Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
- 1 -
2Q5856~
TRIFLUOROMETHYLKETONE DERIVATIVES,
PROCESSES FOR PREPARATION THEREOF
AND USE THEREOF
This invention relates to new trifluoromethylketone
derivatives.
More particularly, this invention relates to new
trifluoromethylketone derivatives and their
pharmaceutically acceptable salts which have an human
leukocyte elastase-inhibiting activity, to processes for
preparation thereof, and to a pharmaceutical composition
comprising the same and to a method of use thereof.
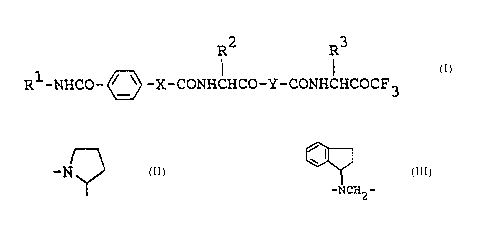
The new trifluoromethylketone derivatives of this
invention are represented by the following formula (I) .
R2 R3
R1-NHCO- ~'~ -X-CONHCHCO-Y-CONHCHCOCF3
~I~
- 2 -
2fl58~f
wherein
R1 is lower alkyl which has one or two substituents
selected from carboxy, esterified carboxy and
di-lower alkylcarbamoyl, phenyl(lower)alkyl, the
phenyl moiety of which may have halogen or nitro or
amino and the alkyl moiety of which may have carboxy
or esterified carboxy, halo-phenyl, morpholino ox
morpholino(lower)alkyl,
R2 and R3 are each lower alkyl,
X is - or -NH- and
I
Y is -N or
-NCH,, -
According to this invention, the new
trifluoromethylketone derivatives (I) and salts thereof
can be prepared by various processes which are illustrated
by the following reaction schemes .
Process 1 .
R2 R3
HOOC- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(II)
R1-NH2 (III)
p2 R3
R1-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF
3
(I)
- 3 -
Process 2 .
R~ R3
R1-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCHCF3
(IV) OH
R2 R3
R1-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(I)
Process 3
R2 R3
Ra-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(Ia)
R2 R3
Rb-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(Ib)
Process 4 .
R2
N02- ~ ~ -CH2NHC0- ~ ~ -CONHCHCON R3
CONHCHCOCF3
(Ic)
R2
NH2- ~ ~ -CH2NHC0- ~ ~ -CONHCHCON R3
CONHCHCOCF3
(Id)
- 4 -
2~s~~s~
Process 5
R2 R3
HOOC-RC-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(Ie)
(Ra)2NH (V)
R2 R3
(Rd)2NC0-RC-NHCO- ~ ~ -X-CONHCHCO-Y-CONHCHCOCF3
(zf)
In the above formulae, R1 is mono- nr ~;- A~tor;~;o,a
a _ _ _ _ _ - ."~,.. ,., ,,, ,", w s. y y cu
carboxy(lower)alkyl and Rb.is mono- or di-
carboxy(lower)alkyl, R~ is lower alkylene, Ra is lower
alkyl, and R1 to R3, X and Y are each as defined above.
A pharmaceutically acceptable salt of the new
trifluoromethylketone derivatives of the formula (I) may
include a salt with an inorganic or organic base such as
an alkali metal salt (e. g. sodium salt, potassium salt,
etc.), an alkaline earth metal salt (e. g. calcium salt,
etc.), ammonium salt, ethanolamine salt, triethylamine
salt, dicyclohexylamine salt or the like, and an acid
addition salt with organic or inorganic acid such as
methane sulfonate, hydrochloride, sulfate, nitrate,
phosphate or the like.
Preferred examples and illustrations of the various
definitions, in the above descriptions, which the present
invention includes within the scope thereof are explained
in detail as follows.
The term "lower" is intended to mean 1 to 6 carbon
atoms, unless otherwise indicated.
Preferred examples of "halogen" is fluorine,
chlorine, bromine and iodine.
- 5 -
Preferred examples of "lower alkyl" may include a
residue of straight and branched alkane having 1 to 6
carbon atoms such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl, t-butyl, pentyl, neopentyl, hexyl and the
like, and preferably the one having 1 to 4 carbon atom(s).
Preferred examples of "esterified carboxy" may
include an alkyl ester, i.e. alkoxycarbonyl such as lower
alkoxycarbonyl (e. g. methoxycarbonyl, ethoxycarbonyl,
propoxycarbonyl, butoxycarbonyl, tert-butoxycarbonyl,
etc.) and a phenyl(lower)alkyl ester, i.e.
phenyl(lower)alkoxycarbonyl such as benzyloxycarbonyl and
a benzoyl(lower)alkyl ester, i.e.
benzoyl(lower)alkoxycarbonyl such as
benzoylmethoxycarbonyl, and the like.
Preferred examples of "lower alkylene" may include
methylene, ethylene, propylene, isopropylene and the like.
Preferred examples of "di-lower alkylcarbamoyl" may
include N,N-dimethylcarbamoyl, N,N-diethylcarbamoyl and
the like.
Processes for preparing the object compound (I) ar
its salts of this invention are explained in detail in the
following.
In the explanation of Processes 1 to 5 as follows,
salts of Compounds (I), (Ia) to (If), and (II) to (V) may
include the same as those exemplified as pharmaceutically
acceptable salt of trifluoromethylketone derivatives (I)
as illustrated hereinbefore.
(1) Process 1 .
Compound (II) + Compound (III) -~ Compound (I)
Compound (I) and its salt can be prepared by reacting
Compound (II) or its salt with a Compound (III) or its
salt.
The reaction of this process can be conducted as
follows .
That is, in one case, as the first step, the carboxy
group of Compound (II) or its salt is usually activated in
a conventional manner, for example, in the form of its
acid halide, azide, acid anhydride or a mixed anhydride,
activated ester, and the like, and is reacted with the
Compound (III) to give Compound (I), and in the other
case, the Compound (II) or its salt is reacted with the
Compound (III) or its salt directly in the presence of a
conventional condensing agent such as
N,N-dicyclohexylcarbodiimide,
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and the
like.
This reaction is preferably carried out in a solvent
such as N,N-dimethylformamide, methylene chloride,
chloroform, tetrahydrofuran, dioxane, ethyl acetate,
methanol, ethanol, water or the like under ice-cooling to
at ambient temperature and the reaction in the presence of
a condensing agent is usually carried out in an anhydrous,
but not critical, conditions.
(2) Process 2 .
Compound (IV) -~ Compound (I)
The Compound (I) and its salt can be prepared by
oxidizing the Compound (IV) or its salt.
The oxidation is carried out by a conventional method
using an oxidizing agent which can be applied to
converting a hydroxymethyl group to a carbonyl group such
as potassium permanganate, chromic compound (e. g. chromic
acid, sodium chromate, dichromic acid, sodium dichromate,
pyridinium chlorochromate, pyridinium dichromate, etc.),
Swern reagent (dimethylsulfoxide and oxalylchloride),
~os$sso
Jones reagent and the like.
The reaction is usually carried out in a conventional
solvent such as water, acetone, dioxane,
dimethylformamide, pyridine or any other organic solvents
which do not adversely influence to the reaction, or a
mixture thereof.
This reaction is preferably carried out under
somewhat milder condition such as under cooling, at room
temperature or under warming.
(3) Process 3 .
Compound (Ia) ~ Compound (Ib)
The Compound (Ib) and its salt can be prepared by
subjecting the Compound (Ia) or its salt to
de-esterification reaction.
The de-esterification reaction is carried out by a
conventional method such as hydrolysis, reduction or the
like, details of which are explained in the following .
1) Hydrolysis .
Hydrolysis is preferably carried out in the presence
of an acid or base.
Suitable acid includes an inorganic acid (e. g.
hydrochloric acid, hydrobromic acid, sulfuric acid, etc.),
an organic acid (e. g. formic acid, acetic acid,
trifluoroacetic acid, propionic acid, benzenesulfonic
acid, p-toluenesulfonic acid, etc.), and the like.
Suitable base includes an inorganic base such as
alkali or alkaline earth metal hydroxide or the
corresponding carbonate or bicarbonate (e. g. sodium
hydroxide, potassium hydroxide, sodium carbonate,
potassium carbonate, sodium bicarbonate, calcium
- 8 -
hydroxide, etc.), ammonium hydroxide or the like;
an organic base such as an alkoxide or phenoxide of the
above metal (e. g. sodium ethoxide, sodium methoxide,
etc.), an amine such as mono-, di or tri- alkylamine (e. g.
methylamine, ethylamine, N,N-dimethyl-1,3-propanediamine,
trimethylamine, triethylamine, etc.) or the like.
The hydrolysis is preferably conducted under somewhat
milder conditions such as under cooling or under warming
in a solvent which does not have adverse influence to the
reaction, e.g, water, a hydrophilic solvent such as
alcohol (e. g. methanol, ethanol, propanol, etc.), acetone,
N,N-dimethylformamide, etc. A liquid abovementioned acid
and base can also be used as a solvent.
2) Reduction .
Reduction, including chemical reduction and
catalytic reduction, is carried out in a conventional
manner.
Suitable reducing agents to be used in chemical
reduction are a metal (e.g. tin, zinc, iron, etc.), or a
combination of such metal and/or metallic compound (e. g.
chromium chloride, chromium acetate, etc.) and an organic
or inorganic acid (e. g, formic acid, acetic acid,
propionic acid, trifluoroacetic acid, p-toluenesulfonic
acid, hydrochloric acid, etc.).
Suitable catalysts to be used in catalytic reduction
are conventional ones such as platinum catalysts (e. g.
platinum plate, spongy platinum platinum black, colloidal
platinum, platinum oxide, etc.), palladium catalysts (e. g.
spongy palladium, palladium black, palladium oxide,
palladium on carbon, colloidal palladium, etc.), or the
like.
The reduction is usually carried out in a solvent
such as water, an alcohol (e. g. methanol, ethanol, etc.)
or the like.
2os~sso
The reduction is preferably carried out under
somewhat milder conditions such as under cooling, at room
temperature or under warming.
(4) Process 4 .
Compound (Ic) -~ Compound (Id)
The Compound (Id) or its salt can be prepared by
reducing the Compound (Ic) or its salt.
The reduction including chemical reduction and
catalytic reduction is carried out in a conventional
manner.
Suitable reducing agents may include the same as
those exemplified in Process 3.
The reduction is usually carried out in a solvent
such as water, an alcohol (e. g. methanol, ethanol, etG.)
or the like.
The reduction is preferably carried out under
somewhat milder conditions such as under cooling, at room
temperature or under warming.
(5) Process 5 .
Compound (Ie) + Compound (V) -~ Compound (If)
Compound (If) and its salt can be prepared by
reacting Compound (Ie) or its salt with Compound (V) or
its salt.
The reaction is carried out by substantially the same
method of that of Process 1.
Pharmaceutically acceptable salts of the
trifluoromethylketone derivatives (I) can be prepared by a
conventional method, i.e., by treating the Compound (I)
with an acid or a base. Suitable examples of the acid or
base may include the same as those exemplified in the
explanation of "Hydrolysis" of Process 3.
- 10 -
~os~ss~
Starting Compounds (II) to (V) each include new
compounds and can be prepared by Preparations as described
hereinafter and by the similar methods thereto.
The object Compound (I) including Compounds (Ia) to
(If) and starting Compounds (II) and (IV) include one or
more isomers due to the asymetric carbon atoms and all of
such isomers are included within the scope of this
invention.
According to this invention, there can be obtained a
mixture of diastereoisomers due to the presence of
compounds bearing both R and S configurations at the
chiral center marked with D of the formula as mentioned
below, and there can also be obtained an optically pure
compound.
It is to be noted that said optically pure compound
changes to a mixture of said diastereoisomers in an
aqueous and/or organic solution.
R2 R3
R1-NHCO- r ~ X-CONHCHCO-Y-CONHCHCOCF3
0
Further, it is to be noted that the object compound
(I) of this invention provides a hydrate form in an
aqueous solution, which is included within the scope of
this invention.
The trifluoromethylketone derivatives (I) and
pharmaceutical acceptable salt thereof have a human
leukocyte elastase-inhibiting activity and is useful as
human leukocyte elastase inhibitors for treating or
preventing degenerative diseases, for example, pulmonary
emphysema, atherosclerosis, rheumatoid arthritis,
arthrosclerosis, osteoarthritis, psoriasis, pancreatitis,
periodontosis, pulmonary fibrosis, cystic fibrosis,
chronic bronchitis, bronchiectasia, diffuse
-l - ~~585~J
panbronchiolitis, respiratory injury, adult respiratory
distress syndrome and the like, and further is useful for
treatment or prevention of asthma, graft rejection
nephritis, hydroa, disseminated intravascular coagulation,
shock, systemic lupus erythematosus, clone disease,
ischemia-reperfusion injury, chronic obstructive pulmonary
disease (COPD), premature rupture of the membrane (PROM),
corneal Barring or fibroblast proliferation (ocular
coagulation, burns, mechanical and chemical injury,
kerato-conjunctivitis, etc.), and sepsis.
In order to illustrate the usefulness of the
trifluoromethylketone derivatives (I) and their
pharmaceutically acceptable salt, pharmacological test
data thereof are shown below.
Test 1. . Protease Inhibition assay (in vitro)
(1) Method .
A buffer used throughout the assay was O.1M HEPE~
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
containing 0.5M NaCl, pH 7.5. Twenty-five microliters of
2 mM methoxysuccinyl-(Ala)2-Pro-Val-p-nitroanilide (100 mM
of dimethyl sulfoxide solution were diluted in the buffer)
and 50 ul of sample (10 ul of sample in organic solvent was
diluted 5-fold in the buffer) were mixed in wells of 96
well-microliter plate. An absorbance of the mixture in
wavelength at 415 nm was measured by a microplate reader
(Corona Electric Co., Ibaraki, Japan). After the measurement,
25 ul of 6 ug/ml human sputum elastase (HSE) was added and the
mixture was allowed to stand for 30 minutes at room
temperature. Then, the absorbance at 415 nm was measuxed.
Percent inhibition by drug was determined by 100 x (1-"r"
inhibitor present/"r" inhibitor absent), where "r" is
absorbance after 30 minutes incubation minus absorbance before
enzyme addition. Effect of inhibitors against porcine pancreas
elastase (Type IV, 5 ug/ml final) was assayed similarly using
N-succinyl-(Ala)3-p-nitroanilide. HSE was obtained from
Elastin Products Company Inc., MO, U.S.A. All other substrates
and protease were purchased from Sigma Chemicals Co.
- 12 -
Inhibitory effect
on several serine
protease activity
IC50 (M)
Test Compound Human sputum Porcine pancreas
(Example No.) elastase elastase
1 4.5 x 10 7 4.4 x 10
6
3 9.8 x 10 7 8.7 x 10-6
4 1.4 x 10 6 2.9 x 10
6
5 3.0 x 10 7 5.1 x 10
6
6 4.3 x 10 7 6.9 x 10
6
7 3.2 x 10 7 3.8 x 10
6
8 8.7 x 10 7 1.6 x 10
5
9 8.7 x 10 7 9.5 x 10-6
10 8.0 x 10 6 3.1 x 10
4
13 7.2 x 10 7 3.3 x 10-6
15 6.1 x 10 7 2.0 x 10
6
16 7.1 x 10 6 3.0 x 10
5
17 1.1 x 10 ~' 3.4 x 10
6
18 6.1 x 10 7 3.8 x 10
6
19 6.8 x 10 7 3.0 x 10
6
2O 8.9 x 10 7 2.4 x 10
6
21 1.2 x 10 6 3.7 x 10
6
22 6.8 x 10 7 4.9 x 10
6
23 8.1 x 10 7 2.4 x 10
6
24 1.4 x 10 6 3.8 x 10
6
27 2.4 x 10 6 1.6 x 10
5
,. ,. _ . _ _ - _
H
~... - 13 - ~QS~~s~l
Test 2. . Determination of the activity in
elastase-induced pulmonary damage.
(1) Method .
Hamsters under pentobarbital anesthesia were used.
Saline or saline-containing human sputum elastase was
instilled intratracheally via a small incision in the
ventral neck region using 1-ml syringe with a 27-gauge
needle. After 3 hours, animals were sacrificed by C0~
asphyxiation, each animal's trachea was reexposed. The
lungs were then laveged using a 2.5-ml aliquot of saline
and then withdrawing the saline, yielding a final volume
of approximately 1.5 ml bronchoalveolar lavage (BAC) fluid
from each animal.
The cells of BAL fluid were collected by
centrifugation and were then diluted with distilled water
to disrupt, and the hemoglobin contents determined
spectrophotometrically at 541 nm.
Test drugs were dissolved in saline or methyl
cellulose, and instilled intratracheally in the same
manner as used to instill elastase, at 5 minutes before
instillation of elastase.
(2) Result .
Inhibitory effect on elastase-induced lung hemorrhage
o ED50
Test Compound 5 min. Hemorrhage inhi-
predose
(Example No.) (ug/site) (~D541 nm) bition (ug/site)
Normal 0.6449~ 0.173*** -
Control 14.66 ~ 1.68 -
13 1 11.63 ~ 1.99 21.6 3.0
10 4.217 ~ 1.02** 74.5
100 0.6384~ 0.222*** 100.0
.w. - 14 - ~~15~,~s0
Normal 0.8352 0.423*** -
Control 12.89 1.44 -
19 1 11.59 1.40 10.8
10 1.663 0.690*** 93.1 2.4
100 0.2141 0.020*** 105.2
Normal 0.8352 0.423*** -
Control 12.89 1.44 -
20 1 10.94 1.35 16.1
10 2.522 0.803*** 86.0 2.5
100 0.2680 0.050*** 104.7
Normal 0.6449 0.173*** -
Control 14.66 1.68 -
21 1 12.76 1.11 13.6
10 5.372 2.06** 66.3 4.5
100 0.6476 0.129*** 100.0
Normal 0.9424 0.403*** -
Control 11.05 1.40 -
23 1 9.435 0.941 16.0
10 3.412 1.31** 75.6 3.2
100 0.7258 0.303*** 102.1
Normal 0.3203 0.159** -
Control 14.11 1.80 -
27 1 10.68 1.25 24.8
10 4.878 0.917** 66.9 3.7
100 0.3451 0.084*** 99.8
Normal 0.3199 0.159*** -
Control 14.11 1.80 -
29 1 12.05 1.95 14.9
10 8.155 1.76* 43.2 17.9
100 4.545 1.49** 69.4
*P<0.05, ** P<0.01, ***P<0.001 (Student' s t-test)
- 15 -
~0'Sg56b
Test 3: Effect on human sputum elastase induced paw
edema in mice.
(1) Materials and methods;
Male C57BL mice at the age of 7-8 weeks were obtained
from Japan Clear Inc,.
Human sputum elastase (HSE) was purchased from
Elastin Products Company, Inc,. The test drug was
administered subcutaneously, and 15 minutes later, HST was
injected into the right hind footpad at the dosage of 20
ug/site, and saline into the left hind footpad as the
control. After 2 hours of HSE injection, the paw edema
was measured with the dial thickness gage, and the
difference of the thickness between right and left hind
footpads was calculated.
(2) Results:
Effect on the elastase induced paw edema in mice
Treatment of
the compound Thickness of footpad %Inhibition
of Example 19 n (x 10 2mm) of
paw edema
(mg/kg)
Control 5 46.8 5.76
1 5 43.8 5.67 6.4
10 5 37.0 13.41 20.9
100 5 27.0 4.66* 42.3
* p<0.05 vs control group (Student's t test)
Test 4: Effect on experimentally induced emphyseman in
hamsters
(1) Materials and methods:
_u _ 16 _ , 20 58'S 60
Male golden Syrian hamsters, weighing approximately
120 g, were obtained from Japan SLC Inc,.
Porcine pancreatic elastase (PPE) was purchased from
Elastin Products Company, Inc,. Dialferin* was purchased
from Japan ROche Inc,.
Hamsters were anesthetized intraperitoneally with
Pentobarbital. The compound of Example 19 was dissolved
in saline. Both prior art compounds A and B were
suspended in 0.5 o methyl cellulose. The drugs were
instilled intratracheally through the oral cavity, 5
minutes before 100 ug/site of PPE in 0.2 ml of saline
instillation. Three weeks after PPE instillation, the
hamsters were anesthetized with Pentobarbital.
Respiratory mechanics were studied in supine hamsters
using a whole body, constant-volume, variable pressure
plethysmograph to measue volume. A water-filled
esophageal catheter was used to estimate pleural pressure.
Quasi-static deflation pressure-volume (P-V) curves were
obtained by intraperitoneally administered Dialferin to
suppress spontaneous breathing inflating the lungs to a
transpulmonary pressure (PL) of 30 cm H20, permitting slow
deflation to a PL of 0 cm H20 and gently aspirating to a
PL of -20cm H20. Quasi-static lung compliance (Cst) was
deffined as the slope of the steep portion of the deflation
P-V curve in the mid-volume range. Vital capacity was
defined as the difference in lung volume between TLC25
(volume at PL=25cm H20) and RV-20 (volume at PL=-20cm
H20).
(2) Results:
Pretreatment with the compound of Example 19
prevented the development of PPE-induced increases in lung
mechanics in a dose dependent manner as shown in the
following table. Considering Cst and VC values, the
3 5 *Trade-mark
B
- 17 20S$S~~
potency of the compound of Example 19 was superior to the
prior art compounds A and B.
Effect on experimentally induced emphysema in
hamsters
Treatment n Cst VC
(~g/site) (ml/cmH20) (ml)
Exp. 1
Normal 8 0.53 0.02*** 4.9 0.1 ***
Control 8 1.54 0.10 7.3 0.2
Compound of
Example 19
1 8 1.39 0.05(150) 7.0 0:1(120)
10 8 0.70 0.04(84x)*** 5.8 0,1(650)***
100 7 0.53 0.02(1000)***5.0 0.1(970)***
Exp. 2
Normal 8 0.51 0.02*** 4.6 0.1 ***
Control 8 1.62 0.12 6.8 0.1
Prior art
Compound A
100 8 1.55 0.11(7%) 6.9 0.2(-l0)
1000 8 1.22 0.15(370) 6.6 0.2(9%)
Exp. 3
Normal 8 0.49 0.02** 4.6 0.2 ***
Control 6 1.18 0.21 7.0 0.2
Prior art
Compound B
32 8 1.95 0.08(32%) 6.1 0.3(39%)*
320 8 0.74 0.05(63%)* 5.8 0.2(490)**
Cst=Quasi static lung compliance, VC=Vital capacity
(o)=Inhibition
*:p<0.05, **:p<0.01, ***:p<0.001 vs Control (Student's t test)
1 a ~~?S'~5~'~
Prior art compound A (Japanese Kokai Tokkyo Koho No.61-218518):
CH3 CH3
CH CH3 CH3
_/ \ ~ \
C1 ~ S02-NHCO~-CONHCH-CON CH
CONHCH-COCF3
Prior art compound B (Japanese Kokai Tokkyo Koho No.2-256657):
CH3 CH3 CH3 CH3
CH I, CH
\
C1~-S02NHC0~ CONHCH-CONCH2CONHCH-COCF3
Pharmaceutical compositions of this invention can be used
in a conventional pharmaceutical forms such as powders, fine
granules, granules, tablets, dragee, injections, inhalations,
microcapsules, capsules, suppository, solution, suspension,
emulsion, syrups and the like. If desired, diluents ar
disintegrators (e. g. sucrose, lactose, starch, crystalline
cellulose, low-substituted hydroxypropyl cellulose, synthetic
aluminum silicate, etc.), binding agents (e. g. cellulose,
methylcellulose, hydroxypropylcellulose,
hydroxypropylmethylcellulose, polypropylpyrrolidone,
polyvinylpyrrolidone, gelatin, gum arabic, polyethyleneglycol,
etc.), coloring agents, sweeting agents, lubricant (e. g.
magnesium stearate, etc.) or the like, may be dispensed with
said composition.
205~5~0
The dosage of said composition of this invention
depends on the patient's age, body weight, condition,
etc., and it is generally administered by the oral or
inhale route at the daily dose level of 1 mg to 1 g a~ the
object compound or its pharmaceutically acceptable salt,
preferably 10 mg to 500 mg on the same basis, at the
interval of 1 to 3 times a day. Typical unit doses may be
5 mg, 10 mg, 20 mg, 50 mg, 100 mg and the like, although
these are only examples and not limitative.
The following Preparations and Examples are given for
the purpose of illustrating this invention.
In the Preparations and Examples, the following
abbreviations are used.
WSCD . 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
HOBT . N-hydroxybenzotriazole
DMF . N,N-dimethylformamide
DMSO . dimethylsulfoxide
30
-20- 2058560
Preparation 1
To a solution of N-(tert-butoxycarbonyl)-L-valine
(10.86 g) and L-proline benzyl ester hydrochloride (12.09
g) in DMF (50 ml) were added HOBT (6.76 g) and WSCD (7.76
g) under ice-bath cooling. After being stirred at room
temperature for 18 hours, the reaction mixture was
concentrated under reduced pressure. The residue was
dissolved in ethyl acetate (400 ml) and washed with 50
aqueous citric acid (200 ml) saturated aqueous sodium
bicarbonate (200 ml). The solution was dried over
magnesium sulfate and evaporated under reduced pressure to
give N-(tert-butoxycarbonyl)-L-valyl-L-proline benzyl
ester (20.06 g) as an oil.
TLC (Kiesel gel*60 F254 silica gel plate, Merck)
(The same meaning in the following Preparations and
Examples, unless otherwise indicated)
Rf . 0.62 (Hexane:AcOEt = 2:1)
The following compounds were prepared by a similar
method to that of Preparation 1.
Preparation 2
4-(Methoxycarbonyl)phenylcarbonyl-L-valyl-L-proline
benzyl ester
oil
TLC Rf . 0.89 (CHCI3:MeOH = 10:1)
Preparation 3
3(RS)-[[4-(Methoxycarbonyl)phenylcarbonyl]-L-valyl-
L-prolyl]amino-1,1,1-trifluoro-2(RS)-hydroxy-4-
methylpentane
mp . 64-67°C
TLC Rf . 0.63 and 0.60 (CHCI3:MeOH = 10:1)
Preparation 4
3(R or S)-[[4-(Methoxycarbonyl)phenylcarbonyl]-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-2(R or S)-hydroxy-4-
methylpentane
*Trade-mark
m
- 21 - ;~osssso
mp . 65-75°C
TLC Rf . 0.65 (CHCI3:MeOH = 10:1)
[a]D2 . -56.23° (C=0.14, MeOH)
Preparation 5
3(R or S)-[[4-(Benzyloxycarbonylmethylaminocarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-2(R
or S)-hydroxy-4-methylpentane
mp . 70-80°C
TLC Rf . 0.65 (CHCI3:MeOH = 10:1)
[a]D2 . -46.59° (C=0.165, MeOH)
Preparation 6
3(R or S)-[[4-(Benzyloxycarbonylmethylamino-
carbonyl)phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-
trifluoro-2(R or S)-hydroxy-4-methylpentane
mp . 80-90°C
TLC Rf . 0.60 (CHCI3:MeOH = 10:1)
[a]r2 . -27.59° (C=0.165, MeOH)
Preparation 7
3(R or S)-[[4-(Methoxycarbonyl)phenylcarbonyl]-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-2(R or S)-hydroxy-
4-methylpentane
mp . 68-85°C
TLC Rf . 0.60 (CHCI3:MeOH = 10:1)
[a]D2 . -42.63° (C=0.175, MeOH)
Preparation 8
N-(tert-Butoxycarbonyl)-L-valyl-L-proline benzyl
ester (20.0 g) was dissolved in 4N hydrogen chloride in
dioxane (30 ml) under ice bath cooling. After being
stirred at room temperature for one hour, the reaction
mixture was evaporated under reduced pressure. The
residue was pulverized with ether to give L-valyl-L-
- 22 -
2Q~~~'6 J
proline benzyl ester hydrochloride (14.56 g).
mp . 66-69°C
TLC Rf . 0.55 (CHCI3:MeOH = 10:1)
Preparation 9
A solution of N-4-(methoxycarbonyl)phenylcarbonyl-L-
valyl-L-proline benzyl ester (18.53 g) in methanol (150
ml) was hydrogenated over lOs palladium on carbon (1.0 g)
at 3 atmosphere pressure of hydrogen for 1.5 hours at room
temperature. After the catalyst was removed by
filtration, the filtrate was evaporated under reduced
pressure to give 4-(methoxycarbonyl)phenylcarbonyl-L-
valyl-L-proline (14.20 g).
mp . 68-71°C
TLC Rf . 0.27 (CHCI3:MeOH = 10:1)
Preparation 10
To a solution of oxalyl chloride (0.82 ml) in
dichloromethane (5 ml) were added dimethylsulfoxide (7,.34
ml) and a solution of 3(RS)-[[4-(4-methoxycarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-
2(RS)-hydroxy-4-methylpentane (2.5 g) in dichloromethane
(10 ml) at -70°C successively. After the mixture was
stirred for one hour at -40°C, triethylamine (2.63 ml) was
added. The mixture was stirred for an additional 30
minutes at the same temperature and washed with 0.5N
hydrochloric acid (15 ml) and 5~ sodium bicarbonate
aqueous solution (15 ml). The organic layer was dried
over magnesium sulfate and evaporated under reduced
pressure. The residue was purified with silica gel (50 g)
column chromatography (chloroform: methanol = 50:1) to give
3(RS)-[[4-(4-methoxycarbonyl)phenylcarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (2.18
g)~
mp . 67-70°C
- 23 - 2i~5$560
TLC Rf . 0.51 (CHCI3:MeOH = 10:1)
Preparation 11
To a solution of 3(RS)-[[4-(4-methoxycarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane (2.1 g) in methanol (40 ml) was added
1N aqueous sodium hydroxide (15 ml) under ice-bath
cooling. After the mixture was stirred at room
temperature for 2 hours, methanol was evaporated. The
concentrate was washed with ether (30 ml), then acidified
to pH 2 with 1N hydrochloric acid. The aqueous solution
was extracted with ethyl acetate (20 ml). The extract was
washed with brine (10 ml), dried over magnesium sulfate
and concentrated under reduced pressure to give
3(RS)-[(4-carboxyphenylcarbonyl)-L-valyl-L-prolyl]amirio-
1,1,1-trifluoro-4-methyl-2-oxopentane (1.94 g).
mp . 215-220°C
TLC Rf . 0.63 (CHCI3:MeOH:AcOH = 8:2:1)
The following compounds were prepared by a similar
method to that of Preparation 11.
Preparation 12
3(R or S)-[[4-(Carboxy)phenylcarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-2(R or S)-hydroxy-4-
methylpentane (0.61 g)
mp . 266-269°C
TLC Rf . 0.42 (Benzene:EtOAc:AcOH = 20:20:1)
[a]n2 . -49.71° (C=0.1, MeOH)
Preparation 13
3(R or S)-[[4-(Carboxy)phenylcarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-2(R or S)-hydroxy-4-
methylpentane
mp . 265-268°C
- 24 - ~~5$~~~
TLC Rf . 0.40 (Benzene:EtOAc:AcOH = 20:20:1)
LalD2 . -64.25° (C=0.16, MeOH)
Preparation 14
A solution of 28~ sodium methoxide in methanol (3.0
ml) was added to a solution of 3(RS)-amino-1,1,1-
trifluoro-2(RS)-hydroxy-4-methylpentane hydrochloride (3.2
g) in ethanol (30 ml) at room temperature. After removal
of the precipitated sodium chloride by filtration,
(2R,3R)-L-tartaric acid (2.3 g) was added to the filtrate.
The mixture was warmed until tartaric acid was dissolved
and filtrated. The filtrate was allowed to stand for 5
hours at room temperature. The precipitated crystalline
solid (1.94 g) was collected by filtration and dissolved
in 1N aqueous sodium hydroxide. The solution was
extracted with ethyl acetate (10 ml) and the extract was
mixed with 4N-hydrogen chloride in ethyl acetate. After
removal of ethyl acetate, the residue was pulverized with
diisopropyl ether (10 ml) to give 3(R or S)-amino-1,1,1-
trifluoro-2(R or S)-4-methylpentane hydrochloride (1.0:3
g)-
mp . 165-170°C
TLC Rf . 0.50 (CHCI3:MeOH = 10:1)
[a]D2 . +11.39° (C=0.13, MeOH)
The following compound was prepared by a similar
method to that of Preparation 14.
Preparation 15
3(R or S)-Amino-1,1,1-trifluoro-2(R or S)-4-
methylpentane hydrochloride
mp . 165-170°C
TLC Rf . 0.55 (CHCI3:MeOH = 10:1)
fa]n2 . -10.56° (C=1.05, MeOH)
.w. ~~58~~~
- 25 -
Preparation 16
To a solution of methyl p-aminobenzoate (0.4 g) in
tetrahydrofuran (10 ml) was added
trichloromethylchloroformate (0.31 g) and the mixture was
allowed to stand overnight at room temperature. L-Vaiine
benzyl ester (0.54 g) was added to the solution and the pH
of the mixture was neutralized with triethylamine. After
being stirred for 30 minutes at room temperature, the
mixture was concentrated under reduced pressure and the
residue was extracted with ethyl acetate (10 ml). The
extract was washed with 1N-hydrochloric acid (10 ml) and
aqueous sodium bicarbonate (10 ml) and concentrated to
dryness to give 4-(methoxycarbonyl)phenylaminocarbony~-L-
valine benzyl ester (1.17 g).
TLC Rf . 0.55 (CHCI3:MeOH = 10:1, V/V)
Preparation 17
4-(Methoxycarbonyl)phenylaminocarbonyl-L-valine (0.83
g) was prepared from 4-(methoxycarbonyl)-
phenylaminocarbonyl-L-valine benzyl ester (1 g) by a
similar method to that of Preparation 9.
TLC Rf . 0.3 (CHCI3:MeOH:H20 = 65:25:4)
Oil
Preparation 18
[4-(Methoxycarbonyl)phenylaminocarbonyl]-L-valyl-L-
proline benzyl ester (1.0 g) was prepared from
4-(methoxycarbonyl)phenylaminocarbonyl-L-valine (0.83 g)
and L-proline benzyl ester hydrochloride (0.65 g) by a
similar method to that of Preparation 1.
TLC Rf . 0.60 (CHCI3:MeOH = 10:1)
Oil
Preparation 19
[4-(Methoxycarbonyl)phenylaminocarbonyl]-L-valyl-
_.
- 26 -
L-proline (0.69 g) was prepared from [4-(methoxycarbonyl)-
phenylaminocarbonyl]-L-valyl-L-proline benzyl ester (1.0
g) by a similar method to that of Preparation 9.
TLC Rf . 0.35 (CHCI3:MeOH:H20 = 65:25:4)
Oil
Preparation 20
3(RS)-[(4-(Methoxycarbonyl)phenylaminocarbonyl]-L-
valyl-L-propyl]amino-l,l,l-trifluoro-2(RS)-hydroxy-4-
methylpentane (1.03 g) was prepared from
[4-(methoxycarbonyl]phenylaminocarbonyl]-L-valyl-L-proline
(0.69 g) and 3(RS)-amino-1,1,1-trifluoro-2(RS)-hydroxy-4-
methylpentane hydrochloride (0.37 g) by a similar method
to that of Preparation 1.
TLC Rf . 0.45 (CHCI3:MeOH = 10:1)
Oil
Preparation 21
3(RS)-[[4-(Methoxycarbonyl)phenylaminocarbonyl]-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-2-oxo-4-methylper~tane
(0.98 g) was prepared from 3(RS)-[[4-(methoxycarbonyl)-
phenylaminocarbonyl]-L-valyl-L-prolyl]amino-1,1,1-
trifluro-2(RS)-hydroxy-4-methylpentane (1.0 g) by a
similar method to that of Preparation 10.
mp . 90-100°C
TLC Rf . 0.50 (CHCI3:MeOH = 10:1)
Preparation 22
3(RS)-[[4-(Carboxy)phenylaminocarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-2-oxo-4-methylpentane (0.2 g)
was prepared from 3(RS)-[[4-(methoxycarbonyl)phenylamino-
carbonyl]-L-valyl-L-prolyl]amino]-1,1,1-trifluoro-2-oxo-4-
methylpentane (0.3 g) by a similar method to that of
Preparation 11.
mp . 125-130°C
- 27 - ~~'SB~~d
TLC Rf . 0.50 (CHCI3:MeOH:H20 = 65:25:4)
Preparation 23
To a solution of N-(tert-butoxycarbonyl)-L-valin~
(4.35 g) and triethylamine (2.13 g) in dry CH2C12 (40 ml)
was added isobutyl chloroformate (2.87 g) at -20°C. After
being stirred at same temperature for 30 minutes, a
solution of N-(2-indanyl)glycine benzyl ester (5.37 g) in
dry CH2C12 (20 ml) was added at -20°C. The reaction
mixture was stirred at -10°C for one hour then at room
temperature for 4 hours. After the reaction mixture was
evaporated under reduced pressure, the residue was
dissolved in ethyl acetate (100 ml) and washed with
saturated aqueous sodium bicarbonate (100 ml x 2). The
organic layer was dried over magnesium sulfate and
evaporated under reduced pressure. The residue was
purified with silica gel (50 g) column chromatography
(CHCI3:AcOEt = 10:1) to give N-(tert-butoxycarbonyl)-L-
valyl-N-(2-indanyl)glycine benzyl ester (2.20 g) as an
oil.
TLC Rf . 0.78 (Hexane:AcOEt = 2:1)
Preparation 24
L-Valyl-N-(2-indanyl)glycine benzyl ester
hydrochloride (1.83 g) was prepared from
N-(tert-butoxycarbonyl)-L-valyl-N-(2-indanyl)glycine
benzyl ester (2.14 g) by a similar method to that of
Preparation 8.
mp . 162-163°C
TLC Rf . 0.58 (CHCI3:MeOH = 10:1)
Preparation 25
N-[4-(Methoxycarbonyl)phenylcarbonyl]-L-valyl-N-(2-
indanyl)glycine benzyl ester (1.24 g) was prepared from
L-valyl-N-(2-indanyl)glycine benzyl ester hydrochloride
- 28 -
iG:~3~~~~~
(1.80 g) and terephthalic acid mono methyl ester (0.86 g)
by a similar method to that of Preparation 1.
mp . 72-76°C
TLC Rf . 0.29 (CHC13)
Preparation 26
N-[4-(Methoxycarbonyl)phenylcarbonyl]-L-valyl-N-(2-
indanyl)glycine (0.83 g) was prepared from
N-[4-(methoxycarbonyl)phenylcarbonyl]-L-valyl-N-(2-
indanyl)glycine benzyl ester (1.20 g) by a similar method
to that of Preparation 9.
mp . 163-164°C
TLC Rf . 0.48 (CHCI3:MeOH = 10:1)
Preparation 27
3(RS)-[[4-(Methoxycarbonyl)phenylcarbonyl]-L-valyl-
N-(2-indanyl)glycyl]amino-1,1,1-trifluoro-2(RS)-hydroxy-4-
methylpentane (1.06 g) was prepared from
N-[4-(methoxycarbonyl)phenylcarbonyl]-L-valyl-N-(2-
indanyl)glycine (0.80 g) and 3(RS)-amino-1,1,1-trifluoro-
2(RS)-hydroxy-4-methylpentane hydrochloride (385 mg) by a
similar method to that of Preparation 1.
mp . 76-78°C
TLC Rf . 0.71 (CHCI3:MeOH = 10:1)
Preparation 28
3(RS)-[[4-(Methoxycarbonyl)phenylcarbonyl]-L-valyl-N-
(2-indanyl)glycyl]amino-1,1,1-trifluoro-4-methyl-2-
oxopentane (0.84 g) was prepared from 3(RS)-[[4-(methoxy-
carbonyl)phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]-
amino-1,1,1-trifluoro-2(RS)-hydroxy-4-methylpentane (1.03
g) by a similar method to that of Preparation 10.
mp . 62-64°C
TLC Rf . 0.74 (CHCI3:MeOH = 10:1)
- 29 -
Preparation 29
3(RS)-[(4-Carboxyphenylcarbonyl)-L-valyl-N-(2-
indanyl)glycyl]amino-1,1,1-trifluoro-4-methyl-2-oxoperitane
(0.76 g) was prepared from 3(RS)-[[4-(methoxycarbonylj-
phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]amino-1,1,1-
trifluoro-4-methyl-2-oxopentane (0.83 g) by a similar
method to that of Preparation 11.
mp . 84-86°C
TLC Rf . 0.15 (CHCI3:MeOH = 10:1)
Example 1
To a mixture of glycine benzyl ester p-toluene-
sulfonate (66 mg) and 3(RS)-[(4-carboxyphenylcarbonyl}-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-4-methyl-2-oxoperltane
(100 mg) in DMF (6 ml) were added HOBT (26 mg) and WSCD
(30 mg) under ice-bath cooling. After being stirred at
room temperature for 4 hours, the mixture was concentrated
under reduced pressure. The residue was dissolved in
ethyl acetate (30 ml) and washed with 5o aqueous citric
acid (20 ml), water (20 ml), 5% aqueous sodium bicarbonate
(20 ml) and brine (20 ml). The solution was dried over
magnesium sulfate and evaporated under reduced pressure to
give 3(RS)-[[4-[(benzyloxycarbonyl)methylaminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane (125 mg).
mp . 77-78°C
TLC Rf . 0.56 (CHCI3:MeOH = 10:1)
The following compounds were prepared by a similar
method to that of Example 1.
Example 2
3(RS)-[[4-[[2-(4-Morpholino)ethyl]aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane
e... - 3 0 -
2~?5~5~~J
mp . 98-102°C
TLC Rf . 0.24 (CHCI3:MeOH = 10:1)
Example 3
3(RS)-[[4-[(3-Benzoylmethoxycarbonyl)propylamino-
carbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-
trifluoro-4-methyl-2-oxopentane
mp . 70-73°C
TLC Rf . 0.71 (CHCI3:MeOH = 10:1)
Example 4
3(RS)-[[4-[(4-Morpholino)aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane
Z5 mp . 191-193°C
TLC Rf . 0.59 (CHCI3:MeOH = 10:1)
Example 5
3(RS)-[[4-[(4-Chlorobenzyl)aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluora-4-
methyl-2-oxopentane
mp . 87-89°C
TLC Rf . 0.43 (CHCI3:Me0H = 10:1)
Example 6
3(RS)-[[4-[(4-Nitrobenzyl)aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane
mp . 94-96°C
TLC Rf . 0.57 (CHCI3:MeOH = 10:1)
Example 7
3(RS)-[[4-[(4-Chlorophenyl)aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro--4-
methyl-2-oxopentane
- 31 -
mp . 71-72°C
TLC Rf . 0.79 (CHCI3:Me0H = 10:1)
Example 8
3(RS)-[[4-[[(1(R)-Benzyloxycarbonyl)-2-phenyl]-
ethylaminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]a~ino-
1,1,1-trifluro-4-methyl-2-oxopentane
mp . 65-67°C
TLC Rf . 0.82 (CHCI3:MeOH = 10:1)
Example 9
3(RS)-[[4-[[(1(S)-Benzyloxycarbonyl)-2-phenyl]eth~yl-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane
mp . 108-110°C
TLC Rf . 0.82 (CHCI3:MeOH = 10:1)
Example 10
3(RS)-[[4-[[1(S),3-bis(Benzyloxycarbonyl)propyl]-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane
mp . 58-60°C
TLC Rf . 0.83 (CHCI3:MeOH = 10:1)
Example 11
To a solution of oxalyl chloride (0.09 ml) in
dichloromethane (2 ml) were added dimethylsulfoxide (0.15
ml) and a solution of 3(R or S)-[[4-
(benzyloxycarbonylmethylaminocarbonyl)phenylcarbonyl]-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-2(R or S)-hydroxy-
4-methylpentane (0.35 g) in dichloromethane (4 ml) at 70°C
successively. After the mixture was stirred for one hour
at -40°C, triethylamine (0.29 ml) was added. The mixture
was stirred for an additional 30 minutes at the same
temperature and washed with 0.5N hydrochloric acid (15 ml)
e~.. -~2-
and 5% sodium bicarbonate aqueous solution (15 ml). The
organic layer was dried over magnesium sulfate and
evaporated under reduced pressure. The residue was
purified with silica gel (20 g) column chromatography
(Chloroform:methanol = 50:1) to give 3(R or S)-[[4-
(benzyloxycarbonylmethylaminocarbonyl)phenylcarbonyl]-L-
valyl-L-prolyl]amino-1,1,1-trifluoro-2-oxo-4-
methylpentane (0.22 g).
mp . 159-161°C
TLC Rf . 0.63 (CHCI3:MeOH = 10:1)
[a]D2 . -39.66° (C=0.105, MeOH)
The following compound was prepared by a similar
method to that of Example 11.
Example 12
3(R or S)-[[4-(Benzyloxycarbonylmethylaminocarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-2-
oxo-4-methylpentane
mp . 141-143°C
TLC Rf . 0.68 (CHCI3:MeOH = 10:1)
[a]D2 . -41.90° (C=0.15, MeOH)
Example 13
A solution of 3(RS)-[[4-[(benzyloxycarbonyl)-
methylaminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]-
amino-1,1,1-trifluoro-4-methyl-2-oxopentane (70 mg) in a
mixture of methanol (10 ml) and water (1 ml) was
hydrogenated over loo palladium on carbon (1.0 g) at 3
atmosphere pressure of hydrogen for 3 hours at room
temperature. After the catalyst was removed by
filtration, the filtrate was evaporated under reduced
pressure to give 3(RS)-[[4-(carboxymethylaminocarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane (61 mg).
_. - 33 -
205560
mp . 99-103°C
TLC Rf . 0.17 (CHCI3:MeOH:AcOH = 8:1:1)
Example 14
To a solution of 3(RS)-[[4-[(3-benzoylmethoxy-
carbonyl)propylaminocarbonyl]phenylcarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (120
mg) in acetic acid (5 ml) was added zinc powder (120 mg)
under ice-bath cooling. After the mixture was stirred at
room temperature for 2 hours, zinc was removed by
filtration. The filtrate was evaporated under reduced
pressure to give 3(RS)-[[4-[(3-carboxypropyl)-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane (95 mg).
mp . 84-86°C
TLC Rf . 0.32 (CHCI3:MeOH:AcOH = 8:1:1)
The following compounds were prepared by a similar
method to that of Example 13.
Example 15
3(R or S)-[[4-(Carboxymethylaminocarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-2-
oxo-4-methylpentane
mp . 75-120°C
TLC (RP-18 WF254S (made by E. Merck)
Rf . 0.55 (MeOH:H20 = 6:5)
[a]D2 . -35.10° (C=0.105, MeOH)
Example 16
3(R or S)-[[4-(Carboxymethylaminocarbonyl)-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro~2-
oxo-4-methylpentane
mp . 90-110°C
TLC (RP-18 WF254S (made by E. Merck)
- 3 4 - ~p58~~p
Rf . 0.50 (MeOH:H20 = 6:5)
[a]D2 . -50.04° (C=0.115, MeOH)
Example 17
3(RS)-[[4-[(1(S),3-Dicarboxypropyl)aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-
4-methyl-2-oxopentane
mp . 74-76°C
TLC Rf . 0.13 (CHCI3:MeOH:AcOH = 8:1:1)
Example 18
A solution of 3(RS)-[[4-[(4-nitrobenzyl)-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane (50 mg) in methanol
(10 ml) was hydrogenated over 10°s palladium on carbon (10
mg) at 4 atmosphere pressure of hydrogen for 2 hours. The
catalyst was removed by filtration and the filtrate was
evaporated under reduced pressure to give 3(RS)-[[4-[(4-
aminobenzyl)aminocarbonyl]phenylcarbonyl]-L-valyl-L-
prolyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (46
mg).
mp . 90-92°C
TLC Rf . 0.44 (CHCI3:MeOH:AcOH = 8:1:1)
Example 19
To a solution of 3(RS)-[[4-(carboxymethylamino-
carbonyl)phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-
trifluoro-4-methyl-2-oxopentane (0.50 g) in water (10 ml)
was added 1N aqueous sodium hydroxide (0.88 ml) at rooms
temperature. The solution was lyophilized to give sodium
salt of starting material (0.52 g).
mp . >230°C
TLC Rf . 0.17 (CHCI3:MeOH:AcOH = 8:1:1)
The following compound was prepared by a similar
-. - 35 -
method to that of Example 19.
Example 20
A sodium salt of 3(RS)-[[4-[(3-carboxypropyl)-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane
mp . 66-69°C
TLC Rf . 0.32 (CHCI3:MeOH:AcOH = 8:1:1)
Example 21
To a solution of 3(RS)-[[4-[[2-(4-morpholino)ethyl]-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane (80 mg) in
1,4-dioxane (1 ml) was added 4N-hydrogen chloride in
dioxane (0.1 ml). The mixture was stirred at room
temperature for 10 minutes, and evaporated to give
3(RS)-[[4-[[2-(4-morpholino)ethyl]aminocarbonyl]-
phenylcarbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-
methyl-2-oxopentane hydrochloride (83 mg).
mp . 64-65°C
TLC Rf . 0.24 (CHCI3:MeOH = 10:1)
Example 22
3(RS)-[[4-(Benzyloxycarbonylmethylaminocarbonyl)-
phenylaminocarbonyl]-L-valyl-L-prolyl]amino-1,1,1-
trifluoro-2-oxo-4-methylpentane (0.25 g) was prepared from
3(RS)-[[4-(carboxy)phenylaminocarbonyl]-L-valyl-L-prolyl]-
amino-1,1,1-trifluoro-2-oxo-4-methylpentane (0.2 g) and
glycine benzyl ester para-toluenesulfonate (0.13 g) by a
similar method to that of Example 1.
mp . 65-70°C
TLC Rf . 0.15 (CHCI3:MeOH = 10:1)
Example 23
3(RS)-[[4-(Carboxymethylaminocarbonyl)phenylamino-
-. - 36 -
carbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-2-oxo-4-
methylpentane (0.14 g) was prepared from
3(RS)-[[4-(benzyloxycarbonylmethylaminocarbonyl)-
phenylaminocarbonyl)]-L-valyl-L-prolyl]amino-1,1,1-
trifluoro-2-oxo-4-methylpentane (0.2 g) by a similar
method to that of Example 13.
mp . 98-128°C
TLC Rf . 0.25 (CHCI3:MeOH:H.,O = 65:25:4)
Example 24
3(RS)-[[4-(Dimethylaminocarbonylmethylaminocarbonyl)-
phenylaminocarbonyl]-L-valyl-L-prolyl]amino-l,l,l-
trifluoro-2-oxo-4-methylpentane (0.03 g) was prepared from
3(RS)-[[4-(carboxymethylaminocarbonyl)phenylamino-
carbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-2-oxo-4-
methylpentane (0.08 g) and dimethylamine hydrochloride
(0.012 g) by a similar method to that of Example 1.
mp . 115-125°C
TLC Rf . 0.20 (CHCI3:MeOH = 10:1)
Example 25
3(RS)-[[4-[(Benzyloxycarbonyl)methylaminocarbonyl]-
phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]amino-1,1,1-
trifluoro-4-methyl-2-oxopentane (250 mg) was prepared from
glycine benzyl ester p-toluenesulfonate (118 mg) and
3(RS)-[(4-carboxyphenylcarbonyl)-L-valyl-N-(2-indanyl)-
glycyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (20~
mg) by a similar method to that of Example 1.
mp . 64-66°C
TLC Rf . 0.81 (CHCI3:MeOH = 10:1)
White powder
Example 26
3(RS)-[[4-(Carboxymethylaminocarbonyl)phenyl-
carbonyl]-L-valyl-N-(2-indanyl)glycyl]amino-1,1,1-
.- - 37 -
20~5$5S~
trifluoro-4-methyl-2-oxopentane (195 mg) was prepared from
3(RS)-[[4-[(benzyloxycarbonyl)methylaminocarbonyl]-
phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane (220 mg) by a
similar method to that of Example 13.
mp . 88-91°C
TLC Rf . 0.25 (CHCI3:MeOH:AcOH = 16:1:1)
Example 27
A sodium salt of 3(RS)-([4-(carboxymethylamino-
carbonyl)phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]-
amino-1,1,1-trifluoro-4-methyl-2-oxopentane (160 mg) was
prepared by a similar method to that of Example 19.
mp . 203-205°C
TLC Rf . 0.25 (CHCI3:MeOH:AcOH = 16:1:1)
Example 28
3(RS)-([4-[(2-(4-Morpholino)ethyl]aminocarbonyl]-
phenylcarbonyl]-L-valyl-N-(2-indanyl)glycyl]amino-1,1,1-
trifluoro-4-methyl-2-oxopentane (180 mg) was prepared from
4-(2-aminoethyl)morpholine (46 mg) and
3(RS)-[(4-carboxyphenylcarbonyl)-L-valyl-N-(2-indanyl)-
glycyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (206
mg) by a similar method to that of Example 1.
mp . 76-80°C
TLC Rf . 0.38 (CHCI3:MeOH = 10:1)
Example 29
A hydrochloride of 3(RS)-[[4-[[2-(4-morpholino)-
ethyl]aminocarbonyl]phenylcarbonyl]-L-valyl-N-(2-indanyl)-
glycyl]amino-1,1,1-trifluoro-4-methyl-2-oxopentane (170
mg) was prepared by a similar method to that of Example
21.
mp . 96-99°C
TLC Rf . 0.38 (CHCI3:MeOH = 10:1)
- 3 s - ~~58~~~0~
Example 30
3(RS)-[(4-(ethoxycarbonyl)methylaminocarbonyl]phenyl-
carbonyl]-L-valyl-L-prolyl]amino-1,1,1-trifluoro-4-methyl-
-2-oxopentane was prepared by a similar method to that of
Example 1.
mp: 96-99°C
Example 31
To a solution of 3(RS)-[[4-(ethoxycarbonyl)methyl-
aminocarbonyl]phenylcarbonyl]-L-valyl-L-prolyl]amino-
1,1,1-trifluoro-4-methyl-2-oxopentane (5.0 g) in
methylene chloride (60 ml), methanol (10 ml) and water (25
ml) was added aqueous sodium hydroxide (NaOH 0.6 g in
water (5 ml)) under ice-bath cooling. After the reaction
mixture was stirred at 0-10°C for 10 minutes, it was
adjusted to pH 9 with 6N hydrochloric acid and then
aqueous solution was washed with methylene chloride (60
ml). To the aqueous solution were added sodium chloride
(5 g) and ethylacetate (60 ml), and then the mixture was
acidified to pH 2 with 6N hydrochloric acid. Ethyl
acetate solution was washed with brine (30 ml), dried over
magnesium sulfate and concentrated under reduced pressure
to the volume of 20 ml. The concentrated solution was
added dropwise to isopropyl ether (225 ml) at room
temperature. Precipitate was filtered and then dried to
give 3(RS)-[[4-(carboxymethylaminocarbonyl)phenyl-
carbonyl]-L-valyl-L-prolyl]amino-1,1,1-trif luoro-4-methyl-
2-oxopentane (3.24 g).
mp: 99-103°C
35