Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
2~7~f;.~
S~C'~ ON
TO Al~ WHOM IT MAY CONC~RN:
3e it known that we, ~ortega MoXhtar-Zadeh, a citizen of
Iran, permanently residinq in the United States of America, at
Charleston, West Virginia, and Rex Eugene ~urray, a citizen of
the United States of America, residing at Charles.on, Wes~
Virginia, have invented a certain new and useful AZ~OTROPIC
ASS~STED TRANSVINYI~TION, of which the follo~ng is a
specification.
2~?7.~
ac~round Or the Invention
. Field of the Tnvention
The presont invention relates to a process ~or preparin~
an alkenyl derivative o~ a Bronsted acid by the
transvinylation reaction o~ an alkenyl derivative of a first
Bronsted acid with a second Bronsted acid, and, more
particularly to such a transvinylation process assisted by
azeotxopic distillation for economically and efficiently
separat-ng the desired transvinylation alXenyl product
derivative from the transvinylation reaction mixture.
2. Desc-iPtion Qf t~e ~rior ~r-
Transvinylation or vinyl interchange technology is ~ ll
known. The reac~ion can be illust_ated by the reaction r 2
vinyl-containing compound (R'C~ ~ Cff.) with an active llyuf~n-
containing compound (RX), as in the ~ollowing:
RX + R C~ 3 C~ ~ RC~--CH. ~ R'X
wherein R is carboxy, amido, aroxy, alkoxy, and the like; X is
hydrogen, h~tdroxyl, alkyl, ar~l, and the like; and R' is
car50xyl, amido, alkyl, substituted alkyl, aryl or suhst-t~
aryl.
Adelman, Journal Organic Chemistry, 14, pp. 1057-lO~,
1949, at p. 105" ter~ed t-ansvinyla~.on "t.~e 'Vinyl
Interchange' ~eaction, to dif rentiate it f_om typical eete-
interchange and ester-acid interchange reac~_ons... n .~.del.;an
noted several advantages ~or preparing vinyl monomerc ~y
transvinylation, including, for example, the very mild
reaction conditions, the low yields o~ by-produc_s and the
relatively higher yield of nonomers of qreatar purity and
ac:ivity compared to monome_s prepared by the reaction o~
acetylene with acids.
Adelman also noted that vinyl 2sters of di~asic acids
were prepared much mor2 easily by vinyl interchange than
through the ace~ylene route, and he demonstrat~d that the
~C". Ol/lj~
X~7;J~ s ~
reaction of vinyl acetate cataly2ed with mercuric salt-~ was
not restricted to car~oxylic acids, but would occur with other
compounds containing active hydrogen, such as aceto~cetic
ester and glycolic esters.
Other researchers have demonstrated the versatility o~
the transvinylation reactian and its applicability to a wide
range of Bronsted acids and derivatives of Bronsted acids,
using a wide variety of di~ferent catalysts. For example,
McKeon, et al., Tet-ahed~on, 28, pp. 227-232 (1972) show the
vinyl interchange reaction ~etween a vinyl ether and an
alcohol using a palladium catalyst. Other sources report the
transvinylation reaction betJeen vinyl chloride and a
car~oxylic acid.
The literature suggests that the prefer-ed catalysts for
t-ansvinylation reac_ions have been mercury and palladium
based compounds. However, Pt~II) and Rh(III) have been
reported bv A. Sabel, J. Smidt, R. Jira and H. Prigge, Che~.
Be_., 102, pp. 2939-295~ (1969), to catalyze the reaction. In
addition, Young, U.S. Patent 3,~55,387, patentqd August 26,
1973, entitled: "A Vapor Phase Transvinylation Process",
claims t~e use of supported Hg, Pd, Pt, Ir, Rh, Ru or Os salt
catalysts in a vapor phase ~ansvinylat_on process. The
experimental portion discloses the use of only palladium or
carbon, copper on car~on, irDn on car~on, palladium/copper on
car~on, palladiumlcopper/iron on silica, mercuric acetate on
carbon, and mercuric chlor~de on car~on. Hg and Pd are cited,
at col. 1, line 67, as th8 preferred metals.
More recently, it has been discovered that ruthenium
compositions are usef~l t:ansvinylation catalysts ~or nume~ol~s
Bronsted acids and ierivatives of 8ronsted acids as disclosed
in U.S. application Serial No. 213,697, filed June 30, 1988,
and assigned to th- assignee of t~e present application. ~he
invention disclosed therein relates to a procass for t~e
t-ansvinyla~ion of a vinyl derivative of a first Bronsted acid
wit~ a second Bronsted acid which comprises providing a liquid
phas~ ~ixture containing th~ vinyl derivativ~ of ths firs~
Bronsted acid and the second Bronsted acid in the presence of
- 3 -
~rC. o~/ljb
2~7~
a rutheniu~ co~pound at a t-~p-ratur- at which t~ansvinylation
occurs, and r-cov~ring a- a product of transvinylation th-
vinyl d-rivativ- o~ th- s-cond 8ron-t-d acid Th- b-n-~icial
u~e oi' ruth-niu~-conta~nlng compound~ as c~taly-~ rO~
transvinylation proces~es overcom-s -v-ral def~ci-ncies not~d
for other catalysts that had been us-d in transvinylation
processe~
Variou~ m-thods hav- been e~ploy-d to s-parate th~
desired vinyl d-riv~tiv- of th~ -cond Bronst-d acid, (vinyl
product d-rivative) frou other conponents o~ the
transvinylation r-action nixture, including the starting
reactants, th- conjugat- acid of th- vinyl derivativ- of the
first Bronst-d acid and oth-r side products produced by the
r~action For xa~pl-, di~tillation, fractional dist,llation,
vacuum distillation and rotary vaporation have been used
For exampl-, in U S Pat-nt No 3~18a~319~ vacuu~ distillation
(Exampl~s 1-3), fractional distillation (Exa~ples 4, 7), and
distillation (Example 13), il~ustrate the use of various
distillation technigues for the r-covery of the ~inyl produc~
de~ivative U X Patent 1,486,443 describes a process for
making hiqh b;oiling point vinyl sters whose separation is
facilitated by f_acsional dist_llation as the reac~_on
progresses Solvent extraction techniques have also been used
for t~e separation and recovery of the vinyl producs
deri~ative
The serious process di''i~ulties of saparating Ihe vinyl
product derivative, particularly for vinyl esters of
ra~oxylic acids, from the ~tarting reactants and sida
products of the reaction has b-en well known for years As
early as 1967 such probl~s were discuss-d in Bear~en, U S
Patent No 3,33~,611 (S-~ Col 1, lls 55-70) ln '.he '611
pat nt, it was recognized t~at conv ntional techniques, such
as distillation, for the separation of t~e vinyl product
d~r;vative from the reactants and by-products of the
transvinylation reaction resulted in high losses of the vin
product deri~ative du- to polyoerization, particularly when
mercury catalysts were employ-d, as well as t~e loss of
~CC. 0~
. ~ :
'
~ ~ ~r~7~J~
starting materials. It was also recogniz-d that losses o~
vinyl product derivative, especially high boiling esters, ~as
intensified in separations involving the distillation Or
mixtures of close boiling materials. The di~iculty ol'
separating close boiling materials was also recognized. The
solution to the separation problem taught in U.S. Patent No.
3,337,611 involves the use of a molar excess of the car~oxylic
acid relative to t~e molar concentration of the vinyl ester
reactant in the transvinylation r~action. Distillation of the
vinyl product derivative remained the method of choice for the
recovery of total ester. See Col. 2, lls. 48-51, Col. 4, lls.
50-39, and Example 1.
Thus, despite the recognition of the advantages of using
transvinylation technology for making vinyl derivatives of
3ronsted acids, t_ansvinylation technology has not achieved
widespread commercial significance because of the inefficient
and economically unattrac-ive methods that have been
conventionally employed to separate and recover the vinyl
product derivative from the transvinylation reaction mixture.
Because fractionation or fractional distillation have
typically been employed to recover the vinyl product
derivative, separat on and recovery of ~he vinyl product
derivative has been found to be particularly difficult wh~re
the vinyl product derivative has a boiling point very close to
the boiling point of the conjuqate acid, i.e., the acid formed
from the vinyl derivative of ~e Brons~ed acid used as .he
starting material during transvinylation, andtor the other
components of the transvinylation reaction mixture. In such
instances, separation by fractional distillation can be
accomplished only by the use of very lar~e, complex, and
expensive distillation columns or not at all. ~he cost of
suc~ columns and the expense of operating them thus makes the
use of tsansvinylation technology co~mercially unattract ve
for t~e product-on o~ many mono~ers.
Thus, there remains a need for a transvinyl~tion process
in which the al~enyl product derivative, ~nd in particular the
vinyl product derivative can be readily, facilely and
_ 5 _
~CC.O1/~jb
2~?7~
economically ~eparat-d fron the oth-r compon-nts of the
transvinylation r-actlon uixtur-, and, $n partlcular, th-
con~ugat- acid of th- alkenyl d-rivativ- of tbe r~r-t Bronst~d
acid A n--d for such a proc-ss $s ~p-cially acut- wh-r- th~
conjugate acid and alk~nyl product derivative ar~ close
boilers, i e , th-y hav- a boiling point clo-- to on- another,
such as on the ord-r of witbin 15 d-grees of on- another,
especially within S degr--s of on- anoth-r Ther- is also a
need in th- art for a transvinylation proc-ss which includes a
means for separating desirable transvinylation reaction
product monom-rs and ~ixturas of mono~ers ~conomically so as
to per~it the comm-rcial production of nany ~onomers and
monomer mixtures that would otherwise not be produced or that
could be produced only at siqnificantly gr~ater expense
There is a ~ore specific need for a tsansvinylation process
that allows for the economical production of alkenyl produc~
derivatives that boil close to t~e boiling point of the
conjugate acid of the aLkenyl derivativ~ of the first Bronsted
acid
~ummarv of t~e Invention
The present invention provides an improved
transvinylation process for the preparat~on of an alkenyl
derivative of a Bronsted acid The improved transvinylation
process of the pr-sent invention includes the use of
azeotropic distillation to assist the t_ansvinylat on process,
to separate the aLkenyl product derivatives from other
components of the transvinylation :eaction ~ixture, including
the starting reactants, and ~specially the conjugate acid of
the alkenyl derivative of the Bronsted acid e~ployed as one of
the starting reactants, and to facilitate recovery of the
aLkenyl product derivative The use of azeotropic
distillation for assisting tsansvinylation processes has not
b~en recognized until this inv~ntion Moreov-r, th b~nefit
to ~e achieved by th~ us- of azeotsopic dist llation to
separate and r-cover t~- alkenyl product derivatives has not
been appr-ciated heretoforQ
_ c _
~ ~CC O~/ljb
2~7,~
It is a primary object Or the present invention to
provide a transvinylation process ~or the preparation Or an
alkenyl product derivative wherein the proces~ is assisted by
azeotropic distillation to effect efficient and cconomical
separation o~ the alkenyl product derivative l'rom the other
components of the transvinylation reaction ~ixture.
It is also an object of the invention to provide a
transvinylation process having improved productivity, lower
equipmen~ costs and lower operating costs.
It is a further object of he invention to provide a
transvinylation process having improved throughput owing to
the simplified separation and recovery of the alkenyl product
derivative.
It is yet another object of the invention to provide a
transvinylation procoss for-the preparation of an alkenyl
product derivative in whic~ the aLkenyl product derivative and
the conjugate acid of the alkenyl derivative of the Bronsted
acid used as the reactant have close boiling points. It is a
related object of the invention to provide a t-ansvinylation
process in which an alkenyl product derivative can be produced
in a single stage reactor where the conjugate acid and the
al~enyl product de-ivative are close boilers.
It is a further object of the present invention to
provide a continuous process for the production o~ vinyl
esters of carboxylic acids which per~its the continuous
re~oval of the vinyl ester of the car~oxylic acid in an
isolated form.
It is yet another o~ject of the present invention to
provide a transvinylation process which is capable of
producing a hishly pure alkenyl product derivative, low in
residual acidity. It is a more specific object of the
invention to provide a transvinylation process ~hich is
capable of pr-~ucing a highly pure vinyl product derivatiVe,
low in acidity.
It is also an ob~ect of th~ invention to provide a
transvinylation process ~hich is-versatile, and is capable of
producing a mixture of alXenyl derivatives.
- 7 -
~CC.O~ b
2~?7.,$;~
These and ot~Qr ob~ects and advantag-s o~ the invent~ on
will be appar~nt ~rom the ~ollowlng d-~cr~ption of the
invention.
~ s used horein th~ term alkenyl product derivative m-an~
and refers to an alkenyl-containing compound having the
formula R(C~,),C~2 - C~ r~sulting ~rom the transvinyla~ion
reaction of an alXenyl derivativ~ of a ~irst Bronsted acid,
and a second 8ronsted acid, wherein R is car~oxy, amido,
aroxy, alkoxy, or the like, n is either zero or one, and R,
and R2 are each individually one o~ hydrogen, alkyl o~ 1 to
about 12 carbon atoms, cycloaLkyl, aryl, alkyl ethe_s, and the
like. Transvinylation as used herein includes both vinyl
interchange and allyl interchange reactions. The novel
process of the present invention applies equally to both types
of interchange reactions. ~
The invention thus provides a process for the preparation
of an alkenyl derivative of a Bronsted acid for~ed by the
transvinylation reaction of an alkenyl derivative of a first
Bronsted acid with a second Bronsted acid ~hich comprises
reacting an alkenyl derivative of the first Bronsted acid with
a second Bronsted acid in the presence of a catalyst capable
of catalyzing the t_ansvinylation reaction, adding to the
transvinylation reaction mixture an azeotropic agent capable
of for~ing an azeotrope with at least the alXenyl product
derivative of the s~cond Bronsted acid and recovering by
azeot-opic distillation as a product of the t:ansvinylat on
reaction an alXenyl derivative of the second Bronsted acid.
The azeotropic agent and the alkenyl produc~ der~va~ive for~
an azeotrope whose boiling point is sufficiently different
from the boiling point of at least the conjugate acid of t~e
alXenyl derivativQ of the first Bronsted acid to permit the
azeotrope and th~ conjugate acid to be separated by
distillation.
The azeotropic assisted trans~inylation process ~n
accordance with the present invention is ~ersatile. The
process may ~Q used to prepare a single monomer o~ relatively
high purity or it may b~ used to produce mixtures o~ monomers.
- 8 -
~C~.O1/l~b
2r~7 J~
The product objectives can be achieved by proper sel-ction of
the azootropic agent and separation ta~ks performed on the
transvinylation reaction ~ixture prior to azeotropic
distillation.
Brief ~escri~tion O~ The Orawinas
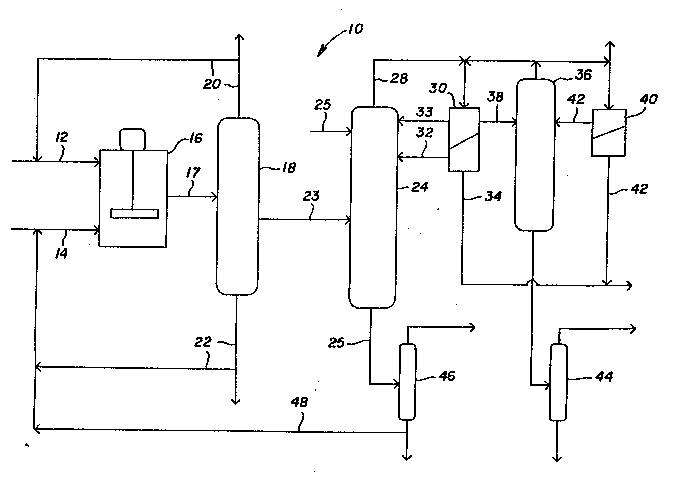
Figure 1 is a schematic illustration o~ the
transvinylation process o~ the present invention, including
recover~ o~ the alkenyl product derivative by azeot_opic
distillation, and Bronsted acid recycle.
~etailed ~escriotion of the Invention
~ he present invention provides a process for the fac-le
preparation and separation of numerous alXenyl deri~atives of
Bronsted acids for~ed by the transvinylation reaction of an
alkenyl derivative of a first Bronsted acid R' (~ C~2 = CR~I,
and a second Bronsted acid (RX1, and assisted by the
azeotropic distillation of the transvinylation reaction
mixture to separate the alkenyl product derivative of the
second Bronsted acid (R(Cq.),C~2 - C.~) from at least the
conjugate acid (R'X) of the alkenyl derivative of the first
Bronsted acid. The process, which may be continuous, semi-
cont~nuous, batch, or se~ atch, may, in the case of an
alkenyl derivativep be illustrated generally as follows:
RX + R' (CF~ 2 ~ C~RI a~ b~ RC~,2 = cgo~l + R~X
wherein R is car~oxy, amido, aroxy, alkoxy, or the liXe; X is
hydrogen, hydroxyl, alkyl, aryl, or the liXe; n is zero or
one; R' is carboxyl, amido, alXyl, substituted alkyl, aryl or
substituted aryl; and R, Rl and R2 ar~ each individually one of
hydrogen, alkyl of 1 t~ a~out 12 car~on atoms, cycloalkyl,
aryl, aIXyl ~thers, and t~.- like.
The transvinylation reaction is an ~quilibrium exchange
reaction ~ith an equilibrium constant of a~out 1.
_ 9 _
~CC.O~ b
2.S?7~ 2
~ccordingly, when equimolar amount~ Or the aLXenyl derivatLvs
of the first Bronsted acid ~R' (CH~),CR2 ' CR~I) and the s~cond
Bronsted acid (RX) are 3110wed to react to equilibrium,
approximately an equimolar mixture oi' the reactants, that is,
the alXenyl derivative of the first Bronsted acid tR(CH~)oC~Z -
CR~RI) and the second Bronsted acid (R'X~, and the
transvinylation products, that is, the aLkenyl product
derivative of the second 3ronsted acid (R(C~ CR2 - C~) and
the conjuqate acid (R'X) of the aIkenyl derivative of t.~e
first 8ronsted acid, are present at the end of the reac_ion.
Since all four components are present in the reaction mixt~re,
the mixture is referred to herein as the transvinylation
reaction mixt~re. As is well known, the equilibrium reaction
may be shifted to favor production of one of the reac~ion
products by appropriate adjust~ent of the reactants and/or by
removal of one or both the reaction products. Accordingly,
the design parameters and product desired will deter~ine
whether the equilibrium is to be shifted and '~he manner in
which it is to be shifted. Nevertheless, it is expected 'Ihat
the transvinylation reaction mixture will include both the
alkenyl product derivative (R(CX.)~C~ - C~) and the conjugate
acld (R'X). As is Xnown, there can also be other reac_i~n
side products formed depending on the starting reactants that
are used.
In accordance with the invention, azeot~opic distillation
is employed to assist the transvinylation process by providing
a relatively simple, facile and economical method for the
recovery of the aIkenyl product derivative, (R(CX~)aC~ ~ CR~I)
produced by the transvinylation reaction. The primary
o~jectivQ of azeotropic distillation is the recovery o~ ~he
alkenyl product derivative or mixtures of the alkenyl product
derivative and the aIkenyl derivative o~ the first 8ronsted
aci~. Stated another way, eithe_ the alkenyl product
derivative or a mixture ~f the aLkenyl product derivative and
the alkenyl deriva~iYe of the first 8ronsted acid are to be
recovered, regardless of the manner in which th~
-- 10 --
~CC.01/ljb
2~:7 ,~
transvinylation react~on mixtur- ~s tr~at-d, i~ at all, prlor
to azeotropic distillation. Accordingly, an az~otropic aqent
i5 added to th- transvinylation reaction mixtur~ to ~orm an
azeotrope with the alkenyl product derivative so that upon
azeotropic distillation, at least the alkenyl product
derivative can be recovered. To that end, the azeotrope has a
boiling point at a temperature su~iciently di~erent ~rom the
boilinq point of at least the conjugate acid (R~X) to allow
the azeotrope to be separated from the transvinylation
reaction mixture by azeot-opic distillation.
Where it is desirable to recover a mixture of the alkenyl
product derivative and the alkenyl derivative Or the ~irst
Bronsted acid; it may or may not b- necessary to also form an
azeotrope with thè alkenyl derivative of the rirst 8rons.ed
acid depending on the relative boilinq point of that aIkenyl
derivative, the alXenyl product derivative azeotrope and other
components in the transvinylation reac_ion mixture. If the
boiling point of the alkenyl derivative of the ~irs. Bronsted
acid is sufficiently different ~rom the boiling point of the
conjugate acid, or other components of the reaction mixtura
from which it is to be separated (or both the conjugate ac-d
and other components in the reaction mixture), it may not be
necessary to ~or~ an azeotrope with the alXenyl derivative of
the first Bronsted acid. It may be possible and even
desirable to separate the alkenyl derivative of the first
Bronsted acid with the azeot.ope. However, if ~he boiling
point of alXenyl derivative of the first Brons~ed acid and the
component of the transvinylation reaction mixture from which
it is to ~e separated are too close, it may be desirable to
select an azeotropic agent or a combination of azeot_opic
agents that form an azeotrope with either or bcth of the
aLXenyl derivative of the first Bronsted acid or the alkenyl
product derivative to allow the derivative mixture to be
separated and recovered.
The azeotropic agent selected must ~e compatible with the
alXenyl product derivative. In addition, the azeotropic agent
forms with the alkenyl product derivative an azeotrope which
~C~.0~ b
:
.
2~?75~;2~
has a boiling po~nt ~ur~ici-ntly d~ rent ~rom th- oth-r
components of the transvinyl~tion r-action mixtur- to p-rmit
the saparation of th~ azeotrope rrOn those components by
a2eotropic distillation in a suitabl- column To that nd, it
is preferr-d that the dl~erenc- in the boiling point o~ the
azeotrope and the boiling point of the oth-r components Or the
transvinylation r-action mixtura ~rom which the azeotrope is
to be separated be on the order of several degrees centigrade,
mos~ preferably about 10 degrees centigrade or more
Separation by azeot-opic distillation i~ generally less
difficult and less expensive the greater the boiling point
differential
It will be appreciated that the azeotropic agent and
alXenyl product derivative may form either a minimum boiling
point azeotrope or a maximum boiling point azeotrope The
azeot_opic agent may for~ either a heterogeneous or a
homogeneous azeotrope with the alkenyl produc_ derivative
Where the azeotroping aqent and the alkenyl product derivative
for~ a heterogeneous azeot-ope, the solubility of the
azeotropic agent and alkenyl product derivative in one another
is minimal ~pon condensation of the azeotrope after
distillation, the azeot_opic agent and alkenyl product
derivative will separate into two phases, so separation of the
a2eotropic agent from the alXenyl product derivative is
facilitated It is thus preferred that the azeot:opic agent
for~s a heterogeneous azeot-ope wit~ the alkenyl product
derivative Use of an azeotropic agent that forms a
- heterogeneous azeot-ope simplifies the se~aration, recovery
and purification of the alXenyl produc~ derivative
The azeotropic distillation assisted transvinylation
process in accordance with the present inYention is very
versatile in that it ~llows for the preparation, separation
and recover! ' the alkenyl produc'_ derivative and of mixtures
of the aIkenyl product derivative a-i the alkenyl derivative
of the ~:rst Bronsted ac d The spec_fic product or product
~ixture to be produced wi l det-r~ine the manner in which the
transvinylation reac_ion products are to ~e trea~ed prior to
- 12 -
~C~.O~
-
2 ~ ~ t~ r~ ~
azeotropic distillation. For example, th- t-ansvinylation
reaction products may be tr~ated to remove all or only a
portion of the alkenyl derivativQ o~ the ~irst Bronsted acid
prior to azeotropic distillation. Where all o~ the aLkenyl
derivative of the ~irst 8ronsted acid is removed prior to
azeotropic distillation, the alkenyl product derivative is
separated from the t-ansvinylation reaction mixture by
azeotropic distillation and is subsequently recovered by
separating the aLkanyl product derivative from the azeot-opic
agent. Where only a portion of the aLkenyl derivative of the
first Brons~ed acid is removed prior to azeot-opic
distillation, a nixture of the ~lkenyl product derivative and
the alkenyl derivative of the first Bronsted acid may be
separated and recovered.
It -~ill be appreciated that while the azeot.opic agent is
capable of for~ing an azeot-ope with the alkenyl produc~
derivative, an azeotropic agent that for~s an azeot_~pe with
both the alkenyl product derivative and the alkenyl derivative
of the first Bronsted acid may also be satisfactorily
employed. Selection of an azeot_opic agent capable of for~ing
an azeotrope with both tbe alkenyl product derivative and with
thè alXenyl derivative of the first Bronsted acid allows a
mixture of those t~o derivatives to be separated and racovered
from the transvinylation reaction mixture~ Of course, it is
also possible to separate and recover a mixture of the alkenyl
derivative of t~e first Bronsted acid and the alkenyl pr~duct
derivative without first for~ing an azeotrope o~ the alXenyl
derivative of the first Bronsted acid so long as the boiling
point of the aIkenyl derivative of the first Bronsted acid and
tha boiling point of the aIkenyl product derivative are either
both higher or both lower than the hoiling point of the
conjugate acid.
~ h~ transvinylation r? ~tion is preferably car~ied out in
the presence of a ~talys~. ~ny of the well Xnown
transvinylat_on a~__tst may ~e employed, including, for
example, ~erc~ry and p2 ~_adium based ~atalysts. Especially
preferrod transvinyla~ion oatalysts are the rutheniu~-~ased
- 13 -
~CC.O1/ljb
z~
catalyst~ disclos-d and clai~-d ln U S application S-rlal No
213,697, ~il d June 30, 1988, ntitl-d Transvinylation
R-action, a-sign-d to the ~a assignee o~ the pres-nt
application
Turning to Figur- 1, th-r- is illustrat d a ~che~atic
r-pre~-ntation o~ th- pr-~-rr-d mbodi nt of th- pres-nt
invention In th- ill~t:at-d process 10, the reactants,
namely th- alkenyl d-rivative of th~ first Bronsted acid, and
the second Bronst-d acid are f-d to r-actor 16 through feed
lines 12 and 14, r-sp-ctiv-ly Tyjpic lly a poly~erization
inhibitor is included to prev-nt und~sirable polyoerization o~
th- vinyl mono~ r R-actor 16 includ-~ a catalyst ~or the
transvinylation reaction Reactor 16 is ~aintained at a
temperature and pressure su~fic ent to effect the
transvinylation reaction After the r-actants are allowed to
react, the transvinylation reaction ~ixture, which contains
concentrations of the alkenyl derivative of the first Bronsted
acid, the second Bronsted acid, the alkenyl product derivative
and conjugate acid of the alkenyl derivative of the first
Bronst-d acid, and possibly catalyst is transferred from
reactor 16 to a separation 20ne 18 t~rough t-ansfer line 1,
In separation zone 18, various separation tasks may be
perfor~ed on the transvinylation r~aetion ~ixture prior to
azeot_opic distillation By way of exampl~, separation zone
18 may b used to separat~ a mixture of the aIkenyl produc~
derivative and the conjugat2 acid fron ~he catalyst and any
unreactQd second Bronsted acid The produet objeetive of the
reaction will determine ~ether the unreacted aIkenyl
derivative of the first Bronsted acid is separated in whole or
in part fro~ the alkenyl product derivative and conjugate acid
in sep~ration zone 18 To produce pure alkenyl product
derivative, ~eparation zone 18 is designed to separate the
unr~ :te~ ~lkenyl d-rivative of Ihe first Bronsted acid and,
op~ ona'ly, to recycle that alken-~ derivative back to t~e
re~-~or by transfer line 20 To produc~ a ~ixture of the
alkenyl product d ivative and the alkenyl derivativ- of the
~ first Bronst~d ac_;, separation zon- 18 is designed to
:'~
- 14 -
C~. 01/~
2~7~
s-parate only a portion of the alXenyl d-rivativ~ (or non- at
all) from the alkenyl product derivativ-. In th- illustrated
embodiment, all, or at least a portion of the alX-nyl
dorivative of the f~rst BronQted acid is r-moved ~rom the
transvinylation reaction ~ixture in separation zone 18 and is
recycled bac~ to the reactor via transf-r lin- 20. The second
Bronsted acid together with catalyst is recovered from
separation zone 18 and is recycled to reactor 16 via tzansfer
line 22.
It will be appreciated that separation zone 18 may be
included after azeotropic distillation, for example, where the
separation zone is used primarily ~or the separation and
recovery of c~talyst after the alkenyl product derivative has
been separated f.om the transvinylation reaction mixture.
The alkenyl product derivative, conjugate acid and,
optionally, unreacted aIkenyl derivative of the first Bronsted
acid are transferred from separation zone 18 to distillation
column 24 via transfèr line 23 for azeot-opic distillation.
The azeotropic agent is capable of forming an azeotrope with
at least the alkenyl product derivative in order to enable the
alkenyl product derivative to be separated from the conjugate
acid. The azeotrope preferably has a boiling point at leas~
10C different than the boiI ng point of the conjugate acid,
although the azeotropic distillation assisted t-ansvinylation
proeess in accordance with the invention is use~ul where the
di~ference in boiling point between the azeot ope and the
conjugate acid (as well as other components of the
transvinylàtion mixt~re from which the alkenyl product
derivative is to be separated) is significantly less. For
example, the present invention is useful even when the boiling
point differential between the azeotrope and the conjugate
acid (or the component) is on the order o~ two or three
degrees centi-~ de, especially where the monomex to be
recovered is :ommercially mportan~.
It may or may not ~e necessary to use an azeotropic agent
to for~ an azeotrope ~_-h the aLkenyl derivatiye of the ~irst
Bronsted acid in order to separate ~he aLkenyl derivative of
-- 15 --
~CC.01/ljb
. .
2~7~
the ~irst BronstQd acid ~rom the conjugate acid depending on
the relative boiling point of thQ alkenyl derivative o~ the
first Bronsted acid, the alkenyl product derivative and the
conjugate acid. In the event the boiling point o~ the alkenyl
derivative of the rirst 8ronsted acid and the ~oiling point of
the conjugate acid are relatively c10s2, i.e., on the order of
less than 15 degrees, then it may well be desirable to for~ an
azeot_ope with the alXenyl de~ivative oS the Sirst Bronsted
acid to effect efficient separation of the alkenyl derivative
o~ the first Bronstad acid from the conjugate acid. However,
if the boilinq point o~ the alkenyl derivative of the first
8ronsted acid and the conjugate acid are not close, and the
boiling points o~ the alkenyl derivative of the first Bronsted
acid and azeotrope of the azeotropic agent and the alkenyl
product derivative are bot~ either lower or higher ~han the
boiling point of the conjugate acid, then the aLkenyl
derivative of the first Bronsted acid may be separated
together with the alXenyl product derivative from the
ccnjugate acid without the use of an azeot-opic agent for the
alXenyl derivative of the first Bronsted acid.
It will be appreciated that it may be desirable to for~
an azeotrope or the alkenyl deriva~ive of the first Bronsted
acid even though the boiling point of the alXenyl derivative
of the first Bronsted acid is sufficiently different from the
boiling point of the conjugatP acid in order to assure that
both the alXenyl derivative of the first Bronsted acid and the
azeotrope of the aIXenyl product derivative boil either higher
or 7 ower than the conjugate acid, and to thereby fur her
assist in the separation of those components from the
conjugate acid. One azeotropic agent capable of forming an
azeotrope with both the alkenyl product derivative and the
alkenyl derivative of the first 3ronsted acid may b~ used. It
is also c~:2mplated ~-at a mixture of azeotropic agents may
be used, one for the aIkenyl product derivative and one for
the aLkenyl deriva~_ve o~ the first Bronsted acid.
In the illu:~rated embodiment, the azeotropic agent is
shown to be fed near t~e top of column 24 through feed line
- 16 -
UCC.O1/ljb
2~7 ~C.,7~
25. Additionally, azeot-opic agent recover~d ~rom the
decanter 30 (as described below) can, and pre~erably is,
recycled to the azeotropic distillation column 24.
Independently of the sourc~ o~ the azeotropic agent, it is
preferred to feed the azeotroping agent near the top of the
azeotropic distillation column to assist in t~e separation o~
undesirable components during distillation. BecausQ the
temperature of the liquid near the top of the column is lower
than it is at other locations, adding the azeotroping agent
near ~he top allows the agent to cool and thereby Xnock down
non-azeotroping components that may be near the top of the
column. Also, where the azeotroping agent is water, there is
less time and opportunity for the water to contact and
possibly hydrolyze the alkenyl product derivative of the
azeot-ope. However, it will be appreciated by thosa skilled
in the art that the azeotropic agent can be fed to
distillat~on column 24 at other points in the column,
including, for example, in the middle, or near the bottom.
The azeotropic agent may also be added to the feed to
distillation column 24.
In the illustrated embodiment, the azeotropic agent and
alXenyl produc~ derivative .orm a heterogeneous minimum
boiling point azeo~.ope. As a result, the azeotrops is drawn
off the top of column 24 through transfer line 28 and
t-ansferred to decanter 30 where the azeotrope separates into
t-~o phases, the alXenyl produc~ derivative and the azeot-opic
agent. Note that if a mixture of alkenyl produc_ derivat.ve
and alkenyl derivative of the first Bronsted acid is separated
from the coniugate acid, then one phase will consict of a
mixture of the aIXenyl product derivative and the alkenyl
derivative of the first Bronsted acid and the other phase will
consist of the azeotropic agent. The azeot~opic agent is
recycled for use in azeotropic distillation column 24 (as
illustrated by transfer line 32~. Some of t~e az~otrcpic
agent may be removed as waste t~rough t_ansfer line 34. ~he
alkenyl product derivative phase may, if desir~d, also be
re~luxed (as, for exampl~, by return to distillation column 24
- 17 -
~CC.OlJljb
2~?7 ,~;,''~
through tran~er line 33) to ~nhance rectlflcat~on
T~e alk-nyl product der~vativ- is trans~erred to drying
column 36 via trans~er lin- 38 Sn drying colu~n 36, the
alkenyl product derivativ- is dr~-d by a2~0tropic drying, the
azeotrope being taken o~ the column and transferred to eithe-
the first decant-r 30 and process-d as describ-d above, or
tsansferred to a second decanter 40, in which phase separation
of the aLkenyl product derivative and azeotropic agent occurs
Alkenyl produc_ derivative from the sec~nd decanter 40 is
returned to drying colu~n 36 via transfe_ line 42, while the
azeotropic agent is processed as waste Azeotropic agent from
decanter 40 may also be recycled for use in distillation
column 24 D~ied alkenyl product deri~ative is transferred to
purification column 44 where alkenyl product derivative is
recovered If desired, the alkenyl product derivative may be
neut~alized with a suitable ~ase, such as, sodium bicar~onate,
to re~ove residual acid
Returning to the azeotropic distillation column 24, the
conjugate acid of the vinyl ester of the first Bronsted acid
~hic~ is produced during tha transvinylation reaction remains
at the column bottom The con~ugate acid is t:ans'er-ed f-om
azeot-opic distillation col = 24 via transfer line 25 to an
acid separation column 46 whera the conjugate acid is
separated and recovered The conjugate acid is recovered for
subsequent use It will ~e appreciated that in the event that
the second Bronsted acid is not completely separated f-om the
conjugate acid and the aIkenyl product derivative in
separation zone 18, the second 3rons.ed ac.d is separated Crom
the azeotrope in azeotropic distillation column 24 and is
passed to acid separation column 46 with the c~njugate acid
In acid separation column 46, the second Bronsted acid is
separated from the conjuqatc acid and recycled via transfer
line 48 to reactor 16
The azeo~ropic distillation column operating temperature
and pressure ar- not particularly critical to the practice of
t~e invention, but they are related, and they ar- highly
dependent on the particular separation to be carried out
- 18 -
~CC 01/ljb
2~?7.,~
Azeotropic distil~ation ~ay be carri-d out at at~o pherlc
pressure or it may be carri-d out under vacuum or under
pressure. The lower lioit on the operating pressure iQ
related to the economical limitations on cond-nser
temperature. The upper pr~ssur~ limit is dopendent on the
limitation of the column bottom temperature which, in turn, is
dependent on the decomposition t~mperature of the components.
Operating temperature, in turn, depends on the condensation
temperature of the colu~n condenser and on the decomposition
te~perature of the components. The low end of the operating
temperature is preferably only about 10C or so hiqher than
the temperature of the cooling mQdia. ~he upper ~nd Or the
operating temperature, on the other hand, is preferably about
10C lower than the deco~position temperature of the mos~
decomposable component in the system. The operating
temperature and pressure are preferably chosen so that the
condenser can be cooled easily and so that none of the
components decompose. ~here water is the azeotropic agent and
the alkenyl product derivative may be susceptible to
hydrolysis (or where the conjugate acid is an unsaturated
acid) it is preferred that azeotropic distillation be car ied
out under a vacuum so that the distillation column can be
~ operated at lower temperature.
- The type of az~otropic distillation column used to car~y
out the azeotropic distillation is not critical. The column
may be a t,ay or a packed column. A packed column is
preferred due to a lower pressure drop across the column.
The rate at which the azeotropin~ agent is fad to the
azeotropic distillation column depends on both the rate at
which the aIkenyl product divative is produced and the
concentration of the alkenyl product derivative in the
azeotrope. For a given alkenyl product derivative and
azeotropic agent, the feed rate of the azeotropic agent is
preferably sufficient to permit all of the aIkenyl product
derivative fed to the column to ~or~ an azeotrope. While some
excess of the azeotroping agent may be employed, it is
preferred to matc~ the feed rate of the azeotroping agent ~ith
- 19 -
UCC.Ol/ljb
2~7~J~'7~
tha production rate of th- alk-nyl product darivativ- Aft-r
the initial charge, pra~erably the azeotropic aqent r-d ~o the
distillation column i9 recover-d Srom the two phas- ~-parat$on
Or the alkenyl product derivativo and az-otropic agent in the
decanter Azeotropic ag~nt from the decanter $s imply
recyclad to the azeotropic distillation column to rlll the
n-eds of the distillation column, upplemented, i~ necessary,
only to the extent that the azeotropic agent is removed ~rom
the system
The azeotsopic distillation assisted t-ansvinylation
process of the present invention can ba used ~or the
preparation of numerous alkenyl product derivatives that do
not contain a vinyl moiety with the appropriate selection of
the azeotroping agent 8y way of illustration, suitable
alkenyl derivatives are compounds of the for~ula
- (C~,~ ,-CR2 ~ C~
wherein n is either zero or one, R, R~ and ~ are each
individually ona of hydrogen, aLkyl of 1 to about 12 car~on
ato~s, cycloalkyl, aryl, alkyl ethers, and the liXe
Illust-:~tive of suitable alXenyl product derivatives 1-
propenyl benzoate (cis and trans), l-propenyl pivalate tc_s
and trans), 2-propenyl benzoate and 2-propenyl pivalata, allyl
pivalate, allyl benzoate, allyl propionate, nethallyl
benzoate, methallyl pivalate, methylallyl propi~nate, allyl
csotonate, allylmethacrylate, allylacrylate, and the like
Azeotropic distillation assisted transvinylation can also
be used for tha preparation of numerous vinyl product
derivatives with the appropriate selection o~ the azeotroping
agent 3y way o~ illustration, the alkenyl product derivative
may ~a any compound in which th e is a vinyl group ~onded to
a Bronsted acid Such compounds may b~ charactari2ed as
vinylatad Bronsted acids Vinyl embracas groups o~ tha
for~ula
RqRIC - CR2_
- 20 -
~CC. Ol/lj~
2~?7.,rJ,'.,~
wherein R, Rl and R~ are each individually one o~ hydrogen,
alkyl of 1 to about 12 car~on atoms, cycloalkyl, aryl, alX~'
ethers, and the like. The Bronsted acid is any species which
can act as a source o~ protons.
Illust-ative o~ suitable alXenyl derivatives o~ a
Bronsted acid for the practice of the invention, are vinyl
acetate, vinyl pivalate, vinyl benzoate, vinyl methacrylate,
vinyl ac~ylate, vinyl propionate, vinyl cinnamate, vinyl
cyclohexanoate, vinyl crotonate, vinyl butyrate, vinyl 2-
methyl butyrate, vinyl isobutyrate, vinyl 2-~et~yl valerate,
vinyl 2-ethylhexanoate, vinyl octanoata, vinyl decanoate,
vinyl cyclohex-3-enoate, vinyl neodecanoate, vinyl
neononanoate, vinyl 2-propyl heptanoate, and other vinyl
esters or neo-esters, N-vinyl pyr-olidinone, N-
vinylsuccinimide, vinyl phenyl ether, vinyl methyl ether,
vinyl ethyl ether, 2-chloroe~hyl viny' ether, ethyl vinyl
ether, 2-vinyloxyethanol, allyl vinyl ether, isopropyl vinyl
ether, propyl vinyl ether, l-vinyloxy-2-propanol, 3-vinyloxy-
l-propanol, butyl vinyl ether, isobutyl vinyl ether, bis(2-
vinyloxyethyl)ether, 2-butylthioethyl vinyl ether, 2-
butoxyethyl vinyl ether, 2-ethoxyethyl-2-vinyl-oxyethyl ethe_,
2-ethylhexyl vinyl ether, 2-~utoxyethyl 2-vinyl-oxyethyl
ether, trimethylnonyl vinyl ether, N-vinyl 2-oxazolidinone, 2-
vinyloxyethyl acetate, 2-vinyloxyethyl pivalate, 2-
vlnyloxyethylacrylate, vinyl chlorid~, ~inyl sulfonamides, and
the like.
Preferred alkenyl derivatives are ~he vinyl esters of
car~oxylic acids and the vinyl alkyl or aryl ethers, mainly
because they are commercially available.
Illustrative of s~ita~le Bronsted acids for the prac ice
of the invention are c~rboxylic acids such as monocarboxylic
and polycarboxylic acids illustrated ~y acatic acid, propionic
acid, butyric acid, isobutyric acid, 2-methyl butyric acid,
crotonic acid, pivalic acid and other neo-acids, stearic acid,
benzoic acid, terephthalic acid, isophthalic acid, phthalic
acid, adipic acid, suc inic acid, malic acid, ~aleic acid,
polyacrylic acids, acrylic acid, methacrylic acid, cinnamic
- 21 -
~CC.O1/ljb
2~?7 J~
acid, 2-ethylhexanoic acid, cyclohexanoic acid, and
cyclohexenoic acid; amides such as 2-pyrrolidinone, 2-
pyrrolidone, ~-caprolactam, 2-oxazolidinone, and succinimide;
alcohols such as methanol, ethanol, n-propanol, isobutanol,
fluorinated alkanols such as 1,1,1,3,3,3-hexafluoro-2-
propanol, monoethanolamine, diethanolamine, and
triethanolamine; phenolic compounds such as phenol,
resorcinol, and 8isphencl A ~2~2-bis(4-hydroxyphenyl)propane];
amino compounds which are suf~iciently acidic such as
secondary aromatic amines, azoles, and the like; hydroxy
esters such as hydroxalkyl acrylates (e.g., 2-hydroxyethyl
ac:ylate, 2-hydroxyethyl methacrylate) and hydroxyalkyl
aIkanoates ~e:g., 2-hydroxye~hyl acetate, 2-hydroxyethyl
pivalate); silanols such as dimethyl silan diol,
t-ime~hylsilane mono-ol, and '~he like.
The prefer~ed 3ro~sted acids are the carboxylic acids,
the alcohols, the amides, the imides, the phenolics, and the
like.
Illust~ative of transvinylation reac-ions that may be
carried out by the process of the invention, are the
following:
- 22 -
UCC.Ollljb
2 ~7.J.~
~kenvl derivative Bronst~d Acld pFoduc~
vinyl acetate ~ pivalic acid - vinyl pivalate
vinyl benzoata I pivalic acid - vinyl pivalate
vinyl acetate + methac~ylic acid - vinyl methac.ylatz
vinyl acetate + acrylic acid - vinyl acrylate
vinyl acetate + benzoic acid - vinyl benzoate
vinyl acetate I propionic acid - vinyl propionatz
vinyl ace~ate + salicyclic acid - vinyl salicylate
vinyl acetate I cinnamic acid - vinyl cinnamate
vinyl propionatz ' + 2-ethylhexanc.ic acid - vinyl
~ 2-ethylhexanoate
vinyl acetate + cyclohexanoic acid - vinyl cyclohexanoate
vinyl acetate 2-pyr-olidinone - N-vinyl
. 2-pyr_olidinone
vinyl pivalate + 2-pyrrolidinone - N-vinyl
2-pyrrolidinone
vinyl pivalate + succinimide - N-vinyl succinimide
vinyl methyl ether - phenol - vinyl phenyl ether
vinyl chloride + methanol - vinyl methyl ether
vinyl methyl ether + ethanol - vinyl ethyl ether
vinyl acetatz + 2-oxazol,dinone - N-vi nyl
- 2-oxazolidinone
vinyl acetate + N-acetyl - N-vinyl
~ e~hyleneurea N-acetylethyleneurea
: vinyl acetate 1 2-hydroxyethyl - 2-vinyloxye~yl
acetate acetate
vinyl pivalate + 2-hydroxyethyl - ~-vinyloxyethyl
pivalate pivalate
vinyl pivalate 1 2-hydroxyethyl- - 2-vinyLhydroxyethyl
acrylate acrylate
As previo~sly set forth, the azeotropic agent may be any
agent that is capable of forming an azeot-ope with the aIkenyl
- 23 -
UCC. Ol/lj~
2~?7
:.
product derivative o~ the transvinylat~on reaction. For
example, the azeotropic agent may ~orm a binary azeotrope with
the alXenyl product derivative or, in the ~vent it is
desirable to separate a mixture of ester , it may ~or~ an
azeotrope with the alkenyl product derivative and with the
alkenyl derivative of tbe rirst Bronsted acid. The boiling
point differential bet~een the azeotrope and the conjugate
acid`(and the alkenyl derivative of the ~ir5t Bronsted acid if
it is not separated from the reaction mixture prior to
azeot-opic distillation, and if an azeot:ope is needed 'or its
separation) is sufficient to allow the desired product to be
separated, and is preferably at least about 10C. By way of
example and ndt in limitation, suitable azeotropic agents
include water, either in the for~ of a liquid or as steam,
cyclohexane, heptane, isopropanol, methanol and alkyl ethe_.
Where the transvinylation reaction produces a vinyl ester of a
carboxylic acid by the reaction of an alkenyl derivative of a
first car~oxylic acid and a second carboxylic acid, water is
especially preferred because it for~s a heterogeneous
azeotrope with both the alkenyl produc~ derivative and with
the alkenyl derivative of the first Bronsted acid. ~pon
condensat~on of the azeot-ope, the water and alkeny' produc:
derivative (and alkenyi derivat_ve of the first Bronsted acid,
if present) separate into two phases which can be separated in
a decanter.
Nater is the preferr~d azeotropic agent in
transvinylation reactions involving an organic acid having
from 1 to 12 car~on atoms, and an alkenyl.derivative of a
first car~oxylic acid of 'he for~ula:
o
R~ - C - Cffl s CH2
where R" is alkyl, aryl, cycloalkyl and has from 1 to about 12
carbon atoms. Water forms a minimum boiling point azeot~ope
with the alkenyl product derivative and with the alkenyl
- 24 -
UCC.01/ljb
2~
derivative of the rirSt carboxylic acid, if present. The
mini~um boiling point azeotrope has a boilinq point greater
than 10C different than the boiling point of the conjugate
acid, the boiling point of the azeotrope being lower than the
boiling point of the conjugate acid.
In accordance with a prsferred embodiment of the present
invention, an alkenyl derivative of a carboxylic acid having
from 1 to 12 carbon atoms is prepared by ~he t.ansvinylation
reaction of vinyl ace~ate and a second carboxylic acid having
o~ from 1 to about 7 car~on atoms in the presenc~ o~ a
ruthenium catalyst as described in copending application
Serial ~o. 213,697. The alkenyl product derivative is
effec_ively and efficiently separated from the transvinylation
reac_ion mixture by azeot.opic dis~illation with wa~er as the
azeotroping agent. Thus, i-n the transvinylation reaction
desc.ibed, acetic acid, which has a boiling point of about
1'6-118C is one of the pr~ducts of the reaction. Water ls
used to produce a minimum boiling point heterogeneous
azeotrope with the alkenyl produc_ derivative. The azeot.ope
has a bciling point lower than the boiling point of acetic
acid. Removal of the al~enyl product derivative from the
othe- componenta is facilitated.
The present invention is particular~y useful for the
preparation of vinyl pivalate, vinyl but~rate, vinyl
propionate and vinyl crotonate by the transvinylation reaction
of vinyl acetat~ with pivalic acid, buty~ic acid, propionic
acid, and crotonic acid, respec-ively. The products of the
transvinylation reaction, i.e., the alkenyl product derivative
and acetic acid have boiling points that are close, lnd would
be very difficult to separate by fractional distillation, as
can be seen in Table I below:
- 25 -
~CC.01/ljb
Table I
B.~.C Aqueous Azsotrope
Vinvl Ester 0~ Dry Ester 3.P.C
vinyl
-propionate 94.9 79 ~3
-butyrate 116.7 87.2 20.4
-crotonate 133.9 92 31
-pivalate 112 86 17
The addition of a suf~icient amount of water ~o ~he
alkenyl product derivative in the azeotropic distillation
apparatus forms an aqueous azeotrope with the vinyl pivalate,
vinyl butyrat~, vinyl propionate and vinyl crotonat-. It has
been discovered that vinyl pivalate and wate_ form an
azeot.ope having a boiling point of about 86~C, while it is
known that vinyl propionate, vinyl butyrate and vinyl
c-ot~na~e for~ azeotropes with wate_ having boiling points,
respectively, of about 94.9C, about 87C, and abou~ 92~. In
all ins.ances, the azeot-ope has a boiling pcint substantially
below the boiling point of acetic acid which allows the vinyl
est~r to be separated readily fr~m acetic ac-d.
Because water for~s a heterogeneous azeot_ope -~ith the
vinyl es~er, upon condensation o~ the azeot_ope, it wil:
separate into t~o phases, namely water and the vinyl es~er,
t.~ereby facilitating simple recovery of the vinyl ester.
Thus, the azeot~opic distilla~ion assisted transvinylation
process described herein can be used to make vinyl pivalate
vinyl butyrate, vinyl crotonate and vinyl propionate direc_ly
from vinyl acetate by transvinylation. The process is more
economical than previously known trans~inylation tPchniques
because of the relative ease with which the vinyl ester c~n be
separated from acetic acid.
The following Examples are illustrative o~, bu~ not in
limitation of, the present invention. These Examples
illustrate the separability of vinyl esters and carboxylic
acids and describe the preparation of a vinyl product ester
- 2S -
~CC.01/ljb
2~7~,2~
rrom carboxylic acids by 'ho azeotropic distillation a-sist-d
transvinylation process o~ th- present inv-ntion.
Ex~ple 1
This Example demonst:ates the feasibility of separat-ng
vinyl acetata, vinyl propionate, acetic acid and propionic
acid. This ~ixture is believed to be typical of the
transvinylation reactIon mixture after catalyst removal that
would result from the preparation of vinyl propionate by the
transvinylation of vinyl acetate and propionic acid. Water
was used as the azeotropic agent.
A mixture consist_ng of 28wt.% vinyl acstate, 30wt.%
vinyl propionate, 18wt.% acetic acid and 24wt.% propionic acid
was charged to a 2 liter kettle. The kettle was equipped with
a 15-t_ay Oldershaw distillation column, a condensing column
connected to the outlet (top) of the distillation oolumn and a
collection flask connected to the outlet end of the condensing
column.
The mixture was heated and an initial reflux ratio of 10
was established. As the temperature of the ke~tle was
inc_eased, it was found that the lower boilinq point vinyl
ace~ate was being separated ,rom the mix~ure. After the vinyl
acetate had been separated f-om the mixture, water was added
to the top of the distillat~on column dropwise lmt-l abou~
15.4g of water had ~een added. An aqueous azeot-ope of vinyl
propionate for~ed, whic~ had a boilinq point (obse~ved) in the
range of about ?8-79C. The azeotrope was analyzed by gas
c.~romatography and found to contain 98.11~ vinyl propionate,
0.6~9% acetic acid and 0.326% propionic acid.
Exammle 2
This Example illustratas the separating of vinyl acetate
and vinyl crotonate from a mixture of vinyl acetate, acetic
acid, vinyl crotonate and crotonic acid by azaotropic
distillation. T~is mixture simulates a transvinylation
reaction mixture, after catalyst separation, that would be
expected to result from the preparation of vinyl crotonate by
- 27 -
~CC.01/ljb
2~7~ ,2~ -
the transvinylation reaction o~ vinyl acetata with c:otonic
acid.
A solution ~8.8 ml) prepared ~rom vinyl acatate (7.0 g),
vinyl crotonate (7.0 gracs), acetic acid (4.0 grams), and
c-otonic acid (2.0 grams) was charged to the kettle of a
single-stage distillation apparatus. Gas chromatography
analysis of this solution is set forth in Table II below:
Tabl~ IT
vinyl vinyl acetic c.otanic ace~al-
crotonat~ acerat~ acid ~i~ dehvde
Area% 46.14 31.22 10.23 10.28 0.074
The ket~le was heated to 75C and 1 ml water was added
via addition funnel. Vinyl acetate/water and vinyl
c-otonate/water azeotropes were distilled at an overhead
temperature ranging f_om 67C-94C. The following f-ac~ions
were collected, and when ~nalyzed by gas chrom~tography were
found to have the composition tarea %) set for~h in Table -II
below:
able T, T
vinyl vinyl acetic crotonic acetal-
overhead c-otonate ace~ate ~i~ 3Si~ dehvde
6~-71C 5.04% 92.81% - - 1.-8%
6~ 7.50% 92.81% - - 0.5
69-73 10.56% 87.4 % 0.39% 0.11% 0./8~
76-84 16.38S 82.3 % - - 0.58%
84-90 42.17% 54.9 % 0.65% - 0.38%
90-93 82.24% 13.8 % 0.~8% 0.19% 0.37%
94O 95.9 % 1.33% 0.60% 0.13% 0.~0%
~ t can ~e seen that vinyl crotonate and vinyl acetate can
be separated and removed from a transvinylation reaction
mixt~re through simpl~ azeotropic distillation and without an
elaborate fractionating column.
- 28 -
~CC.01/ljb
' Z~?7~C~f~
Examole 3
This Example illustra~es the preparation of vinyl
acrylate by azeotropic distillation assist-d transvinylation
o~ vinyl acetate and acrylic acid.
Three separata transvinylation runs were uade. In each
run 1,500 grams of ac:ylic acid, 1.91 grams of rut~enium
catalyst having a for~ula ~RU(co)2ococ~3]n~l~5oo grams of vinyl
acetate, and 6.0 grams of phenothiazine were charged to a 1-
gallon autoclave. The reac~or was pressurized and purged
three times with car~on monoxide (50 psig) and ~hen
pressur~zed to S0 psig. The reactor was heated to 130-136C
for ~ total of 4 hours. The r~actor was cooled, vented, and
the ~ransvinylation mixture, containing ~he catalys~, vinyl
acrylate, acetic acid, acrylic acid, and vinyl acetate, was
discharged from the reactor.
The transvinylation mixtures of each run, respectively,
2,848 grams, 2,938 grams, 2,778 grams, were combined and then
flashed distilled under vacuum to remove the .-ansvinylation
mixture (vinyl acetate, vinyl acrylate, acet-c acid, and some
acrylic acid) from the acrylic acid/ruthenium catalyst
residue. The distillate was ~ecovered.
The flash dis~illed t~ansvinylation mixture was charged
to a fractional distillation apparatus comprising a 12 L
kettle equipped with a 24 tray Oldershaw column and a
fractionating head. ~inyl acetate was removed f.om the flas.h
distilled transvinylation mixture before azeot-Qpic
distillation. During vinyl acetate distillation operations,
phenothiazine inhibitor tdissolved in vinyl acetate) was added
to the ket~le and was fed down the column through '~he use of
an addition funnel. The unreac_ed vinyl acetate (~.p. 72-
73C) was fractionated from the transvinylation mixture at a
reflux ratio of 10/1. The uajority of the residual vinyl
acetate was purged from the column at the high ~10~1) reflux
ratio by gradually and briefly raLsing the head temperature to
- 29 -
~CC.01/ljb
2~7~
82C. The gas chromatography area % analysis of the resulting
mixture is set ~orth in Table rv below:
Table IV
acrylic vinyl vinyl acetic
ac-vlate acetate ~i~acetaLdehYde
34.58~ 45.31~ 0.341 13.208 0.134
The kettle was allowed ~o cool, thereby allowing the
vapors to recede down the column. Water was then charged to
the addition funnel on the top of the dis8illation column and
formed a minimum boiling water~vinyl acrylate azeot-ope having
a boiling point (observed) of 77-78C. Upon condensation, the
water/vinyl acrylate azeotrope separated into an organic
(uppe_) phase and an aqueous (lower) ?hase. The gas
chromatographic analyses (area %) of the azeotropic fraction
(instantaneous samples of the organic overhead~ are set fort~
in Table V below:
able v
overhead vinyl vinyl acetic
(temD oc~ acrtlate acstate acid acetaldehvde
1 (77) 96.013 3.301 0.367 0.059
2 (78) 98.360 1.168 trace* 0.091
3 ~78) 98.541 1.012 trace* 0.098
4 (78-94)** 97.319 0.995 0.489 0.244
~ (94) 9/.1~8 1.072 0.489 0.254
*In overhead samples 2 and 3, the acetic acid content was
below the threshold of the gas chromatogràph integrator.
**Not deter~ined precisely.
The vinyl acrylate fractions were combined,
azeotropically dried (280 mo ~g, ~8-79C) and distilled
through a short path column to give 2 L of product. The
analysis of the refined vinyl acrylate by gas chromatography
is set for~h in Table VI below:
- 30 -
UCC.01/ljb
2~?7~
Table VI
vinyl vinyl acetic
acrvl~te acetato ~i~ acetaldeh~de
98.427 1.247 0.024 0.035
Gas chromatographic analysis revealed ~at a highly pure
product, low in acidity, was obtained using azeotropic
distillation assisted transvinylation. This Exanple thus
demonst~ates that the present invention provides a relatively
simple process for obtaining high purity alkenyl produc~
derivatives that are low in acidity directly and without the
need for ~urther processing to remove ac_d.
rxam~le 4
This Example illustrates the preparation of vinyl
crotonate by azeotropic assisted t-ansvinylation of vir.yl
acetate wit.; crotonic acid.
To a 1-gallon autcclave were charged 1,350 grams of
crotonic acid, 0.9 grams or ruthenium catalyst having the
formula tRU(C~)20COC~3]n~.350 grams of vinyl ac~tate, and 2.7
grams of phenothiaz;ne. The reactor was pressurized and
purged three times with ni.-ogen (50 psig) and then
pressurized to 25 psig. The reactor was heated to 127-133C
for a total of 12 hours. The reactor was cooled, vented and
the transvinylation mixture, containing the catalys~, vinyl
c-otona~e, acetic acid, c_otonic acid, and vinyl acetate, was
discharged from the reactor.
The transvinylation mixture was flashed distilled unde~
vacuum to remove the t-ansvinylation products (vinyl acetate,
vinyl crotonatej acetic acid, and some c,otonic acid) f~om the
crotonic acid/ruthenium catalyst residue.
The flash distilled transvinylation mixture was charged
to a fractional distillation apparatus comprising a 3000 ml
kettle equipped with a 25 tray Oldershaw column and a
fractionating head. During vinyl acetate distillation
operations, phenothiazine inhibitor was added to the kettle
and was fed down the column ~hrough the use of an addition
- 31 -
~CC.01/ljb
2~7.,~
funnel (phenothiazi~o dissolved in vinyl acetate). The
unreacted vinyl acetata (~.p. 72-73C) was ~ractionated from
the transvinylation mixture at a reflux ratio ranging from S/1
(initially) to 10/1 (at the end of the distillation). The
last ~inor amounts of vinyl acetate wers purged from the
column at the high (10/1) reflux ratio by gradually and
briefly raising the head ;emperature to 93C.
The kettle was allowed to cool briefly, thereby allowing
the vapors to recede down the column. Water (saturated with
phenothiazine) was charged to the addition funnel on the top
of the dis~illation column, in the same position as that used
for the inhiDitor feed, and for~ed a minimum boiling
water~'vinyl crotonate azeot_ope having a boiling point
(observed~ of 92C. Upon condensation, the wate_/vinyl
crotonate azeot_ope separated into an organic (upper) phase
and an aaueous (lower) phase. Six azeotropic f~ac ions were
analyzed by gas chromatograpAy. The compositions (by area %)
of each of ~he '-actions is set for~h in Table VII below:
Table vII
Fraction vinyl vinyl acetic c_otonic
No. cr~tonatQ ace-a~_ ac~d ~ acet~ldeh~de
1 98.473~ 0.06,~ 0.452% - 0.473%
2 96.38S 0.014 2.942 - 0.03~
3 95.14S O.g90 3.581 - 0.032
4 99.071 0.159 Q.501 - 0.157
98.410 none 0.734 0.~77 0.041
obser~ed
6 44.985 none 2S.007 14.S94 0.523
observed
In both Lractions 2 and 3, the column flooded and it is
believed the water feed rata was :oo low to comple-ely
azeotrope the vinyl crotonate. After ~raction 3 was comple~e,
the Oldershaw column was replaced with a Vigreux column to
alleviate flooding. Improved separation resulted after the
- 32 -
UCC.01/ljb
2 ~ 7.
column was changed.
S
This Example illustratas the preparation o~ vinyl
pivalate by azeotropic assisted transvinylation o~ vinyl
acetata with pivalic acid.
A roactor was charged with 5,08~ pounds of vinyl acetate,
4,854 pounds of pivallc acid, and ruthenium dicarbonyl acetate
homopolymer (530g). The reactor was pressurized to 25 psi
carbon monoxide ~containing an oxygen atmosphere o~ 500-1000
ppm in the reactor, as required by the inhibit~r
hydroquinone), and heated to 150C for 7 hours. Analysis by
gas chromatography of a reactor sample revealed the reac'_on
had reached e~uilibrium. The reactor was cooled to 50C and
the content~ were flash distilled under vacuum. The
distillation residue, containing catalvst and pivalic acid,
was discharged from the reac~or.
Vinyl acetate was then fractionated f_om the distillate.
After removal of vinyl acetate, the reac_ion mixtur_ was
subjected to azeotropic distillation to se~arate and recover
vinyl pivalate.
The coiumn bot~oms from the vinyl acetate removal step
were distilled by azeotropic distillation. A ~och-Seltzer
column was used in conjunc:ion with a thirl_y gallon decanter.
The column bottoms, which contain csude vinyl pivalate
(preheatad to 90C) was fed 'o the column midpoint and water
was fed to the tOp of the column via a reflux line. A vinyl
pivalate/water azeotrope (b.p. - 86C, 17% by wt. water) was
frac_ionated and distilled overhead. Pivalic acid, ace~ic
acid, and heavies were removed from the botto~ of the column.
The azeotropic product was collected in the decanter wher- it
phase separated. The bottom water layer was returned to ~he
top of the azeotropic dist llation column and the ~op vinyl
pivalat~ layer was collec-a~ for ,^inal drying. During the
operation the crude mater:ai was ed to the column; abou~ 50%
distilled overhead and about 50% was removed from the column
base. Throughout the sep ration, the column top maintained a
- 33 -
UCC.01/ljb
2~7 J~
temperaturs o~ 39.6C-94.5C and the base temperature was
120.8C-126.2C. The yield ~or the azeotropic distillation
was 97.3%.
The composition of the nine ~ractions ~rom the a2eotropic
distillation is set forth in Table VIII below:
Table 'JIII
Fraction vinyl a¢etic vinyl pivalic
~umber acetate acid ~ivalate acid
1 0.480 0.197 99.313
2 O.Og6 0.064 99.813 0.026
3 0.076 0.018 99.875 0.006
4 0.079 0.087 98.863 0.936
O.Ogl 0.123 98.748 1.014
6 0.079 0.011 99.828 0.06~3
7 0.082 0.010 ~.874 0.034
8 0.080 0.013 99.87~ 0.031
9 0.089 0.033 g9.5~9 0.022
It can ~e seen from ~e analysis of fraction numbers 2,
3, 6, 7, 8 and 9 that azeot.opic distillation was par.icularly
effective for the recovery of high purity vinyl pivalate, low
in acid content, and ~t is believed that no fur~lhe- procsssing
of those f-actions to remove acid would be required. During
fractions 4 and 5, the w~ter feed to the azeot~opic
distillation column was undesirably fast. Too rapid a water
feed -esulted i. frac'ions of pivalic acid .hat were higher in
pivalic acid and acetic acid content than were desiraole.
This Example demonstrates the remar.~able advantaqes of
the azeotropic distillation assisted transvinylation process
of the present invention. With the present invention,
separation of vinyl pivalate and acetic acid which have very
close boiling points (112C and 116-118C, respectively) is
much simpler and cheaper, and preparation of vinyl pivalata by
transvinylation of vinyl acetat- and pivalic acid is
economically and commercially att~active.
The present invention thus provides for an im~roved
transvinylation process in which the separation and recovery
- 34 -
-UCC.01/ljb
2~7~J'~.J~
of t~e alXenyl product d-rivative is f~icient and economical.
Azeotropic distillation assi~tad transvinylation has improved
productivity, lower equipment costs and lower operating costs
than conventional Sractionation or distillation duo, at least
in part, to the efficiency and ease of the separat_on and
recovery of the alkenyl product derivative. The process is
capable o~ producing highly pure alkenyl product derivatives,
low in acidity, directly and without the need for further
processing. Moreover, the invention makes economical the
production of alkenyl product derivatlves by transvinylation
in a single stage reactor where the alkenyl product derivative
and conjugate acid of the alkenyl derivative of the first
Bronsted acid are close ~oilers.
- 35 -
UCC.O1/ljb