Note: Descriptions are shown in the official language in which they were submitted.
CA 03193631 2023-03-02
WO 2022/053637 -1-
PCT/EP2021/074978
METHYLATION DETECTION ASSAY
FIELD OF THE INVENTION
The present invention in general relates to the field of DNA methylation
detection. More in
particular the current invention provides a method for DNA methylation
detection using a
combination of methylation-specific restriction enzymes (MSRE) and single
molecule Molecular
Inversion Probes (smMIPs). The present invention also provides uses of said
DNA methylation
detection method such as but not limited to the field of cancer diagnosis
and/or monitoring.
BACKGROUND TO THE INVENTION
Each year, an estimated 8.2 million people die of cancer worldwide. With
appropriate detection
methods and treatment, many of these deaths would be avoidable. Due to the
high incidence
and mortality rates, early and accurate diagnosis is paramount for a quick and
adequate
treatment of patients. Until recently, no truly non-invasive diagnostic
methods for the detection
of cancer existed. An attractive novel method is the detection of abnormally
expressed
biological markers manifested during carcinogenesis in so called "liquid
biopsies". Liquid biopsy
is a technique in which non-solid biological tissues such as urine, stool or
peripheral blood, are
sampled and analyzed for disease diagnosis.
The analysis of Circulating tumor DNA (CtDNA) in liquid biopsy samples of
cancer patients is
not new and has been performed in the past. However, until now, a strong focus
existed on the
detection of tumor specific mutations, which has several limitations. One of
the problems with
liquid biopsy nucleic acid biomarkers is the limited sensitivity for early
detection. Indeed, in early
stages of carcinogenesis, many tumor types have low concentrations of CtDNA.
Sensitivity can
be increased by measuring a multitude of markers simultaneously. The use of
methylation
markers instead of mutation markers has many advantages and is understudied.
However, to
date, no efficient techniques exist allowing multi-region methylation analysis
in plasma or other
types of liquid biopsies.
Most methylation assays currently used are based on bisulfite conversion of
DNA, which allows
for identification of methylated C's, as they are protected from conversion to
T. A major
challenge in bisulfite conversion methods is the degradation of DNA caused by
bisulfite. Even
carefully controlled conditions for complete conversion lead to the
degradation of about 90% of
the incubated DNA. While this is not a problem when large amounts of DNA are
available, it is
.. unacceptable for liquid biopsies, as they yield very limited amounts of
DNA. In addition, careful
control of the reaction parameters is often problematic, which has led to a
poor reputation of
bisulfite conversion in terms of robustness and reliability. Therefore, liquid
biopsy analysis
requires a bisulfite-free analysis.
CA 03193631 2023-03-02
WO 2022/053637 -2-
PCT/EP2021/074978
The alternatives for bisulfite conversion methods are immunoprecipitation
methods and
methods based on methylation-specific restriction enzymes (MSRE).
Immunoprecipitation
methods such as MeDIP or Methyl-cap are based on me-CpG recognizing antibodies
or methyl-
binding proteins. Unfortunately, the antibody or methyl binding proteins are
imperfect, and
introduce false positive results and unwanted bias towards certain regions of
the genome.
Therefore, there is a continuing need in the art for novel methods for DNA
methylation
detection, which are particularly suitable for liquid biopsies containing low
amounts of DNA. In
the present invention, we have surprisingly found that by combining
methylation-specific
restriction enzyme-assisted digestion of DNA for obtaining a set of DNA
fragments, and single
molecule Molecular Inversion Probe-assisted detection of said DNA fragments, a
very powerful
method for DNA methylation detection is obtained. This method cannot only
suitably be used in
the field of cancer diagnostics and/or monitoring, but is applicable to all
fields in which DNA
methylation detection is relevant. In particular, this novel approach has the
potential to increase
sensitivity up to 1000-fold compared to current methylation detection methods,
while reducing
the cost more than a 100-fold.
SUMMARY OF THE INVENTION
In a first aspect, the present invention provides a method for the detection
of DNA methylation
comprising the steps of:
a) providing a sample comprising one or more DNA molecules;
b) digesting said one or more DNA molecules by using one or more methylation-
specific
restriction enzymes (MSRE), thereby obtaining a set of DNA fragments;
c) capturing and amplifying said set of DNA fragments using single molecule
Molecular
Inversion Probe technology (smMIP); thereby obtaining a set of amplified DNA
fragments;
d) detecting said amplified DNA fragments; thereby detecting DNA methylation.
In a further aspect, the present invention provides a method for the detection
of DNA
methylation comprising the steps of:
a) providing a sample comprising one or more DNA molecules;
b) digesting said one or more DNA molecules by using one or more methylation-
specific
restriction enzymes (MSRE), thereby obtaining a set of DNA fragments;
c) capturing and amplifying said set of DNA fragments using one or more single
molecule
Molecular Inversion Probes (smMIPs); each spanning at least one methylation
site in the
DNA; thereby obtaining a set of amplified DNA fragments; wherein each smMIP
comprises one or more specific alignment tag(s);
d) detecting said amplified DNA fragments; thereby detecting DNA methylation.
CA 03193631 2023-03-02
-3-
WO 2022/053637
PCT/EP2021/074978
In a specific embodiment, said smMIP technology comprises the use of one or
more smMIPs
each spanning at least one methylation site in the DNA.
In another particular embodiment, each of said smMIPs (further) comprises:
- a specific extension probe, which specifically hybridizes to a first site
next to a methylation
site;
- a common backbone, which is not capable of hybridizing to said DNA
molecules, and
- a specific ligation probe, which specifically hybridizes to a second site
next to said
methylation site, and
- a specific molecular tag, in particular an alignment tag.
In a specific embodiment, said alignment tag(s) comprise at least four random
nucleotides.
In another embodiment, said alignment tag(s) is/are located between the common
backbone
and the extension probe or between the common backbone and the ligation probe,
or both.
In a particular embodiment, said smMIP comprises two alignment tags each
comprising at least
4 nucleotides located between the common backbone and each of said probes, or
one
alignment tag comprising at least 8 random nucleotides between the common
backbone and
said ligation or extension probe.
In yet a further embodiment, step c) of the method of the present invention
comprises the steps
of:
c1) hybridizing said one or more smMIPs to said DNA fragments;
c2) performing an extension reaction from the extension probe(s) across the
methylation
site(s) in the direction of the ligation probe(s);
c3) performing a ligation reaction using the ligation probe(s), thereby
obtaining a circular
DNA fragment;
c4) performing an exonuclease treatment to digest non-circular DNA fragments;
c5) performing an amplification reaction using common primers capable of
hybridizing to said
common backbone; thereby obtaining a set of amplified DNA fragments.
In yet a further embodiment of the present invention, each of said amplified
DNA fragments
comprises a specific extension probe sequence, a methylation site sequence, a
specific ligation
probe sequence, sequencing adaptors, primer binding sites, and a specific
molecular (in
particular alignment) tag sequence; or the complement thereof.
In a further embodiment, step d) of the method of the present invention
comprises the step of
detecting said amplified DNA fragments using next generation sequencing.
CA 03193631 2023-03-02
-4-
WO 2022/053637
PCT/EP2021/074978
In another particular embodiment, said method comprises the use of at least 2
methylation-
specific restriction enzymes.
In a further embodiment, said methylation-specific restriction enzymes are
only capable of
digesting unmethylated DNA regions.
In another embodiment, said one or more MSRE are selected from the list
comprising: Hpall,
HinP1I, Acil, HpyCH4IV and combinations thereof.
In a further embodiment, the method of the present invention further comprises
one or more
control smMIPs which do not span a CpG region and/or one or more control
smMIPs which
span a CpG region that does not include a restriction site for said MSRE.
In yet a further embodiment, said sample is a solid or liquid biopsy sample
from a subject, in
particular a liquid biopsy.
In a further aspect, the present invention provides the use of a combination
of methylation-
specific restriction enzymes (MSRE) and single molecule Molecular Inversion
Probe technology
(smMIP) in the detection of DNA methylation in a biological sample.
In a further aspect, the present invention provides the use of a combination
of methylation-
specific restriction enzymes (MSRE) to obtain a set of DNA fragments; and
single molecule
Molecular Inversion Probes (smMIPs) to capture and amplify said set of DNA
fragments; in the
detection of DNA methylation in a biological sample.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1: shows the principal of methylation-specific restriction digestion
(MSRE)
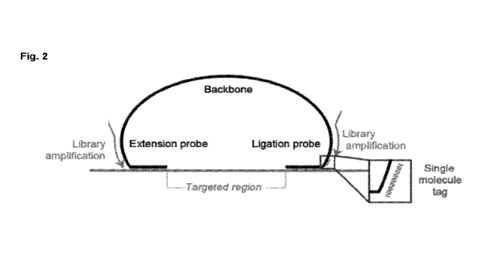
Fig. 2: shows a typical smMIP design according to the present invention.
Fig. 3: shows a schematic overview of the smMIP technology according to the
present
invention.
Fig. 4: Efficiency of the subset of smMIPs. Top 52 smMIPs are 100x more
efficient compared
to all smMIPs, improving the total efficiency of the assay
Fig. 5: Repeatability of the assay. Panel A and B show the samples that were
not digested by
the MSREs (uncut). Panel C and D show the samples that were digested by the
MSREs
(cut). Panel A and C are non-CpG control smMIPs and Panels B and D are CpG
smMIPs.
Samples 1-10 are DNA samples originating from human blood. Sample 11 is DNA
originating from an artificially methylated blood sample and sample 12 is DNA
isolated
from a CRC cell line. The relative coverage of the smMIPS per sample is
calculated as
CA 03193631 2023-03-02
-5-
WO 2022/053637
PCT/EP2021/074978
following: A) the sum of the counts in an uncut sample for non-CpG smMIPs,
divided by
the total counts for all non-CpG smMIPs in the uncut samples B) the sum of the
counts in
an uncut sample for CpG smMIPs, divided by the total counts for all CpG smMIPs
in the
uncut samples C) idem as A, but for cut samples and D) idem as B but for cut
samples.
Fig. 6: Correlation between the relative counts per smMIP for uncut samples of
two
independent runs. The average relative coverage per smMIP is shown for run A
and run
B.
Fig. 7: Correlation between the relative counts per smMIP for cut samples of
two
independent runs. Panel A shows the relative counts per smMIP in a low
methylated,
cut sample and Panel B shows the relative counts per smMIP in a highly
methylated, cut
sample.
Fig. 8: Counts per smMIP in different digested control samples. X-axis: 32 CpG
smMIPs;
Y-axis: absolute counts per smMIP per sample. Example of a nano v2 run on the
MiSeq.
Fig. 9: Results of the qPCR. Different sample conditions for a complete MSRE
digestion were
tested by qPCR. Distinct input amounts of DNA (ng) were digested in distinct
end
volumes (pL). Thereafter, fragmentation was examined by qPCR with primer pairs
around
the MSRE recognition sites. Uncut samples were included as reference. Primer3
was left
out of the analysis as the melt-curves showed this primer is poorly
performing.
Fig. 10: Comparison of normal DNA (healthy blood) and colorectal cancer tissue
DNA.
smMIPs designed for capture of hypermethylated sites are shown in grey. smMIPs
that
capture non CpG sites in black. Blood samples (B) are shown on the left part
of the
figure, colorectal cancer samples (CRC) on the right. Comparing the bars, a
clear
difference between normal and cancer can be made.
Fig. 11: Principal component analysis (PCA) plot. Blood samples (circles) are
clustered on
the left side and colorectal cancer samples (triangles) on the right side.
There is a very
clear distinction between the two groups.
Fig. 12: Absolute read counts for non CpG and CpG smMIPs in MSRE-digested
samples.
In this experiment, all samples were MSRE digested and enriched using a
selection of +/-
800 efficient smMIPS. The absolute read counts are given for gDNA extracted
from
normal blood samples (left) and compared to gDNA extracted from cancer cell
lines
(right). It is clear that a perfect distinction can be made based on the CpG
smMIPs (light
bars). nonCpG smMIPs are also shown (dark bars) and theoretically should not
differ
between cancer and normal samples
Blood = gDNA extracted from whole blood from healthy volunteers. Methylated
blood =
one of these blood samples, in vitro methylated with the CpG methyltransferase
Sss.I
enzyme. CL = gDNA extracted from cell lines. CRC= colorectal cancer, BRCA=
breast
cancer, LUNG= lung cancer, HNSC= head and neck squamous cell cancer, PANC =
pancreatic cancer and PRCA= prostate cancer.
CA 03193631 2023-03-02
WO 2022/053637 -6-
PCT/EP2021/074978
Fig. 13: Principal component plot of the first two PC's.
Based on the experiment displayed in figure 12, a subselection of the 500 most
discriminating smMIPS was performed and the first two principal components
(PCs) were
plotted. Blood samples are shown as circles, cancer samples as triangles and
an
artificially methylated blood sample as a square. The figure shows that a
perfect
separation exists between blood samples and cancer cell lines.
Fig. 14: Proof of principle showing benefits of removing PCR duplicates in
data analysis.
Here, the results of analyses with and without duplicate removal are compared
using the
same raw input data (see equation 1 and 2). Proportion ratios of reads per
sample (Panel
A) or per smMIP (Panel B) are displayed. Proportion ratios between 0.95 and
1.05 are in
white, one interval further in light grey and others dark grey. Each bar
indicates the
number of samples (A) or smMIPs (B) within the specified interval of
proportion ratios.
Intervals are half open, upper limit included..
Fig. 15: Refinement of the proportion analysis; this time comparing ratio per
smMIP per
sample, divided by the reads per smMIP (Panel A) or divided by the reads per
sample (Panel B). This analysis is similar to the one shown in fig. 14, albeit
more refined
(see also equations 3 and 4). The conclusion from this analysis is that a
molecular tag
reduces technical noise within an experiment. Proportion ratios between 0.95
and 1.05
are in white, one interval further in light grey and others in dark grey. Each
bar indicates
the number of smMIPs per sample within the specified interval of proportion
ratios.
Intervals are half open, upper limit included.
CA 03193631 2023-03-02
-7-
WO 2022/053637
PCT/EP2021/074978
DETAILED DESCRIPTION OF THE INVENTION
As already detailed herein above, the present invention relates to a method
for the detection of
DNA methylation comprising a combination of methylation-specific restriction
enzyme-assisted
(MSRE) digestion of DNA thereby obtaining a set of DNA fragments, followed by
single
molecule Molecular Inversion Probe-assisted (smMIP) amplification and
detection of said DNA
fragments.
Accordingly, in a first aspect, the present invention provides a method for
the detection of DNA
methylation comprising the steps of:
a) providing a sample comprising one or more DNA molecules;
b) digesting said one or more DNA molecules by using one or more methylation-
specific
restriction enzymes (MSRE), thereby obtaining a set of DNA fragments;
c) capturing and amplifying said set of DNA fragments using single molecule
Molecular
Inversion Probe technology (smMIP); thereby obtaining a set of amplified DNA
fragments;
d) detecting said amplified DNA fragments; thereby detecting DNA methylation.
More in particular, the present invention provides a method for the detection
of DNA methylation
comprising the steps of:
a) providing a sample comprising one or more DNA molecules;
b) digesting said one or more DNA molecules by using one or more methylation-
specific
restriction enzymes (MSRE), thereby obtaining a set of DNA fragments;
c) capturing and amplifying said set of DNA fragments using one or more single
molecule
Molecular Inversion Probes (smMIPs) each spanning at least one methylation
site in the
DNA; thereby obtaining a set of amplified DNA fragments; wherein each smMIP
comprises one or more specific alignment tag(s);
d) detecting said amplified DNA fragments; thereby detecting DNA methylation.
As used herein and unless otherwise specified, the term "alignment tag" may
also refer to
"alignment sequence", "single molecule tag", "molecular tag", or "tag" and
these terms can be
used interchangeably throughout the application. At any instance, the
'alignment' tag is meant
to be a nucleotide sequence which allows the alignment of amplified DNA
fragments into
particular consensus read sequences. Accordingly, using sequencing tools such
as next
generation sequencing, the alignment tags allow sequence reads containing the
same
alignment tag (and thus originating from the same capture) to be merged into
one consensus
read sequence. Accordingly, duplicate reads can be identified and filtered out
thereby removing
PCR and sequencing artifacts and enabling detection of low-frequency and sub
clonal genetic
variation. This results in an ultra-sensitive targeted sequencing method
exhibiting the specificity
and multiplexing advantage of the MIPs and the quantitation ability of the
"single molecule
tagging" approach. In a particular embodiment, the alignment tag described in
the present
CA 03193631 2023-03-02
WO 2022/053637 -8-
PCT/EP2021/074978
invention does not function as a detection probe. More specifically, tag
sequences as described
in for example US2006292585 or W02012112970 are recognized by an array of tag
probes that
are complementary to the tag sequences in the MIPs and facilitate detection of
PCR amplified
sequences. This is in clear contrast to the alignment tags described herein,
which serve the
purpose of aligning amplified DNA fragments into particular consensus read
sequences
avoiding duplicate reads.
The inventors have thus developed a method to enrich and assess specific DNA
loci for
methylation content using a protocol that combines MSREs and smMIPs. The
technology has
the advantage to increase sensitivity compared to current existing
technologies, while reducing
costs significantly.
Selection of relevant detection biomarkers allows the assay to be either pan-
cancer or cancer
specific. This approach has the potential to increase sensitivity 100 to 1000-
fold compared to
current technologies while reducing the cost more than a 100-fold. The current
invention is a
technique for multiplex analysis of a selected number of methylation sites in
the genome,
allowing sensitive analysis of even small quantities of DNA at an affordable
cost. The field of
liquid biopsies for the early detection of cancer is booming, with large
investments worldwide in
the biotech and pharma sector. The analysis and importance of the methylome is
also an
expanding field, not only in oncology but also well beyond. Methylation of DNA
in particular and
epigenetics in general are linked to a wide range of diseases and health
conditions. As such,
the potential application of this technology is broad, with a first focus on
oncology diagnostics.
This technology has many applications, such as the detection of methylated CpG
signatures in
cancer research. As this technology will be much more sensitive compared to
current protocols,
it has also applications in the detection of methylated DNA fragments in
liquid biopsies.
Accordingly, in a further embodiment, said sample is a solid or liquid biopsy
sample from a
subject, in particular a liquid biopsy. Alternatively, said sample may also be
derived from a cell
line. As stated already, this novel detection assay is specifically suitable
as a cancer detection
assay, which allows for high resolution methylation detection in tissues,
plasma or other
biological matrices (e.g. blood, urine, saliva ...) of cancer patients.
MSREs have been used for a very long time for analysis of methylation in
specific regions of the
genome, and more recently, also for genome wide analysis. In contrast to
antibodies, restriction
.. enzymes are ultimately specific and predictable in their action. In the
presented protocol, we
use MSREs to digest genomic DNA. These MSREs are very specific and cut the DNA
only
when it is unmethylated and not when it is methylated, as illustrated in
figure 1. As used herein,
MSRE may refer to "Methylation-Specific Restriction Enzymes" or "Methylation-
Sensitive
Restriction Enzymes" and the terms can be used interchangeably.
CA 03193631 2023-03-02
-9-
WO 2022/053637
PCT/EP2021/074978
Using MSREs instead of bisulfite, which is a widely used compound used in
methylation
analysis, has strong advantages. The main advantage is that MSREs do not
degrade DNA, in
contrast to bisulfite.
As used herein, the terms "restriction endonucleases" and "restriction
enzymes" refer to
bacterial enzymes each of which cut double-stranded DNA at or near a specific
nucleotide
sequence.
After digestion, specific "a priori" selected DNA loci are enriched and
sequencing libraries are
generated using smMIPs. smMIPs technology is used to capture and enrich
specific DNA
fragments, resulting in DNA libraries that can be sequenced. If used with a
sufficiently high
annealing temperature, smMIPs are highly specific. smMIPs are typically
designed to anneal at
a temperature of about 60 C. However, this may also be altered for example
when a lot of
unwanted 'side products' are generated, wherein the temperature can be
slightly increased.
Accordingly, typical smMIPs have an annealing temperature of about 50 C ¨ 70
C, more in
particular about 55 C ¨ 65 C, such as about 55 C, about 56 C, about 57 C,
about 58 C,
about 59 C, about 60 C, about 61 C, about 62 C, about 63 C, about 64 C,
about 65 C.
In addition, typically smMIPs are allowed to capture the DNA in a reaction at
60 C, for over 20
hours. We have now surprisingly found though that a cycled capture reaction of
5 times 4 hours
at 60 C with an intermediate denaturation step results in an increase of the
percentage of
unique reads. Accordingly, in a particular embodiment, the smMIP reaction
comprises a cycled
capture reaction of x times y hours at a predefined temperature, which is as
discussed above;
wherein x is selected from 3-6, in particular 5; and y is selected from 2-6,
in particular 4.
Moreover, smMIPs are highly multiplexable, so that the combination of both
steps results in the
potential detection of several thousands of CpG methylation sites in a single
sequencing run.
smMIPs based approaches have been used for several applications, in
combination with
various techniques including microarray or next generation sequencing. These
applications
include SNP genotyping, Copy Number Variation quantification, and resequencing
of genomic
regions. smMIPs are extremely suitable for multiplex analyses, with routine
multiplexing up to
50.000 being reported. For smMIPs design, we are routinely using a
bioinformatics pipeline that
was originally developed at the Radboud University in the Netherlands. In this
pipeline,
parameters for smMIPs design can be easily adapted.
Specific smMIPs at carefully chosen informative CpG sites in the genome will
be generated,
and hybridized to the digested DNA that was cut with the methylation-specific
restriction
enzymes. Subsequent extension and ligation of the smMIPs will only take place
if the CpG site
is methylated, because if it is unmethylated, it will be cut by the action of
the restriction enzymes
and no hybridization can take place.
CA 03193631 2023-03-02
WO 2022/053637 - 1 0-
PCT/EP2021/074978
Accordingly, in a specific embodiment, said smMIP technology comprises the use
of one or
more smMIPs each spanning at least one methylation site in the DNA.
In a particular embodiment, each of said smMIPs comprises:
- a specific extension probe, which specifically hybridizes to a first site
next to a methylation
site;
- a common backbone, which is not capable of hybridizing to said DNA
molecules;
- a specific ligation probe, which specifically hybridizes to a second site
next to said
methylation site, and
- a specific molecular tag, in particular an alignment tag.
In a particular embodiment, a set of amplified DNA fragments is obtained using
one or more
smMIPs each spanning at least one methylation site in the DNA; wherein each
smMIP
comprises one or more specific alignment tag(s);
In another particular embodiment, each of said smMIPs further comprises:
- a specific extension probe, which specifically hybridizes to a first site
next to a methylation
site;
- a common backbone, which is not capable of hybridizing to said DNA
molecules, and;
- a specific ligation probe, which specifically hybridizes to a second site
next to said
methylation site.
Such smMIP design in accordance with the present invention is for example
illustrated in figure
2. In the present case, the extension probe hybridizes 5' of the targeted
region (i.e. including the
methylation site), and the ligation probe hybridizes 3' of said targeted
region. Both probes are
attached to each other by means of a non-complementary backbone. Said backbone
is
designed such as to allow hybridization of library amplification primers, for
amplification of the
targeted region. These amplification primers contain P5 and P7 illumina
sequences so that the
resulting PCR product can anneal to the flow cell. Finally, the smMIP
comprises a specific
molecular tag as part of the backbone. While in the present figure the
molecular tag is indicated
to be present between the ligation probe and one of the library amplification
primer sites, it may
alternatively be present anywhere in the smMIP, specifically in the backbone
region. In a
particular embodiment, said molecular tag(s) is/are located between the common
backbone and
the extension probe or between the common backbone and the ligation probe, or
both.
The smMIPs of the present invention are in particular designed such as to have
a relatively
small targeted region (about 50 bp), compared to standard smMIPs (typically at
least 100 bp). In
particular the smMIPs of the present invention are designed to have a targeted
region of about
¨ 60 bp, such as about 40 bp, about 45 bp, about 50 bp, about 55 bp, about 60
bp. This is in
40 particular advantageous in the context of liquid biopsies, containing
fragmented DNA.
CA 03193631 2023-03-02
- -
WO 2022/053637 11
PCT/EP2021/074978
As used herein, the term "primer" refers to an oligonucleotide, whether
occurring naturally as in
a purified restriction digest or produced synthetically, which is capable of
acting as a point of
initiation of nucleic acid sequence synthesis when placed under conditions in
which synthesis of
a primer extension product which is complementary to a nucleic acid strand is
induced, i.e. in
the presence of different nucleotide triphosphates and a polymerase in an
appropriate buffer
(buffer" includes pH, ionic strength, cofactors etc.) and at a suitable
temperature. One or more
of the nucleotides of the primer can be modified for instance by addition of a
methyl group, a
biotin or digoxigenin moiety, a fluorescent tag or by using radioactive
nucleotides.
As used herein, the term "target sequence" refers to a specific nucleic acid
sequence to be
detected and/or quantified in the sample to be analysed.
The smMIP reaction typically comprises 3 separate steps, as shown in figure 3.
During a first
capture reaction (Fig. 3A), the smMIPs are allowed to hybridize to the
fragmented DNA. The
'gap' defined by the targeted region in the smMIP is subsequently filled-in
using a DNA
polymerase, followed by a ligase thereby obtaining a circular smMIP. This step
typically takes
about 16 to 22h.
In a second stage, and in order to 'clean-up' all linear DNA fragments, an exo-
nuclease reaction
is performed, thereby, only circular smMIPs are retained (Fig. 3B). This step
typically takes
about 1h.
Finally, these circular smMIPs are amplified by means of a PCR reaction using
the amplification
primers recognizing sequences in the backbone of the smMIPs (Fig. 3C). The
single molecular
tag can be used during the subsequent detection reaction for identification of
the fragments,
such as for example during multiplex next-generation sequence analysis.
Accordingly, in a further embodiment of the present invention, step c)
comprises the steps of:
c1) hybridizing said one or more smMIPs to said DNA fragments;
c2) performing an extension reaction from the extension probe(s) across the
methylation
site(s) in the direction of the ligation probe(s);
c3) performing a ligation reaction using the ligation probe(s), thereby
obtaining a circular
DNA fragment;
c4) performing an exonuclease treatment to digest non-circular DNA fragments;
c5) performing an amplification reaction using amplification primers capable
of hybridizing to
said common backbone; thereby obtaining a set of amplified DNA fragments.
As used herein the term "PCR" refers to the polymerase chain reaction. The PCR
amplification
process results in the exponential increase of discrete DNA fragments whose
length is defined
by the 5 ends of the oligonucleotide primers.
CA 03193631 2023-03-02
WO 2022/053637 -12-
PCT/EP2021/074978
As used herein, the terms "hybridisation" and "annealing" are used in
reference to the pairing of
complementary nucleic acids.
Following the method of the present invention, in a particular embodiment each
of said amplified
DNA fragments thus comprises a specific extension probe sequence, a
methylation site
sequence, a specific ligation probe sequence, sequencing adaptors, primer
binding sites, and a
specific molecular tag sequence; or the complement thereof.
These amplified DNA fragments can then be detected using any suitable
methodology, such as
by means of next generation sequencing. Accordingly, in a further embodiment,
step d) of the
method of the present invention comprises the step of detecting said amplified
DNA fragments
using next generation sequencing. The molecular tag sequence can be used in
this stage to
align the sequences into particular consensus read sequences. More
specifically, sequence
reads containing the same tag (and thus originating from the same capture) are
merged into
one consensus read sequence which can be detected using next-generation
sequencing. In the
context of the present invention, the tag sequence is a unique barcode that is
incorporated in
each smMIP allowing true molecule counting. Due to the obligatory PCR
amplification step,
which is necessary to obtain enough DNA templates for subsequent (next-
generation)
sequencing, PCR-duplicates will arise. Using this single molecule tag, it is
possible to account
for duplicates and as such, precise quantification is achieved. This accurate
counting would not
be possible when only MIPs are used. Accordingly, in another particular
embodiment, a smMIPs
comprises one or more specific alignment tag(s). The specific tag sequence can
have a
sequence variation as provided herein and may comprise at least 4, 5, 6, 7, 8,
9, 10, ... random
nucleotides.
In a specific embodiment, the alignment tag(s) comprises at least 4 random
nucleotides.
In a particular embodiment, the smMIP comprises two alignment tags, each
comprising at least
4 nucleotides (i.e. 2 separate tags), located near the ligation and extension
probe. Alternatively,
the smMIP comprises one alignment tag comprising at least 8 random nucleotides
located near
the ligation or extension probe. In a preferred embodiment, said alignment
sequence comprises
two tags of at least 4 nucleotides, wherein one of the tags is located between
the common
backbone and the ligation probe, and the other tag is located between the
common backbone
and the extension probe. In a preferred embodiment, the alignment sequence
comprises 8
random nucleotides located between the common backbone and either the ligation
or the
extension probe. Accordingly potential sequence variations erroneously
introduced during the
PCR reaction as well as sequencing artefacts can be easily detected and
ignored. Therefore,
the eventual sequence reads are much more reliable (see last section of the
examples).
It is advantageous and moreover technically feasible in the method of the
present invention to
use multiple MSRE to fragment the DNA. Accordingly, it allows multiple
potential methylation
sites to be detected simultaneously, even if these are cut by different MSRE.
CA 03193631 2023-03-02
WO 2022/053637 -13-
PCT/EP2021/074978
Therefore, in another particular embodiment, the method of the present
invention comprises the
use of at least 2 methylation-specific restriction enzymes.
The MSRE according to the present invention are in particular characterized in
digesting
unmethylated DNA regions. Accordingly, unmethylated DNA regions are digested
and can no
longer bind the smMIPs, thereby leaving only methylated regions to be
detectable, providing an
excellent tool for the identification of such methylated regions. Particularly
suitable MSRE in the
context of the present invention may be selected from the non-limiting list of
Hpall, HinP1I, Acil,
HpyCH4IV and combinations thereof.
Since methylation typically occurs in CpG regions, the present invention may
further comprise
the use of control smMIPs which do not span a CpG region. These regions are
thus typically not
digested by the methylation-specific enzymes and the detection thereof can be
used to assess
and/or monitor the reaction, or be used as a reference quantification marker.
In another
embodiment, the present invention may further comprise the use of control
smMIPs that span a
CpG region that do not include a restriction site for the MSREs (i.e. noRS
smMIPs).
In some circumstances, a sample may contain substances that interfere with a
subsequent
restriction, amplification and/or detection step. In a method according to the
invention, such
interference may be avoided by extracting the DNA from a sample prior to
digestion. Hence, the
invention relates to a method as described above, wherein the DNA is extracted
from the
sample before allowing the DNA to be cut by the MSRE.
In summary, the present invention is particularly directed to the use of a
combination of
methylation-specific restriction enzymes (MSRE) and single molecule Molecular
Inversion Probe
technology (smMIP) in the detection of DNA methylation in a biological sample.
EXAMPLES
Selection of differentially methylated regions (DMRs) for incorporation in a
MSRE-
smMIPS pan-cancer detection panel
We selected a most optimal set of DMRs and developed this set into a MSRE-
smMIPs-seq pan-
cancer assay. Using the pipelines that are used to generate the data, an
analysis for a total of
14 cancer types is performed on the basis of large datasets of 14 common
cancer types,
available from TCGA as well as the Gene Expression Omnibus database, and
common DMRs
are selected.
CA 03193631 2023-03-02
WO 2022/053637 -14-
PCT/EP2021/074978
Subsequently, our smMIPs design bioinformatics pipeline is specifically
adapted towards the
selection of smMIPs with optimal annealing temperatures covering both DMRs and
adapter
sequences, and the higher annealing temperature. We design specific smMIPs for
1000 DMRs
that rank highest in the design pipeline, and these are combined in a single
assay. In a
balancing run for this assay, the performance of each individual smMIP is
evaluated.
Subsequently, a new assay is developed containing 500 well performing smMIPs,
and individual
smMIP concentrations are adapted for uniform performance. A bioinformatics
pipeline for the
analysis of the sequencing data is developed. This pipeline is specifically
tailored towards the
accurate quantification of the methylation status of each DMR/CpG region.
Validation of the MSRE-smMIPs-seq pan-cancer assay in breast, colon and lung
cancer
After generation and optimization of the new MSRE-SMIPS-seq pan-cancer assay,
and final
selection of DMRs, the assay is validated using tissue and finally liquid
biopsy samples from
breast, colon and lung cancer patients as well as in blood samples from
healthy patients in a
first phase. First, the assay is validated in 50 untreated tumor vs normal
tissue samples for the 3
most common cancer types (breast, colon and lung cancer). All samples are
readily available
from the Antwerp tumor bank. Next, the assay is tested on liquid biopsies from
respective
cancer patients (n=50 for each tumor type; also available in Antwerp tumor
bank) and compared
with liquid biopsies from healthy persons (n=50). CtDNA is extracted from all
collected plasma
.. samples using the QIAamp Circulating Nucleic Acid Kit (QiaSymphony) and
analyzed. Data
analysis is performed in-house.
Proof of concept, technical development and optimization of MSRE-smMIPs-seq
technique:
A working principle of the technology has been obtained, and experiments using
the best-
performing smMIPs are performed. Meanwhile, the construction of the
bioinformatics pipeline
for the MSRE-smMIP-seq data analysis is initiated. Clinical samples from
cancer patients
(readily available from the biobank of the UZA) are tested to obtain the proof-
of-principle for the
technique.
.. The combination of different MSREs can efficiently be used for application
in the
methylation sequencing assay
We analyzed the efficiency of combining four different MSREs (Hpall, HinP1I,
Acil and
HpyCH4IV) in a methylation detection assay. The efficiency of the cutting
reaction was proved
by performing a qPCR on a LightCycler 480 machine (Roche) with two control
samples. The
negative control consisted of A phage DNA, which is unmethylated. The positive
control was
artificially methylated A phage DNA. Artificial methylation was performed
using Methyl Sss.I
transferase (New England Biolabs). Primer pairs were designed for the specific
restriction sites
of each MSRE. Both controls were cut and uncut. Uncut samples underwent the
same
CA 03193631 2023-03-02
WO 2022/053637 -15-
PCT/EP2021/074978
experimental conditions (i.e. incubation temperature and timing, buffers) but
without the
MSREs. Results are given in table 1.
Table 1: Results obtained from the qPCR
Sample Mean ct-value
Positive control - methylated DNA - cut 7,5
Positive control - methylated DNA - uncut 6,6
Negative control - unmethylated DNA - cut 22
Negative control - unmethylated DNA - uncut 5
For the positive controls, the mean obtained ct-values are expected to lay
closely together, as
the selected MSREs do not cut methylated DNA fragments. This is clearly the
case, which
proves that methylation effectively blocks the combined MSRE digestion. For
the negative
control, the cut samples should behave like a blanco sample, as it is expected
all DNA is
digested. We obtained a lower ct-value, indicating the DNA was not 100%
digested. However, a
difference in ct-values from cut and uncut samples of 17 was obtained. This
means 1 in 217=
-1 31,000 DNA molecules were not digested, suggesting that the combined MSRE-
cut reaction
is very efficient.
Demonstrating the first working principle and the feasibility of the protocol
using several
control samples
We demonstrated a first working principle and feasibility of the protocol. In
these experiments,
several control samples were used, including DNA extracted from 1) pancreatic
cell lines, 2)
primary lung tumor tissue, 3) human blood samples and 4) non-invasive prenatal
test (NIPT)
samples. The latter samples contained cfDNA and were included to test the
feasibility of the
approach in liquid biopsies. In total, 192 double-tiled designed smMIPs were
used to detect 66
genome-wide differentially methylated regions (DMRs) and 30 non-CpG control
regions. The
control regions do not contain CpG sites and will therefore always be captured
by smMIPs,
irrespective of the MSRE digest. As such, these smMIPs can be used to estimate
total DNA
concentration in the sample.
After library enrichment, next-generation sequencing was performed using the
Miseq reagent v2
nano kit. The data was analyzed with an in-house adapted bioinformatics
pipeline. For each
sample, the number of reads per smMIP is counted. The results that were
obtained in these first
runs showed a high number of reads on tumor, cell line and blood DNA. The NIPT
samples
obtained a lower amount of sequencing reads as expected. NGS run qualities
ranged from 71,6
to 94,6 %Q30.
CA 03193631 2023-03-02
WO 2022/053637 -16-
PCT/EP2021/074978
Interestingly, some smMIPs gave more reads than others across samples and
across
experiments, showing that some smMIPs are more efficient than others. The
efficiency of a
smMIP is considered an inherent property, as it was shown to be independent of
the
composition of the total smMIP pool. A selection of smMIPs was tested both
independently
(capture reaction performed separately) and together in one pool (capture
reaction performed in
the pool). We observed that the effect of one smMIP being more efficient than
another remained
when analyzed separately. As such, only efficient smMIPs can be selected to be
used for
analysis of liquid biopsies.
Optimization of the protocol altering several wet-lab parameters
Currently, a selection of the most efficient smMIPs (32 CpG smMIPs and 20
control smMIPs) is
used to further optimize the technique. Several runs have been performed using
DNA extracted
from human blood samples and a colorectal cell line (HT29, passage 13). One of
the human
blood samples was artificially methylated using the Methyl Sss.I transferase
(New England
Biolabs).
Optimizing the enzymatic DNA cutting reaction
In liquid biopsies, the low concentration of DNA is one of the significant
hurdles to overcome
when designing new technologies. To show that our technology can be used for
low input of
DNA, the enzyme digest reaction was performed using different input
concentrations in different
end-volumes. An overview is given in table 2. For this preliminary experiment,
as in previous
experiments, A phage DNA was used. Uncut phage DNA was used as a positive
control.
Table 2: overview of different sample conditions that were used in the
optimization experiment
Input DNA 1000 ng 500 ng 10Ong 500 ng 100 ng 100 ng 50 ng
10 ng 5 ng
End volume 50 pL 25 pL 10 pL 5pL
A qPCR was performed using primers designed for the specific restriction sites
of each specific
MSRE. Results are given in figure 9. Results of a similar qPCR results are
already described
above. Of interest here is that the enzyme digest performs very similar for
all sample conditions.
It can be concluded that this reaction is very robust, and applicable in
samples with low DNA
input. A DNA amount input of 5 ng was tested successfully and as can be seen
on the figure,
this low input amount performs equally well as a high input of 1pg DNA.
Together, these data
suggests that the enzyme digestion is suitable for liquid biopsies.
ii) Optimizing the efficiency of the assay by selection of a subset
of smMIPs
As previously defined, a selection of 52 smMIPs was made to further optimize
the assay. Figure
4 shows the balancing curve for all smMIPs (blue) compared to the top 52
(red). The x-axis
represents all 192 smMIPs and the y-axis the relative coverage per smMIP, as a
fraction of
counts. The fraction of counts is determined as the absolute counts per smMIP
in a sample
divided by the total number of counts in that sample. As such, samples and
smMIPs with
different numbers of counts can be compared. When all smMIPs are taken into
account, the
CA 03193631 2023-03-02
WO 2022/053637 -17-
PCT/EP2021/074978
most efficient smMIP (=smMIP with highest relative coverage) is 10,000 x more
efficient than
the least efficient smMIP. For the subset of 52 smMIPs, this difference is
reduced to a 100-fold.
In this way, the efficiency of the whole assay was improved.
iii) Optimizing the efficiency of the assay using cycled capture reactions
Another parameter that allows the optimization of the assay, is the capture
reaction. In several
papers, it has been described that smMIPs capture the DNA in a reaction at 60
C, for over 20
hours. We used a cycled capture reaction, meaning we split the 20 hours into 5
times 4 hours. A
denaturation step was implemented in between each cycle of 4 hours. Compared
to a run with
the exact same experimental conditions (except the capture reaction) and the
same samples,
we observed an increase of the percentage of unique reads (5% vs 12%). We are
currently
optimizing the number of cycles and total hours for the capture reaction in
order to obtain as
much unique reads as possible and as such to increase the efficiency of the
assay.
Repeatability of the assay
Preliminary results for the replicability and reproducibility of the assay
have been obtained.
Considering the repeatability, the exact same experiment was conducted by two
independent
researchers. Results are shown in figure 5. The results are given for both cut
and uncut
samples, for the control (=non-CpG smMIPs) and CpG smMIPs.
The experiments were executed independently from each other and in parallel.
Every step,
beginning from the MSRE digest until the analysis was performed separately. In
almost all
samples, the results between two independent researchers (blue vs orange) are
very similar.
These preliminary results suggest the assay is robust, reliable and
replicable. It is also clear
from figure 5D that the relative coverage for CpG smMIPs is high in methylated
samples and
very low in unmethylated samples, as expected.
Reproducibility of the assay
The MSRE-smMIP-seq workflow has been performed several times by independent
.. researchers. Preliminary results for the reproducibility demonstrate that
the assay is
reproducible. Results are given in figures 6 and 7.
Two independent runs (A and B) were executed by the same researcher. The
relative coverage
of the smMIPs is calculated as stated before. The relative counts of run A are
plotted against
the relative counts of run B. In figure 6, the average relative coverage per
smMIP over all uncut
samples was calculated and plotted. The average can be taken since for uncut
(=not digested)
samples, smMIPs behave similarly. There is a very strong correlation of the
average relative
counts in run A and B, with a correlation coefficient r2= 0,9894. The strong
correlation between
the results of the independent runs shows that the MSRE-smMIP method is
reproducible.
.. Figure 7 shows the relative counts per smMIP in a low (A) versus high (B)
methylated sample,
CA 03193631 2023-03-02
WO 2022/053637 -18-
PCT/EP2021/074978
in two independent runs executed by one researcher. For both samples, there is
a strong
correlation between the results of run A and B (r2= 0,9922 in the low
methylated sample, r2=
0,9718 in the high methylated sample). The strong correlation between the
relative coverages of
run A and B again suggests that the method is very likely to be reproducible.
Methylation detection using CpG smMIPs is working effectively in digested
samples
As was already highlighted in the part on repeatability of the assay, the CpG
smMIPs show a
higher number of counts and a higher relative coverage for methylated samples
(samples 12
and 13, figure 5D) compared to human blood DNA samples. This is expected,
since DNA from
human blood samples is only low methylated. The CpG sites in these samples are
digested and
unavailable for capturing by the smMIPs. In the artificially methylated DNA
blood sample and
the DNA extracted from the cell line, the CpG sites are methylated, preventing
digestion by the
MSREs and enabling subsequent smMIP capturing. In figure 8, an example is
given. For all 32
CpG smMIPs, the absolute number of counts per smMIP is given in 3 different
samples (DNA
from CRC cell line, artificially methylated human blood and human blood). The
high methylated
samples show a higher number of absolute counts compared to the low methylated
sample.
Performance of the technology in clinical colorectal cancer tissue DNA
compared to
normal blood DNA
Previous experiments showed a clear difference in highly methylated samples
compared to low
methylated samples. In a follow-up experiment, colorectal cancer tissue and
blood samples
were used to illustrate the ability of the technology to discriminate between
cancer and normal
samples.
For this experiment, DNA was extracted from 14 blood samples and 13 colorectal
cancer tissue
(FFPE-material). The earlier described combination of 52 smMIPs was used
again. 32 of these
smMIPs are designed to capture hypermethylated sites in tumor cells that were
previously
defined using TCGA data. This database provides epigenomic data through 450K
micro arrays.
Differential methylation is defined based on the R-values (output of a 450K
micro array). In the
original smMIP design, R-values with a maximum of 0.25 were selected for
normal tissue. For
cancer, R-values of at least 0.5 were selected. To discriminate high (hyper)
and low (hypo)
methylation, the largest difference in methylation R-values of normal adjacent
tissue and cancer
tissue was used, with a minimal difference of 0.25. By using the smMIPs
designed for these
CpG sites, we expect to find a high number of counts for CpG smMIPs in cancer
tissue (as they
capture the hypermethylated sites), while a low number of counts for CpG
smMIPs is expected
for normal tissue. For this experiment, normal blood samples were used instead
of normal
adjacent tissue.
CA 03193631 2023-03-02
WO 2022/053637 -19-
PCT/EP2021/074978
In figure 10, the result of the experiment is shown. All samples have been
treated with MSRE.
Blood samples (B) are shown on the left side and colorectal cancer tissue
(CRC) on the right
side. As expected, the CRC tissue samples show high read counts for the CpG
specific smMIPs
while the blood samples have lower counts for these smMIPs. The data prove
that a very clear
distinction can be made between normal and cancer using the technology. This
difference is
also demonstrated in figure 11.
The counts for all CpG smMIPs were used in a principal component analysis
(PCA) (figure 11).
In this plot, the preliminary data are plotted on the first two principal
components, that account
for 87% of the variance in the data. Blood samples (circles) and CRC samples
(triangles) can
clearly be distinguished from one another. Blood samples are clustered more
closely together
than CRC samples, which can be explained due to the inherent variability in
cancers. The
sample type was not considered while performing the principal components
analysis, indicating
that the visualized difference between the blood and CRC samples is inherent.
Performance of the technology in several cell line DNA samples compared to
normal
blood DNA samples
In figure 12, the absolute read counts are given for gDNA extracted from
normal blood samples
(left) and compared to gDNA extracted from cancer cell lines (right). From
this graph, it is clear
that a perfect distinction can be made based on the CpG smMIPs (light bars).
nonCpG smMIPs
are also shown (dark bars) and theoretically should not differ between cancer
and normal
samples, which is the case here. In this experiment, a selection (based on
efficiency) of +1- 800
CpG smMIPs were used.
A principal component analysis was then performed on the 500 best
discriminating smMIPs. In
figure 13, the first two principal components are shown. It is again clear
that a perfect
separation is found between blood samples (circles) and cancer samples,
including the
methylated blood sample (triangles and square). Interestingly, the blood
samples are all
grouped very closely, while the cancer samples are much more spread out.
Taken together, these results are a clear proof-of-concept that the technology
is working as
theoretically predicted. Taken all data into account, we are confident our
technology will be
applicable in liquid biopsies as well.
Proof of principle for single molecule Molecular Inversion Probe (smMIP) in
removing
PCR artefacts
To demonstrate the benefit of using single molecule tags to reduce technical
noise caused by
PCR amplification, we compared the results of analyses with and without
duplicate removal
using the same raw input data. The analysis without duplicate removal
simulates an analysis
CA 03193631 2023-03-02
WO 2022/053637 -20-
PCT/EP2021/074978
without single molecule tag since the tag is not considered when duplicates
are not removed.
First, the proportion of the reads per sample compared to the total amount of
reads was
compared between the analyses. Equation 1 below gives the example for sample
X:
q counts
t sm sa samplep samples
s ww/ /oduplicates'odupl at e s (counts smMIP N w/o duplicates
counts smMIPsw/o duplicates)
eq. 1 RatiOsample = ______ X with d I" t eq. 2 Ratiosmwo =
(counts sample Inn up ica es counts smMIP N with
duplicates
counts samples with duplicates' (Z counts smMIPs with
duplicates)
Equation 1 (left): Example of proportion ratio per sample. All reads
associated with sample X are
counted and divided by the total number of reads with and without duplicates.
Then, the proportion without
duplicates is divided by the proportion with duplicates to get the proportion
ratio. Equation 2 (right):
Example of proportion ratio per smMIP. All reads associated with smMIP N are
counted and divided by
the total number of reads with and without duplicates. Then, the proportion
without duplicates is divided by
the proportion with duplicates to get the proportion ratio.
If the single molecule tag would have no effect, it would be expected that for
each sample, the
proportion ratio approximately equals one. In figure 14; Panel A, we can see
the proportion per
sample grouped in bins of 0.1 around 1. The largest number of samples is
indeed in the group
from 0.95 to 1.05; however, there are some samples that are incorrectly
represented when not
accounting for duplicates.
Next, this proportion analysis was repeated, this time comparing the
proportion of reads per
smMIP in both analyses (equation 2). Again, for many smMIPs the proportion is
close to one,
but now there are significantly more smMIPs that have a different count
proportion when taking
duplicates into account (figure 14; Panel B). Additionally, this distribution
is skewed, so smMIPs
with a proportion over one deviate more than smMIPs with a proportion under
one.
Lastly, the proportion analysis was performed more fine-grained, this time
looking into the
proportion of reads per smMIP per sample, compared to the total reads per
smMIP and per
sample (equations 3 and 4 respectively):
counztscos umnMtsIPsmN m,s lap f w/
sample Xowdu/opldicuaptleicsates)
eq. 3 Ratiosample, smMIP N = (counts smMIP N, sample X with duplicates\
counts smMIP N with duplicates "
(counztscosumnMtsIPsaNm,spalemlwe /X w/o duplicates
oduplicates)
eq. 4 Ratiosmmip, sample X = (counts smMIP N, sample X with duplicates\
counts sample X with duplicates .. "
Equation 3 (upper): Example of proportion ratio per smMIP per sample, divided
by the reads per
smMIP. All reads associated with smMIP N, sample X are counted and divided by
the total number of
reads of smMIP N with and without duplicates. Then, the proportion without
duplicates is divided by the
proportion with duplicates to get the proportion ratio. Equation 4 (bottom):
Example of proportion ratio
per smMIP per sample, divided by the reads per sample. All reads associated
with smMIP N, sample X
CA 03193631 2023-03-02
WO 2022/053637 -21-
PCT/EP2021/074978
are counted and divided by the total number of reads of sample X with and
without duplicates. Then, the
proportion without duplicates is divided by the proportion with duplicates to
get the proportion ratio.
Here, the differences are even more prominent. In figure 14 Panel A and B, it
is clear that there
are a lot of smMIPs in samples that diverge when correcting for duplicates.
Interestingly, when
comparing per sample, this effect is more prominent. Both distributions are
also slightly skewed.
From this comparison, we can conclude that using a single molecule tag clearly
reduces the
technical noise in an experiment. This technical noise, caused by PCR
amplification before
sequencing to acquire the needed DNA concentration for sequencing, becomes
more apparent
when analysing the data in more detail. Additionally, overrepresentation of
certain smMIPs
because of a PCR bias is also fixed.