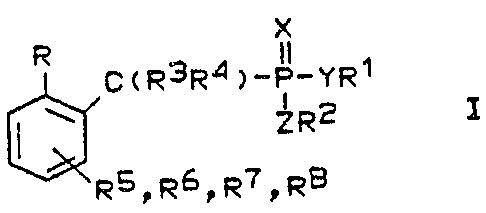Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~~;.~~ ~,~
HOECHST ARTIENGESELLSCHAFT HOE 89/F 398 Dr. WN
Description
2-Formylbenzylphosphonic acid derivatives, their prepar
ation and their use for the treatment of diseases caused
by viruses
The invention relates to novel 2-formylbenzylphosphonic
acid derivatives, to processes fox the preparation of
these compounds, to pharmaceutical agents which contain
the active compounds according to the invention and to
30 their use as medicaments, in particular for the treatment
of diseases caused by viruses.
In order to treat diseases caused by viruses, various
preparations have hitherto been employed, such as, for
example, nucleoside analogs, amantadine, pyrophosphate
analogs or immunomodulators (2d.J. Wood, A.PrI. Geddes, The
Lancet, 1987, 17.89). Some phosphonic acid derivatives are
known which exhibit antiviral activity. These include
compounds such as phosphonoformic acid (PFA), phosphono-
acetic acid (PAA), methylenediphosphonic acid (NlDP) and
tetrazolephosphonic acids (S. M. Roberts, NATO ASI Ser.,
Ser. A 143, 1988, 37; D.W. Hutchinson, M. Naylor, Nucleic
Acids Res., 13, 1985, 8519). PFA has a wide antiviral
spectrum, but causes some toxic side effects, which have
hitherto prevented development to the antiviral medi-
cament (~I.J. Wood, A.M. Geddes, The Lancet, 1987, 1189).
It is known of ortho-phosphonyloxy-acetophenor~s deriv-
atives that they are especially active against picorna-
viruses (EP 21,000).
Diana et al. (J. kIed. Chem. 27, 1984, 691; DOS 2,922,054)
rep-~rt on a class of compound of the type
~___________0
H
- 2 -
in which A is an aromatic ring and C is a phosphonate or
a p-ketophosphonate in which A and C are separated from
one another by means of a bridge of 3-8 methylene
groups (B). From this class of compound, arylalkylphos-
phonic acids having methylene bridges of more than 5
carbon atoms showed antiviral activity against herpes-
viruses. However, arylalkylphosphonic acids having
methylene bridges of less than 5 carbon atoms do not show
antiviral activity. The substitution of the aromatic
radical of these compounds is carried out, in Diana
et al., essentially by means of a 2-chloro-, 4-methoxy-
or 4-carbethoxyphenoxy group.
Benzylphosphonic acids have hitherto not been described
as active antiviral compounds (J.C.H. Mao et al., Anti-
microb. Agents Chemother. 27, 1985, 197).
Surprisingly, it has now been found that 2-formylbenzyl-
phosphonic acid derivatives have antiviral activity.
The invention therefore relates to a compound of the
formula I
x
R C(R3R4)-P-YR~
ZR2 I
~RSDR61R7DR6
in which
R is an aldehyde group or a group which can be converted
into an aldehyde,
R1 and RZ, which may be the same or different, are a
straight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight-chain or branched alkenyl or
alkynyl group having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to 8 carbon atoms, hydrogen, sodium, potassium,
calcium, magnesium, aluminum, lithium, ammonium or
triethylammonium or R1 and R2 tagether form a cyclic
diester having 2 to S carbon atoms in the ring,
/S>~'~fl~ r~
- 3
R3 and R", which may be the same or different, are a
straight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight.-chain or branched alkynyl or
alkenyl group having 2 to 20 carbon atoms, a cycloalkyl
group having 3 to 8 carbon atoms, an alkoxy group having
1 to 4 carbon atoms, hydrogen, fluorine, chlorine,
bromine or iodine,
Rs, R6, R' and R8, which may be identical or different, are
a straight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight-chain or branched alkenyl or
alkynyl group having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to 8 carbon atoms, an alkoxy group having 1 to 4
carbon atoms, hydrogen, fluorine, chlorine, bromine,
iodine, a cyanide, hydroxyl or phenyl group or the
radical of the formula Ia
0
~- 0-R19 I a
Rl9 is a straight-chain or branched alkyl group having 1
to 20 carbon atoms, a straight-chain or branched alkenyl
or alkynyl group having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to B carbon atoms, hydrogen, sodium, potassium,
calcium, magnesium, aluminum, lithium, ammonium or
triethylammonium and
X, Y and Z, which may be identical or different, are
oxygen or sulfur
or a prodrug form of the compound of the formula 1.
~, compound of the formula 1 in which
R1 and RZ are an alkyl group having Z to 10 carbon atoms,
a..n alkenyl or alkynyl group having 2 to 10 carbon atoms,
hydrogen or an aralkyl group having 7 to 16 carbon atoms,
R3 and R4 are an alkyl group having 1 to 4 carbon atoms,
an alkenyl or alkynyl group having 2 to 4 carbon atoms or
hydrogen,
R5, R6, R' and RB are chlorine, bromine, methoxy or
hydrogen and
~~n3.r 3 a
t~
X, Y and Z are oxygen,
is preferred.
By the term "prodrug form of the compound of the
formula I", compounds are meant which are converted into
a compound of the formula I in which R is an aldehyde
group, en route to the site of action. Tn the article by
H. Bundgaard (Design of Prodrugs, x.985, pp. 1 - 92,
Elsevier-Verlag), the term "Prodrug form" is defined and
illustrated by examples.
The notation alkyl group having 1 to 10 carbon atoms is
to be understood as meaning, for example, the following
radicals: methyl, ethyl, propyl, isopropyl, n-butyl,
sec.-butyl, tert.-butyl, 2,2-dimethyl-1-propyl, n-pentyl,
n-hexyl, n-heptyl, n-octyl, n-nonyl and n-decyl. By the
natation alkenyl group having 2 to 10 carbon atoms, the
following compounds, for example, are meant: ethenyl,
propenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl,
nonenyl or decenyl. By the notation alkynyl group having
2 to 10 carbon atoms, the following compounds are meant,
for example: ethynyl, propynyl, butynyl, pentynyl,
hexynyl, heptynyl, nonynyl, octynyl or decynyl. lAn
aralkyl group having 7 to 16 carbon atoms is understood
as meaning the following radicals, for examples phenyl-
methyl, phenylethyl, phenylbutyl, phenylpropyl, phenyl-
pentyl, phenylhexyl, phenylheptyl, phenyloctyl, phenyl-
nonyl or phenyldecyl. A cycloalkyl group having 3 to
8 carbon atoms is understood as meaning radicals ~euch as
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclo-
heptyl or cyclooctyl. Alkoxy groups having 1 to 4 carbon
atoms are radicals such as a~ethoxy, ethoxy, propoxy,
isopropoxy, n-butoxy, s~:c.-butoxy or tert.-butoxy.
The invention furthermore relates to a process for the
preparation of the compound of the formula I in which R
is an aldehyde group, which comprises reacting the
compound of the formula II
..~ ~ r_7 re a a
- 5 -
0 t°I
R
w C R4 1 I
~RS,R6'R''RB
in which
R3 and R~, which may be the same or different, are a
stxaight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight-chain or branched alkynyl or
alkenyl group having 2 to 20 carbon atoms, a cycloalkyl
group having 3 to 8 carbon atoms, an alkoxy group having
1 to 4 carbon atoms, hydrogen, fluorine, chlorine,
bromine or iodine,
R5, R6, R' and Re, which may be identical or different, are
a straight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight-chain or branched alkenyl or
alkynyl group having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to 8 carbon atoms, an alkoxy group having 1 to 4
i5 carbon atoms, hydrogen, fluorine, chlorine, bromine,
iodine, a cyanide, hydroxyl or phenyl group or the
radical of the formula Ia
C_ Q_Rlg Ia
R18 is a straight-chain or branched alkyl group having 1
to 20 carbon atoms, a straight-chain or branched alkenyl
or alkynyl group having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to ~ carbon atoms, hydrogen, sodium, potassium,
calcium, magnesium, aluminum, lithium, ammonium or
triethylammonium and
T is chlorine, bromine, iodine, methylsulfonate,
phenylsulfonate or tosylsulfonate,
with the compound of the formula III
- 6 - ~~.1;~ ~~x r~~
X-R~
P Y-R~ III
' Z-R2
in which,
R1 and R2, which may be the same or different, are a
straight-chain or branched alkyl group having 1 to 20
carbon atoms, a straight-chain or branched alkenyl or
alkynyl graup having 2 to 20 carbon atoms, an aralkyl
group having 7 to 20 carbon atoms, a cycloalkyl group
having 3 to B carbon atoms, hydrogen, sodium, potassium,
calcium, magnesium, aluminum, lithium, ammonium or
triethylammonium or R1 and RZ together form a cyclic
diester having 2 to 6 carbon atoms in the ring,
R9 is a straight-chain, or branched alkyl group having 1
to ~I carbon atoms and
X, Y and Z, Which may be identical or different, are
oxygen or sulfur.
Z5 The invention furthermore relates to a process for the
preparation of the compound of the formula T in which R
is a group which can be converted into an aldehyde, which
comprises reacting the compound of the formula I in which
R is an aldehyde in such a way that a group which can be
converted into an aldehyde is formed.
The term "group which can be converted into an aldehyde"
is understood as meaning radicals which are converted
into an aldehyde en route to the site of action
(H. ~undgaard, Design of I?rodrugs, 1985, pp. 1 - 92,
Rlsevier-Verlag).
In particular, the aldehyde group can be derivatixed in
such a way that the compound of the formula I is formed
in which R is a group, which can be converted into an
aldehyde, of the formula Ib, Ic or Id
9( r% i !" I
LD
- ? -
R14 R15
R10 R11 R13~_~R15 My
0\ ~0 R12_ 1
CH CI-~ CH
i
Ib Ic Id
in which R1° and R11, which may be identical or different,
are a straight-chain or branched alkyl group having 1
to 10 carbon atoms or R1° and R1~ together form a cyclic
acetal having 2 or 3 carbon atoms in the ring,
R12 to R'°, which may be identical or different, are a
straight-chain or branched alkyl group having 1 to
carbon atoms or an aryl group having 6, 10 or
14 carbon atoms,
V is oxygen or sulfur,
10 M is a hydroxyl group, a straight-chain or branched alkyl
group having 1 to 10 carbon atoms, an aralkyl group
having 7 to 20 carbon atoms, an aryl group having 6, 10
or 14 carbon atoms or a radical of the formula Ie or If
v 0
_~H_C_R17 oC_C_R18
Ia if
in which Rl' is a straight-chain or branched alkyl group
having 1 to 10 carbon atoms, an amino, pyridine, or an
aryl group having ~, 10 or 14 carbon atoans and
R18 is an amino group, a pyridine group, a straight-chain
or branched alkyl group having l to 10 carbon atoms, an
aryl groug having 6, 10 or 14 carbon atoms or an aralkyl
group having ? to 20 carbon atoms.
The synthesis of the compound of the formula I in which
R is an aldehyde group is carried out by reacting the
compound of the formula II with the compound of the
formula III, expediently at temperatures between 100 end
250°C, preferably between 120 and iS0°C (US 4,299,b15;
Houben-t~eyl, Methoden der Org. Chemie (Methods of Organic
Chemistry), Vol. ~LII/1, page 423, Thieme-Verlag,
Stuttgart; Iiouben-bdeyl, Methoden der Org. Chemie (Methods
ll~~j,~..,.)
~liJ:. t~ ~N
- g -
of Organic Chemistry), Vol. E2, page 300). The reaction
can be carried out in a suitable solvent, such as hexa-
methylphosphoramide (HI~A), dimethylylformamide (DMF),
dimethyl sulfoxide (DMSO), N,N'-dimethyl-N, N'-propylene-
urea (DMPU) or N,N'-dimethyl-N, N'-ethyleneurea (DMEU).
The reaction can also be carried out without solvent.
Purification is carried out by generally customary
methods, preferably by chromatography on silica gel using
suitable eluents, by distillation or by recrystallization
from suitable solvents.
The compounds of the formula II and III can be pregared
in a manner known per se. The conversion of the phos-
phonic acid diesters into their monoesters, and also into
the corresponding free acids or their salts is carried
out, for example, by boiling with dilute hydrochloric
acid (Houben-Weyl, Methoden der Organischen Chemie
(Methods of Organic Chemistry), Vol. BII/1, 1963), or by
reaction with trimethylbromosilane (C. E. McRenna,
J. Schmidhauser, J.C.S. Chem. Commun., 1979, 739).
Purification is carried out by recrystallization in
suitable solvents or by chromatographic methods, prefer-
ably by ion exchange chromatography using suitable
eluents. The desired salt forms can also be obtained by
ion exchange chromatography.
The synthesis of a prodrug form of the compound of the
formula I can be carried out, for example, by derivatiz-
ing the aldehyde group in the compound of the formula I
in such a way that compounds such as oximes, thiosemi-
carbazones, carboxylic acid hydrazones, Schiff's bases,
oxazolidines, thiazolidines or acetals are formed. For
this purpose, the compound of the fonaula I sn ~ahich R is
an aldehyde gxoup ca:~ be reacted with ~:he compound of the
formula IVa, IVb and/or IVc, IVd or IVe
- 9 -
OH
HO-(CH2)-CH2 R10-OH R11-OH
1 Vo I Vb 1 Vc
R15 R1A
R16 ~~R13 M-NHS
HV R12-NH
IVd~ IV~
in which R1° to R18, M and v have the meaning mentianed and
n is 1 or 2.
Other prodrug forms are foamed in an analogous manner by
the methods described in Bundgaard. The compounds of the
formula T derivatized on the aldehyde group can be
converted in vitro and in vivo into the active, anti-
. virally active form (aldehyde form) (H. Bundgaard, Design
of Prodrugs, 1985, 1 - 92, ~lsevier-verlag). The convey
a sion into the active form can be carried out, for ex
ample, by hydrolysis in aqueous solution or by enzymatic
catalysis in or en route to the sil~e of action.
A test for activity of chemotk~erapeutics for HILT infer--
tions in man causes difficulties, since rno infection
model in laboratory anim~is yet exists. Infection with
other retroviru~es therefore has to be resorted to fox
testing chemotherapeutics. ~n this ~a~e, the infection of
the mouse with the griend leukemia virus has been chosen.
I»or this purpose, normal I lalaoratory mice
(N~!tRT = Naval Medical Research Institute) were infected
by intravenous injection with mouse serum containing
Friend leukemia virus. zn the untreated control animals,
a distinct enlargement of the spleen and liver developed
as a symptom of the infection in the course of 2 weeks.
Treatment was carried out over 10 days, starting 48 hours
after the infection. On the 14th day of the experiment,
the animals were sacrificed and dissected. The spleen was
6', 3 r'a r. y~ y o
~° 3s
r/ ~J r~ n,! 5 3
- 10 -
removed and weighed. As a measurement parameter of the
therapeutic activity, the weight of the spleen of the
treated animals was related to that of the untreated
infection control.
In the case of uninfected adult laboratory mice (20 -24 g
body weight), the spleen weighed about 1 $ of the body
weight or less, while in the case of infected animals,
the spleen attained about 10 $ of the body weight at the
end of the experiment.
The compound of the formula I in which R is an aldehyde
group possesses useful pharmacological properties, in
particular an antiviral action and in particular against
diseases caused both by DNA and RNA viruses, particularly
against diseases which are caused by Herpes simplex
virus (HSV I), myxoviruses, Friend leukemia virus (FLV)
or human immunodeficiency virus (HIV). The compounds
according to the invention are therefore suitable for
combating various diseases caused by viruses, such as
respiratory tract disease, diseases of the skin, the'
eyes, the central nervous system, AIDS and AIDS-related
conditions, such as AIDS-related complex (ARC), genera-
lized lymphadenopathy (GL), AIDS-related neuralgic
conditions (such as mental deficiency or trophic para-
peresis), anti-HIVantibody-positive conditions, Raposi
sarcoma or thrombopenic purpura.
The compound of the formula I and/or its prodrug form can
either be used as a pharmaceutical alone or mixed with
physiologically tolerable auxiliaries or excipients in
effective amounts. It can be administered, for example,
orally in a dose ~f 1 to 500 mg/kg/day, preferably 5 to
50 .vg/kg!day. The administration for parentei~al~ rectal
or topical use or as an aerosol is carried out, for
example in an amount of 0.5 to 500 mg/kg/day, preferably
of 2 to 100 mg/kglday. The compound of the formula I
and/or its prodrug form are expediently administered in
dosage units which contain at least the effective amount
~di !~~ ~ 1t %~
~~ C% %~1 9~ 9
- 11
of the compounds according to the invention, preferably
25 to 6000 mg, particularly preferably 100 to 1000 mg.
These values relate to an adult human having a weight of
75 kg. These dosage units can also be administered
several times per day. The dosage can also be increased
in severe cases. In many cases, however, lower amounts
axe also sufficient. For combating diseases which are
caused by RNA or nNA. viruses, the following are suitable
in particular
diethyl 2-formylbenzylphosphonate,
2-formylbenzylphosphonic acid di(triethylammonium) salt,
monoethyl 2-formylbsnzylphosphonate triethylammonium
salt,
diethyl 2-formylbenzylphosphonate thiosemicarbazone,
diethyl 2-formylbenzylphosphonate nicotinic acid
hydrazone or
diethyl 2-(3,4-dimethyl-5-phenyloxazolidin-2-yl)benzyl-
phosphonate.
The compound of the formula I according to the invention
and/or its prodrug form can also be administered in
combination with other substances, in particular anti-
viral agents and immunostimulators, such as interferons.
The compound of the formula I and/or its prodrug form are
referred to as the active compound in the following.
The invention furthermore includes the use of the active
compound in the preparation of pharmaceuticals which are
employed for the treatment and prophylaxis of the above
mentioned diseases. The invention furthermore relates to
pharmaceuticals which contain one or more active
compounds.
The pharmaceuticals are prepared by processes which are
known per se and familiar t~ those skilled in the art. As
a pharmaceutical, the active compound is either employed
as such or preferably in combination with suitable
pharmaceutical auxiliaries or excipients in the form of
tablets, coated tablets, capsules, suppositories,
f' 'r~
t cf
- 12 -
emulsions, suspensions or solutions, the content of
active compound being up to about 95 $, advantageously
between 10 and ?5 ~.
In addition to solvents, gel-forming agents, suppository
bases, tablet auxiliaries and other active compound
carriers, suitable auxiliaries or excipients for the
desired pharmaceutical formulation are also, for ~xample,
antioxidants, dispersants, emulsifiers, defoaming agents,
flavor modifiers, preservatives, solubilizers or
colorants.
The active compound can be administered orally, parenter-
ally, intravenously or rectally, intranasal administra-
tion as an aerosol being preferred in particular in
addition to oral administration.
For a form for oral use, the active compound is mixed
with the additives suitable for this purpose such as
excipients, stabilizers ~r inert diluents and brought
into a suitable form for administration, such as tablets,
coated tablets, hard gelatin capsules, and aqueous or
oily solutions by the customary methods. Inert excipients
which can be used are, for example, gum arabic, magnesia,
magnesium carbonate, potassium phosphate, lactose,
glucose or starch, in particular cornstarch. In this
case, preparation can be carried out both as dry and as
moist granules. Oily excipients or solvents which are
suitable are, for example, vegetable or anianal oils, such
as sunflower oil or cod-liver oil.
For subcutaneous or intravenous administration, the
active compound is brought into solution, suspension or
emulsion with the substances suitable for this purpose,
such as solubilizers, emulsifiers or other auxiliaries.
suitable solvents are, for example, physiological saline
solution, alcohols, for example ethanol, propanol,
glycerol, sugar solutions such as glucose or mannitol
solutions or a mixture of solvents.
j9!j<b Js
i
- 13 -
The following examples serve to illustrate the invention
further.
Example 1
Preparation of diethyl 2-formylbenzylphosphonate (A)
47.9 g (0.31 mol) of 2-chloromethylbenzaldehyde were
heated to 160°C together with 51.5 g (0.31 mol) of
triethyl phosphate. Ethyl chloxide distilled off during
the course of this . The product was purified by fraction-
al distillation.
Yield: 64.5 g (81 ~); b.p.: 130°C/0.3 mm;
1H-NMR (270 HFiz, CDC1~/TMS) : d - 1.23 (t, 6H, P-0-CH2-
CH3 ) , 3 . 78 ( d, 2H, CHZ-P ) JP_a - 24 Hz, 4 . 04 ( dq, 4H,
P-0-CHI-GH3), 7.19 - 7.97 (m,4H,Ar-H)
Example 2
Preparation of 2-formylbenzylphosphonic acid di(triethyl-
ammonium) salt (B) and
monoethyl 2-formylbenzylphosphonate triethylammonium
salt (C)
100 ml of 6 M HC1 were added to 5.0 g (20 mmol) of
diethyl 2-formylbenzylphosphonate and the mixture was
boiled under reflex for 6 h. Water and HC1 were distilled
off in vacuo, and the residue was co-evaporated three
times with toluene. The remaining brown material was
chromatographed on silica gel (CHaCla/methanol/triethyl-
amine: 75/24/1). Compounds B and C were obtained as oily
products. xt was possible to separate them by chromato-
graphy on diethylaminoethyl 'Sephadex A25 (Et3NH+ form,
Pharmacies, F'reiburg, West Germany). They diffex in their
elution behavior (Rf value) . Elution wa.s carried out with
a triethylammonium bicarbonate gradient of from 0.3 -
1.0 M.
(B): Rf = 0.1; yield: 2.4 g (30 $); m.p.:
1H-NMR (270 ~iFiz, DPSSO/TMS): 8 = 1.07 (t,lBH,N-CH2-CH3),
2 . 8 6 ( q,12H, N-CHZ-CH3 ) , 3 . 2 3 ( d, 2H, CHa-P ) ,7y_H - 2 3 Hz ,
~) !
N ,% 'r~ i,I ~ a
14 -
7.24 - 7.78 {m,4H,Ar-H), 10.31 (s,lH,CHO).
(C): Rf = 0.3; yield: 1.4 g (21 ~);
1H-NI~IR ( 270 P2Eiz, DMSO/TNIS ) : d - 1. 01 - 1.17 (m,12H,
PI-CHZ-CH3 & P-0-CHz-CH3 ) , 2 . 8 8 ( q, 6H, N-CH2-CH3 ) , 3 . 2 7
( d, 2H, CHZ-P ) JP_~ - 23 Hz, 3 . 68 ( { dq, 2H, P-O-CHZ-CH3 ) ,
7.18 - 7.78 (m,4H,Ar-H), 10.31 (s,lH,CHO).
Example 3
Preparation of 2-formylbenzylphosphonic acid di(triethyl-
ammonium) salt (7B)
3.2 g (21 mmol) of trimethylsilyl bromide were added
dropwise to 2.0 g (8 mmol) of the compound A in 10 ml of
absolute dioxane, and the reaction mixture was heated to
50°C and stirred at this temperature for 6 h. The mixture
was evaporated, water was added several times and the
solution was lyophilized. The crude product was purified
by chromatography as in Example 2.
Yield: 1.98 g (62 ~).
Ezample 4
Diethyl 2-formylbenzylphosphonate thiosemicarbazone (D)
2.0 g (8 mmol) of the compound A and 0.73 g of thiosemi
carbazide were dissolved or suspended in 200 ml of
absolute ethanol. 2 ml of acetic acid were added and the
mixture was boiled under reflex for 3 h. In the course of
the slow cooling, the product D precipitated in crystal
line form.
Yield: 1.8 g (68 ~); m.p.~ 195 to 197°C;
1H-~1MR (270 I~iHz, CDC13/TMS) a d _ 1.16 (t,6H,CH2-CH3), 3.38
( d, 2H, CH2-P, JP_~ - 2 3 Hz ) , 3 . 9 4 ( dq, 4H, CHZ-CHI ) ,
7.23 -.7.39 (m,3H,Ar-H), 8.40 (s,lH,~r-H), 11.37
(s,lH,Ar-CH=N).
f. '7
a.
15 _ ~,.1 e.~~~~i~
Example 5
Diethyl 2-formylbenzylphosphonate nicotinic acid
hydrazone (E)
2.0 g (8 mmol) of the compound A and 1.07 g (8 mmol) of
nicotinic acid hydrazide were dissolved in 30 ml of
absolute ethanol. After adding 1 ml of acetic acid, the
mixture was boiled under reflex for 8 h. The solvent was
removed by rotary evaporation and the residue was chrom-
atographed an silica gel (eluent CHZC12/EtOH 9.5/0.5; Rf
- 0.45). The product E was obtained in crystalline form.
Yield: 2.2 g (73 $); m.p.a 136 to 140°C;
1H-NMR 270 Ma3z, CDC13/TMS ) : d = 1.14 - 1. 37 (m, 6H, CH3) ,
3.61 - 3.79 (d,2H,CHz-P), Jp_$ - 24 Hz, 3.87 - 4.16
(m,4H,CH2-CH3), 7.11 - 7.49 (m,4H,Ar-H), 7.70 - 9.23
(m,SH,Py-H), 10.17 & 11.25 (each s, ratio 1 : 3, 1H,NH).
Ezample 6
Diethyl 2-(3,4-dimethyl-5-phenyloxazolidin-2-yl)benzyl-
phosphonate (F)
2.0 g (8 mmol) of the compound A and 1.32 g (8 mmol) of
(-)-ephedrine were dissolved in 100 ml of benzene and
heated under reflex in a water separator for 24 h. The
solvent was then removed by rotary evaporation and the
residue was chromatographed on silica gel (eluent
CHaCl2/ethanol 9.5/0.5; Rf - 0.55). The product F was
obtained as an oil.
Yield: 2.4 g (75 $);
1H-NMR (270 MHz, CDCl~/TPiS); d = 0.80, (d,3H,CH-CH3), 1.25
( m, 6H, 0-CH2-CH3 ) , 2 . 2 7 ( s , 3H, N-CH3 ) , 3 .18 ( dq,1H, CH~CH~ ) ,
3.20 & 3.28 (dd,lH,Ph-CH-CH), 3.59 - 3.7s (m,2H,CH2-P);
4.01 (m,4H,o-cH~-cH3), 5.19 (s,lH,Ar-cH(o-) (r~~), 7.1~ --
7.45 (m,8H,Ar-H), 7.89 - 7.98 (m,lH,Ar-H).
Example 7
Pathogen-free NMRI mine having a weight of about 15 g
were infected intraperitoneally with Herpes simplex
is - ~ ~~~~~'
Type 1 and then treated intraperitoneally, orally or
subcutaneously with the compounds mentioned in Table 1.
The treatment was carried aut twice daily over the course
of 2.5 days, starting after infection. The result of
treatment was determined on the basis of the course of
the disease and the survival rate compared to the un-
treated infection controls. The controls received a
water-soluble methylhydraxyethylcellulose (viscosity
300 Pa. s, administered in 2 ~ strength solution) instead
of the compounds to be tested. The experiments were
carried out using groups of 5 mice each per preparation.
The chemotherageutic action of the compound A can be seen
from Table 1.
Table 1 Herpes
simplex
Z
Preparation Dosage Surviving Average survival
(mg/kg) animals time (days)
Control sc 0 L 6.7
A sc 2.5 2 9.0
A sc 25 0 7.2
A sc 250 0 8.4
Control po 0 1 8.0
A po 2.5 4 8.0
A po 25 5 ---
A po 250 4 8.0
Control sc 0 1 8.3
C sc 3 1 8.5
C sc ZO 3 8.0
C sc 30 4 10.0
Control po 0 1 7.8
C po 3 4 9.0
C po 10 3 8.0
C po 30 1 8.3
Control sc 0 1 8.3
B sc 3 2 7.8
B sc 10 3 7.0
B sc 30 3 6.5
~~e'~~~t"'J
. ../ ',~ r.~ r~6 .y 5
- 17 -
Table l, continuation Herpes simplex
1
Preparation Dosage Surviving A v a r a
g a
animals survival
(mg/3cg) t~ (~ys)
Control po 0 1 7.8
B po 3 3 7.0
B po 10 2 8.3
B po 30 3 6.0
Control ig 0 1 8.3
A ip 3 0 8.0
A ip 10 4 9.0
A ip 30 3 6.5
po = orally
sc = subcutaneously
ip = intraperitoneally
Hxample 8
Cell cultures of Hela and Vero cells were inoculated into
microtitre plates and infected with myxoviruses
(influenza A2). 2 Hours after infection, the compounds B
and C were added to the infected cell cultures in various
dilutions. 48 to 72 hours after infection, the result of
treatment was determined microscopically and photometric
ally by neutral red absorption (color test according to
Finter) (Finter, N.B. Tnterferons, 1966) on the basis of
the cytopathogenic effect. The minimum concentration at
which about half the infected cells do not show a cyto-
pathogenic effect is regarded as the minimum inhibitory
concentration (MIC). The results are summarized in
Table 2.
7rr-~ ~= fi
- ,~,. 4~ >I ~ r~ ' ~
Table 2 Influenza A2
Substance MIC (~glm1) MTD (~g/ml)
C 44.4 > 400
B 4.94 > 400
MzC = minimum inhibitory concentration
MTD = maximum tolerated dose
Bxample 9
Pathogen-free l~tT mice having a weight of about 16 g
were infected intranasally with influenza A2 and then
treated subcutaneously and orally with the compounds
mentioned in Table 3. The compounds were administered to
the animals under slight ether anesthesia using one drop
of virus suspension in each of the nostrils. The treat-
ment was carried out twice daily over the course of
2.5 days, starting after infection. Amantadine was always
used as the comparison. The success of the treatment was
determined on the basis of the course of the disease and
the survival rate compared to the untreated infection
controls. The controls received a water-soluble methyl-
hydroxyethylcellulose (viscosity 300 Pa. s, administered
in 2 ~ strength solution) instead of the compounds to be
tested. The expera.anents were carried out using groups of
5 mice each per preparation.
The chemotherapeutic action is shown in Table 3.
< ~;~3s v~r s
i9 - ~~:~~ ~ ~~
Table 3 influenza
A2
Preparation Dosage Number of Average survival
(mg/kg) surviving time (days)
animals
Control po 0 2 7.5
Amantadine po 80 5 --
A po 2.5 ~ 9.0
A po 3 ~ 8.0
A po 10 4 7.0
A po 25 2 6.3
A po 30 2 7.3
Control sc 0 0 6.6
Amantadine sc 80 5 -_
A sc 2.5 2 6.7
A sc 25 3 7.0
Control sc 0 2 6.7
Amantadine sc 80 5 --
B sc 1.5 1 8.5
B sc 15 4 8.0
Control po 0 1 6.5
Aman~adine po 80 4 8.0
B po 1.5 3 8.5
B po 15 5 --
B po 150 3 7.0
Control sc 0 0 6.8
C sc 0.25 5 ~-
C sc 2.5 5 __
C sc 25 3 7.5
Control po 0 0 7.2
C po 0.25 5 --
C po 2.5 2 6.0
C po 25 5 --
Control po 0 0 6.6
p po 0.25 4 8.0
D po 2.5 1 7.0
D po 25 2 7.3
- 20 - ~~ai~~:~~~~
Table 3, continuatiAn
Influenza A2
Preparation Dosage Number of Average
(mg/kg) Surviving time
animals (days)
Control po 0 0 6.6
E po 0.25 4 8.0
E po 2.5 3 6.7
E po 25 3 6.0
Control po 0 1 7.0
F po 0.25 3 6.5
F po 2.5 3 6.5
F po 25 2 7.0
Z5 sc = subcutaneously
po = orally
Eacannple 10
yaboratory mice (NMRI, female, weight 20 - 24 g) were
infected intravenously with Friend leukemia virus (F~V)-
containing mouse serum. The treatment was started 48 h
after infection. The mice were treated over the course of
10 days with the substances indicated in Table 4. The
substances indicated were administered orally or intra-
peritoneally once a day. 14 days after infection, the
animals were sacrificed by dislocation and the spleens
were removed. The weight of the spleens was determined.
As a measurement parameter of the therapeutic activity,
the weight of the spleen of the animals which had been
treated with compounds A and D was related to.that of the
untreated infection control.
Suramin and azidothymidine (AZT) were used as standard
substances. The action of the preparations is shown in
Table 4.
21
~.~ .~
Table 4 Friend leukemia virus
Preparation Dosage Survival Relative weight
rate $ of the spleen
$ of body weight
Control po 0 100 12.36
AZT po 15.5 100 3.26
A po 17.5 100 7.74
A po 81 100 5.67
Control ip 0 100 10.64
AZT ip 50.0 100 7.60
D ip 50.0 100 7.60
Control ip 0 100 12.36
Suramin ip 50 70 6.06
A ip 25 50 6.27
A ip 50 100 6.52
ip = intraperitoneally
po = orally