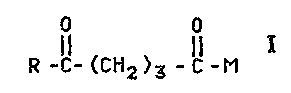Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
2~~~.94~
1
TITLE OE THE INVENTION
Coupling of an anti-tumor to an antibody using glutaraldehyde
preactivated anti-tumor agent.
BACKGROUND OI~ THE IN~IENTION
Chemotherapeutic agents currently used for antitumour
therapy are selected for their toxicity towards rapidly proliferating
cells. Most of them cause undesirable systemic effects such as cardiac
or renal toxicity, marrow aplasia, alopecia, nausea and vomiting.
During the last few years, many authors have tried to eliminate these
1 0 side effects by increasing the availability of the drug to the tumour
site. Enzymes, radioisotopes, DNA, toxins, various macxomolecules,
and antibodies against fibrin or against tumour-specific surface
antigens were bound to drugs in an attempt to increase the selectivity
of the chemotherapeutic agents, or to decrease their toxic effects on
1 5 normal cells ~Rubens R.D., hancet, 2 1974, pp.498-499; Gregoriadis G.
et al., Res. Commun.Chem. 1'athol. Pharm., TO, 1977, pp.351-362).
The targeting of drugs to a tumour by antibodies to surface
antigens may have considerable implications by increasing the
therapeutic index.
2 0 It is recognized that the ideal antineopiastic drug would destroy
cancer cells without adverse effects or toxicities on normal cells, but
no such drug exists. I-Towever, despite the narrow therapeutic index
of many drugs, treatment and even cure are possible in some patients.
Dactinomycin, doxorubicin and daunorubicin are all given
2 5 rapidly intravenously and all cause tissue necrosis if extravasation
2
occurs. When doxorubicin and daunorubicin are given rapidly
intravenously, there is rapid dispersement throughout tissues and
plasma. The r7 t1/2 is 30 min, with detectable plasma levels of
doxorubicin up to 15 h. Both doxorubicin and daunorubicin are
extensively metabolized by the liver, yielding active and inactive
metabolites.
Dactinomycin, doxorubicin and daunorubicin have limited
antitumor activity. Dactinomycin is effective in testicular carcinoma
and sarcomas. Daunorubicin is effective in treating acute leukemia.
1 0 In contrast, doxorubicin is one of the most active antineoplastics ever
identified. In fact it is used to treat acute leukemia, Hodgkin's disease
and non-Hodgkin's lymphomas, small cell and non-small cell lung
cancer, cancers of the breast, ovaries, stomach, thyroid, and bladder,
osteogenic and soft tissue sarcomas, and malignant melanoma. The
1 5 side effects include nausea, vomiting, alopecia, rnyelosuppression,
and dose-dependent cardiotoxicity (>550 mg/m2).
IZelyveld, LJ~P 4,625,019, describes an autopolyrnerized
antitumor agent, that is, daunorubicin is brought in contact with a
bifunctional crosslinking agent, such as glutaraldehyde. A form of
2 0 polymeric product is obtained, which is insoluble in aqueous media
but which, on being resuspended in an aqueous medium in the
absence of glutaraldehyde, gradually releases the antitumor agent in a
soluble farm. This, method mainly consist of mixing together
daunorubicin, an antibody and glutaraldehyde, which can combine in
2 5 three different ways. The conjugates obtained can be any of the
followings:
2~2~.~~~
3
1- 33% Antibody - glutaraldehyde - Daunorubicin
2- 33% Antibody - glutaraldehyde - Antibody
3- 33% Daunorubicin - glutaraldehyde - Daunorubicin
and which only the Antibody - glutaraldehyde - Daunorubicin
conjugate is active. Furthermore, these three possible conjugates can
be linked together by the excess glutaraldehyde in solution to form an
agglomerate, which makes it difficult to isolate the active conjugate.
This is the reason why we refer to an autopolymerized antitumor
agent in this patent.
1 0 This method is not readily reproducible and give an unstable
conjugate product. Unfortunately, this autopolymerized antitumor
agent has the disadvantage of being insoluble in water and thus looses
its specific activity against tumor cells. This insoluble product can
not be used intravenously far a systemic treatment since it is taken up .
1 5 by phagocytic cells such as rnonocytes, macrophage or cells. This
product is not very stable and do not have a very long shelf life.
The pxoblems posed by the administration of antitumor agents
or cytostatic agents are made particularly difficult by the nature of the
illness and vexy high toxicity of the active products.
2 0 It would be highly desirable if the efficiency of the use of
arttitumor agents could be improved so as to allow their gradual
release in the organism, while elearly improving their efficiency and
the patient's eomfort. It would also be highly desirable, if there could
be such an antitumor agent which would be easily produce,
2 S substantially pure, which would also not have a tendency to
polymerized and hence have a long shelf life.
4
~(JMMAR~( ~E TIDE IIOIVEN~I0141
Surprisingly and in accordance with the present invention,
there is provided antitumor agents which overcome the drawback of
the prior art. The already reported techniques for coupling
anthracycline drugs to antibody either cause polymerization or yield a
product which is considerably less active than the free drug. ~LJsing
the method of the present invention, a wide variety of monoclonal
antibodies specific for various tumors are conjugated to other carriers
that could be used for drug targeting.
l 0 In accordance with the present invention, there is provided
novel compounds of the formula I;
0 0
R -IC- (CH2 )3 - C, -M ;
wherein,
- M is selected from the following group consisting of an
1 S hydrogen atom , a peptide residue and a protein residue linked to the
carbon atom via the amino residue of ~-lysine present therein, and
R is an antitumor agent residue such as daunorubicin,
doxorubicin, or epirubicin.
When M is a protein, it can be an antibody which is used to
2 0 target the antitumor agent to the malignant cells and thereby
improving the conditions of such anti-cancer treatments.
The compounds of the present invention are easily produced
and are devoid of significant polymerization since they are
substantially pure. The compounds of the present invention have
5
the ability to provide the full pharmacological activity of the
antitumor agent without the disadvantage normally associated with
said antitumor agent. The improved coupling procedure of the
present invention involved in the production of these compounds of
farmula I is readily reproducible and the resulting compounds are
substiantially stable at 25°C.
Other advantages of the present invention will be readily
illustrated by referring to the following description.
Ih1 THE DIiAi~VIIyIGS
1 0 Figure 1 shows the Cytotoxicity of Equimolar Concentrations of
Free or Monoclonal AntiCEA Bound Daunorubicin on Human
Colon Adenocarcinoma Cells (LoVo).
Figure 2 shows the Cytotoxicity of Equimolar Concentrations of
Free or Monoelonal AntiCEA Bound Daunorubicin on Human MG-
1 5 3 Osteosarcoma Cells.
Figure 3 shows the Cytotoxicity of Equimolar Concentrations of
Free or Monoclonal AntiCEA Bound Daunorubicin on Human
Amnion Cells.
Figure 4 shows the Cytotoxicity of Equimolar Concentrations of
2 0 Free or Monoclonal AntiCEA Bound Daunorubicin on Human
Embryonic Intestine Cells (CCL-6).
~a~~.9~2
6
DETAILED DESCRIPTION OF TI-iE INVENTION
The compounds o'. the present invention correspond to the
general formula I:
0 0
R -IC- (CHI )3 - CI -M I
wherein R and M are as defined previously.
The products of the present invention are prepared as follows:
An antitumor agent R is first reacted with an excess of
glutaraldehyde, which gives an intermediate R-glutaraldehyde of
formula (I), wherein M is an hydrogen atom. This intermediate
1 0 reaction product has a terminal aldehyde group.
The intermediate reaction product is extracted with a solvent
such as dichlorornethane to yield a purified activated R-
glutaraldehyde having a terminal aldehyde group. This activated R-
glutaraldehyde product is then dried on Na-sulfate and dissolved in a
1 5 solvent such as dimethylsulfoxide (DMSO) and then is reacted with
an ~-lysine containing protein or peptide to yield tile final conjugate
R-glutaraldehyde-M. This conjugate is finally obtained by a simple gel
filtration.
Results obtained with activated daunorubiein using this new
2 0 procedure show that the pharmacological activity of the drug could be
saved while limiting the undesirable polymerization of the antibody
normally encountered with bivalent coupling agents. This procedure
is easy and reproducible and reagents are readily available
commercially. The activation of the drug can be accomplished in less
~2~,~~2
7
than 2 hours and the activated drug remains active for a week at
room temperature.
It has also been found that the activated drug can retain its
activity for many months if stored in liquid nitrogen.
All the improvements of the R-glutaraldehyde-M conjugate of
the present invention can be seen from the following reaction
schemes. The method of the present invention is readily
reproducible and gives a R-glutaraldehyde-M conjugate, which does
not have a tendency to polymerize.
1 0 COUPLING OF ANTITUMOR AGENT (R) TO PROTEIN (M)
OLD METHOD PRESENT' METHOD
R + M R + Glutaraldehyde
~, Glutaraldehyde
Separation of R-glutaraldehyde-M R-glutaraldehyde
f 5 conjugate by gel filtration
Diehloromethane
extraction
R-glutaraldehyde-M Purified activated R-glutaraldehyde
Dry on Na-sulfate
2 0 Dissolve in DMSO
Activated R-glutaraldehyde + Peptide or Protein
Separation
by gel filtration
R-glutaraidehyde-M
8
Cell Lines
The cell lines were only used as targets to show that the
conjugate can be directed to the desired sites. As cell lines there may
be used : human embryonic intestine cells (CCL-6), human amnion
cells (CCL-25), human osteosarcoma cells (CRL-1427), human ovarian
carcinoma (CRL-1572), human hepatoma cells (HS-703-T), Mause
melanoma (CRL-6323) and LoVo human adenocarcinoma cells (CCL-
229). These cell lines are readily available from the American Type
Culture Callection under the numbers shown in 'brackets, except for
1 0 the human hepatoma (HS-703-T) which can be obtained from Dr.
Williams C. Parks at Michigan State University, East Lansing,
Michigan, U.S.A.
The LoVo cells produce carcinoembryonic antigen in culture.
The human ovarian carcinoma cells produce alphafoetoprotein. All
cell lines are routinely cultured in RPMI-1640~ medium
supplemented with 10% foetal bovine serum and 100 ug per ml of
streptomycin and 100 ug per ml of penicillin.
Antibody and Peptide
The antibadies and peptides were only used in order to direct
2 0 the conjugates to the desired cell lines used. The antibodies were
obtained through standard monoclonal antibody production
procedures using the above-mentioned cell lines. As antibodies there
may be used . anti-carcinoembryonic manoclonal, anti-
carcinaembxyonic polyclonal antibody, anti-alphafetoprotein
2 5 monoclonal, anti-alphafetoprotein polyclonal antibody, anti-
9
embryonic pre-albumine monoclonal antibody. As a peptide there
may be used: human transferin and lys-bombesin.
°In vitro' Cytotoxicity
In oxdex to evaluate the efficiency of the compounds of formula
~I), the following procedure is used and other methods of in vitro
cytotoxicity could have been used.
The conjugate solution is adjusted to 2% bovine serum
albumin in 0,05 M ammonium acetate buffer. The solution is then
freeze dried and gamma radiated wtih 16 000 rads. Before the assay,
1 0 the dry conjugate is taken up in Dulbecco~ phosphate buffer saline
and added at various concentrations to culture medium.
The cytotoxic activity of daunorubicin and antiCEA conjugate
on the various cell lines is evaluated by inhibition of colony
formation as described in Fmond et al., Anthracyclines, 1983, Ed. G.
1 5 Mathe, Masson Publish N.Y., U.S.A., 105. Briefly, 2,500 cells are added
to 1 ml of RPMI 1640 medium supplemented with 10% foetal
bovine serum in 24 well plates: Cells are allowed to attach for 24
hours, medium is removed and replaced by various test compounds
diluted in growth medium. The tested drugs are incubated with the
2 0 cells for four days in complete growth medium. Each assay is
performed in quadruplicate.
After the growth period, medium is decanted, colonies are fixed
with. formol and stained with crystal violet as reported in Belles-Isles
et al., Brit. J. Cancer, 1980, 41 840.
~0~~.~42
Following the procedures of the present invention, the
following compounds have been obtained:
3t M
_ - hydrogen
- daunorubicin
5 - doxorubicin - hydrogen
- epirubicin - hydrogen
- daunorubicin - antiCEA
- doxorubicin - antiCEA
- epirubicin - antiCEA
1 0 - daunorubicin - antiAFP
- doxorubicin - antiAFP
- epirubicin - antiAFP
- daunorubicin - antiCA-225
- doxorubicin - antiCA-125
1 5 - epirubicin - antiCA-125
- daunorubicin -lys-bombesin
- doxorubicin -lys-bombesin
- epirubicin -lys-bombesin
- daunorubicin - antiEPA
2 0 - doxorubicin - antiEPA
epirubicin - antiEPA
Cytotoxicity of conjugates
Results obtained b~ inhibition of colony formation on the
cytotoxicity of free or antiCEA bound daunorubicin are shown on
2 5 Figures 1 to 4. For any cell line used, we find a dose-response
relationship is determined. The cytotoxicity of antiCEA conjugate for
LoVo cells is 250 ng/ml as compared to 400 ng/ml for the free drug
(Figure 1); the cytotoxicity of antiCEA conjugate for human
osteosarcoma (CRL-1427) cells is significantly higher than the one of
3 0 the free drug (Figure 2).
11
The conjugate is more cytotoxic for normal human amnion
cells and human embryonic intestine (CCL-6) than the free drug
(Figures 3 and 4).
'The LDSQ (lethal dose to kill 50% of the malignant cells) of free
and bound antitumor agent fox the various cell lines is reported in
Table 1 below.
12
TABLE ~
C~~,otoxicitx 1LD5~g _/ml)
of Free
and
Bound
Daunorubicin
For
the
'VariousCell Lanes
%a i.D~
CELL LINES FREE DRUG CONJUGATE decrease
daunorubicin Anti-CEA-daunorubicin
human colon adenocarcinoma 250 37
400
human osteosarcoma 75 24 68
human amnion 77 29 62
1 0 human embryonic intestine142 83 42
Anti-EPA-datinorubicin
human osteosarcoma 160 91 43
Anti-alphafoetoprotein-
daunorubicin
1 $ human amnion 57 49 24
human hepatoma 41 31 24
human osteosarcoma 30 12 60
It can be seen that the dosage required to inhibit 50% of the
malignant Bells for the compounds of the present invention is much
2 0 lower than for the free drug itself. The lowex the LDSO the better the
drug targetting is and hence less sides effects are observed. The LD~o
% decrease is found between 14% to &8% depending on the drug or
the cell line used. Compounds with such an ability to target
antifumor agents without substantially lowering their pharmaceutical
2 5 activity were long waited for.
2~21~4~
13
The method of the present invention for coupling an anti-
tumor agent to an antibody provides molar ratios of anti-tumor to
antibody varying from 0.5:1 to '13:1 as desired. The preferred molar
ratios for coupling anti-tumor agent to antibody being 5:1 to 7:1.
The glutaraldehyde being used is a 25% aqueous solution of
glutaraldehyde in a stoichiometric amount.
The present invention will be more readily understood by
referring to the following Examples which are given to illustrate the
invention rather than to limit its scope.
1 0 Example I
A- Glaxtaraldehyde Activated Daunorubicin
One mg of daunorubicin hydrochloride is dissolved in 4 ml of
0.05 M phosphate buffer at pH 7.5 and 60 ul of 25% glutaraldehyde is
added (grade II, Sigma Chemicals, St. Louis, USA). The mixture is
1 5 stirred for 15 minutes at room temperature and 1 ml of distilled water
is added. The mixture is extracted twice with 5 m1 of
dichloromethane; the organic phases are pooled and treated four
times with an equal volume of 5% lVaHC03 solution containing 15%
glycine: The organic phase is dried with anhydrous sodium sulphate,
2 0 filtered, and the solvent is evaporated under a stream of nitrogen at
room temperature. The dried product is taken in a minimum
volume of dimethylsulfoxide (DMSO).
~02~.~~~
14
1%
The coefficient of extinction is E 495 = 176 in 10% dimethyl-
sulfoxide in 0.05 M phosphate buffer at pH 7.2 for the activated
derivative.
B-Antibody Purification
Monoclonal antiCEA 341-~6-36 antigen is purified from mouse
ascitic fluid by precipitation with 50°lo saturated ammonium sulphate.
The antibody precipitate is dissolved in the original volume of
phosphate buffer at pH 7.2 and dialyzed for 24 hours at 4°C against
1 0 Dulbeccoo phosphate buffer saline. The antibody concentration is
then measured with the Lowry method (Tsukada, Kato, Umemoto,
Takeda, Hara and Hirai, H:J. Nat. Cancer Inst.,19$4, 73, 721).
C- Conjugation Procedure
The glutaraldehyde activated daunorubicin derivative is dissolved
1 5 in 100 ul of dimethylsulfoxide and added with stirring to 500 ug of
monoclonal antiCEA antibody dissolved in 400 ul of phosphate
buffer. The reaction mixture is incubated for 60 minutes at 37°C and
the protein conjugate is separated on Sephadex G-25~ on a PD-10~
column (Pharmacia, Canada), equilibrated with 0.05 M ammonium
2 0 acetate at pH 6.5 containing 0.3 rnM glycine. The conjugate appeaxs in
the void volume of the column. ~'lte conjugation ratio is
determined by spectrophotometry taking an
%D 1%Q
E 495 =176 for daunorubicin and E 28p = 4.5 for monoclonal antibody.
2 5 There is obtained the Daunorubicin- Glutaraldehyde -AntiCE A
monoclonal antibody conjugate.
15
Exaxn lp a II
Proceding as in example I but using antiAFP antibody instead of
the antiCEA monoclonal antibody in step B, there is obtained
antiAFP- daunorubicin conjugate.
$ Example III
Proceding as in example I but using antiCA-125 antibody instead
of the antiCEA monoclonal antibody in step B, there is obtained
antiCA-125- daunorubicin conjugate.
Example IV
1 0 Proceding as in example I but using antiEPA antibody instead of
the antiCEA monoclonal antibody in step B, there is obtained
antiEPA- daunorubicin conjugate.
Example V
Proceding as in example I but using lys-bombesin antibody
1 5 instead of the antiCEA monoclonal antibody in step B, there is
obtained lys-bombesin- daunorubicin conjugate.
Example "VI
Proceding as in example I but using doxorubicin instead of
daunorubicin in step B, there is obtained antiCEA-doxorubicin
2 0 conjugate.
16
Exam ~ VII
Proceding as in example II but using doxorubicin instead of
daunorubicin in step B, there is obtained antiAFP-doxorubicin
conjugate.
Example VIII
Proceding as in example III but using doxorubicin instead of
daunorubicin in step B, there is obtained antiCA-125-doxorubicin
conjugate.
Example IX
1 0 Proceding as in example IV but using doxorubicin instead of
daunorubicin in step B, there is obtained antiEPA-doxorubicin
conjugate.
Example X
Proceding as in example V but using doxorubicin instead of
1 5 daunorubicin in step B, there is obtained lys-bombesin-doxorubicin
conjugate.
Examph XI
Proceding as in example I but using epirubicin instead of
daunorubicin in step B, there is obtained antiCEA-epirubicin
2 0 conjugate.
Example XII
Proceding as in example II but using epirubicin instead of
daunorubicin in step B, there is obtained antiAFP-epirubicin
conjugate.
I7
Exam Ip a ?CIIi
Proceding as in example III but using epirubicin instead of
daunorubicin in step B, there is obtained antiCA-125-epirubicin
conjugate.
Example XIV
Proceding as in example iV but using epirubicin instead of
daunorubicin in step B, there is obtained antiEPA-epirubicin
conjugate.
Example %V
I 0 Proceding as in example V but using epirubicin instead of
daunorubicin in step B, there is obtained lys-bombesin-epirubicin
conjugate.