Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
`` ~h3~ 5~3
IMPROVED HYDROMETALLURGICAL ARSENOPYRITE PROCESS
FIELD OF THE INVENTION
This invention is directed to an improved process
for leach treating gold and silver bearing pyritic and
arsenopyritic concentrates and oxes. More particularly,
the improved process avoids the necessity of adding re-
cycled neutralized solution to the leach solution, thereby
alleviating difficulties in maintaining acid levels in the
leach solution and provides for bleeding solutions contain-
ing dissolved arsenic, iron and sulphate from the process
without the loss of oxidized nitrogen species.
BACKGROUND OF THE INVENTION
In our copending application Serial No. 797,838,
filed November 14, 1985, which has matured as U.S. patent
No. 4,647,307, March 3, 1987, we disclose a hydro-
metallurgical process for the recovery of pr~cious metalfrom a concentrate or ore containing at least some arseno-
pyrite or pyrite wherein at least some of the precious
metal is occluded in the arsenopyrite or pyrite. The
hydrometallurgical process involves forming in a common
~olume space a gas phase and a liquid slurry which com-
prises the ore or concentrate as the solid phase and acid
and water as the liquid phase of the slurry. An oxidation-
reduction reaction having a standard potential between
about 0.90 and about 1.20 volts on the hydrogen scale is
effected in the slurry between the arsenopyrite or pyrite
and an oxidized nitrogen species in which the nitrogen has
a valence of at least plus 3. This solubilizes in the
liquid phase the arsenic, iron and sulphur in the arseno-
pyrite, or the iron and sulphur in the pyrite, all as the
oxidation products, and produces in the liquid phase nitric
oxide in which the nitrogen has a valence of plus 2, as the
- 1 -
,
~3~
reduction product. At least part of the nitric oxide
from -the liquid phase is released into the gas phase.
The nitric oxide in the gas phase, in which a signifi-
cant oxygen partial pressure is maintained by continuous
addition of an oxygen containing gas, is oxidized to
form an oxidized nitrogen species in which the nitrogen
has a valence of at least plus 3. The total amount of
oxygen that is added is at least in an amount stoichio-
metrically required for solubilization in the liquid
phase of the arsenic, iron and sulphur in the arseno-
pyrite, or the iron and sulphur in the pyrite. The
oxidized nitrogen species is absorbed into the slurry
wherein the oxidized nitrogen species become available
for the oxidation-reduction reaction as described. In
this way, the nitrogen, in its oxide forms, functions as
a catalyst for the transport of oxygen from the gas
phase to the oxidation-reduction reactions in the
slurry. This permits the total of the oxidized nitrogen
species and nitric oxide in the system to be substan-
tially less than a stoichiometric balance that would berequired for the oxidation of the arsenic, iron and
sulphur. The slurry is then subjected to a solid-
liquid separation to produce a solid residue and a
liquid fraction. Precious metal is recovered from the
solid residue.
While the process described has a number of
unique features and important advantages, which are
fully discussed in the co-pending application, the
process has three minor shortcomings which are briefly
described below:
1. A bleed solution must be utilized in order
to remove impurities which build up in the solution~
Unfortunately, this bleed solution removes not only
impurities but also valuable catalyst from the process.
- 2 -
~3~
2. The recycle solution in the process is
satura-ted in dissolved calcium (calcium nitrate). When
this solution is recycled to the leach, and leaching
proceeds, the sulphate which is formed in solution tends
to combine with the calcium from the recycled solution
to form calcium sulphate. Such calcium sulphate in-
creases the weight of solids leaving the reaction auto-
clave and thereby increases the burden on the downstream
cyanide circuit. The tendency to form calcium sulphate
in solution causes the build-up of this material on heat
exchanger surfaces, which reduces process efficiency
over time.
3. Since the recycle solution is neutral
(with regard to pH) the introduction of this recycle
stream into the leach can cause difficulties in main-
taining the leach solution at a sufficient acid concen-
tration.
SU~ARY OF THE INVENTION
The invention is directed to an improved
hydrometallurgical process for recovering valuable
metals from pyritic and arsenopyritic concentrates and
ores involving decomposing the arsenopyrite or pyrite
concentrates and ores in acidic solution in a common
~5 volume space which contains a gas phase and a liquid
slurry (which comprises a liquid phase and a solid
phase) through the action of higher valence oxidized
nitrogen species in which the nitrogen has a valence of
at least plus 3. The active oxidized nitrogen species
is regenerated in the same common volume space by an
~; oxygen containing gas. The improvement comprises intro-
ducing the concentrate or ore into a denitrating step,
` along with the solution from the leach. The oxidized
nitrogen species present in the leach solution react
with the concentrate or ore to produce nitric oxide
- 3 -
,
i
59
which is released into the gas phase. The liquid and
solid products of the denitrating step are then sub-
jected to a solid-liquid separation. The solids from
the separation are transported to a leaching vessel
where the solids are treated with oxygen and nitric
oxide taken from the denitrating step. The liquid is a
bleed to the process.
~ pecifically, the process which is intended
for the recovery of precious metal from an ore or con-
centrate containing at least some arsenopyrite orpyrite, wherein at least some o~ the precious metal is
occluded in arsenopyrite or pyrite, comprises: (a)
introducing pyrite or arsenopyrite concentrate or ore
into a denltrating vessel where it is reacted with a
recycled solution which is derived from the process
- after gold and silver has been precipitated, (b) contin-
uously removing nitric oxide gas which is generated in
the gas phase; (c) continuously withdrawing the treated
concentrate or ore ~rom the denitrating vessel and
subjecting it to a solid-liquid separation; (d) trans-
; porting the solids from the solid-liquid separation to a
leaching vessel; (e~ forming in a common volume space a
gas phase and a liquid slurry comprising the ore or
concentrate as the solid phase, acid and water as the
liquid phase oF the slurry and nitric oxide gas from
step (b) above and oxygen as the gas phase; (f) ef~ect-
ing in the slurry between the arsenopyrite or pyrite and
an oxidized nitrogen species in which the nitrogen has a
valence of at least plus 3 an oxidation-reduction reac-
tion having a standard potential between about 0.90 andabout 1.20 volts on the hydrogen scale, thereby solu-
bilizing in the liquid phase the arsenic, iron and
sulphur in the arsenopyrite, or the iron and sulphur in
the pyrite, all as the oxidation products, and producing
in the liquid phase nitric oxide in which the nitrogen
-
~3~ 9
has a valence of plus 2, as the reduction product; (g)
releasing at least part of the nitric oxide generated in
the liquid phase into the gas phase; (h) oxidizing the
nitric oxide in the gas phase, in which a significant
oxygen partial pressure is maintained by continuous
addition of an oxygen containing gas, to form an oxi-
dized nitrogen species in which the nitrogen has a
valence of at least plus 3, the total amount of oxygen
added being at least in an amount stoichiometrically
required for solubilization in the liquid phase of the
arsenic, iron and sulphur in the arsenopyrite or the
iron and sulphur in the pyrite; (i) absorbing the
oxidized nitrogen species into the slurry wherein the
oxidized nitrogen species become available for the
oxidation-reduction reaction of step (f) above whereby
the nitrogen, in its oxide forms, functions as a cata-
lyst for the transport of oxygen from the gas phase to
; the oxidation-reduction reactions in the slurry, thereby
permitting the total of the oxidized nitrogen species
and nitric oxide in the system to be substantially less
than a stoichiometric balance required for the oxidation
of the arsenic, iron and sulphur; (j) subjecting the
slurry to a solid-liquid separation to produce a solid
residue and a liquid fraction; and (k) recovering
precious metal from the solid residue.
~ ;
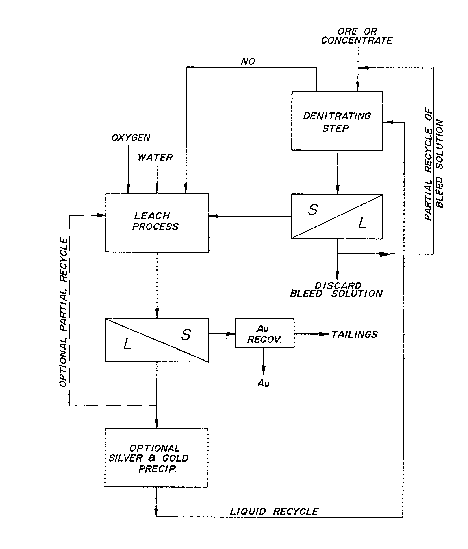
DRAWINGS
In the drawing which discloses a specific
embodiment of the improved process:
Figure l illustrates a graphic flowsheet of
the improved process of the invention.
- 5 -
i9
DETAILED DESCRIPTION OF A SPECIFIC
-
EMBODIMENT OF THE INVENTION
The improved hydrometallurgical process is
basically intended for the recovery of precious metal
from an ore or concentrate containing arsenopyrite or
pyrite wherein at least some of the precious metal is
occluded in the arsenopyrite or pyrite. The improvement
comprises first introducing the concentrate or ore into
a denitrating step, along with the solution from the
- 10 leach. The oxidized nitrogen species present in the
leach solution react with the concentrate or ore to
produce nitric oxide which is released into the gas
phase. The liquid and solid products of the denitrating
step are subjected to a liquid-solid separation. The
resultant solids from the separation are introduced into
the leaching vessel where the solids are treated with
oxygen and nitric oxide Erom the denitrating step. -~
A gas phase and a liquid slurry are formed in
the leaching vessel. The liquid is a bleed to the
process. The slurry is comprised o-E -the ore or concen-
trate as a solid phase and acid and water as a liquid
phase. An oxidation-reduction reaction having a
~ standard potential between about 0.90 and about 1.20
; ~ volts on the hydrogen scale is effected in the slurry
between the arsenopyrite or pyrite and an oxidized
nitrogen species in which the nitrogen has a valence of
at least plus 3. Arsenic, iron and sulphur in the
arsenopyrite, or iron and sulphur in the pyrite, are
solubilized in the liquid phase as oxidation products.
Nitric oxide in which the nitrogen has a valence of plus
2 is produced as a reduction product in the liquid
phase. At least part of the nitric oxide is released
from the liquid phase into the gas phase. The nitric
oxide in the gas phase, in which a significant oxygen
partial pressure is maintained by the continuous addi-
::
~ - 6 -
' ' , , ~' ' ,' ' ~ ` .
- '
' '
.. . .
`
. - ~ .,, . ., .. , ,,.
~3~59
tion of an oxygen containing gas, is oxidized to form an
oxidized nitrogen species in which the nitrogen has a
valence of at least plus 3. The total amount of oxygen
added is at least in an amount that is stoichiometri-
cally required for solubilization in the liquid phase ofthe arsenic, iron and sulphur in the arsenopyrite, or
the iron and sulphur in the pyrite. The oxidized
nitrogen species are absorbed into the slurry wherein
the oxidized nitrogen species become available for the
oxidation-reduction reaction. The nitrogen, in its
oxide forms, functions as a catalyst for the transport
of oxygen from the gas phase to the oxidation-reduction
reactions in the slurry. This permits the total of the
oxidized nitrogen species and nitric oxide in the system
to be substantially less than a stoichiometric balance
required for the oxidation of the arsenic, iron and
sulphur. The slurry is removed from the common volume
space and is subjected to a solid-liquid separation to
produce a solid residue and a liquid fraction. Precious
metal is recovered from the solid residue.
The arsenopyrite and pyrite are decomposed by
the oxidation-reduction reaction in acid solutions in
the slurry where the pH is less than about 1.0 to about
3 by the action of oxidized nitrogen species where the
nitrogen has a valence of plus 3 or greater. These
oxidized nitrogen species include nitrous acid and
; nitrogen dioxide. The oxidized nitrogen species are
present in sufficient concentration in the liquid
fraction (typically about 0.25 Molar (M~ to about
4.0 Molar (M), calculated on a nitric acid basis) to
provide an adequate rate of dissolution (typically
within a residence time of about 2 minutes to about 60
minutes) at the reaction temperature used (typically
about 60C to about 119C for arsenopyrite concentrate
and about 60C to about 180C for pyrite concentrate or
-- 7 --
''
59
ore). Normally, the lower oxidized nitrogen species
concentrations and longer residence times are used when
treating ore while the higher oxidized nitrogen species
concentrations and shorter residence times are used when
treating concentrates.
The main products from the oxidation-reduction
reaction are soluble ferric iron species, soluble arsen-
ate species, soluble sulp~ate species, minor amounts of
elemental sulphur and nitric oxide.
Insoluble gangue minerals and elemental sulfur
remain as solids in the slurry. The slurry is subjected
to a solid-liquid separation to yield a solid residue
and a liquid fraction. A major portion of the gold or
other precious metal contained in the concentrate or ore
remains in the solid residue. Some of the gold or other
precious metal appears in the leach solution, and can be
recovered with activated carbon.
Almost all of the silver present in the con-
centrate will usually remain in the liquid fraction.
The silver can be recovered from the liquid fraction by
using a thiocyanate compound such as sodium thiocyanate,
potassium thiocyanate, or ammonium thiocyanate. Arsenic
and iron can be optionally removed from the silver- Eree
separated liquid fraction by elevating the temperature
~5 to precipitate ferric arsenate. In the case of pyrite,
iron can be removed from the liquid fraction.
In general terms, the process of the invention
can be operated at a standard potential between the
arsenopyrite or pyrite and the oxidized nitrogen species
of about 0.90 volts and about 1.20 volts on the hydrogen
scale. At potentials below about 0.9 volts, arseno-
pyrite or pyrite will not decompose efficiently. At
potentials above about 1.2 volts, no significant oxida-
; tion of the nitrogen species will take place because
~,
-- 8 --
5;9
oxygen per se has a potential of about 1.23 volts on thehydrogen scale.
On the standard oxidation-reduction potential
scale, the reduction of nitrous acid to nitric oxide has
a standard potential of about 0.996 volts. The reduc-
-tion of nitrate to nitrous acid has a sta~dard potential
of about 0.94 volts. Thus the former couple has a
higher driving force than the latter in decomposing
sulphide minerals such as arsenopyrite and pyrite.
Preferably, the process of the invention is
operated at a potential greater than about 0.94 volts up
to about 1.0 volts on the hydrogen scale.
The process can typically be conducted within
a residence time range of about 2 minutes to about 60
minutes calculated on a plug flow basis. A process
which is completed in a time less than about 2 minutes
is difficult to control and basically impractical. On
the other hand, a process which takes more than about 60
minutes to complete is too slow and thus uneconomical.
The process has been conducted experimentally
at initial temperatures from above the freezing point of
the slurry to temperatures of several hundred degrees
~; Celsius. However, temperatures falling in the range of
about 60C to about 180C are preferred for economical
reasons. Similarly, the process has been conducted at
pH ranges of less than about 1.0 to as high as about
; 3Ø In situations where silver is not present, and the
formation of basic iron sulphate or jarosite can be
tolerated in the process, the process can be conducted
at a pH of about 3Ø However, silver is usually
present and therefore it is preferable to operate the
process at lower pH ranges. Typically, a pH of about
1.0 or below is preferred because it is desirable to
keep the iron and arsenic in solution. Also, the pro-
:
_ g _
59
cess is more rapid and economical at a pH range of lessthan about 1Ø
In the process, the oxidized nitrogen species
in a sense act as a transporter of oxygen. The basic
process may be regarded as an oxygen leach rather than
an oxidized nitrogen species or nitric acid leach. The
oxidized nitrogen species serves as a carrier for the
oxygen as the oxidized nitrogen species is cycled
between the gas phase and the liquid phase o the slurry
of the common volume space. It follows that the rate at
which the reaction proceeds is proportional to the
number of oxidized nitrogen species carriers that are in
the process.
Sufficient oxygen must be supplied to the
leaching vessel in order to completely decompose the
arsenopyrite and pyrite in the slurry. Nitric oxide
derived from the denitrating step is also introduced
into the leach vessel. If insufficient oxygen is sup-
plied, then the pressure of the nitric oxide generated
increases and ultimately the reaction stops because
there are no oxidized nitrogen species left in the
liquid phase of the slurry.
In order to overcome the limitations of uti-
lizing a bleed which removes valuable catalyst from the
process, and introducing into the leach vessel a neutral
recycled solution which promotes calcium sulphate forma-
tion and causes difficulties in maintaining the leach
solution at appropriate acid levels, while at the same
time retaining and utilizing all of the unique and
advantageous features of our basic arsenopyrite and
pyrite treating process, we have developed a variation
of the basic process which does not introduce the
concentrate in the leach. The improved process is
described in association with the flow sheet which is
illustrated in Figure 1.
-- 10 --
: ' ' ' '
.
q~
In the process as illustrated in the flowsheet
o~ Figure 1, the concentrate is introduced into a pre-
liminary denitrating stage along with a recycled solu-
tion which is derived from the leach after gold and
silver precipitation. The liquid and solids of the
denitrating stage are subjected to a solid-liquid
separation. The solids are transported to the leach
vessel. Nitric oxide from the denitratil~g stage is also
introduced into the leach vessel. The nitrate in the
leach solution reacts with the arsenopyrite and pyrite
concentrate according to the following general process
criteria and leaching reactions:
A. Mineral Oxidations
(1) Fe AsS -~ Fe+3(aq) + 1/2As2S2 + 3e
~2) FeAsS + 3H20 -~ Fe+3 + H3As03 + SO
+ 3H+ + 6e
(3) FeAsS + 4H20 -~ Fe+3 + H3AsO4 -~ SO
+ 5H+ + 8e
(4) FeAsS + 8H20 -~ Fe+3 + H3As04 + S04=
+ 13H~ + 14e
(5) FeS2 -~ Fe+3(aq) + 2SO + 3e
(6) FeS2 + 8H20 -~ Fe+3 + 2S04= + 16H+
+ 15e
B. Oxidized Nitrogen Species Reduction
(7) HN03 + 3H~ + 3e -~ NO(g) + 2H20
(8) HN02 ~ H+ + e -3 NO(g) + H20
:
In the oxidation of arsenopyrite, it has been
found that 60-90% of the mineral's sulphur is converted
to soluble sulphate species. In the oxidation of
pyrite, the degree of conversion is 80-100%.
Equations A(l) and A(5) will, in principle,
take place at potentials above about 0.6 volts on the
hydrogen scale; however, since 1/~ (~s2S2) on
arsenopyrite and 2S0 on pyrite have molar volumes
larger than FeAsS and FeS2 respectively, the first
submicroscopic layers of these leach products protect
the mineral from further oxidation, and no substantial
: reaction is observed. At potentials above about 0.94
volts on the hydrogen scale, reactions A(4) and A(6)
taXe place, and the protective layers of As2S2 and
S0 are eliminated by oxidation.
In the denitrating step, reaction B(7) absorbs
electrons at a standard potential of 0.94 volts on the
hydrogen scale, just barely adequate to remove electrons
from arsenopyrite and pyrite to drive reactions A(4) and :~
A(6) at a feasible rate (as in Queneau). Reaction B(8),
which takes place in the leach process, absorbs elect-
: rons a~ a standard potential of 0.996 volts on the
hydrogen scale, which is high enough to drive reactions
A(4) and A(6) rapidly at temperatures as low as 60C.
The active nitrogen oxides are required only
to act as a sink for electrons which are released by
decomposition of the minerals in the concentrate or ore.
The oxidized nitrogen species should be present in
sufficient concentration in the solution (typically
about 0.25 M to about 3.0 M or 4.0 M) to provide an
:~ adequate rate of dissolution (typically within a
residence time of about 2 minutes to about 60 minutes)
at the reaction temperature used (typically about 60C
to about 119C for arsenopyrite concentrate, and about
60C to about 180DC for pyrite concentrate or ore).
:
.:
:- :
: .
'
59
Sulphuric acid may be used to form the soluble ferric
iron species and under certain circumstances is produced
in situ.
In the following reaction, nitrous acid is the
decomposition agent for arsenopyrite with sulphuric acid
~- present.
(9) FeAsS + 1/2H2S04 -t 14HN02 -~
1/2Fe2(S04)3 + H3As04 + 14NOtg) + 6H2
Sufficient sulphuric acid was supplied with
arsenopyrite and was consumed to form soluble ferric
iron species. Without such acid, compounds will
precipitate from solution.
In the reaction detailed below, the sulphuric
acid is generated from the decomposition of pyrite.
(10) FeS2 + 15HN02 -~ 1/2Fe2(S04)3 +
1/2H2S04 ~ 15NO(g) + 7H20
In the preceding reactions, the active nitro-
gen oxides are reduced to nitric oxide which may then be
regenerated by an oxidant. ~ useful oxidant is oxygen
which reacts with nitric oxide in the presence of water
to form nitrogen dioxide, nitrous acid and nitric acid
as shown in the reactions set forth below.
(11) ~0 + 1/2 2 ~- N2
(12~ NO + N02 ~ H20 ~- 2HN2
(13) 3HN02 ~ HN03 + H20 -~ 2NO
While the leach process is in continuous
operation, the generation of nitric acid (reaction (13~)
:
~:~
- 13 -
is not desirable and is to be avoided. This is accomp-
lished by conducting reactions A(4) and A(6), B(8) and
reactions (11) and (12) in a common volume space where
the nitrous acid can be readily consumed by reactions
(9) and (10) so as not to form nitric acid according to
reaction (13). The regeneration of nitric oxide to the
higher valence states is done concurrently with the
decomposition of pyrite in the common volume space.
It is clear from equations (11) to (13) that
HNO2 is the principal dissolved oxidized nitrogen
species arising from the gas phase oxidation of NO and
dissolution of the resulting NO2. Reaction (13) is
rather slow, and HNO2 is therefore the principal
dissolved oxidized nitrogen species that is able to
react with the oxidizable minerals (reactions (9) and
(10)). Oxygen is used for nitrogen oxide regeneration.
The rate of regeneration varies directly with oxygen
partial pressure. Any oxygen partial pressure above
ambient is adequate, but oxygen partial pressures of
about 50 psig to about 100 psig are preferred. The
regeneration step is carried out with an oxygen
containing gas concurrently with the decomposition
reaction(s) (reactions A(4) and A(6)). The overall
stoichiometry of arsenopyrite reacting with sulphuric
acid and oxygen utilizing the oxidized nitrogen species
as a catalyst (transporter) is illustrated by the
reaction below.
(14) FeAsS + 1/2 H2SO4 ~ 1/2 Fe2(SO4)3 +
+ 7/2 2 + H2O H3AsO~ + 14 HNO2
+ 14 HNO2
Since the active oxidized nitrogen species are
regenerated during the decomposition step in the common
- 14 -
volume space, the quantity of these species present at
any time may be quite small.
It is apparent from equation calculations that
the H~03 concentrations initially added are far too
low to completely decompose so much arsenopyrite. If
the initially present HN03 were the only oxidant, and
remained the only oxidant, stoichiometric calculations
would show that a minimum of 5 moles of HN03 would
have been required to completely decompose the 1 mole of
arsenopyrite. This is evidenced by the following
equation:
(15) FeAsS ~ 5H~03 ~' 1/3FE2(S04)3
~ 1/3FE(N03)2 + H3As04 + H20 + 5NO(g)
Yet, the mineral has been found to be completely decom-
posed by as little as 0.5 M HN03, or 1/10 of the
stoichiometric requirement, for example, oxidized nitro-
gen species cycled ten times. This illustrates the
highly catalytic property of the oxidized nitrogen
specles.
At oxidized nitrogen species concentrations of
0.25 M or less, the decomposition rate is too slow to be
a practical consideration. At oxidized nitrogen species
concentrations of about 3.0 M, the reaction rate is ver~
; rapid and hence sufficient for most purposes. Greater
concentrations than about 3.0 M do not provide greatly
improved reaction rates.
The mineral decomposition and oxidized nitro-
gen species regeneration steps are both exothermic.Thus, in conducting the reactions, the slurry in the
~ common volume space and the denitrating step must be
; cooled in order to maintain a constant operating temper-
ature.
- 15 -
.
' ~ :
,
The decomposition leach can be carried out
over a wide range of solid-liquid ratios. The solid-
liquid ratio in the denitrating step is dictated by the
mass balance of the overall process.
In the denitrating step, the objective is to
treat the liquid recycle from the silver precipitation
with feed concentrate or ore, in the absence of oxygen,
in order to produce a bleed solution free of oxidized
nitrogen species. This is accomplished by some of the
leaching reactions which have been discussed above.
Temperatures and pressures are generally similar except
that the pressure in the denitrating step should be
slightly greater than in the leaching step, eg. 20 to 50
psig greater, in order to drive the generated nitric
oxide over to the leaching process in the common volume
space. The time duration of the denitrating step is
similar to the leaching step.
Following the denitrating step, a solid-
liquid separation is effected. The partially leached
solids from this separation step are transported to the
lsach vessel in order to be reacted according to the
reactions which are fundamental to our basic leach
process as discussed above. A part of the nitrate-free
solution forms the partial recycle of the bleed solution
which is added to the concentrate or ore to for~ a
slurry which is pumped into the denitrating step. The
remainder of the nitrate-free solution forms the bleed
solution of the process which can be discarded or
treated. The nitrate-free solution is neutralized with
limestone and/or lime in order to reject the waste
materials according to the normal procedures of our
basic process.
Our improved process, among other things, has
the advantage that any desired amount of solution can
form a bleed to the process. The quantity of solution
::~
- 16 -
which is treated in the denitrating step relative to the
amount of solution which is recycled directly to the
leach as the optional recycle will depend on factors
such as the iron and arsenic content of the concentrate.
Another advantage of the preferred process, ie. no
precipitation of iron, arsenic or sulphate from the
liquid recycle, is that no neutralized solution is
introduced into the leach.
The choice of oxidized nitrogen species
concentration, decomposition temperature and time for
leaching is governed by the nature of the material to be
leached. Convenient initial sources of the oxidized
nitrogen species are nitric oxide gas or nitric acid.
The solids are decomposed in a single pass and no
recycle of solids is required. When the decomposition
reactions are complete, a solid-liquid separation is
carried out to produce a solid residue containing the
majority of the gold and a clarified liquid fr~ction
which may contain some of the gold and silver.
Our process offers the option of producing
high purity arsenic trioxide. The conditions of the
leach can be varied to maximize the presence of the
extracted arsenic as soluble arsenite. Arsenic trioxide
can then be precipitated by cooling the filtered decom-
position solution. By using a low decomposition temper-
ature (70C) and a low concentration of oxidized nitro-
gen species (0.5 M HN03 for 1.25 M FeAsS) and then
cooling the filtered decomposition solution to 10C, it
wa5 found that 35 percent of the extracted arsenic was
recovered as As203. ~ormally, however, when arsenic
trioxide production is not required, process conditions
are chosen so as to maximize to oxidiation of arsenic to
the arsenate state.
The separation of silver from the acidic
liquid fraction which contains iron, arsenic, sulphate
- 17 -
`"` ~3~ i9
and oxidized nitrogen species represents another inven-
tive aspect of the process.
A portion of the silver present in the concen-
trate or ore reports to the liquid fraction. The silver
may be recovered as a thiocyanate compound with the
addition of one mole of thiocyanate per mole of silver.
The reaction time involved is very short, typically
about one minute. Thiocyanate compounds which can be
used are sodium -thiocyanate, potassium thiocyanate or
ammonium thiocyanate~
At high solution temperatures, thiocyanate is
oxidized by the oxidized nitrogen species present in the
solution. In a solution which is three molar in nitrate
ions, oxidation of the thiocyanate occurs at an in-
creased rate at temperatures in excess of about 80C.Therefore, if the leach is conducted at a temperature of
lOO~C, for example, the liquid fraction should be cooled
to about 80C or lower, eg., down to 60nC, in order to
avoid decomposing the thiocyanate. An important and
unique feature of the silver removal process is that the
thiocyanate added in excess of that required for silver
removal reacts with the ferric iron present to form
soluble ferric thiocyanate complexes which have an
intense red colour. The presence of this red colour
acts as an indicator to show that sufficient thiocyanate
has been added. A solid and liquid separation is car-
ried out to recover the silver thiocyanate precipitate.
The silver can be recovered from the precipitate by
smelting or by conventional hydrometallurgical treat-
ment. The liquid separated is suitable for recycle tothe denitrating step. Although the inclusion of the
denitrating step eliminates the need to remove dissolved
arsenic, iron and suplhate, removing these dissolved
substances enables the process to be operated with less
- 18 -
~ J~ ~
make-up water being added. This could be an important
feature in water conservation areas.
Dissolved arsenic can be removed from solution
with dissolved iron in the form of ferric arsenate. The
following reaction shows the formation of ferric
arsenate from ferric nitrate and arsenic acid
(arsenate)0
tl3) Fe(N03)3 + H3As04~` 3 HNO3 + FeAsO4 2H20
~ 2 H20
Ferric arsenate is produced, virtually quanti-
tatively from an equimolar solution of ferric nitrate
and arsenate at all temperatures above ambient. How-
ever, the rate can be controlled by temperature regula-
tion and by the addition of nucleating a~ents. With an
-~ unnucleated solution at room temperature, complete
precipitation (> 95 percent removal of iron and arse-
nate) requires several months, at 100C, precipitation
requires several hours; and at 200C, precipitation
occurs in less than one hour. When nucleated by fine
ferric arsenate, the rates become more rapid.
The ferric arsenate produced is a crystalline
solid which shows the X-ray diffraction pattern of
FeAsO4 2H20. The solubility of this material,
when mixed with water, is very low (less than 1 ppm
arsenic). The crystalline ferric arsenate is unique in
that it precipitates from a strong nitric acid solùtion.
For example, a ferric arsenate precipitate has been
produced in 5 M HNO3.
The crystalline ferric arsenate obtained from
this process is distinctly different from the ferric
arsenate that is produced from the neutralization of
acidic ferric nitrate and arsenate solutions. The
latter material is colloidal and shows no X-ray diffrac-
,
- 19 -
`
, , :
- ,.....
~3~
tion pattern. ~en mixed with water, the solubility of
the amorphous ferric arsenate is in excess of 20 ppm
arsenic. The amorphous ferric arsenate is difficult to
filter and can contain ferric hydroxide which also tends
to be colloidal and hence difficult to filter.
It has been discovered that the presence of
sulphate in solution hampers the formation of crystal-
line ferric arsenate. Sulphate must be removed from
solution prior to ferric arsenate precipitation. A
solution which is 1 M in ferric nitrate and arsenate is
stable at 100C in the presence of 0.8 M sulphate as
H2S04 .
A calcium-bearing substance such as calcium
oxide, calcium hydroxide or calcium carbonate, or a
barium-bearing substance such as barium carbonate, can
be used to remove sulphate in order to facilitate
crystalline ferric arsenate precipitation. The calcium
and barium bearing substances also partially neutralize
the solutions, however amorphous ferric arsenate is not
produced if the rate of addition of the neutralizing
agent is slow. The mixture of crystalline ferric
arsenate and calcium or barium sulphate filters very
well.
Because of the inhibiting effect of sulphate
on the formation of ferric arsenate, the rate of ferric
arsenate precipitation is dependent on the rate of
calcium sulphate precipitation. At high temperature,
e.g. over 150C, 95 percent of the iron and arsenic is
removed in less than one hour. At 100C, while some
sulphate is present, in the absence of a nucleation
agent, the rate of iron and arsenic removal is slower,
i.e. 95 percent removal requires in excess of 12 hours.
At 100C, when sulphate removal is complete, and nucle-
ation is provided by recycling previously formed ferric
arsenate, 95 percent removal can be achieved in one
- 20 -
hour. Arsenic removal proceeds at a satisfactory rate
at temperatures below 100C when sulphate removal i5
complete and a nucleation agent is provided.
When treating pyritic concentrates, ferric
iron can be removed from solution by the formation of
insoluble iron compounds e.g. ferric hydroxide or basic
iron sulfate through the neutralization of the solution.
The tendency for silver to be bound up with
jarosite results in silver losses if jarosite precipi-
tates are formed during the decomposition step of theprocess. E~owever, jarosites do not form promptly from
supersaturated solutions since they are a crystalline,
filterable solid that nucleates very slowly. A high
acid level suppresses the formation of jarosite. The
applicants have found that it is possible with the
process to conduct the decomposition step in such a way
that all the iron, and arsenic, and most of the sulphur,
are dissolved long before the precipitation of jarosite
becomes rapid. It is also possible to complete the
decomposition step, separate the gold bearing solid
residues, precipitate any dissolved silver, and then
reheat the liquid fraction (without necessarily addi-
tional neutralization) to precipitate jarosite free of
~; precious metals.
Various trace elements such as copper, mag-
nesium, zinc, bismuth or tellurium may be present in the
concentrate being treated. While some of these trace
elements will report to the solid residue or waste
precipitation residues, some may build up in the liquid
phase or the liquid fraction and have to be bled-off.
-~ When trace elements are present in sufficient concen-
tration, their recovery may be economically justified.
The process is effective in treating arseno-
pyritic and pyritic ores which contain carbonaceous
material. Some of this carbonaceous material may be
~;
- 21 -
- , ,
,
'
-
active and thus interfere with precious metal recovery.
The process renders such carbonaceous material inactive
so that the material does not interfere with subsequent
gold recovery.
The practice of the invention is not limited
to the treatment of gold and silver bearing pyritic and
arsenopyritic concentrates and ores. Kunda ~U.S. Patent
~o. 4,331,469) discloses a process for the recovery of
silver values from silver - containing material which
also contains iron and arsenic. Kunda's process uti-
lizes a nitric acid solution to leach the concentrate.
His process suffers from the fact that the solutions can
not be recycled and thus represents a loss of valuable
reagent. The liquor leaving the process has a high
content of ammonium nitrate and ammonium sulphate and
therefore requires the plant to be associated with a
fertilizer plant.
Utilizing our process, the nitric acid will
react with the concentrate to Eorm nitric oxide. In the
oxidation leach this nitric oxide will be regenerated
with oxygen to provide for complete oxidation of the
sulphide and arsenide minerals. In the denitrating step
the nitrates are recycled to the oxidation leach. A
nitrate-free solution is produced for the recovery of
dissolved metals and neutralization
Example 1
A batch test was done to demonstrate the use
of the denitrating step for the removal of oxidized
nitrogen species from the leach solution by contact with
the feed concentrate.
A leach discharge solution was prepared by
reacting a pyritic concentrate with a nitric acid
solution in an autoclave. Oxygen was introduced into
the autoclave as the leach proceeded in order to oxidize
- 22 -
the nitric oxide so that the oxidized nitrogen species
would be regenerated. After completion of the leaching
the solution contained 12,000 mg per litre oxidized
nitrogen species.
500 ml of this leach solution was placed in an
autoclave with 100 g of the pyritic concentrate. The
autoclave was sealed and then heated to 100C. The
denitrating leach was allowed to proceed until the
pressure in the autoclave was at 170 psig due to nitric
oxide production. At this point, the nitric oxide gas
was vented from the autoclave to maintain an operating
pressure of 160 psig. The test was continued for 30
minutes during which time the pressure was decreased to
120 psig as the nitric oxide gas was vented.
Following this denitrating leach, the solution
was analyzed and found to contain 94.6 mg per litre
oxidized nitrogen species. This demonstrates 99.2%
removal of nitrogen species from the leach solution.
This solution was further sparged with air to
remove dissolved nitric oxide. After air sparging the
solution contained 30 mg per litre oxidized nitrogen
species indicating an overall removal of 99.7%.
Example 2
A batch test was conducted to demonstrate the
various steps in the process. The concentrate used for
this sequence contained pyrite and arsenopyrite and had
the following composition:
Fe = 40.8%, As = 11.1%, S = 40.6%
3,300 g of this concentrate was leached in an autoclave
according to the normal practice of our invention with
22 litres of solution containing 42,000 mg per litre of
oxidized nitrogen species as nitric acid and sufficient
oxygen to satisfy the requirements of the sulphide
- 23 -
`: , : :
'L~ 9
oxidation reactions. The solution from this leach had
the following composition:
56.5 g/l Fe, 15.5 g/l As, 160 g/l SO4
6,900 g of this leach solution together with 1,050 g of
water were com~ined with 1,050 g of fresh concentrate.
A denitrating leach was done for 30 minutes at lOO~C
with venting of the nitric oxide gas to maintain an
operating pressure of 150 psig.
The discharge slurry from the denitrating
leach was filtered to produce a partially leached
residue and a solution. The solution had the following
composition:
60.6 g/l Fe, 18.2 g/l As,
163.5 g/l So4, 480 mg/l nitrates
Following purging with nitrogen for 5 minutes
the nitrate content of the solution was reduced to 180
mg/l. This demonstrates 99.5% removal of oxidized
~ nitrogen species in the denitrating leach.
; The residue from this denitrating leach had
the following composition:
Weight = 823 g
F3 = 38.3%, As = 8.6%, S - 43.9%
300 g of this residue was leached with 1,700 g
of solution containing 3 m/l oxidized nitrogen species
as nitric acid. Leaching was done in an autoclave under
normal conditions for the process with oxygen being
supplied to satisfy the oxidation requirements. The
~ residue from this leach was analyzed as follows:
; Weight = 35.3 g
Fe = 6.6%, As = 9.8%
The overall iron extraction from the concen-
trate through the denitrating step plus oxidizing leach
step was 98.5%. The arsenic extraction was 91.9%.
The above results demonstrate the sequence of
steps in the process flowsheet. While the example
.
- 24 -
:
.
~ 3~
utilized a nitric acid solution to leach the residue
from the denitrating step, it has previously been
demonstrated that under continuous or batch conditions,
such leaching can be done utilizing the nitric oxide gas
from the denitrating step together with oxygen.
As will be apparent to those skilled in the
art in the light of the foregoing disclosure, many
alterations and modifications are possible in the
practice of this invention without departing from the
spirit or scope thereof. Accordingly, the scope of the
invention is to be construed in accordance with the
substance defined by the following claims.
- 25 -
,,. . ,;, , .
.
' '