Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
13(10(1 (~7
IMMUNOASSAY METHODS AND REAGENTS
AND METHODS FOR PRODUCING THE LATTER
The present invention relates to immunoassay
methods and reagents employing fluorescent chelates of
lanthanide metal ions.
The use of such chelates has numerous advantages.
However, the known reagents and methods have not been as
convenient and as effective as is desirable.
In applicant's Canadian patent application 488,513
filed August 12, 19~5, (European patent application
85305477.3, filed July 31, 1985~ applicant has
disclosed assays wherein immunoreactive bodies are
bound to a solid phase, the bodies having connected to
them as label~ or marker a moiety of a ligand which forms
a fluorescent chelate with lanthanide metal ion. In
order to measure the fluorescence of the chelate, the
complex is dissociated with a dissociating solution to
cause migration of the ligand from the solid phase to
; ~ the bulk of the solution. The measurement of the
fluorescence in solution results in loss of sensitivity,
since the presence of liquid phase causes significant
absorption and scattering of the excitation beam and of
the emitted light.
Applicant is also aware of prior assay methods in
which immunocomplexes are formed bound to a solid phase
comprising bodies having as marker or label ligands
binding lanthanide metal ion, especially europium. In
order to determine the quantity of europium which is
bound, an enhancement solution is added which releases
the europium from the ligand. The solution contains a
further ligand which forms a fluorescent chelate with
the europium in solution. In addition to the
disadvantages mentioned above, this method has the
drawback that it is vulnerable to europium contamination
from the environment of the laboratory or other
surroundings, which gives an artificially elevated
fluorescence reading in the presence of an excess of the
development solution.
In accordance with an aspect of the invention, an
. ~
, `~
~300007
immunoassay method comprises immobilizing immunoreactive
bodies on a solid phase. Complementary immunoreactive
bodies are reacted with the immobilized immunoreactive
bodies. The complementary immunoreactive bodies have
connected thereto moieties of a ligand forming with
lanthanide metal ion, a fluorescent chelate label
wherein the metal ion is stably retained. The
complementary immunoreactive bodies bind to the
immobilized immunoreactive bodies. Any excess
complementary immunoreactive bodies are removed which
are not bound to the immobilized bodies. The
fluorescence of the chelate label on bound complementary
immunoreactive bodies is measured in the presence of the
metal ion stably retained with the chelate. A value is
obtained indicative of the quantity of the complementary
immunoreactive bodies bound to the immobilized
immunoreactive bodies.
This method avoids the need for a step of
dissociation of a labelled complex once formed on the
solid phase, thus considerably simplifying the
procedure. Since the fluorescence can be measured in
the presence of an excess of lanthanide metal ion, the
method is invulnerable to lanthanide metal ion
contamination.
Preferably, the chelate is dried before its
fluorescence is measured, thus alleviating problems of
absorption or scattering of the excitation beam and of
the emitted light. The method can be employed in
sandwich or in competitive assay form.
In one preferred form, the complementary
immunoreactive body has connected to it, covalently or
non-covalently (e.g. through a biotin-avidin or
-streptavidin linkage), a moiety of a 1,10-
phenanthroline-2,9-dicarboxylic acid disclosed in
applicant's abovementioned Canadian and European
applications. The preferred moieties are represented as
follows 1,lO~phenanthroline-2,9-dicarboxylic acid
compound selected from the group
~311~
consisting o~ compounds of formula I:
Rl
2 ~ ~ COOH
3 ~
R4 r
R5
¦ COOH
R6
wherein:
each R1 to R6 group is independently hydrogen,
X~R7tn or Rg_,
wherein X is ~SO3-M+ wherein M is metal ion or is a
functional group which couples covalently with proteins
or a group readily convertible to a functional group
which couples covalently with proteins, R7 is a divalent
aliphatic residue having 1 to 12 carbons, or a divalent
carbocyclic or heterocyclic residue having 3 to 12
carbons, and n is O or 1, and
wherein R8 is an aliphatic group having 1 to 12
: carbons, or a carbocyclic or heterocyclic group having 3
to 12 carbons or one or more pairs of adjacent R1 to R6
groups form together with the carbons to which they are
substituents (a) a carbocyclic or heterocyclic ring
containing 3 to 12 carbons, (b) an X-substituted
carbocyclic or heterocyclic ring o~ the general formula
: Ia:
(X~m Ia
~ ~
wherein C ~ is a divalent carbocyclic or
residue having 1 to 12 carbons, X has the signification
given above, and m is an integer from 1 to 4,
or (c) an orthoquinone linkage:
O O
`\~/
with the proviso that at least one of Rl to R6 is
X~R7tn, wherein X, R7, and n have the significations
given above, or at least one pair of adjacent R1 to R6
groups form a ring of the formula Ia given above or an
; orthoquinone linkage, and trihalomPthyl forms, salts,
esters and acid halides thereof which are readily
hydrolyzed to form the acid of formula I.
Applicant has found that such moieties of the 4,7-
diphenyl-l,lOphenanthroline-2,9-dicarboxylic acid form
highly stable fluorescent chelates with lanthanide metal
ion, preferably with europium metal ion, such that the
metal ion remains stably attached to the solid phase and
allows the assay to be conducted by measurement of
fluorescence of the chelate bound to the solid phase.
The invention is, however, by no means limited to use of
the above-mentioned compounds. The suitability of other
fluorescent chelates for use in the present method can
be determined by trial and experiment having regard to
the above principles and the detailed disclosure below.
Immunoreactive bodies, especially antigens and
; 30 antibodies, which are often desired to form the basis of
reagents for immunoassay, are labile molecules. When
they are reacted to introduce the residue of a ligand,
they tend to become inactivated and to lose their
properties of immunoreaction.
In a second aspect, the invention provides
reagents for immunoassay and methods for maturing and
using them whereby loss of immunoreactivity may be
avoided or mitigated.
According to the said second aspect there is
~3~07
provided a reagent for immunoassay comprising a residue
of an immunoreactive body linked to a residue of a
protein or polypeptide, the protein or polypeptide being
labelled by substitution with a moiety of a ligand
5 forming a fluorescent chelate with lanthanide metal ion.
With the reagents of the invention inactivation of
the immunoreactive body is avoided ~;ince the ligand-
introducing reactant does not need t.o react directly
with it.
Usually, in making the reagente: of the invention,
such reactant reacts with amino groups present on the
protein or polypeptlde. It ha~ been found somewhat
surprisingly that when a plurality of ligand moieties
are introduced in each molecule of the protein or
polypeptide, amplification of the fluorescent light
emission can be achieved, as long as the Stokes shift of
the fluorescent chelate (the difference between the
wavel~ngths of maximum absorption of excitation
radiation and of emitted light) is at least about 200
nm. It is suggested that with lower Stokes shifts,
emitted light is reabsorbed by adjacent chelate
moieties.
It is appreciated that the assay system of this
invention can be adapted to detect a variety of
biologically reactive matter. Tha system is
particularly suited to detect a variety of haptens,
antigen protein and antibody protein; for example, the
immunoreactive body may be a hapten, such as cortisol,
cortisol amine, thyroxine, digoxin or biotin. It may be
an antigen protein such as ~-fetoprotein (AFP), human
chorionic gonadotropin (hCG), ferritin, thyrotropin
(TSH), follitropin (FSH), lutropin (LH), thyroxine
binding globulin (TBG), growth hormone or prolactin. It
may be an antibody protein, such as monoclonal antibody
to cortisol, antibody to AFP, antibody to hCG, or
antibody to RubeIla virus.
The protein or polypeptide of the reagent for the
assay may comprise a carrier protein for example bovine
serum albumin (BSA), thyroglobulin, polylysines of
~3~ ()7
molecular weight 4,000 to 400,000, lysine copolymers
with one or more of alanine, phenylalanine, serine,
tyrosine, tryptophan and glutamic acid, having pendent
amino groups on the polymer chain, hemocyanin, myosin,
ferritin, catalase and reduced forms thereof.
The abovementioned immunoreact:ive bodies may be
linked to the abovPmentioned proteills and polypeptides
by, for example, using a bifunctional reagent having one
end group reactive toward groups wh:ich are present on
one of the immunoreactive body and the protein or
polypeptide and not on the other, and a second end group
which is reactive toward groups present on the protein
or polypeptide but not on the immunoreactive body. For
example, the reagent may comprise sulfosuccinimidyl
4-(N-maleimidomethyl) cyclohexane-l-carboxylate
(sulfo-SMCC) containing an amino group reactive
N-hydroxy succinimide moiety at one end and a sul~hydryl
group reactive maleimide moiety at the other. It may
therefore be reacted with immunoreactive bodies such as
proteins, e.g. antibodies or antigens containing -NH2
groups and free from -SH groups. To introduce maleimide
moieties theretoj the resulting conjugate immunoreactive
bodies may be reacted with, for example, BSA which has
been exhaustively reacted with a marker containing -NH2
reactive groups so that all --NH2 groups have been
converted to ligand moieties and which has been reduced
to create -SH groups from its intramolecular disulfide
linkages. The -SH groups then react with the maleimide
moieties.
In other procedures, the immunoreactive body is
reacted with a biotinylating agent to form a
biotinylated derivative. A marker compound (for example
as described in applicant's abovementioned patent
applications) is reacted with avidin or streptavidin to
provide it with pendent moieties of the ligand. The
labelled avidin or steptravidin can then be reacted with
the biotinylated body under conditions allowing
biotin-avidin or biotin-streptavidin association before
or after an immunoassay is preformed with the
~31)i~(~7
biotinylated body.
In an especially preferred form, avidin or
streptavidin is employed having a plurality of the
marker moieties on its molecule, thus achieving the
light amplification effect referred to above.
~ mplification may also be achieved in that a
plurality of biotin moieties may be attached to the
immunoreactive body, whereby a plurality of the
substituted avidin or streptavidin molecules will bind
to the immunoreactive body when the biotinylated
component is reacted therewith.
Modifications and combinations of the above
techniques may be employed. For example avidin or
streptavidin may be linked to an immunoreactive body
through a bifunctional reagent, and a labelled caxrier
protein such as those mentioned above may be
biotinylated and the biotin-avidin or
biotin-streptavidin association may be formed therefrom.
A biotinylated and labelled carrier protein may be
employed, and in use may be linked through a sandwich
type linkage, employing avidin or streptavidin as a
central linking entity, to a biotinylated immunoreactive
body.
According to another aspect of the invention,
processes are provided for making the above reagents
which include the bifunctional linking reagent or the
biotin/avidin or streptaviding linking reaction.
Preferably the marker labels employed are those
described in applicants abovementioned patent
application, but it will be appreciated that other
markers may be employed.
The following Examples will be discussed with
reference to the drawings wherein:
Figures la and lb show graphs plotting absorbance
against 1/dilution for titres of polyclonal anti-
; cortisol antibodies and monoclonal anti-cortisol
antibodies, respectively, after modification with sulfo
SMCC;
Figures 2a and 2b show graphs plotting fluorescence
l3a~
and absorbance against fraction number;
Figures 3a and 3b show graphs plotting standard
curves for cortisol assay by TRFIA;
Figure 4 shows a dose-response curve, and precision
profile of an hCG assay;
Figure 5 shows a correlation between an assay in
accordance with the invention and a shown assay (LKB) on
74 patient samples measured in duplicate;
Figure 6 shows a correlation between an assay
lo method in accordance with the invention and a known
assay method (Bio-Rad) on 64 patient samples measured in
duplicate;
Figures 7a, 7b and 7c are diagrams illustrating
various labelling techniques according to preferred
embodiments of this invention for labelling
immunoreactive bodies; and
Figure 8 is a curve generated by TRFIA for a
competition assay directly labelled cortisol antibodies.
Before discussing in detail the following Examples,
the various types of labelling ~ystems, according to
aspects of this invention, are shown in Figure 7.
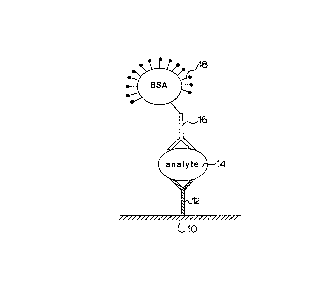
Figure 7a shows a solid support 10 having bound thereto
an immobilized immunoreactive body 12. The
immunoreactive body may be an antibody specific to the
analyte 14. A second immunoreactive body 16 specific to
another site of the analyte 14 binds to the analyte.
The second immunoreactive body may be a second antibody.
The second antibody 16 has directly attached thereto the
chelated fluorophors 18.
According to the embodiment of Figure 7b, the
second antibody 16 has a bovine serum albumin (BSA)
molecule linked to the tail of the antibody 16. The
linking element may be the reactant (sulfo-SMCC). The
BSA molecule is, in turn, directly labelled with a
plurality of the chelated fluorophors 18.
According to the embodiment of Figure 7c, the
second antibody 16 has a plurality of biotin molecules
20 attached thereto. A plurality of streptavidin
molecules 22 have directly attached thereto, a plurality
0~7
of fluorophors 18. The streptavidin molecules are
therefore linked to the antibody 16 by the biotin
molecules 20.
In each of these preferred arrangements of the
assay system, the second immunoreactive body is labelled
with stable fluorescent fluorophors 18 which, when
subjected to stimulating energy, fluoresces to indicate
the presence of the diagnostic immunoreactive body.
Examples of immunoassay methods, reagents and
methods of producing them are given below.
Example 1 - Reaqent and Assav for Cortisol
A carrier protein, bovine serum albumin (BSA) was
first exhaustively labelled with a chelate, and then the
labelled protein was cross linked to a molecule
of an antibody with a bifunctional reagent. A
heterogenous competition assay was then carried out for
serum cortisol, with ovalbumin-cortisol conjugate
immobilized in microtitration wells as the solid-phase.
Materials
Bovine serum albumin (BSA,RIA grade), ovalbumin,
hydrocortisone 21-hemisuccinate and hydrocortisone 21-
hemisuccinate BSA conjugate (cortisol-21-BSA) were
purchased from Sigma Chemical Co., St. Louis MO 63178.
Gelatin (EIA purity) was obtained from Bio-Rad
Laboratories (Canada) Ltd., Mississauga, Ontario L4X 2C8
and Sephadex G25 medium mesh from Pharmacia (Canada)
Ltd., Dorval, Quebec H9P lH6. A radioimmunoassay (RIA)
kit for cortisol, Cort-A-Count (trade mark) was
purchased from Diagnostic Products Corp., Los Angeles
CA 90045. 4,7-bis(chlorosulfophenyl)1,10-phenanthroline
2,9-dicarboxylicacid (Eurofluor S (trade mark) and
hereinafter BCPDA) was synthesized according to the
procedure described in applicants abovementioned patent
application. Monoclonal antibody to cortisol was
purchased from Medix Biotech Inc., Foster City CA 94404
and polyclonal rabbit anti cortisol (against
cortisol-21-BSA) was purchased from Western Chemical
Research Corp., Ft. Collins CO 80522. Protein
concentration was carried out by centrifugation using
~L3~ 7
Centricon (trade mark) 30 miroconcentrakors from Amicon
Canada Ltd., Oakville, Ontario L6H 2B9.
Enzyme immunoassay (EIA) microtitration plates were
obtained from Flow Laboratories, Inc., McLean VA 22102
and read on a EL309 microplate reader, Bio-'rek
Instruments Inc., Winooski VT 05404. Fluoroimmunoassay
(FIA) microtitration plates, Microfluor (trade mark) W,
white opaque 96-well plates were purchased from Dynatech
Laboratories, Inc., Alexandria VA 22314 and read on the
CyberFluor (trade mark) 615 Fluorometer.
High performance liquid chromatography (HPLC) was
carried out using a BioSil TSK 400~ size exclusion column
from Bio-Rad Laboratories on a model 600 Gradient System
equipped with a 490 variable wavelength detector [Waters,
a division of Millipore (Canada) Ltd. Mississauga,
Ontario L4V lM5).
Example lta)
Preparation of cortisol-ovalbumin con-iuaate. Cortisol
ovalbumin was prepared by the mixed anhydride method. 50
mg (0.1 mmoles) of hydrocortisone 21-hemisuccinate were
dissolved in lO mL dixane and 0.1 mL tri-n-butylamine
added. The solution was cooled to 10C, 0.02 mL isobutyl
chloroformate added and the mixture stirred for 30 min.
Then, 500 mg of ovalbumin (0.01 mmoles) dissolved in 10
mL of water adjusted to pH 9 with NaOH were added to the
reaction mixture and stirred 24 h at 4C. After the
reaction was completed, a precipitate vas present. The
mixture was dialyzed 36 h against water and the
precipitate was removed by centrifugation. Urea was
added to the supernatant to achieve a concentration of 6M
and this solution was again dialyzed exhaustively against
water. The protein concentration was determined by the
Bio-Rad Protein Assay.
Conju~ation of BCPDA to BSA. Two mL 0.5 M sodium
carbonate buffer of pH 9.1 were added to 250 mg of BSA
dissolved in 2 m~ of water. 100 mg of BCPDA (50x molar
excess) dissolved in 400 ~ dimethylformamide (DMF) were
added in 5 portions over a 15 min period. Unreacted
ll
BCPDA was removed by exhaustive dialysis against 0.1 M
NaHC03 and the labelled BSA (BSA-BCPDA) stored at 4C.
Reduction of BSA-BCPDA: Reduction was carried out just
prior to conjugation with antibody. To BSA-BCPDA (30
mg/mL in 0.1 M NaHC03) solid urea, dithiothreitol (DTT)
and Tris~ base were added to achieve a concentration of
6 M, 50 mM and 0.1 M respectively. The pH measured was
9: The mixture was incubated for 1 h at 37C.
Exam~le l(b)
Con~uqation of antibody and reduced BSA-BCPDA:
Conjugation with sulfosuccinimidyl 4-(N-
maleimidomethyl)-cyclohexane-l-carboxylate (sulfo-SMCC)
was carried out by a modification of the method of
Yoshitake et al, European Journal of Biochemistry 1979,
Vol. 101, pp 395 to 399. 0.5 mL of anti-cortisol
antibody solution (1 my/mL) was dialyzed overnight
against 0.1 M sodium phosphate buffer of pH 7.0 14.5 ~L
of a 5 mg/mL solution of sulfo-SMCC dissolved in the same
buffer was added and the solution shaken for 1 h at room
temperature. Unreacted sulfo-SMCC was removed by
desalting on a 25 mL column of Sephadex G~ 25 using 0.1 M
sodium phosphate of pH 6.2 containing 5mM ethylenedi-
aminetetraacetate (EDTA) as the elution buffer. The
fractions containing protein were combined and
conc~ntrat~d to 0.1-0.2 mL by centrifugation in a
Centricon 30 device. 0.35 mL of reduced BSA-BCPDA
containing 9 mg of protein was also desalted to remove
excessive reducing agent on a 25 m~ Sephadex G25 column
in the same phosphate buffer of pH 6.2, containing 5 ~M
EDTA, to retard disulfide formation. The protein
containing fractions were combined and concentrated by
centrifugation in a Centricon 30 device, to 0.6-0.8 mL.
It is important to complete the reactions and the
separation and concentration steps as quickly as possible
to prevent hydrolysis of the maleimide group in the
antibody and reformation of the disulfide bonds in the
reduced BSA. The derivatized antibody and reduced BSA
were combined and incubated 20 h at 4C (BSA to IgG ratio
is #30-40 fold). Prior to purification of the IgG-BSA
.~
130a~0~7
12
conjugate, excess SH groups on the reduced BSA molecule
(36 SH groups exist per reduced BSA molecule) were
blocked by addition of a two-fold excess of N-
ethylmaleimide in DMF and incubation for 1 h at room
temperature.
Isolation of BSA-con~uqat~d antibody: The conjugated ~nd
unconjugated antibody and unconjugated labelled BSA were
separated by size exclusion HPLC on a BioSil TSK 400
column (300 x 7.5 mm) eluted at 1 mL/min with 50 mM
Na2SO4l 20 mM sodium phosphate pH 6.8 and fractions of 0.5
mL were coll~cted automatically. 3 or 4 injections of
250 ~L each were required per preparation to complete the
isolation. Fractions were analyzed for total
fluorescence, fluorescent antibody, and total antibody
concentration, as described below.
Total ~luorescence was measured by addition of 200
~L of 10 5M Eu3~ solution in 50 mM Tris buffered saline pH
7.8 ~TBS) to 5 ~L of each fraction and determining the
fluorescence of the solution after 5 minutes, on an
'ARCUS'~ gated fluorometer (LKB Wallac, Turku, Finland~.
To measure the BSA-conjugated antibody
concentration, 100 ~L of fractions diluted 1/20 and 1/200
in 1% BSA in TBS, were added to the wells of coated
Microfluor W plates. Coating was done overnight at 4C
with 100 ~L per well of a 5 ~g/mL solution of
cortisol-BSA conjugate in 0.1 M sodium bicarbonate and
afterwards the plates were blocked for 1 hour at room
temperature with 1% BSA in the same buffer. The plates
were incubated 1 hour at 37C and then washed 3-4 times
- 30 with water~ 100 ~L of lx10-5M Bu3~ solution in TBS was
thsn added. After 5 min, the plates were washed once
with water and dried in a stream of cool air. Surface
fluorescence of the antibody-BSA-BCPDA Eu3~ conjugate
which was bound to the immobilized cortisol-BSA was
measured in a CyberFluor 615 fluorometer.
The total antibody activity (conjugated to BSA and
unconjugated) was measured using a second antibody ~anti
rabbit IgG or anti mouse IgG for polyclonal or monoclonal
anticortisol, respectively) coupled to horseradish
~3a~ 7
13
peroxidase (HRP) in an ELISA assay. EIA plates were
coated with cortisol-BSA conjugate and blocked as above.
HPLC fractions were diluted, added to the EIA plate and
in~-ubated at 37C as above. After washing with water,
100 ~L horseradish peroxidase labelled goat anti rabbit
or anti mouse IgG diluted 1/500 with 50 mM sodium
phosphate pH 8.5 containing 1% NaCl and 1% BSA were added
and incubated a further hour. The plates were washed
with water and the enzymatic activity of the bound
antibody-anti-IgG- HRP complex bound to immobilized
cortisol-BSA was measured by adding 100 ~L of substrate
(1 mg/ml of ABTS in 50mM Na2~PO4, 25mM citric acid, 0.03%
H202). The optical density was determined on a microplate
reader after 5-10 minutes.
Example l(c)
Cortisol assays: A solid phase competition assay was
used with immobilized antigen to measure cortisol.
Microfluor W plates were coated overnight at 4C with 100
~L of 2 ~g/ml cortisol-ovalbumin conjugate in 0.1 M
NaHC03. The plates were rinsed once with water and
blocked for 1 hour at room temperature with 0.1% gelatin,
O.1% Tween 20~, 50 mM phosphate buffer of pH 7.4 and
stored in the same solution at 4C.
Before the assay, a plate was washed twice with
0.05% Tween 20~ in saline (Tween/NaCl) and twice with
water. Antibody (labelled BSA-conjugated antibody
prepared as in Example l(b~) was diluted to 1/400 with
respect to the starting material (1 mg/mL) in TBS
containing 0~3 M trichloroacetic acid (TCA), 1% BSA and
150 ~g/mL BSA-BCPDA to prevent possible fluor-serum
interactions. 10 ~L of serum was pipetted into the wells
and 100 ~L of antibody solution added. The plate was
briefly shaken and incubated 1 h at 37C. The plate was
washed three times with Tween/NaCl and twice with water,
then 100 ~L of lxlO 5M Eu3 in TBS was added. After fi~e
minutes, the plate was rinsed once with deionized water,
dried with a stream of cool air and sur~ace fluorescence
of the dry solid determined on
~'
13~
1~
a CyberFluor 615 fluorometer.
Results:
Con~u~ation of Antibodies
In the coupling scheme which is used to conjugate
BSA-BCPDA ~o antibody, the antibody, which has no free
sulfhydryl groups, is reacted with a bifunctional
coupling reagent containing an amino reactive N-hydroxy
succinimide moiety at one end and a sulfhydryl reactive
maleimide group at the other end. The sulfonic acid
group renders the reagent water soluble. The reagent is
reacted first with the antibody to introduce maleimide
groups (step 1). ~he BSA which has been exhaustively
reacted with BCPDA (step 2) is reduced with DTT to
create free sulfhydryl groups from intramolecular
disulfide bonds in the native molecule (step 3). The
exposed SH groups are then free to react with the
maleimides introduced into the antibody (step 4).
Since it is possible to get inactivation of
antibody at high concentrations of coupling reagent, the
effect of the concentration of coupling reagent in step
l on the activity of the final con~ugated antibody
preparation was studied using both a polyclonal and a
monoclonal anti cortisol antibody. Antibodies were
reacted for 1 hour at varying molar excess of sulfo-SMCC
in step l. The titre of the modified but unconjugated
antibody was measured by on the HPLC fractions ELISA,
using an HRP conjugated anti IgG in microtitration
plates as described in methods (isolation of conjugated
antibody). As shown in Figure 1 the monoclonal antibody
preparation was highly active evan after derivatizing
with 500-fold excess of sulfo-SMCC, the polyclonal
antibody inactivation was evident even at 25-fold molar
excess of sulfo-SMCC (Fig. lb). The monoclonal, was
therefore treated with a 50-fold molar excess of
sulfo-SMCC while the polyclonal was treated with a
20-~old excess, in step 1. Because of the possibility
of cross linking networks in step 4 due to the reaction
of one BSA molecule bearing many SH groups with multiple
molecules of derivatized antibody and also, of one
~3101~
derivatized antibody molecule with multiple molecules of
reduced BSA, the reduced BSA/derivatized antibody molar
ratio was selected to be very high ~> 25 foldj so as to
promote formation of IgG(BSA)n rather than BSA(IgG)n.
After conjugation, the coniugated and unconjugated
antibody were separated by gel filtration on HPLC. The
fractions were monitored for optical density, total
fluorescence, fluorescent antibody binding to
cortisol-BSA coated plate and total antibody binding to
cortisol-B5A coated plates, using a second peroxidase
conjugated anti IgG antibody in an ELISA assay as
described in Material and Methods. The results of a
typical monoclonal preparation is shown in Figs. 2a and
2b. There is a continuous spectrum of fluorescent
antibodies present with different molecular weights,
with a large fraction in the void volume. There are
three peaks of total antibody activity as measured by
the ELISA technique (Fig. 2b), the void volume (peak A),
an included peak also corresponding to coupled antibody
(peak B) and an included peak corresponding to uncoupled
antibody (peak C). Only peak A and the first fractions
of peak B were combined for use in the assays so as to
ensure that essentially all uncoupled antibody is
excluded. Assuming that the binding of the second
antibody HRP-conjugated anti IgG is similar for all the
fractions of anti cortisol, irrespective of the degree
of conjugation to BSA, 30-50% of the antibody is
recovered in the conjugated antibody fractions combined
and used for the immunoassay.
Assay optimization
An outline of the cortisol competition assay is
shown in Flgures 3a and 3b. The calibration curves for
the cortisol assays were plotted with the ratios of the
fluorescence of the standards, B, to the ~luorescence of
the zero standard, Bo~ expressed as a percentage,
(B/Box100%) were plotted vs the log of the cortisol
concentration. Figure 3a shows the results for a 20 ~L
sample volume, whereas Figure 3b shows the results for a
~L sample volume per well.
~L3001(1~7
16
Variation in the amount of coating in the
microtitration wells from 50 to 500 ng of cortisol-
ovalbumin conjugate had little effest on the overall
shape of the calibration curve when this is plotted as
(B/Bo) but the total antibody bound to the plate
(proportional to Bo) reached its maximum ~t a coating of
200 ng/well. It was found that calibration curves with
the conjugated polyclonal antibody became ~lat with no
further change in (B/BoxlO0) when this value reached
30~, regardless of increasing cortisol concentration
whereas the curves with conjugated monoclonal continued
to drop until a value of (B/BoxlO0) close to zero was
obtained. Continued experimentation was thus conducted
exclusively with the monoclonal antibody.
Since >80~ of the cortisol in serum is bound to
transport proteins, a suitable dissociation reagent is
required to release the cortisol before measurement by
immunoassay. Using monoclonal antibody, two reagents
were investigated: 8-anilino-1-naphthalene sulfonic
acid, ANS, at a concentration of 5~ in the final assay
mixture and trichloroacetic acid, TCA as disclosed in
Eskola et al, Clin. Chem. 1985 31, pp 1731-1734, at a
concentration of 0.3 M. TCA gave better results than
ANS and thus it was preferred in the final assay design.
The volume of serum required in order to obtain
sensitive calibration curves was tested. This
parameter affected the shape of the calibration curve
dramatically. The normal range of cortisol in human
serum is 5-30 ~g/dL. Using 20 ~L of serum, it was found
that the curves were too shallow at concentrations
greater than 20 ~g/dL. A better calibration curve is
obtained with 10 ~L sample volume. With 10 ~L serum
(Fig. 3b) good sensitivity was achieved in the whole
range of clinically important values, ~1-50 ~g/dL).
Assay Performance
A typical calibration curvé is shown in Fig. 3b.
The steepest part of the curve is in the normal range
where maximum accuracy and precision is usually needed.
The within run precision for four patient samples
~;~0~ 7
had C.V.'s of 2-10%. This kind of precision is typical
for well established cortisol assays currently available.
To test the linearity of the method patient samples
were diluted with the zero standard and reassayed. For
samples with cortisol concentration less than 50 ~g/dL,
there was a linear relationship between the measured
concentration and the dilution. Above 50 ~g/dL, the
assay cannot accurately measure samples, giving readings
between 50 and 60 ~g/dL regardless of the concentration.
This underestimation of concentration was also true for
the RIA~ assay. After the first two-fold dilution of
samples with concentration >50 ~g/dL, which brings the
concentration to within the sensitivity of the standard
curve, diluting the sample further also resulted in a
linear relationship indicating that this was not a serum
effect.
The recovery of added cortisol was measured in five
different samples. The recovery of cortisol varied from
91% to 1~5~, with a mean of 102%.
27 patient samples were measured by a commercial RIA
kit (Diagnostic Products Corp.) as well as with the
present method with conjugated fluorescent monoclonal
antibody. The samples were selected to cover the whole
range of the assay from 1 to 50 ~g/d~ and included clear,
cloudy, lipemic, and haemolytic specimens. The
correlation between the two assays was good. The
coefficient of correlation was 0.98 with a slope of 1.007
and an intercept of 0.51 ~g/dL.
Example 2
Methods and reagents for a sensitive noncompetitive
sandwich immunofluorometric assay (IFMA) for the
determination of AFP in serum and amniotic fluid.
A monoclonal catching antibody was non-covalently
immobilized in a microtiter strip (or plate) well. A
biotinylated affinity purified antibody was used as the
detection antibody and streptavidin labelled with
BCPDA, was used as the fluorescent label. The complex
~3~
18
consisting of monoclonal antibody-AFP-polyclonal
antibody-biotin-streptavidin-BCPDA-Eu3+ was quantitated
on the dry surface of the well by excitation with a
nitrogen laser beam and monitoring the specific delayed
fluorescence at about 615 nm.
Example 2(a)
Biotinylation of antibody
Affinity-purified goat anti-AFP antibody (Atlantic
Antibodies, Scarborough, ME 04074, cat. no. 077-06) was
dialyzed twice against 5 L of saline and then diluted in
0~1 mol/L carbonate buffer, pH 9.0 to a final
concentration of 500 ~g/mL. To 1 mL of this solution, a
500-fold molar excess of sulfosuccinimidyl
6-~biotinamido) hexanoate (NHS-LC-biotin, Pierce
Chemical Co., Rockford, IL 61105) dissolved in 100 ~L of
dimethylsulfoxide (DMS0) was added. After mixing and
incubation for 1 h at room temperature, the reaction
mixture was dialyzed twice at 4C against 5 L of 0.1
mol/L carbonate buffer, pH 8.3 containing 0.025% (w/v)
sodium azide.
The biotinylated antibody solution was diluted
1:300 in 10 mmol/L Tris-HC1 buffer, pH 7.8 containing
per liter 400 mmols KCl, lOg BSA, O.lg sodium azide and
O.lg thimerosal before use.
Exam~le 2(b)
Preparation of labelled streptavidin
Five mg of streptavidin (Sigma) were dissolved in
33 mL of 0.1 mol/L carbonate buffer, pH 9.1. Seven mg
of BCPDA dissolved in 200 ~L dimethylformamide were
added to the streptavidin solution with stirring, at
room temperature. Alternatively, 200 ~L of absolute
ethanol may be used as the solvent. In this
circumstance, only 2 mg of BCPD is required. After 1 h,
the reaction mixture was dialyzed three times against 5
L of Q.1 mol/L solution of NaHC03, containing 0.025%
(w/v) sodium azide.
The labelled streptavidin solution was diluted 1:50
in 50 mmol/l Tris-HCl buffer, pH 7.8 containing per
liter lOg BSA, 9g NaCl, O.lg sodium azide, O.lg
~3(~ 7
lg
thimerosal and Eu3+ at a final concentration of l0-5M
before use.
Example 2(c)~
Immunoassay
AFP standards
Human AFP (InterMedico, Toronto, Canada) was
calibrated against the international reference standard
(72/227) for AFP. AFP standards covering a range from 1
to 1000 IU/mL were prepared in the standards diluent
solution.
Patient samples and controls
Sera from pregnant women at various gestational
ages, amniotic fluids and sera from patients with liver
and testicular tumors were obtained from the Toronto
General Hospital. Quality control human based sera were
Tri-level ligand controls from Ortho Pharmaceuticals,
Toronto, Canada. Amniotic fluid was diluted 100 fold in
standards diluent solution before analysis.
Comparative methods
A commercially available radioimmunoassay procedure
(Amersham Corp., Arlington Heights, IL 60005) and a
time-resolved immunofluorometric procedure (DELFIA~ hAFP
kit, LKB Wallac, Turku, Finland) were used. Both
procedures were carried out using the manufacturer's
instructions
Preparation of microtiter wells (8 or 12 - well strips or
96-well plates). Polystyrene white microtiter wells
(MicroFluor, Dynatech Laboratories, Alexandria, VA 22314)
were coated with 200 ng/100 ~L/well purified monoclonal
anti-AFP antibody (Medix Biotech Inc., Foster City, CA
94404, cat. no. A-013-01) dissolved in 50 mmol/L
carbonate buffer, pH 9.6, for 18-20 h at 4C. After
coating, the wells were washed manually two times with
wash solution. Tris-buffered saline (TBS), pH 7.5,
containing 0.05~ (v/v) Tween 20. The wells were then
blocked with 200 uL/well of blocking solution (0.1 mol/L
solution bicarbonate, pH 8.3, containing 1% (w/v) bovine
serum albumin (BSA, RIA grade Sigma Chemical Co., St.
Louis, MO 63178), 2% (w/v) sucrose and 0.05% ~w/v)
'.~ ~
1~000~
sodium azide) for 1 h at room temperature. The wells
were washed again as described above and stored dry at
4C.
Immunoassay procedure
Twenty ~L of standards or samp:Les (duplicate or
preferably triplicate measurements) were added to each
well followed by the addition of 100 ~L standards
diluent buffer. After the wells were incubated for 45
min at 37C (air oven), the wells were washed twice with
the wash solution. 100 ~L/well of the 1:300 dilution of
biotinylated anti-AFP antibody solution were then
added, the wells incubated for another 45 min at 370C
and then washed as above. 100 ~L/well of the mixed
labelled streptavidin- Eu3+ working solution were then
added and the wells further incubated for 30 min at
37C. The wells were then washed as above and dried
using a forced air plate dryer. The fluorescence was
measured on the solid phase in a CyberFluor 615
time-resolved fluorometer/analyzer using an excitation
wavelength of 337.1 nm (nitrogen laser source) and an
emission wavelength of 615 + 5 nm (interference filter).
RESULTS
Incubation time and temperature
The effect of the incubation time and temperature
on the performance of the AFP assay were investigated.
The quality of the calibration curve, the precision of
the assay and the accuracy of measurements were
monitored by analyzing a series of 20 clinical samples
previously assayed for AFP by RIA. It was found that a
precise and accurate assay could be established if the
incubation times were fixed at 45, 45 and 30 min, at
37C. With this choice, an assay run could be completed
in less than 3 h.
Sensitivity and precision
~he assay had a dynamic range of 1-1000 IU/mL
(O.g7-970 ng/mL) and a detection limit of 0.1 IU/mL, as
calculated from the mean fluorescence +3 standard
deviations o~ the zero standard. At concentrations of
analyte ~1000 IU/mL, the curve of a log-log plot is
~l3~ 7
relatively flat and cannot be used for analytical
purposes.
Precision studies were performed using tri-level
commercial control sera. As shown in Table I, the
intra-assay coe~ficients of variation ~CVs) for ~FP
levels of 21.5, 58 and 170 IU/mL were 7.6, 5.4 and 6.2%,
respectively. Inter-assay CVs for the same controls
were 3.9, 8.6 and 1.7~, respectively. Day-to-day
precision was also determined over a one month period
for the same controls and found to be 7.2, 7.3 and 8.7~,
respectively.
Recovery and linearity
To assess the recovery of the assay, spiked serum
samples were prepared by adding various concentrations
of AFP to 9 pooled serum specimens (9 different
additions in each, 100 or 200 IU/mh). The analytical
recovery of AFP ranged from 72 to 125% with a mean of
104 ~ 17~.
To evaluate the linearity of the assay, serial
dilutions of 3 different samples were tested and the
amount of AFP in each sample determined. The
concentration of AFP decreased linearity with
increasing dilution, and values obtained were those
expected, if the value of the undiluted sample is taken
as a true value. This finding confirms that the assay
is free of any serum matrix effects.
Correlation studies
A comparison study with a commercial RIA kit of 104
maternal serum and 20 amniotic fluid samples from women
at various gestational ages was undertaken. Correlation
plots were made for maternal serum AFP and amniotic
fluid AFP respectively. Correlation coefficients of
r=0.95 and r=0.92 were achieved for serum and amniotic
fluid, respectively. In addition, the present assay was
evaluated with a commercial time-resolved
fluoroimmunoassay kit (TR-FIA). Good correlation
(r=0.90) was observed when 90 serum samples were tested
in both assays.
~300(~7
22
Table I
Precision Studies with ~-Fetoprotein Assay using
commercial human serum-based quality control sera.
Intra-assay
5AFP (IU/mL)
SampleMean SD %CVReplicates
I 21.5 1.6 7.6 21
II 58.0 3.1 5.4 21
III 170.0 10.5 6.2 21
Inter-assay
SampleMean SD %CVReplicates
I 21.0 0.8 3.9 4
II 68.0 5.9 8.6 4
III 179.0 3~0 1.7 4
Day-to-dav
SampleMean SD %CVReplicates
I 19.1 1.4 7.2 11
II 68.4 5.0 7.3 10
III 171.7 14.9 8.7 10
Example 3
An immunofluorometric assay for the determination
of hCG in serum. The assay was based on the sandwich
principle, and was performed in microtiter wells coated
with a monoclonal antibody to hCG beta-subunit as the
solid phase, and biotin-labelled monoclonal to intact
hCG as the detection antibody. The degree of binding of
the biotinylated antibody to captured hCG molecules was
determined by a bridge reaction with BCPDA which has
been covalently attached to streptavidin. The
fluorescence of the finaI complex formed (Antibodyl-hCG-
Antibody-2-Biotin-Streptavidin-BCPDA-Eu3 ) is then
quantitated, in the dried solid phase, hy pulsed
fluorescence measurements using the CyberFluor 615 gated
fluorometer/analyzer.
Materials and Methods
1. Reagents and Buffers
Human TSH (specific activity, 6.6 IU/mg), human FSH
tspecific activity, 6200 IU/mg), and human LH (specific
~3~00~t7
23
activity, 5000 IU/mg), were obtained from Sigma Chemical
Co., St. Louis, M0 63178. Bovine serum albumin, bovine
globulin and streptavidin ~cat. no. S-4762) were also
from Sigma.
The coating buffer was 0.01 mol/I, Tris, pH 8.25,
containing 0.1 mol/L NaCl. The blocking buffer was 0.1
Mol/L carbonate, pH 8.3, containing 1% BSA and 0.05%
sodium azide. The assay buffer was 0.05 mol/L Tris, pH
7.8, containing 0.15 mol/L NaCl, 0.05% sodium azide, 0.5%
BSA, 0.05% bovine globulin and 0.01% Tween 40. The
streptavidin-europium buffer was 50 mmol/L Tris, pH 7.8,
containing 0.15 mol/L NaCl, 1% BSA, and 0.5% sodium
azide. The wash solution was 0.15 mol/L NaCl, containing
0.05% Tween 20, and 0.05% sodium azide.
2. hCG Standards
Purified hCG (specific activity, 3310 IU/mg in terms
of the first IRP, 75/537, for immunoassay) was obtained
from Calbiochem, Behring Diagnostics, La Jolla, CA 92037.
The preparation was reconstituted according to the
manufacturers recommendation, and diluted in hCG-free
human serum (Chemicon International Inc., El Segundo, CA
90245) to give the desirable standard concentrations.
3. Monoclonal Antibodies
Monoclonal antibody to beta-subunit of hCG (cat. no.
H-298-12), and to intact hCG molecule (cat. no.
H-296-01), where DEAE~-column chromatography purified
immunoglobulin fractions, purchased from Medix Biotech
Inc., Foster City, CA 94404. The degree of cross-
reactivity of these antibodies, as determined by the
manufacturer in a conventional RIA, were: hCG 100%, hCG
beta 100%, hCG alpha 0.0%, hLH 0.3%, and hTSH 0.02~ for
the antibody to the beta-subunit of hCG; and hCG 100%,
hCG beta 0.0%, hCG alpha 0.0%, hLH 90%, hTSH 85%, and
hFSH 16% for the antibody to intact hCG molecules. A
monoclonal antibody to human LH(hLH) was also obtained
from Medix Biotech (Cat. no. L-461-01).
4. Specimens
Human ~erum samples containing various
~3~)00017
24
concentrations of hCG were provided by Hospital Incommon
Laboratory, Toronto, Canada. These samples were from
pregnant and non-pregnant females, and had been stored at
20C. ~o exclude any possible effect of storage on hCG
concentrations, the samples were concu:rrently tested with
the present method and the comparative kits. Specimens
exceeding 500 IU hCG/L were diluted with hCG-free serum
to bring them within the measurement range of the assay.
Lyphochek~ immunoassay control serum (human) levels I, II
and III were from Bio-Rad~ Clinical Division, Richmond,
CA 94801.
5. Comparative Met`hods
Two commercially available kits were used as
comparative methods. The EchoClonal~ hCG Assay (Bio-Rad)
was a sandwich type immunoradiometric (IRMA) procedure.
It uses solid-phase monoclonal anti-hCG antibodies bound
to immunobeads, and Iodine-125~ labelled monoclonal anti-
hCG antibodies as the tracer. This assay is a single
reagent procedure, combining the solid phase and the
tracer in a tracer/immunobead reagent. The counting of
radioactivity and data reductions were performed with the
LKB-Wallac~ (Turku, Finland) 1275 Minigamma counter.
The DELFIA hCG Assay (LKB-Wallac) was a time-
resolved immunofluorometric procedure. As the solid
phase it uses a monoclonal antibody to hCG beta-chain
immobilized into wells of microtiter strips. The tracer
is a monoclonal anti-hCG alpha-subunit labelled with
europium. The measurement of fluorescence was performed
on a LKB 1230 Arcus fluorometer.
Roth kits were calibrated against the World Health
Organization (WHO) first international reference
preparation (IRP 75/537) for immunoassay. Procedures
recommended by the manufacturers were followed for
duplicate measurements of the specimensO
Example 3(a~
Biotinylation of Antibody
The monoclonal antibody against hCG intact molecule
(Anti-hCG) was biotinylated according to the following
procedure. A 0.~ mL aliquot of the antibody solution
., ,
~a3a~ )7
solution (1 mg/mL in 0.015 mol/L potassium phosphate
buffer pH 7.2 containing 0.15 mol/L NaCl and 0.1% NaN3)
is mixed with 0.5 mL of a carbonate~bicarbonate buffer
(o.1 mol/L, pH 9.0). To the mixture is then added a
500-fold molar excess of NHS-LC-Biotin dissolved in 100
~L of distilled water (d~20), and incubated at room
temperature for 30 minutes. The unconjugated biotin is
then removed by dialysis in tubing with Mr-10000 cutoff
at 4C for 24 hours against several changes of 0.1 mol/L
sodium bicarbonate pH 8.3 containing 0.05% sodium
azide. The biotin- anti-hCG conjugate was then titrated
to determine the optimal concentration for the assay,
and stored at 4C. Prior to use the antibody is diluted
with the assay buffer to give a working dilution of 1 to
100.
Example 3(b)
abellinq of Streptavidin with BCPDA
Affinity purified streptavidin (Sigma) was
dissolved in 0.1 mol/L carbonate/bicarbonate buffer, pH
9.1, to obtain a concentration of 0.15 mg/mL. To an
ali~uot of this preparation was then added a 50-fold
molar excess of BCPDA dissolved in N,N-dimethyl-
formamide (Sigma) at a concentration of 35 mg/mL. As
exemplified in Example 2(b), the alternative solvent,
25 absolute ethanol, may be used resulting in the use of
less BCPDA. After lh incubation at room temperature,
the mixture was dialyzed against three changes of 0.1
mol/L sodium bicarbonate, pH 8.3, containing 0.05%
sodium azide. This preparation was stored at 4C.
30 Example 3(c)
Assay
Immobilization of Anti-hCG B-Subunit
The monoclonal antibody to hCG B-subunit
(anti-hCG-beta) was immobilized by adsorption onto wells
35 of microtiter plates or strips. The coating was
prepared by adding 100 ~L per wéll of anti-hCG-beta
monoclonal antibody prepared at a concentration o~ 5
c ~g/mL in the coating buffer. After overnight incubation
at 4C, the wells were washed 5 times with the wash
" .
130~0~)7
26
solution, and then 2 times with H20. To block the
remaining active sites, 200 ~L of the blocking buffer
were then added and allowed to incubate at room
temperature for one hour followed by storage at 4C.
Prior to use the plates were washed 2 times with wash
solution.
Assay Procedures
Biotinylated monoclonal antihody was diluted with
the assay buffer to a concentration of 5 ~g/mL. 50 ~L
duplicates of hCG standards or serum samples and 50 ~L
of biotinylated antibody solution were then pipetted
into microtiter wells. After shaking the wells for
three minutes by means of an automatic shaking device,
they were allowed to incubate at room temperature for
two hours. The reaction mixture was then removed, and
the wells were washed four times with the wash solution
using a multi-channel pipette. Binding of biotinylated
antibody to hCG was determined by addition of 100 ~L per
well of the indicator reagent containing BCPDA labelled
streptavidin (1.5 ~g/mL) and EUC13 (10 5 mol/~). The
indicator reagent was prepared by diluting ~1:100) the
stock preparations of BCPDA conjugated streptavidin
(0.15 mg/mL) and of europium (lmmol/L in 0.01 N HCl)
with the strepavidineuropium buffer. After 45 minutes
incubation at room temperature, wells were washed twice
with washing solution, dried for 5 minutes by means of
a forced air plate dryer, and the fluorescence at the
bottom of the dried wells was measured by the CyberFluor
615 fluorometer.
Results
Detection Limit and Dynamic Ranqe of the Assay
A typical dose response curve obtained with the
assay is presented in Figure 4. Points represent mean
fluorescence values of duplicate measurements from
which the mean value of zero standard is subtracted. A
linear relationship between the response and standard
dose exists in the 1 to 500 IU hCG/L concentration
range.
Using the value corresponding to the mean plus
~IL3~)00~:)7
27
three standard deviations (SD) of the zero standards,
the detection limit of the assay appeared to be less
than 1 IU/L. However, when calculated as 3 SD of the
mean for 15 determinations of the zero hCG standard, the
detection limit was about 1.5 IU/L. The background
fluorescence of the zero standard i6 about 6.5 + 0.35
units.
The working range of the assay was established
according to a precision profile derived for twelve
replicate measurements of each standard concentration.
Taking a 10% coefficient of variation (CV) as the upper
limit of imprecision, the working range of the assay is
1 to 500 IU hCG/L serum. However, as the profile shows
(Fig. 4), the CV in the linear range of the assay is
better than 5~.
As shown in Figure 4, the fluorescence intensity,
after reaching a relatively prolonged plateau, begins to
decline with increasing hCG concentration. This is
presumably due to the high dose hook effect which is
often observed with sandwich assays.
Precision
To evaluate the precision of the assay, three
levels of human serum controls for immunoassay (Bio-Rad)
and a pooled human serum sample were used. Within-run
precision was determined by analyzing 21 replicates of
each sample in the same run. Assessment of between-run
precision was by repeat analysis, in duplicate, of the
control samples in five successive runs. The day-to-day
precision was estimated by determining, in duplicate,
the hCG concentration of the same samples on 12
different occasions during a 30 day period (Table 1).
Dilution Linearity
Linearity was assessed by serial dilution of three
different patient samples with the zero standard serum.
Assaying for hCG, the concentration in the undiluted
samples were used to calculate the expected values of
the diluted samples. As shown in Table 3, there was a
linear relationship between the expected and measured
hCG values, with excellent correlation between them.
13000~
28
Analytical RecoverY
To determine the analytical recovery of the assay,
hCG at three different concentrations was added to three
serum pools. Assays were performed on each sample
before and a~ter the addition. Measured and recovered
concentrations are shown in Table 4. Reco~ery ranged
from 92 to 105%, with an average value of 101.5%.
Cross-reactivity
Interference from other hormones was tested by two
experiments. In one, the response of the assay to
increasing concentrations of hTSH, hFSH and hLH in the
absence of hCG was measured. Of the concentrations of
hTSH (50-200 mIU/L), and hFSH ~50~200 IU/L) tested, none
produced a response significantly different than the two
SD range of the hCG negative sample. The hCG response
of the sample was significantly increased with hLH at
concentrations greater than 100 IU/L with a maximum
cross-reactivity of about 18% (Table 5).
In the second experiment, the same concentrations
of these hormones were added to serum samples containing
a fixed concentration of hCG (100 IU/L). This was done
to evaluate the possibility that at high concentrations,
these hormones might compete with hCG for binding to the
biotinylated anti-hCG-alpha monoclonal antibody, thus
producing a false negative effect. With high
concentrations of hTSH and hFSH present, the hCG
response did not change more than the two SD range of
the expected value (i.e. 100 IU/L~. Similarly, hLH did
not appear to have any false negative effect on hCG
detection, but rather generated a false positive
interference. The degree of cross-reaction was the same
as that noted in the previous experiment,
cross-reacting at a maximum of about 17%.
Because of the significant contribution of hLH to
the assay response at a concentration range that
corresponds to physiological as well as pathological
levels (about 200-400 IU hLH/L), the possibility of
including a scavenger anti-hLH antibody to the system
was investigated. A monoclonal antibody against the
;
~L30~
29
indicating that hLH, even at pathological concentrations,
would not lead to any false po~itive or negative results
in the hCG assay.
Correlation with~Immunoradiometric and Immunofluorometric
Assay~_
hCG concentrations in serum samples from pregnant
and non-pregnant females were assayed, in duplicate, by
the present method (TRFIA) and the comparative
immunoradiometric (Bio-Rad) and immunofluorometric (LKB)
procedures. There was a good agreement between the
values obtained with the test method and the comparative
kits. The correlation coefficients with the LKB and
Bio-Rad~ assays were R=0.99 and R=0.97 respectively,
indicating the reliability of the present procedure
(Figs. 5 and 6).
TABLE 2
WITHIN-RUN, BET~ y~-~D DAY-TO-DAY
PRECISION OF THE ASSAY
Within-Run (n~=21) Between-Run (n=16) Day-to-Day (n=12)
hCG(IU/L hCG IU/LhCG,IU/L
Mean SD %/CV Mean SD ~/CV Mean SD ~/CV
14.5 0.62 4.33 14.52 0.90 6.2 13.45 1.30 9.7
52 2.74 5.26 57.06 3.41 5.9 54.09 2.80 5.2
210 9.03 4.3 225.6 18.2 8.0 212.2 15.3 7.2
400 24~3 6.08 --- --- --- 454.4 31.5 6.9
~.,
L. ~
~3~ 7
TABLE 3
RESULTS OF DILUTION OF SERA WITH HIGH hCG CONCENTRATION
Sample 1~ Sa~ple 2~ Sam~le 3c
Dilution E~ ~d Measured E~px*~d M~red E~ ~d M~red
None --- 440 -- 450 - 310
V2 220 210 225 220 155 160
V4 110 100 112 105 78 79
15 V8 55 49 56 52 39 40
1/16 28 25 28 26 19 21
V32 14 13 14 14 10 lO
a, R=1.00, Slope=0.9567, y=-2.1464
b, R=1.00, Slope=0.9778, y=-1.9091
c, R=1.00, Slope 1.0281, y=0.2552
TABLE 4
a
RECOVERY OF hCG ADDED TO THREE SERUM POOLS-
hCG, IU/I
35 hCG added Measured Recovered%Recoveryb
Sample 1
0-0 1.7 --_ ___
49.5 47 45.3 92
99 105 103.3 104
198 210 203.3 105
Sample 2
0.0 17 ---
49.5 ~7 50 101
99 118 101 102
198 215 198 100
Sample 3
98 ___ _
49.5 150 52 105
99 200 102 103
198 300 202 102
a, 300 ~.L aliquots of each standard hCG preparation was
13~310(~
31
added to 3 ml aliquots of each serum pool~
b, % Recovery - (Concentration recovered/Concentration
added)
TAB~E 5
CRQSS-REACTIVITY WITH hLH
LH addeda hoG Equivalent % crossre~ction hoG Equivalent %Cx~sreaction
loO 12 12 15 15
15 200 30 15 32 16
500 90 18 86 17
LE addedb
20 100 1.0 1.0 0.0 0.0
200 1.7 0.8 0.0 0.0
500 14 2.8 5.0 1.0
a, in the absence of anti-hLH
b, in the presence of anti-hLH
Example 4
Labellinq of Monoclonal Antibodies
Generally, selected monoclonal antibodies may be
directly labelled with BCPDA. For example, a monoclonal
antibody specific to cortisol can be labelled as
follows. 0.20 mg/ml of the monoclonal antibody in O.lM
carbonate buffer of pH 9.1 was mixed with a freshly
prepared absolute ethanolic solution of ~CPDA by adding
the solution in 4 aliquots at 1 minute intervals while
continually vortexing the solution. The excess of BCPDA
added over the amino group concentration on the
monoclonal was 0.5 fold. The working monoclonal
antibody solution had a concentration of 2 ~g/ml. The
; diluent for the working solution was 50 mM Tris buffer
of pH 7.80 containing 9 g NaCl, 10 g BSA and 0.5 g
sodium azide per liter.
Preparation of thyroglobulin-cortisol coniugate
A standard procedure to prepare conjugates as
described in Erlanger et al (1959) "Steroid-Protein
Conjugates: Preparation and Characterization of
~3~)00~7
32
Conjugates of BSA with Progesterone, Deoxycorticosterone
and Esterone" J. Biol. Chem. 234, 1090 was modified as
described by Elder et al (1987) "An Enzyme-
Linked/Immunosorbent Assay (ELISA) for Plasma
Progesterone: Immobilized Antigen Approach" Clin. Chem.
Acta. 162, 199 to prepare the desired conjugate. In
accordance with the modified method, cortisol 21-
hemisuccinate was used instead of progesterone-3-o-
carboxymethyloxime~
Coatinq of Microtitration Strips
The coating buffer was a 0.1 M carbonate solution
of pH 9.5. The blocking buffer was a 50 mM sodium
phosphate solution of pH 7.4 containing 9 NaCl, 1 g
gelatin and 1 mL polyoxyethylenesorbitan monolaurate
(Tween 20) per liter. The wash solution was a 9 g/L
NaCl solution containing 0.5 mL Tween 20 and 0.5 g
sodium azide per liter.
The strips were coated overnight at room
temperature with 100 ~L of a 6 ~g/mL solution of
cortisol-thyroglobulin conjugate in the coating buffer.
After coating, the strips were rinsed once with the wash
solution, blocked for 1 h at room temperature with 200
~L of the blocking buffer, washed twice and dried
overnight at room temperature, preferably under reduced
pressure. Stored in sealed plastic bags at 4C with
desiccant, they are stable for several weeks.
Monoclonal antibody cortisol assay procedure
Before initiating the assay, the cortisol-
thyroglobulin coated strips were washed twice with the
wash solution. Twenty ~L of standards were pipetted
into each well and 100 ~L of the BCPDA-labelled
monoclonal antibody working solution containing 10 5 M
EUC13 was added. The strips are then briefly shaken and
incubated for 1 h at 37~C. ~hs strips are washed three
times with the wash solution and dried with a stream o~
air. Surface fluorescence of the complex,
thyroglobulin-cortisol-antibody-BCPDA-Eu3+ was measured
on the CyberFluor 615 Immunoanalyzer.
The monoclonal anti-cortisol antibody that was
o~
33
labelled at the optimal molar ratio of 18 BCPDA per
protein molecule was used to generate a standard curve
for a competition type assay using aqueous cortisol
standards as show in Figure 8.
Example 5
A second polyclonal antibody specific to the
monoclonal antibody of Example 4 may be used to analyze
for the presence of the analyte-cortisol. The
polyclonal antibody, which is specific to the
monoclonal, may be a polyclonal affinity purified goat
anti-mouse IgG. The polyclonal is labelled as per the
method of Example 4. The working solution of the
polyclonal antibody is at a concentration of 20 ~g/ml.
The thyroglobulin-cortisol conjugate and
microtitration strips of Example 4 were used.
The polyclonal antibody assay procedure of Example
4 was followed where the monoclonal antibody as labelled
was used in the first step. After incubation and
washing, 100 ~L of the working (20 ~g/ml) labelled
- 20 polyclonal; i.e. second antibody solution containing 10
5 M EUC13 was added and incubated for 30 min at 37C.
-~ The wells were then washed and the assay completed by
proceeding as above. In this case, the fluorescent
complex on the dry solid-phase is thyroglobulin-
cortisol-antibody-second antibody-BCPDA-Eu3 .
This ~ompetition assay system produced a
calibration curve similar to that of Figure 8 thereby
indicating the utility of this system as a viable form
of immunoassay.
- 30 Example 6
As with Examples 4 and 5, it is possible to
directly label other monoclonal antibodies. The ~-
~ fetoprotein (AFP) monoclonal antibody may be labelled
- with BCPDA as per the method of Example 4. AFP
- 35 standards were pipetted into AFP antibody coated
microtitration wells. 100 ~L of the directly labelled
antibody solution (5 ~g/ml in antibody containing 10 5 M
Eu3~) were added and incubated for 90 min at 37C. The
wells were washed three times with the wash solution,
.
"~ ,
'
13~007
34
dried for 3 min with cool air and the ~luorescence of
the labelled monoclonal was measured on the CyberFluor
Model 615 (trade mark~ Immunoanalyzer.
The solid phase sandwich type assay demonstrated
the utility of the labelled monoclonal antibody. By
loading a variety of concentrations of AFP into the
wells, a calibration curve of fluorescent intensity vs.
concentration of AFP can be developed.
In the above Examples, the preferred lanthanide
metal ion used in the fluorescent chelate is europium.
It is appreciated that other lanthanide metal ions are
usable, such as, terbium, gadolinium, samarium and
dysprosium.
Although preferred embodiments of the invention
have been described herein in detail, it will be
understood by those skilled in the art that variations
may be made thereto without departing from the spirit of
the invention or the scope of the appended claims.