Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
-
LACTAM DERIVATIVES 1 3 3 9 n 2 2
This invention relates to lactam derivatives, to processes for
their preparation, to pharmaceutical compositions containing them and
to their medical use.
In particular the invention relates to compounds which are potent
and selective antagonists of 5-hydroxytryptamine (5-HT) at 5-HT
receptors of the type located on terminals of primary afferent nerves.
Receptors of this type are now designated as 5-HT 3 receptors and are
also present in the central nervous system. 5-HT occurs widely in the
neuronal pathways in the central nervous system and disturbance of
these 5-HT containing pathways is known to alter behavioural syndromes
such as mood, psychomotor activity, appetite and memory.
Compounds having antagonist activity at 5-HT 3 receptors have been
described previously.
Thus for example published UK Patent Specification No. 2153821A
lS and published European Patent Specifications Nos. 191562, 219193 and
210840 disclose 3-imidazolylmethyltetrahydrocarbazolones which may be
represented by the general formula:
O R4 R3
Il I /
Q . . ... _.
~\ /\/\l- I
t ~o N r
-
\\ / \N/ \ / R2
Rl
25 wherein Rl represents a hydrogen atom or a group selected fromCl_lOalkyl, C3-6 alkenyI, C3_10 alkynyI, C3_7 cycloalkyl,
C3_7cycloalkylCl_4alkyl, phenyl or phenylCl 3alkyl, and in the case
where Q represents a hydrogen atom, R 1 may also represent -CO 2R 5,
-CoR5, -CoNR5R6 or -502R5 (wherein R5 and R6, which may be the same or
30 different, each represents a hydrogen atom, a Cl_6 alkyl or
C3_7cycloalkyl group, or a phenyl or phenylCl_4alkyl group, in which
the phenyl group is optionally substituted by one or more Cl_4 alkyl,
Cl_4alkoxy or hydroxy groups or halogen atoms, with the proviso that
- 2 - l 339n22
R5 does not represent a hydrogen atom when Rl represents a group
-Co2R5 or -502R5);
one of the groups represented by R2, R3 and R4 is a hydrogen atom or a
Cl_6 alkyl, C3_7 cycloalkyl, C2_6 alkenyl, or phenylCl_3alkyl group,
and each of the other two groups, which may be the same or different,
represents a hydrogen atom or a Cl_6 alkyl group;
Q represents a hydrogen atom or a halogen atom or a hydroxy,
Cl_4alkoxy, phenylCl_3alkoxy or Cl_6 alkyl group or a group -NR7R8 or
-CoNR7R8 (wherein R7 and R8, which may be the same or different, each
represents a hydrogen atom or a C1-4 alkyl or C3_4 alkenyl group, or
together with the nitrogen atom to which they are attached form a
saturated 5 to 7 membered ring);
and physiologically acceptable salts and solvates thereof.
We have now found a novel group of compounds which differ in
structure from those described previously, and which are potent
antagonists of the effect of 5-HT at 5-HT3 receptors.
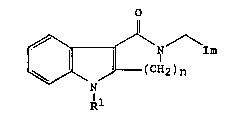
The present invention provides a tricyclic lactam of the general
formula (I):
i
--
// \ / \ / \Im
_- N
1~
R 1
wherein Im represents an imidazolyl group of the formula :
~ ~ 3 ~ 1 339~2~
/R4 /R4
N~ /NR3 or R31\ !yN
R2 R2
and R1 represents a hydrogen atom or a group selected from
Cl_6alkyl, C3_6alkenyI, C3_10alkynyI, C3_7cycloalkyl,
C3_7cycloalkylC1_4 alkyl, phenyl, phenylC1_3alkyl,
phenylmethoxymethyI, phenoxyethyI, phenoxymethyl, -C02R5, -CoR5,
-CûNR5R6 or -502R5 (wherein R5 and R6, which may be the same or
different, each represents a hydrogen atom, a C1_6 alkyl or C3_7
cycloalkyl group, or a phenyl or phenylC1_4alkyl group, in which the
phenyl group is optionally substituted by one or more C1_4 alkyl, C1_4
lS alkoxy or hydroxy groups or halogen atoms, with the proviso that R5
does not represent a hydrogen atom when Rl represents a group -Co2R5
or -502R5);
one of the groups represented by R2, R3 and R4 is a hydrogen atom
or a C1_6alkyl, C3_7cycloalkyl, C3_6alkenyl, phenyl or phenylC1_3alkyl
group, and each of the other two groups, which may be the same or
different, represents a hydrogen atom or a C1_6 alkyl group;
n represents 2 or 3;
and physiologically acceptable salts and solvates thereof.
According to one aspect, the invention provides compounds of
formula (I) wherein R1 represents a hydrogen atom or a group selected
from C1_6 alkyl, C3-6 alkenyl, C3_10 alkynyl, C3_7cycloalkyl,
C3_7cycloalkylC1_4 alkyI, phenyl or phenylC1-3 alkyl (n and Im being
as defined in formula (I)).
Suitable physiologically acceptable salts of the compounds of
general formula (I) include acid addition salts formed with organic or
inorganic acids for example, hydrochlorides, hydrobromides, sulphates,
alkyl- or arylsulphonates (e.g. methanesulphonates or
p-toluenesulphonates), phosphates, acetates, citrates, succinates,
tartrates, fumarates and maleates. The solvates may, for example, be
hydrates.
1 339n22
-- 4
All optical isomers of compounds of general formula (I) and their
mixtures including the racemic mixtures thereof, and all the geometric
isomers of compounds of formula (I), are embraced by the invention.
Referring to the general formula (I), an alkyl group may be a
straight chain or branched chain alkyl group, for example, methyl,
ethyl, n-propyl, prop-2-yI, n-butyl, but-2-yl, 2-methylprop-2-yl,
n-pentyI, pent-3-yl or n-hexyl. A C3-6 alkenyl group may be, for
example, a propenyl or butenyl group. When Rl represents a
C3_6alkenyl or C3_l0alkynyl group, or R3 represents a C3_6alkenyl
group, or R7 or R8 represents a C3_4alkenyl group, the double or
triple bond may not be adjacent to the nitrogen atom. A
phenylCl_3alkyl group may be, for example, a benzyl, phenethyl or
3-phenylpropyl group. A C3_7cycloalkyl group may be, for example, a
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl
group-
A preferred class of compounds of formula (I) is that wherein R
represents a hydrogen atom or a Cl_6 alkyl (e.g. methyl, ethyl,
n-propyl, prop-2-yl), C3_4 alkenyl (e.g. prop-2-enyl~, C3_4 alkynyl
(e.g. prop-2-ynyl), C5_6cycloalkyl (e.g. cyclopentyl),
C5_6cycloalkylmethyl (e.g. cyclopentylmethyl~, phenylC1-2 alkyl (e.g.
benzyl), phenylmethoxymethyl, N~N-diCl-3alkylcarboxamido (e.g. N,N-
dimethylcarboxamido) or Cl_3alkylsulphonyl (e.g. methylsulphonyl)
group. More preferably Rl represents a Cl_4 alkyl (e.g. methyl or
n-propyl), C3_4alkynyl (e.g. prop-2-ynyl~, C5_6cycloalkyl (e.g.
cyclopentyl), C5_6cycloalkylmethyl (e.g. cyclopentylmethyl),
phenylCl_2 alkyl (e.g. benzyl~, phenylmethoxymethyI, or N,N-
diCl_3alkylcarboxamido (e.g. N,N-dimethylcarboxamido) group.
Another preferred class of compounds of formula (I) is that
wherein R2 represents a hydrogen atom or a Cl-3 alkyl (e.g. methyl)
group, more preferably a hydrogen atom.
Another preferred class of compounds of formula (I) is that
wherein R3 represents a hydrogen atom or a Cl_3 alkyl (e.g. methyl)
group, more preferably a hydrogen atom.
A further preferred class of compounds of formula (I) is that
wherein R4 represents a hydrogen atom or a Cl-3 alkyl (e.g. methyl or
n-propyl) group. Most preferably R4 represents a methyl group.
1 339022
-- 5
When R2 and R3 represent hydrogen atoms, R4 is preferably
Cl_6alkyl, C3_7 cycloalkyl, C3-6 alkenyl or phenylCl-3alkyl, more
particularly Cl_6 alkyl.
A further preferred class of compounds of formula (I) is that
wherein n represents 2.
A preferred group of compounds of formula (I) is that wherein R
represents a hydrogen atom or a Cl_4 alkyl, C3_4alkenyl, C3_4alkynyl,
C5_6cycloalkyl, C5_6cycloalkylmethyI, phenylCl_2 alkyl,
phenylmethoxymethyl, N,N-diCl_3alkylcarboxamido or Cl_3slkylsulphonyl
group; R2 represents a hydrogen atom; and R3 and R4 each represent a
hydrogen atom or a Cl_3 alkyl group.
A particularly preferred group of compounds of formula (I) is
that wherein Rl represents a methyl, n-propyl, prop-2-ynyl,
cyclopentyI, cyclopentylmethyl, benzyl or N, N-dimethylcarboxamido
group; R2 and R3 each represent a hydrogen atom; and R4 represents 8
methyl group.
Within the above preferred and particularly preferred groups of
compounds, an especially important group of compounds is that in which
n represents 2.
Preferred compounds according to the invention are:
2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one;
2,3,4,5-tetrahydro-5-(phenylmethyl)-2-[(5-methyl-lH-imidazol-4-yl)-
methyl]-lH-pyrido[4,3-b]indol-1-one;
5-cyclopentyl-2,3,4,5-tetrahydro-2-~(5-methyl-lH-imidazol-4-yl)-
methyl]-lH-pyrido[4,3-b]indol-1-one;
2,3,4,5-tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-propyl-lH-
pyrido[4,3-b]indol-1-one;
5-(cyclopentylmethyl)-2,3,4,5-tetrahydro-2-[(5-methyl-lH-imidazol-4-
yl)methyl]-lH-pyrido[4,3-b]indol-1-one;
3,4,5,6-tetrahydro-6-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
azepino[4,3-b]indol-1(2H)-one;
2,3,4,5-tetrahydro-N,N-dimethyl-2-[(5-methyl-lH-imidazol-4-yl)-
methyl]-l-oxo-5H-pyrido[4,3-b]indole-5-carboxamide;
2,3,4,5-tetrahydro-2-[(5-methyl-lH-imid~zol-4-yl3methyl]-5-(2-
propynyl)-lH-pyrido[4,3-b]indol-1-one;
1 339022
- 6
and their physiologically acceptable salts and solvates.
The potent and selective antagonism of 5-HT at 5-HT3 receptors by
compounds of the invention has been demonstrated by their ability to
inhibit 3-(5-methyl-lH-imidazol-4-yl)-1-[1-(methyl-t3)-lH-indol-
3-yl]-1-propanone binding in rat entorhinal cortex homogenates
(following the general procedure described by G. Kilpatrick et al. in
Nature, 1987, 330, 746), and/or by their ability to inhibit the
5-HT-induced depolarisation of the rat isolated vagus nerve
preparation.
In addition to their activity as potent and selective antagonists
of 5-HT at 5-HT3 receptors, certain compounds according to the
invention have the advantage of an extended duration of action.
A particularly preferred compound on account of both its potency
and duration of action is 2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-
lH-imidazol-4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one and its
physiologically acceptable salts and solvates. Preferred salts of
this compound are the hydrochloride and maleate.
Compounds of formula (I), which antagonise the effect of 5-HT at
5-HT3 receptors, are useful in the treatment of conditions such as
psychotic disorders (e.g. schizophrenia and mania); anxiety; and
nausea and vomiting, particularly that associated with cancer
chemotherapy and radiotherapy. Compounds of formula (I) are also
useful in the treatment of gastric stasis; symptoms of
gastrointestinal dysfunction such as occur with dyspepsia, peptic
ulcer, reflux oesophagitis, flatulence and irritable bowel syndrome;
migraine; and pain. Compounds of formula (I) may also be used in the
treatment of dependency on drugs and substances of abuse, depression,
and dementia and other cognitive disorders.
According to another aspect, the invention provides a method of
treatment of a human or animal subject suffering from a psychotic
disorder such as schizophrenia or mania; or from anxiety; nausea or
vomiting; gastric stasis; symptoms of gastrointestinal dysfunction
such as dyspepsia, reflux oesophagitis, peptic ulcer, flatulence and
irritable bowel syndrome; migraine; or pain, which comprises
administering an effective amount of a
` ~ _ 7 _ 1 339n22
compound of formula (I) or a physiologically acceptable salt or
solvate thereof.
Accordingly, the invention also provides a pharmaceutical
composition which comprises at least one compound selected from
compounds of the general formuls (I), and their physiologically
acceptable salts and solvates (e.g. hydrates), for use in human or
veterinary medicine, and formulated for administration by any
convenient route.
Such compositions may be formulated in conventional manner using
one or more physiologically acceptable carriers and/or excipients.
Thus the compounds according to the invention may be formulated
for oral, buccal, parenteral or rectal administration or in a form
suitable for administration by inhalation or insufflation (either
through the mouth or nose).
For oral administration, the pharmaceutical compositions may take
the form of, for example, tablets or capsules prepared by conventional
means with pharmaceutically acceptable excipients such as binding
agents (e.g. pregelatinised maize starch, polyvinylpyrrolidone or
hydroxylpropyl methylcellulose); fillers (e.g. lactose,
microcrystalline cellulose or calcium hydrogen phosphate); lubricants
(e.g. magnesium stearate, talc or silica); disintegrants (e.g. potato
starch or sodium starch glycollate); or wetting agents (e.g. sodium
lauryl sulphate).- The tablets may be coated by methods well known in
the art. Liquid preparations for oral administration may take the
form of, for example, solutions, syrups or suspensions, or they may be
presented as a dry product for constitution with water or other
suitable vehicle before use. Such liquid preparations may be prepared
by conventional means with pharmaceutically acceptable additives such
as suspending agents (e.g. sorbitol syrup, cellulose derivatives or
hydrogenated edible fats); emulsifying agents (e.g. lecithin or
acacia); non-aqueous vehicles (e.g. almond oiI, oily esters, ethyl
alcohol or fractionated vegetable oils); and preservatives (e.g.
methyl or propyl-p-hydroxybenzoates or sorbic acid). The preparations
may also contain buffer salts, flavouring, colouring and sweetening
agents as appropriate.
1 339022
- 8
Preparations for oral administration may be suitably formulated
to give controlled release of the active compound.
For buccal administration the compositions may take the form of
tablets or lozenges formulated in conventional manner.
The compounds of the invention may be formulated for parenteral
administration by bolus injection or continuous infusion.
Formulations for injection may be presented in unit dosage form e.g.
in ampoules or in multi-dose containers, with an added preservative.
The compositions may take such forms as suspensions, solutions or
emulsions in oily or aqueous vehicles, and may contain formulatory
agents such as suspending, stabilising and/or dispersing agents.
Alternatively, the active ingredient may be in powder form for
constitution with a suitable vehicle, e.g. sterile pyrogen-free water,
before use.
The compounds of the invention may also be formulated in rectal
compositions such as suppositories or retention enemas, e.g.
containing conventional suppository bases such as cocoa butter or
other glycerides.
In addition to the formulations described previously, the
compounds of the invention may also be formulated as depot
preparations. Such long acting formulations may be administered by
implantation (for example subcutaneously or intramuscularly) or by
intramuscular injection. Thus, for example, the compounds of the
invention may be formulated with suitable polymeric or hydrophobic
materials (for example as an emulsion in an acceptable oil) or ion
exchange resins, or as sparingly soluble derivatives, for example, as
a sparingly soluble salt.
For administration by inhalation the compounds according to the
invention are conveniently delivered in the form of an aerosol spray
presentation from pressurised packs or a nebuliser, with the use of a
suitable propellant, e.g. dichlorodifluoromethane,
trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or
other suitable gas. In the case of a pressurised aerosol the dosage
unit may be determined by providing a valve to deliver a metered
amount. Capsules and cartridges of e.g. gelatin for use in an inhaler
or insufflator may be formulated containing a powder mix of a compound
` _ _ 9 _ 1 339n22
of the invention and a suitable powder base such as lactose or
starch.
For intranasal administration, the compounds according to the
invention may be formulated as solutions for administration via a
suitable metered or unit dose device or alternatively as a powder mix
with a suitable carrier for administration using a suitable delivery
device.
The compounds of formula (I) may also be administered in
combination with other therapeutic agents. Thus, for example, in the
treatment of gastric stasis, symptoms of gastrointestinal dysfunction
and nausea and vomiting, the compounds of formula (I) may be
administered in combination with antisecretory agents such as
histamine H2-receptor antagonists (e.g. ranitidine, sufotidine,
l-methyl-5-[[3-[3-(1-piperidinylmethyl)phenoxy]propyl]amino]-lH-1,2,4-
triazole-3-methanol, cimetidine, famotidine, nizatidine or
roxatidine) or H+K+ATPase inhibitors (e.g. omeprazole).
A proposed dose of the compounds of the invention for
administration to man (of approximately 70kg body weight) is 0.001 to
lOOmg, preferably 0.01 to 50mg, more preferably 0.1 to 20mg of the
active ingredient per unit dose expressed as the weight of free base,
which could be administered, for example, 1 to 4 times per day. It
will be appreciated that it may be necessary to make routine
variations to the dosage, depending on the age and condition of the
patient. The dosage will also depend on the route of administration.
Compounds of general formula (I) and physiologically acceptable
salts or solvates thereof may be prepared by the general methods
outlined hereinafter. In the following descriptio~, the groups R
and Im are as defined for compounds of general formula (I) unless
otherwise stated.
According to a first general process (A), a compound of general
formula (I) may be prepared by alkylating a compound of formula (II):
- 1 339022
-- 10
1l
! ._. NH
N (II)
with a compound of formula (III):
LCH2-Im (III)
or a protected derivative thereof, wherein L represents a leaving atom
or group, such as a halogen atom (e.g. chlorine, bromine or iodine),
or an acyloxy group (e.g. trifluoroacetyloxy or acetoxy), or a
sulphonyloxy group (e.g. trifluoromethanesulphonyloxy,
p-toluenesulphonyloxy or methanesulphonyloxy), followed where
necessary by removal of any protecting groups. L is preferably a
halogen atom (e.g. a chlorine atom).
The reaction may be carried out in an inert solvent such as an
ether (e.g. dimethoxyethane, diglyme or tetrahydrofuran), a
substituted amide (e.g. dimethylformamide or N-methylpyrrolidone), an
aromatic hydrocarbon (e.g. toluene), a ketone (e.g. acetone), or
dimethyl sulphoxide, at a temperature between ambient and 100C, in
the presence of a base. Suitable bases include alkali metal hydrides
(e.g. sodium hydride), alkali metal carbonates (e.g. sodium
carbonate), alkali metal amides (e.g. sodium amide), alkali metal
alkoxides (e.g. potassium t-butoxide) or alkali metal hydroxides (e.g.
sodium or potassium hydroxide).
- 1 339022
11
According to another general process (B), a compound of general
formula (I) wherein n represents 2, may be prepared by hydrogenation
of a compound of formul (IV):
11
// \ / \ /-\ (IV)
~ N Im
11 11
-
N
R 1
or a protected derivative thereof, followed where necessary by removal
of any protecting groups.
Hydrogenation according to general process (~) may be effected
using conventional procedures, for example using hydrogen in the
presence of a noble metal catalyst (e.g. palladium, Raney nickel,
platinum or rhodium). The catalyst may be supported o~, for example,
charcoal or alumina, or alternatively a homogeneous catalyst such as
tris(triphenylphosphine)rhodium chloride may be used. The
hydrogenation will generally be effected in a solvent such as an
alcohol (e.g. methanol or ethanol), an ether (e.g. dioxan), or an
ester (e.g. ethyl acetate), or in a mixture of an alcohol and either
a hydrocarbon (e.g. toluene) or a halogenated hydrocarbon (e.g.
- 12 - 1 339022
dichloromethane), at a temperature in the range -20 to +100C, and at
a pressure of from 1 to 10 atmospheres.
According to another general process (C), a compound of general
formula (I) may be prepared by cyclising a compound of formula (V):
0
/;\ /W /11, /-
\ / \ y/ 2 n (V)
1~
wherein W represents a hydrogen atom and Y represents the group NH, or
W represents a halogen atom and Y represents a bond, or a salt or
protected derivative thereof, followed where necessary by removal of
any protecting groups.
According to one embodiment (a) of process (C), the reaction is
effected with a compound of formula (V) wherein W represents a
hydrogen atom and Y represents the group NH, and the cyclisation may
be carried out in aqueous or non-aqueous media, in the presence of an
acid catalyst.
It will be appreciated that these compounds of formula (V) may
exist in the corresponding enol hydrazone tautomeric form.
When an aqueous medium is employed this may be water or a mixture
of water and a organic solvent such as an alcohol (e.g. methanol,
ethanol or isopropanol) or an ether (e.g. dioxan or tetrahydrofuran).
The acid catalyst may be, for example, an inorganic acid such as
concentrated hydrochloric or sulphuric acid. In some cases the acid
catalyst may also act as the reaction solvent. In an anhydrous
reaction medium, which may comprise one or more alcohols or ethers
(e.g. as described above), carboxylic acids (e.g. acetic acid) or
esters (e.g. ethyl acetate), the acid catalyst may alternatively be
a Lewis acid such as boron trifluoride, zinc chloride or magnesium
chloride. The cyclisation reaction may conveniently be carried out at
temperatures of from 20 to 200C, preferably 20 to 125C.
Alternatively the cyclisation according to embodiment (a) of
process (C) may be carried out in the presence of polyphosphate ester
in a reaction medium which may comprise one or more organic solvents,
- 1 339022
- 13
preferably halogenated hydrocarbons such as chloroform,
dichloromethane, dichloroethane, dichlorodifluoromethane, or mixtures
thereof. Polyphosphate ester is a mixture of esters which may be
prepared from phosphorus pentoxide, diethyl ether and chloroform
according to the method described in 'Reagents for Organic Synthesis~,
tFieser and Fieser, John Wiley and Sons, 1967).
According to another embodiment (b) of process (C~, the reaction
is effected with a compound of formula (V) wherein W represents a
halogen atom, for example, a chlorine atom or, more preferably, a
bromine or iodine atom, Y represents a bond, and the cyclisation is
effected photochemically.
The reaction may conveniently be effected by irradiating with a
mercury lamp, preferably a medium or high pressure mercury lamp.
Suitable solvents include nitriles (e.g. acetonitrile~, chlorinated
hydrocarbons (e.g. carbon tetrachloride) and cyclic ethers (e.g.
tetrahydrofuran or dioxan) and mixtures thereof. The reaction may
conveniently be effected in the presence of a base such as a tertiary
amine (e.g. triethylsmine).
According to another general process (D), a compound of general
formula (I) wherein R3 represents a hydrogen atom, may be prepared by
the reaction of a compound of formula (VI):
o
-
//\ /\~'~
~ N ~---u^
l ll ll 1 1l 1 (VI)
;\ /-\ /~--(CH2) ~-\ //-
or a protected derivative thereof, with formamide, at a temperature in
the range of 15û to 200C, followed where necessary by removal of any
protecting groups.
According to another general process (E), a compound of general
formula (I) may be prepared by reacting a compound of formula (VII):
- 14 - 1 33~22
. G
//\ /
~ --
11 11
--
\\ / Nl (CH2) NHCH2Im (VII)
Rl
wherein G represents a hydrogen atom, or a protected derivative
thereof, with phosgene in the presence of a Lewis acid; or by reacting
a compound of formula (VII) wherein G represents an iodine or a
bromine atom, or a protected derivative thereof, with carbon monoxide
in the presence of a palladium (II) salt, followed where necessary by
removal of any protecting groups.
According to one embodiment of process (E), a compound of formula
(VII), wherein G represents a hydrogen atom, is reacted with phosgene
in the presence of a Lewis acid such as anhydrous aluminium
trichloride or stannic chloride. The reaction may conveniently be
effected in an inert solvent such as an aromatic hydrocarbon (e.g.
toluene) or a halogenated hydrocarbon (e.g. dichloromethane), or
mixtures thereof, and at a temperature between ambient and 100C.
According to another embodiment of process (E), a compound of
formula (VII), wherein G represents an iodine or a bromine atom, is
reacted with carbon monoxide in the presence of a palladium (II) salt
(e.g. palladium acetate or palladium chloride) and preferably in the
presence of triphenylphosphine. The reaction may conveniently be
effected in a solvent such as a tertiary amine (e.g.
tri-n-butylamine), optionally in the presence of a co-solvent such as
an ether (e.g. tetrahydrofuran) or an aromatic hydrocarbon (e.g.
toluene), at a temperature in the range 100 to 150C, and at
atmospheric pressure.
According to another general process (F~, a compound of general
formula (I) may be converted into another compound of formula (I)
using conventional techniques. Such conventional techniques include
hydrogenation, alkylation and acylation using protection and
deprotection where necessary.
Thus, according to one embodiment of the interconversion process
(F), hydrogenation may be used to convert an alkenyl or an alkynyl
- 15 - 1 339022
substituent into an alkyl substituent, or an alkynyl into an alkenyl
substituent. Hydrogenation may also be used to replace a
phenylmethoxymethyl group by a hydrogen atom. Hydrogenation according
to general process (F) may be effected using conventional procedures,
for example, using hydrogen in the presence of a catalyst, as
described above in general process (B).
The term 'alkylation' according to general process (F) includes
the introduction of groups such as cycloalkyl, alkenyl or phenalkyl
groups.
Thus, for example, a compound of formula (I) in which Rl
represents a Cl_6 alkyl, C3-6 alkenyl, C3_l0 alkynyl, C3_7 cycloalkyl,
C3_7cycloalkylCl_4 alkyl, phenyl Cl_3 alkyl, phenylmethoxymethyl,
phenoxyethyl or phenoxymethyl group may be prepared by alkylating a
compound of formula (I) in which Rl represents a hydrogen atom, or a
compound in which R3 represents a Cl_6alkyl, C3_7cycloalkyl,
C3_6alkenyl or phenylCl_3alkyl group may be prepared by alkylating the
corresponding compound of formula (I) in which R3 represents a
hydrogen atom, using conventional procedures, for example as described
in published European Patent Specification No. 242973. Thus the
reactions may be effected using an appropriate alkylsting agent of
formula R7Z (where R7 is the group to be introduced and Z is a leaving
atom or group), preferably in the presence of a base.
According to another embodiment of general process (F), a
compound of formula (I) wherein Rl represents -Co2R5, -COR5,
-CoNR5R6 or -502R5 may be prepared by acylating or sulphonylating as
appropriate, a compound of formula (I) wherein Rl represents a
hydrogen atom. The acylation/sulphonylation reactions may be effected
using an appropriate acylating/sulphonylating agent according to
conventional procedures, for example, as described in published
European Patent Specification No. 210840.
It should be appreciated that in the above transformations it may
be necessary or desirable to protect any sensitive groups in the
molecule of the compound in question to avoid undesirable side
reactions. For example, it may be necessary to protect the indole
and/or imidazole nitrogen atoms, for example with an arylmethyl (e.g.
trityl), arylmethoxymethyl (e.g. phenylmethoxymethyl), alkyl (e.g.
t-butyl), alkoxymethyl (e.g. methoxymethyl), acyl (e.g.
- 1 339n22
- 16
benzyloxycarbonyl) or a sulphonyl (e.g. N,N-dimethylaminosulphonyl or
p-toluenesulphonyl) group.
~ Thus according to another general process (G), a compound of
general formula (I) may be prepared by the removal of any protecting
groups from a protected form of a compound of formula (I).
Deprotection may be effected using conventional techniques such as
those described in 'Protective Groups in Organic Synthesis' by T. W.
Greene (John Wiley and Sons, 1981).
For example, an arylmethoxymethyl N-protecting group may be
cleaved by hydrogenolysis in the presence of a catalyst (e.g.
palladium on charcoal). A trityl group may be cleaved by acid
hydrolysis (e.g. using dilute hydrochloric or acetic acid). An
alkoxyalkyl group may be removed using a mineral acid (e.g. dilute
hydrochloric or hydrobromic acid). An acyl group may be removed by
hydrolysis under acidic or basic conditions (e.g. using hydrogen
bromide, dilute hydrochloric acid or sodium hydroxide). A sulphonyl
group may also be removed by alkaline or acidic hydrolysis, and an N,
N-dimethylaminosulphonyl group may also be removed (e.g. from an
imidazole nitrogen atom) by photolysis.
Compounds of formula (II) may be obtained by a Beckmann
rearrangement of an oxime of formula (VIII):
HO\
Il
/; \ / \
. (VIII)
!~ ,., /~tcH2)m
Rl
wherein m represents 1 or 2, or a protected derivative thereof. The
Beckmann rearrangement may be effected using conventional methods, for
example by using an acid (e.g. polyphosphoric or sulphuric acid,
or a mixture of hydrochloric acid, acetic anhydride and acetic acid)
in an inert solvent such as an ether (e.g. dioxan), an amide (e.g.
dimethylformamide) or a hydrocarbon (e.g. toluene or cyclohexane), at
an elevated temperature of, for example, 50 to 120C. Alternatively,
the hydroxy group of the oxime of formula (VIII), may be converted
into a leaving group such as a chloride (using, for example,
1 339022
- 17
phosphorus pentachloride) or a hydrocarbylsulphonate (e.g. a mesylate
or a tosylate) or a trifluoroacetate group (using conventional
acylation methods). Subsequent heating at a temperature of, for
example, 20 to 150C, in an inert solvent as described above, gives a
compound of formula (II).
Compounds of formula (VIII) may be prepared from the
corresponding tricyclic ketone of formula (IX):
ll
(IX)
CH )
Nl 2 m
Rl
wherein m represents 1 or 2, or a protected derivative thereof using
conventional methods, for example by using hydroxylamine hydrochloride
in a solvent such as pyridine.
Compounds of formula (IV) may be prepared, for example, by
reacting a compound of formula (X):
O
//\ / \
~. INH (X )
-
N
11 '
or a protected derivative thereof, with a compound of formula (III)
wherein L is as deFined previously, or a protected derivative thereof,
using the conditions described in process (A).
Compounds of formula (X) may be prepared by heating a compound of
formula (II) wherein n represents 2, with a noble metal catalyst suchas palladium, palladium oxide, platinum or nickel, at a temperature
of, for example, 300 to 350C. The catalyst may be supported on, for
example, charcoal or alumina, and the reaction may optionally be
1 339022
- 18
carried out in the presence of an inert solvent such as an aromatic
hydrocarbon (e.g. p-cymene)
Compounds of formula (V) wherein W represents a hydrogen atom and
Y represents the group NH may be prepared by the reaction of a
compound of formula (XI):
!".
! 11 (XI)
NRlNH
or a salt thereof, with a compound of formula (XII):
o
i N Im (XII)
~CH2)
// n
or a protected derivative thereof, in a suitable solvent such as an
aqueous alcohol (e.g. methanol), and at a temperature of, for example,
from 20 to 100C.
A protected derivative of a compound of formula (XII) may for
example have the keto carbonyl group protected (e.g. as an enol
ether). It will be appreciated that when a compound of formula (XII)
is used in which the keto carbonyl group is protected, it may be
necessary to remove the protecting group in order for reaction to
occur with the compound of formula (XI). Deprotection may be carried
out by conventional methods, for example by acidic hydrolysis (e.g.
using dilute sulphuric or hydrochloric acid). If desired,
deprotection may be effected in situ.
Compounds of formula (XII) may be prepared, for example, by
reacting a compound of formula (XIII):
- 1 339n22
-- 19
NH (XIII )
~ (CH2)
0
or a protected derivative thereof, with a compound of formula (III)
wherein L is as defined previously, or a protected derivative thereof,
using the conditions described in process (A).
Compounds of formula (V) wherein W represents a halogen atom and
Y represents a bond may be prepared, for example, by reacting a
compound of formula (XIV):
o
W
! . . NH
i ll ll I (XIV)
; ~(CH2)
N
Rl
wherein W represents a halogen atom, or a protected derivative
thereof, with a compound of formula (III) wherein L is as defined
previously, or a protected derivative thereof, using the conditions
described in process (A).
Compounds of formula (XIV) may be prepared by reacting a compound
of formula (XV):
/;\ /w
i1 (X V )
NHRl
with a compound of formula (XIII), at an elevated temperature.
Compounds of formula (VI) may be prepared, for example, by
reacting a compound of formula (II), or a protected derivative
thereof, with a compound of formula (XVI):
~ - 20 - l 339022
~/ \~
~ (XVI)
R4/ \N//
wherein L is as defined previously, using the conditions described in
process (A).
Compounds of formula (YII) wherein G represents a halogen atom
may be prepared, for example by rescting a compound of formula (VII)
wherein G represents a hydrogen atom, or a protected derivative
thereof, with an appropriate halogen and alkali metal halide (e.g.
iodine and potassium iodide), in a suitable solvent such as an aqueous
alcohol (e.g. aqueous ethanol).
Compounds of formula (VII) wherein G represents a hydrogen atom
may be prepared, for example, by reacting a compound of formula
(XVII):
// \
11 li (X VI I )
-
\\ / \~/ \(CH ) N~
R
or a protected derivative thereof, with a compound of formula (III)
wherein L is as defined previously, or a protected derivate thereof,
using the conditions described in process (A).
Compounds of formula (III) and protected derivatives thereof, are
either known, or may be prepared, for example, by the methods
described in German Offenlegungsschrift No. ~740352 published on June 9,
1988 and corresponding to Canadian patent application serial number
552,963 filed November 27, 1987.
Compounds of formula (IX) may be prepared, for example, by the
method or methods analogous to that described by H. Iida et al. in J.
~rg. Chem., 1980, 45, 2938.
Compounds of formulae (XI), (XIII), (XV), (XVI) and (XVII) are
either known, or may be prepared from known compounds by conventional
procedures.
Where it is desired to isolate a compound of the invention as a
salt, for example a physiologically acceptable salt, this may be
~ - 21 - 1 339022
achieved by reacting the compound of formule (I) in the form of the
free base with sn sppropriate acid, preferably with an equivalent
amount, in a suitsble solvent such as an alcohol (e.g. ethanol or
methanol), an aqueous alcohol (e.g. aqueous ethanol), a halogenated
hydrocarbon (e.g. dichloromethane), an ester (e.g. ethyl acetate) or
an ether (e.g. tetrahydrofuran).
Physiologically acceptable salts may also be prepared from other
salts, including other physiologically acceptable salts, of the
compound of formula (I) using conventional methods.
Individual enantiomers of the compounds of the invention may be
obtained by resolution of a mixture of enantiomers (e.g a racemic
mixture) using conventional means, such as an optically active
resolving acid; see for example 'Stereochemistry of Carbon Compounds'
by E.L.Eliel (McGraw Hill, 1962) and 'Tables of Resolving Agents' by
S. H. ~ilen.
-- The methods described above for preparing the compounds of the
invention may be used for the introduction of the desired groups at
any stage in the stepwise formation of the required compounds, and it
will be appreciated that these methods can be combined in different
ways in such multi-stage processes. The sequence of the reactions in
multi-stage processes should of course be chosen so that the reaction
conditions used do not affect groups in the molecule which are desired
in the final product.
The invention is further illustrated by the following
Intermediates and Examples. All temperatures are in C. Thin layer
chromatography (t.l.c.) was carried out on silica, and flash column
chromatography (FCC) on silica (Merck*9385). Solvent System A as used
for chromatography denotes dichloromethane:ethanol:û.88 ammonia
solution. Organic extracts were dried, where indicated, over
magnesium sulphate or sodium sulphate. The following abbreviations
are used: DMF - dimethylformamide; THF - tetrahydrofuran; DME -
dimethoxyethane. lH-N.m.r. spectra were obtained at 250MHz for dilute
solutions in d6-dimethyl sulphoxide.
*Trade Mark
- 22 _ 1 339022
Intermediate 1
4-(Chloromethyl)-l-(triphenylmethyl)-lH-imidazole
Thionyl chloride (0.829) was added over 1 min. to a stirred suspension
of l-(triphenylmethyl)-lH-imidazole-4-methanol (1.39) in a mixture of
dichloromethane (50mR) and DMF (l.OmQ) at 23. The solution so
obtained was stirred for 15 min. and extracted with 8~ sodium
bicarbonate solution (80mQ). The organic phase was washed with water
(50mR), dried and evaporated to give an oil which solidified. The
solid was slurried in hexane and filtered to give the title compound
(1.289), m.p. 139-141.
Intermediate 2
4-Formyl-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide
Dimethylsulphamoyl chloride (0.67mQ) was added to a stirred solution
of 5-propyl-lH-imidazole-4-carboxaldehyde (860mg) and triethylamine
(0.87mQ) in dry dichloromethane (lOmQ) under nitrogen. The solution
was heated at reflux for 24h, allowed to cooI, poured into water
(50mQ) and extracted with dichloromethane (3x25mR). The combined,
dried organic extracts were evaporated to give an oil (1.99) which was
purified by FCC eluting with ethyl acetate:hexane (1:1) to give the
title compound (500mg), m.p. 57-58.
Intermediate 3
4-(Hydroxymethyl)-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide
Sodium borohydride (139mg) was added to a stirred solution of
4-formyl-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide (450mg)
in absolute ethanol (5mQ) under nitrogen. After 3h the mixture was
poured into water (30mR) and extracted with dichloromethane (3xl5mR).
The combined, dried organic extracts were evaporated to give a solid
(425mg) which was triturated with ether (2xlOmR) to give the title
compound (350mg), m.p. 86-88.
Intermediate 4
4-(Chloromethyl)-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide
A solution of thionyl chloride (0.12mR) in dry dichloromethane (1.2m~)
was added dropwise to a cold (0) stirred solution of
- - 23 - 1 339022
4-(hydroxymethyl)-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide
(340mg) in dry dichloromethane (7.5mQ) under nitrogen. After 1.5h the
solution was washed with 8~ sodium bicsrbonate solution (2xl5mQ) and
the aqueous phase was extracted with dichloromethane (2xlOmQ). The
combined organic extracts were washed with water (15mQ), dried and
evaporated to give the title compound (180mg) as an oil, t.l.c. (ethyl
acetate) Rf 0.68.
Intermediate 5
3,4-Dihydro-4-methylcyclopent[b]indol-1(2H)-one oxime
3,4-Dihydro-4-methylcyclopent[b]indol-1(2H)-one (1.79) and
hydroxylamine hydrochloride (1.9259) in pyridine were heated at 60
for 18h and cooled. The reaction mixture was evaporated in vacuo to a
residue to which was added 8~ sodium bicarbonate (150mR). Extraction
with ethyl acetate (300mQ) produced a suspension in the organic layer;
this layer and associated solid was separated from the aqueous layer.
The aqueous layer was re-extracted with ethyl acetate (250mQ). The
combined organic extracts (and suspended solid) were evaporated to a
residue, boiled with a mixture of ethanol (150mQ) and methanol (150mQ)
and cooled to ca. 50. The residue was adsorbed from this solution on
to FCC silica and applied to an FCC column. Elution with ethyl
acetate/3-10~ methanol provided the title compound (1.699), m.p.
219-224 (decomp.).
Intermediate 6
2,3,4,5-Tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-one
3,4-Dihydro-4-methylcyclopent[b]indol-1(2H)-one oxime (1.539),
polyphosphoric acid (409) and dioxan (15mQ) were heated at 110-120
for 2.2h under nitrogen. The reaction mixture was cooled, and treated
with 2N sodium carbonate solution (lQ). The suspension was extracted
with ethyl acetate (4x400mQ) and the combined extracts were dried.
Evaporation gave a solid (1.439) which was recrystallised from ethyl
acetate/cyclohexane. This solid was purified by FCC, eluting with
System A (200:10:1) to give a solid (1.26 9) which was recrystallised
from ethanol to provide the title compound (960 mg), m.p. 234-238.
~ - 24 - 1 339022
Intermediate 7
3,4,5,6-Tetrahydro-6-methylazepino[4,3-b]indol-1(2H)-one
1,2,3,9-Tetrahydro-9-methyl-4H-carbazol-4-one oxime (249) and
polyphosphoric acid (6009) in dioxan (500mR) were treated according to
the method described for Intermediate 6. The solid (229) obtained by
evaporation of the organic extracts was recrystallised from ethyl
acetate (300mQ) to give a solid (19.2 9). This was purified by FCC
eluting with System A (200:8:1) to give the title compound (5.59~,
m.p. 212-215.
Intermediate 8
5,6-Dihydro-4-(phenylamino)-1(2H)-pyridinone
A mixture of 2,4-dioxopiperidine (1.139) and aniline (930mg) was
heated at 120 under a stream of nitrogen for 15 min. The resultant
solid was triturated with ether and filtered off to give the title
compound (1.749), m.p. 235-238.
Intermediate 9
2,3,4,5-Tetrahydro-lH-pyrido[4,3-b]indol-1-one
A solution of 5,6-dihydro-4-(phenylamino)-1(2H)-pyridinone (1.59) and
palladium acetate (150mg) in dry DMF (50mR) was treated with cupric
acetate (3.29) and the resulting mixture was heated under nitrogen at
120-130 for 1.5 h. The mixture was then concentrated in vacuo to
give a solid which was triturated with 2N hydrochloric acid (250mQ).
The acid was decanted, and the remaining solid was extracted with
ethyl acetate for 18h. The decanted acid was basified with 2N sodium
hydroxide and extracted with ethyl acetate (3xlOOmR). These organic
extracts were combined with the previous ethyl acetate extracts and
adsorbed onto silica. Purification by FCC eluting with System A
(100:8:1) gave the title compound (874mg), m.p. 212-215.
Intermediate 10
2,3,4,5-Tetrahydro-5-[(phenylmethoxy)methyl]-lH-pyrido[4,3-b]indol-1-
one
A solution of 2,3,4,5-tetrahydro-lH-pyrido[4,3-b]indol-1-one (1.129)
in dry DMF (60mR) was treated with sodium hydride (60~ dispersion in
. 1 339n22
- 25
oil; 480mg) and the resulting mixture was stirred under nitrogen until
effervescence ceased. The mixture was then cooled to 0 and benzyl
(chloromethyl) ether (10~ w/v solution in DMF; 0.835ml) was added over
10 min. Stirring was continued for a further 5 min and then water
(lOm~) was added. The reaction mixture was concentrated in vacuo to
give an oil which was dissolved in ethyl acetate (lOOmR) and washed
with water (3xlOOmQ). The organic phase was dried and adsorbed onto
FCC silica. Purification by FCC eluting with System A (150:8:1) gave
the title compound (1.19), m.p. 133-135.
Intermediates 11 to 14 were prepared in a similar manner to
Intermediate 10, i.e. by treating 2,3,4,5-tetrahydro-lH-pyrido[4,3-
b]indol-l-one with sodium hydride followed by an appropriate
alkylating agent. Isolation and purification of the products were as
described for Intermediate 10 unless otherwise stated.
Intermediate 11
5-Ethyl-2,3,4,5-tetrahydro-lH-pyrido[4,3-b]indol-1-one
2,3,4,5-Tetrahydro-lH-pyrido[4,3-b]indol-1-one (931mg) was treated
with sodium hydride (60~ dispersion in oil; 400mg) and was then
stirred with ethyl iodide ¢10~ v/v solution in DMF; 4ml) to give the
title compound (758mg), m.p. 203-204.5.
Intermediate 12
2,3,4,5-Tetrahydro-5-(1-methylethyl)-lH-pyrido[4,3-b]indol-1-one
2,3,4,5-Tetrahydro-lH-pyrido[4,3-b]indol-1-one (931mg) was treated
with sodium hydride (73~ dispersion in oil; 328 mg) and was then
stirred with 2-bromopropane (615mg) at room temperature for 72 h.
Purification by FCC eluting with System A (200:8:1) gave a foam
(324mg) which was further purified by recrystallisation from ethyl
acetate: hexane (1:1) to give the title compound (294mg~, t.l.c.
(System A, 100:8:1) Rf 0.58.
Intermediate 13
2,3,4,5-Tetrahydro-5-(phenylmethyl)-lH-pyrido[4,3-b]indol-1-one
2,3,4,5-Tetrahydro-lH-pyrido[4,3-b]indol-1-one (559mg) was treated
with sodium hydride (73~ dispersion in oil; 197mg) and was then
stirred with benzyl bromide (513 mg) at room temperature for 30 min.
Purification by FCC eluting with dichloromethane: ethanol (80:1) gave
the title compound (347 mg), m.p. 209-212.
_ - 26 ~ 1 339n22
Intermediste 14
5-(Cyclopentylmethyl)-2,3,4,5-tetrahydro-lH-pyridot4,3-b]indol-1-one
2,3,4,5-Tetrahydro-lH-pyrido[4,3-b]indol-1-one (950mg) was treated
with sodium hydride (60~ dispersion in oil; 408mg) and was then
stirred with cycloPentanemethanol, methanesulphonate (909mg) at room
temperature for 7 days. The solid (570mg) obtained by FCC was further
purified by slow evaporation from a solution in methanol to give the
title compound, m.p. 179-181.
Intermediate 15
2,3,4,5-Tetrahydro-2-t[5-methyl-1-(triphenylmethyl)-lH-imidazol-4-
yl]methyl]-lH-pyridot4,3-b]indol-1-one
A solution of triphenylmethyl chloride (3.369) in dry DMF (40mR) was
added dropwise to a stirred solution of 2,3,4,5-tetrahydro-2-
[(5-methyl-lH-imidazol-4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one (2.89)
in dry DMF (50m~) containing triethylamine (1.529). When addition was
complete the mixture was stirred overnight. The mixture was then
poured into water (lOOOml) and the resulting suspension was extracted
with ethyl acetate (3 x 300mR). The combined organic extracts were
washed with water (2 x 500mQ), dried and concentrated onto silica.
FCC eluting with System A (100:8:1) gave the title compound (4.39~,
m.p. 235-236.
Intermediate 16
2,3,4,5-Tetrahydro-5-methyl-2-[[1-(triphenylmethyl)-lH-imidazol-
4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one
A mixture of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1- one
(0.39) and sodium hydride (80~ dispersion in oil; 0.059) in dry DMF
(5m~) was stirred under nitrogen at 50 until hydrogen evolution
ceased (ca. 0.5h). The mixture was cooled to 40 and a solution of
4-(chloromethyl)-1-(triphenylmethyl)-lH-imidazole (0.53 9) in dry THF
(3m~) was added. The mixture was stirred at 40 to 23 over 2h,
poured into water (lOOm~) and extracted with dichloromethane
(3xlOOmR). The dried organic phase was evaporated to give a semi-
solid which was purified by FCC eluting with dichloromethane:ethyl
acetate:triethylamine (50:50:1) to give a solid. This was slurried in
hexane and filtered to give the title compound (0.379), m.p. 205-210
(decomp.).
~ - 27 _ 1 339n22
Intermediate 17
2,5-Dihydro-5-methyl-lH-pyrido[4,3-b]indol-1-one
A mixture of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-
one (500mg) and 10~ palladium oxide on carbon catalyst (50~ aqueous
paste; 250mg) was heated at 320 for lû min. The cooled solid was
triturated with ethanol (ca. lOOmR), filtered and the resulting
filtrate was evaporated to give the title compound (470mg), m.p.
242.5.
Intermediate 18
2,5-Dihydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one maleate
Sodium hydride (73~ dispersion in oil; 80mg) was added to a stirred
suspension of 2,5-dihydro-5-methyl-lH-pyrido[4,3-b]indol-1-one (440mg)
in dry dimethoxyethane (25mR) under nitrogen and the mixture was
heated at 50 for 6h. 4-(Chloromethyl)-5-methyl-1-(triphenylmethyl)-
lH-imidazole (910mg) was then added, and stirring was continued at 50
for 20h. Water (4.5mQ) and acetic acid (4.5mR) were added and the
solution was heated at reflux for 5h. The mixture was poured into 8
sodium bicarbonate solution (80mQ) and extracted with
dichloromethane:ethanol (10:1; 3x40mR). The combined, dried organic
extracts were evaporated to give a solid (ca. 1.59) which was purified
by FCC eluting with System A (200:10:1) to give the free base of the
title compound as a solid (384mg). A sample of this solid (lOOmg) was
dissolved in absolute ethanol (20mR) and treated with a solution of
maleic acid (40mg) in absolute ethanol (lmQ). The solvent was removed
in vacuo and the residue was triturated with dry ether (3x20mR) to
give a solid (115mg) which was re-crystallised from methsnol-ethyl
acetate to give the title compound (40mg~, m.p. 166-168.
- 28 - 1 339022
Intermediate 19
5,6-Dihydro-4-methoxy-1-[[5-methyl-1-(triphenylmethyl)-lH-imidazol-4-
yl]methyl]-2(lH)-pyridinone
Sodium hydride (80~ dispersion in oil; 360mg) was suspended in dry
DME (50mQ) under nitrogen and 5,6-dihydro-4-methoxy-2(1H)-pyridinone
(1.279) in dry DME (20mQ) was added slowly. The resulting suspension
was stirred at 20 for lh. 4-(Chloromethyl)-5-methyl-1-
(triphenylmethyl)-lH-imidazole (3.729) in dry DME (50mR) was added,
and after the initial reaction had subsided the mixture was heated to
50 for 4h and then cooled. Methanol (5mR) was added dropwise, and
the solvent was removed in vacuo. 8v Aqueous sodium bicarbonate
solution (300mR) was added to the residue and the resulting solution
was extracted with dichloromethane (2x300mR), dried and evaporated in
vacuo to leave an oil which was purified by FCC eluting with System A
(200:8:1) to give the title compound (2.849), m.p. 181-184.
Intermediate 20
2,4-Dioxo-1-[(5-methyl-lH-imidazol-4-yl)methyl]piperidine
To a solution of 5,6-dihydro-4-methoxy-1-[[5-methyl-1-
(triphenylmethyl)-lH-imidazol-4-yl]methyl]-2(1H)-pyridinone (500mg) in
THF (4mR) was added hydrochloric acid (5M; lmR), and the mixture was
stirred at 50 for lh. The solvent was removed in vacuo, triethylamine
(lmQ) was added, and the mixture was again evaporated to dryness. FCC
of the residue eluting with ethyl acetate:methanol:triethylamine
(8:4:1) gave the title compound (139mg), m.p. 100-106 (decomp.).
Intermediate 21
5,6-Dihydro-1-[(5-methyl-lH-imidazol-4-yl)methyl]-4-(2-methyl-2-
phenylhydrazino)-2(1H)-pyridinone
2,4-Dioxo-1-[(5-methyl-lH-imidazol-4-yl)methyl]piperidine (20mg) was
dissolved in ethanol (2mR) and N-methylphenylhydrazine (26mg) was
added. The mixture was stirred for lh and the solvent was removed in
vacuo. The residue was purified by FCC eluting with System A (75:8:1)
29 1 339022
to give the title compound (24mg) as a solid, t.l.c. (System A,
75:8:1) Rf 0.27.
Intermediate 22
N,N,5-Trimethyl-4-[[(trimethylsilyl)oxy]methyl]-lH-imidazole-l-
sulphonamide
A suspension of 4-(hydroxymethyl)-5-methylimidazole hydrochloride
(14.99) in dry dichloromethane (5ûOmQ) containing triethylamine (509)
was treated with trimethylsilyl chloride (21.79) and the reaction
mixture was stirred at room temperature overnight. Dimethylsulphamoyl
chloride (14.39) was added and the reaction mixture was again stirred
at room temperature overnight. The resulting suspension was filtered
and the collected solid was washed with dichloromethane (lOOmR). The
filtrate was concentrated onto silica and purification by FCC eluting
with hexane:ether (4:1) gave the title compound as an oil (7.29),
t.l.c. (ether) Rf 0.5.
Intermediate 23
4-(Hydroxymethyl)-N,N,5-trimethyl-lH-imidazole-l-sulphonamide
A solution of N,N,5-trimethyl-4-[[(trimethylsilyl)oxy]methyl]-lH-
imidazole-l-sulphonamide (2.599) in dry THF (50mR) was treated with a
solution of tetrabutylammonium fluoride (lM solution in THF; lOml).
and the THF was immediately removed in vacuo. The residue was
partitioned between water (lOOmR) and dichloromethane (lOOmR) and the
aqueous layer was extracted with dichloromethane (lOOmR). The
combined, dried organic fractions were concentrated to give the title
compound (1.639) as a solid, m.p. 134-136.
Intermediate 24
4-(Chloromethyl)-N,N,5-trimethyl-lH-imidazole-l-sulphonamide
A suspension of 4-(hydroxymethyl)-N,N,5-trimethyl-lH-imidazole-l-
sulphonamide (2.869) in dry dichloromethane (200mR) containing DMF
(0.5mR) was treated dropwise with a solution of thionyl chloride
(1.1789) in dichloromethane (lOmR). The reaction mixture was cooled
in ice during the addition and blanketed with nitrogen. When addition
was complete (ca. 5min), stirring was continued at 0 for a further 30
1 339022
_ 30
min. Water (200mQ) was then added snd the organic phase was
separated, washed with 8~ sodium bicarbonate (lOOmR), dried and
concentrated to give the title compound (2.39) as a solid, m.p.
115-118.
Intermediate 25
5,6-Dihydro-4-[(2-iodophenyl)methylamino]-2(lH)-pyridinone
A mixture of 2-iodo-(N-methyl)sniline (1.179) and 2,4-dioxopiperidine
(565mg) was heated under a stream of nitrogen for 7h at 110-120.
After cooling the reaction mixture was dissolved in methanol and the
solution was adsorbed onto FCC silica. Purification by FCC eluting
with System A (150:8:1) gave the title compound (1.039), m.p.
163-164.
Intermediate 26
N,N,5-Trimethyl-4-[I,2,3,6-tetrahydro-4-[(2-iodophenyl)-
methylamino]-6-oxo-1-pyridinyl]methyl-lH-imidazole-l-sulphonsmide
A suspension of 5,6-dihydro-4-[(2-iodophenyl)methylamino]-2(1H)-
pyridinone (984mg) in dry DME (50mQ) was treated with sodium hydride
(60~ dispersion in oil; 140mg), and the mixture was stirred under
nitrogen for 6h. 4-(Chloromethyl)-N,N,5-trimethyl-lH-imidazole-l-
sulphonamide (832mg) was then added and the resulting mixture was
stirred at 60 overnight. After cooling the reaction mixture was
poured into water (lOOmQ), and the mixture was extracted with ethyl
acetate (2x50mQ). The combined, dried organic extracts were
concentrated, and the resultant solid was purified by FCC eluting with
System A (150:8:1) to give the title compound (712mg), t.l.c. (System
A, 150:8:1) Rf 0.41.
Intermediate 27
N,N,5-Trimethyl-4-[2,3,4,5-tetrahydro-5-methyl-1-oxo-1H-pyrido[4,3-
b]indol-2-yl)methyl]-lH-imidazole-l-sulphonamide
A solution of dimethylsulphamoyl chloride (0.107mQ) in dry
dichloromethane was added to a stirred solution of 2,3,4,5-tetrahydro-
5-methyl-2-t(5-methyl-lH-imidazol-4-yl)methyl]-lH-pyrido[4,3-b]indol-
l-one (0.2949) and triethylamine (0.2mQ) in dry dichloromethane (30mQ)
,~ ~. ~.. . .
` ~ - 31 - 1 339n22
under nitrogen, and the mixture was heated at reflux for ca. 24h.
After cooling the solution was concentrated onto FCC silica and
purified by FCC eluting with System A (150:8:1) to give an oil. This
oil was triturated with ether to give a solid which was further
purified by slow evaporation from a solution in ethyl acetate to give
the title compound (122 mg), m.p. 194-196, t.l.c. (System A,
100:8:1) Rf 0.43.
Intermediate 28
Phenylmethyl 5-methyl-4-[(2,3,4,5-tetrahydro-5-methyl-1-oxo-lH-
pyrido[4,3-b]indol-2-yl)methyl]-lH-imidazole-l-carboxylate
A solution of benzyl chloroformate (0.28mQ) in dichloromethane (lOmQ)
was added to a stirred solution of 2,3,4,5-tetrahydro-5-methyl-2-
[(5-methyl-lH-imidazol-4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one
(294mg) and triethylamine (0.4mQ) in dichloromethane (30mQ) at 20
under nitroge~, and the mixture was stirred overnight. It was then
concentrated onto FCC silica and purified by FCC eluting with System A
(200:8:1) to give the title compound (62mg~, t.l.c. (System A,
100:8:1) Rf 0.50.
Intermediate 29
2,3,4,5-Tetrahydro-2-[[1-(methoxymethyl)-5-methyl-lH-imidazol-4-yl]-
methyl]-5-methyl-lH-pyrido[4,3-b]indol-1-one and 2,3,4,5-Tetrahydro-2-
[[l-(methoxymethyl)-4-methyl-lH-imidazol-5-yl]methyl]-5-methyl-lH-
pyrido[4,3-b]indol-1-one
A solution of chloromethyl methyl ether (0.26mQ) in dichloromethane
(lOmQ) was added to a stirred solution of 2,3,4,5-tetrahydro-5-methyl-
2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one
(500mg) and triethylamine (0.49mQ) in dichloromethane (50mQ) at 20
under nitrogen, and the solution was stirred for 4 days. It was then
partitioned between dichloromethane (50mQ) and sodium bicarbonate
solution (2x50mQ). The organic extract was dried, concentrated onto
FCC silica, and then purified by FCC eluting with System A (100:8:1)
to give the title compounds (139mg). A portion of the title compounds
(64mg) was taken up in hot ethyl acetate and purified by slow
evaporation from ethyl acetate to give the title compounds.
- - 32
Analysis Found: C,67.3; H,6.9; N,16.5; 1 3 3 9 0 2 2
ClgH22N402 requires C,67.4; H,6.6; N,16.6~.
Intermediate 30
2,3,4,5-Tetrahydro-5-methyl-2-[(4-methyloxazol-5-yl)methy~ H-pyrido[4~3-b]
indol-l-one
Sodium hydride (60~ dispersion in oil; 600mg) was added to a stirred
suspension of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-one
(1.59) in dry DME (150m~) and then the mixture was stirred at 60 for
5h under nitrogen. 5-Chloromethyl-4-methyloxazole (1.2q) was added and
the mixture was stirred overnight. A further quantity of sodium
hydride (60~ dispersion in oil; 600mg) was added and the mixture was
stirred at 60 for 4h, then cautiously treated with water (lOOmR).
The mixture was extracted with dichloromethane containing methanol
lS (ca. 1~) (3xlOOmR) and the combined extracts were evaporated. The
residue was purified by FCC eluting with System A (100:8:1) to give
the title compound (300mg) as a solid, t.l.c. (System A, 100:8:1) Rf
0.4.
Intermediate 31
N-[(l-Methyl-lH-indol-2-yl)ethyl]trifluoroacetamide
A solution of 2-(1-methyl-lH-indol-2-yl)ethanamine (3.489) in dry
dichloromethane (50mQ) containing triethylamine (2.539) was cooled in
en ice bsth, and trifluoroacetic anhydride (5.259) was added dropwise
over 15 min. The mixture was then allowed to warm to room temperature
and stirred for an additional 3h. After this time the reaction mixture
was poured into water (lOOmQ), the organic phase was separated, and
the aqueous phase was washed with dichloromethane (2x50mQ). The
combined, dried organic extracts were concentrated onto FCC silica and
purification by FCC eluting with ether gave the title compound (4.29)
as a solid. A sample of this compound was further purified by slow
eveporation from a solution in dichloromethane, m.p. 124-126.
1 339n22
- 33
Intermediate 32
N,N,5-Trimethyl-4-[[(1-methyl-lH-indol-2-yl)-N-trifluoroacetylamino]-
ethyl]imidazole-l-sulphonamide
A solution of N-[(l-methyl-lH-indol-2-yl)ethyl]trifluoroacetamide
(2.79) in dry DMF (lOOmQ) was treated with sodium hydride (60~
dispersion in oil; 480mg), and the mixture was stirred at room
temperature for 30 min. 4-(Chloromethyl)-N,N,5-trimethyl-lH-
imidazole-l-sulphonamide (2.379) was then added and the mixture was
stirred at room temperature overnight. After this time the reaction
mixture was poured into water (500mR) and the resulting suspension was
extracted with ethyl acetate (2xlOOmR). The combined organic extracts
were washed with water (5x250mQ), dried and adsorbed onto FCC silica.
Purification by FCC eluting with System A (150:8:1) gave the title
compound (1.99), m.p. 156-158.
Intermediate 33
4-[[[(1-Methyl-lH-indol-2-yl)ethyl]amino]methyl]-N,N,5-trimethyl-lH-
imidazole-l-sulphonamide
A mixture of N,N,5-trimethyl-4-[[(1-methyl-lH-indol-2-yl)-N-
trifluoro-acetylamino]ethyl]imidazole-l-sulphonamide (260mg), methanol
(lOmR) and saturated aqueous potassium carbonate solution (5mR) was
heated to 60 for 1.5h. After cooling the mixture was poured into
water (50mR) and the mixture was extracted with ethyl acetate
(2x50mR). The combined, dried organic extracts were concentrated onto
FCC silica and purified by FCC eluting with System A (150:8:1) to give
the title compound (143mg) as an oil, t.l.c. (System A, 100:8:1) Rf
0.51.
Intermediate 34
4-[[[(3-Iodo-l-methyl-lH-indol-2-yl)ethyl]trifluoroacetylamino]-
methyl]-N,N,5-trimethyl-lH-imidazole-l-sulphonamide
A solution of 4-[[[(1-methyl-lH-indol-2-yl)ethyl]amino]methyl]-N,N,5-
trimethyl-lH-imidazole-l-sulphonamide (471mg) in methanol (25mR)
containing potassium carbonate (138mg) was treated with a solution of
iodine (254mg) and potassium iodide (166mg) in water (30mQ) over
30 min. When addition was complete the reaction mixture was stirred
for 8 further 2h. After this time additional methanol was removed in
- - 1 339n22
- 34
vacuo and the resulting suspension was extracted with ethyl acetate
(3x25mR). The combined organic extracts were concentrated onto FCC
silica and purified by FCC eluting with System A (150:8:1) to give the
title compound (367mg), m.p. 141-143.
Intermediate 35
4-[[t(3-Iodo-l-methyl-lH-indol-2-yl)ethyl]amino]methyl]-N,N,5-
trimethyl-lH-imidazole-l-sulphonamide
4-[[[(3-Iodo-l-methyl-lH-indol-2-yl)ethyl]trifluoroacetylamino]-
methyl]-N,N,5-trimethyl-lH-imidazole-l-sulphonamide (199mg) was
deprotected according to the method described in Intermediate 33 to
give the title compound (50mg) as an oil, t.l.c. (System A, 150:8:1)
Rf 0.51.
_ 35 _ 1 339022
Example 1
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one maleate
A mixture of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-
one (0.69) and ca. 78 sodium hydride dispersion in mineral oil
(0.1099) in dry DMF (15mR) was stirred under nitrogen at 50 until
hydrogen evolution ceased (ca. 1.5h). The mixture was cooled to 40
and a solution of 4-(chloromethyl)-5-methyl-1-(triphenylmethyl)-lH-
imidazole (1.129) in dry THF (15mQ) was added. The reaction was then
stirred at 40 for 3h, at 20 for 16h and a further portion of 4-
(chloromethyl)-5-methyl-1-(triphenylmethyl)-lH-imidazole (1.129) in
dry THF (15mQ) was added. The resulting mixture was heated at 40 for
3h, quenched with water (20mQ) and acetic acid (20mQ), and heated at
lû0 for 2h. The mixture was then concentrated in vacuo to ca. 60m Q,
diluted with lM hydrochloric acid (40mR) and washed with ethyl acetate
(3 x 50mQ). The organic phase was discarded and the acidic aqueous
phase was basified (pH9) with potassium carbonate and extracted with
ethyl acetate: ethanol (20:1, 3 x lOOmQ). The extracts were
combined, dried and evaporated to give a brown gum (ca. 19). This
gum was adsorbed onto silica and purified by FCC eluting with System A
(100:8:1) to give a pale brown solid (0.89) m.p. 238-240 (decomp).
This solid was dissolved in a mixture of hot ethanol and methanol
(1:1; lOOmQ) and treated with an ethanolic solution of maleic acid
(318 9). The resulting solution was concentrated to ca. 20mQ and
diluted with dry diethyl ether (ca. 8mQ) to precipitate the title
compound (0.759) as an off-white solid, m.p. 160-162 .
Analysis Found: C,61.6;H,5.5;N,13.6;
C17H18N4-C4H44 requires C,61.5;H,5.4jN,13.8,o
Example 2
3,4,5,6-Tetrahydro-6-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
azepino[4,3-b]indol-1(2H)-one maleate
3,4,5,6-Tetrahydro-6-methylazepino[4,3-b]indol-1(2H)-one (0.649)
was treated with sodium hydride (ca. 75-80 dispersion in oil; 0.1089)
- - 36 - 1 339022
and was then stirred with 4-(chloromethyl)-5-methyl-1-
(triphenylmethyl)-lH-imidazole 8S described in Example 1. The
reaction mixture was then poured into water (300mR) and extracted with
dichloromethane (4x250mQ). The combined, dried organic extracts were
evaporated to give a semi-solid (ca. 1.89) which was purified by FCC
eluting with System A (200:8:1) to give a gum (0.79). This gum (0.79)
was dissolved in a mixture of acetic acid, THF and water (1:1:1; ca.
70mQ) and heated on a steam bath for lh. Work-up as described in
Example 1 gave a gum (0.229) which was purified by FCC eluting with
System A (200:8:1) to give a solid (0.119). Maleate formation gave a
gum which was dried in vacuo to give a foam which was triturated with
a mixture of ether and ethanol (50:1; ca. 25mR) to give the title
compound (0.1459) as a solid, m.p. 132-133.
lH-N.m.r. indicated 0.39mol of ethanol present.
Water Analysis Found 0.583~ w/w_0.14mol H20.
Analysis Found: C,61.4; H,5.7; N,12.6;
Cl8H20N40. C4H404Ø39EtOH. 0.14H20 requires C,61.4; H,6.0; N,12.6~.
Example 3
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-[(phenyl-
methoxy)methyl]-lH-pyrido[4,3-b]indol-1-one maleate
A suspension of 2,3,4,5-tetrahydro-5-[(phenylmethoxy)methyl]-lH-
pyrido[4,3-b]indol-1-one (920mg) in dry DME (75mQ) was treated with
sodium hydride (60~ dispersion in oil; 180mg) under nitrogen and the
reaction mixture was stirred at 60 for 6 h.
4-(Chloromethyl)-5-methyl-1-(triphenylmethyl)-lH-imidazole (1.119) was
then added and the mixture was stirred at 60 overnight. Acetic acid
(lOmR), water (lOmR) and THF (lOmR) were then added and the resulting
solution was heated at reflux for 6 h. After cooling, 2N sodium
hydroxide (lOOmR) was added and the resulting suspension was extracted
with dichloromethane (3 x lOOmR). The combined, dried organic
extracts were adsorbed onto FCC silica, and FCC eluting with System A
(150:8:1) gave the free base of the title compound (1.089) as a
foam. A small amount of this compound (200mg) was dissolved in
methanol (30mR) and the resulting solution was treated with maleic
acid (58mg). The solution was heated for 10 min., cooled, and dry
1 339022
ether was added to precipitate the title compound (170 mg), m.p.
165-168.
Water Analysis Found 0.22~ w/w - 0.06 mol H20.
Analysis Found: C,64.5; H,5.6; N,10.7;
C24H24N40.C4H404. 0.06 H20 requires C,65.0; H,5.5; N,10.8~.
Examples 4 to 7 were prepared in a similar manner to Example 3.
Example 4
5-Ethyl-2,3,4,5-tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one maleate
5-Ethyl-2,3,4,5-tetrahydro-lH-pyrido[4,3-b]indol-1-one (500mg) was
treated with sodium hydride (60~ dispersion in oil; 138mg) and was
then stirred with 4-(chloromethyl)-5-methyl-1-(triphenylmethyl)-lH-
imidazole (927.5mg) to give the free base of the title compound
(320mg) as a solid. Maleate formation gave the title compound
(380mg), m.p. 175.5-177.
Analysis Found: C,62.1; H,5.7; N,13.0;
C18H20N40.C4H404 requires C,62.2; H,5.7; N,13.2~.
Example 5
2,3,4,5-Tetrahydro-5-(1-methylethyl)-2-[(5-methyl-lH-imidazol-
4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one maleate
2,3,4,5-Tetrahydro-5-(1-methylethyl)-lH-pyrido[4,3-b]indol-1-one
(228mg) was treated with sodium hydride (60~ dispersion in oil; 60mg)
and was then stirred with 4-(chloromethyl)-5-methyl-1-(triphenyl-
methyl)-lH-imidazole (371mg) to give the free base of the title
compound (180mg) as a solid. Maleate formation gave the title
compound (172mg), m.p. 203-205.
Analysis Found: C,62.6; H,6.0; N,12.6;
C19H22N40.C4H404 requires C,63.0;H,6.0; N,12.8~.
Example 6
2,3,4,5-Tetrahydro-5-(phenylmethyl)-2-[(5-methyl-lH-imidazol-4-yl)-
methyl]-lH-pyrido[4,3-b]indol-1-one maleate monohydrate
2,3,4,5-Tetrahydro-5-(phenylmethyl)-lH-pyrido[4,3-b]indol-1-one
" - 1 339n22
-
- 3~ _
(960mg) was treated with sodium hydride (73~ dispersion in oil; 132mg)
and was then stirred with 4-(chloromethyl)-5-methyl-1-
(triphenylmethyl)-lH-imidazole (1.39). The free base of the title
compound (571mg) was obtained as a solid by FCC eluting with System A
(175:8:1). Maleate formation gave the title compound (420mg), m.p.
198-200, t.l.c. (System A, 100:8:1) Rf 0.3.
Example 7
5-(Cyclopentylmethyl)-2,3,4,5-tetrahydro-2-[(5-methyl-lH-imidazol-
4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one maleate
5-(Cyclopentylmethyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-b]indol-1-one
(200mg) was treated with sodium hydride (60~ dispersion in oil; 60mg)
and was then stirred with 4-(chloromethyl)-5-methyl-1-
(triphenylmethyl)-lH-imidazole (280mg). The free base of the title
compound was obtained as a solid (96mg) by FCC eluting with System A
(200:8:1). Maleate formation gave the title compound (60mg), m.p.
81-83, t.l.c. (System A, 100:8:1) Rf 0.20.
Example 8
2,3,4,5-Tetrahydro-5-methyl-2-[(5-propyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one maleate
Sodium hydride (60~ dispersion in oil; 25mg) was added to a stirred
suspension of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-one
(124mg) in dry DME (5mQ) under nitrogen. The mixture was heated at
50 for 7h and then treated with a solution of
4-(chloromethyl)-N,N-dimethyl-5-propyl-lH-imidazole-l-sulphonamide
(165mg) in dry DME (3mQ) and stirring was continued at 50 for 20h.
2N Hydrochloric acid (5mQ) was added and the solution was heated at
reflux for 6h. The solution was poured into 8~ sodium bicarbonate
solution (50mQ) and extracted with dichloromethane (3x25mQ). The
combined, dried organic extracts were evaporated to give a solid
(200mg) which was purified by FCC eluting with System A (200:10:1) to
give the free base of the title compound (58mg) as a solid. This was
dissolved in warm absolute ethanol (5mQ) and treated with a solution
of maleic acid (21mg) in ethanol (0.5mQ). The solvent was removed in
1 339022
- 39
vacuo and the residue was crystallised from ethanol:ether to give the
title compound (58mg), m.p. 137-138.
- Analysis Found: C,62.7; H,5.9; N,12.4;
ClgH22N40.C4H404 requires C,63.0; H,6.0; N,12.8V.
Example 9
2,3,4,5-Tetrahydro-N,N-dimethyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
l-oxo-5H-pyrido[4,3-b]indole-5-carboxamide maleate
A solution of 2,3,4,5-tetrahydro-2-[[5-methyl-1-(triphenylmethyl)-lH-
imidazol-4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one (261mg) in dry DMF
(25mQ) was treated with sodium hydride (60~ dispersion in oil; 30mg)
snd the mixture was stirred at room temperature under nitrogen for 15
min. N,N-Dimethylcarbamoyl chloride (lM solution in DMF; lml) was
then added and the solution was stirred at room temperature for an
additional 15 min. Water (lmR) was cautiously added, and the reaction
mixture was then poured into water (lOOmQ). The resulting mixture was
extracted with ethyl acetate (2 x 50mQ) and the combined organic
extracts were washed with water (2 x lOOmQ) and concentrated to give
an oil. The oil was dissolved in a mixture of water (lOmQ), glacial
acetic acid (lOmR) and THF (lOmR) and the solution was heated at
reflux for 1.5 h. After cooling the solution was basified by addition
of 2N sodium hydroxide (lOOmQ), and the resulting mixture was
extracted with ethyl acetate (2 x 75mQ). The combined, dried organic
extracts were adsorbed onto FCC silica and the free base of the title
compound (llOmg) was obtained by FCC eluting with System A (100:8:1)
as a solid. This was dissolved in dry methanol (lOmQ) and heated with
maleic acid (36mg) on a steam bath for 5 min. On cooling, dry ether
(3mQ) was added to precipitate the title compound (105mg), m.p.
161-163.
Water Analysis Found 1.85~ w/w _ 0.49 mol H20.
Analysis Found: C,57.8; H,5.4; N,14.3;
Cl9H2lNso2-c4H4o4- 0 49 H20 requires C,68.0; H,5.5; N,14.7~.
Examples 10, 11 and 12 were prepared in a similar manner to Example 9
unless otherwise stated.
- - 1 339n22
- 40
Example 10
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-
(methylsulphonyl)-lH-pyrido[4,3-b]indol-1-one maleate
2,3,4,5-Tetrahydro-2-[[5-methyl-1-(triphenylmethyl)-lH-
imidazol-4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one (261mg) was treated
with sodium hydride (60~ dispersion in oil; 30mg) and was then stirred
with methanesulphonyl chloride (lM solution in dry DMF; lml) for 45
min. Deprotection, work-up and purification gave the free base of the
title compound (60mg) as a solid. Maleate formation gave the title
compound (57mg), m.p. 152-155.
Analysis Found: C,53.2; H,4.7; N,11.7;
Cl7Hl8N403S.C4H404 requires C,53.2; H,4.7; N,11.8~.
Example 11
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-(2-
propynyl)-lH-pyrido[4,3-b]indol-1-one maleate
A suspension of 2,3,4,5-tetrahydro-2-~[5-methyl-1-(triphenylmethyl)-
lH-imidazol-4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one (522mg) and
potassium carbonate (276mg) in dry acetone (75mQ) was treated with
propargyl bromide (lM solution in acetone; 2ml) and the mixture was
heated at reflux overnight. After cooling~ excess acetone was removed
in vacuo to give an oil which was partitioned between water (lOOmQ)
and ethyl acetate (lOOmQ). The aqueous phase was washed with ethyl
acetate (50mQ) and the combined organic extracts were concentrated in
vacuo. Deprotectio~, work-up and purification gave the free base of
the title compound (lOOmg) as a solid. Maleate formation gave the
title compound (89mg~, m.p. 202-205, t.l.c. (System A, 100:8:1) Rf
0.29.
Example 12
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-(2-
propenyl)-lH-pyrido[4,3-b]indol-1-one maleate
2,3,4,5-Tetrahydro-2-~[5-methyl-1-(triphenylmethyl)-lH-imidazol-4-
yl]methyl]-lH-pyrido[4,3-b]indol-1-one (1.09) was treated with sodium
hydride (60~ dispersion in oil; 114mg) and was then stirred with allyl
bromide (460mg) for 1 h. Deprotection, work-up and purification gave
- - 41 - 1 339n22
the free bsse of the title compound (380mg) as a solid. Msleate
formation gave the title compound (160mg), t.l.c. (System A, 100:8:1)
Rf 0.3.
Analysis Found: C,63.2; H,5.5; N,12.5;
Cl9H20N40.C4H404 requires C,63.3; H,5.5; N,12.8~.
Example 13
5-Cyclopentyl-2,3,4,5-tetrahydro-2-t(5-methyl-lH-imidazol-4-
yl)methyl]-lH-pyrido[4,3-b]indol-1-one maleate
A solution of 2,3,4,5-tetrahydro-2-[[5-methyl-1-(triphenylmethyl)-lH-
imidazol-4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one (523mg) in dry DMF
(30ml) was treated with sodium hydride (60~ dispersion in oil; 46mg)
and stirred for 15 min. at 21 under nitrogen. Cyclopentyl bromide
(298mg) was then added dropwise, snd the mixture W8S stirred for lh
snd then heated at reflux for 4h. The solution was left st 21 for 2
dsys, and then treated with a mixture of acetic acid (7mR), water
(7mR) and THF (8mR). The resulting solution was heated at reflux for
4h, then basified with 2N sodium hydroxide and extracted with
dichloromethane (3x25mR). The combined extracts were washed with
water (2x50mQ), concentrated in vacuo and purified by FCC eluting with
System A (100:8:1) to give the free base of the title compound (42mg)
as a solid. Maleate formation gave the title compound (38mg), m.p.
180 (decomp.), t.l.c. (System A, 100:8:1) Rf 0.3.
Example 14
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-5-propyl-lH-
pyrido[4,3-b]indol-1-one maleate
A solution of 2,3,4,5-tetrahydro-2-t(5-methyl-lH-imidazol-4-
yl)methyl]-5-(2-propenyl)-lH-pyrido[4,3-b]indol-1-one (248mg) in a
mixture of ethanol (20mR) and 2N hydrochloric acid (0.5mR) was
hydrogenated at room temperature and atmospheric pressure over a
pre-reduced 10~ palladium oxide on carbon catalyst (50~ aqueous paste;
50mg). The mixture was filtered and evaporated in vacuo. The residue
was basified with 2N sodium hydroxide (lOmR) and extracted with
dichloromethane (3x20mR). The combined organic extracts were washed
with water (30mR) and evaporated to give the free bsse of the title
-1 339022
_ - 42
compound (258mg) as a solid. Maleate formation gave the title
compound (345mg), t.l.c. (System A, 100:8:1) Rf 0.4.
Water Analysis Found 1.13~ w/w - 0.28mol H20.
Analysis Found: C,62.1; H,5.9; N,12.5;
Cl9H22N40.C4H404 0.28H20 requires C,62.2; H,6.0; N,12.6%.
Example 15
2,3,4,5-Tetrahydro-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-pyrido-
[4,3-b]indol-1-one maleate
A suspension of 2,3,4,5-tetrahydro-2-[(5-methyl-lH-imidazo1-4-yl)-
methyl]-5-[phenyl(methoxymethyl)]-lH-pyrido[4,3-b]indol-1-one (400mg)
in ethanol (20mR) and glacial acetic acid (5mR) was hydrogenated
overnight at room temperature and atmospheric pressure over a
pre-reduced 10~ palladium oxide on carbon catalyst (50~ aqueous paste;
lOOmg). The reaction mixture was filtered and the residue was washed
with ethanol (lOOmR). The filtrate was concentrated in vacuo to give
an oil, to which was added 2N sodium hydroxide (50mQ). The resulting
suspension was extracted with dichloromethane (2x50mR) and the
combined, dried organic extracts were evaporated to give a solid.
This was purified by FCC eluting with System A (75:8:1) to give the
free base of the title comoound as a solid (240mg) which was then
dissolved in dry methanol (50mR). Maleate formation gave the title
compound (261mg), t.l.c. (System A, 75:8:1) Rf 0.2.
Analysis Found: C,60.3; H,5.2; N,13.8;
Cl6Hl6N40.C4H404 requires C,60.6; H,5.1; N,14.1~.
Example 16
2,3,4,5-Tetrahydro-5-methyl-2-[(I,5-dimethyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one maleate
Sodium hydride (73~ dispersion in oil; 40mg) was added to a stirred
suspension of 2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-
4-yl)methyl]-lH-pyrido[4,3-b]indol-1-one (300mg) in dry DMF (3mQ)
under nitrogen. After 30 min. the suspension was cooled to 0 and
iodomethane (0.076mR) was added dropwise. The mixture was allowed to
reach room temperature, stirred for 1.5h, then poured into water
(30mR) and extracted with dichloromethane (3xl5mR). The combined,
~ 43 1 339022
dried organic extracts were evaporated to give an oil (ca. 545mg)
which was purified by FCC eluting with System A (200:8:1) to give a
solid ~95mg). A portion of this material (90mg) was dissolved in
absolute ethanol (3mQ) and treated with a solution of maleic acid
(35mg) in absolute ethanol (lmQ). The solvent was removed in vacuo
and the residue was triturated with dry ether (3x5mQ) to give the
title compound (122mg), m.p. 178-180.
Analysis Found: C,62.1; H,5.7; N,13.1;
Cl8H20N4-C4H44 requires C~62.3; H,5.7; N,13.2
Example 17
2,3,4,5-Tetrahydro-2-[(lH-imidazol-4-yl)methyl]-5-methyl-lH-
pyrido[4,3-b]indol-1-one dimaleate
A solution of 2,3,4,5-tetrahydro-5-methyl-2-[[1-(triphenylmethyl)-lH-
imidazol-4-yl]methyl]-lH-pyrido[4,3-b]indol-1-one (0.229) in a mixture
of acetic acid, THF and water (1:1:1; lOmR) was heated on a steam bath
for 30 min. The suspension so obtained was diluted with lM
hydrochloric acid (20mQ) and washed with ethyl acetate (3x20mQ). The
acidic aqueous phase was basified with solid sodium carbonate and
extracted with dichloromethane:methanol (9:1; 3x20mQ). The combined,
dried organic extracts were evaporated to give a foam which was
dissolved in methanol (5mQ) and treated with a solution of maleic acid
(0.159) ln methanol (5mQ). The clear solution was evaporated to give
a gum which on trituration with ether afforded the title compound
(0.179) as a solid, m.p. 117-118.
Analysis Found: C,56.1; H,4.3; N,10.5;
Cl6Hl6N40.2C4H404 requires C,56.2; H,4.7; N,10.9
Example 18
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one hydrochloride
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one (1.009) was suspended in ethanol (40ml) and
concentrated hydrochloric acid (l.OOml) was added. The mixture was
warmed to 40 and charcoal (0.259) was added. The resulting
" l-33sn22
- 44
suspension was stirred and warmed for 5 min. and then filtered. The
filtrate was evaporated in vacuo to ca. 20ml and was allowed to cool
to 20. Ether (40ml) was added with stirring over 5 min., end the
mixture was stored at 4 overnight. The resulting precipitate was
filtered off, washed with ether (2xlOml), dried in vacuo at room
temperature for 2h and then at 70 for 7 h to give the title compound
(0.959~, m.p. 288-291.
Analysis Found: C,61.4; H,5.8; N,16.7; Cl, 10.7;
Cl7Hl8N40.HCl requires C,61.7; H,5.8; N,16.9; Cl, 10.7~.
Example 19
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one sulphate
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one (0.819) was suspended in absolute ethanol
(6ml) and was warmed at 50 with concentrated sulphuric acid (0.15ml).
More ethanol (4ml) was added and the mixture was stirred with charcoal
(0.19). The suspension was then filtered and the collected solid was
washed with ethanol (ca. 3ml). The resulting filtrate was stirred for
ca. 1 h at room temperature, tert-butyl methyl ether (lOml) was added
slowly and the mixture was stirred for 20 min. The precipitate was
filtered off, washed with ethanol:tert-butyl methyl ether (1:1;6ml),
then with tert-butyl methyl ether (6ml), and dried in vacuo at 40 for
4 days to give the title compound (0.49), m.p. 205-209.
Analysis Found: C,49.5; H,5.6; N,13.5; S,8.4;
C17H18N4- l lH2504 requires C,49-9; H,5.4; N,13.3; S 8 4
1 339022
- 45
Example 20
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A suspension of 2,3,4,5-tetrahydro-5-methyl-lH-pyrido[4,3-b]indol-1-
one (400mg) in dry DME (50m~) was treated with sodium hydride (60~
dispersion in oil; lOOmg), and the mixture was stirred at 60 under
nitrogen for 6h. 4-(Chloromethyl)-5-methyl-1-(triphenylmethyl)-lH-
imidazole (474mg) was added and the reaction mixture was stirred at
60 under nitrogen overnight. 2N Hydrochloric acid (lOmR) and water
(lOmR) were then added, and the mixture was heated at reflux for 6h.
After cooling, the mixture was basified with 2N sodium hydroxide end
the resulting mixture was extracted with ethyl acetate (2x50m~). The
combined, dried organic extracts were concentrated onto FCC silica
and purified by FCC eluting with System A (150:8:1) to give the title
compound (352mg) as a solid, t.l.c. (System A, 100:8:1) Rf 0.28.
H-N.m.r.: o 2.2 (3H,s), 3.04 (2H,t), 3.62 (2H,t), 3.72 (3H,s~, 4.53
(2H,s), 7.1-7.28 (2H,m), 7.43 (lH,s), 7.47-7.55 (lH,dd), 7.94-8.03
(lH,dd).
- 1 339022
- 46
Example 21
2,3,4,5-Tetrshydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one
A mixture of 2,5-dihydro-5-methyl-2-[[(5-methyl-lH-imidazol-4-yl)-
methyl]-lH-pyrido[4,3-b]indol-1-one (50mg) and 10~ palladium oxide on
carbon catalyst (50~ aqueous paste; 25mg) in absolute ethanol (lOm~)
was heated at 80 in a hydrogen atmosphere at 80 p.s.i. for 24h. The
suspension was filtered and the filtrate was evaporated to give an
oil (49mg) which was purified by short path column chromatography on
silica gel (Merck*7739) eluting with System A (150:10:1) to give the
title compound (8mg) as a solid, t.l.c. (System A, 150:10:1) Rf 0.36.
The lH-n.m.r. data obtained for this material were consistent with
those obtained for the product of Example 20.
Exsmple 22
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one
A solution of 2,3,4,5-tetrahydro-2-[[5-methyl-1-(triphenylmethyl)-lH-
imidazol-4-yl]methyl]-lH-pyrido~4,3-b]indol-1-one (261mg) in dry DMF
(25m~) was treated with sodium hydride (60~ dispersion in oil; 30mg)
and the mixture was stirred at room temperature under nitrogen for 15
min. Iodomethane (0.5M solution in DMF; 2m~) was then added and
stirring was continued for a further 15 min. The reaction mixture was
then poured into water (lOOmR) and the resulting suspension was
extracted with ethyl acetate (2 x 50mQ). The combined organic
extracts were washed with water (2 x lûOm~), dried and concentrated to
give a solid. This was dissolved in a mixture of water (lOmQ), THF
(lOm~) and glacial acetic acid (lOmQ) and heated at reflux for 2 h.
After cooling, residual THF was removed in vacuo and the remaining
3G solution was basified (to pH14) by addition of 2N sodium hydroxide.The resulting suspension was extracted with ethyl acetate (2 x 50mQ)
and the combined, dried organic extracts were concentrated onto silica
(Merck*7385). FCC eluting with System A (100:8:1) gave the title
compound (81mg) as a solid. The lH-n.m.r. and t.l.c. data obtained
for this material were consistent with those obtained for the product
of Example 20.
*Trade Mark
1 33~022
- 47
Example 23
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one
5,6-Dihydro-1-[(5-methyl-lH-imidazol-4-yl)methyl]-4-(2-methyl-2-
phenylhydrazino)-2(1H)-pyridinone (20.0mg) was dissolved in 98~
sulphuric acid (lmQ) and the solution was stirred at 25 for 5 min.
The mixture was cautiously poured into 8X aqueous sodium bicarbonate
solution (60mQ) and extracted with 10~ methanol:dichloromethane
(2x60mR). The combined, dried organic extracts were evaporated in
vacuo to leave an oil which was purified by FCC eluting with System A
(100:8:1) to give the title compound (13.5mg) as a solid. The
lH-n.m.r. and t.l.c. data obtained for this material were consistent
with those obtained for the product of Example 20.
Example 24
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one
A solution of N,N,5-trimethyl-4-~[1,2,3,6-tetrahydro-4-[(2-
iodophenyl) methylamino]-6-oxo-l-pyridinyl]methyl]-lH-imidazole-
l-sulphonamide (264mg) in a mixture of dioxane and acetonitrile (2:1;
200mR) containing triethylamine (2mQ) was irradiated in a Pyrex*
immersion well with a medium pressure 125W mercury lamp at room
temperature for 24h. The reaction mixture was then concentrated onto
FCC silica and purified by FCC eluting with System A (150:8:1) to give
the title compound (87mg) as a solid. The lH-n.m.r. and t.l.c. data
obtained for this material were consistent with those obtained for the
product of Example 20.
Example 25
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-
lH-pyrido[4,3-b]indol-1-one
A solution of N,N,5-trimethyl-4-[2,3,4,5-tetrahydro-5-methyl-1-oxo-1H-
pyrido[4,3-b]indol-2-yl)methyl]-lH-imidazole-l-sulphonamide (86mg) in
2N hydrochloric acid (lûmR) and absolute ethanol (2mR) was heated at
100-110 for 4h. The reaction mixture was then cooled and 2N sodium
hydroxide (50mQ) was added. The resulting solution was extracted with
*Tr a de Mar k
~ ,~
`` - 1 3~9û~
- 48
dichloromethane (2x50mQ) and the combined, dried organic extracts
were concentrated onto FCC silica and purified by FCC eluting with
System A (100:8:1) to give a solid (36mg). This was taken up in hot
ethyl acetate and purified by slow evaporation to give the title
compound (12mg). The lH-n.m.r. and t.l.c. data obtained for this
material were consistent with those obtained for the product of
Example 20.
Example 26
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido~4,3-b]indol-1-one
A solution of N~N~5-trimethyl-4-[2~3~4~5-tetrahydro-5-methyl-l-oxo-lH-
pyrido[4,3-b]indol-2-yl)methyl]-lH-imidazole-l-sulphonamide (401mg) in
a mixture of dioxane (150mR) and acetonitrile (150mR) containing
triethylamine (lmQ) was irradiated at room temperature with a medium
pressure mercury lamp for 24h. The reaction mixture was then
concentrated in vacuo onto FCC silica and purified by FCC eluting with
System A (100:8:1) to give the title compound (203mg) as a solid. The
lH-n.m.r. and t.l.c. data obtained for this material were consistent
with those obtained for the product of Example 20.
Example 27
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A solution of phenylmethyl 5-methyl-4-[(2,3,4,5-tetrahydro-5-methyl-1-
oxo-lH-pyrido[4,3-b]indol-2-yl)methyl]-lH-imidazole-l-carboxylate
(134mg) in a mixture of absolute ethanol and 2N hydrochloric acid
(2:1; 30mR) was heated on 8 steam bsth for 15 min. After cooling, the
solution was concentrated in vacuo to ca. 20mR and diluted with water
(40mR). The mixture was then washed with ethyl acetate (2x40mR) and
the acidic aqueous layer was basified with potassium carbonate
solution. The solution was then extracted with ethyl acetate (3x50mR)
and the combined, dried organic extracts, were concentrated onto FCC
silica and purified by FCC eluting with System A (150:8:1) to give a
solid. This was dissolved in hot methanol and triturated with ether
to give the title compound (69mg). The lH-n.m.r. and t.l.c. data
r~: ~' J'',
" .,. ~ .
-- 1 339022
- 49
obtained for this material were consistent with those obtained for the
product of Example 20.
Example 28
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A solution of 2,3,4,5-tetrahydro-2-[[1-(methoxymethyl3-5-methyl-lH-
imidazol-4-yl]methyl]-5-methyl-lH-pyrido[4,3-b]indol-1-one and
2,3,4,5-tetrahydro-2-[[1-(methoxymethyl)-4-methyl-lH-imidazol-5-
yl]methyl]-5-methyl-lH-pyrido[4,3-b]indol-1-one (34mg) in 49~
hydrobromic acid (2mR) was heated on a steam bath for ca. 3h. After
cooling, the resction mixture was basified by addition of potassium
carbonate solution and extracted with ethyl acetate (3x50m~). The
combined, dried organic extracts were concentrated in vacuo to give
the title compound (6mg) as a solid. The lH-n.m.r. and t.l.c. data
obtained for this material were consistent with those obtained for the
product of Example 20.
Example 29
2,3,4,5-Tetrahydro-5-methy~-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A mixture of 2~3~4~5-tetrahydro-5-methyl-2-[(4-methyloxazol-5-yl)methyl]-1H
pyrido[4,3-b]indol-1-one (lOOmg) in formamide (20m~) was heated at
180 for 24h. The mixture was then cooled, diluted with water (lOOmQ)
and extracted with dichloromethane (3xlOOm~). The combined organic
extracts were concentrated in vacuo and the residue was purified by
FCC eluting with System A (100:8:1) to give the title compound (40mg)
as a solid. The lH-n.m.r. and t.l.c. data for this material were
consistent with those obtained for the product of Example 20.
Example 30
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A solution of 4-[[[(1-methyl-lH-indol-2-yl)ethyl]amino]methyl]-N,N,5-
trimethyl-lH-imidazole-l-sulphonamide (140mg) in dry dichloromethane
(15mR) was cooled to 5 and the mixture was stirred under nitrogen
while phosgene (12~ w/w solution in toluene; lm~) was added dropwise.
,j
- 1 339~22
- 50
The reaction mixture was stirred at room temperature for 2h, aluminium
trichloride (60mg) was powdered and added, and stirring was continued
overnight. After this time methanol (lmQ) was added and the reaction
mixture was adsorbed onto FCC silica and purified by FCC eluting with
System A (150:8:1) to give the protected derivative of the title
compound (42mg), as a solid, identical (by t.l.c. and m.p.) to the
product of Intermediate 27. Deprotection as described in either of
Examples 25 or 26 gives the title compound.
Example 31
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
A mixture of 4-[[[(3-iodo-1-methyl-lH-indol-2-yl)ethyl]amino]methyl]-
N,N,5-trimethyl-lH-imidazole-l-sulphonamide (70mg), triphenylphosphine
(52mg) and palladium acetate (22mg) in tri-n-butylamine (5mQ) and dry
THF (lmR) was heated under an atmosphere of carbon monoxide at 120
for lh. After cooling the reaction mixture was poured into 2N
hydrochloric acid (50mR) and the resulting mixture was extracted with
ethyl acetate (2x50mQ; discarded). The acidic solution was then
basified with 2N potassium carbonate and the resulting basic
suspension was extracted with ethyl acetate (2x50mR). The combined,
dried organic extracts were concentrated in vacuo to give an oil and
residual tri-n-butylamine was removed by distillation to leave a
solid. This was adsorbed onto FCC silica and purified by FCC eluting
with System A (150:8:1) to give the protected derivative of the title
compound (21mg) as a solid, identical (by t.l.c. and m.p.) to the
product of Intermediate 27. Deprotection as described in either of
Examples 25 or 26 gives the title compound.
Example 32
2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl-lH-imidazol-4-yl)methyl]-lH-
pyrido[4,3-b]indol-1-one
5,6-Dihydro-1-[(5-methyl-lH-imidazo1-4-yl)methyl]-4-(2-methyl-2-
phenylhydrazino)-2(lH)-pyridinone (60mg) was dissolved in glacial
acetic acid (4mR). Anhydrous zinc chloride (600mg) was added, and the
mixture was heated at 85 for 1.5h. The cooled mixture was poured into
1 33qO22
8~ aqueous sodium bicarbonate solution (lOOmR) and extracted with
ethyl acetate:methanol (10:1) (2xlOOmR). The combined, dried organic
extracts were evaporated in vacuo to leave a solid which was purified
by FCC eluting with System A (100:8:1) to give the title compound
(26mg). The lH-n.m.r. and t.l.c. data obtained for this material were
consistent with those obtained for the product of Example 20.
1 339022
- 52
The following exam,oles illustrate pharmaceutical formulations
according to the invention, containing 2,3,4,5-tetrahydro-5-methyl-2-
[(5-methyl-lH-imidazol-l-yl)methyl]-lH-pyrido[4,~-b]indol-l-one as the
active ingredient. Physiologically acceptable salts and/or solvates
of this compound, and other compounds of formuls (I) and their
physiologically acceptable salts and/or solvates may be formulated in
a similar manner.
TABLETS FûR ORAL ADMINISTRATION
Tablets may be prepared by the normal methods such as direct
compression or wet granulation.
The tablets may be film coated with suitable film forming
materials, such as hydroxypropyl methylcellulose, using standard
techniques. Alternatively the tablets may be sugar coated.
Direct Compression
Tablet mg/tablet
Active Ingredient û.50
Calcium Hydrogen Phosphate BP* 87.25
Croscarmellose Sodium NF 1.80
Magnesium Stearate BP û.45
Compression weight 90.0û
* of a grade suitable for direct compression.
The active ingredient is passed through a 6û mesh sieve, blended
with the calcium hydrogen phosphate, croscarmellose sodium and
magnesium stearate. The resultant mix is compressed into tablets
using a Manesty*F3 tablet machine fitted with 5.5mm, flat bevelled
edge punches.
*Trade Mark
~, j~,.
- 1 339022
Sub-Lingual Tablet mg/tablet
Active Ingredient 0.5
Compressible Sugar NF - 64.5
Magnesium Stearate BP 0.5
S
Compression Weight 65.0
The active ingredient is sieved through a suitable sieve, blended
with the excipients and compressed using suitable punches. Tablets of
other strengths may be prepared by altering either the ratio of active
ingredient to excipients or the compression weight and using punches
to suit.
Wet Granulation
Conventional Tablet mg/tablet
Active Ingredient 0.5
Lactose BP 153.5
Starch BP 30.0
Pregelatinised Maize Starch BP 15.0
Magnesium Stearate BP 1.5
Compression Weight 200.0
The active ingredient is sieved through a suitable sieve and
blended with lactose, starch and pregelatinised maize starch. Suitable
volumes of purified water are added and the powders are granulated.
After drying, the granules are screened and blended with the magnesium
stearate. The granules are then compressed into tablets using 7mm
diameter punches.
Tablets of other strengths may be prepared by altering the ratio
of active ingredient to lactose or the compression weight and using
punches to suit.
1 339022
_ 54
Sub-Lingual Tablet mg/tablet
Active Ingredient 0.5
Mannitol BP 58.5
Hydroxypropylmethylcellulose 5.0
Magnesium Stearate BP 1.0
- Compression Weight 65.0
The active ingredient is sieved through a suitable sieve and
blended with the mannitol and hydroxypropylmethylcellulose. Suitable
volumes of purified water are added and the powders are granulated.
After drying, the granules are screened and blended into tablets using
suitable punches.
Tablets of other strengths may be prepared by altering the ratio
of active ingredient to mannitol or the compression weight and punches
to suit.
CAPSULES mg/capsule
Active Ingredient 0.5
* Starch 1500 98.5
Magnesium Stearste BP 1.0
Fill Weight 100.0
* a form of directly compressible starch.
The active ingredient is sieved and blended with the excipients.
The mix is filled into size No. 2 hard gelatin capsules using suitabled
machinery. Other doses may be prepared by altering the fill weight an
if necessary changing the capsule size to suit.
_ - 55 ~ I 339022
SYRUP
This may be either a sucrose or sucrose free presentation.
A. Sucrose Syrup mg/5ml dose
Active Ingredient0.5
Sucrose BP 2750.û
Glycerine BP 50û.0
Buffer
Flavour
Colour ) as required
Preservative )
Purified Water BP to 5.Oml
The active ingredient, buffer, flavour, colour and preservative
are dissolved in some of the water and the glycerine is added. The
remainder of the water is heated to dissolve the sucrose and is then
cooled. The two solutions are combined, adjusted to volume and mixed.
The syrup is clarified by filtration.
B. Sucrose-Free mg/5ml dose
Active Ingredient 0.5
Hydroxypropylmethylcellulose USP
(viscosity type 4000) 22.5
Buffer
Flavour
Colour ) as required
Preservative )
Sweetener
Purified Water BP to 5.Oml
The hydroxypropylmethylcellulose is dispersed in hot water,0
cooled and then mixed with an aqueous solution containing the active
ingredient and the other components of the formulation. The resultant
solution is adjusted to volume and mixed. The syrup is clarified by
filtration.
5
1 339û22
-- 56
INJECTION FOR INTRAVENOUS ADMINISTRATION
mq/mQ
Active ingredient 0.05 0.5
Sodium Chloride BP as required as required
Water for Injection BP to l.Om~ l.Om~
Sodium chloride may be added to adjust the ton icity of the
solution and the pH may be adjusted, using acid or alkali, to that of
optimum stability and/or facilitate solution of the active ingredient.
Alternatively, suitable buffer salts may be used.
The solution is prepared, clarified and filled into appropriate
size ampoules sealed by fusion of the glass. The injection is
sterilised by heating in an autoclave using one of the acceptable
cycles. Alternatively, the solution may be sterilised by filtration
and filled into sterile ampoules under aseptic conditions. The
solution may be packed under an inert atmosphere of nitrogen or other
suitable gas.0
METERED DOSE PRESSURISED AEROSOL
Suspension Aerosol mg/metered dose Per can
Active Ingredient micronised 0.050 12.ûmg
Oleic Acid BP 0.020 4.80mg
Trichlorofluoromethane BP 23.64 5.679
Dichlorodifluoromethane BP 61.25 14.709
The active ingredient is micronised in a fluid energy mill to a
fine particle size range. The oleic acid is mixed with the trichloro-
fluoromethane at a temperature of 10-15C and the micronised drug is
mixed into the solution with a high shear mixer. The suspension is
metered into aluminium aerosol cans and suitable metering valves,
delivering 85mg of suspension are crimped onto the cans and the
dichlorodifluoromethane is pressure filled into the cans through the
valves.
_ - 57 - I 339022
Solution Aerosol
mg/metered dose Per can
Active Ingredient 0.05 12.0mg
Ethanol 8P 7.500 1.809
Trichlorofluoromethane BP 18.875 4.539
Dichlorodifluoromethane BP 48.525 11.659
Oleic acid BP, on a suitable surfactant e.g. Span*85 (sorbitan
trioleate) may also be included).
The active ingredient is dissolved in the ethanol together with
the oleic acid or surfactsnt if used. The alcoholic solution is
metered into suitable aerosol containers followed by the
trichlorofluoromethane. Suitable metering valves are crimped onto the
containers and dichlorodifluoromethane is pressure filled into them
through the valves.
Inhalation Cartridges
mg/cartridqe
Active Ingredient (micronised) 0.05
Lactose BP to 25.00
The active ingredient is micronised in a fluid energy mill to a
fine particle size range prior to blending with normal tabletting
grade lactose in a high energy mixer. The powder blend is filled into
No. 3 hard gelatin capsules on a suitable encapsulating machine. The
contents of the cartridges are administered using a powder inhaler.
*Trade Mark
: - 5B - 1 339022
SUPPOSITORY
Active Ingredient 0.5mg
* Witepsol H15 to 1.09
s
* Witepsol+H15 is a proprietsry grade of Adeps Solidus Ph. Eur.
A suspension of the active ingredient is prepsred in the molten
Witepsol and filled, using suitable machinery, into 19 size
suppository moulds.
+Trade Mark
"1 .,