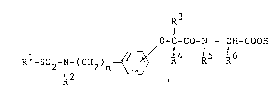Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~32~7~
The present invention is concerned with phenoxy-
alkylcarboxylic acid amides substituted on the phenyl
moiety, with processes for the preparation thereof and
with pharmaceutical compositions which conO~ain these
compounds.
Published Federal Republic of Germany Patent
Specification No. 2809377 describes phenoxyalkyl-
carboxylic acids and some amides with lipid-sinking
and thrombocyte aggregation-inhibiting action which are
substituted in the 4-position.
Published Federal Republic of Germany Patent
Specification No. 36 10 643 describes phenoxyalkyl-
carboxylic acids and some amides with lipid-sinking
and thrombocyte aggregation-inhibiting action which are
substituted in the 2- and 3-position.
Surprisingly, it has now been found that sub~-titutqd
phenoxyalkylcarboxylic acid amides in which the amine
component represents an amino acid display an excellent
lipid-sinking and thromboxane-A2-antagonistic action.
Therefore, according to the present invention,
there are provided new phenoxyalkylcarboxylic acid
amides of the general formula:-
R3
0-C-CO-N-CH-COOH
Rl-SO2-N-(CH2)n ~ R4 R5R6
R2
-2- 2, ~ ~ 2 3 7 3
in which the sulphonylaminoalkyl radical is in the
ortho-, meta- or para-position to the phenoxyalkyl-
carbonamide radical and in which Rl is an aryl,
aralkyl or aralkenyl radical, the aryl moiety o~ which
can, in each case, be substituted one or more times by
halogen, cyano, alkyl, trifluoromethyl or alkoxy, R2
is a hydrogen atom or an alkyl or acyl radicalg R3 and
R4, which can be the same or different, are hydrogen
atoms or lower alkyl radic~ s, _ is 1, 2 or 3, R6 is a
hydrogen atom or an alkyl radical containing 1 to 4
carbon atoms which is optionally terminally substi~uted
by carboxyl, aminocarbonyl or alkoxycarbonyl, by
alkylthio, hydroxyl, phenyl or by imidazol-4-yl and
R5 is a hydrogen atom or, together with R6, forms an
alkylene chain containing 3 or 4 carbon a~oms.
Thus, the phenoxycarboxylic acid amides according
to the present invention are compounds with amino acids
as amine components. Apart fr.om the amide-acids, the
present invention also includes physiologically compatible,
pharmaceutically acceptable sales, esters and amides thereof.
Since the amine components of the phenoxycarboxylic acid
amides contain asymmetrical carbon atoms, the present
invention also includes the pure optical isomers
~enantiomers) and mixtures (racemates) thereof.
The new compounds of general formula I show an
excellent antagonistic action towards thromboxane A2,
as well as towards prostaglandin endoperoxides. They.
_3_ ~32;~73
inhibit the activation of blood pla~elets and of other
blood cells and prevent the constriction of the smooth
musculature of bronchi and blood vessels 9 as well as
the contraction of mesangium cells and similar cells
with contractile properties.
This action makes them valuable therapeutic
agents for the treatment of cardiovascular diseases,
such as acute heart and brain infarct, cerebral and
coronary ischaemia, migrain~ and peripheral arterial
occlusion diseases, as well as venous and arterial
thromboses. Furthermore, an early use thereof can
favourably influence the appearance of organ damages
in shock patients. In addition, they can be used for
the prevention of thrombocyte and leukocyte decrease
in the case of interventions with extracorporeal
circulation and in the case of haemodialysis. An
addition ~hereof to thrombocyte concentrates stabilises
the blood platelets and thus increases the storability
of the preserved materials.
Since thromboxane, in the case of asthma
bronchiale, is a mediator of the inflammatory reaction,
by means of the use of these thromboxane receptor
blockers, the hyperreactivity characteristic for
chronic asthma can be weakened or even overcome.
Furthermore, the new thromboxane receptor
blockers are protectively active in the case of
gastritis and a tendency to ulcers and can thus be used
-4~ 32~7~
for the recidivity prophylaxis thereof. In a model
of experimental acute pancreatitis, the course thereof
can be improved by the use of a thromboxane
antagonist. It is thus to be expected that at least
certain~forms of acute pancreatitis in humans can be
improved in their prognosis by the use of these new
thromboxane antagonists.
In the case of pathological pregnancy, a
disturbance of the equilib~ium of the prostaglandins
is regarded as being the cause. Therefore, by means
of a blockade of the thromboxane and PGF2 alpha
receptors, especially the premature labour pain
activity can be interrupted and, in the case of
pregnancy gestosis and eclampsia, a more favourable
course can be achieved. In addition, the prosta-
glandin-caused symptoms of dysmenorrlloea and of the
premenstrual syndrome can be therapeutically treated.
As aryl radical, alone or in combination with
an alkyl or alkenyl chain, is to be understood, in all
cases, to be an aromatic hydrocarbon radical containing
6 to 14 carbon atoms, especia:Lly a phenyl, biphenylyl,
naphthyl or fluorenyl radical. These aryl radicals
can be substituted one, two or three times in all
possible positions, the substituents being, for
example, halogen, Cl-C6-alkyl, Cl-C6-alkoxy, trifluoro-
methyl or cyano. The phenyl radical is preferred which
can be substituted by halogen, preferably chlorine and
~032'~7~
bromine, methoxy, methyl or trifluoromet~yl.
Of the alkyl and alkoxy substituents in the
aryl, aralkyl and aralkenyl radicals, those are
pre~erred which contain 1 to 4 carbon atoms, espec-
ially the methyl, ethyl, isobutyl and tert.-butyl
radicals, as well as the methoxy radical.
As aralkyl radicals Rl, those are preferred in
which the straight-chained or branched alkylene moiety
contains 1 to 5 carbon ato~,s. Preferred aralkyl
radicals Rl include the phenethyl and 4-chlorophenethyl
radical.
By aralkenyl radicals Rl are to be understood
those in which the alkenylene moiety contains 2 or 3
carbon atoms, ~he styryl and 4-chlorostyryl radical
here being preferred.
By halogen is ~o be understood, in all cases,
fl~lorine, clllorine or bromine.
The alkyl groups R2 are straight-chained or
branched and contain 1 to 16 carbon atoms, methyl and
octyl radicals being preferred.
The acyl radicals R2 are derived from aliphatic
carboxylic acids containing 2 to 16 carbon atoms, from
araliphatic acids and from aromatic carboxylic acids.
Preferred acyl radlcals include acetyl, isobutyroyl,
cinnamoyl, benzoyl, 4-chlorobenzoyl and 4-aminobenzoyl
radicals, as well as n-octanoyl and n-hexadecanoyl
radicals.
__
-6- 2~32~7~
The lower alkyl radicals R3 and R4 can be
9 traigh~-chained or branched and contain 1 to 6,
pre~erably 1 to 4 carbon atoms, the methyl and e-thyl
radicals bein~ preferred.
n`is preferably the number 2.
R5 is preferably a hydrogen atom or, together
with R6, forms an alkylene chain containing 3 or 4
carbon atoms, i.e. a 5- or 6-membered ring is present
which contains a nitrogen a~tom, compounds with a 5;
membered ring here being preferred.
R6 is pre~erably a hydrogen atom or is an alkyl
chain containing 1 to 4 carbon atoms which is optionally
terminally substituted. Preferred substituents
include carboxyl, aminocarbonyl, di-Cl-C6-alkylamino-
carbonyl, 4-substituted piperazin-l-ylcarbonyl (the
4-positioned substituent being benzyl or 4-chloro-
benzyl), Cl-C6-allcoxycarbonyl, Cl-C6-alkylthio,
Cl-C14-aryl-Cl-C4-alkylthio, phenylthio, hydroxyl,
phenyl and 4-imidazolyl. The phenyl nuclei possibly
contained in these substituent:s can also contain 1 or
2 halo~en atoms and preferably chlorine atoms.
Especially preEerred substituents include
carboxyl, aminocarbonyl, diethylaminocarbonyl, 4-benzyl-
and 4-(4-chlorobenzyl)-piperazin-1-ylcarbonyl, methoxy-
and ethoxycarbonyl, methylthio, benzyl- and 4-chloro-
benzylthio, phenyl- and 4-chlorophenylthio, hydroxyl,
phenyl, 4-chlorophenyl and 4-imidazolyl.
_7~ 2 ~ 7 3
In the gro~lp -N - C~l-COOH, those radicals R5 and
R5 R6
R6 are especially preferred, the meaning of which gives
the structure of an essential amino acid. These also
include all possible isomers and the mixtures ~hereof.
The amino acids include, in particular, alanine,
arginine, asparagine, aspartic acid, cysteine, cystine 7
glutamine, glutamic acid, glycine, histidine, isoleucine,
leucine, lysine, methionine~ phenylalanine, proline,
serine, threonine, tryptophane, tyrosine, valine,
homocysteine, homoserine, hydroxylysine, hydroxy-
proline, ornithine, sarcosine, norvaline and 2-amino-
butyric acid.
The esters of the carboxylic acids of the general
formula I can be those with lower monohydroxy alcohols,
for example methanol and ethanol, and those with poly-
hydroxy alcohols, for example glycerol, as well as
those with alcohols which contain other functional
groups, for example ethanolamine.
The amides of the carboxylic acids of general
formula I are those in which the amine component is,
for example, ammonia, a lower dialkylamine, such as
diethylamine, or a hydroxyalkylamine, such as ethanol-
amine or diethanolamine. Other possible amine
components include aikyl-, aralkyl- and aryl-
piperazines.
The present invention also includes the pureoptical isomers (enantiomers) of the compounds of
231~`2~7~
general formula I, as well as mixtures (racemates)
thereof.
Especially preferred compounds of the formula I
are those in which Rl is a phenyl radical or a phenyl
radical substituted once or twice by halogen, methyl,
methoxy or trifluoromethyl, R2, R3 and R4 are hydrogen
atoms 3 n is 2 and the group -N - CH-COOH is the residue
R5 R6
of an essential ami.no acid er an optical isomer or
mixture thereof, the sulphonylaminoalkyl radical there-
by preferably being in the meta-position to the phenoxy-
alkylcarbonylamide radical.
The phenoxycarboxylic acid amides of general
formula I can be prepared
a) by reacting an amine of the general formula:-
HN - (CH2)n ~ (II)
0~1 .
in whîch R2 and n have the above-given meanings,
optionally with intermediate protection of the amino
or hydroxyl group, in per se known manner in any
desired sequence with a sulphonic acid of the general
formula:-
Rl-SO2OH (III)
in which Rl has the above-given meaning, or with a
derivative thereof and with an optionally optically-
active co~pound of the gèneral formula:-
_
~t~2~ 7~;~
R3
X-C-CON - CH-Y (IV)
R4 R5 R6
or.-with a derivative thereof. In general formula IV,
R3 and R4 have the above-given meanings, X is a
reactive group and under Y is a radical -CooR7
(wherein R7 is a hydrogen atom or an equivalent of a
metal ion or a lower alkyl, aralkyl or silyl radical)
or an acid amide radical. ~owever, Y can also be a
radical which, after condensation has taken place, is
converted into an acid amide group or into a -CooR7
radical, whereafter a particular R7 is optionally
converted in per se known manner into ano~her substit-
uent R7 and the compounds obtained are opti.onally converted
into pharmacologically compatible, pharmaceutically acceptable s~lts.
The process according ~o the present invention
is preferably carried out in t.wo steps. The
condensation oE the compounds of general formula II
with sulphonic acids of general formula III or
20 derivatives thereof, on the one hand, and with com- -
pounds of general formula IV, on the other hand., is
preferably so carried out by first blocking one of
the two reactive groups of the compound of general
fo-rmula II with a protective group which can easily
be split off, reacting the compound obtained with a
sulphonic acid of general formula III or with a
derivative ~hereof or with a compound of the general
2 ~ 2 ~i 7 ~3
--10--
formula IV, again splitting off the protective group
and subsequently reacting this reactive intermediate
product with the not yet used compound of general
fo~mula IV or III. A process route is preferred in
which the compound protected on the amino group (i.e.
the compound V) is first reacted with a compound of
general formula IV. After splitting off the
protective group, there then takes place the reaction
with a sulphonic acid of ge~eral formula III or with
a derivative thereof:
R3
Z-N-(CH2)n- ~ ~ , , !
(V) (IV)
Z-W-(CH2)n ~ R3
R 0 - C - CON - CH - Y
R4 R5 R6
(VI)
.
~(CH2)n~ ~ ~\ R3
R
O - C - CON - CH - Y
i4 R5 R6
(VII)
7 a
Rl-SO20H R -S02N-(CH2)n ~ R3
(III) R 0- C - CON - CH - Y
R4 R5 ~6
(VIII)
The symbols R2 n X R3 R4 R5 R6 a d Y d
in the above general formulae have the above-given
meanings and Z is a group suitable for the protection
of amino groups. The preparation of the intermediate
compound of general formul~ VI can also be carried out
in several partial steps: a compound of general
formula V is first reacted with a carboxylic acid of
general formula IX, the carboxyl function of which is
preferably protected by derivative formation:
R3
Z~N~(cH2)n ~/ ~ + X-C-COOH
R2 OH R4
(V) (IX)
Z-N-(cH2) - ~ \\ 3
R 0-C-COOH
R4
(X)
The phenoxyalkylcarboxylic acid of general
formula X is now condensed (possibly after previous
splitting off o~ the carboxyl protective group) with
an optionally optically-active amine of the general
formula :-
~:'` ' ' '' ' .
'
oc~2~7a
HN - CH - Y (XI)
R5 R6
or with a salt o~ the amine. Instead of the free
caxboxylic acids, reactive derivatives are preferably
reacted. The resultan~ compound of general formula VI
is then, as described, freed from the amine protective
group and reacted with a sulphonic acid of general
formula III.
When R3 and R4 are l~wer alkyl radicals, the
phenoxyalkylcarboxylic acids of general formula X can
al~so be prepared by reacting a phenol of general
formula V with a mixture of an aliphatic ketone,
chloroform and an alkali metal hydroxide. This
variant is preferably used for the preparation of
phenoxyisobutyric acid derival:ives:
Z-N-(CH2)n ~ ~ + C;0 ~ CHC13 + M~OH
(V)
_ , Z-~-(cH2)n ~ \\ K,3
R 0-C-COOH
R4
(X)
This process can also be used when Z has already
been replaced by an Rl-SO2- radical.
3 ~ 3
b) A further possibility for the preparation of
compounds of general formula I, of the salts thereof,
as well as of the esters and amides thereof, consists
in reacting a sulphonamide of the general formula:-
Rl-SO2NH (XII)
R2
with an optionally optically-active compound of the
general formula:- ~
X~(CH2)n ~ `~ R3
I (XIII)
0 - C - CON - CH - Y
R4 R5 R6
and possibly converting the group Y of the resultant
compound of general formula VIII into a carboxyl
function or another desired function.
When R~ is to be an acyl radical, the subsequent
acylation of a compound of general formula I is
preferably carried out in which R2 is a hydrogen atom
and the carboxyl function of which is esterified. The
acylation agents used are reactive derivatives of
carboxylic acids and preferably acid chlorides.
The reactive derivatives of sulphonic acids of
general formula III are preferably the halides, as
well as esters. The reaction of the sulphonic acid
halides with compounds of the general formulae II or
VII preferably takes place with the addition of an
~ ?~j 7 .
-14-
acid-binding agent, for example an alkali metal acetate,
sodium hydro~en carbonate, sodium carbonate, sodium
phosphate, calcium oxide, calcium carbonate or
magnesium carbonate. However, this function can also
be undertaken by an organic base, for example pyridine
or triethylamine, in which case, as inert solvent,
there can be used, for example 9 diethyl ether 9 benzene,
methylene chloride, dioxan or an excess of the tertiary
amine. When using an inorg~nic acid binder, as
reaction medium there can be used, for example, water,
aqueous ethanol or aqueous dioxan.
For the reaction of the compound of general
formula II with a compound of general formula IV~ it
has proved to be advantageous first to convert the
lS amino group of the compound of general ormula II into
a protected amino group. Especially preferred are
radicals, such as the benzyloxycarbonyl radical, known
from peptide chemis~ry whlch are easy ~o remove, for
example by hydrogenation. There can also be used
protective radicals such as the phthalimido radical
which, after condensation has taken place between IV
and V, can easily be split off again in known manner
bv the use of hydroxylamine. Sometimes, the splitting
off again can be completely omitted by initially
introducing an Rl-502- radical. ~s reactive compounds
of general formula IV, IX or XIII, those are especially
preferred in which X is the anion of a strong acid,
-15- ~ 3 ~ 5 7 ~
for example of a hydrogen halide or sulphonic acld.
The reactions of these reactive compounds can be very
favoured when using the reaction components in
question in the form of their salts (thus: V in the
form of the sodium or potassium phenolate; XII in the
form of the sodium or potassium salt of the sulphon-
amide). The reaction of the reactive compounds of
general formula IV, IX or XIII with the sodium or
potassium salts of the comp~ounds of general formulae
V and XII takes place in a solvent, for example toluene,
methyl ethyl ketone, dimethylformamide or dimethyl
sulphoxide, preferably with warming.
For the reaction oE carboxylic acids of general
formula X with the amines of general formula XI, it is
preferable to use the carboxylic acids in the form of
reactive derivatives. As such, there can be used acid `
halides, anhydrides, imidazolides, mixed anhydrides of
the carboxylic acid and a chloroformic acid ester,
active esters, for example nitrophenyl esters or
hydroxyphthalimide or hydroxysuccinimide esters. Also
favourable is the condensation between a free
carboxylic acid of general formula X and an amine
component of general formula XI in the presence of an
agent splitting off water9 such as dicyclohexylcarbo-
diimide. In many cases, the acid function of the aminocomponent can be present in salt form but often it is
more preferable to start from the esterified amino-
-16- 2~32~
carboxylic acid of general formula XI, the use of
trialkylsilyl esters thereby having proved to be quite
especially preferable. In the case of the prepar-
ation of trialkylsilyl esters, the amino group is
possibly simultaneously silylated so that N-trialkyl-
silylamino acid-trialkylsilyl esters are obtained.
These can be condensed with,the activated carboxylic
acids of general formula X in the same way as amino
acids not silylated on the ~itrogen. The amino group
of the amino acids or of their esters can possibly also
be reacted in the salt form. As solvents, there can be
used water-alcohol mixtures (e.g. for the reaction of
hydroxysuccinimide esters of the carboxylic acids of
general formula X with the sodium salts of the amino
acids of general formula XI (Y - -COONa)); methylene
chloride, and, in the case of low solubility, dimethyl
sulpho~ide.
c) Compounds of general formula I can also be obtained
by reacting compounds of general formula XIV or their
reactive derivatives
R -SO2-N-(CH2)n ~ \ R3
R2 ~ (XIV)
0-C-COOH
R4
in which Rl, R2, R3, R4 and n have the above-given
meaningsj with an amino acid of general formula XI. -
-17~ ~c-~2~7~3
Some of the compounds of general formula XIV are
described in published Federal Republic of Germany
Patent Specifications Nos. 36 10 643 and 28 09 377 or
can be,prepared according to the processes described
therein.
Reactive derivatives of the compounds of general
formula XIV are preferably acid halides, imida~olides
or mixed anhydrides. Acid chlorides are obtained in
the usual way from the free~acids by reaction with,
for example, thionyl chloride.
, For the preparation of compounds of general
formula VIII by alkylation of a sulphonamide of general
formula XII with a compound of general formula XIII,
those compounds of general formula XIII are preferably
used in which X represents a halogen atom, i~e. a
chlorine or bromine atom. A reaction process is
preferred in which two moles of a sulphonamide of
general formula XI are evaporated to dryness with one
mole of a sodium alcoholate solution. The mixture
obtained is then reacted with one mole of the alkyl
halide of general formula XIII. In this way9 the
formation of dialkylated sulphonamides-i,s substantially
avoided.
The possible subsequent N-alkylation of a
compound of general formula VIII, in which R2 is a
hydrogen atom, can be carried out according to known
_ methods, preferably by reacting it with an alkyl
7 ~
halide or a dialkyl sulphate in the presence of an
acid-binding agent, for example potassium carbonate.
The introduction of an acyl radical R2 into a
sulphonamide of general formula VIII (R2 = H) takes
place under conditlons.such as are usual for the acyl-
ation of amines: reaction with an active carboxylic
acid derivative, for example an acid halide, a mixed
anhydride or an active ester, in an inert solvent in
the presence of a base. As~inert solvent, there can
be used, ~or example, methylene chloride3 benzene,
di,methylformamide and the like.
As substituents Y in compounds of general
~ormula VIII which can be converted into a -CooR7
radical, there can be used, ~or example, a nitrile,
carbaldehyde, hydroxymethyl, aminomethyl or ~ormyl
radical.
The conversion o~ the substituent R7 possibly
to be carried out subsequent to the condensation,
takes place, for example, b~ saponification or hydrolysis of a
carboxylic acid ester (R7 = alkyl) to the correspond-
ing carboxylic acid (R7 = hydrogen) with a Mineral
acid or an alkali metal/alkaline earth metal hydroxide
in a polar solvenc, ~or example water, aqueous
methanol, aqueous ethanol or aqueous dioxan~ at ice-
bath temperature or at a temperature o~ up to 40C.The particular conditions used depend upon the
saponifiability or hydrolyzability of the amide bond
present in the molecule.
-'9- 2~2~7~
On the other hand, however, the carboxylic acids
(R7 = H) can also be esterified in the usual way or
es~ers with a particular radical R7 can be converted
by transesterification into an ester with a different
radical R7. The esterification of the carboxylic acids
is preferably carried out in the presence of an acidic
catalyst, for example hydrogen chloride, sulphuric
acid, ~-toluenesulphonic acid or a strongly acidic ion
exchanger resin. On the other hand, transesterific-
ations require the addition of a small amount of abasic substance, for example o an alkali metal or
alkaline earth metal hydroxide or of an alkali metal
alcoholate. For the esterification of the carboxyl
group or for a transesterification, in principle all
alcohols can be used. Preferred are the lower mono-
hydroxy alcohols, for example methanol, ethanol or
propanol, as well as polyhydroxy alcohols, for example
glycerol, as well as alcohols with other functional
groups, for example ethanolamine and glycerol ethers.
The amides according to the present in~ention
derived from carboxylic acids of general for~ula I are
preferably prepared according to known methods from the
carboxylic acids or reactive derivatives thereof, for
example carboxylic acid halides, esters, azides,
anhydrides or mixed anhydrides, by reaction with amines.As amino components, there can be used, for example,
ammonia, alkylamines and dialkylamines but also amino-
-20- ~ 2~
alcohols, for example, eLhanolamine and 2-amino-pro-
panol. Other valuable amine components include alkyl ,
aralkyl- and arylpiperazines, for example, benzylpiper-
azine.
For the preparation of salts with pharmacologically
or physiologically compatible, pharmaceutically accept-
able organic or inorganic bases, for example, sodium
hydroxide, potassium hydroxide, calcium hydroxide,
ammonium hydroxide, methyl~ucamine, morpholine or
ethanolamine, the carboxylic acids can be reacted with
the corresponding bases. Mixtures of the carboxylic
acids with an appropriate alkali metal carbonate or
hydrogen carbonate can also be considered.
In the specification it will be understood that the
qualification that the salts be "pharmaceu-tically accept-
able" means that the salts have the necessary physical
characteristics, for example, stability, to render them
suitable for formulation into pharmaceutical compositions.
The qualiEication that the sa].ts be "physiologically com-
patible" is to be understood, as extending to salts ofnon-toxic inorganic or organic cations or base components
which have no adverse effects to the extent that such
salts would be unsuitable for administration to living
bodies.
Salts of compounds of formula (I) which are not
pharmaceutically acceptable and physiologically compat-
ible form a useful aspect of the invention of the novel
derivatlves, inasmuch as they can be readily con-
verted, by conventional means, to different salts having
~3257~
-21-
the required physical and chemical charac-teristics to
make them suitable for administration in pharmaceutical
compositions to living bodies.
The pure enantiomers of compounds of general
formula (I) can be obtained by racemate resolution (via
salt formation with optically-active bases) or by
using optically pure amino acids in the syntheses
according to processes a) to c).
For the preparation of~pharmaceutical compositions,
compounds of general formula (I) are mixed in known
manner with appropriate pharmaceutical carrier
substances, aroma, flavouring and colouring materials
and formed, for example, into table-ts or dragees or,
with the addition of appropriate adjuvants, suspended
or dissolved in water or an oil, for example, olive oil.
The compounds of general formula (I) can be
administered orally or parenterally in liquid or solid
~orm. As .injection med.ium, water is preferably used
which contains the stabilising agents, solubilising
agents and/or bufEers usual in the case of in2ectl~on
solutions. Such additives include, for example,
tartrate and borate buffers, ethanol, dimethyl
sulphoxide, comple~ formers (such as ethylenediamine-
tetraacetic acid), high molecular weight polymers
(such as liquid polyethylene oxide) for viscosity
regulation and polyethylene derivatives of sorbit
anhydrides.
Solid carrier materia~s include, for example,
starch, lactose, mannitol, methyl cellulose, talc,
hgihly dispersed silicic acid, high molecular weight
fatty acids (such as stearic acid), gelatine, agar-
agar, calcium phosphate, magnesium stearate, animal
and vegetable fats and solid high molecular weight
polymers (such as polyethylene glycols). Compositions
suitable for oral administration can, if desired,
contain flavouring and sweetening materials.
The administered dosage depends upon the age,
the state of health and the weight of the recipient,
the extent of the disease, the nature of further
treatments possibly carried out simultaneously, the
frequency of the treatments and the nature of the
desired action. Usually, the daily dosage of the
- active compound amounts to 0.1 to 50 mg./kg. of body
25 weight. Normally, 0.5 to 40 and preferably l.0 to
- 20 mg./kg./day in one or more administrations per day
are effec-tive in order to obtain the desired results.
-
~2~
-23-
The physiological activity of the substituted
phenoxyalkylcarboxylic acid amides (I) is demonstrated
and illustrated as follows.
1. TX ~ntagonis-tic Effect of Human Erythrocytes
. _ _ _ ... . _ .
Method
The thrombocyte aggregation is investigated by the
method of Born and Cross (J. Physiol. 168, 178 (1963) in
platelet-rich plasma of healthy blood donors. To inhibit
clotting, the blood is mixe~ with 3.2~ citrate in a
ra-tio by volume 1:9.
, To induce thrombocyte aggregation, U 46619 (Upjohn
~ Co., Kalamazoo, U.S.A.), which is a stable analog
of the prostaglandin endoperoxide PHG2, is used. U 6619
was characterized as a selec-tive thromboxane mimetic
(Coleman et al, Brit. J. Pharmacol. 68, 127 P., 1980).
The aggregation tes-t is carried out in a 4-channel
aggregometer (Profiler( ), of t:he Bio/Data Co., U.S.A.).
The course of the aggregation is followed over a period
of S minutes. At the end of the test, the degree of
aggregation attained is printed ou-t. These values,
which are obtained in the presence of different con-
centrations of the substance to be tested, are used
for the determination of the IC50 for the TX
antagonistic effect. The effectiveness varies
inversely with the IC50 value.
~,;?.:373
-2~-
2. Prevel1tin~ tt~e U 'l6619-Induced Pulmonary Embolism
ethod
Male NMRI mice, with a body weight of 25g, are
used. The test substance is suspended in 1~ methyl-
cellulose and administered to the experimental animals
with the help of a stomach tube. The provocation test
consists of injecting the lethal dose (800-1000 ~ug/kg)
of the thromboxane mimetic (U 46619 of the Upjohn Co.)
rapidly into the tail vein.~l The duration of the
specific antagonistic effect is tested by pretreating
the animals with 25 or 1 mg/kg of the different test
substances and injecting U 46619 after 4 hours. The
survival rate indicates how many of the animals used
have survived the injection of the thromboxane
mimetic. The results are given in the Table below.
-25- ~2~7
~o
- 'O ~ ~ o o ~r o o
a) In ~ er ~
O h O ~1 ~ ~1
E-~ I¢ H
.C
P;
a) ~
r~ tn ~ o o o o o
~ ~ ~ o~ ~ ~0 ~ 00 ~D
~
o o o o o
O O O O O
~rl O ~ ~1 ~I r~l ~I r-l
o~O
O ~ ,,~
~ Lr CO ~
R (~ ,_1 ") ~ u~ Lo
::~ X
~ 5~2.
Preferred in the meaning of the present
invention are, apart from the compounds of general
formula I mentioned in the following Examples, as well
as the,esters, amides and salts thereof, also the
following compounds:
1. N-[3-[2-(phenylsulphonylamino)-ethyl]~phenoxy-
acetyl]-glycine
2. N-[2-[2-(4-chlorophenylsulphonylamino) ethyl]-
phenoxyacetyl]-alanine ~
3. N-[3~[2-(4-methylphenylsulphonylamino)-ethyl]-
~ phenoxyacetyl]-alanine
4. N-[3-[2-(3- or 4-trifluoromethylphenylsulphonyl-
arnino)-ethyl]-phenoxyacetyl]-2-aminobutyric acid
5. N-[3-[2-(4-chlorophenylsulphonylamino)-ethyl]-
phenylacetyl]-alanine 4-be.nzylpiperazide
6. N-[3-[2-(4-methoxyphenylsulphonylamino)-ethyl]-
phenoxyacetyl]-alanine ethyl ester
7. N-[3-[2-(2,4- or 2,5-dichlorophenylsulphonylamino)-
ethyl]-phenoxyacetyl]-alanine
8. N-[4-~2-(3,5-dichlorophenylsulphonylamino)-ethyl]-
phenoxyacetyl]-L-alanine
9. N-[4-[2-(4-trifluoromethylphenylsulphonylamino)-
ethyl]-phenoxyacetyl]-L-alanine
10, N-[4-[2-(4-chlorophenylsulphonylamino)-ethyl]-
phenoxyacetyl]-L-serine
11. N-~4-[2-(4-trifluoromethylphenylsulphonylamino)-
ethyl]-phenoxyacetyl]-L-serine
, ~ . . . .
' ;'
-~7- 2 ~32 ~ 7 ?
12. N-~4-[2-(4-methylphenylsulphonylamino)-ethyl]-
phenoxyacetyl]-glycine.
The following Examples show some of the numerous
prQcess variants which can be used for the synthesis
S of the new compounds according to the presen~ invention.
However, they are not to represent a limitation of
the subject matter of the present inven~ion.
Example l.
N-[3-[2-(4-Chlorophenylsulphonylamino)-eth~l]-
ph~noxYacetyl]-~lycine.
a)'3-[2-(4-Chlorophenylsulphonylamino)-ethyl]-
phenoxyacetyl chloride.
A mixture of 30 g. (81 mmole) 3-~2-(4-~hloro-
phenylsulphonylamino)-ethyl]-phenoxyacetic acid,
23.5 ml. (0.32 mole) thionyl chloride and 3 drops of
dimethylformamide is stirred for 2 hours at 6GC.~
whereafter excess thionyl chloride is distilled off in
a vacuum. The residue obtained is dissolved in
anhydrous diethyl ether and clarified with activated
carbon. After evaporation, an oil is obtained which
slowly crystallises through. It is stirred with iso-
hexane, filtered off with suction and dried. Yield
26.9 g. (94% of theory); m.p. 75 - 77C.
b) Title compound.
To an ice-cold solution of 0.8 g. (lO mmole)
glycine and 15 ml. 2N aqueous sodium hydroxide solution
- is slowly added dropwise a solution of 3.52 g. of the
-28- ' 2 ~2'?~
acid chloride obtained according to a) in 40 ml.
methylene chloride and subseq~lently allowed to post-
react ~or 2 hours a-t 0C. The reaction mix~ure is then
acidif~,ed with dilute hydrochloric acid and the weakly
acidic phase extracted with ethyl acetate. The
organic phase is extracted twice with lN hydrochloric
acid, washed with water and dried with anhydrous sodium
sulphate. The crude product obtained after evaporation
is recrystallised from nitromethane. Yield 2.7 g.
(63% o~ theory); m.p. 134 - 135C.
Example 2.
N-[3-[2~(4-Bromophenylsulphonylamino)-ethyl]-
phenoxyacetyl]-L-alanine.
a) N-Tritnethylsilyl-L-alanine trimethYlsilyl ester.
To a suspension o~ 7.12 g. (80 mmole) L-alanine
in 120 ml. anhydrous tnethylene chloride is added drop-
wise, with stirring, 20.8 ml. trirnethylchlorosilane,
followed by brie~ly heating to reflux temperature, a,
clear solution thereby being obtained. Then, with
gentle cooling, 22.4 ml. triethylamine are added drop-
wise thereto, with stirring, in such a man~ler that the
reaction mixture does not boil too vigorously. Subse-
quently, the reaction mixture, a thick suspension, is
heated for 5 minutes to re~lux temperature, cooled,
filtered through a pressure filter and subsequently
washed with dry methylene chloride. The product
- obtained is dried in a current of nitrogen and further
-29- ~ 7 ~
worked up,in crude form.
b) Title compound.
To a solution of 9.28 g. (20 mmole) 3-[2-(4-
bromophenylsulphonylamino)-ethyl]-phenoxyacetyl-
imidazole in 75 ml. anhydrous tetrahydrofuran is addedat -5C., with the'exclus~on of moisture, 5.14 g.
(22 mmole) of the trimethylsilyl compound obtained
according to a). The reaction mixture is allowed to
come to ambient temperatur~ in the course of 3 hours
and subsequently the tetrahydrofuran is distilled off
in a vacuum. The residue is stirred with 2N hydro-
chloric acid at ambient temperature and then extracted
several times with methylene chloride. The combined
or~anic phases are extracted with an amount of 2N
aqueous sodium hydrogen carbonate solution suf~icient
for salt formation and the ext:ract is acidified. The
precipitated product is filte~ed oEE with suction,
dried and recrystallised from nitromethane. Yield
6.2 g. (60% of theory); m.p. 191 - 192C.
Example 3.
N-[3-[2-(4-Chlorophenylsulphonylamino)-ethyl]-
phenoxyacet~l]-L-alanine.
a) N-[3-[2-(Benzyloxycarbonylamino)-ethyl]-
phenoxyacet~l]-L-alanine.
To a 40C. warm solution of 5.0 g. (15.8 mmole)
3-[2-(benzyloxycarbonylamino)-ethyl]-phenoxyacetic
,acid in 25 ml. anhydrous dimethyl sulphoxide are added
-3~- 2 g~ ~3~'3 7 ~
2.56 g. (15.8 mrnole) carbonyl-bis-imidazole, followed
by stirring for a further 30 minutes at 40C. Subse-
quently, 1.7~ gA (15.8 mmole) L-alanine sodium salt
is added thereto, the reaction mixture is kept for
3 hours at 60C., cooled, stirred into a mixture of
ice and so much hydrochloric acid that the mixture
reaches a pH value of about 2. The precipitated oil
is extracted with methylene chloride. After washing
the methylene chloride phas~e with an aqueous solution
of sodium hydrogen carbonate and water, it is dried
with anhydrous magnesium sulphate and evaporated~
There are obtained 5.21 g. (82% of theory) of product
in the form o~ a colourless oil.
Alternative synthesis for this compound:
~ mixture of 22.0 g. (81 mmole) 3-[2-(benzyloxy-
carbonylamino)-ethyl]-phenol, 200 ml. butanone and
33.5 g. very finely pulverised dry potassium carbonate
is maintained ~or 1 hour at reElux temperature, 300 mg.
potassium iodide and 17.3 g. ~89.3 mmole) N-(2-chloro-
acetyl)-L-alanine ethyl ester are then added thereto
and the reaction mixture is maintained at reflux
temperature for 16 hours. Subsequently, the reaction
mixture is filtered off with suction and the filter
cake is then washed with hot butanone. The combined
butanone phases are evaporated, the oily residue is
taken up in methylene chloride, the methylene chloride
phase is washed with water, dried over anhydro~s
-31- ~3~70
magnesium sulphate and evaporated in a vacuum. Yield:
practically quantitative.
b) Title compound.
~mixture of 8.0 g. (20 mmole) o~ the benzyloxy-
carbonyl compound obtained according to a), 75 ml.
methanol, 11.0 ml. 2N hydrochloric acid an~ about 2 g.
10% palladium-carbon is hydrogenated for 12 hours at
ambient temperature and 6 ~ar pressure until the
necessary amount of hydrogen has been taken up. The
catalyst is then filtered off with suction and the
methanol is distilled off in a vacuum. To the residue,
which consists of the crude hydrochloride of N-[3-(2-
aminoethyl)-phenoxyacetyl]-L-alanine, are added 75 ml.
of water and so much 2N aqueous sodium hydroxide
solution that a pH of 10 i.s reached. With stirring at
0C., there are now added thereto 3.16 g. 4-chloro-
benzenesulphochloride in small portions and, by the
dropwise addition of further dilute aqueous sodium
hydroxide solution, care is taken that a pH of 10 is
maintained. Subsequently, thc reaction mixture is
further stirred for 2 hours a-t 35C., acidified,
filtered off with suction and the product is dried and
recrystallised from nitromethane. Yield 4.7 g. (71%
of theory); m.p. 175 - 177C.
Exam~le 4.
_-[3-[2-(4-Chlorophenylsulphonylam~no)-ethyl]-
~ phenoxyacetyl]-L-al nlne.
-32-
~257~
a) N-[3-[2-(~-chlorophenylsulphonylamino)-ethyl]-
phenoxyacetyl]-L-alanine ethyl ester~
To an ice-cold mixture of 2.2 g. (14 mmole) L-
alanine ethyl ester hydrochloride, 4.3 g. triethylamine
and 100 ml. methylene chloride is slowly added dxopwise,
with stirring, a solution of 5.0 g. (14 mmole) 3-[2-
(4-chlorophenylsulphonylamino)-ethyl]-phenoxyacetyl
chloride in 25 ml. methylene chloride. Subsequently,
the reaction mixture is sti~red for 1 hour at 0C. and
then for 1 hour at 25C., shaken out with dilute hydro-
chloric acid and water, dried over anhydrous sodium
sulphate and finally evaporate.d. Yield 5.1 g. (77% of
theory) of a colourless oil.
b) Title compound.
A mixture of 2.8 g. t6 mmole) of the ester
obtained according to a), 25 ml. ethanol and 15 ml. 2N
aqueous sodium hydroxide solution i8 S tirred for 5
minutes at ice-bath temperature, thereafter diluted
with 25 ml. ice water and acidified with dilute hydro-
chloric acid. After filtering off with suction, drying
and recrystallising from ni~romethane, the yield is
1.9 g. (72% oE theory); m.p. 175 - 177C. The
product is identical with that obtained according to
Example 3.
Example 5.
N-[3-[2-(~-Chlorophenylsulphonylamino)-ethyl]
phe oxyacetyll-L-methionine.
2 ~ 7 ~
To a solution of 8.0 g. (20 mmole) 3-[2-(4-
chlorophenylsulphonylamino)-ethyl]-phenoxyacetic acid
in 35 ml. dimethyl sulphoxide is added at 40C., with
stirring, 3.5 g. (20 mmole) car~onyl-bis-imidazole,
foam formation thereby taking place. After stirring
for 1 hour at 40C., 4.07 g. (24 mmole) L-methionine
sodium salt are added thereto and the reaction mixture
then maintained for 18 hours at 60C. It is then
cooled, poured on to an ice~hydrochloric acid mix~ure
and the greasy product obtained taken up in ethyl
ac,etate. The ethyl acetate phase is extracted with an
aqueous solution of sodium carbonate and the latter
again washed with ethyl acetate. The aqueous phase is
acidi~ied, ~iltered o~f with suction and the product
is washed with water, dried and recrystallised from
nitromethane. Yield 8.3 g. (77% of theory); m.p.
138 - 139C.
In analogous way, from 3-[2-~4-chlorophenyl-
sulphonylamino)-ethyl]-phenoxyacetic acid, there are
obtained the.following compounds:
2. with L-histidine sodium salt:
N-[3-[2-(~-chlorophenylsulphonylamino)-ethyl]-phenoxy-
acetyl]-L-histidine hydrochloride.
yield 34%; m.p. 100 - 103C.
The oily product separating out after pouring into
an ice-hydrochloric acid mixture is here triturated
with an acetone-ether mixture (1:12 v/v) until it
-3~- 2~ 2~7~
crystallises. ~t is tl~en quickly filtered off with
suction (hygroscopic) and dried over phosphorus
pentoxide.
3. with L-2-aminobutyric acid sodium salt:
S N-~3-[2-(4-chlorophPnylsulphonylamino)-ethyl]-phenoxy-
acetyl]-L-2-aminobutyric acid.
Yield 61% of theory; m.p. 147 - 150C. (recrystallised
from nitromethane).
4. with L-asparagine sodiu~ salt:
N-[3-[2-(4-chlorophenylsulphonylamino)-ethyl]-phenoxy-
acetyl]-L-asparagine.
- Yield 62% of theory, m.p. 133 - 135C. (recrystallised
~rom nitromethane).
5. with L-phenylalanine sodium salt:
N-[3-[2-(4 chlorophenylsulphol~ylamino)-ethyl]-phenoxy-
acetyl]-L-phenylalanine.
Yield 63% of theory; m.p. 158 - 159C. (recrystallised
from aqueous ethanol).
6. with L-proline sodium salt:
N-[3-[2-(4-chlorophenylsulphonylamino)-ethyl]-pheno~y-
acetyl]-L-proline.
Yield 93% of theory; m.p. 114 - 116C. (recrystallised
from ethyl acetate).
7. with L-serine sodium salt:
N-[3-[2-(4-chlorophenylsulphonylamino)-ethyl]-phenoxy-
acetyl]-L-serlne~
Yield 49% of theory; m.p. 146 - 148C. (recrystallised
from nitromethane).
-35- 2~3~7 ~
8. with L-nGrvaline sodium salt:
N-[3-~2-(4-chlorophenylsulphonylamino)-ethyl]-phenoxy-
acetyl]-L-norvaline~
Yield 72% of theory; m.p. 126 - 128C. (recrystallised
from nitromethane).
Example 6.
N-[3-[2-(4-Chlorophenylsulphon~lamino~-ethyl]-
pheno~yacetyl]-D-alanine.
Into a 4QC. warm sol)~tion of 10.0 g. (27 mmole)
3-[2-(4-chlorophenylsulphonylamino)-ethyl]-phenoxy-
ac,etic acid in 30 ml. dimethyl sulphoxide are intro-
duced 4.3~ g. (27 mmole) carbonyl-bis-imidazole. The
mixture is allowed to react for 10 minutes, 3176 g.
(27 mmole) 4-nitrophenol are added thereto, ~ollowed
by stirring for 10 minutes. 3.0 g. (27 mmole) D-
alanine sodium salt are then introduced, ~ollowed by
st irr ing for 3 hours at 60C. Afte r c o o 1 ing, the
reaction mixture is stirred into an ice-hydrochloric
acid mixture. The precipitated oil is taken up in
ethyl acetate, the organic phase is washed with water,
dried over anhydrous sodium sulphate and evaporated.
The oily residue obtained crystallises after the
addition of isohexane. Yield 8.0 g. (67% of theory);
m.p. 163 - 166C.
Example 7.
In a process method analogous to Example 6,
there were prepared ~rom 4-[2-(phenylsulphonylamino)-
-36- ~332~7~
ethyl]-phenoxyacetic acid
a) and L-alanine:
N-[4-[2-(phenylsulphonylamino)-ethyl]-phenoxyacetyl]-
L-alanine
Yield '62~ of theory; m.p. 164 - 165C.
b) and D-alanine:
N-[4-[2-(phenylsulphonylamino)-ethyl]-phenoxyacetyl]-
D-ala~ine
Yield S4% oE theory; m.p. 152 - 153C.
The Patent Specifications referred to herein are
more fully identified below:
Federal Republic of Germany Offenlegungsschrift
tLaid Open Patent Specification) 36 10 643~ E.-C.
Witte et al, published (Laid Open) October 1, 1987,
assigned to Boehringer Mannheim GmbH.
Federal Republic of Germany Offenlegungsschrift
(Laid Open Patent Specification) 28 09 377, published
(Laid Open) Setpember 13, 1989, E.-C. Witte et al~
assigned to soehringer Mannheim GmbH.
O