Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
-1- 2 0 ~ 7
PROCESS FOR THE DEHALOGENATION OF ORGANIC COMPOUNDS
~qeldofTheI~ llion
The present invention relates to a process for the dehalogenation of
organic compounds. More particularly, the invention relates to the
degradation and detoxification of organic compounds cont~ining
halogen atoms.
B~fl~rvu~ld of The Invention
Organic halogenated compounds are obtained in relatively large
amounts as by-products of various industrial processes. Representative
- but not limitative - examples of such compounds are chloro- or bromo-
aromatic compounds, such as polychlorinated and polybrominated
biphenyls (PCBs and PBBs), polychloro heterocyclic compounds, such as
p-hexachlorocyclohexane, and organic solvents such as chlorobenzene.
These products are toxic and hazardous, and must be disposed of in an
effect*e manner.
Disposal of PCBs by incineration is expensive, due to the thermal
stability of these compounds and it is complicated because highly toxic
substances, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin may be emitted
during the process. Only a few specialized incinerators are licensed to
handle such dangerous materials, and the facilities in which these
processes are carried out are accused of causing environmental
pollution [New Scientist, 14.10.89]. Because of these problems, many
efforts have been made in the art to develop effective and safe processes
' - -2- 2024~07
for the chemical degradation of halogenated organic compounds,
especially PCBs.
The PriorArt
Many processes have been provided in the art, including processes for
the chemical treatment and reclamation of oils and liquids cont~ining
various quantities of halogenated hydrocarbons. Processes of this type
can be divided into two main categories. The first type of process
includes the reductive dehalogenation, wherein the organic substances
are treated with hydrogen gas (e.g., US 4,840,721, US 4,818,368, EP
306,164 and EP 299,149), or with other hydrogen donating compounds
such as alkali hydride (GB 2,189,804), hypophosphite (US 4,618,686),
sodium borohydride (US 6,794,928). These processes present several
severe drawbacks, because they usually involve either complicated
hydrogenation processes using explosive gases at high temperatures
and pressures, which must be performed in specially designed reactors,
or they involve the use of special reagents which are unfavored in
industry for economical and safety reasons. Furthermore, HCl is
produced in the process, which, as will be apparent to a skilled chemist,
represents an added complication.
The second type of dehalogenation processes involves the reactions of
metals, alkali earth metals, alkali metals, or compounds of these metals
which are chemically capable of causing the degradation of a carbon-
halogen bond, and which lead to the transformation of the organic
halogen into an inorganic halogen bonded to the metal. Some examples
of such processes are the use of metal or metals compounds such as tin,
21~24~07
-3-
lead, aluminum, chloroaluminates, titanium, aluminum oxide, etc. (EP
277,868, EP 184,342 and US 4,435,379). The most used compounds are
alkali metals and alkali metal compounds such as sodium/sodium
hydroxide (US 4,755,628, CA 1,186,265 and EP 99,961), sodium
naphthalene, sodium polyethylene glycol (EP 140,999 and EP 60,089),
sodium carbonate, bicarbonate, alcoholates, etc. (US 4,631,183 and EP
306,398).
Processes of this type also present considerable drawbacks. For the less
reactive metals, dehalogenation usually involves high temperatures, in
the order of 500-1000~C, which are needed for the cleavage of the stable
carbon-chlorine bond, and for the purpose of bringing the metal into
contact with the organic compound in the form of molten salt, fine
dispersion, etc.
Active metallic compounds, on the other hand, may react at lower
temperatures, in the order of 300-600~C. However, a large excess of
expens*e reagents are needed, and the process involves separation and
purification steps which render it both complicated and expensive.
Metallic compounds capable of inducing the dehalogenation at low
temperatures are very reactive, and therefore their handling and use
are limited by the need for rigorous anhydrous conditions and inert
atmosphere, which are required to avoid the danger of uncontrolled
exothermic decomposition of these compounds. These processes,
therefore, are highly hazardous and expensive.
21~L107
-4-
It is therefore clear that it would be highly desirable to provide a process
for the dehalogenation of waste organic compounds which is both simple
and inexpensive.
SIJMMARY OF THE INVENTION
It is an object of the present invention to provide such a process, which
overcomes the drawbacks of the prior art, which does not require
specially designed equipment, which is simple, inexpensive and non-
hazardous.
The process for the dehalogenation of organohalides according to the
invention comprises reacting an organohalide or a mixture of two or
more organohalides with an alkali hydroxide in an alcoholic solution
and in the presence of a catalytically effective amount of a heterogeneous
transfer hydrogenolysis catalyst.
Detailed De~ ion of The Invention
Preferably, the alcohol found in the alcoholic solution is a lower alcohol.
The preferred alkali hydroxide is sodium or potassium hydroxide,
although of course other hydroxides may be employed.
As to the catalyst, any transfer hydrogenolysis catalyst may be
employed, as long as a catalytically effective amount is provided. A
preferred catalyst would be, e.g., palladium-on-carbon. This catalyst is
usually provided as 5% or 10% palladium-on-carbon.
- 2~24~07
The process of the invention is very convenient as far as temperatures
are concerned. Preferred reaction temperatures are comprised between
50~ and 150~C. Although higher temperatures could be employed, this is
generally not required. Likewise, the reaction can proceed at low
pressures, e.g., atmospheric pressure in an open vessel. Normally it
will be preferred to carry out the reaction in a closed reactor at pressures
lower than 3-4 atmospheres. This, as will be apparent to a skilled
person, is a considerable advantage over the prior art, which requires
considerably higher temperatures and pressures.
Furthermore, the process of the invention does not require anhydrous
conditions and may be conveniently carried out in the presence of high
water concentrations (e.g., 25~o). This is an additional advantage of the
invention, since anhydrous conditions require efforts and expenses.
Preferably, the concentration of- the organohalides in the reaction
mixture is comprised between 0.1-10% of the reaction mixture, and the
alkali hydroxide is present in a stoichiometric excess over the
organohalides. Usually, the concentration of organohalides remz3ining
in the reaction mixture under normal conditions is lower than the
detection limits.
The catalyst used in the reaction can be quantitively recovered after
completion of the reaction, washed with water, and reused in a
subsequent reaction. Therefore, this process is highly efficient also from
the point of view of catalyst usage.
~2~
- -6-
The invention also encompasses a process for the purification and the
reclamation of fluids which are cont~min~ted with organohalides,
which process comprises contacting the fluid to be purified with a
stoichiometric excess of an alkali hydroxide, with respect to the
organohalide, in an alcoholic solution and in the presence of a
catalytically effective amount of a heterogeneous transfer hydrogenolysis
catalyst. Examples of such cont~min~ted fluids are, e.g., mineral oils,
silicon oils, lube oils, gas oils, transformation oils, which may be
contaminated, e.g., with chlorinated organic compounds in a
concentration range of about 0.1-60%.
The above and other characteristics and advantages of the invention will
now be better understood through the following illustrative and non-
limitative examples of preferred embodiments thereof. In the following
examples a commercial dielectric liquid "Pyralene" was used to
determine the effectiveness of the' process. "Pyralene" is a trade name
for a dielectric fluid produced by "Progil Fabrique-France". Pyralene
contains about 40% by weight trichlorobenzene and 60% PC~Bs mixture.
Total chlorine contents in Pyralene is approximately 60%.
Quantification of total PCB contents in Pyralene was performed
according to the method of A. Kuchen, O. Blaster and B. Marek
[Fresenius Z. Anal. Chem., 326, 747 (1987)], using sodium aluminum
hydride for analytical reductive dehalogenation. A value of 22% by
weight of dehalogenated biphenyl was obtained.
-7- 2024~0~
Ex~n};~le 1
0.2 ml, 286 mg Pyralene, 780 mg sodium hydroxide (19.5 mmol) and 30
mg palladium on carbon 10% (0.03 mA palladium) were placed in a
glass reactor and 2.5 ml methanol were added. The reactor was purged
twice with nitrogen, sealed and heated to 100~C for 16 hours. At the
conclusion of the reaction, the catalyst was separated by filtration or
centrifugation, washed with tetrahydrofuran (THF) and methanol, and
the combined filtrates were subjected to GC and HPLC analysis.
No observable remainder of Pyralene were detected. Organic products
were mainly benzene and biphenyl (68 mg, 24.5% weight of starting
Pyralene) indicating total dehalogenation of PCBs, based on
dechlorination quantification. The dehalogenated reaction mixture was
subjected to GC analysis using EC detector. The chromatogram (Fig. 1)
reveals that none of the components of the starting Pyralene remained
in any detectable amount after thè dehalogenation. The chromatogram
of 12 ppm solution of Pyralene (Fig. 2) consists of 8-10 components with
retention times of 43-206 min. Taking into account that 10% of these
components would still be observable, one can conclude that the
concentration of Pyralene components dropped from 120,000 ppm to less
than 1.0 ppm, which means over 99.999% decomposition.
~,m~le 2
Example 1 was repeated but without introduction of catalyst. No change
in the starting Pyralene was observed in GC-EC analysis and no
biphenyl was detected, as observed in GC-FID and HPLC analysis.
-- 2024~û7
-8-
~?~mDle 3
Example 1 was repeated but without nitrogen purging. No residual
Pyralene was observed, indicating less than 1.0 ppm PCBs contents.
Biphenyl (24.5% weight) was determined by GC and HPLC, indicating
total hydrogenolysis of PCBs.
~mple 4
Fx~mple 1 was repeated but 0.25 ml water was introduced in addition to
the methanol. Biphenyl (25% weight) was determined after the reaction
was concluded. GC analysis revealed that no residual Pyralene
components were left. A sole product with low retention time (20 min.)
was detected in a concentration scale 1/10,000 lower than the starting
Pyralene.
le 5
1 ml (1.475 gr) of Pyralene, 3.6 gr sodium hydroxide (90 mmol) and 50 mg
palladium on carbon 10% were placed in a 100 ml flask provided with a
magnetic stirrer and a reflux condenser. 8 ml methanol and 2 ml water
were added and the mixture was heated with stirring to 80~C for 18
hours. At the conclusion of the reaction the catalyst was separated and
the filtrate was analyzed by GC.
No observable remainders of Pyralene were detected by GC-EC detector.
Traces of products with lower retention times were detected. After
completion of the reaction, 385 mg of biphenyl (26%) were found in the
mixture by GC analysis.
9 2024107
~am~le 6
Catalyst from example 5 was washed with water and with THF and then
dried under vacuum at 100~C to constant weight (67 mg). This catalyst
was added together with 1.54 gr Pyralene, 3.6 gr sodium hydroxide, 10
ml methanol and 2 ml water into the reaction flask. The mixture was
heated to 80~C for 18 hours.
At the conclusion of the reaction 308 mg biphenyl (20% weight) were
determined in the mixture, indicating that the recycled catalyst is
effective.
~?lmDle 7
RRclamation of Mineral Oil
~mple 1 was repeated but 0.5 ml mineral oil cont~min~ted with 0.2 ml
(280 mg) Pyralene were added to the dehalogenation mixture. After
completion of the reaction, the oil was separated from the methanol by
means of phase separation. The solid was washed with methanol and
the combined methanol fractions were subjected to GC and HPLC
analysis. The oil phase was dissolved in THF and was subjected to GC
and HPLC analysis.
No observable remainders of Pyralene were detected in the solutions.
Organic products contain mainly benzene and biphenyl (68.6 mg), 24.5%
weight of starting Pyralene.
2~24~07
-10-
Exam~les ~20
Various halogenated compounds were dehalogenated according to the
following procedure.
Halogenated compound (1 mmol), 0.72 gr sodium hydroxide (18 mmol),
and 10 mg 10~o palladium on carbon (0.01 mAtom Pd) were placed in a
glass reactor, and 2.5 ml of methanol were added to this mixture. The
reactor was purged twice with nitrogen, sealed and heated to 100~C for
16 hours.
The results of these reactions are sllmm~rized in Table I below.
Example 21
F~mple 1 was repeated, but with 1 gr (18 mmol) of potassium hydroxide
as a base. After the conclusion of the reaction, no residual Pyralene was
detected by GC (EC detector) analysis. Biphenyl (70.5 mg, 24.5% weight)
was determined by GC and HPLC, indicating a highly efficient
dehalogenation reaction.
mI~le 22
Example 1 was repeated but with 2.6 ml of ethanol as a hydrogen donor
and solvent. After the conclusion of the reaction no observable
remainders of Pyralene were detected in the solution, using GC (EC
detector) analysis. Biphenyl (70.0 mg, 24.8 weight %) and benzene were
the main organic products in the GC and HPLC analysis. An
additional, unidentified minor organic product was eluted at lower
retention time (24 min.) in GC analysis.
2024~0~
-11 -
~,m~le 23
Defluorination of fluoroaromatic compounds also takes place using
similar reaction conditions. For example, 190 mg (1 mmol) 4,4'-
difluorobiphenyl was subjected to the reaction conditions described for
Examples 8 - 20. However, a longer reaction time was needed. When
the reaction was continued for 70 hr., no starting difluorobiphenyl was
detected in the solution. 4-Fluorobiphenyl (17 mg, 0.1 mmol, 10%) and
biphenyl (123 mg, 0.8 mmol) were determined by GC as sole products in
the reaction.
In the described process the environmental considerations are satisfiedwith regard to high efficiency of PCBs destruction and also to the
recycling or disposal of all other reagents involved in the process.
Dehalogenated organic products may be used as a source of heat and
contribute to an additional energy credit of the process. Inorganic
products are harmless salts such as sodium chloride and sodium
formate. The latter is a useful and saleable product, and the resulting
revenue may reduce operating costs.
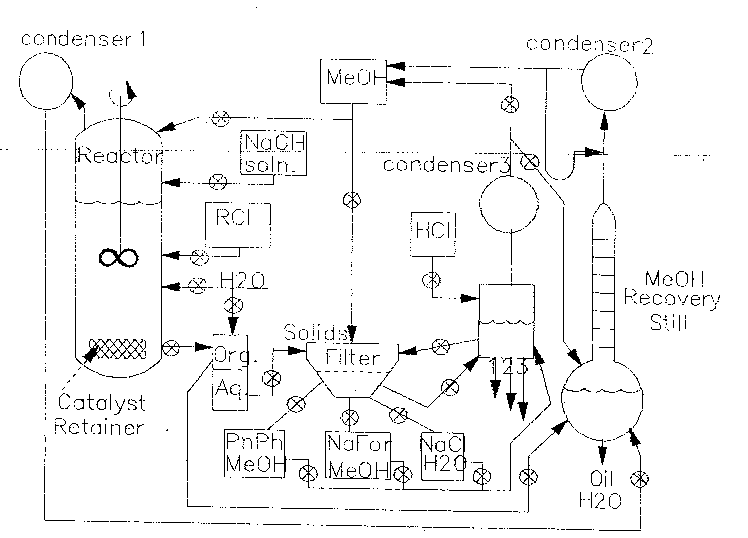
A schematic flow diagram for a dechlorination unit, according to one
process of the invention, is shown in Fig. 3. The work-up process after
the conclusion of the reaction starts with the evaporation of the solvents
through condenser (1) and recycling the methanol using a solvent still
and condenser (2). The non-volatile residue is washed with water into a
liquid-liquid extraction unit, useful for the recovery of purified oils. The
2 0 ~ ~ ~ 0 7
-12-
basic aqueous solution may be reused in the following dehalogenation
process or may be neutralized with hydrochloric acid, followed by
evaporation of water to dryness. Methanol is then added, allowing
separation of soluble sodium formate from sodium chloride, which is
disposed to waste.
The above description and examples have been provided for the purpose
of illustration and are not meant to limit the invention. Many
modifications can be effected in the process of the invention: for
instance, various compounds can be dehalogenated using different
catalysts, solvents and hydroxides, different reaction conditions can be
used, or different fluids can be decont~min~ted, all without exceeding
the scope of the invention.
- -13- 2C2~07
Table I
Example compound untreated treated sample detection
sample/ppm ppm limits/ppm
starting dihalo monohalo
com}?. deriv. deriv.
8 C6H~Cl 45,000 n . d. -- n . d . 10
9 1 ,2-c6H4cl2 60,000 n . d. n . d. 100 10
1,3-C6H4Cl2 60,000 n.d. n.d. n.d. 10
11 1 ,4-c6H4cl2 60,000 n. d. n . d. n . d. 10
12 1,2,3-C6H3Cl3 180,000 n.d. n.d. n.d. 10
13 1,2,4-C6H3Cl3 180,000 n.d. n.d. n.d. 10
14 1,3,5-C6H3Cl3 180,000 n.d. n.d. n.d. 10
hexachloro
cyclohexane 116,000 n.d. -- -- 10
16 1 ,2,3-C6H3Cl3
in mineral oil
(0.5 ml) 180,000 n.d. n.d. n.d. 10
17 1-chloro-
naphthalene 66,000 n.d. -- n.d. 1.0
18 4,4'-dichloro-
biphenyl 86,000 n . d. n. d. n . d . 1.0
19 1 ,4-C6H4Br2 95,000 n . d. n . d . n . d . 10
4,4'-dibromo-
biphenyl 125,000 n.d. n.d. n.d. 1.0
n.d. = not detectable