Note: Descriptions are shown in the official language in which they were submitted.
WO 2022/263673 PCT/EP2022/066633
1
MULTISTEP PROCESS FOR CULTURING TUMOR-INFILTRATING LYMPHOCYTES FOR
THERAPEUTIC USE
FIELD
The present invention is targeted towards depleting suppressive cells,
including regulatory T cells,
and/or reinvigorating exhausted tumor infiltrating lymphocytes (TILs) in vitro
by co-culturing excised
TIL-containing tumor fragments (or tumor digest) with Tumor Microenvironment
(TME) Stimulators,
such as Immune Checkpoint Inhibitors (ICIs), Cytokines/interleukins, and/or
inhibiting the effect of
factors secreted by suppressive cells, including regulatory T cells (such as
IL-10) thereby creating
a favorable tumor microenvironment where inhibitory T cells and/or signals are
removed so that
exhausted T cells can expand faster, to higher numbers, and are more potent
than currently
established TIL expansion protocols. A time lapse of the use of TME
stimulators is of interest.
BACKGROUND
Tumor infiltrating lymphocytes are associated with improved prognosis and
progression-free
survival in cancer patients undergoing immunotherapy such as the use of immune
checkpoint
inhibitors (leis) against CTLA-4 and PD-1/PD-L1.
However, still only a fraction of patients has a durable long-term response to
such therapies as
many other factors seem to be involved in the tumor microenvironment in the
down regulation of
the immune response. One of the key factors seems to be exhaustion of T cells
resulting in the
physical elimination and/or dysfunction of antigen-specific T cells. Factors
involved in this
exhaustion phenomenon involve surface markers expressed on tumor cells,
lymphoid and
mononuclear cells and soluble molecules secreted from regulatory T cells and
NK cells in the
tumor microenvironment (TME). But, also the lack of stimulatory factors such
as interferon gamma
and IL-2 is evident in the TME.
Reversal of T-cell exhaustion is a key target in the development of new
classes of las either as a
monotherapy or in combination with already established therapies. However, as
these targets often
are also responsible for inducing immune tolerance avoiding autoimmune
responses, systemic
administration of inhibitors can cause serious side effects. In addition,
administering T-cell
stimulatory molecules such as IL-2 can also cause serious and sometimes fatal
side effects and
therefore needs to be managed by skilled clinicians. Some approaches have been
taken to
administer drug candidates locally into the tumor thereby possibly avoiding
systemic side effects.
However, as cancer cells are distributed all over the body in many metastatic
patients, the
likelihood of this approach to be successful under such circumstances can be
questioned.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
2
The use of tumor infiltrating lymphocyte (TIL) therapy has shown significant
clinical benefit with
objective response rates of up to 55% and complete responses in up to 20% of
patients in various
malignancies such as metastatic melanoma, head and neck and cervical cancer.
In short, this kind
of therapy leverages the in vitro expansion of autologous T lymphocytes by
stimulating fragments
from the excised tumor with IL-2, anti-CD3 antibodies and feeder cells and
thereby growing these
cells to billions before re-administering the T cells in back to the
lymphodepleted patient followed
by IL-2 infusion where after regression of the tumor is promoted.
TIL therapy is costly and takes time. It would therefore be advantageous to
optimize the current
methods and identify ways to shorten the duration for expansion of the TILs,
increase the
expansion rate, and also achieve more favorable phenotypes and functionality.
SUMMARY
The present invention relates to a method for promoting regression of a cancer
in a mammal by
expanding tumor infiltrating lymphocytes (TILs) into a therapeutic population
of TILs comprising: (a)
culturing autologous T cells by obtaining a first population of TILs from a
tumor resected from a
mammal, (b) performing a depletion of suppressive cells, including regulatory
T cells, and/or
blocking negative signals by the addition of TME stimulators from the group of
"Inhibitors" with or
without cytokines (c) performing a first expansion by culturing the depleted
population of TILs in a
cell culture medium comprising one or more of the "cytokine" group and/or one
or more of the
substances from the "Stimulator" group to produce a second population of TILs;
(d) performing a
second expansion by supplementing the cell culture medium of the second
population of TILs with
additional IL-2 and/or other cytokines from the "cytokine" group, anti-CD3
antibodies, and antigen
presenting cells (APCs), to produce a third population of TILs, wherein the
third population of TILs
is a therapeutic population; and (e) after administering nonmyeloablative
lymphodepleting
chemotherapy, administering to the mammal the therapeutic population of T
cells, whereupon the
regression of the cancer in the mammal is promoted.
DETAILED DESCRIPTION
The present invention is targeted towards depleting suppressive cells,
including regulatory T cells,
and/or reinvigorating exhausted tumor infiltrating lymphocytes (TILs) in vitro
by co-culturing excised
TIL-containing tumor fragments with for example checkpoint inhibitors,
stimulating the TILs with
other cytokines/interleukins known to revert T-cell exhaustion, and/or
inhibiting the effect of factors
secreted by regulatory T cells (such as IL-10) thereby creating a favorable
TME where exhausted T
cells can expand faster, to higher numbers, and are more potent than currently
established TIL
expansion protocols.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
3
This approach is possibly advantageous to systemically administered therapies
as the in-vitro
stimulation can be performed using dose levels that are much higher than would
be tolerated in
vivo. As an example, current TIL protocols utilize IL-2 at a concentration at
3-6,000 IU per mL,
which is 5-10 times higher than the systemically recommended dose.
In addition, as T cells that have a higher affinity to tumor antigens might
have an increased
tendency to get exhausted, targeted in-vitro reinvigoration might help
expanding T-cell clones with
higher affinity that more aggressively can target the tumor thereby eventually
leading to an
improved clinical outcome of this novel approach to TIL therapy.
The present inventors have found that a depletion of suppressive cells,
including regulatory T cells,
and/or blocking negative signals by the addition of one or more TME
stimulators from the group of
"Inhibitors" to obtain a depleted population of TILs followed by performing a
first expansion by
culturing the depleted population of TILs in a cell culture medium comprising:
one or more TME
stimulators from the group of "cytokines", and/or one or more of the TME
stimulators from the
"Stimulator" group to produce a second population of TILs leads to:
= Higher proportion/total cell number of favorable TILs (CD8+, T cells with an
effector
memory phenotype etc.),
= Faster growth rate,
= Higher reactivity of the cultured T-cells (measured by "potency assay"),
= Higher total number of reactive T-cells,
= Higher number of tumor specificities,
= Higher frequency of tumor-specific T cells, and/or
= Higher T-cell repertoire (based on TCR sequencing).
Thus, the present invention relates to expanded tumor infiltrating lymphocytes
(TILs) for use in
treating a subject with cancer, the treatment comprising the steps of: a)
culturing autologous T cells
by obtaining a first population of TILs from a tumor resected from a mammal,
b) performing a
depletion of suppressive cells, including regulatory T cells, and/or blocking
negative signals by the
addition of one or more TME stimulators from the group of "Inhibitors" to
obtain a depleted
population of TILs with or without the addition of "cytokines", c) performing
a first expansion by
culturing the depleted population of TILs in a cell culture medium comprising:
one or more TME
stimulators from the group of "cytokines", and/or one or more of the TME
stimulators from the
"Stimulator" group to produce a second population of TILs, d) performing a
second expansion by
supplementing the cell culture medium of the second population of TILs with
additional IL-2 and/or
other cytokines from the "cytokine" group, anti-CD3 antibody, and antigen
presenting cells (APCs),
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
4
to produce a third population of TILs, wherein the third population of TILs is
a therapeutic
population; and e) after administering nonmyeloablative lymphodepleting
chemotherapy,
administering to the mammal the therapeutic population of T cells, wherein the
T cells administered
to the mammal, optionally followed by IL-2 infusion, whereupon the regression
of the cancer in the
mammal is promoted.
By "tumor infiltrating lymphocytes" or "TILs" herein is meant a population of
cells originally obtained
as lymphocytes that have left the bloodstream of a subject and migrated into a
tumor. TILs include,
but are not limited to, CD8+ cytotoxic T cells (lymphocytes), Th1 and Th17
CD4+ T cells (CD4+
helper cells), natural killer cells, dendritic cells and MI macrophages. TILs
include both primary and
secondary TILs. "Primary TILs" are those that are obtained from patient tissue
samples as outlined
herein (sometimes referred to as "freshly harvested"), and "secondary TILs"
are any TIL cell
populations that have been expanded or proliferated as discussed herein. TILs
can generally be
defined either biochemically, using cell surface markers, or functionally, by
their ability to infiltrate
tumors and induce tumor cell killing. TILs can be generally categorized by
expressing one or more
of the following biomarkers: CD4, CD8, TCR ab, CD27, CD28, CD56, CCR7, CD45Ra,
CD95, PD-
1, LAG-3, TIM-3, CD69, CD103, CD107a, TNFa, IFNg, CD3, and CD25. Additionally,
and
alternatively, TILs can be functionally defined by their ability to infiltrate
solid tumors upon
reintroduction into a patient. TILs may further be characterized by potency -
for example, TILs may
be considered potent if, for example, interferon (IFN) release is greater than
about 50 pg/mL,
greater than about 100 pg/mL, greater than about 150 pg/mL, or greater than
about 200 pg/mL or
interferon (IFN), tumor-necrosis-factor (TNF) production can be detected
intracellularly and
CD107a on the cell surface upon stimulation with coated beads or tumor cells
(tumor cell lines or
tumor digest). Functionality of these stimulated TILs can for example be
further characterized by
classification into single-, double-, or triple positivity for TNF and/or IFN
and/or CD107a, whereby
triple positive TILs are considered the most functional. Potency of the TIL
product can be further
characterized by analyzing direct tumor cell killing by detection of apoptosis
and/or proliferation of
tumor cells.
Number of specificities of the CD8+ T cells and their frequency in the TIL
product can be defined
by staining TILs with MHC multimer complexes displaying tumor peptides of
interest that can be
recognized by CD8+ T cells and/or by sequencing the T cell receptor
repertoire.
The present inventors have found a higher percentage of CD8 T cells expressing
markers
associated with tumor-specificity (exhaustion markers), when performing the
methods of the
present invention, especially in JAB TD (time delay ¨ see for example figure 7
and figure 9). An
increase in % CD8 cells with TME stimulators has been repeatedly associated
with a better
response to ACT. Thus, a time delay (TD) can yield a higher percentage of CD8
T cells expressing
markers associated with tumor-specificity (exhaustion markers), as shown in
e.g. figure 7 and
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
figure 9. An embodiment of the present invention relates to a method for
expanding tumor
infiltrating lymphocytes (TILs) into a therapeutic population, and the medical
uses of this
therapeutic population, according to the methods and uses described here,
wherein in step c) the
one or more TME stimulators added given in time delay (as described herein).
This leads to a
5 higher percentage of CD8 T cells expressing markers associated with tumor-
specificity (exhaustion
markers). A preferred embodiment relates to the use of JAB in combination for
steps b) and c), as
described herein. A and B can be added in step b) while J is added in step c).
An embodiment of
the present invention relates to a product comprising a clinically relevant
number of TILs with a
higher percentage of CD8 T cells expressing markers associated with tumor-
specificity (exhaustion
markers). This can be measured by either pMHC tetramer/multimer staining as
number and
frequencies of cancer-specific populations (tetramer staining as done in
figures 17-19) or by t cell
receptor sequencing. These methods can be used to confirm that the product has
a higher
percentage of CD8 T cells expressing markers associated with tumor-
specificity. The present data
shows for example that JAB/JAB TD samples have >1 population whereas the two
IL-2 samples
have 0 or 1.
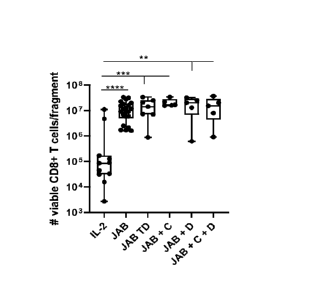
The present inventors have found increased viable CD3 and CD8 cells with TME
stimulators, as
can be seen in figure 8. The latter has been repeatedly associated with a
better response to
adoptive cell therapy (ACT). The culture conditions result in similar numbers
of CD4 T cells
compared to traditional IL2 stimulation. Thus, when step b) and step c) are
performed in time
lapse, increased viable CD3 and CD8 cells are produced in the methods of the
present invention.
A preferred embodiment relates to the use of JAB in combination for steps b)
and c), as described
herein. A and B can be added in step b) while J is added in step c).
Figure 9 shows CD8 T cells with higher expression of particular activation
markers, also called
exhaustion markers (shown in A-C), have been associated to tumor-specific
cells. Although similar
% of cells expressing LAG3 was observed, there was a tendency of higher
citoTIM3 with TME
stimulators compared to IL2 and increased %BTLA+ cells in JAB and JAB TD
conditions, which
may indicate an increase in % tumor-specific cells. BTLA expressing cells were
associated with
better outcome of ACT. Of note, JAB TD conditions tend to have higher %BTLA+
cells. Thus, these
effects are seen when step b) and step c) are performed in time lapse. A and B
can be added in
step b) while J is added in step c).
Figure 10 shows more Tern cells when TME stimulators are used (see especially
with JAB and
JAB+C+D TD conditions). The Tern subset has been repeatedly associated with a
favorable
outcome to ACT. The combination of JAB, and especially A and B in step b) and
J in step c) is
therefore favourable.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
6
(Krishna, S etal., Stem-like CD8 T cells mediate response of adoptive cell
immunotherapy against
human cancer, Science 370, 1328-1334 (2020)).showed that responders to ACT had
higher
numbers of stem-like 0D39- CD69- CD8 T cells in the infusion products, whereas
non-responders
tended to have more CD39+ CD69+ CD8 T cells. With TME stimulators, especially
in time delay
(TD) conditions the present inventors are able to increase the stem-like CD8 T
cells and decrease
the CD39+ CD69+ cells. Increased % of stem-like C039- 0D69- CD8 T cells with
TME stimulators
was also observed. Increased number of these stem-like CD8 T cells
particularly in JAB TD
condition (It was shown that responders to ACT had higher numbers of stem-like
CD39- CD69-
CD8 T cells in the infusion products). Thus, these effects are seen when step
b) and step c) are
performed in time lapse. A and B can be added in step b) while J is added in
step c) to give the
effect.
The time delay results in the examples of the present disclosure shows
favorable results with more
T and CD8 T cells, increased %CD28+ CD8 T cells, less % CD39+ CD69+ and more %
CD39-
CD69- CD8 T cells.
The examples show increased number of reactive CD8 T cells with TME
stimulators. Also,
increased number of triple positive CD8 T cells with TME stimulators (higher
cytotoxic ¨ tumor
killing ¨ capacity).
Figure 12 shows more % of CD28+ CD8 T cells with TME stimulators, and
especially in JAB TD
conditions, and tendency of higher /0CD28+ in TD conditions in general
(indication of more cells
capable of responding to further stimulation).
Figure 13 shows cells triple positive for reactivity markers, thatmight show a
higher expression of
costimulatory and effector molecules, increasing their cytotoxic capacity.
Higher total numbers of
reactive, specifically triple+ T cells might be beneficial for ACT outcome.
CD8 T cells cultures with
the TD tend to have higher percentage of reactive and triple+ cells, as well
as higher total
numbers.
Figure 14 shows cells triple positive for reactivity markers might show a
higher expression of
costimulatory and effector molecules, increasing their cytotoxic capacity.
Higher total numbers of
reactive, specifically triple+ T cells might be beneficial for ACT outcome.
CD8 T cells cultured with
the TD tend to have higher percentage of reactive and triple+ cells, as well
as higher total
numbers.
Figure 16 shows increased reactivity with TD conditions in some cases (ie:
0V7), suggesting that it
might be beneficial to include TD in certain cases. Figures 18 and 19 show
TILs grown with JAB or
JAB TD exhibit a higher number of T cells specific for cancer-associated
antigens Especially donor
Ce4 shows an increase of these T cell populations when TME stimulators were
added, compared
to the IL-2 condition. It has been shown that a higher number and frequency of
(neo)antigen-
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
7
specific T cells in the infusion product is correlated with a better survival
after TIL infusion (Heeke,
C. etal., Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive
cell therapy with
tumor-infiltrating lymphocytes in melanoma. J Clin Invest; 132(2)).
Figures 20-22 show that adding JAB with a time delay of 96h instead of 48h or
no time delay leads
to similar total numbers of cells and also to similar higher frequencies and
numbers of CD3+ and
CD8+ T cells compared to the other TME stimulator combinations, which shows
that the time delay
of 96h can be applied without compromising cell expansion.
Figure 23 shows that the JAB TD of 96h leads to an increased percentage of
CD8+ T cells
expressing activation/exhaustion markers BTLA, LAG3 and TIM3 compared to the
other TME
stimulator conditions while CD28 expression stays similar, which indicates the
expansion of more
activated but maybe exhausted cells.
Figure 24 shows that by adding TME stimulators with a 96h time delay, the
population of Temra
cells increases again to levels in the IL-2 samples, which can point towards
more activated but
exhausted cells in these samples.
Figure 25 shows that the frequencies and numbers of the C039-CD69- population
is similar in all
TME stimulator samples.
A further aspect of the present invention relates to expanded tumor
infiltrating lymphocytes (TILs)
for use in promoting regression of a cancer in a subject with cancer, the
regression comprising the
steps of: a) culturing autologous T cells by obtaining a first population of
TILs from a tumor
resected from a mammal, b) performing a depletion of suppressive cells,
including regulatory T
cells, and/or blocking negative signals by the addition of one or more TME
stimulators from the
group of "Inhibitors" to obtain a depleted population of TILs with or without
the addition of
"cytokines", c) performing a first expansion by culturing the depleted
population of TILs in a cell
culture medium comprising: one or more TME stimulators from the group of
"cytokines", and/or one
or more of the TME stimulators from the "Stimulator" group to produce a second
population of TILs,
d) performing a second expansion by supplementing the cell culture medium of
the second
population of TILs with additional IL-2 and/or other cytokines from the
"cytokine" group, anti-CD3
antibody, and antigen presenting cells (APCs), to produce a third population
of TILs, wherein the
third population of TILs is a therapeutic population; and e) after
administering nonmyeloablative
lymphodepleting chemotherapy, administering to the mammal the therapeutic
population of T cells,
wherein the T cells administered to the mammal with or without IL-2 treatment,
whereupon the
regression of the cancer in the mammal is promoted.
The terms "treatment", "treating", "treat", and the like, refer to obtaining a
desired pharmacologic
and/or physiologic effect. The effect may be prophylactic in terms of
completely or partially
preventing a disease or symptom thereof and/or may be therapeutic in terms of
a partial or
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
8
complete cure for a disease and/or adverse effect attributable to the disease.
"Treatment", as used
herein, covers any treatment of a disease in a mammal, particularly in a
human, and includes: (a)
preventing the disease from occurring in a subject which may be predisposed to
the disease but
has not yet been diagnosed; (b) inhibiting the disease, i.e., arresting its
development or
progression; and (c) relieving the disease, i.e., causing regression of the
disease and/or relieving
one or more disease symptoms. "Treatment" is also meant to encompass delivery
of an agent in
order to provide for a pharmacologic effect, even in the absence of a disease
or condition. For
example, "treatment" encompasses delivery of a composition that can elicit an
immune response or
confer immunity in the absence of a disease condition e.g., in the case of a
vaccine.
The term "anti-CD3 antibody" refers to an antibody or variant thereof e.g., a
monoclonal antibody
and including human, humanized, chimeric or murine antibodies which are
directed against the
CD3 receptor in the T-cell antigen receptor of mature T cells. Anti-CD3
antibodies include OKT3,
also known as muromonab. Anti-CD3 antibodies also include the UHCT1 clone,
also known as T3
and CD3e. Other anti-CD3 antibodies include, for example, otelixizumab,
teplizumab, and
visilizumab. In an embodiment, the cell culture medium comprises OKT3
antibody. In some
embodiments, the cell culture medium comprises about 30 ng/mL of OKT3
antibody. In an
embodiment, the cell culture medium comprises about 0.1 ng/mL, about 0.5
ng/mL, about 1 ng/mL,
about 2.5 ng/mL, about 5 ng/mL, about 7.5 ng/mL, about 10 ng/mL, about 12
ng/mL, about 15
ng/mL, about 20 ng/mL, about 25 ng/mL, about 30 ng/mL, about 35 ng/mL, about
40 ng/mL, about
50 ng/mL, about 60 ng/mL, about 70 ng/mL, about 80 ng/mL, about 90 ng/mL,
about 100 ng/mL,
about 200 ng/mL, about 500 ng/mL, and about 1 pg/mL of 0KT3 antibody. In an
embodiment, the
cell culture medium comprises between 0.1 ng/mL and 1 ng/mL, between 1 ng/mL
and 5 ng/mL,
between 5 ng/mL and 10 ng/mL, between 10 ng/mL and 20 ng/mL, between 20 ng/mL
and 30
ng/mL, between 30 ng/mL and 40 ng/mL, between 40 ng/mL and 50 ng/mL, and
between 50
ng/mL and 100 ng/mL of OKT3 antibody. In some embodiments, the cell culture
medium does not
comprise OKT3 antibody. Cytokines can be added in 0,1 ng/mL-10 ng/mL, 1 ng/mL-
100 ng/mL, or
in 1-100 ng/mL.
The term "IL-2" (also referred to herein as "IL2") refers to the T-cell growth
factor known as
interleukin-2, and includes all forms of IL-2 including human and mammalian
forms, conservative
amino acid substitutions, glycoforms, biosimilars, and variants thereof.
After preparation of the tumor fragments, the resulting cells (i.e.,
fragments) are cultured in media
containing IL-2 under conditions that favor the growth of TILs over tumor and
other cells. In some
embodiments, the tumor digests are incubated in e.g. 2 mL wells in media
comprising inactivated
human AB serum (or, in some cases, as outlined herein, in the presence of aAPC
cell population)
with 6000 IU/mL of IL-2. This primary cell population is cultured for a period
of days, generally from
6 to 14 days, resulting in a bulk TIL population, generally about 1 x 106 to 1
x 108 bulk TIL cells. In
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
9
some embodiments, the growth media during the first expansion comprises IL-2
or a variant
thereof. In some embodiments, the IL-2 is recombinant human IL-2 (rhIL-2). In
some embodiments
the IL-2 stock solution has a specific activity of 20-30 x 106 IU/mg for a 1
mg vial. In some
embodiments the IL-2 stock solution has a specific activity of 20 x 106 IU/mg
for a 1 mg vial. In
some embodiments the IL-2 stock solution has a specific activity of 25 x
10611.1/mg for a 1 mg vial.
In some embodiments the IL-2 stock solution has a specific activity of 30x 106
IU/mg for a 1 mg
vial. In some embodiments, the IL- 2 stock solution has a final concentration
of 4-8 x 106 IU/mg of
IL-2. In some embodiments, the IL- 2 stock solution has a final concentration
of 5-7x 106 IU/mg of
IL-2. In some embodiments, the IL- 2 stock solution has a final concentration
of 6 x 106 IU/mg of IL-
2. In some embodiments, the IL-2 stock solution is prepare as described in the
examples. In some
embodiments, the first expansion culture media comprises about 10,000 IU/mL of
IL-2, about 9,000
IU/mL of IL-2, about 8,000 IU/mL of IL-2, about 7,000 IU/mL of IL-2, about
6000 IU/mL of IL-2 or
about 5,000 IU/mL of IL-2. In some embodiments, the first expansion culture
media comprises
about 9,000 IU/mL of IL-2 to about 5,000 IU/mL of IL-2. In some embodiments,
the first expansion
culture media comprises about 8,000 IU/mL of IL-2 to about 6,000 IU/mL of IL-
2. In some
embodiments, the first expansion culture media comprises about 7,000 IU/mL of
IL-2 to about
6,000 IU/mL of IL-2. In some embodiments, the first expansion culture media
comprises about
6,000 IU/mL of IL-2. In an embodiment, the cell culture medium further
comprises IL-2. In some
embodiments, the cell culture medium comprises about 3000 IU/mL of IL-2. In an
embodiment, the
cell culture medium further comprises IL-2. In a preferred embodiment, the
cell culture medium
comprises about 3000 IU/mL of IL-2. In an embodiment, the cell culture medium
comprises about
1000 IU/mL, about 1500 IU/mL, about 2000 IU/mL, about 2500 IU/mL, about 3000
IU/mL, about
3500 IU/mL, about 4000 IU/mL, about 4500 IU/mL, about 5000 IU/mL, about 5500
IU/mL, about
6000 IU/mL, about 6500 IU/mL, about 7000 IU/mL, about 7500 IU/mL, or about
8000 IU/mL of IL-2.
In an embodiment, the cell culture medium comprises between 1000 and 2000
IU/mL, between
2000 and 3000 IU/mL, between 3000 and 4000 IU/mL, between 4000 and 5000 IU/mL,
between
5000 and 6000 IU/mL, between 6000 and 7000 IU/mL, between 7000 and 8000 IU/mL,
or about
8000 IU/mL of IL-2.
IL-2, IL-4, IL-7, IL-12, IL-15, and/or IL-21 can also be added to step (b)
and/or (c) of the present
methods. Sometimes this is also done in step (e). A preferred embodiment
relates to IL-2 to be
added to step (b) and/or (c) of the present methods. These are part of the
definition "cytokines" and
part of the group of "cytokines" mentioned herein. The term "IL-4" (also
referred to herein as "IL4'')
refers to the cytokine known as interleukin 4, which is produced by Th2 T
cells and by eosinophils,
basophils, and mast cells. IL-4 regulates the differentiation of naive helper
T cells (Th0 cells) to
Th2 T cells. The term "IL-7" (also referred to herein as "IL7'') refers to a
glycosylated tissue-derived
cytokine known as interleukin 7, which may be obtained from stromal and
epithelial cells, as well as
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
from dendritio cells. The term "IL-15" (also referred to herein as "IL15")
refers to the T cell growth
factor known as interleukin-15, and includes all forms of IL-15 including
human and mammalian
forms, conservative amino acid substitutions, glycoforms, biosimilars, and
variants thereof. The
term "IL-21" (also referred to herein as "IL21") refers to the pleiotropic
cytokine protein known as
5 interleukin-21, and includes all forms of IL-21 including human and
mammalian forms, conservative
amino acid substitutions, glycoforms, biosimilars, and variants thereof.
Interleukin 12 (IL-12) is an
interleukin that is naturally produced by dendritic cells, macrophages,
neutrophils, and human B-
Iymphoblastoid cells (NC-37) in response to antigenic stimulation. The term
"IL-12" (also referred to
herein as "IL12") refers to the pleiotropic cytokine protein known as
interleukin-12, and includes all
10 forms of IL-12 including human and mammalian forms, conservative amino
acid substitutions,
glycoforms, biosimilars, and variants thereof.
Another aspect of the present invention relates to a method for expanding
tumor infiltrating
lymphocytes (TILs) into a therapeutic population of TILs comprising: a)
culturing autologous T cells
by obtaining a first population of TILs from a tumor resected from a mammal,
b) performing a
depletion of suppressive cells, including regulatory T cells, and/or blocking
negative signals by the
addition of one or more TME stimulators from the group of "Inhibitors" to
obtain a depleted
population of TILs with or without the addition of "cytokines", c) performing
a first expansion by
culturing the depleted population of TILs in a cell culture medium comprising:
one or more TME
stimulators from the group of "cytokines", and/or one or more of the TME
stimulators from the
"Stimulator" group to produce a second population of TILs, d) performing a
second expansion by
supplementing the cell culture medium of the second population of TILs with
additional IL-2 and/or
other cytokines from the "cytokine" group, anti-CD3 antibody, and antigen
presenting cells (APCs),
to produce a third population of TILs, wherein the third population of TILs is
a therapeutic
population.
The one or more TME stimulators from the group of "cytokines" can be added in
step b). IL-2, IL-4,
IL-7, IL-12, IL-15, and/or IL-21, as defined above are "cytokines".
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" that function by antagonizing/inhibiting
receptors expressed on T
cells (or their ligands) known to cause T-cell
downregulation/deactivation/exhaustion. The group of
"Inhibitors" can be selected from the group consisting one or more of: A)
substances that act
through the PD-1 receptor on T cells or its ligands PD-L1 or PD-L2, B)
substances that act through
the CTLA-4 receptor on T cells, C) substances that act through the LAG-3
receptor on T cells, D)
substances that act through the TIGIT/CD226 receptor on T cells, E) substances
that act through
the KIR receptor on T-cells, F) substances that act through the TIM-3 receptor
on T cells, G)
substances that act through the BTLA receptor on T cells, and H) substances
that act through the
A2aR receptor on T-cells.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
11
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are selected from the group consisting one
or more of: A)
substances that act through the PD-1 receptor on T cells, B) substances that
act through the
CTLA-4 receptor on T cells, C) substances that act through the LAG-3 receptor
on T cells, and D)
substances that act through the TIGIT/CD226 receptor on T cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein group of "Inhibitors" are: A: substances that act through the PD-1
receptor on T cells, and
B: substances that act through the CTLA-4 receptor on T cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are: A: substances that act through the PD-1
receptor on T cells,
B: substances that act through the CTLA-4 receptor on T cells, and C)
substances that act through
the LAG-3 receptor on T cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are: A: substances that act through the PD-1
receptor on T cells,
B: substances that act through the CTLA-4 receptor on T cells, and D)
substances that act through
the TIGIT/CD226 receptor on T cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are: A) substances that act through the PD-1
receptor on T cells,
B) substances that act through the CTLA-4 receptor on T cells, C) substances
that act through the
LAG-3 receptor on T cells, and D) substances that act through the TIGIT/CD226
receptor on T
cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are selected from the group consisting one
or more of: P)
substance that act through the molecule IDO, Q) substances that act through
the 1GH3 molecule
TGFP receptor on T cells, R) substances that act through the IL-10 molecule or
IL-10 receptor on T
cells, and S) substances that act through the IL-35 molecule or IL-35 receptor
on 1-cells. This
group work by "Soluble inhibition" by antagonizing/inhibiting soluble
molecules and cytokines and
their receptors known to cause T-cell downregulation/deactivation/exhaustion.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Inhibitors" are selected from the group consisting of
one or more of: T)
cyclophosphamides, U) TKIs, V) substances that act through aCD25, and X)
IL2/Diphteria toxin
fusions. This group works by adding factors known to downregulate and/or
deplete regulatory T
cells thereby favoring ex-vivo effector T-cell expansion.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
12
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Stimulator" are selected from the group consisting one
or more of: I)
substances that act through the 0X40/CD134 receptor on T cells, J) substances
that act through
the 4-1BB/CD137 receptor on T cells, K) substances that act through the CD28
receptor on T cells,
L) substances that act through the ICOS receptor on T cells, M) substances
that act through the
GITR receptor on T cells, N) substances that act through the CD4OL receptor on
T cells, 0)
substances that act through the CD27 receptor on T cells, and W) substances
that act through CD-
3. These are "Stimulators" that work by agonizing/stimulating receptors
expressed on T cells
known to cause T-cell upregulation/activation/reinvigoration
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the group of "Stimulator" is: J) substances that act through the 4-
1BB/CD137 receptor on
T cells.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein: the group of "Inhibitors" in step b) are: A: substances that act
through the PD-1 receptor
on T cells, and B: substances that act through the CTLA-4 receptor on T cells,
and wherein the
group of "Stimulator" in step c) is: J) substances that act through the 4-
1BB/CD137 receptor on T
cells. One or more cytokines can be added to steps b) and/or c). These
embodiments are shown
experimentally in examples 1-2.
An aspect of the present invention relates to including a time lapse between
steps b) and c), i.e.
where the two steps are performed with a certain time period apart.
Thus, embodiment of the present invention relates to the uses and methods of
the present
invention, wherein the step b) and step c) are performed 1-2 days apart. An
embodiment of the
present invention relates to the uses and methods of the present invention,
wherein the step b) and
step c) are performed 1-3 days apart. An embodiment of the present invention
relates to the uses
and methods of the present invention, wherein the step b) and step c) are
performed 1-4 days
apart. An embodiment of the present invention relates to the uses and methods
of the present
invention, wherein the step b) and step c) are performed 1-5 days apart. An
embodiment of the
present invention relates to the uses and methods of the present invention,
wherein the step b) and
step c) are performed 1-6 days apart. An embodiment of the present invention
relates to the uses
and methods of the present invention, wherein the step b) and step c) are
performed 1-7 days
apart.
An embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 1-8 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 2-8 days apart. An embodiment of the present invention relates
to the uses and
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
13
methods of the present invention, wherein the step b) and step c) are
performed 3-8 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 4-8 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 5-8 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 6-8 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 7-8 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 2-7 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 3-7 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 4-7 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 5-7 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 6-7 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 2-6 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 3-6 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 4-6 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 5-6 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 2-5 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 3-5 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed 4-5 days apart. An embodiment of
the present
invention relates to the uses and methods of the present invention, wherein
the step b) and step c)
are performed 2-4 days apart. An embodiment of the present invention relates
to the uses and
methods of the present invention, wherein the step b) and step c) are
performed 3-4 days apart. An
embodiment of the present invention relates to the uses and methods of the
present invention,
wherein the step b) and step c) are performed on two consecutive days. This
means that step b)
for example can be performed during a given day (for example a workday), and
then step c) is
performed the next day (for example the next workday). This means that the
time delay (TD) or
time lapse can be less than 1 day, for example 20 hours, 18-24 hours, or 14-20
hours.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
14
Experimental findings indicate that lymphodepletion prior to adoptive transfer
of tumor-specific T
lymphocytes plays a key role in enhancing treatment efficacy by eliminating
regulatory T cells and
competing elements of the immune system ("cytokine sinks"). Accordingly, some
embodiments of
the invention utilize a lymphodepletion step (sometimes also referred to as
"immunosuppressive
conditioning") of the patient prior to the introduction of the TILs of the
invention.
The methods of the present invention, from step (a) to step (d), can be
performed in a closed
system. The term "closed system" refers to a system that is closed to the
outside environment. Any
closed system appropriate for cell culture methods can be employed with the
methods of the
present invention. Closed systems include, for example, but are not limited to
closed G-Rex
containers.
The term "TME stimulators" relates to substances (or agents) that have the
ability to create a
favorable microenvironment within the tumor where exhausted T cells can be
reinvigorated in order
to expand many fold and restore their anti-tumor functionality. Thus, in one
or more embodiments,
the one or more TME stimulators are selected from the groups consisting of:
(x) one or more
substances that are capable of antagonizing and/or inhibiting receptors
expressed on T cells (or
their ligands) known to cause T-cell downregulation, deactivation and/or
exhaustion, (y) one or
more substances that are capable of agonizing and/or stimulating receptors
expressed on T cells
known to cause T-cell upregulation, activation, and/or reinvigoration, (z) one
or more substances
that are capable of antagonizing and/or inhibiting soluble molecules and
cytokines and their
receptors known to cause T-cell downregulation, deactivation, and/or
exhaustion, and (v) one or
more substances that are capable of downregulating and/or depleting
suppressive cells, including
regulatory T cells, thereby favoring ex-vivo effector T-cell expansion, and
(w) one or more
substances from the groups (x), (y), (z) and/or (v). Group (w) can be one, two
or three of the
substances from (x), (y), (z) and/or (v). In one or more embodiments, (w) is
one or two of the
substances from (x). In one or more embodiments, (w) is one or two of the
substances from (y). In
one or more embodiments, (w) is one or two of the substances from (z). In one
or more
embodiments, (w) is one or two of the substances from (v).
In one or more embodiments, the substances that are capable of antagonizing
and/or inhibiting
receptors expressed on T cells (or their ligands) known to cause T-cell
downregulation,
deactivation and/or exhaustion are selected from the groups consisting of: A:
substances that act
through the PD-1 receptor on T cells, B: substances that act through the CTLA-
4 receptor on T
cells, C: substances that act through the LAG-3 receptor on T cells, D:
substances that act through
the TIGIT/CD226 receptor on T cells, E: substances that act through the KIR
receptor on T cells, F:
substances that act through the TIM-3 receptor on T cells, G: substances that
act through the
BTLA receptor on T cells, and H: substances that act through the A2aR receptor
on T cells. It is to
be understood that the definition of substances that act through a given
receptor also can cover the
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
same receptors ligand. This means e.g. that for the PD-1 receptor, substances
that target the PD-
L1 or PD-L2 can also be covered. Group A can therefore cover substances that
act through the
PD-1 receptor on T cells as well as its ligand(s).
The substances of the present invention can be an antibody. The substances of
the present
5 invention can be a peptide. The substances of the present invention can
be a small molecule.
The term "antibody" refers to an antibody or variant thereof e.g., a
monoclonal antibody including
human, humanized, chimeric, or murine antibodies, or F(ab')2 or Fab fragment,
or Nanobody.
In one or more embodiments, the substance of group A is an antibody selected
from one or more
from the group consisting of pembrolizumab, nivolumab, cemiplimab, sym021,
atezolizumab,
10 avelumab, durvalumab, Toripalimab, Sintilimab, Camrelizumab,
Tislelizumab, Sasanlimab, and
Dostarlimab. In one or more embodiments, the substance of group A is a small
molecule selected
from one or more from the group consisting of MAX-10181, YPD-29B, IMMH-010,
INCB086550,
GS-4224, DPPA-1, TPP-1, BMS-202, CA-170, JQ1, eFT508, Osimertinib,
PlatycodinD, PD-
LYLSO, Curcumin, and Metformin. In one or more embodiments, the substance of
group A is
15 selected from one or more from the group consisting of pembrolizumab,
nivolumab, cemiplimab,
5ym021, atezolizumab, avelumab, durvalumab, Toripalimab, Sintilimab,
Camrelizumab,
Tislelizumab, Sasanlimab, Dostarlimab, MAX-10181, YPD-29B, IMMH-010,
INCB086550, GS-
4224, DPPA-1, TPP-1, BMS-202, CA-170, JQ1, eFT508, Osimertinib, PlatycodinD,
PD-LYLSO,
Curcumin, and Metformin. In one or more embodiments, the substance of group A
is
pembrolizumab. In one or more embodiments, the substance of group A is
nivolumab. In one or
more embodiments, the substance of group A is cemiplimab. In one or more
embodiments, the
substance of group A is sym021. In one or more embodiments, the substance of
group A is
atezolizumab. In one or more embodiments, the substance of group A is
avelumab. In one or more
embodiments, the substance of group A is durvalumab. In one or more
embodiments, the
substance of group A is Toripalimab. In one or more embodiments, the substance
of group A is
Sintilimab. In one or more embodiments, the substance of group A is
Camrelizumab. In one or
more embodiments, the substance of group A is Tislelizumab. In one or more
embodiments, the
substance of group A is Sasanlimab. In one or more embodiments, the substance
of group A is
Dostarlimab. In one or more embodiments, the substance of group A is MAX-
10181. In one or
more embodiments, the substance of group A is YPD-29B. In one or more
embodiments, the
substance of group A is IMMH-010. In one or more embodiments, the substance of
group A is
INCB086550. In one or more embodiments, the substance of group A is GS-4224.
In one or more
embodiments, the substance of group A is DPPA-1. In one or more embodiments,
the substance of
group A is TPP-1. In one or more embodiments, the substance of group A is BMS-
202. In one or
more embodiments, the substance of group A is CA-170. In one or more
embodiments, the
substance of group A is JQ1. In one or more embodiments, the substance of
group A is eFT508. In
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
16
one or more embodiments, the substance of group A is Osimertinib. In one or
more embodiments,
the substance of group A is PlatycodinD. In one or more embodiments, the
substance of group A is
PD-LYLSO. In one or more embodiments, the substance of group A is Curcumin. In
one or more
embodiments, the substance of group A is Metformin.
In one embodiment group A is a substance that acts through the PD-1 receptor
by blocking the
interaction with its ligand including PD-L1/PD-L2. In one embodiment group A
is a substance that
acts through the PD-L1/L2 by blocking the interaction with its receptor
including PD-1. In one
embodiment group A is a substance that blocks the interaction between PD-1 and
PD-L1/PD-L2. In
one embodiment group A is a substance that blocks the signaling of the PD-1
receptor and/or its
downstream signaling pathways. In one embodiment group A is a substance that
downregulates
the expression of PD-L1/PD-L2. In one embodiment group A is a substance that
promotes
degradation of PD-L1/PD-L2.
In one or more embodiments, the substance of group B is selected from one or
more antibodies
from the group consisting of ipilimumab and tremelimumab. In one or more
embodiments, the
substance of group B is ipilimumab. In one or more embodiments, the substance
of group B is
tremelimumab.
In one embodiment group B is a substance that acts through the CTLA4 receptor
by blocking the
interaction with its ligand including B7-1 or B7-2. In one embodiment group B
is a substance that
acts through the B7-1 or B7-2 by blocking the interaction with its receptor
including CTLA4. In one
embodiment group B is a substance that blocks the interaction between CTLA4
and B7-1 or B7-2.
In one embodiment group B is a substance that blocks the signaling of the B7-1
or B7-2 receptor
and/or its downstream signaling pathways. In one embodiment group B is a
substance that blocks
the signaling of the CTLA4 receptor and/or its downstream signaling pathways.
In one embodiment
group B is a substance that induces cell death upon binding the CTLA4
receptor. In one
embodiment group B is an antibody with antibody-dependent cellular
cytotoxicity (ADCC). In one
embodiment group B is a substance that depletes CTLA4 expressing cells. In one
embodiment
group B is a substance that depletes regulatory T cells through binding CTLA4
and killing the cell.
In one embodiment group B is a substance capable of blocking the interaction
of CTLA4 and its
ligand, and mediating cell specific cytotoxicity through binding to CTLA4.
In one or more embodiments, the substance of group C is selected from one or
more from the
group consisting of relatlimab, eftilagimo alpha, sym022, BMS-986016, and
GSK28-31781.
In one embodiment group C is a substance that acts through the LAG3 receptor
by blocking the
interaction with its receptor. In one embodiment group C is a substance that
blocks the signaling of
the LAG3 receptor and/or its downstream signaling pathways.
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
17
In one or more embodiments, the substance of group D is tiragolumab or
Liothyronine. In one or
more embodiments, the substance of group D is tiragolumab. In one or more
embodiments, the
substance of group D is Liothyronine.
In one embodiment group D is a substance that acts through the TIGIT receptor
by blocking the
interaction with its receptor. In one embodiment group D is a substance that
blocks the signaling of
the TIGIT receptor and/or its downstream signaling pathways. In one embodiment
group D is a
substance that induces cell death upon binding the TIGIT receptor. In one
embodiment group D is
an antibody with antibody-dependent cellular cytotoxicity (ADCC). In one
embodiment group D is a
substance capable of blocking the interaction of TIGIT and its ligand, and
mediating cell specific
cytotoxicity through binding to TIGIT.
In one or more embodiments, the substance of group E is lirilumab. In one or
more embodiments,
the substance of group F is sym023. In one or more embodiments, the substance
of group G is
40E4 and PJ196.
In one or more embodiments, the substances that are capable of agonizing
and/or stimulating
receptors expressed on T cells known to cause T-cell upregulation, activation,
and/or reinvigoration
are selected from the groups consisting of: I: substances that act through the
0X40/C0134
receptor on T cells, J: substances that act through the 4-1BB/CD137 receptor
on T cells, K:
substances that act through the CD28 receptor on T cells, L: substances that
act through the ICOS
receptor on T cells, M: substances that act through the GITR receptor on T
cells, N: substances
that act through the CD4OL receptor on T cells, and 0: substances that act
through the CD27
receptor on T cells.
In one or more embodiments, the substance of group J is selected from one or
more antibodies
from the group consisting of urelumab and utomilumab. In one or more
embodiments, the
substance of group J is selected from one or more peptides from the group
consisting of BCY7835,
and BCY7838 from Bicycle Therapeutics. In one or more embodiments, the
substance of group J
is selected from one or more from the group consisting of BCY7835, BCY7838,
urelumab and
utomilumab. In one or more embodiments, the substance of group J is urelumab.
In one or more
embodiments, the substance of group J is utomilumab. In one or more
embodiments, the
substance of group J is BCY7835. In one or more embodiments, the substance of
group J is
BCY7838.
In one embodiment group J is a substance that act through the 4-1BB/0D137
receptor. In one
embodiment group J is a substance that act through the 4-1BB/CD137 receptor
and stimulates the
growth of T cells. In one embodiment group J is a substance that act through
the 4-1BB/CD137
receptor and stimulates antigen presenting cells (ARC).
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
18
The group J substances can be used in combination with an anti-CD3 substance
such as OKT-3.
One combination can therefore be urelumab and OKT-3 (urelumab/OKT-3). Another
combination
can be utomilumab and OKT-3 (utomilumab /OKT-3). An anti-CD3 substances, such
as OKT-3,
belongs to group W as defined herein. In one embodiment group W is a substance
that binds and
activates CD3 on T cells. In one or more embodiments, the substance of group K
is theralizumab.
In one or more embodiments, the substance of group 0 is valilumab.
In one or more embodiments, one or more of the substances of group A can be
combined with one
or more of the substances of group B. In one or more embodiments, one or more
of the
substances of group A can be combined with one or more of the substances of
group B, and with
one or more of the substances of group J. These combinations are shown to be
effective in the
examples of the present disclosure. This means that one or more substances of
group A selected
from one or more from the group consisting of pembrolizumab, nivolumab,
cemiplimab, sym021,
atezolizumab, avelumab can be combined with one or more of the substances of
group B which is
selected from one or more from the group consisting of ipilimumab and
tremelimumab. These can
then be combined with one or more substances of group J which is selected from
one or more from
the group consisting of urelumab and utomilumab. The group J substances can be
used in
combination with an anti-CD3 substance such as OKT-3. One combination can
therefore be one or
more substances of group A selected from one or more from the group consisting
of
pembrolizumab, nivolumab, cemiplimab, sym021, atezolizumab, avelumab combined
with
ipilimumab from group B and urelumab from group J. A specific selection can be
pembrolizumab
combined with ipilimumab from group B and urelumab from group J, with or
without an anti-CD3
substance such as OKT-3.
In one or more embodiments, the substances that are capable of antagonizing
and/or inhibiting
soluble molecules and cytokines and their receptors known to cause T-cell
downregulation,
deactivation, and/or exhaustion are selected from the groups consisting of: P:
substances that act
through the ID01/2 receptor on T cells, Q: substances that act through the
TGR3 receptor on T
cells, R: substances that act through the IL-10 receptor on T cells, and S:
substances that act
through the IL-35 receptor on T cells.
In one or more embodiments, the substance of group P is epacedostat. In one or
more
embodiments, the substance of group Q is linrodostat. In one or more
embodiments, the substance
of group R is galunisertib.
In one or more embodiments, the substances that are capable of downregulating
and/or depleting
suppressive cells, including regulatory T cells, thereby favoring ex-vivo
effector 1-cell expansion
are selected from the groups consisting of: T: cyclophosphamides, U: TKIs, V:
substances that act
through aCD25, and X: IL2/Diphteria toxin fusions.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
19
In one or more embodiments, the substance of group U is sunitinib. In one or
more embodiments,
the substance of group V is selected from one or more from the group
consisting of sorafenib,
imatinib and daclizumab. In one or more embodiments, the substance of group X
is dinileukin
diftitox.
Using the approaches presented herein allows for dose levels that are much
higher than would be
tolerated in vivo. The concentrations can therefore be at least twice as high
as the maximum
allowed dose tolerated in vivo. The concentration can be even higher such as 5-
10 times as high
as the maximum allowed dose tolerated in vivo. Thus, in one or more
embodiments, the
concentration of substance in is 0.1 pg/mL to 300 pg/mL. The concentration can
also be 1 pg/mL
to 100 pg/mL. The concentration can also be 10 pg/mL to 100 pg/mL. The
concentration can also
be 1 pg/mL to 10 pg/mL.
In one or more embodiments, the therapeutic population of T cells is used to
treat a cancer type
selected from the groups consisting of: 1: solid tumors, 2: ICI naïve tumors,
3: MSI-H tumors, 4:
Hematological tumors, 5: Hyper-mutated tumors (such as POL-E and POL-D mutated
tumors), and
6: virus-induced tumors.
In one or more embodiments, the therapeutic population of T cells is used to
treat a cancer type
selected from the groups consisting of breast cancer, renal cell cancer,
bladder cancer, melanoma,
cervical cancer, gastric cancer, colorectal cancer, lung cancer, head and neck
cancer, ovarian
cancer, Hodgkin lymphoma, pancreatic cancer, liver cancer, and sarcomas.
In one or more embodiments, the therapeutic population of T cells is used to
treat a breast cancer.
In one or more embodiments, the therapeutic population of T cells is used to
treat renal cell cancer.
In one or more embodiments, the therapeutic population of T cells is used to
treat bladder cancer.
In one or more embodiments, the therapeutic population of T cells is used to
treat melanoma. In
one or more embodiments, the therapeutic population of T cells is used to
treat cervical cancer. In
one or more embodiments, the therapeutic population of T cells is used to
treat gastric cancer. In
one or more embodiments, the therapeutic population of T cells is used to
treat colorectal cancer.
In one or more embodiments, the therapeutic population of T cells is used to
treat lung cancer. In
one or more embodiments, the therapeutic population of T cells is used to
treat head and neck
cancer. In one or more embodiments, the therapeutic population of T cells is
used to treat ovarian
cancer. In one or more embodiments, the therapeutic population of T cells is
used to treat Hodgkin
lymphoma. In one or more embodiments, the therapeutic population of T cells is
used to treat
pancreatic cancer. In one or more embodiments, the therapeutic population of T
cells is used to
treat liver cancer. In one or more embodiments, the therapeutic population of
T cells is used to
treat sarcomas.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
In one or more embodiments, steps (a) through (c), (d) or (e) are performed
within a period of
about 20 days to about 45 days. In one or more embodiments, steps (a) through
(c) or (d) are
performed within a period of about 20 days to about 40 days. In one or more
embodiments, steps
(a) through (c) or (d) are performed within a period of about 25 days to about
40 days. In one or
5 more embodiments, steps (a) through (c) or (d) are performed within a
period of about 30 days to
about 40 days. In one or more embodiments, steps (a) through (b) are performed
within a period of
about 10 days to about 28 days. In one or more embodiments, steps (a) through
(b) are performed
within a period of about 10 days to about 20 days.
In some embodiments, the depletion step (step (b)), can be performed for a
period of 1-7 days.
10 This period can also be 1-3 days, 1-2 days, or 4-7 days.
In some embodiments, the first TIL expansion can proceed for 1 day, 2 days, 3
days, 4 days, 5
days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, or
14 days. In some
embodiments, the first TIL expansion can proceed for 1 day to 14 days. In some
embodiments, the
first TIL expansion can proceed for 2 days to 14 days. In some embodiments,
the first TIL
15 expansion can proceed for 3 days to 14 days. In some embodiments, the
first TIL expansion can
proceed for 4 days to 14 days. In some embodiments, the first TIL expansion
can proceed for 5
days to 14 days. In some embodiments, the first TIL expansion can proceed for
6 days to 14 days.
In some embodiments, the first TIL expansion can proceed for 7 days to 14
days. In some
embodiments, the first TIL expansion can proceed for 8 days to 14 days. In
some embodiments,
20 the first TIL expansion can proceed for 9 days to 14 days. In some
embodiments, the first TIL
expansion can proceed for 10 days to 14 days. In some embodiments, the first
TIL expansion can
proceed for 11 days to 14 days. In some embodiments, the first TIL expansion
can proceed for 12
days to 14 days. In some embodiments, the first TIL expansion can proceed for
13 days to 14
days. In some embodiments, the first TIL expansion can proceed for 14 days. In
some
embodiments, the first TIL expansion can proceed for 1 day to 11 days. In some
embodiments, the
first TIL expansion can proceed for 2 days to 11 days. In some embodiments,
the first TIL
expansion can proceed for 3 days to 11 days. In some embodiments, the first
TIL expansion can
proceed for 4 days to 11 days. In some embodiments, the first TIL expansion
can proceed for 5
days to 11 days. In some embodiments, the first TIL expansion can proceed for
6 days to 11 days.
In some embodiments, the first TIL expansion can proceed for 7 days to 11
days. In some
embodiments, the first TIL expansion can proceed for 8 days to 11 days. In
some embodiments,
the first TIL expansion can proceed for 9 days to 11 days. In some
embodiments, the first TIL
expansion can proceed for 10 days to 11 days. In some embodiments, the first
TIL expansion can
proceed for 11 days.
In one or more embodiments, step (c) is performed within a period of about 6
days to about 18
days. In one or more embodiments, step (c) is performed within a period of
about 7 days to about
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
21
14 days. In one or more embodiments, step (c) is performed within a period of
about 7 days to
about 10 days. In one or more embodiments, step (c) is performed within a
period of about 6 days
to about 12 days.
In one or more embodiments, step (d) is performed within a period of about 12
days to about 18
days. In one or more embodiments, step (d) is performed within a period of
about 10 days to about
28 days. In one or more embodiments, step (d) is performed within a period of
about 10 days to
about 20 days. In one or more embodiments, step (d) is performed within a
period of about 12 days
to about 18 days.
In some embodiments, the transition from the first expansion to the second
expansion occurs at 1
day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days,
11 days, 12 days, 13
days, or 14 days from when fragmentation occurs. In some embodiments, the
transition from the
first expansion to the second expansion occurs 1 day to 14 days from when
fragmentation occurs.
In some embodiments, the first TIL expansion can proceed for 2 days to 14
days. In some
embodiments, the transition from the first expansion to the second expansion
occurs 3 days to 14
days from when fragmentation occurs. In some embodiments, the transition from
the first
expansion to the second expansion occurs 4 days to 14 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 5 days
to 14 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 6 days to 14 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 7 days
to 14 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 8 days to 14 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 9 days
to 14 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 10 days to 14 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 11 days
to 14 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 12 days to 14 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 13 days
to 14 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 14 days from when fragmentation
occurs. In some
embodiments, the transition from the first expansion to the second expansion
occurs 1 day to 11
days from when fragmentation occurs. In some embodiments, the transition from
the first
expansion to the second expansion occurs 2 days to 11 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 3 days
to 11 days from when fragmentation occurs. In some embodiments, the transition
from the first
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
22
expansion to the second expansion occurs 4 days to 11 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 5 days
to 11 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 6 days to 11 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 7 days
to 11 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 8 days to 11 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 9 days
to 11 days from when fragmentation occurs. In some embodiments, the transition
from the first
expansion to the second expansion occurs 10 days to 11 days from when
fragmentation occurs. In
some embodiments, the transition from the first expansion to the second
expansion occurs 11 days
from when fragmentation occurs.
One of the key findings has been that more TILs can be generated faster. This
has high value
because there is a certain amount of cells that are needed in order to be
relevant for medical
treatment. More cells faster will drive down the costs for production and also
provide treatment to
the patient faster. In one or more embodiments, step (c) results in 1 x 106 to
lx 107 cells, such as 2
x 106to 5x 106 cells. In one or more embodiments, step (c) results in 5 x
106to lx 107 cells. In one
or more embodiments, step (c) results in 1 x 106 to 5x 107 cells. In one or
more embodiments, step
(c) results in 1 x 10' to 5x 10' cells. In one or more embodiments, step (c)
results in 1 x 107 to lx
1012 cells, such as 1 x 108 to 5x 109 cells, such as 1 x 109to 5x 109 cells,
such as 1 x 108to 5x 1010
cells, such as 1 x 109 to 5x 1011 cells. In one or more embodiments, step (c)
results in an at least
104 fold increase as compared to the number of cells after the expansion in
step (c), such as at
least 103 fold increase, such as at least 102 fold increase, such as at least
10 fold increase. In one
or more embodiments, step (d) results in 1 x 10' to lx 1010 cells. In one or
more embodiments, step
(d) results in 1 x 107 to 1x 109 cells. In one or more embodiments, step (d)
results in 1 x 10' to lx
108 cells. In one or more embodiments, step (d) results in lx 1010 to lx 1011
cells. In one or more
embodiments, step (d) results in 1 x 1011 to 2x 1011 cells. In one or more
embodiments, step (d)
results in at least lx 1011 cells. In one or more embodiments, step (d)
results in at least 2x 1011
cells.
In some embodiments, the antigen-presenting feeder cells (APCs) are PBMCs. In
some
embodiments, the antigen-presenting feeder cells (APCs) are allogeneic feeder
cells. In some
embodiments, the antigen-presenting feeder cells are artificial antigen-
presenting feeder cells. In
an embodiment, the ratio of TILs to antigen-presenting feeder cells in the
second expansion is
about 1 to 25, about 1 to 50, about 1 to 100, about 1 to 125, about 1 to 150,
about 1 to 175, about
1 to 200, about 1 to 225, about 1 to 250, about 1 to 275, about 1 to 300,
about 1 to 325, about 1 to
350, about 1 to 375, about Ito 400, or about 1 to 500. In an embodiment, the
ratio of TILs to
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
23
antigen-presenting feeder cells in the second expansion is between 1 to 50 and
1 to 300. In an
embodiment, the ratio of TILs to antigen-presenting feeder cells in the second
expansion is
between 1 to 100 and 1 to 200. In one or more embodiments, the APCs are
artificial APCs
(aAPCs).
In an embodiment all of the TILs obtained in step c are transferred to step d
and co-cultured with a
fixed number of APCs. For example if 10-100 or 1-200 million cells are
obtained in step c, all of
them are transferred to step d and cultured with 4 billion APC. The ratio or
cells to APCs can be
1:200. It could also be in increments, 10-50 million TILs are co-cultured with
4 billion APC's. 50-
100 are co-cultured with 10 billion APC's.
In an embodiment the TILs obtained in step c, are cultures with APCs in a
concentration of about
0,1-1 million cells/cm2, of about 1 to 2 million cells/cm2, of about 1 to 3
million cells/cm2, of about 1
to 5 million cells/cm2, of about 1 to 10 million cells/cm2, of about 1 to 20
million cells/cm2, of about 1
to 50 million cells/cm2. The APCs can also be in a concentration of 1, 2, 3, 4
or 5 million cells/cm2.
In an embodiment, TILs expanded using APCs of the present disclosure are
administered to a
patient as a pharmaceutical composition. In an embodiment, the pharmaceutical
composition is a
suspension of TILs in a sterile buffer. TILs expanded using PBMCs of the
present disclosure may
be administered by any suitable route as known in the art. In some
embodiments, the T cells are
administered as a single intra-arterial or intravenous infusion, which
preferably lasts approximately
30 to 60 minutes. Other suitable routes of administration include
intraperitoneal, intrathecal, intra-
tumoral, and intralymphatic. In one or more embodiments, the therapeutic
population of TILs are
infused into a patient.
In one or more embodiments, the cells are removed from the cell culture and
cryopreserved in a
storage medium prior to performing step (d).
In one or more embodiments, the method further comprises the step of
transducing the first
population of TILs with an expression vector comprising a nucleic acid
encoding a chimeric antigen
receptor (CAR) comprising a single chain variable fragment antibody fused with
at least one
endodomain of a T-cell signaling molecule.
In one or more embodiments, step (c) further comprises a step of removing the
cells from the cell
culture medium.
In one or more embodiments, step (a) further comprises processing of the
resected tumor into
multiple tumor fragments, such as 4 to 50 fragments, such as 20 to 30
fragments. In one or more
embodiments, the fragments have a size of about 1 to 50 mm3. In one or more
embodiments, the
fragments have a size of about 5 to 50 mm3. In one or more embodiments, the
fragments have a
size of about 0.1 to 10 mm3. In one or more embodiments, the fragments have a
size of about 0.1
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
24
to 1 mm3. In one or more embodiments, the fragments have a size of about 0.5
to 5 mm3. In one or
more embodiments, the fragments have a size of about 1 to 10 mm3. In one or
more embodiments,
the fragments have a size of about 1 to 3 mm3.The terms "fragmenting",
"fragment," and
"fragmented", as used herein to describe processes for disrupting a tumor,
includes mechanical
fragmentation methods such as crushing, slicing, dividing, and morcellating
tumor tissue as well as
any other method for disrupting the physical structure of tumor tissue.
In one or more embodiments, the mammal is a human. In some embodiments, the
TILs are
obtained from tumor fragments. In some embodiments, the tumor fragment is
obtained by sharp
dissection. In some embodiments, the tumor fragment is between about 0.1 mm3
and 10 mm3. In
some embodiments, the tumor fragment is between about 1 mm3 and 10 mm3. In
some
embodiments, the tumor fragment is between about 1 mm3 and 8 mm3. In some
embodiments, the
tumor fragment is about 1 mm3. In some embodiments, the tumor fragment is
about 2 mm3. In
some embodiments, the tumor fragment is about 3 mm3. In some embodiments, the
tumor
fragment is about 4 mm3. In some embodiments, the tumor fragment is about 5
mm3. In some
embodiments, the tumor fragment is about 6 mm3. In some embodiments, the tumor
fragment is
about 7 mm3. In some embodiments, the tumor fragment is about 8 mm3. In some
embodiments,
the tumor fragment is about 9 mm3. In some embodiments, the tumor fragment is
about 10 mm3. In
some embodiments, the tumors are 1-4 mm x 1-4 mm x 1-4 mm. In some
embodiments, the
tumors are 1 mm x 1 mm x 1 mm. In some embodiments, the tumors are 2 mm x 2 mm
x 2 mm. In
some embodiments, the tumors are 3 mm x 3 mm x 3 mm. In some embodiments, the
tumors are 4
mm x 4 mm x 4 mm. Currently fairly large fragment sizes are needed (more than
5 mm3). The
present invention allows for the use of smaller fragments because the cells
grow in a more
optimized way reaching the cell count needed for treatment faster. The use of
smaller fragments
means that patients that until now have not been treatable because e.g.
because their tumor has
been too small or because it only has been possible to obtain a small tumor
sample, now can be
treated. The size of the fragments used in the methods of the present
invention can therefore be
important.
In some embodiments, the tumor fragmentation is performed in order to maintain
the tumor internal
structure. In some embodiments, the tumor fragmentation is performed without
preforming a
sawing motion with a scalpel. In some embodiments, the TILs are obtained from
tumor digests. In
some embodiments, tumor digests were generated by incubation in enzyme media,
for example
but not limited to RPM! 1640, 2 mM GlutaMAX,10 mg/mL gentamicin, 30 U/mL
DNase, and 1.0
mg/mL collagenase, followed by mechanical dissociation (GentleMACS, Miltenyi
Biotec, Auburn,
CA). After placing the tumor in enzyme media, the tumor can be mechanically
dissociated for
approximately 1 minute. The solution can then be incubated for 30 minutes at
37 C in 5% CO2 and
it then mechanically disrupted again for approximately 1 minute. After being
incubated again for 30
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
minutes at 37 C in 5% CO2, the tumor can be mechanically disrupted a third
time for
approximately 1 minute. In some embodiments, after the third mechanical
disruption if large pieces
of tissue were present, 1 or 2 additional mechanical dissociations were
applied to the sample, with
or without 30 additional minutes of incubation at 37 C in 5% CO2. In some
embodiments, at the
5 end of the final incubation if the cell suspension contained a large
number of red blood cells or
dead cells, a density gradient separation using Ficoll can be performed to
remove these cells.
In one or more embodiments, the cell culture medium is provided in a container
selected from the
group consisting of a G-Rex container and a Xuri cellbag.
An aspect relates to a population of tumor infiltrating lymphocytes (TILs)
obtainable by a method of
10 any of the previous claims.
A further aspect relates to expanded tumor infiltrating lymphocytes (TILs) for
use in treating a
subject with cancer, the treatment comprising the steps of: culturing
autologous T cells by
obtaining a first population of TILs from a tumor resected from a mammal
performing a first
expansion by culturing the first population of TILs in a cell culture medium
comprising IL-2 and one
15 or more TME stimulators to produce a second population of TILs;
performing a second expansion
by supplementing the cell culture medium of the second population of TILs with
additional IL-2
and/or other cytokines from the "cytokine" group, anti-CD3, and antigen
presenting cells (APCs), to
produce a third population of TILs, wherein the third population of TILs is a
therapeutic population;
and after administering nonmyeloablative lymphodepleting chemotherapy,
administering to the
20 mammal the therapeutic population of T cells, wherein the T cells
administered to the mammal,
whereupon the regression of the cancer in the mammal is promoted.
In an embodiment, the invention includes a method of treating a cancer with a
population of TILs,
or use of the TILs to treat cancer, wherein a patient is pre-treated with non-
myeloablative
chemotherapy prior to an infusion of TILs according to the present disclosure.
In an embodiment,
25 the non-myeloablative chemotherapy is cyclophosphamide 60 mg/kg/d for 2
days (days 7 and 2
prior to TIL infusion) and fludarabine 25 mg/m2/d for 5 days (days 5 to 1
prior to TIL infusion). In an
embodiment, after non-myeloablative chemotherapy and TIL infusion (at day 0)
according to the
present disclosure, the patient receives an intravenous infusion of IL-2
intravenously at 720,000
IU/kg every 8 hours to physiologic tolerance.
In an embodiment, the non-myeloablative chemotherapy is cyclophosphamide 500
mg/m2/day iv.
for 3 days on day -4, -3, -2 and fludarabine 30 mg/m2/day i.v. for 2 days on
day -4, -3 followed by
TIL infusion on day 0. In an embodiment, after non-myeloablative chemotherapy
and TIL infusion
(at day 0) according to the present disclosure, the patient receives an
intravenous infusion of IL-2
intravenously at 720,000 IU/kg every 8 hours to physiologic tolerance.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
26
In some embodiments, the present disclosure provides a method of treating a
cancer with a
population of tumor infiltrating lymphocytes (TILs) comprising the steps of
(a) obtaining a first
population of TILs from a tumor resected from a patient; b) performing a
depletion of suppressive
cells, including regulatory T cells, and/or blocking negative signals by the
addition of one or more
TME stimulators from the group of "Inhibitors" to obtain a depleted population
of TILs with or
without the addition of "cytokines", (cc) performing an initial expansion of
the first population of TILs
in a first cell culture medium to obtain a second population of TILs, wherein
the second population
of TILs is at least 5-fold greater in number than the first population of
TILs, and wherein the first
cell culture medium comprises IL-2 and one or more TME stimulators; (d)
performing a rapid
expansion of the second population of TILs using a population of myeloid
artificial antigen
presenting cells (myeloid aAPCs) in a second cell culture medium to obtain a
third population of
TILs, wherein the third population of TILs is at least 50-fold greater in
number than the second
population of TILs after 7 days from the start of the rapid expansion; and
wherein the second cell
culture medium comprises IL-2 and anti-CD3; (e) administering a
therapeutically effective portion of
the third population of TILs to a patient with the cancer. In some
embodiments, the present
disclosure a population of tumor infiltrating lymphocytes (TILs) for use in
treating cancer, wherein
the population of TILs are obtainable by a method comprising the steps of b)
performing a
depletion of suppressive cells, including regulatory T cells, and/or blocking
negative signals by the
addition of one or more TME stimulators from the group of "Inhibitors" to
obtain a depleted
population of TILs with or without the addition of "cytokines", (c) performing
an initial expansion of a
first population of TILs obtained from a tumor resected from a patient in a
first cell culture medium
to obtain a second population of TILs, wherein the second population of TILs
is at least 5-fold
greater in number than the first population of TILs, and wherein the first
cell culture medium
comprises IL-2; (d) performing a rapid expansion of the second population of
TILs using a
population of myeloid artificial antigen presenting cells (myeloid aAPCs) in a
second cell culture
medium to obtain a third population of TILs, wherein the third population of
TILs is at least 50-fold
greater in number than the second population of TILs after 7 days from the
start of the rapid
expansion; and wherein the second cell culture medium comprises IL-2 and anti-
CD3; (e)
administering a therapeutically effective portion of the third population of
TILs to a patient with the
cancer. In some embodiments, the method comprises a first step (a) of
obtaining the first
population of TILs from a tumor resected from a patient. In some embodiments,
the IL-2 is present
at an initial concentration of about 3000 IU/mL and anti-CD3antibody is
present at an initial
concentration of about 30 ng/mL in the second cell culture medium. In some
embodiments, first
expansion is performed over a period not greater than 14 days. In some
embodiments, the first
expansion is performed using a gas permeable container. In some embodiments,
the second
expansion is performed using a gas permeable container. In some embodiments,
the ratio of the
second population of TILs to the population of aAPCs in the rapid expansion is
between 1 to 80
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
27
and 1 to 400. In some embodiments, the ratio of the second population of TILs
to the population of
aAPCs in the rapid expansion is about 1 to 300.
A further aspect relates to a population of tumor infiltrating lymphocytes
(TILs) obtainable by a
method comprising: culturing autologous T cells by obtaining a first
population of TILs from a tumor
resected from a mammal performing a first expansion by culturing the first
population of TILs in a
cell culture medium comprising IL-2 and one or more TME stimulators to produce
a second
population of TILs; and performing a second expansion by supplementing the
cell culture medium
of the second population of TILs with additional IL-2 and/or other cytokines
from the "cytokine"
group, anti-CD3, and antigen presenting cells (APCs), to produce a third
population of TILs,
wherein the third population of TILs is a therapeutic population.
A further aspect relates to a therapeutic population of TILs comprising IL-2
and one or more TME
stimulators.
A further aspect relates to a therapeutic population of TILs comprising IL-2,
one or more TME
stimulators, IL-2, anti-CD3, and antigen presenting cells (APCs).
Tables
Table 1 ¨ Flow cytometry antibodies
Marker clone company
CD3 UCHT1 BD Biosciences
CD4 SK3 BD Biosciences
CD8 RPA-T8 BD Biosciences
IFN-g B27 BD Biosciences
TNF-a Mabl 1 BD Biosciences
CD107a H4A3 BD Biosciences
FVS780 BD Biosciences
CD56 B159 BD Biosciences
CCR7 2-L1-A BD Biosciences
CD45RA HI100 BD Biosciences
LAG-3 T47-530 BD Biosciences
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
28
PD-1 MIH4 BD Biosciences
TIM-3 7D3 BD Biosciences
BTLA J168-540 BD Biosciences
CD57 NK-1 BD Biosciences
CD28 CD28.2 BD Biosciences
CD27 L128 BD Biosciences
CD69 FN50 BD Biosciences
CD39 TU66 BD Biosciences
Dynabeads
Human T- Life
Activator technologies
CD3/CD28/CD137
Protein transport
inhibitor, Golgi BD Biosciences
Plug
Foxp3 /
Transcription
Ebioscience
Factor Staining
Buffer Set
Brilliant Stain
BD Biosciences
buffer
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
29
Table 2:
Target Name Manufacturer
Group A PD-1 Pembrolizumab Merck
Group B CTLA-4 Ipilimumab Bristol-
Meyers Squibb
Group C LAG-3 Relatlimab Creative
Biolabs
Biosimilar
Group D TIGIT Tiragolumab Creative
Biolabs
Biosimular
Group J 4-1BB (CD137) Urelumab Creative
Biolabs
Biosimular
Table 3:
"Inhibitors" by antagonizing/inhibiting receptors expressed on T-cells (or
their ligands)
known to cause T-cell downregulation/deactivation/exhaustion:
Group Receptor on T-cell
antagonist examples
A PD-1
Pembrolizumab
A PD-1 Nivolumab
A PD-1
Cemiplimab
A PD-1 Sym021
A PD-1
Toripalimab
A PD-1
Sintilimab
A PD-1
Camrelizumab
A PD-1
Tislelizumab
A PD-1
Sasanlimab
A PD-1
Dostarlimab
A PD-L1
Durvalumab
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
A PD-L1 Avelumab
A PD-L1
Atezolizumab
A (SMI) PD-1 inhibitor MAX-10181
A (SMI) PD-1/PD-L1 interaction YPD-
29B
A (SMI) PD-1/PD-L1 interaction IMMH-
010
A (SMI) PD-L1 inhibitor
INCB086550
A (SMI) PD-1/PD-L1 interaction DPPA-1
A (SMI) PD-1/PD-L1 interaction TPP-1
A (SMI) PD-1/PD-L1 interaction BMS-
202
A (SMI) PD-1/PD-L1 interaction CA-170
A (SMI) PD-L1 expression JQ1
A (SMI) PD-L1 expression eFT508
A (SMI) PD-L1 expression
Osimertinib
A (SMI) PD-L1 expression
PlatycodinD
A (SMI) PD-L1 degradation PD-LYLSO
A (SMI) PD-L1 degradation Curcumin
A (SMI) PD-L1 degradation
Metformin
B
B CTLA-4
Tremelimumab
C LAG-3
Relatlimab
C
C LAG-3 Sym022
C(SMI) LAG-3 BMS-
986016
C(SMI) LAG-3 GSK28-
31781
D TIGIT/CD226
Tiragolumab
D(SMI)
Liothyronine
E KIR
Lirilunnab
F TIM-3 Sym023
G BTLA 40E4 and
PJ196
H A2aR
"Stimulators" by agonizing/stimulating
receptors expressed on T-cells known to
cause T-cell
upregulation/activation/reinvigoration:
Group Receptor on T-cell agonist
examples
I 0X40/CD134
J 4-1BB Urelumab
J 4-1BB
Utomilumab
J (Peptide)
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
31
J (Peptide) 4-1BB BCY7838
K CD28
Theraluzimab
L ICOS
M GITR
N CD4OL
O CD27
Varlilumab
W CD3 OKT-3
W CD3 UCHT1
W CD3 HIT3a
"Inhibitors" by antagonizing/inhibiting
soluble molecules and cytokines and their
receptors known to cause T-cell
downregulation/deactivation/exhaustion: PD-1
Dostarlimab
Soluble
Group factor/cytokine
antagonist examples
P ID01/2
Epacadostat
P ID01/2
Linrodostat
Q TGFB
Galunisertib
R IL-10
S IL-35
"Inhibitors" by adding factors know to
downregulate and/or deplete regulator T-
cells thereby favoring ex-vivo effector T-
cell expansion: PD-L1 inhibitor GS-4224
Soluble
Group factor/inhibitor
T cyclophosphamide
U TKIs
sunitinib
U TKIs
sorafenib
U TKIs
imatinib
/ aCD25
Daclizumab
IL2/Diphteria toxin
X fusion
Dinileukin Diftitox
"Cytokines" PD-LYLSO
Group
Z1 IL-2
Z2 IL-7
Z3 IL-12
Z4 IL-15
Z5 IL-21
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
32
C LAG-3
Relatlimab
C LAG-3
Eftilagimo alpha
C LAG-3 Sym022
C(SMI) LAG-3 BMS-
986016
C(SMI) LAG-3 GSK28-
31781
D TIGIT/CD226
Tiragolumab
D(SMI)
Liothyronine
E KIR Lirilumab
F TIM-3 Sym023
G BTLA 40E4 and
PJ196
H A2aR
"Stimulators" by agonizing/stimulating receptors expressed on T-cells known to
cause
T-cell upregulation/activation/reinvigoration:
Group Receptor on T-cell agonist
examples
I 0X40/CD134
J 4-1BB Urelumab
J 4-1BB
Utomilumab
J (Peptide) 4-1BB BCY7835
J (Peptide) 4-1BB BCY7838
K CD28
Theraluzimab
L ICOS
M GITR
N CD4OL
O CD27
Varlilumab
W CD3 OKT-3
W CD3 UCHT1
W CD3 HIT3a
"Inhibitors" by antagonizing/inhibiting soluble molecules and cytokines and
their receptors
known to cause T-cell downregulation/deactivation/exhaustion:
Soluble
Group factor/cytokine
antagonist examples
P ID01/2
Epacadostat
P ID01/2
Linrodostat
Q TGFI3
Galunisertib
R IL-10
S IL-35
"Inhibitors" by adding factors know to downregulate and/or deplete regulator T-
cells
thereby favoring ex-vivo effector T-cell expansion:
Soluble
Group factor/inhibitor
T cyclophosphamide
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
33
TKIs sunitinib
TKIs sorafenib
TKIs imatinib
V aCD25
Daclizumab
IL2/Diphteria toxin
X fusion
Dinileukin Diftitox
"Cytokines"
Group
Z1 IL-2
Z2 IL-7
Z3 IL-12
Z4 IL-15
Z5 IL-21
Table 4: Reagents, antibodies and streptavidin conjugates for tetramer
staining
Reagent Number, clone company
CD3-FITC 561807, UCHT1 BD Biosciences
CD8-BV480 566121, RPA-T8 BD Biosciences
Dasatinib #D-3307 LC Laboratories
NIR live/dead #15550745 FisherScientific
HLA-A0201 DTU-produced
monomer
Top30 Peptides Pepscan, custom order
APC-SA #405207 Biolegend
PE-SA #405204 Biolegend
PerCP-ef710-SA #46-4317-82 FisherScientific
PE-CF594-SA #562284 BD Biosciences
PE-Cy7-SA #557598 BD Biosciences
APC-R700-SA #565144 BD Biosciences
BUV737-SA #612775 BD Biosciences
BV786-SA #563585 BD Biosciences
BV650-SA #564876 BD Biosciences
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
34
Brilliant Stain BD Biosciences
#566349
buffer
Table 5: Peptide sequences and their origin used in tetramer screen
No. HLA Protein Sequence cancer antigen
class
1 A2 MAGE-A2 LVHFLLLKY Cancer/Testis
2 A2 gp100 / Pme117 IMDQVPFSV Melanoma
Differentiation
3 A2 CDKN1A GLGLPKLYL Overexpressed
4 A2 gp100 / Pme117 YLEPGPVTA Melanoma
Differentiation
A2 Melan-A / MART-1 (WT) EAAGIGILTV Melanoma Differentiation
6 A2 MAGE-C2 KVLEFLAKL Cancer/Testis
7 A2 MAGE-A10 SLLKFLAKV Cancer/Testis
8 A2 gp100 / Pme117 KTWGQYWQV Melanoma
Differentiation
9 A2 STEAP1 MIAVFLPIV Overexpressed
A2 Telomerase RLFFYRKSV Overexpressed
11 A2 LAGE-1 MLMAQEALAFL Cancer/Testis
12 A2 MAGE-A2 YLQLVFGIEV Cancer/Testis
13 A2 MAGE-C2 LLFGLALIEV Cancer/Testis
14 A2 STAT1-alpha/13 KLQELNYNL Overexpressed
A2 TAG-1 SLGWLFLLL Cancer/Testis
16 A2 MC1R TILLGIFFL Melanoma
Differentiation
17 A2 Melan-A / MART-1 ILTVILGVL Melanoma
Differentiation
18 A2 TRP-2 FVWLHYYSV Melanoma
Differentiation
19 A2 KIF20A AQPDTAPLPV Overexpressed
A2 MAGE-A10 GLYDGMEHL Cancer/Testis
21 A2 p53 RMPEAAPPV Overexpressed
22 A2 SSX-2 KASEKIFYV Cancer/Testis
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
23 A2 STEAP1 FLYTLLREV Overexpressed
24 A2 TRP-2 SVYDFFVVVL Melanoma
Differentiation
25 A2 GnTV VLPDVFIRCV Overexpressed
26 A2 Livin (ML-IAP) SLGSPVLGL Overexpressed
27 A2 MAGE-Al YLEYRQVPV Cancer/Testis
28 A2 Meloe-1 TLNDECWPA Overexpressed
29 A2 NY-ESO-1 / LAGE-2 SLLMWITQC Cancer/Testis
30 A2 TRAG-3 ILLRDAGLV Cancer/Testis
Table 6: Patient list
Name Diagnosis Gender Age Ethnicity
Pathological Description Stage Prior Treatment
MM 1 Melanoma Female 76 Caucasian Nodular
melanoma II treatment naive
MM2 Melanoma Female 48 Caucasian Malignant
melanoma, NOS IIIB treatment naive
MM3 Melanoma Male 56 Caucasian Spindle
cell melanoma, NOS IIB treatment naive
Head and neck
HN1 Male 64 Caucasian Squamous cell carcinoma, NOS III
treatment naive
cancer
Head and neck
HN2 Male 69 Caucasian Squamous cell carcinoma, NOS III
treatment naive
cancer
Head and neck
HN3 Male 67 Caucasian Squamous cell carcinoma, NOS IV
treatment naive
cancer
Colorectal
CC1 Female 88 Caucasian Mucinous adenocarcinoma IIA
treatment naive
carcinoma
Colorectal
CC2 Male Si Caucasian Adenocarcinoma, NOS I treatment
naive
carcinoma
0C1 Ovarian carcinoma Female 56 Caucasian
Adenocarcinoma, NOS IIIC treatment naive
0C2 Ovarian carcinoma Female 52 Caucasian
Adenocarcinoma, NOS IV treatment naive
0C3 Ovarian carcinoma Female 54 Caucasian
Serous adenocarcinoma IIIA treatment naive
LC1 NSCLC Male 72 Caucasian Squamous
cell carcinoma, 5105 IB treatment naive
LC2 NSCLC Male 62 Caucasian Squamous
cell carcinoma, NOS IB treatment naive
Ce1 Cervical cancer Female 64 Caucasian
Squamous cell carcinoma, NOS IB treatment naive
CeZ Cervical cancer Female 59 Caucasian
Squamous cell carcinoma, NOS IB treatment naive
Ce3 Cervical cancer Female 47 Caucasian
Squamous cell carcinoma, NOS IIIB treatment naive
Ce4 Cervical cancer Female 62 Caucasian
Squamous cell carcinoma, NOS IIB treatment naive
Ce5 Cervical cancer Female 46 Caucasian
Squamous cell carcinoma, NOS IB treatment naive
NY-01 Kidney cancer Female 70 N/A
Clear cell renal cell carcinoma N/A treatment naive
NY-02 Kidney cancer Male 77 N/A
Clear cell renal cell carcinoma N/A treatment naive
NY-03 Kidney cancer Male 64 N/A
Clear cell renal cell carcinoma N/A treatment naive
Trastuzumab for
NY-04 Kidney cancer Female 59 N/A
Clear cell renal cell carcinoma N/A
mammacancer
NY-05 Kidney cancer Male 81 N/A
Clear cell renal cell carcinoma N/A treatment naive
NY-06 Kidney cancer Male 57 N/A
Clear cell renal cell carcinoma N/A treatment naive
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
36
Unclassified renal cell
Axitinib 32 series until
NY-08 Kidney cancer Male 59 N/A
N/A
carcinoma April
2021
NY-11 Kidney cancer Female 77 N/A
Clear cell renal cell carcinoma N/A treatment naive
NY-12 Kidney cancer Male 50 N/A
Clear cell renal cell carcinoma N/A treatment naive
NY-13 Kidney cancer Male 51 N/A
Clear cell renal cell carcinoma N/A treatment naive
OvCa-
Ovarian cancer Female 72 N/A High grade
serous carcinoma N/A treatment naive
01
OvCa-
Ovarian cancer Female 76 N/A High grade
serous carcinoma N/A treatment naive
02
OvCa- Ovarian cancer Female 74 N/A
High grade serous carcinoma N/A treatment naive
04
OvCa-
06 Ovarian cancer Female 45 N/A
High grade serous carcinoma N/A treatment naive
OvCa-
Ovarian cancer Female 62 N/A High grade
serous carcinoma N/A treatment naive
07
OvCa-
Ovarian cancer Female 78 N/A High grade
serous carcinoma N/A treatment naive
08
OvCa-
Ovarian cancer Female 59 N/A High grade
serous carcinoma N/A treatment naive
09
LC3 Lung cancer Female 48 N/A
Adenocarcinoma I N/A
LC4 Lung cancer Male 77 N/A Pulmonary
adenocarcinoma IA3 N/A
LC5 Lung cancer Male 65 N/A Non-small
cell lung cancer 1118 N/A
Genera/
It should be understood that any feature and/or aspect discussed above in
connections with the
compounds according to the invention apply by analogy to the methods described
herein.
The following figures and examples are provided below to illustrate the
present invention. They are
intended to be illustrative and are not to be construed as limiting in any
way.
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
37
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 shows frequency of CD8+ and CD4+ T cells expressing for all cancer
types using IL-2 +/-
TME-S. Shown is the median + 95% Cl interval. Refer to table 2 for the
specific stimulator
of each group.
Figure 2 shows total number of viable CD8+ T cells per fragment for all cancer
types using IL-2 +/-
TME-S. Shown is the median + 95% Cl interval. Refer to table 2 for the
specific stimulator
of each group. Statistics performed by two-tailed Mann-Whitney U test.
p>0.05 was considered non-significant. *p<0.05, **p<0.01,
***p<0.001,****p<0.0001.
Figure 3 shows total number of CD8+ T cells per fragment expressing either
one, two or three of
the cytotoxic degranulation marker CD107a, or the cytokines IFNg and TNFa for
cervical cancer
using IL-2 +/- TME-S. Shown is the median + 95% Cl interval. Refer to table 2
for
the specific stimulator of each group.
Figure 4 shows total number of CD8+ T cells per fragment expressing either
one, two or three of
the cytotoxic degranulation marker CD107a, or the cytokines IFNg and TNFa for
cervical cancer
using IL-2 +/- TME-S. Shown is the median + 95% Cl interval. Refer to table 2
for
the specific stimulator of each group.
Figure 5 shows total number of CD8+ T cells per fragment expressing either
one, two or three of
the cytotoxic degranulation marker CD107a, or the cytokines IFNg and TNFa for
cervical cancer
using IL-2 +/- TME-S. Shown is the median + 95% Cl interval. Refer to table 2
for
the specific stimulator of each group. Statistics performed by two-tailed Mann-
Whitney U
test. p>0.05 was considered non-significant. The numbers on the figure are the
calculated p
values.
Figure 6 shows time in culture and success rate for TIL cultures. TIL cultures
were established with
renal cell carcinoma, ovarian cancer, cervical cancer and lung cancer
fragments. The cultures were
maintained and harvested as described in Example 6. Figure A shows the time
from culture start
plotted against the number of viable cells per fragments at harvest. The
dotted line represents
100.000 viable cells per fragment. The minimum required cells for a clinical
scale TIL product.
Figure B shows the success rate for the different conditions defined as an
expansion to more than
100.000 viable cells per fragment.
Figure 7 shows T and NK cells and T cell subsets in TIL cultures with TME
stimulators. TIL cultures
were established with kidney, ovarian, cervical and lung cancer fragments.
Cultures with indicated
conditions were established. Scatter plots showing (A) % T and NK cells of
live (B) % CD4 and
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
38
CD8 cells of T cells. Data are presented as median with 95% Cl. *ID < 0.05,
**P < 0.01 by 2way
AN OVA.
Figure 8 shows T cell expansion in cultures treated with TME stimulators. TIL
cultures were
established with kidney, ovarian, cervical and lung cancer fragments. TIL
cultures with indicated
conditions were established. Scatter plots showing viable cells per fragment
for (A) CD3+ T cells
(B) CD8+ T cells (C) CD4+ T cells. Data are presented as median with 95% Cl.
< 0.05, **P <
0.01, ***P < 0.001 by Mann-Whitney test.
Figure 9 shows expression of activation markers by CD8 T cells from TIL
cultures with TME
stimulators. TIL cultures were established with kidney, ovarian, cervical and
lung cancer fragments.
Cultures with indicated conditions were established. Scatter plots showing AD
of (A) BTLA+ (B)
LAG3+ (C) TIM3+ (D) CD28+ (E) CD28+ (F) 0D57+ CD8 T cells. Data are presented
as median
with 95% Cl. *P < 0.05, **P < 0.01 by Mann-Whitney test.
Figure 10 shows CD8 T cell differentiation in TIL cultures with TME
stimulators. TIL cultures were
established with kidney, ovarian, cervical and lung cancer fragments. Cultures
with indicated
conditions were established. Summary of CD8 T cell subsets Tem (CCR7- CD45RA-
), Temra
(CCR7-, CD45RA+), Tcm (CCR7+, CD45RA-), Tnaive (CCR7-F, CD45RA+). Data are
presented as
median with 95% Cl. < 0.05 by 2way ANOVA.
Figure 11 shows stem-like 0D8 T cells in TIL cultures with TME stimulators.
TIL cultures were
established with kidney, ovarian, cervical and lung cancer fragments. Cultures
with indicated
conditions were established. (A) Summary of CD8 T cell subsets based on CD39
and 0069
expression. Scatter plots showing % of (B) CD39+ CD69+ and (C) CD39- CD69- CD8
T cells. (D)
Scatter plot showing the number of stem-like (CD39- CD69-) CD8 T cells in the
cultures. Data are
presented as median with 95% Cl. *10 < 0.05, **P < 0.01, ***P < 0.001, ****P <
0.0001 by Mann-
Whitney test.
Figure 12 shows a summary of the effect of time delay (TD) compared to non
time delay (non TD)
in TIL cultures with TME stimulators. TIL cultures were established with
kidney, ovarian, cervical
and lung cancer fragments. Data from JAB and JAB + C + D conditions were
pooled as non TD
and data from JAB TD and JAB + C + D TD conditions were pooled as TD. Scatter
plots showing
(A) viable CD3+ T cells per fragment (B) viable CD8+ T cells per fragment (C)
% of TIM3+ CD8 T
cells (D) % of CD28+ CD8 T cells (E) % of 0D39+ CD69+ CD8 T cells (F) % of
0D39- 0D69- CD8
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
39
T cells. Data are presented as median with 95% Cl. *P < 0.05, **P < 0.01, ***P
< 0.001, ****P <
0.0001 by Mann-Whitney test.
Figure 13 shows a higher frequency of CD8 T cells with high cytotoxic capacity
in TIL cultures with
TME stimulators. TIL cultures were established with kidney and ovarian cancer
fragments with
indicated conditions and young TILs stimulated with dynabeads coated with
aCD3, aCD28 and a4-
1BB and then stained for reactivity markers IFNy, TNFa and CD107a. (A)
Proportion of CD8+ T
cells either single-, double- or triple positive for all combinations of IFNy,
TNFa and CD107a. Total
% indicates the total fraction of reactive CD8+ cells.
Figure 14 shows CD8 T cells with high cytotoxic capacity in TIL cultures with
TME stimulators. TIL
cultures were established with kidney and ovarian cancer fragments with
indicated conditions and
young TILs stimulated with dynabeads coated with aCD3, aCD28 and a4-1 BB and
then stained for
reactivity markers IFNy, TNFa and CD107a. (A) Scatter plot showing the total
number of reactive
CD8+ T cells in the different cultures. Data are presented as median with 95%
Cl. (B) Scatter plot
showing the number of triple-positive CD8+ T cells in the different cultures.
Data are presented as
median with 95% Cl. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by
Mann-Whitney test.
Figure 15 shows CD8 T cells with high cytotoxic capacity in TIL cultures with
TME stimulators with
Time Delay. TIL cultures were established with kidney and ovarian cancer
fragments with indicated
conditions and young TILs stimulated with dynabeads coated with aCD3, aCD28
and a4-1BB and
then stained for reactivity markers IFNy, TNFa and CD107a. (A) Scatter plot
showing the number
of reactive CD8+ T cells in the different cultures. Data are presented as
median with 95% Cl. (B)
Scatter plot showing the number of triple-positive CD8+ T cells in the
different cultures. Data are
presented as median with 95% Cl. *P < 0.05, **P < 0.01, *"*P < 0.001, "*"*P <
0.0001 by Mann-
Whitney test.
Figure 16 shows time delay increases the proportion of reactive 1-cells for
some patient samples
TIL cultures were established with kidney and ovarian cancer fragments as
described in example
7. Cultures with indicated conditions were established and young TILs
stimulated with dynabeads
coated with aCD3, aCD28 and a4-1BB and then stained for reactivity markers
IFNy, TNFa and
CD107a. The proportion of reactive cells (single-, double- or triple positive
for combinations of
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
IFNy, TNFa and CD107a) were plotted against the number of viable CD8+ cells
obtained in each
culture. Closed figures represent cell cultures from 0V7 and open figures
samples from patient
0V9. Square represents the IL-2 cultures. Circle cultures stimulated with JAB
or JAB+C+D and
triangles represents cultures stimulated with JAB or JAB+C+D with a time
delay. *P < 0.05, **ID <
5 0.01, ***P < 0.001, ****P < 0.0001 by Mann-Whitney test.
Figure 17 shows detected CD8+ T cell populations specific for cancer-
associated antigens with
pMHC tetramers. TIL cultures were established with cervical cancer fragments.
Cultures with
indicated conditions were established and young TILs stained with a library of
30 cancer/testis
10 pMHC tetramers to identify T cell specificities. (A) List of identified
specific CD8+ T cells
populations across the three Cervical cancer patient samples Ce1, Ce3 and Ce4.
Cultures were
grown from five tumor fragments, unless otherwise indicated. The number
indicates the % of
tetramer-positive cells within the CD8+ population.
15 Figure 18 shows detected CD8+ T cell populations specific for cancer-
associated antigens with
pMHC tetramer across cervical cancer patients. TIL cultures were established
with cervical cancer
fragments. Cultures with indicated conditions were established and young TILs
stained with a
library of 30 cancer/testis pMHC tetramers to identify T cell specificities.
(A)+(B) Cultures were
grown from five tumor fragments, unless otherwise indicated. Plot showing the
number and sum of
20 frequencies of reactive CD8+ T cell populations in the different
cultures. Each line represents one
Cervical cancer patient. Triangles represent samples made from ten tumor
fragments instead of
five.
Figure 19 shows number and frequency of detected CD8+ T cell populations
specific for cancer-
25 associated antigens are higher in TD samples than in non TD samples. TIL
cultures were
established with cervical cancer fragments. Cultures with indicated conditions
were established
and young TILs stained with a library of 30 cancer/testis pMHC tetramers to
identify T cell
specificities. (A)+(B) Plot showing the number and sum of frequencies of
reactive CD8+ T cell
populations in the different cultures.
Figure 20 shows viable cells per fragment for TIL cultures cultured in IL2,
JAB, JAB TD (48h) and
JAB TD (96h) TIL cultures were established with kidney, ovarian and cervical
cancer fragments (1
Ce, 2 kidney and 3 Ovarian). The cultures were maintained and harvested as
described in
Example 7.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
41
Figure 21 shows T and NK cells and T cell subsets in TIL cultures with TME
stimulators. TIL
cultures were established with kidney, ovarian, cervical and lung cancer
fragments. Cultures with
indicated conditions were established. Scatter plots showing (A) % T and NK
cells of live (B) %
CD4 and CD8 cells of T cells. Data are presented as median with 95% Cl. ****P
<0.0001 by 2way
ANOVA.
Figure 22 shows T cell expansion in cultures treated with TME stimulators. TIL
cultures were
established with kidney, ovarian, cervical and lung cancer fragments. TIL
cultures with indicated
conditions were established. Scatter plots showing viable cells per fragment
for (A) CD3+ T cells
(B) CD8+ T cells (C) CD4+ T cells. Data are presented as median with 95% Cl.
*P < 0.05 by Mann-
Whitney test.
Figure 23 shows expression of activation markers by CD8 T cells from TIL
cultures with TME
stimulators. TIL cultures were established with kidney, ovarian, cervical and
lung cancer fragments.
Cultures with indicated conditions were established. Scatter plots showing %
of (A) BTLA+ (B)
LAG3+ (C) TIM3+ (D) CD28+ (E) CD28+ (F) CD57+ CD8 T cells. Data are presented
as median
with 95% Cl. *P < 0.05, **P < 0.01 by Mann-Whitney test.
Figure 24 shows CD8 T cell differentiation in TIL cultures with TME
stimulators. TIL cultures were
established with kidney, ovarian, cervical and lung cancer fragments. Cultures
with indicated
conditions were established. Summary of CD8 T cell subsets Tern (CCR7- CD45RA-
), Temra
(CCR7-, CD45RA+), Tcm (CCR7+, CD45RA-), Tnaive (CCR7+, CD45RA+). Data are
presented as
median with 95% Cl. ****P < 0.0001 by 2way ANOVA.
Figure 25 shows stern-like 0D8 T cells in TIL cultures with TME stimulators.
TIL cultures were
established with kidney, ovarian, cervical and lung cancer fragments. Cultures
with indicated
conditions were established. (A) Summary of CD8 T cell subsets based on CD39
and C069
expression. Scatter plots showing % of (B) CD39+ CD69+ and (C) CD39- CD69- CD8
T cells. (D)
Scatter plot showing the number of stem-like (CD39- CD69-) CD8 T cells in the
cultures. Data are
presented as median with 95% Cl. *P <0.05 by Mann-Whitney test.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
42
EXAMPLES
Example 1¨ "Young" tumor-infiltrating lymphocytes (TILs) with TME stimulators
This example demonstrated the generation of "young" tumor-infiltrating
lymphocytes (TILs) with
TME stimulators.
Tumor material of various histologies were obtained from commercial sources.
eighteen
independent patient tumors or tumor digests were obtained (3 ovarian cancer, 3
metastatic
melanoma, 3 head and neck cancer, 2 lung cancer, 2 colorectal cancer, 5
cervical cancer). The
cervical cancer samples were shipped fresh in sterile transport media. The
rest of the tumor
samples were cryopreserved samples and were shipped to Cbio A/S in sterile
freezing medium.
The tumor material was handled in a laminar flow hood to maintain sterile
conditions.
TILs were prepared as previously described in detail (Friese, C. etal., CTLA-4
blockade boosts the
expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian
cancer. Sci Rep 10,
3914 (2020); Jin, J. et at, Simplified Method of the Growth of Human Tumor
Infiltrating
Lymphocytes in Gas-permeable Flasks to Numbers Needed for Patient Treatment,
Journal of
Immunotherapy, 35- Issue 3 (2012)). Briefly, TIL cultures were set up using
tumor fragments or
tumor digest. The tumors were divided into 1-3 mm3 fragments and placed into a
G-Rex 6-well
plate (WilsonWolf; 5 fragments per well) with 10 ml complete medium (CM)
supplemented with
6000 IU/mL IL-2 (6000 1U/ml, Clinigen) only (baseline) or in combination with
TME stimulators of
each of the PD-1/PD-L1 antagonists (group A), CTLA-4 antagonist (group B), LAG-
3 antagonist
(group C), TIGIT antagonist (group D) and 4-1BB agonist (group J) in
combination with anti-CD3, in
a humidified 37 C incubator with 5% CO2 at the same time or with a time delay
or time lapse. CM
and IL-2 was added every 4-5 days until a total volume of 40 ml was reached.
Subsequently, half
of the medium was removed and replaced with CM and IL-2 every 4-5 days. TIL
cultures from
tumor digest were initiated by culturing single-cell suspensions (5x105/m1)
obtained by overnight
enzymatic digestion in flat-bottom 96-well plates in 250 pL CM and IL-2 (6000
IU/ml, Clinigen) in a
humidified 37 C incubator with 5% CO2. Half of the medium was removed and
replaced with CM
and IL-2 every 2-3 days.
CM consisted of RPMI1640 with GlutaMAX, 25 mM HEPES pH 7.2 (Gibco), 10% heat-
inactivated
human AB serum (Sigma-Aldrich), 100 U/mL penicillin, 100 pg4-nL streptomycin
(Gibco), and 1.25
pg/ml Fungizone (Bristol-Myers Squibb).
This example demonstrated the generation of "young" tumor-infiltrating
lymphocytes (TILs) with
TME stimulators having an age of 10-28 days.
Example 2¨ Phenotype analysis of "young" TIL cultures with TME stimulators
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
43
This example demonstrates the phenotype analysis of "young" TIL cultures with
TME stimulators.
When cultures designated for young TIL generation were harvested, their
phenotype was
assessed by flow cytometry. TIL phenotype was determined by assessment of the
viability and the
CD3+ subset, the CD3+CD8+ subset and the CD3+CD4+ subset in both frequency and
absolute
cell count.
TIL Panel: CD3, CD4, CD8, Live Dead Marker
Briefly, about 0.5x106 young TILs were washed and then incubated with titrated
antibodies (BD
Biosciences, Table 1) and Brilliant Stain Buffer (BD Biosciences) for 30 min
at 4 C. Cells were
washed twice with PBS and directly analyzed by flow cytometry (CytoFLEX,
Beckman Coulter).
This example demonstrated the phenotype analysis of "young" TIL cultures with
TME stimulators.
Example 3¨ TME-stimulators in combination enhance the frequency and number of
CD8+ T
cells and reduce the frequency of CD4+ T cells
Example 3 illustrated in figure 1-2 demonstrated that adding a combination of
TME stimulators from
group J (4-1BB inhibitors and ligand)in combination with anti-CD3, group A
(including inhibitors of
P01 and its ligand PD-L1), group B (inhibitors of CTLA-4 and ligand), group C
(LAG-3 antagonist)
and group D (TIGIT antagonist) at the same time or group A (including
inhibitors of PD1 and its
ligand PD-L1), group B (inhibitors of CTLA-4 and ligand) on day 0 and group J
(4-1BB stimulators)
in combination with anti-CD3with a time delay or time lapse of 2 days to the
standard young TIL
protocol performed as described in example 1 and staining T cells using anti-
CD3, anti-CD4 and
anti-CD8 flow cytometry antibodies as described in example 2 significantly
enhanced CD8+ T-cell
growth which resulted in a significantly increased frequency (figure 1) and
total number (figure 2) of
CD8+ T cells compared to IL-2 alone and to a decreased frequency of CD4+ T
cells (figure 1). This
frequency of CD8+ T cells was even more increased when TME stimulator of group
J in
combination with anti-CD3 was added on day 2, compared to TME stimulators of
group A, B and J
in combination with anti-CD3 added at the same time.
This was illustrated using a representative number of tumor fragments from
various solid cancers
including ovarian, head and neck, colorectal, melanoma, cervical, colorectal,
and lung cancer.
Summing up this example, adding TME stimulators with a time delay or time
lapse to the young TIL
processing step provided a novel improvement over the existing standard TIL
protocol that allowed
for generation of a TIL product containing an increased total number and
frequency CD8+ T cells
and a reduced frequency CD4+ T cells.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
44
Example 4 ¨ Cytotoxic potential analysis of "young" TIL cultures with TME
stimulators
This example demonstrates the analysis of the cytotoxic potential of "young"
TIL cultures with TME
stimulators performed as described in example 1.
When cultures designated for young TIL generation were harvested, their
reactivity and cytotoxic
potential was assessed by flow cytometry. Reactivity was assessed by
stimulation of young TILs
with CD3/CD28/0D137 coated beads and subsequent staining of cytotoxic
degranulation marker
CD107a on the cell surface and cytokines INFg and TNFa intracellularly.
Characterization of T cell
subsets was additionally analyzed using following markers:
TIL cell surface: CD107a, CD3, CD4, CD8
TIL intracellularly: INFg, TNFa
Briefly, about 2x106 young TILs per sample were thawed and rested overnight in
a 24-well plate in
RPM! + 10% inactivated human AB serum and 1% Pen/Strep. The next day, cells
were harvested
and counted. 1x105 TILs were transferred to a 96 well plate in triplicates and
stimulated with
CD3/0D28/CD137 dynabeads with a bead-to-cell ratio of 1:10 for six hours in
presence of
aCD107a antibody and Golgi Plug.
After six hours, cells were washed and then incubated with titrated surface
antibodies (BD
Biosciences, Table 1) and PBS for 30 min at 4 C. Cells were washed twice with
PBS + 0.5% BSA
and then fixed overnight at 4 C with fixation buffer (FoxP3 Staining Buffer
Set, ebioscience, Table
1). The next day cells were washed twice with Permeabilization buffer (FoxP3
Staining Buffer Set
ebioscience, Table 1) and then stained for intracellular cytokine antibodies
(BD biosciences, Table
1) and PBS for 30 min at 4 C. Cells were washed twice with Permeabilization
buffer (FoxP3
Staining Buffer Set, ebioscience, Table 1), resuspended in PBS + 0.5% BSA and
directly analyzed
by flow cytometry (CytoFLEX, Beckman Coulter).
This example demonstrates the reactivity and functionality analysis of "young"
TIL cultures with
TME stimulators.
Example 5¨ TME-stimulators in combination added with or without time delay or
time lapse
enhance the total number of CD8+ T cells with cytotoxic potential
Example 5 illustrated in figure 3, 4 and 5 demonstrated that adding a
combination of TME
stimulators from group J (4-1BB stimulator) in combination with anti-CD3,
group A (including
inhibitors of PD1 and its ligand PD-L1), group B (inhibitors of CTLA-4 and
ligand), group C (LAG-3
antagonist) and group D (TIGIT antagonist) at the same time or group A
(including inhibitors of
PD1 and its ligand PD-L1), group B (inhibitors of CTLA-4 and ligand) on day 0
and group J (4-1 BB
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
stimulators) in combination with anti-CD3 with a time delay or time lapse of 2
days to the standard
young TIL protocol performed as described in example 1 and stimulating T cells
using aCD3,
aCD28 and a41BB coated beads followed by an (intra-)cellular staining with
aCD107a, aCD3,
aCD4, aCD8, alFNg and aTNFa as described in example 4 resulted in an increased
total number
5 of CD8+ T cells expressing either one, two or three of the cytotoxic
markers (figure 3, 4) and
resulted in a significantly higher number of reactive CD8+ T cells in total,
compared to the IL-2
condition (figure 5), whereas adding the TME stimulators of group J in
combination with anti-CD3,
group A and group B with a time delay or time lapse led to a more significant
increase of the total
number of cytotoxic CD8+ T cells compared to adding the stimulators without
time delay or time
10 lapse (figure 5).
This was illustrated using a representative number of tumor fragments from
cervical cancer. The
combination of TME stimulators of group J in combination with anti-CD3, group
A, group B, group
C and group D with or without time delay or time lapse seems to increase the
number of CD8 T
cells with a cytotoxic potential compared to the standard protocol with IL-2.
Example 6¨ "Young" tumor-infiltrating lymphocytes (TILs) with TME stimulators
This example demonstrated the generation of "young" tumor-infiltrating
lymphocytes (TILs) with
TME stimulators as described in Example 1 with following changes:
Tumor material of various histologies were obtained from commercial sources or
collaborations
with Odense University Hospital. 27 independent patient tumors (7 ovarian
cancer, 10 renal cell
carcinoma, 5 Cervical, 5 Lung Cancer, Table 6). Fresh tumor material was
shipped to Cbio NS in
sterile transport media. The tumor material was handled in a laminar flow hood
to maintain sterile
conditions.
The tumors were divided into 1-3 mm3 fragments and placed into a G-Rex 6-well
plate
(VVilsonWolf; 5 fragments per well unless otherwise indicated) with 5 ml
complete medium (CM)
supplemented with 6000 IU/mL IL-2 (6000 IU/ml, Clinigen) only (baseline) or in
combination with
TME stimulators of each of the PD-1/PD-L1 antagonists (group A), CTLA-4
antagonist (group B),
LAG-3 antagonist (group C), TIGIT antagonist (group D) and 4-1BB agonist
(group J) in
combination with anti-CD3, in a humidified 37 C incubator with 5% CO2 at the
same time or with a
time delay or time lapse of 2 days. TME stimulation combinations are called
corresponding to the
stimulator groups J, A, B, C, D, without or with time delay of 2 days (TD).
Example 7¨ Culturing TILs with TME Stimulators increases cell number and
success rate
while reducing culture time
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
46
Example 7 illustrated in Figure 6 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol performed reduces
days in culture to
reach higher numbers of expanded TILs, as shown in Figure 6 A. Figure 6 B
illustrates that adding
the different combinations of TME stimulators significantly enhanced success
rate of expanding
young TILs from tumor fragments of ovarian cancer, renal cell carcinoma,
cervical cancer and lung
cancer compared to the standard IL-2 conditions from 48% to 96% for cultures
with stimulators
from group J (4-1 BB inhibitors and ligand), group A (including inhibitors of
PD1 and its ligand PD-
L1), group B (inhibitors of CTLA-4 and ligand) added at the same time (JAB)
and to 100% for JAB
TD, JAB+C+D and JAB+C+D TD.
This was illustrated using a representative number of tumor fragments from
ovarian cancer,
cervical cancer, lung cancer and renal cell carcinoma.
Example 8¨ Phenotype analysis of "young" TIL cultures with TME stimulators
This example demonstrates the phenotype analysis of "young" TIL cultures with
TME stimulators.
When cultures designated for young TIL generation as described in Example 6
were harvested,
their phenotype was assessed by flow cytometry. TIL phenotype was determined
by assessment of
the viability and the CD3+ subset, the CD3-CD56+ subset, the CD3+CD8+ subset
and the
CD3+CD4+ subset in both frequency and absolute cell count, and frequencies of
CD8+ T cells
expressing the phenotypic markers CD27, CD28, CD39, CD57, CD69, BTLA, LAG3,
1IM3,
CD45RA, CCR7.
TIL Panel: CD3, CD4, CD8, CD56, BTLA, LAG3, TIM3, CD28, CD27, CD57, CD39,
CD69,
CD45RA, CCR7, Live Dead Marker
Briefly, about 0.5x106 young TILs were washed and then incubated with titrated
antibodies (BD
Biosciences, Table 1) and Brilliant Stain Buffer (BD Biosciences) for 30 min
at 4 C. Cells were
washed twice with PBS and directly analyzed by flow cytometry (CytoFLEX,
Beckman Coulter).
This example demonstrated the phenotype analysis of "young" TIL cultures with
TME stimulators of
ovarian cancer, renal cell carcinoma, cervical and lung cancer fragments.
Example 9 ¨ TME stimulators in combination enhance the frequency and number of
CD8+ T
cells
Example 9 illustrated in Figure 7 and Figure 8 demonstrated that adding a
combination of TME
stimulators as described in Example 6 to the standard young TIL protocol and
staining T cells
using anti-CD3, anti-CD4 and anti-CD8 flow cytometry antibodies as described
in example 8
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
47
significantly enhanced T cell growth which resulted in similar frequencies of
CD3 and NK cells
(Figure 7 A) and a significantly increased frequency of CD8+ T cells (Figure 7
B) and total number
of CD3+ (Figure 8 A) and CD8+ (Figure 8 B) T cells in TME stimulator samples
compared to IL-2
alone. No change in CD4+ T cell frequency or total number was detected in TME
stimulator
samples compared to IL-2.This was illustrated using a representative number of
tumor fragments
from ovarian cancer, cervical cancer, lung cancer and renal cell carcinoma.
Higher numbers of T cells, specifically CD8+ T cells, has been repeatedly
shown to be associated
with better outcome of adoptive TIL transfer (Radvanyi, 2012).
Summing up this example, adding TME stimulators without or with a time delay
of 2 days to the
young TIL processing step provided a novel improvement over the existing
standard TIL protocol
that allowed for generation of a TIL product containing an increased total
number and frequency
CD8+ T cells.
Example 10¨ TME stimulators in combination enhance the frequency of BTLA+ and
CD28+
CD8+ T cells
Example 10 illustrated in Figure 9 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol and staining T cells
using anti-CD27,
anti-CD28, anti-CD57, anti-BTLA, anti-LAG3 and anti-TIM3- flow cytometry
antibodies as described
in example 8 results in a significantly higher frequency of BTLA expressing
CD8 T cells when
adding TME stimulator combinations, with the tendency towards a higher
percentage in JAB TD
samples, as shown in Figure 9A. Similarly, there was the tendency towards a
higher frequency of
TIM3+ CD8+ T cells in all samples grown with TME stimulators, but especially
in JAB TD samples,
as shown in Figure 9 C.
Both markers have been described to be expressed on activated and cytotoxic
CD8+ T cells,
representing the tumor specific T cells fraction.
Additionally, a significantly higher frequency of CD28+ CD8+ T cells was
detected in JAB TD and
JAB+C+D TD samples (Figure 9D), pointing towards a higher proportion of cells
expressing the
coactivation marker and therefore representing cells that are able to respond
to stimulation and are
capable of tumor recognition.
The other markers LAG3 (Figure 9 B), 0D27 (Figure 9 E) and CD57 (Figure 9 F)
do not show large
differences in the TME stimulator samples compared to the standard IL-2
condition or between
each other.
This was illustrated using a representative number of tumor fragments from
ovarian cancer,
cervical cancer, lung cancer and renal cell carcinoma.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
48
These results point towards an expansion of cells with a more activated and
tumor-specific
phenotype. Especially BTLA has been associated with better outcome of TIL
infusion (Radvanyi,
2012). Expression of CD28 is mostly retained in TME stimulator expanded TILs
or even
significantly increased in JAB TD expanded TILs compared to the standard IL-2
condition, which
points towards TILs that still express costimulatory molecules and can
therefore be activated upon
antigen recognition.
Example 11¨ TME stimulators in combination enhance the frequency of CD8+ T
cells with
an effector memory phenotype
Example 11 illustrated in Figure 10 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol and staining T cells
using anti-CD3,
anti-CD8, anti-CD45RA and anti-CCR7 flow cytometry antibodies as described in
example 8
resulted in an increase of CD8 TILs with an effector memory (CD54RA-, CCR7-)
phenotype in TILs
expanded with TME stimulators compared to the IL-2 conditions. Especially TILs
expanded with
JAB or JAB+C+D TD showed a significant increase in effector memory T cells.
This was illustrated using a representative number of tumor fragments from
ovarian cancer,
cervical cancer, lung cancer and renal cell carcinoma.
The effector-memory phenotype has repeatedly been associated with a favorable
outcome of
Adoptive Cell Therapy (ACT).
Example 12¨ TME stimulators in combination enhance the frequency and total
number of
CD8+ T cells that are negative for CD39 and C069 with a stem-cell like
phenotype
Example 12 illustrated in Figure 11 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol and staining T cells
using anti-CD3,
anti-CD8, anti-CD39 and anti-0069 flow cytometry antibodies as described in
example 8 results in
an increase of the frequency of C039-CD69- CD8 TI Ls and a decrease of
CD39+CD69+ CD8+ T
cells compared to the standard IL-2 condition, as shown in Figure 11 A, 11 B
and 11 C. This effect
was most pronounced for TILs expanded with JAB stimulators added with a time
delay (TD)..
This was illustrated using a representative number of tumor fragments from
ovarian cancer,
cervical cancer, lung cancer and renal cell carcinoma.
CD39-CD69- cells have been shown to be correlated with response to ACT in
melanoma patients,
as especially higher numbers of double negative cells are significantly higher
in patients that
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
49
respond to therapy. These cells were shown to exhibit a stem-like phenotype
characterized by self-
renewal capacity to be able to reconstitute the cytotoxic effector cell
population upon stimulation
(Krishna, S etal., Stem-like CD8 T cells mediate response of adoptive cell
immunotherapy against
human cancer, Science 370,1328-1334 (2020).
Example 13¨ TME stimulators added with a time delay of 2 days lead to a
product with a
favorable phenotype
Example 13 illustrated in Figure 12 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol and staining T cells
using anti-CD3,
anti-CD8, anti-CD39 and anti-0069, anti-TIM3, anti-CD28 flow cytometry
antibodies as described
in example 8 and comparing TIL samples expanded with IL-2, and TME stimulators
with or without
TD, resulted in significantly higher numbers of CD3+ and CD8+ TILs per
fragment in samples
expanded with TME stimulators added with a time delay of 2 days compared to
TME stimulators
added without time delay (Figure 12 A, 6). There was a tendency towards higher
1IM3 expression
as well as a significantly higher proportions of cells expressing the
coactivation marker CD28 in TD
samples, which points towards cells that are activated and tumor-specific
(Figure 12 C, D).
Frequencies of CD39+CD69+ cells were significantly lower in TD than in non TD
samples, whereas
the frequency of C039-CD69- cells had the tendency to be higher in TD samples
compared to non
TD samples (Figure 12 E, F).
Summarizing, comparing the phenotype of TILs expanded with TME stimulators of
group A and B
or A, B, C, D and adding J with a time delay of 2 days resulted in higher
numbers of relevant,
tumor specific TILs with a favorable phenotype.
Example 14¨ Cytotoxic potential analysis of "young" TIL cultures with TME
stimulators
This example demonstrates the analysis of the cytotoxic potential of "young"
TIL cultures with TME
stimulators performed as described in example 6.
When cultures designated for young TIL generation were harvested, their
reactivity and cytotoxic
potential was assessed by flow cytometry. Reactivity was assessed by
stimulation of young TILs
with aCD3/aCD28/aCD137 coated beads and subsequent staining of cytotoxic
degranulation
marker CD107a on the cell surface and cytokines INFg and TNFa intracellularly.
Characterization
of T cell subsets was additionally analyzed using following markers:
TIL cell surface: CD107a, CD3, CD4, CD8, Live-Dead
TIL intracellularly: INFg, TNFa
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
Briefly, about 2x106 young TILs per sample were thawed and rested overnight in
a 24-well plate in
RPM! + 10% inactivated human AB serum and 1% Pen/Strep. The next day, cells
were harvested
and counted. 1x106 TILs were transferred to a 96 well plate in triplicates and
stimulated with
aCD3/aCD28/aCD137 dynabeads with a bead-to-cell ratio of 1:20 for six hours in
presence of
5 aCD107a antibody and Golgi Plug.
After six hours, cells were washed and then incubated with titrated surface
antibodies (BD
Biosciences, Table 1) and PBS for 30 min at 4 C. Cells were washed twice with
PBS + 0.5% BSA
and then fixed overnight at 4 C with fixation buffer (FoxP3 Staining Buffer
Set, ebioscience, Table
1). The next day cells were washed twice with Permeabilization buffer (FoxP3
Staining Buffer Set
10 ebioscience, Table 1) and then stained for intracellular cytokine
antibodies (BD biosciences, Table
1) and PBS for 30 min at 4 C. Cells were washed twice with Permeabilization
buffer (FoxP3
Staining Buffer Set, ebioscience, Table 1), resuspended in PBS + 0.5% BSA and
directly analyzed
by flow cytometry (CytoFLEX, Beckman Coulter).
This example demonstrates the reactivity and functionality analysis of "young"
TIL cultures with
15 TME stimulators.
Example 15¨ TME-stimulators in combination added with time delay enhance the
frequency
and total number of TILs triple-positive for cytotoxic markers
Example 15 illustrated in Figure 13, 14 and 15 demonstrated that adding a
combination of TME
20 stimulators as described in Example 6 to the standard young TIL protocol
with or without a time
delay of 2 days and stimulating T cells using aCD3, aCD28 and a41 BB coated
beads followed by
an (intra-)cellular staining with aCD107a, aCD3, aCD4, aCD8, alFNg and aTNFa
as described in
example 14 resulted in an increased frequency of CD8+ T cells expressing all
three of the cytotoxic
markers, especially in TILs expanded with JAB TD and JAB+C+D TD (Figure 13).
Figure 14 A
25 shows, that the total number of reactive cells was significantly
increased in all samples expanded
with TME stimulators but especially in TD samples. A similar tendency could be
seen for the total
number of triple-positive TILs, shown in Figure 14 B.
The effect of the time delay is again illustrated in Figure 15 with a direct
comparison between TME
stimulators added with or without time delay. There was a clear tendency
towards a higher number
30 of total reactive CD8+ T cells and a higher number of triple positive
CD8+ T cells per tumor
fragment, when stimulators are added with a time delay of 2 days.
Summarized, TILs expanded with TME stimulators added with a 2-day time delay
showed a higher
frequency and total number of cells that are activated upon bead stimulation
and specifically higher
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
51
number of cells expressing all three markers IFNg, TNFa and CD107a, shown to
exhibit a higher
capacity for antigen recognition and cytotoxicity.
This was illustrated using a representative number of tumor fragments from
ovarian cancer and
renal cell carcinoma.
Example 16¨ TME stimulators in combination added with time delay clearly
enhances the
frequency of reactive cells in selected patient samples
Example 16 illustrated in Figure 16 demonstrated that adding a combination of
TME stimulators as
described in Example 6 to the standard young TIL protocol with or without a
time delay of 2 days
and stimulating T cells using aCD3, aCD28 and a41BB coated beads followed by
an (intra-)cellular
staining with aCD107a, aCD3, aCD4, aCD8, alFNg and aTNFa as described in
example 14,
resulted in a higher frequency of reactive cells in ovarian cancer patient 0V7
in TILs expanded with
JAB TD compared to TILs expanded with JAB without time delay. This difference
was not
associated with a decrease in expansion. The difference in reactivity was not
seen for 0V9, where
reactivity was similarly high in JAB and JAB TD samples.
In summary, this example shows, that adding TME stimulators with a time delay
of 2 days can
result in a higher reactivity after unspecific bead stimulation for some
patients, whereas it does not
make a difference in other patients, Therefore, adding TME stimulators with a
time delay seems to
be advantageous to increase cytotoxic potential of the TIL product while
retaining proliferation of
TILs.
Example 17¨ Analysis of CD8+ T cells specificities with a panel of 30 cancer-
associated
pMHC tetramers
This example demonstrates the analysis of the T cell specificities within the
different TIL products
expanded with and without TME stimulators as described in Example 6 to the
standard young TIL
protocol with or without a time delay of 2 days.
This was illustrated using a representative number of tumor fragments from
three cervical cancer
patients that are positive for the HLA allele A0201. Of two patients, IL-2
samples were available. All
samples were expanded from five tumor fragments unless otherwise indicated.
Tetramers represent a selection of 30 peptides bound to an HLA 0201 molecule,
the majority of
peptides derived from Cancer-Testis proteins well known to be expressed in
numerous tumor
entities and recognized by T cells (Table 5). Other peptides are derived from
proteins found to be
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
52
overexpressed in some cancer entities while a small fraction is derived from
melanocytic peptides
that play a role in melanoma progression and are therefore not relevant in
cervical cancer.
When cultures designated for young TIL generation were harvested, their
specificities were
assessed by staining with a panel of 30 different pMHC tetramers color-coded
with unique
combinations of two fluorophores each, and subsequently analyzed by flow
cytometry. Reactivity
was defined by > 0.001%,> 10 specific CD8+ cells and by inspecting tetramer+
populations.
Characterization of T cell subsets was additionally analyzed using following
markers:
CD3, CD8, Live-Dead
In short, HLA A0201 monomers were incubated with the library of 30 cancer-
associated peptides
to load peptides onto the HLA molecules. Subsequently, pMHC monomers were
labeled with two
different streptavidin-fluorophores in separate wells with unique combinations
for each individual
pMHC combination (Table 4). After incubation, the combi-coded pMHC tetramer
library was mixed
and TIL samples were stained with the tetramer library followed by a surface
antibody staining with
anti-CD3, anti-CD8 and Live-Dead. Antigen-specific cells were identified by
gating on double
positive cells for each relevant combination AND negative for other
fluorophores.
Example 18¨ Cancer antigen specific CD8+ T cells are present in cervical
cancer patients
with a higher number and frequency in TILs expanded with JAB TD
Example 18 illustrated in Figure 17 demonstrated that by adding a combination
of TME stimulators
as described in Example 6 to the standard young TIL protocol with or without a
time delay of 2
days and staining with a tetramer library as described in example 17, T cell
populations specific for
cancer-testis and overexpressed antigens can be identified in TIL samples
expanded from cervical
cancer patients. Figure 17 shows seven different peptides recognized by CD8+ T
cells across the
three selected cervical cancer patients and the corresponding TIL samples
grown with TME
stimulators with or without time delay or the IL-2 condition. The number
represents the frequency of
the specific population and the number in the bottom the sum thereof.
Figure 18 A illustrates the total number of specific CD8+ T cell populations
and Figure 18 B the
sum of frequencies of all present populations per patient. TILs expanded with
IL-2 showed one
(Ce4) or no (Ce1) specific CD8+ T cell population. In contrast, TILs expanded
with JAB showed
five and three populations, respectively. The JAB TD sample from Ce4 showed an
increase to
seven populations. Ce3 showed the same number of populations across all
available samples, an
IL-2 sample was not available due to lack of cells to analyze.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
53
JAB+C+D samples exhibited some T cell populations but in general to a lower
number and
frequency compared to JAB and JAB TD. JAB+C+D TD samples were not available
for these
patients.
Of note, most (five) peptides derive from cancer/testis antigens, while STEAP1
and KIF20A
represent overexpressed antigens, of which KIF20A has been described to be
overexpressed in
cervical cancer (Zhang, W etal., High Expression of KIF20A Is Associated with
Poor Overall
Survival and Tumor Progression in Early-Stage Cervical Squamous Cell
Carcinoma, PLoS
11(12):e0167449 (2016))
Figure 19 summarizes the number and frequency of specific CD8 T cells
populations of non TD
and TD samples, compared to IL-2 samples, illustrating higher total numbers
and frequencies of
cancer-testis and overexpressed antigen specific CD8 T cells in both non TD
and TD samples, with
the tendency towards higher numbers and specifically higher frequencies in TD
samples.
It has previously been shown that responders of adoptive cell therapy showed a
significant higher
number and frequency of neo-antigen specific T cells and that a high number
correlated with better
survival. It therefore seems to be crucial to broaden the T cell repertoire in
the TIL product (Heeke,
C. etal., Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive
cell therapy with
tumor-infiltrating lymphocytes in melanoma. J Clin Invest; 132(2)).This data
showed that cancer
specific CD8+ T cells can be detected in cervical cancer patients and by
adding TME stimulators,
especially with a time delay of 2 days, the number and frequency of these
populations can be
increased, therefore broadening the T cell repertoire and potentially making
adoptive cell therapy
more successful.
Example 19¨ "Young" tumor-infiltrating lymphocytes (TILs) with TME stimulators
This example demonstrated the generation of "young" tumor-infiltrating
lymphocytes (TILs) with
TME stimulators as described in Example 6 with following changes:
The fresh or frozen tumors were divided into 1-3 mm3 fragments and placed into
a G-Rex 6-well
plate (WilsonWolf; 5 fragments per well) with 5 ml complete medium (CM)
supplemented with 6000
IU/mL IL-2 (Clinigen) only (baseline) or in combination with TME stimulators
of each of the PD-
1/PD-L1 antagonists (group A), CTLA-4 antagonist (group B), and 4-1 BB agonist
(group J) in
combination with anti-CD3, in a humidified 37 C incubator with 5% CO2 at the
same time or with a
time delay or time lapse of 48h or 96h. TME stimulation combinations are
called corresponding to
the stimulator groups J, A, B, without or with time delay (TD) and relevant
time delay in hours.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
54
Example 20¨ Culturing TILs with TME Stimulators increases cell number and
success rate
while reducing culture time
Example 20 illustrated in Figure 20 demonstrated that adding TME stimulators
as described in
Example 19 to the standard young TIL protocol leads to similar numbers of
viable cells per
fragment. Surprisingly, adding J with a TD of 96 hours leads to similar number
of cells compared to
the 48h TD, although slightly lower compared to the 48h time delay or no time
delay. This could be
due to the fact that TILs were expanded from frozen fragments, whereas other
conditions derived
from fresh tumor fragments.
This was illustrated using a representative number of tumor fragments from
ovarian cancer, renal
cell carcinoma and cervical cancer.
Example 21 ¨ Culturing TILs with TME stimulators and an increased time delay
does not
change the composition of the TIL product
Example 21 illustrated in Figure 21 and 22 demonstrated that adding TME
stimulators as described
in Example 19 to the standard young TIL protocol with or without a time delay
of 48 or 96 hours
and stained with anti-CD3, anti-CD4, anti-CD56 and anti-CD8 antibodies as
described in Example
8 led to similar or slightly higher frequencies of CD3+ cells and decreased
frequencies of NK cells
across all samples with TME stimulators compared to the IL-2 condition, with
no differences
between the 96h and 48h time delay (Figure 21 A). The frequencies of CD4+ T
cells were
decreased and CD8 frequencies increased compared to the IL-2 condition with
again only slight
differences between the different TME stimulator conditions (Figure 21 B).
Figure 22 A illustrates that total numbers of CD3+ T cells in samples expanded
with TME
stimulators were consistently higher than in the IL-2 condition, although not
significant. Numbers of
total CD8+ T cells tend to be slightly lower in samples receiving JAB with a
96h time delay
compared to JAB or JAB TD 48h whereas numbers of CD4+ T cells do not show any
difference
across all samples (Figure 22 B, C)
This was illustrated using a representative number of tumor fragments from
ovarian cancer, renal
cell carcinoma and cervical cancer.
In total, this data showed that adding TME stimulators with a time delay of
96h leads to a high cell
expansion without compromising on CD8+ T cell numbers.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
Example 22¨ Culturing TILs with TME stimulators and an increased time delay
leads to an
increased expression of activation markers BTLA, LAG3 and TIM3 while CD28
expression is
retained
Example 22 illustrated in Figure 23 demonstrated that adding TME stimulators
as described in
5 Example 19 to the standard young TIL protocol with or without a time
delay of 48 or 96 hours and
stained with anti-CD3, anti-CD8, anti-BTLA, anti-LAG3, anti-TIM3, anti-CD28,
anti-CD27 and anti-
CD57 antibodies as described in Example 8 led to an increased expression of
activation markers
BTLA, LAG3 and TIM3 in TIL samples expanded with TME stimulators with most
significant
differences in samples expanded with a 96h time delay (Figure 23 A, B, C).
Expression of CD28 is
10 elevated in all TIL samples expanded with TME stimulators but seemed to
be slightly higher in TILs
expanded with the 96h time delay (Figure 23 D).
At the same time, expression of CD27 and CD57 remained unchanged in the JAB TO
96h samples
compared to JAB or JAB TD (Figure 23 E, F).
This was illustrated using a representative number of tumor fragments from
ovarian cancer, renal
15 cell carcinoma and cervical cancer.
These differences point toward the expansion of more tumor specific and
activated cells, that retain
the expression of costimulatory molecules like CO28 and therefore beneficial
for tumor recognition.
Example 23¨ Culturing TILs with TME stimulators and an increased time delay
leads to an
20 increased population of terminally differentiated cells (Temra)
Example 23 illustrated in Figure 24 demonstrated that adding TME stimulators
as described in
Example 19 to the standard young TIL protocol with or without a time delay of
48 or 96 hours and
stained with anti-CD3, anti-CD8, anti-CCR7 and antiCD45RA antibodies as
described in Example 8
led to a significantly bigger population of CCR7- CD45RA+ CD8+ Temra cells in
the TIL samples
25 expanded with JAB and a 96h time delay compared to samples without or
48h time delay.
This was illustrated using a representative number of tumor fragments from
ovarian cancer, renal
cell carcinoma and cervical cancer.
Terminally differentiated Temra cells are usually more cytotoxic but might be
more exhausted and
not as proliferative as effector-memory cells, that are predominantly present
in the other TME
30 stimulator samples. Adding the JAB TME stimulators at day 0 reduces the
frequency of Temra cells
compared to IL-2 alone. The time delay of 48 hours seems to further reduce
this Temra population,
whereas the 96 hour time delay reverts this trend mimicking the IL-2 alone
data.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
56
Example 24¨ Culturing TILs with TME stimulators and an increased time delay
leads to an
expansion of CD8+ T cells that are negative for CD39 and CD69 with a stem-cell
like
phenotype
Example 24 illustrated in Figure 25 demonstrated that adding TME stimulators
as described in
Example 19 to the standard young TIL protocol with or without a time delay of
48 or 96 hours and
stained with anti-CD3, anti-CD8, anti-CD39 and anti-CD69 antibodies led to a
decrease in the
CD39-1- CD69+ CD8+ T cell population in samples expanded with TME stimulators
and an increase
in the double negative fraction compared to the IL-2 samples. There were no
significant differences
between JAB, JAB TD 48h and 96h. Also the total numbers of CD39-CD69- cells
were mostly
similar although the 96h time delay seemed to lead to slightly lower total
numbers of this
population which might be due to a slightly lower expansion in total.
Summarized, this data shows that as discussed in Example 12, adding TME
stimulators to the TIL
cultures led to an increased expansion of CD39-CD69- cells that have been
described to have a
stem cell like phenotype, that seemed to be clinically relevant in ACT trials.
Summarizing Examples 20-24, expanding TILs with TME stimulators with a time
delay of 96h led to
a similar expansion of desired CD8+ T cells compared to the shorter 48h time
delay. These CD8+
T cells show a favorable activated and potentially tumor specific phenotype.
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
57
Items
1. Expanded tumor infiltrating lymphocytes (TILs) for use in treating a
subject with cancer, the
treatment comprising the steps of:
- a) culturing autologous T cells by obtaining a first population of TILs
from a tumor
resected from a mammal,
- b) performing a depletion of suppressive cells, including regulatory T
cells, and/or
blocking negative signals by the addition of one or more TME stimulators from
the group of
"Inhibitors" to obtain a depleted population of TILs,
- c) performing a first expansion by culturing the depleted population of TILs
in a cell
culture medium comprising:
- one or more TME stimulators from the group of "cytokines", and/or
- one or more of the TME stimulators from the "Stimulator" group to produce
a
second population of TILs,
- d) performing a second expansion by supplementing the cell culture medium of
the
second population of TILs with additional IL-2 and/or other cytokines from the
"cytokine"
group, anti-CD3 antibody, and antigen presenting cells (APCs), to produce a
third
population of TILs, wherein the third population of TILs is a therapeutic
population; and
- e) after administering nonmyeloablative lymphodepleting chemotherapy,
administering to
the mammal the therapeutic population of T cells, wherein the T cells
administered to the
mammal, whereupon the regression of the cancer in the mammal is promoted.
2. Expanded tumor infiltrating lymphocytes (TILs) for use in promoting
regression of a cancer in a
subject with cancer, the regression comprising the steps of:
- a) culturing autologous T cells by obtaining a first population of TILs
from a tumor
resected from a mammal,
- b) performing a depletion of suppressive cells, including regulatory T
cells, and/or
blocking negative signals by the addition of one or more TME stimulators from
the group of
"Inhibitors" to obtain a depleted population of TILs,
- c) performing a first expansion by culturing the depleted population of
TILs in a cell
culture medium comprising:
- one or more TME stimulators from the group of "cytokines", and/or
- one or more of the TME stimulators from the "Stimulator" group to produce
a
second population of TILs,
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
58
- d) performing a second expansion by supplementing the cell culture medium
of the
second population of TILs with additional IL-2, anti-CD3 antibody, and antigen
presenting
cells (APCs), to produce a third population of TILs, wherein the third
population of TILs is a
therapeutic population; and
- e) after administering nonmyeloablative lymphodepleting chemotherapy,
administering to
the mammal the therapeutic population of T cells, wherein the T cells
administered to the
mammal, whereupon the regression of the cancer in the mammal is promoted.
3. A method for expanding tumor infiltrating lymphocytes (TILs) into a
therapeutic population of
TILs comprising:
- a) culturing autologous T cells by obtaining a first population of TILs from
a tumor
resected from a mammal,
- b) performing a depletion of suppressive cells, including regulatory T
cells, and/or
blocking negative signals by the addition of one or more TME stimulators from
the group of
"Inhibitors" to obtain a depleted population of TILs,
- c) performing a first expansion by culturing the depleted population of TILs
in a cell
culture medium comprising:
- one or more TME stimulators from the group of "cytokines", and/or
- one or more of the TME stimulators from the "Stimulator" group to produce
a
second population of TILs,
- d) performing a second expansion by supplementing the cell culture medium of
the
second population of TILs with additional IL-2, anti-CD3 antibody, and antigen
presenting
cells (APCs), to produce a third population of TILs, wherein the third
population of TILs is a
therapeutic population.
4. The uses and methods of items 1-3, wherein one or more TME stimulators from
the group of
"cytokines" are added in step b).
5. The uses and methods of items 1-4, wherein the group of "cytokines" are
selected from the
group consisting of IL-2, IL-7, IL-12, IL-15, and IL-21.
6. The uses and methods of items 1-5, wherein the group of "Inhibitors" are
selected from the
group consisting one or more of:
- A) substances that act through the PD-1 receptor on T-cells,
- B) substances that act through the CTLA-4 receptor on T-cells,
- C) substances that act through the LAG-3 receptor on T-cells,
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
59
- D) substances that act through the TIGIT/CD226 receptor on T-cells.
- E) substances that act through the KIR receptor on T-cells,
- F) substances that act through the TIM-3 receptor on T-cells,
- G) substances that act through the BTLA receptor on T-cells, and
- H) substances that act through the A2aR receptor on T-cells.
7. The uses and methods of items 1-5, wherein the group of "Inhibitors" are
selected from the
group consisting one or more of:
- A) substances that act through the PD-1 receptor on T-cells,
- B) substances that act through the CTLA-4 receptor on T-cells,
- C) substances that act through the LAG-3 receptor on T-cells, and
- D) substances that act through the TIGIT/CD226 receptor on T-cells.
8. The uses and methods of items 1-7, wherein the substance of group A is
selected from one or
more from the group consisting of pembrolizumab, nivolumab, cemiplimab,
sym021, atezolizumab,
avelumab, and durvalumab.
9. The uses and methods of items 1-8, wherein the substance of group B is
selected from one or
more from the group consisting of ipilimumab and tremelimumab.
10. The uses and methods of items 1-9, wherein the substance of group C is
selected from one or
more from the group consisting of relatlimab, eftilagimo alpha, and sym022.
11. The uses and methods of items 1-10, wherein the substance of group D is
tiragolumab.
12. The uses and methods of items 1-11, wherein the group of "Inhibitors" are:
- A: substances that act through the PD-1 receptor on T-cells, and
- B: substances that act through the CTLA-4 receptor on T-cells.
13. The uses and methods of items 1-12, wherein the group of "Inhibitors" are:
- A: substances that act through the PD-1 receptor on T-cells,
- B: substances that act through the CTLA-4 receptor on T-cells, and
- C) substances that act through the LAG-3 receptor on T-cells.
14. The uses and methods of items 1-13, wherein the group of "Inhibitors" are:
- A: substances that act through the PD-1 receptor on T-cells,
- B: substances that act through the CTLA-4 receptor on T-cells, and
CA 03223030 2023- 12- 15
WO 2022/263673
PCT/EP2022/066633
- D) substances that act through the TIGIT/CD226 receptor on T-cells.
15. The uses and methods of items 1-14, wherein the group of "Inhibitors" are:
- A) substances that act through the PD-1 receptor on 1-cells,
- B) substances that act through the CTLA-4 receptor on T-cells,
5 - C) substances that act through the LAG-3 receptor on T-cells, and
- D) substances that act through the TIGIT/CD226 receptor on T-cells.
16. The uses and methods of items 1-15, wherein the group of "Inhibitors" are
selected from the
group consisting one or more of:
- P) epacadostat,
10 - Q) substances that act through the TGFP receptor on T-cells,
- R) substances that act through the IL-10 receptor on 1-cells, and
- S) substances that act through the IL-35 receptor on 1-cells.
17. The uses and methods of items 1-16, wherein the group of "Inhibitors" are
selected from the
group consisting one or more of:
15 - T) cyclophosphamides,
- U) TKIs,
- V) substances that act through aCD25, and
- X) IL2/Diphteria toxin fusions.
18. The uses and methods of items 1-17, wherein the group of "Stimulator" are
selected from the
20 group consisting one or more of:
- I) substances that act through the 0X40/CD134 receptor on 1-cells,
- J) substances that act through the 4-1BB/CD137 receptor on T-cells,
- K) substances that act through the CD28 receptor on T-cells,
- L) substances that act through the ICOS receptor on 1-cells,
25 - M) substances that act through the GITR receptor on T-cells,
- N) substances that act through the CD4OL receptor on 1-cells,
- 0) substances that act through the 0D27 receptor on T-cells, and
- W) substances that act through CD3 on T cells.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
61
19. The uses and methods of item 18, wherein the group of "Stimulator" is:
- J) substances that act through the 4-1BB/CD137 receptor on T-cells.
20. The uses and methods of item 11, wherein the substance of group J is
selected from one or
more from the group consisting of urelumab and utomilumab.
21. The uses and methods of items 1-20, wherein:
the group of "Inhibitors" in step b) are:
- A: substances that act through the PD-1 receptor on 1-cells, and
- B: substances that act through the CTLA-4 receptor on T-cells, and
wherein the group of "Stimulator" in step c) is:
- J) substances that act through the 4-16B/CD137 receptor on T-cells.
22. The uses and methods of items 1-21, wherein step b) and step c) are
performed in time lapse,
i.e. one day apart, or such as 2, 3, 4, 5, 6 or 7 days apart.
23. The uses and methods of item 22, wherein the step step b) and step c) are
performed 1-2 days
apart.
24. The uses and methods of item 22, wherein the step step b) and step c) are
performed 1-3 days
apart.
25. The uses and methods of item 22, wherein the step b) and step c) are
performed 1-4 days
apart.
26. The uses and methods of item 22, wherein the step b) and step c) are
performed 1-5 days
apart.
27. The uses and methods of item 22, wherein the step b) and step c) are
performed 1-6 days
apart.
28. The uses and methods of item 22, wherein the step b) and step c) are
performed 1-7 days
apart.
29. The uses and methods of item 22, wherein the step b) and step c) are
performed 2-4 days
apart.
30. The uses and methods of item 22, wherein the step b) and step c) are
performed 4-8 days
apart.
31. The uses and methods of items 1-30, wherein the concentration of the
substance is 0.1 pg/mL
to 300 pg/mL, such as 1 pg/mL to 100 pg/mL, such as 10 pg/mL to 100 pg/mL,
such as 1 pg/mL to
10 pg/mL, such as 2-20 pg/mL.
CA 03223030 2023- 12- 15
WO 2022/263673 PCT/EP2022/066633
62
32. The uses and methods of items 1-31, wherein steps (a) through (b) are
performed within a
period of about 7 days to about 28 days.
33. The uses and methods of items 1-32, wherein step (c) is performed within a
period of about 7
days to about 21 days.
34. The uses and methods of items 1-33, wherein the therapeutic population of
T cells is used to
treat a cancer type selected from the groups consisting of breast cancer,
renal cell cancer, bladder
cancer, melanoma, cervical cancer, gastric cancer, colorectal cancer, lung
cancer, head and neck
cancer, ovarian cancer, Hodgkin lymphoma, pancreatic cancer, liver cancer, and
sarcomas.
35. The uses and methods of items 1-34, wherein step (c) results in 1 x 107to
lx 1012 cells, such
as 1 x 108 to 5x 109 cells, such as 1 x i0 to 5x 109 cells, such as 1 x 108 to
5x 1010 cells, such as 1
x 10 to 5x 1011 cells.
36. The uses and methods of items 1-35, wherein the anti-CD3 antibody is OKT3.
37. The uses and methods of items 1-36, wherein the mammal is a human
individual.
38. The uses and methods of items 1-37, wherein the antibody is selected from
the group
consisting of a monoclonal antibody, a human antibody, a humanized antibody, a
chimeric
antibody, a murine antibody, a F(ab )2 or Fab fragment, and a Nanobody.
39. The uses and methods of items 1-38, wherein group A is selected from one
or more from the
group consisting of pembrolizumab, nivolumab, cemiplimab, sym021,
atezolizumab, avelumab,
durvalumab, Toripalimab, Sintilimab, Camrelizumab, Tislelizumab, Sasanlimab,
Dostarlimab, MAX-
10181, YPD-29B, IMMH-010, INCB086550, GS-4224, DPPA-1, TPP-1, BMS-202, CA-170,
JC21,
eFT508, Osimertinib, PlatycodinD, PD-LYLSO, Curcumin, and Metformin.
40. The uses and methods of items 1-39, wherein group B is selected from one
or more antibodies
from the group consisting of ipilimumab and tremelimumab.
41. The uses and methods of items 1-39, wherein the substance of group J is
selected from one or
more from the group consisting of urelumab, utomilumab, B0Y7835, and BCY7838.
42. A population of tumor infiltrating lymphocytes (TILs) obtainable by a
method of any of the
previous items.
43. A population of tumor infiltrating lymphocytes (TILs) comprising a
clinically relevant number of
TILs with a higher percentage of CD8 T cells expressing markers associated
with tumor-specificity
(exhaustion markers).
CA 03223030 2023- 12- 15