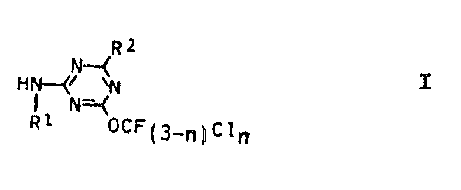Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~~ ;i f.> r'~; .., ;~
. ..
O.Z. 0050/41799
2-Amino-4-fluoroalkoxy-1 3 5-triazines
and the preparation thereof
The present invention relates to novel 2-amino-
4-fluoroalkoxy-1,3,5-triazines of the formula I
Rz I
HN-(~~
R1 OCF~3_~~Cl~
where
R1 is hydrogen, C1--C4-alkyl, C3-C6-alkenyl or C3-C4-
alkynyl;
RZ is hydrogen, halogen, C1-C4-haloalkyl, trifluorometh-
oxy or chlorodifluoromethoxy and
n is 0 or 1.
The present invention also relates to a process
for preparing the 1,3,5-triazines I by reacting 2-halo-
4-fluoroalkoxy-1,3,5-triazines with amines.
The 1,3,5-triazines I are valuable intermediates
for organic syntheses, especially for preparing crop
protection agents, eg, herbicidal sulfonylurea deriva-
tives.
There are many indications in the literature that
fluoroalkyl and fluoroalkoxy groups are equivalent to
halogen in that they have similar electronic properties.
pKa measurements (Proc. Natl. Acad. Sci. USA 134 (1960)
1207, J. Azn. Chem. Soc. 83 (1961) 4860) demonstrate that,
for example, fluoroalkoxy groups attract electrons by
induction and, conversely, also act as electron donors
because of their resonance capacity. Taking all the
effects together, the trifluoromethoxy group is in fact
more strongly deactivating than the halogens, so that the
term super-halogens is also used (J. Am. Chem. Soc. 83
(190'1) 4860). .This applies in the same way to their
ability to be substituted by nucleophiles. Chemical
Abstracts 87, 53396 demonstrates, for example, that when
2,4-bis(trichloromethyl)-6-trifluoromethyl-1,3,5-triazine
is stirred with basic amines in benzene there is
~,
~ ~~ y~:~ ": =;
- 2 - O.Z. 0050/41799
replacement of both haloalkyl groups. The ability of the
trifluoromethoxy radical to act as leaving group is also
utilized, for example, in sugar chemistry (CA. 105,
115325; 107, 96978).
These facts explain why to date no process for
preparing 2-amino-4-fluoroalkoxy-1,3,5-triazines has been
disclosed. The only disclosure has been in the pyrimidine
series where 4-difluoromethoxy-2-halo compounds were
converted with toxic methyl mercaptan into 2-methyl-
thiopyrimidines which were then oxidized to 2-methyl-
sulfonyl derivatives, followed by nucleophilic displace-
ment with amine (US-A 4 542 216).
We have now found that the novel 2-amino-4-
fluoroalkoxy-1,3,5-triazines of the formula I
NCR z I
i
OCF~3_n~Cln
where
R1, RZ and n have the abovementioned meanings, are
obtained in an advantageous manner when 2-halo-1,3,5-
triazines of the formula II
~x
hat-(~~'N II
N=
OCF~3_~~Clrt
where Hal is fluorine, chlorine, bromine or iodine, and
RZ and n have the abovementioned meanings, axe reacted
with an amine of the formula III
H-NH-R1 III
where Rl has the abovementioned meaning.
The reaction of 2,4-difluoro-6-trifluoromethoxy-
1,3,5-triazine with ammonia can be represented as
follows:
c ~ ,~- .,.; d ~ ;~.~, '?
- 3 -- a.Z. 0050/4179
F F
F-(N~ * NH3 aHF HZN°-~N=(~y
OCFj OCF3
The process provides in a straightforward and
economic manner novel 2-amino-4-fluoroalkoxy-1,3,5-
triazines in high yield and purity. Unexpectedly, fluoro-
alkoxy groups are not replaced under the reaction condi-
tions. In view of the prior art, all these advantageous
properties are surprising.
Preferred products I and, accordingly, preferred
starting materials II are those where R1 is hydrogen,
C1-C4-alkyl such as methyl, ethyl, n-propyl, i-propyl, n
butyl, sec-butyl, i-butyl, tert-butyl; C3-C4-alkenyl such
as 2-propenyl, 2-methylethenyl, 2-butenyl, 3-butenyl, 1-
methyl-2-propenyl or 2-methyl-2-propenyl; C3-C4-alkynyl
such as propargyl, 2-butynyl, 3-butynyl or 1-methyl-2-
propynyl; RZ is hydrogen, fluorine, chlorine, bromine,
trichloromethyl, dichlorofluoromethyl, chlorodifluoro-
methyl, trifluoromethyl, 1,1-dichloro-2,2,2-trifluoro-
ethyl, 1,1,2,2,2-pentafluoroethyl or 1,1,2,2,2-penta-
chloroethyl, also trifluoromethoxy or chlorodifluoro-
methoxy, and n is 0 or 1.
Among the amines which can be employed, the
following should be mentioned: ammonia, methylamine,
ethylamine, n-propylamine, isopropylamine, n-butylamine,
isobutylamine, sec--butylamine,tert-butylamine, 2-propen-
ylamine, 2-methylethenylamine, 2-butenylamine, 3-butenyl-
amine, 1-methyl-2-propenylamine, 2-methyl-2-propenyl-
amine, propargylamine, 2-butynylamine, 3-butynylamine and
1-methyl-2-propynylamine.
The 2-halo-1,3,5-triazines II can be reacted with.
the amines III.in an aprotic polar solvent at from -80 to
40°C, either employing the amine III in an excess rela
tive to II or using an additional organic base.
The reaction of the 2-halo-1, 3, 5-triazine TI with
the amine III can be carried out in the absence or,
J
r. ~'-~'~':'.':..i
Y ~ '.. v.. ,. ~ .a
- 4 - o.Z. 0050/41799
advantageously, in the presence of a solvent.
Particularly suitable solvents are the following:
ethers such as methyl tert-butyl ether, diethyl ether,
ethyl propyl ether, n-butyl etPiyl ether, di-n-butyl
ether, diisobutyl ether, diisoamyl ether, diisopropyl
ether, cyclohexyl methyl ether, tetrahydrofuran, 1,2-
dimethoxyethane, diethylene glycol dimethyl ether and
anisol.e, esters such as ethyl acetate, n-butyl acetate
and isobutyl acetate, and chlorohydrocarbons such as
methylene chloride, 1,1,2,2-tetrachloroethane, 1,1-
dichloroethylene, 1,2-dichloroethane, chlorobenzene, 1,2-
dichlorobenzene and 1-chloronaphthalene, and mixtures
thereof.
The solvent is expediently used in an amount of
from 100 to 2000$, preferably 400 to 1200, of the weight
of the starting material II.
It is advantageous to add from 1.8 to 2.5, in
particular 1.95 to 2.2, mole equivalents of the amine
III, based on the starting material II, over the course
of from 0.5 to 2 hours to a mixture of starting material
II and one of the abovementioned solvents at -80 to 40°C,
preferably -70 to 25°C, to stir until the reaction is
complete (after about 3 hours) and then to warm to 25°C
for the working up.
If only approximately the stoichiometric amount
of the amine III is used, it is expedient to use an
additional organic base in order to trap the hydrogen
halide which is formed. Suitable bases for this purpose
are trimethylamine, triethylamine, ethyldiisopropylamine,
triisopropylamine, N,N-dimethylaniline, N,N-dimethyl
cyclohexylamine, N-.methylpyrrolidine, pyridine, quino
line, cx-, ~- and ~y-picoline, 2, 4- and 2, 6-lutidine and
triethylenediamine. It is generally sufficient to add
from 0.9 to l.i equivalents of the base relative to the
starting material II.
The reaction can be carried out under atmospheric
or superatmospkzeric pressure, continuously or batchwise.
t' d' y Si j ~ ~~>.
i., ~;J va i .' ...~ W ! >'
- 5 - O.Z. 0050/41799
Working up can be carried out in a conventional
manner, eg. the reaction mixture is extracted with water
to remove the salts, and the ox-ganic phase is dried and
purified by, for example, chromatography. However, it is
also possible to concentrate the organic phase directly
and to stir the residue with a solvent.
With a view to their further processing to give
herbicidal substances, eg. sulfonylurea derivatives, the
following 1,3,5-triazines of the formula I are particu
larly preferred:
2-amino-4-chloro-6-trifluoromethoxy-1,3,5-triazine,
2-amino-4-fluoro-6-trifluoromethoxy-1,3,5-triazine,
2-amino-4-chloro-6-chlorodifluoromethoxy-1,3,5-triazine,
2-amino-4-fluoro-6-chlorodifluoromethoxy-1,3,5-triazine,
2-amino-4,6-bis-trifluoromethoxy-1,3,5-triazine,
2-amino-4,6-bis-chlorodifluoromethoxy-1,3,5-triazine,
2-amino-~-trifluoromethoxy-6-trifluoromethyl-1,3,5-
triazine,
2-amino-4-chlorodifluoromethoxy-6-trifluoromethyl-1,3,5-
triazine,
2-methylamino-4-chloro-6-trifluoromethoxy-1,3,5-triazine,
2-methylamino-4-fluoro-6-trifluoromethoxy-1,3,5-triazine,
2-methylamino-4-chloro-6-chlorodifluoromethoxy-1,3,5-
triazine,
2-methylamino-4-fluoro-6-chlorodifluoromethoxy-1,3,5-
triazine,
2-methylamino-4,6-bis-trifluoromethoxy-1,3,5-triazine,
2-methylamino-4,6-bis-chlorodifluoromethoxy-1,3,5-
triazine,
2-methylamino-4-trifluoromethoxy-6-trifluoromethyl-1,3,5-
triazine,
2-methylamino-4-chlorodifluoromethoxy-6-trifluoromethyl-
1,3,5-triazine,
2-allylamino-4-chloro-6-trifluoromethoxy-1,3,5-triazine,
2-allylamino-~-fluoro-6-trifluoromethoxy-1,3,5-triazine,
2-allylamino-4-chloro-6-chlorodifluoromethoxy-1,3,5
triazine,
~~r?.~t r.
- 6 - O.Z. 0050/41799
2-allylamino-4-fluoro-6-chlorodifluoromethoxy-1,3,5-
triazine,
2-allylamino-4,6-bis-trifluorome~thoxy-1,3,5-triazine,
2-allylamino-4,6-bis-chlorod.ifluoromethoxy-1,3,5-
tri.azine,
2-allylamino-4-trifluoromethoxy-6-trifluoromethyl-1,3,5-
triazine,
2-allylamino-4-chlorodifluoromethoxy-6-trifluoromethyl-
1,3,5-triazine.
The 2-halo-1,3,5-triazines required as starting
materials II can be prepared by the process described in
Japanese paid-Open Application 17 039 ('63), Chemical
Abstracts 60, 2986 d in tetrachloromethane. However, a
considerably more advantageous process is that described
in German Application P 40 24 755 (O.Z. 0050/41798) of
the same date, which is represented by the following
scheme:
R2 R~ R2
Ha1--(N~N Cl2 N"'~ a) SbF /SbCl N
N~ =HC'~-~ Hal--(i 'N ~ Hal°(~ 'N
N=~ b) HF(SbCIS) N=
OCHg OCC13 . OCF(3_~)C1~
I V II
The chlorination of the methoxy-1,3,5-triazine
IV with chlorine to give the trichloromethoxy-1,3,5-
triazine V is carried out, for example, at from 100 to
180°C.
Suitable for the chlorination are elemental
chlorine or chlorine-releasing substances such as
sulfuryl chloride or phosphorus pentachloride. Chlorine
can also be prepared in situ by oxidation of hydrogen
chloride, for example with hydrogen peroxide.
The chlorination can be carried out in the
presence of an inert solvent, for example a chloro
hydrocarbon such as chlorobenzene, 1,2-, 1,3- or 1,4
dichlorobenzene, a nitro compound such as nitrobenzene,
a carboxylic acid such as acetic acid or propionic acid,
an anhydride such as acetic anhydride, an acid chloride
Ja -~ ~ ~ '-:w,~
- 7 - O.Z. 0050/41799
such as chloroacetyl chloride, a-chloro-propionyl
chloride or a,a'-dichloropropion~~l chloride, an inorganic
acid halide such as phosphorus trichloride or phosphorus
oxychloride or, preferably, without solvent in a melt of
the starting material.
The reaction rate may be increased by the use of
a radical initiator, those suitable being irradiation
with light, preferably W light, or addition of
a,a'-azoisobutyronitrile, expediently in an amount of
from 0.2 to 7 mol ~ based on the starting material. The
reaction rate can also be increased by adding a catalyst;
suitable for this is phosphorus pentachloride,
expediently in an amount of from 0.5 to 7 mol ~ based on
starting material IV. In this case, the starting material
IV and catalyst are mixed and then the chlorination is
started. In place of phosphorus pentachloride it is also
possible to add starting components which form the latter
under the reaction conditions, eg. phosphorus trichloride
or yellow phosphorus, and then to start the chlorination.
Starting material IV can be reacted with chlorine
in approximately the stoichiometric amount or, prefer-
ably, in excess, advantageously from 3.1 to 11, in
particular 3.3 to 5, moles of chlorine per methoxy
equivalent in the starting material TV. The reaction is
carried out at from 100 to 180°C, advantageously from 120
to 150°C, under atmospheric or superatmospheric pressure,
continuously or batchwise.
If the chlorination is carried out under 1 bar,
it is expedient to use from 3.3 to 5 males of gaseous
chlorine per methoxy equivalent in the starting material
IV, which corresponds to a chlorine conversion of from 91
to 60~. The chlorine conversion can be increased by
appropriate measures, eg. by use of moderate pressure,
expediently from 1 to 10 bar, or by using a bubble
column. It is advantageous to allow the gaseous chlorine
to stay in contact with the organic phase for as long as
possible by, for example, vigorously stirring the latter
- 8 - O.Z. 0050/41799
or forcing the chlorine to pass through a thick layer of
the organic phase. The reaction generally takes from
about 0.5 to 12 hours.
The procedure in a preferred embodiment of the
process is to pass the required amount of gaseous chlor
ine into the vigorously stirred liquid starting material
IV over the course of from 0.5 to 12 hours, preferably 1
to 10 hours, starting at from 120 to 130°C and raising
the temperature continuously, where appropriate by
utilizing the exothermic nature of the reaction, until it
is at from 135 to 150°C toward the end of the reaction.
It is obvious that for larger batches the exothermic
nature of the reaction must be taken into account by
external cooling or suitable metering of the chlorine;
when the reaction subsides the cooling bath is removed
and, where appropriate, heat is then applied.
The products can be worked up and isolated in a
conventional manner. For example, residual hydrogen
chloride, chlorine or catalyst can be driven out of the
hot organic phase using an inert gas; this leaves behind
a high yield of crude product which is already rather
pure. It can be further purified by distillation or
chromatography, or else used immediately for further
reactions.
The halogen replacement on the trichloromethoxy-
1,3,5-triazine V is carried out, for example, at from 0
to 180°C.
Suitable for the halogen replacement are antimony
trifluoride in the presence or absence of catalytic
amounts of an antimony(V) salt, eg. antimony(V) chloride
or hydrogen fluoride. It is expedient to use an excess of
from 1 to 200, preferably 5 to 25, mol ~ of antimony
trifluoride per trichloromethyl equivalent. The catalytic
amount of antimony(V) salt is from 1 to 20, preferably 5
to 18, mol ~ per trichloromethyl equivalent. The starting
material V is preferably metered at from 90 to 130°C into
the mixture containing the agent for halogen replacement,
~r ,~~ ,,,i ;,~ ~3 -; ;?
,;;:.
9 - O.Z. 0050/41799
and the mixture is then heated at from 110 to 180°C for
from 10 to about 240 minutes. Subsequent working up is by
distillation.
However, the reaction can also be carried out
continuously, adding the starting material V at from 110
to 180°C over the course of from 10 to about 240 minutes
and, at the same time, removing the lower boiling product
II by distillation under reduced pressure. Traces of
antimony salts which are carried over can be removed by
extraction with concentrated hydrochloric acid.
The halogen replacement remains at the chlorodi-
fluoromethoxy stage if no antimony(V) salt is used for
catalysis or if only small amounts, eg. from 0.5 to 5 mol
~, are employed and the amount of antimony trifluoride is
reduced to from 60 to 90 mol $ per trichloromethyl
equivalent.
The halogen replacement can also be carried out
with hydrogen fluoride, in place of antimony trifluoride,
at from 0 to 150°C, preferably 40 to 120°C. For this
purpose, an excess of from 300 to 700, preferably 350 to
400, mol ~ of hydrogen fluoride per trichloromethyl
equivalent is added to the starting material V in an
autoclave, and the mixture is then stirred for from 10
minutes to about 10 hours. The reaction rate can be
increased in the same way as described fox the use of
antimony trifluoride, ie. by addition of a catalyst such
as antimony pentachloride. A reaction time of about
4 hours is generally sufficient. After release of
pressure and removal of volatile constituents, working up
is carried out as described.
The novel 2-amino-4-fluoroalkoxy-1,3,5-triazines
I are valuable intermediates for preparing crop protec-
tion agents. They can be subjected to the process
described in German Application P 40 24 754 (O. Z.
0050/41800) of the same date, for example 2-amino-4-
fluoro-6-trifluoromethoxy-1,3,5-triazine can be reacted
with methanol to give 2-amino-6-rnethoxy-4-
~o~..~~~):~
- 10 - O.Z. 0050/41799
trifluoromethoxy-1,3,5-triazine which reacts with
2-carbomethoxybenzenesulfonyl isocyanate to give
herbicidal sulfonylureas. However, they can also be
reacted directly with the said isocyanate to give
herbicidal sulfonylureas.
EXAMPLES
I. Examples of the preparation of the precursors (cf.
German Application P 40 24 755 (O.Z. 0050/41798) of
the same date).
EXAMPLE I.1
2,4-Difluoro-6-trichloromethoxy-1,3,5-triazine
A stream of gaseous chlorine was passed into a
mixture of 300 g (2.041 mol) of 2,4-difluoro-6-methoxy-
1,3,5-triazine and 0.3 g of a,a'-azoisobutyronitrile at
130°C with UV irradiation in such a way that the tempera-
ture reached 140 - 145°C within 2 hours. The progress of
the reaction was checked by NMR spectroscopy and then
chlorine was passed in at 135 - 140°C (external heating)
for a further 3 hours.
The precipitate was removed by filtration with
suction and the filtrate was distilled under reduced
pressure to yield 444 g (87~ of theory) of the title
compound of boiling point 40 - 46°C/0.3 mbar.
EXAMPLE I.2
2,4-Difluoro-6-trifluoromethoxy-1,3,5-triazine
Half of 210 g (0.838 mol) of 2,4-difluoro-6-
trichloromethoxy-1,3,5-triazine was added to a stirred
mixture of 187.4 g (1.048 mol) of antimony trifluoride
and 35.2 g (0.117 mol) of antimony pentachloride in such
a way that the initial temperature of 110°C rose to
125°C; when reflex ceased, external heating was necessary
while addition was continued. The mixture was stirred at
125 - 130 °C for one hour, and a fraction boiling at 100 -
105°C was removed by distillation through a 25 cm packed
column. After the reaction subsided, the remaining half
of the trichloromethoxy compound was added dropwise
within 30 minutes, and the fraction boiling at
- 11 - o.z. 0050/41799
100 - 105°C was continuously distilled out. The total
reaction time was 3 hours. 134.4 g (79.8 of theory) of
the title compound were obtained with np° = 1.3650.
EXAMPLE I.3
6-Chlorodifluoromethoxy-2,4-difl.uoro-1,3,5-triazine
210 g (0.838 mol) of 2,4-difluoro-6-trichloro-
methoxy-1,3,5-triazi.ne were added within 10 minutes to
110 g (0.615 mol) of antimony trifluoride while stirring
at 110°C. After addition of 3/4 of 9.38 g (0.0313 mol) of
antimony pentachloride, the mixture was heated to 145°C
and stirred for 1 hour. The remaining catalyst was added,
and the mixture was stirred for a further 2 hours while
a fraction boiling at 95 - 105°C was obtained through a
30 cm packed column: 20 g (11.8 of theory) of 2,4-
difluoro-6-trifluoromethoxy.-1,3,5-triazine. The residue
was distilled without a column to yield 94.8 g (52$ of
theory) of the title compound of boiling point 125 -
130°C; noa = 1.4042.
EXAMPLE I.4
2,4-Dichloro-6-trifluoromethoxy-1,3,5-triazine
52 g (0.183 mol) of 2,4-dichloro-6-trichlorometh-
oxy-1,3,5-triazine were added within 5 minutes to a
stirred mixture of 40.9 g (0.229 mol) of antimony tri-
fluoride and 7.03 g (0.0234 mol) of antimony pentachlor-
ide at 90°C, during which the temperature rose to 180°C.
The mixture was then stirred at 170 - 180°C for
20 minutes, after which the crude product was distilled
out at 90 - 103°C/70 mbar. Another distillation yielded
32.3 g (75.5$ of theory) of the title compound of boiling
point 165 - 173°C.
II. Preparation of the compounds I according to the
invention
EXiIMPLE II.1
2-Amino-4-fluoro-6-trifluoromethoxy-1,3,5-triazine
4.4 g (0.259 mol) of gaseous ammonia were passed
over the course of 45 minutes into a stirred mixture of
~~~~~~r~
- 12 - O.Z. 0050/41799
26 . 0 g ( 0 . 1293 mol ) of 2, 4-c~.if:Luoro-6-trifluoromethoxy-
1,3,5-triazine and 100 ml of tetrahydrofuran at -70 to
-65°C. The mixture was then stirred for 2 hours at -70°C
and overnight while warming to 22°C. The residue from
concentration under reduced pressure was stirred with
water, filtered off with suction and washed. Drying
yielded 22 g (85.9 of theory) of the title compound of
melting point 138 - 139°C.
EXAMPLE II.2
2,4-Bismethylamino-6-trifluoromethoxy-1,3,5-triazine and
2-methylamino-4-fluoro-6-trifluoromethoxy-1,3,5-triazine
5.9 g (0.189 mol) of methylamine were passed over
the course of 30 minutes into a stirred mixture of 19.0 g
(0.0945 mol) of 2,4-difluoro-6-trifluoromethoxy-1,3,5
triazine and 100 ml of diethyl ether at -70°C. The
mixture was stirred for 2 hours at -70°C and overnight
while warming to 22°C. The residue from concentration
under reduced pressure was taken up in methylene chloride
and washed with water. The solution was dried and chroma-
tographed through a silica gel column. The first two
fractions contained 5.0 g (25$ of theory) of 2-methyl-
amino-4-fluoro-6-trifluoromethoxy-1,3,5-triazine of
melting point 68 - 72°C, and fractions 4 - 7 yielded
10.7 g (51~ of theory) of less soluble 2,4-bismethyl-
amino-6-trifluoromethoxy-1,3,5-triazine of melting point
150 - 152°C.
EXAMPLE II.3
2-Amino-4-chlorodifluoromethoxy-6-fluoro-1,3,5-triazine
and 2,4-diamino-6-chlorodifluoromethoxy-1,3,5-triazine
7.8 g (0.46 mol) of ammonia were passed over the
course of 45 minutes into a stirred mixture of 50.0 g
(0.23 mol)oft,4-difluoro-6-chlorodifluoromethoxy-1,3,5-
triazine and 150 ml of tetrahydrofuran at -70°C. The
mixture was stirred for 2 hours at -70°C and overnight
while warming to 22°C. It was concentrated under reduced
pressure, washed with water and dried. The product was
then loaded in methylene chloride onto a silica gel
:..: . . ,: ,,
- 13 - 0.2. 0050/417~~
column and eluted with the same solvent. Fractions 1 - 8
yielded 21.5 g (43.6 of theory} of 2-amino-4-fluoro-6-
chlorodifluoromethoxy-1,3,5-triazine of melting point 131
- 133°C.
Washing with ethyl acetate then yielded in
fractions g - 14 the less soluble 2,4-diamino-6-chlorodi-
fluoromethoxy-1,3,5-triazine (11.2 g, 23~ of theory) of
melting point 114°C.
EXAMPLE II.4
2-Chlorodifluoromethoxy-4-fluoro-6-methylamino-1,3,5-
triazine and2,4-bismethylamino-6-chlorodifluoro-methoxy-
1,3,5-triazine
5.2 g (0.166 mol) of methylamine were passed over
the course of 20 minutes into a stirred mixture of 18.1 g
(0.083 mol) of 4-difluorochloromethoxy-2,6-difluoro
1,3,5-triazine and solvent at -70°C. The mixture was
stirred fox 2 hours at -70°C and overnight while warming
to 22°C. It was concentrated under reduced pressure,
taken up in methylene chloride, washed with water and
dried. Chromatography on silica gel yielded in the
initial fractions 5.5 g (2~$ of theory) of 2-
chlorodifluoromethoxy-4-fluoro-6-methylamino-1,3,5-
triazine of melting point 62-64°C. Subsequent fractions
yielded 8.7 g (44~ of theory) of 2,4-bismethylamino-6-
chlorodifluoromethoxy-1,3,5-triazine of melting point 118
- 120°C.
III. Conversion of the 1,3,5-triazines into herbicidal
sulfonylurea derivatives
EXAMPLE III.1
2-Amino-4-methoxy-6-trifluoromethoxy-1,3,5-triazine
9.1 g (0.0505 mol) of 30~s strength sodium
methylate were added over the course of 15 minutes to a
stirred mixture of 10 g (0.0505 mol) of 2-amino-4-fluoro-
6-trifluoromethoxy-1,3,5-triazine and 100 ml of methanol
at 0°C. The mixture was stirred at 0°C for one hour and
then concentrated under reduced pressure, taken up in
methylene chloride and extracted with water. Drying and
9 y'
., '.'
- 14 - O.Z. 0050/41799
concentration yielded 10.5 g (99~ of theory) of the title
compound of melting point 96 - 101°C.
EXAMPLE I7:I.2
2-Amino-4-chlorodifluoromethoxy-6-methoxy-.1,3,5-triazine
8.4 g (0.0466 mol) of 30~ strength sodium methy-
late were added over the course of 15 minutes to a
stirred mixture of 10 g (0.0466 mol) of 2-amino-4-chloro-
difluoromethoxy-6-fluoro-1,3,5-triazine and 100 ml o~
methanol at 0°C. The mixture was stirred at 0°C fox one
hour and then concentrated under reduced pressure, taken
up in methylene chloride and extracted with water. Drying
and concentration yielded 10.4 g (98.5 of theory) of the
title compound of melting point 109 - 111°C.
EXAMPLE III.3
2-Amino-4-ethoxy-6-trifluoromethoxy-1,3,5-triazine
2.3 g (0.093 mol) of 97$ sodium hydride were
added a little at a time to 300 ml of ethanol at 20 -
35°C and dissolved by stirring for 15 minutes. Then,
while stirring at 0°C, 18.5 g (0.093 mol) of 2-amino-4-
fluoro-6-trifluoromethoxy-1,3,5-triazine were added over
the course of 10 minutes, and the mixture was stirred at
0°C for 1 hour and at 22°C overnight. The residue from
concentration under reduced pressure caas taken up in
methylene chloride, extracted with water and dried.
Concentration yielded 17.9 g (85.9 of theory) of the
title compound of melting point 69 - 71°C.
EXAMPLE III.4
2-Amino-4-chlorodifluoromethoxy-6-ethoxy-1,3,5-triazine
1.2 g (0.0466 mol) of 97~ sodium hydride were
added a little at a time to 150 ml of ethanol at 20 -
35°C and diss~ilved by stirring for 15 minutes. Then,
while stirring at 0°C, 10.0 g (0.046 mol) of 2-amino-4-
chlorodifluoromethoxy-6-fluoro-1,3,5-triazine were added,
and the mixture was stirred at 0°C for 1 hour and at 22°C
overnight. The residue from concentration under reduced
f~
7~S ~. f ~~ i.o
,~ ~t' ~ (~~'i _
- 15 - O.Z. 0050/41799
pressure was taken up in methy3_ene chloride, extracted
with water and dried. Concentration yielded 10.6 g (94.6
of theory) of the title compound of melting point 63 -
65°C.
E~A~iPLE III.5
2-Amino-4-methylamino-6-trifluoromethoxy-1,3,5--criazine
3.5 g (0.111 mol) of methylamine were passed over
the course of 20 minutes into a stirred solution of 11 g
(0.055 mol) of 2-amino-4-fluoro-6-trifluoromethoxy-1,3,5-
triazine in 150 ml of tetrahydrofuran at 0°C. The mixture
was stirred at 0°C for one hour and at 22°C overnight. It
was then concentrated under reduced pressure, stirred
with water and dried. 10.8 g (93.1 of theory) of the
title compound of melting point 155 - 157°C (decomposi
tion) were obtained.
EXAMPhE III.6
2-.Amino-4-chlorodifluoromethoxy-6-methylamino-1,3,5-
triazine
2.9 g (0.0932 mol) of methylamine were passed
over the course of 20 minutes into a stirred solution of
10 g (0.0466 mol) of 2-amino-4-chlorodifluoro-methoxy-6-
fluoro-1,3,5-triazine in 150 ml of diethyl ether at 0°C.
The mixture was stirred at 0°C for one hour and at 22°C
overnight. Washing with water, drying and concentration
yielded 9.4 g (89.5 of theory) of the title compound of
melting point 143°C (decomposition).
EXAMPhE III.7
2-Amino-4-dimethylamino-6-trifluoromethoxy-1,3,5-triazine
5.0 g (0.111 mol) of dimethylamine were passed
over the course of 20 minutes into a stirred solution of
11 g (0.055 mol) of 2-amino-4-fluoro-6-trifluoro-methoxy-
1,3,5-triazine~in 150 ml of tetrahydrofuran at 0°C. The
mixture was stirred at 0°C for one hour and at 22°C
overnight. Concentration, washing with water and drying
yielded 9.9 g (80.7 of theory) of the title compound of
melting point 114 - 118°C (decomposition).
~~~~:u~?~,
- 16 - O.Z. 0050/41799
EXAMPLE III.8
2-Amino-4-chlorodifluoromethox_Y-6-dimethylamino-1,3,5-
triazine
4.2 g (0.093 mol) of dimethylamine were passed
over the course of 20 minutes into a stirred solution of
g (0.0466 mol) of 2-amino-4-c:hlorodifluoro-methoxy-6-
fluoro-1,3,5-triazine in 150 ml of diethyl ether at 0°C.
The mixture was stirred at 0°C for one hour and at 22°C
overnight. Washing with water, drying and concentration
10 yielded 9.8 g (87.8 of theory) of the title compound of
melting point 130 - 133°C (decomposition).
EXAMPLE III.9
Methyl 2-(4-methoxy-6-trifluoromethoxy-1,3,5-triazin-2-
ylaminocarbonylaminosulfonyl)benzoate
3.6 g (0.015 mol) of 2-carbomethoxybenzene-
sulfonyl isocyanate in 4 ml of 1,2-dichloroethane were
added over the course of 5 minutes to a stirred mixture
of 3.15 g (0.015 mol) of 2-amino-4-methoxy-6-trifluoro-
methoxy-1,3,5-triazine and 150 ml of 1,2-dichloroethane
at 22°C, and the mixture was stirred at 22°C for
12 hours. It was then concentrated under reduced pressure
and crystallized using 1 . 1 methyl tert-butyl ether/
petroleum ether, and the product was filtered off with
suction and washed with petroleum ether to yield 5.1 g
(75.4 of theory) of the title compound of melting point
149°C (decomposition).
EXAMPLE III.10
Sodium salt of methyl 2-(4-methoxy-6-trifluoromethoxy-
1,3,5-triazin-2-ylaminocarbonylaminosulfonyl)benzoate
1. 8 g ( 0 . 004 mol ) of the compound from Example
III.9 were suspended in 30 ml of methanol and, while
stirring at 10 - 15°C, 0.72 g (0.004 mol) of 30~ strength
sodium methylate solution was added. The clear solution
was stirred for 10 minutes and then concentrated under
reduced pressure, resulting in 1.9 g (100 of theory) of
the title compound of melting point 118°C (decom-
position).
~~'s ~"~=~;
a;r;vra
- 17 - O.Z. 0050/41799
EXAMPLE III.11
Ethyl 2-(4-methylamino-6-trifluoromethoxy-1,3,5-triazin-
2-ylaminocarbonylaminosulfonyl)benzoate
3.1 g (0.012 mol) of 2-carboethoxybenzene-
sulfonyl isocyana.te in 3 ml of methylene chloride were
added over the course of 10 minutes to a stirred mixture
of 2.5 g (0.012 mol) o.f. 2-amino-4-methylamino-6-
trifluoromethoxy-1,3,5-triazine and 150 ml of methylene
chloride at 22°C, and the mixture was stirred at 22°C for
30 hours. It was then concentrated under reduced
pressure, stirred with methyl tent-butyl ether and
filtered off with suction. Further washing with methanol
and drying resulted in 3.8 g (67.4 of theory) of the
title compound of melting point 182 - 184°C
(decomposition).