Note: Descriptions are shown in the official language in which they were submitted.
WO 2022/148856
PCT/EP2022/050323
1
SELF-CLEANING CO2 REDUCTION SYSTEM AND RELATED
METHODS
TECHNICAL FIELD
The present techniques generally relate to self-cleaning of a CO2 reduction
system, and more
particularly to a self-cleaning system and methods involving application of an
unsteady
electrochemical forcing.
BACKGROUND
The reduction of carbon dioxide (CO2) emissions is essential to mitigate
climate change driven
environmental damage. The rapidly decreasing cost of renewable electricity,
coupled with the need
for energy storage from these intermittent sources, has motivated
electrochemical pathways for the
CO2 reduction reaction (CO2RR) to valuable chemicals and fuels.
Gas diffusion electrodes facilitate effective CO2 mass transport to the
cathode catalyst (figure 1),
enabling electrolyzers to operate at the current densities required for
industrial deployment, e.g.,
in excess of 100 mA cm-2. Alkali metal cations, typically potassium, are
implemented broadly in
aqueous electrolytes to reduce ohmic losses and improve the CO2RR current
density and
selectivity. Performing CO2 electrolysis at high current densities inevitably
produces large
quantities of hydroxide ions on the cathode, driving up the local pH and thus
encouraging the
chemical reaction of dissolved CO2 with these hydroxide ions to produce
bicarbonate ions on route
to carbonate ions (figure 2), as mentioned in the studies of Lu X.. et al.
entitled "In Situ
Observation of the pH Gradient near the Gas Diffiision Electrode of CO2
Reduction in Alkaline
Electrolyte" (J. Am. Chem. Soc. 2020, 142 (36), 15438-15444) and Zhong H. et
al ., entitled "Effect
of CO2 Bubbling into Aqueous Solutions Used .1br Electrochemical Reduction of
CO21br Energy
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
2
Conversion and Storage" (J. Phys. Chem. C 2015, 119 (1), 55-61). The negative
potential on the
cathode forms an interfacial electric field that attracts cations from the
electrolyte to the cathode
outer Helmholtz plane (see the study of Singh M. R, et al., entitled
"Hydrolysis of Electrolyte
Cations Enhances the Electrochemical Reduction of CO, over Ag and Cu" - J. Am.
Chem. Soc.
2016, 138 (39), 13006-13012). At steady state conditions, potassium and
carbonate ions are
present in excess of the solubility limit, resulting in the formation of solid
potassium carbonate
salts. This effect is not expected to be unique to potassium carbonate;
carbonates of other
commonly used alkali metal cations will have more salt formation issues due to
their lower
solubility limits (table 1).
Table 1: Solubility and cost comparison for alkali metal cations and their
carbonate salts.
Alkali metal Solubility* Price**
cation OH- (M) HCO3- (M) C032- (M) ($ CAD / kg
carbonate salt)
Li + 5.22 0.18 303.00
Na+ 25.00 1.23 2.90 189.00
21.57 3.62 8.03 161.20
Rb+ *** 16.88 7.92 9.66 3900.00
Cs + *** 20.01 10.78 8.01 760.00
* See CRC Handbook of Chemistry and Physics: A Ready-Reference Book of
Chemical and
Physical Data. Choice Rev. Online, 2010, 47(07), 47-3553-47-3553.
**Prices are from MilliporeSigma Canada Co. for ACS reagent ( 2%99.0%) purity
*** These cations are rarely used in CO2 electrolyzers due to their high
price.
These salts precipitate within the catalyst and gas diffusion layers,
progressively reducing CO2
mass transport until the pores are completely blocked and CO2RR is eliminated.
Salt precipitation
- inevitable at steady state conditions - precludes stable CO2RR.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
3
The conventional approach to mitigate the effects of carbonate salt formation
has been to rinse the
electrode with water, either by disassembling the cell or injecting water
periodically into the CO2
supply during operation, as mentioned in the studies of Nwabara U. 0. et al.,
entitled "Durable
Cathodes and Electrolyzers fbr the Efficient Aqueous Electrochemical Reduction
of CO2"
(ChemSusChem 2020, 13 (5), 855-875) and of Verma S. et al., entitled "Insights
into the Low
Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an
Alkaline Flow
Electrolyzer" (ACS Energy Lett. 2018, 3 (1), 193-198). The addition of water
content hampers
CO2 transport to the catalyst layer, thereby encouraging hydrogen (H2)
generation and lowering
CO2 electrolysis efficiency during and immediately after the washing cycle.
Systems using rinsing-
based approaches have achieved only small enhancements in stability (< 10
hours total duration)
and struggle to maintain a stable current density, as mentioned in the studies
of Verma S. et al.
(cfr. supra), of EndrOdi B. et al, entitled "Alultilayer Electrolyzer Stack
Converts Carbon Dioxide
to Gas Products at High Pressure with High Efficiency" (ACS Energy Lett. 2019,
4 (7), 1770-
1777) and of De Mot. B., et al., entitled "Direct Water Injection in Catholyte-
Free Zero-Gap
Carbon Dioxide Electrolyzers" (ChemElectroChem 2020, 7 (18), 3839-3843). Salt
precipitation
occurs deep in the microporous layer of the gas diffusion electrode and once
formed, is very
difficult to remove.
The present electrochemical techniques address at least some of these
challenges to reduce salt
formation during conversion of CO2 into value-added products in comparison to
known techniques
in the field.
SUMMARY
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
4
As will be explained below in relation to various example implementations, the
present techniques
relate to prevention of salt formation by alternating an applied cell voltage
between an operational
voltage and a lower regeneration voltage.
In a first aspect, the present disclosure relates to a method for reducing CO2
in an electrolytical
system and/or for self-cleaning a gas diffusion electrode in an electrolytical
system operating CO2
reduction, the method comprising:
providing an electrolytical system;
applying an operational voltage to the electrolytical system to operate CO2
reduction for
a first period of time defining an operation cycle, thereby forming carbonate
ions at a
cathode side of the electrolytical system and having a local carbonate ion
concentration;
and
subsequently applying a regeneration voltage to the electrolytical system for
a second
period of time defining a regeneration cycle to force electromigration of the
formed
carbonate ions to an anode side of the electrolytical system;
remarkable in that the regeneration voltage is lower than the operational
voltage.
For example, the present disclosure relates to a method for self-cleaning a
gas diffusion electrode
in an electrolytical system operating CO2 reduction, the method comprising:
providing an electrolytical system;
applying an operational voltage to the electrolytical system to operate CO2
reduction for
a first period of time defining an operation cycle, thereby forming carbonate
ions at a
cathode side of the electrolytical system and having a local carbonate ion
concentration;
and
subsequently applying a regeneration voltage to the electrolytical system for
a second
period of time defining a regeneration cycle to force electromigration of the
formed
carbonate ions to an anode side of the electrolytical system;
remarkable in that the regeneration voltage is lower than the operational
voltage.
For example, the regeneration voltage is more negative than the operational
voltage.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
Advantageously, the duration of the operation cycle is chosen to maintain the
local carbonate ion
concentration at the cathode side below a carbonate salt solubility limit With
preference, the local
carbonate ion concentration being determined by solubility calculation, for
example via computer
simulation (e.g., COMSOL).
5 In some implementations, the duration of the operation cycle can be
chosen to maintain the local
carbonate ion concentration at the cathode side below a carbonate salt
solubility limit.
For example, the first period of time is between 1 second and 1200 seconds,
preferably between
60 seconds and 300 seconds.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 80 % via
electromigration to the anode
side.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 90 % via
electromigration to the anode
side.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 99 % via
electromigration to the anode
side.
For example, the second period of time is between 1 second and 60 seconds,
preferably between
30 seconds and 60 seconds.
Advantageously, said method further comprises repeating the operation cycle
and the regeneration
cycle by alternating a voltage applied to the electrolytic system between the
operational voltage
and the lower regeneration voltage.
With preference, each operation cycle is performed for the same duration
and/or each regeneration
cycle is performed for the same duration.
For example, the duration of each operation cycle varies between 1 second and
1200 seconds,
preferably between 60 seconds and 300 seconds.
For example, the duration of each regeneration cycle varies between 1 second
and 60 seconds,
preferably between 30 seconds and 60 seconds.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
6
Advantageously, the regeneration voltage is chosen to obtain a CO2 reduction
rate below 1 mA. cm-
Advantageously, the operational voltage is between -3.0 and - 4.5 V,
preferably between -3.2 and
- 4.0 V. For example, the operational voltage is -3.6 V.
Advantageously, the regeneration voltage is between - 2.5 V and -5.0 V, or
between -2.5V and -
4.0V, preferably between - 2.1 V and -3.5 V. For example, the regeneration
voltage is -2.0 V.In a
preferred embodiment, the electrolytical system is a membrane electrode
assembly (MEA)
comprising a gas diffusion electrode serving as a cathode.
In an alternate embodiment, the electrolytical system is a flow cell system
comprising a liquid
catholyte and a gas diffusion electrode serving as a cathode.
Whichever the chosen embodiment, the cathode comprises a metal layer deposited
on substrate,
for example a carbon paper substrate or a PTFE substrate. For example, the
cathode comprises a
silver layer deposited on a carbon paper substrate and/or the cathode
comprises a copper layer
deposited on a PTFE substrate.
1 5 Advantageously, the electrolytical system comprises an anolyte. For
example, the anolyte is an
aqueous solution of one or more alkaline compounds, said one or more alkaline
compounds
comprising one alkali metal cations selected from lithium, sodium, potassium,
rubidium, cesium
and any combination thereof
In a second aspect, the present disclosure relates to the use of the method
according to the first
aspect in a an electrolytical system comprising a gas diffusion electrode
wherein at an applied cell
voltage carbonate ions are formed when the electrolytical system is operating
CO2 reduction;
wherein the use comprises self-cleaning the gas diffusion electrode
In a third aspect, the present disclosure relates to a self-cleaning
electrolytical system for CO2
reduction into C2 products, the electrolytical system comprising: a cathode;
an anode; an
electrolyte; an ion-exchange membrane separating the anode and cathode; an
electrical energy
source applying a voltage to the electrolytical system; the self-cleaning
electrolytic system is
remarkable in that it further comprises a controller in operative
communication with the electrical
energy source to alternate the applied voltage between an operational voltage
and a lower
regeneration voltage, thereby imposing an operation cycle in alternate with a
regeneration cycle.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
7
With preference, the controller is a control amplifier that is programmed or
manually actuated.
Advantageously, the control amplifier and the electrical energy source are
combined in a
potentiostat.
In one aspect, there is provided a method for reducing CO2 in an
electrolytical system. The method
comprises:
applying an operational voltage to the electrolytical system to operate CO2
reduction for a
first period of time defining an operation cycle, thereby forming carbonate
ions at a cathode
side of the electrolytical system and having a local carbonate ion
concentration; and
subsequently applying a regeneration voltage to the electrolytical system for
a second
period of time defining a regeneration cycle to force electromigration of the
formed
carbonate ions to an anode side of the electrolytical system;
wherein the regeneration voltage is lower than the operational voltage.
In some implementations, the duration of the operation cycle can be chosen to
maintain the local
carbonate ion concentration at the cathode side below a carbonate salt
solubility limit.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 80 % via
electromigration to the anode
side.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 90 % via
electromigration to the anode
side.
In some implementations, the duration of the regeneration cycle can be chosen
to reduce the local
carbonate ion concentration at the cathode side by at least 99 % via
electromigration to the anode
side.
In some implementations, the first period of time can be between 60 seconds
and 300 seconds.
In some implementations, the second period of time can be between 30 seconds
and 60 seconds.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
8
In some implementations, the method can further comprise repeating the
operation cycle and the
regeneration cycle by alternating a voltage applied to the electrolytic system
between the
operational voltage and the lower regeneration voltage.
In some implementations, the duration of each regeneration cycle can be chosen
to sufficiently
reduce the local carbonate ion concentration at the cathode side to remain
under the carbonate salt
solubility limit during a subsequent operation cycle.
In some implementations, each operation cycle can be performed for the same
duration.
In some implementations, each regeneration cycle can be performed for the same
duration.
In some implementations, the duration of each operation cycle can vary between
60 seconds and
300 seconds.
In some implementations, the duration of each regeneration cycle can vary
between 30 seconds
and 60 seconds.
In some implementations, the number of operation cycles can be chosen to
operate CO2 reduction
during at least 150 hours, while maintaining a CO2RR selectivity towards C2
products of at least
80%.
In some implementations, a total duration of all operation cycles and
regeneration cycles can be
236 hours for an operation duration of 157 hours.
In some implementations, the regeneration voltage can be chosen to obtain a
CO2 reduction rate
below 1 mA.cm-2.
In some implementations, the operational voltage can be between -3.0 and - 4.5
V. For example,
the operational voltage can be -3.6 V.
In some implementations, the regeneration voltage can be between - 2.5 V and -
5.0 V. For
example, the regeneration voltage can be -2.0 V.
In some implementations, the electrolytical system can be a membrane electrode
assembly (MEA)
comprising a gas diffusion electrode serving as a cathode.
In some implementations, the electrolytical system can be a flow cell system
comprising a liquid
catholyte and a gas diffusion electrode serving as a cathode.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
9
In some implementations, the cathode can include a metal layer deposited on a
substrate, for
example a carbon paper substrate or a PTFE substrate.
In some implementations, the cathode can include a copper layer deposited on a
PTFE substrate.
In other implementations, the cathode can include a silver layer deposited on
a carbon paper
substrate.
In some implementations, the electrolytical system can include an electrolyte
liberating alkali
metal cations that form alkali metal carbonate salts with the carbonate ions,
when above the
corresponding carbonate salt solubility limit. For example, the alkali metal
cations can include
lithium, sodium, potassium, rubidium, caesium ions, or any combinations
thereof.
In another aspect, there is provided a method for self-cleaning a gas
diffusion electrode in an
electrolytical cell operating CO2 reduction at an applied cell voltage and
forming carbonate ions,
the method including alternating the applied cell voltage between an
operational voltage and a
lower regeneration voltage.
In some implementations, alternating the applied cell voltage between the
operational voltage and
the lower regeneration voltage can include applying the operational voltage
for an operation
duration maintaining a local carbonate ion concentration at the gas diffusion
electrode below a
carbonate salt solubility limit. For example, the operation duration can be at
most 1200 seconds.
In another example, the operation duration can be between 60 seconds and 300
seconds.
In some implementations, alternating the applied cell voltage between the
operational voltage and
the lower regeneration voltage can include applying the regeneration voltage
for a regeneration
duration that results in electromigration of at least 80 % of the carbonate
ions that are formed at
the gas diffusion electrode. Optionally, alternating the applied cell voltage
between the operational
voltage and the lower regeneration voltage can include applying the
regeneration voltage for a
regeneration duration that results in electromigration of at least 90 % of the
carbonate ions that are
formed at the gas diffusion electrode. Further optionally, alternating the
applied cell voltage
between the operational voltage and the lower regeneration voltage can include
applying the
regeneration voltage for a regeneration duration that results in
electromigration of at least 99 % of
the carbonate ions that are formed at the gas diffusion electrode.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
In some implementations, alternating the applied cell voltage between the
operational voltage and
the lower regeneration voltage can include applying the regeneration voltage
for a regeneration
duration that results in the removal of an amount of carbonate ions allowing
remaining under a
carbonate salt solubility limit during the subsequent application of the
operational voltage. For
5 example, the regeneration duration is at most 60 seconds. In another
example, the regeneration
duration can be between 30 seconds and 60 seconds.
In some implementations, alternating the applied cell voltage between the
operational voltage and
the lower regeneration voltage can be performed during 236 hours comprising a
total operation
duration of 157 hours, while maintaining a CO2RR selectivity towards C2
products of at least 80%.
10 In some implementations, the regeneration voltage can be chosen to
obtain a CO2 reduction rate
below 1 mA. cm-2.
In some implementations, the operational voltage can be between -3.0 and - 4.5
V. For example,
the operational voltage can be -3.6 V.
In some implementations, the regeneration voltage can be between - 2.5 V and -
5.0 V. For
example, the regeneration voltage can be -2.0 V.
In some implementations, the gas diffusion electrode can serve as a cathode in
a membrane
electrode assembly (MEA). In other implementations, the gas diffusion
electrode can serve as a
cathode in a flow cell system.
In some implementations, the gas diffusion electrode can include a silver
layer deposited on a
carbon paper substrate. In other implementations, the gas diffusion electrode
can include a copper
layer deposited on a PTFE substrate.
In some implementations, the electrolytical cell can include an electrolyte
liberating alkali metal
cations that form alkali metal carbonate salts with the carbonate ions, when
above a corresponding
carbonate salt solubility limit. For example, the alkali metal cations can
include lithium, sodium,
potassium, rubidium, caesium ions, or any combinations thereof
In another aspect, there is provided a self-cleaning electrolytical system for
CO2 reduction into C2
products. The electrolytical system comprises:
a cathode;
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
11
an anode;
an electrolyte;
an ion-exchange membrane separating the anode and cathode;
an electrical energy source applying a voltage to the electrolytical system;
and
a controller in operative communication with the electrical energy source to
alternate the
applied voltage between an operational voltage and a lower regeneration
voltage, thereby
imposing an operation cycle in alternate with a regeneration cycle.
In some implementations, the controller can be configured to apply the
operational voltage via the
electrical energy source for a duration that maintains a local carbonate ion
concentration at a
cathode side of the system below a carbonate salt solubility limit.
In some implementations, the controller can be configured to apply the
regeneration voltage via
the electrical energy source until at least 80 % of carbonate ions that are
formed at the cathode
cross the ion-exchange membrane. Optionally, the controller can be configured
to apply the
regeneration voltage via the electrical energy source until at least 80 % of
carbonate ions that are
formed at the cathode cross the ion-exchange membrane. Further optionally, the
controller can be
configured to apply the regeneration voltage via the electrical energy source
until at least 99 % of
carbonate ions that are formed at the cathode cross the ion-exchange membrane.
In some implementations, the controller can be configured to maintain the
regeneration voltage
during each regeneration cycle to remove an amount of carbonate ions from the
cathode side that
is sufficient to remain under a carbonate salt solubility limit during the
subsequent operation cycle.
In some implementations, the controller can be configured to maintain each
operational cycle for
at most 1200 seconds, or between 60 seconds and 1200 seconds.
In some implementations, the controller can be configured to maintain each
regeneration cycle for
at most 60 seconds, or between 30 seconds and 60 seconds.
In some implementations, the controller can be configured to perform each
operation cycle for the
same duration.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
12
In some implementations, the controller can be configured to perform each
regeneration cycle for
the same duration.
In some implementations, the controller can be configured to perform a number
of operation cycles
that allow CO2 reduction during at least 150 hours, while maintaining a CO2RR
selectivity towards
C2 products of at least 80 %. For example, a total duration of all operation
cycles and regeneration
cycles can be 236 hours for an operation duration of 157 hours.
In some implementations, the regeneration voltage can be chosen to obtain a
CO2 reduction rate
below 1 mA. cm-2.
In some implementations, the operational voltage can be between -3.0 and - 4.5
V. For example,
the operational voltage can be -3.6 V.
In some implementations, the regeneration voltage can be between - 2.5 V and -
5.0 V. For
example, the regeneration voltage can be -2.0 V.
In some implementations, the electrolytical system can be a membrane electrode
assembly (MEA)
comprising a gas diffusion electrode serving as the cathode. In other
implementations, the
electrolytical system can be a flow cell system comprising a gas diffusion
electrode serving as the
cathode, wherein the electrolyte is a catholyte and the system further
comprises an anolyte in which
the anode is immersed.
In some implementations, the cathode can include a silver layer deposited on a
carbon paper
substrate. In other implementations, the cathode can include a copper layer
deposited on a PTFE
substrate.
In some implementations, the controller can be a control amplifier that is
programmed or manually
actuated. For example, the control amplifier and the electrical energy source
can be combined in a
potentiostat.
In some implementations, the electrolyte can comprise alkali metal cations
that form alkali metal
carbonate salts with the carbonate ions, when above a corresponding carbonate
salt solubility limit.
For example, the alkali metal cations can include lithium, sodium, potassium,
rubidium, caesium
ions, or any combinations thereof.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
13
The various aspects, implementations and features of the present techniques
are further described
herein, including in the claims, figures and following description.
BRIEF DESCRIPTION OF THE DRAWINGS
The figures describe various aspects and information regarding the techniques
described and
claimed herein.
Figure 1: Schematic of the MEA CO? electrolyzer.
Figure 2: CO2 conversion to bicarbonate and carbonate during regular
electrolyzer operation.
Figure 3: Carbonate migration during cell operation at the regeneration
voltage.
Figure 4: Strategy to mitigate carbonate formation by cycling between
operational and
regeneration cell voltages.
Figure 5: COMSOL Multiphysics simulation of pH for different operational times
with -3.8 V
continuous operation. Salt crystal formation is predicted where salt
concentrations in the model
exceed the solubility limit (indicated by the dashed line).
Figure 6: COMSOL Multiphysics simulation of CO2 concentration for different
operational times
with -3.8 V continuous operation. Salt crystal formation is predicted where
salt concentrations in
the model exceed the solubility limit (indicated by the dashed line).
Figure 7: COMSOL Multiphysics simulation of HCO3- concentration for different
operational
times with -3.8 V continuous operation. Salt crystal formation is predicted
where salt
concentrations in the model exceed the solubility limit (indicated by the
dashed line).
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
14
Figure 8: COMSOL Multiphysics simulation of IC+ concentration for different
operational times
with -3.8 V continuous operation. Salt crystal formation is predicted where
salt concentrations in
the model exceed the solubility limit (indicated by the dashed line).
Figure 9: COMSOL Multiphysics simulation of pH for different regeneration
times (regeneration
voltage = -2.0 V).
Figure 10: COMSOL Multiphysics simulation of CO2 concentration for different
regeneration
times (regeneration voltage = -2.0 V).
Figure 11: COMSOL Multiphysics simulation of HCO3- concentration for different
regeneration
times (regeneration voltage = -2.0 V).
Figure 12: COMSOL Multiphysics simulation of pH for different total times when
applying the
alternating voltage strategy (periodic 60 s of operating and 30 s of
regeneration voltage).
Figure 13: COMSOL Multiphysics simulation of CO2 concentration for different
total times when
applying the alternating voltage strategy (periodic 60 s of operating and 30 s
of regeneration
voltage).
Figure 14: COMSOL Multiphysics simulation of HCO3- concentration for different
total times
when applying the alternating voltage strategy (periodic 60 s of operating and
30 s of regeneration
voltage).
Figure 15: Carbonate concentrations within the MEA at different operational
times for continuous
operation at -3.8 V (current density of 172 mA cm-2).
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
Figure 16: Carbonate concentrations within the MEA at different regeneration
times (regeneration
voltage = -2.0 V) after 60 seconds of continuous operation.
Figure 17: Current density of the different regeneration voltage (cyclic -3.8
V operational voltage
for 60 s and regeneration voltage for 30 s).
5 Figure 18: The net carbonate ion growth rate averaged during the first
60s of simulated operation
at -3.8 V. The solid red line is the generation rate; the solid blue line is
rate of species transport,
including convection, diffusion and electromigration; the solid black line is
the difference between
the generation and reduction lines thereby describing the net change of
carbonate ion
concentration.
10 Figure 19: Carbonate concentrations within the MEA and comparison of
electromigrative and
concentration-driven diffusive effects.
Figure 20: COMSOL Multiphysics simulation of hydroxide concentration for
different total times
when applying the alternating voltage strategy (periodic 60 seconds of
operating and 10 seconds
of regeneration voltage, periodic 60 seconds of operating and 20 seconds of
regeneration voltage).
15 Figure 2L COMSOL Multiphysics simulation of carbonate concentration for
different total times
when applying the alternating voltage strategy: periodic 60 seconds of
operating and 10 seconds
of regeneration voltage.
Figure 22: COMSOL Multiphysics simulation of carbonate concentration for
different total times
when applying the alternating voltage strategy: periodic 60 seconds of
operating and 20 seconds
of regeneration voltage.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
16
Figure 23: Electrochemical performance of silver catalyst on carbon paper:
stability of
continuously operated sample at -3.6 V.
Figure 24: Electrochemical performance of silver catalyst on carbon paper:
stability of alternating
operation sample (60 seconds at operational voltage and 30 seconds at
regeneration voltage of -
2.0 V).
Figure 25: Electrochemical performance of silver catalyst on carbon paper:
selectivity of
alternating operation sample at different operational voltages.
Figure 26: Electrochemical performance of silver catalyst on carbon paper:
selectivity of
continuous operation sample at different operational voltages.
Figure 27: Electrochemical performance of silver catalyst on carbon paper:
stability of
continuously operated sample at -3.4 V.
Figure 28: Electrochemical performance of silver catalyst on carbon paper:
stability of alternating
operation sample (60 seconds at the operational voltage of -3.6 V and 30
seconds at regeneration
voltage of -2.0 V) that has the same average current density with -3.4 V
continuously operated
test.
Figure 29: Back side of the copper on PTFE electrode after continuous
operation at -3.8 V during
long-term operation.
Figure 30: Electrochemical performance of copper catalyst on PTFE electrode:
selectivity of
continuously operated sample at -3.8 V during long-term operation.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
17
Figure 31: Electrochemical performance of copper catalyst on PTFE electrode:
current density of
continuous operation during long-term operation.
Figure 32: Raman analysis of the solid phase salt precipitates taken from the
continuously operated
copper on PTFE electrode..
Figure 33: Electrochemical performance of silver catalyst on PTFE electrode:
selectivity and
current density of continuous operation during long-term operation at -3.8 V.
Figure 34: Electrochemical performance of silver catalyst on PTFE electrode:
post-experiment
CO? gas stream cathode channel.
Figure 35: Electrochemical performance of silver catalyst on PTFE electrode:
backside of the post-
experiment PTFE electrode sample.
Figure 36: Back side of the copper on PTFE electrode after alternating
operation (60 seconds at
operational voltage of -3.8 V and 30 seconds at regeneration voltage of -2.0
V).
Figure 37: Electrochemical performance of copper catalyst on PTFE electrode:
selectivity of
alternating operation sample (60 seconds at operational voltage of -3.8 V and
30 seconds at
regeneration voltage of -2.0 V) during long-term operation.
Figure 38: Electrochemical performance of copper catalyst on PTFE electrode:
current density of
alternating operation sample during long-term operation.
Figure 39: Electrochemical performance of copper catalyst on PTFE electrode:
magnified early
view of current density and late view of current density.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
18
Figure 40: Ex-situ X-ray photoelectron spectroscopy characterization of a
copper on PTFE
electrode before electrolysis. Copper (I) oxide, copper (II) oxide, and
metallic copper were
detected, suggesting that the sputtered copper catalyst was oxidized in
ambient air prior to the
experiment.
Figure 41: Ex-situ X-ray photoelectron spectroscopy characterization of a
copper on PTFE
electrode after electrolysis. The copper catalyst was predominantly in
metallic form, suggesting
that the copper (I) oxide and copper (II) oxide were reduced to metallic
copper at the beginning of
operation. The small amount of copper (I) oxide was likely caused by oxidation
during reactor
disassembly and transport.
Figure 42: Electrochemical performance of copper catalyst on PTFE electrode:
selectivity of
alternating operation sample at different operational voltages.
Figure 43: Electrochemical performance of copper catalyst on PTFE electrode:
selectivity of
continuous operation sample at different operational voltages.
Figure 44: Electrochemical performance of copper catalyst on PTFE electrode:
energy expended
on regeneration and operational modes
Figure 45: Schematics of ID MEA configuration
Figure 46: TOC graphic
Figure 47: Carbonate concentrations within the MEA: different total times when
applying the
alternating voltage strategy (periodic 60 seconds of operational voltage and
30 seconds of
regeneration voltage). Salt crystal formation is predicted where salt
concentrations in the model
exceed the solubility limit (indicated by the dashed line).
DETAILED DESCRIPTION
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
19
The descriptions, examples, methods and materials presented in the claims and
the specification
are not to be construed as limiting but rather as illustrative only.
Meanings of technical and scientific terms used herein are to be commonly
understood as by one
of ordinary skill in the art to which the invention belongs, unless otherwise
defined.
It is understood that whether the term "about" is used explicitly or not,
every quantity given herein
is meant to refer to an actual given value, and it is also meant to refer to
the approximation to such
given value that would reasonably be inferred based on the ordinary skill in
the art, including
approximations due to the experimental and/or measurement conditions for such
given value. It is
commonly accepted that a 10% precision measure is acceptable and encompasses
the term "about".
Although various implementations of the invention may be described in the
context of a single
embodiment, these implementations may also be provided separately or in any
suitable
combination. Conversely, although the invention may be described herein in the
context of
separate embodiments for clarity, the implementations of the techniques
described herein may also
be implemented in a single embodiment, unless incompatible.
Any publications, including patents, patent applications and articles,
referenced or mentioned in
this specification are herein incorporated in their entirety into the
specification, to the same extent
as if each individual publication was specifically and individually indicated
to be incorporated
herein. In addition, citation or identification of any reference in the
description of some
embodiments of the invention shall not be construed as an admission that such
reference is
available as prior art to the present invention.
The present techniques relate to self-cleaning of a gas diffusion electrode in
an electrolytical cell
operating CO2 reduction at an applied cell voltage where carbonate ions are
formed. The self-
cleaning techniques involve alternating the applied cell voltage between an
operational voltage
and a lower regeneration voltage. An operational cycle is defined by
application of the operational
voltage for an operational duration, and the regeneration cycle is defined by
application of the
regeneration voltage for a regeneration duration. Duration of each operational
cycle and
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
regeneration cycle can be tailored to reduce or avoid carbonate salt
precipitation at the gas
diffusion electrode side (e.g., cathode side for CO2RR) of the electrolytical
cell. Carbonate ions
that are formed at the cathode side during the operational cycle can be
transferred to an anode side
of the electrolytical cell via electromigration during the subsequent
regeneration cycle. Once
5 migrated to the anode side, the carbonate ions are further changed to
CO2. The techniques proposed
herein can be referred to as an alternating voltage approach, an alternating
approach, an alternating
voltage strategy, an alternating strategy or an unsteady electrochemical
forcing strategy.
Different alternating voltage and pulsed electrolysis strategies have been
employed in CO2
electrolyzers with a range of duty cycles. Depending on the specific
conditions, these strategies
10 can be used to adjust the surface CO:H2 ratio (see Kumar B., et al. -
ACS Catal. 2016, 6(7), 4739-
4745), increase C2+ production (see Aran-Ais, R M. et al. - Nat. Energy 2020,
5 (4), 317-325) ,
and decrease H2 generation (see Kimura K. W. et al. - ChemSu,sChem 2018, //
(11), 1781-1786).
Computational modelling was used to illustrate that steady state operation of
electrolyzers for CO?
reduction can yield high carbonate concentrations, which further lead to
inevitable salt formations.
15 The present salt formation prevention strategy includes avoiding
reaching the steady state
conditions. To do so, the present techniques include varying the applied cell
voltage between two
values, and more specifically, applying cyclically an operation voltage for an
operation duration,
and a regeneration voltage for a regeneration duration. The resulting
regeneration potential lowers
the reaction rate to nearly 0 mA cm-2, eliminating hydroxide formation, while
maintaining a
20 sufficiently negative polarization at the cathode to transport carbonate
ions to the anode under
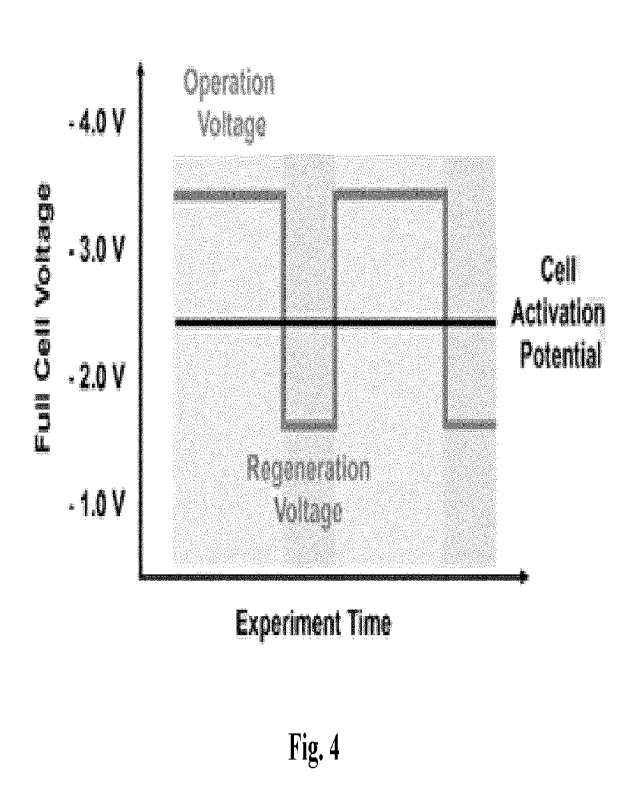
electromigration (figure 3). As better seen in figure 4, the applied cell
voltage can be varied in a
step-like manner between the operational voltage and the regeneration voltage.
In other
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
21
implementations, the applied cell voltage can be gradually varied to
reversibly reach the
operational voltage or the regeneration voltage.
Based on experimentation using carbon paper and PTFE-based electrodes for
silver and copper
catalysts, respectively, CO2 electrolysis was performed in a membrane
electrode assembly (MEA)
electrolyzer, using the present alternating voltage approach. A similar
product distribution to that
of constant voltage operation was obtained, but demonstrated enhanced
stability. The copper-
PTFE electrodes were able to sustain the product distribution when operated
alternatively for 157
hours of operation over 236 hours of total duration, as compared to ¨10 hours
of operation when
the same copper-PTFE electrodes were operated continuously.
In some implementations, selection of a duration for each operation cycle and
regeneration cycle
is based on the variation of a local carbonate ion concentration at the
cathode side. To avoid any
salt precipitation, the local carbonate ion concentration can be maintained
below the carbonate salt
solubility limit during operation. Additionally, the local carbonate ion
concentration can be
reduced sufficiently (via electromigration), e.g., by at least 80%, during the
regeneration cycle to
ensure that the local carbonate ion concentration will not reach the carbonate
salt solubility limit
during a subsequent operation cycle. For example, selecting the duration for
each operation cycle
and regeneration cycle can include simulating the local carbonate ion
concentration variation
history for a specific voltage application scenario.
To better understand the present salt prevention strategy, a computational
model of CO2RR was
developed to assess concentration profiles of key species during operation
(figures 5 to 14) of CO2
electrolysis. When a constant voltage of -3.8 V was applied continuously, a
local carbonate
concentration reached a potassium carbonate solubility limit (based on a
solubility product
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
22
constant of 2073 at 20 C, see Solubility Calculation below and CRC Handbook
of Chemistry and
Physics: A Ready-Reference Book of Chemical and Physical Data. Choice Rev.
Online 2010, 47
(07), 47-3553-47-3553) within 1200 seconds (figure 15). Salt crystal formation
is expected where
the computational model predicts salt ions in excess of the solubility limit
(indicated in figure 15).
The steady state conditions were reached after 4000 seconds with the local
potassium and
carbonate ionic concentration on the cathode well above the solubility limit.
These results
confirmed that steady state conditions cannot be achieved without the local
concentration of
carbonate ions exceeding saturation, and thus salt precipitation is inherent
and inevitable in these
systems on the timescale of minutes. However, experimentation also shown that
after the first 60
seconds of operation, the carbonate ions concentration was only 2.1 M, well
below the potassium
carbonate solubility limit.
Another series of simulations, including the use of multiple regeneration
periods during which a
regeneration voltage of -2.0 V was applied, allowed to analyze concentration
changes immediately
after 60 seconds of operation (figure 16). The regeneration voltage was chosen
as the highest cell
voltage which could obtain a near-zero current density (below 1 mA cm' on
average, figure 17),
thereby maximizing an electric field strength while minimizing
hydroxide/carbonate generation.
These results demonstrate that increasing the regeneration time significantly
reduce the carbonate
ions concentration at the cathode. Applying a 30-second regeneration period
lowered the carbonate
ions concentration ¨ 2000-fold, to ¨ 10-3 M from its pre-regeneration level of
2.1 M, indicating an
elimination of > 99.9% of carbonate ions at the cathode. During operation,
also referred to as the
operational period, carbonate ions can travel to the anode but the rate of
carbonate generation
exceeds the rate of carbonate ions migration (figure 18). To verify that
electromigration, not
thermodynamic diffusion, was responsible for these lower carbonate ions
concentrations,
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
23
electromigrative effects were temporarily removed from the model. Without
electromigration, the
carbonate concentrations in the cathode catalyst layer were at least an order
of magnitude higher
(figure 19) and the hydroxide concentrations were also substantially higher
(figure 20). These
findings suggest that a regeneration step can maintain carbonate
concentrations below the
solubility limit and thereby prevent carbonate salt formation.
A cycle with 60-second operation followed by 30-second regeneration (figure
47) was simulated.
The highest carbonate concentration reached in the alternating simulation was
3.4 M, well below
the solubility limit. This limit was reached at ¨ 2000 seconds (22 cycles)
after which the peak
species concentrations did not increase further with the highest carbonate
concentration reaching
only 3.4 Mat this time. Simulations were also performed with shorter
regeneration times per cycle,
namely 10 and 20 second variants, but the peak carbonate concentrations were
much closer to the
solubility limit (e.g., the 20-second regeneration time had a peak carbonate
concentration of ¨6 M,
figures 21 and 22). It should be noted that, although hydroxide and
bicarbonate ions can also form
salt precipitates with potassium cations, the peak concentrations of hydroxide
and bicarbonate ions
in the model were much lower than their respective solubility limits,
suggesting that carbonate is
the dominant salt precipitate in this system (figure 5 to 14). The alternating
voltage strategy
maintains a stable carbonate concentration below the carbonate salt solubility
limit.
To further demonstrate that the alternating strategy was successful in
reducing carbonate salt
formation, a cathode was fabricated by spraying a carbon gas diffusion layer
with silver
nanoparticles on a substrate and carbon monoxide (CO) was produced from CO2 in
a CO2RR MEA
electrolyzer including the fabricated cathode. The anolyte was 0.1 M potassium
bicarbonate and
the anode was an iridium-based catalyst that was used to perform oxygen.
Referring to figure 23,
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
24
when performing CO2RR at a constant operational voltage of -3.6 V, the CO
selectivity dropped
from 98% to 76% after just 12 hours of operation. During the operational
period, the H2 selectivity
increased by a complementary amount, while the current density decreased
slightly from 170 mA
cm-2 to 160 mA cm-2. This behavior is considered as characteristic of salt
formation in the MEA
electrolyzer and associated blockage of CO2 reactant (see inset of figure 23).
In order to apply the present alternating strategy, also referred to as an
unsteady electrochemical
forcing strategy, the system was cyclically operated with the application of
the same operational
voltage of -3.6 V for an operation duration of 60 seconds, and further
application of a regeneration
voltage of -2.0 V for a regeneration duration of 30 seconds (figure 24). For a
direct comparison
with the above detailed continuously operated system, the alternating system
was operated for 12
hours (18 hours total duration including 6 hours regeneration). Unlike the
continuously operated
MEA electrolyzer, which operated at the same current density for the same
amount of operational
time, the cyclically operated MEA electrolyzer had no visible salt formation
and sustained a high
CO selectivity. Comparing operational voltages over short time scales, the
alternating sample
(figure 25, table 2) exhibited similar selectivities and current densities to
that of the sample
operated continuously (figure 26).
Table 2: Product distribution for alternating voltage experiments with silver
and copper
cathodes.
Faradaic Efficiency (%)
Full cell
Potential
N- Acetalde
Sample (V)
H2 CO CH4 C2H4 Formate Acetate Ethano propano
hyde
Acid Acid 1
1
-3.40 4.2 92.1 0.2 0.1 0.7
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
+2.9 +4.6 +0.0 +0.0 +0.3
-3.60 4.3 93.5 0.2 0.1 0.9
+1.7 +5.7 +0.1 +0.0 +0.2
Carbon paper
sprayed with -3.80 4.7 93.2 0.4 0.2 0.7
silver
+2.0 +5.1 +0.1 +0.0 +0.2
nanoparticles
-4.00 8.9 88.7 0.5 0.2 1.2
+5.7 +3.7 +0.2 +0.1 +0.4
-4.20 19.2 77.4 0.7 0.2 1.4
+6.1 +10.0 +0.2 +0.0 +0.2
-3.40 6.1 54 0.2 25.2 4.1 1.1
7.2 0.6 0.4
+3.2 +9.1 +0.1 +3.9 +2.0 +0.1 +0.9 +0.2 +0.0
-3.60 6.3 36.2 0.3 37.1 3.2
2.9 10.9 0.9 0.7
PTFE with +2.9 +5.4 +0.1 3.7 +1.1
+0.4 +3.7 +0.2 +0.2
sputtered
copper & -3.80 5.9 10.6 0.7 56.7 1.9 4.7
15.6 1.8 1.4
sprayed copper
nanoparticles +2.1 3.4 0.3 5.5 0.2
+1.4 +3.1 0.9 +0.4
-4.00 13.2 9 2.6 43 1.8 7.5 16.2 2.1 1.6
+5.8 +1.9 +0.9 +7.2 +0.7 +1.3 +1.9 +0.6 +0.4
-4.20 17.9 6.1 4.9 32.1 2.1
9.2 17.1 2.4 1.8
+12.0 +1.7 +0.7 +6.1 +0.5 +2.4 +5.7 +0.7 +0.5
The test was stopped after 18 hours (total duration) for direct comparison
with the continuously
operated system.
To validate that enhancement of the stability was due to the use of a
regeneration period as per the
5 proposed method, and not from the lower average current density,
another series of tests was
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
26
performed including operation of a silver cathode sample at a slightly lower
constant operational
voltage (-3.4 V shown in figures 27 and 28). After 18 hours of continuous
operation, the effects of
salt precipitation were again major; the CO selectivity had decreased to 83%
and salt precipitates
half-filled the gas channels. In comparison, based on the same time-averaged
current density and
total charge passed, the alternating strategy yielded stable performance and
no detectable carbonate
salt.
To demonstrate the versatility of the alternating strategy, another series of
tests was performed
using an electrode including a copper-based catalyst on a PTFE-based substrate
as reported in the
experimental section (see study of Gabardo et al., entitled "Continuous Carbon
Dioxide
Electroreduction to Concentrated Multi-Carbon Products Using a Membrane
Electrode
Assembly" (Joule 2019, 3 (11), 2777-2791). It was noted that despite the
change in both the
catalyst material and electrode substrate, the stability was maintained. When
the copper electrode
was operated continuously, there was much salt precipitation visible after 48
hours (figure 29)
which in turn caused the CO2RR selectivity to decrease to 72% (figure 30) and
the current density
to decline (figure 31). Raman analysis of the cathode salt precipitates
confirmed potassium
carbonate to be the dominant precipitate (figure 32). Operating a silver
sample on PTFE yielded
similar salt precipitation, confirming that CO2RR products were not the cause
of precipitation on
these PTFE electrodes (figures 33, 34 and 35). Unsteady forcing, with 60
seconds of operation at
-3.8 V followed by 30 seconds of regeneration at -2.0 V, yielded a stable
CO2RR selectivity for
157 operational hours (236 hours of total duration) with no detectable
evidence of salt formation
(figure 36) and no degradation in performance prior to shutting down the
experiment (figure 37).
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
27
The current density of the copper-PTFE system fluctuated during the 236-hour
experiment (figure
38). Early in the experiment, there was a gradual increase in current density
from 110 to 250 mA
cm-2 during the 60 seconds of operation, as the electrolyzer cycled back to
the operational voltage
(top part of figure 39). However, after 2000 cycles (50 hours total duration),
the response of the
current density was immediate upon application of the operational voltage,
jumping to 175 mA
cm-2 and remaining constant for the 60-second operational period (bottom part
of figure 39). This
change in temporal response suggests that the capacitance of the system
decreased over the total
duration such that the electrical double layer responded quickly to the
application of the higher
voltage. In battery and water electrolyzer applications, a similar decay in
capacitance is observed
for copper and iridium-based catalysts when cycled over long periods (see
studies of Lai C. M. et
al. (.I. Power Sources 2018, 379 (January), 261-269), Hsu Y.-K., et al.
(Electrochim. Acta 2014,
139, 401-407) and Malleo D., et al. (Rev. Sci. Instrum. 2010, 81 (1), 016104).
After this initial
warm-up period, the alternating system achieved a fast current density
response to the voltage
change to maintain a uniform reaction rate during the operational periods. Ex-
situ XPS analysis of
a copper sample suggests that the catalyst was in metallic form during
operation (figures 40 and
41).
When comparing figure 42¨ table 2 with figure 43, one can see that the current
density and product
selectivity were nearly identical for the alternating and continuous
operational strategies over short
time scales and at different operational voltages. The stability towards C7
products reported here
appears to be the longest in existing literature amongst CO2 electrolyzers
operating at industrially
viable current densities (table 3).
CA 03203665 2023- 6- 28
WO 2022/148856 PCT/EP2022/050323
28
Table 3: The copper on PTFE electrode presented in this work is the longest
demonstration in
existing literature operated at industrially relevant current densities.
iC2 Operation
Catalysts (mA cm-2) al Time CI FR Cell type
References
1 CAINP/Cu/PTFE 138 157 h 81% MEA
This work
2 3D catalyst ¨120 60 h ¨60 "A MEA Science 367,
661 666 (2020)
3 Cu3N 101 20 h 60% Flow cell Alum) Lett.
19, 8658-8663 (2019)
Molecular tuned ¨79 180 h 70% MEA
Nature. 577 (7791), 509-513
4 ¨
Cu (2020)
Graphite/NP/Cu/
¨ 79 100 h ¨ 80% MEA Joule. 3, 2777-2791 (2019)
PTFE
Graphite/NP/Cu/
6 ¨ 55 150 h 83% Flow cell Science. 360, 783-787 (2018)8
PTFE
Ordered Angew. Chemie.
129, 10980-
7 ¨ 0.15 24 h 77/o H cell
Mesoporous N-C 10984 (2017)
An activation voltage refers herein to the voltage required to reach an onset
potential for both
5 cathodic and anodic reactions, thereby generating a current density in
accordance with an
activation energy of the triggered redox event. The regeneration voltage is
selected to be below
the activation voltage, and thus the regeneration period operates at a
negligible current density,
which is a much lower current density than during the operational period.
Therefore, there is
minimal additional energy required to power the regeneration period since the
regeneration period
can consume less than 1% of the system energy requirements (figure 44). The
alternating system
also reduces the addition of new electrolyte salts, new catalyst materials,
and catalyst replacement
downtimes, combining for a significant operational advantage in comparison to
continuously
operated systems.
In summary, when CO? electrolysis is performed at industrially relevant
current densities, the
steady state alkaline conditions lead, inevitably, to carbonate salt
formation. The self-cleaning CO2
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
29
reduction method implementations that are proposed herein can circumvent
steady state by cycling
the applied voltage between an operational voltage and a regeneration voltage.
The regeneration
voltage is applied during the regeneration period in order to maintain an
electric field for carbonate
ions to migrate to the anode, thereby lowering carbonate ions concentrations
at the cathode and
avoiding damaging of the cathode via salt formation and plugging. The
alternating approach was
applied to silver and copper catalysts on carbon paper and PTFE based
electrodes, respectively.
The product selectivity resulting from the cyclically operated system was
shown to be similar to
that of the continuously operated system, with the advantage that alternating
operation with
regeneration yielded no detectable carbonate formation. More specifically,
using the alternating
strategy, the copper-PTFE sample in a MEA-based electrolyzer was operated in
alternate for 157
hours (236 hours total duration), while maintaining a C2 product selectivity
of 80% and a C2 partial
current density of 138 mA cm-2 with a cost of < 1% additional system energy
input.
Test and determination methods
Raman Spectroscopy
Potassium carbonate (John Wiley & Sons, I. SpectraBase Compound ID=DepkjwUOQKb
SpectraBase Spectrum ID=.1XEQ5H3aIck https : //spe ctrabas e.
com/spectrum/JXEQ5H3aIck
(accessed Dec 19, 2020) and potassium bicarbonate (John Wiley & Sons, I. S.
SpectraBase
Compound ID=DBxdA3hF csM SpectraBase Spectrum
ID=E0IHi W8 W Wv5
https://spectrabase.com/spectrum/E0IHiW8WVVv5 (accessed Dec 19, 2020) were
both detected,
but potassium carbonate had a much higher intensity.
X-Ray Photoelectron Spectroscopy
X-Ray Photoelectron Spectroscopy (XPS) measurements were performed with a
Thermo Fisher
ESCALAB 250 Xi XPS.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
Experimental parts
The following part includes information related to the COMSOL Multiphysics
simulation results
and model mechanism; current density plots of the different regeneration
voltages; current density
5 and selectivity plots of continuous operation of silver and copper
catalysts; electrochemical
performance comparison between continuous and alternating voltage with the
same average
current density; current density and selectivity of continuous operation of
silver catalyst; electrode
preparation; operation of the electrochemical MEA cell; and product analysis.
Solubility Calculation
10 The solubility product constant of potassium carbonate (Ksp) describes
the equilibrium between
the solid and its constituent ions in a solution. The value of the constant
identifies the degree to
which the compound can dissociate in water. The K5p value of potassium
carbonate is 2073 at 20
C.1
K2CO3(s) .=` 2K+ (aq) + C032- (aq)
(El)
Ksp = [K12[C032-]1
(E2)
Applying the solubility product constant of potassium carbonate equation (E2)
into the 1D MEA
15 COMSOL model, the simulation time of the continuous operation run
reached Ksp = 2073 at 1200s
of continuous operation at -3.8 V. where [C032] = 7.8 M, [K+] = 16.6 M.
Moreover, due to the charge neutrality of the local cathode electrolyte, the
concentrations of the
constituent ions can be expressed in E3. The basic condition around the
cathode (pH - 14), the
concentrations of the [1-1],[HCO3-] and [011] were relatively small and
negligible, as compared
20 to [K+] and [C032-] . Therefore, the concentrations of [1( ] and [C032-]
maintained the
approximate ratio of 2:1.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
31
11( 1 + 1111 = 111CO3-1 + 10111 + [C0321
(E3)
Electrode Preparation
The carbon paper - silver gas diffusion electrode (GDE) was prepared by
airbrushing catalyst inks
with a nitrogen carrier gas. The catalyst silver ink was prepared with 12 mL
ethanol (Greenfield
Global Inc., >99.8%), 150 p.L Nafion (Fuel Cell Store D521 Alcohol-based 1100
EW, 5 wt%), and
mg silver nanoparticles (Sigma-Aldrich 576832-5G, <100 nm particle size). The
catalyst ink
mixtures were sonicated for two hours, and then sprayed on a gas diffusion
carbon paper (Fuel
Cell Store Sigracet 39 BC, with a microporous layer) with a spray density of
0.15 mL cm-2. After
airbrushing, the GDE was dried for 24 hours at room temperature (-20 C). The
10 polytetrafluoroethylene (PTFE) based copper electrode used was prepared
by plasma sputtering
and then airbrushing catalyst inks with a nitrogen carrier gas. Approximately
300 nm of copper
catalyst was sputtered onto the PTFE substrate using an AJA International ATC
Orion 5 Sputter
Deposition System (Toronto Nanofabrication Centre, University of Toronto). An
additional copper
layer was sprayed on top of the sputtered layer. The copper ink was prepared
with 12 mL ethanol,
15 150 pL Nafion, and 15 mg of copper nanoparticles (Sigma-Aldrich 774081-
5G, 25 nm particle
size). Catalyst inks were sonicated for two hours and then sprayed on the
sputtered PTFE sample
with a spray density of 0.15 mL cm-2. After airbrushing, the GDE was dried for
24 hours at room
temperature (-20 C). A Sustainion anion exchange membrane (Dioxide Materials
Sustainion 37)
was used in the electrolyzer. The anode electrode was prepared by spraying
iridium chloride (Alfa
Aesar, IrC13.xH20 99.8%) on a titanium support (Fuel Cell Store 592795-1,
Titanium Felt). The
coated electrode was treated by a thermal decomposition methodm.
Operation of the electrochemical MEA cell
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
32
All electrochemical experiments were performed in an anion exchange membrane-
based MEA
electrolyzer (Fuel Cell Store, 72500322, AEM Water Electrolyzer - 5cm2). The
electrolyte was
pumped through the cell by a peristaltic pump. The CO2 inlet gas flow rate was
approximately 80
standard cubic centimeters per minute (sccm). The constant voltage
electrochemical tests were
performed by running one fresh cathode sample at multiple voltages of interest
sequentially (-3.4
V, -3.6 V, -3.8 V, -4.0 V, and -4.2 V). The alternating voltage
electrochemical tests were performed
using the same sequential operational voltage above for 60 seconds, followed
by a 30 second -2.0
V regeneration voltage. The voltages reported are full cell voltages with no
iR compensation.
Product analysis
The gas products from CO2 reduction were analyzed in 1 mL volumes using a gas
chromatograph
(PerkinElmer Clarus 680), possessing a thermal conductivity detector (TCD) and
a flame
ionization detector (FID). Using argon as the carrier gas (Praxair, 99.999%),
the gas
chromatograph was equipped with a Molecular Sieve 5A capillary column and a
packed Carboxen-
1000 column. The flow rate of the gas was measured before each 1 mL volume was
collected.
The gas sample was collected by water displacement for one operational and
regenerational
iteration for alternating voltage tests. Then, we used the integration of
total charge passing over
the iteration to calculate the gas product Faradaic efficiency.
The liquid products were quantified using nuclear magnetic resonance
spectroscopy (NMR). 1H
NMR spectra of freshly acquired samples were collected on an Agilent DD2 500
spectrometer
using water suppression mode with dimethyl sulfoxide (DMSO) as an internal
standard.
1D MEA COMSOL Multiphysics Model:
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
33
The one-dimensional was modelled by COMSOL Multiphysics version 5.5,
incorporating both the
carbon dioxide reduction reaction (CO2RR) on the cathode and the oxygen
evolution reaction
(OER) on the anode in 0.1M KHCO3 anolyte. An anion exchange membrane (AEM) was
sandwiched between the cathode and anode. The major focus of this study was to
compare the
local carbonate concentration with and without the alternating voltage salt
prevention strategy. The
Secondary Current Distribution and Transport of Diluted Species physics
modules within
COMSOL were used to model the chemical reactions between aqueous CO2, HCO3-,
C032-, H+,
011- and K+ in a time-dependent study. This model was a modified version of
previous reports,
see for example the study of McCallum C., et al., entitled "Reducing the
crossover of carbonate
and liquid products during carbon dioxide electroreduction" (Cell Reports
Physical Science, 2021,
2, 100522). There were several general assumptions for this simulation.
Firstly, a constant
concentration of CO2 was supplied at the humidified GDE/CL interface, and
constant
concentrations of chemical species were set at the right-hand boundary of the
anolyte layer.
Secondly, a Cu/Nafion layer was directly deposited on top of the porous Cu
catalyst layer to serve
as a current collector. Thirdly, the cathode and anode were separated by an
AEM, and an
electrolyte was distributed through the porous media.
The geometry (figure 45) consisted of a gas diffusion electrode (GDE), a
cathode catalyst layer
(CL), a current collector layer (CCL), an anionic exchange membrane (AEM), an
iridium oxide
(IrOx) anode catalyst layer and an anolyte layer. An electrical potential was
applied at the left-
hand boundary GDE layer. The ground was applied at the anode catalyst/anolyte
interface. A CO2
concentration at the GDE/CL interface was specified to be equal to the maximum
Solubility in
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
34
0.1M KHC 03 electrolyte. The equilibrium values were specified at the right-
hand boundary of the
anolyte layer.
CO2 Solubility in 0.1M KTIC03 Electrolyte:
The CO2 Solubility in pure water was determined by Henry's Law (E4 E5).
Solubility in water
depends on the temperature and pressure. 16'17
[CO2] aq,0 = K0 [CO2]9
(E4)
100 T
(E5)
inKo 93.4517(--) - 60.2409 + 23.3585/n(¨)
100
Where K0 is the Henry volatility constant, which can be influenced by
temperature T. However,
due to the "Salting out- effect as explained by the Sechenov Equation,18 the
Solubility of CO2 in
a 0.1M KHCO3 electrolyte decreases as the salt concentration increases (E6 -
E8). As such, CO2
Solubility can be calculated using the sets of equations are shown below.
log(
[C 2]aq,0) =
(E6)
[C 0 daq
K, = E(hion + hG)
(E7)
hG = hG,0 + hT(T - 298.15)
(E8)
The K, represents the Sechenov constant, and Cs is the molar concentration of
the electrolyte
solution. The Solubility is determined based on IC+, HCO3, C032- and OH- ions
concentration
and the specific parameters which are shown in table 4.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
Table 4: Corresponding Sechenov constants in 0.1M KHCO3 electrolyte - see
study of
Weisenberger S., et al., entitled "Estimation of gas solubilities in salt
solutions at temperatures
from 273K to 363 K (AlChE J., 1996, 42 (1), 298-300.
Ion hion
K 0.0922
OH- 0.0839
C032- 0.1423
HCO3- 0.0967
hG,0 for CO2 -0.0172
hT for CO2 -0.000338
5 Catalyst Electrochemical Reactions:
Electrochemical reactions were applied within the respective catalyst layers
(E9 - E12): CO2
reduction to CO , ''2, C2 H4 , C2 H5 0 H on the cathode and oxygen evolution
on the anode catalyst
layer (E13).
CO2RR:
2H20 + 2e- H2 20H-
(E9)
CO2 + H20 + 2e- CO + 20H-
(E10)
2CO2 + 8H20 + 12e- C2/14 + 120H-
(Ell)
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
36
2CO2 + 9H20 + 12e- ¨> C2H5OH + 120H-
(E12)
OER:
2H20 ¨> 02 4H+ + 2e-
(E13)
Ohm's Law and Poisson Equation:
The electrode and electrolyte potentials were governed by Ohm's Law (E14). The
electromigration
of the charged species (J-1CO3, C032-, H , OH- and K) (El 5) was controlled by
the electrolyte
potential and the combination of electroneutrality and induced space charge
for ion-exchange
membrane, which is governed by the Poisson equation (E16).
Ocp
(E14)
i = ¨o--
ax
V = clit +
(E15)
az')
(E16)
606r - =
dx FL ZC + Paem
Where o- was the electrical conductivity of different media as listed in Table
5. 0/ was the
electrolyte potential. was the combination of electroneutrality and induced
space charge for the
ion-exchange membrane. eoand Er were the permittivity of vacuum and the
relative permittivity
of water, respectively. pa, was the space charge for the membrane that exists
exclusively in the
membrane domain. The detailed values for AEM are listed in Table 6.
Table 5: Electrical conductivity of different domains. (see study of Gabardo
C. M. et al., efr
supra)
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
37
Domain electrical conductivity
[S/m]
Copper cathode catalyst 0. 8 e5
Current collector 1.7e6
Anion exchange membrane 8.0
IrOx Anode catalyst 1.4e7
Anolyte 4.56
Table 6: Parameters for AEM.
Parameters Value (unit)
Permittivity of vacuum 8.8542e-12 (F/m)
Relative permittivity of water 80 (1)
Membrane space charge 1 (M)
Porous Medium Effective Diffusion
All layers except the electrolyte diffusion boundary layer were considered as
a porous medium.
The effective diffusivity was governed by the Bruggeman model. The porosity
was 0.6 in the Cu
cathode catalyst and current collector. The porosity was 0.9 in the IrOx Anode
catalyst. The
porosity was 0.1 for the AEM with a 90% reduction in diffusion coefficients
for the cations (see
studies of Dinh C. T. et al., entitled "CO2 Electroreduction to Ethylene via
Hydroxide-Mediated
Copper Catalysis at an Abrupt Interface" (Science, 2018, 360 (6390), 783-787)
and of Singh M.
R. et al., entitled "Mechanistic Insights into Electrochemical Reduction of
CO) over Ag Using
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
38
Density Functional Theory and Transport Models" (Proc. Natl. Acad. Sci., 2017,
114 (42), E8812¨
E8821).
Butler-Volmer Equations:
The electrode kinetics of CO2 reduction and water oxidation were modelled by
the Butler-Volmer
equation (E17 ¨E21).
(E17)
ic, H2 = i13,H2 eXP{ac,H2 F RT (77)}
=ac,coF (E18)
tc,co = io,coexPt (n)1
RT
fac,C2H4F (E19)
iC,C2 H4 ¨ 1:0,C2 exP RT (77)1
=
ca ,ETOHF (E20)
1c,Et0H = 1:0,Et0HeXPi _______________________________ RT (71)1
=
aa,02F (E21)
= to,o2exPf (n))
RT
= Vapp E0,i
(E22)
The exchange current density (i0j) and charge transfer coefficient (ac,i) were
obtained from
experimental results, determined in the same way as previous works (see
Burdyny T. et al - ACS
Sustain. Chem. Eng. 2017, 5 (5), 4031-4040). The overpotential (77) was
determined by the
difference between the applied voltage Vap p and the equilibrium voltage
(E0,i) (E22). The kinetics
constants are listed in Table 7.
CA 03203665 2023- 6- 28
WO 2022/148856
PCT/EP2022/050323
39
Table 7: Experimental electrode kinetics for CO2RR and HER.
Reaction io,i (A m-2) E0,1 (V vs RHE)
COER 9.7e7 -0.51
0.136
C2H4ER 1. le6 -0.33
0.17
HER 5.3e6 -0.41
0.136
Et0HER 5.4e5 -0.32
0.119
OER le-1 0.82
1.02
Species transport:
In the reaction-diffusion model, the species transport equations (E23 - E24)
were governed by the
Nernst-Planck equations. Diffusion and electromigration terms were considered
for the
transportation of chemical species.
act
= ER
(E23)
¨ - - ¨
at ox
= _________________________________________________ F
Diaci aV
(E24)
c- ¨
Ox RT ax
Ci, Di and zi represent the species concentration, diffusion coefficient, and
charge number,
respectively. The diffusion coefficient and charge number are listed below in
Table 8.
Table 8: Diffusion coefficients and charge in the IVIEA system (see Vanysek, P
- CRC Handb.
Chem. Phys. 1996, 96 (73), 5-98).
CA 03203665 2023- 6- 28
WO 2022/148856 PCT/EP2022/050323
Species Diffusion coefficient Charge
number
(m2s-1)
CO2 1.91e-9 0
C032- 0.923e-9 -2
HC 03- 1.185e-9 -1
9.31e-9 +1
OH- 5.26e-9 -1
K 1.96e-9 +1
Carbonate Equilibrium Equation:
The model predicted a steady-state equilibrium between aqueous CO2, HCO3-,
C032-, 11+ ,and
OH- by considering several chemical reactions in alkaline conditions (E25 -
E28). Water
5 dissociation (E29) was also considered in this system. The reaction
rate constants were determined
by the temperature and salinity4. The corresponding equations are listed
below.
CO2 + H20 <--->H + HCO
(E25)
HCO3 <--> 11+ + CO
(E26)
CO2 + OH- <-->CO
(E27)
HCO + OH- 4--> CO V + H2O
(E28)
H20 <--> H + OH-
(E29)
CA 03203665 2023- 6- 28