Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
-~ W091tlO~ PCr/EP90/02156
,; . .~, ~. ...
"Cycloalkyl-substituted glutaramide diuretic agents
Ihis invention relates to a series of eyel ~ l~substitNted
glutaramide derivatives which are diuretic ager~ts having utility
in a variety of therapeutie areas ineluding the treatment of
various eardiovascular disorders such as hyFe~t~sion, heart
failure and ranal insuffieiency.
Ascording to the specification of our Eurcpean patent
applica~ions E$-A-0274234 and EP-A-0343911 we describe and claim
ce~tain cycloalkyl-substituted glutaramide derivatives as diuretic
agents. The present invention pr w ides further rela~ed ca~çounds
having a 2,3-dihydroindene substituent.
Ihe c=wpcunds are inhibitors of the zinc-depe ment, neutral
endopeptidase E.C.3.4.24.11. This enz~me is invol~ed in the
breakdown of several peptide hormones, including atrial
natriuretic factor (ANF), which is secreted by the heart and which
has potent vascdilatory, diuretic and natriuretic activity. Thus,
the cc~pcunds of the invention, ~y inhibiting the neutral
endo~epkidase E.C.3.4.24.11, can pokentiate the biologi~Al effects
of ANF, and in particular the c~w}Y~nds are diuretic agents having
utility in the treatment of a n~c~er of disorders, including
hypertension, hsart failure, an~ina, renal insufficiency,
pre=enstrual sy~drcme, cyclical oedema, Menieres disease,
hyperaldosteronism (primary and secondary) pulmonary ~edema,
ascites, and hypercalciuria. In addition, because of their
ability to potentiate the effects of ANF the ccr~ccunds have
utility in the treatment of glauc~ma. As a further result of
their ab;lity to inhibit the ~eutral endopepkidase E.C.3O4.24.11
, . . r
~, : . ' '. ,' :' `
' ': '
WO 91t10644 PCT/EP90/02156
~ 212`~
the ccnpc~nds of the m vention ~ay have activity in cther
therapeutic areas including for example the tre~tment of asth~a,
inflammation, pain, epilepsy, affective disorders, dementia and
geriatric confusion, obesity and gastr~in~estinal disorders
(especially diarrhoea and irritable kowel syndrome), the
modulation of gastric acid secretion and the treatment of
hyperreninaemia and leukaemia.
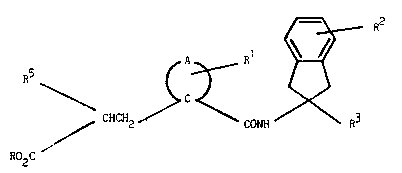
Ihe ccmçoonds of the present invention are of the formula:
/9~ R
~ CHCH2~ B
R02C
(I)
~herein A co~pletes a 4 to 7 mem~ered carbocyclic rLng which may
be saturated or mono-unsaturated and which may
optionally be fused to a further saturated or
: unsaturated 5 or 6 m~mbered carbocyclic ring ;
R is H, Cl-C6 alkyl, benzyl or an altErnative biolabile
ester-formung group;
Rl is H or ~ -C4 aIkyl;
R2 is H, OH, Cl-C~ alkyl, Cl-C4 alkoxy, halo or CF3;
,
, . .
:, . ~': ' : . .
~. WO 91/10644 ~ ~ 2 ~ 7~ 2 ~ 2 ~ PCI`/EP90/02156
3 . 4
R is CH2~1 or C~02R ~ere~n R is as previously defined
for R;
and R5 is C1~6 a~yl~ C2 6 al~l~ C2~6 a~l,
C3-C7 cyc:lo~l, or C3-C7 cycloaL~ryl,
or R5 is Cl-C6 alkyl substituted ~y halo, hydro~, Cl C:6
aL~coxy, Cl-C6 alkoxy(Cl-C6)a~, C3~7 cycloalJ~l,
C3~7 cycloalk~yl, aryl, arylo~, hetero~lyl, -~R6R,
--NR8COR, --~R S02R , {X~R6R7 or R6R7N--(C~ a~co~;
or R5 is Cl{:6 allcyl substituted by a grol~p of the :Eorn~lla:
Rl~
_NRll l R13
R14
R12
NR SO2 C R or
R14
{~Rll_C_R
R15
: " - . . ~
' ~ ~ i. .
W O 91/10644 2 0 7 21~ ~ PCT/EP90/02156 ~
4 .
wherein R and R7 are each in1ependently H, Cl-C4 alkyl, C
cycloalkyl, aryl, aryl(Cl-C4)alkyl, C2~C6 alk ~ l,
or heterccyclyl; or the two grcup6 R6 and R7 are taken
together with the nitrogen to which they are at~ched to
form a pyrrolidinyl, piperidino, morpholino, piperazinyl
or N-(Cl-C4)alkyl-piperazinyl group;
R is H or Cl-C4 alkyl;
R is Cl--C4 aIkyl~ CF3, aryl, aryl(Cl-C4)alkyl~
aryl(Cl-C4)alkoxy, heterccyclyl, ~ -C4 alkoxy or NR R
wherein R6 and R7 are as previGusly defined;
R10 is Cl-C4 alkyl, C3-C7 cycloalkyl, aryl or
heterocyclyl;
Rll is H, Cl-C6 alkyl, aryl or C3-C7 cycloalkyl;
R is RllcoNRll_, RllsO NRll_ R1 ~17N (CH
Rl10-, wherein each * l is as previausly defined above;
R13 and R14 are each independently H or Cl-C6 alkyl; or
R13 is H and R14 is Cl-C6 alkyl which is substituted by
OH, ~ -C4 aIkoxy, SH, SCH3, N~ , aryl(Cl-C6)aIkYl :
OCONH-, NH2CO-, 002H, guanidino, aryl, or heterocyclyl;
or the two groups R13 and R14 are joined together to
form, with the car~on atom to which they are attached, a
5 or 6 n~ red carbocyclic ring which may 'ae saturated
or monG-unsaturated and which may opkionally be
,,;
WO 91/10644 2 b~ 2 1 2 6 PCI/EP90/02156
.. .
substituted ~y C~{~4 a~l or fus~d to a :Eur~er 5 or 6
memb~ saturated or unsaturated car~ocyclic rir~;
or R13 ~.s H a~3. R12 and. R14 are lir~ced to form a
2- (N{r)Rll-4-arir~r~lidinyl) grwp;
R15 is R16R17Ncx~_ RlloCI~_, Rlloc~2_ or he~clyl,
~erein Rll is as previously defined abave;
R16 a~ *7 are ea~h ~epe~ently H or C1~6 al~l;
and p is O or an integer of f m m 1 to 6;
and pbarmaceutically acceptable ~lts thereof ~nd bicprec~rsors
therefor.
In the above definition, unless ctherwise indicated, alkyl
groqpe having three or more carbon atLms may be straight or
~ranched-chain. The term aryl as used herein mYans an arcmatic
h~h3x~lrbon group such as pbenyl, naphthyl or bip enyl which may
opkionally be substitu~ed with one or more OH, CN, CF3, ~ -C4
alkyl, ~ -C4 aIkDxy groups or halo atoms. Halo means fluor~,
chloro, bramo or iodo.
.~ . . . . . ... .
:. ~.. . .
, : , ,., , ., ., : , ,
,, :-, , ,: . - ,,
W O 91/10644 2 0 7 ~1 ~ 6 PCT/EPgO/02156
m e term heterocyclyl means a 5 or 6 membered nitrogen,
oxygen or sulphur contaim ng heterocyclic group which, unless
otherwise stated, may be saturated or unsaturated an~ which may
optionally include a further oxygen or one to t~rnee nitrogen atoms
in the ring and which may optionally be kenzo ~1 or s~bstituted
with for example, one or more halo, ~ -C4 alkyl, hydroxy,
c ~ l, benzyl, oxo, am m o or mono or di~ C4 alkyl)amuno or
~ -C4 a ~ l)amono grcups. Parti~l æ examples of heterocycles
mclude pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, pyrrolyl,
imidazolyl, pyrazolyl, triazolyl, tetrazolyl, furanyl,
te~rahydrofuranyl, tetrahyoLopyranyl, dioxanyl, thienyl, oxazolyl,
isoxazolyl, thiazolyl, indolyl, isoindolinyl, quinolyl,
qum oxal myl, quinazolinyl and benzimidazolyl, each bleing
optionally substituted as previously defined.
The cowpcunds of formula ~I) may contain several asymmetric
centres and thus they can exist as enantio~ers and diastere~mYrs.
m e invention includes both mixtures and the separated individual
is ~ s.
Ihe pbarma oeutically acceptable salts of the c~DFIurds of
formula (I) containing an acidic centre are those formed with
bases which ~orm non-toxic salts. Examples include the alkali
m~etal salts such as the sodi~m, potassium or calcium salts or
salts with amines such as diethylamine. CcnlYunds having a ba~sic
centre can also form acid addition ~lts with pharma oe utically
aoceptable acids. Examples include the hydrochloride
hydrobromide, sulphate or bisulphate, phosphate or hydrogen
phosphate, a oetate, citrate, fumarate, gluconate, lactate,
. ~ .. ..
,
.. . . . . .
: :
:
.~ W 0 91/10644 2~0:~ 2 1 2 6 PCT/EP90/02156
: :,
maleate, succinate and tartrate salts.
The term bioprecursor in the above defLnition means a
pharma oe utically accEptable biologically degradable derivative of
the compcund of formula tI) which, upon admini~ration to an
c~nimal or human being, is converted in the body to produ oe a
ocmpound of the formLla ~I).
A preferred grcup of ccnpounu`s of the for~l1a (I) are those
wherein A is ( ~ )4 and Rl and R2 are H, i.e. ccmpGunds of the
formula (II) below wherein R, R3 and R5 are as previcusly defi~ed
for formula (I):
R Q
CHCH2 CONH R3
/
R02C
(II)
: Also preferred are those a=poY n~s of formulae (I) and (II)
wherein R and R4 (when P~3 is 002R4) are both H (diacids) as well
as biolabile no .~n~ di-ester derivatives thereof wherein one or
~oth of R and R4 is a biolabile ester-forming group.
'.~ 'rhe term biolabile ester-forming group is well understood in
the art as m~aning a group which provides an ester which can be
readily cleaved in the body to likerate the corresponding diacid
of formwla (I) wh~rein R and R4 are both H. A number of such
/
.. ..
... . ..
,, . , ,. ,.~ .
W O 91/10644 2 0 i ~ PCT/EP90/02156 .~.
""' 1.
ester grcups are well kncwn, for example in the penicillin area or
in the case of the ACE-inhibitor antihypextensive agents.
In the case of the ccmpcunds of formulae (I) and (II) such
biolabile prordrug esters are particularly adva~tagecus in
providing compcAnds of the form~la (I) suitable for oral
ad=uristration. The suitability of any parti~llar ester-formung
~ p can be assessed by conventional animal or in v tr~ enzyme
hydrolysis studies. muS, desirably for optinn~o effect, the ester
shculd only be hydrolysed after absorption; accordingly, the ester
should be resistant to hydrolysis before absorption by digestive
enz~mes but should be readily hydrolysed by, for example, liver
enzymes. In this way the active diacid is released into the
blccKb~ naam following oral absorpkion.
In aA~;tion to lcwer alkyl esters (particul æly ethyl) and
benzyl esters, suitable biolabile esters include alkannyloxyalkyl
esters, including alkyl, cyclc~lkyl and aryl substituted
derivatives thereof, aryloxyalkyl esters, arcyloxyalkyl ester.s,
arylalkyloxyalkyl esters, arylesters, aralkylesters, and haloaIkyl
esters wherein said alkanoyl grcups have from 2 to 8 carbon atoms
and said aIkyl groups have ~rc~ l to 8 carbon atams and are
~branche~ or straight chain and said aryl grcups are phenyl,
naphthyl or indanyl optionally substituted with one or more ~ -C4
alkyl or Cl-C4 aIkoxy groups or halo atcms.
~muS examples of R and R4 when they are biolabile
: ester-forming grcups okher than ethyl and ~enzyl include: ¦
l-(2,2-diethyIbutyryloxy)ethyl, 2-ethylpropionyloxymethyl,
.-1-(2-ethylprcpionyloxy)ethyl, l-(2,4~dimethylbenzoyloxy)ethyl,
207~2~
;~ W O 91/10644 ~ PCTlEPgO/021~6
l-(benzoyloxy)benzyl, l-(benzoylcxy~ethyl, 2-methyl-l-
prcpionyloxyprcpyl, 2,4,6-trimethylben20ylo~nnethyl, l-(2,4,6-
trimethyl-benzyloxy)ethyl, pivaloyloxymethyl, phenethyl,
phenpropyl, 2,2,2-trifluor~ethyl, l- or 2-naphthyl, 2,4-
dimethylphenyl, 4-t-butyl-phenyl, 5-(4-methyl-l,3-dioxalyny1-
2-ony1)methyl and S-inlanyl.
Crmpounds of the formulae (I) and (II) wh ~ R is benzyl or
t-butyl and R4 is ethyl are valuable Lntermediates for the
preparation of the diacids wherein R and R4 are bcth H.
In a fur~her preferred group of o~mpcunds R5 is methylene
substituted by a gr~up of the formLla -NHCCCR ~ 13R14'
particularly where R12 is NH2, RllCONH- or RllSO NH R13
R14 is -(CH2)4 ~ . Particularly preferred are such gro~ps derived
fram S-lysine; thus especi~ preferred R5 suksti~utents of ~his
type include S-lysyl-amincnm*hyl, N2-a oetyl-S-lysyl ~ ethyl and
N2~tha~1phonyl-S-lysyl-z i~e~yl.
In further groups of preferred oa pounds R5 is ~ -C6 alkyl,
or cl-c6 allcyl substituted }~ -c6 aL~y, partiallarly
methoxyethyl; or R5 is ~ -C6 alkyl substituted by phenyl.
Particularly pre~erred individual o~mpoun~s o~ the mvention
include:
2-~1-[2(S) car ~ xy-3-(S-lysylam~ propyl] ~ ~ l-
carbonylamino~-2,3-dihydr3indene-2-carboxylic acid,
~ 2~ [2(S)-carboxy-3-(~ -methanesulphonyl-S-lysylami~o)-
::~ p ~ pyl]cyclo ~ lcar~onylamino}-2,3 dihydroir~en ~ 2 car~oxylic
acid, and
,~
:
:, ::;, ~ :
.: .
.
WO 91/10644 2 ~ 7 2 i 2 ~ PCT/EP90/02156
2~ [2(S)-car~oxy-3-(N2-methanesulphonyl-S-lysylamuno)-
propyl]c~clopentylcarbonylamino~-2-hydroxymethy:L-2,3-dihydroindene
and biolabile ester derivatives thereof.
The cocFcLnds of formula (I) are prepared ]Dy a number of
different processes. The basic prooedure involves the synthesis
of a partially pr ~ d cycloaIkyl-substitutad glutaric acid
derivative which is coupled to an amine to gi~e the desired
glutaramide. ~he carboxylic acid group in the amine, if free, or
any reactive groups in R , may ~ protection during the
coupling step and such protecting groups are removed in the final
stages of the prccess.
The synthetic ro~te is illustrated in Scheme 1 wherein A, Rl
and R2 are as previously defined, R5 is as defined for R5 with .
any reacti~e graup therein prokected if necsssary, R18 is as
defined for R excluding H, or is a conventional carboxylic acid
protecting grcup, and R3 is either CH2OH or 002Rl9 wherein R19 is
as previously defined for R4 excluding H or is a conventional
car~oxylic acid protecting gr w p:
.
'...................... ' '
WO 91/10644 2 a 7 212 6 Pcr/EP9o/a2l56
. I . . . ~ ' .
11
Sc~l~me I
CUCU2 / \ C0211 + H,,N ~R
R O2C (III) (IV)
\ C~C~z~ \CONa ~ R3
R O2C (V)
, ~1, .
(I)
.
Ihe reaction of the ccmpconds of formula (III) and (IV) is
achieved using conventional amide ccuplLng techniques. Ihus in
one process the reaction is achieved with the reactants dissolved
in an organic solvent, e.g. dichlorcmethane, us mg a diimide
condensing agent, for example l-ethyl-3-(dimethylamlnopropyl)-
carbcdiimide, or N,N'-dicyclohexylcarbodiimide, advantageously in
the presence of l-hydroxyben2otriazole and an o ~ c kase such as
4-methylm~rpholine. The reaction is generally complete after a
period of fr~m 12 to 24 hours at roam temperature and the product
is then isolated by conventional proo dures, i.e. by washi~g with
water or fil~ration to rem~ve the urea by-product and evaporation
of the solvent. The product may ~e further purified by
.. . .. .
wo gl/10~44 2 0 7 212 6 PCT/EP90/02156
12
crystallisation or chrcmatography, if ne~essary. m e ccnpoonds of
formula (V) mclude ccmpcon~s of formLla (I) wherein R and R are
-C6 alkyl or benzyl.
In scme cases the coupled product, in prx~ted form, may be
subjected to conventional chemical transformati.on reactions to
allow preparation of further compounds of formula ~V). Thus for
example ccmpc~nds of formula ~V) wherein R5 contains an ester
gr3up may be hy~rolysed or hydrogenated to generate the carboxylic
acid which may be further reacted, for example with an amune, to
give amide deri~atives.
Similarly oompoands wherein R5 contains a substituted or
protected amino group (for example a benzylam mo, dibenzylam mo,
benzyloxycarbonylamino or t-butyloxycarbonylamino group) may be
converted to the free am m es by hydrogenation or protonolysis as
apprDpriate. qhe amines produced may be further reacted, thus for
example reaction with a sulphonyl halide yields the c~rresponding
sulphonamides, acylation with an acid chloride or anhydride yields
the correspond~^ng amides, reaction with an isocyanate yields urea
derivatives and reaction with a chloroformate yields the carbamate
products respectively. All these transformations are entirely
conventional and appropriate conditions and reagents for their
performance will be well kncwn to those skilled in the art as will
o~her variations and possibilities.
The diesters of formula (V) wherein R3 is 0~2Rl9 may be
further reacted to give the monoester of diacid derivatives of
formula (I) wherein one or both of R and R4 are H. The conditions
used will depend on the precise nature of the groups R18 a~d R
present in the compcund of formLla (V) and a number of variations
., ' ' ~ :
~ WO 91tlO644 ~ 2 0 7 212 6 PCT/EP90/02156
13
are possible. Thus for example when bcth of R and R19 are
benzyl, hydroge~ation of the product will yleld the diacid of
formula (I) wherein R3 is 002R4 and R and R are both H.
Alternatively if one of R18 and Rl9 is benzyl and the other is
alkyl, hydrogenation will yield a monoester product. This can be
hydr3lysed, if desired, again to yield the diacid product. When
one of R18 and Rl9 is t-butyl treatment of the compound of
formula (V) with trifluoroacetic acid yields the correspondi~g
acid. The diester product wherein R18 and Rl9 are benzyl or lcwer
alkyl can also be treated with trimethylsilyl iodide to produce
the dicarboxylic acid product. If some other carboxylic acid
protecting group is used for R18 or Rl9 then clearly appropriate
conditions for its removal must be employed in the final step to
give the ester or diacid product of formula (I). In the case
where R3 is ~ OH a single deprokection step is requlred to
produce the ccmpcunds of formLla (I) using deprotection ~ethods as
app m priate to the particular R18 group present in the compound of
form~la (V). In the case where the ring A or the substituent R5
is unsaturated, the deprotection must be eff~cted by non-reductive
meth~, thus for example if either of R and R is benzyl, they
may be remcved by treatment with trimethylsilyl iodide.
As well as removing any protect m g group which may be present
in R5 , a number of chemical transforma~ion reactions are possible
on the final mono-ester or diacid pro~ucts as previcusly
de æ ribed. In each case the product ~ay be o~btained as the free
carboxylic acid or it may be neutralised with an appropriate base
and isolated in c~lt form.
- ,: ,
t ' ' ~ ' " :' ,' ' . .
wo gl/10644 - `2 0 7 2 i 2 6 PCTIEP90/02156 ._
14
In a variant of the above procedure, o~om ds of the formula
(I) wherein RS is ~ -C6 alkyl substituted by -NR OOR , -NR S02R
NRllcocR12R13Rl4 or _NRllso2CRl2Rl3Rl are prepared by a procesS
w~Lich involves acylating or sulFhonylating a compound of the
formula: ~ ~ R2
~ ~Y '''
~ R20NH Y / \ U ~ 3'
., CHCH2
R 02C / ~ I~
wherein R20 is as defined for R8 or Rll, R18 and R3 are as
previcusly defined and Y is a ~ -C6 alkyl group; by reaction with
an acid of the formula R9Oo2H, A OSO3H, R12Rl3Rl4CCo2H~ or
R12R13 * 4CSo3H, or an activated derivative thereof. The resulting
amide or ~-1phonamide product is then deprotEcted if required and
the monc- or diester pro*uct cleaved to yield the carbcxylic acids
of formula (I) wherein R is H and R3 is CH2OH or ~02H as
previcusly d~scribed.
The corpcunds of formula (VI) are prepared following the
procGdures shown in Scheme l but using a compcund of formula (III)
having R5 as a protected amune derivative. Thus, for example R5
can contain a bis-[(lS)-phenylethyl}amuncmethyl substituent.
Hydrogenation of the coupled product gives the corresponding free
amune of formLla (VI) wherein R20 is H and Y is ~ . mis r3ute
is of partic~lar value for the pr2paration of carpounds having
2(S) stereochemistry in the glutaramide backbone.
... . . .
:: , ~` ~ '' ':
W O 91/10644 i . PCT/EP90/02156
~t 2~72~26 i; '
The starting cycloaIkyl-substituted glutaric acid m~no esters
of formula III may be pxepared as described in our ~ n ~patent
applications EP-A-0274234, 89305180.5 and 8~304698.7.
m e amines of formula ~IV) are generally 'hn3wn ccnpcunds
or they are prepared by appropriate synthetic ]prcoeduras in
acoordance with literature precedents. Ihus ~n one p ~ e the
cYmpcunds of formula (IV) wherein R is CH20H maY be preparsd by
ion of the corresponding acid, or lower alXyl ester for
example using sodium b~rchydride.
~ xc~priate ca~plir~ and pxatectir~ methods for all of 'che
above steps and alternative variations arx~ proo~ures will be well
}~awn to those ~dcilled in t~e art by ref~rence to stan~ard text
booXs and to the ex~les provided hereafter.
As previa~sly n~Ttiaa~d, the co~rds of the inverltion are
pctent inh~bitors of the neutral endcpeptid~se (E.C.3.4.24.11).
l~is enzyme is involved in the breakda~ of a ~ of peptide
hormones anld, in particular, it is iTn~olved in the breahdc~n of
atrial natriuretic factor (ANF). I~is hormone consists of a
family of related natriuretic peptides, secreted by the heart, of
which the major circulatLng form in humans is known to be the 28
amino,acid peptide referrad to as alpha-hANP. Thus, by preventLng
the degradation of ANF, by end~peptidase E.C.3.4.2~.11, the
ccnçounds of the invention can potentiate its biolugical effects
and the crnpcunds are thus diuretic and natriure~ic agents of t
utility in a ~ r of disorders as previously described.
Activity aga mst neutral endcpeptidase E.C.3.4.24.11 is
a ~ using a prooedure based on the assay described by J.T.
Gafford, R.A. Skidgel, E.G. Erdos and L.B. Hersh, Bioche~Lstry,
.
,~ , .
:.
'
W O 91/10644 PCT/EP90/02156
16
1983, 32, 3265-3271. The methcd mvolves determ mQrg the
concentration of ccmpound required to reduoe by 50% ~he rate of
release of radiolabelled hippuric acid fram hippuryl-Ir
phenylalanyl-~,arginine by a neutral endopept.idase preparation
from rat kidney.
Ihe activity of the ccmpcon~s as diuretic agents is
determined by measuring their ability to increase urine cutput and
.cn~;um ion excretion in saline loaded conscious mloe. In thLis
test, male mioe (Charles River CDl, 22-28 g) are acclimatised and
starved overnight in metab3wls. The mioe are dosad intravenKusly
via the ta;l ve m, with the test compound dissolved in a volu~e of
saline solution equivalent to 2.5% of body weight. Urine samples
are collected each hour for two hours in pre-weighed tubes and
analysed for electrolyte concentration. Urine volume and sodium
ion c~ncentration from the test a ~ s are ccmpared to a control
group which receiYed only saline.
For admlnistration to man in the curative or prcphylactic
treatm~nt of hypertension, congestive heart failure or ren21
insufficiency, oral dosages of the ccmpcurds will generally be in
the range of from 4-800 mg daily for an average adult patient (70
kg). Thus for a typical adh~t patient, individual tablets or
capsules contain from 2 to 400 mg of active compound, in a
suitable pharma oeutically acceptable vehicle or carrier for
adminuatration singly, or in mLltiple doses, once or several tLmes
a day. Do6ages for intravenous ad~inist~ation w~uld typically be
within the range 1 to 400 mg per single dose as required. In
practi oe the physician will determ m e the actual dosage which will
be mLst suit3ble for an individual patient and it will ~aly with
.
~_ W O 91/10644 PCT/EP90/02156
17
the age, weight and response of the parti~llar patient. Ihe above
dosages are exemplary of the average case but there can, of
ccurse, be indivi~lal in=tan oes where higher or lower dk~tge
ranges are merited, and stlch are within the so3pe of this
invention. -
- For human use, the conpcunds of the for~Lla (I) can be
admd~L~i~ereI alone, but will generally be adn~ ered in
: admixture with a pharmaceutical carrier selected with regard to
.~ the intended route of admdristration and ~tandard p ~ acY~ttical
practi oe. For example, they ~ay be administered orally Ln the
~- form of tablets contaimng such excipients as starch or lactose,
or in capsules or ~vules either alone or in admixture with
excipients, or in the form of elixirs or suspensions containing
flavcuring or oolouring agents. qhey may be injected
p~rerterally, for example, intravencusly, mtr~m=sc=larly or
sL~x~rt mecusly. For p~renteIa1 administration, they are best used
in the form of a sterile aqueous solu~ion which may contain cther
mL~d~n~es, for ~xample, enough salts or gluc~se to make the
solution isotonic with blood.
The c~cpw nds may be administered alone ~ut may also be
administered t~gether with such other agents as the physician
shall direct to optimise control of blood pressu~e or to treat
congestive heart failure, renal insufficiency or ~ther disor~ers
in any particular patient in accordance with established mYdical
practi oe . muS the cccpcun~s can ~e co-acrcuIustered with a
variety of cardiovascular agenks, for example with an A OE
inhibitor such as captopril or enalapril to facilitate the contr~l
of blood pressure m treatment of hypertension; or with digitalis,
W O 91t10644 PCT/EP90/02156 ~
..
18
or ancther cardiac stimLlant, or with an ACE inhibitor, ~or the
treatment of congestive heart failure. Okher possibilities
m clude co-administration ~ith a calcium antagonist (e.g.
nifedipine, amlodcpine or d;ltiazem) a beta-kllocker (e.g.
atenolol) or an alpha-blocker (e.g. prazosin or doxazosin) as
shall be detçrmined by the physician as a~prc~riate for the
treatment of the particular patient or c~ndition involved.
In addition to the above, the cccpcunds may also be
adnu~L~ibered in conjunction with exogenous ANF, or a derivative
thereof or related peptide or peptide fragment having
diuretic/natriuretic activity or with other ANF-gene related
peptides (e.g. as described by D. L. Vesely et al, Biochem.
Biophys. Res. Ccmm., 1987, 143, 186).
Thus in a further aspect the invention provides a
pharma oe utical cc~o6ition ccmprising a compound of the formLla
(I), or a pharmaoe utically acceptable calt thereof or bic~recursor
therlefor, together with a pharma oe uti~lly acceptable diluent or
carrier.
m e invention also include~ a ccmpcund of the fornula (I), or
a pharmaceutically aooeptable salt thereof or bioprecursor
therefor, for use in mdicine, partia~larly for use as a diuretic
agent for the treatment of hypertension, congestive heart failure
or renal lnsufficiency in a human being.
The invention further includes the use of a compcund of the
fornLla (I) for the manufacture of a nedicament for the treatment
of hypertension, heart failure, angina, renal insufficiency,
premln=tIual syndrome, cyclical oedema, M~niëres disease,
hyperaldost2r~nism, pulmanary oedema, ascites, hypercalciuria,
- ~ .. . :.,, . : -
,. :: ~ . . ~: . . . . ; 1
:. .,
~ W O 91/10644 2 ~ ~ 21% 6 PCT/EP90/02156.
19
glaucGma, asthma, inflammatio~l, pain, epilepsy, affective
disoxders, de~entia ~ d geriatric confusion, ~esity,
gastrointes*inal disDrders (including diarrhoeia), hyperTeninaemia,
leuXaemia, and the m~dulation of gastric acid secretion.
m e preparation of the compcunds of the m vention will now be
more particularly illustrated by reference to the following
experimental Examples. Ihe purity of ccmpcun~s was rcutinely
moni~ored by thin layer chromatcgraphy using Merck Kieselgel 60
F254 plates. ~-Nuclear magnetic r2asonance spectra were recorded
using a Nicolet QE-300 spYctrcmeter and were in all cases
consistent with the prcpos2d structures.
.. ,., ~ . : :
. , ~
W O 91/10644 2 0 7 212 6 PCT/EP90/02156 ~ ~
~. ,
2-~1 r 2(R~S)-CarboxY-4-Phen~IkUtyl~c~ClC~nty_carbonylamm o~-
2 3-dihydroindene-2 ~ oxy1ic acid
(a) 2-Amino-2 3-dihvdr~indene-2-carboxy1ic ac:id, benzyl ester
A mixture of 2-amuno,2,3-dihydr~indene-2-~rboxylic acid
hydrochloride (R. M. Pinder, B. H. Butcher, D. A. Buxt~n and D J
Hc~ells, J. M~d. Chem., 1971, 14, 892), (11.33 g, 0.053 m),
benzyl alcx~hol (27.5 ml, 0.27 m), para-toluenesulphonic acid
monchydrate (12.1 g, 0.064 m) and benzene (150 ml) ~as koiled
under reflux wi~h continuous removal of water usLng a Dean Stark
trap. After 48 hours, further quantities of benzyl alc~hol (27.5
ml, 0.27 m) ~nd benzene (100 ml) were added, and the reaction
allowed to oontinue under reflux for a further 72 hours. ~he cool
raaction mixture was diluted wi~h diethyl ether and the ~esulting
white precipitate collected by filtration and washed wi~h diethyl
ether. The crude tosylate salt was then dissolved in water and
this solution kasified with IM aqueous sodi~m hydroxide solution,
then extrac~ed with ethyl aoetate. The combined extracts were
washed with saturated br~e, dried (anhy~ra2s Na2S04) and
filtered. Evaporation under vacu~n of the f-ltrate gave an oil
(7.6 g, 53.6 %) which solidified at room ~erature over-night.
~rystallisation of a san~le fmm hexane afforded th~ pure pr~duct
as a ~hite solid, m.p. 55-55.5 &. Found: C,75.98; H,6.38; N,5.18.
C17H17N02 re~uires C,76.38; H,6.41: N,5.24%.
i
; . : . . , :
. WO 91/10644 2 ~17 212 6 PCr/EP9OtO2156
21
(b) 2~ 2(R.S)-Ethox~or~ 4-pher~l lcycl~pentyl-
car~onyla~nino)-2,3 dihv~roindene 2 car~oxy1ic acid, b~zvl ester
C~alyl chloride (470 ~, 3.74 mnol) was ac~ded at ro~n
t~tl~re to a stirred solution of 1-~2(R,S~--etho~ycar~onyl-4-
pheny~utyl]~ycl~ta~ car~oxylic acid (600 ~, 1.87 mm:~l) in
dry dichlor~neff~ar~ (10 ml) contain~ng dry dimethylform~nide (2
drops). After 2 hGurs the solvent was removed under vacuum and
the residLal oxalyl chloride evap~rat0d azeotropically using dry
dichlorRmet ~ (3 x 10 ml).
The crude acid chloride was dissolved in dry dichloro~ethane
(15 ml), then the resulting solution added dropwise to a stirred,
ice-cold solution of the product frcm step (a) (500 mg, 1.87 mmol)
and dry triethylamune (210 mg, 2.06 mmol) in dry dichlorome~hane
(25 ml); stirring was continued for 1 hcur at 0C, then for 16
hours at rocm temperature. me reaction ~ e was dilu~ed with
dichloromethane (60 ml), washed suocessively with wa~r, IM
hydrochloric acid, water, saturated aqueous sodi~m bicarbonate
solution and watex, dried (anbydrous Na25Q4) and filtered,
Evaporation under vacuum of the filtrate afforded a brswn gum (880
mg) which was purified by chromatography on silica gel using an
ethyl a oe tate in hexane elution gradient ~0-25%). hvaporation
under vacuum of the appropriate fractions provided the re~uired
product as a clear oil, which subsequently formed a white waxy
solid; crystallisation fr~m diethyl ether-hexane gave the pure
product (437 mg, 41.1%), m.p. 89-90 C. Found: C,76.21; H,7.22;
36H41N05 requires C,76.16; H,7.28; N,2.47%
::
., ., : .
W O 9l/l0644 2 q 7 2 i ~ 6: PCT/EP9U/02156
2-11-r2(R S)-CarboxY-4-PhenYIbutvllcycloPentvlcarbonYlaminol-
2,3-dihydroindene-2-carkoxy1ic acid
(c) A IM aguecus solution of sodium hydro~ide (5 ml, 5 mmol) was
added at roum temperature to a stirred sol~tion of the above
product (430 mg, 0.76 mmole) in 1,4-dioxan (10 ml) and methanol
(2.5 ml). After 5 days, the reaction mixt~re was diluted with
water (40 ml~, its pH adjusted to 7 with 2M hY~rcchloric acid, and
the orga~ic solvents re~oved by evaporation under vacuum. The
result m g ~ e was washed with diethyl ether, acidified to pH 2
with 2M hydrochloric acid, then extracted with ethyl acetate. q~e
ccmbined 2xtracts were washed with saturated brine, dried
(anhydr~us Na2S04), filtered, and evaporated under vacuum.
Crystallisation of the residue fram ethyl acetate-hexane gave the
required product as a white solid (200 mg, 56.6%). Fcund:
C,71.30; H,6.88; N.3.25. C2 ~ 1NO5; 0-17 CH3oO2C2H5 requires
C,71.59; H,7.02; N,3.02%.
EX~MP$E 2
2-fl- r 2fR~s~-carboxy-4-methoxybutyl~cyclGpentylcarbon
amino~-2,3-d~hydrGindene-2-carboxylic acid
~a) 2-~l-r2fR,S)-Benzvloxycarbonvl-4-m~thoxYbutvllcvclopentyl-
carbonylamunol-2,3-dihvdroinclene-2-carboxvlic acid, benzyl ester
qhe procedure of Example l(b) was followed using 1-[2(R,S)-
ben2yloxycarbonyl-4-methoxybutyl~cyclcpe~tane carboxylic acid (334
mg, 1 mmole) and 2-amlno-2,3-dihydroindene-2-carboxy1ic acid
~enzyl ester (267 mg, 1 mmol), to furnish the reglired diester
(430 mg, 72.5%). Found: C,72.95; H,7.03; N,3.08. C36H41N06; 0.5
0 requlres C,72.75; H,7.14; N,2.36%.
i
,~
..
' ,' . ~. . :
: , ,
: :: .. :..... . . . .
~ W O 91/10644 2 ~ ~ 21;2 ~ PCT/EP90/02156
23
(b) 2~ [2(R,S~-Carboxy-4-methoxybutyllcYclo~entylcarbonvl-
amino~-2L3-dihydroindene-2-carboxylic acid
A solution of the above pro*uct (390 mg, 0.67 mmol) Ln
ethanol (20 ml) was hydrogenated over 10% palladium on c ~
~39 m~) a~ 15 p.s.i. (1 ~ar) and room ~ ablre for 1 hour. The
catalyst was removed by filtration thraugh a pad of Px~ccel
(upper) and Hyflo (lower), then the filtrate evaporatd un~er
vac~m, and residual ethanol removed azectrcpically with
dichloromethane to provide the r ~ d product as a white solid
(235 mg, 87.2%), m.p. 142-144C. Found: C,65.17; H,~.29; N,3.32.
C22~ gNO6 requires C~65.49; H,7.25; N,3.47%.
EX~MPLE 3
2~ r2tR,S)-CarboxY-3-~S-lysvlamino)proPyllcyclop~t
carbonYlaminol-2,3-dihydroinBene-2-carkoxYlic acid
(a) 2-Amino-2,3-dihydroindene-2-carbox~1ic acid, ethYl ester,
hydrochloride
A stirred, ice-oold solution of 2-amino-2,3-dihydroindene-2-
carboxYlic acid hYdroLhloride ~2.48 g, 11.6 mmol) in absDlute
ethanol was satuxated with dry hYdrogen chloride. The resulting
s ~ ion was stirred at r w m temperat~re overni~ht, then warmed
to 40C and treated further with dry hydrogen chloride until a
cl OE solution was obtained (2 hours). After a further 4 hcurs at
40 & , the reaction mixture was evaporated to dryness under vacuum,
then the residue crystallised frcm e~hanol-diethyl ether to give
the pure product (2.41 y, 85.~%), m.p. 189-193 &. Found: C,59.74;
H,6-69; N,5-76- C12H1 ~ 2; HCl reguires C,59.63; H,6.67; N,5.79%.
- . .. .":
,: , ,.:, :. .,
: . ~ .. : '' ,:,',' "'. ~'
. ~ ~ . :, .
wo gl/10644 ~ 6 ` PCT/EPgO/02156
24
(b) 2-~lr2(R,S)-tert-ButoxvcarbonY1-3-(dibenzYlamlno)propyl
cycl~e~tvlcarbonvlamlno~-2.3-dihYd-r-oindene-2-{arboxylic acid,
ethyl ester
To a stirred, ice-cold solution of 2(R,S)-tert-butoxycarbonyl-
3-(dibenzylamuno)propylcyclopentane carboxylic acid h~dLcchloride
~7.84 g, 0.016 m), l-hydroxybenzokriazole (2.16 g, 0.016 m) and
4-methylmorpholLne (4.86 g, 0.048 m) in dry di ~Lorcmethane (lO0
nL'L) was added 1-(3-dimethylamuncpropyl)-3-ethylcarbodii~ide
hydrcchloride (6.13 g, 0.032 m); stirring was contLnued for 1 hour
a~ o&, then for 16 hours at roGm temperatuxe. The
dic~iLoromethane was removed by evaporation under vacuum c~t roam
temperature, and the residue partitioned between diethyl ether c~nd
water. The ether phase was separated, washed with water, dried
(anhydrcus N ~S04) and P~L~ered; subsequent evaporation un~er
vacuum at room temperature of the filtrate provided the crude
activated ester (9.33 g, 95.4%) ~ a hemi-solvate with
dic~loro~ethane, of sufficient purity for further pr ~ i~n.
To a stirred solution of this activated ester (3.24 g, 5.3
1) in dry dic~loromethane (40 nl) at 0C was added
2-amuno-2,3-dihydroindene-2-carboxylic acid ethyl ester (1.09 g,
5.3 mmol) and 4-dimethylaminopyridine (0.71 g, 5.8 mmol). After
0.5 hc~rs the solvent was removed by evaporation under vacuum and
the resulting ViscGus oil allowed to stand at room temperature for
3 days before partitio m ng between diethyl ether and water. m e
ether p~a~e was separated, washed with water, dried (anyh1m us
MgS04) and f;ltered. Evaporation under vacuum of the filtra~e
furnished an oil which was purified by chromatograFhy on silica
gel using a dichlorcmethane in hexane elution gradient (20-100%).
'. ~
:,. . ~' :,,
~ `, :~ . ., -
~ 7 ~ ~
W O 91/10644 ' . PCT/EP90/02156
Evaporation of the apprcpriate fractions prcvided the required
product ~2.10 g, 61.2%). Fcund: C,73.92; ~,7.88; N,4.55.
C40HsoN2os 0 5 H20 ~ C,74.15; H,7.94; N,4.33%.
(c) 2~ r3-p~2 (R~s)-~-~oxy~m~:Lp~-cvcl~tvl-
carbonvlamino~-2.3-dih~droindene-2-carboxylic acid, ethyl ester
A solution of the above diester t2.7 g, 4.2 mmol) in a
mixture of ethanol (6 ~L) and water (0.5 ~L) was hy*rogenated over
20% paLl~d;um hydraxide on ch2u~xx~L (270 mg) at 50 p.s.i. (3.45
bar) and room temperat~re for 16 hcurs. Ihe catalyst was removed
by f~LLL~tion through a pad of ~ L (upper) and Hyflo (lower),
then the filtrate evaporated under vacuum, and resi~ ~1 solvents
rem~ved azeotrcpically with dichlorcmethane, to affor~ the
required product (1.92 g, 99.6%). Found: C,67.64; H,8.44; N,5.89.
C2 ~38N205 requires C,68.09; H,8.35; N,6.11%.
(d) 2-~1-r2(R.S)-tert-Butyloxycarbonyl--3-~ , N6--di-benzyloxy-
carbon~l-S-lysyl-aminoLer~Eyllcyclcpent~1carbony1ami~o~-2,3-
dihYdroindene-2-carboxy1ic acid. ethyl ester
1-(3-Dimethyl ~ r~py1)-3-ethylcarbodiimide hydrochloride
(944 mg, 4.92 mmol) was added to a stirred solution of the above
amine (1.13 g, 2.46 mmol), l-hydroxybenzotriazole t322 mg, 2.46
mmol), N2, N6-di-kenzyloxycar~onyl-S-lysine (1.02 g, 2.46 mmol)
and 4-methylmorpholine ~746 mg, 7.38 mmol) in dry ,dichloromethane
(25 ml) at 0C. Sti~ring was continued for 0.5 hGurs at o&, then
for 16 hcurs at room temperature, before ~ of the solvent
under vacuum. Ihe resi*ue was partitiuned between diethyl ether
and w~ter, then the ether phase washRd with lM hydrochloric acid
and water, dried (anhydrcus Na2S04) and filtera~. Evap~ration
under v,cuum of the filtrate, followed by chromatcgraphy of ~e
, : . :
:, .: , .
:
,
W O 91/10644 ? ~ ~ 2 i 2 ~ PCT/EP90/02l$6 _ i
. 26
residual oil on silica gel ~ing a diethyl ether in hexane elution
gradient (0-50%), gave the required product tl.30 g, 61.8%).
FOund c,67.31; H,7.40; N,6-09- C48H62N410 neqUIres C'67 42;
H,7.31; N,6.55%.
(e) 2~ r2(R.S~-Carbo~v-3-(N2, N6-di-benzyloxycarbonyl-S-ly~yl-
amino)proEyl~ycloEentylcarbonylamino~-2.3-dih~droindene-2-
carboxylic acid, ethYl ester
Trifluoroa oetic acid (5 ml) was added dropwise to a stirred
solution of the above pro*uct (1.28 g, 1.5 mmol) in dry
dichloromethane (10 ml) at 0C. The ice-kath was removed,
stirring continued for 1 hour at room temperature, then the
reaction mixbure evaporated under vacuulm. The crude product was
dissolved in ethyl a oe tate and nesidual trifluoroacetic acid
removed by washing th2 solution with sa~urated aqueous sodium
bicarbonate solution. Drying (anhydrcus Na2S04), f;ltration a~d
evaporation under vacuum of the ethyl acetate solution, followed
by azeotropic remcNal of residual solvent with dichlom methane,
affor,ded the required prcduct (1.12 g, 90.4%). Found: C,63.85;
H~6-66; N~6-59- c44H54N40l0; 1-5 H2~ reqUlres C,63 ss; H,6.g6;
N,6.78%.
~f) 2-~1-[2(R.S) ~ v-3-(S-lvsylamino~PropYllcYclcpe~tyl-
carbonYlaminol-2,3-dihydroindene-2-carb~xylic acid, e~h~l ester
A solution of the abcve product (1.10 g, 1.33 mmol) in a
n~aure of ethanol (7 ml) and water (0.5 ml) was hy*rogenated over
10% palladium on charccal (110 mg) at 60 p.s.i. (4.1 bar) an~ rocm
temperature for 16 hours. Wbrk-up as described above for Exawple
2 (b) pr wided the required product as a beige foam (740 mg,
98~9%). Foun~: C,59.98; H,7.56; N,9.52. C28H42N406; 1.75 H20
requires C,59.82; H,8.16; N,9.97%.
: . . :~ , .
- `` WO 91/10644 2 0 7 212 ~ PCI/EP90/02156
27
(g) 2~ r2!R,S)~ar~oxy-3-~S-lysYlamino)pr~llcyclcpe
car~lamino~-2,3 dihydroin~ 2 car~cy1ic acid
A solution of the above ester (700 m~, 1.24 ~ ) in lM
aq~ s sodium hy~roxide solution (7.5 ml, 7.5 ~1) was allcr~ed
to stand at n~n t~nperature for 16 halrs, t:hen loaded onto a
col~ of strorP~ly acidic iorre~a~e resin. ~e col~ was
aqueous pyridine. Evaporation un~er vacuum of the appropriate
fractior~ gave a glass whi~h was dissolved in distilled water;
freeze drying of this aqueous solution provided t~e required
product (370 mg, 55.4%). Found: C,57.58; H,7.95; N,10.31.
c26H38N4o6; 2H2 ~ es C,57.97; H,7.86; N,10.40%.
EX~MPLE 4
2~ r 3- (N -Asetyl-S-lys~la~ino)-2(R,S)-carboRvpeoRyllcyclo-
entvlcarbonvlamino~-2,3-dihYdroindene-2-carbox~lic acid
m e procedure of EXample 3 was followed but using
N2-aoe~l-N6-be~loxyOE~o~l-S-lysioe ~ step (d)o Deprs~tection
gave the title ccmpound. Fcund: C,58.30; H,7.58; N,10.07.
C28H40N4o7; 1-75 H2 ~ C,58.36; H,7.61; N,9.72%.
EX~MPLE 5
2-11- r 2 (R, S) ~oxv-3- (N2-metharl~lPhorlvl-s~ amino) -
prc~yllcvclo~entvlcarbonvlam m o~-2,3-dihvdroindene-2-carboxylic
acid
The procedure of Example 3 was followed ~ut using N6-
yloxycarbonyl-N2-methanesulphonyl-S-lysine in step (d).
Deprokection gave the title compound. Found C,53.99; H,7~13;
N,9.31. C27H40N~08S; ~ 0 requires C,54.17; H,7.07; N,9036%-
,. . : :. . ,:
. :, .:
,
W O 91/10644 2 0 7 2 i 2 ~ PCT/EP90/02156 _ I
28
~ 6
2-f 1-r2(S~ 3-rS-l~vlaminolp~ vcl~l-
OE bonvlamino)-2,3-dihy~rnindene-2-carboxylic acid
(a) 2-(l-r2(S~-tert-Butoxycarbonyl-3-lLs~s)-allpha~ alFhal_
dimethvldibenzYlamino~prcPvllcyclopentYlcarbonvlamlno~-2,3-
d~h ~ dene-2-carboxv1ic acid, ethyl ester
The p ~ e of Example 3 was followed but using 1-~2(S)-
terk-butoxycarbony1-3-[(S,S)-alpha, alphal-l-dimethyldibenzyl-
amino)prDpyl]cyclcpentane carboxylic acid m stqp (b) to give the
title pr~duct, in 79.1% yield. Fcund: C, 75.45; H,8.30; N,4.01.
42 54 2 5 ~ s C,75.64; H,8.16; N,4.20%.
(b) 2-~1-r3-Aminc-2tS~-tert-butoxYcarbon~lprcpyllcyclopentYl-
ca~bonylamino)-2,3-dihy~roindene-2-carbox~lic acid, ethYl ester
~ ydrogenation of the above product as described in Example
3(c) gave the 2(S) iscmer of the amone in 99.1% yield. Rf
(silica) 0.50 (dichloromet ~ methanol, 9:1).
(c) 2-(1-r2(S) ~ oxy-3-tS-lys~lam~no)p ~ pyllcvcl ~ entyl-
carbonylamino~-2.3-dihv~roindene-2 ~ oxYlic acid
The abave amine was ccupled to ~ , N6-di-benzyloxYcarbonyl-S-
lys m e following the prooedure of Example 3(d) and the product
deprotected as described in EXample 3 steps (e) t~ (g) to give the
title compcund. Fw nd: C,56.71; H,7.48; N,9.16. C26H38N406; 2.5
H20 requires C,57.02; H,7.91; N,10.23%.
. . WO 91/10644 ~ 0 7 ? 12 6 PCr/EP90/02156
. .
29
EX~MPLE 7
2-f 1- r 2 (S) ~box~-3- (N2-met:ha~onyi S-lysYlamino) -
pr ~ l]cyclc~pentylcar~orvlamino!- ~ indene 2 ~ xylic
acid
me p ~ e of EXample 6 was followed hut using N6~
benzyloxy-car~onyl-N2-methanesulphonyl-S-lys~e m the cGupling
step to give the title compound. Found: C,53.60; H,7.27; N,8.82.
C27H40N48S; 1-5 ~2 ~ C,53.36; H,7.13; N,9.22%.
EX~MPIE 8
2-~ 1-[2(R,S)-Carboxypentvl]cYclcPentylcarbonYlamino~-2-
hy~r~xymethyl-2,3-dihydr3indene
(a) 2-Amino-2-h ~ ethyl-2 3-dihvdroindene
A solution of 2-am m o-2,3-dihydroindene-2-carboxylic, ethyl
ester, hy~rochloride (1.7 g, 7.03 mmol) in 50% aqueous ethanol (15
ml), was added dropwise to a stirred, ice-cold solution of sodium
borohydride (1.11 ~, 29.4 mmol) in 50% aqueuus ethanol (35 ml),
then the reaction mixture heated under reflux for 3 hours.
The buIk of the ethanol wa~ rem3ved by evaporation under
vacuum, and the residLal suspension saturated with sodium chloride
then extracted with ethyl a oetate. The combined extracts were
washed with saturated brine, dried (anhydrous MgS04), filtered anl
evaporated under vacuum. Crystallisation of the resulting white
solid (1.05 g) from ethyl acetate-hexane afforded the reguired
product (0.99 g, 84.8%), m~p. 89.5-90.5 &. Fcund: C,72.32;
H,8.02; N,8.17. ~ oH13NO; 0.15 H20 requlres C,72.39; H,8.08;
N,8.44%.
(b) 2-~l[(R S)-BenzyloxycarbonylFentyl1cycloDentylcarbon~lam m o)-
2-hyd3~yme~hyl-2 ? 3-dih~noindene
: ~ . .. . i
: . ~ ~ - . . .. .
.
wo gl/10644 2 Q 7 ~1~ 6 PCT/EP90/02l56
1-(3-Dimethylanunoprcpyl)-3-ethylcarbodilmide hydrochlor.ide
(767 mg, 4 mmol) was add~d to a stirred, ice-oold n~bure of 1-
[2(R,S)-b~nzyloxycartonylpentyl]cyclopentane carha~ylic acid
(637 mg, 2 mmol), l-hydroxyben20triazole (270 mg, 2 mmol),
4-methylmorpholine (202 mg, 2 ~mol), the product from step (a)
above (332 mg, 2 mmol) and dry dichloromethane (10 ml)0 Stirring
was continued for 0.5 hours at 0C, then for 24 hcurs at rcom
temperat~re.
Ihe dichlorcmethane was removed by evaporation under vacuum,
and the residue partitioned between diethyl ether and water. m e
ether phase was separated, washed with water, lM hydrcchloric
acid, water, saturated aqueous sodium bicarbonate solution and
water, then dried (anhydrous M~S04) and filtered. Evaporation
under vac~um of the filtrate furniah d an oil which was p~rified
by chromatography on silica gel using diethYl ether-hexane (1:1)
as eluent. Evaporation under vacuum of the appr~priate fractions
provided the required prcduct (180 mg, 19.0%). Fo~nd: C,73.75;
H,7-84; N,3-06. C29H37N04; 0.5 ~ o ~ s C,73.69; H,8.10;
N,2.96%.
(c) 2-(1-r2~R.S~-CarboxYpentYllcycloPentYlcarbonylamino~-2-
hYdroxvn~thyl-2,3-dihYdroindene
A solution of the above pro*uct (170 m3, 0.36 mmol) in a
mixture of ethanol (20 ml) and water (1 ml), was hydrogenated over
5% palladium on c ~ at 50 p.s.i. (3.45 bar) and room
temperature, t~ give the title prc~uct as a white solid ~115 mg,
83.5%), m.p. 126-128 & , after trituration with diethyl ether-
hexane (1:1). Found: C,69.04; H,8.15; N,3.63. C22H31N04; 0.5 ~ 0
requires C,69.08; H,8.43; N,3.66%.
~-. W 0 91/10644 2 0 7 212 6 PCT/EP90/0215b
31
~ _9
2~ 2(R,S!~o~ 4~1b~11~nb~1~ylam~no~-
2-hydrDxymethyl-2,3-dihydroindene
(a) 2-~1-r2fR,S)-BenzyloxycarbonYl-4-ph y ~utyl~cvclcpentyl
carbonvlamino~-2-h ~ ethvl-2,3-dihvdroi~ne
Ihe prooedure of Example 8(b) was followed using 1-[2(R,S)-
benzyloxycarkonyl-4-phenyIbutyl]cyclcF(rldooYaaraoxylic acid as
starting material and allowing the reaction mixture to stand at
room temperature for a further 5 days. The ~ d product was
oktained as an oil (19.9%). Rf (silica) 0.15 (diethyl ether-
hexane, 1:1).
(b) 2-(1- r 2fR.S)-Carboxv-4-Phenylbutyllc~clopentYlcar~on~lamin~)-
2-hvdroxYmethyl-2,3-dihydroindene
Hydrogenation of the above product as described in Example
8(c), followed by chromQtograFhy on silica gel using ethyl aoetate
as eluent and evaporation under vacuum of the apprcpriate
fractions, followed by trituration of the residue with diethyl
ether-hexane (1:4), gave the required product (62.4%) as a white
solid, m.p. 140-143C. Fo~n~: C,73.76; H,7.62, N,3.12.
C2 ~33N4; V4 H20 eqyIres C,73.69; H,7.67; N,3.18%.
EX~MPIE lQ
2-~1-r2~R,SI ~ ox~-3-~S-lvsvlamino!Propyl~cyclopentvl-
/
carbonylamino~2-hydroxymethyl-2.3-dihydroindene
(a~ 2-~1-r2(R S)-tert-Butox~carbonyl-3-(d~benzylamino)prceyll
cyclocentylcarbonylamuno~-2-hydr~xvmethyl-2~3-dihydroind~ne
4-M~thylmorphol m e (1.78 g, 17.6 ~mol) was added to a stirred
solution of the N-hydrcxyberzotriazole-derived activated ester
,~
''
, . .
.. . .
W 0 91/t0644 .~ PCT/EP90/021~6 ,~
hemi-solvate with dichloromethane of 2(R,S)-tert-butoxycar~ony1-3-
( ~ ylamino)propylcyclopentane carhoxylic acid (Ex2mple 3b)
(9.78 g, 16 1) and 2-amino-2-hydroxymethyl-2,3-dihydroindene
(2.65 g, 16 1) m dry dichloromethane (50 ml) at room
temperature. After 2 hours the solvent was removed by evaporation
under vacuum and the residue allowed to s ~ d a~ room te~perature
for 2 days be~ore being partitioned between~diethyl ether and
water. Ihe ether pbase was separated, washed with water, dried
(anhydrcus Mg504) and filtered. Evaporation under vacuum of the
filtrate, followeol ~y purification of the residue by
chr~matography on silica gel using a dichlorcmethane in hexane
elution gradient (20-100%), afPorded the required product (3.30 g,
33.6%). Found: C,75.09; H,8.02; N,4.69. C38H48N204, 0.2 CH2C12
requires C,74.75; H,7.95; N,4.57%.
(b) 2~ 3-Amino-2(R.S!-~ert-butoxycarbonYlprcpYl~cyclopent
carbonYlaminol-2-hYdroxvmethyl-2.3-dihydroindene
Ihe abcve product was h~drogenated as described in Example
3(c) to give the title amine in 98.2% yield. Found: C,68.96;
H,8-88: N,6-63- C24H36N204 reguires C,69.20; H,8.71; N,6.73%.
(C) 2-fl-r2 ~,S)-te~t-ButoxYcar~on~yl-3-(N2~ N6-di-b ~ ylox~
carbonyl-S-lysylamino)pro~y~lcyclo~entYlcarbonylamunol-2-hydroxy-
m~thyl-2 3-dih~lroindene
m e above amine was coupled to N2, N6~dibenzyloxycarbonyl-S-
lysine following the prooedure of Example 3(d) but using a
gradient of diethyl ether-hexane (1:8 to 1:1) followed by eth~l
a oe tate as eluents for chromatographic purification to give the
product in 62.0% yield. Found: C,67.59; H,7.53; N,6.75.
C46H6~M409 requires C,67.36; H,7.44; N,6.89%.
,
' ' ' ' ' ' . .
~- ~ wo gl/10644 2 0 ~ 2 ~ 2 ~ PCT/EP9OtO2156
~ ~.
33
(d) 2~ r 2(R,S)-Car~oxy-3-t~ , N6-di-ben2Yloxycarbonyl-S-lysyl-
amino~propyl]cyclcpentylcarbonylamInol-2-hydr3xymethyl-2,3-
dihydroindene
A stirred, ice-cold solution of the above product (1.24 g,
1.52 mmol) in dry dichloromethane (20 ml) was saturated with dry
hydrogen chloride. After a further 2 hours at 0C, the re~ction
~ e was evaporated under vacuum and the residue azeotroped
with dichlor~methane to provide the title prcduct as a white fo~m
(1.05 g, 85.3%). Fcun~: C,64.28; H,6.83; N,7.21. C42HS2N409; 1.5
H20 requires C,64.3S; H,7.07; N,7.15%.
(e) 2~ 2(R,S)-Carkoxv-3-(S-lysYl ~ )proPvllc~cloPentyl-
car~onylamino~-2-h,vdroxymeth~rl-2,3-dih~vdroindene
The above product was hydrogenated following the prooedure of
Example 3(f) to give the required title acid as a white foam
(79.7%). Fcund: C,61~20; H,8.14; N,10.41. C26H40N405; 1.25 H20
requires C,61,09; H,8.38; N,10.96%.
' ~ME~E 11
2 - ~ 1- r 2 ~ s ! -Carbo~y-3-(N2-~ethanesulphonyl-S-lYsvlamlno)-
,,
DropyllcvclopentYlcarbonvlamln3)-2-hY ~ ~hvl-2.3-dihvdro mdene
m e prooedure of Example 10 was followed but using the
N-hYdroxy~enzokriazole-derived activated ester of 2(S)-tert-
butyloxycaxbonyl-3-[(S,S)-alpha, alphal-dimethyldiben~yla D o]-
propylcyclopentane carboxylic acid as startLng material in step
(a) and ooupling with N6-tert-~utyloxycarbonyl-~ -methane-
sulphonyl-S-lysine in step (c). Deprotection as previously
.
described gave the product which was dissolved in a little
distilled water and freeze dried to give the title produc~ as a
white solid. ~ound: C,51.50; H,7.38; N,8.67. C27H42N407S; HCl;
1.5 ~ O requires C,51.46; H,7.36; N,8.89%.