Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
2016765
BACKGROUND OE THE INVENTION
Field of the Invention
The present invention concerns highly water soluble,
stable, crystalline antiviral (including antiretroviral) salts
of 2',3'-dideoxyinosine, 2',3'-dideoxy-2',3'-didehydrothymi-
dine and 2'3'-dideoxy-2'-fluoroinosine, particularly sodium
salts thereof.
Backqround Information
Infectious viral diseases are recognized as an important
medical problem. Progress against infectious viral disease
requires the development of drugs with selective antiviral
activity, while remaining benign to normal cell lines. A
number of antiviral agents currently under study which seem to
possess some selectivity are nucleoside analogs. In general,
these compounds are structural analogs of th~ naturally
occurring nucleosides. Structural modification in either the
purlne or pyrimidine base nucleus and/or the saccharide
component results in a synthetically modified nucleoside
derivative which, when incorporated into a viral nucleic acid
forming process, acts to disrupt further synthesis of viral
nucleic acid.
Effectiveness of these antiviral agents depends on
selective conversion by viral enzymes, but not by host
enzymes, to the corresponding nucleotide analog which is then
-- 1 --
converted to the triphosphate followed by incorporation into
viral nucleic acid. A problem with this antiviral strategy
has been the emergence of certain viral strains whose enzymes
poorly promote phosphorylation of the nucleoside analogs. To
circumvent this problem, intact nucleotide analogs appear to
be potentially quite useful as antivirals for incorporation
into viral nucleic acid.
PCT application WO87/012~4 to Mitsuya and Broder de-
scribes the use of 2',3'-dideoxyinosine for use against AIDS.
EP 273,277 to Lin and Prusoff discloses the use of 2',3'-
dideoxy-2',3'-didehydrothymidineintreatingpatientsinfected
with a retrovirus.
Erik De Clercq, "Potential Drugs for the Treatment of
AIDS", Journal of Antimicrobial Chemotherapv, 23, Suppl. A,
35-46, (1989), describes the use of dideoxynucleoside ana-
logues to inhibit the infectivity and cytopathic effect of
human immunodeficiency virus (HIV).
EP 287,313 discloses 2',3'-dideoxy-2'-fluoroinosine (F-
ddI) and its activity against HIV.
SUMMARY OF THE INVENTIO~
The present invention concerns very highly water soluble,
stable, crystalline salts of2 ,3'-dideoxy-2',3'-didehydrothy-
-- 2
j
~0~67~;S
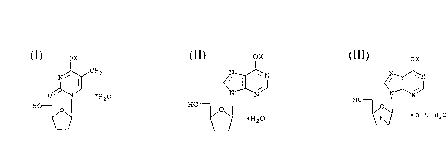
midine ("D4T") of the formula
ox
N~CH3
O~N ~~20
HO
(I)
2 ,3 -dideoxyinosine ("DDI")
ox
<~X~N .
~ E~20 (II)
and 2',3 -dideoxy-2'-fluoroinosine (F-ddI)
H~ ( I I I )
~ O . 5 H20
, wherein X is a cation, e.g., Na, K or Mg.
The present invention also concerns pharmaceutical
compositions comprising an antiviral effective amount of such
salts and a solid, liquid or gaseous physiologically accept-
able diluent.
67~
Still further, the present invention relates to a method
of treating a warm blooded animal, for example, a human,
comprising administering to such warm blooded animal an
antiviral effective amount of one of the aforesaid salts.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 depicts an IR for sodium-2',3'-dideoxyinosine.
Eig. 2 depicts a TGA for sodium-2',3'-dideoxyinosine.
Fig. 3 depicts a DSC for sodium-2-,3'-dideoxyinosine.
Fig. 4 depicts a NMR for sodium-2',3'-dideoxyinosine.
Fig. 5 depicts an IR for sodium-2',3'-dideoxy-2',3'-
didehydrothymidine.
Fig. 6 depicts a TGA for sodium-2 ,3'-dideoxy-2',3'-
didehydrothymidine.
Fig. 7 depicts a DSC for sodium-2',3'-dideoxy-2',3'-
didehydrothymidine.
Fig. 8 depicts X-ray data for sodium-2',3'-dideoxy-2',3'-
didehydrothymidine.
Fig. 9 depicts an NMR for sodium-2',3'-dideoxy-2',3'-
didehydrothymidine.
Fig. 10 depicts an IR for sodium-2',3'-dideoxy-2'-
fluoroinosine.
Fig. 11 depicts an NMR for sodium-2',3'-dideoxy-2'-
fluoroinosine.
-- 4 --
~L676~
Fig. 12 depicts a DSC for sodium-2',3'-dideoxy-2'-
fluoroinosine.
Fig. 13 depicts a TGA for sodium-2',3'-dideoxy-2'-
fluoroinosine.
DETAILED DESCRIPTION OF THE INVENTION
As indicated above, the present invention pertains to
pharmaceutically acceptable non-toxic salts of 2',3'-dideoxy-
inosine, 2',3'-dideoxy-2',3'-didehydrothymidine and 2',3'-
dideoxy-2'-fluoroinosine containing, for example, Na~, Li~, K~,
Ca~ and Mg+'. Such salts may include organic cations and also
those containing an appropriate cation such as an alkali or
alXaline earth metal ion or an ammonium or a ~uaternary amino
ion. The preferred salt is the sodium salt. Metal salts can
be prepared by reacting the metal hydroxide with 2',3'-
dideoxyinosine, 2',3'-dideoxy-2',3'-didehydrothymidine or
2',3'-dideoxy-2'-fluoroinosine. A less soluble metal salt can
be precipitated from the solution of a more soluble salt by
addition of the suitable metal ion.
The compounds o~ this invention have desirable antiviral
activity. They exhibit activity against viruses, for example,
Herpes Simplex virus I, Herpes Simplex virus II, cytomegalo-
virus, Varicella Zoster virus, influenza virus, vaccinia,
polio, rubella, smallpox, cowpox, Epstein-Barr virus, measles
virus, human respiratory virus, papillomavirus and Sinbis
virus, just to mention a few and also against retroviruses,
for example, virus of human ; ~nodeficiency (HIV).
As mentioned above, the compounds of the present inven-
tion are useful active ingredients in human and veterinary
medicine for the treatment and prophylaxis of diseases caused
2~ 61~
by retroviruses. Examples of fi.elds of indication in human
medicine regarding retroviruses are as follows:
(1) the treatment or prophylaxis of human retrovirus
infections;
(2) the treatment or prophylaxis of diseases causad hy
HIV (virus of human immunodeficiency; previously called HTLV
III/LAV or AIDS) and the stages associated therewith such as
ARC (AIDS related complex) and LAS (lymph adenopathy syndrome)
and the immune weakness and encephalopathy caused by this
retrovirus;
(3) the treatment or prophylaxis of HTLV I infection or
HTLV II infection;
(4) the treatment or prophylaxis of the AIDS carrier
state (AIDS transmitter state); and
(5) the treatment or prophylaxis of diseases caused by
hepatitis B virus.
Examples of indications in veterinary medicine are as
follows:
(1) Maedivisna (in sheep and goats),
(2) progressive pneumonia virus (PPV) (in sheep and
goats),
(3) caprine arthritis encephalitis virus (in sheep and
goats),
(4) Zwoegerziekte virus (in sheep),
(5) infectious virus of anemia (of the horse), and
(6) infections caused by cat leukemia virus.
For use against viral infections the compounds of this
invention can be formulated into pharmaceutical pr~parations.
Such preparations are composed of one or more of the inventive
compounds in association with pharmaceutically acceptable
carriers. The reference Reminqton s Pharmaceutical Sciences~
17th Edition, A.R. Gennaro, editor (Mack Publishing Company,
1985) discloses typical carriers and methods of preparation.
The compounds of the invention are administered systemi-
cally to warm blooded animals, e.g., humans. By systemic
administration is intended oral, rectal, and parenteral (i.e.,
intramuscular, intravenous, subcutaneous and nasal) routes.
Generally, it will be found that when a compound of the
present invention is administered orally, a larger quantity of
the reactive agent may be required to produce the same effect
as the smaller quantity given parenterally. In accordance
with good clinical practice, it is preferred to administer the
instant compounds at a concentration level that will produce
an effective antiviral effect without causing any harmful or
untoward side effects.
Therapeutically and prophylactically the instant com-
pounds are usually given as pharmaceutical compositions
comprised of an effective antiviral amount of a compound
according to the'invention and one or more pharmaceutically
acceptable carriers, as stated hereinabove. Pharmaceutical
compositions for effecting such treatment will contain a major
or minor amount, e.g., from 100 to 0.5% of at least one
compound of the present invention in combination with a
pharmaceutical carrier, the carrier comprising one or more
solid, semi-solid, or liquid diluents, fillers and formulation
-- 7 --
adjuvants which are non-toxic, inert and pharmaceutically
acceptable.
Such pharmaceutical compositions are preferably in dosage
unit form; i.e., physically discrete units containing a
predetermined amount of the drug corresponding to a fraction
or multiple of the dose which is calculated to produce the
desired therapeutic response. Other therapeutic agents can
also be present.
Pharmaceutical compositions providing from about 50 mg to
2 grams of the active ingredient per unit dose are preferred
and are conventionally prepared as tablets, lozenges, cap-
sules, powders, granules, aqueous or oily suspensions, syrups,
elixirs, and aqueous solutions. Preferred oral compositions
are in the form of tablets or capsules and may contain
conventional excipients such as binding agents (e.g., syrup,
acacia, gelatin,sorbitol,tragacanth or polyvinylpyrrolidone),
fillers (e.g., lactose, sugar, corn starch, calcium phosphate,
sorbitol, or glycine), lubricants (e.g., magnesium stearate,
talc, polyethylene glycol or silica), disintegrants (e.g.,
starch), buffering agents (inorganic or organic) and wetting
agents (e.g., sodium lauryl sulfate). Solutions or suspen-
sions of an inventive compound with conventional pharmaceuti-
cal vehicles are employed for parenteral compositions, such as
an aqueous solution for intravenous injection or an oily
suspension for intramuscular injection. Such compositions
having the desired clarity, stability and adaptability for
parenteral use are obtained by dissolving from 0.1% to 35% by
weight of an active inventive compound in water or a vehicle
comprising a polyhydric aliphatic alcohol such as glycerine,
propylene glycol, and polyethylene glycol or mixtures thereof.
The polyethylene glycols comprise a mixture of non-volatile,
usually li~uid, polyethylene glycols which are soluble in both
~01~
water and organic liquids and have molecular weights from
about 200 to 1500.
The inventive compounds are prone to acid hydrolysis.
For optimum bioavailability, tablets, capsules and granules
required for oral dosage should be enteric coated, strongly
buffered against gastric acid, or both.
The salts of this patent application are stable, crystal-
line and of very high water solubility (> 300 mg/ml), ideally
suited for high concentration, low volume IV-IM injectables.
Most importantly, the high ~lk~lin;ty (pH 9.5-11) of these new
and novel salts allow for self-buffering against gastric acid.
Thus, less buffer could be required in oral dosage forms.
The salts of the invention are reversibly "immobilized"
in the enolic form, and thus, can be considered as intermedi-
ates for synthesis of varied potentially bio-active deriva-
tives as enol esters (prodrugs) and enol ethers. The esters
and ethers could also function as protecting (blocking)
groups, allowing for synthetic work on other functions or
moieties of the molecules.
Considering the biological activities possessed by the
compounds of the instant invention, it can be seen that these
compounds have antiviral properties, particularly suited to
their use in combating viral infections. Thus, another aspect
of the instant invention concerns a process for treating viral
(including retroviral~ infections in a mammal in need of such
treatment which comprises systemic aAm; n; stration to such
~a~- 1 of an effective dose of an inventive compound.
On the basis of testing, an effective dose could be
expected to be from about 0.1 to about 5 mg/kg body weight
with about 1 to about 4 mg/kg body weight a preferred dosage
_ g _
~; ~ . . - . .
20~6~
range. It is envisioned that for clinical antiviral applica-
tion, compounds of the instant invention will be administered
in the same manner as for the reference drug zidorudine (AZT).
For clinical applications, however, the dosage and dosage
regimen must in each case be carefully adjusted, utilizing
sound professional judgement and consideration of the age,
weight and condition of the recipient, the route of adminis-
tration and the nature and gravity of the illness. Generally
a daily oral dose will comprise from about 150 mg to about 5
grams, preferably 50-1500 mg of an inventive compound adminis-
tered from one to three times a day. In some instances, a
sufficient therapeutic effect can be obtained at lower doses,
while in others, larger doses will be required.
Description of the SDecific Embodiments
The compounds which constitute this invention and their
methods of preparation will appear more fully from a consider-
ation of the following examples which are given for the
purpo~e of illustration only and are not to be construed as
limiting the invention in sphere or scope. All temperatures
are under~tood to be in degrees C when not specified. All
compounds gave satisfactory elemental analyses.
Exam~le 1
Pre~aration of Sodi~m-DDI Monohydrate
ONa
N~J
HO
H20
-- 10 --
i7~
MW: DDI = 236.14
MW: NaOH = 40
1 q DDI = X = 169 mq NaOH = 4.3 ml of lN-NaOH is
236.14 40 40 e~uivalent to 1 g of
DDI for a 1/1 molar
ratio.
1. Add 1 g of DDI to 4.51 ml of a~ueous lN-NaOH (1.05 molar
equivalents with moderate stirring at 10-25~C over a 5
minute interval. A solution (pH 9.5 ~ , or near
solution is obtained.
2. If re~uired, pass the solution through the e~uivalent of
a 0.2 - 0.45 micron Gelman HT-Tuffryn filter to clarify.
Steps 1 and 2 should be completed within 1.5 hours.
3. Add the Na-DDI solution, over a 10-15 minute interval to
50-75 ml of very vigorously stirred isopropanol (alterna-
tively, acetone could be used~. Crystals form. Continue
vigorous stirring (closed system) for 2 hours.
4. Collect the crystals via a suitable vacuum filtration
procedure. Do not draw excess air through the filter-
cake. Tamp the crystalline filter-cake (under vacuum) to
remove any cracks or fissures which may form. Do not
draw excess air through the cake.
5. Wash the filter-cake with three separate 15 ml portions
of isopropanol and then with three separate 20 ml
portions of acetone. Do not draw excess air through the
filter-cake between washings. Remove any cracks or
fissures which may form between washings by tamping.
~016~
6. High vacuum dry the crystals at 24-35~C for 24 hours.
Expected yield: 1-l.lg.
7. If desired, acetone can be totally substituted for the
preferred isopropanol and the modes of addition reversed,
if desired viz, the solvents added to the stirring
aqueous DDI solution over a half-hour interval.
KOH and/or other strong organic and inorganic bases can
be substituted for NaOH to form the corresponding salts.
- 12 -
20167~X
Chemical and Physical Properties Obtained for Na-DDI-H20
1. Water Solubility: >300 mg/ml tpH 9.5-11)
2. Aqueous Solution Stability: 5% loss for 1 week at 50~C;
10% loss for 1 week at 70~C
3. Solid State Stability: No loss for 5 weeks at 70~C
4. Elemental Analysis: ClffHl1N4O3Na H2O (ClOH13N4O4Na)
MW - 276.26
Theory Found
% C 43.5 43.18
% H 4.7 4.6
% N 20.3 20.16
% Na (Ash) 8.3 8.26
% ~2~ KF 6.5 (for monohydrate) 6.57
5. IR: See Fig. 1
6. TGA: See Fig. 2
7. DSC: See Fig. 3
8. X-Ray (film):
X-Ray d-Lines
DDI Na-DDI H2O
d I _ d I/Io
14.57 -100 8.75 - 60
8.39 - 80 7.82 - 80
7.46 - 20 6.21 - 30
6.60 - 10 5.81 - 20
6.00 - 20 5.28 - 10
- 13 -
20:~6~
DDI Na-DDI H20
d I/Io d . I~Io
5.50 - 20 5.00 - 10
5.14 - 20 4.65 - 40
4.79 - 30 4.22 - 50
4.46 - 30 3.89 - 100
3.54 _ go 3.49 - 20
3.21 - 40 3.25 - 20
3.04 - 30 3.09 - 10
2.92 - 50
2.77 - 10
2.61 - 20
9. NMR: See Fig. 4 (no solvents present)
Table I
HPLC Assav Method
Column: IBM Phenyl, S micron, 4.5 x 150 nm
Mobile Phase: 98% 0.015M NH4H2P04 in Milli-Q Water,
pH 7.1 with concentrated NH40H/2%
acetonitrile
Flow Rate: 0.1 mg/ml in water
Approximate Retention
Times: ddI: 10 minutes
Hypoxanthine: 3 minutes
- 14 -
, . .
- . - - .
- . -
':
, - :
,
;~167~
Example 2
Preparation of Sodium-D4T Hydrate
ONa
~3~CH3
.H20
HO~
MW: D4T = 224.2
MW: NaOH = 40
1 g D4T = X = 178.4 mg NaOH = 4.46 ml of lN-NaOH is
224.21 40 40 equivalent to 1 g of D4T
for a 1/1 molar ratio.
1. Add 1 g of D4T to 4.7 ml of aqueous lN-NaOH (1.05 molar
equivalents) with moderate stirring at 15-25~C over a
five minute interval. A solution (pH 9.5 - 11) or near
solution is obtained.
2. Repeat and follow Steps 2-7 as described in Example 1 for
the preparation of Na-DDI H2O.
3. Expected yield of D4T-~2O = 1 - 1.1 g.
Chemical and Physical Parameters Obtained for Na-D4T-H2_
1. Water Solubility: > 300 mg/ml (pH 9.5 - 11)
2. Aqueous Solution Stability: 1 week at 50~C-20% rPm~;n;ng
3. Solid State Stability: No loss for 1 week at 70~C
4. Elemental Analysis: ClOHllNzO4Na H2O (ClOHl3N2O5Na)
MW: 264.22
Theory Found
% C 45.5 45.05
% H 5.0 4.89
- 15 -
2~ 7~
% N 10.6 10.37
% Na 8.7 8.43
% H2O KF 6.8 (for monohydrate) 6.9
5. IR: See Fig. 5
6. TGA: See Fig. 6
7. DSC: See Fig. 7
8. X-RaY: See Fig. 8
HPLC Assay Method
Column: IBM Phenyl, 5 micron, 4.5 x 150 nrn
Mobile Phase: 98% 0.015M NH4H2PO4 in Milli-Q Water,
pH 7.1 with concentrated NH4OH/2%
acetonitrile
Flow Rate: 0.1 ml/minute
Injection Volume: 25 microliter
Run Time: 20 minutes
Wavelength: 254 nm
Temperature: ambient, 20-30~C
Sample Conc.: approximately 0.1 mg/ml in water
Approximate Retention
Times: d4T: 8 minutes
Thymine: 3 minutes
Example 3
Preparation of Sodium F-ddI Hemihydrate
ONa
N~
Ho~
1/~~1 .
0.5 H2O
MW: FDDI = 254.24
MW: NaOH = 40
-- 16 --
. . ~ '
'
- ~ . .
436 mq FDDI = x = 68.6 mq NaOH = 1.72 ml NaOH is
254.24 40 40 equivalent to 436 mg
of FDDI for a l:1
molar ratio
1. FDDI (426 mg) was slurried in 1.8 ml of lN NaOH (256
mg/ml NaFDDI). A pH 9.9 solution was obtained in 2 minutes.
2. Two ml of isopropanol was added. The solution remained
clear.
3. Isopropanol was slowly added with slurring. Heavy
needle-like crystallization started when 15 ml of isopropanol
was added. The mixture was slurried for 10 minutes.
4. Fifteen ml of acetone was added and the mixture was
slurried an additional 15 minutes.
5. The crystals were removed by vacuum filtration, washed
with 15 ml of acetone and 20 ml of ether, and vacuum-dried
(P205 at ~0~C for 2 hours and then at 24~C for 24 hours).
Yield: 430 mg.
A 21 mg sample readily dissolved in 0.05 ml of water (z
400 mg/ml; pH 10.0).
Elemental Analysis: C10HlON4FO3Na
Theory Found
% C 42.0 41.19
% H 3.9 3.73
% N 19.6 18.8
% F 6.6 6.13
% Na 8.0 8.11
% H20 KF3.23 (hemihydrate)3.23
7~;~
X-Ray tfilm):
X-Ray d-Lines
d I/Io
16.20 50
9.40 50
6.91 50
5.81 50
5.40 100
NMR: See Fig. 11. Data consistent with proposed structure.
No acetone or ether present. No a-isomer present.
IR: See Fig. 10.
TGA: See Fig. 13.
DSC: See Fig. 12.
It will be appreciated that the instant specification and
claims are set forth by way of illustration and not limita-
tion, and that various modifications and changes may be made
without departing from the spirit and scope for the present
invention.
- 18 -