise a
complete
development plan to support the use of AT in patients treated with
etrolizumab.
Example 3 ¨ Comparable Pharmacokinetics, Safety, and Tolerability of
Etrolizumab Administered via Prefilled Syringe or Autoinjector in a Randomized
Trial in Healthy Volunteers
Summary
Etrolizumab is a novel, dual-action anti-07 integrin antibody being studied in
several phase 3 trials in patients with inflammatory bowel disease. An
autoinjector (AI)
device is being developed in parallel to complement the prefilled syringe
(PFS) with
needle safety device (NSD) used for subcutaneous administration in these
trials. This
Example demonstrates the comparability of pharmacokinetics (PK), tolerability,
and
safety of both devices.
This randomized, open-label, 2-part study in healthy participants evaluated
the
comparability of etrolizumab exposure between the AT and PFS-NSD. Part 1
(pilot)
involved a small number of participants, and initial results were used to
finalize the study
design of the larger part 2 (pivotal). In both parts, participants were
randomly assigned to
receive a single dose of subcutaneous (SC) etrolizumab 105 mg via AT or PFS-
NSD.
Randomization was stratified by body weight. The primary PK outcomes were
Cmax,
AUCIast, and AUCo-inf.
One hundred and eighty healthy participants (part 1: n = 30, part 2: n = 150)
received a single SC dose of etrolizumab via AT or PFS-NSD. Primary PK results
from
part 1 supported modification of the part 2 study design. Results from part 2
demonstrated that etrolizumab exposure was equivalent between devices, with
geometric
mean ratios (GMRs) between AT and PFS-NSD of 102% for Cmax, 98.0% for AUCIast,
and 97.6% for AUCo_inf. Median T. and mean terminal ti/2 were also similar
between
devices. The GMRs and 90% confidence intervals of all primary PK parameters
were
fully contained within the predefined equivalence limits (80% to 125%).
This PK study demonstrated that single SC injections of etrolizumab 105 mg
using an AT or PFS-NSD result in equivalent etrolizumab exposure and similar
safety
and tolerability in healthy participants. Taken together, these results
support the use of an
AT for etrolizumab administration.
Introduction
54
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
The PK comparability study (NCT02996019) presented in this Example aimed to
demonstrate the comparability of etrolizumab exposure following SC
administration
using the AT and PFS-NSD, and to evaluate the safety and tolerability of
etrolizumab
following SC injection using the 2 devices. Part 1 of the study was an
exploratory pilot
cohort used to evaluate the geometric mean ratio (GMR) and variability of PK
parameters for etrolizumab administration with the AT versus PFS-NSD. Those
results
informed the study design including sample size and study duration for part 2
(the
pivotal cohort). In part 2, the study aimed to demonstrate exposure
comparability
between a single dose of etrolizumab administered via AT or PFS-NSD. The PK
comparability study presented in this Example leveraged the exploratory PK
results from
the tolerability study presented in Example 2 to refine the study design and
final
protocol.
Methods
Study Design and Procedures. This study was a randomized, multicenter, open-
label, parallel-group study conducted in healthy participants at three
clinical sites within
the United States (see Figure 18). This 2-part study consisted of a pilot
cohort (part 1)
and a pivotal cohort (part 2) with a sample size sufficient for 80% power to
detect the
exposure difference (if any) between the 2 device groups. In both parts,
healthy
participants were randomly assigned 1:1 to receive a single dose of
etrolizumab 105 mg
SC via either AT (test device) or PFS-NSD (reference device). Etrolizumab was
administered by a health care professional (HCP) into the participant's
abdomen.
Randomization in both cohorts was stratified by body weight (< 79.9 vs > 80
kg).
Participants. Eligible healthy participants included men and women between 18
and 55 years of age with a body mass index (BMI) between 18.0 and 30.0 kg/m2.
Based
on results from the pilot cohort, it was also required that participants in
the pivotal cohort
have a body weight within the range of 60 to 100 kg (inclusive) at the time of
study
entry. Participants must have been in good health (no clinically significant
findings from
medical history, physical examination, 12-lead electrocardiogram, or vital
signs).
Participants with any prior exposure to immunosuppressants or anti-integrin
therapies
(including etrolizumab) were excluded.
Assessments and Outcome Measures. Blood samples for determination of
etrolizumab serum concentrations in both part 1 and part 2 were collected
predose and 6
hours postdose on day 1, then on days 2, 4, 6, 8, 11, 15, 29, 43, 57, and 71
(study
completion). The geometric mean ratio (GMR) and variability of the maximum
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
etrolizumab concentration (C.), area under the concentration-time curve from
the time
of drug administration to the last measurable concentration (AUCIast), AUC
extrapolated
to infinity (AUC0-inf), as well as the ratio of AUCIast to AUC0-inf (AUCR) of
etrolizumab,
were measured in part 1. For part 2, Cmax, AUClast, and AUC0-inf were measured
as
primary endpoints. Secondary PK parameters included the time to maximum
concentration of etrolizumab (t.), the terminal elimination half-life (t1/2),
and AUCR.
Blood samples for determination of antidrug antibodies (ADAs) in parts 1 and 2
were
collected before dosing on day 1, and on days 29, 57, and 71. Data from all
ADA-
positive participants were included in the final PK statistical analysis
unless the
participant met the PK analysis predefined exclusion criteria. Safety and
tolerability
assessments included the incidence, nature, and severity of adverse events,
graded
according to National Cancer Institute Common Terminology Criteria for Adverse
Events, version 4.03. The incidence of injection-site reactions, changes in
vital signs,
physical examination findings, clinical laboratory results, and the incidence
of ADAs
were also assessed.
Bioanalytical methods. Etrolizumab concentrations were measured using a
validated immunoassay (ICON). This Gyrolab fluorescence immunoassay utilized a
minimum required dilution (MRD) of 1/100, with a minimum quantifiable
concentration
of 80 ng/mL etrolizumab in UC, CD, and healthy volunteer sera. Anti-
etrolizumab
antibodies in serum were detected using a validated assay. This colorimetric
ELISA
used a 1/20 MRD and a monoclonal anti-etrolizumab control antibody. Relative
sensitivity of the method was determined to be 12.0 ng/mL in healthy volunteer
serum.
Drug tolerance of the assay was established: 28 ng/mL of the positive control
ADA
could be detected in the presence of 501.tg/mL etrolizumab.
Sample Size Determination. Enrollment of up to 30 healthy participants in part
1
was planned to ensure at least 12 participants in each arm (24 total) had
evaluable PK
profiles to enable estimation of the GMR and coefficient of variation (CV%)
for PK
parameters (Cmax, AUClast, and AUC0-inf). Part 2 planned for enrollment of 146
participants. Assuming a dropout rate or non-evaluable PK profiles from
approximately
10% of participants through day 71, a total of approximately 131 healthy
participants
with evaluable full PK profiles were expected to provide at least 80% power to
demonstrate exposure comparability for C., AUCIast, and AUCof based on the GMR
and PK variability outcomes from the part 1 pilot cohort.
56
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Pharmacokinetic analyses. PK parameters were determined from the serum
etrolizumab concentrations using noncompartmental methods (NCA) and were
performed using Phoenix WinNolin (Centara USA, Inc., Version 6.4),
Statistical Analyses. The analysis population consisted of all participants
who
received an SC injection of etrolizumab and who had an evaluable PK profile,
which was
defined as having sufficient samples available to accurately determine key PK
parameters. In particular, participants with early termination on or before
day 15 were
considered not having an evaluable PK profile. Participants with no sample
available to
determine the concentration of day 71 were excluded from statistical analysis
of AUCIast;
participants with < 3 available samples among days 28, 43, 51, and 71 were
excluded
from statistical analysis of AUCof. Participants with predose concentration >
5% of
Cmax may be excluded from statistical analysis of all PK parameters at the
discretion of
the study and sponsor clinical pharmacokinetist/biostatistician. Descriptive,
exploratory
analysis of PK parameters was carried out with data from part 1 (pilot), with
a focus on
evaluating GMRs, CV%, and distribution of AUCR to inform the part 2 (pivotal)
sample
size and final study design. Only data from the pivotal cohort (part 2) were
included in
comparability statistical analysis. In part 2, an analysis of variance,
including treatment
as the fixed effect, was performed to assess comparability of Cmax, AUClast,
and AUCo-inf
between the Al and PFS-NSD groups. Data for C., AUCIast, and AUCof were
natural
log (1n)-transformed before analysis, and the 90% confidence intervals (CIs)
of the
GMRs for the AT group relative to those from the PFS-NSD group were calculated
by
taking the antilog of the corresponding 90% CIs for the differences between
the means
(log scale). Exposure between the AT and PFS-NSD groups met PK comparability
criteria if the 90% CIs of the GMRs for C., AUCIast, and AUCof were all within
80%
to 125%.
Results
Pharmacokinetics _________ Pilot Study (Part 1). All 30 participants enrolled
and
randomized in part 1 received a single 105 mg SC dose of etrolizumab via AT (n
= 15) or
PFS-NSD (n = 15). Twenty-seven participants completed the study; 2
participants (1 in
each arm) discontinued earlier because of loss of follow-up and 1 discontinued
because
of a serious AE (seizure) 32 days following administration of etrolizumab (PFS-
NSD
arm); this AE was not considered related to etrolizumab treatment. Participant
characteristics are shown in Table 3.
Table 3. Participant characteristics
57
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
Pilot cohort (part 1) Pivotal cohort (part 2)
AI PFS-NSD AI PFS-NSD
n = 15 n = 15 n = 74 n = 76
Mean age (min, max),
42(28, 54) 34(20, 53) 35 (21, 55) 37 (19, 55)
years
Mean weight (min, max), 76.3 (55.90,
79.7 (65.1, 108.8) 74.3 (50.8, 92.1) 76.2 (61.0, 99.4)
kg 97.5)
Mean height (min, max), 172.0 (154.3, 173.5 (154.6, 170.3
(151.2, 170.7 (151.5,
cm 190.8) 194.9) 193.4) 189.0)
Mean BMI (min, max), 26.2 (19.2,
26.8 (23.2, 29.9) 24.5 (20.6, 29.2) 26.3 (20.4, 30.0)
kg/m2 29.9)
Sex, n (%)
Male 9(60.0) 10 (66.7) 40 (54.1) 40 (52.6)
Female 6 (40.0) 5 (33.3) 34 (45.9) 36 (47.4)
Race, n (%)
Asian 0 0 2 (2.7) 1(1.3)
Black/African American 5 (33.3) 4(26.7) 31 (41.9) 23 (30.3)
White 10 (66.7) 11 (73.3) 40 (54.1) 51 (67.1)
Unknown 0 0 1(1.4) 1(1.3)
Ethnicity, n (%)
Hispanic or Latino 6(40.0) 3 (20.0) 14 (18.9) 14 (18.4)
Not Hispanic or Latino 9(60.0) 12 (80.0) 60 (81.1) 62 (81.6)
JCV antibody-positive, n
7(46.7) 8(53.3) 38 (51.4) 46 (60.5)
1%)
Al autoinjector. Billibody mass index. JCV John Cunningham virus. PFS-NSD
prefilled syringe with needle safety
device.
In the overall cohort, most participants were male (63.3%) and white (70.0%).
The body weight and age of participants were not well-balanced between
treatment arms;
the mean body weight was 79.7 kg in the AT arm and 74.3 kg in the PFS-NSD arm,
and
the mean age was 42 years in the AT arm and 34 years in the PFS-NSD arm. Since
body
weight appears to impact etrolizumab exposure, especially AUC0-in1 (see Figure
19), the
imbalance in body weight between treatment arms could potentially bias
assessment of
GMR values of primary PK parameters and PK variability. Therefore,
participants in the
pilot cohort with a body weight lower than 60 kg (n=4) or higher than 100 kg
(n=1) were
excluded from the exploratory analysis aiming to evaluate GMR and CV% of PK
parameters which were used to guide the determination of final sample size for
pivotal
study (part 2). The PK evaluable population meeting the body weight
restriction of 60 to
100 kg in part 1 included 14 participants in the AT arm and 11 participants in
the PFS-
NSD arm. As expected, when body weight range between the two arms was balanced
via restricting body weight to a 60-100kg range, AT versus PFS-NSD group
primary PK
parameter GMR values were 0.95, 1.02, and 1.02 for Cmax, AUClast, and AUCo-
inf,
respectively (Table 4).
Table 4. Summary of PK parameters in cohort 1 participants meeting the body
weight
restriction of 60 to 100 kg
58
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
Al PFS-NSD GMR (%) Pooled CV%
(test) (reference)
Parameter
Geometric
na Geometric meanb
meanb
C.. ( g/mL) 14 10.6 11 11.1 0.955 30.1
AUCiast (day. g/mL) 12 273.3 9 267.9 1.020 34.1
AUCo-inf 1.024 34.0
14 284.6 10 278
(day. g/mL)
Al autoinjector. ANOVA analysis of variance. AUCo-,nf AUC extrapolated to
infinity. AUChw area under the
concentration-time curve from the time of drug administration to the last
measurable concentration (Last is Day 71 for
all available data). C,, maximum concentration. GMR geometric mean ratio. PFS-
NSD prefilled syringe with needle
safety device.
aNumber of observations in each treatment eligible for analysis.
bGeometric means are based on the least-squares means for C. and AUC
parameters from ANOVA, calculated by
transforming the natural log means back to the linear scale.
Hence, the sample size for pivotal cohort was calculated based on these GMR
and CV% values, and a body weight restriction was added for the part 2 study
as an
inclusion criterion. Furthermore, all participants had AUCR values > 80% (data
not
shown), which meets the requirement for bioequivalence study stated in the US
Food and
Drug Administration (FDA) guideline (US Department of Health and Human
Services.
Guidance for industry: statistical approaches to establishing bioequivalence.
Accessed
March 4, 2020), and hence resulted in a modification of the last day of the
pivotal study
from originally planned day 85 to day 71.
Pharmacokinetics ________________________________________________________
Pivotal Study (Part 2). In the pivotal cohort, 150 (100%) of
the enrolled and randomized participants received a single SC 105 mg dose of
etrolizumab via AT (n = 74) or PFS-NSD (n = 76). Eight participants
discontinued the
study; 5 because of loss of follow-up (3 in the AT arm and 2 in the PFS-NSD
arm), 1
because of a protocol violation in not meeting body weight criteria (PFS-NSD),
and 2 (1
in each arm) because of participant withdrawal. In the overall part 2
(including both
arms), 53.5% of participants were male, 60.7% were white, and 36.0% were
African
American. Treatment arms were well-balanced for body weight and age; mean body
weight was 76.2 kg in the AT arm and 76.3 kg in the PFS-NSD arm, and mean age
was
35 years in the AT arm and 37 years in the PFS-NSD arm (Table 3).
Etrolizumab serum concentrations over time for the AT and PFS-NSD groups are
shown in Figure 20; PK parameters are summarized in Table 5. GMRs (90% CIs)
between the AT and PFS-NSD groups were 102% (94.2-111%) for Cmax, 98.0% (89.3-
107%) for AUCIast, and 97.6% (88.6-107%) for AUC0-inf (Table 6).
Table 5. Summary of PK parameters for etrolizumab in the pilot and pivotal
studies
59
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
Pilot cohort (part 1)a Pivotal cohort (part 2)
Parameter
AI PFS-NSD AI PFS-
NSD
(n = 14) (n = 11) (n = 76) (n =
74)
C.a. (gg/mL) 14 10.6(28.3%) 10 11.0(35.3%) 73 12.1
(32.0%) 73 12.2(28.3%)
3.98 (2.90' 73 5.04 (2.98' 73 6.97 (3.00,
tma.b (day) 14 5.00 (2.95' 10
9.94) 6.97) 14.0) 14.0)
AUCiast
13 272 (39.8%) 9 268(27.0%) 69 319
(35.3%) 72 325 (33.1%)
(day.m/mL)
AUCo-inf
(day. g/mL) 13 284 (41.6%) 10 278 (27.0%) 69 329
(36.0%) 72 337 (35.2%)
t1/2 (day) 13 12.2 (4.90) 10 11.4 (4.04) 69 11.8
(3.85) 72 12.2 (4.39)
CL/F (L/day) 13 0.369 (41.6%) 10 0.378(27.0%) 69 0.319
(36.0%) 72 0.311 (35.2%)
AUCI2d 13 0.957 (3.5%) 9 0.975 (2.6%) 67
0.966 (3.4%) 72 0.964 (3.7%)
<80% 0 0 0 1(1%)
>80% 13 (100%) 9 (100%) 67
(100%) 71(99%)
Al autoinjector. AUC0f AUC extrapolated to infinity. AUCIast area under the
concentration-time curve from the time
of drug administration to the last measurable concentration. AUCR ratio of
AUCiast to AUCo-inf. C, maximum
concentration. PFS-NSD prefilled syringe with needle safety device. PK
pharmacokinetic. ty2 terminal elimination
half-life. tmax time to maximum concentration.
.. Geometric mean (geometric CV%) data are presented unless otherwise
indicated.
aIncludes only participants who met body weight restrictions (60 to 100 kg) in
the pilot cohort.
bMedian (min, max) presented for tm.. 'Arithmetic mean (SD) presented for tin.
dn (%) presented.
Table 6. Outcomes of ANOVA to assess comparability of PK parameters for AT
versus
PFS-NSD (pivotal cohort)
AI PFS-NSD GMRe GMR
90% CPI
(test) (reference) (%) (%)
Parameter
Geometric Geometric
na na
meanb meanb
Cmax (gg/mL) 73 12.5 73 12.2 102 94.2-
111
AUCiast 98.0 89.3-
107
69 319 72 325
(day. g/mL)
AUCo-inf 97.6 88.6-
107
69 329 72 337
(day. g/mL)
Al autoinjector. ANOVA analysis of variance. AUCo-,,f AUC extrapolated to
infinity. AUCIast area under the
concentration-time curve from the time of drug administration to the last
measurable concentration. Cm maximum
concentration. GMR geometric mean ratio. PFS-NSD prefilled syringe with needle
safety device.
aNumber of observations in each treatment eligible for analysis.
bGeometric means are based on the least-squares means for C. and AUC
parameters from ANOVA, calculated by
transforming the natural log means back to the linear scale.
'Geometric mean ratio for test/reference ratio of parameter means for natural
log-transformed parameter (expressed as
a percent). Natural log-transformed ratios transformed back to the linear
scale,
d90% confidence interval for ratio of parameter means of natural log-
transformed parameter (expressed as a percent).
.. Natural log-transformed confidence limits transformed back to the linear
scale.
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
The 90% CIs of the GMRs for each of these primary PK parameters were within
the predefined equivalence limits of 80% to 125%, which meets the predefined
comparability criteria and supports equivalent exposure of etrolizumab between
2 device
groups. The results also demonstrated similar median time to maximum observed
concentration (tmax, 5.04 vs 6.97 days) and mean terminal elimination half-
life (t1/2, 11.8
vs 12.2 days) between AT and PFS-NSD groups.
Safety. The overall incidence of treatment-emergent ADAs among post-baseline
evaluable participants was 20.7% (6/29) in the pilot cohort and 29.7% (44/148)
in the
pivotal cohort, and was similar when comparing the AT and PFS-NSD groups in
both the
pilot cohort (3/15 [20%] vs 3/14 [21.4%]) and the pivotal cohort (20/73
[27.4%] vs 24/75
[32.0%]). PK profiles of ADA+ subjects appear to be similar to those ADA
negative
subjects (see Figure 21). Treatment-emergent adverse events (TEAEs) were
experienced
by 53% of participants in the pilot cohort and 35% in the pivotal cohort, and
most were
mild in severity (Table 7).
Table 7. Treatment-Emergent Adverse Events (TEAE)
Pilot cohort (part 1) Pivotal cohort (part 2)
Overall
(parts 1
PFS- Total PFS- Total
AI AI and 2)
NSD n = 30 NSD n = 150
n = 15 n = 74 n =
180
n = 15 n = 76
16 (53.3) 49 (32.7) 65
(36.1)
Any TEAE 9 (60.0) 7 (46.7) 22 (29.7) 27 (35.5)
Number of TEAEs 22 7 29 32 45 77 106
Any SAE 0 1 (6.7) 1(3.3) 0 0 0
1(0.6)
Any ISR-related TEAE 5(33.3) 4(26.7) 9(30.0) 1(1.4) 0
1 (0.7) 10 (5.6)
Suspected to be caused
by
Study drug 6(40.0) 3 (20.0) 9(30.0) 7(9.5) 10
(13.2) 17 (11.3) 26 (14.4)
Other causes 7(46.7) 2(26.7) 11 (36.7) 18 (24.3) 21
(27.6) 39 (26.0) 50 (27.8)
Concurrent Illness 4 (26.7) 0 4 (13.3) 7 (9.5) 14
(18.4) 21 (14.0) 25 (13.9)
Concomitant 0 3 (2.0) 3
(1.7)
0 0 2 (2.7) 1 (1.3)
medication
Procedures 3 (20.0) 1(6.7) 4 (13.3) 0 1(1.3)
1(0.7) 5 (2.8)
Other 2(13.3) 3 (20.0) 5 (16.7) 10 (13.5)
6(7.9) 16 (10.7) 21 (11.7)
Missing 0 0 0 1(1.4) 0 1(0.7)
1(0.6)
AE by most severe NCI
CTCAE grade
Grade 1 8 (53.3) 4 (26.7) 12 (40.0) 19 (25.7) 27
(35.5) 46 (30.7) 58 (32.2)
Grade 2 1(6.7) 2(13.3) 3 (10.0) 3 (4.1) 0 3
(2.0) 6(3.3)
Grade 3 0 1(6.7) 1(3.3) 0 0 0
1(0.6)
Total 9(60.0) 7 (46.7) 16 (53.3) 22 (29.7) 27
(35.5) 49 (32.7) 65 (36.1)
61
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
AE adverse event; Al autoinjector, ISR injection site reaction, NCI CTCAE
National Cancer Institute Common
Terminology Criteria for Adverse Events, PFS-NSD prefilled syringe with needle
safety device, SAE serious adverse
event, TEAE treatment-emergent adverse event, Data are reported as n (%)
unless otherwise specified.
One participant in the PFS-NSD arm of the pilot cohort had a serious AE (grade
3
seizure) approximately 32 days following injection that led to study
discontinuation and
was not considered related to treatment with etrolizumab. Nine participants (5
with AT
and 4 with PFS-NSD) in the pilot cohort and 1 participant (AI arm) in the
pivotal cohort
had injection site reactions that were reported as TEAEs. Treatment-related
TEAEs in
the pilot cohort were experienced by 6 (40.0%) participants in the AT arm and
3 (20.0%)
in the PFS-NSD arm, and in the pivotal cohort by 7 (9.5%) in the AT arm and 10
(13.2%)
in the PFS-NSD arm.
Discussion
The pivotal cohort in this study confirmed comparable etrolizumab exposure
between AT and PFS-NSD groups following a single dose of SC etrolizumab in
healthy
participants. The GMRs observed with each of the primary PK parameters were
between
98% and 102%, with 90% CIs within the prespecified equivalence limits. These
GMRs
for all primary exposure parameters are impressive, given the GMRs obtained
when
comparing other AIs and PFS-NSDs in healthy participants. For example, a
recent study
comparing AT and PFS devices for the SC administration of an adalimumab
biosimilar
(SB5) demonstrated GMRs of 102%, 107%, and 110% for Cmax, AUClast, and AUCinf,
respectively, with 90% CIs within equivalence limits of 80% to 125%. In a
similar study
of the adalimumab biosimilar BI 695501 they were 100% (90% CI 82.1-122.3%) for
AUC0-inf and 110% (90% CI 96.8-125.4%) for Cmax, the latter of which was above
the
equivalence upper 90% CI equivalence limit of 125% (Voltaire -AI study)
(Ramael et
al., Rheumatol Ther. . 2018;5(2):403-21; Shin et al., Drug Des Devel Ther. .
2018;12:3799-805). The pilot cohort was valuable to inform the final design of
the
pivotal cohort. The pilot cohort mainly evaluated GMRs and the variability of
PK
parameters, which are the key assumptions used in the sample size estimation.
To
minimize the risk of underpowering the pivotal PK comparability study, a small
pilot
cohort was added to the original study protocol with the intent of gaining
certainty
around GMR values of the primary PK parameters and PK variability following
administration of etrolizumab by AT or PFS-NSD. The GMR and PK variability
values
obtained from the pilot cohort (see Table 4) provided added confidence in
estimating the
sample size for the pivotal cohort. Although body weight was stratified at
62
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
randomization, the final body weight distribution range was still imbalanced
in the pilot
cohort, which may have biased the final GMR outcome. To minimize such bias,
only
data from participants with a body weight within the range of 60 to 100 kg (a
common
body weight range for both arms within the pilot cohort) were used for the
estimation of
GMR and PK variabilities. This body weight restriction of 60 to 100 kg was
also
implemented in the pivotal cohort as an inclusion criterion. The pilot cohort
also
evaluated a study duration of 10 weeks (70 days), 2 weeks shorter than the
original study
planned study duration as suggested by the FDA. As expected, all evaluable
participants
in the pilot cohort had AUCR values > 80%, the value required by the FDA for
PK
comparability studies. This result suggested that the 10-week study duration
is long
enough to capture more than 80% of AUCinf and hence the duration of the
pivotal study
could be shortened from 12 weeks to 10 weeks without the risk of missing the
requirement of < 20% extrapolated AUC for the calculated AUCinf.
Immunogenicity
was relatively high in this study compared with other studies using
etrolizumab. The rate
of ADAs was > 20% in both the pilot and pivotal cohorts, whereas 5% (2/38) of
patients
with UC who received single or multiple etrolizumab doses in a phase 1 trial
(Rutgeerts
et al., Gut. 2013;62(8):1122-30) and 5% (4/81) of patients with UC who
received
multiple etrolizumab doses in a randomized phase 2 trial (Vermeire et al.,
Lancet.
2014;384(9940):309-18) had detectable ADAs following etrolizumab treatment.
Immunogenicity incidence rate in the pilot and pivotal cohorts was equivalent
in both
PFS-NSD and AT groups. The higher ADA rate in the current study may have been
the
result of differences in study design: the current study had a single SC dose
regimen and
a study population of healthy participants not taking immunosuppressive drugs.
In
comparison, patients in prior trials of etrolizumab had insufficiently
controlled,
moderate-to-severe UC and were commonly being treated with immunosuppressive
agents. Although the incidence of ADAs after a single SC injection of
etrolizumab was
relatively high (>20%) in this study, the impact of ADA positivity on PK
appears to be
minimal, given that the PK profiles of ADA-positive participants largely
overlapped with
those observed in ADA-negative participants (Figure 21). Moreover, the
variability
(CV%) of AUCo_inf or AUCInst values ranged from 33% to 36% in the pivotal part
2
study, which was in line with those observed for other monoclonal antibodies
(Ramael et
al., Rheumatol Ther. . 2018;5(2):403-21; Anumolu et al., Clin Pharmacol Drug
Dev.
2018;7(8):829-36). Such a small exposure variability further confirms that the
total
exposure (AUCof) between ADA-positive and ADA-negative participants was very
63
CA 03190109 2023-01-24
WO 2022/026699 PCT/US2021/043690
similar. Data from all evaluable participants, regardless of their ADA status,
were
included in the comparability statistical assessment, and the final outcome
showed a high
degree of exposure similarity between groups. The bioequivalence testing of
the AT and
PFS-NSD groups and sample size determination in the pivotal study design
following a
single SC dose of etrolizumab were based on data from a subset of participants
within a
body weight range of 60 to 100 kg. Although our finding of comparable
etrolizumab
exposure between the AT and PFS-NSD was based on data from participants within
a
defined body weight range, it can be applied to individuals outside this body
weight
range, given that the current study identified no exposure difference solely
due to the
drug delivery device. Furthermore, a similar study for another therapeutic
antibody (that
did not restrict by body weight) suggests that this may be a fair assumption
(Ramael et
al., Rheumatol Ther. . 2018;5(2):403-21). In that study, a similar
relationship between
drug exposure and body weight was observed, but when comparing AT and PFS-NSD
devices for adalimumab SC administration, bioequivalence was still achieved
regardless
of body weight. Clearance of etrolizumab is known to be significantly impacted
by body
weight (Tang et al., Aliment Pharmacol Ther. . 2018;47(11):1440-52), a finding
confirmed in the pilot cohort of this study. In these healthy participants, a
single SC dose
of etrolizumab was generally safe and well-tolerated when administered using
either an
AT or PFS-NSD. Treatment-related TEAEs were comparable with the AT versus the
PFS-
NSD in the pivotal cohort (9.5% vs 13.2%). The majority of TEAEs were mild or
moderate in severity, and mostly resolved by the end of the study. One serious
AE
(seizure) led to study discontinuation in the PFS-NSD arm of the pilot cohort;
however,
this was not considered to be related to treatment with etrolizumab.
Conclusion
Results from this PK comparability study demonstrate that etrolizumab exposure
is similar when administered Sc via either Al or PFS-NSD, with 90% Cl for
exposure
parameters (AUCIast, cm, AUCinf) contained within bioequivalence limits (80-
125%)
between devices. In addition, single SC doses of etrolizumab administered via
AT were
generally well-tolerated in healthy participants and were not associated with
increased
adverse events compared with PFS-NSD injection. This study also highlights the
value
of a pilot cohort in facilitating the design of a pivotal PK comparability
study, as data
from the pilot cohort increased confidence in the study design and minimized
assumption
bias.
64
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
Example 4
This Example provides details relating to PFS-NSD injection forces.
There are multiple force definitions during the injection experience for the
user.
The user actions are: 1) remove the needle cover, and 2) press the plunger
until the
needle safety is activated. Figure 22 shows the different force definitions.
Table 8 shows
the forces and their limits. Table 9 shows that the injection forces are
within limits for
both lmL and 2.25mL configurations.
Table 8
Force Lirnit
Average Injection Force (aka Glide Force) <26 Newton (N)
Break Loose Force <33N
Maximum Injection Force <33N
Table 9
Attribute Real time aging (2-8 C) High Temperature
(25
Time is shown in months below C)
T=initial T=6 T=12, T=3 T=6
24
Glide Force Within limits Within limits
Break Loose Within limits Within limits
Force
Max Injection Within limits Within limits
Force
There is a spring mechanism that pushes the liquid out for the autoinjector.
Therefore, injection time is relevant attribute. In certain embodiments the
injection time
is between about 0.4 seconds and about 9 seconds. In certain embodiment, the
injection
time is about 5 seconds.
The autoinjector used for the presently disclosed formulation provides
advantageous injection times and extended stability.
Embodiments of the presently disclosed subject matter
From the foregoing description, it will be apparent that variations and
modifications may be made to the presently disclosed subject matter to adopt
it to
various usages and conditions. Such embodiments are also within the scope of
the
following claims.
The recitation of a listing of elements in any definition of a variable herein
includes definitions of that variable as any single element or combination (or
sub-
combination) of listed elements. The recitation of an embodiment herein
includes that
CA 03190109 2023-01-24
WO 2022/026699
PCT/US2021/043690
embodiment as any single embodiment or in combination with any other
embodiments or
portions thereof.
All patents and publications mentioned in this specification are herein
incorporated by reference to the same extent as if each independent patent and
publication was specifically and individually indicated to be incorporated by
reference.
66