Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
~lr~,t7
AND THEIR THERAPEUTIC APPLICATION
The present invention relates to compounds which
are 2-(1-piperidyl)ethanol derivatives or salts thereof, their
preparation, their therapeutic application and to an
intermediate useful in the preparation of the compounds.
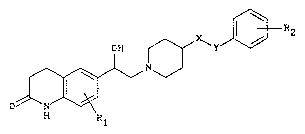
The present invention provides a compound which is
a 2-(1-piperidyl)ethanol derivative of the general ~ormula (I)
lo o~ 3
H R
in which
R1 represents hydrogen, halogen atom or methyl
R2 represents one or two substituents independently chosen from
halogen, (~1-4)alkyl, hydroxyl, (C14~alkoxy, trifluoromethyl,
acetylamino and methylsulphonylamino, and
either X represents a CH2 group and Y represents a CH2, (CH2)2
or CO group,
or X represents a CO group and Y represents a CH2 group,
provided that R1 and R2 are not both hydrogen when X and Y each
represent a CH2 group, or a pharmaceutically acceptable acid
addition salt thereof. The present invention also provides a
process for the preparation of the 2~ piperidyl)ethanol
derivatives and the salts thereof, an intermediate useful in
the synthesis of them and therapeutic application oE them. The
compound of formula (I) in which R1 and R2 each represent a
~¢?i7,~
2 - .
hydrogen atom and X and Y each represent a CH2 group is
described in EP-A-481853.
In a preferred embodiment R1 represents hydrogen,
fluorine or methyl and R2 represent~ one or two ~ubstituents
independently chosen from fluorine, chlorine, methyl, hydroxy,
methoxy, trifluoromethyl, acetylamino and methylsulphonylamino.
The pharmaceutically acceptable acid is preferably
the fumarate or oxalate.
Examples of specific compounds of the present
invention are:
(+)-6-[1-hydroxy-2-[4-[2-(4-fluorophenyl)ethyl]-1-piperidyl]-
ethyl]-3,4-dihydroquinolin-2(lH)-one;
(+)-6-[1-hydroxy-2-[4-[2-(4-chlorophenyl)ethyl]-1-piperidyl]-
ethyl]-3,4-dihydroquinolin-2(1H)-one;
(+)-8-fluoro-6-[1-hydroxy-2-[4-[2-(4-fluorophenyl)ethyl~
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one;
~ 6-[1-hydroxy-2-[4-(3-phenylpropyl)-1-piperidyl]ethyl]-3,4-
dihydroquinolin-2(1H)-one;
(+)-6-[1-hydroxy-2-[4-(2-phenyl-2-oxoethyl)-1-piperidyl]ethyl]-
3,4-dihydroquinolin-2(1H)-one;
(+)-6-[1-hydroxy-2-[4-[2-(4-fluorophenyl)-1-oxoethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one;
(+)-8-fluoro-6-[1-hydroxy-2-[4-(2-phenylethyl)-1-piperidyl]-
ethyl]-3,4-dihydroquinolin-2(lH)-one;
(+)-8-fluoro-6-[1-hydroxy-2-[4-[2-(3-fluorophenyl)ethyl]~
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one;
(+)-6-[1-hydroxy-2-[4-[2-[4-(trifluoromethyl)phenyl]ethyl]-
1-piperidyl]ethyl~-3,4-dihydroquinolin-2(1H)-one;
- 3 -
(-)-6-~1-hydroxy-2-[4-[2-[4-(trifluoromethyl)phenyl]ethyl~-
1-piperidyl]ethyl]~3,4-dihydroquinolin-2(lH) one;
(+)-6-[1-hydroxy-2-[4-t2-[4-(trifluoromethyl?phenyl]ethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(lH)-one;
(+)-6-[1-hydroxy-2~[4-[2-t4-(acetylamino)phenyl]ethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.;
(+)-6-~1-hydroxy-2-t4-(2-phenylethyl~-1-piperidyl]ethyl]-
7-fluoro-3,4-dihydroquinolin-2(lH)-one;
(+)-6-[1-hydroxy-2-[4-[2-(2-methylphenyl)ethyl]-1-piperidyl]-
ethyl]-8-methyl-3,4-dihydroquinolin-2(lH)-one; and
(+)-6-[1-hydroxy-2-[4-[2-(3,5-difluorophenyl)ethyl]-
l-piperidyl]ethyl] 3,4-dihydroquinolin-2(lH)-one.
Given that the molecule represented by the general
formula (I) possesses an asymmetric carbon atom, the compounds
of the invention may exist in the form of pure enantiomers or a
racemic mixture. The compounds of the invention ars in the
form of free bases or addition salts with pharmaceutically
acceptable acids.
The compounds of the invention may be prepared
according to scheme 1 on the next page.
- 4 - 2~
Sch~me 1
3~V~ ~3 R2 (v)
3~ A~ ~,J3R2 1 II )
Hh ll Cl
(III)
H Rl
O ~ ~"~P~2 (IV)
~Rl I
OH ~ ~R2 (Ia)
~... !
~R2 (I)
OH ~ Y
N~J
- 5 ~ ?
A compound of general formula (II) in which
either A represents a CH2 group and B represents a CH2 or (CH2) 2
group~
or A or B represents a CH2 group and the other represents a
1,3-dioxolan-2,2-diyl group,
is reacted with a compound of general formula (III), in which
R~ is as defined above, to give a compound of general formula
(IV).
The ketone functional group of the compound (IV) is
then reduced with a reducing agent such as potassium
borohydride.
If A represents a CH2 group and B represents a CH2 or (CH2) 2
group, the compound of formula (Ia) thus obtained corresponds
to the general formula (I) -in which X represents a CH2 group
and Y represents a CH2 or ~CH2) 2 group.
If A or B represents a CH2 group and the other represents a
1,3-dioxolan-2,2-diyl group, the compound of general formula
(Ia) is hydrolysed in acidic medium, and a compound of general
formula (I) in which X or Y represents a CH~ group and the
other represents a CO group is thus obtained. If desired, the
compound of formula (I) thus obtained is converted into a
pharmaceutically acceptable acid addition salt.
The starting compounds of general formula (II) are
known or alternatively they can be prepared according to
methods similar to methods which are known or which are
described in the literature, for example in J. Org. Chem., 22,
1376 (1957); C.A., 52, 8138a (1958); C. R. Acad. Sci., 255, 956
(1962); US-P-4254127; EP-A-12643 and EP-A-481853: FR-A-2459795.
Thus, for example, a compound of general formula
- 6 - ~r~ ~b~
(V), in which V or W represents a CH2 group and the other
represents a C0 group, may be used as starting material.
In order to obtain a compound of general formula (II~ in which
A and B each represent a CH2 group, the C0 group of the
compound (V) is reduced by means of hydrazine in the presence
of a base, and then the nitrogen of the piperidine ring is
deprotected by debenzylation.
In order to obtain a compound of general formula (II) in which
A or B represents a CH2 group and the other represents a
1,3-dioxolane-2,2-diyl group, the C0 group of the compound (V)
is converted by ketalisation by means of ethylene glyc~l in the
presence of 4-methylbenzenesulphonic acid, and then the
nitrogen of the piperidine is deprotected by debenzylation~
The compounds of general formula (II) in which A
and B each represent a CH2 group and R2 is as defined above
except hydrogen are new and form part of the invention as
synthesis intermediates. They may be prepared according to the
processes illustrated by Schemes 2 and 3 below.
Scheme 2 illustrates a similar process to that
described in C~ R. Acad. Sci., 255, 956 (1962); first, an ester
of general formula (VI) in which Alk represents a (C~ 4) alkyl
group, is reacted with a benzeneacetonitrile of general formula
(VII) in which R2 is as defined above, in the presence of
sodium amide. An enol of general formula (VIII) is obtained and
treated in acidic medium in order to obtain a ketone of general
formula (IX) which is reduced for example by means of hydrazine
hydrate in the presence of potassium hydroxide and triethylene
glycol in order to obtain a compound of general formula (X),
and finally this compound is subjected to debenzylation by
- 7 ~ ..J ~
catalytic hydrogenation or by chemical deprotection by reackion
with trichloroethyl chloroformate followed by treatment of the
resultant product with zinc in acetic acid in order to obtain a
compound of general formula (II') which corresponds to the
general fo~mula (II) when A and B each represent a CH2 group.
Scheme 3 illustrates a process similar to that
described in J. Org. Chem~, 22, 1376 (1957); first,
4-methylpyridine of formula (XI) i6 reacted with a benzaldehyde
of general formula (XII) in which R2 is as defined above, in
acetic anhydride. A compound of general formula (XIII) is
obtained, and this compound is subjected to catalytic
hydrogenation.
- 8 ~ . 7
Scheme 2
O- Alk
~0 (VI )
CJ~3 R2 (~ I I )
( V I I I )
CN
3~/ (IX)
I
~ z ( X)
HN ~ R2 (II ~ )
wg~ a ~
Scheme 3
~ C~3
IJ~ (XI)
~ HC ~ (XII)
lo
~ R2
~ f (XIII)
j R2 (II')
H~
The starting compounds of general formulae (VI),
(VII), (XI) and (XII) are available commercially.
Details of the preparation of certain specific
compounds are given in the examples below.
The starting compounds of general formula (III) are
also known or may be prepared according to methods similar to
methods which are known or which are described in the
literature, for example in EP-A-0351282 and in J. Med. Chem.,
29, 2433 (1986).
Thus, for example, a quinolin-2(lH)-one which is suitably
substituted by R1 may be used as starting material, and is
- 10 --
~ubjected to catalytic hydrogenation in order to obtain the
corresponding 3,4-dihydroquinolin-2(1H)-one, and the compound
of general formula (III) is obtained by reaction with
chloroacetic acid chloride in the presence of aluminium
chloride.
Finally, the enantiomers of a compound according to
the invention may be prepared from the racemate according to
conventional methods, for example by formation of an additior
salt with an optically pure chiral acid, fractional
crystallisation, and release of the base.
The following examples illustrate in detail the
preparation of some compounds of the invention. Microanalyses
and IR and NMR spectra confirm the structures of the compounds
obtained. The compound numbers indicated in brackets in the
titles correspond to those in the tables which are given
further on.
The example and compound numbers given in Roman numerals
correspond to the starting 4-(2-phenylethyl)piperidines of
general formula (II'), and the example and compound numbers
given in Arabic numerals correspond to final compounds of
general formula (I).
Example I (Compound No. III)
4-[2-(4-Fluorophenyl)ethyl]piperidine, hydrochloride.
I.l. 2-(4-Fluorophenyl)-3-hydroxy-3-[1-(phenylmethyl)-4-
piperidyl]prop-2-enenitrile.
13.5 g (0.1 mole) of 4-fluorobenzeneacetonitrile,
50 ml of toluene and 4.4 g of sodium amide are introduced into
2~ ~
a soo-ml round bottom ~lask. The mixture is stirred at room
temperature for 30 minutes and then 27.2 g (0.11 mole) of ethyl
l-(phenylmethyl)-4-piperidinecarboxylate are added by means of
a dropping funnel. After the addition is completed, the mixture
is heated at 80C for 3 hours. It is then cooled in an ice bath
and 300 ml of water are added, the mixture is stirred for
30 minutes and 15 ml of acetic acid are added. A yellow
precipitate is obtained which is separated by filtration,
washed with water and dried in the presence of phosphorus
pentoxide. 17.6 g of compound are obtained which are used as
such in the next stage.
I.2. 2-(4-Fluorophenyl)-l-[l-(phenylmethyl)-
4-piperidyl]ethanone.
25 ml of water, 50 ml of concentrated sulphuric
acid and 50 ml of acetic acid are introduced into a 500-ml
round bottom flask placed in a water and ice bath and 24.4 g
(0.072 mole~ of 2-(4-fluorophenyl)-3-hydroxy-
3-[1-phenylmethyl)-4-piperidyl]prop-2-enenitrile are added. The
mixture is refluxed for 12 hours. The round bottom flask is
cooled in a water and ice bath. 200 ml of ice are introduced
into a 1-litre Erlenmeyer flask and the mixture prepared above
is slowly added; 200 ml of ethyl acetate are added together
with 200 ml of ammonium hydroxide in order to adjust the pH to
9. The mixture is decanted, the organic phase is recovered and
the aqueous phase is extracted three times with 150 ml of ethyl
acetate. The organic phases are pooled and they are washed
three times with 150 ml of water until a pH of 7 to 8 is
obtained. The organic phase is dried over sodium sulphate,
,V~.V~
- 12 -
filtered and the ethyl acetate is evaporated under reduced
pressure. 20.7 g of oily compound are obtained which are used
as such in the next stage.
I.3. 4-[2-(4-Fluorophenyl)ethyl]-l-(phenylmethyl)piperidine.
62 g (0.199 mole) of 2-(4-fluorophenyl)-
l-[1-(phenylmethyl)-4-piperidyl]ethanone in solution in 400 ml
of ethanol are introduced into a l-litre round bottom flask.
11.6 ml (0.23 mole) of hydrazine hydrate are added and the
mixture is refluxed for 2.5 hours. It is cooled in an ice bath
and then the solution is concentrated to half the volume. The
product crystallises. 400 ml of water are then added, the
mixture is stirred and the white precipitate is filtered. This
precipitate is drained and dried in a desiccator in the
presence of phosphorus pentoxide. 73.3 g of intermediate
hydrazone are obtained. 64.8 g (about 0.199 mole) are taken up
in 300 ml of triethylene glycol and 30 g of potassium hydroxide
are added. The mixture is rapidly heated to 200C for 1 hour.
It is rapidly cooled in ice. 600 ml of water are then added and
the mixture is extracted three times with ethyl acetate. The
organic phase is washed with a saturated aqueous solution of
sodium chloride. It is dried over sodium sulphate and then the
solvent is evaporated under reduced pressure. A dark chestnut-
coloured oil is obtained which is purified by chromatography on
a silica gel column. 36.68 g of a pale oil are obtained which
are used as such in the next stage.
I.4. 4-[2-(4-Fluorophenyl)ethyl]piperidine, hydrochloride.
9.8 g (about 0.033 mole) of
- 13 ~
4-[2-(4-fluorophenyl)ethyl]-1-(phenylmethyl)piperidine in
solution in 100 ml of Pthanol and 10 ml of a normal aqueous
solution of hydrochloric acid are introduced into a Parr
apparatus. 1 g of 10 ~ palladium on carbon is added.
Hydrogenation is carried out at 0.35 MPa and at 50C for
6 hours. The catalyst is removed by filtration, it is washed
with water and ethanol, and 2 ml of concentrated hydrochloric
acid are added. The mixture is evaporated and azeotropically
dried with an ethanol/toluene mixture. It is again evaporated
and the residue is taken up in 100 ml of tetrahydrofuran. The
mixture is stirred for 15 minutes and the onset of
precipitation is observed. 20 ml of diisopropyl ether are added
and the mixture is further stirred for 15 minutes. The solid is
rapidly filtered and then dried in a desiccator in the presence
of phosphorus pentoxide. 5 g of compound are obtained.
Melting point: 131~132C.
Example II (Compound No. IV)
4-[2-(4-Chlorophenyl)ethyl]piperidine, acetate.
~0
II.l. 2-(4-Chlorophenyl)-3-hydroxy-3-[1-(phenylmethyl)-4-
piperidyl]prop-2-enenitrile.
30.3 g (0.2 mole) of 4-chlorobenzeneacetonitrile,
100 ml of toluene and 8.8 g of sodium amide are introduced into
a 500-ml round bottom flask. The mixture is stirred at room
temperature for 30 minutes and then 49.4 g (about 0.2 mole) of
ethyl l-(phenylmethyl)-4-piperidinecarboxylate diluted in 40 ml
of toluene are added by means of a dropping funnel. After the
addition is completed, the mixture is heated at 80DC for
- 14 ~ r~
3 hours. It is then cooled in an ice bath and 800 ml of water
are added, the mixture is stirred for 30 minutes and 15 ml of
acetic acid are added. A precipitate is obtained which is
separated by filtration, washed with water and dried in the
presence of phosphorus pentoxide. 65.67 g of compound are
obtained which are used as such in the next stage.
II.2. 2-(4-Chlorophenyl)-l-[l-(phenylmethyl)-
4-piperidyl]ethanone.
50 ml of water, 100 ml of concentrated sulphuric
acid and then 100 ml of acetic acid are introduced into a
500-ml round bottom flask placed in a water and ice bath and
30.21 g (0.086 mole) of 2-(4-chlorophenyl)-3-hydroxy-
3-tl-(phenylmethyl~-4-piperidyl]prop-2-enenitrile are added
while stirring. The mixture is refluxed for 12 hours. 200 ml of
ice are introduced into a l-litre Erlenmeyer flask and the
mixture prepared above is slowly added, and 400 ml of ammonium
hydroxide are added in order to adjust the pH to between 8 and
9. The mixture is stirred in an ice bath and the precipitate
formed is separated by filtration, washed with water and dried
in the presence of phosphorus pentoxide. 28.75 g of compound
are obtained which are used as such in the next stage.
II.3. 4-[2-(4-Chlorophenyl)ethyl]-l-(phenylmethyl)-
piperidine.
26.26 g (0.08 mole) of 2-(4-chlorophenyl)--
l-[l-(phenylmethyl)-4-piperidyl]ethanone in solution in 160 ml
of ethanol are introduced into a 500-ml round bottom flask.
4.7 ml (0.093 mole) of hydrazine hydrate are added and the
mixture is refluxed for 1.5 hours. It is cooled in an ice bath
and then the solution is concentrated to nalf the volume. The
product crystallises. 160 ml of water and 200 ml of ethyl
acetate are then added, the mixture is stirred and the organic
phase is separated; the aqueous phase is extracted once more,
the organic phase is washed with a saturated aqueous solution
of sodium chloride and dried over sodium sulphate, the solvent
is evaporated under reduced pressure, and 28.3 g of
intermediate hydrazone are obtained in the form of an oil which
crystallises. It is taken up in 130 ml of triethylene glycol
and 14 g of potassium hydroxide are added. The mixture is
rapidly heated to 200C for 1 hour. It is rapidly cooled in
ice. 260 ml of water are added and the mixture is extracted
five times with ethyl acetate. The organic phase is washed with
a saturated aqueous solution of sodium chloride. It is dried
over sodium sulphate and then the solvent is evaporated under
reduced pressure. 27.07 g of a chestnut-coloured oil are
obtained which are used as such in the next stage.
II.4. 4-[2-(4-Chlorophenyl)ethyl]piperidine, acetate.
10.7 g (0.034 mole) of 4-[2-(4-chlorophenyl)ethyl]-
1-(phenylmethyl)piperidine, 14.4 g (0.06~ mole) of
trichloroethyl chloroformate, 0.5 g of potassium carbonate and
100 ml of toluene are introduced into a 500-ml round bottom
flask. The mixture is refluxed for 5 hours and allowed to cool,
200 ml of water are added and the mixture is stirred, the
aqueous phase is separated and extracted with ethyl acetate,
the organic phase is washed with a saturated aqueous solution
of sodium chloride and dried over sodium sulphate and the
16 ~ AJ~r~
solvent is evaporated under reduced pressure. 21.7 g of an oil
are obtained which are introduced into a 500-ml round bottom
flask, 150 ml of acetic acid and 9.13 g ~0.139 mole) of
powdered zinc are added, and the mixture is stirred overnightO
The zinc is separated by filtration and the filtrate is
evaporated under reduced pressure; the residue is triturated
with diethyl ether and the solid i~ separated by filtration,
washed with diethyl ether and dried in the presence of
phosphorus pentoxide. 7.6 g of crude product are finally
isolated.
Melting point: 155C.
Example III (Compound No. VII)
4-[2-(4-Methoxyphenyl)ethyl]piperidine, hydrochloride.
III.l. 4-[2-(4-Methoxyphenyl)ethenyl]piperidine.
46.56 g, equivalent to 48.6 ml (0.5 mole) of
4-methylpyridine, 122.5 g, equivalent to 109.~ ml (0.9 mole) of
4-methoxybenzaldehyde and 50 ml of acetic anhydride are
introduced into a round bottom flask and the mixture is heated
~t 180~C for 3.5 hours. It is allowed to stand at room
temperature overnight and then concentrated under reduced
pressure. The residue is washed several times with diethyl
ether and dried in the presence of phosphorus pentoxide. 35 g
of compound are obtained which are used as such in the next
stage.
Melting point: 122-123C.
III.2. 4-[2-(4-Methoxyphenyl)ethyl]piperidine, hydrochloride.
35 g ~0.166 mole) of 4-t2-(4-methoxyphenyl)-
ethenyl]piperidi~e, a mixture of 175 ml of ethanol and 175 ml
of 1 N hydrochloric acid and 3 g of '0 ~ palladised carbon are
introduced into a Parr flask and hydrogenation is carried out
at about 0.35 Mpa, at 50C for 14 hours. The catalyst is
separated by filtration, by washing it with ethanol, and the
solvent is evaporated under reduced pressure. Traces of water
are removed from the residue by azeotropic entrainment with
toluene and the residue is washed with diethyl ether and dried
in the presence of phosphorus pentoxide. 36.08 g of compound
are obtained. 3 g of it are recrystallised from 2-propanol and
the product is dried at 80UC in the presence of phosphorus
pentoxide. 2.53 g of compound are obtained.
Melting point: 178-179~C.
Example IV (Compound No. VIII)
4-[2-(4-Trifluoromethylphenyl)ethyl]piperidine, hydrochloride.
IV.l. 4-[2-(4-Trifluoromethylphenyl)ethenyl]piperidine.
46.56 g, equivalent to 48.6 ml (0.5 mole) of
4-methylpyridine, 104.47 g, equivalent to 81.9 ml (0.6 mole) of
4-trifluoromethylbenzaldehyde and 50 ml of acetic anhydride are
introduced into a round bottom flask and the mixture is heated
at 1~0C for 7.25 hours. It is concentrated under reduced
pressure. The residue is triturated in 100 ml of isopropyl
ether, drained and dried in the presence of phosphorus
pentoxide. 73.65 g of compound are obtained which are used as
such in the next stage.
Melting point: 72C.
s ~ r~
~ 18 ~
IV.2. 4-t2-~4-Trifluoromethylphenyl3ethyl~piperidine,
hydrochloride.
73.15 g (0.293 mole) of 4-[2-(4-trifluoro-
methylphenyl)ethenyl]piperidine, a mixture of 350 ml of ethanol
and 350 ml of 1 N hydrochloric acid and 5 g of 10 % palladised
carbon are introduced into a Parr fla~k and hydrogenation is
carried out at about 0.35 Mpa, at 50C for 8 hours. The
catalyst is separated by filtration, washed with ethanol and
the solvent is evaporated under reduced pressure, and the
residue is washed with diethyl ether and dried in the presence
of phosphorus pentoxide. 37.2 g of compound are obtained. 3 g
of it are recrystallised from 30 ml of toluene and the compound
is taken up in 30 ml of diethyl ether, drained and dried at
80C in the presence of phosphorus pentoxide. 2.72 g of
compound are obtained.
Melting point: 166-167C.
Example V (Compound No. XIV)
N-[4-[2-(4-Piperidyl)ethyl]phenyl]acetamide, hydrochloride.
V.l. 4-[2-(4-Nitrophenyl)ethenyl]pyridine.
15.11 g (0.1 mole) of 4-nitrobenzaldehyde, 9.3 g
(0~1 mole) of 4-methylpyridine and 15 ml of acetic anhydride
are introduced into a round bottom flask and the mixture is
heated in an oil bath (bath temperature: 180C) for 7 hours. It
is concentrated under reduced pressure and the residue is taken
up in petroleum ether and triturated. The solid is separated by
filtration and dried.
-- 19 --
V.2. 4-[2-(4-Pyridyl)ethyl]benzeneamine.
The product from the preceding stage, 140 ml of
ethanol, lO0 ml of 1 N hydrochloric acid and 3 g of 10
palladised carbon are introduced into a Parr flask and
hydrogenation is carried out at 0.28 MPa for 8 hours, at room
temperature. The catalyst is separated by filtration, the
filtrate is evaporated and the residue is dried by entrainment
with toluene and triturated in diethyl ether. 10.3 g of product
are obtained after drying.
Melting point: 108-110C.
V.3. N-[4-[2-(4-Pyridyl)ethyl]phenyl]acetamide.
5 g (0.025 mole) of ~-[2-(4-pyridyl)ethyl]-
benzeneamine and 25 ml of dichloromethane are introduced into a
round bottom flask and the mixture is stirred for 5 minutes at
room temperature in order to obtain a solution; 10 ml of acetic
anhydride are added and the mixture is stirred for 2 hcurs at
room temperature.
The precipitate is separated by filtration, taken up in 50 ml
of water and diluted ammonium hydroxide is added, the mixture
is stirred for 30 minutes and the white precipitate is
separated by filtration, washed with water and dried in the
presence of phosphorus pentoxide.
5.35 g of compound are obtained.
Melting point: 178C.
V.4. N-[4-[2-(4-Piperidyl)ethyl]phenyl]acetamide,
hydrochloride.
5.25 g of N-[4-[2-(4-pyridyl)ethyl]phenyl]acetamide
- 20 ~ t~ ~- ~
in solution in a mixture of 50 ml of ethanol and 25 ml of 1 N
hydrochloric acid are introduced into a Parr flask and 0.5 g of
10 % palladised carbon are added and hydrogenation is carried
out at 0.35 MPa at 50C for 16 hours. The catalyst is separated
by filtration, the solvents are evaporated and the residue is
taken up in a mixture of ethanol and diethyl ether and
triturated.
5.2 g of product are obtained after filtration and drying in
the presence of ph~sphorus pentoxide.
1 g of it is recrystallised from propanol containing 1 %
concentrated hydrochloric acid.
0.68 g of compound are obtained.
Melting point: 256-258C.
Example VI (Compound No. XI)
4-[2-(3,5-Difluorophenyl)ethyl]piperidine, hydrochloride.
VI.l. 4-[2-(3,5-Difluorophenyl)ethenyl~pyridine.
25 g (0.176 mole) of 3,5-difluorobenzaldehyde,
16.37 g (0.176 mole) of 4~methylpyridine and 20 ml of acetic
acid are introduced into a round bottom flask and the mixture
is heated ln an oil bath (bath temperature: 180C) for 7 hours.
The solvent is evaporated under reduced pressure and an oil is
obtained which crystallises on cooling. This residue is taken
up in 50 ml of diisopropyl ether, the mixture is triturated and
the solid is separated by filtration, washed with diisopropyl
ether and dried in the presence of phosphorus pentoxide.
30.94 g of product are obtained.
Melting point: 114C.
- 21 ~r~ "
VI.2. 4-[2-(3,5-difluorophenyl)ethyl~piperidine,
hydrochloride.
30.90 g (0.142 mole~ of 4-[2-(3,5-difluoro-
phenyl)ethenyl]pyridine, 140 ml of ethanol, 140 ml of 1 N
hydrochloric acid and 2.5 g of 10 % palladised carbon are
introduced into a Parr flask and hydrogenation is carried out
at 0. 35 MPa for 14 hours.
The catalyst is separated by filtration, the filtrate is
evaporated and the residue is dried by entrainment Wit]l toluene
and then in the presence of phosphorus pentoxide.
30.88 g of compound are obtained. 1.5 g of it are purified by
recrystallisation from 60 ml of toluene, washed with diethyl
ether and then dried in the presence of phosphorus pentoxide.
1.28 g of pure hydrochloride are finally isolated.
Melting point 161-162C.
Table 1 below illustrates the chemical structures
and the physical properties of some compounds of general
formula (II').
In the "salt" column, HCl denotes a hydrochloride and AcOH
denotes an acetate; in the "m.p. ~C)" column, denotes a
crude product, that is to say unpurified and whose structure
has as yet not been confirmed by elemental microanalysis.
Table 1
4-(2-Phenylethyl)piperidines of general formula (II')
- 2~ r, ~3
,~ R2
/
No . R2 Salt m . p ( C
I 2-F HCl 151-1S2
II 3-F HC1 171-172
10III 4-F HCl 131-132
IV 4 -Cl AcOH 15 5
V 4-CH3 HCl 160-161
VI 3-CH3 HCl 119-12 0
Vl I 4 -OCH3 HCl 17 8 -17 9
15VIII 4-CF3 HCl 166-167
IX 2-CF3 BCl lS6 187
X 3-CF3 HCl 73-74
XI 3 ~ 5-F2 BCl 161-162
XII 2,4-F2 HCl 130-131
20XIII 3 ~ 4-F2 HCl 137-138
XIV 4 -NHCOCH3 HCl 256-258
- 23 - ~r~"-
Example 1 (Compound No. 3)
(~)-6-[1-Hydroxy-2-[4-[2-(4-fluorophenyl)ethyl]-
l-piperidyl]ethyl]- 3,4-dihydroquinolin-2(lH~-one.
4.5 g (0.02 mole3 of 6-(chloroacetyl3-
3,4-dihydroquinolin~2(lH)-one, 100 ml of ethanol and 20 ml of
water are placed in a round bottom flask. 4.9 g (0.02 mole) of
4-[2-(4-fluorophenyl)ethyl~piperidine hydrochloride and then
4 g (about 0.04 mole) of sodium carbonate are added. The
mixture is refluxed for 1.25 hours. It is allowed to cool and
then 9 g of potassium borohydride are added. It is further
stirred for 2 hours and then the mixture is allowed to stand
overnight at room temperature. 200 ml of water are then added,
the mixture is stirred for 1 hour and the precipitate formed is
filtered. It is thcroughly washed with water and then drained
and dried in a desiccator in the presence of phosphorus
pentoxide. The product obtained is purified by chromatography
on silica gel, eluting with a 9/1 dichloromethane/methanol
mixture, and then it is recrystallised from 90 ml of ethanol.
It is dried in a desiccator in the presence of phosphorus
pentoxide and then under vacuum at 80~C. 5.33 g of compound are
obtained.
Melting point: 159-160C.
Example 2 (Compound No. 4)
(~3-6-[1-Hydroxy-2-[4-[2-(4-chlorophenyl)ethyl]-
1-piperidyl]ethyl]-3,~-dihydroquinolin-2(1H)-one.
2.1. 4-[2-(4-Chlorophenyl)ethyl]piperidine, hydrochloride.
Z5.14 g (0.08 mole) of 4-[2-(4-chlorophenyl)ethyl]-
l-(phenylmethyl)piperidine in solution in lOo ml of anhydrous
2 4 ~ ~ r ~ ~
dichloromethane and 8.4 ml (0.088 mole) of ethyl chloroformate
are introduced into a round bottom flask. The mixture is
refluxed for 3.5 hours. The solvent is evaporated and the
residue is purified by chromatography on a silica gel column,
eluting with isopropyl ether. A pal~ oil i5 recovered to which
50 ml of an aqueous solution of sodium hydroxide at 30 % by
volume and 100 ml of ethanol are added. The mixture is heated
at 80C for 26 hours. It is evaporated to dryness and then the
residue is taken up in a solution of 100 ml of water and 100 ml
of a saturated aqueous solution of sodium chloride. The mixture
is extracted three times with ethyl acetate and then the
organic phase is washed with a saturated aqueous solution of
sodium chloride until a neutral pH is obtained. 5 ml of
concentrated hydrochloric acid are added to the organic phase
and it is then evaporated to dryness: a product crystallises.
An azeotropic entrainment is carried out with ethanol, the
precipitate is taken up in ether, the mixture is filtered and
the precipitate is washed with ether and then dried in a
desiccator in the presence of phosphorus pentoxide. 8.35 g of
hydrochloride are obtained which are used as such in the next
stage.
2.2 (+)-6-[1 Hydroxy-2 [4-[2-(4-chlorophenyl)ethyl]-
1-piperidyl]ethyl]-3,4-dihydroquinolin-2(1}l)-one.
3.35 g (0.015 mole) of 6-chloroacetyl-
3,4-dihydroquinolin-2(1~1) one, 3.90 y (0.015 mole) of
4-[2-(4-chlorophenyl)ethyl]piperidine hydrochloride and 4.24 g
(0.04 mole) of sodium carbonate in a solution of 80 ml oE
ethanol and 20 ml of water are placed in a 500-ml round bottom
- 25 ~ 3,~
flask. The mixture is refluxed for 1 hour. It is allowed to
cool to room temperature and then 7.25 g of potassium
borohydride are added. The mixture is stirred and then allowed
to stand overnight. 160 ml of water are then added, the mixture
is stirred and the precipitate is collected by filtration. It
is washed with water, drained and dried in a desiccator in the
presence of phosphorus pentoxide. The product obtained is
recrystallised from 350 ml of ethanol and then it is again
recrystallised from 80 ml of propanol. 4.12 g of compound are
obtained.
Melting point: 189--190C.
Example 3 (Compound No. 18)
(+)-8-Fluoro-6-[1-hydroxy-2-[4-[2-(4-fluorophenyl)ethyl]-
1-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
3.6 g ~0.015 mole~ of 6-(chloroacetyl~-8-fluoro-
3,4-dihydroquinolin-2(1H)-one, 3.65 g (0.015 mole~ of
4-[2-(4-fluorophenyl)ethyl]piperidine hydrochloride and 3 g
(about 0.03 mole) of sodium carbonate in a mixture of 80 ml of
ethanol and 20 ml of water are placed in a round bottom flask.
The mixture is refluxed for 1.25 hours. It is cooled and then
7 g of potassium borohydride are added. The mixture is stirred
for 3 hours and then allowed to stand overnight. 160 ml of
water are added, the mixture is stirred for 30 minutes and then
the precipitate is collected by filtration. It is washed with
water, dried in a desiccator in the presence of phosphorus
pentoxide and then purified by chromatography on a silica gel
column, eluting with a 9/1 dichloromethane/methanol mixture.
The product is recrystallised from 70 ml of 2-propanol and then
-- 26 ~
dried at 80C in the presence of potassium hydroxide. 2.93 g of
compound are obtained.
Melting point: 163-164C.
Example 4 (Compound No. 10)
(+)-6-[1-Hydroxy-2-[4-(3-phenylpropyl)-1-piperidyl]ethyl~-3,4-
dihydroquinolin-2(lH)-one.
3.35 g (0.015 mole) of 6-(chloroacetyl)~
3,4-dihydroquinolin-2(lH)-one in solution in 100 ml of ethanol
and 20 ml of water, 3.05 g (0.015 mole) of
4-(3-phenylpropyl)piperidine and 2.12 g (0.02 mole) of sodium
carbonate are placed in a 500-ml round bottom flask. The
mixture is refluxed for 1 hour. It is allowed to cool to room
temperature and then 6.4 g of potassium borohydride are added.
The mixture is stirred and then allowed to stand overnight at
room temperature. 200 ml of water are added and the mixture is
stirred; the precipitate formed is filtered, thoroughly washed
with water and dried in a desiccator in the presence of
phosphorus pentoxide and then recrystallised from ethanol.
4.04 g of compound are obtained after drying.
Melting point: 164-165C.
Example 5 (Compound No. 11)
(+)-6-[1-Hydroxy-2-[4-(2-phenyl-2-oxoethyl)-1-piperidyl]ethyl]-
3,4-dihydroquinolin-2(1H)-one.
5.1. 1-Phenyl-2-[1-tphenylmethyl)-4-piperidyl]ethanone.
2.35 g ~0.0966 at.-g) of magnesium turnings and
40 ml of diethyl ether are introduced into a 500-ml round
- 27 ~
bottom flask and the mixture is stirred slowly ~nd 1 ml of
bromobenzene and then an iodine crystal are added. The stirring
is increased and 9.2 ml of bromobenzene (equivalent to a total
of 0.0966 mole~ in 20 ml of diethyl ether are added at reflux
temperature. The reflux is maintained for 30 minutes and then
the mixture is allowed to cool to room temperature. A solution
of 10 g (0.0466 mole) of 1-(phenylmethyl)
4-piperidineacetonitrile in 10 ml of diethyl ether is then
gently added while heating so as to maintain a slight reflux.
The reflux is maintained for 3.25 hours. The mixture is cooled
in an ice-cold bath and 100 ml of water and then 50 ml of 5 N
hydrochloric acid are slowly added and the mixture is stirred
for 1.5 hours at room temperature. A precipitate is for~ed
which is recovered by filtration and washed with water. After
drying in the presence of phosphorus pentoxide, 15.8 g of
product are obtained which are used as such in the next stage.
5.2. 4-[(2-Phenyl-1,3 dioxolan-2-yl)methyl]-
l-(phenylmethyl)piperidine.
15.8 g (0.0466 mole) of l-phenyl-
2-[1-(phenylmethyl)-4-piperidyl]ethanone in solution in 150 ml
of toluene are introduced into a round bottom flask. 30 g
(27 ml) of ethylene glycol and 16 g of 4-methylbenzenesulphonic
acid are added. The mixture is refluxed for 3.5 hours using a
Dean-Stark apparatus. The toluene is evaporated under reduced
pressurP and 150 ml of ethyl acetate and then 50 ml of a
solution of ammonium hydroxide at 30 % by volume are added. The
mixture is stirred, decanted and extracted a second time with
ethyl acetate and the organic phases are pooled, washed with a
- 28 -
saturated solution of sodium chloride, dried over sodium
sulphate and the solvent i~ evaporated under reduced pressure.
14.56 g of an oil are obtained which are used as such in the
next stage.
5.3. 4-[(2-Phenyl-1,3-dioxolan-2-yl)methyl]piperidine.
14.56 g (0.043 mole) of 4-[(2-phenyl-1,3-dioxolan-
2-yl)methyl]-1-(phenylmethyl)piperidine in solution in 100 ml
of ethanol and 20 ml of water are introduced into a Parr
apparatus. 1 g of 10 ~ palladium on carbon is added. The
mixture is hydrogenated at 0.35 MPa and at 50C for 5 hours.
The catalyst is separated by filtration and washed with water
and with ethanol: the filtrate is recovered and the solvent
evaporated under reduced pressure. 10.26 g of an oil are
obtained which are used as such in the next stage.
5.4. (+)-6~ Hydroxy-2-[4-[(2-phenyl-1,3-dioxolan-
2-yl)methyl]-1-piperidyl]ethyl]-3,4-dihydroquinolin-
2(lH)-one.
5.13 g (0.0207 mole) of 4-[(2-phenyl-1,3-dioxolan-
2-yl)methyl]piperidine in solution in 100 ml of ethanol and
20 ml of water, 4.64 g (0.0207 mole) of 6-(chloroacetyl)-
3,4-dihydroquinolin-2(1~1)-one and 2.2 g (0.0207 mole) of sodium
carbonate are placed in a 500-ml round bottom flask. The
25 mixture is heated at 80~C for 1.25 hours. It is allowed to cool
to room temperature and then 9.8 g of potassium borohydride are
added. The stirring is continued at room temperature and then
the mixture is allowed to stand overnight. 200 ml of water are
then added. The mixture is stirred and then the beige
~ ?~
-- 29 --
precipitate formed is filtered~ This precipitate is washed with
water and drained. 7.54 g of a moist compound are obtained
which are used as such ~n the next stage.
5.5. (+)-6-[1-Hydroxy-2-[4-(2-phenyl-2-oxoethyl)-1-piperidyl]-
ethyl]-3,4-dihydroquinolin-2(lH)-one.
7.54 g (0.0173 mole) of 6-[1-hydroxy-
2-[4-[(2-phenyl-1,3-dioxolan-2-yl)methyl]-1-piperidyl]ethyl]-
3,4-dihydroquinolin-2(1H)-one are introduced into a 500-ml
round bottom flask. The compound is suspended in 150 ml of a
normal aqueous solution of hydrochloric acid, 50 ml of ethanol
are added and the mixture is heated at 80C for 1 hour and then
at 100C for 1 hour. It is evaporated to dryness and a gummy
residue is obtained which is taken up in a mixture of 25 ml of
ethanol, 25 ml of ethyl acetate and 150 ml of an ammoniated
solution (26 to 2~ % of ammonium hydroxide by volume). The
mixture is stirred at room temperature for 1 hour and then the
chestnut coloured precipitate obtained is filtered. It is dried
in a desiccator in the presence of phosphorus pentoxide and
then purified by chromatography on a silica gel column, eluting
with a 9/1 dichloromethane/methanol mixture. The product
obtained is recrystallised from 45 ml of propanol. 1.45 g of
compound are obtained.
Melting point: 186-187C.
Z5
Example 6 ~Compound No. 13)
(+)-6-[1-Hydroxy-2-[4-[2 (4-fluorophenyl)-1-oxoethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1}l)-one~
- 30 ~l~r~7 ?~
6.1. 2-(4-Fluorophenyl)-3~hydroxy-3-[1-(phenylmethyl)-
4-piperidyl]prop-2-enenitrile.
13.5 g (0.1 mole) of (4-fluorophenyl)acetonitrile,
50 ml of toluene and 4.~ g of sodium amide are introduced into
a 500-ml round bottom flask. The mixture is stirred at room
temperature for 30 minutes and then 27.2 g (o.11 mole) of ethyl
1-(phenylmethyl~-4-piperidinecarboxylate are added by means of
a dropping funnel. After the addition is completed, the mixture
is heated at 80OC for 3 hours. It is then cooled in an ice bath
and 300 ml of water are added, the mixture is stirred for
30 minutes and 15 ml of acetic acid are added. A yellow
precipitate i5 obtained which is separated by filtration,
washed with water and dried in the presence of phosphorus
pentoxide~ 17.6 g of compound are obtained which are used as
such in the next stage.
6.2. 2-(4-Fluorophenyl)-l-[l-(phenylmethyl)-
4-piperidyl]ethanone.
25 ml of water, 50 ml of concentrated ~ulphuric
acid and 50 ml of acetic acid are introduced into a 500-ml
round bottom flask placed in a water and ice bath and 24.4 g
(0.072 mole) of 2-(4-fluorophenyl)-3-hydroxy-
3-[1-phenylmethyl)-4-piperidyl]prop-2-enenitrile are added. The
mixture is refluxed for 12 hours. The round bottom flask is
cooled in a water and ice bath. 200 ml of ice are introduced
into a l-litre Erlenmeyer flask and the mixture prepared above
is slowly added; 200 ml of ethyl acetate are added together
with 200 ml of ammonium hydroxide in order to adjust the pH to
9. The mixture is decanted, the organic phase is recovered and
the aqueous phase is extracted three times with 150 ml of ethyl
acetate. The organic phases are pooled and they are washed
three times with 150 ml of water until a pH of 7 to 8 is
obtained. The organic phase is dried over sodium sulphate and
filtered and the ethyl acetate is evaporated under reduced
pressure. 20.7 g oE oily compound are obtained which are used
as such in the next stage~
6.3. 4-[2-[(4-Fluorophenyl~methyl]-1,3-dioxolan-2-yl]-1-
(phenylmethyl)piperidine.
10.35 g (0.033 mole) of 2-(4-fluorophenyl)-
l-[1-(phenylmethyl)-4-piperidyl]ethanone, 100 ml of toluene,
20 g of ethylene glycol and 10.35 g of 4-methylbenzenesulphonic
acid are introduced into a 500-ml round bottom flask equipped
with a Dean-Stark apparatus and the mixtur~ is refluxed for
1.75 hours. The toluene is evaporated and the residue is taken
up in 200 ml of ethyl acetate. 100 ml of a 30 ~ solution of
ammonium hydroxide are added and the organic phase is decanted,
washed three times with 100 ml of water and dried and the
solvent is evaporated under reduced pressure. 13.4 g of an oily
product are obtained which are used as such in the next stage.
6.4. 4-[2-[(4-Fluorophenyl)methyl]-1,3-dioxolan-2-yl]-
piperidine.
13.4 g (0.033 mole) of 4-[2-[(4-fluoro-
phenyl)methyl]-1,3-dioxolan-2-yl]-1-(phenylmethyl)piperidine in
solution in 100 ml of ethanol and 20 ml of water are introduced
into a Parr apparatus and 2 g of 10 % palladium on carbon are
added and hydrogenation is carried out at 50C at a hydrogen
- 32 ~ ~v~
pressure of 0.35 MPa for 5 hours. The catalyst is separated by
filtration and washed with water and with ethanol, the filtrate
is evaporated under reduced pressure and the residue is dried
under vacuum. 9.74 g of an oily compound are obtained which
crystallise and which ar~ used as such in the next stage.
6.5. (+)-6-[1-Hydroxy-2-[~-[2-(4-fluorophenyl)-1,3-dioxolan-
2-yl]-1-piperidyl]ethyl]-3,4-dihydroquinolin-2(1}l)-one.
4.37 g (0.0165 mole) of 4-[2-[(4-fluoro-
phenyl)methyl]-1,3-dioxolan-2-yl]piperidine in solution in
~0 ml of ethanol and 20 ml of water are introduced into a
500-ml round bottom flask. 3.69 g (0.0165 mole) of 6-
(chloroacetyl)-3,4-dihydroquinolin-2(1H) one and 2 g (about
0.02 mole) of sodium carbonate are added. The mixture is
refluxed for 50 minutes. The mixture is cooled and then 8.56 g
of potassium borohydride are added. The stirring is continued
at room temperature and then the mixture is allowed to stand
overnight at room temperature. 160 ml of water are added and
the mixture is stirred for 30 minutes. The precipitate is
separated by filtration and washed with water. 8.14 g of moist
product are obtained which are used as such in the next stage.
6.6. (+)-6-[1-Hydroxy-2-[4-[2-(4-fluorophenyl)-1-oxoethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
7.5 g (0.0165 mole) of 6-[1-hydroxy--2-[4-[2-(4-
fluorophenyl)-1,3-dioxolan-2-yl]-1-piperidyl]ethyl]-
3,4-dihydroquinolin-2(1H)-one in suspension in 150 ml of a
normal aqueous solution of hydrochloric acid are introduced
into a 500-ml round bottom flaskO 50 ml of ethanol are added
- 33 -
and the mixture is stirred at 80C for 1 hour. It is allowed to
cool and then evaporated to dryness. The crystallised residue
is taken up in a mixture of 25 ml of ethanol, 25 ml of ethyl
acetate and 150 ml of an ammoniated aqueous solution (26 to
28 % ammonium hydroxide by volume). The mixture is stirred at
room temperature for 2 hours and filtered and the residue is
taken up in ethanol. The mixture is stirred, a small amount of
water is added and the insoluble matter i~ separated by
filtration. The product is dried in a desiccator in the
presence of phosphorus pentoxide and then purified by
chromatography on a silica gel column, eluting with a 70/15/15
dichloromethane/methanol/ethyl acetate mixture. The product is
recrystallised from lO ml of propanol and dried. 0.8 g of
compound is obtained.
Melting point: 171-~72C.
Example 7 (Compound No. 15)
(+)-8-Fluoro-6-[l-hydroxy-2-[4-(2-phenylethyl)-1-piperidyl]-
ethyl]-3,4-dihydroquinolin-2(1H)-one.
7.1. N-(2-Fluorophenyl)-3-phenylprop-2-enamide.
A round bottom flask containing a solution of
33.3 g (0.3 mole) of 2-fluorobenzeneamine in 300 ml of toluene
is introduced into an ice bath. 25.5 ml of pyridine are added
followed dropwise by a solution of 50 g (0.3 mole) of
phenylprop-2-enoyl chloride in 300 ml of toluene. The mixture
is stirred for 1 hour while maintaining the round bottom flask
in the ice bath, and then the mixture is allowed to stand
overnight at room temperature. The precipitate formed is
- 34 ~ ~ f .~
filtered, washed with water and then with petroleum ether and
dried in a desiccator in the presence of phosphorus pentoxide.
36.2 g of compound are obtained.
Melting point: 116C.
7.2. 8-Fluoroquinolin-2(lH)-one.
34 g (0.141 mole) of N-(2-fluorophenyl)-
3-phenylprop-2-enamide are suspended in 180 ml of
chlorobenzene. 93 g (0.7 mole) of aluminium chloride are added,
with stirring, in small fractions. The mixture is then refluxed
for 3.5 hours. It is slightly cooled and slowly poured into
1 litre of a water/ice mixture while stirring. The precipitate
obtained is washed with water and then with petroleum ether. It
is dried in a desiccator in the presence of phosphorus
pentoxide. 22.5 g of compound are obtained.
Melting point: 193C.
7.3. 8-Fluoro-3,4-dihydroquinolin-2~lH)-one.
22 g (0.134 mole) of 8 fluoroquinolin-2(lH)-one are
introduced into a mixture of 100 ml of ethanol and 50 ml of a
normal aqueous solution of hydrochloric acid containing 2 g of
10 % palladium on carbon, and hydrogenation is carried out at
50C at a pressure of 0.35 MPa for 10 hours. The suspension is
filtered in the heated state and the catalyst is washed with
hot ethanol. A white mass crystallises from the filtrate. It is
evaporated to dryness and the residue is triturated with a
small amount of petroleum ether. The mixture is filtered,
drained and then dried in a desiccator. 17.85 g of compound are
obtained.
~ q- ~ ~3
Melting point: 140C.
7.4. 6-(Chloroacetyl)-8-fluoro-3,4-dihydroquinolin-2(1H)-one.
42.4 g (0.318 mole) of aluminium chloride, 30 ml of
dichloromethane and 23 D 9 g, equivalent to 17 ml (0.212 mole) of
chloroacetyl chloride are placed in a 500-ml round bottom
flask. The mixture is stirred at room temperature for
30 minutes and then 17.5 g (0.106 mole) of 8-fluoro-
3,4-dihydroquinolin-2(11~)-one are added rapidly and in small
fractions. It is heated at 80~C for 3.5 hours. A blackish
solution is obtained which is poured slowly into 1 litre of a
water/ice mixture. The mixture is stirred for 15 minutes and
the precipitate is collected by filtration, thoroughly washed
with water, drained and dried in a desiccator. 25 g of compound
are obtained.
Melting point: 194~C.
7.5. (+)-8-~luoro-6-[1-hydroxy-2-[4-(2-phenylethyl)-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
3.62 g (0.015 mole) of 6-(chloroacetyl)-8-fluoro-
3,4-dihydroquinolin-2(1H)~one, 3.38 g (0.015 mole) of
4-(2-phenylethyl)piperidine hydrochloride and 4 g (about
0.04 mole) of sodium carbonate are placed in a round bottom
flask. 80 ml of ethanol and 20 ml of water are added. The
mixture is refluxed for one hour. It is cooled and then 7 g of
potassium borohydride are added all at once. The mixture is
stirred at room temperature for 2.5 hours. 150 ml of water are
added, the mixture is stirred and then the precipitate formed
is filtered. It i5 dried, recrystallised from 50 ml of
- 3 6 ~ r ~
2-propanol and dried. 2.5 g of solid are obtained.
Melting point: 138-140C.
Example 8 (Compound No. 17)
(+)-8-Fluoro-6-[1-hydroxy-2-[4 [2-(3-fluorophenyl)ethyl]-
1-piperidyl]ethyl]-3,4-dihydroquinolin-2(lH)-one.
3.6 g (0.015 mole) of 6-(chloroacetyl)-8-fluoro-
3,4-dihydroquinolin-2(1H)-one, 3.6 g (0.015 mole) of
4-[2-(3-fluorophenyl)ethyl]piperidine hydrochloride, 3 g (about
0.03 mole) of sodium carbonate and a mixture of 80 ml of
ethanol and 20 ml of water are placed in a round bottom flask.
The mixture is refluxed for 1.5 hours. It is cooled and then
7 g of potassium borohydride are added. The mixture is stirred
overnight at room temperature, poured into 160 ml of water and
stirred for 1 hour. The precipitate formed is then filtered,
washed with water, drained and dried in a desiccator in the
presence of phosphorus pentoxide. 4.9 ~ of product are obtained
which are purified by chromatography on a silica yel column,
eluting with a 9/1 dichloromethane/methanol mixture. 4 g of an
oily product are obtained which crystallise and which are
recrystallised from 30 ml of 2-propanol. 2.4 g of pure compound
are obtained after drying.
Melting point: 116-117C~
Example 9 lCompound No. 9, 9a and 9b)
(+)-6-[1-Hydroxy-2-[4-[2-[4-(trifluoromethyl)phenyl~ethyl]--
l-piperidyl]ethylJ-3,4-dihydroquinolin-2(lH)-one and its (-)
and (+) enantiomers.
37 -
9.1. (+)-6-[1-~ydroxy-2-~4-[2-[4-(trifluoromethyl)phe~yl]-
ethyl~-l-piperidyl]ethyl]-3,4-dihydroguinolin-2(1H)-one.
22.3 g (0.1 mole) of 6-(chloroacetyl)-
3,4-dihydroquinolin-2(1H)-one , 29.3 g (0.1 mole) of
S 4-[2-[4-(trifluoromethyl)phenyl]ethyl]piperidine hydrochloride,
21.2 g (0.2 mole) of sodium carbonate, 400 ml of ethanol and
100 ml of water are introduced into a l-litre round bottom
flask and the mixture is heated in an oil bath (bath
temperature: 120C) for 1 hour. It is cooled using a cold water
bath, poured into 800 ml of water and the chestnut-coloured
precipitate is separated by filtration and washed with wa'cer,
and 4 g of this intermediate ketone are collected for other
uses.
The rest is taken up in 400 ml of ethanol and 100 ml of water,
80 g of potassium borohydride are added and the mixture is
stirred for two days at room temperature.
The mixture is poured into 800 ml of water and stirred for
30 minutes and the precipitate is filtered, washed with water
and dried in the presence of phosphorus pentoxide.
34.64 g of product are obtained which are recrystallised from
440 ml of 2-propanol and 31.18 g of product are obtained after
drying in the presence of phosphorus pentoxide.
20 g of it are collected for separation of the enantiomers and
the remaining 11.18 g are recrystallised for the last time from
120 ml of 2-propanol.
9.94 g of purified compound are finally isolated after drying
in the presence of phosphorus pentoxide.
Melting point: 163-164C.
3 8 ~ `r,? ~ ~,~! ,J3
9.2. (-)-6-[1-Hydroxy-2-[4-t2-[4-(trifluoromethyl)phenyl~-
ethyl]-l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
20 g (0.0448 mole) of (+)-6-t1-hydroxy-
2-[4-[2-[~-(trifluoromethyl)phenyl]ethyl]-1-piperidyl]ethyl]-
3,4-dihydroquinolin-2(1H)~one, 6.8 g (0.0448 mole) of L(+)-
mandelic acid and 100 ml of ethanol are introduced into an
Erlenmeyer flask. The mixture i6 ~tirred, thus formin~ into a
mass, it is refluxed and ethanol is added until the salt
dissolves, equivalent to about 1500 ml of ethanol.
The mixture is filtered in the heated state and the filtrate is
heated in order to redissolve the precipitate; 60 ml of ethanol
are added in order to obtain a clear solution which is allowed
to stand, without stirring, at room temperature for 4 hours.
The crystals formed are filtered, washed with a small amount of
ethanol and dried in the presence of phosphorus pentoxide.
14.56 g of product are obtained which are recrystallised two
times from ethanol thus leaving 7.09 g of salt.
Melting point: 234-234.5C.
The base is released in a conventional manner in
dichloromethane and using aqueous ammonium hydroxide. After
separation of the organic phase and evaporation of the solvent,
the base is recrystallised from 50 ml of propanol and dried in
the presence of phosphorus pentoxide.
4.34 g of compound are finally isolated.
Melting point: 172-173C.
[~20 = _34.0 (c = 1.0; CHCl3).
9.3. (+)-6-[1-Hydroxy-2-[4-t2-[4-(trifluoromethyl)phenyl]-
ethyl]-l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1}l)-one.
3 9 -- ~ ~ ~' '~ ~' ''~' '~
All the mother liguors from the preceding stage are
pooled, the solvent is evaporated, dichloromethane and ammonium
hydroxide are added to the evaporation residue, the organic
phase is separated and the solvent is evaporated.
15 g (0.0336 mole) of base are obtained which are suspended in
100 ml of ethanol, 5.11 g of D(-)-mandelic acid are added, the
mixture is refluxed and ethanol is added until the salt
dissolves, equivalent to about 1300 ml of ethanol.
The mixture is filtered in the heated state, the filtrate is
lo heated in order to redissolve the precipitate and the solution
is allowed to stand, without stirring, at room temperature for
4 hours.
The crystals formed are filtered, washed with a small amount of
ethanol and dried in th~ presence of phosphorus pentoxide.
10.96 g of product are obtained which are recrystallised twice
from ethanol thus leaving 6.11 g of salt.
Melting point: 234-235~C.
The base is released in a conventional manner in
dichloromethane and using aqueous ammonium hydroxide. After
separation of the organic phase and evaporation of the solvent,
the base is recrystallised from 50 ml of propanol and dried in
the presence of phosphorus pentoxide.
3.78 g of compound are finally isolated.
Melting point: 174-175~C.
[~20 = +35.0~ (c = 1.0; CHCl3).
~ 40 - 2~ 7 .~
Example_10 ~Compound No. 51)
(+~-6-[l-Hydroxy-2-[d~-[2~ (acetylamino)phenyl]ethyl]~
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
3.35 g (0.015 mole) of 6-(chloroacetyl)-
3,4-dihydroquinolin-2~1~)-one, 4.23 g (0.015 mole) of N-[4-~2-
(4-piperidyl)ethyl]phenyl] acetamide hydrochl oride, 3 . lg g
(0.03 mole) of sodium carbonate, 80 ml of ethanol and 20 ml o:e
water are introduced into a round bottom flask and the mixture
is refluxed for 2 hours.
It is allowed to cool and 7 g of potassium borohydride are
added and the mixture is stirred overniyht at room temperature.
160 ml of water are added, the mixture is stirred for
30 minutes and the precipitate is separated by filtration,
washed with water and dried.
5.5 g of product are obtained, which are recrystallised from
propanol and dried at 90~.
3.g g of compound a~e finally obtained.
Melting point: 205-206C.
Example 11 (Compound No. 39)
(+)-6-[1-Hydroxy-2-[4-(2-phenylethyl)-1-piperidyl]ethyl~-
7-fluoro-3,4-dihydroquinolin-2(lH)-one.
]1~1. N-(3-Fluorophenyl)-3 phenylprop-2-enamide.
30 g (0.27 mole) of 3-fluorobenzenamine, 300 ml of
toluene and 25 ml of pyridine are introduced into an Erlenmeyer
flask, cooled using an ice bath, and then a solut:ion of 45 g
(0.27 mole) of 3-phenylprop-2-enoyl chloride in 300 ml of
toluene is added dropwise without allowing the temperature to
-- 41 2 ~ ~ ~k -~v ~3
exceed 10C.
The mixture is stirred for another 15 minutes in an ice bath
and then overnight at room temperature.
A precipitate is removed by filtration and 50 ml of water are
added to the filtrate a~d the mixture is vigorously stirred for
1 hour. The precipitate is separated by filtration, washed with
water and dried in the presenc~ of phosphorus pentoxide.
51 g of product are obtained.
Melting point: 115C.
11.2. 7-Fluoro~uinolin-2(lH)-one.
51 g (0.211 mole) of N-(3-fluorophenyl)-
3-phenylprop-2-enamide and 200 ml of chlorobenzene are
introduced into a round bottom flask followed by 140 g
(1.05 mole) of aluminium chloride in small fractions, the
mixture is refluxed for 4 hours and allowed to stand overnight.
It is poured into a mixture of water and ice and filtered, and
the solid is washed with water and then with petroleum ether
and allowed to dry in the open air.
34 g of compound are obtained.
11.3. 7-Fluoro-3,4-dihydroquinolin-2(1}l)-one.
34 g (0.208 mole) of 7-fluoroquinolin-2(1ll)-one,
150 ml of ethanol, 50 ml of 1 N hydrochloric acid and 3 g of
10 % palladised carbon are introduced into a Parr flask and
hydrogenation is carried out at 0.42 M~a at 50C for 16 hours.
The catalyst is separated by filtration, the filtrate is
evaporated and the white residue is triturated with 50 ml of
ethanol, separated by filtration and dried in the presence of
- 4~ -
phosphorus pentoxide. 27.03 g of product are obtained which are
used as such in the next stage.
11.4. 6-(Chloroacetyl~-7-fluoro-3,4-dihydroquinolin
2(1H)-one.
65 g (0.49 mole~ of aluminium chloride, 50 ml of
dichloromethane and 26 ml (0.327 mole~ of chloroacetyl chloride
are introduced into a 500-ml round bottom flask and the mixture
is stirred at room temperature for 30 minutes.
27 g (0.163 mole) of 7-fluoro-3,4-dihydroquinolin-2(1H)-one are
then added in small fractions and the mixture i6 refluxed for
4 hours and allowed to stand overnight at room temperature.
It is slowly poured into 300 ml of ice-cold water, the mixture
is stirred for 10 minutes and the precipitate is separated by
filtration, washed with water and then with hexane, drained and
dried in the presence of phosphorus pentoxide.
38 g of product are ob ained which are used as such in the next
stage.
0 11.5. (+)-6-[1-Hydroxy-2-t4-(2-phenylethyl)-1-piperidyl]-
ethyl]-7-fluoro-3,4-dihydroquinolin-2(lH)-one.
3.6 g (0.015 mole) of 6-(chloroacetyl)-7-fluoro-
3,4-dihydroquinolin 2(1H)-one, 3.38 g of
4-(2-phenylethyl)piperidine hydrochloride, 3.18 g (0.03 mole)
of sodium carbonate, 160 ml of water, 40 ml of water and 50 ml
of N N-dimethylformamide are introduced into a round bottom
flask and the mixture is stirred at room temperature for
3 days.
7 g of potassium borohydride are added and the mixture is
~ r~ D
- 43 -
stirred overnight at room temperature. It is poured into 350 ml
of water, the mixture is stirred for 80 minutes and the
precipitate is separated by filtration, dried and purified hy
chromatography on a silica gel column, eluting with a 9~1
dichloromethane/ methanol mixture, recrystallised from 60 ml of
propanol and dried in the presence of phosphorus pentoxide.
1.48 g of compound are finally obtained.
Melting point: l9g-200UC.
Example 12 (Compound No. 36)
(+)-6-[l-Hydroxy-2-[4-[2-(2-methylphenyl)ethyl]-
l-piperidyl]ethyl]-8-methyl-3,4-dihydroquinolin-2(1H)-one.
12.1. N-(2-Methylphenyl)-3-phenylprop-2-enamide.
32.15 g (0.3 mole~ of 2-methylbenzeneamine, 300 ml
of toluene and 25.5 ml of pyridine are introduced into a
2-litre round bottom flasX cooled using an ice bath, and then a
solution of 50 g (0.3 mole) of 3-phenylprop-2-enoyl chloride in
300 ml of toluene is added dropwise~
The ice bath is removed and then the mixture is stirred for
3.5 hours at room temperature and allowed to stand overnight
A white precipitate is obtained which is separated by
filtration, washed several times with water and then with
diethyl ether and dried in the presence of phosphorus
pentoxide.
69.47 g of product are obtained.
Melting point: 166~C.
12.2. 8-Methyl~linolirl--2(1}l)-one.
-- 44 ~
67.5 g (0~284 mole~ of N-(2-methylphenyl)
3-phenylprop-2-enamide and 370 ml of chlorobenzene are
introduced into a 2-litre round bottom flask; the mixture is
stirred and 188.1 g (1.409 mole) of aluminium chloride are
added in small fractions. The mixture i8 refluxed for 4 hours
and allowed to stand overnight.
It is slowly poured into 1 litre of water and ice and the
chestnut~coloured precipitate is separated by filtration,
washed several times with water and then with hexane, and dried
in the presence of phosphorus pentoxide.
42.76 g of compound are obtained.
Melting point: 224C.
12.3. 8-Methyl-3,4-dihydroquinolin-2(lH)-one.
42.76 g (0.266 mole) of 8-methylquinolin-2(1H)-one,
200 ml of ethanol, 100 ml of 1 N hydrochloric acid and 4.2 g of
10 % palladised carbon are introduced into a Parr flask and
hydrogenation is carried out at 0.25 MPa at 50C for 15 hours.
The catalyst is separated by filtration in the heated state, by
washing it with hot ethanol, the filtrate is evaporated and the
residue is washed with petroleum ether and dried in the
presence of phosphorus pentoxide. 38.15 g of product are
obtained which are used as such in the next stage.
Melting point: 131-132C.
12.4. 6-(Chloroacetyl)-8-methyl-3,4-dihydroquinolin-
2(lH)-one.
57.21 g ~0.42~ mole) of aluminium chloride, 45 ml
of dichloromethane and 32032 g, equivalent to 22.8 ml,
- 45 -
(0.286 mole) of chloroacetyl chloride are introduced into a
500-ml round bottom flask and the mixture is stirred at room
temperature for 30 minutes. 23O02 g (0.286 mole) of 8-methyl-
3,4-dihydroquinolin-2(1H)-one are then added in small fractions
and the mixture is refluxed for 3.5 hours.
It is slowly poured into 1 litre of ice-cold water, the mixture
is stirred for 10 minutes and the chestnut-coloured precipitate
is separated by filtration, washed several times with water and
then with petroleum ether, drained and dried in the presence of
phosphorus pentoxide.
33.45 g of product are obtained which are used as such in the
next stage.
12.5. (+)-6-[1-Hydroxy-2-[4-[2-(2-methylphenyl)ethyl]-
1-piperidyl]ethyl]-8-methyl-3,4-dihydroquinolin-
2(lH~-one.
3.56 g (0.015 mole) of 6-(chloroacetyl)-8-methyl-
3,4-dihydroquinolin-2(1H)-one, 3.6 g (0.015 mole) of 4-[2-
(2-methylphenyl)ethyl]piperidine hydrochloride, 4.24 g
(0.04 mole) of sodium carbonate, 100 ml of ethanol and 20 ml of
water are introduced into a 500-ml round bottom flask and the
mixture is refluxed for 1 hour.
The mixture is cooled, 7.16 g of potassium borohydride are
added and the mixture is stirred overnight at room temperature.
200 ml of water are added, the mixture is stirred for
30 minutes and the precipitate is separated by filtration,
dried and purified by chromatography on a silica gel column,
eluting with a 9/1 dichloromethane/ methanol mixture. 4.09 g of
product are obtained which are recrystallised from 130 ml of
- 46 ~
propanol and dried in the presence of phosphorus pentoxide.
3.68 g of product are obtained which are recrystallised for a
second time from 325 ml of ethanol.
3.29 g of compound are finally obtained.
Melting point: 185-186C.
Example 13 (Compound No. 40)
(+)-6-[1-Hydroxy-2-[4 [2-(3,5-difluorophenyl)ethyl]-
l-piperidyl]ethyl]-3,4-dihydroquinolin-2(1H)-one.
3.35 g (0.015 mole) of 6-(chloroacetyl)-
3,4-dihydroquinolin-2(1}l)-one, 3.9 g (0.015 mole) of
4-[2-(3,5-difluorophenyl)ethyl]piperidine hydrochloride, 80 ml
of ethanol and 20 ml of water are introduced into a round
bottom flask and the mixture is refluxed for 2 hours.
It is allowed to cool, 7 g of potassium borohydride are added
and the mixture is stirred overnight at room temperature.
160 ml of water are added, the mixture is stirred for
15 minutes and the solid is saparated by filtration, washed
with water and then with hexane, dried and purified by
chromatography on a silica gel column, eluting with a 9/1
dichloromethane/methanol mixture.
3.7 g of product are obtained which are recrystallised from
60 ml of ethanol. 3.08 g of compound are Einally isolated after
drying.
Melting point: 173-174C.
The following table illustrates the chemical
structures and the physical properties of some compounds of
general formula (I).
- 47 -
All the compounds are in the racemic state except compounds No.
9a and No. 9b which are the laevorotatory and dextrorotatory
enantiomers of compound No. 9, respectively.
In the "salt" column, "-" denotes a compound in the form of a
base, "fum." denotes a compound in the form of a fumarate and
"ox." denotes a compound in the form of an oxalate.
~ ~ C-- ~
- 48 -
Table 2
Compound~ of general formula (I)
J~ R2
OH ~ Y
/\
H R1
~o. R1 R2 X Y Salt m.p. ~C)
5_ _ I . I _ _
1 H 2 -F CH2 CH2 . 161-162
2 H 3-F CH2 CH2 165-166
3 H 4 - F CH2 CHz 159 - 160
4 H 4 - ClCH2 CH2 189 - 190
1 05 H 3- CH3CH2 CH2 163 -164
6 H 4 - CH3 CH2 CH2 174 - 176
7 H 4 - OHCH2 CH2 225 - 226
8 H 4-OCH3CH2 CH2 192-193
9 H 4 ~ CF3 CH2 CH2 163 - 164
lS9a laevorotatory enantiomer 172-173
9b dextrorotatory enantiomer 174-175
H H CH2 CH2CH2 164 - 165
11 H H CH2 CO 186 - 187
12 H H CO CH2 183-184
2 O13 H 4 - F CO CH2 - 171- 172
.~ _ ___ _.
-- 49 --
. . ~
No . Rl R2 X Y Salt m. p . ( C)
. _
14 H 4 - CH3 CO CH2 199 - 200
8 - F H CH2 CH2 138 - 140
16 8-F 2-F CH2 CH2 139-140
17 8-F 3-F CHz CH2 116-117
18 8 - F 4 - F CH2 CH2 163 - 164
19 8 - F 4 - OCH3 CH2 CH2 166 - 167
1 0 20 8 - F 4 - CF3 CH2 CH2 172 - 173
21 8-F 4-Cl CH2 CH2 fum. 215-216
22 H 3-Cl CH2 CH2 ox . 219-220
23 H 4-F CH2 CO 192-193
24 8 - F 4 - F CH2 CO 176 - 177
8-F 2-C;13CH2 CH2 139-140
26 8 - CH3 H CH2 CH2 195 - 196
27 8-CH3 3-CF3 CHz CH2 164-165
28 H 2 - CF3 CH2 CH2 172 - 173
29 8 - CH3 2 - F CH2 CH2 166 - 167
2 0 30 H 2 - CH3 CH2 CH2 161 - 162
31 8 - CH3 4 - F CH2 CH2 180 - 181
32 8-CH3 4 CF3CH2 CH2 190-191
33 8 - CH3 4 - CH3 CH2 CH2 206 - 207
34 8 - F 2 - CF3 CH2 CH2 123 - 124
2 5 35 8 - CH3 2 - CF3 C~2 CH2 172 - 173
36 8 - CH3 2 - CH3 CH2 CH2 185 - 186
37 8-CH, 3-CH3 CH2 CH, - I186-187
-- 50 --
l 11~. R,- ~X ~ ~ r ~-c~
38 8-CH3 4-F CHz CO -178-179
39 7 -F H CH2 CH2 199 - 200
40 H 3,5 - F2 CH2 CH2 173 - 174
41 8 - F 3,5 - F2 CH2 CH2 146 - L47
42 8 - CH3 3,5 - F2 CH2CH2 162 - 163
43 7 -F 2 -F CH2 CH2 193 - 199
1 0 44 H 2,4 - Fz CH2 CH2 171 - 172
45 H 3,4 - F2 CH2 CH2 L63.5 - 164.5
46 7 -F 2 -CH3 CH2 CH2 199-200
47 8 - CH3 2,4 - F2 CH2CH2 161 - 152
48 8 - CH3 3,4 -F2 CH2CH2 156 - 157
49 8-F 3,4-~2 CH2 CH2 148-149
50 8 - F 2,4 - F2 CH2 CH2 164 - 165
51 H 4 - NH COCH3 CH2 CH2 205 - 206
52 H 4 - CF3 CH2 CO 192 - 193
53 H 4 -NHSO2CH3 CH2 CH2 208 - 209
2 0 54 8 - F H CH2 CH2 CHz 131 - 132
, 55 B-CH3 H ~'`B~CB2 ~ 152 153
The c~mpounds of formula (I) and the
pharmaceutically acceptable acid addition salts thereof have
been the subj ect of pharmacological trials which have
demonstrated their usefulness as active substances of
neuroprotective medicinal products.
From herein the compounds of formula (I) and the
pharmaceutically acceptable acid addition salts thereof are
referred to as the "active compounds of the invention"~
Thus, in particular, the neuroprotective activity
of the active compounds of the invention has been snown in a
focal ischaemia ~odel by ligature of the middle cerebral artery
in mice according to a method similar to that described in
Brain Research, 522, (1990), 290-307.
Six days after occlusion of the middle cerebral artery by
electrocoagulation under halothane anaesthesia, the mice are
again anaesthetised and the cerebral cortex ipsilateral to the
occlusion is removed. After homogenisation of the tissue, the
extent of the cerebral infarction is evaluated by measuring the
increase in the density of peripheral benzodiazepine sites (~3)
using the compound [3H]PK 11195 from New England Nuclear. The
treatments are curatively administered via the intraperitoneal
route at the following times: 5 minutes, 3 hours, 6 hours,
18 hours and 24 hours.
Some active compounds of the invention decrease the density of
the peripheral benzodia7.epine sites by about 70 % at a dose of
10 mg/kg.
The active compounds of the invention have also
been the subject of a test of inhibition of the binding of
[3H]ifenprodil to rat cerebral cortex polyamine-sensitive
~ ~r: 7~
- ~2 -
receptors, according to the procedure described by Schoemaker
et al., Eur J Pharmacol , 176, 249-250, (1990).
The 150- to 230-g male Sprague-Dawley rat is sacrificed and the
cerebral cortex is homogenised in 20 volumes of ice-cold 50 mM
Tris-HCl buffer (pH = 7.4 at ooc)~ by means of an
Ultra-TurraxT~ (Ikawerk) or PolytronTH (Kinematica) apparatus.
The homogenate is washed twice by centrifuging for 10 minutes
at 45000 x g, tne pellet being resuspended in fresh buffer. The
final pellet is taken up in 20 volumes of the same buffer.
A lOo-~l aliquot of this suspe~sion is incubated in a final
volume of 1000 ~1 with 1 nM of [3H]ifenprodil (specific
activity: 30 to 35 Ci/mmol) for 120 minutes at 0c, in the
presence of 3 ~M of GBR 12909 (Research Biochemicals Inc.,
Natick, MA, USA), in the absence or in the presence of
competing substance.
After incubation, the mixture is diluted with 5 ml of ice-cold,
50 mM Tris-HCl buffer (pH = 7.4 at 0C) and the membranes are
recovered by filtration on Whatman GF/BT~ filters pretreated
with 0.05 % polyethylenimine, and then washed with two times
5 ml of ice-cold buffer.
Nonspecifis binding is determined using 10 ~M ifenprodil, the
data are analysed according to the usual methods and the ICso
concentration, which inhibits the binding of [3H]ifenprodil by
50 %, is calculated.
The ICso values range from 2 nM to 10 ~M (2 x 10-9 to
1 x 10-5 ~.
The active compounds of the invention were also the
subject of a test of inhibition of the binding of
[3H]ifenprodil to rat cerebral cortex sigma receptors
r~ t~
- 53 -
(Schoemaker et al., Eur J Pharmacol , a83, 1670, (l990).
The 150- to 230-g male Sprague-Dawley rat is sacrificed and the
cerebral cortex is homogenised in 20 volumes of ice-cold 50 mM
Tris-HCl buffer (pH -- 7.4 at 0C), by means of an
Ultra-TurraxTM (Ikawerk) or PolytronT~ (Xinematica) apparatus.
The homogenate is washed twice by centrifuging for 10 minutes
at 45000 x g, the pellet being resuspended in fresh buffer. The
final pellet is taken up in 20 volumes of the same buffer.
A 100-~l aliquot of this suspension is incubated in a final
volume of 1000 ~l with 0.5 nM of [3H]ifenprodil (specific
activity: 30 to 35 Ci/mmol) for 30 minutes at 37C`, in the
absence or in the presence of competing substance. After
incubation, the m~mbranes are recovered by filtration on
Whatman GF/BT~ filters pretreated with 0.05 ~ polyethylenimine,
and then washed with two times 5 ml of ice-cold buffer.
Nonspecific binding is determined using 10 ~M ifenprodil, the
data are analysed according to the usual methods and the IC50
concentration, which inhibits the bindiny of [3H]ifenprodil by
50 %, is calculated.
The ICso values of the active compounds of the invention range
from 2.5 to 800 nM (2.5 x 10-9 to 8 x 10-7 M).
Finally, the active compounds of the invention were
tested with respect to their activity towards maximum
convulsions induced in mice by supermaximal electroshock.
The procedure for this test is described by E.A. Swinyard and
J.H. Woodhead in Antieplleptlc Drugs Raven Press, New York,
111-126 (1982).
30 minutes after intraperitoneal administration of the test
compound, the num~er of mice with convulsions (extensions of
? r~
- 54 ~
the hind legs) immediately after application of an electric
current (0.4 s, 60 mA, 50 Hz) using transcorneal electrodes, is
noted. The results are expressed as the ADsD, the dose which
protects 50 ~ of the animals, calculated according to the
method of J.T. Lichtfield and F. Wilcoxon (J. Pharm. Exp. Ther.,
96, 99-113 (1949)) from 3 to 4 doses each administered to a
group of 8 to 10 mice.
The AD50 values of the most active compounds in this test are
of the order of 10 mg/kg by the intraperitoneal route.
The results of the tests carried out on the active
compounds of the invention suggest that thev can be used for
the treatment and prevention of cerebral disorders such as
those resulting for example from an ischaemic attack, cardiac
or respiratory failure, cerebral thrombosis or embolism, for
the treatment of dementia resulting from multiple infarctions
for the treatment of senile dementia, for example Alzheimer's
disease or Pick's disease, for the treatment of
olivopontocerebellar atrophy and other neurodegenerative
diseases such as Huntington's chorea, for the treatment of
schizophrenia, for the treatment of cranial or spinal traumas,
for the treatment of convulsive states, as antiemetics during
the treatment of certain cancers with cisplatin, and for the
treatment of AIDS (see Sclence ~50, 1593 (1990)).
For this purpose, they may be for-mulated as
pharmaceutical compositions, in which they are the active
ingredient.
To this effect, the active compounds of the
invention may be provided in all pharmaceutical forms which are
suitable for enteral or parenteral administration, in
- 55 -
combination with appropriate excipients, for example in the
form of tablets, sugared pills, gelatin capsules, capsules,
suppositories or in solutions or suspensions to be taken orally
or injected, containing doses which permit a daily
administration of 1 to 1000 mg of active substance.