Note: Descriptions are shown in the official language in which they were submitted.
USE OF HETEROCYCLIC COMPOUND IN TREATING DISEASES RELATED TO
KINASE DRUG-RESISTANT MUTATION AND METHOD THEREFOR
Technical Field
The present disclosure relates to the use of a heterocyclic compound in the
preparation of a
drug for the treatment or adjuvant treatment of a disease or condition
associated with an RET
(Rearranged during transfection) drug-resistant mutation in an individual and
a treatment method
using the heterocyclic compound.
Background
Protein kinases are a class of enzymes catalysing protein phosphorylation
reactions. By
mediating the process of cell signal transduction, protein phosphorylation
regulates the
physiological activities of cells, such as cell survival, proliferation,
differentiation, apoptosis, and
metabolism. The dysfunction of the protein kinases is closely associated with
many diseases,
including tumours, autoimmune diseases, inflammatory reactions, central
nervous system diseases,
cardiovascular diseases, diabetes, and the like.
As a protooncogene, RET encodes an RET protein which is a transmembrane
receptor type
tyrosine protein kinase consisting of a cysteine-rich cadherin-like
extracellular domain (for binding
to ligands), a transmembrane domain, and an intracellular domain with tyrosine
kinase activity.
The activated RET protein can activate multiple downstream signal pathways,
including
RAS/RAF/ERK pathway, PI3K/Akt pathway, and J NK pathway, thereby resulting in
cell
proliferation, migration, and differentiation. The alteration (mutation or
fusion) of the RET gene
and the abnormal expression of wild-type RET gene lead to abnormal activation
of RET proteins,
such that the signal pathways are overactive, which is one of the main
mechanisms of
carcinogenesis. Abnormally activated RET proteins are involved in the
proliferation and invasion
of different tumour cells through a variety of signal pathways, thereby
affecting the occurrence and
development of tumours. The alteration of the RET gene has a more significant
effect on
1
CA 03210109 2023- 8- 28
downstream cascade reactions, where the mutation of the RET gene is mainly
associated with
medullary thyroid cancer and papillary thyroid cancer, and the fusion of the
RET gene is mainly
associated with non-small cell lung cancer and chronic myeloid leukaemia.
Therefore, it is of great
medical value to inhibit RET activity (Nature Reviews Cancer, 2014, 14 (3):
173-186).
RET inhibitors have great potential for the treatment and prevention of a
variety of diseases
(such as tumours, irritable bowel syndrome, and the like). At present, two
compounds
(Selpercatinib (also known as LOX0-292) and Pralsetinib (also known as BLU-
667)) have been
approved for marketing, and multiple compounds are in clinical trials.
However, clinical studies
have shown that long-term administration of RET inhibitors can lead to RET
drug-resistant
mutations (such as G810R, G810S, G810C, Y806C, Y806N, and V738A), which can
easily lead
to decreased efficacy, relapse, and poor prognosis 0 ournal of Thoracic
Oncology, 2020, 15(4),
541-549; Annals of oncology: official journal of the European Society for
Medical Oncology,
2021, 32(2), 261-268; Annals of oncology: official journal of the European
Society for Medical
Oncology, 2020, 31(12), 1599-1600; Annals of oncology: Official Journal of the
European Society
for Medical Oncology, 2020, 31(12), 1725-1733, etc.). Therefore, the
development of compounds
effective for diseases related to RET drug-resistant mutations has a good
application prospect.
Summary
In one aspect, the present disclosure provides the use of a compound of
Formula I, a
stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof, an N-
oxide thereof, a
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or a stable isotope
derivative, metabolite, or prodrug thereof in the preparation of a drug for
the treatment or adjuvant
treatment of a disease or condition associated with an RET drug-resistant
mutation (e.g., one or
more mutations selected from the group consisting of G810R, G810S, G810C,
Y806C, Y806N,
and V738A) in an individual:
2
CA 03210109 2023- 8- 28
H
N.R,
V, NI
Ni
N
(---')
N
R3
Formula I
wherein:
R1 is selected from 4-10-membered heterocyclyl and 5-10-membered heteroaryl,
each
optionally substituted with one or more substituents independently selected
from the group
consisting of hydroxyl, halogen, CN, NO2, Ci_4 alkyl, C1-4 haloalkyl, C14
hydroxyalkyl, C14
haloalkoxy, C1-4 heteroalkyl, C3_6 cycloalkyl and C3_6 cycloalkoxy;
R2 is selected from the group consisting of halogen, C1_6 alkyl, C1-6
haloalkyl, C1-6 heteroalkyl,
4-10-membered heterocyclyl and 5-10-membered heteroaryl, wherein the alkyl,
heteroalkyl,
heterocyclyl and heteroaryl are each optionally substituted with one or more
substituents
independently selected from the group consisting of hydroxyl, halogen, CN,
NO2, C1-4 alkyl, C1-4
haloalkyl, C1_4 hydroxyalkyl, C1_4 haloalkoxy, C1-4 heteroalkyl, C3-6
cycloalkyl and C3-6
cycl oal koxy;
R3 is selected from the group consisting of H, halogen, C1_6 alkyl, C1_6
haloalkyl, C1-4
hydroxyalkyl, C1-4 heteroalkyl and C3_6 cycloalkyl; and
Xl, X2 and X3 are each independently selected from CH and N.
In some preferred embodiments, the present disclosure provides the use of a
compound 1, a
stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof, an N-
oxide thereof, a
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or a stable isotope
derivative, metabolite, or prodrug thereof in the preparation of a medicine
for the treatment or
3
CA 03210109 2023- 8- 28
adjuvant treatment of a disease or condition associated with an RET drug-
resistant mutation (e.g.,
one or more mutations selected from the group consisting ot G810R, G810S,
G810C, Y 806C,
Y 806N, and V738A) in an individual, wherein the compound 1 has the following
structure:
N N
e,
= N
F 1
For example, the pharmaceutically acceptable salt at the compound 1 is a
tumarate ot the
compound 1.
In another aspect, the present disclosure provides the compound ot Formula I,
the stereoisomer,
tautomer, or mixture of stereoisomer and tautomer thereof, the N-oxide
thereof, the
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or the stable isotope
derivative, metabolite, or prodrug thereof for use in the treatment or
adjuvant treatment of a disease
or condition associated with an RET drug-resistant mutation (e.g., one or more
mutations selected
tram the group consisting of G810R, G8105, G810C, Y 806C, Y 806N, and V738A)
in an
individual.
In another aspect, the present disclosure provides a method for the treatment
or adjuvant
treatment of a disease or condition associated with an RET drug-resistant
mutation (e.g., one or
more mutations selected from the group consisting of G810R, G810S, G810C, Y
806C, Y 806N,
and V738A) in an individual, wherein the method comprises administering to the
individual an
effective amount of the compound of Formula I, the stereoisomer, tautomer, or
mixture of
stereoisomer and tautomer thereof, the N-oxide thereof, the pharmaceutically
acceptable salt,
eutecticum, polymorph or solvate thereof, or the stable isotope derivative,
metabolite or prodrug
thereof.
In another aspect, the present disclosure provides the use of the compound of
Formula I, the
4
CA 03210109 2023- 8- 28
stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof, the N-
oxide thereof, the
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or the stable isotope
derivative, metabolite, or prodrug thereof in the preparation of a drug for
the modulation (e.g.,
reduction or inhibition) of abnormal RET activity associated with an RET drug-
resistant mutation
(e.g., one or more mutations selected from the group consisting of G810R,
G810S, G810C, Y 806C,
Y806N, and V738A).
In another aspect, the present disclosure provides the compound of formula I,
the stereoisomer,
tautomer, or mixture of stereoisomer and tautomer thereof, the N-oxide
thereof, the
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or the stable isotope
derivative, metabolite, or prodrug thereof for the modulation (e.g., reduction
or inhibition) of
abnormal RET activity associated with an RET drug-resistant mutation (e.g.,
one or more
mutations selected from the group consisting of G810R, G810S, G810C, Y 806C,
Y806N, and
V738A).
In another aspect, the present disclosure provides a method for the modulation
(e.g., reduction
or inhibition) of abnormal RET activity associated with an RET drug-resistant
mutation (e.g., one
or more mutations selected from the group consisting of G810R, G810S, G810C,
Y806C, Y806N,
and V738A), wherein the method comprises administrating an effective amount of
the compound
of Formula I, the stereoisomer, tautomer, or mixture of stereoisomer and
tautomer thereof, the N-
oxide thereof, the pharmaceutically acceptable salt, eutecticum, polymorph or
solvate thereof, or
the stable isotope derivative, metabolite or prodrug thereof.
The compound has good inhibition effect on RET drug-resistant mutations, and
features good
pharmacokinetic, safety and other properties.
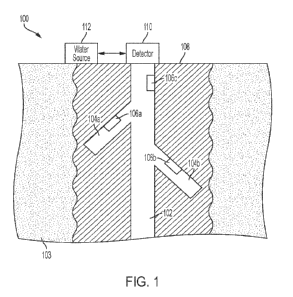
Brief Description of the Drawings
FIG. 1: In vivo efficacy of Compound 1-A and control compound in Ba/F3 KIF5B-
RETG81 R
subcutaneous xenograft tumour model.
Detailed Description of the Invention
Definitions
5
CA 03210109 2023- 8- 28
Unless otherwise defined in the context, all technical terms and scientific
terms used herein are
intended to have the same meaning as commonly understood by those skilled in
the art. The
reference to a technology used herein is intended to refer to a technology
generally understood in
the art, including those technological alterations or equivalent technological
replacements that are
obvious to those skilled in the art. While it is believed that the following
terms are well understood
by those skilled in the art, the following definitions are still set forth to
better explain the present
disclosure.
The term "including," "comprising," "having," "containing," or "relating to"
and additional
variations thereof herein are inclusive or open-ended, and do not exclude
additional unlisted
elements or method steps, although additional unlisted elements or method
steps do not necessarily
exist (i.e., these terms also encompass the terms "substantially consisting
of" and "consisting of").
As used herein, the term "alkyl" is defined as a linear or branched saturated
aliphatic
hydrocarbon. In some embodiments, alkyl has from 1 to 12, e.g., from 1 to 6,
carbon atoms. For
example, as used herein, the terms "C1_6 alkyl" and "C1_4 alkyl" refer to a
linear or branched group
having from 1 to 6 carbon atoms and a linear or branched radical having from 1
to 4 carbon atoms
respectively (e.g., methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-
butyl, tert-butyl, n-
pentyl, isopentyl, neopentyl, or n-hexyl), which are optionally substituted
with one or more (e.g.,
from 1 to 3) suitable substituents, e.g., halogen (in this case, the radical
is termed "haloalkyl") (e.g.,
CH2F, CHF2, CF3, CCI3, C2F5, C2CI5, CH2CF3, CH2CI, or -CH2CH2CF3). The term
"C1_4 alkyl"
refers to a linear or branched aliphatic hydrocarbon chain having from 1 to 4
carbon atoms (i.e.,
methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, or tert-
butyl).
As used herein, the term "heteroalkyl" refers to an optionally substituted
alkyl radical that has
one or more backbone chain atoms selected from atoms other than carbon, such
as oxygen,
nitrogen, sulphur, phosphorus, or combinations thereof. A numerical range
(e.g., C1_6 heteroalkyl)
that may be given refers to the number of carbons in a chain, including from 1
to 6 carbon atoms
in this example. For example, -CH2OCH2CH3 group is termed C3 heteroalkyl. Its
attachment to the
rest of the molecule may be through a heteroatom or carbon atom in the
heteroalkyl chain.
6
CA 03210109 2023- 8- 28
As used herein, the term "haloalkyl" refers to an alkyl radical substituted
with one or more
(e.g., from 1 to 3) same or different halogen atoms, and the term "C1_6
haloalkyl" and "C1_4
haloalkyl" refer to a haloalkyl radical having from 1 to 6 carbon atoms and a
haloalkyl radical
having from 1 to 4 carbon atoms respectively, such as -CF3, -C2F5, -CHF2, -
CH2F, -CH2CF3, -
CH2CI, or -CH2CH2CF3.
As used herein, the term "hydroxyalkyl" refers to a group formed by
substituting hydrogen
atom(s) in an alkyl radical with one or more hydroxy, e.g., Ci_4 hydroxyalkyl
or Ci_3 hydroxyalkyl,
and its examples include, but are not limited to, hydroxymethyl, hydroxyethyl,
hydroxypropyl,
hydroxybutyl, -CH(OH)CH3, -C(CH3)20H, and the like.
As used herein, the term "alkoxy" refers to a group formed by inserting an
oxygen atom into
any reasonable position of an alkyl radical (as defined above), and is, for
example, C1_6 alkoxy, C1-
4 alkoxy, or C1_3 alkoxy. Representative examples of Ci_6 alkoxy include, but
are not limited to,
methoxy, ethoxy, propoxy, isopropoxy, n-propoxy, isopropoxy, n-butoxy,
isobutoxy, tert-butoxy,
pentyloxy, hexyloxy, -CH2-0CH3, and the like, and the alkoxy is optionally
substituted with one
or more (e.g., from 1 to 3) same or different substituents.
As used herein, the term "condensed ring" or "fused ring" refers to a ring
system formed by
two or more ring structures sharing two adjacent atoms with each other.
As used herein, the term "Spiro ring" refers to a ring system formed by two or
more ring
structures sharing one ring atom with each other.
As used herein, the term "bridged ring" refers to a ring system formed by two
or more ring
structures sharing two atoms (that are not directly connected) with each
other.
As used herein, the term "cycloalkyl" refers to a saturated or unsaturated non-
aromatic
monocyclic or multicyclic (such as bicyclic) hydrocarbon ring radical,
including but not limited to
monocycloalkyl (such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl,
cyclooctyl, and cyclononyl) and bicycloalkyl, including a spiro ring,
condensed (fused) ring, or
bridged ring system (i.e., spirocycloalkyl, condensed (fused) cycloalkyl, and
bridged cycloalkyl,
such as bicyclo[1.1.1]pentyl, and bicyclo[2.2.1]hepty1). In the present
disclosure, a cycloalkyl
7
CA 03210109 2023- 8- 28
radical is optionally substituted with one or more (e.g., from 1 to 3) same or
different substituents.
A carbon atom on a cycloalkyl radical is optionally substituted with an oxo
group (i.e., forming
C=0). The term "C3_6 cycloalkyl" refers to a cycloalkyl radical having from 3
to 6 ring-forming
carbon atoms, which may be a monocycloalkyl radical, such as cyclopropyl,
cyclobutyl,
cyclopentyl, cyclohexyl, cycloheptyl, or cyclooctyl, and may also be a
bicycloalkyl radical, such
as C5_8 spirocycloalkyl, C5_8 bridged cycloalkyl, C5_8 fused cycloalkyl, C5_6
spirocycloalkyl, C5-6
bridged cycloalkyl, or C5-6 fused cycloalkyl.
As used herein, the term "cycloalkoxy" means -0-cycloalkyl, where the
cycloalkyl is as defined
above. Representative examples of cycloalkoxy radicals include, but are not
limited to,
cyclopropoxy, cyclobutoxy, cyclopentoxy, cyclohexoxy, and the like.
As used herein, the term "heterocyclyl" or "heterocyclic ring" refers to a
monocyclic or
multicyclic (for example, condensed, spiro, or bridged cyclic) radical having
2 or more than 2 (e.g.,
3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, or 14) carbon atoms, and one or more (for
example, 1, 2, 3, or 4)
heteroatoms, wherein the heteroatoms include, but are not limited to, an
oxygen atom, a nitrogen
atom, and a sulphur atom, and the carbon atoms and heteroatoms on the
heterocyclyl are optionally
substituted with an oxo group (for example, forming C=0, S(=0), or S(=0)2).
As used herein, the term "4-10-membered heterocyclyl" means a heterocyclyl
radical
containing from 4 to 10 ring atoms, including but not limited to, 4-9-membered
heterocyclyl, 4-8-
membered heterocyclyl, 4-7-membered heterocyclyl, 5-6-membered heterocyclyl, 3-
8-membered
heterocyclyl, 3-7-membered heterocyclyl, 4-7-membered nitrogen-containing
heterocyclyl, 4-7-
membered oxygen-containing heterocyclyl, 4-7-membered sulphur-containing
heterocyclyl, 5-6-
membered nitrogen-containing heterocyclyl, 5-6-membered oxygen-containing
heterocyclyl, 5-6-
membered sulphur-containing heterocyclyl, and the like. The "nitrogen-
containing heterocyclyl,"
"oxygen-containing heterocyclyl," and "sulphur-containing heterocyclyl" each
optionally further
contain one or more additional heteroatoms selected from oxygen, nitrogen, and
sulphur. Examples
of 4-10-membered heterocyclyl include, but are not limited to, oxiranyl,
aziridinyl, azacyclobutyl,
8
CA 03210109 2023- 8- 28
oxacyclobutyl, tetrahydrofuranyl, pyrrolidinyl, pyrrolidonyl (e.g.,
---) ), imidazolidinyl,
pyrazolidinyl, tetrahydropyranyl, piperidinyl, morpholinyl, dithianyl,
thiomorpholinyl,
piperazinyl, and trithianyl.
As used herein, the term "heteroaryl" or "heteroaromatic ring" refers to a
monocyclic or
multicyclic aromatic group comprising one or more same or different
heteroatoms, including a
monocyclic heteroaryl radical and a bicyclic or multicyclic ring system
comprising at least one
heteroaromatic ring (an aromatic ring system comprising at least one
heteroatom), which may have
5, 6, 7, 8, 9, 10, 11, 12, 13, or 14 ring atoms, for example, 5, 6, 7, 8, 9,
or 10 ring atoms. The
heteroatom may be oxygen, nitrogen, or sulphur. A carbon atom and a heteroatom
on the heteroaryl
are optionally substituted with an oxo group (for example, forming C=0, S(=0),
or S(=0)2).
As used herein, the term "5-10-membered heteroaryl" or "5-10-membered
heteroaromatic ring"
means a heteroaryl radical (heteroaromatic ring) comprising from 5 to 10
(e.g., from 5 to 6) ring
atoms, including 5-10-membered nitrogen-containing heteroaryl, 5-10-member
oxygen-containing
heteroaryl, 5-10-membered sulphur-containing heteroaryl, 5-6-membered nitrogen-
containing
heteroaryl, 5-6-membered oxygen-containing heteroaryl, 5-6-membered sulphur-
containing
heteroaryl, and the like. The "nitrogen-containing heteroaryl," "oxygen-
containing heteroaryl," and
"sulphur-containing heteroaryl" each optionally comprise one or more
additional heteroatoms
selected from oxygen, nitrogen, and sulphur. Examples thereof include, but are
not limited to,
thienyl, furanyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl,
isoxazolyl, isothiazolyl,
triazolyl, tetrazolyl, oxadiazolyl, thiadiazolyl, etc., or pyridyl,
pyridazinyl, pyrimidinyl, pyrazinyl,
triazinyl, etc., and a 5-10-membered condensed ring group comprising these
radicals.
As used herein, the term "heteroaryl" encompasses a condensed ring structure,
and the point of
attachment of the condensed ring structure to additional radicals may be on
any ring of the
condensed ring structure. Therefore, heteroaryl radicals of the present
disclosure further include,
but are not limited to, (mono)heteroaryl-(mono)heteroaryl, (mono)heteroary1-
(monocyclo)aryl,
(mono)heteroary1-(mono)heterocyclyl, and (mono)heteroary1-(mono)cycloalkyl,
such as 5-6-
9
CA 03210109 2023- 8- 28
membered (mono)heteroary1-5-6-membered (mono)heteroaryl, 5-6 membered
(mono)heteroaryl-
phenyl, 5-6-membered (mono)heteroary1-5-6-membered (mono)heterocyclyl, or 5-6-
membered
(mono)heteroaryl-C4_6(mono)cycloalkyl (e.g., 5-6-membered heteroaryl-
cyclobutyl, 5-6-
membered heteroaryl-cyclopentyl, or 5-6-membered heteroaryl-cyclohexyl).
Examples of
heteroaryl include, but are not limited to, indolyl, isoindolyl, indazolyl,
benzimidazolyl, quinolinyl,
1
)ss'r
NH ss',,11
I
isoquinolinyl,NH NH N 1\(
N 1\1\
, and the like.
As used herein, the term "halo" or "halogen" radical is defined to encompass
F, Cl, Br, or I.
The term "substitution" means that one or more (e.g., one, two, three, or
four) hydrogens on a
specified atom are replaced by a selection from the indicated radicals,
provided that the normal
valence of the specified atom in the present case is not exceeded and the
substitution leads to a
stable compound. A combination of substituents and/or variables is permissible
only when such a
combination leads to a stable compound.
If a substituted radical is described as "optionally.. substituted," the
substituted radical may be
(1) unsubstituted, or (2) substituted. If a carbon of a substituted radical is
described as being
optionally substituted with one or more substituents in a list of
substituents, one or more hydrogens
(to the extent of any present hydrogens) on the carbon may be independently
and/or together
replaced with independently selected optional substituents. If a nitrogen of a
substituted radical is
described as being optionally substituted with one or more substituents in a
list of substituents, one
or more hydrogens (to the extent of any present hydrogens) on the nitrogen may
each be replaced
with independently selected optional substituents.
If a substituent is described as being "independently selected from" a group,
each substituent
is selected independently of the others. Therefore, each substituent may be
the same as or different
from another (other) substituent(s).
CA 03210109 2023- 8- 28
As used herein, the term "one or more" means 1 or more than 1, such as 2, 3,
4, 5, or 10, under
reasonable conditions.
Unless otherwise specified, as used herein, the points of attachment of a
substituent may be
from any suitable positions at the substituent.
When a bond of a substituent is shown as a bond connecting two atoms through a
ring, such a
substituent may bond to any ring-forming atom in the substitutable ring.
The present disclosure further includes all pharmaceutically acceptable
isotopically-labelled
compounds, which are the same as the compounds of the present disclosure,
except that one or
more atoms are replaced with atoms which have the same atomic number, but an
atomic mass or
mass number different from the atomic mass or mass number predominantly found
in nature.
Examples of isotopes suitable for inclusion in the compounds of the present
disclosure include, but
are not limited to, isotopes of hydrogen (e.g., deuterium (2H), tritium (3H));
isotopes of carbon
(e.g., nc, 13C, and 14C); isotopes of chlorine (e.g., 36CI); isotopes of
fluorine (e.g., 18.-Ar) ;
isotopes of
iodine (e.g., 1231 and 1251); isotopes of nitrogen (e.g., 13N and 15N);
isotopes of oxygen (e.g., 150,
170, and 180); isotopes of phosphorus (e.g., 32P); and isotopes of sulphur
(e.g., 35S). Certain
isotopically labelled compounds (e.g., those incorporating a radioisotope) of
the present disclosure
are useful in drug and/or substrate tissue distribution study (e.g.,
analysis). Radioisotopes tritium
(i.e., 3H) and carbon-14 (i.e., 14C) are particularly preferred for their ease
of incorporation and
detectability. Substitutions with positron emission isotopes (e.g., 11C, 18F,
15u,-,, and 13N) may be
used to test substrate receptor occupancy in positron emission tomography
(PET) studies. The
isotopically-labelled compounds of the present disclosure may be prepared by
methods analogous
to those described in the accompanying routes and/or examples and preparations
by replacing a
non-isotopically labelled reagent with an appropriate isotopically labelled
reagent.
Pharmaceutically acceptable solvates of the present disclosure include those
in which the
crystallisation solvent may be substituted with an isotope, for example, D20,
acetone-d6, or
DMSO-d6.
The term "stereoisomer" means an isomer formed due to at least one asymmetric
centre. In a
11
CA 03210109 2023- 8- 28
compound with one or more (e.g., one, two, three, or four) asymmetric centres,
its exo/meso
mixtures, single enantiomers, and diastereomer mixtures and individual
diastereomers may be
produced. Specitic individual molecules may also exist as geometric isomers
(cis/trans). Similarly,
the compound at the present disclosure may exist in a mixture at two or more
structurally ditterent
forms (commonly referred to as tautomers) in rapid equilibrium. Representative
examples of
tautomers include keto-enol tautomers, phenol-keto tautomers, nitroso-oxime
tautomers, imine-
enamine tautomers, and the like. For example, nitroso-oximes may exist in
equilibrium in the
tol lowing tautomeric torms in solution:
NO
It should be understood that the scope of the present disclosure encompasses
all such isomers
or mixtures thereof in any proportions (for example, 60%, 65%, 70%, 75%, 80%,
85%, 90%, 95%,
96%, 97%, 98%, 99%).
A solid line (
), a solid wedge ( --"`"), or a dashed wedge ( ¨"II") may be used
herein to depict chemical bonds ot the compounds ot the present disclosure.
The use of solid lines
to depict bonds to asymmetric carbon atoms is intended to indicate that all
possible stereoisomers
at that carbon atom (e.g., specitic enantiomers or racemic mixtures) are
included. The use ot solid
or dashed wedges to depict bonds to asymmetric carbon atoms is intended to
indicate that the
stereoisomers shown exist. When present in a racemic mixture, solid and dashed
wedges are used
to define relative stereochemistry, rather than absolute stereochernistry.
Unless otherwise
specified, the compound of the present disclosure is intended to exist in the
form of stereoisomers
(including cis and trans isomers, optical isomers (such as R and S
enantiomers), diastereomers,
geometric isomers, rotamers, contormational isomers, atropisomers, and
mixtures thereof). The
compound ot the present disclosure may exhibit more than one type of isomerism
and be composed
of mixtures thereof (e.g., racemic mixtures and diastereomeric pairs).
The present disclosure encompasses all possible crystalline forms or
polymorphs of the
compound of the present disclosure, which may be a single polymorph or a
mixture of more than
12
CA 03210109 2023- 8- 28
one polymorph at any ratio.
Eutecticum refers to the fact that the active molecules of a drug and
additional physiologically
acceptable molecules of acids, bases, salts, and non-ionic compounds are
connected by hydrogen
bonds, 1C-IE stacking, van der Waals forces, and additional non-covalent bonds
to be combined in
the same crystal lattice.
It should also be understood that some compounds of the present disclosure may
exist in free
form for treatment, or, where appropriate, in the form of pharmaceutically
acceptable derivatives
thereof. In the present disclosure, pharmaceutically acceptable derivatives
include, but are not
limited to, pharmaceutically acceptable salts, esters, solvates, N-oxides,
metabolites, or prodrugs,
which, after being administered to patients in need thereof, can directly or
indirectly provide the
compound of the present disclosure or metabolites or residues thereof.
Therefore, when the
"compound of the present disclosure" is referred to herein, it is also
intended to encompass the
above various derivative forms of the compound.
Pharmaceutically acceptable salts of the compound of the present disclosure
include both acid
addition salts and base addition salts, for example, hexafluorophosphate, and
meglumine salt. For
a review of suitable salts, see Stahl and Wermuth, "Handbook of Pharmaceutical
Salts: Properties,
Selection, and Use" (Wiley-VCH, 2002).
As used herein, the term "ester" means an ester derived from the compound of
each general
formula in the present disclosure, which includes physiologically hydrolysable
esters
(hydrolysable under physiological conditions to release the compound of the
present disclosure in
the form of free acid or alcohol). The compound of the present disclosure
itself may also be an
ester itself.
The compound of the present disclosure may exist in the form of solvate
(preferably hydrate),
where the compound of the present disclosure comprises a polar solvent as a
structural element of
the crystal lattice of the compound, especially, for example, water, methanol,
or ethanol. The
amount of the polar solvent, especially water, may be stoichiometric or non-
stoichiometric.
Those skilled in the art will understand that, since available lone pairs of
electrons are required
13
CA 03210109 2023- 8- 28
to oxidize nitrogen to form oxides, not all nitrogen-containing heterocyclic
rings can form N-
oxides. Those skilled in the art can recognize nitrogen-containing
heterocyclic rings that can form
N-oxides. Those skilled in the art can also recognize that tertiary amines can
form N-oxides. The
synthesis methods for the preparation of N-oxides of heterocyclic rings and
tertiary amines are well
known to those skilled in the art, including but not limited to the use of
peroxyacids such as
peroxyacetic acid and m-chloroperoxybenzoic acid (MCPBA), hydrogen peroxide,
alkyl
hydroperoxides such as tert-butyl hydroperoxide and sodium perborate, and
dioxirane such as
dimethyl dioxirane to oxidize heterocyclic rings and tertiary amines. These
methods for the
preparation of N-oxides have been widely described and summarized in
literature, for example, T.
L. Gilchrist, Comprehensive Organic Synthesis, vol. 7, pp 748-750; A. R.
Katritzky and A. J.
Boulton, Eds., Academic Press; and G. W. H. Cheeseman and E. S. G. Werstiuk,
Advances in
Heterocyclic Chemistry, vol. 22, pp 390-392, A. R. Katritzky and A. J .
Boulton, Eds., Academic
Press.
The present disclosure further includes, within its scope, metabolites of the
compound of the
present disclosure, i.e., substances formed in vivo from the administered
compound of the present
disclosure. Such products may be produced by, for example, oxidation,
reduction, hydrolysis,
amidation, deamidation, esterification, enzymolysis, and the like of the
administered compound.
Therefore, the present disclosure includes metabolites of the compound of the
present disclosure,
including compounds prepared by exposing a mammal to the compound of the
present disclosure
for a period of time sufficient to produce its metabolites.
The present disclosure further includes, within its scope, the prodrugs of the
compound of the
present disclosure, which are certain derivatives of the compound of the
present disclosure that
may themselves have less pharmacological activity or no pharmacological
activity, and that may
be converted into the compound of the present disclosure having the desired
activity by, for
example, hydrolytic cleavage when administered into or onto the human body.
Generally, such
prodrugs are functional group derivatives of the compound, which are easily
converted into the
desired therapeutically active compound in vivo. Additional information on the
use of prodrugs
14
CA 03210109 2023- 8- 28
may be found in "Pro-drugs as Novel Delivery Systems", Volume 14, ACS
Symposium Series (T.
Higuchi and V. Stella). The prodrugs of the present disclosure may be prepared
by, for example,
substituting appropriate functional groups present in the compound of the
present disclosure with
certain moieties known to those skilled in the art as "pro-moiety (for
example, as described in
"Design of Prodrugs", H. Bundgaard (Elsevier, 1985))".
The present disclosure further includes the compound of the present disclosure
comprising
protective groups. In any process of preparing the compound of the present
disclosure, protection
of sensitive groups or reactive groups on any related molecules may be
necessary and/or desirable,
thereby forming a chemically protected form of the compound of the present
disclosure. This may
be achieved by conventional protective groups, for example, those protective
groups as described
in T. W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis, J ohn
Wiley & Sons,
1991. These references are incorporated herein by reference. The protective
groups may be
removed at an appropriate subsequent stage using methods known in the art.
The term "about" means within 10%, preferably within 5%, more preferably
within 2%, of
said value.
"Pharmaceutically acceptable carriers" in the present disclosure refer to
diluents, adjuvants,
excipients, or vehicles which are administered together with a therapeutic
agent, and are, within
the scope of sound medical judgement, suitable for contact with the tissues of
human beings and/or
other animals without excessive toxicity, irritation, allergic reaction, or
other problems or
complications commensurate with a reasonable benefit/risk ratio.
Pharmaceutically acceptable carriers useful in the pharmaceutical composition
of the present
disclosure include, but are not limited to, sterile liquid. Examples of
suitable pharmaceutically
acceptable carriers are as described in Remington's Pharmaceutical Sciences
(1990).
The compound of the present disclosure or a pharmaceutical composition
comprising it may
act systemically and/or locally. For this purpose, they may be administered
through a suitable route.
For these administration routes, the compound of the present disclosure or a
pharmaceutical
composition comprising it may be administered in a suitable dosage form.
CA 03210109 2023- 8- 28
The term "effective amount" as used herein refers to an amount of a compound
that, after being
administered, will relieve one or more symptoms of the disease being treated
to a certain extent.
The dosage regimen may be adjusted to provide the optimal desired response.
For example, a
single bolus may be administered, several divided doses may be administered
over time, or the
dose may be proportionally reduced or increased as indicated by the exigencies
of the treatment. It
should be noted that the dose may vary with the type and severity of the
condition to be alleviated,
and may include single or multiple doses. It should be further understood that
for any particular
individual, the specific dosage regimen should be adjusted over time according
to the needs of the
individual and the professional judgement of the person administering the
composition or
supervising the administration of the composition.
The dosage of the compound of the present disclosure to be administered will
depend on the
individual being treated, the severity of the disease or condition, the rate
of administration, the
disposal of the compound, and the judgement of the prescribing physician. In
general, the effective
dosage ranges from about 0.0001 to about 50 mg per kg body weight per day. In
some cases, dosage
levels not higher than the lower limit of the aforesaid range may be adequate,
while in other cases
still larger doses may be employed without causing any harmful side effect,
provided that such
larger doses are first divided into several smaller doses for administration
throughout the day.
The content or amount of the compound of the present disclosure in the
pharmaceutical
composition may be from about 0.01 mg to about 1,000 mg.
Unless otherwise specified, the term "treating" as used herein means
reversing, alleviating,
inhibiting the progress of, or preventing the disorder or condition to which
such term applies, or
one or more symptoms of such disorder or condition.
The "individual" as used herein includes human or non-human animals. Exemplary
human
individuals include human individuals suffering from diseases (such as the
diseases described
herein) (referred to as patients) or normal individuals. In the present
disclosure, "non-human
animals" include all vertebrates, for example, non-mammals (such as birds,
amphibians, reptiles)
and mammals, for example, non-human primates, livestock, and/or domesticated
animals (such as
16
CA 03210109 2023- 8- 28
sheep, dogs, cats, cows, and pigs).
In some embodiments, the compound of the present disclosure may be
administered in
combination with one or more additional therapeutic agents or prophylactic
agents (for example,
additional drugs for treating a cancer or neoplastic disease). In some
embodiments, the method of
the present disclosure may also include the administration of one or more
additional therapeutic
agents or prophylactic agents (e.g., additional drugs for treating a cancer or
a neoplastic disease).
Pharmaceutical Use and Treatment Method
In some embodiments, the present disclosure provides the use of a compound of
Formula I, a
stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof, an N-
oxide thereof, a
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or a stable isotope
derivative, metabolite, or prodrug thereof in the preparation of a drug for
the treatment or adjuvant
treatment of a disease or condition associated with an RET drug-resistant
mutation (e.g., one or
more mutations selected from the group consisting of G810R, G810S, G810C,
Y806C, Y806N,
and V738A) in an individual:
H
X3y, N
k---)
NI
Ra X'
Formula!
wherein:
R1 is selected from 4-10-membered heterocyclyl and 5-10-membered heteroaryl,
each
optionally substituted with one or more substituents independently selected
from the group
consisting of hydroxyl, halogen, CN, NO2, Ci_4 alkyl, C1-4 haloalkyl, C1-4
hydroxyalkyl, C14
haloalkoxy, C1-4 heteroalkyl, C3_6 cycloalkyl and C3_6 cycloalkoxy;
17
CA 03210109 2023- 8- 28
R2 is selected from the group consisting of halogen, C1_6 alkyl, C1_6
haloalkyl, C1_6 heteroalkyl,
4-10-membered heterocyclyl and 5-10-membered heteroaryl, wherein the alkyl,
heteroalkyl,
heterocyclyl and heteroaryl are each optionally substituted with one or more
substituents
independently selected from the group consisting of hydroxyl, halogen, CN,
NO2, C1-4 alkyl, C1-4
haloalkyl, C1-4 hydroxyalkyl, C1_4 haloalkoxy, C1_4 heteroalkyl, C3_6
cycloalkyl and C3-6
cycl oal koxy;
R3 is selected from the group consisting of H, halogen, C1_6 alkyl, C1_6
haloalkyl, C14
hydroxyalkyl, C1_4 heteroalkyl and C3_6 cycloalkyl; and
X', X2 and X3 are each independently selected trom CH and N.
In some preferred embodiments, in the compound of Formula I, a stereoisomer,
tautomer, or
mixture of stereoisomer and tautomer thereof, an N-oxide thereot, a
pharmaceutically acceptable
salt, eutecticum, polymorph, or solvate thereof, or a stable isotope
derivative, metabolite, or
prodrug thereot as related to in the present disclosure, the compound of
Formula I is a compound
1:
N
N N
r
c.
N
1
For example, the pharmaceutically acceptable salt of the compound is a
fumarate of the
compound 1.
In some embodiments, the present disclosure provides the compound ot Formula
I, the
stereoisomer, tautomer, or mixture ot stereoisomer and tautomer thereot, the N-
oxide thereot, the
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or the stable isotope
derivative, metabolite, or prodrug thereof for use in the treatment or
adjuvant treatment of a disease
or condition associated with an RET drug-resistant mutation (e.g., one or more
mutations selected
18
CA 03210109 2023- 8- 28
from the group consisting of G810R, G810S, G810C, Y806C, Y 806N, and V738A) in
an
individual.
In some embodiments, the present disclosure provides a method for the
treatment or adjuvant
treatment of a disease or condition associated with an RET drug-resistant
mutation (e.g., one or
more mutations selected from the group consisting of G810R, G810S, G810C,
Y806C, Y 806N,
and V738A) in an individual, wherein the method comprises administering to the
individual an
effective amount of the compound of Formula I, the stereoisomer, tautomer, or
mixture of
stereoisomer and tautomer thereof, the N-oxide thereof, the pharmaceutically
acceptable salt,
eutecticum, polymorph or solvate thereof, or the stable isotope derivative,
metabolite or prodrug
thereof.
In some embodiments, the disease or condition associated with an RET drug-
resistant mutation
is a drug-resistant disease, preferably a drug-resistant cancer or tumour or
irritable bowel
syndrome; the caner or tumour is, for example, an advanced cancer or tumour or
a metastatic cancer
or tumour; the cancer or tumour is preferably lung cancer (e.g., non-small
cell lung cancer), breast
cancer, head and neck cancer, rectal cancer, liver cancer, lymphoma, thyroid
cancer (e.g.,
medullary thyroid cancer or papillary thyroid cancer), colon cancer, multiple
myeloma, melanoma,
glioma, cerebroma or sarcoma.
In some embodiments, the drug-resistant disease is a disease resistant to
Selpercatinib and/or
Pralsetinib.
In some embodiments, the drug-resistant disease is non-small cell lung cancer
(e.g., RET
fusion-positive non-small cell lung cancer), medullary thyroid cancer (e.g.,
advanced or metastatic
medullary thyroid cancer) or thyroid cancer (e.g., advanced or metastatic RET
fusion-positive
thyroid cancer) resistant to Selpercatinib and/or Pralsetinib.
In some embodiments, the present disclosure provides the use of the compound
of Formula I,
the stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof,
the N-oxide thereof,
the pharmaceutically acceptable salt, eutecticum, polymorph, or solvate
thereof, or the stable
isotope derivative, metabolite, or prodrug thereof in the preparation of a
drug for the modulation
19
CA 03210109 2023- 8- 28
(e.g., reduction or inhibition) of abnormal RET activity associated with an
RET drug-resistant
mutation (e.g., one or more mutations selected from the group consisting of
G810R, G810S,
G810C, Y 806C, Y 806N, and V738A).
In some embodiments, the present disclosure provides the compound of formula
I, the
stereoisomer, tautomer, or mixture of stereoisomer and tautomer thereof, the N-
oxide thereof, the
pharmaceutically acceptable salt, eutecticum, polymorph, or solvate thereof,
or the stable isotope
derivative, metabolite, or prodrug thereof for the modulation (e.g., reduction
or inhibition) of
abnormal RET activity associated with an RET drug-resistant mutation (e.g.,
one or more
mutations selected from the group consisting of G810R, G810S, G810C, Y 806C, Y
806N, and
V738A).
In some embodiments, the present disclosure provides a method for the
modulation (e.g.,
reduction or inhibition) of abnormal RET activity associated with an RET drug-
resistant mutation
(e.g., one or more mutations selected from the group consisting of G810R,
G810S, G810C, Y 806C,
Y 806N, and V738A), wherein the method comprises administrating an effective
amount of the
compound of Formula I, the stereoisomer, tautomer, or mixture of stereoisomer
and tautomer
thereof, the N-oxide thereof, the pharmaceutically acceptable salt,
eutecticum, polymorph or
solvate thereof, or the stable isotope derivative, metabolite or prodrug
thereof.
In some embodiments, the RET drug-resistant mutation is one or more mutations
selected from
the group consisting of G810R, G810S, and G810C.
In some embodiments, the RET drug-resistant mutation is selected from one or
more of RET
solvent front mutation and RET hinge mutation.
In some embodiments, the drug-resistant mutation is RET solvent front
mutation.
In some embodiments, the RET drug-resistant mutation is an 810 mutation, such
as G810
mutation.
In a preferred embodiment, the RET drug-resistant mutation is one or more
mutations selected
from the group consisting of G810R, G810S, and G810C.
In some embodiments, the compound has a structure represented by Formula I-A:
CA 03210109 2023- 8- 28
N'T;71r
N N
,R2
Fonnuia I -A
wherein R1, R2, R3, Xl and X2 are as defined above for Formula I.
In some embodiments, the compound has a structure represented by Formula I-B:
I W
X2L
N
R2
Formula I - B
wherein Rl, R2, R3, Xl and X2 are as defined above for Formula I.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R1 is 5-10-membered heteroaryl which is
optionally
substituted with one or more substituents independently selected from the
group consisting of
hydroxyl, halogen, CN, Ci_4 alkyl, C1-4 haloalkyl, C3.6 cycloalkyl and C3_6
cycloalkoxy.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R1 is 5-6-membered heteroaryl which is
optionally
substituted with one or more substituents independently selected from the
group consisting of
halogen, C1_3 alkyl, and C3-6 cycloalkyl.
21
CA 03210109 2023- 8- 28
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R1 is pyrazolyl which is optionally
substituted with one or
more substituents independently selected from the group consisting of halogen,
C1_3 alkyl (e.g.,
methyl) and C3_6 cycloalkyl (e.g., cyclopropyl).
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, IR1 is pyrazolyl substituted with methyl
(e.g., 5-methyl-IN-
pyrazol-3-y1 or 1-methyl-1H-pyrazol-4-y1) or pyrazolyl substituted with
cyclopropyl (e.g., 5-
cycl opropyl-1 H -pyrazol-3-y1).
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R2 is selected from the group consisting
of halogen (e.g., Cl),
C1_6 alkyl (e.g., methyl) and 5-6-membered heteroaryl, wherein the alkyl and
heteroaryl are each
optionally substituted with one or more substituents independently selected
from the group
consisting of halogen, C1_4 alkyl, C1-4 hydroxyalkyl, C1-4 heteroalkyl, C3-6
cycloalkyl and C3-6
cycloalkoxy.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R2 is 5-6-membered heteroaryl which is
optionally
substituted with one or more substituents independently selected from the
group consisting of
halogen, C1_4 alkyl, C1-4 hydroxyalkyl, C3_6 cycloalkyl and C3_6 cycloalkoxy.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R2 is 5-membered heteroaryl which is
optionally substituted
with one or more substituents independently selected from the group consisting
of halogen, C1-4
alkyl, C1-4 hydroxyalkyl, C3-6 cycloalkyl and C3-6 cycloalkoxy.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R2 is pyrrolyl, pyrazolyl, imidazolyl,
furanyl or thiazolyl,
which is optionally substituted with one or more substituents independently
selected from the
OH
group consisting of F, Cl, methyl, , cyclopropyl and -0-cyclopropyl.
22
CA 03210109 2023- 8- 28
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R3 is selected from H, C1_6 alkyl and
C3_6 cycloalkyl.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R3 is selected from H and C1_6 alkyl.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, R3 is H or methyl.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula I-
B provided by the present disclosure, Xl is CH or N; and preferably Xl is N.
In some embodiments, in the use of the compounds of Formula I, Formula I-A and
Formula l-
B provided by the present disclosure, X2 is CH or N; and preferably, X2 is CH.
In some embodiments, in the use of the compounds of Formula I provided by the
present
disclosure, X3 is CH or N; and preferably, X3 is N.
The present disclosure encompasses any combination of the above embodiments.
In some embodiments, the compounds include, but are not limited to:
H H H H
N....... IN N\CNµNH ''',In:N \Cõ(--N,.... , p, Pr. IN NµC-NINH
'N'ir-Y1 __N Y''''''r[1.11 -CNõc N--:,-51 Nr,cNH
,Nh N N , NH
eir2,,cr , "NH iµj IN c,NN .j i,ij
N .., N.f..)-
N .---
*
/51,
N *I N
k'l
N
N
H
N C,114_N -11 Lt511.,
r4,- )N, thli N 10 0 _N
1.' Me l'ICI
1 F 2
H
N H
I -N
=====,,,c.,,....N N
NN "NH 'il'Cl-NJNH -firf:77"" 'nr)-411NNH NIrr-NNH NHNN
C:NH
l! N.....- 1.i,,' 1 ---- N
....1:I
N N
N
ICLI CCII ,1 tq, ti: N, C)*
N_N, ,.1
NN \ N
.= µ,- -F -\-q --'-C 1,....
13 NF 14 N-
e a 10 F 11 N- 12
23
CA 03210109 2023- 8- 28
H H H H
F
1,'T''.....r-N NN \C--,NH NYPII-N)----,N, N -\C-II H , N,
N, N ,',_. 'NH ...1 N 1
6--. N NH -1' -N, N, N f,,, NH N N N
.,.._ NH
if '''... '.---- y,
N- =-=..
----(
1 il...
N .., NW- N ,==== N ....,
õ....,
'I ...%. N oN
<-;
N
0 , N
,N
NO-F
Nq
.i...._,
1 / N- /Th
15 18 17 18 F 19 20
F
H
H H H A "... õ.1,-.%\rN \_,,
H H II
---.
1- \C11NH 1.1.....):
Nt7NH Nnri NZ-NNH N,,N tr-_, NH ---....,..^-rN _,._._ NI,
Yi.N),--,1',10_ N., N NH
õ
--c' N, N 't..NH N, N I
7
N,..5., 1 .,...,.. 115 ---
NI ......
II = N N.T.,--
9
N N
..-
N
N
N N N
ti....`N. N
1
NN\ N
,N I 'N til
Ir'N
H
No --- IC3... 40H
_rs
,... \ L271 1
21 22 23-1 23-2 24 25
28 6
H
F
,y7r.11_____N, .,.,I.r.11 H\c:N
-,--N, "nr-^')-N, N-..1
tõ,...NH
NI
N., N (NH , N ,., NH NIA L)/NH 14-õ,N
Gõ,...,(NH
1\
, I
r5
I N NI .õ.,
isr1,1)
...,
..õ..i)
,N,
N" N
N
N N
L' i 'N LiCi 'N
i I
F ' 101 r),INC
I 4.0H N
F
Nõ...,..1,' N...N
27
.., N /)
1õ.,..... - ...-' 0 IR
\ /
F 28 29 30 F .
In some embodiments, the compound is:
I-
_N,
r ,IN
1,----)1
N
L'aN,
'-'"---(.
1 F
=
In some embodiments, the pharmaceutically acceptable salt is a tumarate.
In some embodiments, the pharmaceutically acceptable salt ot the compound is:
24
CA 03210109 2023- 8- 28
N
II NH
N N
I ,N
HOOC cooEi
N"
F =
In some embodiments, the compound of the present disclosure may be prepared by
the method
described in WO 2020/168939 Al.
Examples
The present disclosure will be further described below in combination with
examples, but the
provision of these examples is not intended to limit the scope of the present
disclosure.
The abbreviations as used herein have the following meanings:
Abbreviat Hour
Meaning
ion
High-performance
liquid
ally! Allyl HPLC chromatography
2 2'-bis(diphenylphosphino)- Potassium
KHM DS B I NAP 11'-binaphthyl bis(tri
methylsilyl)amide
B I NOL 1,1'-bi naphthol KOAc Potassium acetate
B2(pin)2 Bis(pinacolato)diboron LC-MS Liquid
chromatography-mass
spectrometry
CD3OD Deuterated methanol
LDA Lithium
diisopropylamide
DCE 1,2-dichloroethane Lithium
Li H M DS DCM/CH2 Dichloromethane
bis(trimethylsilyl)amide
Cl2 Me Methyl
DI EA/DI P
N,N-diisopropylethylamine Medium
pressure liquid
EA MPLC chromatography
DMA/DM AC Dimethylacetamide MS Mass spectrometry
DM E Dimethoxyethane min Minute
DMF N,N-dimethylformamide Sodium
NaHMDS bis(trimethylsilyl)amide
DMSO-d6 Deuterated dimethyl sulfoxide
NIS N-iodosuccinimide
DMSO Dimethyl sulfoxide
NM P N-
methylpyrrolidone
1,1-bis
Dppf (diphenylphosphine)ferrocene NMR Nuclear magnetic
resonance
EA/Et0Ac Ethyl acetate PCy3
Tricyclohexylphosphine
Et Ethyl Pd Palladium
CA 03210109 2023- 8- 28
Pd(acac)2 Bis(acetylacetone)palladium rt. Room temperature
Bis(dibenzylideneacetone)pall 2-
dicyclohexylphosphino-
Pd(dba)2 adium RuPhos 2',6'-
diisopropoxy-1,1'-
Tris(dibenzylideneacetone)dip diphenyl
Pd2(dba)3 alladium SPh 2-
dicyclohexylphosphino-
1,1- os 2',6'-dimethoxy-
biphenyl
Pd(dppf)C bis(diphenylphosphino)ferroce TEA Triethylamine
12 ne dichloropalladium
Pd(OAc)2 Palladium acetate TFA Trifluoroacetic
acid
, Tetrakis(triphenylphosphine)p THF Tetrahydrofuran
Pd(PPh3)4 alladium TLC Thin-layer
chromatography
PE Petroleum ether
ntPhos 4,5-bisdiphenylphosphino-9,9-
a X
Prep- Preparative high-performance dimethylxanthene
HPLC liquid chromatography XPh 2-
dicyclohexylphosphino-
os
RET Rearranged during transfection 2',4',6'-
triisopropylbiphenyl
The compound of the present disclosure was separated and purified by
preparative TLC, silica
gel column chromatography, Prep-HPLC, and/or Flash column chromatography, and
its structure
was validated by 11d NM R and/or MS. The reaction was monitored by TLC or LC-
MS.
A Bruker superconducting nuclear magnetic resonance spectrometer (model AVACE
III HD
400 MHz) was used for 1H NM R spectroscopy.
Aglient 1260 I nfinity/Aglient 6120 Quadrupole was used for LC/MS.
Silica gel GF 254 was used as the stationary phase for TLC.
200-300 mesh silica gel (Qingdao Marine) was generally used as the stationary
phase for
column chromatography.
A Biotage flash column chromatograph was used for flash column chromatography.
Agilent 1260, Waters 2489, and GeLai 3500 chromatographs were used for Prep-
HPLC.
A Biotagelnitiator microwave reactor was used for microwave reaction.
In the following examples, unless otherwise specified, the reaction
temperature was room
temperature (15-30 C).
Reagents used in the present disclosure were purchased from Acros Organics,
Aldrich
Chemical Company, or Terbo Chemical and the like.
Example 1: 2-(6-(6-((6-4-chloro-1H-pyrazol-1-
yppyridin-3-yl)methyl)-3,6-
26
CA 03210109 2023- 8- 28
diazabicyclo[3.1.1]heptan-3-yppyridin-3-0-6-methyl-N-(5-methyl-1H-pyrazol-3-
yllpyrimidin-4-amine (Compound 9)
Br
H
N
DIASO. ir:: l'*
iggc Step 1
N /
N Pd !dpoIC :VW
C1 "C KOAc 1oxans . Q= =.... N
.., I
Step 2 N
F c'-')
Id 1 El uc N
1c
la
W
.\T-3'-ir:146c41.11-1 1- ri
N,,,,t, ::
)... j .TI
:c.NH , Y '' I N
.Y. -- riH
6 N N
1bp
PthrippliCVDCLI
Cs:C01. 1,4-clionne ..H20. 40 *C N , I TPA DM r z
=
Step 3 N Step 4
A. k
k4
% '
l'rrITI144
1'
"
1,
,r.5...,
roscoq. TEA
\ P + I-41A N= K CO Inkl r 8" f: C -El
El
Step 5 1
.._N'
¨
\--1 Aso Step 6
1 h 1 1. N
IA
k,..
ID
Step 1: Tert-butyl 3-(5-bromopyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-
carboxylate
(Compound lc)
Compound la (1.50 g) and Compound lb (1.77 g) were successively added to a 100
mL single-
necked flask, and then DMSO (20.0 mL) and K2CO3 (5.83 g) were successively
added. The mixture
was heated to 90 C, and stirred under the protection of nitrogen at this
temperature for 20 h. After
completion of the reaction, the reaction mixture was cooled to room
temperature, diluted with
water (100 mL), and extracted with EA. The organic phases were combined,
washed with saturated
brine, dried over anhydrous sodium sulphate, filtered, concentrated under
reduced pressure, and
separated and purified by flash silica gel column chromatography (PE:EA=5:1),
to provide
Compound lc (2.03 g). MS m/z (ESI ): 354.1 [M +H]t
27
CA 03210109 2023- 8- 28
Step 2: Tert-butyl 3-(5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-y1) pyridin-
2-y1)-3,6-
diazabicyclo [3.1.1] heptan-6-carboxylate (Compound id)
Compound lc (2.03 g), B2(Pin)2 (4.01 g), KOAc (1.55 g), 1,4-dioxane (15.0 mL),
and
Pd(dppf)C12=DCM (644.67 mg) were successively added into a 100 mL single-
necked flask, and
were heated to 90 C for reaction under the protection of nitrogen. After
completion of the reaction,
the reaction mixture was cooled to room temperature, diluted with water (30
mL), and extracted
with EA (40 mLx3). The organic phases were combined, washed with saturated
brine, dried over
anhydrous sodium sulphate, filtered, concentrated to dryness under reduced
pressure, and separated
and purified by flash silica gel column chromatography (DCM:Me0H=15:1), to
provide
Compound id (2.11 g). MS m/z (ESI ): 402.3 [M +H]+.
Step 3: Tert-butyl 3-(5-(4-methyl-64(5-methyl-1H-pyrazol-3-y1)-amino)
pyrimidin-2-y1)
pyridin-2-y1)-3,6-diazabicyclo [3.1.1] heptan-6-carboxylate (Compound if)
Compound le (950 mg) was dissolved in 1,4-dioxane (50.0 mL), Compound id (2.11
g),
Cs2CO3 (3.15 g), and water (5.0 mL) were successively added, and then
Pd(dppf)C12=DCM (477.83
mg) was added. The mixture was heated to 90 C, and kept for reaction under the
protection of
nitrogen at this temperature for 14 h. After completion of the reaction, the
reaction mixture was
cooled to room temperature, diluted with water (100 mL), and extracted with EA
(60 mL x3). The
organic phases were combined, washed with saturated brine, dried over
anhydrous sodium
sulphate, filtered, and then concentrated to dryness under reduced pressure,
to provide Compound
if (587.0 mg). MS m/z (ESI): 463.3 [M +H]t
Step 4: 2-(6-(3,6-diazabicyclo [3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-
(5-methy1-
1H-pyrazol-3-y1) pyrimidin-4-amine (Compound 1g)
Compound if (1.36 g) was dissolved in DCM (20.0 mL), TFA (20.0 mL) was then
added, and
the mixture was kept for reaction under the protection of nitrogen at room
temperature. After
completion of the reaction, the reaction mixture was concentrated to dryness
under reduced
pressure, and separated and purified by Prep-H PLC to provide trifluoroacetate
of Compound 1g
(587.0 mg). MS m/z (ESI ): 363.3 [M +H]t
28
CA 03210109 2023- 8- 28
Step 5: 6-(4-chloro-1H-pyrazol-1-y1) nicotinaldehyde (1j)
Compound 1h (150 mg), Compound 1i (99 mg) and K2CO3 (334 mg) were added into
DM F (4
mL), heated to 80 C to react for 14 h, and the products were detected by LC-
MS. After completion
of the reaction, the reaction mixture was cooled to room temperature, diluted
with water, and
extracted with ethyl acetate (25 mL x 3). The organic layers were combined and
washed with water
three times, washed with saturated brine, dried over anhydrous sodium
sulphate, and filtered. The
filtrate was concentrated to dryness under reduced pressure, and separated and
purified by silica
gel plate chromatography (DCM:Me0H=10:1) to provide the target compound lj (23
mg). MS
m/z (ESI ): 208.0 [M+H].
Step 6: 2-(6-(64(6-(4-chloro-1H-pyrazol-1-y1) pyridin-3-y1) methyl)-3,6-
diazabicyclo
[3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyrimidin-4-amine
(Compound 9)
Trifluoroacetate of Compound 1g (30 mg) and Compound 1j (23 mg) were dissolved
in
methanol (0.5 mL), and then Et3N (8 mg) and NaBH3CN (26 mg) were successively
added. The
reaction mixture was kept for reaction at room temperature for 20 h, and then
concentrated to
dryness under reduced pressure, separated and purified by Prep-HPLC to provide
Compound 9 (5
mg). MS m/z (ESI): 554.2 [M+H]t
11-1 NM R (400 MHz, DMSO-d5) 8 11.98 (s, 1H), 9.65 (s, 1H), 9.13 (d, J = 2.0
Hz, 1H), 8.81 -
8.74 (m, 1H), 8.47-8.41 (m, 2H), 8.03-7.97 (m, 1H), 7.96-7.92 (m, 1H), 7.86
(d, J = 8.4 Hz, 1H),
6.84 (br, 1H), 6.78 (d, J = 8.8 Hz, 1H), 6.32 (br, 1H), 3.82 - 3.67 (m, 4H),
3.66 - 3.51(m, 4H),
2.60 - 2.53 (m, 1H), 2.33 (s, 3H), 2.26 (s, 3H), 1.60 (d, J = 8.4 Hz, 1H).
Example 2: 2-(6-(6-(4-(5-cyclopropy1-1H-pyrazol-1-yll benzyI)-3,6-diazabicyclo
[3.1.1]
heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyri midi n-4-a mine
(Compound 14)
29
CA 03210109 2023- 8- 28
Cu! NLY-dimethy1-1,2-
ediy1eriediaraine
c,=
* C-j)¨ab c$CC :AP. l'!
Step I =
+
>'?
'42
C.;)-01¨V
rilf water r. 1.4j
14: - Step 2 =4e
TEA
-t¨cA - =
step 3
14
Step 1: 1-(4-(1,3-dioxolan-2-y1) phenyl)-3-cyclopropy1-1H-pyrazole (14c) and
14441,3-
dioxolan-2-y1) phenyl)-5-cyclopropy1-1H-pyrazole (14d)
To a round-bottom flask, 14a (472 mg), 14b (1 g), Cul (831 mg), N,N'-dimethy1-
1,2-
ethylenediamine (384 mg), Cs2CO3 (4.27 g) and DM F (10 mL) were successively
added, heated to
115 C and kept for reaction for 3 h at this temperature under the protection
of N2. After the raw
materials were completely converted as monitored by LC-MS, the reaction
mixture was cooled to
room temperature, diluted with 20 mL of water, and extracted with ethyl
acetate (25 mL x 3). The
organic layer was washed with water three times and dried over anhydrous
sodium sulphate, and
filtered. The filtrate was concentrated under reduced pressure to give a
mixture of Compounds 14c
and 14d (1.1 g), which was used directly without further purification for the
next step of reaction.
ESI-MS (m/z): 257.1 [M+H].
Step 2: 4-(5-cyclopropy1-1H-pyrazol-1-yll benzaldehyde (14e) and 4-(3-
cyclopropy1-1H-
pyrazol-1-yll benzaldehyde (14f)
The mixture of 14c and 14d from the previous step (930 mg) was dissolved in a
mixed solvent
of THF (20 mL) and water (20 mL), and then a 37% HCI solution (15 mL) was
added dropwise.
The mixture was kept for reaction at room temperature for 18 h, and the raw
materials were
completely converted as monitored by LC-MS. Part of the solvent was evaporated
under reduced
pressure, and the reaction mixture was adjusted to pH of about 9 with
saturated NaHCO3, and
extracted with ethyl acetate (20 mL x 3). The organic layers were combined and
dried over
CA 03210109 2023- 8- 28
anhydrous sodium sulphate, and filtered. The filtrate was concentrated under
reduced pressure,
separated and purified by silica gel plate chromatography (DCM:Me0H=10:1) to
give Compound
14e (less polar compound in TLC) (400 mg) and Compound 14f (more polar
compound in TLC)
(20 mg). ESI-MS (m/z): 213.2 [M+H].
Step 3: 2-(6-(6-(4-(3-cyclopropy1-1H-pyrazol-1-y1) benzyI)-3,6-diazabicyclo
[3.1.1]
heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyrimidin-4-amine
(Compound 14)
Trifluoroacetate of Compound lg (30 mg) and Compound 14e (26 mg) were
dissolved in
methanol (0.5 mL), and then Et3N (8 mg) and NaBH3CN (26 mg) were successively
added.
Thereafter, the reaction mixture was kept for reaction at room temperature for
16 h. After
completion of the reaction, the reaction mixture was concentrated to dryness
under reduced
pressure, and separated and purified by Prep-H PLC to provide Compound 14 (22
mg). MS m/z
(ESI): 559.3 [M+H].
3+1 NM R (400 MHz, DMSO-d6) 8 11.97 (s, 1H), 9.65 (s, 1H), 9.12 (di = 2.4 Hz,
1H), 8.43
(dd, J = 9.2, 2.4 Hz, 1H), 8.28 (d, J = 2.4 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H),
7.41 (d, J = 8.4 Hz,
2H), 6.84 (br, 1H), 6.78 (d, J = 8.8Hz, 1H), 6.30 (br, 1H), 6.21 (d, J =
2.4Hz, 1H), 3.82 - 3.65 (m,
4H), 3.64- 3.47(m, 4H), 2.59- 2.53 (m, 1H), 2.33 (s, 3H), 2.26 (s, 3H), 2.00 -
1.92 (m, 1H), 1.59
(d, J = 8.4 Hz, 1H), 0.95 - 0.88 (m, 2H), 0.75 - 0.69 (m, 2H).
Example 3: 2-(6-(6-(4-(3-cyclopropy1-1H-pyrazol-1-y1) benzyI)-3,6-diazabicyclo
[3.1.1]
heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1) pyrimidin-4-
amine
(Compound 15)
Trifluoroacetate of Compound 1g (30 mg) and Compound 14f (20 mg) were
dissolved in
methanol (0.5 mL), and then Et3N (8 mg) and NaBH3CN (26 mg) were successively
added.
Thereafter, the reaction mixture was kept for reaction at room temperature for
16 h. After
completion of the reaction, the reaction mixture was concentrated to dryness
under reduced
pressure, and separated and purified by Prep-H PLC to provide Compound 15(12
mg). MS m/z
(ESI): 559.3 [M+H].
31
CA 03210109 2023- 8- 28
3+1 NMR (400 MHz, DMSO-d6) 8 11.79 (br, 1H), 9.66 (s, 1H), 9.12 (d, J = 2.4
Hz, 1H), 8.44
(dd, J = 8.8, 2.4 Hz, 1H), 7.59-7.45 (m, 5H), 6.83(br, 1H), 6.78 (d, J =
9.2Hz, 1H), 6.28 (br, 1H),
6.09 (d, J = 1.6 Hz, 1H), 3.85-3.72 (m, 4H), 3.66 - 3.53 (m, 4H), 2.62-2.52
(m, 1H), 2.33 (s, 3H),
2.25 (s, 3H), 1.88-1.77 (m, 1H), 1.60 (d, J = 8.4 Hz, 1H), 0.98 - 0.92 (m,
2H), 0.75 - 0.69 (m, 2H).
Example 4: Fumarate of 2-(6-(64(6-(4-fluoro-1H-pyrazol-1-y1) pyridin-3-y1)
methyl)-3,6-
diazabicyclo [3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-
pyrazol-3-y1)
pyrimidin-4-amine (Compound 1-A)
N N NN -r- NH
II NH
N N
HOOC.õ00H
THF HOOCcOoH
,N
N N N
I \F 1-A 'F
Compound 1 (5 g) was added to 200 mL of THF and stirred until it was
completely dissolved.
Then 1.56 g of fumaric acid was added slowly, and the reaction mixture was
kept for reaction at
20-45 C for 46 h. After completion of the reaction, the mixture was filtered
and washed with THF
to obtain 7.06 g of a wet solid, which was then dried under vacuum at 40-45 C
to give 4.78 g of
solid powder.
3+1 NMR (400 MHz, DMSO-d6) 8 12.43 (br, 311), 9.66 (s, 1H), 9.13 (d, J = 2.4
Hz, 1H), 8.60
(d, J = 4.4 Hz, 1H), 8.44 (dd, J = 8.8, 2.4 Hz, 1H), 8.41 (d, J = 1.2 Hz, 1H),
7.97 (dd, J = 8.4, 2.0
Hz, 1H), 7.91 (d, J = 4.4 Hz, 1H), 7.86(d, J = 8.4 Hz, 1H), 6.81 (br, 1H),
6.76 (d, J = 8.8 Hz, 1H),
6.62(s, 2H), 6.31 (br, 1H), 3.79-3.76 (m, 4H), 3.68-3.58 (m, 4H), 2.62-2.56
(m, 1H), 2.33 (s, 3H),
2.25 (s, 3H), 1.61 (d, J = 8.4 Hz, 1H).
Example 5: 2-(1-(54(3-(5-(4-methyl-64(5-methyl-1H-pyrazol-3-y1) amino)
pyrimidin-2-
yl) pyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-y1) methyl) pyridin-2-y1)-
1H-pyrazol-3-y1)
propan-2-ol (Compound 24)
32
CA 03210109 2023- 8- 28
1zI
(-1N
'r1,1: µr.: ',-
-:_.- -
..
cr¨b NN-dinathyletb-
ertediamin, 1; i.
.% ...I Ci., CO . :"W Sitr,O= 1-F fiC1 1-; ^.*-
.k:..::ii,li G.-...)
,
Step 1 g, ... Step 2 %.õ Step 3 µ
r1 Step 4 __ ..
\
L-CLN
Step 1: Methyl 1-(5-(1,3-dioxolan-2-y1) pyridin-2-y1)-1H-pyrazol-3-carboxylate
(Compound 24b)
Compound 16a (2.53 g) and Compound 24a (1.52 g) were dissolved in DM F (10
mL), and then
N,N'-dimethylethylenediamine (379.29 mg), Cs2CO3 (7.01 g) and Cul (409.74 mg)
were
successively added. The reaction mixture was heated to 90 C and kept for
reaction at this
temperature under the protection of nitrogen for 2 h. After completion of the
reaction, the reaction
mixture was cooled to room temperature, quenched with water (60 mL) and
extracted with EA (30
mL x 3). The organic phases were combined, washed with saturated saline, dried
over anhydrous
sodium sulphate, filtered, and concentrated under reduced pressure. The
filtrate was separated and
purified by silica gel column chromatography (PE:EA = 50:1 to 5:1) to obtain
Compound 24b
(1.59 g). MS m/z (ESI ): 276.0 [M+H]t
Step 2: 2-(1-(5-(1,3-dioxolan-2-y1) pyridin-2-y1)-1H-pyrazol-3-y1) propan-2-ol
(Compound 24c)
Compound 24b (200 mg) was added to dried THF (10 mL) and allowed to cool in a
dry ice
ethanol bath for 15 min. Then, a solution of MeMgBr in Et20 (3 N, 0.65 mL) was
slowly added
dropwise. The mixture was allowed to react at this temperature for 15 min, and
then warmed to
room temperature to further react for 4 h. After completion of the reaction,
the reaction mixture
was quenched with a saturated aqueous solution of ammonium chloride (1 mL),
diluted with water
(30 mL), and extracted with EA (30 mL x 3). The organic phases were combined,
washed with
saturated saline, dried over anhydrous sodium sulphate, filtered, and
concentrated to give
33
CA 03210109 2023- 8- 28
Compound 24c (200 mg). MS m/z (ES1): 276.1 [M+H]t
Step 3: 6-(3-(2-hydroxyprop-2-y1)-1H-pyrazol-1-yll nicotinaldehyde (Compound
24d)
Compound 24c (200 mg) was added to THF (5 mL), then hydrochloric acid (2 N,
4.5 mL) was
added, and the reaction mixture was kept for reaction at 25 C for 12 h. After
completion of the
reaction, a saturated aqueous solution of NaHCO3 was added to the reaction
mixture to adjust pH
to 7-8 and the mixture was extracted with EA (30 mL x 3). The organic phases
were combined and
washed with saturated saline, dried over anhydrous sodium sulphate, and
filtered. The filtrate was
concentrated under reduced pressure to afford Compound 24d (165 mg). MS m/z
(ES1): 232.1
[M+H]t
Step 4: Preparation of 2-(1-(54(3-(5-(4-methyl-64(5-methyl-1H-pyrazol-3-y1)
amino)
pyrimidin-2-y1) pyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-y1) methyl)
pyridin-2-y1)-1H-
pyrazol-3-y1) propan-2-ol (Compound 24)
Trifluoroacetate of Compound 1g was purified by Flash column chromatography
under basic
conditions (purification conditions: C18 reversed-phase column; mobile phase:
0.5% aqueous
solution of ammonium bicarbonate, and CH3CN; gradient: 0% CH3CN for elution
for 5 min, 0%-
100% CH3CN for gradient elution for 25 min, and 45%-65% CH3CN for product
collection),
concentrated, and lyophilized to obtain free amine 1g.
Compound 24d (50.5 mg) and Compound 1g (30 mg) were then added to DMA (3 mL)
and
stirred under the protection of nitrogen at room temperature for 1 h.
NaBH(OAc)3 (101 mg) was
then added and the reaction was held at room temperature overnight. After
completion of the
reaction, water (60 mL) was added to the reaction mixture to quench the
reaction and the mixture
was extracted with EA (30 mL x 3). The organic phases were combined and washed
with saturated
saline, dried over anhydrous sodium sulphate, and filtered. The filtrate was
concentrated under
reduced pressure, and separated and purified by Prep-H PLC to afford Compound
24 (12 mg). MS
m/z (ES1): 578.3 [M+H]t
3+1 NM R (400 MHz, DM SO-d6) 8 11.97 (br, 1H), 9.67 (s, 1H), 9.12 (cl, J = 2.0
Hz, 1H), 8.48-
8.41 (m, 2H), 8.37 (d, J = 1.6 Hz, 1H), 7.94 (dd, J = 8.4, 2.4 Hz, 1H), 7.82
(d, J = 8.4 Hz, 1H),
34
CA 03210109 2023- 8- 28
6.82 (br, 1H), 6.79 (d, J = 8.8 Hz, 1H), 6.52 (d, J = 2.8 Hz, 1H), 6.31 (br,
1H), 5.10 (s, 1H), 3.82-
3.70 (m, 4H), 3.68-3.55 (m, 4H), 2.61-2.54 (m, 1H), 2.33 (s, 3H), 2.26 (5,
3H), 1.60 (d, J = 8.4 Hz,
1H), 1.49 (5, 6H).
Example 6: 2-(6-(6-((6-(3-fluoro-5-methyl-1H-pyrazol-1-y1) pyridin-3-y1)
methyl)-3,6-
diazabicyclo [3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-
pyrazol-3-y1)
pyrimidin-4-amine (Compound 28)
leg
N,N-dinethylethylerlediaithiLe '1- BA N-fhlocobenzette
sulfonamide
Fi F
Ny CS2C.ca. 1:05gF..
Step 1 L01-11¨ Step 2
.213a 28b n
niN croi
_N
HCh:COn TA% NaBH;OAci3. MAC c:L.)
Step 3 191 F Step 4
2Eld
LC1.1
--
28
Step 1: 5-(1,3-dioxolan-2-y1)-2-(5-methyl-1H-pyrazol-1-y1) pyridine (Compound
28b)
Compound 28a (785 mg), Compound 16a (2000 mg), N,N'-dimethylethylenediamine
(766 mg),
Cul (1660 mg), and Cs2CO3 (8500 mg) were added to DM F (50 mL), and heated to
110 C to react
for 12 h under the protection of nitrogen. After completion of the reaction,
the reaction mixture
was cooled to room temperature, quenched with water and extracted with EA. The
organic phases
were combined, washed with water, dried over anhydrous sodium sulphate, and
filtered. The
filtrate was concentrated under reduced pressure, and separated and purified
by silica gel column
chromatography (PE:EA=7:3) to afford Compound 28b (2010 mg). MS m/z (ESI ):
232.1 [M+H].
Step 2: 5-(1,3-dioxolan-2-y1)-2-(3-fluoro-5-methyl-1H-pyrazol-1-y1) pyridine
(Compound
28c)
Compound 28b (600 mg) was added to anhydrous THF (30 mL), cooled to -70 C, and
1.6 mL
of a 2.5 M solution of n-butyl lithium in THF was added dropwise under the
protection of nitrogen,
CA 03210109 2023- 8- 28
and the reaction was allowed to continue at -70 C for 1 h. Then, a solution of
N-
fluorobenzenesulfonamide in THF (30 mL) was added dropwise, and thereafter the
reaction was
allowed to continue at -70 C for 12 h. After completion of the reaction, water
was added to the
reaction mixture to quench the reaction, and the mixture was extracted with
EA. The organic phases
were combined, washed with water, dried over anhydrous sodium sulphate, and
filtered. The
filtrate was concentrated under reduced pressure, and separated and purified
by silica gel column
chromatography (PE:EA=17:3) to obtain Compound 28c (80 mg). MS m/z (ESI):
250.1 [M +H].
Step 3: 6-(3-fluoro-5-methyl-1H-pyrazol-1-y1) nicotinaldehyde (Compound 28d)
Concentrated hydrochloric acid (12 N, 2 mL) was added dropwise to a solution
of Compound
28c (80 mg) in a mixed solvent of THF (2 mL) and water (2 mL) to react at room
temperature for
8 h. Then, the reaction mixture was adjusted to pH of about 10 with a
saturated solution of K2CO3,
and then extracted with EA. The organic phases were combined, dried over
anhydrous sodium
sulphate, and filtered. The filtrate was concentrated under reduced pressure,
and separated and
purified by silica gel column chromatography (DCM:Me0H=9:1) to afford Compound
28d (55
mg). MS m/z (ESI): 206.1 [M+H]t
Step 4: 2-(6-(64(6-(3-fluoro-5-methy1-1H-pyrazol-1-y1) pyridin-3-y1) methyl)-
3,6-
diazabicyclo [3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-
pyrazol-3-y1)
pyrimidin-4-amine (Compound 28)
Compound 1g (30 mg) and Compound 28d (20 mg) were dissolved in DMAC (1 mL),
allowed
to react at room temperature for 1 h, then NaBH(OAc)3 (70 mg) was added, and
the reaction was
allowed to continue at room temperature for 12 h. After completion of the
reaction, the reaction
mixture was diluted with ethyl acetate. The organic phase was washed with an
aqueous solution of
K2CO3, dried over anhydrous sodium sulphate, and filtered. The filtrate was
concentrated under
reduced pressure, and separated and purified by Flash column chromatography to
obtain
Compound 28 (8 mg). MS m/z (ESI): 552.3 [M+H].
3+1 NMR (400 MHz, DM SO-d6) 8 11.98 (s, 1H), 9.66 (s, 1H), 9.12 (d, J = 2.4
Hz, 1H), 8.45-
8.42 (m, 2H), 7.97 (dd, J = 8.4, 2.4 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 6.84
(br, 1H), 6.78 (d, J =
36
CA 03210109 2023- 8- 28
9.2 Hz, 1H), 6.32 (br, 1H), 6.08 (d, J = 5.2 Hz, 1H), 3.78-3.70 (m, 4H), 3.65-
3.56 (m, 4H), 2.60-
2.54 (m, 1H), 2.33 (s, 3H), 2.25 (s, 3H), 2.21 (s, 3H), 1.59 (d, J = 8.4 Hz,
1H).
Example 7: 2-(6-(64(6-(furan-2-y1) pyridin-3-y1) methyl)-3,6-diazabicyclo
[3.1.1] heptan-
3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1) pyrimidin-4-amine
(Compound
29)
rf:
-
\La PclIPPr,.)., 9
Q_cCt 6. t_Q_4,Q3Ji C .
Step 1 Step/
24s 1I
29
Step 1: 6-(furan-2-y1) nicotinaldehyde (29b)
To a round-bottom flask, 29a (312 mg), 1h (200 mg), Na2CO3 (341 mg,),
Pd(PPh3)4 (62 mg),
1,4-dioxane (10 mL), and water (2.5 mL) were successively added, and heated to
95 C under the
protection of nitrogen to react for 2 h. After completion of the reaction, the
reaction mixture was
cooled to room temperature, and separated and purified by Flash silica gel
column chromatography
(PE:EA=87%:13%) to give Compound 29b (150 mg). MS m/z (ESI): 174.2 [M+H].
Step 2: Preparation of 2-(6-(64(6-(furan-2-y1) pyridin-3-y1) methyl)-3,6-
diazabicyclo
[3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyrimidin-4-amine
(Compound 29)
Compound 1g (30 mg) and Compound 29b (21 mg) were added to DMA (1 mL) to react
at
room temperature for 2 h. Then, NaBH(OAc)3 (70 mg) wa s added, and the
reaction was allowed
to continue at room temperature for 16 h. After completion of the reaction,
the reaction mixture
was separated and purified directly by Prep-HPLC to afford Compound 29(30 mg).
MS m/z (ESI):
520.3 [M+H]t
11-1 NM R (400 MHz, DMSO-c15) 11.98 (s, 1H), 9.66 (s, 1H), 9.12 (d,J = 2.0 Hz,
1H), 8.52 (d,
J = 1.6 Hz, 1H), 8.44 (dd, J = 9.2, 2.4 Hz, 1H), 7.87 - 7.78 (m, 2H), 7.67 (d,
J = 8.0 Hz, 1H), 7.07
37
CA 03210109 2023- 8- 28
(dd, J = 3.2, 0.4 Hz, 1H), 6.83 (br, 1H), 6.78 (d, J = 9.2 Hz, 1H), 6.64 (dd,
J = 3.2, 2.0 Hz, 1H),
6.31 (br, 1H), 3.82 - 3.69 (m, 4H), 3.65-3.47 (m, 4H), 2.59 - 2.53 (m, 1H),
2.33 (s, 3H), 2.26 (s,
3H), 1.59 (d, J = 8.4 Hz, 1H).
Example 8: Fumarate of 2-(4-fluoro-1-(54(3-(5-(4-methyl-64(5-methy1-1H-pyrazol-
3-y1)
amino) pyrimidin-2-y1) pyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-y1)
methyl) pyridin-2-
yll-1H-pyrazol-3-y1) propan-2-ol (Compound 30-A)
10 IF th ONO.. PUMP-. =,=:==
"0. GP'S. , BD 41, C,C , Dif C2.41 35 2 = ¨0-
4,c _J
SteP I F Step 2 Step 3
3/.
tOd 37t1 r". 331.
14111'4 "T.:1(
HD HD
PieVje=r 71-F, -76 ri NsC a Mis.C.Ac-=
Ce:H% I
SteP 4
3CL :0=F
ht440,..
3C F
FOC; =,,,ccoe
Step 7
N
30-A
Step 1: 4-fluoro-3-iodo-1H-pyrazole (30b)
Compound 30a (3.0 g) was dissolved in chloroform (50 mL), and then NIS (8.36
g) was added.
The mixture was heated to 80 C and allowed to react overnight under the
protection of nitrogen.
After completion of the reaction, the reaction mixture was cooled to room
temperature, water (60
mL) was added to quench the reaction, and the reaction mixture was extracted
with EA (30 mL x
3). The organic phases were combined, washed with saturated saline, dried over
anhydrous sodium
sulphate, filtered, concentrated, and separated and purified by silica gel
column chromatography
(PE:EA=7:1) to afford Compound 30b (720 mg). MS m/z (ESI): 213.0 [M +H].
Step 2: 5-(1,3-dioxolan-2-y1)-2-(4-fluoro-3-iodo-1H-pyrazol-1-y1) pyridine
(30c)
38
CA 03210109 2023- 8- 28
Compound 16a (705 mg) and Compound 30b (500 mg) were dissolved in DM F (10
mL), and
then Cs2CO3 (1.75 g) was added. The mixture was heated to 110 C and allowed to
react for 16 h
under the protection of nitrogen. After completion of the reaction, the
reaction mixture was cooled
to room temperature, water (60 mL) was added to quench the reaction, and the
reaction mixture
was extracted with EA (30 mL x 3). The organic phases were combined, washed
with saturated
saline, dried over anhydrous sodium sulphate, filtered, concentrated, and
separated and purified by
silica gel column chromatography (PE:EA=9:1) to afford Compound 30c (720 mg).
MS m/z (ESI):
361.9 [M+H].
Step 3: Methyl 1-(5-(1,3-dioxolan-2-y1) pyridin-2-y1)-4-fluoro-1H-pyrazol-3-
carboxylate
(30d)
Compound 30c (250 mg) was dissolved in DCM (15 mL), and then M e0Na (160 mg),
methyl
formate (721 mg), and Pd(PPh3)2Cl2 (83 mg) were successively added. The
mixture was heated to
35 C and allowed to react for 2 h under the protection of nitrogen. After
completion of the reaction,
the reaction mixture was cooled to room temperature, directly concentrated,
and separated and
purified by silica gel column chromatography (PE:EA=5:1) to obtain Compound
30d (150 mg).
MS m/z (ESI): 294.1 [M+H]t
Step 4: 2-(1-(5-(1,3-dioxolan-2-yll pyridin-2-y1)-4-fluoro-1H-pyrazol-3-y1)
propan-2-ol
(30e)
Compound 30d (180 mg) was dissolved in dried THF (15 mL), cooled in a dry ice
ethanol bath
at -78 C for 15 min, and then a solution of M eM gBr in ether (3 N, 0.95 mL)
was slowly added
dropwise. Thereafter, the reaction was kept for 15 min, and then allowed to
warm to room
temperature to react for 4 h. After completion of the reaction, a saturated
aqueous solution of
ammonium chloride (1 mL) was added to quench the reaction, the reaction
mixture was diluted
with water (30 mL), and extracted with EA (30 mL x 3). The organic phases were
combined,
washed with saturated saline, dried over anhydrous sodium sulphate, and
filtered. The filtrate was
concentrated under reduced pressure to give crude Compound 30e (145 mg), which
was used
directly in the next step of reaction without further purification. MS m/z
(ESI): 294.2 [M +H]t
39
CA 03210109 2023- 8- 28
Step 5: 6-(4-fluoro-3-(2-hydroxypropan-2-y1)-1H-pyrazol-1-y1) nicotinaldehyde
(30f)
Compound 30e (300 mg) was dissolved in THF (5 mL), and then hydrochloric acid
(2 N, 15
mL) was added. The mixture was allowed to react in an ice bath for 2 h. Then,
to the reaction
mixture, a saturated aqueous solution of NaH CO3 was added to adjust pH to 7-
8, and the mixture
was extracted with EA (30 mL x 3). The organic phases were combined, washed
with saturated
saline, dried over anhydrous sodium sulphate, and filtered. The filtrate was
concentrated under
reduced pressure, and separated and purified by silica gel column
chromatography (PE:EA=3:1)
to afford Compound 30f (200 mg). MS m/z (ESI ): 250.1 [M +H].
Step 6: 2-(4-fluoro-1-(54(3-(5-(4-methyl-64(5-methyl-1H-
pyrazol-3-y1) amino)
pyrimidin-2-yll pyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-y1) methyl)
pyridin-2-y1)-1H-
pyrazol-3-y1) propan-2-ol (Compound 30)
Compound 1g (46 mg) and Compound 30f (45 mg) were dissolved in DMA (1 mL) and
allowed
to react at room temperature for 1 h under the protection of nitrogen. Then,
NaBH(OAc)3 (101 mg)
was added, and the mixture was allowed to react overnight at room temperature.
After completion
of the reaction, water (60 mL) was added to quench the reaction, and the
reaction mixture was
extracted with EA (30 mL x 3). The organic phases were combined, washed with
saturated saline,
dried over anhydrous sodium sulphate, and filtered. The filtrate was
concentrated under reduced
pressure, and separated and purified by Prep-H PLC to give Compound 30 (10
mg). MS m/z (ESI):
596.3 [M+H]t
Step 7: Fumarate of 2-(4-fluoro-1-(54(3-(5-(4-methyl-64(5-methy1-1H-pyrazol-3-
y1)
amino) pyrimidin-2-y1) pyridin-2-yI)-3,6-diazabicyclo [3.1.1] heptan-6-y1)
methyl) pyridin-2-
y1)-1H-pyrazol-3-y1) propan-2-ol (Compound 30-A)
Compound 30 (117 mg, 0.19 mmol) was dissolved in THF (4.0 mL), and then
fumaric acid (46
mg, 0.39 mmol) was added. The mixture was allowed to react with stirring
overnight at room
temperature under the protection of nitrogen. After completion of the
reaction, the mixture was
filtered, washed with THF, and dried under vacuum to obtain 115 mg of the
fumarate.
lld NM R (400 MHz, DM SO-d6) 8 12.73 (br, 2H), 9.66 (br, 111), 9.12 (d,f = 2.4
Hz, 1H), 8.56
CA 03210109 2023- 8- 28
(d, J = 4.8 Hz, 1H), 8.44 (dd, J = 8.8, 2.4 Hz, 1H), 8.37 (d, J = 2.0 Hz, 1H),
7.96 (dd, J = 8.4, 2.4
Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 6.83 (br, 1H), 6.78 (d, J = 8.8 Hz, 1H),
6.62 (s, 2H), 6.30 (br,
1H), 5.24 (s, 1H), 3.79-3.72 (m, 4H), 3.64-3.58 (m, 4H), 2.59- 2.54 (m, 1H),
2.33 (s, 3H), 2.26 (s,
3H), 1.60 (d, J = 8.4 Hz, 1H), 1.54 (s, 6H).
Example 9: 2-(6-(64(5-(4-fluoro-1H-pyrazol-1-y1) pyrazin-2-y1) methyl)-3,6-
diazabicyclo
[3.1.1] hept-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyrimidin-4-amine
(Compound 31)
õ
N r
st=c% 1-=
N
N,L14T-44:
q')a %.1
,o6-t0/4013 Me.0 Cszt
31 a Step 1 ("--) Step 2 I
Cr
3 '
31h
Step 1: 2-(6-(6((5-chloropyrazin-2-yll methyl)-3,6-diazabicyclo [3.1.1] heptan-
3-y1)
pyridin-3-y1)-6-methyl-N-(5-methy1-1H-pyrazol-3-y1) pyrimidin-4-amine (31b)
Compound 1g (150 mg) and Compound 31a (71 mg) were dissolved in DMAC (5 mL) to
react
at room temperature for 1 h, then NaBH(OAc)3 (351 mg) was added, and the
reaction was allowed
to continue at room temperature for 12 h. After completion of the reaction,
the reaction mixture
was diluted with ethyl acetate. The organic phase was washed with an aqueous
solution of K2CO3,
dried over anhydrous sodium sulphate, and filtered. The filtrate was
concentrated under reduced
pressure, and separated and purified by Prep-TLC (DCM:Me0H=15:1) to obtain
Compound 31b
(110 mg). MS m/z (ESI ): 489.2 [M+H]t
Step 2: 2-(6-(6-((5-(4-fluoro-1H-pyrazol-1-y1) pyrazin-2-y1) methyl)-3,6-
diazabicyclo
[3.1.1] heptan-3-y1) pyridin-3-y1)-6-methyl-N-(5-methyl-1H-pyrazol-3-y1)
pyrimidin-4-amine
(Compound 31)
Compound 31b (30 mg) and Compound 30a (106 mg) were dissolved in DM F (1.5
mL),
41
CA 03210109 2023- 8- 28
Cs2CO3 (60 mg) was added, and the mixture was heated to 90 C to react with
stirring for 1 h. After
completion of the reaction, the reaction mixture was cooled to room
temperature, diluted with ethyl
acetate, and washed with water. The organic phase was dried over anhydrous
sodium sulphate, and
filtered. The filtrate was concentrated under reduced pressure, and separated
and purified by Prep-
HPLC to obtain Compound 31 (30 mg). MS m/z (ESI): 539.3 [M+H]t
11-1 NM R (400 MHz, DMSO-d5) 11.99 (s, 1H), 9.66 (s, 1H), 9.12 (d,./ = 2.4 Hz,
1H), 9.09 (d,
J = 1.6 Hz, 1H), 8.72 (dd, J = 4.8, 0.8 Hz, 1H), 8.56 (d, J = 1.2 Hz, 1H),
8.44 (dd, J = 8.8, 2.0 Hz,
1H), 8.03 (d, J = 4.0 Hz, 1H), 6.85 (br, 1H), 6.78 (d, J = 9.2 Hz, 1H), 6.33
(br, 1H), 3.87-3.75 (m,
6H), 3.65-3.52 (m, 2H), 2.57-2.52 (m, 1H), 2.33 (s, 3H), 2.26 (s, 3H), 1.61
(d, J = 8.4 Hz, 1H).
Separation Method:
Except that Compound 28 of Example 6 was separated and purified using Biotage
M PLC, Prep-
HPLC purification of compounds of other examples was carried out using Aglient
1260, Waters
2489 or GeLai 3500 HPLC at a column temperature of 25 C and a detection
wavelength of 214
nm, 254 nm or 280 nm respectively, with other separation conditions shown in
the table below:
Example Compound Separation column
Mobile phase and gradient
Flow 9te
model
(mL/min)
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep solution of ammonium
1 9 C18 OBD (19 mmx150
bicarbonate 28.0
mmx5.0 gm) Gradient: 0 min 40% A, 60%
B
16 min 90% A, 10% B
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep solution of ammonium
2 14 C18 OBD (19 mmx150
bicarbonate 28.0
mmx5.0 m) Gradient: 0 min 30% A, 70%
B
16 min 90% A, 10% B
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep solution of ammonium
3 15 C,8 OBD (19 mmx150
bicarbonate 24.0
mmx5.0 gm) Gradient: 0-2 min 20% A,
80% B
16 min 80% A, 20% B
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep
solution of formic acid
5 24 OBD (19 mmx150
Gradient:% B
30.0
mmx5.0 1.1m) 16 min: 60% A, 40% B
42
CA 03210109 2023- 8- 28
Example Compound Separation column
Mobile phase and gradient
Flow rte
model (mL/min)
A: MeCN; B: 0.05% aqueous
C18 spherical 20-35 solution of ammonium
6 28 bicarbonate
16.0
i.im 100A , 4g Gradient: 0 min: 0%A, 100%
B
20 min: 100% A, 0% B
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep solution of ammonium
7 29 C18 OBD (19 mmx150 bicarbonate
28.0
mmx5.0 m) Gradient: 0-4 min 30% A,
70% B
24 min 90% A, 10% B
A: MeCN; B: 0.05% aqueous
Waters SunFire Prep
solution of formic acid
8 30 C18 OBD (19
30.0
Gradient: 0 min: 10% A, 90% B
mmx150 mmx5.0 Am)
16.0 min: 70% A, 30% B
A: MeCN; B: 0.05% aqueous
Waters XBridge Prep solution of ammonium
9 31 C18 OBD (19 bicarbonate
28.0
mmx150 mmx5.0 ttm) Gradient: 0 min 30% A, 70% B
16 min: 90% A, 10% B
Biological Evaluation
Experimental Example 1: RET Inhibition Experiment
Experimental method: According to the instructions of HTRF KinEASE-TK kit
(Cisbio), the
inhibitory effect of the compound of the present disclosure on the activity of
drug-resistant mutant
RET enzymes (RET-G81OR, RET-G810S and RET-G810C) was determined. The three
drug-
resistant mutant RET enzymes were each pre-incubated with different
concentrations of test
compounds (in the inhibition rate test of the compound: for RET-G81OR and RET-
G810C, the
compound concentration was 300 and 1,000 nM; for RET-G810S, the compound
concentration
was 30 and 300 nM. In the IC50 test of the compound: for RET-G81OR and RET-
G810C, the
compound concentration was 1-10,000 nM; for RET-G810S, the compound
concentration was 0.1-
10,000 nM, 9 concentrations in each case). After pre-incubation for 30 min at
room temperature,
the substrate and adenosine triphosphate (ATP) were added to start the
reaction. For RET-G81OR
and RET-G810S, incubation was carried out at room temperature for 90 min; for
RET-G810C,
incubation was carried out at room temperature for 120 min. TK antibody-
cryptate and
streptavidin-XL665 were added, and the test was performed after incubation at
room temperature
for 60 min. With the solvent group (DM SO) as the negative control and the
buffer group (without
43
CA 03210109 2023- 8- 28
RET enzyme) as the blank control, the relative inhibitory activity percentage
(i.e. inhibition rate)
of compounds with different concentrations was calculated as per the following
formula:
Relative inhibitory activity percentage = 1 - (compound group of different
concentrations -
blank control) / (negative control - blank control)* 100%
The relative inhibitory activity percentages of the compounds with different
concentrations
were plotted with respect to the compound concentrations, and the curve was
fitted according to a
four-parameter model to calculate the I Cso value as per the following
formula:
y = min + (max - min) 1(1 + (x/IC5or(-H illslope))
where y is the relative inhibitory activity percentage, max is the maximum
value of the fitted
curve, min is the minimum value of the fitted curve, x is the logarithmic
concentration of the
compound, and Hillslope is the slope of the curve.
The experimental results are shown in Table 1-7.
Table 1. Inhibition rates of the compounds of the present disclosure at a
concentration of 300
nM on the activity of mutant RET-G81OR enzyme
Compound No. Inhibition rate on RET-G81OR
(%)
2 70.41 4.57
3 72.49 3.15
4 78.98 0.24
6 77.16 3.87
9 78.59 4.50
10 72.78 1.58
12 69.44 0.31
Table 2. 1050 (nM) for inhibition of RET-G81OR enzyme by the compounds of the
present
disclosure
Compound No. ICso (nM) for inhibition of RET-
G81OR
5 185.30
18 132.40
As can be seen from Table 1 and Table 2, the compounds of the present
disclosure showed
significant inhibition on RET-G81OR enzyme.
Table 3. Inhibition rates of the compounds of the present disclosure at a
concentration of 300
nM on the activity of mutant RET-G810S enzyme
Compound No. Inhibition rate on RET-
44
CA 03210109 2023- 8- 28
G810S (%)
4 98.89 0.18
5 97.89 0.57
6 83.01 1.42
7 91.80 0.98
8 83.73 3.16
9 97.64 0.19
10 88.46 2.23
11 90.87 1.13
12 99.19 0.55
13 97.21 1.01
14 92.56 0.08
15 97.22 0.39
16 85.27 0.37
17 90.64 0.68
19 80.98 7.32
20 96.67 0.03
25 97.26 0.72
26 97.83 0.32
28 90.81 1.24
29 93.21 0.91
30-A 99.01 0.15
31 99.69 0.14
Table 4. I C50 (nM) for inhibition of RET-G810S enzyme by the compounds of the
present
disclosure
Compound No. IC50 (nM) for inhibition of RET-G810S
18 15.94
21 6.61
24 57.21
27 23.11
As can be seen from Table 3 and Table 4, the compounds of the present
disclosure showed
significant inhibition on RET-G810S enzyme.
Table 5. Inhibition rates of the compounds of the present disclosure at a
concentration of 300
nM on the activity of mutant RET-G810C enzyme
Compound No. Inhibition rate on RET-G810C (%)
12 69.10 5.56
19 82.80 3.66
20 86.86 1.73
22 84.26 6.82
27 82.66 2.47
31 74.13 1.28
Table 6. I C50 (nM) for inhibition of RET-G810C enzyme by the compounds of the
present
disclosure
CA 03210109 2023- 8- 28
Compound No. ICso (nM) for inhibition of RET-
G810C
18 36.34
21 172.90
30-A 124.90
As shown in Table 5 and Table 6, the compounds of the present disclosure
showed significant
inhibition on RET-G810C enzyme.
Table 7. 1050 (nM) for inhibition of RET mutant enzymes by the compounds of
the present
disclosure
IC50 (nM) for ICso (nM) for IC50 (nM)
for
Compound No, inhibition of RET- inhibition of
RET- inhibition of RET-
G81OR G810S G810C
BLU-667 168.45 0.45 1.77 0.27
48.81 3.82
LOX0-292 73.97 16.40 5.66 0.22
151.30 30.70
1-A 19.62 0.67 2.69 0.56
53.16 10.67
As can be seen from Table 7, Compound 1-A of the present disclosure remarkably
inhibited
RET-G81OR enzyme, RET-G810S enzyme, and RET-G810C enzyme.
Example 2: Inhibitory Effect of Compound on Proliferation of Three RET (G810)
Mutants
Experimental purpose: To evaluate the inhibitory effect of the compound on
proliferation of
three BaF3 cell lines (Ba/F3 KIF5B-RETG81 R, Ba/F3 K I F5B-RETG81 s, Ba/F3 K I
F5B-RETG819
in vitro.
Experimental method: Three Ba/F3 cell lines (Ba/F3 KIF5B-RETG81 R, Ba/F3 KIF5B-
RETGKos, Ba/F3 KIF5B-RETG810C) in logarithmic phase were respectively
collected and
inoculated into 96-well plates, with 2000 cells/95 pl/well. To the 96-well
plate, 5 IA of solutions of
BL U-667, Loxo-292 and Compound 1-A were added respectively (final
concentration: 1.52-10000
nM). In addition, negative control wells (without the test compound) and blank
control wells
(without cells) were set. The cells were cultured at 37 C and 5% CO2 for 3
days, then Cell Titer
Glo reagent was added at 50 [d/well to lyse the cells, the Luminescence signal
value was detected
by a nnicroplate reader, and the inhibition rate % was calculated.
Inhibition rate % = [1 - (average signal value of compound group - average
signal value of
blank control) / (average signal value of negative control - average signal
value of blank control)]
46
CA 03210109 2023- 8- 28
* 100%; the inhibition rate % was fitted using the four-parameter equation
"log(agonist) vs
response-variable slope (four parameters)" in GraphPad Prism 6.0, and the I
Cso was obtained by
calculation.
Experimental results: See Table 8. The following results show that Compound 1-
A is superior
to BL U-667 and Loxo-292 in inhibiting the proliferation of Ba/F3 K I F5B-
RETG81 R, Ba/F3
K I F5B-RETG810S, and Ba/F3 K I F5B-RETG810C cel Is.
Table 8. Inhibition I Cso (nM) of the compounds on the proliferation of the
three cell lines
Cells BL U-667 Loxo-292 1-A
Ba/F3 KI F5B-RETG81 R 181.50 80.02 116.30 1.15
26.28 5.76
Ba/F3 K I F5B-RETG81 s 28.96 4.47 87.83 0.75
20.21 0.35
Ba/F3 KI F5B-RETG81' 146.40 3.40 477.00 4.40
65.30 1.00
Example 3: Efficacy Test of Compound in Mice
Experimental purpose: To study the in vivo efficacy of the compound in BALB/c
Nude mouse
model with Ba/F3 KIF5B-RETG81 R subcutaneous tumour.
Drug preparation: Compound 1-A, BL U-667 and Loxo-292 were respectively mixed
with 0.5%
methyl cellulose (MC) as the solvent to prepare homogenous suspensions for
administration (PO
(IG), BID).
Tumour measurement: The tumour diameter was measured with a vernier caliper
twice a week.
The formula for calculation of tumour volume is: V = 0.5xaxb2, where a and b
represent the long
and short diameters of the tumour respectively. The antitumour effect of the
compound was
evaluated by TGI (%). TGI (%) represents the tumour growth inhibition rate.
TGI (%) = [(1-
(average tumour volume at the end of administration in the treatment group -
average tumour
volume at the beginning of administration in the treatment group)]/(average
tumour volume at the
end of treatment in the solvent control group - average tumour volume at the
beginning of treatment
in the solvent control group)] x 100%. The results are shown in Table 9 and
FIG. 1.
Statistical analysis: Prism Graphpad 8.0 software was used for statistical
analysis based on the
relative tumour volume at the end of the experiment. The comparison between
two groups and
multiple groups was analysed by two-way AN OVA, and p<0.05 represented
significant difference.
Experimental results: No mouse died in the experiment. In the Ba/F3 KIF5B-
RETG81 R model,
47
CA 03210109 2023- 8- 28
the tumour growth inhibition rate (TGI (%)) of Compound 1-A and the control
compounds BL U-
667 and Loxo-292 at the same dose of 30 mg/kg was 72.37%, 29.78% and 2.69%,
respectively,
indicating that the tumour inhibition effect of Compound 1-A is better than
that of the control
compounds BLU-667 and Loxo-292, with p<0.05 in both cases.
Table 9. Analysis of efficacy of compound on Ba/F3 KI F5B-RETGnm tumour-
bearing mouse
model (based on tumour volume)
Number Dosage Vp.1 (mm, Vp12 (mm, TGI p
value (vs
solvent
Group of mice regimen meani-SEM) mean SEM) (%)
group)
Solvent group
1 7 (0.5% MC), 101.26 4.78 743.62 91.16 -
PO BIDx12
BLU-667, 30
2 7 mg/kg, P0, 101.82 5.93 552.89 29.7
29.78 <0.05
BIDx12
Loxo-292,30
3 7 mg/kg, P0, 100.47 4.52 725.55 54.84
2.69 >0.05
BIDx12
1-A, 30
4 7 mg/kg, P0, 101.08 4.75 278.53 51.13
72.37 <0.001
BIDx12
Note: Vp_i: tumour volume one day before administration; Vp12: tumour volume
on Day 12 of
administration.
The above examples do not limit the embodiment of the present disclosure in
any way. In
addition to those described herein, various modifications of the present
disclosure will be apparent
to those skilled in the art based on the foregoing description. Such
modifications are also intended
to fall within the scope of the appended claims. The references cited in the
present disclosure
(including all patents, patent applications, journal articles, books, and any
other publications) are
incorporated herein by reference in their entirety.
48
CA 03210109 2023- 8- 28