Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
20~771~
. `V092/034~ PCT/EP91/01491
THERAPEUTIC AGENTS
This invention relates to indane and
cyclopentanothiophene derivatives which are potent,
orally active antagonists of platelet activating factor
and as such have clinical utility for treating allergic
and inflammatory conditions such as asthma and
arthritis respectively.
Platelet activating factor ~PAF, l-O-al~yl-2-
acetyl-sn-gl~cQryl-~-phospho-ylcholinQ) is an ether
phospholipid whose structure was first elucidated in
1979. It is produced by, released from and interacts
with many pro-inflammatory cells, platelets and the
kidney. In addition to potent platelet aggregating
activity, PAF exhibits a wide spectrum of biological
activities elicited either directly or via the release
of other powerful mediators such as thromboxane A2 or
the leukotrienes. In vitro, PAF stimulates the
. movement and aggregation of neutrophils and the release
therefrom of tissue-damaging enzymes and oxygen
radicals. These activities contribute to actions of
PAF i~ YiVo consistent with it playing a significant
role in inflammatory and allergic responses. Thus,
intradermal PAF has been shown to induce an
inflammatory response, with associated pain,
accumulation of inflammatory cells and increased
'
.
... . . . .
W092/034~ ~0~ ~ 71 ~ PCT/EP91/01491
vascular permeability, comparable with the allergic
skin reaction following exposure to allergen.
Similarly, both the acute broncho-constriction and
chronic inflammatory reactions elicited by allergens in
asthma can be mimicked by intratracheal administration
of PAF. Accordingly agents which antagonise the
actions of PAF and, consequently also prevent mediator
release by PAF, will have clinical utility in the
treatment OL a variety o^ allergic and infla~matory
conditions such as asthma and arthritis, respectively.
In addition to the above, PAF has been implicated
as being involved in a number of other medical
conditions. Thus in circulatory shock, which is
characterised by systemic hypotension, pulmonary
hypertension and increased lung vascular permeability,
the symptoms can be mimicked by infusion of PAF. This
coupled with evidence showing that circulating PAF
levels are increased by endotoxin infusion indicate
that PAF is a prime mediator in certain forms of shock.
Intravenous infusion of PAF at doses of 20-200 pmol
-1 -1
kg min into rats results in the formation of
extensive haemorrhagic erosions in the gastric mucosa
and thus PAF is the most potent gastric ulcerogen yet
described whose endogenous release may underlie or
contribute to certain forms of gastric ulceration.
Psoriasis is an inflammatory and proliferative disease
~092/034~ ~ ~ 7 ~ A u PCT/EPgl/01491
characterised by skin lesions. PAF is pro-inflammatory
and has been isolated from lesioned scale of psoriatic
patients indicating PAF has a role in the disease of
psoriasis. And finally, increasing evidence supports a
potential pathophysiological role for PAF in
cardiovascular disease. Thus recent studies in angina
patients show PAF is rel2ased during atrial pacing and,
in pigs, intracoronary injection of PAF induces a
prolonged decr~ase in coronary riow wnile in guinea pig
hearts it induces regional shunting an~ ischaemia. PAF
has also been shown to initiate thrombus formation in a
i mesenteric artery preparation both when administered
exogenously and when released endogenously. More
recently PAF has been shown to play a role in brain
ischaemia induced in animal models of stroke.
Thus compounds of the invention, by virtue of
their ability to antagonise the actions of PAF, could
well be of value in the treatment of any of the above
conditions.
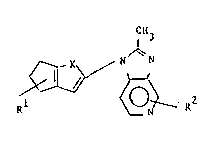
According to the invention there are provided
compounds of formula (I):
CH3
R~
W092/0~ ~ ~ J ~3 PCT/EP91/01491
wherein X is -CH-CH- or S;
Rl is -CooR3~ -CN, -CoNR4R5 or -NR6CoR3
where R3 is H, Cl-C6 alkyl, C3-C7 cycloalkyl, or
Cl-C4 alkyl substituted by phenyl;
:~ R4 and R5 are each independently H, Cl-c6
alkyl which may optionally be substituted by
a hydroxyl or Cl-C~ alkoxy group, C3-C7
cycloalkyl, phenyl or pyridyl/ said phenyl
: and pyridyl groups being optionally
substituted by up to three groups
independently selected from halo, hydroxy,
-CF3, Cl-C4 alkyl and Cl-C4 alkoxy, or R4 and
R5 together with the nitrogen atom to which
they are attached represent a pyrrolidine,
piperidine or morpholine ring; R6 is H or
Cl-C4 alkyl;
and R2 is H, halo, or Cl-C4 alkyl;
and their pharmaceutically acceptable salts.
In the definitions given herein the term halo
means fluoro, chloro, bromo or iodo. Alkyl and alkoxy
groups of 3 or more carbon atoms may be straight or
branched-chain.
R5, when present, is preferably H and Rl is
preferably at the 2-position of the indane nucleus
(when X is -CH=CH-) or the 5 position of the
cyclopentanothiophene nucleus (when X is -S-).
;,' '
~092/0~34 2 ~3 8 ~ f 1 v~ PCT/EP91/01491
Examples of Rl groups are cyano, carboxy,
ethoxycarbonyl, t-butylcarbonylamino and group -CoNR4R5
where R4 and R5 together form a piperidino or
morpholino group or R4 and R5 are independently
selected from H, t-butyl, cyclopentyl, cyclohexyl,
methyl, ethyl, propyl, isopropyl, pyridyl and pyridyl
substituted by a methyl group. A particularly
preferred compound is 2-(2-met~.ylimid2-o[~,5-c]pyrid-
l-yl)-5-(N-pyrid-2-yl-carbamoyl)-cyclopentano[b]-
thiophene. Other particula ly preferred c_mpounds ar2:
2-(N,N-dipropylcarbamoyl)-5-(2-methylimidazo-
[4,5-c]pyrid-l-yl)indane and 2-(N,N-dicyclopentyl-
carbamoyl)-5-(2-methylimidazo[4,5-c]pyrid-l-yl)indane.
The compounds of formula (I) contain at least one
asymmetric centre and will therefore exist as one or
more pairs of enantiomers, and such pairs of individual
isomers may be separable by physical methods, e.g. by
fractional crystallisation or chromatography of the
parent compounds or of a suitable salt or derivative
thereof. Alternatively, particular isomers may be
prepared using the corresponding optical isomers of the
precursors used in preparation of compounds of the
invention. The invention includes all the enantiomers
of the compounds of formula (I) whether separated or
not.
The pharmaceutically acceptable acid addition
sa1ts of the compounds of formu1a (I) are those formed
'' '
W092/0~ ~ 2 ~ PCT/EP91/01491
from acids which form non-toxic acid addition salts,
for example the hydrochloride, hydrobromide, sulphate
or bisulphate, phosphate or acid phosphate, acetate,
~` citrate, fumarate, gluconate, lactate, maleate,
succinatP, tartrate, methanesulphonate,
benzenesulphonate and p~toluenesulphonate.
~ hen ~ is -C~=CH- and Rl is CN, the compound of
formula (I) may oe o~talned by cyclising the
corresponding compo-~nd of formula (I~), for example by
treating i~ h ac2_i- arhydrid2 in the presence of
acetic acid:
~ (CH~CO)20 ~ CH3
CN N 3 2H CN N N
R2 ~ NH2 ~ R2
(II)
This reaction may be carried out by heating the
compound (II) with acetic anhydride and glacial acetic
acid, followed by separation of t~e product by
conventional methods.
' `
" , ~.
`~092/0~ ~ ~ PCT/EPg1/01491
: The corresponding compound (III) may be obtained
by hydrolysing compound (I) in which Rl is CN and X i5
-CH=CH-, generally in the presence of an alkali,
followed by acidification:
C ~ C ~R
( II I )
Compound (III) may be esterified by conventional
.. methods to produce corresponding compounds in which R3
: is Cl-C6 alkyl, C3-C7 cycloalkyl or Cl-C4 alkyl
substituted by phenyl.
:, When Rl is CoNR4R5 and X is -CH=CH- the compound
of formula (I) may be prepared from the acid (III) by
converting the latter to the corresponding carbonyl
chloride, for-example by treatment with oxalyl
chloride, and treating the product with the appropriate
amine, or sodium salt of the amine:
' "
:' :
WO 92/03434 2 ~ ~ 1 7 i ~ PCr/EP9l/0l491
3 ~ /~N
CO2H ~R COCl ~,~
NHP~4?~5 or ~a NR R
(III)
~ ~)\ N N
R~ R
~
~N~
When R1 is NR6CoR3 the acid (III) may first be
converted to a corresponding compound (IV~ in which
is NH2, for example by treatment with
diphenylphosphorylazide:
` '
,: , .
2~3~
V092/0~ ~ PCT/EP91/01491
N N R2
~ ~
(III)
(Ph) 2P (o)h3
: .
~ ~ 3
NH2 N N
~ N
(IV)
.~ The amine (IV) may then be converted to the
~, carbonylamino compound (V) in which R6 is H by reaction
: `
with the appropria~e carbonyl chloride: ~
.
''.',
.
. ~ :
., .
: .,
WO 92~03434 2 ~ ~ 7 7 ~L ~ PCr/EPgl/0l491
NH2 N N
(I~l) }o/~
"3COCl
/--~1
R -- C--N N
O )--J~ R
~)~
N
Compounds in which R6 is alkyl may be made by
alkylating compounds of formula (V) in known manner.
The novel intermediates (IV) constitute a further
aspect of the present invention.
The compounds of formula (II) may be prepared from
the corresponding cyanoindanes by the synthesis shown
in Scheme 1. This synthesis entails nitration of the
cyanoindane, for example with fuming nitric acid in
acetic anhydride, followed by reduction of the nitro
group, suitably with hydrogen in the presence of a
.
.: ;
. .
;, ' '"' " ' ' "' '" ~ ' ''
:.
2 ~ ~3 r~
:-~092/034~ PCTtEP91/Ot491
palladium/carbon catalyst. The amine so formed may
then be reacted with the appropriate
chloro-nitropyridine derivative and the nitro group in
the compound produced is reduced catalytically to give
the compound of formula (II).
Scheme l
~ r~ ~ 1 HNO3 ~ NO H2 Pd/C
CN
NH2 ~, CN 2 ~`'`~2 ~ ï
R ~J R ~0
'
` `;
(Tl)
;~11
;i~ C~ 2
~ ,
.
~g~7~ ~
W092/0~ ~ PCT/EP91/01491
12
When X is -S- and Rl is -CooR3 the cyclopentano
[b]thiophenes of formula (I) may be made firstly by
reacting a 3-cyclopentanone carboxylic ester with
sulphur and a cyanoacetate to form a cyclopentano[b]
thiophene substituted by amino and carboxylate groups
of formula (VI):
R 02C ~ O + S + NC5H"CO,,R
~ ~ C02R
R302C~ S ~
In the above formulae group R7 is an optionally
substituted alkyl group such as ethyl or t-butyl. The
reaction may be conducted by heating the reagents
together in the presence of a solvent such as
dimethylformamide and a base such as triethylamine.
The compound of formula tVI) may then be reacted
with 4-chloro-3-nitropyridine in the presence of a base
such as sodium bicarbonate, followed by reduction with
hydrogen and a palladium catalvst to form a compound of
formula (VII) below, ring closure with acetic anhydride
and removal of the carboxylate group on the thiophene
ring for example if R7 = t-butvl b~ treatment with a
.. ~
~V092/034~ ~ PCT/EP91/01491
strong acid such as trifluoroacetic acid followed by
heating with copper in the presence of quinoline.
These process steps are illustrated by Scheme 2
below:
Scheme 2
R 02C ~ ' Z ~ / 2,
(vI)
~ C02R C02R
R302C Y li2, Pd/CR 02C ~ SN ~ N
~ No2 (VII)
~ / Nii2
(Cll CO) o
3 COOI~
~ 7
R302C ~ ~ ~
.: ,)~ ,/' /
_ N
' .
~ . .
2~87'f ~ ~
WO 92/03434 PCI'/EP91/01491
14
C0211 ~R o2c~ 3
N 1~ Cu t quin~ c \-J
- : ' , ., ,. ~ , . ,' . , :
~092/034~ ~ 3 i 7 ~ J PCT/EPgl/014gl
The corresponding compounds of formula (I) in
which R3 is H may be obtained by hydrolysing the ester
produced by the synthesis o~ Scheme 2 by conventional
methods. The compounds of formula (I) in which X is
-S- and Rl is CoNR4R5 or NR6CoR3 may be prepared from
those in which R1 is C02H by methods analogous to those
described above for compounds in which X is -CH=CH-.
The acid in which Rl is C02H and X is S may be
converted to the corresponding nitrile by conventional
methods.
In the above synthesis of compound (VI) the Co2R3
group may be attached to the cyclopentane ring of
compound (VI) at either the 4- or the 5-position of the
cyclopentano~b]thiophene and in practice a mixture of
both isomers is produced. These isomers may be
separated by conventional methods, alternatively the
further synthetic steps for obtaining compounds of
formula (I) may be carried out on the mixture and the
mixture of end products separated.
The activity of the compounds of the invention is
shown by their ability to inhibit the platelet
aggregating activity of PAF ~n vitro. Testing is
performed as follows:
Blood samples are taken from either rabbit or man
into 0.1 vol disodium ethylenediamine tetraacetic acid
buffer and the samples centrifuged for 15 minutes to
"
. :" '
:, ............................................................ '
20~7'Y:~
W092/03434 PcT/Ep9l/ol49
16
obtain platelet rich plasma. The plasma is ~urther
centrifuged to give a platelet pellet which i9 washed
with a buffer solution (4mM KH2P04, 6mM Na2HP04, 100 mM
NaC1, 0.1% glucose and 0.1% bovine serum albumin, pH
7.25) and finally resuspended in buf~er solution to a
concentration of 2 x 1o8 platelets/ml. A sample (0.5
ml) is pre-incubat~d fcr two minut2s at 37C in a Paton
aggregometer with stirring, eithe_ with v~hlc~e alone,
or with vehicle containing the particular compound
under test. PAF is add~d at a su~^ ci~n~ ~oncen-rGtion
to give a maximum aggregating response in the absence
of test compound (lo 8 to 10 9 molar), and the platelet
aggregation is measured by following the increase in
light transmission of the solution. The experiment is
repeated in the presence of test compound at a range of
- concentrations and the concentration of compound
required to reduce the response to 50% of its maxiumum
value is recorded as the IC50 value.
The activity of the compounds of formula (I) is
also demonstrated in v'vo by their ability to protect
mice from the lethal effect of an injection of PAF. A
mixture of PAF (50 ~g~kg) and DL-propranolol (5 mg/kg)
in 0.9% w/v sodium chloride is injected tO.2 ml) via a
tail vein immediately prior to the PAF/propranolol
injection or administered orally by gavage two hours
earlier. The compounds are tested at several doses in
groups of 5 mice and the dose which reduc2s mortality
to 50% is recorded as the PD50 value.
.
' ' ' ~ ' .' ..
.
..
~092~03434 2 ~3 3 7 ~ PCT/EP91/01491
For therapeutic use the compounds of the formula
(I) will generally be administered in admixture with a
pharmaceutical carrier selected with regard to the
intended route of administration and standard
pharmaceutical practice. For example, the~ may be
administered orally in the form of tablets containing
such excipients as starch or lactose, or in capsules or
ovules either alone CL in admi~t~ ith excipients, or
in the form of elixirs or suspensions containing
flavouring or colouring agents. The~ may be injec.eà
parenterally, for example, intravenously,
intramuscularly or subcutaneously. For parenteral
j administration, they are best used in the form of a
sterile aqueous solution which may contain other
substances, for example, enough salts or glucose to
make the solution isotonic with blood.
For administration to man in the curative ar
` prophylactic treatment of allergic bronchial conditions
and arthritis, oral dosage of the compounds will
generally be in the range of from 2-lO00 mg daily for
an average adult patient (70 kg). Thus for a typical
adult patient, individual tablets or capsules contain
from l to 500 mg of active compound, in a suitable
pharmaceutically acceptable vehicle or carrier.
Dosages for intravenous administration would typically
be within the range l to lO mg per single dose as
required. For the -reatment of a ergic and bronchial
,.. . .
,,,, ,j
W092/0~ ~ 2 0 ~ ~ 7 ~ ~ PCT/EP91/01491
18
hyper-reactive conditions, inhalation via a nebuliser
or aerosol may be the preferred route of drug
administration. Dose levels by this route would be
within the range O.l to 50 mg per single dose as
required. In practice the physician will determine the
actual dosage which will be most suitable for an
individual patient and it will vary with age, weight
and response of the particular patient. The above
dosages are exemplary of the average case but there
can, o. course, be individual instances where higher or
lower dosage ranges are merited, and such are within
the scope of this invention.
Thus in a further aspect the invention provides a
pharmaceutical composition comprising a compound of the
formula (I) or a pharmaceutically acceptable salt
thereof, together with a pharmaceutically acceptable
diluent or carrier.
The invention also includes a compound of the
formula (I) or a pharmaceutically acceptable salt
thereof, for use in medicine, in particular in the
treatment of allergic and inflammatory conditions in a
human being.
The preparation of the compounds of the invention
is ~urther illustrated by the following Examples.
~.
.
.. . . . ..
.' ' , , ,:
~ ~ ~ 7 7 ~ ~ PCr/EP91/01491
19
-ExamPle 1
2-cyano-5-~2-methylimidazo[4!5--c]pyri-d-l-yl~L~3
~(~> ~0 N~> I'd/C J~/
1~2N
Cl
`;~i . H2N~'~ ~ C~
E~OH N '
N~ CN
Ac 2 0 \tJ~,
~ N J
(a) Fuming nitric acid (40 ml, 1.5 g/ml) was added
dropwise with stirring to acetic anhydride (80 ml)
keeping the temperature of the ~ixture at 0C. After
the addition was complete 2-cyanoindane (11.33 g, 79.5
mmol, J. Chem. Soc. (B), (1969), 1197) was added
dropwise with stirring over 30 minutes maintaining the
reaction temperature between -5 and 0C. The mixture
was stirred for a further 15 minutes then poured onto
ice. The mixture was extracted with dichloromethane (4
x 150 ml~ and the extracts were washed with saturated
aqueous sodium bicarbonate t3 x 150 ml), dried (MgS04)
and concentrated under reduced pressure. The residue
was recrystallised from ethanol to give
2-cyano-S-nitro-indane (12.09 g, 81%) as a yellow
solid, m.p. 80-82C.
W092/03434 2 ~ 3 ~ 7 ~ ~ PCT/EP91/01491
H NMR (300 MHz, CDC13) ~ = 3.32-3.53 (5H, complex),
7.43 (lH,d, J =9Hz), 8.15 (lH,s), 8.17 (lH,d,J = 9Hz)
P.p.m.
(b) A solution of 2-cyano-5-nitro-indane (11.90 g,
63.3 mmol) in methanol/dichlorometnane = 1:1 (200 ml)
was hydrogenated over 10% palladium on charcoal (1.2 g)
at 30 p.s.i. and 20C for 5 hours. Tne ca~al ~st was
filtered off and the filtrate was concentrated under
reduced pressure to gi ve 5-amlno-2-cyano-ind2ne (10.4
g, ca quantitativ~) which was used ~ r~tly ^sr t~o
next reaction. A portion, recrystallised from ethanol,
formed pinkish needles, m.p. 73-76C.
H NMR (300 MHz, CDC13) ~ = 3.11-3.30 (SH, complex),
3.65 (2H, br s), 6.58 tlH,d,J=8Hz), 6.61 (lH,s), 7.03
(lH,d,J=8Hz~ p.p.m.
(c) 4-Chloro-3-nitropyridine (11.46 g, 72.3 mmol) was
added to a suspension of 5-amino-2-cyano-indane
(10.4 g, 65.7 mmol) in ethanol (150 ml) at room
temperature. The mixture was stirred overnight at room
temperature and then poured into excess ice-cold
aqueous ammonia. The yellow solid was filtered off,
partially digested in hot ethanol (150 ml), cooled, and
re-filtered to give 2-cyano-5-(3-nitropyrid-4-ylamino)-
indane (13.61 g, 74%).
~ , , , .:, :
'', '
~09~/0~ ~ ~ PC~/EP91/01491
21
H NMR (300 MHz, CDC13) ~ = 3.31-3.48 (5H, complex),
6.92 (lH,d,J=5Hz), 7.15(1H,d,J=6Hz), 7.19(1H,s),
7.37(1H,d,J=6Hz), 8.28(1H,d,H=5Hz), 9.31(1H,s),
9.64(lH, br s) p.p.m.
- (d) 2-Cyano-5-(3-nitropyrid-4-ylamino)indane (12.46
gm, 44.5 mmol) was suspended in methanol/
dichloromethane = 1:1 (750 ml) and hydrogenated at 20C
and 30 p.s.i. over 10% palladiu~ on charc~al (1.25 g)
for 2 hours. The catalys~ W25 filto-od CfC and the
filtrate was concentrated under reduced pressure to
give 5-(3-aminopyrid-4-ylamino)-2-cyano-indane (12.28
g, ca quantitative) as a yellow solid, m.p. 98-100C.
H NMR (300 MHz, dmso-d6) ~ = 3.03-3.35(4H,m), 3.54
(lH,p,J=7Hz), 4.10(1H, br s), 4.64(2H, br s),
6.84(1H,d,J=5Hz), 6.92(1H,dd,J=7 and 2Hz),
7.00(1H,d,J=2Hz), 7.18(1H,d,J=7Hz), 7.60(2H,d,J=5Hz),
7.84(lH,s)p.p.m.
-
(e) A mixture of 5-(3-aminopyrid-4-ylamino)-2-
cyano-indane (12.28 g, ca 44.5 mmol from step (d)
above), acetic acid (70 ml) and acetic anhydride (70
ml) was heated at reflux under nitrogen for 1.75 hours,
cooled, and concentrated under reduced pressure. The
residual brown gum was dissol~ed in 2N hydrochloric
2 ~ r! ~ ~ '
W092/0~ ~ PCT/EPg1/01491
acid (40 ml) and washed with ethyl acetate (50 ml).
The aqueous layer was rendered basic by the addition of
2N aqueous sodium hydroxide, and the product was
extracted into dichloromethane (4 x 50 ml). The
combined extracts were washed with water (50 ml), dried
(MgS04) and concentrated under reduced pressure.
The residue was purified by column chromatography
(silica gel, 60-200~), eluting with ethyl acetate/
methanol=7:1. Fractions containing product were
combined and concentrated to give a brown sum, which
was recrystallised from ethyl acetate/methanol, to give
the title compound as an off-white powder (9.674 g,
79%), ~.p. 174-176C.
Analysis %:-
Found; C,74.40; H,5.17; N,20.72.
C17H14N4 requires C,74.43; H,5.14; N,20.42.
'
Example 2
S-f2-Methylimidazo~4.5-clpyrid-1-yl)indan-2-
carboxylic acid
' ~ ô,l~ ~, NaOII ~ 11()2C~ ) I 3
- . ~
.~ .
~V092/0~ ~ 2 ~ ~ 7 7 1 ~ PCT/EP91/01491
23
A mixture of 2-cyano-5-(2-methylimidazo-
[4,5-c]pyrid-l-yl)-indane (Example 1, 739 mg, 2.70
mmol), 50% aqueous sodium hydroxide (l ml) and methanol
(6 ml) was heated at reflux under nitrogen for 9 hours,
cooled, poured onto ice, and the pH of the solution was
adjusted to pH5 by the addition of dilute hydrochloric
acid. The product precipitated as a white solid,
(426mg, 54%) which was filtered off and dried in vacuo
to sive the title compound, m.p. 264-267C.
Analysis %:-
Found: C,68.91; H,5.08; N,14.08.
Cl7H15N302.l/5 H20 requires C,68.76; H,5.23; N,14.15.
Example 3
Ethyl 5-~2-Methylimidazo[4.5-c~pyrid-1-yl)indane-
2-carboxylate
:,
C~3
EtO2C ~ N N
2 ~
WO 92/03434 PCT/EP91/0]491
24
Oxalyl chloride (1.3ml, 14.9mmol) was added
dropwise over 5 min to a stirred suspension of
5-(2-methylimidazo[4,5-c]pyrid-1-yl)indane-2-
carboxylic acid (Example 2, 1.46g, 5.0 mmol) in dry
dichloromethane (20 ml) under nitrogen at room
temperature. AftPr being stirred for lh, the resulting
solution was concentra.od unde- rPduced pressure,
redissolved in drv dichlorome~h2ne (~0 ml) ~n~ 2
mixture of ethanol (0.352 ml, 6.0 ~.ol) and
triethylamine ~0.69~ ml, 5.0 ~Lm_l) ~2S a~ec' at rsom
temperature. After a further lh, the solution was
washed with saturated aqueous sodium bicarbonate (30
ml), dried (MgS04), and concentrated under reduced
pressure. The residue was purified by flash
chromatography, eluting with ethyl acetate:methanol
=4:1 to give the title compound as a colourless gum,
1.4g (88%). The fumarate salt had m.p. 193-194C
(methanol).
:,,
Ar~alysis g~:-
Found: C,63.17; H,5.38; N,9.47
C1gH19N302.C4H402 requires; C,63.15; H,5.30; N,9.61
,: . :
.:
. - . . ' , ' , . :,,
~092/0~ ~ ~ ~ 7 ~ } !3 PCT/EP91/01491
Exam~le 4
2-(N-tert-Butvlcarbamoyl)-5- r 2-methylimida~o-
r4 5-clpyrid-1-yllindane
li O~C ~) CH~ ~ Ch ~ \ N)~'
C ~
5-~2-Nethylimidazo[4,5-c]pyrid-1-yl]indan-2-
carboxylic acid (Example 2, 587 mg, 2 mmoles) was
suspended in dry dichloromethane (15 ml). Oxalyl
chloride (760 mg, 6 mmoles) was added followed by
N,N-dimethylformamide (lO ~l). The mixture was stirred
at room temperature for 1 hour to give a pale yellow
: solution. The solvent was removed under reduced
;:.l pressure and the resulting 5-~2-methylimidazo~4,5-c]-
pyrid-l-yl]indan-2-carbonyl chloride was redissolved in
dichloromethane (15 ml). tert-Butylamine (438 mg, 6
mmoles) in dichloromethane (3 ml) was added over 5
minutes and the reaction mixture was stirred at room
temperature for l hour.
,
.` . :
~ .. , ,. : , . . -
2 ~ 8 1 ~ ~ ~
W092/0~ ~ PCT/EP91tO1491
26
Saturated sodium bicarbonate (20 ml) was added and
the mixture stirred at room temperature for 1 hour.
The dichloromethane layer was separated, dried over
magnesium sulphate and the solvent removed under
pressure. The residual foam was chromatographed over
silica gel (40-63 ~), eluting with 15% methanol in
ethyl acetate. Fractions containing product were
evaporated to give a foam (500 mgJ which was dissolved
in ether/ethyl acetate = 3:1 (20 ml), filtered and
allowed to crystallise. The precipitated solid was
filtered and dried in vacuo yielding the title compound
as a white solid (345 mg, 50%), m.p. lS2-154C.
Analysis %:
Found: C,71.99: H,7.07; N,15.82.
C21H24N40 requires C,72.39; H,6.94; N,16.08.
` .
ExamDles 5-14
The compounds of Table 1 were made by the method
of Example 3 using the appropriate nucleophile, and the
acid chloride derived from the acid described in
Example 2.
. . . ::.
. ! ;~ ................. , ;~.. . , :
'~ ', ' ' . , ', ' ' . ' '
' O 92/03434 '~ 7 7 1 ~ PCI/EP91/01491
27
Table 1
;,
~ CU3
2 (\~(
:,
~.,
.,
:i
., .
"'~ . ..
. ~
WO 92/03434 ~ ~ `3 ~ r3) i i~ PCI'/EP91/01491
28
. _
o ~ ~ C~ ~` 1 o ~ ,1
U~ ~ ~ L~ ~ O L'`, L')
~Z ~ ~ ~ O ~ ~1 ~/
U~
. ~ ~ a~ ~r L') t`~ q~ ~ ~
3 ~ L~) L) L. Li ~ .D L) It.
L'' ~ C: L'~ ~D ~ ;~
Y li~ L') r--1 ~J L~ ~ O Gl
1~ C-' ~3 ~ ~D ~3 '~ D ~
' .~ o'~ o`~ ~ o
~~ --
:` ~
~ L'~ ~D ~` CO
' ~ .~
', :~ ~ .
~ ,
, .
`
: .,
YO 92/03434 2 9 ~ o ~ 7 rl ~ ~3 PCI`/EP91/01491
` ' :
N ('7 ~ ~ N
~ D 1~ _1 ~15 ~ 1 Il~ r~ ~/
,.~, ~ ~ '.D ~D ~O ~D ~O 11~ ~
J~ ~ 5 L') ~ O I~ CO ~ ~ 1~
:` ,t: ~-i ~i C O L'~ L''5 -' ~1 O O
t~ r- ~ ~ ~o ~D ~ ~O ~
...__ _._ 0~
~ U.5 ~
~ ~ ~ ~ 0~ ~ Z~ ~ 0~
~ ~ ~ Z ~ ~ ~ _~
,.~ ~ ~ ;~ ~ ~ C~ C~ (~ t~ '
., ~ ~ ~ U ~ 1~
, :~5 ~ ~ ~ ~1 ~ ~
.~ .a~ 00 a~ ~o o r~
~ ~1
L~ ,~ N j ,.~ ~
. .
, ~` ' ~ ' '
. ' ~ ' . .
W O 92/03434 2 a ~ 7 7 1~ PC~r/EP91/01491
11 g '--I
N ~ j ~ _
_ _ _ _
N
:~' _ _
~' ~ ~
.~ ____
~ ~r
._ ._ ._ . _ ._ ._
~:" ~ ~
[~ ~
' ' ~
:' ' `' :'` ':
2~'7 1~
V092/0~34 PCT/EP91/01491
Exam~le 15
(2R,2's~ and (2s.2's) N-(2-Hydroxy~ y~pentyl)
-5-t2-methvlimidazo~4.5-c]pyrid-1-yl)indane-2-
carboxamide
~ 0~_ C
A solution of 5-(2-methylimidazo[4,5-c]pyrid-
l-yl)indane-2-carbonyl chloride (Example 4, 0.93g, 3.0
mmol) in dry dichloromethane (20 ml) was prepared. A
mixture of triethylamine (0.42 ml, 3.0 mmol) and
S-l-amino-2-hydroxy-4-methylpentane (350 mg, 3.0 mmol)
was added with stirring at room temperature. After
13h, the mixture was treated with saturated aqueous
sodium bicarbonate (30 ml) and the products were
extracted into dichloromethane (3 x 30 ml). The
combined extracts were dried (MgS04), concentrated
under reduced pressure and purified by flash
chromatography (gradient elution with ethyl
acetate/methanol/diethylamine) to afford two main
product fractions.
:.
,
.
.
.,- ,
w092/o3434 ~3~7 ~ '3 PCr/EP91/01491 ~
32
The faster running diast~ aomer (Rf = 0.25,
EtOAc/MeOH = 3:1) was a light brown foam, 360 mg (31%),
; m.p. 67C.
H NMR (300 ~Hz, CDC13) ~ = 0.87 (3H,d,J=6Hz), 0.91
(3H,d,J-6Hz), 1.22 (lH,m), 1.39 (lH,m), 1.71 (lH,m~,
2.48 (3H,s), 3.10 (lH,m), 3.26 (5H,m), 3.53 (lH,m),
3.80 (lH,m), 6.11 (l-X, br s), 7.02 (lH,d,J=2~Z), 7.06
(lH,d,J=8Hz), 7.10 (lH,s), 7.35 (lH,d,J=3Hz), 8.30
(lH,d,J=5Hz), 8.97 ~ln~s~ p.p.m.
Th~ slower runnin~ diastereomer (~f = 0.06,
EtOAc/MeOH = 3:1) was repurified by flash
chromatography tgradient elution with dichloromethane/
methanol) to give a brown solid (324mg, 28%), m.p.
102-C.
H NMR (300 MHz, CDC13) ~ = 0.86 (3H,d,J=6Hz), 0.90
(3H,d,J=6Hz), 1.22 (lH,m), 1.66 (2H,m), 2.47 (3H,s),
3.06-3.48 (7H, complex), 3.79 tlH,m), 7.06 (3H,m), 7.35
(lH,d,J=8Hz), 8.30 (lH,d,J=5Hz), 8;97 (lH,s), p.p.m.
~' : ': . : -
"
-W092/0~ ~ ~ ~ 7 r ~ ~ PCT/EP91/01491
- 33
Example 16
N-CYclohexyl-N-(2-propYl~-5-(2-methylimidazo[4 5-c]
~vrid-1-Yl)indane-2-carboxamide
A mixture of cyclohexylisopropylamine (0.56g, 4.0
mmol) and sodium hydride (163mg, 60% dispersion in oil,
4.0 mmol) in 8ml dry tetrahydrofuran was stirred at
room temperature under nitrogen for 15 mins. A
suspension of 5-(2-methylimidazo[4,5-c]pyrid-1-yl)
indane-2-carbonyl chloride ~Example 4, 0.64g, 2.0 mmol)
in dry tetrahydrofuran (8ml) was added, and the mixture
was stirred at room temperature for 2h. The solvent
was removed under reduced pressure, and the residue
partitioned between dichloromethane and saturated
aqueous sodium bicarbonate. The organic layer was
separated, dried ~MgS04), and concentrated under
reduced pressure. The residue was purified by flash
.. . .
.: .
.
,~ .
' ~
.
W092/034~ ~ 3 ~ PCT/EP91/01491
chromatography (gradient elution with dichloromethane/
methanol) followed by chromatography on silica gel
(20-40~), eluting with ethyl acetate: methanol:conc.
aqueous ammonia = 94:5:1, to give the title compound,
35 mg (4~). m.p. (fumarate salt) 100C.
H NMR (300 MHz, dmso-d6) ~ = 0.95-1.39 (4H,m), 1.13
(3H,d,J=6Hz), 1.23 (3H,d,J=6Hz), 1.45-1.70 (6H,m), 2.39
(3H,s), 3.02-3.4S (5H,m), 3.63 (2H,m), 6.52 (2H,s),
7.09 (lH,d,J=5Hz), 7.23 (lH,d,J=8Hz), 7.31 (lH,s), 7.37
(lH,d,J=8Hz), 8.20 (lH,d,J=5Hz), 8.81 (lH,s) p.p.m.
Analysis % -
Found: C, 64.78; H, 6.48; N, 8.80
C27H32N40.3/4 fumarate C, 64.80; H, 6.42; N, 9.16%
H20 requires:
Example 17
N.N-Dicyclopentyl-5-(2-methylimidazo r 4.5-c~pyrid-1-yl)-
indane-2-carboxamide
,
, .
,
':
Y092/034~ 2 ~ !~ 7 7 ~ ~ PCT/EP91/01491
A mixture of dicyclopentylamine (306 mg, 2.0 mmol)
and sodium hydride (80 mg, 60% dispersion in oil, 2.0
mmol) in 8 ml of dry tetrahydrofuran was stirred under
nitrogen at room temperature for lS minutes. A
suspension of 5-(2-methylimidazo[4,5-c]pyrid-l-
yl)indane-2-carbonyl chloride (Example 4, 312 mg, l.0
mmol) in dry tetrahydrofuran (4 ml) was added and the
mixture was stirred for 18 hours. The solvent was
removed under reduced pressure, and the residue was
dissolved in dichloromethane (50 ml) and washed with
saturated aqueous sodium bicarbonate (30 ml). The
organic solution was dried (MgS04) and concentrated
under reduced pressure. The residue was purified by
flash chromatography (eluting with ethyl
acetate/methanol = 4:l) followed by preparative
h.p.l.c. (Cl8 silanised silica, eluting with
methanol/water = 9:l). The product was isolated as its
fumarate salt, a white solid (38 mg, 9%) m.p. 133C.
Analysis %:-
Found: C,64.78; H,6.48; N,8.80;
; C27H32N40.3~2 C4H404.l/2 H20: C,64 80; H,6.42; N,9.16.
~,,
.~ .
~: .
' ' `
.
wo g2/03434 ; ~ 3 I r~ PCI/EP91/014gl
36
Example 18
(a) 2-Amino-5-(2-methylimidaZo[4~5-c~pyrid-l-ylLiDaan~
;
N a > 1(,~ ~J ~
C~ N
~O~
5- [ 2-Methvlimi dazo~4,5-c]~r~_d-l~yl~indan-2-
carboxylic acid (Example 2, 1.17 g, 4 mmoles) was
suspended in toluene (8 ml). Triethylamine (444 mg,
4.4 mmoles) and diphenylphosphoryl azide (1.21 g, 4.4
mmoles) were added and the mixture was heated at 100C
for 20 minutes. The reaction mixture was cooled and
treated with lM sodium hydroxide (30 ml). The aqueous
phase was separated and washed with toluene (30 ml),
neutralised (pH7) with 2M hydrochloric acid and
extracted with dichloromethane (10 x 60 ml). The
extracts were combined and dried (MgS04) and the
solvent removed 'n vacuo. The product was
chromatographed over silica gel t40-63~) eluting with
diethylamine:methanol:ethyl acetate = 5:10:85.
Fractions containing product were evaporate~, yielding
the title compound as an off-white foam, (870 mg, 82%).
.
''," '' '' ' ''' ''`' ' ', " ' , '
~ ~ ~ 7 rl 1~
` V092/034~ PCT/EPg1/01491
lH NMR (300 MHz), CDC13), ~ = 2.00(2H,br),
2.54(3H,s), 2.82(2H,m), 3.21(2H,m), 3.96(lH,m),
7.05-7.21(3H,m), 7.39(1H,d,J=6Hæ), 8.36(1H,d,J-4Hz),
9.06(lH,s), p.p.m.
,...
(b) 2-ftert-Butvlcar~onvlamino)-5-(2-methylimidazo-
r 4 5-clpvrid-1-yl)indane
'tC~,_ N ~--~--) ca3
~ N ~)
2-Amino-5-(2-methylimidazo[4,5-c]pyrid-1-yl]indane
(264 mg, 1 mmole) was dissolved in dichloromethane (5
ml) and pivaloyl chloride (132 mg, 1.1 mmole) and
triethylamine (111 mg, 1.1 mmole) were added. The
reaction was stirred at room temperature for 2 hours.
The dichloromethane solution was washed with saturated
sodium ~icarbonate tl ml), dried (MgS04), and the
solvent removed In vacuo. The crude product was
chromatographed over silica gel (~0-63 ~) eluting with
diethylamine:methanol:ethyl acetate = 3:3:94, and the
fractions containing product were evaporated.
20~771~
W092/03434 PCT/EP91/01491
38
The foam obtained (210 mg) was dissolved in
methanol (5 ml), fumaric acid (70 mg) added and the
solvent removed in vacuo. The resulting white foam was
then sonicated in a mixture of ethyl acetate/ether =
1:3 (10 ml). The white solid was filtered off and
dried, yielding the title compound as the fumarate salt
(92 mg, 20~), m.p. 110-120C.
Analvsis %:-
Found C,64.32; H,6.42; N,11.71;
C21H24N40.C4H404 re~uires C,64.64; H,6.08; N,12.06.
. .
~;
. ~ ~
.
,
2~ i'7~
:'092/034~. PCr/EP91/01491
39
Example 19
5-Ethoxvcarbonyl-2-(2-methvlimidazor4,5-cl~Yrid-1-Yl)
-cvclopentanorblthiophene and 6-ethoxvcarbonY1-2-(2-
methylimidazo-r4,5-cl~vrid-1-yl)cYclo~entano!bl
thiophene
O CH3
~ ~ ' EtO2C ~ CH3
CO2~CH3
~NH 3
,, 2Et
Cl
~,~2 CH3 ~CH3
'. 2) H," Pd/C ~( C02Et
3) HOAc Ac2O
1~
.
''~
W092/034~ ~ PCT/EPgl/01491
` 40
(a) Ethyl 3-cyclopentanonecarboxylate (10 g, 65 mmol),
t-butyl cyanoacetate (9.4 g, 65 mmol), sulphur ~2.15 g,
65 mmol) and triethylamine (6.58 g, 65 mmol) were
heated togeiher in dimethyl-formamide at 100C for 4
; hours. The solvent was removed under reduced pressure,
the residue was dissolved in ethyl acetate (50 ml),
washed with watPr (~ x 20 ml), drieà (MgS04) and
concentrated under reduced pressure. The residue was
purified by flash chromatograph~ ~gr2di~nt ~lution with
hexane/ethvl acetat~ ~o give a ~.ixtu~e of 2-amino-3-t-
butoxycarbonyl-5-ethoxycarbonyl-cyclopentano[b]-
thiophene and 2-amino-3-t-butoxycarbonyl-6-ethoxy-
carbonylcyclopentano~b~thiophene (5.0 g, 25%).
'''
lH NMR (300 MHz, CDC13) ~ = 1.15(3H,t,J = 7Hz),
1.5(9H,s), 2.6-3.4 (5H,m), 4.2(2H,q,J = 7Hz) p.p.m.
,
: .
` (b) The aminothiophenes (5.0 g, 16 mmol) from (a)
above, 4-chloro-3-nitropyridine (2.55 g, 16 mmol) and
:,,
sodium bicarbonate (1.35 g, 16 mmol) were stirred
together in ethanol (50 ml) under nitrogen for 48
hours. The mixture was concentrated under reduced
pressure and partitioned between ethyl acetate and
water. The organic phase was separated, dried (MgS04)
and concentrated under reduced pressure to give a
residue which was purified by flash chromatography
,..... ~
2 OJ 7 i i ~
V092/03434 PCT/EP91/01491
` 41
(gradient elution with dichloromethane/methanol) to
give a mixture of 3-t-butoxycarbonyl-5-ethoxycarbonyl-
2-(3-nitropyrid-4-ylamino)cyclopentano~b]thiophene and
3-t-butoxy-carbonyl-6-ethoxycarbonyl-2-(3-nitropyrid-
4-ylamino)cyclopentano~b]thiophene (2.6 g, 37%).
H N~R (300 ~Hz, CDC13)
5 isomer: ~ = 1.3(3H,t,J=7~7), 1.6(9H,s), 3.4(4H,m),
3.7(1H,m), 4.25(2X,q,J = 7X-), 7.6(1~,d,J = 5Hz),
8.5(1H,d,J = 5~ ), 9.~ ,s)p.p.m.
~,
4 isomer: ~ = 1.3(3H,t,J = 7Hz), 1.6(9H,s), 2.7(2H,q),
3.0(2H,m), 4.1(1H,t,J = 5Hz), 7.6(1H,d,J = 5Hz),
8.5(1H,d,J = 5Hz), 9.4(1H,s) p.p.m.
(c) The aminonitropyridines (2.5 g, 5.8 mmol) (from
(b) above) were dissolved in ethanol (50 ml) and
hydrogenated at 50 p.s.i. and 20C over 10% palladium
on carbon (250 mg) for 3 hours. The catalyst was
filtered off and the filtrate concentrated under
reduced pressure to give a mixture of 2-(3-aminopyrid-
4-ylamino)-3-t-butoxycarbonyl-5-ethoxy-carbonylcyclo-
pentano~b]thiophene and 2-(3-aminopyrid-4-ylamino)-
3-t-butoxycarbonyl-6-ethoxycarbonyl-cyclopentano-
~b~thiophene (2.1 g, 90%).
. .
.. . .
.. . .
~ 17~i
W092/034~ PCT/EP91/01491
H NMR (300 MHz, CDC13).
5-isomer: ~ = 1.3(3H,t,J = 7Hz), 1.6(9H,s), 1.8(2H,br
s), 3.1-3.4(4~,m), 3.65(1H,m), 4.2(2H,q), 7.4~1H,d,J =
5Hz), 8.1(1H,d,J = 5Hz), 8.13(1H,s), 10.06(1H, br s)
P-p.m.
4-isomer: ~ = 1.3(3H,t,J = 7Hz), 1.60(9H,s),
2.70(2H,m), 3.00(2H,m), 3.56(2H,br s), 4.05(1H,m),
4.20(2H,q J = 7Hz), 7.42(1H,d,J = 5Hz), 8.12(1H,d, J =
5HZ), 8.13(lH,s), 10.12(1H, br s~.
(d) The diaminopyridines (2.1 g, 5.2 mmol) (from (c)
above), were heated in a mixture of acetic acid (S0 ml)
and acetic anhydride (50 ml) at reflux for 1 hour. The
~; excess of reagents was removed under reduced pressure
and the residue was dissolved in ethyl acetate (50 ml)
amd washed with saturated aqueous sodium bicarbonate (2
~ x 30 ml). The organic layer was separated, dried
i (MgS04~, and concentrated under reduced pressure and
:;
the residue was purified by flash chromatography
~3 (gradient elution with dichloromethane/methanol) to
give a mixture of 3-t-butoxycarbonyl-5-ethoxycarbonyl-
- 2-(2-methylimidazo[4,5-c]pyrid-1-yl)cyclopentano-
~b~thiophene and 3-t-butoxycarbonyl-6-ethoxycarbonyl-
2-(2-methyl-imidazo~4,5-c]pyrid-1-yl)cyclopentano~b]
thiophene (1.5 g, 68%).
2~r~ 7 ~
V092/034~ PCT/EP91/01491
43
lH NMR (300 MHz, CDC13).
5 isomer: ~ = l.OO(9H,s~, 1.28(3H,t,J = 7Hz),
2.48(3H,s), 3.40(5H,m), 4.2(2H,q,J = 7Hz), 7.05(1H,d,J
= 5Hz), 8.32(1H,d,J = 5Hz), 8.95(1H,s)p.p.m.
4 isomer: ~ = l.OO(9H,s), 1.28(3H,t,J = 7Hz),
2.45(3H,s), 2.7-3.2(4H,m), 4.2(2H,q,J = 7Hz, and lH,m),
7.00(1H,d,J = SHz), 8.35(1H,d,J = SHz),
8.95(1H,s)p.p.m.
; (e) The t-butylesters (1.5 g, 3.5 mmol) (from (d)
above) were dissolved in trifluoroacetic acid and
stirred at room temperature for 1 hour. The solvent
was removed under reduced pressure, and the residue
partitioned between ethyl acetate and saturated aqueous
sodium bicarbonate. The aqueous phase was washed with
ethyl acetate and acidified with acetic acid, and
extracted with ethyl acetate. The combined extracts
were dried (MgS04) and concentrated under reduced
pressure to give a mixture of 5-ethoxycarbonyl-2
(2-methylimidazo~4,5-c]pyrid-l-yl)cyclopentano~b]
thiophene-3-carboxylic acid and 6-ethoxycarbonyl-2-(2-
methylimidazo[4~5-c]pyrid-l-yl)cyclopentano~b]
thiophene-3-carboxylic acid (1.1 g, 84~).
.. . . .
,
.. . .
~ & ~ 7 '~
W092/034~ PCT/EPgl/01491
- 44
H N~R (300 MHz, CDC13).
5 isomer: ~ = 1.3(3H,t,J = 7Hz), 2.62(3H,s),
3.40(4H,m), 3.75(1H,m), 7.41(1H,d), 8.40(1~,d),
8.82(lH,s) p.p.m.
4 isomer: ~ = 1.3(3H,t,J = 7Hz), 2.60(3H,s),
2.80(2H,m), 3.2(7H,m), ~.2(2;~,q,J = 7~z and lH,m),
7.43(1H,d), 8.40(1X,d!, 8.82(1H,s~ p.p.m.
(f) The carbcxylic 2s~ds (lOO mg, 0.~? m~ol) (from (e)
above), were dissolved in quinoline (1 ml) and copper
powder (50 mg) was added. The mixture was heated under
reflux for 1 hour, cooled, poured into water (10 ml)
and extracted with ethyl acetate (3 x 15 ml). The
combined extracts were washed with concentrated aqueous
ammonia (20 ml) and water (20 ml), dried (MgSO4) and
concentrated under reduced pressure. The residue was
purified by flash chromatography (gradient elution with
dichloromethane/methanol) to give a mixture of
5-ethoxycarbonyl-2-(2-methylimidazo[4,5-c]pyrid-1-yl)-
cyclopentano[d]thiophene and 6-ethoxycarbonyl-2-
(2-methyl-imidazo~4,5-c]pyrid-1-yl)cyclopentano~b]
thiophene (40 mg, 45%).
The hydrochloride salt had m.p. 210-220C.
~'~092/0~ ~ ~1 3 7 7 ~ ~ PCT/EP9ltO1491
Analysis %:
Found: C,55.28; H,5.19; N,11.24.
C17H17N302S.HCl.1/4 H20 requires C,55.43; H,5.02;
N,11.41%.
lH NMR (DMSO-d~)
5-isomer; ~ = 1.3(3X,t), 2.5t3H,s), 3.2(4H,m),
`~ 3.8(1H,m), 4.15(4H,q~, 7.8(1H,d!, 8.6!1H,d), 9.4(1H,s)
P-p-m-
4-isomer: ~ = 1.3(3H,t), 2.6(3H,s), 3.0(2H,m),
2.8(2H,m), 4.15(4H,q), 4.40(1H,t), 7.8(1H,d),
8.6(1H,d), 9.4(1H,s) p.p.m.
`, '
Example 20
2- r 2-Methylimidazo(4 5-c)pyrid-1-vll
cvclo~entano~blthio~hene 5 and 6-car~oxvlic acids
CH CH OC ~ ~
3 2 SN N NaOH ~ CH3
aq ethanol N ~ N
N
+ ~ 3
'r: ` ~ C ~
wo 92/03434 ~ 7 ~ ~ PCT~EP91/01491
46
A mixture of 5-ethoxycarbonyl-2-(2-methylimidazo-
[4,5-c]-pyrid-1-yl)cyclopentano[b]thiophene and
6-ethoxycarbonyl-2-(2-methylimidazo[4,5-c]pyrid-1-yl)-
cyclopentano[b]thiophene (Example 18, 3S0 mg, 1.07
mmoles) was dissolved in a mixture of ethanol (3 ml)
and sodium hydroxide (80 mg, 2.0 mmole) in water (2 ml)
was added. The mixture was stirred for 3 hours at room
temperature then evaporated to low volume ln vacuo and
acidified to pH 4.5 with acetic acid. Continuous
extraction with CH2C12 gave the product (300 mg, 93%),
m.p. 320C.
Analysis %:-
Found: C,60.58: H,4.21: N,ll.01.
C15H13N302S requires C,60.20: H,4.35: N,10.70.
Example 21
2-(2-Methylimidazo r 4.5-clpyrid-1-yl)-5-t4-morpholino-
carbonvl)cyclo~entanorb~thiophene and 2-~2-
methylimidazor4.5-clpyrid-1-yl)-6-(4-mor~holino
carbonyl)cyclopentano~b]thiophene
A mixture of 2-~2-methylimidazo[4,5-c]pyrid-1-yl)-
cyclopentano~b]thiophene-5-carboxylic acid and
2-(2-methylimidazo[4,5-c]pyrid-1-yl)cyclopentano[b]
thiophene-4-carboxylic acid (100 mg, 0.33 mmol) was
stirred together with a solution of oxalyl chloride (50
~1, 0.57 mmol) and dimethyl formamide (1 drop) in dry
dichloromethane (5 ml) at room temperature for 1 hour.
~ ~ 3 ;~ r~
V092/034~ PCT/EP9l/01491
The solution was concentrated under reduced pressure
and the residue dried in vacuo. The acid chloride thus
formed was redissolved in dry dichloromethane ~3 ml)
and added to a solution of morpholine (300 mg, 3.45
mmol) in dry dichloromethane (5 ml) at 0C. The
mixture was allowed to warm to room temperature and was
stirred for a further 1 hour. The mixture was poured
into water (20 ml), and the aqueous phase was extracted
with dichloromethane (3 x 10 ml). The combined
extracts were dried (MgS04), concentrated under reduced
pressure, and the residue was purified by flash
chromatography (gradient elution with
dichloromethane/methanol) to give the title compounds
(45 mg, 37~)-
Analvsis %:- (as fumarate salt, m.p. 232-240C).
Found: C,56.96: H,5.13; N,11.45.
C1gH2oN402S.C4H404 requires C,57.02; H,4.96; N,ll.S7.
Exam~les 22 and 23
The compounds of Table 2 were made by the method
of Example 21 using diethylamine and 2-aminopyridine
instead of morpholine.
WO 92/03434 2 ~ 3 ~ ri ~ ' PCI'/EP91/01491
4~
il ~ r ~ ~l ~n
.