Note: Descriptions are shown in the official language in which they were submitted.
WO 2022/074397
PCT/GB2021/052609
1
MODIFICATION OF A NANOPORE FORMING PROTEIN OLIGOMER
Field
The present invention relates to methods of chemically modifying proteins and
polynucleotides, and to the modified proteins and polynucleotides themselves.
The
methods are particularly useful for producing chemically modified protein
nanopores.
Background
Nanopore sensing is an approach to analyte detection and characterization that
relies on the observation of individual binding or interaction events between
the analyte
molecules and an ion conducting channel. Nanopore sensors can be created by
placing a
single pore of nanometre dimensions in an electrically insulating membrane and
measuring
voltage-driven ion currents through the pore in the presence of analyte
molecules. The
presence of an analyte inside or near the nanopore will alter the ionic flow
through the
pore, resulting in altered ionic or electric currents being measured over the
channel. The
identity of an analyte is revealed through its distinctive current signature,
notably the
duration and extent of current blocks and the variance of current levels
during its
interaction time with the pore.
Polynucleotides are important analytes for sensing in this manner. Nanopore
sensing of polynucleotide analytes can reveal the identity and perform single
molecule
counting of the sensed analytes, but can also provide information on their
composition
such as their nucleotide sequence, as well as the presence of characteristics
such as base
modifications, oxidation, reduction, decarboxylation, deamination and more.
Nanopore
sensing has the potential to allow rapid and cheap polynucleoti de sequencing,
providing
single molecule sequence reads of polynucleotides of tens to tens of thousands
bases
length.
Known methods of nanopore sensing exploit the use of biological protein
nanopores. Such nanopores are typically multimeric proteins made up of
multiple
monomeric subunits. For example, the known pore-forming toxin ct-hemolysin
assembles
into a heptameric pore. Other pore stoichiometries are known for different
pore-forming
proteins.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
2
It is often desirable to modify protein nanopores to lead to effects which are
not
seen, or are seen to a lesser extent, in unmodified pores. Various
modifications have been
disclosed to achieve a wide variety of different outcomes. For example, it is
known that a
nanopore can be chemically modified to comprise attachment points for a
molecular
adapter such as a cyclodextrin, with such adapters finding particular use in
sensing small
analytes such as individual nucleotides. Nanopores can also be modified to
introduce
specific modifications to alter the properties of naturally or artificially
occurring
constrictions within the pore channel. Such constrictions, which are sometimes
known as
"reading heads", typically interact with analytes as the analyte interacts
with the pore and
by altering the properties of the reading head the analyte detection signal
can be altered
accordingly. In yet another example, nanopores can be modified to include
membrane
anchors such as cholesterol. Such anchors can interact with bilayers in order
to promote
the stable localisation of the nanopore in the membrane.
All of these modification strategies can involve chemical modification of the
nanopore. However, current methods of modifying nanopores and related
proteins,
discussed below, are associated with problems
One known method of modifying a protein such as a protein monomer of a protein
nanopore is to rely on modifications that can be generated by a cell
expressing the protein.
For example, routine molecular biology techniques such as those discussed in
Sambrook et
al., Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor
Press,
Plainsview, New York (2012), and Ausubel et al., Current Protocols in
Molecular Biology
(Supplement 114), John Wiley & Sons, New York (2016), can be used to alter,
introduce
or delete amino acids in a protein. Once expressed, the modified protein can
be purified
for use. However, the range of chemical modifications that can be made in such
a way is
extremely limited as it is determined by the range of chemical species that
can be
processed by the expressing cell, and by the existence of cellular machinery
to process
such species. Such techniques are insufficient to allow a protein to be
modified with an
extensive array of chemical moieties. Furthermore, such techniques are not
universally
applicable: different cloning and expression strategies are typically needed
for different
proteins and expression in a given system for a first protein may not be
successful for a
second protein.
A second known method of modifying a protein such as a protein monomer of a
protein nanopore is to express the unmodified protein in a routine manner, and
then to seek
to modify the expressed protein. In essence, there are two alternative
approaches that can
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
3
be taken. In a first approach, the expressed protein is modified prior to
being purified, and
attempts are then made to purify the modified protein. In a second approach,
the expressed
protein is purified, and then the purified protein is modified. Both of these
approaches,
however, are associated with difficulties. As explained below, the origin of
these
difficulties is that chemical modification of expressed proteins is rarely
efficient, and
typically even the most efficient modification strategies result in only
around 95%
modification, with ¨5% or more protein molecules remaining unmodified.
In the first approach, the protein of interest is subjected to chemical
modification
prior to being purified. As explained above, even modification strategies
generally
considered efficient typically result in no more than 95% of the available
protein molecules
being modified, with 5% or more remaining unmodified. Attempts can then be
made to
purify the modified protein molecules from the unmodified molecules (and other
impurities). Unfortunately, it is typically difficult or impossible to isolate
exclusively
modified protein molecules from unmodified analogs. Modifications may
typically have a
small impact on the overall mass of the protein molecule, meaning that
separation
techniques that depend on distinguishing proteins by mass may not be
effective_
Modifications may not alter the charge of the protein and so techniques
relying on
distinguishing proteins based on charge interactions may similarly fail The
consequence
is that in many cases purification strategies fail to separate modified
proteins from their
unmodified counterparts, and the resulting output from the purification
process contains a
mixture of modified and unmodified protein molecules in proportions determined
by the
efficiency of the modification chemistry used.
In the second approach, the protein of interest is subjected to chemical
modification
after being purified. The purification protocol used may be efficient in
separating the
unmodified protein of interest from impurities present from the expression
medium.
However, problems again arise when the modification chemistry is conducted on
the
purified protein. As the modification chemistry is not 100% efficient, the
output of the
modification process is again a mixture of modified and unmodified protein
molecules in
proportions determined by the efficiency of the modification chemistry used.
The presence of even relatively low levels (-5%) unmodified protein in a
population of otherwise modified protein can be problematic. This is
particularly the case
when the protein is a monomer of an oligomeric protein pore. For example, the
presence
of some unmodified monomers in a population of otherwise modified monomers
means
that a distribution of assembled pores typically arises. For example, for a
heptameric pore
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
4
such as ct-hemolysin being assembled from a mixture of both modified and
unmodified
monomers, assembled pores may comprise only modified monomers, only unmodified
monomers, or a mixture of modified and unmodified monomers. Whilst the
distribution of
such pores can be statistically calculated, characterisation of an individual
pore assembled
into a membrane for use e.g. in nanopore sensing is far from straightforward.
This further
leads to issues in interpreting data obtained from the pore in a functional
apparatus, as
signals need to be interpreted on the basis that the population of pores used
to generate the
apparatus is inherently non-homogeneous. In some cases, this difficulty can
lead to
otherwise functional pores being abandoned, data being lost or convoluted,
artefacts in
obtained data being introduced by the pore; and/or a loss of reproducibility
when a given
homogenous sample is subjected to analysis using a non-homogenous sample of
pores.
Accordingly, there is a pressing need for new strategies for chemically
modifying
proteins such as monomers of protein nanopores. The methods of the present
disclosure
overcome some or all of the problems discussed above.
St] m ma ry
The disclosure relates to a method of chemically modifying a polypepti de or
polynucleotide monomer, typically a monomer of an oligomeric protein nanopore.
The
monomer is contacted with a multifunctional molecule comprising (i) a reactive
group for
reacting with the monomer, (ii) a chemical modifying group for providing the
chemical
modification; and (iii) a cleavable purification tag for purifying the
monomer. The reactive
group of the multi-functional molecule is allowed to react with the monomer,
thereby
attaching the chemical modifying group and cleavable purification tag to the
monomer to
form a chemically modified tagged monomer. The chemically modified monomer is
contacted with a support such as a chromatography matrix or magnetic beads,
and the
purification tag is allowed to bind to the support. The binding of the
purification tag to the
support results in binding the chemically modified tagged monomer to the
support. The
purification tag is then cleaved by any suitable means. Cleavage of the
purification tag
releases the chemically modified monomer from the support. A key advantage of
the
method is the coupled modification and purification strategy, which avoids or
minimises
issues associated with inefficiencies in either step as discussed above.
Accordingly, provided herein is a method of chemically modifying a polypeptide
or
polynucleotide monomer (e.g. a monomer of an oligomeric protein nanopore);
comprising:
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
a) contacting the monomer with a multifunctional molecule, wherein the multi-
functional molecule comprises (i) a reactive group; (ii) a chemical modifying
group
and (iii) a cleavable purification tag;
b) allowing the reactive group of the multi-functional molecule to react with
the
5 monomer thereby attaching the chemical modifying group and cleavable
purification tag to the monomer to form a chemically modified tagged monomer;
c) contacting the chemically modified tagged monomer formed in step (b) with a
support;
d) allowing the purification tag to bind to the support thereby binding the
chemically
modified tagged monomer to the support; and
e) cleaving the purification tag thereby releasing the chemically modified
monomer
from the support.
In some embodiments the multifunctional molecule is of Formula (I) or Formula
(II):
A-B-D-C A -D-C
Formula (I) Formula (II)
wherein:
A is a reactive group;
11 is a chemical modifying group; and
D-C forms a cleavable purification tag;
preferably wherein D comprises a cleavable linker and C comprises a support-
binding group.In some embodiments the wherein the reactive group comprises the
chemical modifying group. In some embodiments the multifunctional molecule is
of
Formula (III) as defined herein.
In some embodiments the monomer comprises a reactive functional group and step
(b) comprises allowing the reactive group of the multifunctional molecule to
react with the
reactive functional group of the monomer.
In some embodiments the reactive group of the multifunctional molecule
comprises
an amine-reactive group; a carboxyl-reactive group; a sulfhydryl-reactive
group or a
carbonyl-reactive group; preferably a cysteine-reactive group. In some
embodiments the
reactive group comprises a maleimide, an azide, a thiol, an alkyne, an NHS
ester or a
haloacetamide.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
6
In some embodiments the chemical modifying group introduces hydrophilic,
hydrophobic, positively charged, negatively charged, hydrogen-bonding,
supramolecular
associations or zwitterionic properties to the protein monomer. In some
embodiments the
chemical modifying group comprises an amino acid, a nucleotide, a polymer, a
hydrogen-
bonding group, a membrane anchor, a sugar, a dye, a chromophore, a fluorophore
or a
molecular adapter. In some embodiments the chemical modifying group comprises
a
natural or unnatural amino acid, a polypeptide, a nucleotide or nucleotide
analog, an
oligonucleotide or oligonucleotide analog, a polysaccharide, a lipid, a
polyethylene glycol,
a cyclodextrin, a DNA intercalator, an aptamer or an analyte binding domain.
In some embodiments the support comprises a chromatography matrix, preferably
an agarose or sepharose resin; one or more beads, preferably magnetic beads;
or a solid
surface, preferably a glass, silica, polymer or ceramic surface. In some
embodiments the
support is functionalised for binding to the purification tag. In some
embodiments the
purification tag comprises a biotin group and the support comprises
streptavidin,
neutravidin or avidin, preferably streptavidin.
In some embodiments the cleavable linker is cleaved by physical or chemical
means. In some embodiments the cleavable linker comprises a UV photocleavable
nitro-
benzyl moiety.
In some embodiments, in step (e) of the method cleaving the purification tag
comprises exposing the support and/or the tagged monomer to light; preferably
UV light.
In sonic embodiments, in step (e) cleaving the purification tag comprises
exposing the
support and/or the tagged monomer to a change in pH. In some embodiments, in
step (e)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
a chemical reagent; preferably a reducing reagent. In some embodiments, in
step (e)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
a enzyme; preferably a protease.
In some embodiments the monomer is a polypeptide having a mass of from about
10 kDa to about 1 MDa. In some embodiments the monomer is a monomer of an
oligomeric protein pore, preferably a lysenin pore, a y-hemolysin pore, an a-
hemolysin
pore; a NetB pore; a CytK pore or a leukocidin pore; or a homolog or paralog
thereof In
some embodiments the oligomeric protein pore is a multi-component pore.
In some disclosed embodiments the monomer is a polynucleotide; preferably a
monomer of a DNA origami pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
7
In some embodiments the monomer is a monomer of an oligomeric pore; and the
multifunctional molecule reacts with a reactive functional group located on
the monomer at
a surface-exposed position when the monomer is oligomerised to form the pore.
In some
embodiments the surface-exposed position is located at the surface of the
channel through
the pore; or on the exterior surface of the pore. In some embodiments the
multifunctional
molecule reacts with a reactive functional group located on the monomer at a
position
located at or near a constriction of the channel through the pore when the
monomer is
oligomerised to form the pore.
In some embodiments the method comprises, prior to step (a), the steps of (i)
expressing the monomer in a cellular expression system or a cell-free
expression system;
and (ii) isolating and/or purifying the monomer.
In some embodiments, step (d) of the method further comprises the step of
removing unmodified monomer(s) and/or unreacted multifunctional molecule(s),
if
present, from the support.
In some embodiments the method further comprises the step of:
0 oligom eri sing the chemically modified monomer to form a
chemically modified
oligomer, e.g. to form a chemically modified oligomeric protein nanopore.
In some embodiments the monomer is a protein monomer and step (f) comprises
forming
an oligomeric protein. In some embodiments step (f) comprises oligomerising
two or more
chemically modified monomers to form a homooligomer, e.g. to form an
oligomeric
protein nanopore. In sonic embodiments said monomers are protein monomers and
said
homooligomer is a homooligomeric protein pore.
In some embodiments step (f) comprises oligomerising one or more chemically
modified monomers with one or more unmodified or differently-modified monomers
to
form a heterooligomer, e.g. to form an oligomeric protein nanopore. In some
embodiments
said monomers are protein monomers and said heterooligomer is a
heterooligomeric
protein pore.
In some embodiments step (f) comprises oligomerising one or more chemically
modified first monomers with one or more chemically modified second monomers
to form
a heterooligomer, e.g. to form an oligomeric protein nanopore; wherein the
chemical
modification made to the first monomer is the same or different to the
chemical
modification made to the second monomer. In some embodiments said monomers are
protein monomers, said first monomer has a different amino acid sequence to
said second
monomer, and the heterooligomer is a heterooligomeric protein pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
8
Also provided is a method of producing a homooligomeric protein, e.g. a
hornooligomeric protein nanopore; comprising
i) producing a plurality of chemically modified protein monomers in a
method as
described herein; and
ii) oligomerising two or more of the chemically modified protein monomers
obtained in step (i) to form a homooligomeric protein.
In some embodiments said homooligomeric protein is a homooligomeric protein
pore.
Also provided is a method of producing a heterooligomeric protein, e.g. a
heterooligomeric protein nanopore; comprising
i) producing one or more chemically modified first protein monomers in a
method
as described herein; and
ii) producing one or more chemically modified second protein monomers in a
method as described herein; and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein.
In some embodiments said heterooligomeric protein is a heterooligomeric
protein pore
Also provided is a method of producing an oligomeric protein, e.g an
oligomeric
protein nanopore; comprising
i) producing one or more chemically modified first protein monomers in a
method
as described herein; and
ii) providing one or more unmodified second protein monomers, and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein.
In some embodiments said heterooligomeric protein is a heterooligomeric
protein pore.
Also provided is a chemically modified monomer, e.g. a chemically modified
monomer of an oligomeric protein nanopore, obtainable by carrying out a method
as
described herein.
Also provided is a homogeneous population comprising a plurality of chemically
modified monomers; wherein at least 95% of the monomers in the population are
chemically modified with a chemical modifying group; e.g. the monomers may be
monomers of one or more oligomeric protein nanopores (e.g. of one or more
types of
protein nanopore). In some embodiments the chemically modified monomers are as
described in more detail herein.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
9
Also provided is a chemically modified oligomer, e.g. an oligomeric protein
nanopore, obtainable by carrying out a method as described herein.
Also provided is a homogeneous population comprising a plurality of chemically
modified oligomers, e.g. of oligomeric protein nanopores; wherein at least 95%
of the
oligomers in the population comprise a defined number of monomers chemically
modified
with a chemical modifying group. In some embodiments the chemically modified
monomers are as described in more detail herein.
Also provided is a method of characterising an analyte, comprising:
i) producing a chemically modified oligomeric pore in a method as described
herein; or providing a chemically modified oligomeric pore as described
herein;
and
ii) taking one or more measurements as the analyte moves with respect to
the pore,
wherein the one or more measurements are indicative of one or more
characteristics of the analyte, and thereby characterising the analyte as it
moves
with respect to the pore.
In some embodiments the analyte is a polynucleotide, a polypeptide or a
polysaccharide
Also provided are related systems, methods and uses.
Brief Description of the Figures
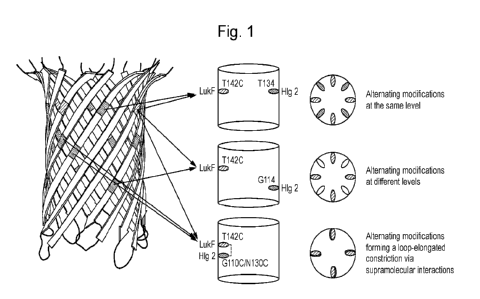
Figure 1. A schematic showing how the disclosed methods can be used to
introduce
multiple modifications into the barrel of a bi-component pore-founing toxin
such as
gamma-hemolysin. Specific residues are depicted for illustration only and are
non-
limiting.
Figure 2. UV cleavage products of photocleavable multifunctional molecules
discussed in
the examples (e.g. see Example 1).
Figure 3. Schematic showing chemical modification of cysteine mutants of
monomers
using multifunctional molecules as discussed in the examples. (see Example 2)
Figure 4. Gel showing successful modification of Lysenin mutant Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/598C/C272A/C283A) in accordance with
the disclosed methods, as discussed in Example 2. Lane 1 ¨ Unmodified protein
monomer
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
(Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C/C272A/C283A)); Lane 2 ¨
Crude reaction mixture - Maleimide-Isoleucine adduct with Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C/C272A/C283A); Lane 3 ¨ Flow
through after equilibrating the modified protein monomers on StrepTactin beads
(flow
5 through indicates unmodified protein monomers unbound to StrepTactin
beads); Lane 4/5
¨ Further washings of the modified monomer bound StrepTactin beads to remove
unmodified protein monomer (gel indicates no unmodified monomer eluting in
further
washes); Lane 6 ¨ Modified Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-
Maleimide-Icoleucine/C272A/C283A) eluted from StrepTactin beads after UV
cleavage of
10 the linker.
Figure 5. Gel showing successful modification of Lysenin mutant Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/163C/C272A/C283A) in accordance with
the disclosed methods, as discussed in Example 2. Lane 1 ¨ Unmodified protein
monomer
(Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C/C272A/C283A))
Lane 2 ¨ Crude reaction mixture - Maleimide-Isoleucine adduct with Lys-
(E84Q/E85 S/E92Q/E94D/E97 S/T106K/D126G/T63C/C272A/C283 A); Lane 3 ¨ Fl ow
through after equilibrating the modified protein monomers on StrepTactin beads
(flow
through indicates unmodified protein monomers unbound to StrepTactin beads);
Lane 4/5
¨ Further washings of the modified monomer bound StrepTactin beads to remove
unmodified protein monomer (gel indicates no unmodified monomer eluting in
further
washes); Lane 6 ¨ Modified Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C-
Maleimide-Icoleucine/C272A/C283A) eluted from StrepTactin beads after UV
cleavage of
the linker.
Figure 6. Gel showing successful modification of a mutant of the F component
of the
Gamma-Hemolysin pore, LukF-T142C-Del(E1-K15) in accordance with the disclosed
methods, as discussed in Example 2. Lane 1 ¨ Unmodified protein monomer, LukF-
T142C-Del(E1-K15); Lane 2 ¨ Crude reaction mixture - Maleimide-Isoleucine
adduct
with LukF-T142C-Del(E1-K15); Lane 3 ¨ Flow through after equilibrating the
modified
protein monomers on StrepTactin beads (flow through indicates unmodified
protein
monomers unbound to StrepTactin beads); Lane 4/5 ¨ Further washings of the
modified
monomer bound StrepTactin beads to remove unmodified protein monomer (gel
indicates
no unmodified monomer eluting in further washes); Lane 6¨ Modified LukF-T142C-
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
11
Maleimide-Alanine-Del(E1-K15) eluted from StrepTactin beads after UV cleavage
of the
linker
Figure 7. Gel showing successful modification of a mutant of the S component
of the
Gamma-Hemolysin pore, H1g2-G114C-De1(E1 -G10) in accordance with the disclosed
methods, as discussed in Example 2. Lane 1 ¨ Unmodified protein monomer, H1g2-
G114C-Del(E1-G10); Lane 2 ¨ Crude reaction mixture - Maleimide-Isoleucine
adduct
with I-Eg2-G114C-Del(E1-G10); Lane 3 ¨ Flow through after equilibrating the
modified
protein monomers on StrepTactin beads (flow through indicates unmodified
protein
monomers unbound to StrepTactin beads); Lane 4/5 ¨ Further washings of the
modified
monomer bound StrepTactin beads to remove unmodified protein monomer (gel
indicates
no unmodified monomer eluting in further washes); Lane 6 ¨ Modified H1g2-G114C-
Maleimide-Isoleucine-Del(E1-G10) eluted from StrepTactin beads after UV
cleavage of
the linker.
Figure 8 Gel showing successful oligomerisation of modified Lysenin ¨ [Lys-
(E84Q/E85 S/E92Q/E94D/E97 S/T106K/D 126G/S98C-Mal eimi de-
Icoleucine/C272A/C283 A)] to form a nonameric pore. Results discussed in
Example 3.
Lane 1 ¨ Modified protein monomer, Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A), Lane 2 ¨ Crude reaction mixture of Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A) with 1:1 Sphingomyelin after overnight storage.
Reaction
mixture shows oligomeric pore formation (upper band) and unreacted modified
monomer
Lys-(E84Q/E85 S/E92Q/E94D/E97 S/T106K/D126G/S98C-Maleimi de-
Icoleucine/C272A/C283A); Lane 3 ¨ Supernatant after centrifuging to separate
pore-
sphingomyelin pellet. Some pores are seen in supernatant as well.; Lane 4 ¨
Modified
[Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A)]9 pore;
Figure 9. Gel showing successful oligomerisation of Lysenin ¨ [Lys-
(E84Q/E85 S/E92Q/E94D/E97 S/T106K/D 126G/T63 C-Mal eimi de-
PNA(Thymine)/C272A/C283A)] to form a nonameric pore. Results discussed in
Example
3. Lane 1 ¨Modified protein monomer, Lys-
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
12
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/163C-Maleimide-PNA(Thymine)
/C272A/C283A); Lane 2 ¨ Crude reaction mixture of Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/163C-Maleimide-PNA(Thymine)
/C272A/C283A) with 1:1 Sphingomyelin after overnight storage. Reaction mixture
shows
oligomeric pore formation (upper band) and unreacted modified monomer Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A); Lane 3 ¨ Supernatant after centrifuging to separate
pore-
sphingomyelin pellet. Some pores are seen in supernatant as well.; Lane 4 ¨
Modified
[Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C-Maleimide-PNA(Thymine)
/C272A/C283A)]9 pore
Figure 10. Gel showing successful oligomerisation of various gamma-hemolysin
mutants
described in Example 4. Lane 1 ¨ Unmodified (LukF-T142C-Del(E1-K15)) monomer ¨
guide to see where the monomer would appear on the gel; Lane 2 ¨ Unmodified
(H1g2-
G114C-Del(El-G10)) monomer ¨ guide to see where the monomer would appear on
the
gel; Lane 3 ¨ modified pore oligomer [(LukF-T142C-Maleimide-Isoleucine-Del(E1 -
K15))4 (I-11g2-G114C-Maleimide-Alanine -Del(E1-G10))4] ¨ top band. Also, can
be seen
the non-oligomerised modified monomer bands.; Lane 4 ¨ modified pore oligomer
[(LukF-T142C-Maleimide-Isoleucine-Del(El-K15))4 (H1g2-G114C-Maleimide-
Isoleucine-Del(E1-G10))4] ¨ top band. Also, can be seen the non-oligomerised
modified
monomer bands., Lane 5 ¨ modified pore oligomer [(LukF-T142C-Maleimide-Alanine-
Del(El-K15))4 (H1g2-G114C-Maleimide-Alanine -De1(El-G10))4] ¨ top band. Also,
can
be seen the non-oligomerised modified monomer bands.; Lane 6 ¨ modified pore
oligomer
[(LukF-T142C-Maleimide-Isoleucine-Del(E1-K15))4 (H1g2-G114C-Maleimide-Alanine-
Del(El-G10))4] ¨ top band. Also, can be seen the non-oligomerised modified
monomer
bands.
Figure 11. Characterisation data for the polynucleotide analyte of SEQ ID NO:
20 when
characterised using a panel of chemically modified lysenin pores produced in
accordance
with the disclosed methods. Two lysenin pores:
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C/T106K/D126G/C272A/C283A) and
Lys-(T63 C/E84Q/E85 S/E92Q/E94D/E97S/T106K/D126G/C272A/C283 A)
were modified with 5 different chemical molecules and the effect of the
modification was
analysed by comparing the open pore current level of the modified pore against
the
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
13
unmodified pore when the 3.6 lambda DNA is passing through each pore. Top
level
within each channel represents the open pore current level. Bottom level
within each
channel represents the current level observed when the DNA is passing through
the pore.
Figure 12. Changes in the signal of the 3.6Kb Lambda DNA translocating through
lysenin
mutant pores as a function of the modifier molecule. Panel A shows data for
unmodified
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C/T106K/D126G/C272A/C283A) and panel B
shows data for the modified pore
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
Isoleucine/T106K/D126G/C272A/C283A).
The left panel shows an event of the entire 3.6Kb DNA passing through the
pore. The right
panel shows the expanded version of the squiggle for 0.4 seconds.
Figure 13. Changes in the GGAA region of the 3.6Kb Lambda DNA translocating
through lysenin mutant pores as a function of the modifier molecule. Panel A
shows data
for unmodified
Lys-(E84Q/E85 S/E92Q/E94D/E97S/S98C/T106K/D126G/C272A/C283 A). Panel B
shows data for the modified pore Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
Isoleucine/T106K/D126G/C272A/C283A); and Panel C shows data for the modified
pore
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
PNA(Thymine)/T106K/D126G/C272A/C283A).
Figure 14. Gel showing successful modification of a mutant of the F component
of the
Gamma-Hemolysin pore, LukF-T142C-Del(E1-K15) in accordance with the disclosed
methods, as discussed in Example 2. A: Modification with maleimide-asparagine.
B:
Modification with maleimide-CH2-NH2. C: Modification with maleimide-arginine.
D:
Modification with maleimide-isoleucine. E: Modification with maleimide-
asparatic acid.
For each of A-E, Lane 1 = Unmodified protein monomer, LukF-T142C-Del(E1-K15);
Lane 2 = monomer modified with cleavable complex; Lane 3 = Flow through after
equilibrating the modified protein monomers on StrepTactin beads (flow through
indicates
unmodified protein monomers unbound to StrepTactin beads); Lane 4 = Modified
LukF-
T142C-Del (El -K15)-Maleimi de-[modifi cation] eluted from StrepTactin beads
after UV
cleavage of the linker.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
14
Figure 15. Gel showing successful modification of a mutant of the S component
of the
Gamma-Hemolysin pore, H1g2-N130C-De1(E1 -G10) in accordance with the disclosed
methods, as discussed in Example 2. A: Modification with maleimide-asparagine.
B:
Modification with maleimide-CH2-NH2. C: Modification with maleimide-arginine.
D:
Modification with maleimide-isoleucine. E: Modification with maleimide-
asparatic acid.
For each of A-E, Lane 1 = Unmodified protein monomer, H1g2-N130C-Del(El-G10);
Lane 2 = monomer modified with cleavable complex; Lane 3 = Flow through after
equilibrating the modified protein monomers on StrepTactin beads (flow through
indicates
unmodified protein monomers unbound to StrepTactin beads); Lane 4 = Modified
H1g2-
N130C-Del(El-G10)-Maleimide1modification] eluted from StrepTactin beads after
UV
cleavage of the linker.
Figure 16. Gel showing successful oligomerisation of (LukF-T142C-Maleimide-
Isoleucine-Del(El-K15)) with wild-type (WT) H1g2 monomer. Lane 1 - Modified
(LukF-
T142C-Maleimide-Isoleucine-Del(El-K15)) monomer; Lane 2 ¨ unmodified wild-type
(WT) H1g2 monomer. Lane 3 ¨ modified pore oligomer [(LukF-T142C-Maleimide-
Isoleucine-Del(E1 -K15))4 (I-11g2-WT)4] ¨ band indicated with arrow.
Figure 17. Characterisation data for the polynucleotide analyte of SEQ ID NO:
20 when
characterised using gamma-hemolysin pores modified in accordance with the
disclosed
methods. A. representative characterisation data for the translocation of SEQ
ID NO. 20
when characterised using (left) unmodified Gamma-Hemolysin ¨ [(Luk,F-T142C-
Del(E1-
K15))4 (H1g2-WT)4] pores produced and oligomerised as discussed herein, as
compared to
corresponding modified Gamma-Hemolysin ¨ [(LukF-T142C-Maleimide-Aspartate-
Del(El-K15))4 (H1g2-WT)4] pores produced and oligomerised as discussed herein.
B:
Changes in the GGAA region of the 3.6Kb Lambda DNA (residues 38-41 and 52-55
of
SEQ ID NO: 20) as it translocates through the pores.
Figure 18. Gel showing successful modification of an alpha-hemolysin (a-HL)
monomer
and the successful oligomerisation of the modified monomer, in accordance with
the
disclosed methods. A: Successful modification of a-HL M1 13C monomer (Lane 1 ¨
Unmodified protein monomer, a-HL-M113C; Lane 2 ¨ Crude reaction mixture -
Maleimide-Isoleucine (Mal-Ile) adduct with a-HL-M113C; Lane 3 Flow through
after
equilibrating the modified protein monomers on StrepTactin beads (flow through
indicates
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
unmodified protein monomers unbound to StrepTactin beads); Lane 4/5 ¨ Further
washings of the modified monomer bound StrepTactin beads to remove unmodified
protein monomer (gel indicates no unmodified monomer eluting in further
washes); Lane
6 ¨ Modified a-HL-M113C with Maleimide-Isoleucine (Mal-Ile)). B: Successful
5 oligomerisation of the modified a-HL monomer. (Lane 1 ¨ Modified protein
monomer,
a-HL-M113C-Mal-Ile; Lane 2 ¨ Crude reaction mixture of a-HL-M113C-Mal-Ile with
1:1
Sphingomyelin after overnight storage. Lane 3 ¨ Supernatant after centrifuging
to separate
pore-sphingomyelin pellet. Some pores are seen in supernatant as well.; Lane 4
¨ Modified
(a-HL-M113C-Mal-Ile)7 pore.
Figure 19. Characterisation data for the polynucleotide analyte of SEQ ID NO:
20 when
characterised using alpha-hemolysin pores modified in accordance with the
disclosed
methods. A: representative characterisation data for the translocation of SEQ
ID NO: 20
when characterised using (left) unmodified a-HL Ml 13C pores produced and
oligomcriscd
as discussed above, as compared to corresponding modified a-HL [a-HL-M113C-Mal-
Ile)7] pores produced and oligomerised as discussed above B. Changes in the
translocation (squiggle) data from (A). C: Changes in the translocation
squiggle in the
first 0.3 s following the characteristic sp18 signal of the polynucleotide
analyte.
Figure 20. Gel showing successful modification of cytotoxin-K (Cyt-K) monomers
and
the successful oligomerisation of the modified monomers, in accordance with
the disclosed
methods. A: Successful modification of Cyt-K WT-Q123S/K129C/E140S/Q146S
monomer (Lane 1 ¨ Unmodified protein monomer, Cyt-K(WT-
Q123 S/K129C/E140S/Q146S); Lane 2 ¨ Crude reaction mixture - Maleimide-
Isoleucine
(Mal-Ile) adduct with Cyt-K(WT-Q1235/K129C/E140S/Q1465); Lane 3 ¨ Flow through
after equilibrating the modified protein monomers on StrepTactin beads (flow
through
indicates unmodified protein monomers unbound to StrepTactin beads); Lane 4/5
¨
Further washings of the modified monomer bound StrepTactin beads to remove
unmodified protein monomer (gel indicates no unmodified monomer eluting in
further
washes); Lane 6 ¨ Modified Cyt-K(WT-Q123S/K129C/E140S/Q1465) with Maleimide-
Isoleucine (Mal-Ile)). B: Successful oligomerisation of the modified Cyt-K
monomer.
(Lane 1 ¨ Modified protein monomer, CytK-(WT-Q123S/K129C-Mal-Ile/E140S/Q146S;
Lane 2 Crude reaction mixture of CytK-(WT-Q123S/K129C-Mal-Ile/E140S/Q1465 with
1:1 Sphingomyelin after overnight storage. Lane 3 ¨ Supernatant after
centrifuging to
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
16
separate pore-sphingomyelin pellet. Some pores are seen in supernatant as
well.; Lane 4 ¨
Modified (CytK-(WT-Q123S/K129C-Mal-Ile/E140S/Q146S)7 pore.).
Figure 21. Characterisation data for the polynucleotide analyte of SEQ ID NO:
20 when
characterised using Cyt-K pores modified in accordance with the disclosed
methods. A:
representative characterisation data for the translocation of SEQ ID NO: 20
when
characterised using (left) unmodified Cyt-K WT-Q1235/K1295/E1405/Q1465 pores
produced and oligomerised as discussed above, as compared to corresponding
modified
Cyt-K WT-Q123S/K129C-Maleimide-isoleucine/E140S/Q146S pores produced and
oligomerised as discussed above. B: Changes in the translocation (squiggle)
data from
(A). C: Representative changes in the translocation squiggle in the first 0.3
s following the
characteristic sp18 signal of the polynucleotide analyte.
Figure 22. Gel comparing the modification methods of the invention with
conventional
methods for modifying proteins. Results obtained for lysenin (Lys-
(/E84Q/E85K/E92Q/E94D/E97S/S98C/T106K/D126G/C272A /C283 A)) modified with
maleimide-isoleucine both without a cleavable purification tag (conventional
methods, lane
2) and in accordance with the disclosed methods (lanes 3 and 6). Results
described in
Example 9.
Detailed Description
The present invention will be described with respect to particular embodiments
and
with reference to certain drawings but the invention is not limited thereto
but only by the
claims. Any reference signs in the claims shall not be construed as limiting
the scope. Of
course, it is to be understood that not necessarily all aspects or advantages
may be achieved
in accordance with any particular embodiment of the invention. Thus, for
example those
skilled in the art will recognize that the invention may be embodied or
carried out in a
manner that achieves or optimizes one advantage or group of advantages as
taught herein
without necessarily achieving other aspects or advantages as may be taught or
suggested
herein.
The invention, both as to organization and method of operation, together with
features and advantages thereof, may best be understood by reference to the
following
detailed description when read in conjunction with the accompanying drawings.
The
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
17
aspects and advantages of the invention will be apparent from and elucidated
with
reference to the embodiment(s) described hereinafter. Reference throughout
this
specification to "one embodiment" or "an embodiment" means that a particular
feature,
structure or characteristic described in connection with the embodiment is
included in at
least one embodiment of the present invention. Thus, appearances of the
phrases "in one
embodiment" or "in an embodiment" in various places throughout this
specification are not
necessarily all referring to the same embodiment, but may. Similarly, it
should be
appreciated that in the description of exemplary embodiments of the invention,
various
features of the invention are sometimes grouped together in a single
embodiment, figure,
or description thereof for the purpose of streamlining the disclosure and
aiding in the
understanding of one or more of the various inventive aspects. This method of
disclosure,
however, is not to be interpreted as reflecting an intention that the claimed
invention
requires more features than are expressly recited in each claim. Rather, as
the following
claims reflect, inventive aspects lie in less than all features of a single
foregoing disclosed
embodiment.
It should be appreciated that "embodiments" of the disclosure can be
specifically
combined together unless the context indicates otherwise. The specific
combinations of all
disclosed embodiments (unless implied otherwise by the context) are further
disclosed
embodiments of the claimed invention.
In addition as used in this specification and the appended claims, the
singular forms
"a", "an", and "the" include plural referents unless the content deadly
dictates otherwise.
Thus, for example, reference to -a polynucleotide" includes two or more
polynucleotides,
reference to "a motor protein" includes two or more such proteins, reference
to "a
helicase" includes two or more helicases, reference to "a monomer" refers to
two or more
monomers, reference to "a pore- includes two or more pores and the like.
All publications, patents and patent applications cited herein, whether supra
or
infra, are hereby incorporated by reference in their entirety.
Definitions
Where an indefinite or definite article is used when referring to a singular
noun e.g.
"a" or "an", "the", this includes a plural of that noun unless something else
is specifically
stated. Where the term "comprising" is used in the present description and
claims, it does
not exclude other elements or steps. Furthermore, the terms first, second,
third and the like
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
18
in the description and in the claims, are used for distinguishing between
similar elements
and not necessarily for describing a sequential or chronological order. It is
to be
understood that the terms so used are interchangeable under appropriate
circumstances and
that the embodiments of the invention described herein are capable of
operation in other
sequences than described or illustrated herein. The following terms or
definitions are
provided solely to aid in the understanding of the invention. Unless
specifically defined
herein, all terms used herein have the same meaning as they would to one
skilled in the art
of the present invention. Practitioners are particularly directed to Sambrook
et al.,
Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Press,
Plainsview,
New York (2012); and Ausubel et al., Current Protocols in Molecular Biology
(Supplement 114), John Wiley & Sons, New York (2016) for definitions and terms
of the
art. The definitions provided herein should not be construed to have a scope
less than
understood by a person of ordinary skill in the art.
"About" as used herein when referring to a measurable value such as an amount,
a
temporal duration, and the like, is meant to encompass variations of 20 % or
10 %,
more preferably 5 %, even more preferably 1 %, and still more preferably
0 1 %
from the specified value, as such variations are appropriate to perform the
disclosed
methods.
"Nucleotide sequence", "DNA sequence" or "nucleic acid molecule(s)" as used
herein refers to a polymeric form of nucleotides of any length, either
ribonucleotides or
deoxyiibonucleotides. This Leon refers only to the primary structure of the
molecule. Thus,
this term includes double- and single-stranded DNA, and RNA. The term -nucleic
acid" as
used herein, is a single or double stranded covalently-linked sequence of
nucleotides in
which the 3' and 5' ends on each nucleotide are joined by phosphodiester
bonds. The
polynucleotide may be made up of deoxyribonucleotide bases or ribonucleotide
bases.
Nucleic acids may be manufactured synthetically in vitro or isolated from
natural sources.
Nucleic acids may further include modified DNA or RNA, for example DNA or RNA
that
has been methylated, or RNA that has been subject to post-translational
modification, for
example 5'-capping with 7-methylguanosine, 3'-processing such as cleavage and
polyadenylation, and splicing. Nucleic acids may also include synthetic
nucleic acids
(XNA), such as hexitol nucleic acid (HNA), cyclohexene nucleic acid (CeNA),
threose
nucleic acid (TNA), glycerol nucleic acid (GNA), locked nucleic acid (LNA) and
peptide
nucleic acid (PNA). Sizes of nucleic acids, also referred to herein as
"polynucleotides" are
typically expressed as the number of base pairs (bp) for double stranded
polynucleotides,
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
19
or in the case of single stranded polynueleotides as the number of nucleotides
(nt). One
thousand bp or nt equal a kilobase (kb). Polynucleotides of less than around
40 nucleotides
in length are typically called "oligonucleotides" and may comprise primers for
use in
manipulation of DNA such as via polymerase chain reaction (PCR).
The term "amino acid" in the context of the present disclosure is used in its
broadest sense and is meant to include organic compounds containing amine
(NIL) and
carboxyl (COOH) functional groups, along with a side chain (e.g., a R group)
specific to
each amino acid. In some embodiments, the amino acids refer to naturally
occurring L ct-
amino acids or residues. The commonly used one and three letter abbreviations
for
naturally occurring amino acids are used herein: A=Ala; C=Cys; D=Asp; E=G1u;
F=Phe;
G=Gly; H=His; I=Ile; K=Lys; L=Leu; M=Met; N=Asn; P=Pro; Q=G1n; R=Arg; S=Ser;
T=Thr; V=Val; W=Trp; and Y=Tyr (Lehninger, A. L., (1975) Biochemistry, 2d ed.,
pp.
71-92, Worth Publishers, New York). The general term "amino acid" further
includes D-
amino acids, retro-inverso amino acids as well as chemically modified amino
acids such as
amino acid analogues, naturally occurring amino acids that are not usually
incorporated
into proteins such as norleucine, and chemically synthesised compounds having
properties
known in the art to be characteristic of an amino acid, such as 13-amino
acids. For example,
analogues or mimetics of phenylalanine or proline, which allow the same
conformational
restriction of the peptide compounds as do natural Phe or Pro, are included
within the
definition of amino acid. Such analogues and mimetics are referred to herein
as "functional
equivalents" of the respective amino acid. Other examples of amino acids are
listed by
Roberts and Vellaccio, The Peptides: Analysis, Synthesis, Biology, Gross and
Meiehofer,
eds., Vol. 5 p. 341, Academic Press, Inc., N.Y. 1983, which is incorporated
herein by
reference.
The terms "polypeptide, and "peptide- are interchangeably used herein to refer
to
a polymer of amino acid residues and to variants and synthetic analogues of
the same.
Thus, these terms apply to amino acid polymers in which one or more amino acid
residues
is a synthetic non-naturally occurring amino acid, such as a chemical analogue
of a
corresponding naturally occurring amino acid, as well as to naturally-
occurring amino acid
polymers. Polypeptides can also undergo maturation or post-translational
modification
processes that may include, but are not limited to: glycosylation, proteolytic
cleavage,
lipidization, signal peptide cleavage, propeptide cleavage, phosphorylation,
and such like
A peptide can be made using recombinant techniques, e.g., through the
expression of a
recombinant or synthetic polynucleotide. A recombinantly produced peptide it
typically
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
substantially free of culture medium, e.g., culture medium represents less
than about 20 A,
more preferably less than about 10 A, and most preferably less than about 5 %
of the
volume of the protein preparation.
The term "protein" is used to describe a folded polypeptide having a secondary
or
5 tertiary structure. The protein may be composed of a single polypeptide,
or may comprise
multiple polypepties that are assembled to form a multimer. The multimer may
be a
homooligomer, or a heterooligmer. The protein may be a naturally occurring, or
wild type
protein, or a modified, or non-naturally, occurring protein. The protein may,
for example,
differ from a wild type protein by the addition, substitution or deletion of
one or more
10 amino acids.
A "variant" of a protein encompass peptides, oligopeptides, polypeptides,
proteins
and enzymes having amino acid substitutions, deletions and/or insertions
relative to the
unmodified or wild-type protein in question and having similar biological and
functional
activity as the unmodified protein from which they arc derived. The term
"amino acid
15 identity" as used herein refers to the extent that sequences are
identical on an amino acid-
by-amino acid basis over a window of comparison Thus, a "percentage of
sequence
identity" is calculated by comparing two optimally aligned sequences over the
window of
comparison, determining the number of positions at which the identical amino
acid residue
(e.g., Ala, Pro, Ser, Thr, Gly, Val, Leu, Ile, Phe, Tyr, Trp, Lys, Arg, His,
Asp, Glu, Asn,
20 Gln, Cys and Met) occurs in both sequences to yield the number of
matched positions,
dividing the number of matched positions by the total number of positions in
the window
of comparison (i.e., the window size), and multiplying the result by 100 to
yield the
percentage of sequence identity.
For all aspects and embodiments of the present invention, a "variant" has at
least
50%, 60%, 70%, 80%, 90%, 95% or 99% complete sequence identity to the amino
acid
sequence of the corresponding wild-type protein. Sequence identity can also be
to a
fragment or portion of the full length polynucleotide or polypeptide. Hence, a
sequence
may have only 50 % overall sequence identity with a full length reference
sequence, but a
sequence of a particular region, domain or subunit could share 80 A, 90 %, or
as much as
99 % sequence identity with the reference sequence.
The term "wild-type" refers to a gene or gene product isolated from a
naturally
occurring source. A wild-type gene is that which is most frequently observed
in a
population and is thus arbitrarily designed the "normal" or "wild-type" form
of the gene. In
contrast, the term "modified-, "mutant- or "variant- refers to a gene or gene
product that
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
21
displays modifications in sequence (e.g., substitutions, truncations, or
insertions), post-
translational modifications and/or functional properties (e.g., altered
characteristics) when
compared to the wild-type gene or gene product. It is noted that naturally
occurring
mutants can be isolated; these are identified by the fact that they have
altered
characteristics when compared to the wild-type gene or gene product. Methods
for
introducing or substituting naturally-occurring amino acids are well known in
the art. For
instance, methionine (M) may be substituted with arginine (R) by replacing the
codon for
methionine (ATG) with a codon for arginine (CGT) at the relevant position in a
polynucleotide encoding the mutant monomer. Methods for introducing or
substituting
non-naturally-occurring amino acids are also well known in the art. For
instance, non-
naturally-occurring amino acids may be introduced by including synthetic
aminoacyl-
tRNAs in the IVTT system used to express the mutant monomer. Alternatively,
they may
be introduced by expressing the mutant monomer in E. coil that are auxotrophic
for
specific amino acids in the presence of synthetic (i.e. non-naturally-
occurring) analogues
of those specific amino acids. They may also be produced by naked ligation if
the mutant
monomer is produced using partial peptide synthesis_ Conservative
substitutions replace
amino acids with other amino acids of similar chemical structure, similar
chemical
properties or similar side-chain volume. The amino acids introduced may have
similar
polarity, hydrophilicity, hydrophobicity, basicity, acidity, neutrality or
charge to the amino
acids they replace. Alternatively, the conservative substitution may introduce
another
amino acid that is aromatic or aliphatic in the place of a pre-existing
aromatic or aliphatic
amino acid. Conservative amino acid changes are well-known in the art and may
be
selected in accordance with the properties of the 20 main amino acids as
defined in Table 1
below. Where amino acids have similar polarity, this can also be determined by
reference
to the hydropathy scale for amino acid side chains in Table 2.
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
22
Table 1 - Chemical properties of amino acids
Ala aliphatic, hydrophobic,
neutral Met hydrophobic, neutral
Cys polar, hydrophobic, neutral
Asn polar, hydrophilic, neutral
Asp polar, hydrophilic,
charged (-) Pro hydrophobic, neutral
Glu polar, hydrophilic,
charged (-) Gln polar, hydrophilic, neutral
Phe aromatic, hydrophobic,
neutral Arg polar, hydrophilic, charged (+)
Gly aliphatic, neutral Ser polar,
hydrophilic, neutral
His aromatic, polar, hydrophilic, Thr polar,
hydrophilic, neutral
charged (+)
Ile aliphatic, hydrophobic,
neutral Val aliphatic, hydrophobic, neutral
Lys polar, hydrophilic,
charged(+) Trp aromatic, hydrophobic, neutral
Leu aliphatic, hydrophobic,
neutral Tyr aromatic, polar, hydrophobic
Table 2 - Hydropathy scale
Side Chain Hydropathy
Ile 4.5
Val 4.2
Leu 3.8
Phe 2.8
Cys 2.5
Met 1.9
Ala 1.8
Gly -0.4
Thr -0.7
Ser -0.8
Trp -0.9
Tyr -1.3
Pro -1.6
His -3.2
Glu -3.5
Gln -3.5
Asp -3.5
Asn -3.5
Lys -3.9
Arg -4.5
As described in more detail herein, a mutant or modified protein, monomer or
peptide can be chemically modified in any way and at any site. A mutant or
modified
monomer or peptide is preferably chemically modified by attachment of a
molecule to one
or more cysteines (cysteine linkage), attachment of a molecule to one or more
lysines,
attachment of a molecule to one or more non-natural amino acids, enzyme
modification of
an epitope or modification of a terminus. Suitable methods for carrying out
such
modifications are well-known in the art. The mutant of modified protein,
monomer or
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
23
peptide may be chemically modified by the attachment of any molecule. For
instance, the
mutant of modified protein, monomer or peptide may be chemically modified by
attachment of a dye or a fluorophore.
Chemically modifying monomers
The disclosure relates to a method of modifying a monomer such a monomer of a
protein nanopore. As explained in more detail below, the method has the
advantage of
improving the production of modified proteins such as nanopores. Populations
of modified
nanopores thus produced typically have improved properties compared to
populations of
nanopores produced by prior techniques.
In nanopore sensing it is particularly important that the population of
nanopores
used in any sensing application is homogeneous. If the population is non-
homogeneous
then it is problematic to know if any variance in the signal obtained when a
sample is
analysed derives from the nanopore that is used to detect the analyte, or the
analyte itself
This can reduce the efficiency at which an analyte is characterised due to
increased need
for data processing, or at a worst case can result in otherwise good data
being discarded.
Accordingly, it is necessary that the population of nanopores used in sensing
apparatuses is
of known homogeneity and that any deviation in the reproducibility at which
the nanopores
themselves are produced is minimised.
The problem is particularly acute when chemically modified nanopores are used.
There are many reasons why chemical modification of nanopores may be required:
for
example, in order to modify the properties of the signal obtained when an
analyte interacts
with the pore, to alter the pore stability, or to alter the physical
interaction of the analyte
with the pore (e.g. by altering the kinetics at which the analyte interacts
with the pore). For
protein nanopores, chemical modification can be effected by known routes.
However, as
explained above, a problem is that the modification methods used are typically
not highly
efficient, with a significant percentage of protein molecules not being
modified.
Accordingly, it is often difficult or impossible to obtain a homogeneous
population of
chemically modified nanopores.
A further difficulty lies in the separation of chemically modified proteins
such as
chemically modified nanopores from analogs which have not been chemically
modified. If
the modification results in a small change in the overall mass, charge or
conformation of
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
24
the protein it may not be possible to separate modified from unmodified
protein in an
efficient manner.
These difficulties combine to impede production of homogenous populations of
proteins such as protein nanopores. If the modification is undertaken before
purification
then limitations in purification techniques means that a homogeneous
population often
cannot be generated. If purification is undertaken prior to modification then
inefficiencies
in modification chemistries means that a homogeneous population again often
cannot be
generated.
The methods of the present disclosure address these issues. By combining the
modification and purification steps together the inefficiencies in these
processes are
"cancelled out". The methods involve modifying the monomer of interest with a
moiety
which combines both the desired chemical modification and a purification tag.
Only those
monomers which are successfully modified have the purification tag, and so
only these
monomers bind to a purification substrate such as a chromatography matrix.
Once bound,
a cleavable linker between the chemical modification and the purification tag
is cleaved,
thus releasing the modified monomer from the purification substrate Because
only
modified monomers have the required tag to bind to the purification substrate,
and because
only monomers which are successfully cleaved are released from the
purification substrate,
the yield of modified monomer can be very pure, with monomer populations
obtained
typically greater than 95% pure, and often up to 100% pure. The methods of the
present
disclosure thus have significant advantages compared to knovvii methods in
which
modification and purification strategies are decoupled.
Accordingly provided herein is a method of chemically modifying a polypeptide
or
polynucleotide monomer; the method comprising:
a) contacting the monomer with a multifunctional molecule, wherein the multi-
functional molecule comprises (i) a reactive group; (ii) a chemical modifying
group
and (iii) a cleavable purification tag;
b) allowing the reactive group of the multi-functional molecule to react with
the
monomer thereby attaching the chemical modifying group and cleavable
purification tag to the monomer to form a chemically modified tagged monomer;
c) contacting the chemically modified tagged monomer formed in step (b) with a
support;
d) allowing the purification tag to bind to the support thereby binding the
chemically
modified tagged monomer to the support; and
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
e) cleaving the purification tag thereby releasing the chemically modified
monomer
from the support.
Typically the monomer is a monomer of an oligomeric protein nanopore.
5 In some embodiments, the monomer is a polynucleotide monomer. For
example,
the monomer may be a monomer of a DNA origami pore, or an origami pore formed
from
an analog of DNA such as PNA. In some embodiments the monomer is a polypeptide
such
as a protein. For example the monomer may be a monomer of a protein nanopore,
such as
a monomer of a monomeric protein nanopore or, more typically, the monomer of
an
10 oligomeric protein nanopore. Monomers of nanopores which are
particularly amenable to
being modified in accordance with the claimed methods are discussed below.
The methods involve contacting the monomer with a multifunctional molecule. As
used herein, a multifunctional molecule is a molecule comprising at least
three
components: (i) a reactive group for reacting with an appropriate site on the
monomer,
15 such as reactive functional group on the monomer; (ii) a chemical
modifying group which
corresponds to the chemical modification being made to the monomer; and (iii)
a cleavable
purification tag. The cleavable purification tag typically comprises (i) a
cleavable linker;
and (ii) a purification tag. The purification tag is suitable for binding to a
purification
support such as a chromatography matrix, thus allowing the modified monomer to
be
20 purified. The cleavable linker allows the modified monomer to be
released from the
purification support. This is described in more detail below.
The method thus comprises allowing the reactive group on the multifunctional
molecule to react with the monomer. Typical reactive groups suitable for use
in this way
are described in more detail below.
25 Once the reactive group has reacted with the monomer and the monomer
has thus
been modified with the multifunctional molecule, the modified monomer is
contacted with
a support, typically a purification support. The purification tag on the
multifunctional
molecule binds to the support thereby binding the modified monomer to the
support. The
bound monomer can then be washed or otherwise subjected to purification
techniques to
eliminate impurities including unmodified monomer and/or unreacted
multifunctional
molecules.
The method further comprises cleaving the purification tag. As explained
herein,
this releases the chemically modified monomer from the support. Methods for
cleaving
cleavable linkers are discussed in more detail herein.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
26
Multifitnctional Molecule
The provided methods comprise the use of a multifunctional molecule as
discussed
above.
The multifunctional molecule may be in some embodiments represented by
Formula (I) or Formula (II) below:
A -B-D-C A -D-C
Formula (I) Formula (II)
wherein:
A is a reactive group;
B is a chemical modifying group; and
D-C forms a cleavable purification tag.
In some embodiments D comprises a cleavable linker as discussed herein. In
some
embodiments C comprises a support-binding group as discussed herein.
A multifunctional molecule according to Formula (I) provides an in-line
design. In
this way the chemical modifying group is "hidden" by the cleavable linker and
the reactive
group and is exposed by the cleaving of the cleavable linker. Such designs can
be useful
when the chemical modifying group is reactive under the conditions of the
binding of the
multifunctional molecule to the purification support, for example
A multifunctional molecule according to Formula (II) provides a "branched"
design. The modification group is extrinsic and is typically accessible
throughout the steps
of the claimed method. Such designs can be useful when the desired chemical
modification is incompatible with the chemistry of the cleavable linker, for
example.
Cleavage of the cleavable linker can in some cases leave a residue from the
linker.
In some embodiments the residue if present once the linker has been cleaved
corresponds
to a chemical modifying group as used herein. This is particularly the case
when the
multifunctional molecule is according to Formula (I). In other embodiments any
residue
from the cleavable linker is separate to the chemical modifying group. This is
particularly
the case when the multifunctional molecule is according to Formula (II).
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
27
Thus in some embodiments the reactive group comprises the chemical modifying
group. For example, in some embodiments the multifunctional molecule is of
Formula
(III) below:
A-D-C
Formula (III)
wherein:
A is a reactive group comprising a chemical modifying group; and
D-C forms a cleavable purification tag.
Reactive group
The reactive group (A) of the multifunctional molecule is suitable for
reacting with
the monomer being subjected to the methods disclosed herein. Any suitable
reactive group
can be used. For example, the reactive group may be an amine-reactive group; a
carboxyl-
reactive group; a sulfhydryl-reactive group or a carbonyl-reactive group. In
some
embodiments the reactive group of the multi-functional molecule comprises a
cysteine-
reactive group. In some embodiments the reactive group comprises a maleimide,
an azide,
a thiol, an alkyne, an NHS ester or a haloacetamide.
In some embodiments the reactive group may be a group capable of reacting with
a
non-natural amino acid such as 4-azido-L-phenylalanine (Faz) and any one of
the amino
acids numbered 1-71 in Figure 1 of Liu C. C. and Schultz P. G., Annu. Rev.
Biochem.,
2010, 79, 413-444. Such groups are particularly useful when corresponding non-
natural
amino acids are comprised in the monomer.
In some embodiments the reactive group is a click chemistry group. Click
chemistry is a term first introduced by Kolb et al. in 2001 to describe an
expanding set of
powerful, selective, and modular building blocks that work reliably in both
small- and
large-scale applications (Kolb HC, Finn, MG, Sharpless KB, Click chemistry:
diverse
chemical function from a few good reactions, Angew. Chem. Int. Ed. 40 (2001)
2004-
2021). They have defined the set of stringent criteria for click chemistry as
follows: "The
reaction must be modular, wide in scope, give very high yields, generate only
inoffensive
by-products that can be removed by non-chromatographic methods, and be
stereospecific
(but not necessarily enantioselective). The required process characteristics
include simple
reaction conditions (ideally, the process should be insensitive to oxygen and
water), readily
available starting materials and reagents, the use of no solvent or a solvent
that is benign
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
28
(such as water) or easily removed, and simple product isolation. Purification
if required
must be by non-chromatographic methods, such as crystallization or
distillation, and the
product must be stable under physiological conditions".
Suitable examples of click chemistry include, but are not limited to, the
following:
(a) copper-free variant of the 1,3 dipolar cycloaddition reaction, where an
azide
reacts with an alkyne under strain, for example in a cyclooctane ring;
(b) the reaction of an oxygen nucleophile on one linker with an epoxide or
aziridine reactive moiety on the other; and
(c) the Staudinger ligation, where the alkyne moiety can be replaced by an
aryl
phosphine, resulting in a specific reaction with the azide to give an amide
bond.
Any reactive group may be used in the methods. The reactive group may thus be
one that is suitable for click chemistry, particularly when a complementary
group is present
on the monomer. The reactive group may be any of those disclosed in WO
2010/086602,
particularly in Table 4 of that application.
In some embodiments the reactive group is a haloacetamide, for example,
iodoacetamide, brom oacetemi de or chloroacetamide
In some embodiments the reactive group is selected from a vinyl group, TCO,
tetrazine and a strained alkyne; DB C 0 ; an activated acid e.g. an acid
chloride; and
piperazine and reactive amines.
In some embodiments the reactive group is a polynucleotide or polynucleotide
analog, e.g. PNA. In some embodiments the reactive group of the
multifunctional molecule
comprises a nucleotide analog such as a PNA base or PNA polymer which can
interact
non-covalently via supramolecular associations and/or hydrogen bonding with
the
monomer, e.g. with a monomer containing the complementary PNA base or polymer.
In some embodiments the polynucleotide or polynucleotide analog binds to a
complementary polynucleotide or polynucleotide analog on the monomer.
Host ¨guest chemistry can also be used to provide the reaction between the
reactive
group and the monomer. For example, in some embodiments the monomer comprises
a
ligand for binding to a metal complex, and the reactive group comprises a
metal complex.
Thus, in some embodiments the reactive group of the multifunctional molecule
comprises
a metal complex which can interact non-covalently via chelation or
supramolecular
association with the monomer containing a site that can act as a ligand to
complex with the
modifier molecule by forming a stable association.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
29
The reactive group may be any of those disclosed in Sakamoto and Hamachi,
"Recent progress in chemical modification of proteins", Anal. Sci 2019 (35) 5-
27; or
McKay and Finn, "Click chemistry in complex mixtures: bioorthogonal
bioconjugation",
Chem. Biol. 2014, 21(9) 1075-1101, both of which are hereby incorporated by
reference in
their entirety.
The methods disclosed herein are particularly amenable to the use of
multifunctional molecules comprises thiols or maleimide groups for reaction
with cysteine
residues on the monomer; NHS-ester groups for reaction with amine groups on
the
monomer; or an azide or alkyne for participating in a click chemistry reaction
with the
corresponding group on the monomer.
The reactive group may therefore react with a reactive functional group on the
monomer. In other words, in some embodiments the monomer comprises a reactive
functional group and step (b) comprises allowing the reactive group of the
multifunctional
molecule to react with the reactive functional group of the monomer.
The reactive functional group may be present naturally in the monomer or may
be
introduced, e.g. by genetic manipulation or by chemical modification of the
monomer.
The reactive functional group may originate from a non-natural amino acid
incorporated
into the monomer during its synthesis or expression, e.g. during cell-free
expression, e.g.
via in vitro transcription/translation.
Chemical modiffing group
The chemical modifying group of the multifunctional molecule provides the
chemical modification desired. Any suitable chemical modifying group can be
used in the
provided methods.
In some embodiments the chemical modifying group has a molecular mass of at
most kDa, such as at most 500 Da, e.g at most 400 Da, such as at most 300 Da,
e.g. at
most 200 Da. Typical amino acids have an approximate mass of around 110 Da and
are
exemplary chemical modifying groups for use in the methods disclosed herein,
although
the methods disclosed herein are not limited to such groups.
In some embodiments the chemical modifying group is an aliphatic group such as
an alkane or alkene. The chemical modifying group may be a linear or branched
alkane
e.g. comprising from 1 to 20 carbon atoms, e.g. from 2 to 10 carbon atoms. The
chemical
modifying group may be a linear or branched alkene e.g. comprising from 1 to
20 carbon
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
atoms, e.g. from 2 to 10 carbon atoms. The chemical modifying group may be an
alkyl,
alkenyl; alkynyl; or alkoxy group. The chemical modifying group may be a
cyclic group
such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl groups;
piperazine,
piperidine, morpholine, 1,3-oxazinane, pyrroli dine, imidazoli dine, and
oxazolidine.
5 In some embodiments the chemical modifying group is an aromatic group
such as a
Co to Clo aromatic ring (e.g. benzene/phenyl); or a 5- to 10-membered
heteroaromatic
group, e.g. pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyridine,
pyridazine,
pyrimidine, and pyrazine.
In some embodiments the chemical modifying group comprises an amino acid, a
10 nucleotide; a polymer; a hydrogen-bonding group; a membrane anchor; a
sugar, a dye, a
chromophore, a fluorophore or a molecular adapter. In some embodiments the
chemical
modifying group comprises a natural or unnatural amino acid; a polypeptide; a
nucleotide
or nucleotide analog; an oligonucleotide or oligonucleotide analog; a
polysaccharide; a
lipid; a polyethylene glycol; a cyclodextrin; a DNA intercalator; an aptamer
or an analyte
15 binding domain.
In some embodiments the chemical modifying group introduces hydrophilic,
hydrophobic, positively charged, negatively charged, hydrogen-bonding,
supramolecular
associations or zwitterionic properties to the protein monomer.
In some embodiments the chemical modifying group is or comprises an amino
acid.
20 The amino acid may be a natural or unnatural amino acid. A plurality of
amino acids may
be comprised in the chemical modifying group, e.g. the chemical modifying
group may
comprise a peptide.
In some embodiments the chemical modifying group is or comprises a nucleotide
or polynucleoti de. Analogs of naturally occurring nucleotides/polynucleotides
are also
25 included. For example, the chemical modifying group may comprise RNA,
PNA, LNA, or
BNA.
In some embodiments the chemical modifying group may comprise a saccharide or
polysaccharide such as dextrose, maltose, glucose, etc.
In some embodiments the chemical modifying group may comprise a dye such as
30 an anthraquinoine or phthalocyanine. In some embodiments the chemical
modifying group
may comprise a fluorophore such as hydroxycoumarin, aminocoumarin,
methoxycoumarin, fluorescein, X-Rhodamine, Texas Red, Cy5, Cy7 etc.
In some embodiments, the chemical modifying group is an adaptor which is a
compound which has an effect on the physical or chemical properties of a
nanopore once
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
31
the monomer has assembled into the pore. Typically an adapter improves the
interaction
of the pore with an analyte such as a nucleotide or polynucleotide. The
adaptor may alter
the charge of the barrel or channel of the pore or specifically interact with
or bind to the
nucleotide or polynucleotide thereby facilitating its interaction with the
pore.
An adapter may be a cyclic molecule. An adaptor may comprise one or more
chemical groups that are capable of interacting with a nucleotide or
polynucleotide e.g. by
hydrophobic interactions, hydrogen bonding, Van der Waal's forces, 7c-cation
interactions
and/or electrostatic forces.
An adapter may comprise one or more amino groups. The amino groups can be
attached to primary, secondary or tertiary carbon atoms. The adaptor may
comprise a ring
of amino groups, such as a ring of 6, 7, 8 or 9 amino groups. Alternatively or
additionally
an adapter may comprise one or more hydroxyl groups. The hydroxyl groups can
be
attached to primary, secondary or tertiary carbon atoms. The hydroxyl groups
may form
hydrogen bonds with uncharged amino acids in the pore.
Suitable chemical modifying groups include, but are not limited to,
cyclodextrins,
cyclic peptides and cucurbiturils. The chemical modifying group may be any of
those
disclosed in Eliseev, A. V., and Schneider, H-J. (1994)]. Am. Chem. Soc. 116,
6081-6088.
The adaptor may be heptakis-6-amino-O-cyclodextrin (am7-I3CD), 6-monodeoxy-6-
monoamino-f3-cyclodextrin (ami-f3CD) or heptakis-(6-deoxy-6-guanidino)-
cyclodextrin
(gu7-I3CD). The guanidino group in gu7-I3CD has a much higher pKa than the
primary
amines in am7-f3CD and so it more positively charged. The adapter may be a y-
cyclodextrins
In some embodiments the chemical modifying group is selected from (i)
Maleimides including diabromomaleimides such as: 4-phenylazomaleinanil, 1.N-(2-
Hydroxyethyl)maleimide, N-Cyclohexylmaleimide, 1.3-Maleimidopropionic Acid,
1.1-4-
Aminopheny1-1H-pyrrol e,2,5,dione, 1.1-4-Hydroxypheny1-1H-pyrrole,2,5,dione, N-
Ethylmaleimide, N-Methoxycarbonylmaleimide, N-tert-Butylmaleimide, N-(2-
Aminoethyl)maleimide , 3-Maleimido-PROXYL , N-(4-Chlorophenyl)maleimide, 1-[4-
(dimethylamino)-3,5-dinitropheny1]-1H-pyrrole-2,5-dione, N-[4-(2-
Benzimidazolyl)phenylimaleimide, N44-(2-benzoxazolyl)phenylimaleimide, N-(1-
naphthyl)-maleimide, N-(2,4-xylyl)maleimide, N-(2,4-difluorophenyl)maleimide ,
N-(3-
c hl oro-para-toly1)-maleimide, 1-(2-amino-ethyl)-pyrrole-2,5-dione
hydrochloride, 1-
cyclopenty1-3-methy1-2,5-dihydro-1H-pyrrole-2,5-dione, 1-(3-aminopropy1)-2,5-
dihydro-
1H-pyrrol e-2,5-di one hydrochloride, 3 -methy1-1-[2-oxo-2-(piperazi n-l-
ypethyl] -2,5 -
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
32
dihydro-1H-pyrrole-2,5-dione hydrochloride, 1-benzy1-2,5-dihydro-1H-pyrrole-
2,5-dione,
3-methyl-I -(3,3,3 -trifluropropy1)-2,5-dihydro-1H-pyrrole-2,5-dione, 1-[4-
(methylamino)cyclohexyl]-2,5-dihydro-1H-pyrrole-2,5-dione trifiuroacetic acid,
SMILES
0=C1C=CC(=0)N1CC=2C=CN=CC2, SMILES 0=C1C=CC(=0)N1CN2CCNCC2, 1-
benzy1-3-methy1-2,5-dihydro-1H-pyrrole-2,5-dione, 1-(2-fluoropheny1)-3-methyl-
2,5-
dihydro 1H-pyrrole-2,5-dione, N-(4-phenoxyphenyl)maleimide , N-(4-
nitrophenyl)maleimide (ii) Iodocetamides such as :3-(2-Iodoacetamido)-proxyl,
N-
(cyclopropylmethyl)-2-iodoacetamide, 2-iodo-N-(2-phenylethyl)acetamide, 2-iodo-
N-
(2,2,2-trifluoroethyl)acetamide, N-(4-acetylpheny1)-2-iodoacetamide, N-(4-
(aminosulfonyl)pheny1)-2-iodoacetamide, N-(1,3-benzothiazol-2-y1)-2-
iodoacetamide, N-
(2,6-diethylpheny1)-2-iodoacetamide, N-(2-benzoy1-4-chloropheny1)-2-
iodoacetamide,
(iii) Bromoacetamides: such as N-(4-(acetylamino)pheny1)-2-bromoacetamide , N-
(2-
acetylpheny1)-2-bromoacetamide , 2-bromo-n-(2-cyanophenyl)acetamide, 2-bromo-N-
(3-
(trifluoromethyl)phenyl)acetamide, N-(2-benzoylpheny1)-2-bromoacetamidc , 2-
bromo-N-
(4-fluoropheny1)-3-methylbutanamide, N-Benzy1-2-bromo-N-phenylpropionamide, N-
(2-
bromo-butyryl )-4-chl oro-benzenesulfonami de, 2-Bromo-N-m ethyl -N-phenyl
acetami de,
2-brom o-N-ph en ethyl -acetami de, 2-adam antan-l-yl -2-b rom o-N-cycl ohexyl
-acetami de, 2-
bromo-N-(2-methylphenyl)butanami de, Monobromoacetanili de, (iv) Disulphides
such as:
aldrithio1-2 , aldrithio1-4 , isopropyl disulfide, 1-(Isobutyldisulfany1)-2-
methylpropane,
Dibenzyl disulfide, 4-aminophenyl disulfide, 3-(2-Pyridyldithio)propionic
acid, 3-(2-
Pyridyldithio)propionic acid hydrazide, 3-(2-Pyridyldithio)propionic acid N-
succinimidyl
ester, am6amPDP1-f3CD and (v) Thiols such as: 4-Phenylthiazole-2-thiol,
Purpald,
5,6,7,8-tetrahydro-quinazoline-2-thiol.
Cleavable purification tag
The multifunctional molecule comprises a cleavable purification tag for
binding the
chemically modified monomer to a purification support during the claimed
methods.
Typically the cleavable purification tag comprises a cleavable linker and a
purification tag.
Any suitable cleavable linker can be used in the disclosed methods. The linker
may
comprise a short chain oligopeptide or oligonucleotide containing e.g. from
about 1 to
about 20 amino acids or nucleotides. The linker may comprise a polymer such as
a
polyethylene glycol or a saccharide containing from about 1 to about 20 repeat
units. For
example, the linker may comprise PEG2, PEG3 or PEG 4.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
33
In some embodiments the cleavable linker is cleaved by physical or chemical
means. Any suitable means can be used.
The cleavable moiety of the cleavable linker may be e.g. a pH sensitive group;
a
redox sensitive group; a light-sensitive group; a temperature sensitive group
or a chemical-
sensitive group which is sensitive to cleavage by reaction of the group with a
specific
chemical.
In some embodiments the cleavable linker is cleavable by exposure to light;
i.e. it is
photocleavable. Thus, in some embodiments of the disclosed method, in step (e)
cleaving
the purification tag comprises exposing the support and/or the tagged monomer
to light;
preferably UV light. Photocleavable linkers include nitobenzyl moieties. Such
groups are
cleavable under UV irradiation.
In some embodiments the cleavable linker is cleavable by exposure to a change
in
pH. Thus, in some embodiments of the disclosed method, in step (e) cleaving
the
purification tag comprises exposing the support and/or the tagged monomer to a
change in
pH. pH-sensitive cleavable linkers include hydrazones and cis-aconityl. An
example of a
cleavable hydrazone linker is shown below:
N
0
0
wherein the wave lines represent the points of attachment to the rest of the
multifunctional
molecule. Those skilled in the art will appreciate that the PEG linker shown
above can be
replaced with other linkers including those discussed herein; and similarly
the attachment
chemistry shown can be exchanged for other appropriate attachment chemistry
(e.g. the
amide linkage shown could be replaced with an ester linkage).
In some embodiments the cleavable linker is cleavable by exposure to a
chemical
reagent. Thus, in some embodiments of the disclosed method, in step (e)
cleaving the
purification tag comprises exposing the support and/or the tagged monomer to a
chemical
reagent; preferably a reducing reagent. Chemical-sensitive cleavable linkers
include
disulphides. Disulphide bonds are susceptible to cleavage by addition of a
reducing agent
such as DTT and beta-mercaptoethanol. An example of a cleavable disulphide
linker is
shown below:
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
34
N
0 0
wherein the wave lines represent the points of attachment to the rest of the
multifunctional
molecule. Those skilled in the art will appreciate that the PEG linker shown
above can be
replaced with other linkers including those discussed herein; and similarly
the attachment
chemistry shown can be exchanged for other appropriate attachment chemistry
(e.g. the
ester linkage shown could be replaced with an amide linkage)
In some embodiments the cleavable linker is cleavable by exposure to an enzyme
such as a protease or nuclease. Thus, in some embodiments of the disclosed
method, in
step (e) cleaving the purification tag comprises exposing the support and/or
the tagged
monomer to an enzyme; preferably a protease.
Enzyme-sensitive linkers include protease-sensitive peptide linkers comprising
recognition sequences for one or more endo- and/or exo-proteases. Examples
include the
sequences DDDDK (SEQ ID NO: 24; cleaved by enteropeptidase from E. coli and S.
cerevisiae); LVPRGS (SEQ ID NO: 25; cleaved by thrombin and factor Xa);
ENLYFQG
(SEQ ID NO: 26; cleaved by TEV protease) and LEVLFQGP (SEQ ID NO: 27; cleaved
by
Rhinovirus 3C protease). B-glucuronide linkers can be cleaved by lysosomal 13-
glucuronidase.
An example of an enzyme-cleavable linker is shown below:
H
0
I C
0 - = N
H
, , Re
I
0
= NH.
H =N 0
In some embodiments the cleavable linker is of the form Cl-Lk, Lk-C1 or Lk-C1-
Lk
wherein Cl is a cleavable moiety and Lk is a linker.
In some embodiments the cleavable linker comprises a polymer and a
photocleavable moiety. In some embodiments the cleavable linker comprises a
PEG linker
and a nitobenzyl moiety. In some embodiments the cleavable linker comprises a
structure
of the form:
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
0
0
0
NH2
0
wherein the wavy lines represent the points of attachment to the purification
tag and
chemical modifying group (either directly or via a linker), respectively.
5
Any suitable purification tag can be used in the cleavable purification tag
comprised in the multifunctional molecule. For example, the purification tag
may
comprise or consist of biotin. Biotin is particularly suitable for use in the
disclosed
methods as it forms a strong non-covalent attachment with streptavidin and
related proteins
10 (neutravidin, avidin, etc)
Other purification tags include peptide purification tags suitable for IMAC
(immobilised metal affinity chromatography) chemistry. For example, the
purification tag
may comprise a poly-His tag (e.g. HHEIH, HHHHHH or HHHHHHHH; SEQ ID NOs: 28-
30). Such tags are suitable for binding to a purification support comprising a
metal such as
15 nickel or cobalt. Still other purification tags include peptide
tags such as Strep
(WSHPQFEK; SEQ ID NO: 31), FLAG (DYKDDDDK; SEQ ID NO: 32), Human
influenza hemagglutinin (HA) (YPYDVPDYA; SEQ ID NO: 33), Myc (EQKLISEED;
SEQ ID NO: 34), and V5 (GKPIPNPLLGLDST; SEQ ID NO: 35), etc.
Other suitable purification tags include: Biotin-carboxy carrier protein
(BCCP);
20 Calmodulin binding peptide (CBP); Chitin binding domain (CBD);
Histidine affinity tag
(HAT); Polyarginine (Arg-tag); Polyaspartate (Asp-tag); Polylysine (Lys-tag);
Polyphenylalanine (Phe-tag); Streptavadin-binding peptide (SBP); Tetrazine
tag; TCO tag;
Azide tag; and DBCO / Alkyne tag.
In some embodiments the cleavable purification tag comprises a cleavable
linker
25 comprising a polymer and a photocleavable moiety; and biotin. In
some embodiments the
cleavable purification tag comprises a PEG linker and a nitobenzyl moiety and
biotin. In
some embodiments the cleavable purification tag comprises a structure of the
form:
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
36
0
H N )NN H
o
0
(Tcji
N
0 0 0
N H2
0
wherein the wavy line represents the point of attachment to the chemical
modifying group
(either directly or via a linker).
Support
The disclosed methods comprise allowing the monomer, once functionalised with
the multifunctional molecule, to bind to a support for purification.
Any suitable support can be used.
In some embodiments the support comprises a chromatography matrix, preferably
an agarose or sepharose resin. Such resins are commercially available from
suppliers such
as Sigma Aldrich.
In some embodiments the support comprises beads (i.e. one or more beads).
Magnetic beads are preferred as such beads allow for facile purification e.g.
using washing
with buffer. Functionalised magnetic beads are commercially available with a
variety of
functionalisations from suppliers such as Sigma Aldrich and Bio-Rad.
In some embodiments the support comprises a solid surface. Any suitable
material
can be used. Suitable materials include glass, silica, polymers such as
polyester, and
ceramics such as hydroxyapatite.
In some embodiments the support is functionalised for binding to the
purification
tag. Those skilled in the art will appreciate that the support can be
functionalised
depending on the purification tag comprised in the multifunctional molecule
that is used.
Alternatively, the purification tag can be chosen depending on the support
material to be
used. Thus, the choice of purification tag and support material is an
operational parameter
which can be determined by the user of the disclosed methods.
In some embodiments the support comprises streptavidin, neutravidin or avidin,
or
a derivative of streptavidin, neutravidin or avidin such as traptavidin. Such
supports are
particularly useful when the multifunctional molecule comprises a purification
tag
comprising biotin.
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
37
In some embodiments the support comprises a metal such as nickel or cobalt.
The
metal ion may be provided with a suitable chelator such as nitriloacetic acid
(NTA) or
iminodiacetic acid (IDA) For example, the support may comprise Ni-NTA. Such
supports
are particularly useful when the multifunctional molecule comprises a
purification tag
comprising a His tag.
In some embodiments the support comprises streptactin. Such supports are
particularly useful when the multifunctional molecule comprises a purification
tag
comprising a Strep tag.
In some embodiments the support comprises an antibody for a sequence such as
FLAG, HA, Myc or V5 as discussed above.
In some particular embodiments of the disclosed methods, the purification tag
comprises a biotin group and the support comprises streptavidin, neutravidin
or avidin,
preferably streptavidin.
In some embodiments the cleavable purification tag comprises a structure of
the
form:
HN)LNH
oI
0
NH2
0
wherein the wavy line represents the point of attachment to the chemical
modifying group
(either directly or via a linker) and the support comprises streptavidin; e.g
the support may
comprise an agarose or sepharose resin comprising streptavidin or streptavidin-
coated
magnetic beads.
Monomer
The provided methods comprise the modification of a monomer. The discussion
herein focusses primarily on the modification of protein monomers of protein
nanopores,
and particularly the in
of inonomers of multicornponent protein nanopores.
However, the disclosed methods are not limited to such monomers and are useful
for the
chemical modification of monomeric proteins, polynucleotides, components of
polynucleotide origami (e.g. DNA origami) structures such as origami pores,
enzymes
(including motor proteins as discussed herein), and the like.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
38
In some embodiments the monomer is a polypeptide having a mass of from about
kDa to about 1 MDa.
In embodiments of the invention which relate to monomers of a nanopore, any
suitable nanopore can be used. In one embodiment a nanopore is a transmembrane
pore.
5 A transmembrane pore is a structure that crosses the membrane to some
degree. It
permits hydrated ions driven by an applied potential to flow across or within
the
membrane. The transmembrane pore typically crosses the entire membrane so that
hydrated ions may flow from one side of the membrane to the other side of the
membrane.
However, the transmembrane pore does not have to cross the membrane. It may be
closed
10 at one end. For instance, the pore may be a well, gap, channel, trench
or slit in the
membrane along which or into which hydrated ions may flow.
The monomer may be a monomer of a biological or artificial nanopore. Suitable
pores include, but are not limited to, protein pores and polynucleotide pores.
In one embodiment the monomer is a monomer of a polynucleotide pore. For
example, a polynucleotide pore may be a DNA origami pore (Langecker et al.,
Science,
2012; 338. 932-936) Suitable DNA origami pores are disclosed in W02013/083983
A
monomer of a polynucleotide origami pore is typically a polynucleotide of
between 50 nt
and 1000 kb; such as between 100 nt and 100 kb, e.g. between 1000 nt (1 kb)
and 10 kb.
The monomer assembles into a structure permitting ion transport from one
chamber to
another. One or more monomers may assemble into such a structure. Typically a
plurality
of monomers assemble into the structure, an origami pore is typically
oligomeric. The
monomers that assemble into an origami pore may be of the same type (i.e. the
pore may
be homooligomeric) or may be of two or more different types (i.e. the pore may
be a
multicomponent heterooligomeric pore).
More often, the monomer is a monomer of a transmembrane protein pore. A
transmembrane protein pore is a polypeptide or a collection of polypeptides
that permits
hydrated ions, such as polynucleotide, to flow from one side of a membrane to
the other
side of the membrane. In the methods provided herein, the transmembrane
protein pore
typically is capable of forming a pore that permits hydrated ions driven by an
applied
potential to flow from one side of the membrane to the other. The
transmembrane protein
pore preferably permits polynucleotides to flow from one side of the membrane,
such as a
triblock copolymer membrane, to the other. The transmembrane protein pore
allows a
polynucleotide to be moved through the pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
39
In one embodiment, the monomer is a monomer of a monomeric nanopore; i.e. the
monomer forms a transmembrane protein pore. In one embodiment the monomer is a
monomer of an oligomeric pore. The monomer may be a monomer of a pore made up
of
several repeating subunits, such as at least 6, at least 7, at least 8, at
least 9, at least 10, at
least 11, at least 12, at least 13, at least 14, at least 15, or at least 16
subunits. In some
embodiments the monomer is a monomer of a pore comprising at least 20
subunits, at least
30 subunits, at least 40 subunits or at least 50 subunits.
For example, members of the MACPF superfamily form large transmembrane
pores and pore complexes. For example, pleurotolysin (PlyAB) from Pleurotus
ostreatus
consists of two distinct monomeric components. Pleurotolysin A (PlyA) and
Pleurotolysin
B (PlyB). PlyA binds on the membrane and acts as a scaffold to recruit the
second
component PlyB which spans the membrane to form the transmembrane channel.
PlyB
monomers can be modified with the said method to introduce different chemical
groups
within the channel. Accordingly, in some embodiments the monomer is a monomer
of
PlyA or Ply B. PlyAB and its use in detecting proteins are discussed by Huang
et al,
"Electro-osmotic vortices promote the capture of folded proteins by PlyA B
nanopores",
Nano Letters 2020, 20(5), 3819-3827, the contents of which are hereby
incorporated by
reference in their entirety.
The monomer may be a monomer of a hexameric, heptameric, octameric or
nonameric pore. The pore may be a homo-oligomer or a hetero-oligomer.
A transmembrane protein pore typically comprises a bairel or channel through
which the ions may flow. The subunits of the pore typically surround a central
axis and
contribute strands to a transmembrane 13-barrel or channel or a transmembrane
cc-helix
bundle or channel.
Typically, the barrel or channel of a transmembrane protein pore comprises
amino
acids that facilitate interaction with an analyte, such as a target
polynucleotide (as
described herein). These amino acids are preferably located near a
constriction of the
barrel or channel. A transmembrane protein pore typically comprises one or
more
positively charged amino acids, such as arginine, lysine or histidine, or
aromatic amino
acids, such as tyrosine or tryptophan. These amino acids typically facilitate
the interaction
between the pore and nucleotides, polynucleotides or nucleic acids.
In one embodiment, the monomer is a monomer of a transmembrane protein pore
derived from Pp-bait-el pores or cx-helix bundle pores. 13-barrel pores
comprise a barrel or
channel that is formed from 13-strands. Suitable 13-barrel pores include, but
are not limited
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
to, (3-toxins, such as a-hemolysin, anthrax toxin and leukocidins, outer
membrane
phospholipase A and other pores, such as lysenin, NetB and CytK. a-helix
bundle pores
comprise a barrel or channel that is formed from a-helices. Suitable a-helix
bundle pores
include, but are not limited to, inner membrane proteins and a outer membrane
proteins.
5 In one embodiment the monomer is a monomer of a transmembrane pore
derived
from or based on a-hemolysin (a-HL), lysenin, or haemolytic protein
fragaceatoxin C
(FraC).
In one embodiment the monomer is a monomer of an actinporin. Actinporins
include Equinatoxin II (EqtII) from Actinic' equina and Fragaceatoxin C (FraC)
from
10 Actinia.fragacea.
In one embodiment the monomer is a monomer of a homolog or paralog of any one
of the protein pores discussed herein.
In one embodiment, the monomer is a monomer of a transmembrane pore derived
from lysenin, or a paralog or homolog thereof. Examples of suitable pores
derived from
15 lysenin are disclosed in WO 2013/153359.
In one embodiment, the monomer is a monomer of a transmembrane pore derived
from or based on a-hemolysin (a-HL), or a paralog or homolog thereof. The wild
type a-
hemolysin pore is formed of 7 identical monomers or sub-units (i.e., it is
heptameric). An
a-hemolysin pore may be a-hemolysin-NN or a variant thereof The variant
preferably
20 comprises N residues at positions El 11 and K147.
In one embodiment the monomer is a monomer of a transmembrane pore derived
from or based on NetB, or a paralog or homolog thereof. NetB is a pore-forming
toxin
produced by Clostridium perfringens.
In one embodiment the monomer is a monomer of a transmembrane pore derived
25 from or based on CytK, or a paralog or homolog thereof. CytK is a pore-
forming toxin
produced by Bacillus cerius.
In one embodiment the monomer is a monomer of a gamma-hemolysin pore, or a
paralog or homolog thereof
In some embodiments the monomer is a monomer of a multi-component pore. As
30 used herein, a multi-component pore is a pore which comprises two or
more different
monomers. For example, gamma-hemolysin is a multicomponent pore comprising two
different types of monomer: in other words it is a bicomponent pore. The
disclosed
method is particularly suitable for modifying multicomponent pores because it
can be used
to inctroduce different modifications into the different components of the
pore. Thus, a
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
41
first modification can be made in a first component, a second modification can
be made in
a second component, etc. By making different modifications in different
components of a
multi-component pore, the functionality of the chemical modification can be
precisely
located at multiple locations (e.g at two or more locations) within the pore,
e.g. within the
channel of the pore. For example, modifications can be made to introduce or
modify two
recognition sites within the channel of a multifunctional pore allowing for
improved
characterisation of analytes such as polynucleotides. An example of this
strategy is shown
in Figure 1, which depicts how the disclosed methods could be used to
introduce multiple
modifications into a bicomponent pore such as gamma-hemolysin.
As explained above, the monomer may be a monomer of any of the nanopores
discussed above. The methods provided herein also relate to methods of
producing a
chemically modified nanopore. The nanopore used in such methods may be any of
the
nanopores discussed in the context of monomers above.
The chemical modification in accordance with the disclosed methods can be made
at a specific position within the monomer. The specific position can be
determined based
on the structure of the free monomer or the structure adopted by the monomer
when the
monomer forms an oligomer (if applicable). For monomers which oligomeri se,
the
structure used to determine the positioning of the modification is typically
the three-
dimensional structure of the oligomer formed from the monomer. For example,
the
structure may be the 3D structure of an oligomeric protein pole. 3D X-ray
crystal
structures are known for many oligomeric pores, and/or can determined by
computational
modelling.
In some embodiments the modification can be made to place the chemical
modifying group at a set position in the assembled pore. In some embodiments
the
modification is such that the modification is positioned at a solvent-
accessible position on
a surface of the pore. In some embodiments the modification is positioned on
an external
surface of the pore. In some embodiments the modification is positioned to
interact with a
membrane when the pore is assembled in the membrane, e.g. to improve anchoring
of the
pore to the membrane. In some embodiments the modification is positioned at or
near an
opening of a pore; e.g. at or near the cis or trans opening of a channel
running through the
pore. In some embodiments the modification is positioned within the channel of
the pore.
In some embodiments the modification is at an internal-facing residue within
the channel
or barrel of the pore. In some embodiments the modification is positioned at
or near a
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
42
constriction within the channel of the pore. In some embodiments the
modification is
positioned to increase a constriction within the channel of the pore. In some
embodiments
the modification is positioned to introduce a constriction within the channel
of the pore.
In some embodiments the modification alters the properties of a channel
through the pore,
e.g. by introducing chemical functionality to the channel.
In some embodiments the monomer can be modified to ensure the presence of an
appropriate number of modification sites for reaction with the reactive group
of the
multifunctional molecule. In some embodiments the monomer can be modified to
introduce one or more modification sites. In some embodiments the monomer can
be
modified to remove or delete one or more modification sites. In some
embodiments the
monomer can be modified to delete one or more sites which would otherwise
react with the
reactive group of the multifunctional molecule and also to introduce one or
more
modification sites for reacting with the reactive group of the multifunctional
molecule; i.e.
the monomer can be modified to replace one or more modification sites with one
or more
different modification sites.
One or more modification sites can be introduced into the monomer by any
suitable
means. For example, in some embodiments the monomer is a polypeptide monomer
of an
oligomeric protein pore and one or more modification sites are introduced by
mutation of
the native amino acid sequence of the polypeptide monomer. In some embodiments
the
one or more modification sites are introduced by insertion of one or more
residues into the
native sequence. In some embodiments the one of mole modification sites are
introduced
by substitution of one or more residues of the native sequence.
For example, one or more cysteine residues may be introduced into the native
sequence of the monomer. Cysteine residues may be used e.g. when the
multifunctional
molecule comprises a sulfhydryl-reactive group as the reactive group, e.g.
when the
multifunctional molecule comprises a thiol or maleimide group as the reactive
group. One
or more cysteine residues may be introduced e.g. by substitution of one or
more non-
cysteine residues in the monomer. One or more cysteine residues may be
introduced e.g.
by insertion of one or more amino acids into the native amino acid sequence of
the
monomer.
For example, one or more amino-containing residues, e.g. lysine, may be
introduced into the native sequence of the monomer. Amino-containing residues
may be
used e.g. when the multifunctional molecule comprises a amino-reactive group
as the
reactive group, e.g. when the multifunctional molecule comprises an NHS-ester
group as
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
43
the reactive group. One or more amino-containing residues, e.g. lysines, may
be
introduced e.g. by substitution of one or more residues in the monomer. One or
more
amino-containing residues, e.g. lysines, may be introduced e.g. by insertion
of one or more
amino acids into the native amino acid sequence of the monomer.
Amino acid insertions, deletions and substitutions may be made in the native
amino
acid sequence of the monomer using techniques known in the art; e.g. those
described in
Sambrook et al., Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring
Harbor
Press, Plainsview, New York (2012); and Ausubel et al., Current Protocols in
Molecular
Biology (Supplement 114), John Wiley & Sons, New York (2016).
One or more reactive non-natural amino acids may be introduced into the native
sequence of the monomer. Reactive non-natural amino acids may be used e.g.
when the
multifunctional molecule comprises a reactive group for reacting with such non-
natural
amino acids, such as a click reagent. One or more non-natural amino acids may
be
introduced as described herein e.g. by including synthetic aminoacyl-tRNAs in
the IVIT
system used to express the mutant monomer or by expressing the mutant monomer
in a
suitable bacterial expression system (e.g. E. coil) comprising bacteria that
are auxotrophic
for specific amino acids in the presence of synthetic (i.e. non-naturally-
occurring)
analogues of those specific amino acids. They may also be produced by naked
ligation if
the mutant monomer is produced using partial peptide synthesis.
In some embodiments, the monomer is modified to comprise one or more
modification sites. In some embodiments the monomer is modified to comprise 1,
2, 3, 4
or 5 modification sites. In some embodiments the monomer is modified to
comprise
exactly one or two modification sites, such as exactly one modification site.
Accordingly, in some embodiments of the disclosed methods, the monomer is a
monomer of an oligomeric pore; and the multifunctional molecule reacts with a
reactive
functional group located on the monomer at a surface-exposed position when the
monomer
is oligomerised to form the pore. In some embodiments the surface-exposed
position is
located at the surface of the channel through the pore. In some embodiments
the surface-
exposed position is located on the exterior surface of the pore.
In some embodiments the multifunctional molecule reacts with a reactive
functional group located on the monomer at a position located at or near (e.g.
within 10
nm, e.g. within 5 nm, such as within 3 nm, e.g. within 2 nm, e.g. within 1 nm)
a
constriction of the channel through the pore when the monomer is oligomerised
to form the
pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
44
In some embodiments the monomer is a monomer of an aerolysin-like pore, such
as
aerolysin, lysenin, epsilon toxin (E-Toxin) type B, parasporin-2, LSL,
monalysin,
enterotoxin or haemolytic lectin, or a paralog or homolog thereof In some
embodiments
the monomer is a monomer of a leukocidin-like pore, such as gamma-hemolysin,
(HlgAB
or H1gCB), LukAB/HG, LukED, Panton-Valentine leukocidin (LukSF-PV/PVL), or
LukIVIF', or a paralog or homolog thereof In some embodiments the monomer is a
monomer of a hemolysin-like pore, such as VCC, CytK, NetB or alpha-hemolysin,
or a
paralog or homolog thereof In some embodiments the monomer is a monomer of an
AB
toxin-like pore, such as anthrax toxin protective antigen, or a paralog or
homolog thereof
In some embodiments the monomer is a monomer of the LukF subunit of gamma-
hemolysin (SEQ ID NO: 10) or a variant, paralog or homolog thereof. The
structure of
gamma-hemolysin has been deposited at the protein data bank (PDB) under
accession code
3B07. For example, in some embodiments the monomer is a variant having at
least 60%,
e.g. at least 70%, e.g. at least 80%, for example at least 85%, typically at
least 90%, such
as at least 95%, e.g. at least 96%, at least 97%, at least 98%, at least 99%
or at least 99.5%
or at least 99.9% sequence identity or sequence homology to SEQ ID NO: 10.
In some embodiments the monomer is modified at one or more of the positions
corresponding to K146, N144, T142, S140, A138, N136, N134, G132, S130, G128,
S126,
S124, D122, G120, T118, G116, T114, Q112, Q110, or E108 of SEQ ID NO: 10.
These
positions correspond to positions within the barrel of the gamma-hemolysin
pore (once
assembled) and may in some embodiments be modified in order to alter, e.g. to
improve,
interaction of an analyte such as a polynucleotide with the pore.
In some embodiments the monomer is modified at one or more of the positions
corresponding to K43, D44, K45, S46, Y47, D48 or K49 of SEQ ID NO: 10. These
positions correspond to positions in the cap domain of the gamma-hemolysin
pore (once
assembled) and may in some embodiments be modified in order to alter, e.g. to
improve,
interaction of an analyte such as a polynucleotide with the pore.
In some embodiments the monomer is modified at one or more of the positions
corresponding to Y145, 1143, E141, F139, T137, G135, L133, G131, L129, N127,
1125,
1123, G121, F119, Y117, L115, N113, V111 or F109 of SEQ ID NO: 10. These
positions
correspond to positions outside of the barrel of the gamma-hemolysin pore
(once
assembled) and may in some embodiments be modified in order to alter, e.g. to
improve
anchoring of the pore, e.g. by altering or improving interaction of the pore
with a
membrane.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
In some embodiments the monomer is modified at one or more of the positions
corresponding to A196, G197, R198, Q199, S200, S201, A202, Y203, W257, N258,
G259,
F260, Y261 or W262 of SEQ ID NO: 10. These positions correspond to positions
in the
rim domain of the gamma-hemolysin pore (once assembled) and may in some
5 embodiments be modified in order to alter, e.g. to improve anchoring of
the pore, e.g. by
altering or improving interaction of the pore with a membrane.
Those skilled in the art will appreciate that corresponding positions in other
monomers (e.g. other monomers disclosed herein) can be modified in accordance
with the
disclosed methods. Corresponding positions can be determined e.g. by sequence
alignment
10 and/or structure modelling.
In some embodiments the monomer is a monomer of the H1g2 subunit of gamma-
hemolysin (SEQ ID NO: 11) or a variant, paralog or homolog thereof. For
example, in
some embodiments the monomer is a variant having at least 60%, e.g. at least
70%, e.g. at
least 80%, for example at least 85%, typically at least 90%, such as at least
95%, e.g. at
15 least 96%, at least 97%, at least 98%, at least 99% or at least 99.5% or
at least 99.9%
sequence identity or sequence homology to SEQ ID NO: 11 In some embodiments
the
monomer is modified at one or more of the positions corresponding to N138,
S136, T134,
S132, N130, S128, S126, G124, S122, P121, Q118, S119, N116, G114, N112, G110,
K108, S106, D104, or S102 of SEQ ID NO: 11. These positions correspond to
positions
20 within the barrel of the gamma-hemolysin pore (once assembled) and may
in some
embodiments be modified in order to alter, e.g. to improve, interaction of an
analyte such
as a polynucleotide with the pore.
In some embodiments the monomer is modified at one or more of the positions
corresponding to K37, D38, K37, K39, K40, Y41, N42, K43 or D44 of SEQ ID NO:
11.
25 These positions correspond to positions in the cap domain of the gamma-
hemolysin pore
(once assembled) and may in some embodiments be modified in order to alter,
e.g. to
improve, interaction of an analyte such as a polynucleotide with the pore.
In some embodiments the monomer is modified at one or more of the positions
corresponding to Q139, Y137, 1135, K133, Y131, F129, G127, G125, 1123, P121,
S119,
30 F117, G115, 1113, Y111, L109, Q107, V105 or A103 of SEQ ID NO: 11. These
positions
correspond to positions outside of the barrel of the gamma-hemolysin pore
(once
assembled) and may in some embodiments be modified in order to alter, e.g. to
improve
anchoring of the pore, e.g. by altering or improving interaction of the pore
with a
membrane.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
46
In some embodiments the monomer is modified at one or more of the positions
corresponding to Q180, D181, P182, T183, G184, P185, T241, R242, H243 or R244
of
SEQ ID NO: 11. These positions correspond to positions in the rim domain of
the gamma-
hemolysin pore (once assembled) and may in some embodiments be modified in
order to
alter, e.g. to improve anchoring of the pore, e.g. by altering or improving
interaction of the
pore with a membrane.
Those skilled in the art will appreciate that corresponding positions in other
monomers (e.g. other monomers disclosed herein) can be modified in accordance
with the
disclosed methods. Corresponding positions can be determined e.g. by sequence
alignment
and/or structure modelling.
In some embodiments the monomer is a monomer of lysenin (SEQ ID NO: 12) or a
variant, paralog or homolog thereof The structure of lysenin has been
deposited at the
protein data bank (PDB) under accession code 5EC5. For example, in some
embodiments
the monomer is a variant having at least 60%, e.g. at least 70%, e.g. at least
80%, for
example at least 85%, typically at least 90%, such as at least 95%, e.g. at
least 96%, at least
97%, at least 98%, at least 99% or at least 99.5% or at least 99.9% sequence
identity or
sequence homology to SEQ ID NO: 12. In some embodiments the monomer is
modified at
one or more of the positions corresponding to D35, K37, T39, T41, G43, K45,
V47, S49,
T51, T53, T55, T57, S59, G61, T63, S65, G67, A69, 172, S74, E76, S78, S80,
S82, Q84,
S86, V88, M90, Q92, D94, Y96, S98, V100, E102, T104 or K106 of SEQ ID NO: 12.
In some embodiments the monomer is a monomer of Cytotoxin K from Bacillus
cereus (CytK) (SEQ ID NO: 13) or a variant, paralog or homolog thereof For
example, in
some embodiments the monomer is a variant having at least 60%, e.g. at least
70%, e.g. at
least 80%, for example at least 85%, typically at least 90%, such as at least
95%, e.g. at
least 96%, at least 97%, at least 98%, at least 99% or at least 99.5% or at
least 99.9 A
sequence identity or sequence homology to SEQ ID NO: 13. In some embodiments
the
monomer is modified at one or more of the positions corresponding to E113,
T115, T117,
S119, S121, Q123, G125, S127, K129, S131, T133, G136, S138, E140, G142, T144,
Q146, T148, S150, S152, S154, or K156 of SEQ ID NO: 13.
In some embodiments the monomer is a monomer of aerolysin (SEQ ID NO: 14) or
a variant, paralog or homolog thereof The structure of aerolysin has been
deposited at the
protein data bank (PDB) under accession code 5JZT. For example, in some
embodiments
the monomer is a variant haying at least 60%, e.g. at least 70%, e.g. at least
80%, for
example at least 85%, typically at least 90%, such as at least 95%, e.g. at
least 96%, at least
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
47
97%, at least 98%, at least 99% or at least 99.5% or at least 99.9% sequence
identity or
sequence homology to SEQ ID NO: 14. In some embodiments the monomer is
modified at
one or more of the positions corresponding to G214, D216, T218, R220, D222,
A224,
N226, S228, T230, T232, G234, S236, K238, T240, T242, K244, K246, P248, V250,
E252, E254, S256, E258, A260, N262, S264, A266, Q268, G270, S272, S274, S276,
S278,
S280, R282 or T284 of SEQ ID NO: 14.
In some embodiments the monomer is a monomer of NetB (SEQ ID NO: 15) or a
variant, paralog or homolog thereof. The structure of NetB has been deposited
at the
protein data bank (PDB) under accession code 4H56. For example, in some
embodiments
the monomer is a variant having at least 60%, e.g. at least 70%, e.g. at least
80%, for
example at least 85%, typically at least 90%, such as at least 95%, e.g. at
least 96%, at least
97%, at least 98%, at least 99% or at least 99.5% or at least 99.9% sequence
identity or
sequence homology to SEQ ID NO: 15. In some embodiments the monomer is
modified at
one or more of the positions corresponding to 1112, K114, D116, S118, S120,
G122, S124,
G126, N128, S130, E132, T135, G137, G139, N141, S143, N145, Q147, T149, E151,
S153 or P155 of SEQ ID NO: 15.
In some embodiments the monomer is a monomer of alpha-hemolysin (SEQ ID
NO: 16) or a variant, paralog or homolog thereof. The structure of alpha-
hemolysin has
been deposited at the protein data bank (PDB) under accession code 7AHL. For
example,
in some embodiments the monomer is a variant having at least 60%, e.g. at
least 70%, e.g.
at least 80%, for example at least 85%, typically at least 90%, such as at
least 95%, e.g. at
least 96%, at least 97%, at least 98%, at least 99% or at least 99.5% or at
least 99.9%
sequence identity or sequence homology to SEQ ID NO: 16. In some embodiments
the
monomer is modified at one or more of the positions corresponding to E111,
M113, T115,
T117, G119, N121, N123, T125, D127, D128, T129, G130, K131, G133, L135, G137,
N139, S141, G143, T145 or K147 of SEQ ID NO: 16.
In some embodiments the monomer is a monomer of VCC (SEQ ID NO: 17) or a
variant, paralog or homolog thereof The structure of VCC has been deposited at
the
protein data bank (PDB) under accession code 3044. For example, in some
embodiments
the monomer is a variant having at least 60%, e.g. at least 70%, e.g. at least
80%, for
example at least 85%, typically at least 90%, such as at least 95%, e.g. at
least 96%, at least
97%, at least 98%, at least 99% or at least 99.5% or at least 99.9% sequence
identity or
sequence homology to SEQ ID NO: 17. In some embodiments the monomer is
modified at
one or more of the positions corresponding to E281, K283, V285, G287, E289,
G291,
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
48
T293, G295, E297, S299, K304, K306, E308, R310, S312, T314, S316, W318, T320
or
N322 of SEQ ID NO: 17.
In some embodiments the monomer is a monomer of Anthrax PA (SEQ ID NO: 18)
or a variant, paralog or homolog thereof. The structure of Anthrax protective
antigen has
been deposited at the protein data bank (PDB) under accession code 3J9C. For
example,
in some embodiments the monomer is a variant having at least 60%, e.g. at
least 70%, e.g.
at least 80%, for example at least 85%, typically at least 90%, such as at
least 95%, e.g. at
least 96%, at least 97%, at least 98%, at least 99% or at least 99.5% or at
least 99.9%
sequence identity or sequence homology to SEQ ID NO: 18. In some embodiments
the
monomer is modified at one or more of the positions corresponding to D276,
S278, Q280,
T282, S284, T286, T288, S290, N292, S294, S296, T298, T300, E302, H304, N306,
E308,
H310, S312, D315, G317, S319, S321, G323, S325, S327, S329, T331, A333, D335,
S337,
S339, A341, E343, T345, A347, T349 or G351 of SEQ NO: 18.
In some embodiments the monomer is a monomer of e-Toxin (SEQ ID NO: 19) or
a variant, paralog or homolog thereof The structure of E-Toxin type B has been
deposited
at the protein data bank (PDB) under accession code 6RB9. For example, in some
embodiments the monomer is a variant having at least 60%, e.g. at least 70%,
e.g. at least
80%, for example at least 85%, typically at least 90%, such as at least 95%,
e.g. at least
96%, at least 97%, at least 98%, at least 99% or at least 99.5% or at least
99.9% sequence
identity or sequence homology to SEQ ID NO: 19. In some embodiments the
monomer is
modified at one or more of the positions corresponding to S90, T92, K94, T96,
T98, T100,
T102, T104, T106, G108, S110, Q112, 114, K116, T118, E123, G125, S127, S131,
S133,
A135, T137, T139, T141, S143, E145, T147 or P151 of SEQ ID NO: 19.
In some embodiments the monomer is a monomer of Fragaceatoxin C (FraC) from
Actinia.fragacear (SEQ ID NO: 21) or a variant, paralog or homolog thereof The
structure
of FraC from Actinia .fragacea has been deposited at the protein data bank
(PDB) under
accession code 3W9P. For example, in some embodiments the monomer is a variant
having at least 600A, e.g. at least 70%, e.g. at least 80%, for example at
least 85%, typically
at least 90%, such as at least 95%, e.g. at least 96%, at least 97%, at least
98%, at least
99% or at least 99.5% or at least 99.9% sequence identity or sequence homology
to SEQ
ID NO: 21.
As mentioned above, in some embodiments the monomer is a monomer of PlyA
(SEQ ID NO: 22) or PlyB (SEQ ID NO: 23) from Pleurotus ostreatus or a variant,
paralog
or homolog thereof. The structure of PlyAB has been deposited at the protein
data bank
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
49
(PDB) under accession code 4V2T. For example, in some embodiments the monomer
is a
variant having at least 60%, e.g. at least 70%, e.g. at least 80%, for example
at least 85%,
typically at least 90%, such as at least 95%, e.g. at least 96%, at least 97%,
at least 98 43, at
least 99% or at least 99.5% or at least 99.9% sequence identity or sequence
homology to
SEQ ID NO: 22. In some embodiments the monomer is a variant having at least
60%, e.g.
at least 70%, e.g. at least 80%, for example at least 85%, typically at least
90%, such as at
least 95%, e.g. at least 96%, at least 97%, at least 98%, at least 99% or at
least 99.5% or at
least 99.9% sequence identity or sequence homology to SEQ ID NO: 23.
In some embodiments the monomer is a monomer of a leukocidin pore such as
LukF/H1gB (PDB code 1LKF), the S component of Panton-Valentine leukocidin (PDB
code 1T5R) and the F component of Panton-Valentine leukocidin (PDB code 1PVL);
an
AB toxin such as Anthrax toxin protective antigen; an Aerolysin-like pore such
as
Parasporin-2 (PDB code 2ZTB), Clostridium perfringens epsilon toxin (PDB code
6RB9),
Hemolytic Lectin (PDB code 1W3A); and pesticidal proteins of Bacillus
thuringiensis
such as Cry and Cyt proteins.
In some embodiments the monomer is a monomer of one of the six known
leukocidins of S. aureus: gamma hemolysins HlgAB and H1gCB; LukAB/HG; LukED;
Panton-Valentine leukoci din (LukSF-PV/PVL) and LukMF'.
As mentioned above, in some embodiments the monomer is a monomer of an
enzyme, such as an oligomeric enzyme. In one embodiment the monomer is a
monomer of
an oligomeric helicase. These and other enzymes suitable for modification in
accordance
with the invention can, in some embodiments, be motor proteins as described in
more
detail herein.
Further method steps
Those skilled in the art will appreciate that additional steps can be included
in the
disclosed methods
For example, in some embodiments, the method comprises, prior to step (a), the
steps of (i) expressing the monomer in a cellular expression system or a cell-
free
expression system; and (ii) isolating and/or purifying the monomer. The
monomer may be
expressed in any suitable expression system. Suitable expression systems
include bacterial
expression systems such as those which use strains of E. col/ to express the
monomer from
a suitable vector. Other suitable expression systems include expression in
insect cells or
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
yeast. Expression methods are well known to those skilled in the art as
discussed in
reference texts such as Sambrook et al., Molecular Cloning: A Laboratory
Manual, 4th ed.,
Cold Spring Harbor Press, Plainsview, New York (2012); and Ausubel et al.,
Current
Protocols in Molecular Biology (Supplement 114), John Wiley & Sons, New York
(2016).
5 Cell-free expression systems (e.g. in vitro transcription/translation,
IVTT) systems are well
known in the art and are commercially available from suppliers such as
Promega. IVTT
has been used to prepare monomers of oligomeric protein pores.
In some embodiments, step (d) of the method can further comprise the step of
removing unmodified monomer(s) and/or unreacted multifunctional molecule(s),
if
10 present, from the support. Any suitable technique can be used. For
example, the bound
modified monomer can be washed using an aqueous or non-aqueous solved, e.g. a
wash
buffer, in order to remove unbound or weakly bound impurities from the
support.
Methods of producing oligomers
In some embodiments the methods of the present disclosure are used to produce
oligomers.
In some embodiments the method disclosed herein further comprises the step of:
oligomerising the chemically modified monomer to form a chemically modified
oligomer.
In some embodiments the monomer is a protein monomer and step (f) comprises
forming
an oligomeric protein. The monomer may be a monomer of an oligomeric protein
nanopore and step (f) may comprise forming an oligomeric protein nanopore.
In some embodiments step (f) comprises oligomerising two or more chemically
modified monomers to form a homooligomer. In such cases the two or more
monomers
are the same; i.e. the two or monomers are of the same monomeric structure and
have been
chemically modified in the same way. In some embodiments the two or more
monomers
are protein monomers and said homooligomer is a homooligomeric protein pore.
In some embodiments step (f) comprises oligomerising one or more chemically
modified monomers with one or more unmodified or differently-modified monomers
to
form a heterooligomer. In some embodiments the two or more monomers are
protein
monomers and said homooligomer is a homooligomeric protein pore.
The one or more different monomers can be unmodified monomers which are of
the same type (e.g. the same sequence) as the chemically modified monomer but
have not
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
51
been modified. The one or more different monomers can be unmodified monomers
which
are of a different type (e.g. are of a different sequence) as the chemically
modified
monomer and have not been modified. The one or more different monomers can be
chemically modified monomers which are of the same type (e.g. the same
sequence) as the
first chemically modified monomer but have been modified with a different
chemical
modification. The one or more different monomers can be modified monomers
which are
of a different type (e.g. are of a different sequence) as the first chemically
modified
monomer and have not been modified.
The one or more first monomers can comprise two or more, e.g. 2, 3, 4, 5, 6,
7, 8, 9
or 10 first monomers. The one or more second monomers can comprise two or
more, e.g.
2, 3, 4, 5, 6, 7, 8, 9 or 10 second monomers.
Thus the disclosure also provides a method of producing a homooligomeric
protein;
comprising
i) producing a plurality of chemically modified protein monomers in a
method as
described herein; and
ii) oligomeri sing two or more of the chemically modified
protein monomers
obtained in step (i) to form a homooligomeric protein.
In some embodiments the homooligomeric protein is a homooligomeric protein
pore, such
as a protein pore described herein.
The disclosure also provides a method of producing a heterooligomeric protein;
comprising
i) producing one or more chemically modified first protein monomers in a
method
as described herein; and
ii) producing one or more chemically modified second protein monomers in a
method as described herein; and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein.
In some embodiments the heterooligomeric protein is a heterooligomeric protein
pore.
The disclosure further provides an oligomeric protein; comprising
i) producing one or more chemically modified first protein
monomers in a method
as described herein; and
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
52
ii) providing one or more unmodified second protein monomers; and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein.
In some embodiments the heterooligomeric protein is a heterooligomeric protein
pore.
Monomers and oligomers provided in the present disclosure
The present disclosure also provides the products of the claimed methods.
Thus, the disclosure provides a chemically modified monomer obtainable by
carrying out a method as described herein. In some embodiments the monomer is
a
monomer as described herein. In some embodiments the monomer is chemically
modified
by one or more modifications as described herein. In some embodiments the
monomer is a
monomer of a protein nanopore as described herein.
The disclosure also provides a chemically modified oligomer obtainable by
carrying out a method as described herein. In some embodiments the oligomer is
an
oligomer comprising two or more monomers as described herein. In some
embodiments
the oligomer is chemically modified by one or more modifications as described
herein. In
some embodiments the oligomer is an oligomeric protein nanopore as described
herein.
The disclosure also provides populations of chemically modified monomers. The
populations of such monomers are characterised inter alia by their
homogeneity. In some
embodiments the disclosure thus provides a homogeneous population comprising a
plurality of chemically modified monomers; wherein at least 95% of the
monomers in the
population are chemically modified with a chemical modifying group. In some
embodiments at least 96%, such as at least 97%, e.g. at least 98%, for example
at least
99%, e.g. at least 99.5% such as at least 99.9% or at least 99.99% of the
monomers in the
population are chemically modified with a chemical modifying group. In some
embodiments 100% of the monomers in the population are modified with a
chemical
modifying group. As explained above, previously known methods are typically
not
capable of providing such homogeneous populations as modification chemistry is
typically
not 100% efficient, and modified monomers typically cannot be readily
separated or
purified from impurities. In some embodiments the population is a population
of
monomers of a protein pore, e.g. a protein pore as described herein.
The disclosure also provides populations of chemically modified oligomers,
such as
oligomeric protein pores. The populations of such oligomers are characterised
inter alia by
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
53
their homogeneity. In some embodiments the disclosure thus provides a
homogeneous
population comprising a plurality of chemically modified oligomers; wherein at
least 95%
of the oligomers in the population are chemically modified with a chemical
modifying
group. In some embodiments at least 96%, such as at least 97%, e.g. at least
98%, for
example at least 99 A, e.g. at least 99.5% such as at least 99.9% or at least
99.99% of the
oligomers in the population are chemically modified with a chemical modifying
group. In
some embodiments 100% of the oligomers in the population are modified with a
chemical
modifying group. As explained above, previously known methods are typically
not
capable of providing such homogeneous populations as modification chemistry is
typically
not 100% efficient, and modified monomers typically cannot be readily
separated or
purified from impurities in order to provide highly homogeneous populations of
oligomers.
In some embodiments the population is a population of oligomeric protein
pores, e.g.
protein pores as described herein.
Methods of using the monomers and oligomers provided herein
The chemically modified monomers and oligomers provided herein are useful in a
variety of ways.
Chemically modified proteins find general utility in the fields of
biotechnology and
medicine. For example, modified enzymes can have enhanced or altered
functionality
compared to their native counterparts.
In some embodiments the monomers are monomers of a nanopore; and/or the
oligomer is an oligomeric nanopore. Such nanopores can be used in the
detection and
characterisation of analytes such as polynucleotides.
Accordingly, provided herein is a method of characterising an analyte,
comprising:
i) producing a chemically modified oligomeric pore in a method as described
herein; and
ii) taking one or more measurements as the analyte moves with respect to
the pore,
wherein the one or more measurements are indicative of one or more
characteristics of the analyte, and thereby characterising the analyte as it
moves
with respect to the pore.
Also provided is a method of characterising an analyte, comprising:
i) providing a chemically modified oligomeric pore as
described herein, and
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
54
ii) taking one or more measurements as the analyte moves
with respect to the pore,
wherein the one or more measurements are indicative of one or more
characteristics of the analyte, and thereby characterising the analyte as it
moves
with respect to the pore.
In some embodiments the analyte is a polynucleotide as described in more
detail herein.
Characterising analytes
The disclosed characterisation methods may be carried out using any apparatus
that
is suitable for investigating a membrane/pore system in which a pore is
inserted into a
membrane. Membranes are described in more detail herein.
The characterisation method may be carried out using any apparatus that is
suitable
for transmembrane pore sensing. For example, the apparatus may comprise a
chamber
comprising an aqueous solution and a barrier that separates the chamber into
two sections.
The barrier may have an aperture in which a membrane containing a
transmembrane pore,
e.g. a chemically modified transmembrane pore as described herein, is formed
Transmembrane pores are described herein.
The characterisation methods may be carried out using the apparatus described
in
WO 2008/102120, WO 2010/122293 or WO 00/28312.
The binding of a molecule (e.g. a target polynucleotide) in the channel of the
pore
will have an effect on the open-channel ion flow through the pore, which is
the essence of
-molecular sensing" of pore channels. The characterisation methods may thus
involve
measuring the ion current flow through the pore, typically by measurement of a
current
(for example, WO 2000/28312 and D. Stoddart et al., Proc. Natl. Acad. Sci.,
2010, 106,
7702-7 or WO 2009/077734). Alternatively, the ion flow through the pore may be
measured optically, such as disclosed by Heron et al: J. Am. Chem. Soc. 9 Vol.
131, No. 5,
2009. Therefore the apparatus may also comprise an electrical circuit capable
of applying a
potential and measuring an electrical signal across the membrane and pore. The
characterisation methods may be carried out using a patch clamp or a voltage
clamp. The
characterisation methods preferably involve the use of a voltage clamp.
For the accurate determination of individual nucleotides, the reduction in ion
flow
through the channel is advantageously correlated to the size of the individual
nucleotide
passing through the constriction (or "reading head"). Accordingly,
modification of the
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
reading head of a nanopore in accordance with the claimed methods can be
useful in tuning
this interaction and improving the characterisation signal that can be
obtained.
The characterisation methods may be carried out on a silicon-based array of
wells
where each array comprises 128, 256, 512, 1024, 2000, 3000, 4000, 6000, 10000,
12000,
5 15000 or more wells.
The characterisation methods may involve the measuring of a current flowing
through the pore. The method is typically carried out with a voltage applied
across the
membrane and pore. The voltage used is typically from +2 V to -2 V, typically -
400 mV to
+400mV. The voltage used is preferably in a range having a lower limit
selected
10 from -400 mV, -300 mV, -200 mV, -150 mV, -100 mV, -50 mV, -20mV and 0 mV
and an
upper limit independently selected from +10 mV, + 20 mV, +50 mV, +100 mV, +150
mV,
+200 mV, +300 mV and +400 mV. The voltage used is more preferably in the range
100
mV to 240mV and most preferably in the range of 120 mV to 220 mV. It is
possible to
increase discrimination between different nucleotides by a pore by using an
increased
15 applied potential.
The characterisation methods are typically carried out in the presence of any
charge
carriers, such as metal salts, for example alkali metal salts, halide salts,
for example
chloride salts, such as alkali metal chloride salt. Charge carriers may
include ionic liquids
or organic salts, for example tetramethyl ammonium chloride, trimethylphenyl
ammonium
20 chloride, phenyltrimethyl ammonium chloride, or 1-ethyl-3-methyl
imidazolium chloride.
In the exemplary apparatus discussed above, the salt is present in the aqueous
solution in
the chamber. Potassium chloride (KC1), sodium chloride (NaCl) or caesium
chloride
(CsC1) is typically used. KC1 is preferred. The salt may be an alkaline earth
metal salt
such as calcium chloride (CaCl2). The salt concentration may be at saturation.
The salt
25 concentration may be 3M or lower and is typically from 0.1 to 2.5 M,
from 0.3 to 1.9 M,
from 0.5 to 1.8M, from 0.7 to 1.7M, from 0.9 to 1.6M or from 1 M to 1.4M. The
salt
concentration is preferably from 150 mM to 1 M. The characterisation method is
preferably carried out using a salt concentration of at least 0.3 M, such as
at least 0.4 M, at
least 0.5 M, at least 0.6 M, at least 0.8 M, at least 1.0 M, at least 1.5 M,
at least 2.0 M, at
30 least 2.5 M or at least 3.0 M. High salt concentrations provide a high
signal to noise ratio
and allow for currents indicative of binding/no binding to be identified
against the
background of normal current fluctuations.
The characterisation methods are typically carried out in the presence of a
buffer.
In the exemplary apparatus discussed above, the buffer is present in the
aqueous solution in
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
56
the chamber. Any suitable buffer may be used. Typically, the buffer is HEPES.
Another
suitable buffer is Tris-HC1 buffer. The methods are typically carried out at a
pH of from
4.0 to 12.0, from 4.5 to 10.0, from 5.0 to 9.0, from 5.5 to 8.8, from 6.0 to
8.7 or from 7.0 to
8.8 or 7.5 to 8.5. The pH used is preferably about 7.5.
The characterisation methods may be carried out at from 0 C to 100 C, from
15
C to 95 C, from 16 C to 90 C, from 17 C to 85 C, from 18 C to 80 C, 19
C to 70 C,
or from 20 C to 60 C. The characterisation methods are typically carried out
at room
temperature. The characterisation methods are optionally carried out at a
temperature that
supports enzyme function, such as about 37 C.
Membrane
In embodiments of the invention which comprise the use of a transmembrane
nanoporc, the transmcmbrane nanoporc is typically present in a membrane. Any
suitable
membrane may be used in the system.
The membrane is preferably an amphiphilic layer. An amphiphilic layer is a
layer
formed from amphiphilic molecules, such as phospholipids, which have both
hydrophilic
and lipophilic properties. The amphiphilic molecules may be synthetic or
naturally
occurring. Non-naturally occurring amphiphiles and amphiphiles which form a
monolayer
are known in the art and include, for example, block copolymers (Gonzalez-
Perez et at.,
Langmuir, 2009, 25, 10447-10450). Block copolymers are polymeric materials in
which
two or more monomer sub-units that are polymerized together to create a single
polymer
chain. Block copolymers typically have properties that are contributed by each
monomer
sub-unit. However, a block copolymer may have unique properties that polymers
formed
from the individual sub-units do not possess. Block copolymers can be
engineered such
that one of the monomer sub-units is hydrophobic (i.e. lipophilic), whilst the
other sub-
unit(s) are hydrophilic whilst in aqueous media. In this case, the block
copolymer may
possess amphiphilic properties and may form a structure that mimics a
biological
membrane. The block copolymer may be a diblock (consisting of two monomer sub-
units), but may also be constructed from more than two monomer sub-units to
form more
complex arrangements that behave as amphipiles. The copolymer may be a
triblock,
tetrablock or pentablock copolymer. The membrane is preferably a triblock
copolymer
membrane.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
57
Archaebacterial bipolar tetraether lipids are naturally occurring lipids that
are
constructed such that the lipid forms a monolayer membrane. These lipids are
generally
found in extremophiles that survive in harsh biological environments,
thermophiles,
halophiles and acidophiles. Their stability is believed to derive from the
fused nature of
the final bilayer. It is straightforward to construct block copolymer
materials that mimic
these biological entities by creating a triblock polymer that has the general
motif
hydrophilic-hydrophobic-hydrophilic. This material may form monomeric
membranes that
behave similarly to lipid bilayers and encompass a range of phase behaviours
from vesicles
through to laminar membranes. Membranes formed from these triblock copolymers
hold
several advantages over biological lipid membranes. Because the triblock
copolymer is
synthesised, the exact construction can be carefully controlled to provide the
correct chain
lengths and properties required to form membranes and to interact with pores
and other
proteins.
Block copolymers may also be constructed from sub-units that arc not classed
as
lipid sub-materials; for example a hydrophobic polymer may be made from
siloxane or
other non-hydrocarbon based monomers The hydrophilic sub-section of block
copolymer
can also possess low protein binding properties, which allows the creation of
a membrane
that is highly resistant when exposed to raw biological samples. This head
group unit may
also be derived from non-classical lipid head-groups.
Triblock copolymer membranes also have increased mechanical and environmental
stability compared with biological lipid membranes, for example a much higher
operational temperature or pH range. The synthetic nature of the block
copolymers
provides a platform to customise polymer based membranes for a wide range of
applications.
In some embodiments, the membrane is one of the membranes disclosed in
International Application No. W02014/064443 or W02014/064444.
The amphiphilic molecules may be chemically-modified or functionalised to
facilitate coupling of the polynucleotide. The amphiphilic layer may be a
monolayer or a
bilayer. The amphiphilic layer is typically planar. The amphiphilic layer may
be curved.
The amphiphilic layer may be supported.
Amphiphilic membranes are typically naturally mobile, essentially acting as
two
dimensional fluids with lipid diffusion rates of approximately 10-8 cm s-1.
This means that
the pore and coupled polynucleotide can typically move within an amphiphilic
membrane.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
58
The membrane may be a lipid bilayer. Lipid bilayers are models of cell
membranes
and serve as excellent platforms for a range of experimental studies. For
example, lipid
bilayers can be used for in vitro investigation of membrane proteins by single-
channel
recording. Alternatively, lipid bilayers can be used as biosensors to detect
the presence of
a range of substances. The lipid bilayer may be any lipid bilayer. Suitable
lipid bilayers
include, but are not limited to, a planar lipid bilayer, a supported bilayer
or a liposome.
The lipid bilayer is preferably a planar lipid bilayer. Suitable lipid
bilayers are disclosed in
WO 2008/102121, WO 2009/077734 and WO 2006/100484.
Methods for forming lipid bilayers are known in the art. Lipid bilayers are
commonly formed by the method of Montal and Mueller (Proc. Natl. Acad. Sci.
USA.,
1972; 69: 3561-3566), in which a lipid monolayer is carried on aqueous
solution/air
interface past either side of an aperture which is perpendicular to that
interface. The lipid
is normally added to the surface of an aqueous electrolyte solution by first
dissolving it in
an organic solvent and then allowing a drop of the solvent to evaporate on the
surface of
the aqueous solution on either side of the aperture. Once the organic solvent
has
evaporated, the solution/air interfaces on either side of the aperture are
physically moved
up and down past the aperture until a bilayer is formed. Planar lipid bilayers
may be
formed across an aperture in a membrane or across an opening into a recess.
The method of Montal & Mueller is popular because it is a cost-effective and
relatively straightforward method of forming good quality lipid bilayers that
are suitable
for protein pore insertion. Other common methods of bilayer formation include
tip-
dipping, painting bilayers and patch-clamping of liposome bilayers.
Tip-dipping bilayer formation entails touching the aperture surface (for
example, a
pipette tip) onto the surface of a test solution that is carrying a monolayer
of lipid. Again,
the lipid monolayer is first generated at the solution/air interface by
allowing a drop of
lipid dissolved in organic solvent to evaporate at the solution surface. The
bilayer is then
formed by the Langmuir-Schaefer process and requires mechanical automation to
move the
aperture relative to the solution surface.
For painted bilayers, a drop of lipid dissolved in organic solvent is applied
directly
to the aperture, which is submerged in an aqueous test solution. The lipid
solution is
spread thinly over the aperture using a paintbrush or an equivalent. Thinning
of the solvent
results in formation of a lipid bilayer. However, complete removal of the
solvent from the
bilayer is difficult and consequently the bilayer formed by this method is
less stable and
more prone to noise during electrochemical measurement.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
59
Patch-clamping is commonly used in the study of biological cell membranes. The
cell membrane is clamped to the end of a pipette by suction and a patch of the
membrane
becomes attached over the aperture. The method has been adapted for producing
lipid
bilayers by clamping liposomes which then burst to leave a lipid bilayer
sealing over the
aperture of the pipette. The method requires stable, giant and unilamellar
liposomes and
the fabrication of small apertures in materials having a glass surface.
Liposomes can be formed by sonication, extrusion or the Mozafari method (Colas
et al. (2007) Micron 38:841-847).
In some embodiments, a lipid bilayer is formed as described in International
Application No. WO 2009/077734. Advantageously in this method, the lipid
bilayer is
formed from dried lipids. In a most preferred embodiment, the lipid bilayer is
formed
across an opening as described in W02009/077734.
A lipid bilayer is formed from two opposing layers of lipids. The two layers
of
lipids are arranged such that their hydrophobic tail groups face towards each
other to form
a hydrophobic interior. The hydrophilic head groups of the lipids face
outwards towards
the aqueous environment on each side of the bilayer. The bilayer may be
present in a
number of lipid phases including, but not limited to, the liquid disordered
phase (fluid
lamellar), liquid ordered phase, solid ordered phase (lamellar gel phase,
interdigitated gel
phase) and planar bilayer crystals (lamellar sub-gel phase, lamellar
crystalline phase).
Any lipid composition that forms a lipid bilayer may be used. The lipid
composition is chosen such that a lipid bilayer having the required
properties, such surface
charge, ability to support membrane proteins, packing density or mechanical
properties, is
formed. The lipid composition can comprise one or more different lipids. For
instance,
the lipid composition can contain up to 100 lipids. The lipid composition
preferably
contains 1 to 10 lipids. The lipid composition may comprise naturally-
occurring lipids
and/or artificial lipids.
The lipids typically comprise a head group, an interfacial moiety and two
hydrophobic tail groups which may be the same or different. Suitable head
groups include,
but are not limited to, neutral head groups, such as diacylglycerides (DG) and
ceramides
(CM); zwitterionic head groups, such as phosphatidylcholine (PC),
phosphatidylethanolamine (PE) and sphingomyelin (SM); negatively charged head
groups,
such as phosphatidylglycerol (PG); phosphatidylserine (PS),
phosphatidylinositol (PI),
phosphatic acid (PA) and cardiolipin (CA); and positively charged headgroups,
such as
trimethylammonium-Propane (TAP). Suitable interfacial moieties include, but
are not
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
limited to, naturally-occurring interfacial moieties, such as glycerol-based
or ceramide-
based moieties. Suitable hydrophobic tail groups include, but are not limited
to, saturated
hydrocarbon chains, such as lauric acid (n-Dodecanolic acid), myristic acid (n-
Tetradecononic acid), palmitic acid (n-Hexadecanoic acid), stearic acid (n-
Octadecanoic)
5 and arachidic (n-Eicosanoic); unsaturated hydrocarbon chains, such as
oleic acid (cis-9-
Octadecanoic); and branched hydrocarbon chains, such as phytanoyl. The length
of the
chain and the position and number of the double bonds in the unsaturated
hydrocarbon
chains can vary. The length of the chains and the position and number of the
branches,
such as methyl groups, in the branched hydrocarbon chains can vary. The
hydrophobic tail
10 groups can be linked to the interfacial moiety as an ether or an ester.
The lipids may be
mycolic acid.
The lipids can also be chemically-modified. The head group or the tail group
of the
lipids may be chemically-modified. Suitable lipids whose head groups have been
chemically-modified include, but arc not limited to, PEG-modified lipids, such
as 1,2-
15 Diacyl-sn-Glycero-3-Phosphoethanolamine-N 4Methoxy(Polyethylene glycol)-
2000];
functionalised PEG Lipids, such as 1,2-Di stearoyl-sn-Gl ycero-3
Phosphoethanolamine-N-
[Biotinyl(Polyethylene Glycol)2000]; and lipids modified for conjugation, such
as 1,2-
Di ol eoyl-sn-Glycero-3-Phosphoethanolamine-N-(succinyl) and 1,2-Dipalmitoyl-
sn-
Glycero-3-Phosphoethanolamine-N-(Biotiny1). Suitable lipids whose tail groups
have
20 been chemically-modified include, but are not limited to, polymerisable
lipids, such as 1,2-
bis(10,12-iiicosadiynoy1)-sn-Glycei o-3-Phosphocholine, fluorinated lipids,
such as 1-
Palmitoy1-2-(16-Fluoropalmitoy1)-sn-Glycero-3-Phosphocholine; deuterated
lipids, such as
1,2-Dipalmitoyl-D62-sn-Glycero-3-Phosphocholine; and ether linked lipids, such
as 1,2-
Di-O-phytanyl-sn-Glycero-3-Phosphocholine. The lipids may be chemically-
modified or
25 functionalised to facilitate coupling of the polynucleotide.
The amphiphilic layer, for example the lipid composition, typically comprises
one
or more additives that will affect the properties of the layer. Suitable
additives include, but
are not limited to, fatty acids, such as palmitic acid, myristic acid and
oleic acid; fatty
alcohols, such as palmitic alcohol, myristic alcohol and oleic alcohol;
sterols, such as
30 cholesterol, ergosterol, lanosterol, sitosterol and stigmasterol;
lysophospholipids, such as
1-Acy1-2-Hydroxy-sn- Glycero-3-Phosphocholine; and ceramides.
In another embodiment, the membrane comprises a solid state layer. Solid state
layers can be formed from both organic and inorganic materials including, but
not limited
to, microelectronic materials, insulating materials such as Si3N4, A1203, and
SiO, organic
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
61
and inorganic polymers such as polyamide, plastics such as Teflon or
elastomers such as
two-component addition-cure silicone rubber, and glasses. The solid state
layer may be
formed from graphene. Suitable graphene layers are disclosed in WO
2009/035647. If the
membrane comprises a solid state layer, the pore is typically present in an
amphiphilic
membrane or layer contained within the solid state layer, for instance within
a hole, well,
gap, channel, trench or slit within the solid state layer. The skilled person
can prepare
suitable solid state/amphiphilic hybrid systems. Suitable systems are
disclosed in WO
2009/020682 and WO 2012/005857. Any of the amphiphilic membranes or layers
discussed above may be used.
Motor Proteins
In disclosed methods which comprise characterising an analyte as the analyte
moves with respect to a nanoporc, a motor protein can be used to control said
movement.
In addition, the methods of chemically modifying a monomer as disclosed herein
are also
applicable to the chemical modification of such a motor protein (or a monomer
thereof)
whether for use in a disclosed method or in any other method.
As used herein, a motor protein is any protein that is capable of binding to a
polynucleotide and controlling its movement with respect to a nanopore, e.g.
through the
pore.
In one embodiment, a motor protein is or is derived from a polynucleotide
handling
enzyme. A polynucleotide handling enzyme is a polypeptide that is capable of
interacting
with and modifying at least one property of a polynucleotide. The enzyme may
modify the
polynucleotide by cleaving it to form individual nucleotides or shorter chains
of
nucleotides, such as di- or trinucleotides. The enzyme may modify the
polynucleotide by
orienting it or moving it to a specific position.
In one embodiment, the motor protein is derived from a member of any of the
Enzyme Classification (EC) groups 3.1.11, 3.1.13, 3.1.14, 3.1.15, 3.1.16,
3.1.21, 3.1.22,
3.1.25, 3.1.26, 3.1.27, 3.1.30 and 3.1.31.
Typically, the motor protein is a helicase, a polymerase, an exonuclease, a
topoisomerase, or a variant thereof.
In some embodiments, a motor protein can be modified to prevent the motor
protein
disengaging from a polynucleotide or polynucleotide adapter. For example,
modification
of a motor protein in order to prevent it from disengaging from a spacer on a
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
62
polynucleotide adapter is discussed in WO 2014/013260, which is hereby
incorporated by
reference in its entirety, and with particular reference to passages
describing the
modification of motor proteins such as helicases in order to prevent them from
disengaging
from polynucleotide strands. For example, a motor protein can be modified by
treating
with tetramethylazodicarboxamide.
For example, the motor protein may have a polynucleotide-unbinding opening
e.g.
a cavity, cleft or void through which a polynucleotide strand may pass when
the motor
protein disengages from the strand. The motor protein may be modified by
closing the
polynucleotide-unbinding opening. Closing the polynucleotide-unbinding opening
may
therefore prevent the motor protein from disengaging. For example, the motor
protein may
be modified by covalently closing the polynucleotide-unbinding opening. In
some
embodiments, a preferred motor protein for addressing in this way is a
helicase.
In one embodiment, a motor protein is an exonuclease. Suitable enzymes
include,
but arc not limited to, exonuclease I from E. coil (SEQ ID NO: 1), exonuclease
III enzyme
from E. coil (SEQ ID NO: 2), RecJ from T thernzophilus (SEQ ID NO: 3) and
bacteriophage lambda exonuclease (SEQ ID NO. 4), TatD exonuclease and variants
thereof. Three subunits comprising the sequence shown in SEQ ID NO: 3 or a
variant
thereof interact to form a trimer exonuclease.
In one embodiment, a motor protein is a polymerase. The polymerase may be
PyroPhage 3173 DNA Polymerase (which is commercially available from Lueigen
Corporation), SD Polymerase (commercially available from Biorone), Klenovv
from NEB
or variants thereof In one embodiment, the enzyme is Phi29 DNA polymerase (SEQ
ID
NO: 5) or a variant thereof Modified versions of Phi29 polymerase that may be
used in
the invention are disclosed in US Patent No. 5,576,204.
In one embodiment a motor protein is a topoisomerase. In one embodiment, the
topoisomerase is a member of any of the Moiety Classification (EC) groups
5.99.1.2 and
5.99.1.3. The topoisomerase may be a reverse transcriptase, which are enzymes
capable of
catalysing the formation of cDNA from a RNA template. They are commercially
available
from, for instance, New England Biolabs and Invitrogen .
In one embodiment, a motor protein is a helicase. Any suitable helicase can be
used in accordance with the methods provided herein. For example, the or each
motor
protein used in accordance with the present disclosure may be independently
selected from
a He1308 helicase, a RecD helicase, a TraI helicase, a TrwC helicase, an XPD
helicase, and
a Dda helicase, or a variant thereof Monomeric helicases may comprise several
domains
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
63
attached together. For instance, TraI helicases and Trai subgroup helicases
may contain
two RecD helicase domains, a relaxase domain and a C-terminal domain. The
domains
typically form a monomeric helicase that is capable of functioning without
forming
oligomers. Particular examples of suitable helicases include He1308, NS3, Dda,
UvrD,
Rep, PcrA, Pifl and TraI. These helicases typically work on single stranded
DNA.
Examples of helicases that can move along both strands of a double stranded
DNA include
FtfK and hexameric enzyme complexes, or multisubunit complexes such as RecBCD.
He1308 helicases are described in publications such as WO 2013/057495, the
entire
contents of which are incorporated by reference. RecD helicases are described
in
publications such as WO 2013/098562, the entire contents of which are
incorporated by
reference. XPD helicases are described in publications such as WO 2013/098561,
the
entire contents of which are incorporated by reference. Dda helicases are
described in
publications such as WO 2015/055981 and WO 2016/055777, the entire contents of
each
of which are incorporated by reference.
In one embodiment a helicase comprises the sequence shown in SEQ ID NO: 6
(Trwc Cba) or a variant thereof, the sequence shown in SEQ ID NO: 7 (He1308
Mbu) or a
variant thereof or the sequence shown in SEQ ID NO: 8 (Dda) or a variant
thereof.
Variants may differ from the native sequences in any of the ways discussed
herein. An
example variant of SEQ ID NO: 8 comprises E94C/A360C. A further example
variant of
SEQ ID NO: 8 comprises E94C/A360C and then (AM1)G1G2 (i.e. deletion of M1 and
then addition of G1 and G2).
In some embodiments a motor protein (e.g. a helicase) can control the movement
of
polynucleotides in at least two active modes of operation (when the motor
protein is
provided with all the necessary components to facilitate movement, e.g. fuel
and cofactors
such as ATP and Mg2') and one inactive mode of operation (when the motor
protein is not
provided with the necessary components to facilitate movement).
When provided with all the necessary components to facilitate movement (i.e.
in
the active modes), the motor protein (e.g. helicase) moves along the
polynucleotide in a 5'
to 3' or a 3' to 5' direction (depending on the motor protein). In embodiments
in which the
motor protein is used to control the movement of a polynucleotide strand with
respect to a
nanopore, the motor protein can be used to either move the polynucleotide away
from (e.g.
out of) a pore (e.g. against an applied field) or the polynucleotide towards
(e.g. into) a pore
(e.g. with an applied field). For example, when the end of the polynucleotide
towards
which the motor protein moves is captured by a pore, the motor protein works
against the
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
64
direction of the field resulting from the applied potential and pulls the
threaded
polynucleotide out of the pore (e.g. into the cis chamber). However, when the
end away
from which the motor protein moves is captured in the pore, the motor protein
works with
the direction of the field resulting from the applied potential and pushes the
threaded
polynucleotide into the pore (e.g. into the trans chamber).
When the motor protein (e.g. helicase) is not provided with the necessary
components to facilitate movement (i.e. in the inactive mode) it can bind to
the
polynucleotide and act as a brake slowing the movement of the polynucleotide
when it is
moved with respect to a nanopore, e.g. by being pulled into the pore by a
field resulting
from an applied potential. In the inactive mode, it does not matter which end
of the
polynucleotide is captured, it is the applied field which determines the
movement of the
polynucleotide with respect to the pore, and the motor protein acts as a
brake. When in the
inactive mode, the movement control of the polynucleotide by the motor protein
can be
described in a number of ways including ratcheting, sliding and braking.
In the active mode, motor proteins typically consume fuel molecules Fuel is
typically free nucleotides or free nucleotide analogues. The free nucleotides
may be one or
more of, but are not limited to, adenosine monophosphate (AMP), adenosine
diphosphate
(ADP), adenosine triphosphate (ATP), guanosine monophosphate (GMP), guanosine
diphosphate (GDP), guanosine triphosphate (GTP), thymidine monophosphate
(T1VIP),
thymidine diphosphate (TDP), thymichne niphosphate (TTP), uridine
monophosphaie
(UMP), uridine diphosphate (UDP), uridine triphosphate (UTP), cytidine
monophosphate
(CMP), cytidine diphosphate (CDP), cytidine triphosphate (CTP), cyclic
adenosine
monophosphate (cAMP), cyclic guanosine monophosphate (cGMP), deoxyadenosine
monophosphate (dAMP), deoxyadenosine diphosphate (dADP), deoxyadenosine
triphosphate (dATP), deoxyguanosine monophosphate (dGMP), deoxyguanosine
diphosphate (dGDP), deoxyguanosine triphosphate (dGTP), deoxythymidine
monophosphate (dTMP), deoxythymidine diphosphate (dTDP), deoxythymidine
triphosphate (dTTP), deoxyuridine monophosphate (dUMF'), deoxyuridine
diphosphate
(dUDP), deoxyuridine triphosphate (dUTP), deoxycytidine monophosphate (dCMP),
deoxycytidine diphosphate (dCDP) and deoxycytidine triphosphate (dCTP). The
free
nucleotides are usually selected from AMP, TMP, GMP, CMP, UMP, dAMP, dTMP,
dGMP or dCMP. The free nucleotides are typically adenosine triphosphate (ATP).
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
A cofactor for a motor protein is a factor that allows the motor protein to
function.
The cofactor is preferably a divalent metal cation. The divalent metal cation
is preferably
mn2 , Ca' or Co'. The cofactor is most preferably Mg'.
5 Polynucleotide analytes
In embodiments of the disclosed methods which comprise detecting or
characterising an analyte, the analyte is typically a polynucleotide.
A polynucleotide, such as a nucleic acid, is a macromolecule comprising two or
10 more nucleotides. A polynucleotide can be single-stranded or double-
stranded. A double-
stranded polynucleotide is made of two single stranded polynucleotides
hybridised
together. The target polynucleotide can be a single-stranded polynucleotide or
a double-
stranded polynucleotide.
A polynucleotide may comprise any combination of any nucleotides. The
15 nucleotides can be naturally occurring or artificial.
A nucleotide typically contains a nucleobase, a sugar and at least one
phosphate
group. The nucleobase and sugar form a nucleoside.
The nucleobase is typically heterocyclic. Nucleobases include, but are not
limited
to, purines and pyrimidines and more specifically adenine (A), guanine (G),
thymine (T),
20 uracil (U) and cytosine (C).
The sugar is typically a pentose sugar. Nucleotide sugars include, but are not
limited to, ribose and deoxyribose. The sugar is preferably a deoxyribose. The
polynucleotide preferably comprises the following nucleosides: deoxyadenosine
(dA),
deoxyuridine (dU) and/or thymidine (dT), deoxyguanosine (dG) and deoxycytidine
(dC).
25 The nucleotide is typically a ribonucleotide or deoxyribonucleotide.
The nucleotide
typically contains a monophosphate, diphosphate or triphosphate. The
nucleotide may
comprise more than three phosphates, such as 4 or 5 phosphates. Phosphates may
be
attached on the 5' or 3' side of a nucleotide Nucleotides include, hut are not
limited to,
adenosine monophosphate (AMP), guanosine monophosphate (GMP), thymidine
30 monophosphate (TMP), uridine monophosphate (UMP), 5-methylcytidine
monophosphate,
5-hydroxymethylcytidine monophosphate, cytidine monophosphate (CMP), cyclic
adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP),
deoxyadenosine monophosphate (dAMP), deoxyguanosine monophosphate (dGMP),
deoxythymidine monophosphate (dTMP), deoxyuridine monophosphate (dUMP),
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
66
deoxycytidine monophosphate (dCMP) and deoxymethylcytidine monophosphate. The
nucleotides are preferably selected from AMP, TMP, GMP, CMP, UMP, dAMP, dTMP,
dGMP, dCMP and dUMP.
A nucleotide may be abasic (i.e. lack a nucleobase). A nucleotide may also
lack a
nucleobase and a sugar (i.e. is a C3 spacer).
The nucleotides in the polynucleotide may be attached to each other in any
manner.
The nucleotides are typically attached by their sugar and phosphate groups as
in nucleic
acids. The nucleotides may be connected via their nucleobases as in pyrimidine
dimers.
The polynucleotide can be a nucleic acid, such as deoxyribonucleic acid (DNA)
or
ribonucleic acid (RNA). The polynucleotide can comprise one strand of RNA
hybridized
to one strand of DNA. The polynucleotide may be any synthetic nucleic acid
known in the
art, such as peptide nucleic acid (PNA), glycerol nucleic acid (GNA), threose
nucleic acid
(TNA), locked nucleic acid (LNA), bridged nucleic acid (BNA) or other
synthetic
polymers with nucleotide side chains. The PNA backbone is composed of
repeating N-(2-
aminoethyl)-glycine units linked by peptide bonds. The GNA backbone is
composed of
repeating glycol units linked by phosphodi ester bonds The 'TNA backbone is
composed of
repeating threose sugars linked together by phosphodiester bonds. LNA is
formed from
ribonucleotides as discussed above having an extra bridge connecting the 2'
oxygen and 4'
carbon in the ribose moiety.
The polynucleotide is preferably DNA, RNA or a DNA or RNA hybrid, most
preferably DNA. A DNA/RNA hybrid may comprise DNA and RNA on the same strand.
Preferably, the DNA/RNA hybrid comprises one DNA strand hybridized to a RNA
strand.
The backbone of the polynucleotide can be altered to reduce the possibility of
strand scission. For example, DNA is known to be more stable than RNA under
many
conditions. The backbone of the polynucleotide strand can be modified to avoid
damage
caused by e.g. harsh chemicals such as free radicals.
DNA or RNA that contains unnatural or modified bases can be produced by
amplifying natural DNA or RNA polynucleotides in the presence of modified NTPs
using
an appropriate polymerase.
The nucleotides in the polynucleotide may be modified. The nucleotides may be
oxidized or methylated. One or more nucleotides in the polynucleotide may be
damaged.
For instance, the polynucleotide may comprise a pyrimidine dimer. Such dimers
are
typically associated with damage by ultraviolet light and are the primary
cause of skin
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
67
melanomas. One or more nucleotides in the polynucleotide may be modified with
a label
or a tag.
A single-stranded polynucleotide may contain regions with strong secondary
structures, such as hairpins, quadruplexes, or triplex DNA. Structures of
these types can be
used to control the movement of the polynucleotide with respect to the
nanopore. For
example, secondary structures can be used to pause the movement of the
polynucleotide
through a nanopore, as described in more detail herein. Each successive
secondary
structure along the strand pauses the movement of the strand with respect to
the nanopore
as it is unwound and translocated. The polynucleotide may reform secondary
structures
after it has translocated through the nanopore. Such secondary structures can
be used to
prevent the polynucleotide from moving back through the nanopore under low or
no
applied negative voltages (applied to the trans side of the nanopore) and
therefore assist in
controlling the movement of the polynucleotide so it only occurs in a
controlled manner in
the relevant steps of the methods provided herein.
As used herein, a double stranded polynucleotide may comprise single stranded
regions and regions with other structures, such as hairpin loops, triplexes
and/or
quadruplexes. Such secondary structures can be useful as described above in
the context
of single-stranded polynucleotides.
The two strands of a double-stranded molecule may be covalently linked, for
example at the ends of the molecules by joining the 5' end of one strand to
the 3' end of
the oilier with a hairpin structure.
A target polynucleotide can be any length. For example, the target
polynucleotide
can be at least 10, at least 50, at least 100, at least 150, at least 200, at
least 250, at least
300, at least 400 or at least 500 nucleotides or nucleotide pairs in length.
The target
polynucleotide can be 1000 or more nucleotides or nucleotide pairs, 5000 or
more
nucleotides or nucleotide pairs in length or 100000 or more nucleotides or
nucleotide pairs
in length or 500,000 or more nucleotides or nucleotide pairs in length, or
1,000,000 or
more nucleotides or nucleotide pairs in length, 10, 000,000 or more
nucleotides or
nucleotide pairs in length, or 100,000,000 or more nucleotides or nucleotide
pairs in
length, or 200,000,000 or more nucleotides or nucleotide pairs in length, or
the entire
length of a chromosome.
A target polynucleotide may be an oligonucleotide. Oligonucleotides are short
nucleotide polymers which typically have 50 or fewer nucleotides, such 40 or
fewer, 30 or
fewer, 20 or fewer, 10 or fewer or 5 or fewer nucleotides. The target
oligonucleotide is
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
68
preferably from about 15 to about 30 nucleotides in length, such as from about
20 to about
25 nucleotides in length. For example, the oligonucleotide can be about 15,
about 16,
about 17, about 18, about 19, about 20, about 21, about 22, about 23, about
24, about 25,
about 26, about 27, about 28, about 29 or about 30 nucleotides in length.
The target polynucleotide may be a fragment of a longer polynucleotide. In
this
embodiment, the longer polynucleotide is typically fragmented into multiple,
such as two
or more, shorter polynucleotides.
The target polynucleotide may comprise the products of a PCR reaction, genomic
DNA, the products of an endonuclease digestion and/or a DNA library.
The target polynucleotide may be naturally occurring. The target
polynucleotide
may be secreted from cells. Alternatively, the target analyte can be an
analyte that is
present inside cells such that the analyte must be extracted from the cells
before the
method can be carried out.
The target polynucleotide may be sourced from common organisms such as
viruses,
bacteria, archaea, plants or animals. Such organisms may be selected or
altered to adjust
the sequence of the target polynucleotide, for example by adjusting the base
composition,
removing unwanted sequence elements, and the like. The selection and
alteration of
organisms in order to arrive at desired polynucleotide characteristics is
routine for one of
ordinary skill in the art.
The source organism for the target polynucleotide may be chosen based on
desired
characteristics of the sequence. Desired characteristics include the ratio of
single-stranded
vs double-stranded polynucleotides produced by the organism; the complexity of
the
sequences of polynucleotides produced by the organism, the composition of the
polynucleotides produced by the organism (such as the GC composition), or the
length of
contiguous polynucleotide strands produced by the organism. For example, when
a
contiguous polynucleotide strand of around 50 kb is required, lambda phage DNA
can be
used. If longer contiguous strands are required, other organisms can be used
to produce
the polynucleotide; for example E. coil produces around 4.5 Mb of contiguous
dsDNA.
The target polynucleotide is often obtained from a human or animal, e.g. from
urine, lymph, saliva, mucus, seminal fluid or amniotic fluid, or from whole
blood, plasma
or serum. The target polynucleotide may be obtained from a plant e.g. a
cereal, legume,
fruit or vegetable. The target polynucleotide may comprise genomic DNA. The
genomic
DNA may be fragmented. The DNA may be fragmented by any suitable method. For
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
69
example, methods of fragmenting DNA are known in the art, Such methods may use
a
transposase, such as a MuA transposase. Often the genomic DNA is not
fragmented.
In some embodiments the polynucleotide is synthetic or semi-synthetic. For
example, DNA or RNA may be purely synthetic, synthesised by conventional DNA
synthesis methods such as phosphoramidite based chemistries. Synthetic
polynucleotides
subunits may be joined together by known means, such as ligation or chemical
linkage, to
produce longer strands. In some embodiments internal self-forming structures
(e.g.
hairpins, quadruplexes) can be designed into the substrate e.g. by ligating
appropriate
sequences. Synthetic polynucleotides can be copied and scaled up for
production by
means known in the art, including PCR, incorporation into bacterial factories,
and the like.
In some embodiments, the polynucleotide may have a simplified nucleotide
composition. In some embodiments the polynucleotide has a repeating pattern of
the same
subunit. For example, a repeating unit may be (AmGn)q, wherein m, n and q are
positive
integers. For example, m is often from 1 to 20, such as from 1 to 10 e.g. from
1 to 5, e.g. 1,
2, 3, 4 or 5. n is often from 1 to 20, such as from 1 to 10 e.g. from 1 to 5,
e.g. 1, 2, 3, 4 or
5 m and n may be the same or different q is often from 1 to about 100,000 A
typical
repeating unit may be for example (AAAAAAGGGGGG)q (SEQ ID NO: 36). Repeating
polynucleotides can be made by many means known in the art, for example by
concatenating together synthetic subunits with sticky ends that enable
ligation. In some
embodiments the polynucleotide may therefore be a concatenated polynucleotide.
Methods of concatenating polynucleotides are described in PCT/GB2017/051493.
Polyinickotide adapter
A target polynucleotide assessed in a method provided herein may comprise a
polynucleotide adapter. A polynucleotide adapter can be used to load e.g. a
motor protein
onto a polynucleotide in order to control the movement of the polynucleotide
with respect
to a nanopore. For example, WO 2015/110813 describes the loading of motor
proteins
onto a target polynucleotide such as an adapter, and is hereby incorporated by
reference in
its entirety.
An adapter typically comprises a polynucleotide strand capable of being
attached to
the end of a target polynucleotide. A polynucleotide adapter may be added to
both ends of
the target polynucleotide. Alternatively, different adapters may be added to
the two ends
of the target polynucleotide. An adapter may be added to just one end of the
target
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
polynucleotide. Methods of adding adapters to polynucleotides are known in the
art.
Adapters may be attached to polynucleotides, for example, by ligation, by
click chemistry,
by tagmentation, by topoisomerisation or by any other suitable method.
An adapter may be synthetic or artificial. Typically, an adapter comprises a
5 polymer as described herein. In some embodiments, the adapter comprises a
polynucleotide. In some embodiments an adapter may comprise a single-stranded
polynucleotide strand. In some embodiments an adapter may comprise a double-
stranded
polynucleotide. A polynucleotide adapter may comprise DNA, RNA, modified DNA
(such
as a basic DNA), RNA, PNA, LNA, BNA and/or PEG. Usually, the adapter comprises
10 single stranded and/or double stranded DNA or RNA.
An adapter may be a Y adapter. A Y adapter is typically double stranded and
comprises (a) at one end, a region where the two strands are hybridised
together and (b), at
the other end, a region where the two strands are not complementary. The non-
complementary parts of the strands form overhangs. The hybridised stem of the
adapter
15 typically attaches to the 5' end of a first strand of a double-stranded
polynucleotide and the
3' end of a second strand of a double-stranded polynucleotide; or to the 3'
end of a first
strand of a double-stranded polynucleotide and the 5' end of a second strand
of a double-
stranded polynucleotide. The presence of a non-complementary region in the Y
adapter
gives the adapter its Y shape since the two strands typically do not hybridise
to each other
20 unlike the double stranded portion. A motor protein or polynucleotide
binding protein may
bind to an overhang of an adapter such as a Y adapter. In another embodiment,
a motor
protein or polynucleotide binding protein may bind to the double stranded
region. In other
embodiments, a motor protein or polynucleotide binding protein may bind to a
single-
stranded and/or a double-stranded region of the adapter. In other embodiments,
a first
25 motor protein or polynucleotide binding protein may bind to the single-
stranded region of
such an adapter and a second motor protein or polynucleotide binding protein
may bind to
the double-stranded region of the adapter.
In some embodiments, one of the non-complementary strands of a polynucleotide
adapter such as a Y adapter may comprise a leader sequence, which when
contacted with a
30 transmembrane pore is capable of threading into a nanopore. The leader
sequence
typically comprises a polymer such as a polynucleotide, for instance DNA or
RNA, a
modified polynucleotide (such as abasic DNA), PNA, LNA, polyethylene glycol
(PEG) or
a polypeptide. In some embodiments, the leader sequence comprises a single
strand of
DNA, such as a poly dT section. The leader sequence can be any length, but is
typically
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
71
to 150 nucleotides in length, such as from 20 to 120, 30 to 100, 40 to 80 or
50 to 70
nucleotides in length.
In one embodiment, a polynucleotide adapter is a hairpin loop adapter. A
hairpin
loop adapter is an adapter comprising a single polynucleotide strand, wherein
the ends of
5 the polynucleotide strand are capable of hybridising to each other, or
are hybridized to
each other, and wherein the middle section of the polynucleotide forms a loop.
Suitable
hairpin loop adapters can be designed using methods known in the art.
Typically, the 3'
end of a hairpin loop adapter attaches to the 5' end of a first strand of a
double-stranded
polynucleotide and the 5' end of the hairpin loop adapter attaches to the 3'
end of a second
10 strand of a double-stranded polynucleotide; or the 5' end of a hairpin
loop adapter attaches
to the 3' end of a first strand of a double-stranded polynucleotide and the 3'
end of the
hairpin loop adapter attaches to the 5' end of a second strand of a double-
stranded
polynucleotide.
A polynucicotide or polynucleotide adapter may comprise one or more spacers,
e.g.
from one to about 10 spacers, e.g. from 1 to about 5 spacers, e.g. 1, 2, 3, 4
or 5 spacers.
The spacer may comprise any suitable number of spacer units A spacer typically
provides an energy barrier which impedes movement of a polynucleotide binding
protein.
For example, a spacer may impede movement of a motor protein or polynucleotide
binding
protein by reducing the traction of the protein, e.g. using an abasic spacer.
A spacer may
physically block movement of the protein, for instance by introducing a bulky
chemical
group to physically impede the movement of the polynucleotide binding protein.
In some embodiments, one or more spacers are included in the polynucleotide or
in
a polynucleotide adapter to provide a distinctive signal when they pass
through or across a
nanopore. One or more spacers may be used to define or separate one or more
regions of a
polynucleotide; e.g. to separate an adapter from the target polynucleotide.
In some embodiments, a spacer may comprise a linear molecule, such as a
polymer,
e.g. a polypeptide or a polyethylene glycol (PEG). Typically, such a spacer
has a different
structure from the target polynucleotide. For instance, if the target
polynucleotide is DNA,
the or each spacer typically does not comprise DNA. In particular, if the
target
polynucleotide is deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), the
or each
spacer preferably comprises peptide nucleic acid (PNA), glycerol nucleic acid
(GNA),
threose nucleic acid (TNA), locked nucleic acid (LNA) or a synthetic polymer
with
nucleotide side chains. In some embodiments, a spacer may comprise one or more
nitroindoles, one or more inosines, one or more acridines, one or more 2-
aminopurines,
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
72
one or more 2-6-diaminopurines, one or more 5-bromo-deoxyuridines, one or more
inverted thymidines (inverted dTs), one or more inverted dideoxy-thymidines
(ddTs), one
or more dideoxy-cytidines (ddCs), one or more 5-methylcytidines, one or more 5-
hydroxymethylcytidines, one or more 2'-0-Methyl RNA bases, one or more Iso-
deoxycytidines (Iso-dCs), one or more Iso-deoxyguanosines (Iso-dGs), one or
more C3
(0C3H60P03) groups, one or more photo-cleavable (PC) [OC3H6-C(0)NHCH2-C6H3NO2-
CH(CH3)0P03] groups, one or more hexandiol groups, one or more spacer 9 (iSp9)
[(OCH2CH2)30P03] groups, or one or more spacer 18 (iSp18) ROCH2CH2)60P031
groups; or one or more thiol connections. A spacer may comprise any
combination of
these groups. Many of these groups are commercially available from 1DT
(Integrated
DNA Technologies ). For example, C3, iSp9 and iSp18 spacers are all available
from
IDT . A spacer may comprise any number of the above groups as spacer units.
In some embodiments, a spacer may comprise one or more chemical groups, e.g.
one or more pendant chemical groups. The one or more chemical groups may be
attached
to one or more nucleobases in a polynucleotide adapter. The one or more
chemical groups
may be attached to the backbone of a polynucleotide adapter. Any number of
appropriate
chemical groups may be present, such as 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or
more. Suitable
groups include, but are not limited to, fluorophores, streptavidin and/or
biotin, cholesterol,
methylene blue, dinitrophenols (DNPs), digoxigenin and/or anti-digoxigenin and
dibenzylcyclooctyne groups.
In sonic embodiments, a spacer may comprise one or more abasic nucleotides
(i.e.
nucleotides lacking a nucleobase), such as 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12
or more abasic
nucleotides. The nucleobase can be replaced by ¨H (idSp) or ¨OH in the abasic
nucleotide. Abasic spacers can be inserted into target polynucleotides by
removing the
nucleobases from one or more adjacent nucleotides. For instance,
polynucleotides may be
modified to include 3-methyladenine, 7-methylguanine, 1,N6-ethenoadenine
inosine or
hypoxanthine and the nucleobases may be removed from these nucleotides using
Human
Alkyladenine DNA Glycosylase (hAAG). Alternatively, polynucleotides may be
modified
to include uracil and the nucleobases removed with Uracil-DNA Glycosylase
(UDG). In
one embodiment, the one or more spacers do not comprise any abasic
nucleotides.
Suitable spacers can be designed or selected depending on the nature of the
polynucleotide or polynucleotide adapter, the motor protein and the conditions
under
which the method is to be carried out.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
73
Tags
In some embodiments a polynucleotide or polynucleotide adapter may comprise a
tag or tether. For example, a polynucleotide can bind to a tag on a nanopore,
e.g., via its
adaptor, and release at some point, e.g., during characterization of the
polynucleotide by
the nanopore.
A tag or tether may be uncharged. This can ensure that the tags or tethers are
not
drawn into the nanopore under the influence of a potential difference.
One or more molecules that attract or bind a polynucleotide or adaptor may be
linked to a nanopore, e.g. a nanopore as described herein. Any molecule that
hybridizes to
the adaptor and/or target polynucleotide may be used. The molecule attached to
the pore
may be selected from a PNA tag, a PEG linker, a short oligonucleotide, a
positively
charged amino acid and an aptamer. Pores having such molecules linked to them
are
known in the art. For example, pores having short oligonucleotides attached
thereto arc
disclosed in Howarka et al (2001) Nature Biotech. 19: 636-639 and WO
2010/086620, and
pores comprising PEG attached within the lumen of the pore are disclosed in
Howarka et al
(2000) J Am. Chem. Soc. 122(11): 2411-2416.
A short oligonucleotide attached to the pore, comprising a sequence
complementary
to a sequence in the leader sequence or another single stranded sequence in an
adaptor may
be used to enhance capture of the target polynucleotide in the methods
described herein.
A tag or tether may comprise or be an oligonucleotide as described herein,
e.g. of
length 10-30 nucleotides or about 10-20 nucleotides. Such an oligonucleotide
can have at
least one end (e.g., 3'- or 5'-end) modified for conjugation to other
modifications or to a
solid substrate surface including, e.g., a bead. The end modifiers may add a
reactive
functional group which can be used for conjugation. Examples of functional
groups that
can be added include, but are not limited to amino, carboxyl, thiol,
maleimide, aminooxy,
and any combinations thereof. The functional groups can be combined with
different
length of spacers (e.g., C3, C9, C12, Spacer 9 and 18) to add physical
distance of the
functional group from the end of the oligonucleotide sequence.
In some embodiments, the tag or tether may comprise or be a morpholino
oligonucleotide. The morpholino oligonucleotide can have about 10-30
nucleotides in
length or about 10-20 nucleotides in length. The morpholino oligonucleotides
can be
modified or unmodified. For example, in some embodiments, the morpholino
oligonucleotide can be modified on the 3' and/or 5' ends of the
oligonucleotides. Examples
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
74
of modifications on the 3' and/or 5' end of the morpholino oligonucleotides
include, but are
not limited to 3' affinity tag and functional groups for chemical linkage
(including, e.g., 3'-
biotin, 3'-primary amine, 3'-disulfide amide, 3'-pyridyl dithio, and any
combinations
thereof); 5' end modifications (including, e.g., 5'-primary ammine, and/or 5'-
dabcyl),
modifications for click chemistry (including, e.g., 3'-azide, 3'-alkyne, 5'-
azide, 5'-alkyne),
and any combinations thereof.
A tag or tether may further comprise a polymeric linker, e.g., to facilitate
coupling
to a detector e.g. a nanopore. An exemplary polymeric linker includes, but is
not limited to
polyethylene glycol (PEG). The polymeric linker may have a molecular weight of
about
500 Da to about 10 kDa (inclusive), or about 1 kDa to about 5 kDa (inclusive).
The
polymeric linker (e.g., PEG) can be functionalized with different functional
groups
including, e.g, but not limited to maleimide, NHS ester, dibenzocyclooctyne
(DBCO),
azide, biotin, amine, alkyne, aldehyde, and any combinations thereof. In some
embodiments, the tag or tether may further comprise a 1 kDa PEG with a 5'-
maleimide
group and a 3'-DBCO group. In some embodiments, the tag or tether may further
comprise
a 2 kDa PEG with a 5'-maleimi de group and a 3'-DBCO group In some
embodiments, the
tag or tether may further comprise a 3 kDa PEG with a 5'-m al eimide group and
a 3'-D13CO
group. In some embodiments, the tag or tether may further comprise a 5 kDa PEG
with a
5'-maleimide group and a 3'-DBCO group.
A tag can be included on a nanopore using the disclosed modification methods.
In
some embodiments, the tag or tether may be attached directly to a nanopore or
via one or
more linkers. The tag or tether may be attached to the nanopore using the
hybridization
linkers described in WO 2010/086602. Alternatively, peptide linkers may be
used.
Peptide linkers are amino acid sequences. The length, flexibility and
hydrophilicity of the
peptide linker are typically designed such that it does not to disturb the
functions of the
monomer and pore. Preferred flexible peptide linkers are stretches of 2 to 20,
such as 4, 6,
8, 10 or 16, serine and/or glycine amino acids. More preferred flexible
linkers include
(SG)I, (SG)2, (SG)3, (SG)4, (SG)5 and (SG)8 wherein S is serine and G is
glycine.
Preferred rigid linkers are stretches of 2 to 30, such as 4, 6, 8, 16 or 24,
proline amino
acids. More preferred rigid linkers include (P)12 wherein P is proline.
Anchor
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
In one embodiment, a polynucleotide or polynucleotide adapter may comprise a
membrane anchor or a transmembrane pore anchor. In one embodiment the anchor
assists
in the characterisation of a target polynucleotide in accordance with the
methods disclosed
herein. For example, a membrane anchor or transmembrane pore anchor may
promote
5 localisation of the selected polynucleotides around a nanopore.
The anchor may be a polypeptide anchor and/or a hydrophobic anchor that can be
inserted into the membrane. In one embodiment, the hydrophobic anchor is a
lipid, fatty
acid, sterol, carbon nanotube, polypeptide, protein or amino acid, for example
cholesterol,
palmitate or tocopherol. The anchor may comprise thiol, biotin or a
surfactant.
10 In one aspect the anchor may be biotin (for binding to streptavidin),
amylose (for binding
to maltose binding protein or a fusion protein), Ni-NTA (for binding to poly-
histidine or
poly-histidine tagged proteins) or peptides (such as an antigen).
In one embodiment, the anchor comprises a linker, or 2, 3, 4 or more linkers.
Preferred linkers include, but arc not limited to, polymers, such as
polynucleotides,
15 polyethylene glycols (PEGs), polysaccharides and polypeptides. These
linkers may be
linear, branched or circular. For instance, the linker may be a circular
polynucleotide. The
adapter may hybridise to a complementary sequence on a circular polynucleotide
linker.
The one or more anchors or one or more linkers may comprise a component that
can be cut
or broken down, such as a restriction site or a photolabile group. The linker
may be
20 functionalised with maleimide groups to attach to cysteine residues in
proteins. Suitable
linkers are described in WO 2010/086602.
In one embodiment, the anchor is cholesterol or a fatty acyl chain. For
example,
any fatty acyl chain having a length of from 6 to 30 carbon atom, such as
hexadecanoic
acid, may be used. Examples of suitable anchors and methods of attaching
anchors to
25 adapters are disclosed in WO 2012/164270 and WO 2015/150786.
Further embodiments
Also provided are multifunctional molecules suitable for use in the disclosed
30 methods. A provided multifunctional molecule is typically a
multifunctional molecule as
described in more detail herein.
Kit
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
76
Also provided are kits for modifying a monomer. In one embodiment the kit
comprising a multifunctional molecule as disclosed herein and a support for
binding the
purification tag of the multifunctional molecule. In one embodiment the kit
further
comprises buffer solutions for carrying out the binding and/or any washing
steps. Those
skilled in the art will appreciate that any of the multifunctional molecules
and supports
described herein can be used in the disclosed kits.
System
Also provided are systems for characterising comprising a chemically-modified
monomer or chemically modified oligomer as described herein. In one embodiment
the
system comprises a chemically modified monomer or oligomer as described
herein,
together with a motor protein for controlling the movement of a analyte, such
as a
polynucleotide analytc, with respect to a pore formed from the monomer or
oligomer.
In some embodiments the system comprises a plurality of monomers, typically a
homogeneous population of chemically modified monomers as described herein In
some
embodiments the system comprises a plurality of oligomers, typically a
homogeneous
population of chemically modified monomers as described herein. In some
embodiments
the system comprises a plurality of nanopores, typically a homogeneous
population of
chemically modified nanopores as described herein. In some embodiments the
population
is 100% homogeneous. ie all of the monomers/oligomers/pores in the population
are
chemically modified. In some embodiments the plurality of
monomers/oligomers/pores
are present on a chip for analysing (e.g. characterising) an analyte.
In some embodiments the system further comprises an analyte such as a
polynucleotide analyte and/or other components such as a membrane; membrane
anchor;
tether, etc as described herein.
Further Aspects
The following are numbered aspects of the invention.
1. A method of chemically modifying a polypepti de or
polynucleotide monomer;
comprising:
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
77
a) contacting the monomer with a multifunctional molecule, wherein the multi-
functional molecule comprises (i) a reactive group; (ii) a chemical modifying
group
and (iii) a cleavable purification tag;
b) allowing the reactive group of the multi-functional molecule to react with
the
monomer thereby attaching the chemical modifying group and cleavable
purification tag to the monomer to form a chemically modified tagged monomer;
c) contacting the chemically modified tagged monomer formed in step (b) with a
support;
d) allowing the purification tag to bind to the support thereby binding the
chemically
modified tagged monomer to the support; and
e) cleaving the purification tag thereby releasing the chemically modified
monomer
from the support.
2. A method according to aspect 1, wherein the
multifunctional molecule is of
Formula (I) or Formula (II):
A-B-D-C
Formula (I) Formula (II)
wherein:
A is a reactive group;
B is a chemical modifying group; and
D-C forms a cleavable purification tag;
preferably wherein D comprises a cleavable linker and C comprises a support-
binding
group.
3. A method according to aspect 1, wherein the reactive group comprises the
chemical
modifying group.
4. A method according to any one of the preceding aspects,
wherein the monomer
comprises a reactive functional group and step (b) comprises allowing the
reactive group
of the multifunctional molecule to react with the reactive functional group of
the monomer.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
78
5. A method according to any one of the preceding aspects, wherein the
reactive
group of the multifunctional molecule comprises an amine-reactive group; a
carboxyl-
reactive group; a sulfhydryl-reactive group or a carbonyl-reactive group;
preferably wherein the reactive group of the multi-functional molecule
comprises a
cysteine-reactive group.
6. A method according to any one of the preceding aspects, wherein the
reactive
group comprises a maleimide, an azide, a thiol, an alkyne, an NHS ester or a
haloacetamide.
7. A method according to any one of aspects 1 and 3 to 6, wherein the
chemical
modifying group introduces hydrophilic, hydrophobic, positively charged,
negatively
charged, hydrogen-bonding, supramolccular associations or zwitterionic
properties to the
protein monomer;
preferably wherein the chemical modifying group comprises (i) an amino acid, a
nucleotide, a polymer, a hydrogen-bonding group, a membrane anchor, a sugar, a
dye, a
chromophore, a fluorophore or a molecular adapter; or (ii) a natural or
unnatural amino
acid, a polypeptide, a nucleotide or nucleotide analog, an oligonucleotide or
oligonucleotide analog, a polysaccharide, a lipid, a polyethylene glycol, a
cyclodextrin, a
DNA inteicalatoi, an aptamer or an analyte binding domain.
8. A method according to any one of the preceding aspects, wherein the
support
comprises a chromatography matrix, preferably an agarose or sepharose resin;
one or more
beads, preferably magnetic beads; or a solid surface, preferably a glass,
silica, polymer or
ceramic surface.
9. A method according to any one of the preceding aspects, wherein the
support is
functionalised for binding to the purification tag.
10. A method according to any one of the preceding aspects, wherein the
purification
tag comprises a biotin group and the support comprises streptavi din, neutravi
din or avi din,
preferably streptavi din.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
79
11. A method according to any one of the preceding aspects, wherein the
cleavable
linker is cleaved by physical or chemical means;
preferably wherein the cleavable linker comprises a UV photocleavable nito-
benzyl
moiety.
12. A method according to any one of the preceding aspects, wherein in step
(e)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
light; preferably UV light.
13. A method according to any one of the preceding aspects, wherein in step
(e)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
a change in pH.
14. A method according to any one of the preceding aspects, wherein in step
(c)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
a chemical reagent; preferably a reducing reagent
15. A method according to any one of the preceding aspects, wherein in step
(e)
cleaving the purification tag comprises exposing the support and/or the tagged
monomer to
an enzyme; preferably a protease.
16. A method according to any one of the preceding aspects, wherein the
monomer is a
polypeptide having a mass of from about 10 kDa to about 1 MDa.
17. A method according to any one of the preceding aspects, wherein the
monomer is a
monomer of an oligomeric protein pore, preferably a lysenin pore, a y-
hemolysin pore, an
a-hemolysin pore; a NetB pore; a CytK pore or a leukocidin pore; or a homolog
or paralog
thereof.
18. A method according to aspect 16, wherein the oligomeric protein pore is
a multi-
component pore.
19. A method according to any one of aspects 1 to 15 wherein
the monomer is a
polynucleotide; preferably wherein the monomer is a monomer of a DNA origami
pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
20. A method according to any one of the preceding aspects, wherein the
monomer is a
monomer of an oligomeric pore; and wherein the multifunctional molecule reacts
with a
reactive functional group located on the monomer at a surface-exposed position
when the
5 monomer is oligomerised to form the pore;
preferably wherein the surface-exposed position is located at the surface of
the
channel through the pore; or on the exterior surface of the pore.
21. A method according to aspect 20, wherein the multifunctional molecule
reacts with
10 a reactive functional group located on the monomer at a position located
at or near a
constriction of the channel through the pore when the monomer is oligomerised
to form the
pore.
22. A method according to any one of the preceding aspects, wherein the
method
15 comprises, prior to step (a), the steps of (i) expressing the monomer in
a cellular expression
system or a cell-free expression system; and (ii) isolating and/or purifying
the monomer.
23. A method according to any one of the preceding aspects, wherein step
(d) further
comprises the step of removing unmodified monomer(s) and/or unreacted
multifunctional
20 molecule(s), if present, from the support.
24. A method according to any one of the preceding aspects, further
comprising the
step of:
oligomerising the chemically modified monomer to form a chemically modified
25 oligomer;
wherein preferably the monomer is a protein monomer and step (f) comprises
forming an
oligomeric protein.
25. A method according to aspect 24, wherein step (f) comprises
oligomerising two or
30 more chemically modified monomers to form a homooligomer,
wherein preferably said monomers are protein monomers and said homooligomer is
a homooligomeric protein pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
81
26. A method according to aspect 24, wherein step (f) comprises
oligomerising one or
more chemically modified monomers with one or more unmodified or differently-
modified
monomers to form a heterooligomer,
wherein preferably said monomers are protein monomers and said heterooligomer
is a heterooligomeric protein pore.
27. A method according to aspect 24, wherein step (f) comprises
oligomerising one or
more chemically modified first monomers with one or more chemically modified
second
monomers to form a heterooligomer; wherein the chemical modification made to
the first
monomer is the same or different to the chemical modification made to the
second
monomer;
wherein preferably said monomers are protein monomers, said first monomer has
a
different amino acid sequence to said second monomer, and the heterooligomer
is a
heterooligomeric protein pore.
28 A method of producing a homooligomeric protein; comprising
i) producing a plurality of chemically modified protein monomers in a
method
according to any one of aspects 1 to 23; and
ii) oligomerising two or more of the chemically modified protein monomers
obtained in step (i) to form a homooligomeric protein;
wherein said homooligomeric protein is preferably a homooligomei ic protein
pole.
29. A method of producing a heterooligomeric protein;
comprising
i) producing one or more chemically modified first protein monomers in a
method
according to any one of aspects 1 to 23; and
ii) producing one or more chemically modified second protein monomers in a
method according to any one of aspects 1 to 23; and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein;
wherein said heterooligomeric protein is preferably a heterooligomeric protein
pore.
30. A method of producing an oligomeric protein, comprising
i) producing one or more chemically modified first protein
monomers in a method
according to any one of aspects 1 to 23; and
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
82
ii) providing one or more unmodified second protein monomers; and
iii) oligomerising said one or more first monomers and said one or more
second
monomers to form a hetero-oligomeric protein;
wherein said heterooligomeric protein is preferably a heterooligomeric protein
pore.
31. A chemically modified monomer obtainable by carrying out a method
according to
any one of aspects 1 to 23.
32. A homogeneous population comprising a plurality of chemically modified
monomers; wherein at least 95% % of the monomers in the population are
chemically
modified with a chemical modifying group;
wherein the chemically modified monomers are preferably as defined in any one
of
aspects 5 to 7 or 16 to 21.
33. A chemically modified oligomer obtainable by carrying out a method
according to
any one of aspects 24 to 30
34. A homogeneous population comprising a plurality of chemically modified
oligomers; wherein at least 95% of the oligomers in the population comprise a
defined
number of monomers chemically modified with a chemical modifying group;
wherein the chemically modified monomers are preferably as defined in any one
of
aspects 5 to 7 or 16 to 21.
35. A method of characterising an analyte, comprising:
i) producing a chemically modified oligomeric pore in a method according to
any
one of aspects 24 to 30; or providing a chemically modified oligomeric pore
according to aspect 33; and
ii) taking one or more measurements as the analyte moves
with respect to the pore,
wherein the one or more measurements are indicative of one or more
characteristics of the analyte, and thereby characterising the analyte as it
moves
with respect to the pore;
wherein preferably the analyte is a polynucleoti de, a polypeptide or a
polysaccharide.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
83
It is to be understood that although particular embodiments, specific
configurations as well
as materials and/or molecules, have been discussed herein for methods
according to the
present invention, various changes or modifications in form and detail may be
made
without departing from the scope and spirit of this invention. The following
examples are
provided to better illustrate particular embodiments, and they should not be
considered
limiting the application. The application is limited only by the claims.
EXAMPLES
These examples describe the modification of a polypeptide monomer in
accordance with
the claimed methods and the subsequent use of oligomeric pores produced from
such
monomers in the characterising of analytes.
Example 1
General synthesis of modifier molecule with UV cleavable linker site and amino
acid
as modification (B)
jyx
-ka,z
2-1EtivilarnItio Amino add amino add a,klutt
maleirnicle X. %i(k: than
=
Phritodevabfa linker with
terminal biotin tag.
4
, X
hvAi
1):3
Final modifier molecule
2-Ethylamino maleimide was reacted with the ¨COOH end of the preferred amino
acid to
form the maleimide - amino acid adduct. This was purchased as a custom
synthesised
molecule from Cambridge Research Biochemicals. Biotin tag with photocleavable
linker
was purchased from Broadpharm USA (cat number - BP-24161 / PC Biotin-PEG3-NHS
carbonate ester). 0.04 mmol of maleimide-amino acid adduct was dissolved in 1
ml of
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
84
DMF and was added 1 eq. (mol/mol) of the Biotin photocleavable linker. 1.5 eq.
(mol/mol)
TEA to the mixture and the mixture was stirred overnight. Formation of the
molecule was
confirmed by mass spectrometry and was used in the modification of the
protein/ peptides
without further purification.
1 ¨ Isoleucine as the chemical modifying group
Maleimide-Isoleucine adduct (MW. ¨ 253.2 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
photocleavable linker was purchased from Broadpharm USA (Cat No. - BP-24161 /
PC
Biotin-PEG3-NHS carbonate ester M.W. -840.9 g/mol). 0.04 mmol of maleimide-
Isoleucine adduct was dissolved in 1 ml of DMF and was added 1 eq. (mol/mol)
of the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
used in the modification of the protein/ peptides without further
purification.
2¨ Alanine as the chemical modifying group
Maleimide-Alanine adduct (MW. ¨ 211.2 g/mol) was purchased as a custom
synthesised
molecule from Cambridge Research Biochemicals. Biotin tag with photocleavable
linker
was purchased from Broadpharm USA (Cat No. - BP-24161 / PC Biotin-PEG3-NHS
carbonate ester M.W. - 840.9 g/mol). 0.04 mmol of maleimide-alanineadduct was
dissolved in 1 ml of DMF and was added 1 eq. (mol/mol) of the Biotin
photocleavable
linker. 1.5 eq. (mol/mol) TEA to the mixture and the mixture was stirred
overnight.
Formation of the molecule was confirmed by mass spectrometry and was used in
the
modification of the protein/ peptides without further purification.
3 ¨Arginine as the chemical modifying group
Maleimide-Arginine adduct (MW. ¨ 296.3 g/mol) was purchased as a custom
synthesised
molecule from Cambridge Research Biochemicals. Biotin tag with photocleavable
linker
was purchased from Broadpharm USA (Cat No. - BP-24161 / PC Biotin-PEG3-NHS
carbonate ester M.W. - 840.9 g/mol). 10 mg (0.04 mmol) of maleimide-arginine
adduct
was dissolved in 1 ml of DMF and was added 1 eq. (mol/mol) of the Biotin
photocleavable
linker. 1 5 eq. (mol/mol) TEA to the mixture and the mixture was stirred
overnight.
Formation of the molecule was confirmed by mass spectrometry and was used in
the
modification of the protein/ peptides without further purification.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
4 ¨Aspartic acid as the chemical modifying group
Maleimide-Aspartic acid adduct (M.W. ¨ 255.2 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
5 photocleavable linker was purchased from Broadpharm USA (Cat No. - BP-
24161 / PC
Biotin-PEG3-NHS carbonate ester M.W. -840.9 g/mol). 10 mg (0.04 mmol) of
maleimide-
aspartic acid adduct was dissolved in 1 ml of DMF and was added 1 eq.
(mol/mol) of the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
10 used in the modification of the protein/ peptides without further
purification.
5 ¨Asparagine as the chemical modifying group
Maleimide-Asparagine adduct (M.W. ¨ 254.2 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
15 photocleavable linker was purchased from Broadpharm USA (Cat No. - BP-
24161 / PC
Biotin-PEG3-NTIS carbonate ester MW. - 840.9 g/mol)_ 10 mg (0 04 mmol) of
maleimi de-
asparagine adduct was dissolved in 1 ml of DMF and was added 1 eq. (mol/mol)
of the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
20 used in the modification of the protein/ peptides without further
purification.
6 ¨Glutamine as the chemical modifying group
Maleimide-Glutamine adduct (M.W. ¨ 266.2 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
25 photocleavable linker was purchased from Broadpharm USA (Cat No. - BP-
24161 / PC
Biotin-PEG3-NHS carbonate ester M.W. -840.9 g/mol). 10 mg (0.04 mmol) of
maleimide-
glutamine adduct was dissolved in 1 ml of DMF and was added 1 eq. (mol/mol) of
the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
30 used in the modification of the protein/ peptides without further
purification.
7 ¨Phenylalanine as the chemical modifying group
Maleimide-Phenylalanine adduct (M.W. 287.3 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
86
photocleavable linker was purchased from Broadpharm USA (Cat No. - BP-24161 /
PC
Biotin-PEG3-NHS carbonate ester M.W. -840.9 g/mol). 0.04 mmol of maleimide-
phenylalanine adduct was dissolved in 1 ml of DMF and was added 1 eq.
(mol/mol) of the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
used in the modification of the protein/ peptides without further
purification.
8¨ Lysine as the chemical modifying group
Maleimide-Lysine adduct (MW. ¨ 268.3 g/mol) was purchased as a custom
synthesised
molecule from Cambridge Research Biochemicals. Biotin tag with photocleavable
linker
was purchased from Broadpharm USA (Cat No. - BP-24161 / PC Biotin-PEG3-NHS
carbonate ester M.W. - 840.9 g/mol). 0.04 mmol of maleimide-lysine adduct was
dissolved
in 1 ml of DIVIF and was added 1 eq. (mol/mol) of the Biotin photocleavable
linker. 1.5 eq.
(mol/mol) TEA to the mixture and the mixture was stirred overnight. Formation
of the
molecule was confirmed by mass spectrometry and was used in the modification
of the
protein/ peptides without further purification
9¨ PNA(Thymine) as the chemical modifying group
Maleimide-PNA(Thymine) adduct (MW. ¨ 406.3 g/mol) was purchased as a custom
synthesised molecule from Cambridge Research Biochemicals. Biotin tag with
photocleavable linker was purchased from Broadphann USA (Cat No. - BP-24161 /
PC
Biotin-PEG3-NHS carbonate ester M.W. -840.9 g/mol). 10 mg (0.04 mmol) of
maleimide-
PNA(thymine) adduct was dissolved in 1 ml of DMF and was added 1 eq. (mol/mol)
of the
Biotin photocleavable linker. 1.5 eq. (mol/mol) TEA to the mixture and the
mixture was
stirred overnight. Formation of the molecule was confirmed by mass
spectrometry and was
used in the modification of the protein/ peptides without further
purification.
10 ¨ CH2-NH2 as the chemical modifying group
Maleimide-CH2-NH2 was conjugated to a biotin tag with a photocleavable linker
in
accordance with the preceding examples and used in the modification of the
protein/
peptides as described.
The cleavage of the multifunctional molecules described in Examples 1 to 10 is
shown in
Figure 2.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
87
Example 2
General modification of protein monomers with the modifier molecules
Chemical Modification of. Cysteine Mutants (General ¨ e.g. Leukocidin and
Lysenin)
Cys mutants of the protein monomers were stored with a reducing agent (e.g. ¨
DTT -
Dithiolthreitol) to avoid di-sulfide bond formation. Reducing agent was
removed from the
purified monomer solutions by buffer exchange in a 7K MWCO desalting column
using
50mM Tris-HC1, 150mM NaC1, pH7. A linker of choice was added to each sample
and left
at room temperature (RI) for 16h to allow the monomers to bind to the chemical
modifiers
via maleimide-cysteine (thiol) chemistry. Unreacted linker molecules were
removed from
the solution by centrifugation at 1500rcf for 2 minutes through a 7K MWCO
desalting
column. The flow through was added to equilibrated StrepTactinTm Sepharose
High
Performance resin (GE Healthcare Life Sciences, Cat No. 28935600) and left on
a rotator
for 2h at 23 C, allowing modified monomers to bind to the beads via their
biotin tag
Unbound/unmodified protein monomers were removed via centrifugation at 1500rcf
for 2
minutes in three wash steps using buffer. After washing, the same buffer was
added to the
StreplactinTM resin and this was subjected to UV light in two 30 second
intervals to cleave
the photo-cleavable linker, releasing modified monomers from the beads which
were
collected in the flow through.
This general modification is illustrated in Figure 3.
1 - Chemical modification of Lysenin mutant, Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C/C272A/C283A)
Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C/C272A/C283A) TEV cleaved
to remove thioredoxin-strep moiety was used as the starting material. The
monomer was
buffered exchanged once to 50mM Tris-HC1, 150mM NaCl, pH7 to remove DTT
solution.
3u1 of 10mg/mL maleimide-Isoleucine adduct in DMF was added to 120 uL of 0.6
mg/ml
buffer exchanged Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C/C272A/C283A). The solution was
left at RI for 16h. Unreacted linker molecules were removed from the solution
by
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
88
centrifugation at 1500rcf for 2 minutes through a 7K MWCO desalting column.
The flow
through was added to equilibrated StrepTactinTm Sepharose High Performance
resin (GE
Healthcare Life Sciences, Cat No. 28935600) and left on a rotator for 2h at 23
C, allowing
modified monomers to bind to the beads via their biotin tag.
Unbound/unmodified protein
monomers were removed via centrifugation at 1500rcf for 2 minutes in three
wash steps
using buffer. After washing, the same buffer was added to the StrepTactinTm
resin and this
was subjected to UV light in two 30 second intervals to cleave the photo-
cleavable linker,
releasing modified monomers from the beads which were collected in the flow
through as
the modified monomer - Lys-(E84Q/E85 S/E92Q/E94D/E97S/T106K/D126G/S98C-
Maleimide-Icoleucine/C272A/C283A). Successful modification was checked by SDS-
PAGE electrophoresis on a 4-20% gel (Figure 4).
2 - Chemical modification of Lysenin mutant Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C/C272A/C283A)
Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C/C272A/C283A) 1EV cleaved
to remove thioredoxin-strep moiety was used as the starting material The
monomer was
buffered exchanged once to 50mM Tri s-HC1, 150mM NaC1, pH7 to remove DTT
solution.
3u1 of 10mg/mL maleimide-Isoleucine adduct in DMF was added to 120 uL of 0.6
mg/ml
buffer exchanged Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/163C/C272A/C283A). The solution was
left at RT for 16h. Unreacted linker molecules were removed from the solution
by
centrifugation at 1500rcf for 2 minutes through a 7K MWCO desalting column.
The flow
through was added to equilibrated StrepTactinTm Sepharose High Performance
resin (GE
Healthcare Life Sciences, Cat No. 28935600) and left on a rotator for 2h at 23
C, allowing
modified monomers to bind to the beads via their biotin tag.
Unbound/unmodified protein
monomers were removed via centrifugation at 1500rcf for 2 minutes in three
wash steps
using buffer. After washing, the same buffer was added to the StrepTactinTm
resin and this
was subjected to UV light in two 30 second intervals to cleave the photo-
cleavable linker,
releasing modified monomers from the beads which were collected in the flow
through as
the modified monomer - Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C-
Malemide-PNA(Thymine)/C272A/C283A). Successful modification was checked by SDS-
PAGE electrophoresis on a 4-20% gel. (Figure 5)
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
89
3- Chemical modification of Gamma-Hemolysin mutant, LukF-T142C-De1(E1-K15)
LukF-T142C-Del(E1-K15) TEV monomer was buffered exchanged once to 50mM Tris-
HC1, 150mM NaC1, pH7 to remove DTT solution. 3u1 of 10mg/mL maleimide-Alanine
adduct in DMF was added to 120 uL of 0.6 mg/ml buffer exchanged LukF-T142C-
Del(E1-
K15). The solution was left at RT for 16h. Unreacted linker molecules were
removed from
the solution by centrifugation at 1500rcf for 2 minutes through a 7K MWCO
desalting
column. The flow through was added to equilibrated StrepTactinTm Sepharose
High
Performance resin (GE Healthcare Life Sciences, Cat No. 28935600) and left on
a rotator
for 2h at 23 C, allowing modified monomers to bind to the beads via their
biotin tag.
Unbound/unmodified protein monomers were removed via centrifugation at 150Orcf
for 2
minutes in three wash steps using buffer. After washing, the same buffer was
added to the
StrepTactinTm resin and this was subjected to UV light in two 30 second
intervals to cleave
the photo-cleavable linker, releasing modified monomers from the beads which
were
collected in the flow through as the modified monomer - LukF-T142C-Maleimide-
Al anine-Del(E1 -K15) Successful modification was checked by SDS-PAGE
electrophoresis on a 4-20% gel. (Figure 6)
4 - Chemical modification of Gamma-Hemolysin mutant, H1g2-G114C-Del(El-G10)
H1g2-G114C-Del(E1-G10) monomer was buffered exchanged once to 50mM Tris-HC1,
150mM NaC1, pH7 to remove DTT solution. 3u1 of 10mg/mL maleimide-Isoleucine
adduct
in DMF was added to 120 uL of 0.6 mg/ml buffer exchanged H1g2-G114C-Del(E1-
G10).
The solution was left at RT for 16h. Unreacted linker molecules were removed
from the
solution by centrifugation at 1500rcf for 2 minutes through a 7K MWCO
desalting column.
The flow through was added to equilibrated StrepTactinTm Sepharose High
Performance
resin (GE Healthcare Life Sciences, Cat No. 28935600) and left on a rotator
for 2h at
23 C, allowing modified monomers to bind to the beads via their biotin tag.
Unbound/unmodified protein monomers were removed via centrifugation at 150Orcf
for 2
minutes in three wash steps using buffer. After washing, the same buffer was
added to the
StrepTactinTm resin and this was subjected to UV light in two 30 second
intervals to cleave
the photo-cleavable linker, releasing modified monomers from the beads which
were
collected in the flow through as the modified monomer - H1g2-G114C-Maleimide-
Isoleucine-Del(E1-G10). Successful modification was checked by SDS-PAGE
electrophoresis on a 4-20% gel. (Figure 7)
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
5¨ Further chemical modifications of Gamma-Hemolysin mutant, LukF-T142C-
Del(E1-K15) and H1g2-N130C-Del(E1-G10)
Monomers of LukF and H1g2 mutant subunits of gamma-hemolysin were modified
with
5 maleimide-asparagine, maleimide-CH2-NH2, maleimide-arginine, maleimide-
isoleucine
and maleimide-aspartic acid in accordance with the procedures set out above.
Successful
modification was confirmed by SDS-PAGE electrophoresis (Figures 14 and 15).
Example 3
Oligomerisation of modified lysenin monomers to form an Oligomeric pore
1 ¨ Lysenin ¨ ILys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Nlaleimide-
lcoleucine/C272A/C283A)19
A custom 5-lipid mix designed to replicate rabbit blood cells was ordered from
Encapsula Nanosciences with the following composition: Phosphatidylserine
(0.325mg/m1),
POPE (0.55mg/m1), Egg PC (0.9mg/m1), Sphingomyelin (0.275mg/m1), Cholesterol
(0.45mg/m1). Modified monomer, Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A) solution and five lipid mix were combined in a 1.1
ratio, mixed
well and left at 23 C for 16h. The mixture was spun in a centrifuge at
21,000rcf for 10
minutes to pellet the sphingomyelin/pore complex. The supernatant was removed,
the
pellet resuspended in buffer (50mM Tris, 150mM NaCl, 2% SDS, pH9) and the
mixture
heated for 2h at 60 C to release the formed pore from the lipid-protein
complex. The tube
was allowed to cool down to room temperature before centrifugation at
21,000rcf for 10
minutes. The supernatant was collected and diluted 10-fold in 50mM Tris, 150mM
NaC1,
pH9 to give a final SDS concentration of 0.2% in the final pore solution.
Successful
oligomerisation was checked by SDS-PAGE electrophoresis on a 4-20% gel.
(Figure 8)
2¨ Lysenin ¨ [Lys-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/T63C-Maleimide-
PNA(Thymine)/C272A/C283A)19
A custom 5-lipid mix designed to replicate rabbit blood cells was ordered from
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
91
Eneapsula Nanosciences with the following composition: Phosphatidylserine
(0.325mg/m1),
POPE (0.55mg/m1), Egg PC (0.9mg/m1), Sphingomyelin (0.275mg/m1), Cholesterol
(0.45mg/m1). Modified monomer, Lys-
(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G/S98C-Maleimide-
Icoleucine/C272A/C283A) solution and five-lipid mix were combined in a 1:1
ratio, mixed
well and left at 23 C for 16h. The mixture was spun in a centrifuge at
21,000rcf for 10
minutes to pellet the sphingomyelin/pore complex. The supernatant was removed,
the
pellet resuspended in buffer (50mM Tris, 150mM NaCl, 2% SDS, pH9) and the
mixture
heated for 2h at 60 C to release the formed pore from the lipid-protein
complex. The tube
was allowed to cool down to room temperature before centrifugation at
21,000rcf for 10
minutes. The supernatant was collected and diluted 10-fold in 50mM Tris, 150mM
NaC1,
pH9 to give a final SDS concentration of 0.2% in the final pore solution.
Successful
oligomerisation was checked by SDS-PAGE electrophoresis on a 4-20% gel.
(Figure 9)
Example 4
Oligomerisation of modified gamma-hemolysin monomers to form an oligomeric
pore
Generic oligomerisation protocol for modified Gamma-Hemolysin pore
Monomer solutions were each concentrated to 0.5mg/m1 and LukF and H1g2
components
were combined in a 1:1 ratio. 'Leukocyte' liposomes were added (17% final
concentration)
and the solutions were incubated at 37 C for 16h to allow oligomerisation to
take place.
SDS was added to the oligomer samples (1.7 % final concentration) and were
bath
sonicated for 10 minutes. Successful oligomerisation was checked by SDS-PAGE
electrophoresis on a 4-20% gel.
'Leukocyte' Preparation
To prepare liposomes with the lipid composition of human leukocytes, 965111 of
5mg/m1
Phosphatidyl Choline (38.6%), 1970 of 25mg/m1Phosphatydulethanoamine (33.4%),
18.75 1 of 100mg/m1Phosphatidylserine (15%), 13.10 of 100mg/m1 Sphingomyelin
(10.5%) and 6.5 1 of 25mg/m1 Cardiolipid (1.3%) (all from Avanti Polar Lipids)
in
chloroform were combined in a glass vial and bath sonicated for 15 minutes. A
stream of
nitrogen was used to evaporate the organic solvent and lml of buffer (50mM
HEPES,
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
92
30mM NaC1, pH7.5) was added to the vial to solubilise lipids. The mixture was
vortexted
before being bath sonicated for a further 15 minutes. This was then extruded
through a
0.4ttm filter. Liposomes stored at 4 C.
1 - Gamma-Hemolysin ¨ [(LukF-T142C-Maleimide-Iso1eucine-De1(E1-K15))4 (H1g2-
G114C-Maleimide-Alanine -De1(E1-G10))41
Modified monomer solutions, (LukF-T142C-Maleimide-Isoleucine-Del(El-K15)) and
(H1g2-G114C-Maleimide-Alanine-Del(E1-G10) were each concentrated to 0.5mg/m1
and
LukF and H1g2 components were combined in a 1:1 ratio. 'Leukocyte' liposomes
were
added (17% final concentration) and the solutions were incubated at 37 C for
16h to allow
oligomerisation to take place. SDS was added to the oligomer samples (1.7 %
final
concentration) and were bath sonicated for 10 minutes. Successful
oligomerisation was
checked by SDS-PAGE electrophoresis on a 4-20% gel (Rubi staining) ¨ Figure 10
(lane
3).
2 - Gamma-Ilemolysin ¨ [(LukF-T142C-Maleimide-Isoleueine-Del(El-K15))4 (1-11g2-
G114C-Maleimide-Isoleueine -De1(E1-G10))41
Modified monomer solutions, (LukF-T142C-Mal eimi de-Isol euci ne-Del (El -
K15)) and
(H1g2-G114C-Maleimide- Isoleucine-Del(E1 -G10) were each concentrated to
0.5mg/m1
and LukF and H1g2 components were combined in a 1:1 ratio. 'Leukocyte'
liposomes were
added (17% final concentration) and the solutions were incubated at 37 C for
16h to allow
oligomerisation to take place. SDS was added to the oligomer samples (1.7 %
final
concentration) and were bath sonicated for 10 minutes. Successful
oligomerisation was
checked by SDS-PAGE electrophoresis on a 4-20% gel (Rubi staining) ¨ Figure 10
(lane
4).
3- Gamma-Hemolysin ¨ [(LukF-T142C-Maleimide-Alanine-De1(E1-K15))4 (H1g2-
G114C-Maleimide-Alanine-De1(E1-G10))41
Modified monomer solutions, (LukF-T142C-Maleimide- Alanine -Del(E1-K15)) and
(H1g2-G114C-Maleimide-Alanine-Del(E1-G10) were each concentrated to 0.5mg/m1
and
LukF and H1g2 components were combined in a 1:1 ratio. 'Leukocyte' liposomes
were
added (17% final concentration) and the solutions were incubated at 37 C for
16h to allow
oligomerisation to take place. SDS was added to the oligomer samples (1.7 %
final
concentration) and were bath sonicated for 10 minutes. Successful
oligomerisation was
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
93
checked by SDS-PAGE electrophoresis on a 4-20% gel (Rubi staining) ¨ Figure 10
(lane
5).
4 - Gamma-Hemolysin ¨ [(LukF-T142C-Maleimide-Alanine-Del(El-K15))4 (H1g2-
G114C-Maleimide-Isoleucine-Del(El-G10))41
Modified monomer solutions, (LukF-T142C-Maleimide- Alanine-Del(El-K15)) and
(H1g2-G114C-Maleimide-Iso1eucine-Del(E1-G10) were each concentrated to
0.5mg/m1
and LukF and H1g2 components were combined in a 1:1 ratio. 'Leukocyte'
liposomes were
added (17% final concentration) and the solutions were incubated at 37 C for
16h to allow
oligomerisation to take place. SDS was added to the oligomer samples (1.7 %
final
concentration) and were bath sonicated for 10 minutes. Successful
oligomerisation was
checked by SDS-PAGE electrophoresis on a 4-20% gel (Rubi staining) ¨ Figure 10
(lane
6).
5 - Gamma-Hemolysin ¨ 1(LukF-T142C-Maleimide-holeueine-Del(E1-K15))4 (H1g2-
WT)41
Modified monomer solution (LukF-T142C-Maleimide-Isoleucine-Del(El-K15)) and
wild-
type (WT) H1g2 components were combined and oligomerised as discussed above.
Successful oligomerisation was confirmed by SDS-PAGE as above (Figure 16, lane
3).
Example 5
General methods, Protein synthesis and purification
DNA synthesis (Leukocidin and Lysenin)
All constructs were cloned in the pT7 expression vector and verified by Sanger
Sequencing
(Source Bioscience). The genes encoding point mutations were generated by PCR
mutagenesis using Restriction Endonucleases (NdeI/HindIII). A set of cloning
primers
were designed to introduce the desired mutation on a DNA template. The DNA
template
has been digested at specific recognition sites using Restriction Enzymes
(NdeI/EconI -
HindIII/BamHI). Mutagenic primers were then added to the digested DNA template
and a
PCR reaction was performed using a Q5HotStart DNA Polymerase (NEB, Cat No.
M0494S). Gene encoding the point mutation was generated by in vivo
Recombination,
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
94
using XL10Gold Ultracompetent Cells (Agilent, Cat No. 200315) and ampicillin-
agar
plates. One colony was used to inoculate LB media and grown at 37 C for 16h. A
plasmid
prep of the DNA was made using Qiagen Plasmid Plus Midi Kit (Qiagen, Cat No.
12945).
Protein Expression and Purification (Leukocidin)
H1g2, LukF and their mutants were overexpressed and purified separately as
soluble
monomeric proteins. Transformed Escherichia coli strain Lemo21(DE3) (NEB, Cat
No.
C2528J) harbouring the expression vector encoding the desired LukF or H1g2
monomer
was grown at 37 C in LB media supplemented with 4lug/uL chloramphenicol and
10Oug/uL carbenicillin until logarithmic growth phase was achieved. Expression
of the
desired protein was induced using Isopropyl-13-D-thiogalactoside (IPTG) to a
final
concentration of 0.5mM. The temperature was reduced to 25 C and allowed to
express for
18h. Cells were harvested by centrifugation at 6000xg for 20 minutes at 4 C
and pellets
resuspended in buffer (50mM HEPES, 300mM NaCl, 2mM EDTA, 0.1% DDM,
lxbugbuster, benzonase nuclease, protease inhibitor tablets, pH8) before being
disrupted
by sonication and left to lyse for 4h on a magnetic stirrer. Lysate was
clarified at 39,000xg
for 35 minutes at 4 C. The supernatant was diluted 10-fold with 50mM HEPES and
loaded
onto a HS50 cation exchange column (Poros Media by Applied BioSciences) where
an
increasing NaCl gradient was used for elution. Fractions containing the
desired protein
were further purified on an Superdex 75 10/300 GL size exclusion column (GE
Healthcare
Life Sciences).
Protein Expression and Purification (Lysenin)
Transformed Escherichia coli strain BL21(DE3) (NEB, Cat No. C2527H) harbouring
an
expression vector encoding the desired lysenin monomer with an thioredoxin-
strep moiety
was grown at 37 C in TB media supplemented with 0.1mg/m1 ampicillin until
logarithmic
growth phase was achieved (0D600 0.8). Expression of the desired protein was
induced
using Isopropyl-13-D-thiogalactoside (IPTG) to a final concentration of 0.5mM.
The
temperature was reduced to 18 C and allowed to express for 16h. Cells were
harvested by
centrifugation at 6000xg for 20 minutes at 4 C. Pellets were resuspended in
buffer (50mM
Tris-HC1, 0.5M NaC1, 2mM DTT, benzonase nuclease, protease inhibitors, 2mM
EDTA,
pH 8.0) and left on a magnetic stirrer for 3h before being disrupted by
sonication. Lysate
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
was clarified at 45,000xg for 60 minutes at 4 C. The supernatant was loaded
onto 5m1
StrepTrapTm High Performance columns (GE Healthcare Life Sciences, Cat No.
28907548)
and proteins were eluted with 2mg/m1 desthiobiotin. Fractions containing the
desired
protein were further purified on an Superdex200 increase 10/300 GL size
exclusion
5 column (GE Healthcare Life Sciences, Cat No. 28990944) and analysed by
SDS-PAGE.
Peak fractions were pooled and cleaved with TEV-strep protease to remove the
thioredoxin-strep moiety. The mixture was loaded back onto a StrepTrapTm High
Performance column to gain untagged lysenin monomers in the flow through. The
final
elution was concentrated using an Amicon Ultra-15 Centrifugal Filter Unit
(Merck
10 Millipore, Cat No. UFC901024).
Example 6
Oligomerised chemically modified pores produced in accordance with the
disclosed
15 methods were assembled into nanopore detection devices and used to
characterise a
polynucleotide analyte.
Electrical methods
20 Electrical measurements were acquired from a variety of lysenin
nanopores (chemically
modified in accordance with the disclosed methods, and unmodified) inserted
into block
co-polymer membrane of MinION flow cells (Oxford Nanopore Technologies).
Lysenin
pore samples in 0.2% SDS (V/V) were incubated with Brij58 (final concentration
of 0.1%
(V/V)) for 10 minutes at room temperature before diluting the pore samples
(0.05 ug/mL)
25 in MinION flow cell buffer (25 mM potassium phosphate, 150 mM potassium
ferrocyanide, 150 mM potassium ferricyanide, pH 8.0) for pore insertion. All
pore
experiments were done on MinION Mklb devices (Oxford Nanopore Technologies,
ONT).
MinKNOW core 1.11.5 version software developed and provided by ONT was used to
control scripts during all experiments.
Pore insertion
For insertion of pores, 300 p1. of diluted pore samples were loaded into the
priming port of
the flow cell. The pore insertion script of MinKNOW was used to apply voltage
starting
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
96
from -100 mV, increasing 10 mV every 15 seconds up until -450 mV. 1 mL of flow
cell
buffer was perfused through the priming port to remove any excess pores.
Groups and
positions with single pores were evaluated using the standard flow cell check
protocol
using MinKNOW.
3.6 kb Library / Analyte preparation
A double stranded 3.6 kb DNA analyte (SEQ ID NO: 20) was prepared using
specific
primers and PCR. The PCR product was subjected to NEBNext end repair, NEBNext
dA-
tailing modules (New England Biolabs (NEB)), to generate a 3' A overhangs.
Ligation of Y adapter to analyte
1 pg of 3.6 kb analyte was ligatcd to AMX from Oxford Nanopore Technologies
sequencing kit (LSK-SQK109) in a 100 [IL volume using LNB from (LSK-SQK109)
and
T4 DNA Ligase (NEB). The sample was purified using Agencourt AMPure XP
(Beckman
Coulter) beads, with two washes with LFB/SFB from Oxford Nanopore Technologies
sequencing kit (LSK-SQK109). The ligated substrate was eluted into EB from
(LSK-
SQK109), all following the manufacturer's guidelines. This is also referred to
as "3.6kb
Lambda DNA library".
Flow cell loading
For flowcell loading, 800 [IL of FLB from Oxford Nanopore Technologies
sequencing kit
(SQK-LSK109) prepared with FLT (SQK-LSK109) was flowed through the system, 5
minute wait, then 200 pi- of FLB + FT was flowed through the system with the
SpotON
port opened. 37.5 pL SQB from Oxford Nanopore Technologies sequencing kit (SQK-
LSK109), 12 ..IL of the recovered bead purified 3.6kb Lambda DNA library
(preparation
explained below) and 25.5 1_, of LLB (SQK-LSK109) were mixed. 75 p1_, of the
3.6 kb
sequencing mix was added to a MinION flowcell, using the SpotOn Flowcell Port.
75 [LI,
of the sequencing mix was added to a MinION flowcell, using the SpotOn
Flowcell Port.
The raw data was collected using MinKNOW software (Oxford Nanopore
Technologies)
at -180 mV (4000 kHz acquisition frequency) and helicase-controlled DNA
movement
monitored.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
97
Figure 11 shows characterisation data for the polynucleotide analyte of SEQ ID
NO: 20
when characterised using a panel of chemically modified lysenin pores produced
in
accordance with the disclosed methods. Two lysenin pores:
Lys-(E84Q/E85S/E92Q/E94D/E975/598C/T106K/D126G/C272A/C283A) and
Lys-(T63C/E84Q/E85S/E92Q/E94D/E975/T106K/D126G/C272A/C283A)
were modified with 5 different chemical molecules and the effect of the
modification was
analysed by comparing the open pore current level of the modified pore against
the
unmodified pore when the 3.6 lambda DNA is passing through each pore. The
modifications made to the lysenin pores covered aromatic, aliphatic,
hydrophobic,
hydrophilic, positive charge and H-bonding residues including DNA base
attachment ¨
PNA(Thymine).
Compared to the unmodified pore, the open pore current levels of the modified
pores
change significantly as a result of the change in diameter and change in
chemical
environment within the barrels
Figure 12 shows enhanced details of the signal observed as the 3.6Kb Lambda
DNA
translocates through the modified lysenin mutant pores, as a function of the
modifier
molecule. Figure 12(A) shows data obtained from the unmodified pore
Ly s-(E84Q/E85S/E92Q/E94D/E975/S98C/T106K/D126G/C272A/C283A).
Figure 12(B) shows corresponding data from the modified pore
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
isoleucine/T106K/D126G/C272A/C283A).
For each of Figures 12(A) and 12(B) the left panel shows an event of the
entire 3.6Kb
DNA passing through the pore. As can be seen, the open pore current level
(240pA in A
and 250pA in B) drops down to about 160pA when the DNA is passing through the
pore.
Fluctuations of the current at 160pA indicate different base compositions
passing through
the narrowest region (constriction) of the pore. The right panel shows the
expanded
version of the current-time data "squiggle" for 0.4 seconds. The shape and
range (current
variation within the squiggle level) vary significantly between the unmodified
and
modified pores.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
98
Figure 13 shows changes in the GGAA region of the 3.6Kb Lambda DNA (residues
38-41
and 52-55 of SEQ ID NO: 20) as it translocates through lysenin mutant pores as
a function
of the modifier molecule. The DNA analyte was translocated through an
unmodified
lysenin pore
(Lys-(E84Q/E85S/E92Q/E94D/E97S/598C/T106K/D126G/C272A/C283A); panel A)
and the signal obtained compared to that for pores modified in accordance with
the
methods described herein:
(Lys-(E84Q/E85S/E92Q/E94D/E97S/598C-maleimide-
Isoleucine/T106K/D126G/C272A/C283A); panel B and
Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
PNA(Thymine)/T106K/D126G/C272A/C283A; panel C).
The GGAA signal section is circled in each of the traces. As can be seen, the
G and A
levels of the signal obtained from the modified pore is different in the
modified pores
compared to the unmodified pore, with a particularly significant alteration in
the signal
obtained from the Lys-(E84Q/E85S/E92Q/E94D/E97S/S98C-maleimide-
PNA(Thymine)/T106K/D126G/C272A/C283A pore (panel C)
Figure 17 shows representative characterisation data for the polynucleotide
analyte of SEQ
ID NO: 20 when characterised using (left) unmodified Gamma-Hemolysin ¨ [(LukF-
T142C-Del(E1-K15))4 (H1g2-WT)4] pores produced and oligomerised as discussed
above,
as compared to corresponding modified Gamma-Hemolysin ¨ [(Luk_F-T142C-
Maleimide-
Aspartate-Del(E1-K15))4 (H1g2-WT)4] pores produced and oligomerised as
discussed
above. Panel A shows the unexpanded trace. Compared to the unmodified pore,
the open
pore current levels and translocation characteristics of the polynucleotide
analyte vary
significantly between the unmodified pore and the modified pore, as a result
of the change
in diameter and change in chemical environment within the barrels resulting
from the
modification. Panel B shows changes in the GGAA region of the 3.6Kb Lambda DNA
(residues 38-41 and 52-55 of SEQ ID NO: 20) as it translocates through the
pores. The
GGAA signal section is circled in each of the traces. As can be seen, the G
and A levels of
the signal obtained from the modified pore is different in the modified pores
compared to
the unmodified pore.
CA 03193980 2023- 3- 27
WO 2022/074397 PCT/GB2021/052609
99
Example 7
Modification of alpha hemolysin (a-HL) monomers, oligomerisation, and analyte
characterisation
Modification to alpha hemolysin (a-HL) monomers (Ml 13C) with maleimide-
isoleucine
was carried out in accordance with the general modification method explained
in the
previous sections. Once the modified monomers were obtained they were combined
with
five-lipid mix in 1:1 ratio and left at 23 C for 16h. The mixture was spun in
a centrifuge at
21,000 ref for 10 minutes to pellet the sphingomyelin/pore complex. The
supernatant was
removed, the pellet resuspended in buffer (50mM Tris, 150mM NaCl, pH8) and the
mixture heated for 2h at 60 C to release the formed pore from the lipid-
protein complex.
The tube was allowed to cool down to room temperature before centrifugation at
21,000rcf
for 10 minutes. The supernatant was collected and diluted 10-fold in 50mM
Tris, 150mM
NaCl, p118 to give a final SDS concentration of 0.2% in the final pore
solution. Successful
oligomerisation was checked by SDS-PAGE electrophoresis on a 4-20% gel
Figure 18A shows modification of a-I-IL M113C monomers (Lane 1 ¨ Unmodified
protein
monomer, a-HL-M113C; Lane 2 ¨ Crude reaction mixture - Maleimide-Isoleucine
(Mal-
Re) adduct with a-HL-M113C; Lane 3 ¨ Flow through after equilibrating the
modified
protein monomers on StrepTactin beads (flow through indicates unmodified
protein
monomers unbound to StrepTactin beads); Lane 4/5 ¨ Further washings of the
modified
monomer bound StrepTactin beads to remove unmodified protein monomer (gel
indicates
no unmodified monomer eluting in further washes); Lane 6¨ Modified a-HL-M113C
with
Maleimide-Isoleucine (Mal-Ile)). Figure 18B shows the oligomerisation of
modified a-
HL-113C-maleimide-isoleucine to form a homooligomeric heptameric pore (Lane 1
¨
modified protein monomer, a-HL-M113C-Mal-Ile; Lane 2 ¨ Crude reaction mixture
of
a-HL-M113C-Mal-Ile with 1:1 Sphingomyelin after overnight storage. Lane 3 ¨
Supernatant after centrifuging to separate pore-sphingomyelin pellet. Some
pores are seen
in supernatant as well.; Lane 4 ¨ Modified (I-IL-M113C-Mal-Ile)7 heptameric
oligomerised
pore.).
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
100
The modified a-HL heptameric pores were used to characterise a DNA analyte as
described above and the characterisation data compared with corresponding data
from
unmodified pores. Representative data is shown in Figure 19.
Figure 19A shows electrophysiology data for unmodified ((a-HL-M1 13C)7, left
panel) and
modified ((a-HL-M113C-Ma1-Ile)7, right panel) pores. Open pore current of the
unmodified pores is higher than the modified pores, which (without being bound
by
theory) is considered to arise as the internal diameter of the pore is reduced
after
modification. The drop in current (delta) when DNA is translocating through
the pore is
higher for the unmodified pore (delta ¨35 pA) compared to the modified pore
(delta ¨25
pA). Open pore noise is also higher for the unmodified pore compared to the
modified
pore. These indicate the modified pore has changed the characteristics of the
DNA
squiggle compared to the unmodified version. Figure 19B shows a zoomed-in
region of
the data in Figure 19A. Figure 19C shows traces of the signal focussed on the
first 0.3
seconds after the sp18 signal from the analyte. Differences in the signal can
be clearly
detected arising from the modification in accordance with the methods herein
Example 8
Modification of cytotoxin-K (Cyt-K) monomers, oligomerisation, and analyte
characterisation
Modification to cytotoxin-K (Cyt-K WT-Q123S/K129C/E140S/Q146S-H6(C)) monomers
with maleimide-isoleucine was carried out in accordance as explained under the
general
modification method explained in the previous sections. Once the modified
monomers
were obtained (figure 1 ¨ modified with maleimide Isoleucine group ¨ Mal-Ile),
they were
combined with five-lipid mix in 1:1 ratio and left at 23 C for 16h. The
mixture was spun in
a centrifuge at 21,000 rcf for 10 minutes to pellet the sphingomyelin/pore
complex. The
supernatant was removed, the pellet resuspended in buffer (50mM Tris, 150mM
NaCl,
pH8) and the mixture heated for 2h at 60 C to release the formed pore from the
lipid-
protein complex. The tube was allowed to cool down to room temperature before
centrifugation at 21,000rcf for 10 minutes. The supernatant was collected and
diluted 10-
fold in 50mM Tris, 150mM NaCl, pH8 to give a final SDS concentration of 0.2%
in the
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
101
final pore solution. Successful oligomerisation was checked by SDS-PAGE
electrophoresis
on a 4-20% gel.
Figure 20A shows modification of Cyt-K monomers (Lane 1 ¨ Unmodified protein
monomer, Cyt-K(WT-Q123S/K129C/E140S/Q146S); Lane 2 ¨ Crude reaction mixture -
Maleimide-Isoleucine (Mal-Ile) adduct with Cyt-K(WT-Q123S/K129C/E140S/Q146S);
Lane 3 ¨ Flow through after equilibrating the modified protein monomers on
StrepTactin
beads (flow through indicates unmodified protein monomers unbound to
StrepTactin
beads); Lane 4/5 ¨ Further washings of the modified monomer bound StrepTactin
beads to
remove unmodified protein monomer (gel indicates no unmodified monomer eluting
in
further washes); Lane 6¨ Modified Cyt-K(WT-Q123S/K129C/E140S/Q146S) with
Maleimide-Isoleucine (Mal-Ile)). Figure 20B shows the oligomerisation of
modified Cyt-
K WT-Q123S/K129C-maleimide-isoleucine/E140S/Q146S to form a homooligomeric
pore
(Lane 1 ¨ Modified protein monomer, CytK-(WT-Q123S/K129C-Mal-Ile/E140S/Q146S;
Lane 2¨ Crude reaction mixture of CytK-(WT-Q123S/K129C-Ma1-Ile/E140S/Q146S
with
1.1 Sphingomyelin after overnight storage Lane 3 ¨ Supernatant after
centrifuging to
separate pore-sphingomyelin pellet. Some pores are seen in supernatant as
well.; Lane 4 ¨
Modified (CytK -(WT-Q 123 S/K129C-Mal -Ile/E140S/Q146S)7 pore.).
The modified Cyt-K pores were used to characterise a DNA analyte as described
above
and the characterisation data compared with corresponding data from unmodified
pores.
Representative data is shown in Figure 21.
Figure 21A shows electrophysiology data for unmodified CytK-(WT-
Q123 S/K129S/E140S/Q146S)7 and modified CytK-(WT-Q123S/K129C-Ma1-
Ile/E140S/Q1465)7 pores. Both open pore current and sequencing current (i.e.
squiggle
level) of the unmodified pores is higher than the modified pores, which
(without being
bound by theory) is considered to arise as the internal diameter of the pore
is reduced after
modification. This data indicates the modified pore has changed the
characteristics of the
DNA squiggle compared to the unmodified version. Figure 21B shows
characteristic data
from the poly-T,GGAA region in the 3.6 Kb asymmetric library, zoomed into the
first 0.3 s
of the trace following the sp18 signal from the analyte. Both first and second
poly-T
regions can be clearly seen in the unmodified pore (underlined; poly-T 1 and
2), whereas in
the unmodified pore the first poly-T region cannot be distinguished and the
signal from the
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
102
second poly-T region is more dominant. This data confirms that the modified
pore has
changed the characteristics of the DNA squiggle compared to the unmodified
version.
Example 9
The benefits of the methods provided herein compared to other modification
methods are
illustrated in this example.
As discussed herein, achieving 100% reaction efficiency is typically
impossible in a
reaction. If a modification is effected on a purified protein monomer of an
oligomeric pore
then heterogeneity arises in the pore population, with a mixture of modified
and
unmodified protein being obtained.
This is illustrated in Figure 22 which shows the results of modifying a
lysenin monomer
(Lys-(/E84Q/E85K/E92Q/E94D/E97S/S98C/T106K/D126G/C272A/C283A)) with
maleimide-isoleucine with a cleavable purification tag_ The maleimide-
isoleucine reacts at
position S98C. Lane 1 shows the unmodified starting material. Lane 2 shows
crude
incomplete reaction mixture containing both modified and unmodified protein
monomers.
Oligomerising with this monomer mixture would lead to heterogeneous pore
populations.
The effects of modifications on electrophysiological data obtained from
oligomerised
protein pores are illustrated above with significant differences arising from
modified pores
compared to unmodified pores. If the monomers of the crude reaction mixture of
Lane 2
were to be used to produce pores for electrophysiological experiments then the
data
obtained would depend on the position and number of modifications within the
pore and
hence would be difficult or impossible to interpret. Nor can chromatography be
used to
purify crude reaction mixtures e.g. prior to oligomerisation. The molecular
weight
difference between modified and non-modified pore monomers may be small
preventing
efficient separation meaning that homogeneous pores still cannot be readily of
efficiently
produced. Even if such purification could be achieved in some cases,
exhaustive method
development would be required depending on the monomer and the functionalities
of the
modifier.
By contrast, the methods disclosed herein produce a homogeneous population, as
all
unmodified monomers can be eliminated, e.g. by washing or elution whereas
modified
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
103
monomers bind to a support material. This is illustrated in lane 3 of Figure
22 which
shows that non-modified monomers are eluted. This method is applicable to
modify and
purify the modified pore monomers independent of the substrate without the
requirement
for individual method development.
Protein monomers modified in accordance with the methods disclosed herein can
be
released from the support material cleanly by cleaving the purification tag.
As shown in
Lane 6 of Figure 22, the modified monomers are obtained without contamination
from
unmodified monomers. Furthermore, the methods disclosed herein are amenable to
a wide
variety of proteins without requiring specific purification strategies to be
developed for
each monomer of interest.
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
104
Description of the Sequence Listing
SEQ ID NO: 1 shows the amino acid sequence of (hexa-histidine tagged)
exonuclease I
(EcoExo I) from E. coil.
SEQ ID NO: 2 shows the amino acid sequence of the exonuclease III enzyme from
E. colt.
SEQ ID NO: 3 shows the amino acid sequence of the RecJ enzyme from I: therm
ophilus
(TthRecJ-cd).
SEQ ID NO: 4 shows the amino acid sequence of bacteriophage lambda
exonuclease. The
sequence is one of three identical subunits that assemble into a trimer.
(http://www.neb.com/nebecomm/products/productM0262.asp).
SEQ ID NO: 5 shows the amino acid sequence of Phi29 DNA polymerase from
Bacillus
subtilis phage Phi29.
SEQ ID NO: 6 shows the amino acid sequence of Trwc Cba (Citromicrobium
bathyomarinum) helicase.
SEQ ID NO: 7 shows the amino acid sequence of He1308 Mbu (Methanococcoides
burtonii) helicase
SEQ ID NO: 8 shows the amino acid sequence of the Dda helicase 1993 from
Enterobacteria phage T4.
SEQ ID NO: 10 shows the amino acid sequence of the LukF subunit of gamma-
hemolysin
(Del 1-15).
SEQ ID NO: 11 shows the amino acid sequence of the H1g2 subunit of gamma-
hemolysin
(Del 1-10).
SEQ ID NO: 12 shows the amino acid sequence of a monomer of lysenin-
(E84Q/E85 S/E92Q/E94D/E97 S/T 106K/D 126G).
SEQ ID NO: 13 shows the amino acid sequence of a monomer of cytotoxin K from
Bacillus cereus (CytK).
SEQ ID NO: 14 shows the amino acid sequence of a monomer of aerolysin.
SEQ ID NO: 15 shows the amino acid sequence of a monomer of Necrotic enteritis
toxin B
(NetB) from Clostridium perfringens
ID NO: 16 shows the amino acid sequence of a monomer of alpha-hemolysin (a-
HL).
SEQ ID NO: 17 shows the amino acid sequence of a monomer of Vibrio cholera
cytolysin
(HlyA) / VCC.
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
105
SEQ ID NO: 18 shows the amino acid sequence of a monomer of Anthrax toxin
protective
antigen (Anthrax PA).
SEQ ID NO: 19 shows the amino acid sequence of a monomer of epsilon toxin (s-
Toxin).
SEQ ID NO: 20 shows the polynucleotide sequence of a 3.6 kb lambda DNA analyte
used
in the Examples.
SEQ ID NO: 21 shows the amino acid sequence of a monomer of Fragaceatoxin C
(FraC)
from Actinia fragacea.
SEQ ID NO: 22 shows the amino acid sequence of the PlyA monomer of the
pleurotolysin
PlyAB from Pleurotus ostreatus.
SEQ ID NO: 23 shows the amino acid sequence of the PlyB monomer of the
pleurotolysin
PlyAB from Pleurotus ostreatus.
SEQ ID NOs: 24-27 show the amino acid sequences of various protease-sensitive
peptide
linkers referred to herein.
SEQ ID NOs: 28-35 show the amino acid sequences of various peptide tags
referred to
herein.
SEQ ID NO. 36 shows the amino acid sequence of a polynucleotide repeating unit
referred
to herein.
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
106
SEQUENCE LISTING
SEQ ID NO: 1 - exonuclease I from E. coil
MMNDGKQQSTFLEHDYETEGTHPALDRPAQFAAIRTDSEENVIGEPEVEYCKPADDYLPQ
PGAVLITGITPQEARAKGENEAAFAARIHSLFTVPKTCILGYNNVRFDDEVTRNIFYRNF
YDPYAWSWQHDNSRWDLLDVMRACYALRPEGINWPENDDGLPSFRLEHLTKANGIEHSNA
HDAMADVYATIAMAKLVKTRQPRLFDYLFTHRNKHKLMALIDVPQMKPLVHVSGMFGAWR
GNTSWVAPLAWHPENRNAVIMVDLAGDISPLLELDSDTLRERLYTAKTDLGDNAAVPVKL
VHINKCPVLAQANTLRPEDADRLGINRQHCLDNLKILRENPQVREKVVAIFAEAEPFTPS
DNVDAQLYNGFFSDADRAAMKIVLETEPRNLPALDITFVDKRIEKLLENYRARNFPGTLD
YAEQQRWLEHRRQVFTPEFLQGYADELQMLVQQYADDKEKVALLKALWQYAEEIVSGSGH
HHHHH
SEQ ID NO: 2 - exonuclease III enzyme from E. coli
MKFVSFNINGLRARPHQLEAIVEKHQPDVIGLQETKVHDDMFPLEEVAKLGYNVEYHGQK
GHYGVALLTKETPIAVRRGEPGDDEEAQRRIIMAEIPSLLGNVTVINGYFPQGESRDEPI
KFPAKAQFYQNLQNYLETELKRDNPVLIMGDMNISPTDLDIGIGEENRKRWLRTGKCSFL
PEEREWMDRLMSWGLVDTERHANPQTADRFSWFDYRSKGFDDNRGLRIDLLLASQPLAEC
CVETGIDYEIRSMEKPSDHAPVWATFRR
SEQ ID NO: 3 - RecJ enzyme from T. thermophilus
MFRRKEDLDPPLALLPLKGLREAAALLEEALRQGKRIRVHGDYDADGLTGTAILVRGLAA
LGADVHPFIPHRLEEGYGVLMERVPEHLEASDLELTVDCGITNHAELRELLENGVEVIVT
DHHTPGKTPPPGLVVHPALTPDLKEKPTGAGVAELLLWALHERLGLPPPLEYADLAAVGT
IADVAPLWGWNRALVKEGLARIPASSWVGLRLLAEAVGYTGKAVEVAFRIAPRINAASRL
GEAEKALRLLLTDDAAEAQALVGELHRLNARRQTLEEAMLRKLLPQADPEAKAIVLLDPE
GHPGVMGIVASRILEATLRPVELVAQGKGTVRSLAPISAVEALRSAEDLLLRYGGHKEAA
GFAMDEALFPAFKARVEAYAARFPDPVREVALLDLLPEPGLLPQVFRELALLEPYGEGNP
EPLFL
SEQ ID NO: 4 - bacteriophage lambda exonuclease
MTPDIILQRTGIDVRAVEQGDDAWHKLRLGVITASEVHNVIAKPRSGKKWPDMKMSYFHT
LLAEVCTGVAPEVNAKALAWGKQYENDARTLFEFTSGVNVTESPIIYRDESMPTACSPDG
LCSDGNGLELKCPFTSRDFMKERLGGFEAIKSAYMAQVQYSMWVTRKNAWYFANYDPRMK
REGLHYVVIERDEKYMASFDEIVPEFIEKMDEALAEIGFVFGEQWR
SEQ ID NO: 5 - Phi29 DNA polymerase
MKHMPRKMYSCAFETTTKVEDCRVWAYGYMNIEDHSEYKIGNSLDEFMAWVLKVQADLYE
HNLKFDGAFIINWLERNGFKWSADGLPNTYNTIISRMGQWYMIDICLGYKGKRKIHTVIY
DSLKKLPFPVKKIAKDFKLTVLKGDIDYHKERPVGYKITPEEYAYIKNDIQIIAEALLIQ
FKQGLDRMTAGSDSLKGFKDIITTKKFKKVEPTLSLGLDKEVRYAYRGGFTWLNDRFKEK
EI GEGMVEDVNSLYPAQMYSRLLPYGEP IVFEGKYVWDEDYPLHI QH RCEEE LKEGYI P
TIQI KRSRFYKGNEYLKSSGGEIADLWLSNVDLELMKEHYDLYNVEYI S GLKFKATT GLF
KDFI DKWTYI KT T SEGA I KQLAKLMLN S LYGKFASN P DVTGKVP YLKEN GAL G FRL GEEE
TKDPVYTPMGVFI TAWARYTT I TAAQACYDRI I YCDT DS IH LT GT E I PDVIKDIVDP KKL
GYWAHE ST EKRAKYLRQ KT YI QD YMKEVDGKLVEGS PDDYTDIKFSVKCAGMTDKI KKE
VT FENTKVGFSRKMKPKPVQVPGGVVLVDDT FT I KS GGSAW S H PQ FEKGGGSGGGSGGSA
WS PQ FFK
SEQ ID NO: 6 - Trwc Cba helicase
MLSVANVRSPSAAASYFASDNYYASADADRSGQWIGDGAKRLGLEGKVEARAFDALLRGE
LPDGSSVGNPGQAHRPGTDLTFSVPKSWSLLALVGKDERIIAAYREAVVEALHWAEKNAA
ETRVVEKGMVVTQATGNLAIGLFQHDTNRNQEPNLHFHAVIANVTQGKDGKWRTLKNDRL
WQLNTTLNSIAMARFRVAVEKLGYEPGPVLKHGNFEARGISREQVMAFSTRRKEVLEARR
GPGLDAGRIAALDTRASKEGIEDRATLSKQWSEAAQSIGLDLKPLVDRARTKALGQGMEA
TRIGSLVERGRAWLSRFAAHVRGDPADPLVPPSVLKQDRQTIAAAQAVASAVRELSQREA
AFERTALYKAALDFGLPTTIADVEKRTRALVRSGDLIAGKGEHKGWLASRDAVVTEQRIL
SEVAAGKGDSSPAITPQKAAASVQAAALTGQGFRLNEGQLAAARLILISKDRTIAVQGIA
GAGKSSVLKPVAEVLRDEGHPVIGLAIQNTLVQMLERDTGIGSQTLARFLGGWNKLLDDP
GNVALRAEAQASLKDEVLVLDEASMVSNEDKEKLVRLANLAGVHRLVLIGDRKQLGAVDA
GKPFALLQRAGIARAEMATNLRARDPVVREAQAAAQAGDVRKALRHLKSHTVEARGDGAQ
VAAETWLALDKETRARTSIYASGRAIRSAVNAAVQQGLLASREIGPAKMKLEVLDRVNTT
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
107
REELRHLRAYRAGRVLEVSRKQQALGLFTGEYRVIGQDRKGKLVEVEDKRGKRFRFDPAR
IRAGKGDDNLTLLEPRKLEIHEGDRIRWTRNDHRRGLFNADQARVVEIANGKVTFETSKG
DLVELKKDDPMLKRIDLAYALNVHMAQGLTSDRGIAVMDSRERNLSNQKTFLVTVTRLRD
HLTLVVDSADKLGAAVARNKGEKASATEVTGSVKPTATKGSGVDQPKSVEANKAEKELTR
SKSKTLDFGI
SEQ ID NO: 7 - He1308 Mbu helicase
MMIRELDIPRDIIGEYEDSGIKELYPPQAEATEMGLLEKKNLLAAIPTASGKILLAELAM
IKAIREGGKALYIVPLRALASEKFERFKELAPEGIKVGISTGDLDSRADWLGVNDIIVAT
SEKTDSLLRNGTSWMDEITTVVVDEIHLLDSKNRGPTLEVTITKLMRLNPDVQVVALSAT
VGNAREMADWLGAALVLSEWRPTDLHEGVLFGDAINFPGSQKKIDRLEKDDAVNLVLDTI
KAEGQCLVFESSRRNCAGFAKTASSKVAKILDNDIMIKLAGIAEEVESTGETDTAIVLAN
CIRKGVAFHHAGLNSNHRKLVENGFRQNLIKVISSTPTLAAGLNLPARRVIIRSYRRFDS
NEGMQPIPVLEYKQMAGRAGRPHLDPYGESVLLAKTYDEFAQLMENYVEADAEDIWSKLG
TENALRTHVLSTIVNGFASTRQELFDFFGATFFAYQQDKWMLEEVINDCLEFLIDKAMVS
ETEDTEDASKLFLRGTRLGSLVSMLYTDPLSGSKTVDGEKDICKSTGGNMGSLEDDKCDD
ITVTDMTLLHLVCSTPDMRQLYLRNTDYTIVNEYIVAHSDEFHEIPDKLKETDYEWFMGE
VKTAMLLEEWVTEVSAEDITRHENVGEGDIHALADTSEWLMHAAAKLAELLGVEYSSHAY
SLEKRIRYGSGLDLMELVGIRGVGRVRARKLYNAGFVSVAKLKGADISVLSKLVGPKVAY
NILSGIGVRVNDKHENSAPISSNTLDTLLDKNQKTENDFQ
SEQ ID NO: 8 - Dda helicase
MTEDDLTEGQKNAFNIVMKAIKEKKHHVTINGPAGTGKTTLIKFIIEALISTGETGIILA
APTHAAKKILSKLSGKEASTIHSILKINPVTYEENVLFEQKEVPDLAKCRVLICDEVSMY
DREILFKILLSTIPPWCTIIGIGDNKQIRPVDPGENTAYISPFETHKDFYQCELTEVKRSN
APIIDVATDVRNGKWIYDKVVDGHGVRGFTGDTALRDFMVNYFSIVKSLDDLFENRVMAF
TNKSVDKLNSIIRKKIFETDKDFIVGEIIVMQEPLFKTYKIDGKPVSEIIFNNGQLVRII
EAEYTSTFVKARCVPGEYLIRHWDLTVETYGDDEYYREKIKIISSDEELYKFNLFLGKTA
ETYKNWNKGGKAPWSDEWDAKSQFSKVKALPASTEHKAQGMSVDRAFIYTPCTHYADVEL
AQQLLYVGVTRGRYDVFYV
SEQ ID NO: 10 - LukF (gamma-hemolysin.)
AEGKITPVSVKKVDDKVTLYKTTATADSDKEKTSQILTENETKDKSYDKDTLVLKATGNT
NSGFVKPNPNDYDFSKLYWGAKYNVSISSQSNDSVNVVDYAPKNQNEEFQVQNTLGYTFG
GDISISNGLSGGLNGNTAFSETINYKQESYRTTLSRNTNYKNVGWGVEAHKIMNNGWCPY
GRDSFHPTYGNELFLAGRQSSAYAGQNFIAQHQMPLLSRSNENREFLSVLSHRQDGAKKS
KITVIYQREMDLYQIRWNGFYWAGANYKNEKTRTFKSTYEIDWENHKVKLLDTKETENNK
SEQ ID NO: 11 - H1g2 (gamma-hemolysin).
ENKIEDIGQGAEIIKRTQDITSKRLAITQNIQFDFVKDKKYNKDALVVKMQGFISSRTTY
SDLKKYPYIKRMIWPFQYNISLKTKDSNVDLINYLPKNKIDSADVSQKLGYNIGGNFQSA
PSIGGSGSFNYSKTISYNQKNYVTEVESQNSKGVKWGVKANSFVTPNGQVSAYDQYLFAQ
DPTGPAARDYFVPDNQLPPLIQSGFNPSFITTLSHERGKGDKSEFEITYGRNMDATYAYV
TRHRLAVDRKHDAFKNRNVTVKYEVNWKTHEVKIKSITPK
SEQ ID NO: 12 - lysenin-(E84Q/E85S/E92Q/E94D/E97S/T106K/D126G).
MSAKAAEGYEQIEVDVVAVWKEGYVYENRGSTSVDQKITITKGMKNVNSETRTVTATESIGSTISTGDAFEIG
SVEVSYSHSHQKSQVSMTQTDVYSSKVIEHTIKIPPTSKFTRWQLNADVGGAGIEYMYLIDEVTPIGGTQSIP
QVITSRAKIIVGRQIILGKTEIRIKHAERKEYMTVVSRKSWPAATLGHSKLFKFVLYEDWGGFRIKTLNTMYS
GYEYAYSSDQGGIYEDQGTDNPKQRWAINKSLPLRHGDVVTFMNKYFTRSGLCYDDGPATNVYCLDKREDKWI
LEVVG
SEQ ID NO: 13 - CytK (cytotoxin K from Bacillus cereus).
MQTTSQVVTDTGQNAKTHTSYNTENNEQADNMTMSLKVTFTDDPSADKQTAVINTTGSFM
KANPILSDAPVDGYPIPGASVTLRYPSQYDIAMNLQDNTSRFFHVAFTNAVEETTVTSSV
SYQLGGSIKASVTPSGPSGESGATGQVTWSDSVSYKQTSYKTNLIDQTNKHVKWNVFFNG
YNNQNWGIYTRDSYHALYGNQLFMYSRTYPHETDARGNLVPMNDLPALTNSGFSPGMIAV
VISEKDTEQSSIQVAYTKHADDYTLRPGFTEGTGNWVGNNTKDVDQKTENKSEVLDWKNK KLVEKK
SEQ ID NO: 14 - aerolysin.
AEPVYPDQLRLFSLGQGVCGDKYRPVNREEAQSVKSNIVGMMGQWQISGLANGWVIMGPGYNGEIKPGTASNT
WCYPTNPVTGEIPTLSALDIPDGDEVDVQWRLVHDSANFIKPTSYLAHYLGYAWVGGNHSQYVGEDMDVTRDG
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
108
DGWVIRGNNDGGCDGYRCGDKTAIKVSNFAYNLDPDSFKHGDVTQSDRQLVKTVVGWAVNDSDTPQSGYDVTL
RYDTATNWSKTNTYGLSEKVTTKNKFKWPLVGETELSIEIAANQSWASQNGGSTTTSLSQSVRPTVPARSKIP
VKIELYKADISYPYEFKADVSYDLTLSGFLRWGGNAWYTHPDNRPNWNHTFVD,PYKDKASSIRYQWDKRYIP
GEVKWWDWNWTIQQNGLSTMQNNLARVLRPVRAGITGDFSAESQFAGNIEIGAPVPLAA
SEQ ID NO: 15 - NetB from Clostridium perfringens.
SELNDINKIELKNLSGEIIKENGKEAIKYTSSDTASHKGWKATLSGTFIEDPHSDKKTAL
LNLEGFIPSDKQIEGSKYYGKMKWPETYRINVKSADVNNNIKIANSIPKNTIDKKDVSNS
IGYSIGGNISVEGKTAGAGINASYNVQNTISYEQPDFRTIQRKDDANLASWDIKEVETKD
GYNIDSYHAIYGNQLFMKSRLYNNGDKNFTDDRDLSTLISGGESPNMALALTAPKNAKES
VIIVEYQRFDNDYILNWETTQWRGTNKLSSTSEYNEFMFKINWQDHKIEYYL
SEQ ID NO: 16 - alpha-hemolysin.
ADSDINIKTGTTDIGSNTTVKTGDLVTYDKENGMHKKVEYSFIDDKNHNKKLLVIRTKGTIAGQYRVYSEEGA
NKSGLAWPSAFKVQLQLPDNEVAQISDYYPRNSIDTKEYMSTLTYGENGNVTGDDTGKIGGLIGANVSIGHTL
KYVQPDEKTILESPTDKKVGWKVIENNMVNQNWGPYDRDSWNPVYGNQLFMKTRNGSMKAADNFLDPNKASSL
LSSGESPDFATVITMDRKASKQQTNIDVIYERVRDDYQLHWTSTNWHGTNTKDKWTDRSSERYKIDWEKEEMT
SEQ ID NO: 17 - Vibrio cholerae Cytolysin (HlyA)/VCC.
NINEPSGEAADIISQVADSHAIKYYNAADWQAEDNALPSLAELRDLVINQQKRVLVDFSQISDAEGQAEMQAQ
FRKAYGVGFANQFIVITEHKGELLFTPFDQAEEVDPQLLEAPRTARLLARSGFASPAPANSETNTLPHVAFYI
SVNRAISDEECTFNNSWLWKNEKGSRPFCKDANISLIYRVNLERSLQYGIVGSATPDAKIVRISLDDDSTGAG
IHLNDQLGYRQFGASYTTLDAYFREWSTDAIAQDYRFVFNASNNKAQILKTFPVDNINEKFERKEVSGFELGV
TGGVEVSGDGPKAKLEARASYTQSRWLTYNTQDYRIERNAKNAQAVSFTWNRQQYATAESLLNRSTDALWVNT
YPVDVNRISPLSYASFVPKMDVIYKASATETGSTDFIIDSSVNIRPIYNGAYKEYYVVGAHQSYHGFEDTPRR
RITKSASFTVDWDHPVFTGGRPVNLQLASENNRCIQVDAQGRLAANTCDSQQSAQSFIYDQLGRYVSASNTKL
CLDGEALDALQPCNQNLTQRWEWRKGTDELTNVYSGESLGHDKQTGELGLYASSNDAVSLRTITAYTDVFNAQ
ESSPILGYTQGKMNQQRVGQDHRLYVRAGAAIDALGSASDLLVGGNGGSLSSVDLSGVKSITATSGDFQYGGQ
QLVALTFTYQDGRQQTVGSKAYVTNAHEDRFDLPAAAKITQLKIWSDDWLVKGVQFDLN
SEQ ID NO: 18 - Anthrax toxin protective antigen.
EVKQENRLLNESESSSQGLLGYYFSDLNFQAPMVVTSSTTGDLSIPSSELENIPSENQYFQSAIWSGFIKVKK
SDEYTFATSADNHVTMWVDDQEVINKASNSNKIRLEKGRLYQIKIQYQRENPTEKGLDFKLYWTDSQNKKEVI
SSDNLQLPELKQKSSNSRKKRSTSAGPTVPDRDNDGIPDSLEVEGYTVDVKNKRTFLSPWISNIHEKKCLTKY
KSSPEKWSTASDPYSDFEKVTGRIDKNVSPEARHPLVAAYPIVHVDMENTILSKNEDQSTQNTDSQTRTISKN
TSTSRTHTSEVEGNAEVHASFEDIGGSVSAGESNSNSSTVAIDHSLSLAGERTWAETMGLNTADTARLNANIR
YVNTGTAPIYNVLPTTSLVLGKNQTLATIKAKENQLSQILAPNNYYPSKNLAPIALNAQDDESSTPITMNYNQ
FLELEKTKQLRLDTDQVYGNIATYNFENGRVRVDTGSNWSEVLPQIQETTARTIFNGKDLNLVERRIAAVNPS
DPLETTKPDMTLKEALKIAFGENEPNGNLQYQGKDITEFDFNEDQQTSQNIKNQLAELNATNIYTVLDKIKLN
AKMNILIRDKREHYDRNNIAVGADESVVKEAHREVINSSTEGLLLNIDKDIRKILSGYIVEIEDTEGLKEVIN
DRYDMLNISSLRQDGKTFIDEKKYNDKLPLYISNPNYKVNVYAVTKENTIINPSENGDTSTNGIKKILIFSKK
GYEIG
SEQ ID NO: 19 - s-Toxin.
KASYDNVDTLIEKGRYNTKYNYLKRMEKYYPNAMAYFDKVTINPQGNDFYINNPKVELDGEPSMNYLEDVYVG
KALLTNDTQQEQKLKSQSFTCKNTDTVTATTTHTVGTSIQATAKFTVPFNETGVSLTTSYSFANTNTNTNSKE
ITHNVPSQDILVPANTTVEVIAYLKKVNVKGNVKLVGQVSGSEWGEIPSYLAFPRDGYKFSLSDTVNKSDLNE
DGTININGKGNYSAVMGDELIVKVRNLNTNNVQEYVIPVDKKEKSNDSNIVKYRSLYIKAPGIK
SEQ ID NO: 20 - 3.6 kb lambda DNA
GCCATCAGATTGTGTTTGTTAGTCGCTTTTTTTTTTTGGAATTTTTITTTTGGAATTTITTTITTGCGCTAAC
AACCT CCTGCCGT T T T GCCCGT GCATAT CGGT CACGAACAAAT CT GAT TACTAAACACAGTAGCCT
GGAT T T G
TT CTAT CAGTAAT CGAC CT TATT CC TAAT TAAA TAGA GCAAAT CC CCT TATT
GGGGGTAAGACAT GAAGAT GC
CAGAAAAACAT GACCT GTT GGCCGC CAT T CT CGCGGCAAAGGAACAAGGCAT C GGGGCAAT CCT T
GCGTT T GC
AATGGCGTACCTT CGCGGCAGATATAAT GGCGGT GCGT T TACAAAAACAGTAAT CGACGCAACGAT GT
GCGCC
AT TAT CGCCTAGT T CAT TCGT GACC T T CT CGACT T CGCCGGACTAAGTAGCAAT CT C GCT
TATATAACGAGCG
T GT T TAT CGGCTA CAT C GGTACT GA CT CGAT T GGT T C GCTTAT CAAACGCTT C GCT
GCTAAAAAAGCCGGAGT
AGAAGAT GGTAGAAAT CAATAAT CAAC GTAAGGCGT T CCTCGATATGCT GGC GT GGT
CGGAGGGAACTGATAA
C G GAC GT CAGAAAAC CAGAAAT CAT G GT TAT GAC GT CAT T G TAGG C G GAGAG C TAT T
TACT GAT TACT C C GAT
CACCCT CGCAAAC T T GT CACGCTAAACCCAAAACT CAAAT CAACAGGCGCCGGACGCTACCAGCT T CT
TT CCC
GT T GGT GGGAT GC CTAC CGCAAGCAGCT T GGCCT GAAA GAC T T CT CT CC GAAAAGT
CAGGACGCT GT GGCAT T
CA 03193980 2023- 3- 27
WO 2022/074397
PCT/GB2021/052609
109
GCAGCAGAT TAAGGAGC GT GGCGCT T TACCTAT GAT T GAT C GT GGT GATAT CC GT CAGGCAAT
CGACCGT T GC
AGCAATAT CT GGGCT T CACT GCCGGGCGCT GGT TAT GGT CAGT T C GAGCATAAGGCT GACAGCCT
GAT T GCAA
AATTCAAAGAAGCGGGCGGAACGGT ------------ CAGAGAGAT T GAT GTAT GAG CAGAGT --------
------- CAC C_:GC GAT TAT CT C C GCT CT G
GT TAT CT GCAT CAT CGT CT GC CT GT CAT GGGCT GT TAAT CAT TAC CGT GATAACGCCAT
TACCTACAAAGCCC
AGCGCGACAAAAATGCCAGAGAACT GAAGCT GGCGAACGCGGCAAT TACT GACAT GCAGAT GCGT CAGCGT
GA
T GT T GCT GCGCT C GAT GCAAAATACACGAAGGAGT TAGCT GAT GC TAAAGCT GAAAAT GAT GCT
CT GCGT GAT
GAT GT T GCCGCT GGT C:GT CGT CGGT TGCACATCAAAGC:AGT CT GT CAGT CAGT GCGT
GAAGCCACCACCGCCT
CCGGCGTGGATAATGCAGCCT CCCC CCGACT GGCAGACACC GCT GAACGGGAT TAT T T CACCCT
CAGAGAGAG
GCT GAT CAC TAT GCAAAAACAACT GGAAGGAACCCAGAAGTATAT TAAT GAGCAGT GCAGATAGAGT T
GCC CA
TAT CGAT GGGCAACT CA.T GCAAT TA.T T GT GAGCAATACACACGCGCT T C CAGC GGAGTATAAAT
GCCTAAAGT
AATAAAACCGAGCAAT C CAT T TACGAAT GT T T GCT GGGT TT CT GT T T TAACAACAT T T T
CT GCGCCGCCACAA
AT T T T GGCT GCAT CGACAGTT TT CT T CT GCCCAAT T C CAGAAACGAAGAAAT GAT GGGT
GAT GGT T T CCT T T G
GT GCTACT GCT GC CGGT TT GT TT T GAACAGTAAACGT CT GT TGAGCACATCCT
GTAATAAGCAGGGCCAGCGC
AGTAGCGAGTAGCATTT TT T T CAT CGT GT TAT T CCCGAT GC T T T T T GAAGTT C GCAGAAT
CGTAT GT GTAGAA
AAT TAAACAAACC CTAAACAAT GAGT T GAAAT T T CATAT T GT TAATAT T TAT TAAT GTAT GT
CAGGT GCGAT G
AT CGT CAT T GTAT T CC CGGA_TTAACTA T GT CCA CA GCCCT GACGGGGAA CT T CT CT
GCGGGA GT GT CCGGGA
ATAAT TAAAACGA.TGCA.CACAGGGT T TAGCGCGTACA.CGTAT T GCAT TAT GCCAACGCCCCGGT GCT
GACAC G
GAAGAAACCGGAC GT TA.T GAT TTAGCGTGGAAA.GATT T GT GTAGT &IT CT GAAT GCT CT
CAGTAAATAGTAAT
GAAT TAT CAAAGGTATAGTAATAT C T T T TAT GT T CAT GGATATTT GTAACCCAT CGGAAAACT
CCT GCTT TAG
CAAGAT TT T CCCT GTA.T TGCT GAAAT GT GAT T T CT CT T GAT TT CAACCTAT
CATAGGACGT TT CTATAAGAT
CGT GT T T CT TGAGAATT TAACAT T TACAACCT T T T TAAGT C CT T T TAT TAACACGGT GT
TAT CGT T T T CTAAC
AC GAT GT GAATAT TAT C T GT GGCTAGATAGTAAATATAAT GT GAGACGT T GT GACGT T T
TAGT T CAGAATAAA
ACAAT T CACAGT C TAAAT CT T TT CGCACT T GAT CGAATATT T CT T
TAAAAATGGCAACCTGAGCCATTGGTAA
AACCT T COAT GT GATAC GAGGGCGC GTAGT T T GOAT TAT CGT T T T TAT C GTT T CAAT
CT GGT CT GACCT OCT T
GT GT T T T GT T GAT GATT TAT GT CAAATAT TAGGAAT GT T TT CACT TAATAGTAT T GGT
T GCGTAACAAAGT GC
GGTCCTGCT GGCA.T T CT GGAGGGAAATACAACCGACAGATGTATGTAAGGCCAACGT GCT CAAAT CT T
CATAC
AGAAAGAT T T GAAGTAATAT T TTAAC C GCTAGAT GAAGAGCAAGC G CAT G GAG C GACAAAAT
GAATAAAGAAC
AAT CT GCT GAT GA.T CCC T CCGT GGAT CT GAT T CGT GTAAAAAATAT GCT TAATAGCACCAT
TT CTAT GAGT TA
CCCT GAT GT T GTAAT T GCAT GTATA GAACATAAGGT GT CT C T GGAAGCAT T CAGAGCAAT T
GAGGCAGCGT T G
GT GAAGCAC GATAATAATAT GAAGGAT TAT T CCCT GGT GGT T GAC T GAT CAC CATAACT
GCTAAT CAT T CAAA
CTATT TACT CT GT GACA GAGC CAACACGCAGT CT GT CACT GT CAG GAAAGT GGTAAAACT
GCAACT CAAT TAC
TGCAATGCCCTCGTAAT TAAGTGAATTTACAATATCGTCCT GT T C GGAGGGAAGAAC GCGGGAT GT T
CAT T CT
T CAT CACT T T TAA T T GA T GTATAT GCT CT CT T T T CT GACGT TAGT CT CC
GACGGCAGGCT T CAAT GACCCAGG
CT GAGAAAT TCCCGGACCCTT TT T GCT CAAGAGCGAT GT TAAT T T GT T CAAT CAT T T
GGTTAGGAAAGCGGAT
GTTGCGGGTTGTTGTTCTGCGGGTTCTGTTCTTCGTTGACATGAGGTTGCCCCGTATTCA.GTGTCGCTGATTT
GTATT GT CT GAAGT T GT TT T TACGT TAAGT T GAT GCAGAT CAAT TAATACGATACCT GCGT
CATAAT T GAT TA
TT T GACGT GGTT T GAT GGCCT CCAC GCACGT T GT GATAT GTAGAT GATAAT CAT TAT
CACTTTACGGGTCCTT
TCCGGTGAAAAAAAAGGTACCAAAAAAAACATCGTCGTGAGTAGTGAACCGTAAGC
SEQ ID NO: 21 - FraC from Actinia fragacea
SADVAaAVIDGAGLGFDVLKTVLEALGNVKRKIAVGIDNESGKTWTAMNTYFRSGTSDIV
LPHKVAHGKALLYNGQKNRGPVATGVVGVIAYSMSDGNTLAVLFSVPYDYNWYSNWWNVR
VYKGQKRADQRMYEELYYHRSPFRGDNGWHSRGLGYGLKSRGFMNSSGHAILEIHVTKA
SEQ ID NO: 22 - PlyA from Pleurotus ostreatus
MAYAQWVIIIIHNVGSKDVKIKNLKPSWGKLHADGDKDTEVSASKYEGTVIKPDEKLQIN
ACGRSDAAEGTTGTFDLVDPADGDKQVRHFYWDCPWGSKTNTWTVSGSNTKWMIEYSGQN
LDSGALGTITVDTLKKGN
SEQ ID NO: 23 - PlyB from Pleurotus ostreatus
MEAVLSRQAATAEAIGRFQDSSTSVGLVAGSPSTRIRRQADNVVLKSTSQAGDTLNDVIQ
DPTRRNKLINDNNLLKGTIMGRDGPVPSSRELIVRPDTLRATINNRATIETTTMEAEFTE
TLMESNYNSASVKVSAPFITANSEYSESSSFKNTETEKSMYTSSRYLFPQGRIDFTTPDS
GFDDVIKLSPQFTSGVQAALAKATGTEKREALQNLFQEYGHVERTKVHIGGVLSAHTMET
FSRSENETEVKQDVKAGLEGAVKGWGGGATAGHGNTQGTITTSQNRKLNVKYIVNGGDYT
KIQNTEFWVASTNQSEHWRVIEVTEVTAVADLLPQPIRGQVKDLLKPLLGKWVDVEKVPG
LESLPVSVYRPKGAIPAGWFWLGDTADASKALLVKPTLPARSGRNPALTSLHQGSGMTEQ
PFVDLPQYQYLSTYFGSFAHDTPPGSTLRGLRPDHVLPGRYEMHGDTISTAVYVTRPVDV
PFPEDECFDLKSLVRVKLPGSGNPPKPRSALKKSMVLFDSGEK
CA 03193980 2023- 3- 27