Note: Descriptions are shown in the official language in which they were submitted.
WO 2023/007241
PCT/IB2022/000420
1
COMPOSITIONS AND METHODS RELATED TO TET-ASSISTED PYRIDINE
BORANE SEQUENCING FOR CELL-FREE DNA
CROSS-REFERENCE TO RELATED APPLICATIONS
(00011
This application claims the benefit of U.S. Provisional Application No.
63/203,565
filed July 27, 2021, the contents of which is incorporated herein by reference
in its entirety.
100021
The contents of the electronic sequence listing (sequencelisting.xml; Size:
8,000
bytes; and Date of Creation: July 26, 2022) is herein incorporated by
reference in its entirety.
FIELD
[00031
The present disclosure provides compositions and methods related to TET-
assisted
Pyridine Borane Sequencing (TAPS). In particular, the present disclosure
provides optimized
TAPS for cfDNA (cfTAPS), which provides high-quality and high-depth whole-
genome cell-
free methylomes. The compositions and methods provided herein facilitate the
acquisition of
multimodal information about cfDNA characteristics, including DNA methylation,
tissue of
origin, and DNA fragmentation for the diagnosis and treatment of disease.
BACKGROUND
[00041 Although recent advances in cancer research offer new ways to treat
cancer, early
detection still represents the best opportunity for curing cancer. Early-stage
treatment not only
greatly improves patient survival but also costs considerably less.
Circulating cell-fiee DNA
(cfDNA) - the free-floating DNA in blood plasma originating from cell death in
various healthy
and diseased tissues - holds tremendous potential to develop an early cancer
detection assay.
Genetic information in cfDNA, such as mutations and copy-number variations
(CNVs),
demonstrate potential utility for monitoring cancer progression and treatment.
However,
genetic alterations are challenging to detect given the low fraction of tumor
DNA in early-stage
disease. Furthermore, genetic alterations are weakly informative about the
tissue-of-origin,
which is needed to determine the location of malignancy.
[00051 In contrast, widespread epigenetic changes such as DNA methylation of
both cancer
cells and tumor microerwironment occur early in tumorigenesis. Recent studies
have shown
cfDNA methylation to be one of the most promising biomarkers for early cancer
detection, by
providing thousands of methylation changes that can be combined to overcome
detection
limits, and tissue-of-origin information that allows cancer localisation with
high confidence.
DNA methylation is best determined by a whole-genome, base-resolution, and
quantitative
sequencing method, such as bisulfite sequencing. However, bisulfite sequencing
is DNA
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
2
damaging and expensive; therefore, current cfDNA methylation sequencing is
limited by being
low-depth, targeted, or low-resolution and qualitative enrichment-based
sequencing, thus
imperfectly capturing the cfDNA methylome.
SUMMARY
100061 Embodiments of the present disclosure include a method of obtaining a
methylation
signature. In accordance with these embodiments, the method includes isolating
cell free DNA
(cfDNA) from a sample; preparing a sequencing library comprising the cfDNA;
and
performing TET-assisted Pyridine Borane Sequencing (TAPS) on the sequencing
library to
obtain a methylation signature of the cfDNA. In some embodiments, the
methylation signature
is a whole-genome methylation signature.
100071 In some embodiments, the unique mapping rate resulting from TAPS on the
cfDNA
is at least 80% and/or the unique deduplicated mapping rate is at least 70%.
(0008/
In some embodiments, preparing the sequencing library comprises ligating
sequencing adapters to the isolated cfDNA.
[00091 In some embodiments, carrier DNA is added to the sequencing library
prior to
performing TAPS
NOM In some embodiments, the method further comprises identifying at least one
methylation biomarker from the cfDNA whole-genome methylation signature, and
determining
whether the methy lation biomarker is indicative of cancer.
[00111
In some embodiments, the methylation biomarker comprises a differentially
methylated region (DMR).
[00121 In some embodiments, the method further comprises classifying the
sample based
on the DMR as compared to a reference DMR.
[00131 In some embodiments, the reference DMR corresponds to a non-cancerous
control,
or a cancerous control.
[00141
In some embodiments, the method further comprises identifying at least one
methylation biomarker from the cfDNA whole-genome methylation signature, and
determining
a tissue-of-origin corresponding to the methylation biomarker.
[00151 In some embodiments, the method further comprises classifying the
sample based
on the tissue-of-origin biomarker.
[00161 In some embodiments, the method further comprises identifying a DNA
fragmentation profile, and determining whether the fragmentation profile is
indicative of
cancer.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
3
[00171
In some embodiments, the method further comprises identifying at least one
sequence variant from the cfDNA, and determining whether the sequence variant
is indicative
of cancer.
100181 In some embodiments, performing TAPS on the sequencing library to
obtain the
whole-genome methylation signature comprises identifying 5mC modifications in
the cfDNA
and providing a quantitative measure for frequency of the 5mC modifications.
100191 In some embodiments, performing TAPS on the sequencing library to
obtain the
whole-genome methylation signature comprises identifying 5hmC modifications in
the cfDNA
and providing a quantitative measure for frequency of the 5hmC modifications.
100201 In some embodiments, performing TAPS on the sequencing library to
obtain the
whole-genome methylation signature comprises identifying 5caC modifications in
the cfDNA
and providing a quantitative measure for frequency of the 5caC modifications.
10021/ In some embodiments, performing TAPS on the sequencing library to
obtain the
whole-genome methylation signature comprises identifying 5fC modifications in
the cilDNA
and providing a quantitative measure for frequency of the 5fC modifications.
[00221 Embodiments of the present disclosure also include a method of
determining
whether a subject has cancer using any of the methods described herein. In
some embodiments,
the cancer comprises hepatocellular carcinoma (HCC) or pancreatic ductal
adenocarcinoma
(PDAC).
[00231
Embodiments of the present disclosure also include a method of determining
whether a subject has early stage cancer using any of the methods described
herein. In some
embodiments, the cancer comprises early stage hepatocellular carcinoma (HCC)
or early stage
pancreatic ductal adenocarcinoma (PDAC).
100241
in still other preferred embodiments, the present invention provides
multimodal
methods of analyzing et-DNA in a patient sample comprising: isolating ciDNA
from a patient
sample; converting 5mC and/or 5hmC residues in the sample to DHU residues to
provide a
modified cfDNA sample; sequencing the modified cfDNA sample to identify
methylated
regions in the sample, wherein a cytosine (C) to thymine (T) transition or a
cytosine (C) to
DHU transition in the modified cfDNA sample as compared to an unmodified
reference cfDNA
provides the location of either a 5mC or 5hmC in the cfDNA; and performing one
or more
additional analytical steps on the modified cfDNA selected from the group
consisting of: a)
determining copy number variation of one or more targets in the modified cIDNA
sample; b)
determining the tissue of origin or one or more targets in the modified cfDNA
sample; c)
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
4
determining the fragmentation profile of the modified cfDNA sample; and d)
identifying one
or more single nucleotide mutations in the modified cfDNA sample.
100251 In some embodiments, the step of sequencing the modified cfDNA sample
to
identify methylated regions in the sample comprising identifying at least one
differentially
methylated region (DMR).
[00261
in some embodiments, the multimodal method further comprises classifying
the
sample based on the DMR as compared to a reference DMR.
[00271 In some embodiments, the reference DMR corresponds to a non-cancerous
control,
or a cancerous control.
100281 In some embodiments, the step of determining copy number variation
(CNV) of one
or more targets in the modified cfDNA sample comprises determining the
observed read count
for a target sequence across the genome by dividing the reference genome into
bins and
counting the number of reads in each bin.
[00291 In some embodiments, the presence of copy number aberrations of greater
than 500
kb is indicative of CNV in a patient.
[00301 In some embodiments, the step of determining the tissue of origin or
one or more
targets in the modified cfDNA sample comprises tissue deconvolution of data
obtained from
sequencing the modified cfDNA sample.
10031] In some embodiments, the tissue deconvolution comprises comparing DNA
methylation value identified in the modified cfDNA sample with reference DMRs
from two or
more different tissues.
[00321 In some embodiments, the step of determining the fragmentation profile
of the
modified cfDNA sample comprises classifying the fragment length and
periodicity of
fragments in the modified cfDNA sample.
100331
In some embodiments, classifying the length and periodicity of fragments in
the
modified cfDNA sample further comprises calculating the proportion of cfDNA
fragments of
from 300 to 500 bp in 10 bp length range bins.
[00341
In some embodiments, the step of identifying one or more single nucleotide
mutations in the modified cfDNA sample further comprises distinguishing C to T
SNPs from
5mC or 5hmC at a specific position in the cfDNA by comparing sequencing
results after TAPS,
wherein the presence of a T read at the specific position in a compliment to
the original bottom
strand of the cIDNA is indicative of a C to T SNP and the presence of a C read
at the specific
position in a compliment to the original bottom strand of the cfDNA is
indicative of 5mC or
5hmC .
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
[00351 In some embodiments, two or more of steps a, b, c and d are performed
on the
modified cfDNA.
[00361 In some embodiments, three or more of steps a, b, c and d are performed
on the
modified cfDNA.
[00371 In some embodiments, all of steps a, b, c and d are performed on the
modified
cfDNA.
100381 In some embodiments, the unique mapping rate resulting from the
sequencing step
is at least 80% and/or the unique deduplicated mapping rate is at least 70%.
[00391 In some embodiments, the sequencing step further comprises preparing a
sequencing
library comprising the cfDNA by ligating sequencing adapters to the isolated
cfDNA.
100401 In some embodiments, carrier DNA is added to the cfDNA.
[00411 In some embodiments, the multimodal method provides a cfDNA whole-
genome
methylation signature and the method further comprises identifying at least
one methylation
biomarker from the cfDNA whole-genome methylation signature, and determining
whether the
methylation biomarker is indicative of cancer.
[00421 In some embodiments, the multimodal method further comprises
identifying 5mC
modifications in the cfDNA and providing a quantitative measure for frequency
of the 5mC
modifications.
[00431 In some embodiments, the multimodal method further comprises
identifying 5hmC
modifications in the cfDNA and providing a quantitative measure for frequency
of the 5hmC
modifications.
100441 In some embodiments, the multimodal method further comprises
identifying 5caC
modifications in the cfDNA and providing a quantitative measure for frequency
of the 5caC
modifications.
100451 In some embodiments, the multimodal method further comprises 5fC
modifications
in the cfDNA and providing a quantitative measure for frequency of the 5fC
modifications.
[00461 In some embodiments, the step of converting 5mC and/or 5hmC residues in
the
sample to DHU residues to provide a modified cIDNA sample comprises oxidizing
5mC and/or
5hmC residues to provide 5caC and/or 5fC residues and reducing the 5caC and/or
5fC residues
to DHU residues.
[00471 In some embodiments, the step of oxidizing 5mC and/or 5hmC residues to
provide
5caC and/or 5fC residues comprises treatment of the sample with a Tet enzyme.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
6
[00481 In some embodiments, the step of oxidizing 5mC and/or 5hmC residues to
provide
5caC and/or 5fC residues comprises treatment of the sample with a chemical
oxidizing agent
so that one or more 5fC residues are generated.
100491 In some embodiments, the step of reducing the 5caC and/or 5fC residues
to DHU
residues comprises treatment of the sample with a borane reducing agent.
[00501
Embodiments of the present disclosure also include a method of determining
whether a subject has early stage cancer using any of the multimodal methods
described herein.
BRIEF DESCRIPTION OF THE DRAWINGS
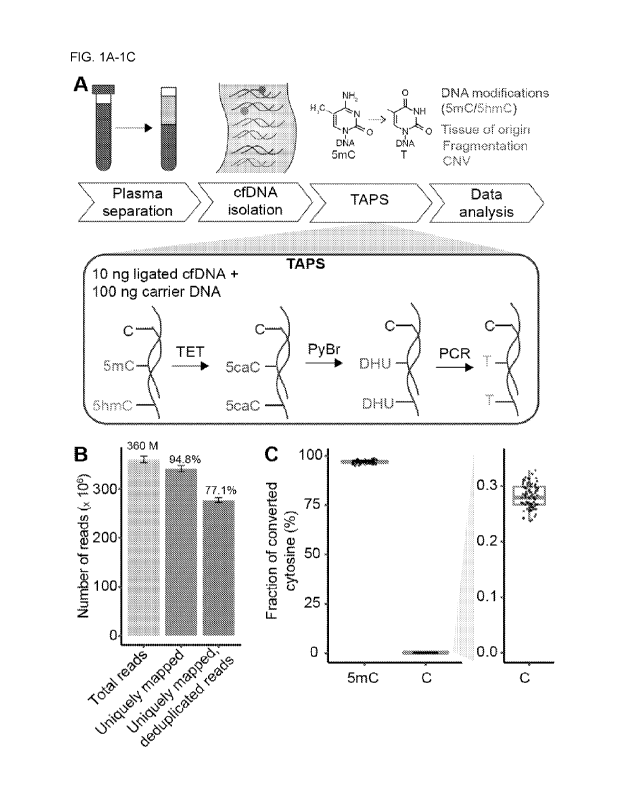
[00511 FIGS. 1A-1C: cfDNA analysis by TAPS. (A) Schematic representation of
the TAPS
approach for cfDNA analysis. CfIDNA is isolated from 1-3 mL of plasma. long of
cfDNA is
ligated to Illumina sequencing adapters and topped up with 100 ng of carrier
DNA.
Subsequently, 5mC and 5hmC in DNA are oxidized by mTet1CD enzyme to 5caC,
reduced by
PyBr to DHU and amplified and detected as T in the final sequencing.
Computational analysis
of TAPS data allows for simultaneous characterization of multiple cfDNA
features including
DNA methylation, tissue of origin, fragmentation patterns and CNVs. (B) Number
of total
reads, uniquely mapped reads and uniquely mapped, PCR deduplicated reads in 87
cfDNA
TAPS libraries. Total number of reads and mean percentage of uniquely mapped
reads and
deduplicated reads compared to total leads are shown above the bars. Error
bars represent
standard error. (C) 5mC conversion rate and false positive rate in 85 cfDNA
TAPS libraries
based on spike-in controls with modified or unmodified cytosines at the known
positions. Each
dot represents an individual sample.
[00521
FIGS. 2A-2I: cfDNA methylation in clinical samples. (A) Cancer stage
distribution
of 21 HCC patients and 23 PDAC patients included in the study. (B) Mean per
CpG genome
modification level in non-cancer controls, HCC and PDAC cfDNA. Each dot
represents an
individual sample. (C) PCA plot of cfDNA methylation in 1 kb genomic windows
in non-
cancer controls and HCC. (D) PCA plot of cfDNA methylation in 1 kb genomic
windows in
non-cancer controls and PDAC. (E) The overrepresentation analysis on the
regions correlated
most with PC2 for HCC and PC1 for PDAC in regulatory regions. (F) Receiver
operating
characteristic (ROC) curve of model classification performance based on
differentially
methylated enhancers in HCC and non-cancer controls (n = 51, HCC = 21, non-
cancer controls
= 30). (G) LOO cancer prediction scores for HCC and non-cancer controls.
Dashed line
represents probability score threshold. Samples with a probability score above
this threshold
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
7
were predicted as HCC. (H) ROC curve of model classification performance based
on
differentially methylated promoters between PDAC and non-cancer controls (n =
53, PDAC
23, non-cancer controls = 30). (I) LOO cancer prediction scores for PDAC and
non-cancer
controls. Dashed line represents probability score threshold. Samples with a
probability score
above this threshold were predicted as PDAC.
[0053j
FIGS. 3A-3E: cfTAPS enables analysis of tissue of origin and fragmentation
patterns
in cfDNA. (A) The mean tissue contribution in non-cancer individuals estimated
by NNLS.
Tissue contributions less than 1.5% are aggregated as 'Other'. (B) Boxplot
showing the
estimated liver cancer contribution within non-cancer, HCC and PDAC group.
Statistical
significance was assessed with a paired t-test. n.s. - not significant. (C)
The length distribution
of cfDNA fragments in the three groups. For each sample, proportion (13) in 10-
base pair
intervals of long cfDNA fragments (300-500 bp) was used as fragmentation
features for PCA
analysis and machine learning. (D) Boxplot showing proportion of short (70-
150bp) and long
(300-500bp) fragments in non-cancer controls, PDAC, and HCC. The Kruskal-
Wallis test was
performed to test differences in fragment size distribution between groups.
Statistically
significant differences are marked with an asterisk (*P value < 0.05, **P
value < 0.01, ***P
value < 0.001, ****P value < 0.0001). (E) PCA plot of cfDNA 10bp-fragment
fraction in non-
cancer controls and HCC (left panel); and non-cancer controls and PDAC (right
panel).
[0054] FIGS. 4A-4C: Integrating multimodal features from cfTAPS enhances multi-
cancer
detection. (A) Heatmap showing individual model performance on multi-cancer
prediction and
the predicted probabilities for each patient. Each vertical column is a
patient. Detection yes/no
means patients being correctly classified or misclassified based on a
particular feature.
Predicted score means the probability of classifying the patients to a
specific group based on a
particular feature. (B) Schematic detailing the method of integrating multiple
features (DNA
methylation, tissue contribution and fragmentation fraction) extracted from
cfrAPS data for
multi-cancer prediction. (C) The actual and predicted patient status
calculated in LOO cross-
validation.
[0055j FIGS. 5A-5D: cfDNA TAPS. (A) Agarose gel of 10 representative cfDNA
TAPS
libraries after post-amplification clean-up. All cfDNA TAPS libraries were
prepared from 10
ng of cfDNA and amplified for 7 PCR cycles. (B) Number of mapped read-pairs
for hg38,
spike-ins and carrier DNA in 87 cfDNA TAPS libraries. Mean percentage of
mapped read-
pairs compared to total read-pairs is shown above the bars. Error bars
represent standard error.
(C) Number of total reads, uniquely mapped reads and uniquely mapped. PCR
deduplicated
reads in cfDNA WGBS (EGAD00001004317) (24). Total number of reads and mean
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
8
percentage of uniquely mapped reads and deduplicated reads compared to the
total reads are
shown above the bars. Error bars represent standard error. (D) Correlation
between technical
replicates of cfDNA TAPS libraries prepared from the same cfDNA samples
sequenced to low
depth 2.6x. Methylation was calculated in 100 kb windows.
100561 FIGS. 6A-61: Global cfDNA methylation patterns in cancer and controls.
(A) Age
and gender distribution of pan creati ti s, cirrhosis, PDAC, HCC and non-
cancer control patients
included in cf-TAPS cohort. (B) Genome-wide distribution of CpG modification
in cfDNA in
non-cancer controls, HCC and PDAC. Bar plots shows distribution of average CpG
modification for each group. Overlaid line plots show CpG methylation
distribution in each
patient. (C-D) Correlation plots of average cfDNA CpG modification level in
HCC patients
and (C) tumor size (mm) and (D) tumor stage. (E-F) Correlation plots for PDAC
patients and
(E) tumor size (mm) and (F) tumor stage. Each dot represents an individual
patient. Dashed
lines represent the linear trend fitted with linear regression. Shaded area
represents 95%
confidence intervals of the fitted model. Pearson correlation coefficients
(cor) and P values are
shown in the plots. (G) Distribution of CpG modification levels over
chromosome 4 in cfDNA
of non-cancer controls, HCC and PDAC. Each line represents an individual
patient. Average
CpG modification value was calculated per 1 Mb windows along chromosome 4 and
Gaussian-
smoothed (smoothing window size 10). (H) Methylation variance in I Mb genomic
windows
in non-cancer controls, HCC and PDAC. (I) PCA plot of cfDNA methylation in 1
kb genomic
windows in non-cancer controls and HCC, non-cancer controls and PDAC (Crohn's
disease
and colitis are coloured in green and yellow respectively).
100571 FIGS. 7A-71: HCC and PDAC prediction based on cfDNA DMRs. (A) Overview
of
the LOO model training and validation approach. Total number of samples is
labelled as n. At
each iteration, the model training set consists of n ¨ 1 samples.
Differentially methylated
enhancers (for HCC) or promoters (for PDAC) were selected for model building.
The
predictive model was evaluated on the held-out test sample in each fold.
Cirrhosis and
pancreatitis samples were not included in DMR identification and model
building. (B) HCC
cancer prediction scores for cirrhosis samples. Each blue dot represents the
predicted score for
an individual LOO model. The Black dot shows average probability score for a
particular
sample. The dashed line represents probability score threshold. Samples with
average
probability score above this threshold were predicted as HCC. (C) Gene
Ontology analysis of
genes related to differentially methylated enhancers based in HCC cIDNA (P
value < 0.002)
using Enrichr against NCI-Nature Pathway Interaction, Top 10 categories
selected based on P
value are shown in the graph. Gene-enhancer interactions were assigned using
GeneHancer
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
9
reference database. (D) Methylation of representative differentially
methylated enhancer in
HCC cfDNA for DLC1 gene (two-tailed t-test P value = 8.765e-06). (E) PDAC
cancer
prediction scores for pancreatitis samples. Each yellow dot represents the
predicted score for
an individual LOO model. The black dot shows the average probability score for
a particular
sample. The dashed line represents probability score threshold. Samples with
average
probability score above this threshold were predicted as PDAC. (F) Gene
Ontology analysis of
the genes nearest to the differentially methylated promoters in PDAC cfDNA (P
value < 0.002)
using Enrichr against NCI-Nature Pathway Interaction. Top 10 categories
selected based on P
value are shown on the graph. (G) Methylation of representative differentially
methylated
promoter in PDAC cfDNA for RB1 gene (two-tailed t-test P value = 0.0017). (H)
HCC cancer
prediction scores for the independent cfDNA WGBS dataset (EGAD00001004317).
Each dot
represents the predicted score for an individual LOO model. Grey dot belongs
to non-cancer
controls and the red dot belongs to HCC. The Black dot shows average
probability score for a
particular sample. The dashed line represents probability score threshold.
Samples with average
probability score above this threshold were predicted as HCC. (I) Percentage
of ref DMRs that
can be detected in down-sampled reads. DMRs that were identified in original
LOU model
training were treated as ref DMRs.
14)0581
FIGS. 8A-8I: cfDNA tissue of origin. (A) t-SNE plot of reference tissue
methylation
atlas. (B) The average tissue contribution in HCC and PDAC individuals. (C)
Boxplot showing
the estimated T cell contribution in non-cancer, HCC and PDAC cfDNA samples.
(D) ROC
curve of model performance using tissue contribution to classify HCC vs. non-
cancer. (E) LOO
cancer prediction scores for HCC and non-cancer controls using classifiers
trained on tissue
contribution. The dashed line represents the probability score threshold.
Samples with
probability score above this threshold were predicted as HCC. (F) Cancer
scores for cirrhosis
samples using HCC vs. non-cancer classifiers. Each blue dot represents the
predicted scores
for an individual model. Black dot shows the average probability score for a
particular sample.
Dashed line represents probability score threshold. Samples with average
probability score
above this threshold were predicted as HCC. (G) ROC curve of model performance
using tissue
contribution to classify PDAC vs control. (H) LOU cancer prediction scores for
PDAC and
non-cancer controls using classifiers built based on tissue contribution.
Dashed line represents
probability score threshold. Samples with probability score above this
threshold were predicted
as PDAC. (I) PDAC Cancer scores for pancreatitis samples using PDAC vs. non-
cancer
classifiers. Each yellow dot represents the predicted scores for an individual
model. Black dot
shows the average probability score for a particular sample. Dashed line
represents probability
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
score threshold. Samples with average probability score above this threshold
were predicted as
PDAC.
1430591 FIGS. 9A-9B: CNVs analysis in cfDNA. (A) CNV estimation heatmap
from cfDNA in 100kb bin. (B) cfDNA samples with CNV larger than 500k.
100601 FIGS. 10A-10G: cfDNA fragmentation patterns for cancer prediction. (A)
Fragment
size distribution of cfDNA in public whole genome bisulfite sequencing data.
Frequency was
calculated as number of fragments of particular length divided by total number
of fragments.
(B) ROC curve of HCC and non-cancer control prediction scores from a
generalized linear
model using proportion of long cfDNA fragments (300-500 bp) in 10 bp bins as
features. (C)
Cancer prediction scores for HCC and non-cancer controls in classifiers
trained using LOO
cross-validation. The dashed line represents the probability score threshold.
Samples with a
probability score above this threshold were predicted as HCC. (D) HCC cancer
prediction
scores for cirrhosis samples in these classifiers. Each blue dot represents
the predicted score
for an individual model. Black dots show average prediction score. The dashed
line represents
probability score threshold: samples with average probability score above this
threshold were
predicted as HCC. (E) ROC curve of PDAC and non-cancer control prediction
scores from a
generalized linear model using proportion of long cfDNA fragments (300-500 bp)
in 10 bp bins
as features. (F) LOO cancer prediction scores for PDAC and non-cancer controls
in classifiers
built based on cfDNA fragments frequency in 10 bp length range. The dashed
line represents
the probability score threshold. Samples with probability score above this
threshold were
predicted as PDAC. (G) PDAC cancer prediction scores for pancreatitis samples
in classifiers
built based on cfDNA fragments frequency in 10 bp length range. Each yellow
dot represents
the predicted score for an individual model. Black dots show average
prediction score. The
dashed line represents probability score threshold: samples with average
probability score
above this threshold were predicted as PDAC.
(00611
FIGS. 11A-11C: Multi-cancer detection with cfTAPS. (A) Methylation, tissue
contribution and fragmentation fraction model performance on three-class
classification. Upper
panel shows the accuracy of each classifier, lower panel shows the actual and
predicted patient
status in LOU cross-validation analysis. (B) Heatmap showing the methylation
status of the
selected genomic region used for cancer-type prediction. (C) Gene Ontology
analysis using
Enrichr against NCI-Nature Pathway Interaction on the nearest genes of the
selected DMRs for
three class classification.
(0062]
FIG. 12: Schematic depiction of different patterns derived from C to T SNPs
and
methylated cytosines in target sequences before and after TAPS. In the diagram
OT means
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
11
Original Top, OB means Original Bottom, CTOT means Complimentary to Original
Top,
CTOB means Complimentary to Original Bottom.
DETAILED DESCRIPTION
10063] Recently, TET-assisted Pyridine Borane Sequencing (TAPS), a bisulfite-
free DNA
methylation sequencing method was developed, as described in International PCT
Appin.
PCT/US2019/012627, filed January 8, 2019, which claims priority to U.S.
Provisional Patent
Appin. Nos. 62/614,798, filed January 8, 2018; 62/660,523, filed April 20,
2018; and
62/771,409, filed November 26, 2018, each of which is incorporated herein by
reference in its
entirety. TAPS is based on the use of mild chemistry to detect DNA methylation
directly and
demonstrated improved sequence quality, mapping rate and coverage compared to
bisulfite
sequencing, while reducing sequencing cost by half The combination of direct
methylation
detection and the non-destructive nature of TAPS makes it useful not only for
DNA
methylation analysis, but also for simultaneous genetic analysis in cfDNA, as
described further
herein, which could enhance non-invasive cancer detection by liquid biopsies.
Embodiments
of the present disclosure include optimized TAPS for cfDNA (cfTAPS) to deliver
high-quality
and high-depth whole-genome methylome from as low as 10 ng cfDNA.
[00641 As described further herein, cfTAPS was applied to hepatocellular
carcinoma (HCC)
and pancreatic ductal adenocarcinoma (PDAC) cfDNA, two cancer types with
particularly poor
prognosis, mostly due to detection at an advanced disease stage. Non-invasive
methods for
early detection of PDAC and HCC are not available, which contributes to their
late diagnosis.
For decades. HCC detection has relied on liver ultrasound, combined with serum
a-fetoprotein
(AFP) measurements. However, these methods have low specificity and
sensitivity. There is
no blood test to detect or diagnose PDAC. Carbohydrate antigen 19-9 (CA19-9)
is used for
monitoring PDAC treatment and development, but its sensitivity and specificity
are too low to
diagnose or screen for PDAC. Therefore, novel approaches for PDAC and HCC
detection are
urgently needed.
(00651 Results provided herein demonstrate that the rich information from
cfTAPS enables
integrated multimodal epigenetic and genetic analysis of differential
methylation, tissue of
origin, and fragmentation profiles to accurately distinguish cfDNA samples
from patients with
HCC and PDAC from controls and patients with pre-cancerous inflammatory
conditions.
Additionally, results provided herein demonstrate the successful optimization
and application
of cfTAPS to characterize whole-genome base-resolution methylome in cfDNA from
HCC,
PDAC and non-cancer controls. Using just 10 ng cfDNA, cfTAPS libraries
demonstrated
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
12
greatly improved sequencing quality and depth compared to previous cfDNA WGBS.
Indeed,
using less cfDNA input than previous studies, cfDNA TAPS generated the most
comprehensive
cell-free methylation to date. The much higher yield of informative reads
allows a-TAPS
to extract more information from a given amount of cfDNA and makes it a viable
option for
large-scale cfDNA methylation studies. The use of TAPS resulted in superior
unique mapping
rates and deduplicated unique mapping rates as compared to other methods. in
some
embodiments, the unique mapping rate is at least 65% and/or the unique
deduplicated mapping
rate is at least 55%. In some embodiments, the unique mapping rate is at least
70% and/or the
unique deduplicated mapping rate is at least 60%. In some embodiments, the
unique mapping
rate is at least 75% and/or the unique deduplicated mapping rate is at least
65%. In some
embodiments, the unique mapping rate is at least 80% and/or the unique
deduplicated mapping
rate is at least 70%. In some embodiments, the unique mapping rate is at least
85% and/or the
unique deduplicated mapping rate is at least 72%. In some embodiments, the
unique mapping
rate is at least 90% and/or the unique deduplicated mapping rate is at least
75%.
[00661
The deep sequencing achieved by cfTAPS enables detailed analysis of the
cell-free
methylome and whole-genome discovery of methylation biomarkers for early
cancer detection.
While significant global hypomethylation was not observed, suggesting that the
fraction of
cfDNA derived from tumor cells is low (as corroborated by the lack of CNVs in
most cancer
patients included herein), results indicated that local methylation signals in
regulatory regions
such as enhancers and promoters contained cancer-specific information that
could accurately
distinguish HCC and PDAC from controls. This is particularly significant
considering the
inflammation-enriched real-world control group used in the patient cohort and
that the HCC
model disclosed herein can correctly identify all HCC and control patients
from a cfDNA
WGBS dataset as an independent validation.
14)0671 Another important advantage of cIDNA methylation for early cancer
detection is the
ability to determine tissue-of-origin information. Using currently available
public WGBS
tissue databases, a whole-genome tissue deconvolution of al-APS data was
performed, and
results indicated increased liver tumor contribution in HCC cfDNA and distinct
immune
signatures in cancer cfDNA. The tissue deconvolution itself can be used for
cancer detection.
Finally, since TAPS converts modified cytosine directly, it maximally retains
the underlying
genetic information compared to other approaches that convert unmodified
cytosines. In the
present disclosure, CNVs and fragmentation information was extracted from
cITAPS, the latter
of which is lost in cfDNA WGBS. Results further demonstrated that an
integrated approach
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
13
combining differential methylation, tissue of origin and fragmentation
profiles could improve
the model performance for multi-cancer detection.
100681 Section headings as used in this section and the entire
disclosure herein are merely
for organizational purposes and are not intended to be limiting.
1. Definitions
(00691 Unless otherwise defined, all technical and scientific
terms used herein have the
same meaning as commonly understood by one of ordinary skill in the art. In
case of conflict,
the present document, including definitions, will control. Preferred methods
and materials are
described below, although methods and materials similar or equivalent to those
described
herein can be used in practice or testing of the present disclosure. All
publications, patent
applications, patents and other references mentioned herein are incorporated
by reference in
their entirety. The materials, methods, and examples disclosed herein are
illustrative only and
not intended to be limiting.
100701 The terms "comprise(s),- "include(s)," "having," "has,-
"can,- "contain(s)," and
variants thereof, as used herein, are intended to be open-ended transitional
phrases, terms, or
words that do not preclude the possibility of additional acts or structures.
The singular forms
-a," -and" and -the" include plural references unless the context clearly
dictates otherwise.
The present disclosure also contemplates other embodiments "comprising,"
"consisting of' and
"consisting essentially of," the embodiments or elements piesented herein,
whether explicitly
set forth or not.
100711 For the recitation of numeric ranges herein, each
intervening number there between
with the same degree of precision is explicitly contemplated. For example, for
the range of 6-
9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the
range 6.0-7.0, the
number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are
explicitly contemplated.
1007211 For the recitation of numeric ranges herein, each
intervening number there between
with the same degree of precision is explicitly contemplated. For example, for
the range of 6-
9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the
range 6.0-7.0, the
number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are
explicitly contemplated.
100731 "Correlated to" as used herein refers to compared to.
100741 As used herein, "methylation" refers to cytosine
methylation at positions C5 or N4
of cytosine, the N6 position of adenine, or other types of nucleic acid
methylation. In vitro
amplified DNA is usually unmethylated because typical in vitro DNA
amplification methods
do not retain the methylation pattern of the amplification template. However,
"unmethylated
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
14
DNA" or "methylated DNA- can also refer to amplified DNA whose original
template was
unmethylated or methylated, respectively.
[00751
Accordingly, as used herein a "methylated nucleotide" or a "methylated
nucleotide
base- refers to the presence of a methyl moiety on a nucleotide base, where
the methyl moiety
is not present in a recognized typical nucleotide base. For example, cytosine
does not contain
a methyl moiety on its pyrimidine ring, hut 5-methylcytosine contains a methyl
moiety at
position 5 of its pyrimidine ring. Therefore, cytosine is not a methylated
nucleotide and 5-
methylcytosine is a methylated nucleotide.
[00761
As used herein, a -methylated nucleic acid molecule" refers to a nucleic
acid
molecule that contains one or more methylated nucleotides.
100771
As used herein, a -methylation state", "methylation profile", "methylation
status,"
and "methylation signature- of a nucleic acid molecule refers to the presence
of absence of one
or more methylated nucleotide bases in the nucleic acid molecule. For example,
a nucleic acid
molecule containing a methylated cytosine is considered methylated (e.g., the
methylation state
of the nucleic acid molecule is methylated). A nucleic acid molecule that does
not contain any
methylated nucleotides is considered unmethylated.
[00781
As used herein, "methylation frequency" or "methylation percent (%)" refer
to the
number of instances in which a molecule or locus is methylated relative to the
number of
instances the molecule or locus is unmethylated. Methylation state frequency
can be used to
describe a population of individuals or a sample from a single individual. For
example, a
nucleotide locus having a methylation state frequency of 50% is methylated in
50% of instances
and unmethylated in 50% of instances. Such a frequency can be used, for
example, to describe
the degree to which a nucleotide locus or nucleic acid region is methylated in
a population of
individuals or a collection of nucleic acids. Thus, when methylation in a
first population or
pool of nucleic acid molecules is different from methylation in a second
population or pool of
nucleic acid molecules, the methylation state frequency of the first
population or pool will be
different from the methylation state frequency of the second population or
pool. Such a
frequency also can be used, for example, to describe the degree to which a
nucleotide locus or
nucleic acid region is methylated in a single individual. For example. such a
frequency can be
used to describe the degree to which a group of cells from a tissue sample are
methylated or
unmethylated at a nucleotide locus or nucleic acid region.
100791 As used herein, the term "whole-genome cfDNA methylation signature"
refers to a
signature obtained through any method that looks across the entire breadth of
the genome for
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
candidate methylation markers, rather than a narrow few candidate sites (as
with an array based
technology).
[00801 As used herein, the term "copy number variation" (abbreviated CNV)
refers to a
circumstance in which the number of copies of a specific segment of DNA varies
among
different individuals' genomes.
[00811
As used herein, the term "unique mapping rate" refers to a metric used in
validation
of sequencing data, and specifically the percentage of sequencing reads that
map to exactly one
location within the reference genome. In some embodiments, the unique mapping
rate may be
calculated as the proportion of reads (e.g., with MAPQ>=1 using bwa align)
with defined
parameters (e.g., 500,120,1000,20) compared to total number of sequenced
reads.
100821
As used herein, the term "unique deduplicated mapping rate" refers to the
percentage
of deduplicated sequencing reads (after removing the duplicates) that map to
exactly one
location within the reference genome. In some preferred embodiments, the
unique deduplicated
mapping rate may be determined by calculating the proportion of properly
mapped reads after
removing PCR duplicates (e.g., with MarkDuplicates (Picard)) compared to total
number of
sequenced reads.
[00831
As used herein, the term "tissue deconvolution" refers to sorting sequenced
cfDNA
in a sample into its tissues of origin, and determining the relative
contribution from the tissues.
In some preferred embodiments, cf13NA methylation is compared to methylation
values in a
reference atlas (e.g., at DMRs). These methods preferably use a regression
method where
ciDNA origin proportions are regression coefficients.
100841
As used herein, the terms -patient" or -subject" refer to organisms to be
subject to
various tests provided by the technology. The term "subject- includes animals,
preferably
mammals, including humans. in a preferred embodiment, the subject is a
primate. in an even
more preferred embodiment, the subject is a human. Further with respect to
diagnostic
methods, a preferred subject is a vertebrate subject. A preferred vertebrate
is warm-blooded; a
preferred warm-blooded vertebrate is a mammal. A preferred mammal is most
preferably a
human. As used herein, the term "subject' includes both human and animal
subjects. Thus,
veterinary therapeutic uses are provided herein. As such, the present
technology provides for
the diagnosis of mammals such as humans, as well as those mammals of
importance due to
being endangered, such as Siberian tigers; of economic importance, such as
animals raised on
farms for consumption by humans; and/or animals of social importance to
humans, such as
animals kept as pets or in zoos. Examples of such animals include but are not
limited to:
carnivores such as cats and dogs; swine, including pigs, hogs, and wild boars;
ruminants and/or
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
16
ungulates such as cattle, oxen, sheep, giraffes, deer, goats, bison, and
camels; pinnipeds; and
horses.
2. TET-assisted Pyridine Borane Sequencing (TAPS)
(0085]
Embodiments of the present disclosure provide a bisulfite-free, base-
resolution
method for detecting 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC)
in a
sequence (TAPS), including for use with circulating cell free DNA. As
disclosed in in
International PCT Appin. PCT/US2019/012627 (filed January 8, 2019, which
claims priority
to U.S. Provisional Patent Appin. Nos. 62/614,798, filed January 8, 2018;
62/660,523, filed
April 20, 2018; and 62/771,409, filed November 26, 2018, each of which is
incorporated herein
by reference in its entirety), TAPS comprises the use of mild enzymatic and
chemical reactions
to detect 5mC and 5hmC directly and quantitatively at base-resolution without
affecting
unmodified cytosine. The present disclosure also provides methods to detect 5-
formylcytosine
(5fC) and 5-carboxylcytosine (5caC) at base resolution without affecting
unmodified cytosine.
Thus, the methods provided herein provide mapping of 5mC, 5hmC, 5fC and 5caC
and
overcome the disadvantages of previous methods such as bisulfite sequencing.
[00861
114-ethods for Identjfting 5mC. In some embodiments, the methods of the
present
disclosure include identifying 5mC in a DNA sample (targeted DNA or whole-
genome), and
providing a quantitative measure for the frequency of the 5mC modification at
each location
where the modification was identified in the DNA. In some embodiments, the
percentages of
the T at each transition location provide a quantitative level of 5mC at each
location in the
DNA. In accordance with these embodiments, methods for identifying 5mC can
include the
use of a blocking group. In other embodiments, methods for identifying 5mC do
not require
the use of a blocking group (e.g., cfTAPS described further below).
100871 When a blocking group is used to identify 5mC in a DNA (e.g., cfDNA)
without
including 5hmC, the 5hmC in the sample is blocked so that it is not subject to
conversion to
5caC and/or 5fC. In some embodiments, the 5hmC in the sample DNA are rendered
non-
reactive to the subsequent steps by adding a blocking group to the 5hmC. In
one embodiment,
the blocking group is a sugar, including a modified sugar, for example glucose
or 6-azide-
glucose (6-azido-6-deoxy-D-glucose). The sugar blocking group can be added to
the
hydroxymethyl group of 5hmC by contacting the DNA sample with uridine
diphosphate
(UDP)-sugar in the presence of one or more glucosyltransferase enzymes. In
some
embodiments, the glucosyltransferase is 14 bacteriophage 13-
glucosyltransferase (I3GT), T4
bacteriophage a-glucosyltransferase (aGT), and derivatives and analogs
thereof. fiGT is an
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
17
enzyme that catalyzes a chemical reaction in which a beta-D-glucosyl (glucose)
residue is
transferred from UDP-glucose to a 5-hy droxymethylcytosine residue in a
nucleic acid.
[00881 Methods for Identifying 5hmC. In some embodiments, the methods of the
present
disclosure include identifying 5mC or 5hmC in a DNA sample (targeted DNA or
whole-
genome). In some embodiments, the method provides a quantitative measure for
the frequency
the of 5mC or 5hmC modifications at each location where the modifications were
identified in
the DNA. In some embodiments, the percentages of the T at each transition
location provide a
quantitative level of 5mC or 5hmC at each location in the DNA. In accordance
with these
embodiments, the method for identifying 5mC or 5hmC provides the location of
5mC and
5hmC, but does not distinguish between the two cytosine modifications. Rather,
both 5mC and
5hmC are converted to DHU. The presence of DHU can be detected directly, or
the modified
DNA can be replicated by known methods where the DHU is converted to T. In
some
embodiments, methods for identifying 5hmC include the use of a blocking group.
In other
embodiments, methods for identifying 5hmC do not require the use of a blocking
group (e.g.,
cfTAPS described further below).
[0089i Methods for Identifying 5mC and/or Identifying 5hmC. The present
disclosure
provides a method for identifying 5mC and identifying 5hmC in a DNA (e.g.,
cfDNA) by
performing the method for identifying 5mC on a first DNA sample, and
performing the method
for identifying 5mC or 5hmC on a second DNA sample. In some embodiments, the
first and
second DNA samples are derived from the same DNA sample. For example, the
first and
second samples may be separate aliquots taken from a sample comprising DNA to
be analyzed
(e.g., cfDNA).
[00901 Because the 5mC and 5hmC (that is not blocked) are converted to 5fC and
5caC
before conversion to DHU, any existing 5fC and 5caC in the DNA sample will be
detected as
5mC and/or 5hmC. However, given the extremely low levels of 5fC and 5caC in
gcnomic DNA
under normal conditions, this will often be acceptable when analyzing
methylation and
hydroxymethylation in a DNA sample. The 5fC and 5caC signals can be eliminated
by
protecting the 5fC and 5caC from conversion to DHU by, for example,
hydroxylamine
conjugation and EDC coupling, respectively. In accordance with these
embodiments, the
method identifies the locations and percentages of 5hmC in the DNA through the
comparison
of 5mC locations and percentages with the locations and percentages of 5mC or
5hmC
(together). Alternatively, the location and frequency of 5hmC modifications in
a DNA can be
measured directly.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
18
[00911 In some embodiments, the step of converting the 5hmC to 5fC comprises
oxidizing
the 5hmC to 5fC by contacting the DNA with, for example, potassium
perruthenate (KRu04)
(as described in Science. 2012, 33, 934-937 and W02013017853, incorporated
herein by
reference); or Cu(II)/TEMPO (copper(II) perchlorate and 2,2,6,6-
tetramethylpiperidine-1-oxyl
(TEMPO)) (as described in Chem. Commun., 2017,53, 5756-5759 and W02017039002,
incorporated herein by reference). The 5fC in the DNA sample is then converted
to DHIJ by
the methods disclosed herein (e.g., by the borane reaction).
[0092i In some embodiments, identifying 5fC and/or 5caC provides the location
of 5fC
and/or 5caC, but does not distinguish between these two cytosine
modifications. Rather, both
5fC and 5caC are converted to DHU, which is detected by the methods described
herein.
100931 Methods for Identifying 5caC In some embodiments, the method includes
identifying 5caC in a DNA sample (targeted DNA or whole-genome), and provides
a
quantitative measure for the frequency the of 5caC modification at each
location where the
modification was identified in the DNA. In some embodiments, the percentages
of the T at
each transition location provide a quantitative level of 5caC at each location
in the DNA. In
accordance with these embodiments, methods for identifying 5caC can include
the use of a
blocking group. In other embodiments, methods for identifying 5caC do not
require the use of
a blocking group (e.g., cfTAPS described further below).
(0094] In some embodiments, when the 5fC is blocked (and 5mC and 5hmC are not
converted to DHU), the identification of 5caC in the DNA can occur. In some
embodiments,
adding a blocking group to the 5fC in the DNA sample comprises contacting the
DNA with an
aldehyde reactive compound including, for example, hydroxylamine derivatives,
hydrazine
derivatives, and hyrazide derivatives. Hydroxylamine derivatives include
ashydroxylamine;
hydroxyl amine hydrochloride; hydroxyl ammonium acid sulfate; hydroxyl amine
phosphate; 0-
methylhydroxylamine; -hexylhy droxylamine; 0-penty lhy droxyl
amine ; 0-
benzylhydroxylamine; and particularly, 0-ethylhydroxylamine (EtONH2), 0-
alkylated or 0-
arylated hydroxylamine, acid or salts thereof Hydrazine derivatives include N-
alkylhydrazine,
N-arylhydrazine, N- benzylhydrazine, N,N-dialkylhydrazine, N,N-
diarylhydrazine, N,N-
dibenzylhydrazine, N,N-alkylbenzylhydrazine, N,N- ary lb enzylhy drazine, and
N,N-
alkylarylhydrazine.
Hydrazide derivatives include -toluenesulfonylhydrazide, N-
acylhydrazide, N,N-alkylacylhydrazide, N,N-benzylacylhydrazide, N,N-
arylacylhydrazide, N-
s ulfony lhy drazi de, N,N-alkylsulfonylhy drazi de, N,N-benzyls ulfony lhy
drazi de, and N,N-
aryls ulfonylhy drazi de.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
19
[00951 Methods for Identifying 5fC. In some embodiments, the method includes
identifying
5fC in a DNA sample (targeted DNA or whole-genome), and provides a
quantitative measure
for the frequency the of 5fC modification at each location where the
modification was identified
in the DNA. In some embodiments, the percentages of the T at each transition
location provide
a quantitative level of 5fC at each location in the DNA. In accordance with
these embodiments,
methods for identifying 5fC can include the use of a blocking group. In other
embodiments,
methods for identifying 5fC do not require the use of a blocking group (e.g.,
cfTAPS described
further below).
[00961 In some embodiments, adding a blocking group to the 5caC in the DNA
sample can
be accomplished by (i) contacting the DNA sample with a coupling agent, for
example a
carboxylic acid derivatization reagent like carbodiimide derivatives such as 1-
ethy1-3-(3-
dimethylaminopropyl)carbodiimide (EDC) or N,N'-dicyclohexylcarbodiimide (DCC),
and (ii)
contacting the DNA sample with an amine, hydrazine or hydroxylamine compound.
Thus, for
example, 5caC can be blocked by treating the DNA sample with EDC and then
benzylamine,
ethylamine, or another amine to form an amide that blocks 5caC from conversion
to DHU (e.g.,
by pic-BH3).
3. TAPS for cfDNA (cfTAPS)
[00971 The present disclosure provides optimized TAPS for cfDNA (cfTAPS) to
provide
high-quality and high-depth whole-genome cell-free methylomes. As described
further below,
in one embodiment of the present disclosure, cfTAPS was applied to 85 cfDNA
samples from
patients with hepatocellular carcinoma (HCC) or pancreatic ductal
adenocarcinoma (PDAC)
and non-cancer controls. From just 10 ng of cfDNA (1-3 mL of plasma), the most
comprehensive cfDNA methylome to date was generated. The results provided
herein
demonstrated that cfTAPS provides multimodal information about cfDNA
characteristics,
including DNA methylation, tissue of origin, and DNA fragmentation. Integrated
analysis of
these epigenetic and genetic features enables accurate identification of early
HCC and PDAC.
Because the methods of the present disclosure utilize mild enzymatic and
chemical reactions
that avoid the substantial degradation of nucleic acids associated with
methods like bisulfite
sequencing, the methods of the present disclosure are useful in analysis of
low-input samples,
such as circulating cell-free DNA and in single-cell analysis.
[0098i
In accordance with these embodiments, the present disclosure provides a
method of
obtaining a methylation signature. In some embodiments, the method includes
isolating cell
free DNA (cfDNA) from a sample; preparing a sequencing library comprising the
cfDNA; and
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
performing TET-assisted Pyridine Borane Sequencing (TAPS) on the sequencing
library to
obtain a methylation signature of the cfDNA. In some embodiments, the
methylation signature
is a whole-genome methylation signature.
100991
In some embodiments, preparing the sequencing library comprises ligating
sequencing adapters to the isolated cf-DNA to facilitate performing a
sequencing reaction. In
some embodiments, carrier nucleic acids or a mix of carrier nucleic acids
(e.g., DNA) are added
to the sequencing library prior to performing TAPS. Carrier nucleic acids can
be any specific
or non-specific DNA molecules (or nucleic acid derivatives thereof) that
enhance one or more
aspects of cfDNA recovery from a sample. In some embodiments, carrier DNA
comprises a
DNA molecule having a specific sequence; and in other embodiments, carrier DNA
comprises
a mix of DNA molecules having different sequences. In some embodiments,
carrier DNA can
include DNA with the following sequence, including any fragments and/or
derivatives thereof:
AGGCAACTTTATGCCCATGCAACAGAAACTATAAAAAATACAGAGAATGAAAAG
AAACAGATAGATTTTTTAGTTCTTTAGGC C C GTAGTC TGC AAATC CTTTTATGATT
TTCTATCAAACAAAAGAGGAAAATAGACCAGTTGCAATCCAAACGAGAGTCTAA
TAGAATGAGGTC GAAAAGTAAAT C GC GC GGGTTTGTTAC T GATAAAGCAGGC AA
GAC CTAAAATGTGTAAAG G G CAAAGTGTATACTTTG G C GT C AC C C C TTACATATT
TTAGGTC TTTTTTTATTGTGC GTAACTAACTT GC CATCTTCAAACAGGAGGGCTGG
AAGAAGCAGAC C GC TAAC ACAGTACATAAAAAAGGAGAC ATGAACGATGAACA
TCAAAAAGTTTGCAAAACAAGCAACAGTATTAACCTTTACTACCGCACTGCTGGC
AGGAGGCGCAACTCAAGCGTTTGCGAAAGAAACGAACCAAAAGCCATATAAGG
AAACATAC GGCATTTC CC ATATTACAC GC CAT GATATGC TGCAAATC C C TGAAC A
GC AAAAAAATGAAAAATATAAAGTTC C TGAGTTC GATTC GTC C AC AATTAAAAA
TATCTCTTCTGC AA A AGGCCTGGACGTTTGGGAC AGCTGGCC ATT AC A A A AC ACT
GAC GGCACTGTC GC AAAC TATC AC GGCTAC CAC ATC GTCTTTGCATTAGCCGGAG
ATC C TAAAAATGC GGATGACACATC GATTTACATGTTC TATCAAAAAGTC GGC GA
AAC TTC TATTGACAGC TGGAAAAAC GC TGGC C GC GTC TTTAAAGACAGCGAC AA
ATTCGATGCAAATGATTCTATCCTAAAAGACCAAAC AC AAG AATG GTC AG GTTC
AGCCACATTTACATCTGACGGAAAAATCCGTTTATTCTACACTGATTTCTCCGGT
AAACATTACGGC AAACAAAC AC TGAC AAC TGC AC AAGTTAAC GTATC AGC ATC A
GACAGCTCTTTGAACATCAACGGTGTAGAGGATTATAAATCAATCTTTGACGGTG
AC GGAAAAAC GTATCAAAATGTACAGCAGTTCATC GATGAAGGCAACTAC AGC T
C AGGCGAC AAC CATAC GCT GAGAGATC C TC ACTAC GTAGAAGATAAAGGC C AC A
AATAC TTAGTATTTGAAGC AAAC AC TGGAAC TGAAGATGGC TAC C AAGGC GAAG
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
21
AATCTTTATTTAACAAAGCATACTATGGCAAAAGCACATCATTCTTCC GTCAAGA
AAGTC AAAAAC TTC TGCAAAGC GATAAAAAAC GC AC GGC TGAGTTAGCAAAC GG
CGCTCTC GGTATGATTGAGCTAAACGATGATTACACACTGAAAAAAGTGATGAA
AC CGC TGATT GCATCTAACACAGTAACAGATGAAATTGAAC GC GC GAAC GTCTTT
AAAATGAAC GGC AAATGGTAC C TGTTC AC TGAC TC C C GC GGATCAAAAATGACG
ATTGACGGCATTACCiTCTAACGATATTTACATGCTTGGTTATGTTTCTAATTCTTT
AACTGGCCCATAC AAGC C GC TGAAC AAAAC TGGC C TTGTGTTAAAAATGGATC TT
GATCCTAACGATGTAACCTTTACTTACTCACACTTCGCTGTACCTCAAGCGAAAG
GAAACAATGTCGTGATTACAAGCTATATGACAAACAGAGGATTCTACGCAGACA
AACAATCAACGTTTGCGCCTAGCTTCCTGCTGAACATCAAAGGCAAGAAAACAT
CTGTTGTCAAAGACAGCATCCTTGAACAAGGACAATTAACAGTTAACAAATAAA
AACGCAAAAGAAAATGCCGATATC CTATTGGCATTGACGGTCTCCAGTAAAGGT
GGATACGGATCCGAATTCGAGCTCCGTCGACAAGCTTGC GGCCGCACTCGAGCA
C CAC CAC CAC C AC CAC TGAGATC C GG C TGC TAAC AAAGC C C GAAAGGAAGCTGA
GTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGG (SEQ ID NO: 1).
[01001
In some embodiments, the use of carrier DNA results in higher library
yields. As
would be recognized by one of ordinary skill in the art based on the present
disclosure, carrier
DNA can be obtained by any means known in the art, including but not limited
to, PCR
amplification from a vector or plasmid template using one or more primers. In
some
embodiments, at least 1 ng of carrier DNA can be used. In some embodiments, at
least 10 ng
of carrier DNA can be used. In some embodiments, at least 25 ng of carrier DNA
can be used.
In some embodiments, at least 50 ng of carrier DNA can be used. In some
embodiments, at
least 100 ng of carrier DNA can be used. In some embodiments, at least 150 ng
of carrier DNA
can be used. in some embodiments, at least 200 ng of carrier DNA can be used.
In some
embodiments, at least 250 ng of carrier DNA can be used. In some embodiments,
at least 500
ng of carrier DNA can be used. In some embodiments, about 1 ng to about 500 ng
of carrier
DNA can be used. In some embodiments, about 1 ng to about 500 ng of carrier
DNA can be
used. In some embodiments, about 50 ng to about 250 ng of carrier DNA can be
used. In some
embodiments, about 75 ng to about 150 ng of carrier DNA can be used. In some
embodiments,
about 50 ng to about 150 ng of carrier DNA can be used. In some embodiments,
about 75 ng
to about 125 ng of carrier DNA can be used.
101911
In some embodiments, and as described herein, the method further comprises
identifying at least one methylation biomarker from the cf1DNA whole-genome
methylation
signature, and determining whether the methylation biomarker is indicative of
cancer. In some
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
22
embodiments, the methylation biomarker comprises a differentially methylated
region (DMR).
In some embodiments, the method further comprises classifying the sample based
on the DMR
as compared to a reference DMR. In some embodiments, the reference DMR
corresponds to a
non-cancerous control, or a cancerous control.
101021 In some embodiments, and as described herein, the method further
comprises
identifying at least one methylation biomarker from the cfDNA whole-genome
methylation
signature, and determining a tissue-of-origin corresponding to the methylation
biomarker. In
some embodiments, the method further comprises classifying the sample based on
the tissue-
of-origin biomarker.
101031 In some embodiments, and as described herein, the method further
comprises
identifying a DNA fragmentation profile, and determining whether the
fragmentation profile is
indicative of cancer. In accordance with these embodiments, DNA fragmentation
profile can
be determined from cfTAPS whole genome sequencing data (e.g., read pair
alignment
positions). In some preferred embodiments, sequenced reads from ciTAPS are
first aligned to
a reference genome. The length of cfDNA fragment is then extracted from
alignment files
produced from the sequencing data. The proportion in 10-bp intervals of cfDNA
fragments is
used as the fragmentation profile of the cell free DNA.
101041
In some embodiments, the method further comprises identifying at least one
sequence variant from the cfDNA, and determining whether the sequence variant
is indicative
of cancer. For example, in some embodiments, cITAPS can also differentiate
methylation from
C-to-T genetic variants or single nucleotide polymorphisms (SNPs), and
therefore, can be used
to detect genetic variants. In some embodiments, methylations and C-to-T SNPs
can result in
different patterns in cfTAPS. For example, methylations can result in T/G
reads in an original
top strand/original bottom strand, and A/C reads in strands complementary to
these. In some
embodiments, C-to-T SNPs can result in T/A reads in an original top
strand/original bottom
strand and strands complementary to these. These different patterns are
illustrated in FIG. 12.
This further increases the utility of cfTAPS in providing both methylation
information and
genetic variants, and therefore mutations, in one experiment and sequencing
run. This ability
of the cfTAPS methods disclosed herein provides integration of genomic
analysis with
epigenetic analysis, and a substantial reduction of sequencing cost by
eliminating the need to
perform standard whole genome sequencing (WGS).
101051
In accordance with the above embodiments, methods of the present disclosure
include the use of cfTAPS to generate information pertaining to methylation
signatures,
methylation biomarkers, DNA fragment profiles, DNA sequence information (e.g.,
variants),
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
23
and tissue-of-origin information in a single experiment to diagnose/detect
cancer in a subject.
As would be recognized by one of ordinary skill in the art based on the
present disclosure,
cfTAPS as disclosed herein can be used to generate any combination of
methylation signatures,
methylation biomarkers, DNA fragment profiles, DNA sequence information (e.g.,
variants),
and tissue-of-origin information to diagnose/detect cancer in a subject. In
some embodiments,
a methylation signature can be obtained, and one or more of a methylation
biomarker, a DNA
fragment profile, DNA sequence information (e.g., variants), and tissue-of-
origin information
can also be obtained and used to diagnose/detect cancer in a subject. In some
embodiments,
the methylation status of a biomarker can be obtained, and one or more of a
methylation
signature, a DNA fragment profile, DNA sequence information (e.g., variants),
and tissue-of-
origin information can also be obtained and used to diagnose/detect cancer in
a subject. In some
embodiments, a DNA fragmentation profile can be obtained, and one or more of a
methylation
signature, a methylation biomarker, DNA sequence information (e.g., variants),
and tissue-of-
origin information can also be obtained and used to diagnose/detect cancer in
a subject. In some
embodiments, a DNA sequence variant can be identified, and one or more of a
methylation
signature, a methylation biomarker, a DNA fragment profile, and tissue-of-
origin information
can also be obtained and used to diagnose/detect cancer in a subject. In some
embodiments,
tissue-of-origin information can be obtained (e.g., from a whole genome cfDNA
methylation
signature), and one or more of the methylation signature, a methylation
biomarker, a DNA
fragment profile, and DNA sequence information (e.g., variants), can also be
obtained and used
to diagnose/detect cancer in a subject.
101061
Accordingly, in some preferred embodiments, the present invention provides
multimodal methods of analyzing cfDNA in a patient sample comprising:
isolating cfDNA
from a patient sample; converting 5mC and/or 5hmC residues in the sample to
DHU residues
to provide a modified cfDNA sample; sequencing the modified cfDNA sample to
identify
methylated regions in the sample, wherein a cytosine (C) to thymine (T)
transition or a cytosine
(C) to DHU transition in the modified cfDNA sample as compared to an
unmodified reference
cfDNA provides the location of either a 5mC or 5hmC in the cfDNA; and
performing one or
more additional analytical steps on the modified cfDNA selected from the group
consisting of:
a) determining copy number variation of one or more targets in the modified
cfDNA
sample;
b) determining the tissue of origin or one or more targets in the modified
cliDNA
sample;
c) determining the fragmentation profile of the modified cfDNA sample; and
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
24
d) identifying one or more single nucleotide mutations in the modified cfDNA
sample.
101071
In some preferred embodiments, the one or more additional step is step a.
In some
preferred embodiments, the one or more additional step is step b. In some
preferred
embodiments, the one or more additional step is step c. In some preferred
embodiments, the
one or more additional step is step d_
101081
In some preferred embodiments, the one or more additional steps is steps a
and b. In
some preferred embodiments, the one or more additional steps is step a and c.
In some preferred
embodiments, the one or more additional steps is steps a and d. In some
preferred embodiments,
the one or more additional steps is steps b and c. In some preferred
embodiments, the one or
more additional steps is steps b and d. In some preferred embodiments, the one
or more
additional steps is steps c and d.
101091
In some preferred embodiments, the one or more additional steps is steps a,
b and c.
In some preferred embodiments, the one or more additional steps is steps a, b
and d. In some
preferred embodiments, the one or more additional steps is steps b, c and d.
[0110i
In some preferred embodiments, the one or more additional steps are all of
steps a,
b, c and d.
101111 In some embodiments, an unmodified reference cfDNA to be compared to a
modified cfDNA sample may comprise any unmodified reference cfDNA, including
for
instance, a publicly available reference cfDNA or an unmodified control sample
from the
patient.
101121 In some embodiments, performing TAPS on the sequencing library to
obtain the
whole-genome methylation signature comprises identifying 5mC modifications in
the cfDNA
and providing a quantitative measure for frequency of the 5mC modifications.
In some
embodiments, performing TAPS on the sequencing library to obtain the whole-
genome
methylation signature comprises identifying 5hmC modifications in the cfDNA
and providing
a quantitative measure for frequency of the 5hmC modifications. In some
embodiments,
performing TAPS on the sequencing library to obtain the whole-genome
methylation signature
comprises identifying 5caC modifications in the cfDNA and providing a
quantitative measure
for frequency of the 5caC modifications. In some embodiments, performing TAPS
on the
sequencing library to obtain the whole-genome methylation signature comprises
identifying
5fC modifications in the cIDNA and providing a quantitative measure for
frequency of the 5fC
modifications.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
[0113i
As would be recognized by one of ordinary skill in the art based on the
present
disclosure, the methods described herein (e.g., cfTAPS) can be used to
diagnose/detect any
type of cancer. Types of cancers that can be detected/diagnosed using the
methods of the
present disclosure include, but are not limited to, lung cancer, melanoma,
colon cancer,
colorectal cancer, neuroblastoma, breast cancer, prostate cancer, renal cell
cancer. transitional
cell carcinoma, cholangi carcinoma, brain cancer, non-small cell lung cancer,
pancreatic
cancer, liver cancer, gastric carcinoma, bladder cancer, esophageal cancer,
mesothelioma,
thyroid cancer, head and neck cancer, osteosarcoma, hepatocellular carcinoma,
carcinoma of
unknown primary, ovarian carcinoma, endometrial carcinoma, glioblastoma,
Hodgkin
lymphoma and non-Hodgkin lymphomas. In some embodiments, types of cancers or
metastasizing forms of cancers that can be detected/diagnosed by the methods
of the present
disclosure include, but are not limited to, carcinoma, sarcoma, lymphoma, germ
cell tumor and
blastoma. In some embodiments, the cancer is invasive and/or metastatic cancer
(e.g., stage II
cancer, stage III cancer or stage IV cancer). In some embodiments, the cancer
is an early stage
cancer (e.g., stage 0 cancer, stage I cancer), and/or is not invasive and/or
metastatic cancer.
[01141
In some embodiments, the methods of the present disclosure (e.g., cfTAPS)
can be
used to determine whether a subject has hepatocellular carcinoma (HCC) or
pancreatic ductal
adenocarcinoma (PDAC). In some embodiments, the method includes determining
whether a
subject has early stage hepatocellular carcinoma (HCC) or early stage
pancreatic ductal
adenocarcinoma (PDAC).
[01151
In accordance with these embodiments, the present disclosure provides
methods for
identifying the location of one or more of 5mC, 5hmC, 5caC and/or 5fC in a
nucleic acid
quantitatively with base-resolution without affecting the unmodified cytosine.
In some
embodiments, the nucleic acid is DNA. In some embodiments, the DNA is cfDNA
(e.g.,
circulating cfDNA). In some embodiments, the nucleic acid is RNA. In some
embodiments, a
nucleic acid sample comprises a target nucleic acid that is DNA or a target
nucleic acid that is
RNA. In some embodiments, the methods are applied to a whole genome, and not
limited to a
specific target nucleic acid.
[01161
The nucleic acid may be any nucleic acid having cytosine modifications
(i.e., 5mC,
5hmC, 5fC, and/or 5 caC). The nucleic acid can be a single nucleic acid
molecule in the sample,
or may be the entire population of nucleic acid molecules in a sample (whole
genome or a
subset thereof). The nucleic acid can be the native nucleic acid from the
source (e.g., cells,
tissue samples, etc.) or can pre-converted into a high-throughput sequencing-
ready form, for
example by fragmentation, repair and ligation with adapters for sequencing.
Thus, nucleic acids
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
26
can comprise a plurality of nucleic acid sequences such that the methods
described herein may
be used to generate a library of target nucleic acid sequences that can be
analyzed individually
(e.g., by determining the sequence of individual targets) or in a group (e.g.,
by high-throughput
or next generation sequencing methods).
101171 A nucleic acid sample can be obtained from an organism from the Monera
(bacteria),
Protista, Fungi, Plantae, and An i mal i a Kingdoms. Nucleic acid samples may
he obtained from
a from a patient or subject, from an environmental sample, or from an organism
of interest. In
some embodiments, the sample is obtained from a human subject/patient,
including but not
limited to, a human with cancer or a human suspected of having cancer. In some
embodiments,
the sample is obtained from a tissue or cell from a human (e.g., obtained from
a biopsy),
including a tissue or cell that is cancerous or suspected of being cancerous.
In some
embodiments, the nucleic acid sample is extracted or derived from a cell or
collection of cells,
a bodily fluid, a tissue sample, an organ, and an organelle. In some
embodiments, the nucleic
acid sample is obtained from a bodily fluid, including but not limited to,
blood (plasma, serum,
whole blood), urine, feces/fecal fluid, semen (seminal fluid), vaginal
secretions, cerebrospinal
fluid (CSF), ascitic fluid, synovial fluid, pleural fluid (pleural lavage),
pericardial fluid,
peritoneal fluid, amniotic fluid, saliva, nasal fluid, otic fluid, gastric
fluid, breast milk, and any
other bodily fluid comprising cfDNA, as well as cell culture supernatants. In
some
embodiments, the sample is obtained from a bodily fluid that is cancerous or
suspected of being
cancerous. Because the methods of the present disclosure utilize mild
enzymatic and chemical
reactions that avoid the substantial degradation of nucleic acids associated
with methods like
bisulfite sequencing, the methods of the present disclosure are useful in
analysis of low-input
samples, such as circulating cell-free DNA and in single-cell analysis.
101181 in some embodiments, the DNA sample comprises pi cogram quantities of
DNA. In
some embodiments, the DNA sample comprises from about 1 pg to about 900 pg
DNA, from
about 1 pg to about 500 pg DNA, from about 1 pg to about 100 pg DNA, from
about 1 pg to
about 50 pg DNA, or from about 1 to about 10 pg DNA. In some embodiments, the
DNA
sample comprises less than about 200 pg, less than about 100 pg DNA, less than
about 50 pg
DNA, less than about 20 pg DNA, less than about 15 pg DNA, less than about 10
pg DNA, or
less than about 5 pg DNA.
101191 In some embodiments, the DNA sample comprises nanogram quantities of
DNA.
The sample DNA for use in the methods of the present disclosure can be any
quantity including,
but not limited to, DNA from a single cell or bulk DNA samples. In some
embodiments, the
methods can be performed on a DNA sample comprising from about 1 to about 500
ng of DNA,
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
27
from about 1 to about 200 ng of DNA, from about 1 to about 100 ng of DNA, from
about 1 to
about 50 ng of DNA, from about 1 to about 10 ng of DNA, from about 2 to about
5 ng of DNA.
In some embodiments, the DNA sample comprises less than about 100 ng of DNA,
less than
about 50 ng of DNA, less than 40 ng of DNA, less than 30 ng of DNA, less than
20 ng of DNA,
less than 15 ng of DNA, less than 5 ng of DNA, and less than 2 ng of DNA. In
some
embodiments, the DNA sample comprises microgram quantities of DNA.
101201 A DNA sample used in the methods described herein may be from any
source
including, for example a bodily fluid, tissue sample, organ, organelle, cell
or collection of cells.
In some embodiments, the DNA sample is obtained from a human subject/patient,
including
but not limited to, a human with cancer or a human suspected of having cancer.
In some
embodiments, the DNA sample is obtained from a tissue or cell from a human
(e.g., obtained
from a biopsy), including a tissue or cell that is cancerous or suspected of
being cancerous. In
some embodiments, the DNA sample is extracted or derived from a cell or
collection of cells,
a bodily fluid, a tissue sample, an organ, and an organelle. In some
embodiments, the DNA
sample is obtained from a bodily fluid, including but not limited to, blood
(plasma, serum,
whole blood), urine, feces/fecal fluid, semen (seminal fluid), vaginal
secretions, cerebrospinal
fluid (CSF), ascitic fluid, synovial fluid, pleural fluid (pleural lavage),
pericardial fluid,
peritoneal fluid, amniotic fluid, saliva, nasal fluid, otic fluid, gastric
fluid, breast milk, and any
other bodily fluid comprising cfDNA, as well as cell culture supernatants. In
some
embodiments, the DNA sample is obtained from a bodily fluid that is cancerous
or suspected
of being cancerous. In some embodiments, the DNA sample is circulating cell-
free DNA (cell-
free DNA or cfDNA), which is DNA found in the blood and is not present within
a cell. As
would be recognized by one of ordinary skill in the art based on the present
disclosure, cfDNA
can be isolated from a bodily fluid using methods known in the art. Commercial
kits are
available for isolation of cfDNA including, for example, the Circulating
Nucleic Acid Kit
(Qiagen). The DNA sample may result from an enrichment step, including, but is
not limited
to
antibody immunoprecipitation, chromatin immunoprecipitati on, restriction
enzyme
digestion-based enrichment, hybridization-based enrichment, or chemical
labeling-based
enrichment.
[01211 The DNA may be any DNA having cytosine modifications (i.e., 5mC, 5hmC,
5fC,
and/or 5caC) including, but not limited to, DNA fragments and/or genomic DNA.
The DNA
can be a single DNA molecule in the sample, or may be the entire population of
DNA molecules
in a sample (whole genome or a subset thereof). The DNA can be the native DNA
from the
source or pre-converted into a high-throughput sequencing-ready form, for
example by
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
28
fragmentation, repair and ligation with adapters for sequencing. Thus, DNA can
comprise a
plurality of DNA sequences such that the methods described herein may be used
to generate a
library of target DNA sequences that can be analyzed individually (e.g., by
determining the
sequence of individual targets) or in a group (e.g., by high-throughput or
next generation
sequencing methods).
[0122j
in accordance with these embodiments, the methods of th e present
disclosure include
the step of converting the 5mC and 5hmC (or just the 5mC if the 5hmC is
blocked) to 5caC
and/or 5fC. In some embodiments, this step comprises contacting the DNA or RNA
sample
with a ten eleven translocation (TET) enzyme. The TET enzymes are a family of
enzymes that
catalyze the transfer of an oxygen molecule to the C5 methyl group on 5mC
resulting in the
formation of 5-hydroxymethylcytosine (5hmC). TET further catalyzes the
oxidation of 5hmC
to 5fC and the oxidation of 5fC to form 5caC. TET enzymes useful in the
methods of the present
disclosure include one or more of human TETI, TET2, and TET3, murine TETI,
TET2, and
TET3; Naegleria TET (NgTET); Coprinopsis cinerea (CcTET); the catalytic domain
of mouse
TETI (mTET1CD); and derivatives or analogues thereof. In some embodiments, the
TET
enzyme is NgTET. In some embodiments, the TET enzyme is human TETI (hTET1). In
some
embodiments, the TET enzyme is mTET1CD.
14)1231
Methods of the present disclosure can also include the step of converting
the 5caC
and/or 5fC in a nucleic acid sample to DHU. In some embodiments, this step
comprises
contacting the DNA or RNA sample with a reducing agent including, for example,
a borane
reducing agent such as pyridine borane, 2-picoline borane (pic-BH3), borane,
sodium
borohydride, sodium cyanoborohydride, and sodium triacetoxvborohydride. In
some
embodiments, the reducing agent is pyridine borane and/or pic-BH3.
101241
The methods of the present disclosure can also include the step of
amplifying the
copy number of a modified nucleic acid by methods known in the art. When the
modified
nucleic acid is DNA, the copy number can be increased by, for example, PCR,
cloning, and
primer extension. The copy number of individual target DNAs can be amplified
by PCR using
primers specific for a particular target DNA sequence. Alternatively, a
plurality of different
modified target DNA sequences can be amplified by cloning into a DNA vector by
standard
techniques. In some embodiments, the copy number of a plurality of different
modified target
DNA sequences is increased by PCR to generate a library for next generation
sequencing
where, e.g., double-stranded adapter DNA has been previously ligated to the
sample DNA (or
to the modified sample DNA) and PCR is performed using primers complimentary
to the
adapter DNA.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
29
[01251 In some embodiments, the method comprises the step of detecting the
sequence of
the modified nucleic acid. The modified target DNA or RNA contains DHU at
positions where
one or more of 5mC, 5hmC, 5fC, and 5caC were present in the unmodified target
DNA or
RNA. DHU acts as a T in DNA replication and sequencing methods. Thus, the
cytosine
modifications can be detected by any direct or indirect method that identifies
a C to T transition
known in the art Such methods include sequencing methods such as Sanger
sequencing,
microarray, and next generation sequencing methods. The C to T transition can
also be detected
by restriction enzyme analysis where the C to T transition abolishes or
introduces a restriction
endonuclease recognition sequence.
101261
Embodiments of the present disclosure also provide kits for identification
of 5mC
and 5hmC in a DNA. Such kits comprise reagents for identification of 5mC and
5hmC by the
methods described herein. The kits may also contain the reagents for
identification of 5caC and
for the identification of 5fC by the methods described herein. In some
embodiments, the kit
comprises a TET enzyme, a borane reducing agent and instructions for
performing the method.
In some embodiments, the TET enzyme is TETI and the borane reducing agent is
selected from
one or more of the group consisting of pyridine borane, 2-picoline borane (pic-
BH3), borane,
sodium borohydride, sodium cyanoborohydride, and sodium triacetoxyborohydride.
In some
embodiments, the TETI enzyme is NgTet1 or murine Teti (e.g., mTet1CD) and the
borane
reducing agent is pyridine borane and/or pic-BH3.
101271 In some embodiments, the kit further comprises a 5hmC blocking group
and a
glucosyltransferase enzyme. In some embodiments, the blocking group added to
5hmC is a
sugar. In some embodiments, the sugar is a naturally-occurring sugar or a
modified sugar, for
example glucose or a modified glucose. In some embodiments, the blocking group
is added to
5hmC by contacting a nucleic acid sample with UDP linked to a sugar, for
example UDP-
glucose or UDP linked to a modified glucose in the presence of a
glucosyltransferase enzyme,
for example, T4 bacteriophage 13-glucosyltransferase (13GT) and T4
bacteriophage a-
glucosyltransferase (aGT) and derivatives and analogs thereof
[01281
In some embodiments, the kit further comprises an oxidizing agent selected
from
potassium perruthenate (KRu04) and/or Cu(II)/TEMPO (copper(II) perchlorate and
2,2,6,6-
tetramethylpiperidine-1-oxyl (TEMPO)). In some embodiments, the kit comprises
reagents for
blocking 5fC in the nucleic acid sample. In some embodiments, the kit
comprises an aldehyde
reactive compound including, for example, hydroxylamine derivatives, hydrazine
derivatives,
and hydrazide derivatives as described herein. In some embodiments, the kit
comprises
reagents for blocking 5caC as described herein. In some embodiments, the kit
comprises
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
reagents for isolating DNA or RNA. In some embodiments the kit comprises
reagents for
isolating low-input DNA from a sample, for example cfDNA from blood, plasma,
or serum.
14)1 29j
In some embodiments, the methods of the present disclosure include treating
a
patient (e.g., a patient with cancer, with early-stage cancer, or who is
suspected of having
cancer). In some embodiments, the methods includes determining a methylation
signature as
provided herein and administering a treatment to a patient based on the
results of determining
the methylation signature. The treatment can include administration of a
pharmaceutical
compound, a vaccine, performing a surgery, imaging the patient, and/or
performing another
test. In some embodiments, the methods of the present disclosure can be used
as part of clinical
screening, a method of prognosis assessment, a method of monitoring the
results of therapy, a
method to identify patients most likely to respond to a particular therapeutic
treatment, a
method of imaging a patient or subject, and a method for drug screening and
development.
161301
In some embodiments, methods of the present disclosure include diagnosing
cancer
in a subject. The terms "diagnosing" and -diagnosis" as used herein refer to
methods by which
the skilled artisan can estimate and even determine whether or not a subject
is suffering from
a given disease or condition or may develop a given disease or condition in
the future. The
skilled artisan often makes a diagnosis on the basis of one or more diagnostic
indicators, such
as for example a methylation biomarker and/or a methylation signature, which
is indicative of
the presence, severity, or absence of the condition (e.g., cancer).
1013 11
Along with diagnosis, clinical cancer prognosis relates to determining the
aggressiveness of the cancer and the likelihood of tumor recurrence to plan
the most effective
therapy. If a more accurate prognosis can be made or even a potential risk for
developing the
cancer can be assessed, appropriate therapy, and in some instances less severe
therapy for the
patient can be chosen. Assessment of a subject based on methylation signature
can be useful to
separate subjects with good prognosis and/or low risk of developing cancer who
will need no
therapy or limited therapy from those more likely to develop cancer or suffer
a recurrence of
cancer who might benefit from more intensive treatments. As such, "making a
diagnosis" or
"diagnosing", as used herein, is further inclusive of making a determination
of a risk of
developing cancer or determining a prognosis, which can provide for predicting
a clinical
outcome (with or without medical treatment), selecting an appropriate
treatment (or whether
treatment would be effective), or monitoring a current treatment and
potentially changing the
treatment, based on the identification and assessment of a methylation
signature, as disclosed
herein.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
31
[01321
In some embodiments, methods of the present disclosure include determining
whether to initiate or continue prophylaxis or treatment of a cancer in a
subject. In some
embodiments, the method comprises providing a series of biological samples
over a time period
from the subject: analyzing the series of biological samples to determine a
methylation
signature as disclosed herein in each of the biological samples; and comparing
any measurable
change in the methylation signatures in each of the biological samples Any
changes in the
methylation signatures over the time period can be used to predict risk of
developing cancer,
predict clinical outcome, determine whether to initiate or continue the
prophylaxis or therapy
of the cancer, and whether a current therapy is effectively treating the
cancer. For example, a
first time point can be selected prior to initiation of a treatment and a
second time point can be
selected at some time after initiation of the treatment. Methylation
signatures can be measured
in each of the samples taken from different time points and qualitative and/or
quantitative
differences noted. A change in the methylation signatures from the different
samples can be
correlated with risk for developing cancer, prognosis, determining treatment
efficacy, and/or
progression of the cancer in the subject. In some embodiments, the methods and
compositions
of the invention are for treatment or diagnosis of disease at an early stage,
for example, before
symptoms of the disease appear. In some embodiments, the methods and
compositions of the
invention are for treatment or diagnosis of disease at a clinical stage.
(0133]
Unless otherwise defined herein, scientific and technical terms used in
connection
with the present disclosure shall have the meanings that are commonly
understood by those of
ordinary skill in the art. For example, any nomenclatures used in connection
with,
and techniques of, cell and tissue culture, molecular biology, immunology,
microbiology,
genetics and protein and nucleic acid chemistry and hybridization described
herein are
those that are well known and commonly used in the art. The meaning and scope
of the terms
should be clear; in the event, however of any latent ambiguity, definitions
provided herein take
precedent over any dictionary or extrinsic definition. Further, unless
otherwise required by
context, singular terms shall include pluralities and plural terms shall
include the singular.
4. Materials and Methods
[0134j Experimental design. Whole blood samples from 30 non-cancer controls
were
obtained from John Radcliffe hospital (Ethical approvals IDs 16/YH/0247 and
18/WM/0237).
Pancreatitis blood samples from 8 patients were obtained from John Radcliffe
hospital. The
study was approved by Oxfordshire REC-A (10/H0604/51) and is registered on the
UK N1HR
portfolio as study number 10776. PDAC patients were consented for this study
via the Oxford
CA 03226747 2024- 1-23
WO 2023/007241
PCT/1B2022/000420
32
Radcliffe Biobank (09/H0606/5+5, project: 19/A177) and whole-blood samples
were collected
from 24 patients. Collection of plasma samples from 21 HCC and 4 cirrhosis
patients was REC
approved (Ethical approval 2/NE/0395, IRAS project ID:116370). No sample-size
calculations
were performed. Sample size was determined based on availability. PDAC, HCC,
pancreatitis
and cirrhosis samples were collected from subjects with clinically diagnosed
disease. Non-
cancer control samples were collected from individuals without cancer
diagnosis at the time of
sample collection or previous history of cancer.
[0135i
The main goal of the study was comprehensive, multidimensional
characterization
of cf1DNA in cancer and controls by whole-genome methylation sequencing using
TAPS.
CfDNA TAPS libraries were constructed and paired-end 150 bp sequenced on a
NovaSeq 6000
sequencer (Illumina). Technical details are described in the sections below.
Samples with 5mC
conversion below 90% calculated based on methylated lambda spike-in control
were excluded
from downstream analysis.
[01361 Collection and preparation of cIDNA samples. Blood was collected into
EDTA-
coated Vacutainers. Plasma was separated from collected blood samples withing
4 h from
collection. Plasma was collected by centrifuging blood at 1600 xg for 10 min
at 4 C and 16000
xg for 10 mm at 4 C and stored at -80 C for ct-DNA purification. cfDNA from
plasma was
extracted using Qiamp Circulating Nuclei Acid Kit (Qiagen). cfliNA was
quantified by Qubit
Fluorometer (Life Technologies).
[01371 Preparation of carrier DNA and spike-in controls. Carrier DNA was
prepared by
PCR amplification of the pNIC28-Bsa4 plasmid (Addgene, cat. no. 26103) in a
reaction
containing 1 ng DNA template, 0.5 !AM primers (Fwd: 5'-
AGGCAACTTTATGCCCATGCAA-3' (SEQ ID NO: 2);
Rev: 5 -
CCAAGGGGTTATGCTAGTTATTGC-3' (SEQ ID NO: 3)) and 1X Phusion High-Fidelity
PCR Master Mix with HF Buffer (Thermo Scientific). The CpG-methylated lambda
DNA and
2kb unmodified spike-in control DNA were prepared as described previously. CpG-
methylated
lambda DNA, carrier DNA and 2 kb unmodified control were fragmented by Covaris
M220
(Peak Incident Power - 50 W, Duty Factor - 20%, Cycles per Burst (cpb) - 200,
time - 150 s)
and size-selected on 0.9-- 1.2x AMPure XP beads to select for 150-250 bp
fragments.
[01381 Preparation of sequencing adopters.
Adapter oligos (5 -
ACACTCTTTCCCTACACGACGCTC TTCCGATCT-3' (SEQ ID NO: 4); 5'-
/5Phos/GATCGGAAGAGCACACGTCT-3' (SEQ ID NO: 5)) were obtained from IDT with
HPLC purification. Adapter oligos were annealed together in a 50 1AL reaction
containing 15
1AM of each oligo, 10 mN4 Tris-Cl (pH = 8.0), 0.1 m1VIEDTA (pH = 8.0) and 50
mM NaCl with
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
33
the following program: 2 min at 95 C, 140 cycles of 20 sec at 95 C (decrease
temperature
0.5 C every cycle) and hold at 4 C. Annealed 15 uM Illumina multiplexing
adapters were then
aliquoted into small single-use vials and stored at -80 C.
101391 mret ICD oxidation. mTet1CD was prepared as described previously. DNA
was
incubated in a 501.11 reaction containing 50 mM HEPES buffer (pH 8.0), 100 JAM
ammonium
iron (II) sulfate, 1 mM ct-ketoglutarate, 2 mM ascorbic acid, 2 rnM
dithiothreitol, 100 mM
NaCl, 1.2 mIVI ATP and 4 uM mTet1CD for 80 min at 37 C. After that, 0.8 U of
Proteinase K
(New England Biolabs) were added to the reaction mixture and incubated for 1 h
at 50 C. The
product was cleaned up on Bio-Spin P-30 Gel Column (Bio-Rad) and 1.8x AMPure
XP beads
following the manufacturer's instruction.
101401 Pyridine borane reduction. Oxidized DNA in 35 al of water was reduced
in a 50 ul
reaction containing 600 mM sodium acetate solution (pH 4.3) and 1 M pyridine
borane (Alfa
Aesar) for 16 hat 37 C and 850 r. p.m. in an Eppendorf ThermoMixer. The
product was purified
using Zymo-Spin columns.
[01411 cIDNA TAPS. 10 ng of cfDNA were spiked-in with 0.15% CpG-methylated
lambda
DNA and 0.015 % unmodified 2 kb control and used for an end-repair and A-
tailing reaction
and ligated to Illumina Multiplexing adapters with KAPA HyperPrep kit
according to the
manufacturer's protocol. Subsequently 100 ng of carrier DNA were added to
ligated libraries
and samples were double-oxidized with mTet1CD and reduced with pyridine borane
according
as described above. Converted libraries were amplified using NEBNext Multiplex
Oligos for
Illumine (96 Unique Dual Index Primer Pairs) with KAPA Hifi Uracil Plus
Polymerase for 7
cycles and cleaned up on 1>< AMPure XP beads. CfDNA TAPS libraries were paired-
end
150 bp sequenced on a NovaSeq 6000 sequencer (Illumina).
101421 TAPS mapping and pre-processing. Raw sequenced reads were processed
with
trim galore (version 0.6.2 www. bioinformatics.babraham. ac. uk/proj
ects/trim_galore/) to trim
adapter and low-quality bases with the following parameters --paired --length
35 --gzip --cores
2. Clean reads were aligned to human reference genome (GRCh38
ftp.nchi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA 000001405. 15_GRCh38/seqs
for
alignment_pipelines.ucsc ids/GCA 000001405.15 GRCh38 no a,lt
analysis_set.fna.gz.)
combining spike-in sequences using bwa mem (version 0.7.17-r1188) with the
following
parameters -I 500,120,1000,20. Reads with MAPQ <1 were excluded from further
analysis.
Picard MarkDuplicates (version 2.18.29-SNAPSHOT) was used to identify
duplicate reads.
MethylDackel extract (version 0.5.0 https://github.com/dpryan79/MethylDackel)
was used for
methylation calling using the following parameters -q 10 -p 13 -t 4 --
mergeContext --OT
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
34
10,140,75,75 --OB 10,140,75,75. CpG sites overlapped with common SNP
(dbSNP153),
blacklisted regions, centromeres, and sex chromosomes were excluded for
further analysis.
101431 cfTWA WGBS analysis. CIDNA WGBS data was downloaded from
EGAD00001004317. Raw sequenced reads were processed with trim galore (version
0.6.2
www. bioinformatics .babraham.ac.uk/proj ects/trim galore): adapter
and low-
quality bases were trimmed with the following parameters --paired --length 35 -
-gzip --cores
2. Clean reads were aligned to human reference genome (GRCh38) using bismark
(Bismark
Version: v0.22.0) with default parameters. deduplicate bismark was used for
deduplication.
Samtools was used to filter the fragments with -q 10, and only reads mapped in
proper pairs
were used for fragmentation analysis. bismark_methylation extractor was used
to extract
methylation from deduplicated barn files with default parameters.
101441 PCA on DNA methylation and feature overrepresentation analysis. The
genome was
binned into lkb windows. Methylation level was calculated using number of
methylated CpGs
divided by the number of total CpGs sequenced. Windows with mean CpG coverage
(number
of total CpG sequenced/ total number of CpG positions) <2 were excluded for
further analysis.
Dimdesc was used with parameter proba = 0.01 to determine the regions that
contribute most
to each principal component obtained by the PCA function (largest eigenvalues
of each
eigenvector). Bedtools fisher was used to test the number of overlaps between
the top 200
contributing regions (sorted by absolute correlation value) and the selected
genomic features.
Selected genomic features included regulatory element from Ensemble
(ftp. ens embl org/pub/release-
97/regulation/homo sapiens/homo sapiens. GRCh38.Regulatory Build. regulatory
features . 2
0190329. gff. gz) and CpG islands from
UCSC
(hgdownl o ad . soe.ucsc. edu/gol denP ath/h g38/datab as e/cpgT sl an dExt.
txt. gz).
14)1451
Two class prediction using DNA methylation signature. Two class prediction
models
were trained and evaluated based on a LOO approach. Briefly, one sample was
held out as the
testing set while the remaining samples were used for model training. DMRs
(promoters for
PDAC and enhancers for HCC) were identified in the training set by t-test (P
value < 0.002,
methylation difference > 0.05). In each leave-one-out fold 443-775
differentially methylated
enhancers and 160-318 differentially methylated promoters were identified in
the HCC vs. non-
cancer control and PDAC vs. non-cancer control feature selection steps,
respectively. In total,
1,521 enhancers, and 531 promoters were selected during the cross-validation
process. The
predictive model was built on selected DMRs using cv. Glmnet and validated on
the test
sample. This procedure was repeated N times, where N = number of samples. ROC
curves
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
were prepared in R based on the predicted scores of held out test samples from
cvglm models.
Cirrhosis patients and cfDNA WGBS data were used as independent validation
sets to evaluate
the performance of HCC model. Pancreatitis patients were used as independent
validation set
to evaluate the performance of PDAC model. Aligned BAM files were down-sampled
from
100M to 200M read pairs using samtools view. For each down-sampled set, the
method
described above was used to detect DMRs. Ref DMR were defined as the total
unique DMR in
the LOO cross-validations. The percentage of ref DMRs were computed by
dividing the
overlapped DMR between down-sampled set and the ref DMR and the total ref DMR.
[01461 GO analysis of DMRs. Genes regulated by differentially methylated
enhancers in
HCC cfDNA were identified using the GeneHancer database. The genes closest to
the
differentially methylated promoters in PDAC were identified as related using
following R
packages: Annot ati onHub (version 2.18.0), TxDb.Hsapiens.UCSC .hg38.knownGene
(version
3.10.0) and org.Hs.eg.db (version 3.10.0). GO analysis was performed on these
identified genes
using Enrichr tool against NCI-Nature Pathway Interaction database.
[01471
Tissue Reference Map. CpG-level tissue methylation data was collated from
six
public sources (sources of public methylation WGBS data for generation of
tissue map are not
included in the present disclosure but can be made available upon request).
After filtering
diseased, sex-specific, and low-coverage samples, 144 healthy, adult tissue
samples were
retained, and grouped into 32 physiologically distinct tissue groups (raw data
pertaining to
cfDNA tissue contribution for each patient in cITAPS cohort are not included
in the present
disclosure but can be made available upon request). 133 out of 144 samples
were already
aligned to hg38; the remaining 11 samples were converted from hg19 to hg38
using the UCSC
hgLiftOver tool.
[01481
About 79,000 enhancers were filtered from En sembl Regulatory Build using a
ti s sue-
specific DMR finding algorithm similar to Moss et al. Specifically, this
algorithm performs
pairwise one-vs-all comparisons for each tissue group in the reference atlas,
selecting the
regions which show the largest median methylation difference and consistent
methylation
across the tissue group in question. As in Moss et al., pairwise tissue group
correlations were
also calculated, and included DMRs that best separated each tissue group from
the first and
second most highly correlated tissue.
[01491
Tissue Deconvolution by Non-negative Least Squares Regression. Tissue
deconvolution was performed using non-negative least squares regression and
implemented
using Scipy's optimize function in Python 3.8. Given a tissue reference matrix
A, and a vector
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
36
of observed methylation ratios ys in a sample s, the tissue contribution x was
estimated by
solving the following minimization problem:
101501 min If A ¨
- = g
101511 subject to x 0.
14)1521 Fragmentation analysis. The length of the DNA fragments was obtained
from
alignment files using Samtools. Fragmentation profiles were calculated as the
fraction of
cfDNA fragments at 10 bp length range bins. PCA analysis and plots were
generated in R.
[01531 For fragmentation-based prediction, proportion of cfDNA
fragments (300 to 500 bp)
in 10 bp length range bins was calculated. Models were built and trained by
leave-one-out
approach using cv. glmnet method. ROC curves were prepared in R based on
prediction scores
from validation.
101541 CNV analysis. Alignment files for each sample were downsampled to 225M
read
pairs with samtools view. QDNAseq package was used for copy number variation
analysis.
The bin annotation was downloaded from
QDNAseq.hg38
(github.com/asntech/QDNAseq.hg38) and bin size 100 kb was used. Regions which
were
blacklisted or have mappability less than 80 were excluded for further
analysis. cutoffs 0.8 and
1.2 were used to define copy number losses and gains respectively in the
callBins function.
Patients which have copy number aberrations with length range bigger than 500
kb were
classified as patients with CNV.
[01551 Three class prediction models. Three class prediction
models were trained and
evaluated based on a LOO approach. For DNA methylation, the candidate features
were
initially narrow down to 824,320 lkb windows encompassing mapping to
regulatory regions
as mentioned previously. The methylation model aims to capture the cancer-type
specific
methylation change by selecting DMRs based on a pairwise comparison using a t-
test. DMRs
were then ranked by P value, and the top 5 DMRs in each pairwise comparison
were selected
for model training. The prediction model was built on DMRs selected among the
training set
using a SVM model implemented in the caret package (train method =
"symLinear2") and
validated on the test sample. This procedure was repeated N times, where N =
number of
samples. For tissue contribution and fragmentation fraction, the raw matrixes
were used to
build models following the same method as for DMRs. These three models were
integrated by
taking the averaged (mean) predictions across the three modalities, where the
selected
prediction in each case was the one with the maximum averaged predicted score.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
37
[01561
It is understood that the foregoing detailed description and accompanying
examples
are merely illustrative and are not to be taken as limitations upon the scope
of the disclosure,
which is defined solely by the appended claims and their equivalents.
101571 Various changes and modifications to the disclosed embodiments will be
apparent
to those skilled in the art. Such changes and modifications, including without
limitation those
relating to the chemical structures, substituents, derivatives, intermediates,
syntheses,
compositions, formulations, or methods of use of the disclosure, may be made
without
departing from the spirit and scope thereof
5. Examples
[01581
It will be readily apparent to those skilled in the art that other suitable
modifications
and adaptations of the methods of the present disclosure described herein are
readily applicable
and appreciable, and may be made using suitable equivalents without departing
from the scope
of the present disclosure or the aspects and embodiments disclosed herein.
Having now
described the present disclosure in detail, the same will be more clearly
understood by
reference to the following examples. which are merely intended only to
illustrate some aspects
and embodiments of the disclosure, and should not be viewed as limiting to the
scope of the
disclosure. The disclosures of all journal references, U.S. patents, and
publications referred to
herein are hereby incorporated by reference in their entireties.
101591
The present disclosure has multiple aspects, illustrated by the following
non-limiting
examples.
Example 1
[0.1601 Adaptation of TAPS for cfDNA sequencing. Experiments were conducted to
optimize the TAPS protocol to work with low input cfDNA (10 ng, purified from
1-3 mL of
plasma). Briefly, 10 ng cfDNA is first ligated to Illwinna adapters and 100 ng
of carrier DNA
is then added to the sample prior to TET oxidation and pyridine borane (PyBr)
reduction steps
(FIG. 1A). It found that the addition of carrier DNA improves the recovery of
cfDNA during
the workflow and results in higher library yields when compared to the
standard TAPS protocol
(FIG. 5A). Subsequently, 5-methylcytosine (5mC) and 5-hydroxymethylcytosine
(5hmC) in
ctIDNA are oxidized by mTet1CD enzyme to 5-carboxylcytosine (5caC) and reduced
to
dihydrouracil (DHU). which is amplified as T in the final PCR step (FIG. 1A).
101611 The cfTAPS was applied to 87 cfDNA samples. Libraries were sequenced to
a mean
of 360M read pairs (11.6x mean depth, range 8.2-22x), and resulted in high
unique mapping
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
38
rate and unique deduplicated mapping rate of 94.8% and 77.1%, respectively
(FIG. 1B, raw
data pertaining to sequencing statistics are not included in the present
disclosure but can be
made available upon request). Among the mapped reads, 99.95% were mapped to
the human
genome (FIG. 5B). In comparison, a recent cfDNA whole-genome bisulfite
sequencing
(WGBS) study sequenced to a similar depth (a mean of 371M read pairs) and
resulted in
significantly lower unique mapping rate (63.6%) and unique deduplicated
mapping rate
(53.9%) (FIG. 5C), even though it used more cfDNA input (from 5 mL plasma).
This
highlights the advantage of cfTAPS to generate higher quality and more complex
data than
cfDNA WGBS while requires less cfDNA input.
101621
Subsequently, the accuracy of cfTAPS for detecting 5mC was assessed based
on
spike-in controls, which have modified and unmodified cytosines in the known
positions. CpG
methylated lambda DNA was used to estimate the conversion of 5mC. Two samples
had a low
conversion rate below 85% and were excluded from downstream analysis (raw data
pertaining
to sequencing statistics are not included in the present disclosure but can be
made available
upon request). The remaining 85 samples had a mean 5mC conversion rate of
97.0% or a false
negative rate (non-conversion rate of 5mC) of 3.0% (FIG. IC). The false
positive rate
(conversion rate of unmodified C), estimated based on unmodified amplicon
spike-in, was
0.28%, which confirms that cfTAPS allows highly sensitive and specific
detection of 5mC in
cfDNA (FIG. IC). High reproducibility of cfTAPS between technical replicates
was further
confirmed (FIG. 5D).
Example 2
[01631 Whole-genome DNA methylation from cfTAPS. Next, experiments were
conducted to characterize the cfDNA methylome in the 85 cfDNA samples that
passed initial
quality control. The cohort included samples from 21 patients with HCC, 23
with PDAC, 30
non-cancer controls, 4 patients with cirrhosis and 7 with pancreatitis (FIG.
6A). Cirrhosis and
pancreatitis are precancerous conditions affecting liver and pancreas
respectively. Most PDAC
and HCC patients in the cohort were at a non-metastatic stage, with 52% of
PDAC patients and
67% HCC patients at stage I and II (FIG. 2A; clinical data pertaining to the
cfTAPS study
cohort are not included in the present disclosure but can be made available
upon request).
Among the 21 HCC patients, only 4(19%) had elevated levels of APF (over 20
ng/mL). Among
the 18 PDAC patients which had CA19-9 measurement, 16 (89%) had elevated
levels of CA19-
9 (over 37 U/mL). However, it is important to note, that CA19-9 level is often
elevated in non-
malignant conditions including inflammatory disease. Of note, the non-cancer
controls were
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
39
collected from an endoscopy clinic and were enriched with gastro-intestinal
inflammatory
conditions such as Crohn's disease and colitis (clinical data pertaining to
the cITAPS study
cohort are not included in the present disclosure but can be made available
upon request). While
distinguishing these non-cancer controls from cancer patients is more
challenging than a
typically healthy control group, this may provide a more real-world comparison
of a diagnostic
test in an aging population.
101641
Global methylation levels of cfDNA in cancer and control samples were
analyzed.
CfDNA methylation displayed atypical bimodal distribution in all groups with
most CpG sites
either fully methylated or unmethylated (FIG. 5B). Average CpG methylation
level in control
samples was 75.5% and was similar in cancer cfDNA (HCC: 74.9%; PDAC: 75.1%).
Previously reported global cfDNA hypomethylation in HCC was only observed in a
few
samples with late stage or large tumor size (FIG. 2B and FIG. 6C-6F). By
contrast, a higher
variance of methylation in 1 Mb genomic windows was observed between cancer
patients
compared to controls (FIG. 6G-6H).
[0.1651 Experiments were then conducted to investigate whether whole-genome
cfDNA
methylation signatures have the potential to discriminate between cancer
patients and non-
cancer controls. Principal Component Analysis (PCA) of cfDNA methylation in 1
kb genomic
windows was performed first. Both HCC (FIG. 2C) and PDAC samples (FIG. 2D)
showed
partial separation from controls in principal component 2 (PC2) and PC1,
respectively. Noted
that the inflammatory patients (Crohn's disease and colitis) do not separate
from the other non-
cancer controls (FIG. 61). Experiments were then conducted to investigate
where the windows
that most contributed to the cancer/control separation were enriched in the
genome. Results
indicated that the top 200 windows with the highest correlation with PC2 for
HCC were
enriched in enhancers (FIG. 2E). Conversely, the 200 windows most highly
correlated with
PC1 for PDAC were highly enriched in promoters (FIG. 2E), suggesting that
different cancer
types have different cfDNA methylation signals.
Example 3
[01661 Differential DNA methylation from elTAPS. Since methylation patterns in
regulatory regions significantly contributed to discrimination between cancer
and controls in
unsupervised analysis, experiments were conducted to investigate the
predictive potential of
cfDNA methylation in enhancer and promoter regions for HCC and PDAC prediction
respectively, using a supervised machine leaming approach with leave-one-out
(L00) cross-
validation. Briefly, in each round of LOO cross-validation, one sample was
used as a validation
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
set and the remaining samples for model training. Within each fold,
differentially methylated
enhancers and promoters were identified for HCC and PDAC, respectively, and
used to train a
regularized generalized linear model classifier (glmnet) to distinguish each
cancer type from
the control samples. This model was then evaluated on the held-out test sample
for each fold
(FIG. 7A). Cirrhosis and pancreatitis samples were not included in model
building but were
used as an independent validation set to evaluate performance of the
classifiers to discriminate
between cancer and pre-malignant conditions.
[0167i
Significant prediction of HCC (AUC = 0.99) was achieved based on
differentially
methylated enhancers (FIG. 2F-2G; raw data pertaining to differentially
methylated enhancers
used for HCC vs. Control predictions are not included in the present
disclosure but can be made
available upon request). Moreover, based on predicted scores, 3 out of 4
cirrhosis samples
could be distinguished from HCC, suggesting that the model is able to detect
cancer-specific
features (FIG. 7B). Gene ontology analysis was then performed on the
differentially methylated
enhancers and found significant enrichment in signalling pathways commonly
affected in liver
cancer, including regulation of RAC1 activity and IL8- and CXCR1-mediated
signalling (FIG.
7C). For example, in cfDNA of HCC patients, significant hypermethylation of
the enhancer
that regulates expression of the DLC1 gene, a tumor suppressor for human liver
cancer
involved in RAC1 and Rho signalling pathways, was observed (FIG. 7D).
(0168] Accurate prediction of PDAC (AUC = 0.98) was achieved based on
differentially
methylated promoters (FIG. 2H-2I; raw data pertaining to differentially
methylated promoters
used for PDAC vs. Control predictions are not included in the present
disclosure but can be
made available upon request). Similarly, the classifier was able to predict 6
out of 7 pancreatitis
samples as non-cancer, despite not being trained on any pancreatitis samples
(FIG. 7E).
Differentially methylated promoters in PDAC cfDNA were enriched in signalling
pathways
affected in PDAC including RB1 regulation and p38 signalling pathways (FIG.
7F). For
instance, results indicated significant hypermethylation in the RBI gene
promoter (FIG. 7G),
a well-studied tumor suppressor gene. Hypermethylation of RBI promoter was
previously
found in human cancers and downregulation of RB I were reported in pancreatic
cancer.
[01691 Finally, the HCC model was validated on an independent dataset from a
recent
cfDNA WGBS study, which contains 4 HCC patients and 4 non-cancer controls.
Results
indicated that the models built on differentially methylated enhancers
identified from a-TAPS
data were able to correctly classify all HCC and non-cancer controls from this
external dataset
(FIG. 7H). It is important to note that the high sequencing depth of cfTAPS is
essential for de
novo differential methylation analysis from cfDNA and the differentially
methylated regions
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
41
(DMRs) identified were significantly decreased when the data was down-sampled
to 100-200M
read pairs (FIG. 71). Taken together, cITAPS enables whole-genome discovery of
DMRs in
ciDNA, and the distinct methylation patterns in regulatory regions enable
accurate prediction
of HCC and PDAC.
Example 4
[01701 cffAPS informs tissue-of-origin. CIDNA methylation has been shown to
provide
tissue-of-origin information. Most approaches use 450K methylation array
tissue data, which
covers less than 1% of CpGs in the human genome, to infer tissue contribution
from cfDNA
methylation. To further utilize the whole-genome information from cfTAPS for
cfDNA
deconvolution, CpG-level methylation data were collated from 144 publicly
available tissue
and blood cell WGBS, and stratified into 32 physiologically distinct tissue
and blood cell types,
including liver tumor tissue (sources of public methylation WGBS data for
generation of tissue
map are not included in the present disclosure but can be made available upon
request). Given
the prevalence of tissue-specific DNA methylation in enhancer regions, an
enhancer-
aggregated reference map of tissue methylation was constructed. The resulting
methylation
reference map displays good clustering of blood and immune cell types, and
even
physiologically related solid tissues (FIG. 8A).
[0171/
Tissue contribution in ciTAPS samples was calculated by performing non-
negative
least squares regression (NNLS). cfDNA tissue contribution was broadly similar
between
cancer and control groups, and in agreement with previous reports, with blood
and immune
cells dominant, and lower proportions of solid tissues (FIG. 3A, FIG. 8B; raw
data pertaining
to cfDNA tissue contribution for each patient in cif/6+PS cohort are not
included in the present
disclosure but can be made available upon request). Importantly, a
significantly increased liver
tumor contribution in HCC alone was observed (FIG. 3B, paired t-test, P value
0.0016), and a
significantly increased memory T cell contribution in PDAC samples was
observed (paired t-
test, P value 0.028) (FIG. 8C). A regularized generalized linear model was
trained based on
tissue contribution, evaluating all samples using LOO cross-validation, and
was demonstrated
to correctly separate the majority of samples in both cancer types (HCC vs non-
cancer control:
AUC = 0.77; PDAC vs non-cancer control: AUC = 0.81). However, these models
perform
worse at distinguishing pancreatitis and cirrhosis compared to methylation-
based models (FIG.
9D-8I). Tissue deconvolution is currently limited by the availability of
public WGBS data.
Nevertheless, these results indicate that cfrAPS provides valuable tissue-of-
origin information
for early cancer detection.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/IB2022/000420
42
Example 5
101721 Fragmentation patterns from cfTAPS. Although the main purpose of ctTAPS
is
DNA methylation sequencing, it only induces base-changes at modified
cytosines, thus keeping
the majority of DNA intact. Additional genetic information can therefore be
extracted from
cfTAPS data to further improve the sensitivity of early cancer detection.
Experiments were
conducted to first investigate the CNVs from cfTAPS data. As expected with the
non-advanced
cancer cohort, CNVs were only predicted in 4 HCC patients and 3 PDAC patients
(FIG. 9A-
9B). Next, experiments were conducted to investigate whether cfTAPS can retain
reliable
cfDNA fragmentation information, which has recently been shown to change
significantly
during cancer development and has therefore been adopted in cancer detection
assays.
[01731 It was first confirmed that cfDNA fragmentation patterns detected with
cfTAPS are
concordant with cfDNA fragmentation pattern generated by whole-genome
sequencing
(WGS), with the dominant peak at 167 bp, a secondary peak at ¨ 320 bp and
smaller peaks
below 167 bp with 10 bp periodicity, reflecting nucleosomal fragmentation
patterns (FIG. 3C;
raw data pertaining to fragment length distribution in each individual are not
included in the
present disclosure but can be made available upon request). By contrast,
fragmentation patterns
were clearly different in previously published cfDNA WGBS, as the 10 bp
oscillations in the
cfDNA fragmentation profile were lost, presumably due to DNA damage (FIG.
10A).
Consistent with previous cfDNA WGS, results indicated that cancer patients
have a higher
frequency of cfDNA fragments below 150 bp (Kruskal-Wallis test, HCC: P value
6.871e-06,
PDAC: P value 0.006731) and a lower proportion of long fragments between 310-
500 bp
(Kruskal-Wallis test, HCC: P value 2.627e-07, PDAC: P value 1.263e-06)
compared to non-
cancer controls (FIG. 3D), further confirming the faithful preservation of
cfDN A fragmentation
information in cfTAPS.
[0741 A new approach was then developed for characterization of cfDNA
fragmentation
profiles using cfTAPS. Briefly, the cfDNA fragmentation distribution was
divided into 10 bp
bins and calculated the proportion of fragments in each 10 bp bin (FIG. 3C).
It was found that
cfDNA long fragments (300-500 bp) length proportion in 10 bp bins separated
PDAC and HCC
from controls in unsupervised analysis by PCA (FIG. 3E). Results further
showed that this
cfDNA fragmentation signature can be used to distinguish HCC and PDAC from non-
cancer
controls with high accuracy (HCC AUC = 0.92, PDAC AUC = 0.84) (FIGS. 10B, 10C,
10E,
and 10F). However, this approach was less accurate at distinguishing cancer
from cirrhosis and
pancreatitis compared to methylation-based classifiers (FIGS. 10D and 10G),
suggesting
fragmentation information is less cancer-specific.
CA 03226747 2024- 1-23
WO 2023/007241
PCT/1B2022/000420
43
Example 6
101751 Multi-cancer detection with c1TAPS. Experiments were then conducted to
investigate the utility of cfTAPS for multi-cancer detection. The top 5 DMRs
of each pairwise
comparison (non-cancer controls versus HCC, non-cancer controls versus PDAC,
HCC versus
PDAC) were selected as features in the multi-cancer differential methylation
model. A Support
Vector Machine (SVM) model was trained to estimate the respective probability
that the blood
sample came from each group. Similar models were built using tissue
contribution and
fragmentation profile. Using LOO cross validation, results indicated that the
methylation model
can achieve an overall accuracy of 0.77, which outperforms the tissue
contribution model and
fragmentation profile model (accuracy 0.62 and 0.46, respectively, FIG. 4A,
FIG. 11A).
[01761
To further enhance the multi-cancer predictive model, a multimodal
classifier was
built that combined differential methylation, tissue contribution and fragment
profile (FIG.
413). This integrated model took the averaged scores across the three
modalities and used the
most confident prediction for each sample. The overall accuracy of the
combined model was
0.86 (64 out of 74 were classified correctly) and the accuracy for
distinguishing controls from
any cancer type is 0.92 (FIG. 4C), which highlights the benefits of
incorporating multimodal
information for cancer type prediction. Finally, the DMRs used for multi-
cancer prediction
were explored (FIG. 11B; data pertaining to methylation features used for HCC,
PDAC, and
Control predictions are not included in the present disclosure but can be made
available upon
request). Interestingly, results indicated that the nearby genes of these
regions were enriched
in Notch and Wnt signalling, and EGFR (ErbB) signalling, which provides
biological support
for these potential multi-cancer biomarkers (FIG. 11C).
CA 03226747 2024- 1-23