Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
1431
4-16170/+/CGC l233
N3-Cyclopentyl-substituted adenine derivstives
The instant invention is directed to certain functionalized
~-cyclopentyl-substituted adenine dervatives as adenosine receptor
ligands, to pharmaceutical compositions thereof, to methods for
their preparation, and to their use in mammals as therapeutically
effective adenosine receptor agonists.
The compunds of the invention are effective as adenosine, particu-
larly adenosine-2 (A-2), receptor ligands which are useful in
mammals as adenosine receptor agonists, particularly as adenosine-2
(A-2) receptor agonists.
Said advantageous propertles render the compounds of the invention
useful for the treatment of conditions in mammals responsive to
selective adenosine receptor stimulation, particularly to adeno-
sine-2 receptor stimulation, e.g. cardiovascular conditions such as
hypertension, thrombosis and atherosclerosis, also central nervous
~ystem conditions comprising psychotic conditions such as schizo-
phrenia, and convulsive disorders such as epilepsy.
The compounds of the invention are structurally related to the
natural product aristeromycin which is cited, e.g. in J. Org.
Chem. 51, 1287-1293 ~1986) and publications referred to therein,
and which is characterized in the literature as a carbocyclic analog
o$ adenosine.
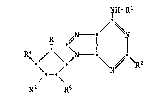
More specificaily, the instant invention is directed to the com-
pounds of the formula I
~8~43~
- 2 - 21489-7299
~IH--Rl
R ~N il/ ~T
\N~ R ( I )
.
R3 Rs
wherein R, R3 and R5 independently represent hydrogen or hydroxy
provided ~hat at least one of R, R3 and R5 represents hydroxy;
R1 represents hydrogen, lower alkyl, C3-C7alkenyl, hydroxy-lower
alkyl, C3-C6cycloalkyl, C3-C6cycloalkyl-lower alkyl, 2-norbornyl,
2-norbornyl-lower alkyl, aryl, aryl-lower alkyl, aryl-
C3-C6cycloalkyl wherein aryl represents thienyl, pyrldyl,
pyrrolyl, lndolyl, naphthyl, phenyl, phenyl substituted by one to
three of halogen, trifluoromethyl, lower alkoxy or lower alkyl, or
phenyl substituted by a substituent -W-Z ln whlch W repxesents a
direct bond, lower alkylene or oxy-lower alkylene and Z represents
cyano, carboxy or carboxy derivatized in the form of a
pharmaceutically acceptable ester or amlde; 9-fluoroenyl or
9-fluoroenyl-lower alkyl; or R1 represents a bicyclic radical
seleated rom the group consisting o~ 1,2,3,4-tetrahydro-2-
naphthyl, 2-lndanyl, 2,3-dihydro-3-benzofuranyl, 2,3-dihydro-3-
benzothiofuranyl, 3,4-dihydro-2H-[I]-3-benzapyranyl and 3,4-
dihydro-2H-[1~-3-benzothiopyranyl, any said bicyclic radicals
: ~ being optlonally substituted on ~he benzo partion by lower alkyl,
: lawer alkoxy or halogsn; R2 represents~hydrogen, halogen, -SR
or -N(R93R1 ln each of whlch R1 has meanlng~as deflned
hereinbe~ore provided that R1 ln -SRl does not represent hydrogen;
R9 repressnts hydrogen or lawer slkyl7 R4 repressnts hydroxymethyl
.
~' . . . .
,: , :
~X~3843~
- 2a - 21489-7299
hydrogen or lower alkyl; or R~ represents lower alkoxymethyl,
lower alkylthiomethyl or -CONHR6 in which R6 represents 10~7er
alkyl, aryl-lower alkyl wherein aryl is as defined above for R1,
C3-C6cycloalkyl or hydroxy-lower alkyl; pharmaceutically
acceptable ester derivatives thereof in which free hydroxy groups
are esterified in form of a pharmaceutically acceptable ester; and
salts thereof.
The invention is even directed ~o the compounds of
formula I wherein all substituents have the meaning given
hereinbefore except that R1 is different fxom C3-C7alkenyl and
hydroxy-lower allsyl and wherein R9 denotes exclusively hydrogen.
-- 3 --
Preferred are the compounds of formula I wherein R and R3 represent
hydrogen or hydroxy; Rs represents hydroxy; Rl represents hydrogen
or lower alkyl; or Rl represents aryl or aryl-lower alkyl wherein
aryl represents thienyl, pyrldyl, naphthyl, phenyl or phenyl
substituted by halogen, trifluoromethyl, lower alkoxy, lower alkyl
or by a substltuent -W-Z ln whlch W represents a direct bond, lower
alkylene, or oxy-lower alkylene, and Z represents cyano, carboxy,
carboxy derivati~ed in the form of a pharmaceutically acceptable
ester or carboxy derivatized in the form of a pharmaceutlcally
acceptable amlde; or Rl represents a substituent of the formula B
(f~ \ (B3
in which A represents methylene, oxygen or sulfur, m represents zero
or one, and Ra represents hydrogen, lower alkyl, lower alkoxy or
halogen; R~ represents hydrogen, halogen, -SRI or -N(R9)RI in whlch
Rl has meanlng as defined herelnbefore except that Rl in SRI does
not represent hydrogen; R9 represents hydrogen or lower alkyl; R4
represents hydroxymethyl provided that R2 does not represent either
hydrogen or -NHRI in which Rl represents hydrogen or lower alkyl; or
R4 represents lower alkoxymethyl, lower alkylthiomethyl, or -CONHR~
in which R~ represents lower alkyl, aryl-lower alkyl, C3-C6cyclo-
alkyl or hydroxy-lower alkyl; pharmaceutically acceptable ester
derivatives thereof in whlch one or more free hydroxy ~roups are
esterified in form of a pharmaceutically acceptable ester; and
pharmaceutically acceptable salte thereof.
The lnvention is even directed to those compounds of tormula I
wherein all substltuents have the meaning given hereinbefore as
being preferred except that Rl is dlfferent from aryl and wherein R9
denotes excluslvely hydrogen.
~ 2~3843~
A particular embodiment of the invention relates to the compounds of
formula I cited hereinabove wherein R4 represents -CONHR6, and R,
Rl, R2, R3, Rs, R~, R9, m and R have meaning as defined above;
pharmaceutically acceptable ester derivatives thereof as defined
above; and pharmaceutically acceptable salts thereof.
Another particular embodi~ent relates to the compounds of formula I
wherein R4 represents hydroxymethyl with the proviso for R2 given
hereinbefore.
Preferred are the compounds of formula II
~Hz
R~ T (Il)
R3 OH
whereln R2 represents hydrogen, halogen, SRl or N(R9~RI; R1
represents aryl. or aryl-lower alkyl in which aryl represents
thienyl, pyridyl, phenyl or phenyl substituted by halogen, tri-
fluoromethyl, lower alkoxy, lower alkyl or by a substituent -W-Z in
which W represents a direct bond, lower alkylene or oxy-lower
alkylene, and Z represents cyano, carboxy, lower alkoxycarbonyl,
carbamoyl, N-mono- or N,N-di-lower alkylcarbamoyl; or Rl represents
a substituent of the formula B'
(B')
\,/ \,~
in which A represents a direct bond, methylene, oxygen or sulfur,
and Ra represents hydrogen, lower alkyl, lower alkoxy or halogen; R3
represents hydrogen or hydroxy; and R~ represents lower alkyl,
C3-C6cycloalkyl or hydroxy-lower alkyl; R9 represents hydrogen or
1;~8843~L
lower alkyl; pharmaceutlcally acceptable prodrug ester deri-vatives
thereof in which free hydroxy groups are esterified in form of a
pharmaceutically acceptable ester; and pharmaceutically acceptable
salts thereof.
The invention is even directed to those compounds of formula II
wherein all substituents have the meaning given hereinbefore as
being preferred except that Rl is different from aryl and wherein
R9 denotes exculsively hydrogen.
Preferred are the compounds of formula II wherein R2 represents
hydrogen, chloro or N(~9)RI; Ra represents hydrogen; R3 represents
hydroxy; R6 represents lower alkyl, C3-C6cycloalkyl or hydroxy-lower
alkyl; and Rl, R9 and A have meaning as defined above; pharma- -
ceutically acceptable prodrug ester derivatives thereof in which
free hydroxy groups are esterified in Eorm of a pharmaceutically
acceptable ester; and pharmaceutically acceptable salts thereof.
Further preferred are the compounds of formula IIa
~7H2
R2 (IIa)
OR7 oR8
wherein R2 represents hydrogen, chloro, N(R9)-(CH2) -Cs or C6cyclo-
alkyl, or -N(R9)-(CH2) -Ar in which n represents zero or the integer
1, 2 or 3, R5 represents Cl-C3alkyl, and Ar represents 2-, 3- or
4-pyridyL~ phenyl or phenyl substituted by halogen, trlfluoromethyl,
lower alkoxy, lower alkyl or by a substi~uent -W-Z in which W
represents a direct bond, straight chain Cl-C4alkylene or oxy-Cl-C3-
alkylene and Z represents cyano, carboxy, lower alkoxycarbonyl,
carbamoyl, N-mono- or N,N-di-lower alkylcarbamoyl; R6 represents
C1-C4alkyl, cyclopropyl or hydroxy-C2-C~alkyl; R7 and R8 represent
12~
hydrogen, lower alkanoyl, lower alkoxy-lower alkanoyl, aroyl,
carbamoyl, N-mono- or N,N-di-lower alkylcarbamoyl; and pharmaceutl-
cally acceptable salts thereof.
The invention is even directed to those compounds of formula IIa
wherein R2 represents hydrogen, chloro or NH(CHz) -Ar in which
n represents the integer 1, 2 or 3, and Ar, R6, R7 and R8 have
meaning as defined hereinbefore as being further preferred.
Particularly prPferred are said compounds of formula IIa wherein R2
represents hydrogen, chloro, NH-CH2CH2-cyclohexyl, N(CH33-CH2CH2-
cyclohexyl, N(CH3)-CH2CHz-Ar or -NH-CH2CHz-Ar in which Ar represents
7- or 3-pyridyl, phenyl or phenyl monosubstituted by a substituent
-CH2CH2-Z in which Z represents cyano, carboxy, lower alkoxycarbo-
nyl, carbamoyl, N-mono- or N,N-di-lower alkylcarbamoyl; R6 repre-
sents ethyl or hydroxyethyl; R7 and R8 represent hydrogen, lower
alkanoyl or lower alkoxy-C2-CI,alkanoyl; and pharmaceutically
acceptable salts thereof.
The invention is even directed to those compounds of formula IIa
wherein R2 represents hydrogen, chloro or NHCH2CH2Ar in which Ar,
R6, R7 and ~8 have meaning as defined hereinbefore as being
particularly preferred.
Most preferred are the compounds of formula IIa wherein R2 repre-
sents hydrogen, 2-phenylethylamino, 2-(p-carboxyethylphenyl)-
ethylamino or 2-(2-pyridyl)ethylamino; R6 represents ethyl; R7
and R8 represent hydrogen; and pharmaceutically acceptable salts
thereof.
A particular preferred embodiment of the invention is also
represented by the compounds of formula IIb
~1.28843~
-- 7 --
~H2
R6~ N - ~\ (IIb)
CH z -~-RI I
OR7 oR8
wherein R6 represents ethyl; R7 and R8 represent hydrogen or lower
alkanoyl; R9 rPpresents hydrogen or methyl; Rl represents hydrogen
or methyl; Rll represents cyclohexyl, pyridyl, phenyl, or phsnyl
monosubstituted by halogen, lower alkoxy or -CHzCH2-Z in which Z
represents carboxy or lower alkoxycarbonyl; and pharmaceutically
acceptable salts thereof.
The general definitions used herein have the following meaning
within the scope of the present invention.
The term "lower" referred to above and hereinafter in connection
with organic radicals or compounds respectively defines such with up
to and including 7, preferably up to and including 4 and advantage-
ously one or two carbon atoms.
Lower alkyl is straight chain or branched and preferably contains
1 to 4 carbon atoms, and represents for example ethyl, propyl,
butyl, and advantageously methyl.
Lower alkoxy i8 straight chain or branched and preferably contains
1 to 4 carbon atoms, and represents for example methoxyj ethoxy9
propoxy.
:
Lower alkylene is straight chain or branched and preferably contains
1 to 4 carbon atoms, and represents for example methylene or
ethylene.
Louer alkenyl represents C~-C7alkenyl, advantageously allyl.
. .
' '
..
3~
-- 8 --
Halogen is preferably chloro, but may also be fluoro, bromo or iodo.
Cycloalkyl represents preferably 3 to 6 ring membered cycloalkyl,
i.e. C3-C6cycloalkyl. C~-C6Cycloalkyl represents cyclopropyl,
cyclobutyl, cyclopentyl or cyclohexyl9 preferably cyclopropyl in the
group R6.
Cycloalkyl-lower alkyl represents preferably (cyclopentyl or
cyclohexyl)-C1-C4alkyl, advantageously 1- or 2-(cyclopentyl or
cyclohexyl)-ethyl, -propyl or -butyl.
Aryl is an optionally substituted carbocy-~lic or heterocyclic
aromatic radical, being preferably phenyl, 1- or 2-naphthyl, or
phenyl substituted by one to three of lower alkyl, lower alkoxy,
halogen or tri~luoron~ethyl, or phenyl substltuted by a substituent
-W-Z in which W represents a direct bond, lower alkylene or
oxy-lower alkylene and Z represents cyano, carboxy or carboxy
derivatized in the form of a phar~aceutically acceptable ester or
amide; or pyridyl; or thienyl; or pyrrolyl; or indolyl.
Aryl-lower alkyl represents preferably aryl-Cl-C4alkyl, e.g. benzyl
or 1- or 2-phenyl-(ethyl, propyl or butyl) each unsubstituted or
substituted on the phenyl ring as defined under aryl above; or 2-,
3- or 4-pyridylmethyl or 2-(2-, 3- or 4-pyridyl)-(ethyl, propyl or
butyl); or 1- or 2-naphthylmethyl or 2-(1- or 2-naphthyl~-(ethyl,
propyl or butyl).
Hydroxy-lower alkyl represents preferably 2-, 3- or 4-hydroxy-C2-C4-
alkyl, advanta~eously hydroxyethyl.
Pyridyl represents 2-, 3- or 4-pyridyl, advantageously 2- or
3-pyridyl. Thienyl represents 2- or 3-thienyl. Pyrrolyl represents
preferably N-pyrrolyl~ Aryl-cycloalkyl reprasents for example
2-phenylcyclohexyl, 2-phenylcyclopropyl or 2-N-pyrrolyl-cyclohexyl.
- 9 -
A bicyclic benzo-fused 5 or 6-membered saturated carbocyclic radical
represents preferably 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl,
each optionally substituted on benzo portion as indicated above for
phenyl under aryl.
A bicyclic benzo-fused 5 or 6-membered saturated heterocyclic
radical represents preferably 2,3-dihydro-3-benzofuranyl,
2,3-dihydro-3-benzothiofuranyl, 3,4-dihydro-2H-[1~-3-benzopyranyl or
3,4-dihydro-2H-[1]-3-benzothiopyranyl, each optionally substituted
on benzo portion as indicated above for phenyl under aryl.
Lower alkoxycarbonyl preferably contains 1-4 carbon atoms in the
alkoxy portion and represents for example mèthoxycarbonyl, propoxy-
carbonyl, isopropoxycarbonyl or advantageously ethoxycarbonyl.
Lower alkanoyl represents preferably straight chain or branched
C1-CI~alkanoyl, e.g. acetyl, isobutyryl, or pivaloyl.
Lower alkoxy-lower alkanoyl represents preferably lower alkoxy-C2-
C4alkanoyl, e.g. methoxyacetyl, or 3-ethoxy-propionyl.
Aroyl represents preferably benzoyl, benzoyl substituted by one to
three of lower alkyl, lower alkoxy, halogen or trifluoromethyl; 2-,
3- or 4-pyridylcarbonyl; or 2- or 3-thienylcarbonyl.
N-Mono- and N,N-di-lower alkylcarbamoyl represents for example
N-methyl-, N-ethyl-, N,N-d~methyl- and N,N-diethylcarbamoyl.
Carboxy esterified in form of a pharmaceutically acceptable ester
repre3ents advantageously esterified carboxy that may be convertible
by solvoIysis or under physiological conditions to free carboxy,
e.g. lower alkoxycarbonyl; (amino, mono- or di-lower alkylamino)-
substituted lower alkoxycarbonyl; carboxy substituted lower alkoxy-
carbonyl, e.g. alpha-carboxy-substituted lower alkoxycarbonyl; lower
alkoxycarbonyl-substituted lower alkoxycarbonyl, e.g. alpha-lower
alkoxycarbonyl-substituted lower alkoxycarbonyl; aryl-substituted
~.. ..
~ Z8~343~
-- 10 --
lower alkoxycarbonyl, e.g. optlonally substituted benzyloxy carbonyl
or pyridylmethoxycarbonyl; (hydroxy, lower alkanoyloxy or low~r
alkoxy)-substituted lower alkoxy carbonyl, e.g. pivaloyloxymsthoxy-
carbonyl; (hydroxy, lower alkanoyloxy or lower alkoxy)-substituted
lower alkoxymethoxycarbonyl; bicycloalkoxycarbonyl-substituted lower
alkoxycarbonyl, e.g. bicyclo[2,2,1]heptyloxycarbonyl-substituted
lower alkoxycarbonyl, especially bicyclo[2,2,1]heptyloxycarbonyl-
substituted methoxycarbonyl such as bornyloxycarbonylmethoxycarbo-
nyl; 3-phthalidoxycarbonyl; (lower alkyl, lower alkoxy, halo)-sub-
stituted 3-phthalidoxycarbonyl; lower alkoxycarbonyloxy-lower
alkoxy-carbonyl, e.g. 1~(msthoxy- or ethoxyc~}bonyloxy)-ethoxy-
carbonyl; aryloxycarbonyl, e.g. phenoxycarbonyl or pheDoxycarbonyl
advantageously substituted at the ortho position by carboxy or
lower alkoxycarbonyl.
Carboxy derivatized in form of a pharmaceutically acceptable amide
represents preferably carbamoyl, N-mono-lower alkylcarbamoyl or
N,N-di-lower alkylcarbamoyl.
The pharmaceutically acceptable ester derivatlves in which one or
more free hydroxy groups are esterified in the form of a pharma-
ceutically acceptable ester are particularly prodrug esters that may
be convertible by solvolysis under physiological conditions to the
compounds of formula I having free hydroxy groups.
Preferred as said prodrug pharmaceutically acceptable esters are
straight chain or branched lower alkanoic acid esters, e.g., the
acetic, isobutyric, pivaloic acid esters, lower alkoxy-lower
alkanoic acid esters, e~g., the methoxyacetic, 3-ethoxypropionic
acid esters; arylcarboxylic acid esters, e.g., the ben~oic, nicoti-
nic acid esters; carbamic and N-mono or N,N-di-lower alkylcarbamic
acid esters (carbamates), e.g. the N-mono- or N,N-di-ethylcarbamic
or N-mono- or N,N-di-methylcarbamic acid esters. Most preferred are
the lower alkanoic acid and lower alkoxyalkanoic acid esters.
~.~88~3~
Pharmaceutically acceptable salts are generally acid addition salts,
which are preferably such of therapeutically acceptable inorganic or
organic acids, such as strong mineral acids, for example hydrohalic,
e.g. hydrochloric or hydrobromic acid; sulfuric, phosphoric or
nltric acid; aliphatic or aromatic carboxylic or sulfonic acids,
~.g. formlc, acetic, propionic, succinic, glycolllc, lactic/ malic,
tartaric, gluconic, citric, maleic, fumaric, pyruvic, phenylacetic,
benzoic, 4-aminobenzoic, anthranilic, 4-hydroxybenzoic, salicylic,
4-aminosalicylic, pamoic, nicotinic, methanesulfonic, ethanesul-
fonic, hydroxyethanesulfonic, benzenesulfonic, p-toluenesulfonic,
naphthalenesul~onic, sulfanilic, cyc:Lohexylsulfamic acid; or
ascorbic acid. ~or compounds having a free carboxy group, salts are
also derived fro~ bases, e.g. the alkali metal salts such as the
sodium salt.
The novel compounds of the invention are active in state of the art
in vitro and in vivo test systems, indicative of adenosine receptor
agonist activity in mammals.
The adenosine receptor agonists of the inventlon are useful in
mammals including man for the treatment of central nervous system
disorders, particularly psychoses such as schizophrenia, and of
cardiovascular disorders, particularly hypertension and thrombosis.
The above-cited properties are demonstrable in in vitro and in vivo
tests, using advantageously mammals, e.g. rats, dogs, monkeys or
isolated organs, tissues and preparations thereof. Said compounds
can be applied in vitro in the form of solutions, e.g. preferably
aqueous solutions, and in vivo either enterally or parenterally
advantageously orally or intravenously, e.g. withln gelatln cap-
~ules, as starch suspenslons or in aqueous solutions. The dosage in
vitro may range between about 10 5 molar and 10 9 molar concentra-
tions. The dosage in vivo may range between about 0.001
and 25 mg/kgJday, preferably between about 0.0025 and 10 mg/kglday
depending on the eompound and the route of administration.
~X~3843~L
- 12 -
Adenosine-2 (A-2) receptor binding properties, indicative of the
adenosine-2 receptor agonist activity of the compounds of the
invention are determined in vitro by determining their ability to
inhlbit the specific binding of 3H-5'-N-ethylcarboxamidoadenosine
(3H-NECA), e.g. essentially as described by R.F. Bruns et al.,
Mol. Pharmacol. 29, 331 (1986), in striatal membrane preparations
from corpus striatum of male Sprague-Dawley rats. The concentration
of a particular compound required to displace the specific binding
of 4 nM 3H-NECA is determined in the presence of 50 nM cyclopentyl-
adenosine.
Adenosine 1 (A-l) receptor binding properties of the compounds of
the invention indicative of adenosine-l-receptor agonist activity
are determined, e.g., essentially according to R.F. Bruns et al. in
Proc. Natl. Acad. Sci. U.S.A 77, 5547 (1980), by deter~ining their
ability to inhibit the specific binding of 3H-cyclohexyladenosine
(3H-CHA) in rat brain membrane preparations from male Sprague-Dawley
rats. The concentration of a particular compound required to
displace the specific binding of 1 nM 3~-CHA is determined.
Selectivity for the adenosine-2 (A2) receptor can be ascertained by
comparing the relative potency ln the two adenosine receptor assays.
Indicative of in vivo adenosina receptor agonist actirity, the
hypotensive activity of the compounds o~ the invention as well
as their effect on heart rate can be measured in normotensive or
spontaneous hypertensive rats on intravenous or oral administration.
Typically, the blood pressure lowering effect in normotellsive rats
can be determined as follows:
Adult male rats weighing 300-400 g are anesthetized using Inactin
(100 mglkg, i.p.) A femoral artery and contralateral vein are cannu-
lated for dlrect blood pressure measurement and i.v. drug adminis-
tration, respectively. Animals are allowed a 15 minute equilibration
period before testing. Veh~cle ( 1 mlJkg, i.v.) is administered over
3~
- 13 ~
a 30 second period followed by a 0.3 ml saline flush administered
over a 30 second period. Changes in diastolic blood pressure are
recorded using a Beckman polygraph while heart rate is recorded as a
derivative of the blood pressure pulse. The test compound is
administered in the same manner as vehicle and a dose response curve
is established. Percent changes in heart rate and blood pressure are
recorded.
The blood pressure lowering effect in the spontansous hypertensive
rat is determined on oral adminlstration.
The compounds of the invention which are selective as adenosine-2
receptor agonists effectively lower blood pressure without any
significant effect on the heart rate.
Antipsychotic activity can be demonstrated e.g. by measuring the
inhibition of one-way conditioned avoidance or Sidman avoidance in
the rat, or by measuring the antagonism of the behavioral stimulant
effects of apomorphine.
Antithrombotic activity can be demonstrated by measuring the
inhibition of collagen induced platelet aggregation.
Illustrative compounds of the invention display ICso values in the
adenosine-2-receptor binding assay in the range of about 5 x 10 6 to
2 x 10 8M; the compounds also display hypotensive activity in the
anesthesized normotensive rat at a dose of about 0.0025
to 0.035 mg/kg i.v. and in the spontaneous hypertensive rat at a
dose of about 3 to 10 mg~kg p.o.
The compounds of the invsntion of formula I and herein-cited
derivatives thereof can be prepared uslng processes which comprise:
a) for compounds of formula I wherein R, Rs, R`2, R3, R~ and ~ havs
meanin8 as defined hereinabove provided that one of R and Rs
represents hydroxy, condensing a compound of the formula III
~.288431
- 14 -
~H--RI
N/ ~T (III)
N,.
H \N ~ R2
wherein Rl and R2 have meaning as defined above, with a compound of
the formula IV
R~/-\
~ ", (IV)
R3
wherein R3 and R4 have meaning as defined above, in the prssence of
a strong base, and separating any resulting isomers if so required;
b) for compounds of formula I wherein R, Rl, R2, R3, R4 and Rs have
meaning as defined herelnabovel condenslng a compound of the
formula V
~N ~ (V)
\R2
R3 Rs
wherein X represents a leaving group; R, R2, R3, R4 and Rs have
meaning as ~ust deflned above, with a compound of the formula VIa
Rl-NH2 ~VIa~
ln which R1 has meaning as defined above;
' . "'
.
.
,
. .
~2~3~4~
- 15 -
c) for compounds of formula I wherein R2 represents -SR1 or
-N(R9)RI, condensing a compound of the formula VII
~H--Rl
R~ T (VII~
C` ,~
R3 Rs
wherein R, R1, R3, R4 and Rs have meaning as defined above, and X
represent6 a leaving group, with either a compound of the for-
mula VIa
Rl-NH2 (VIa)
wherein R1 has meaning as deined above; or with a compound of the
formula VIb
Rl-SH (VIb)
or a reactive alkali metal salt derivative thereof wherein R1 has
meaning as deined above or with a compound of the ormula VIc
R1-N(R9)~-H (VIc)
wherein Rl and R9 have meaning as defined above;
d) for compounds of Eormula I wherein R4 represents -CONHRs as
defined hereinabove, oxidizing a corresponding compound of the
:
formula I wherein R4 represents hydroxymethyl and wherein other
hydroxy groups are in protected form, and derivatizing th~ so
obtained carboxylic acid to a compound of formula I whereln R4
represents -CONHR6; :~
':
~8~343~
- 16 -
e) for compounds of formula I wherein R4 represents lower alkylthio-
methyl condensing a compound of formula I whereln R4 represents
hydroxymethyl with a di-lower alkyl disulfide in the presence of a
tri-lower alkylphosphine or converting a compound of formula I
wherein R4 represents hydroxymethyl to a reactive esterified
derivative thereof and reacting same with a lower alkylmercaptan or
a reactive alkali metal salt derivative thereo~;
f) for compounds of formula I wherein R2 represents -SRl, reacting a
compound of the formula VII wherein X represents -S~ and wherein R,
R1, R3, R4 and Rs have meaning as defined above with an electro-
philic reagent corresponding to the radlcal Rl; and, as further
required in any of the above-cited processes, temporarily protecting
any interfering reactive group(s) in the starting materials and then
subsequently removing the protecting groups to yield a resulting
compound of formula I; and, if desired, converting a resulting
compound of formula I into another compound of the invention, and if
desired, converting a resulting free compound into a salt or a
resulting salt into a free compound or into another salt, and if
requlred, separating any mixture of isomers or racemates obtained
lnto the single isomers or racemates, and if required, resolving a
racemate into the optical antipodes.
A leaving group in the above processes respresentq especially halo,
for example chloro, bromo or iodo, aliphatically or aromatically
substituted sulfonyloxy, for example methylsulfonyloxy or 4-methyl-
phenylsulfonyloxy (tosyloxy), or aliphatically substituted thio, for
example lower al~ylthio such as methylthio.
In starting compounds and intermediates which are converted to the
compounds of the invention in a manner describ2d herein, functional
groups present, such as amino and hydroxy, are optionally protected
by conventional protecting groups that are common in pr parative
organic chemistry. Protected amino and hydroxy groups are those that
.
, .: . .:
''
.
3~
- 17 -
can be converted under mild conditions into free amino and hydroxy
groups without the molecular framework being destroyed or undesired
side reactions taking place.
Well-known protecting groups that meet these conditions and their
introduction and removal are described, for example, in
J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum
Press, London, New York 1973, T.W. Greene, and "Protective Groups in
Organic Synthesis", Wiley, New York 1984.
For example, a hydroxy group may be protected in the form of esters,
e.g. as acyl derivatives such as the lower alkanoyl, benzyloxycarbo-
nyl or lower alkoxycarbonyl esters, or such hydroxy group may bP
protected in the form of ethers, e.g. as the 2- tetrahydropyranyl,
trityl or benzyl ethers.
Hydroxy group~ on adjacent carbon atoms can also be protected e.g.
in the form of ketals or acetals, such 8S isopropylidene or benzyli-
dene derivatives.
In a resulting protected compound of formula I or intermediate, in
which one or more of the functional groups are protected, the
protected functional groups, e.g. hydroxy groups, can be liberated
in a manner known per se, e.g. by means of solvolysis, especially
hydrolysis with acid, or by hydrogenolysis.
The preparation of the compounds o~ the invention according to
process a~ which involves opening of an epoxide ring is preferably
carried out in a polar solvent such as dimethylformamide and at an
elevated temperature, advantageously at a temperature ranglng from
50 to ~25C. The reactive organometallic derivative, P.g. the
lithium, sodium or potassium derivative of the starting material of
formula III is preferably first prepared in situ by reacting a
compound of the formula III with a corresponding strong base such as
~.X8~431
- 18 -
sodium, potassium or lithium hydride or amide in a polar anhydrous
solvent such as dimethylformamide, advantageously at room tempera-
ture.
Process a) is preferred for compounds of formula I wherein R2
represents hydrogen or halogen.
Starting materials of formula III (adenine and derivatives thereof)
can be prepared according to methods known in the art for the
synthesis and der1vatization of purines, e.g. as illustrated in
Barton and Ellis, Comprehensive Organic Chemistry Vol. 4,
pp. 499-518.
Starting materlals of formula IV are either known in the art or are
preferably prepared as illustrated below. A more specific embodiment
relates to the compounds of formula IVa, particularly the compounds
of formula IVa wherein R4 represents -CONHR6 as defined herein.
\ ~ i) R4~ R~ . / \.
~H ~H
(VIII) (IX) (IVa)
i): selenium dioxide ii): m-chloroperbenzoic acid
A cyclopentene derivative VIII wherein R4 has meaning as defined
herein can be hydroxylated, e.g. with selenium dioxlde in organic
solvents such as tetrahydrofuran and dimethoxyethane, preferably at
reflux temperature to give the hydroxy substltuted cyclopentene
derivative of formula IX. Epoxidation of the cyclopentene deriva-
tives of either formula VIII or IX, e.g. with m-chloroperbenzoie
acid in a solvent such as dichloromethane at room temperature,
yl lds a corresponding epoxide of formula IV wherein R3 represents
hydrogen or wherein R3 represents hydroxy (of formula IVa above),
respectively.
J.~88431
-- 19 --
The epoxidation of the cyclopentene derivatives can also be carried
out under Sharpless epoxidation conditions with t-butyl hydroper-
oxide, preferably in the presence of vanadium or titanium catalysts
such as vanadyl acetylacetonate or titanium tetraisopropoxide.
Asymmetric epoxldation for kinetic resolution of the epoxides-into
the optically active isomers can be similarly carried out in the
presence of e.g. a diester of d- or &-tartaric acid, as described in
Pure and Applied Chemistry 55, 589 (1983).
The preparation of the compounds of the invention according to
process bj which involves the displacement of a leaving group X
(e.g. chloro) in a compound of the formula V by an amine of the
formula VIa is preferably carried out at elcvated temperature, e.g.
at a temperature ranging from 75 to 150C, with an excess of the
amine, in the absence or presence of a solvent, particularly a polar
solvent such as methanol or dimethylformamide, or under elevated
pressure, or in the presence of a base such as triethylamine.
The starting materials of formula V wherein R2 represents hydrogen
or halogen can advantageously be prepared by condensing a compound
of the formula X
HzN
(X)
R3 ~5
wherein X, R and R2-Rs have meaning as defined for compounds of
formula V, optionally in partially protected form, with formic acid
or a mixture of formic acid and acetic anhydrlde, with a lower
alkylcarboxylic acid ester of a di-lower alkoxymethanol or with a
tri-lower alkyl orthoformate, and as required, liberating any
protected hydroxy groups.
:
9 ~43~
- 20 -
The condensation is preferably carried out by reacting a compound
of formula X wlth a tri-lower alkyl orthoformate, such as triethyl
orthoformate ln a polar solvent such as dlmethylacetamide in the
presence of an acid such as concentrated hydrochloric acid, prefer-
ably at room temperature.
The intermediates of formula X can be prepared by condensing e.g. a
compound of the formula XI
~1
H2N\ /-~
~ XI)
C~ \Nf/ \R2
wherein R2 has meaning as defined for formula V, with a compound of
the formula XII
R~ NH2 (XII)
~ "
R3 Rs
whereln R, R3, R4 and Rs have meaning as defined above, e.g.
according to general procedures described in J. Am. Chem. Soc. 91,
3075 (1969) and J. Org. Chem. 45, 531 (1980), preferably in the
presence of a base such as triethylamine.
The compounds of formula XI and XII can in turn be prepared accord-
ing to procedures known in the art, s.g. the compounds of for-
mula XII can be prepared accordlng to Tetrahedron Letters 22, 2331
(1981~ or J. Org. Chem. 45, 531 (1980).
843~
- 21 -
Ths starting materials of formula V wherein R~ represents -SRi or
-N(Rq)RI 7 and X represents a leaving group, can be prepared e.g. by
reacting a compound of formula V wherein X represents hydroxy (or as
the oxo tautomer thereof) which a halogenating agent such as
phosphorus oxychloride.
The intermediates of formula V wherein X represents hydroxy can in
turn be prepared by first converting a compound of formula V, or a
protected derivative thereof, wherein X and R~ both represent chloro
to a compound of formula V wherein X represents hydroxy and R2
represents chloro by hydrolysis with acid and subsequently
converting said intermediate, using methodology as described e.g.
for process (b) and (f), to a compound of formula V wherein X
represents hydroxy and R2 represents -SRI or N(Rg)RI.
The preparation of the compounds of the invention according to
process c) which involves the displacement of the leaving group X
(e.g. chloro) in a compound of formula VII by an amine of the
formula VIa or VIc is carried out essentially as described above
under process b). The displacement by a mercaptan of the formula VIb
is carried out preferably in the presence of a strong base e.g. an
alkali metal hydroxide such as sodium hydroxide.
The starting materials of formula VIa, VIb and VIc are either known
or are prepared using methods known in the art, and as described
herein.
The starting materials of formula VII can be prepared e.g. essPn-
tially as described uDder process a), by reacting the correspond-
ingly substituted purine derivatives with an epoxide of the for-
mula IV.
The preparation of compounds of the invention wherein R4 represents
-CONHR6 according to process d) can be carried out by oxidzing the
corresponding compounds wherein R4 represents hydroxymethyl, the
other hydroxy groups in the molecule being in a protected form, with
~ ~8~3at3~L
e.g. potassium permanganate, and converting the so obtained carbo-
xylic acid to a reactive derivative, e.g. the acid chloride, and
condensing said carboxylic acid chloride with an amine of the
formula R6-NH2, under conditions well known in the art.
Th0 preparation of compounds of the invention wherein R4 represents
lower alkylthiomethyl according to process e) can be carried out by
converting the corresponding compounds wherein R4 represents
hydroxymethyl, the other hydroxy groups being preferably in a
protected form, to e.g. the chloro derivative by treatment with e.g.
thionyl chloride in hexamethylphosphorus triamide (HMPT) and
reacting said chloro derivatlve with e.g. the lithium salt of a
lower alkyl mercaptan, for example lithium methylmercaptide, in a
polar solvent such as tetrahydrofuran.
Alternately, a compound wherein R~ is hydroxymethyl is treated with
a di-lower alkyl disulfide in the presence of e.g. tributylphosphine
at elevated temperature in a polar solvent such as dimethylforma-
mide.
The starting compounds for processes d) and e) can be prepared e.g.
as described in process a).
The preparation according to process (f) of compounds of the
invention whereln R2 represents -SRl is carried out according to
procedures well-known in the art, e.g. by displacement of a leaving
group.
The starting materials of formula VII wherein X represents -SH can
be prepared 8 . g. by reacting a compound of formula VII wherein X
represents a leaving group, e.g. chloro, with an alkali metal
hydrogen sulfide such as sodium hydrogen sulfide.
~ ~3~
- 23 -
The compounds of the invention or intermediates leading thereto can
be converted lnto other compounds of the invention or corresponding
intermediates using chemical methodology known in the art and as
illustrated herein.
The compounds of formula I wherein R2 represents halogen, e.g.
chloro~ can be converted according to process c) as described above
to compounds of formula I wherein R2 represents -NHRI.
The conversion of compounds of formula I containing free hydroxy
groups to ester derivatives thereof may be carried out by condensa-
tion with a corresponding carboxylic acid, advantageously as a
reactive functional derivative thereof, according to acylation
(esterification) procedures well-known in the art.
The compounds of formula I wherein R4 represents hydroxymethyl
~amino and any other hydroxy groups in the molecule being in
protected form) can be converted to compounds of formula I wherein
R4 represents lower alkoxymethyl by condensation with an equivalent
amount of e.g. a lower alkyl halide such as a lower alkyl iodide in
the presence of an equivalent amount of a strong base, such as
sodium hydride in a non-aqueous solvent, such as dimethylformamide.
A compound of formula I containing a primary amino group (e.g.
wherein NHRI is NHz) may be converted to a compound of formula I
wherein NHRI represents a secondary amine, e.g. wherein Rl repre-
sents lower alkyl, by treatment with a reactive derivative of the
alcohol corresponding to Rl e.g. with a lower alkyl halide such as a
lower alkyl iodide, accordin~ to methodology well-known in the art
for alkylation of amines.
A compound of formula I containing a lower alkoxycarbonyl group,
e.g. a compound of formula I wherein the substituent Z represents
lower alkoxycarbonyl, can be converted to the corresponding
~.2~4~
- 24 -
carboxyllc acid by hydrolysis of the ester, e.g. with aqueous base,
such as sodium hydroxide or potassium hydroxide solution or with
dilute mineral acid, e.g. hydrohalic acid.
A compound of the invention wherein ~ represents carboxy can be
converted to the corresponding compounds wherein Z represents lower
alkoxycarbonyl according to known esterification procedures, e.g. by
treatment as a reactive functional derivative, such as an acyl
halide or a mi~ed anhydride e.g. derlved from a lower alkyl halo-
carbonate such as ethyl chloroformats, with the appropriate alcohol.
The above-mentioned reactions are carried out according to standard
methods, in the presence or absence of diluents, preferably such as
are inert to the reagents and are solvents thereof, o catalysts,
condensing or said other agents respectlvely and/or inert atmos-
phere~, at low temperatures, room temperature or elevated tempera-
tures preferably near the boiling point of the solvents used, at
atmospheric or superatmospheric pressure.
.
The invention further includes any variant of the present processes,
in which an intermediate product obtainable at any stage thereof is
used as starting material and the remaining ~teps are carried out,
or in which the starting materials are formed under the reaction
conditions, or in whlch the reaction cnmponents are used in the form
of their salts or optically pure antipodes. Whenever desirable, the
above processes are carried out after first suitably protecting any
potentially interfering reactive functional groups, e.g. as
illustrated herein.
Advantageously, those starting materials should be used in said
reactions that lead to the formation of those compounds indicated
above as being preferred.
The invention also relates to novel starting materials and processes
for their manuf~cture.
. . , ~ ~ ., .
~ Z81343~L
- 2~ -
Depending on the choice of starting materials and methods, the new
compounds may be in the form of one of the possible isomers, for
example, as dlastereomers, as optical isomers (antipodes), as
racemates, or as mixtures thereof.
In case diastereomeric mixtures of the above compounds or intermedi-
ates are obtained, these can be separated into the single racemlc or
optically active isomers by methods in themselves known, e.g. by
fractional distillation, crystalllzation or chromatography.
The racemic products of formula I or basic intermediates can be
resolved into the optical antipodes, for example, by separation of
diastereomeric salts thereof, e.g., by the fractional crystalliza-
tion of d- or ~-(tartrate, dibenzoyltartrate, mandelate or camphor-
sulfonate) salts.
Advantageously, the more active of the antipodes of the compounds of
thls inventlon is isolated.
Finally, the compounds of the invention are either obtained in the
free form, or as a salt thereof. For example, any resulting free
base can be converted into a corresponding acid addition salt,
preferably with the use of a pharmaceutically acceptable acid or
anion exchange preparation, or resulting salts can be converted into
the correspondlng free bases, for example, with the use of a
stronger base, such as a metal or ammonium hydroxide, or any basic
salt~ e.g., an alkali metal hydroxide or carbonate, or a cation
exchange preparation. These or other salts, for example, the
picrates, can also be used for purification of the bases obtained;
the bases are then first converted into salts. In view of the close
relationship betwe~n the free compounds and the compounds in the
form of their salts, whenever a compound is rsferred to in this
context, a corresponding salt is also intended, provided such is
possible or appropriate under the circumstances.
- ', . '
.
2~
26 -
The compounds, including their salts, may also be obtained in the
form of their hydrates, or include other solvents used for the
crystallization.
The present invention also relates to the use of the compounds of
the invention for the preparation of pharmaceutical compositions,
especially pharmaceutical compositions having selective adenosine
(particularly adenosine-2) receptor stimulating activity which can
be used for the treatment of e.g. psychotic conditions, such as
schizophrenia, and cardiovascular conditions, such as hypertension,
thrombosis and atherosclerosis.
The pharmaceutical compositions according to the invention are those
suitable for enteral, such as oral or rsctal, transdermal and
parenteral administration to mammals, including man, for the
treatment of diseases responsive to adenosine (particularly adeno-
sine-2) receptor stimulation as given above, such as hypertension,
comprising an effective adenosine-2 receptor stimulating amount of a
compound of the invention, alone or in combination with one or more
pharmaceutically acceptable carriers.
The pharmacologically active compounds of the invention are incor-
porated into pharmaceutical compositions comprising an effective
amount thereof in conjunction or admixture with excipients or
carriers suitable for either enteral or parenteral application.
Preferred are tablets and gelatin capsules comprising the active
lngredient together with a) diluents, e.g. lactose, dextrose,
sucrose, mannitol, sorbitol, cellulose andjor glycine; b~ lubri-
cants, e.g. silica, talcum, stearic acid, its magnesium or calcium
salts and/or polyethylene glycol; for tablets also c? binders, e.g.
magnesium aluminum silicate, starch paste, gelatin, tragacanth,
methylcellulose, sodium carboxymethylcellulose and/or polyvinyl-
pyrrolidone; if desired, d) disintegrants, e.g. starches, agar,
alginic acid or its sodium salt, or effervescent mixtures; and/or
e) absorbents, colorants, flaYors and sweeteners. Injectable
- 27 -
compositions are preferably aqueous isotonic solutions or suspen-
sions, and supposltories are advantageously prepared from fatty
emulsions or suspensions. Said compositions may be sterilized and/or
contain adjuvants, such as preserving, stabilizing, wetting or
emulsifying agents, solution promoters, salts for regulatlng the
osmotic pressure and/or buffers. In addition, the compositions may
also contain other therapeutically valuable substances. Said
compositions are prepared according to conventional mlxing, gran-
ulating or coating methods, respectively, and contain about
0.1 to 75 %, preferably about 1 to 50 %, of the active ingridient.
Suitable formulations for transdermal application include an
effective amount of a compound of the invention with carrier.
Advantageous carriers include absorbable pharmacologically accept-
able solvents to assist passage through the skin of the host.
Characteristically, transdermal devices are in the form of a bandage
comprising a bac~ing member, a reservoir containing the compound,
optionally with carriers, optionally a rate controlling barrier to
deliver the compound to the skin of the host at a controlled and
predetermined rate over a prolonged period of time, and means to
secure the device to the skin.
The present invention also relates to the use of compounds of the
invention having adenosine receptor stimulating properties and
pharmaceutical compositions comprising said compounds for the
treatment in mammals of disorders responsive to selective adenosine
receptor stimulation, particularly psychotic conditions (e.g.
schizophrenia) and cardiova~cular conditions (e.g. hypertension and
thrombosis~.
One aspect relates advantageously to the method of treatment of
cardiovascular disorders in mammals, e.g. such responsive to
adenosine (particularly adenosine-2) receptor stimulation, for
example hypertension, using an effective amount of a compound of the
invention, preferably in the form of above-cited pharmaceutlcal
compositions.
38~3~
- 28 -
The dosage of active compound administered is dependent on the
species of warm-blooded animal (mammal), the body weight, age and
indlvidual condltion, and on the form of administration.
A unit dosage for a mammal of about 50 to 70 kg may contain between
about 1 and 50 mg of the active ingredient.
The following examples are intended to illustrate the invention and
are not to be construed as being limitations thereon. Temperatures
are given in degrees Centigrade. If not mentioned otherwise, all
evaporations are performed under reduced pressure, preferably
between about 2 and 13 k~a. The structure of final products,
intermediates and starting materials ls confirmed by analytical
methods, e.g. ~icroanalysis and spectroscopic characteristics
~e.g. MS, IR, NMR).
The numbering of the positions of the adenine and or purine rings is
as conventionally used in the art (e.g. Merck Index, tenth edition).
Unless otherwise indicated the final products are preferably
isolated as the free base by crystallization from a mixture of an
alcohol (methanol, ethanol or isopropanol) and ethyl ether.
Example 1: To a suspension of 1.2 g sodium hydride (60 %, washed
with dry ether) in 20 ml of dry dimethylformamlde is added 2.58 g
adenine. After 10 minute3 stirring at room temperature the resulting
mixture ls treated with a solution of 4.9 g 2-alpha-hydroxy-3-
alpha-4-alpha-epoxy-cyclopentane-1-~-N-ethylcarboxamide in 30 ml of
dry dlmethylformamide, heated at 105 for 18 hours, then cooled to
room temperature. The reaction mixture is quenched with water and
concentrated under vacuo. The crude product iq purified by reverse
phase chromatography using 120 g of reverse phase octadecylsilane
(C1g)-bonded silica gel to yield a) 2-alpha-4-alpha-dihydroxy-3~-
(9-adenyl)-cyclopentane-1~-N-ethylcarboxamide, m.p. 250-253~C, and
b) 2-alpha-3-alpha-dlhydroxy-4-~-(9-adenyl)-cyclopentane~ -N-
~2~384~L
- 29 -
ethylcarboxamide (the compound of formula IIa wherein R2, R7 and R3
are hydrogen and R6 is ethyl) which is recrystallized from methanol,
m.p. 208.5-209C; NMR(CD30D): 8.36 (lH,s); 8.18 (IH,s); 4.5 (lH,dd~;
4.28 (lH,dd).
The starting material is prepared as follows: To a mixture of 750 ml
o~ dry tetrahydrofuran and 375 ml of dry dimetho~yethane is added
15 g 3-cyclopentene-1-N-ethylcarboxamide, ~ollowed by 12 g selenium
dioxide at room temperature. After the reaction i5 heated to 70C
with mechanical stirring overnight and cooled to room temperature,
the resulting solution is filtered through Celite~. Ths filtrate i8
concentrated under vacuo and the residue is chromatographed on 400 g
silica gel using 4 % methanol in ethyl acetate as eluent to give
2-alpha-hydroxy-3-cyclopentene-1-B N-ethylcarboxamide, m.p. 48-50C.
A solution o~ 4.6 g 2-alpha-hydroxy-3-cyclopentene-1-B-N-ethyl-
carboxamide, 10.23 g m-chloroperbenzoic acid and 70 ml of dichloro
methane is stirred at room temperature for 2 hours. The 601vent is
removed under reduced pressure and the residue is partltioned
between ether and water. The aqueous layer is concentrated in vacuo
to obtain 2-alpha-hydroxy-3-alpha-4-alpha-epoxy-cyclopentane-1-B-
N-ethylcarboxamide, m.p. 83-85C.
Similarly prepared are:
c) 2-alpha-3-alpha-dihydroxy-4-B-19-(2-chloroadenyl)]-cyclopentane-
1-B-N-ethylcarboxamide, m.p. 233-237C; NMR(CD30D~; 8.34 (lH,s);
4.43 (lH,dd); 4.25 (lH,t);
d) 3 alpha-hydroxy-4-B-t9-adenyl)-cyclopentane-1-B-N-ethylcarbox-
amide, m.p. 282-285~C; NMR(CD30D): 8.17 (lH,s); 8.15 (lH,s);
4.7 (l~l,s);
~) 2-alpha-3-alpha-dihydroxy-4-~-(9-adenyl)-cyclopentane-1-B-N-
cyclopropylcarboxamide, m.p. 218-220C; NMR(CD30D~: 8.20 (lH,s);
4.47 (lH,dd); 4.25 (lH,t);
~ 28843~
- 30 -
f) 2-alpha-4-alpha-dihydroxy-3-~-(9-adenyl)-cyclopentane-1-~-N-
cyclopropylcarboxamide, m.p. above 250C;
g) 3-alpha-hydroxy-4-~-[9-(2-chloroadenyl)~-cyclopentane-1-~-N-
ethylcarboxamlde, m.p. 197-200C; NMR(CD30D): 8.1 (lH,s);
4.13 (l~,q).
The starting material for compounds c and d, trans-3,4-epoxy-cyclo-
pentane-1-N-ethylcarboxamide, is prepared by epoxldation of
3-cyclopentene-1-N-ethylcarboxamide with m-chloroperbenzoic acid
followed by chromatographic purificatlon on silica gel (using 2 %
methanol in ethyl acetate as eluent).
Example 2:
a) A stirred mixture of 58 mg of 2-alpha-3-alpha-dihydroxy-4-~-[9-
(2-chloroadenyl)]-cyclopentane-1-~-N-ethylcarboxamide and 1.5 ml of
freshly distilled 2-phenylethylamine is heated to 130C for 14 hours
and cooled to room temperature. Excess 2-phenylethylamine is removed
under vacuo and the residue i9 triturated with ether. The solid
which is obtained after filtration is recrystallized from methanol
to give 2-alpha-3-alpha-dihydroxy-4-~-[2-(2-phenylethylamino)-9-
adenyl]-cyclopentane-1-~-N-athylcarboxamide, the compound of
formula IIa wherein R2 is 2-phenylethylamino, R6 is ethyl, R7 and R3
are hydrogen; m.p. 234-236C; NMR(CD30D): 7.9 (lH,s); 4.46 (lH,t);
4.33 (lH,t).
Similarly prepared are:
.
b) 3-alpha-hydroxy-4-~-[2-(2-phenylethylamino)-9-adenyl]cyclo-
pentane-1-~-N-ethylcarboxa~ide, m.p. 220-224~C; NMR~CD30D): 7.73
(lH,s); 3.22 (2H,g); 2.91 (2H,t); 2.48 (2H,t);
c) 2-alpha-4-alpha-dihydroxy-3-~-[2-(2-phenylethylamino)-9-adenyl]-
cyclopentane~ -N-ethylGa~boxamide, m.p. 224-226C; NMR(CD30D):
7.8 (IH,s); 4.25 (lH,dd);
~L2~
- 31 -
d) 2-alpha-3-alpha-dihydroxy-4-~-[2-(2-pyridylethylamino)-9-adenyl3-
cyclopentane-1-B-N-ethylcarboxamide; NMR(CD30D): 7.93 (lH,s); 4.47
(lH,t); 4.37 (lH,t).
Example 3: To a suspension of 241 mg sodium hydride (60 %, washed
with dry ether) in 8 ml dry dimethylformamide i~ added ~32 mg of
2-chloroadenine. After 10 minutes stirring at room temperature, the
resulting mixture is treated with 800 mg of l-~-hydroxymethyl-Z-
alpha-hydroxy-3-alpha-4-alpha-epoxycyclopentane in 5 ml dry di-
methylformamide and heated at 105C for 18 hours, then cooled to
room temperature. The reaction mixture is quenched with water and
concentrated under vacuo. The crude product is chromatographed on
100 g rever~e phase octadecylsilane (Clg)-bonded silica gel (eluent,
5 % methanol in water) to give a) 2-alpha-4-alpha-dihydroxy-1-~-
hydroxymethyl-3-~-[9-(2-chloroadenyl)]-cyclopentane, m.p. 252-254C,
and b) 2-alpha-3-alpha-dihydroxy-1-~-hydroxymethyl-4-~-[9-(2-chloro-
adenyl)~-cyclopentane, m.p. 236-238C; NMR(CD30D): 8.17 (lH,s);
4.5 (lH,dd); 4.04 (lH,dd).
The starting material i9 preparad as follows: A solution of 4.15 g
of 1-hydroxymethyl-3-cyclopentene in S00 ml of dry tetrahydrofuran
and 250 ml of dimethoxyethane is treated with 4.15 g selenium
dioxide at room temperature. The resulting mixture is heated at 70C
for 18 hours, then cooled to room temperature and filtered through
Celite~. The filtrate i5 concentrated under vacuo and the residue i3
chromatographed on 100 g of silica gel (eluent, 4 % methanol in
ethyl acetate) to give trans-l-hydroxymethyl-2-hydroxy-3-cyclopen-
tene.
A solution of B18 mg trans-1-hydroxymethyl-2-hydroxy-3-cyclopentene,
2.2 g m-chloroper~enzoic acid and 40 ml of dichloromethane is
~tirred at room temperature for 80 =inutes. The solvent ls removed
unter reduced pressure and residue is partitioned ~oetween ether and
~28l~
- 32 -
water. The aqueous layer is concentrated in vacuo to obtain
hydroxymethyl-2-alpha-hydroxy-3-alpha-4-alpha-epoxy-cyclopen
tane.
Example 4: A suspension of 40 mg of sodium hydride (60 %, washed
with ether) in 3 ml of dimethylformamide is treated with 200 mg of
2-(2-phenylethylamino~adenine at room temperature. Stirring is
continued at room temperature until the reaction mixture becomes
homogeneous. The reaction mixture is then treated with 160 mg
2-alpha-hydroxy-3-alpha-4-alpha-epoxy-cyclopentane-l-~-N-ethyl-
carboxamide. The resulting solution is heated at 100C for
14 hours. The reaction mixture is then cooled to room temperature
quenched with water and the solvent is removed under vacuum. The
crude product mixture is first chromatographed in silica gel eluting
with 10 % MeOH~CHzCl2 to 15 % Me0H/CH2Cl2. The restllting product is
then chromatographed on reverse phase octadecylsilane (Clg)-bonded
silica gel eluting with up to 50 % methanol in water; a mixture of
lsomers, 2-alpha-3-alpha-dihydroxy-4-~-[2-(2-phenylethylamino~-
9-adenyl]-cyclopentane-1-~-N-ethylcarboxamide of example 2a and
2-alpha-4-alpha-dihydroxy-3-~-[2-(2-phenylethylamino)-9-adenyl]-
cyclopentane-1-~-N-ethylcarboxamide is obtained.
The starting material 2-(2-phenylethylamino)adenine is prepared by
reacting 2-chloroadenine with excess 2-phenylethylamine at about
1 ~OC .
Example 5: To a stirring solution of 2.7 g of 4-~-(5-amino-6-chloro-
4-pyrimidinylamino)-2-alpha-3-alpha-dimethylmethylenedioxy-cyclopen-
tane-1-~-N-ethylcarboxamide in 40 ml of triethyl orthoformate is
added 0.7 ml of concentrated hydrochloric acid. The solution is
stirred at room temperature for 24 hours. After removal of the
solvent, 4-~-(6-chloro-9-purinyl)-2-alpha-3-alpha-dimethylmethylene-
dioxy-cyclopentane-1-~-N ethylcarboxamide 1s obtained as an oil,
which is used in the next step without further purification.
~2~
- 33 -
A solution o~ 2.2 g of 4-~-(6-chloropurin-9-yl)-2-alpha-3-alpha-di-
methylmethylenedioxy-cyclopentane-~ N-ethylcarboxamide in 30 ml of
saturated methanolic ammonia is heated overnight in a sealed tube.
After removing the solvent, the residue is heated at 55C in 15 ml
lN hydrochloric acid for 1.75 hours. The solvent is removed under
vacuum to give an amorphous solid.
Chromatography on a flash silica gel column eluting using a
methanol/dichloromethane gradient gives 4-~-(9-adenyl)-2-alpha-
3-alpha-dihydroxycylopentane-1-~-N-ethylcarboxamide, identical to
the compound of example lb.
The starting material is prepared as ~ollows: A mixture o~ 10.5 g of
5,6-dimethylmethylenedioxy-2-azabicyclo[2.2.1]heptan-3-one and 50 ml
of ethylamine is heated at 140C in a steel bomb overnight. After
removal of the solvent, the residue is purified by column
chromatography (silica gel) using a gradi~nt of dichloro-
methane/methanol as eluent to give 4-~-amino-2-alpha-3-alpha-
dimethylmethylenedioxy-cyclopentane-1-~-N-ethylcarboxamide.
To a solution of 5.3 g of 4-~-amino-2-alpha-3-alpha-dimethylmethy-
lenedioxy-cyclopentans-l-~-N-ethylcarboxamide in 70 ml of n-butanol
is added 7.5 g of 5-amino 4,6-dichloropyrimidine followed by ~.2 ml
of triethylamine. After the reaction is heated for 24 hours at
150C, the solvent is removed in vacuo. The residue is partitioned
between ethyl acetate and a saturated sodium bicarhonate solution.
The organic layer is dried over sodium sulfate and concentrated to
dryness. The crude solid is chromatographed on a silica gel column
eluting using a hexane/ethyl acetate gradient to yleld 4-~-(5-amino-
6-chloro-4-pyrimidinylamino)-2-alpha-3-alpha-dimethylmethyIenedioxy-
cyclopentane-l-~-N-ethylcarboxamide.
b) Simllarly prepared is 2-alpha-3-alpha-dihydroxy-4-~-[9-(2-chloro-
adenyl)]-cyclopentane-l-~-N-ethylcarboxamide, the compound o~
example lc.
34~
- 34 -
The starting ~aterial is prepared as follows: To a solution of
614 mg 5-amino-2,4,6-trichloropyrimidine and 813 mg 4-~-amino-
2-alpha,3-alpha-dimethylmethylenedioxy-cyclopentane-1-~-N-ethyl-
carboxamide in 15 ml of n-butanol is added 1 ml of dry triethyl-
amlne; the resulting mixture ls heated at reflux for 15 hours and
cooled to room temperature. ~11 the volatiles are evaporated and the
resldue ls partltioned between dichloromettlane and water. The
organic layer is dried over magneslum sulfate and concentrated to an
oil. The crude product is chromatographed on 100 g silica gel using
1:1 hexane and ethyl acetate as eluent to give 4-~-(5-amlno-2,6-
dlchloro-4-pyrlmldlnylamino)-2-alpha-3-alpha-dlmethylmethylene-
dioxy-cyclopentane-1-~-N-ethylcarboxamide as an amorphous solid.
Example_6: A stirred mixture of 26 mg 2-alpha-3-alpha-dihydroxy-1-~-
hydroxymethyl-4-~-19-(2-chloroadenyl)~-cyclopentane and 0.3 ml
freshly distilled 2-phenylethylamine is heated at 130C for 14 hours
and cooled to room temperature. Excess 2-phenylethylamine i3 removed
under vacuo and the residue is triturated with ether. The solid
which is obtained after filtration is purified by flash column
chromatography using a reverse phase octadecylsilane (C1g)-bonded
silica gel packing and eluting with methanol/water (5:3 to 1:1). The
resulting product is then crystallized from ethanol/ether to give
2-alpha-3-alpha-dihydroxy-1-~-hydroxymethyl-4-~-[2-(2-phenylethyl-
amino~-9-adenyl]-cyclopentane, m.p. 118-120C; ~MR(CD30D~:
7.73 (1H,s); 7.18 (5~,m); 2.85 (2H,t~.
Example 7: The following compounds of formula II wherein R3 repre-
sents hydroxy can be prepared substantially according to the general
procedures previously described herein.
~2~3~34~
- 35 -
Compound _ R ~e~
(a, NH(CH2)2-p-C~H4-CH2CH2COOH CH2CH3 235-237C
(b) NH(CH2)2-p-C6H4-OCH2COOH CH2CH3
(c) 3,4-dihydro-5-methoxy-2H[l]benzothio-
pyran-3-ylamino CH2CH3
~d) 2-indanylamino CHzCH3
(e) 1,2,3,4-tetrahydro-2-naphthylamino CH2CH3
(f) 3,4-dihydro-2H-[l]-benzopyran-3-ylamino CH2CH3
(g) H CH2CH20H
(h) SCH2CH2C6Hs CH2CH3
(i) NH(CH2)2-p-C6H4-OCH2CH[2COOH CHzCH3
(;) 2-cyclohexylethylamino CH2CH3 228-232C
(k) 1-naphthylmethylamino CH2CH3 223-225C
(l) N methyl-2-phenethylamino CH2CH3 194-196C
(m) 2-p-methoxyphenyl-1-methyl-ethylamino CH2CH3
(n) 2-phsnylpropylamino CH2CH3 210-212C
(o) NH(CN2)2-p-C6H4-CHzCOOH CHzCH3
(p) NH(C}12)2-p-C6H4-CHzCON(CH3)z CH2CH3
(q) NH(CHz)2-p-C6H4Cl CHzCH3
~r) NHCHzCH2CH20H CH2CH3 173-176C
Compound (a) can be advantageously prepared as follows: p-Bromophenyl-
acetonitrile is first condensed with t-butyl acrylate under conditions
of the palladium acetate catalyzed Heck reaction. The resulting
acrylate is hydrogenated with palladium on charcoal catalyst followed
by reduction (of the cyano group) with sodium borohydride in the
presence of cobalt(II) chloride to yield t-butyl p-(2-aminoethyl)-
phenylpropionate. Condensation with 2-alpka-3-alpha-dihydroxy-4-~-[9-
(2-chloroadenyl)]-cyclopentane~ -N-ethylcarboxamide yi~lds the
t~butyl ester of compound (a) which is hydrolyzed to compound (a) with
aqueous hydrochloric acid.
1288~3~
- 36 -
Compound b) i9 similarly prepared. The starting material for
condensation with the 9-(2-chloroadenyl)cyclopentane derivative i8
prepared by condensation of p-hydroxyphenylacetonitrile with t-butyl
bromoacetate in the presence of potassium carbonate.
The starting material for compound (c) is prepared as follows: To a
cooled mixture of 30.6 g of m-methoxybenzenethiol, 54.4 g of 45 %
potassium hydroxide in 100 ml of dimethylsulfoxide is added 36.0 g
of alpha(bromomethyl)acrylic acid in 25 ml of dimethylsulfoxide at
such a rate as to maintain the reaction temperature at 50-55C.
After 1 hour the reaction mixture is diluted with water and washed
with ether. After acidification, the product is extracted with
ether, the organic layer is dried over magnesium sulfate and th2
solvent is re~oved in vacuo to afford alpha-(3-methoxybenzenethio-
methyl)acrylic acid. This material is dissolved in 570 ml of
o-dichlorobenzene and 7.2 g of triethylamine and heated to 200C for
5 hours. After cooling, the products are extracted with sodium
bicarbonate solution, the aqueous layer is acidified and the
products extracted with ether. After drying over magnesium sulfate,
the solvent i9 removed ln vacuo to afford a mixture of 3,4-dihydro-
5-methoxy-2H-[l]-benzothiopyran-3-carboxylic acid and 3,4-dihydro-
7-methoxy-2H-[11-benzothiopyran-3-carboxylic acid.
This mixture of acids is dissolved in 500 ml of t-butyl alcohol and
treated with 17 g of trlethylamine and 35 ml of diphenylphosphoryl
azide. After 5 hours reflux, the solvent is removed in vacuo and the
residue is dissolved in ether and washed with lN sodium hydroxide
and lN hydrochloric acid. After drying over magnesium sulfate, the
solvent is removed in vacuo and the residue is chromatographed on
silica gel (1 kg) with ether/hexane (1:4) as the eluent to afford in
succession N-t-butoxycarbonyl-3,4-dihydro-5-methoxy-2H-[1]-benzo-
thiopyran-3-amine and N-t-butoxycarbonyl-3,4-dihydro-7-methoxy-2H-
[1]-benzothiopyran-3-amine.
~28~
- 37 -
A solution of 10 g of N-t-butoxycarbonyl-3~4-dlhydro-5-msthoxy-2H~
ll]-benzothiopyran-3-amine in 30 ml of trifluoroacetic acid is kept
at room temperature for 1 hour. The solvent is removed in vacuo, the
residue is treated with lN sodium hydroxide and the product is
extracted with ether. After drying over magnesium sulfate, the
solvent is removed in vacuo to afford 3,4-dihydro-5-methoxy-2H-[1]-
benzothiopyran-3 amine as an oil.
The starting material for compound (o) is prepared as follows: A
mixture of 20 g of p-bromophenylacetic acid, 30 ml of ether, 1 ml of
sulfuric acid and 35 ml of isobutylene is shaken in a sealed bottle
for 24 hours. The reaction mixture is diluted with ether and washed
with sodium hydroxide solution. After drying over magnesium sulfate
the ether is removed in vacuo to afford the t-butyl ester as an oil. A
mixture of 9.6 g of this material is refluxed with a mixture of 6.1 g
of N-vinylphthalimide, 160 mg of palladium acetate, 800 mg of
tri~o-tolylphosphine, 10 ml of acetonitrile and 8 ml diisopropyl-
ethylamlne for 24 hours. The reaction is diluted with water, the
resulting precipitate is collected and recrystallized from
methanoljdichloromethane. The resulting solid is hydrogenated at
400 kPa pressure over 2 g of 10 % palladium on carbon catalyst in
100 ml of ethanol and 100 ml of tetrahydrofuran for 16 hours a$ room
temperature. After removal of the solvent in vacuo the residue is
heated at reflux with 10 ml of hydrazine hydrate and 20 ml of ethanol
for ~ hours. The reaction is diluted with ether and washed with 5 %
potassium hydroxide solution. The ether is dried over magnesium
sulfate solution and the solvent is removed in vacuo. The residue is
chromatographed on silica gel with 5 % ammonia saturated methanol in
dichloromethane as the eluent, to afford p-(t-butoxycarbonylmethyl)-
Z-phenethylamine as an oil.
The starting material for compound (p) is prepared QS follows: A
mixture of 6 g of p-bromophenylacetic acid in 100 ml of dichloro-
methane and 5 ml of oxalyl chloride is stirred at room temperature for
16 hours. After removal of the solvent in vacuo the residue is
dissolved in dichloromethane and treated with excess dimethylamine at
.
~lX8~3~L
- 38 -
room temperature. After 1 hour the reaction mixture ls washed with
water, the organic layer ls d}ied over magnesium sulfate and the
solvent i6 removed in vacuo to afford p-bromo-N,N-dimethyl-phenyl-
acetamide as an oil, which is converted to p-(dimethylaminocarbonyl-
methyl)-2-phenethylamine as described for the starting material above.
Example 8: 2-alpha-3-~lpha-dihydroxy-1-~-hydroxymethyl-4-~-(5-amino-
2,6-dichloro-4-pyrimidinylamino)-cyclopentane (480 mg) i~ treated with
8.0 ml triethyl orthoformate and 0.1 ml concentrated HCl, and the
reaction mixture is stirred at room temperature for one hour. The
reaction mixture is then concentrated under vacuum and the residual
oil is dissolved in 20 ml of saturated methanolic ammonia, the
solution is placed in a sealed tube and heated at 60C for
12-16 hours. The solvent is removed under vacuum and the residue
heated with 30 ml lN HCl at 60C for 2 hours. The solvent is removed
under vacuum and the crude product is separated by flash chromato-
graphy on a reverse phase octadecylsilane (Cl~)-bonded silica gel
packing, eluting first with water, followed by 5 %, lO % and 20 %
methanol in water to yield 2-alpha-3-alpha-dihydroxy~ -hydroxy-
methyl-4-~-(2-chloro-9-adenyl)-cyclopentane (see example 3); NMR
(CD30D): 8.2 (lH,s); 4.50 (lH,dd); 4.03 (lH,dd~; 3.70 (2H,m);
2.46 (lH,m); 2.25 (lH,m); 1.90 (lH,m).
The starting material is prepared as follows: A mixture of 5.2 g of
methyl 4-~-amino-2-alpha-3-alpha-dihydroxycyclopentans-1-~-carboxylate
(Tetrahedron Letters 1981, 2331), 3.4 g of 5-amino-2,4,6-trichloro-
pyrimidine and 5.2 ml of triethylamine in 60 ml n-butanol ii heated to
reflux under nitrogen atmosphere for 16 hours. The reaction mixture is
cooled to room temperature and the solvent removed under vacuum. The
residue is partitioned between ethyl acetate and water. The organi~
soIution i9 extracted with saturated sodium chloride solution, dried
over magnesium sulfate and evaporated to dryness. The crude product i~
purified by flash column chromatography on silica gel, eluting with
ethyl acetate and then 10 % methanol/ethyl acetate to yield methyl
2-alpha-3-alpha-dihydroxy-4-~-(5-amino-2,6-dichloro-4-pyrlmidinyl-
~ ~:88~31
- 39 -
amino)cyclopentane-1-~-carboxylate as a white solid. NMR (CD30D):
4.43 (lH,q); 4.25 (lH,t); 3.95 (lH,t); 3.72 (3H,s); 2.94 (lH,m);
2.60 (lH,m); 1.70 ~lH,m).
Calcium chlorlde (466 mg) and sodium borohydride (320 mg) are combined
in 30 ml tetrahydrofuran at room temperature. The reaction mixture is
stirred at room temperature for one hour, then 700 mg of methyl
2-alpha-3-alpha-dihydroxy-4-~-(5-amino-2,6-dichloro-4-pyrimidinyl-
amino)cyclopentane-1-B-carbox~late in 30 ml tetrahydrofuran is added.
The reaction mixture is stirred at room temperature for two days. The
reaction mixture is treated with 14 ml acetic acid at room temperature
and stirring is continued for two hours. The solvent is removed under
vacuum to afford an amorphous solid. The crude product mixture i8
separated by flash column chromatography on reverse phase octadecyl-
silane (C~ bonded silica packing, eluting wlth methanol/water (first
1:9, then 2:~, then 3:7) to yield 2-alpha-3-alpha-dihydroxy-1-~-
hydroxymethyl-4-~-(5-amino-2,6-dichloro-4-pyrimidinylamino)-cyclo-
pentane; NMR (CD30D): 4.40 (lH,q); 3.9 (2H,dd); 3.6 (2H,dd);
2.4 (l~l,m); 2.15 (lH,m) 1.28 (lH,m).
2-alpha-3-alpha-dihydroxy-1-~-hydroxymethyl-4-~-(2-chloro-
9-adenyl)-cyclopentane (150 mg) i9 treated with 3.0 ml phenethylamine
and the mixture is heated at 130C for 5.5 hours. The residue is
triturat~d with an ethyl etherlwater mixture. The layers are separated
and the aqueous layer i9 combined with the insoluble material and
concentrated to an oil. The crude product mixture i8 separated by
flash column chromatography on reverse phase Clg-octadecylsilane
(C1g)-bonded silica gel packing, eluting first with water, followed by
10 %, 30 %, 50 % methanol in water to yield 2-alpha-3-alpha-
dihydroxy~ -hydroxymethyl-4-~-[2-(2 phenylethylaminoj 9-adenyl]-
cyclopentane as a white solid; m.p. 149-150~C; NMR (d6-DMSO):
7.75 (lH,s); 7.28 (SH,m); 6.7 (2H,bs); 6.22 (lH,t); 5.00 (lH,d);
4.69 ~lH,t); 4.53 (2H,m); 4.37 (2H,t); 3.84 (lH,m).
~I X~38~
- 40 -
Example 10:
a) To 2.5 g of (~)-4-~-(5-amino-2,6-dichloro-4-pyrimidinyl-amino)-
2-alpha-3-alpha-dimethylmethylenedioxy-cyclopentane-l-B-N-ethyl-
carboxamide is added 40 ml triethyl orthoformate and 0.5 ml of
concentratsd hydrochloric acid at room temperature. After stirring at
room temperature for three hours the reaction mixture is concentrated
under high vacuum to afford a yellow oil. The oil i9 dissolved in
100 ml of a saturated solution of ammonia in methanol, and heated at
65-70C in a steel pressurs reactor overnight. The reaction mixture ls
cooled to room temperatu}e and solvent removed under vac~um to afford
crude product. Chromatography on silica gel, eluting with dichloro-
methane followed by dichloromethane containing ~p to 10 % of methanol,
gives (+)-2-alpha-3-alpha-dimethylmethylenedioxy-4-~-(2-chloro-9-
adenyl)-cyclopentane-1-~N-ethylcarboxamide as a white amorphous
solid; ~Ds~ +2.80 (CS1.5~ methanol); NMR (CD30D): 8.25 (lH,s);
5.05 (2H,m~; 3.23 (2H,q); 2.94 (lH,m); 2.54 (2H,t); 1.58 (3H,s);
1.31 (3H,s); 1.12 (3H,t).
A mixture of 1.8 g of (~)-2-alpha 3-alpha-dimethylmethylenedioxy-
4-~-(2-chloro-9-adenyl?-cyclopentane-1-~-N-ethylcarboxamide and 40 ml
of lN hydrochloric acid is heated at 60C for 3 hours. The solvent is
removed under vacu~m and the residue triturated with methanol and
ether, then collected and dried under vacuum to give (-)-2-alpha-
3-alpha-dihydroxy-4-~-(2-chloro-9-adenyl)-cyclopentane-1-~-N-ethyl-
carboxamide hydrochloride; m.p. 190C (dec.); ~D5~ -8.70 (c-0.77,
methanol); NMR (CD30D): 9.29 (lH,s); 4.48 (lH,dt); 4.23 ~lH,dt);
2.90 (lH,m); 2.70 (lH,m?; 2.24 (lH,m); 1.16 (3H,t).
b) Similarly prepared ls (~)-2-alpha 3-alpha-dihydroxy-4-~-(2-chloro-
9-adenyl)-cyclopentane-I-~-N-ethylcarboxamide.
The optically acitve starting materlals are prepared as follows:
Racemic 4-~-amino-2-alpha-3-alpha-dimethylmethylenedioxycyclo-
pentane-l-B-N-ethylcarboxamide (example 5s 5.4 g) is combined with
8.90 g of (-)-dibenzoyl-L-tartaric acid monohydrate in 100 ml boiling
, . ~,,
~884~1
ethanol and allowed to cool slowly to room temperature. The crystals
which form are collected, washed with cold ethanol and dried under
vacuum to yield colorless needles; ~Ds- -66.69 (c=1.09, methanol).
The (-)-4-~-amino-2-alpha-3-alpha-dimethylmethylenedioxy-cyclo-
pentane~ -N-ethylcarboxamide dibenzoyl-D-tartrate salt is dissolved
in water, the solution is treated with excess sodium bicarbonate and
the solvent is removed under vacuum. The filtrate is triturated with
ethyl acetate. The solids are filtered off and the filtrate i5
concentrated under vacuum to give (-)-4-~-amino-2-alpha-3-alpha-di-
methylmethylenedioxy-cyclopentane-l-~-N-ethylcarboxamide as a yellow
oil; ~Ds= -31.15 ~c=2.27, methanol); NMR (CDCl3): 4.81 (lH,dd);
4.39 (lH,d); 3.26 (2H,m); 1.80 (lH,dt~.
The filtrate from the crystallization of the levorotatory salt i~
concentrated under vacuum and the residue treated with excess aqueous
sodium bicarbonate. The solvent is removed under vacuum and the
residue i9 triturated with ethyl acetate. The insoluble material is
collected and the filtrate concentrated under vacuum to give a yellow
oil. The oil is combined with 4.00 g (~)-dibenzoyl-D-tartaric acid
monohydrate in 90 ml boiling ethanol and sllowed to cool slowly to
room temperature. The resulting crystals are collected, washed with
cold ethanol and dried under vacuum to yield the dextrorotatory salt
as colorless plates; ~D5= +68.41 (c=1.14, CD30D); NMR (CDCl3):
6.55 (lH,s); 4.81 (2H,dd); 4.4 (2H,d); 3.5 (lH,s); 3.2~ (2H,q);
2.77 (lH,m); 2.4 (lH,m); 1.8 (lH,t); 1.48 (3H,s); 1.29 (3~,s);
1.13 (3H,t).
(+)-4-~-Amino-2-alpha-3-alpha-dimethylmethylenedioxy-cyclopentane-
~ ethylcarboxamide dibenzoyl-D-tartrate (7.04 g) is dissolved in
water and treated with excess NaHC03. The solvent i5 removed under
vacuum and the residue is triturated with ethyl acetate. The solids
are filtered off and the ~iltrate concentrated under vacuum to afford
4-~-amino-2-alpha-3-alpha-dimethylmethylenedioxy-cyclopentane-
1-~-N-ethylcarboxamide as a yellow oil; ~5= ~32.26 (c=1.52,
methanol); NMR (CDCl3): 4.81 ~lH9dd) 4.41 (lH,d); 3.29 (2H,m);
1.84 (lH,dt).
~I X8843~l
- 42 -
To a solution of 2.016 g of 5-amino-2,4,6-trichloropyrimidine and
2 A 32 g of (+)-4-~-amino-2-alpha-3-alpha-dimethylmethylenedioxy-cyclo-
pentane-1-~-N-ethylcarbox~mide in 40 ml n-butanol i9 added 2.8 ml of
triethylamine and the resulting mixture is heated at reflux under
nitrogen atmosphere overnlght. The reaction mixture is cooled to room
temperature and the solvent removed under vacuum. The residue i8
partitioned between ethyl acetate and water. The ethyl acetate
solution is extracted with saturated sodium chloride solution, dried
over magnesium sulfate and evaporated to dryness to yield a dark
amorphous solid. The crude product ls chromatographed on sllica gel
eluting with ethyl acetate, hexane (1:2 to 4:1) to give
(+)-4-~-(5-amino-2,6-dichloro-4-pyrimidinylamino)-2-alpha-3-alpha-di-
methylmethylenedioxy-cyclopentane-1-~-N-ethylcarboxamide, as a white
amorphous solid; ~Ds= +27.36 (c=1.2, methanol); NMR ~CDCl3):
5.95 (lH,bs); 4.72 (2H,t); 4.51 (lH,d); 3.31 (2H,m); 2.84 (lH,d);
2.55 (lH,m); 1.93 (lH,d); 1.49 (3H,s); 1~29 (3H,s); 1.18 (3H,t).
Similarly, conden~ation of 5-amino-2,4,6-trichloropyrimidine with the
levorotatory amine yields the corresponding levorotatory intermediate.
Example 11:
a) t-Butyl p-(2-aminoethyl)-phenylpropionate (3.12 g) i9 combined neat
with 500 Ing of (-)-2-alpha-3-alpha-dihydroxy-4-~-(2-chloro-9-adenyl)-
cyclopentane~ -N-ethylcarboxamide, ~Ds~ -8.70 (ceO.77, methanol),
and heated at 120C under nitrogen atmosphere for five hours. The
reaction mixture is cooled to room temperature and triturated with
ether. The insoluble material i8 filtered off and dried under ~acuum.
The soIid obtained i5 triturated with water and the insoluble material
is collected and dried under vacuum to yield 2-alpha-3-alpha-di-
hydroxy-4-~-[2-(p-(2-t-butoxycarbonylethyl)-phenethyl-amino)-9-
adenyl]-cyclopentane-l-~-N-ethylcarboxamide as an off-white solid;
m.p. 215C (dec.), NMR (CDCl3): 7.9 (lH,s); 7.13 (4H,q); 4.68 (lH,q);
4.47 (lH,t); 4.32 (lH,t); 3.58 (2H5t); 3.23 (2H,q); 2.82 (4H,m);
2.5 (2H,t); 1.4 (9H,s); 1.14 (3H,t); such being an optical antipode o~
~:88431
- 43 -
the compound of formula IIa wherein R2 represents p-(t-butoxycarbonyl-
ethyl)-phenethylamino, R7 and R8 represent hydrogen and R6 zepresents
ethyl.
b) Similarly prepared is the optical antipode derived from the
correspondlng dextrorotatory starting material.
c) Similarly prepared is the racemic 2-alpha-3-alpha-dihydroxy-
~-~-[2-[p-~t-butoxycarbonylethyl)-phenethylamino]-9-adenyl]-cyclo-
pentane-l-~-N-ethylcarboxamidel m.p. 206-208C, using the racemic
starting material.
t-Butyl p-(2-aminoethyl)-phenylpropionate is prepared as follows:
p-Bromophenylacetonitrile (50 g), t-butyl acrylate (46 ml),
palladium (II) acetate (575 mg) and tri-o-tolylphosphine (3.1 g) are
combined with 125 ml triethylamine in a steel pressure reactor and
heated at 140C for 16 hours. The reaction mixture is cooled to room
temperature and poured into 500 ml 3N hydrochloric acid at 0C. The
solids are extracted into ethyl acetate and the ethyl acetate solution
is extracted with saturated sodium chloride solution and dried over
magnesium sulfate. The crude product is triturated with
ether/hexane (1:1), filtered and dried under vacuum to yield t-butyl
p-(cyanomethyl)-phenyl-acrylate; m.p. 80-82C; NMR (CDCl3):
7.5 (2H,d); 7.32 (2H,d); 6.39 (2H,d); 3.78 (2H,s); 1.52 (9H,s).
t-Butyl p-~cyanomethyl)-phenylacrylate (6.0 g) is combined wlth 600 mg
10 % palladium on carbon in 80 ml isopropanol and 2~ ml lN hydro-
chlorlc acid and treated with hydrogen at room temperature. After
8 hours at 300 kPa pressure, the catalyst is filtered off and the
filtrate concentrated under vacuum. The residue is triturated with
ether, filtered and dried under vacuum to afford t-butyl p-(2-amino-
ethyl)-phenylpropionate hydrochloride as a white solid. The hydro-
chloride salt is partitioned between ethyl acetate and lN sodium
hydroxide. The ethyl acetate extract is washed with a saturated sodium
chloride solution and dried o~er magnesium sulfate. Filtration and
concentration of the filtrate affords t-butyl p-(2-aminoethyl)-
~2~8~
- 44 -
phenylpropionate as a yellow oil; NMR (CDCl3): 7.1 (4H,s);
2.98 (2H,t); 2.89 (2H,t); 2.73 (2H,t); 2.5 (2H,t); 2.02 (2~1,s);
1.42 (9H,s).
Example 12:
a) Optlcally active 2-alpha-3-alpha-dihydroxy-4-~-[2-lp-(t-butoxy-
carbonylethyl)-phenethylamino]-9-adenyl]-cyclopentane-1-~-N-ethyl-
carboxamide of example 11a (160 mg) i8 treated with 20 ml of lN
hydrochloric acid and heated at 60C for one hour. The solvent is
removed under vacuum and the residue triturated with ethanol, filtered
off and dried under vacuum to give (-)-2-alpha-3-alpha-dihydroxy-
4-~-12-(p-(2-carboxyethyl)-phenethylamino)-9-adenyl]-cyclopentane-
l-~-N-ethylcarboxamide hydrochloride as a white solid; m.p. 243-245C;
~Ds= -4.34 (c=0.99, DMS0); NMR (CD30D): 8.11 (lH,s); 7.18 (4H,q);
4.76 (lH,q); 4.5 (lH,m); 4.28 (lH,m); 3.75 (2H,dt); 3.24 (2H,q);
2.94 (2H,t); 2.85 (4H,t); 2.44 (4H,t); 2.3 (lH,m); 1.16 ~3H,t);
representing the levorotatory antipode of the compound of formula IIa
wherein R2 represents p-(carboxyethyl)-phenethylamino, R7 and R3
represent hydrogen and R6 represents ethyl.
b) Similarly prepared is the corresponding dextrorotatory antipode,
m.p. 242-245C, ~Ds= ~3.48 (DMS0).
c) Similarly prepared is also the racemic compound (example 7a),
m.p. 235-237C.
Example 1_: Treatment of (-)-2-alpha-3-alpha-dihydroxy-4-~-[2-(p-2-
carboxyethyl)-phenethylamino)-9-adenyl]-cyclopentane~ N-ethyl-
carboxamide hydrochloride with ethanol and conc. sulfuric acid as
catalyst und~r reflux overnight gives after workup (-)-2-alpha-3-
alpha-dihydroxy-4-~-[2-[2-(p-2-ethoxycarbonylethyl-phenethylamino~-
9-adenyl]-cyclopentane-1-~-N-ethylcarboxamlde; NMR (CD30D):
4.05 (q,2H); 1.29 (t,3H).
8~
- 45 -
Exa_p_e 14: A mixture of 18 mg of 2-alpha-3 alpha-dlhydroxy-1-~-
hydroxymethyl-4-~-(2-chloro-9-adenyl)-cyclopentane (example 3),
100 mg of sodium hydrogen sulfide, and 0.5 ml of N,N-dimethylformamide
i6 stirred at 140~C overnight. The reaction mixture is cooled to room
temperature, neutralized with O.lN hydrochlorlc acid -to pH 6, then
concentrated under reduced pressure to give crude 2-alpha-3-aLpha-
dihydroxy-1-~-hydroxymethyl-4-~-(thio-9-adenyl)-cyclopentane. The
crude product is dissolved in a mixture of 3 ml ethanol, 1 ml 0.25N
sodium hydroxide, O.S ml allyl bromide and the mixture i~ stirred at
room temperature for 20 hours. The reaction mixture is poured into
water, neutralized with O.lN hydrochloric acid to pH 7, then con-
centrated under reduced pressure to yield an amorphous solid. Flash
chromatography of the crude product on a reverse phase G1g-column,
eluting with water/methanol (3:2) affords 2-alpha-3-alpha-dihydroxy-
l-~-hydroxymethyl-4-~-(2-allylthio-9-adenyl)-cyclopentane,
m.p. 126-128C, NMR (CD30D): 8.2 (lH,s); 6.0 (lH,m); 5.3 (lH,dd);
5.1 (lH,dd); 4.75 (lH,m); 4.58 (lH,m); 4.05 (lH,dd); 3.8 (lH,m);
3.67 (lH,m); 2.4 (lH,m); 2.24 (lH,m); 2.02 (lH,m~.
Example 15: Treatment of 2-alpha-3-alpha-diacetoxy-4-~-[2-(2-phenyl-
ethylamino)-6-chloro-9-purinyl]-cyclopentane-1-~-N-ethylcarboxamide
with saturated methanolic ammonia at 100C in a sealed tube yields
2-alpha-3-alpha-dihydr~xy-4-~-[2 (2-phenylethylamino)-9-adenyl]-
cyclopentane-1-~-N-ethylcarboxamide of example 2a.
The starting material is prepared as follows: To a solution of 740 mg
of 2-alpha-3-alpha-dimethylmethylenedioxy-4-~-(5-amino-2,6-dichloro-
4-pyrimidinylamino)-cyclopentane-1-~-N-ethylcarboxamide in 10 ml
triethyl orthoformate is added 0.15 ml concentrated hydrochloric acid
at room temperature. The reaction mixture is stirred for 18 hours,
then concentrated under reduced pressure to give an oil. A solutioD of
the oil in n-butanol is then heated at reflux for 4 hours and cooled.
All the volatiies are evaporated to yield crude product which is then
purified by ~lash chromatography on silica gel. Eluting with 4 %
~L2~ 3~
- 46 -
methanol in dichloromethane gives 2-alpha-3-alpha-dimethylmethylene-
dioxy-4-~-(2,6-dichloro-9-purinyl)-cyclopentane-1-~-N-ethylcarbox-
amide; NMR (CD30D/CDCl3): 8.2 (lH,s); 2.1-2.9 (3~1,m).
A solution of 60 mg of 2-alpha-3-alpha-dimethylmethylenedioxy-4-~-
(2,6-dichloro-9-purinyl)-cyclopentane-1-~-N-ethylcarboxamlde in 5 ml
1 N hydrochloric acid is heated to reflux ~or 5 hours and cooled. The
reaction mixture is concentrated undsr reduced pressure to give crude
product. The crude product is dissolved in 3 ml of methanol, 0.1 ml o~
phenylethylamine is added and the solution is heated under reflux for
5 hours. After the reaction is complete, all Lhe volatiles are
evaporated. The resulting crude solid is purified by chromatography on
a reverse phase C-18 packing, eluting with wat~r/methanol (8:1 to 5:1)
to give 2-alpha-3-alpha-dihydroxy-4-~~[2-(2-phenylethylamino)-6-
hydroxy 9-purinyl]-cyclopentane-1-~-N-ethylcarboxamide; NMR (CD30D):
8.3 (lH,s); 7.2-7.4 (5H,m); 4.85 (lH,q); 4.45 (lH,dd); 4.3 (lH,dd);
3.15 (l~,t); 2.95 (lH,t); 2.84 (lH,m); 2.62 (lH,m); 2.2 (lH,m).
Selectlve acetylation using a procedure as illustrated for a similar
transformation in Can. J. Chem. 59, 2601 (1981) yields 2-alpha-3-
alpha-diacetoxy-4~~-12-(2-phenylethylamino)-6-hydroxy-9-purinyl]-
cyclopentane-l-~-N-ethylcarboxamide.
Chlorination with phosphorous oxychloride and diethylaniline yields
2-alpha-3-alpha-diacetoxy-4-~-[2-(2-phenylethylamino)-6-chloro-9-
ptlrinyl]-cyclopentane-1-~-N-ethylcarboxamide.
Example 16: The following compounds are prepared substantially
according to procedures described in the previous examples:
a) 2-alpha-3-alpha-dihydroxy-4-~-[2-chloro-N6-(2-N-pyrrolylcyclo-
hsxyl)-9-adenyl~-cyclopentane-1-~-N-ethylcarboxamide, m.p.
above 150C (dec.);
b) 3-alpha-hydroxy-4-~-[2-(3-phenylpropylamino)-9-adenyl]-cyclo-
pentane-l-~-N-ethylcarboxamide, m.p. 185-186C;
,
.
~.~8~
- 47 -
c~ 3-alpha-hydroxy~ -hydroxymethyl-4-~-[2-(2-phenylpropylamino~-
9-adenyl]-cyclopentane.
d) 2-alpha-3-alpha-dihydroxy-4-~-~2-anilino-9-adenyl]-cyclo-
pentane-l-~-N-ethylcarboxamide;
e) 2-alpha-3-alpha-dihydroxy-1-~-hydroxymethyl-4-~-[2-(2-phenethyl
thio)-9-adenyl]cyclopentane;
f) 2-alpha-3-alpha-dihydroxy-1-B-hydroxymethyl-4-~-[2-[p-(2-carboxy-
ethyl)-phenethylamino]-9-adenyl]-cyclopentane;
g) 2-alpha-3-alpha-dihydroxy-1-~-hydroxymethyl-4-~-[2-anilino-9-
adenyl]-cyclopentane.
Example 17:
a) Preparation of 10,000 tablets each contalning 10 mg of the active
ingredient:
Formula:
2-alpha-3-alpha-dihydroxy-4-~-(9-adenyl)-
cyclopentane-l-~-N-ethylcarboxamide100.00 g
Lactose 2,400.00 g
Corn starch 125.00 g
Polyethylene glycol 6,000 150.00 g
Magnesium stearate 40.00 g
Purified water q.s.
Procedure:
All the powders are passed through a scresn with openings of 0.6 mm.
Then $he drug substance, lactose, magnesium stearate and half of the
starch are mixed in a suitable mixer. The other hal of the starch
is suspended ln 65 ml of water and the suspension added to the
~'28~
- 48 -
boiling solution of the polyethylene glycol in 260 ml of water. The
paste formed is added to ths powders, which are granulated, i~
necessary, with an additional amount of water. The granulate i8
dried overnight at 35C, broken on a screen with 1.2 mm openings and
compressed lnto tablets, using concave punches upper bisected.
b) Preparation of l,000 capsules each containing 10 mg of the active
ingredient:
~ormula:
2-alpha-3-alpha-dihydroxy-4-~-[2-(2-phenyl-
ethylamino)-9-adenyl]-cyclopentane-1-~-N-
carboxamide 10.00 g
Lactose 207.00 g
Modified starch ~0.00 g
Magnesium stearate 3.00 g
Procedure:
A11 the powders are passed through a screen with openings of 0.6 mm.
Then the drug substance is placed in a suitable mixer and mixed
first with the magnesium stearate, then with the lactose and starch
until homogeneous. No. 2 hard gelatin capsules are filled with
300 mg of said mixture each, using a capsule filllng machine.
c) Similarly prepared are capsules and tablets of the other compounds
disclosed herein, e.g. of 2-alpha-3-alpha-dihydroxy~ [2-(p-carboxy-
ethyl-phenethylamino)-9-adenyl]-cyclopentane-1-~-N-ethylcarboxamida
hydrochloride.