Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
2~3~2~
l-AMINO-5~HALOGENOURACILS, PRO~ESS FOR THEIR PREPARATION,
AND CENTRAL NERVOUS SYSTEM DEPRESSANTS CONTAINING SAME AS
ACTIVE INGREDIENT
TECHNICAL FIELD
The present invention relates to a central nervous
system depressant containing a l-amino-5-halo~enouracil
as an active ingredient, a novel l-amino-5-halogenouracil
useful as a central nervous system depressant, and a
novel process for preparing the 1-amino-5-halogenouracil.
BACKGROUND ART
Stress on human body tends to increase with the
complication of social environment, and thus an increased
number of patients complain of symptoms such as sleep
duration disorder, insomnia and the like, which are
considered to be induced by stress.
Although physiological phenomenon named sleep is
very complicated and its mechanism has not been
elucidated in detail, several nucleic acid-related
compounds which affect sleep have been reported. Such
compounds include the following:
(1) Uridine:
Sleep-promoting effect [see Biomed. Res., 4, 223
(1983); Neurosci. Res., 1, 243 (1984); Proc. Natl. Acad.
Sci. V.S.A., 81, 6240 (1984); Neuroscience Letters, 49,
207 (1984)];
(2) Uracil;
Hexobarbital induced sleep potentiating effect [see
J. Am. Pharm. Assoc., 44, 56 (1955); ibid., 44, 550
(1955)];
~3) N3-Benzyluridine and its derivatives:
Hypnotic effect and/or pentobarbital induced sleep
potentiating effect [see Chem. Pharm. Bull., 33, 4088
(1985); Japanese Patent Laid-Open Publication No.
207218/1987];
t4) N-Allyl or N-benzyl substituted derivati~es of
uracil, thymine or 6-methyluracil:
2~3~24
Hypnotic effect and/or pentobarbital in~uced sleep
potentiating effect [see Chem. Pharm. Bull., 35, 4982
(1987); Abstract of the Proceedings of the 108th Annual
Meeting of Pharmaceutical Society of Japan, page 708
(1988)l.
It has been also reported that the N-allyl
substituted derivative of uracil (Nl,N3-diallyl uracil)
and uridine have anticonvulsive effect ~see Brain Res.,
55, 291 (1973); Chem. Pharm. Bull., 35, 4928 tl987)]-
However, these conventional compounds (i) have no
hypnotic effect though they exhibit sleep-promoting
effect (uridine and uracil), and (ii) must be
administered in large doses ~320-752 mg/kg) to induce
sleep by intraperitoneal injection, and thus they do not
always have satisfactory effects.
An object of the present invention is to provide a
central nervous system depressant which comprises as an
active ingredient a compound exhibiting more potent
central nervous system depressant effects such as
2~ hypnotic effect, sleep-promoting effect, anticonvulsive
effect and the like than the conventional compounds.
DISCLOSURE OF THE INVENTION
We have conducted extensive research in order to
find out a compound which has central nervous system
sedative effects. As a resultl we have found that 1-
amino-5-halogenouracil has excellent central nervous
system sedative effects.
While l-amino-5-fluorouracil has hitherto been known
as only one l-amino-5-halogenouracil, it has not been
reported that the compound has central nervous system
sedative effects [see Sci. Pharm., 52, 46 (1984)].
An object of the present invention is to provide a
central nervous system aepressant which comprises a 1-
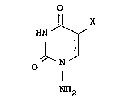
amino-5-halogenouracil represented by the formula:
2034224
HN ~
~ ~ [I]
O N
NH2
wherein X represents a halogen atom, or a
pharmaceutically acceptable salt thereof as an active
ingredient.
Another object of the present invention is to
provide a novel l-amino-5-halogenouracil represented by
the formula:
1~ 0
Il X'
~N ~ ~ ~
1~ , lII]
N
O
NH~
wherein X' represents chlorine, bromine or iodine, or a
salt thereof.
A further object of the present invention is to
provide a process for preparing a l-amino-5-
halogenouracil represented by the formula [I] shown abovewhich comprises reacting a pyrimidine derivative
represented by the formula;
OR
1 X
3U N ~ [III]
1~ J
RO N
wherein X has the same meaning as defined above and R
represents a protective group, with an aminating agent to
aminate the l-position of the pyrimidine derivative, and
removing the protective groups.
4 ~34224
BEST MODE F~R CAR~YING OUT THE INVENTION
The present invention will now be explained in
detail below.
The l-amino-5-halogenouracil which is an active
ingredient of the central nervous system depressant of
the present invention is the compound represented by the
above shown formula ~I] (referred to hereinafter simply
as "active ingredient of the present pharmaceutical
composition").
In the formula [I], the halogen atoms represented by
X include fluorine, iodine, bromine and chlorine. In
this connection, the compounds represented by the formula
[I] except for one in which X represents fluorine are
novel compounds.
The active ingredient of the present pharmaceutical
composition may be in the form of salts and includes acid
addition salts with inorganic acids such as hydrochloric
acid, hydrobromic acid, sulfuric acid or phosphoric acid,
or with organic acids such as citric acid, acetic acid,
succinic acid, maleic acid, methanesulfonic acid or p-
toluenesulfonic acid.
The active ingredient of the present pharmaceutical
composition can be prepared, for example, by the method
in which a pyrimidine derivative represented by the
following formula [III] is used as a starting compound,
which is reacted with an aminating agent (Z-NH2) to give
an intermediate [IV] by the specific amination of the 1-
position of the pyrimidine derivative (referred to
hereinafter as "amination reaction"), and the protective
groups represented by R are removed ~referred to
hereinafter as "deprotection reaction"):
.
2034~24
OR X OR
1~ ~ Z-NH
RO N RO
[III] NH2 [IV]
0
11 X
HN
O N
NH2 ~I]
wherein X has the same meanings as defined above, R
represents a protective group and Z~NH2 represents an
aminating agent.
The halogen atom represented by X in the starting
compound may be selected appropriately so that it
corresponds to X in the desired compound represented by
the formula [I]~ The protective groups represented by R
include silyl groups such as trimethylsilyl,
t r i e t h y 1 s i 1 y 1 , t - b u t y 1 d i m e t h y 1 s i 1 y 1 ,
methyldiisopropylsilyl and triisopropylsilyl, and alkyl
groups such as methyl and ethyl. Particularly, the silyl
groups are advantageously used.
The starting compound can be prepared by introducing
3~ the protective groups represented by R into a 5-
halogenouracil according to a conventional method. For
exampler when a silyl group is used as the protective
group, protection can be accomplished by using 2- to 10-
fold moles of the silylating agent in proportion to 1
mole of the 5-halogenouracil and reacting the mixture in
a reaction solvent such as pyridine, picolinel
diethylaniline, dimethylaminopyridine, dimethylformamide,
6 203~22~
acetonitrile, tributylamine, triethylamine or the like,
which may be used alone or in admixture thereof, at a
reaction temperature in the range of 0 to 50C for 1 to
30 hours.
After introducing the protective groups, the
pyrimidine derivative represented by the above formula
[III] is isolated and purified, if necessary, by a
conventional isolation and purification means for nucleic
acid bases such as distillation, adsorption
chromatography with silica gel or the like, or
recrystallization, and then subjected to amination
reaction as the startin~ compound.
The aminating agent (Z-NH2) used for the amination
reaction includes hydroxylamines. Specifically, there
can be mentioned o-arylsulfonylhydroxylamines such as
o-mesitylenesulfonylhydroxylamine,
o-(2,4,6-triisopropylbenzenesulfonyl)hydroxylamine,
and o-(2-nitrobenzenesulfonyl)hydroxylamine,
o-nitrophenylhydroxylamines such as
o-(2,4-dinitrophenyl)hydroxylamine and
o-picrylhydroxylamine,
o-mesitoylhydroxylamine and the like.
The amination reaction with these aminating agents
can be accomplished by using 1- to 5-fold moles of an
aminating agent, preferably 1- to 1.5-fold moles in
proportion to 1 mole of the starting compound and
reacting the mixture in a reaction solvent (a haloyenated
hydrocarbon such as dichloromethane, dichloroethane, or
chloroform, an ether type sol~ent such as
tetrahydrofuran, or dioxane, or an aromatic hydrocarbon
such as benzene, toluene, or xylene, which may be used
alone or in admixture thereof) at a reaction temperature
in the range of 0 to 50~C, preferably 0 to 30~C for 1 to
10 hours.
After the amination reaction, the intermediate [IV]
is subjected to deprotection reaction to obtain the
203~22~
active ingredient of the present pharmaceutical
composition.
The deprotection reaction may be conducted according
to any conventional method for the deprotection of the
protective group used. For example, when a silyl group
is used as the protective group, it can be removed by
ammonium fluoride treatment or acidic or alkaline
hydrolysis. Also, when an alkyl group such as methyl or
ethyl is used, the alkyl group can be removed by acidic
or alkaline hydrolysis.
The active ingredient of the present pharmaceutical
composition thus obtained can be isolated and purified by
an appropriate combination of conventional isolation and
purification means, for example, chromatography such as
adsorption chromatography or recrystallization which are
applied to the isolation and purification of nucleic acid
bases.
While the dose of the compound represented by the
formula [IJ as the active ingredient of the present
pharmaceutical composition depends on many factors such
as the severity of patients or acceptabilities to the
composition and finally should be determined by the
judgement of doctors, it is generally in the range of
0.05 to 2 g per day for an adult patient, which is
administered once or in portions. The route for
administration of the composition may be any of
appropriate routes such as oral or parenteral
administration.
For oral administration, the compGsition may be in
the form of a solid preparation such as powder, granules,
capsules or tablets or a liquid preparation such as syrup
or elixir. For parenteral administration, the
composition may be in the form of injection, suppository,
agent for external application or for inhalation. These
preparations are prepared according to a conventional
method with the addition of a pharmaceutically acceptable
preparatory aid to the active ingredient of the present
~34~4
pharmaceutical composition. It is also possible to
formulate the composition into a sustained release
preparation by a well-known technique.
In the production of the solid preparation for oral
administration, the active ingredient of the present
pharmaceutical composition is mixed with an excipient
such as lactose, starch, crystalline cellulose, calcium
lactate, calcium monohydrogenphosphate, magnesium
aluminometasilicate or anhydrous silicic acid to give a
powder, or, if necessary, the powder is further mixed
with a binding agent such as white sugar,
hydroxypropylcellulose or polyvinylpyrrolidone, or a
disintegrating agent such as carboxymethylcellulose or
carboxymethylcellulose calcium for wet or dry granulation
to give granules. In the production of tablets, these
powders or granules, if necessary, mixed with a lubricant
such as magnesium stearate or talc may be punched into
tablets. Alternatively, these granules or tablets can be
coated with an enteric bas e su ch a s
hydroxypropylmethylcellulose phthalate or a methyl
methacrylate copolymer to give enteric-coated
preparations, or they can be coated with ethylcellulose,
carnauba wax or a hydrogenated oil to give sustained
release preparations. Further, in order to prepare
capsules, powder or granules may be charged into hard
capsules, or the active ingredient of the present
pharmaceutical composition is first dissolved in
glycerol, polyethylene ~lycol, sesame oil, olive oil or
the like and next coated with a gelatin film to give soft
capsules.
In order to prepare the liquid preparation for oral
administrationl the active ingredient of the present
pharmaceutical preparation and a sweetener such as white
sugar, sorbitol or glycerol may be dissolved in water to
give a clear syrup, or the syrup may be further mixed
with an essential oil or ethanol to give an elixir or
with gum arabic, tragacanth gum, polysorbate 80, or
.
- . ~:
. ~ .
203422~
carboxymethylcellulose sodium to give an emulsion or a
suspension. These liquid preparations may also contain
flavoring agents, colorants, preservatives or the like,
if desired.
In order to pxepare the preparation for injection,
the active ingredient of the present pharmaceutical
composition may be dissolved in distilled water for
injection, if necessary, together with a pH adjusting
agent such as sodium hydroxide, hydrochloric acid, lactic
acid, sodium lactate, sodium monohydrogenphosphate or
sodium dihydrogenphosphate, and an isotonizing agent such
as sodium chloride or glucose, aseptically filtered and
charged into ampoules, or these solutions may be mixed
with mannitol, dextrin, cyclodextrin, or gelatin and
lyophilized under vacuum to give injections which should
be dissolved on use~ Furthermore, the active ingredient
of the present pharmaceutical composition can be mixed
w i t h l e c i t h i n , p o l y s o r b a t e 8 ~ , o r
polyoxyethylenehydrogenated castor oil, and the mixture
is emulsified in water to give an emulsion for injection.
In order to prepare the preparation for rectal
administration, the active ingredient of the present
pharmaceutical composition may be melted by heating
together with a suppository base such as txi-, di- or
mono-glycerides o~ cacao fatty acid or polyethylene
glycol, poured into a mold and cooled, or the active
ingredient of the present pharmaceutical composition may
be dissolved into polyethylene glycol or soybean oil and
coated with a gelatin film.
In order to prepare the preparation for external
application, the active ingredient of the present
pharmaceutical composition is added to white vaseline,
beeswax, liquid parafin or polyethylene glycol and the
mixture is kneaded, i necessary, under heat to give an
ointment, or it is kneaded with an adhesive such as rosin
or an alkyl acrylate polymer and then spread over
20~422~
~onwoven fabrics made of, for example, polyeth~lene to
give a tape preparation.
The present invention will be illustrated below with
reference to Synthesis Examples, Test Examples and
Preparation Examples.
SYnthesis Example 1
l-Amino-5-fluorouracil
5-Fluorouracil was silylated as usual with
hexamethyldisilazane, and then 8.8 g (32 mmole) of 2,4-
ditrimethylsilyloxy-5-fluoropyrimidine thus obtained,
distilled and purified was dissolved in dichloromethane
(48 ml). To the solution was added 7.5 g (35 mmole) of
mesitylenesulfonylhydroxylamine (MSH) under ice-cooling,
and the mixture was reacted with stirring at room
temperature for 4 hours.
After reaction, the reaction solution was
concentrated under reduced pressure. Distilled water
(200 ml) was added to the residue, and oily impurities
were extracted with chloroform (50 ml) from the mixture.
2Q The aqueous phase was neutralized with a weakly basic
resin and then concentrated to dryness under reduced
pressure.
The crude crystal thus obtained was purified by
sublimation under the conditions of 150C and 5 mmHg to
give 2.9 g (yield, 62%) of 1-amino-5-fluorouracil.
The product was further recrystallized from 5Q%
ethanol to give colorless needle crystals.
M.P.: 196-199C (lit. 205-207C)
Elementary analysis for C4H4N3O2F:
Calculated (%): C, 33.11; H, ~.78; ~, 28.96
Found (%): C, 33.15; H, 2.79; N, 28.78
Synthesis Example 2
l-Amino-5-bromouracil
After the silylation of 5-bromouracil conducted in
the same manner as in Synthesis Example 1, 8.8 9 t26
mmole) of 5-bromo-2,4-ditrimethylsilyloxypyrimidine
distilled and purified was dissolved in 50 ml of
2~3~22~
dichloromethane. To the solution was added 6.7 g (31
mmole) of MSH under ice-cooling, and the mixture was
reacted with stirring at room temperature for 4 hours.
After reaction, the reaction solution was
concentrated under reduced pressure. To the residue were
added distilled water (200 ml) and subsequently 2N-sodium
hydroxide to adjust the pH to 6.0, and the solution was
concentrated under reduced pressure.
The crude product thus obtained was collected by
filtration and recrystallized from 50% ethanol to give 3
~ (yield, 55~) of crystalline 1-amino~5-bromouracil.
M.P.: 214-215C
Elementary analysis for C4H4N3O2Br:
Calculated (~): C, 23.32; H, 1.96; N, 20.40
Found (%): C, 23.59; H, 1.97; N, 20.14
NMR spectrum (~, ppm, DMSO-d5):
5.54 (2H, s, NH2, disappeared by the addition of
D20 )
8.14 (lH, s, 6-H)
11.84 (lH, s, NH, disappeared by the addition of
D20 )
Synthesis Example 3
l-Amino-5-chlorouracil
Starting from 5-chlorouracil, 1-amino-5-chlorouracil
was obtained in the same manner as in Synthesis E~ample
~.
M.P.: 224-225C (recrystallized from water)
Elementary analysis for C4H4N3O2Cl:
Calculated (%): C, 29.74; H, 2.50; N, 26.01
30 Found (%): C, 29.83; H, 2.55; N, 25.93
NMR spectrum l~, ppm, DMSO-d6):
5.54 (2H, s, NH2, disappeared by the addition of
D20)
8.09 (lH, s, 6-H)
11.86 (lH, s, disappeared by the addition of D2O)
Synthesis Example 4
l-Amino-5-iodouracil
12
2~3~224
Starting from 5-iodouracil, 1-amino-5-iodouracil was
obtained in the same manner as in Synthesis Example 2.
M.P.: 195-196C (recrystallized from water)
Elementary analysis for C4H4N3O2I:
Calculated (%): C, 18.99; H, 1.59; N, 16.61
Found (%): C, 19.05; H, 1.57; N, 16.51
NMR spectrum (~, ppm, DMSO-d6):
5.51 (2H, s, ~H2, disappeared by the addition of
D20 )
i3.08 (lH, s, 6-H)
11.69 (lH, s, di,sappeared by the addition of D2O)
Test Example 1
Hypnotic effect
The following compounds suspended in a physiological
saline solution containing 0.5% carboxymethylcellulose
were administered to ICR mice (male).
The time from the loss of righting reflex until the
recovery of it (sleeping time) was measured. The results
are shown in Table 1.
Table 1
Dose Route of No. of Mean
Compound (mg/kg) Admini- Animals Sleep ng
1-Amino-5- 100 ip 2 53
fluorouracil
l-Amino-5- 100 P 3 21
fluorouracil _
l-Amino-5- 112 ip 2 80
bromouracil
l-Amino-5- 200 P 1 14
chlorouracil
* ip = intraperitoneal; po = peroral
Test Example 2
Anticonvulsive efect
a) Thiosemicarbazide induced convulsion
:
'~ .
13 203~22~
Thiosemicarbazide (20 mg/kg) was administered
intraperitoneally to ICR mice (male), and 30 minutes
thereafter the following compounds suspended in a
physiological ,saline solution containing 0.5%
carboxymethylcellulose were administered subcutaneously.
The time required for initiating convulsion (initial
convulsion time) was measured. Physiological saline
containing no test compounds was used as a control. The
results are shown in Table 2.
Table 2
: ~
Dose No. of Initial Convulsion
Compound (mg/kg) Animals Time (min)
l-Amino-5-fluorouracil 50 4 80.9 + 12.9*
1-Amino~5-bromouracil 50 8 118.3 + 6~9*~
Control 4 60.3 + 6.8
*: significant at a significant level of 5% or less
**: significant at a significant level of 0.5% or less
b) Picrotoxin induced convulsion
The following compounds suspended in a physiological
saline solution containing 0.5% carboxymethylcellulose
were administered intraperitoneally to ICR mice (male).
Immediately thereafter, picrotoxin was also administered
intraperitoneally in the same manner, and the number of
convulsions per hour and lethality of the animals within
1 hour after the administration of picrotoxin were
examined. Physiological saline containing no test
compounds were used as a control. The results are shown
in Table 3.
14
2~3~22~
Table 3
Compound Dose No. of ~No. of Lethality
(mg/kg) Animals Convulsions (%)
l-Amino-S- 10 1.3 ~ 1.0* 20
bromouracil
l-Amino-5- 20 1.8 + 1.7* 0**
bromouracil
Control _ 8 5.6 + 0.9 75
*: significant at a significant level of 0.1% or less
**: significant at a significant level of 1% or less
Test Example 3
Acute.toxicitY
l-Amino-5-bromouracil was suspended in a
physiological saline solution containing 0.5%
carboxymethylcellulose. 500 mg/kg of the compound was
administered intraperitoneally to 8 ICR mice, and
lethality of the animals was observed for 1 week. As a
result, all of the mice were survived with no lethality.
Preparation Example 1
Tablet
l-Amino-5-bromouracil 10 g
Corn starch 65 g
Carboxymethylcellulose 20 g
Polyvinylpyrrolidone 3 g
Calcium stearate 2 q
Total 100 9
Tablets each weighing 100 mg are prepared in a
conventional manner. Each tablet contains 10 mg of 1-
amino-5-bromouracil.
Preparation Example 2
Powder and Capsule
1-Amino-5-fluorouracil 20 g
Crystalline cellulose 80 q
Total 100 9
15 2034~24
Both the powders are mixed into a powder
preparation. Separately, 100 mg of the preparation is
charged into a No. 5 hard capsule to form a capsule
preparation.
INDUSTRIAL APPLICABILITY
The active ingredient of the present pharmaceutical
compositionr as apparent from the aforementioned Test
~xamples, has the following characteristics as compared
with conventional compounds and is very useful as a
central nervous system depressant.
(i) A little dose of the active ingredient of the
present pharmaceutical composition induces central
nervous system depressant effects such as hypno~ic effect
or anticonvulsive effect.
In order to induce hypnotic effect by
intraperitoneal administration, a dose of 320 to 752 mg
is required for conventional compounds. For example, the
doses of 433 mg, 320 mg and 752 mg per kg body weight
were required for Nl,N3-diallyluracil, Nl-methoxymethyl-
N3-benzyluracil and N3-benzyl-2',3',5'-tri-o-
methyluridine, respectively.
On the other hand, l-amino-5-halogenouracil, the
active ingredient of the present pharmaceutical
composition, can induce sleep with a dose of about 1~0 mg
per kg body weight.
(ii) The active ingredient of the present
pharmaceutical composition can express its activity by
oral administration. Hitherto, there have been reported
no nucleic acid-related substances having hypnotic effect
by oral administration.
Iiii) The active ingredient of the present
pharmaceutical composition is a compound which has an
extremely low toxicity. ~ The simultaneous production of
both the l-amino derivative and the 1,3~diamino
derivative which is a defec~ in the conventional method
[Sci. Pharm., 52. 46 (1984)] is suppressed by the
synthesis method of the present invention~ and the 1-
16 2~3~224
position of pyrimidines can be specifically aminated togive l-amino-5-halogenouracil in a high yield.
. ~ .
.