Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~3 ~ ~3~
METHOD AND APPARATUS FOR COLLECTING
SAMPLES FOR ANALYSIS OF CHEMICAL COMPOSITION
BACKGROUND OF THE INVENTION
On-line analysis of the chemical composition o a
solute in a carrier medium (e.g., a solvent) combination has
been accompiished using various known techniques, such as a gel
permeation chromatograph ~GPC) coupled to an ultraviolet (W)
spectrometer. Generally, the success of such a technique
depends on how well the solutes can be differentiated from the
carrier medium. This differentiation has been relatively easy
in most cases, but there are cases in which the carrier medium
seriously interferes with the on-line analysis of the solute,
rendering a particular analysis technique less useful tha~
desired.
For example, in flow-thru high pxessure liquid
chromatographic (HPLC) systems coupled to a Fourier transform
infrared (FTIR) spectrometer, the chrvmatographis solvent often
interferes with the detection of the solute. Th~refore,
various flow-thru cells (e.g., see C.C~ Johnson and L.T.
Taylor, Anal. che~., 56, 2642-2647 (1984)) as well as
interfaces to eliminate the solvent prior to analysis (e.g.,
see C.M. Conroy, P.R. Griffiths, and R. Jinno, Anal. ChemO, 57,
822-825 (1985)) have been designed. Eliminating low-boiling
point 501vent5 such as hexane is easy to accomplish, but not so
with water or higher-boiling organic solvents. With high
boiling point solvents such as trichlorobenzene (TCB), which is
a common solvent in the analysis of polymers by gel permeation
chromatography tGPC), the situation is one of the worst.
Consequently, on-line polymer composition analysis in a TCB
solvent system by GPC/FTIR has so far remained in the realm of
concept only. Similarly, FTIR as a powerful and versatile
analytical tool for HPLC, GPC and process analyzers has been
limited in its application because o~ the solvent or process
interference problems.
~3~3~
Ideally, to eliminate the solvent-solute interference
problems, whether in a GPC experiment or in continuous process
control, one would like to eliminate the solvent altogether.
If a technique can eliminate TCB, then it readily may be used
to eliminate substantially any other chromatographic or process
solvent.
Overcoming the above mentioned problem in high-
temperature (HT) GPC would benefit studies aimed at determining
composition distributions (CD) in polymers ~e.g.,L. Wild, T.R.
Ryle, D.C. Knobeloch, and I.R. Peat, J._ Polymer Science:
Polymer Physics Edition, 20 441-455 (1982~). CD is the change
in comonomer composition of polymer chains as a function of
their molecular weight (MW). Most CD studies of crystalline or
amorphous polymers have depended on large-scale fractionation
or cross-fractionation, often followed by a slow solvent-
stripping step to prepare the fractions for subse~uent
analytical measurements. Only in favorable cases, such as in
semicrystalline polymers, where a relationship may be
established between comonomer content and melting temperature
has the need for an on-line composition detector not been
essential. In contrast, amorphous polymers cannot benefit from
such empirical relationships. Consequ~ntly, most separations
have depended on solvent/non-solvent fractionations (e.g., H.
Sato et al, Macromolecules, 19, 2613 (1986)). These operations
are very tedious and time-intensive, and although the
individual steps can be automated to reduce manpower
requirements (D.L. Newhouse, R.G. Wheeler, and R.H. Waltz, U.S.
Patent 4,604,363 (1586)), the time-intensive nature of the
analysis still remains a big hurdle to be overcome. Recent
advances in HT-GPC- W have been used for composition analysis
(e.g., S~ Mori and T. Suzuki, J. of Liquid Chrom., 4(10), 1685
(1981)), but the limitations impo~ed on the choice o~ solvent
and the rPquirement that the polymer or solute must have a W -
active group restrict the applicability of this technique.
Although FTIR is a less sensitive tool than W, it is by far
the more powerful structural tool because of its superior
selectivity in terms of chemical species differentiation.
3 ~ 3 ~
Another area where FTIR would be of tremendous
importance is in the area o~ on-line analysis of liquid proc~ss
streams, regardless if the strsam is heterogeneous in nature or
if the solutes are UV~inactive. The essential question is how
to eliminate the solvent on-line for subsequent, automated on-
line or off-line analytical measurements such as by FTIR.
SUMMARY OF THE INVENTION
In accordance with the present invention, methods and
apparatus are provided which are particularly suitabl-e for
determining the compositional distribution (CD~ of unknown
materials such as polymers using a fractionation unit which
employs a high boiling point solvent (such as TCB). To that
end, an interface system is provided between such
fractionation unit (typically a GPC unit) and an analyzer unit
such as a Fourier transform infrared (FTIR) analyzer. The
interface system comprises a vacuum oven, the temperature and
pressure of which is adjusted depending on the boiling
point/vapor pressure characteristics of the solvent or solvent
mixture that is to be eliminated. The oven may be configured
in various ways. One is to equip it with a carousel-type
programmable fraction collector having a discrete number of
plates or hollowed dishes formed of potassium bromide (KBx~.
Another is to us~ a continuous collector which may be used, for
instance, as an on-line plant stream (liquid phase) analyzer.
In that case, the device would be separated into two
differentially pumped chambers; one for collection of solvent-
free samples and the other for on-line analysis (e.g. by FTIR
or near-infrared fiber optic systems). Either way, the
effluent from a fractionator unit is supplied as droplets and,
as each droplet falls on the fraction collector, the solvent is
flashed off immediately, leaving behind a residue. The process
may be repeated as many times as desired before moving to the
next collector position. Either during or after the collection
step, the compositions of the fractions are determined
automatically using a chosen microanalytical technique.
13~9~3~
For safety reasons, it is preferable that the
interface system have no exposed heating elements. Pressure
and temperature are regulated so that when a solution droplet
comes in contact with the collecting medium on a fraction
collector (e.g., infrared window-dish), the solvent flash
evaporates, leaving behind a rPsidue. In the case of hi~h-
boiling point solvents such as trichlorobenzene, an additional
stream of preheated nitrogen or other inert gas blowing over
the collection site hPlps smooth the flash evaporation and
eliminates potential splashing problems during the collection
step.
Splashing could occur, for example, if the oven
temperature is too high or too low for a given pressure. The
droplet size may also play a role, depending on the physical
properties of the droplet such as surface tension. Splashing
may be minimized using different tip geometries to control
droplet size, adjusting the flow and/or temperature of the
inert gas blown onto the collection site, and/or adjusting the
gap between the effluent tip and the collecting plate. Under
such conditions, the gap could be so small that the effluent
flows onto the collecting plate without forming droplets but
the solvent is, nevertheless, flash-evaporated.
A preferable approach to suppress splashing and othsr
problems, however, is to atomize the effluent such as by using
an ultrasonic atomizer, pre~erably of the non-pneumatic and
non-electrostatic type (for safety). If such a nozzle is used,
then the inert gas flow pattern in the vacuum oven preferably
is designed so that the atomi~ed particles are channeled only
toward the collecting plate.
In any case, so long as the droplets are confined to
falling on the collecting plate, smaller droplets are preferred
to enhance evaporation.
The improved system of this invention can be used
with either homogeneous or heterogeneous solutions to study
compositional variations in a complex chemical system as it
emerges from the interfaced unit.
~ 3 ~
Although the exampl~s which will be described
hereinafter are based on fractionations using size exclusion
chromatography, the interface unit can be adapted, with proper
modifications and valvings, to other fractionations based on
crystallinity differences (temperature elution fractionation),
field flow fractionations (FFF), adsorption chromatography, or
other arrangements which may involve homogeneous or
heterogeneous polymer solutions or other mixtures.
DESCRIPTION-OF THE DRAWINGS
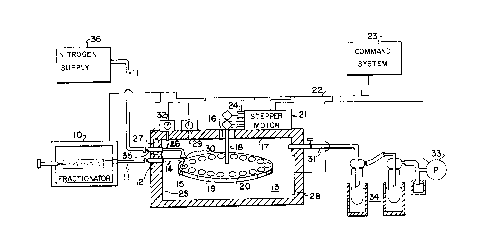
Figure 1 is a schematic diagram, not drawn to scale,
including a fractionator and an interface system constructed in
accordance with the invention; and
Figure 2 is a Fourier Transform Infrared (FTIR)
spectrum of an ethylene-propylene rubber residue collected
utilizing an apparatus of the type shown in Figure 1;
Figure 3 illustrates a CD ~rofile ~upper trace) and
gel permeation chromatogram (lower trace) for one type of
ethylene-propylene rubber (EPR) which was analy~ed employlng
this invention;
Figure 4 illustrates a CD profile (upper trace) and
gel permeation chromatogram (lower trace) for a second EPR
sample collected by the method of this invention;
Figure S illustrates in a cross-sectional view, an
oven and associated apparatus constructed in accordance with
this invention.
DETAILED DESCRIPTION OF.THE INVENTTON
A schemati~ diagram, not drawn to scale, including a
fractionator, and an interface system according to the present
invention, i5 shown in Figure 1. The interface is relatively
enlarged to show its details.
Referring to Figure 1, a fractionator lO such as a
gel permeation chromatograph of a conventional and commercially
available type such as a Waters Type 150-C GPC, is shown.
Fractions of a sample of a material to be analyzed are provided
in the form of a solute carried by a liquid solvent such as
trichlorobenzene (TCB) via an outlet line 11~ The outlet line
11 is connected to a sample inlet port 12 of a vacuum oven 13.
~3~3~
An effluent sample supplying means 14, for example,
stainless steel tubing of 0.009 inches inner
diameter, extends from sample inlet port 12 to a
point within oven 13 adjacent a sample collection
s station 15. Suitable valving (not shown) may be
provided between outlet line 11 and sample inlet
port 12 as needed to insure that liquid samples are
provided at an appropriate rate at the collection
station 15. It is preferable that small droplets be
10 supplied to enhance the desired step of flash
evaporation. Typically, oven 13 may have a capacity
of 0.2 cubic feet and preferably is capable of being
heated to a controlled temperature of the order of
200C. Vacuum ovens which are available
commercially (such as NAPCO ~ Model 5831) may be
modified so as to be suited for such use, provided
there are no heating elements exposed on the inside
of oven 13 or the oven may be constructed
specifically for the present purposes. A hole
20 having a vacuum seal 16 is provided in the center of
the ceiling 17 of the oven 13 through which a shaft
18 is inserted. The shaft 18 is attached to a
fraction collector 19 inside the oven 13. The
fraction collector 19 is a disk of, for example,
25 aluminum of 14.5 centimeters diameter. As will
appear below, such a configuration is particularly
suitable where subsequent FTIR analysis is to be
performed. Holes are provided along the circum-
ference of the disk to hold, for example, sixteen
30 KBr (potassium bromide) plates, cups or vials 20.
The shaft 18 is attached to a stepper motor unit 21
which is mounted externally on top of the oven 13.
A power and logic signal cable 22 is connected from
the stepper motor 21 to a command or control system
35 23 including, for example, a Compaq ~ Computer.
The position of the fraction collector 19 is sensed
- 6a - ~ ~ 9 ~3~
using a position sensor 24 (such as a fixed
photocell and a slotted wheel connected to shaft
18). The position of the fraction collector 19 is
controlled making use of the sensor 24, stepper
s motor 21, and the command system 23 (software
commands) in a conventional closed loop position
control arrangement.
Another hole is provided in the side wall
25 of the oven 13 to introduce a nitrogen line 26
10 which is connected to
- 6a -
an external nitrogen supply 36. The nitrogen line 26 is
associated with a heater 27 so that nitrogen provided via line
26 can maintain the desired temperature at the surface of th~
KBr cups 20, a~ required. Line 26 may be coaxial with tubing
14, (surrounding tubing 14) or may be separate from tubing 14.
In the latter case, line 26 preferably is also made flexible
or movable to adjust the position of a blowing point 30.
A heater 28 is provided outside of oven 13 and a
temperature sensing apparatus 29 (such as a thermocouple) is
provided within oven 13 in the vicinity of collection station
15. The héater 28, temperature sensor 29 and command system 23
are coupled in a closed loop temperature control system. While
the heaters 27 and 28 are shown as separate devices, in
practice they may be parts of a single heater. A single heater
arrangement may be advantageous in that the temperature of oven
13 and that of the blown nitrogen then may readily be
maintained substantially equal. For safety reasons, it is
preferable that the temperature of the hot nitrogen should not
exceed that of oven 13.
Oven 13 is provided with a vacuum line 31 which
extends inside of oven 13, a pressure (vacuum~ sensing
apparatus 32 connected to oven 13, and a vacuum pump 33
arranged to maintain the pressure within oven 13 at a
predetermined level.
A dual cold trap 34 is associated with vacuum line 31
for collection of solvent evaporated within oven 13 as will be
explained below~
The pressure sensing apparatus 32 is coupled to
command system 23 to assist in maintaining the pressure
(vacuum) within oven 13 in a predetermined range. The control
arrangement preferably is configured so that if pressure and/or
temperature exceed predetermined safety limits, warning signals
are generated, power to the oven 13 is shut off and the
chromatographic effluent is diverted away from the oYen 13.
The geometry of the XBr collecting crystal 20 was
found to be an important parameter. Flat KBr plates were found
to be a less suitable collection media when the effluent is
~3~5~
introduced as droplets. In that case, droplets of solution
tend to flow to the edges of the plate 20 before the solvent
evaporates. Consequently the collected sample (e.g.l a
polymer) deposits along the edges of plate 20 and it is
difficult to get a good spectrum from such a sample.
Therefore, the use of KBr cups 20 is o~ten preferred. The
latter act as a small ~vial~ and position the solute at the
center of the cup 20.
If, on the other hand, flat KBr plates are to be
used, then the GPC effluent should be applied to the plate 20
as a fine mist, using such devices as a non-electrostatic,
ultrasonic atomizing nozzle (e.g. a Sono-Tek ultrasonic
nozzle). The fine particle sizes of the atomized spray help
the flash-evaporation, depositing a thin layer of fractionated
materia} on the collection plate 20.
Depending on the particular circumstances, besides
KBr cups, other collection media are possible. One possibility
is the use of metal or glass cups containing suitable powders
for diffuse reflectance measurements using normal accessories
or microsampling devices. Another is to use a metal strip
(with low infrared absorption characteristics) on which the
material is deposited for subsequent surface analysis (e.g.,
using surface analysis techniques or microsampling
accessories). A third possibility is to use tiny metal dishes
for collecting fractions to be analyzed using a mass
spectrometric technique, for example.
The apparatus according to this invention can be
~iniaturized, converted to a continuous on-line process
analyzer using the dual chamber configuration described above,
or used as an interface device for other types of analytical
techniques. In particular, a grid type design, typical of some
fxaction collection devices, is an alternative. If the
interface is to be used for on-line process analysis, then the
details of the oven and that of the collecting medium are
likely to be changed.
During a run, the varuum oven 13 is maintained at a
high enough temperature and vacuum to allow the solvents to
~1 3 ~
flash evaporate as they come in contact with the KBr dish 20.
The effluent to be sampled (e.g. GPC effluent) is directed into
the oven 13 through the stainless steel tubing 14 inserted
through the vacuum-tight opening on the sidewall of the oven
13. Although this tubing 14 may be externally valved for
proper flow-rate control, it was not needed for the experiments
described herein.
The effluent enters the oven 13, and falls on the KBr
cups 20 drop-by-drop at a flow rate of, for example, 0.5
milliliters per minute. In order to assure that each droplet
immediately loses its solvent, the temperature and pressure of
the oven 13 should be adjusted, depending on the boiling
point/vapor pressure characteristics of the solvent to be
eliminated. For example, if TCB is the solvent, the
temperature-pressure relationship given below should be
consulted.
Table: Vapor pressure of TCB
Vapor
pressure 760 400 100 40 10 5
(mn~Ig)
Temp-
erature 213.0 187.7 140.0 114.8 87.7 67.3 38.4
C)
This relationship establishes a minimum temperature
needed for evaporation o a single drop, but actual
temperatures in oven 13 should be maintained above the
theoretical value by, for example, at least 4QC to insure
efficient evaporation of many drops falling on the same cup 20.
This additional temperature increment is required since the
heat of vaporization will drop the temperature of the cup 20
below the boiling point of the solvent (under the given
conditions). To insure the cup 20 is hot enough throughout the
collection stage, the temperature of oven 13 should therefore
be maintained higher than the theoretical boiling point of the
solvent, and/or one must provide an additional source of heat
directly to the cup surface.
With TCB as the chromatographic solvent, a stream of
hot nitrogen blowing over the collecting cup 20 was found to be
~ 3 ~
beneficial, and at the same time effectively eliminated
splashing problems. The heated nitrogen was introduced via the
flexible metal tubing 26. The temperature of the nitrogen is
controlled by means of a thermocouple (not shown) placed after
the heating cartridge 27. The tip of the nitrogen line 26 may
be equipped with a baf~le to control the size of the heated
area. Alternatively, a perforated toroidal (ring-shaped)
nozzle may be employed to provide a unifo~m cylindrical jet of
inert gas at the sample collection station 15.
As droplets fall on a cup 20 at collection station
15, and the solvent immediately flash~s off, the cup 20 will
contain the fractionated residue. The flashed-off solvent is
continuously carried away from the vacuum oven via vacuum line
31 into cold trap 34 where the solvent is condensed. Cold trap
24 is emptied on a regular basis. A~ter a fraction is
collected, the carousel 19 moves to the next position, and so
on. It was found that sample quantities as small as one
microgram were sufficient for subsequent FTIR analysi~. The
residence time at each position is either preset or triggered,
depending on the experimental setup and the distribution
pattern of the chromatographic information ~ie. continuous or
discrete signals)~ If an ult~asonic atomizer is used,
especially of the non-electrostatic type, then the nozzle-to-
plate distance is adjusted, depending on the desired pattern of
the sprayed material. The nozzle geometry and the ultrasonic
frequency can also be adjusted to control the spray pattern or
the spot size. To assure that the atomized mist emerging from
the nozzle is channeled entirely toward the collecting plate
20, the vacuum port 31 preferably is positioned beneath the
carousel 19, and pr~ferably ben~ath plate 20 at collection
station 15. If the latter option i5 chosen, then additional
openings on the carousel 19, in the vicinity of plate 20, will
assure a uniform flow of heated nitro~en around the plate 20.
In addition, a cylindrical jet of hot nitrogen in the direction
of the plake 20, and of a chosen diameter, will further channel
or direct the atomized particles emerging from the nozzla
toward plate 20 only.
~ 3 ~
Af~er the collection is ovPr, the temperature of the
oven 13 is brought down and the vacuum broken. The carousel 19
i5 now ready for composition analysis using selected
microanalytical techniques. Since FTIR was contemplated in
this case, the carousel 19 is detached from shaft 18, removed
from the oven 13 and placed into the auto sampler assembly of
the FTIR spectrometer (into which it is designed to fit) to
automatically record the spectra of the collected samples.
An interface system of the type shown in Figure 1 was
connected to a GPC (Waters, Inc., Type 150-C~ and the effect of
the vacuum on the flow rate and the system pressure was
examined. It was found that the flow rate in the GPC is not at
all affected by the pressure drop in the vacuum oven 13 at 1 mm
Hg as compared to 760 mm Hg.
Experimental details.
In the experiment, first, a 0.6 weight percent
solution of EPR was prepared in TCB and 100 microliters of this
solution was injected into the GPC coupled to the interface
system of the type shown in Figur~ 1. The column temperature
was maintained at 120C, flow rate at 0.5 ml/min. The pressure
of the oven 13 was maintained at 40 mm Hg, while its
temperature was about 150~C, with a gentle flow of N2 through
the system. The operation of ~he interace system was
observed. The effluent was continuously flash evaporated in
the interface oven 13 and a thin film of the EPR residue was
formed on a gBr cup 20 (13x2 mm). The FTIR spectrum of the
sample was recorded and is shown in Figure 2. Similar
experiment~ were performed successfully with ethylene-vinyl
acetats ~EVA) copolymers.
In the FTIR spectrum shown in Figure 2, not only a
C-H stretching region at about 2800-3000 cm~1 but also a
bending region at about 1400 cm~l can clearly be observed.
Based on experiments similar to those described above, the
detectability limit of the method was investigated. It was
found that one microgram of sample is sufficient to permit
quanitatlve analysis. With typical concentrations of the low
11
~ 3 ~
and high ends of the GPC effluent of the order of 10-6 grams
per milliliter, if ten fractions are collected for each sample,
the collection period will be about two minutes per fraction
and the deposition amounts of sample at the extremes will be
about one microgram (minimum). A residue of, for example 6.6
micrograms would display an excellent signal-to-noise ratio.
In two separate experiments, seven fractions were
collected from each of two rubbers. The ov~n temperature was
maintained ~t 130'C, oven pressure was in the vicinity of 5-10
mm Hg, and the nitrogen flow rate was about o.3 cc/minute.
The mov~ments of carousel 19 were timed based on an
analysis of the elution time profile from a previous
chromatogram of the same material utilizing a conventional
differential refractometer (DRI). Durin~ the actual fraction
collection, the DRI detector was bypas~ed.
To adjust the temperature and the pressure of the
oven 13 to assure smooth flash-evaporation and eliminate
splashing problems, an external source of heated nitrogen 36
was introduced into the system and directed at the sample-
collection site 15. Although this additional flow calls for a
slight sacrifice in the vacuum of the oven 13, the loss is
small, since it could bring the vacuum from 1 mm to 5 mm Hg or
hi~her. From standard temperature-pressure tables, it appears
that 1-5 mm Hg vacuum at 50-70~C would be a minimum vacuum to
accomplish the task. Actual oven temperatures should be
maintained 30-50C higher than the theoretical evaporation
point to account for any heat of vaporization at the collector
position 15. The flashed-off solvents were collected in the
dual cold trap 34.
The amount of polymer deposited in each dish 20 will
depend on the particular fractionation and polymer system
studied. For instance, if ethylene-propylene co-polym0rs are
fractionated on a GPC column at a temperature of 130-150C,
using trichlorobe~zene as a solvent, the average weight of the
deposit will be in the 1-50 microgram range. Under these
conditions, good quantitative FTIR spectra are obtained.
13 ~ 5~
The two EPR samples were fractionated using the
apparatus of Figure 1. The spectra Qf the collected fractions
were obtained by placing the carousel 19 into the autosampler
of an FTIR spectrometer. From the spectra, the percent
ethylene of each fraction was calculated. The retention time
vs. percent ethylene profile is shown in the upper trace of
Figure 3 for the first sample. The vertical lines on the GPC
curve show the timing of the fraction collection. The ease of
obtaining such CD information is unprecedented. The higher
molecular weight samples are retrieved earlier (lower trace-
left hand end) while the higher molecular weight samples are
retrieved later.
The Ethylene CD for the second sample is shown in the
upper trace in Figure 4. In this case the percent ethylene
decreases with decreasing k~. The presence of a low-ethylene
content, low-MW fraction is evident from Figure 4 (right hand
end). Although these profiles were obtained from a single GPC
fractionation, the extension to more complex cxoss-
fractionations or solvent/non-solvent fractionations will
readily be apparent to those engaged in this art.
A more accurate representation of an oven 13 and
associated components as explained above is shown in cross
section in Figure 5 wherein the same r~Prence characters are
used as in Figure 1 for corresponding parts.
The concept of flash evaporating such high
boiling solvents as TCB has been successfully demonstrated by
means of the illustrated apparatus. It could be used for
further investigation of structure-property relationships.
While the apparatu~ described above may be
implemented utilizing various arrangements of commercially
available hardware elements or may be custom designed according
to a particular application, one set of readily usable
controller components which was obtained from Cybernetic
Microsystems of San Gregoria, California comprises their ModPl
CY525 Motor Controller, CY750 I/0 Controller, CY232 Serial
Controller, CY300 LCD/Keyboard Controller, CY250 System
Controller and CYB-002 Csntrol Board.
13
14
In general, the sensing and control elements
themselves may be constructed in a conventional manner (See,
for example, the description of similar components in U.S.
Patent No. 4,604,363 referred to abovP).
The novel aspects of the foregoing method and
apparat~s are set forth in the following claims.
1~