Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
Wo 92/08796 PCr/US91/0X442
2~96222
TITl F.
Bifi.n~ion~l Select~hle Fusion Genes
s
PsACK(~.ROUNn OF THF ~NVFNTION
The present invention relates generally to genes expressing selectable
phenotypes. More particularly, the present invention relates to genes capable of co-
e~ s~ g both ~1O. . ~ n~ positive selert~ble and negative select~hle phenoty-pes.
Genes which e~press a select~ble phenotype are widely used in recombinant
DNA technology as a means for idenlirying and i~ ting host cells into which the gene
has been introduced. Typically, the gene e~p~ssing the selectable phenotype is
introduced into the host cell as part of a recombinant expression vector. Positive
selectable genes provide a means to identify and/or isolate cells that have retained
15 introduced genes in a stable foIm, and, in this capacity, have greatly facilitated gene
transfer and the analysis of gene function. Negative selectable genes, on the other
hand, provide a means for el;...i~ ;.-g cells that retain the introduced gene.
A variety of genes are available which confer s~1ect~hle phenotypes on animal
cells. The bacterial neomycin phosphotransferase (neo) (Colbere-Garapin et al., J.
20 Mol. Biol. 150:1, 1981), hy~ cin phosphQtransferase (hph) (Santerre et al., Gene
30:147, 1984), and Y~nthine-guanine phosphoribosyl transferase (gpt) (Mulligan and
Berg, Proc. Natl. Acad. Sci. USA 78:2072, 1981) genes are widely used dominant
positive selectable genes. The Herpes simplex virus type I thymidine kinase (HSV-I
TK) gene (Wigler et al., Cell 11:223, 1977), and the cellular adenine
25 phosphoribosyltransferase (APRT) (Wigler et al., Proc. Natl. Acad. Sci. USA
76:1373, 1979) and hypo~nthine phosphoribosyltransferase (HPRT) genes (Jolly et
al., Proc. Natl. Acad. Sci. USA 80:477, 1983) are co,-..l-only used recessive positive
selectable genes. In general, domin~nt select~ble genes are more versatile than
recessive genes, bec~llse the use of recessive genes is limited to mutant cells deficient in
30 the se~ect~ble function, whereas do~ nt genes may be used in wild-type cells.Several genes confer negative as well as positive selectable phenotypes,
including the HSV-I 1~, HPRT, APRT and gpt genes. These genes encode enzymes
which catalyze the conversion of nucleoside or purine analogs to cytotoxic
il~te. ..~f.l;~es The nucleoside analog GCV is an efficient substrate for HSV-I TK, but
35 a poor substrate for cellular TK, and the.Gfc,l~i may be used for negative selection
against the HSV-I TK gene in wild-type cells (St. Clair et al., Anlimicrob. Agents
Chemother. 31:844, 1987). However, the HSV-I TK gene may only be used
WO 92/08796 PCr/USgl/OX442
20962~2
effectively for positive selectiQn in mutant cells lacking cellular TK activity. Use of the
HPRT and APRT genes for either positive or negative selection is cimil~rly limited to
HPRT- or APRT- cells, le~e.,~ ely (Fenwick, "The HGPRT System", pp. 333-373,
M. Co~ n (ed.), Molecular Cell Gene~ics, John Wiley and Sons, New York, 1985;
S Taylor et al., "The APRT System", pp., 311 -332, M. Gol~ n (ed.), Molecular Cell
Genetics, John Wiley and Sons, New York, 1985). The gpt gene, on the other hand,may be used for both positive and negative selection in wild-t,vpe cells. Negative
selection against the gpt gene in wild-type cells is possible using 6-thiox~nthin~, which
is efficiently converted to a ~;ylul~ic nllGleotid~ analog by the b~çteri~l gpt enzyme, but
not by the cellular HPRT enzyme (Besnard et al., Mol. Cell. Biol. 7:4139, 1987).More recently, attention has turned to select~hle genes that may be incorporatedinto gene transfer vectors ~ecignYl for use in human gene therapy. Gene therapy is a
method for perm~nently curing a he.~li~y genetic disease which results from a defect
in or absence of one or more genes. Collectively, such ~lise~ces result in signi~lc~nt
15 morbidity and mortality. FY .~ples of such genetic ~ice~ces include hellluyhilias A and
B (caused by a deficiency of blood coagulation factors VIII and IX, ~yec~ ely),
alpha-1-antitrypsin deficiency, and adenosine d~...in~se d~filciency. In each of these
particular cases, the mic-sing gene has been identified and its complel.~ .y DNA(cDNA) molecularly cloned (Wood et al., Nature 312:330, 1984; Anson et al., Nature
20 315:683, 1984; and Long et al., Biochemistry 23:4828, 1984; Daddona et al., J. Biol.
Chem. 259:12101, 1984). While palliative therapy is available for some of these
genetic rlice~ces, often in the form of ~lminictration of blood products or blood
transfusions, one way of ~ ....A~enlly curing such genetic flice~ces is to introduce a
repl~çem~nt for the defective or miccing gene back into the somatic cells of the patient,
a process referred to as "gene Ihel~yy" (Anderson, Science 226:401, 1984). Gene
therapy can also be used as a means for ~lg...~ g normal gene function, for ex~mrle~
by introducing a heterologous gene capable of modifying cellular activities or cellular
phenotype, or alternatively, e~ yl~;~sing a drug needed to treat a ~lise~ce
The process of gene therapy typically involves the steps of (1) removing
30 som~tic (non-germ) cells from the patient, (2) introducing into the cells ex vivo a
replaçe.-.c nt gene via an ayyluyliate vector capable of eAyl~s~illg the repl~em~nt gene,
and (3) transplanting or transfusing these cells back into the patient, where the
replaçement gene is e~ ssed to provide some thclayelJIic benefit. Gene transfer into
somatic cells for human gene therapy is yresenlly achieved ex vivo (Kasid et al., Proc.
35 Natl Acad. Sci. USA 87:473, 1990; Rosenberg et al., N. Engl. J. Med. 323:570,1990), and this relatively inefficient process would be facilitated by the use of a
dc....;n~llt positive sel~ct~hle gene for ide"~iryil~g and i~ol~ting those cells into which the
Wo 92/08796 2 0 9 62 22 PCr/USsl/08442
repl ~e~ n~ gene has been introduced before they are lelu"led to the patient. The neo
gene, for example, has been used to identify genetir~lly m~ifi~A cells used in human
gene therapy.
In some in~t~nces, however, it is possible that thé introduction of genetically
5 mt~-lifie(l cells may actually co,l,~lo"lise the health of the patient. The ability to
selectively elimin~te genetir~lly m~lified cells in vivo would provide an additional
_argin of safety for p~tients undergoing gene therapy, by p~.lllilling reversal of the
procedure. This might be ~co,.-l)lished by incorporating into the vector a negative
selectable (or 'suicide') gene that is capable of functioning in wild-type cells.
10 Inco,~,ul~tion of a gene c~pahle of conf~"ing both dominant positive and negative
Sel~-Cts~hle ~he,nOl~ ,S would ensure co-eA~ ion and co-reg~ tion of the positive and
negative select~hle ~helloly~es, and would minimi7e the size of the vector. However,
positive selection for the gpt gene in some instances requires precise selectionconditions which may be ~liffirult to detl- ...ine Moreover, the feasibility of using the
gpt gene for in vivo negative selection has not yet been clearly established. For these
,~asons, co-eA~,~s~ion of a domin~nt positive selectable phenotype and a negative
select~hle phenc.l~l,e is typically achieved by co-t~s~ing two lirr~ t genes which
sepa,alely encode other dc~ -t positive and negative sel~ct~hle functions, rather than
using the gpt gene.
The existing strategies for co-expressing dominant positive and negative
select~ble phenotypes encoded by dirr~"e.~- genes often present complex rh~ nges As
indicated above, the most widely used technique is to co-transfect two pl~mir1~
s~,pa.~lely encoding two phenotypes (Wigler et al., Cell 16:777, 1979). However, the
efficiency of co-transfer is rarely 100%, and the two genes may be subject to
il~ dent genetic or epigPnetir, reg~ tion A second ~te~ ~ is to link the two genes
on a single pl~cmid, or to place two in~epen-l~nt l,a,lscliJ,lion units into a viral vector.
This method also suffers from the disadvantage that the genes may be independently
regula~ed. In retroviral vectors, suppression ûf one or the other independent
t ~ls~ lion unit may occur (Elllel,llan and Temin, Mol. Cell. Biol. 6:792, 1986). In
~d~lition, in some cil~ nces there may be incnfficient space to accnmmod~te two
fimction~l trans~;liptiûn units within a viral vector, although retroviral vectors with
fi~nctiQn~l multiple pç~lllole,~ have been succes~rully made (Overell et al., Mol. Cell.
Biol. 8:1803, 1988). A third strategy is to express the two genes as a bicistronic
mRNA using a single p~,lloter. With this method, however, the distal open reading
frame is often tr~n~l~tYl with variable (and usually reduced) çffisiency (l~-lfm~n et al.,
EMBO J. 6:187, 1987), and it is unclear how effective such an expression strategy
would be in ~lilll~ cells.
Wo 92/08796 - Pcr/ussl/o8442
209~2X2 4
The present invention provides a method for more efficiently and reliably co-
eAplessi,lg a dominant positive selectable phenoly~c and a negative selectable
phenotype encofl~d by dirr~ genes.
~UMMARY OF THF ~NVF.NTION
The present invention provides a select~ble fusion gene co~ g a dol,Jinan
positive select~hle gene fused to and in reading frame with a negative select~ble gene.
The s~le~!; ble fusion gene en~es a single bir~ l fusion protein which is capable
of conferring a ~lomin~nt positive selectable phcn~yl~e and a negative selectable
phenotype on a cellular host. In a l,leÇ~.~d emho lim~-nt, the selectable fusion gene
comrfi~es nucleotide se~luences from the hph gene fused to mlcleotirle sequences from
the HSV-I TK gene, referred to herein as the HyTK select~ble fusion gene (Se~quence
Listing No. 1). The HyTK selectable fusion gene confers both hygromycin B
l~s;c~ ~.-ce (Hmr) for d~ positive selectiQn and ganciclovir sensitivity (GCVS) for
negative selP~;~n
The present invention also provides recombinant eApression vectors, for
eA~llllple, retroviruses, which include the select~hle fusion genes, and cells tr~n~d~lce~
with the recombin~nt ~A~-~,ssion vectors.
The selectable fusion genes of the present invention are expressed and
regulated as a single genetic entity, pc .~ g co-reg~ tiQn and co-expression with a
high degree of effirien~y.
RR~FF nF~CRIPTION OF THF l)R~WIl~l('..~
Figure 1 shows diagrams of the pl~mitl~ tgCMV/hygro, tgCMV/TK and
25 tgCMV/HyTK which contain proviral sl-uclul~es used in the present invention. The
three pl~mi~1~ are id~nt~ except for the genes ins~. ~d ~1~. ~n the HCMV pçolllolcr
(filled bQx) and the SV40 early region pol~ade~rlation signal (h~t~ll~ box).
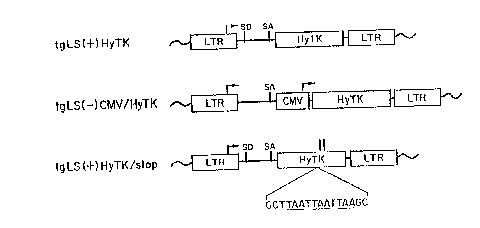
Figure 2 shows diagrams of the proviral ~llu~;lul~;s from the pl~cm; l~ shown inFigure 1. The horizontal arrows in-lic~te transcriptional start sites and direction of
30 transcription. The open box labeled LTR is the retroviral long terminal repeat. The
viral splice donor is labeled SD and the acc~lo sequences are labeled SA. The open
box labeled CMV is the cytomegalovirus l,rollJole.. In tgLS(+)HyTK/stop, the
positions of the two internal initiation codons retained in the HyTK sele~t~ble fusion
gene are inr1ir~ted by vertical arrows. The location at which the universal translation
35 t~min~tr,lr oli~nllrleotide was ins~,ttd is also m~*rA
Figures 3 and 4 are graphs showing the results of a short-term proliferation
assay in which the hy~ul.lycin resistant (Hmr) NIH/3T3 cell pools and Hmr and HAT
Wo 92/08796 2 0 9 6 2 2 2 Pcr/US9l/08442
resistant (HATr) Rat-2 cell pools were tested for g~n~icl(!vir sensitivity (GCVS). Figure
3 shows that GCV inhibits gr~wth of N~3T3 cells transfected with tgCMV/HyTK,
but does not inhibit growth of NI~U3T3 cells L ~ Ç~l~d with tgCMV/Hygro. Figure 4
~ shows that GCV inhibits growth of Rat-2 cells transfected with tgCMVlHyTK (initially
5 sele~ for Hmr or HATr) even at the lowest cUI~C~ Lions of GCV, and also inhibits
growth of Rat-2 cells transfected with tgCMV/TK, although at slightly higher
concentrations. GCV did not inhibit growth of Rat-2 cells transfected with
tgCMV/Hygro.
Figure 5 shows the results of Northern analysis of Hmr and HATr cell pools.
10 Polyadenylated mRNA was e~LI~l;Led from each Hmr and HATr cell pool, and used to
pl~pale Northern blots which were probed with sequences from the hph gene (PanelA), the HSV-I TK gene (Panel B), or the B-actin gene (Panel C) (for mRNA
equivalence). The position~ of the 28S and 18S ribosomal RNAs are in~ir~ted The
mRNA present in each lane was eYtr~rte~l from the following cells: Lane 1, Rat-2 cells
15 Llallsr~L~;d with tgCMV/hygro; Lane 2, Rat-2 cells transfected with tgCMV~rK; Lane
3, Rat-2 cells Lla,lsÇecLed with tgCMV/HyTK and sele~t~i for Hmr; Lane 4, Rat-2 cells
transfected with tgCMV/HyTK and selected for HATr; Lane 5, N~I/3T3 cells
re~;~d with tgCMV/hygro; Lane 6, NlHJ3T3 cells l,al,srecled with tgCMV/HyTK.
Figure 6 shows phologla~hs of stained colonies of unillfecled NIEV3T3 cells
20 (plates a, b and c) and NIH13T3 cells inÇ~led with the tgLS(+)HyTK (plates d and e)
or tgLS(-) CMV/HyTK (plates f and g) retroviruses. The cells were grown in m~Ainm
alone or medium supplçm~-nte~ with GCV, Hm or Hm plus GCV in a long-term
proliferation assay. The data show that uninfected NI~V3T3 cells were resi~t~nt to
GCV and grew to confluence (plate b), but were killed by Hm (plate c). Growth of25 NlH/3T3 cells infected with the tgLS(+)HyTK and tgLS(-) CMV/HyTK retrovirusesand grown in the presence of Hm (plates d and f) was inhibited by GCV (plates e and
g)-
nFTAn Fl) I)F.~CR~PTION OF THF. ~NVli.NTTON
SEQ ID NO: 1 and SEQ ID NO:2 (a~pe~ing imme~ tely prior to the claims)show specific embo l;....-l.t~ of the nucleotide sequence and cc,~ onding amino acid
sequence of the HyTK select~ble fusion gene of the present invention. The HyTK
select~ble fusion gene shown in the Sequence Listing comrri~es sequences from the
35 hph gene (nucl~tides 1-971) linked to sequences from the HSV-I TK gene
(nl1cleoti~es 972-2076).
Wo 92/08796 Pcr/us91/08442
209622~ 6
D~,r.,.;l;ol~
As used herein, the terrn "sP1ect~b1e fusion gene" refers to a nucleotide se~uence
co~ lising a dominant positive select~ble gene which is fused to and in reading frame
with a negative se1çct~hle gene and which encodes a single bifunctional fusion protein
S which is capable of collr.,.liilg a dominant positive select~ble phenotype and a negative
se!ect~hle phenotype on a cellular host. A "do~ positive select~ble gene" refers to
a sequence of nucleotides which encodes a protein conf~lling a dominant positiveselectable phenotype on a cellular host, and is ~iSc~lsse~ and exemplified in further
detail below. A "negative select~ble gene" refers to a sequence of nucleotides which
10 encodes a protein confelliilg a negative select~ble phenotype on a cellular host, and is
also discussed and exemr1ifie~ in further detail below. A "selectable gene" refers
.~. . i.~ .lly to do...;~ positive sçlect~hle genes and negative se1ect~h1e genes.
A se1çct~bkP gene is "fused to and in reading frame with" another se1Pct~hle gene
if the expression products of the selectable genes (i.e., the proteins çncoded by the
15 se1ect~ble genes) are fused by a peptide bond and at least part of the biological activity
of each of the two proteins is ret~ined With reference to the HyTK selectable fusion
gene disclosed herein, for example, the hph gene (encoding hygromycin-B
phosphotransferase, which confers the dominant positive selectable phenotype of
hygromycin resistance (Hmr)) is fused to and in reading frame with the HSV-I TK gene
20 (encoding Herpes Simplex Virus Type I thymidine kinase, which confers a negative
sP1çct~ble phenotype of ganciclovir sensitivity, or (GCVS)) if the hph and HSV-I TK
proteins are fused by a peptide bond and e~lcssed as a single bifunctional fusion
protein. The cG-ll~onent selectable gene sequences of the present invention are
preferably contiguous; however, it is possible to construct sçlect~hle fusion genes in
25 which the coll.pol ent sele~t~ble gene se~uen~e~ are sepa,at~d by internal nontr~n~l~te~
nucleotide sequences, such as introns. For purposes of the present invention, such
noncontiguous selectable gene sequences are considered to be fused, provided that
eA~r~ssion of the se1Pct~ble fusion gene results in a single bifunctional fusion protein in
which the c~ ssion products of the col~poncnt sP,1ect~ble gene sequen~es are fused by
30 a peptide bond.
"Nucleotide sequence" refers to a hetel~l)olymer of deoxyribonucleotides or
ribonucleotides, such as a DNA or RNA sequence. Nucleotide sequences may be in
the form of a s,p~lc ~grnprlt or as a co~ )ol-el-t of a larger consll UC~. Preferably, the
nucleotide sequences are in a quantity or concentration enabling identification,35 manipulation, and recovery of the sequence by standard bioch-Pmic~1 methods, for
ex~nlrle, using a cloning vector. Recombinant nucleotide sequences are the product of
various combin~ion~ of cloning, rcstriction, and ligation steps resulting in a construct
WO 92/08796 2 0 9 6 2 2 2 PCr/USsl/08442
,.
having a ~lluclulal coding se~uçnce ~ n~ich~ble from homologous s~ue"ces found
in natural sy~l~ms. Generally, nucleotide sequences en~ g the structural coding
sequence, for example, the select~ble fusion genes of the present invention, can be
~semhl~ from nucleo~ide fr~ment~ and short oligQnucleotir1e linkers, or from a series
5 of oligonucleotides, to provide a ~ tl.~lic gene which is capable of being cA~l~ssed in
a l~col~bulant transcriptional unit. Such sçquçnces are preferably provided in the form
of an open reading frame u~ u~)led by intern~l nontr~n~l~ted sequences, or introns,
which are typically present in eukaryotic genes. Genomic DNA co..~ ii-g the relevant
selçct~hle gene se~u~ ~-ces is preferably used to obtain ~,lol,liate nucle~ti~le sequences
enco~ling selçct~ble genes; however, cDNA fr~gm~nts may also be used. Sequences of
non-t ~n~l~te~l DNA may be present 5' or 3' from the open reading frame or within the
open reading frame, provided such sequences do not il~telr~lc with manipulation or
eApr~s~ion of the coding regions. Some genes, however, may include introns whichare n~ess~ ~/ for proper eA~l~s~ion in certain hosts, for ex~mrle the HPRT select~ble
gene in~lucles introns which are nf cess~. y for eA~l~;ssion in ~,m~ onal stem (ES) cells.
As suggested above, the nucleotide sequences of the present invention may also
comprise RNA se4u~ nces, for example, where the nucleotide sequences are p~c~ge~as RNA in a retrovirus for infecting a cellular host. The use of retroviral eA~lession
vectors is ~ cucse~l in greater detail below.
The term "recombinant e~-pl~;ssion vector" refers to a replicable unit of DNA orRNA in a form which is capable of being ~ ~1 into a target cell by ~ r~ction or
viral infection, and which codes for the t;A~,lession of a select~ble fusion gene which is
tr~n~ibe~ into mRNA and tr~n~l~t~ into protein under the control of a genetic elc .-ent
or elements having a regulatory role in gene expression, such as ~ scliplion andtr~n~l~tion initi~tion and tçrmin~tion se~lu~ -ces The n~-llbina~ A~ ion vectors of
the present invention can take the form of DNA constructs replicated in bacterial cells
and transfected into target cells directly, for example, by calcium phosphate
pl~,ci~itation, electroporation or other physical transfer metho~s The recombinant
t;A~- ssion vectors which take the form of RNA constructs may, for example, be in the
form of infectious retroviruses packaged by suitable "p~cl~ging" cell lines which have
previously been transfected with a proviral DNA vector and produce a retrovirus
colllainillg an RNA transcript of the proviral DNA. A host cell is infected with the
retrovirus, and the retroviral RNA is replicated by reverse transcription into a double-
yl DNA in~e~ te which is stably integrated into chromosomal DNA of the
host cell to form a provirus. The provirus DNA is then expressed in the host cell to
pl.Alucc polypeptides encoded by the DNA. The l~olllbinant ~Apl~ssion vectors of the
p~senl invention thus include not only RNA constructs present in the infectious
Wo 92/08796 2 0 9 6 2 2 2 PCr/USsl/08442
~'
retrovirus, but also copies of proviral DNA, which include DNA reverse transcripts of
a retrovirus RNA genome stably integlaled into chlc~ so...ql DNA in a suitable host
cell, or cloned copies thereof, or cloned copies of uninlc~dted interm~liqte forms of
retroviral DNA. Proviral DNA includes transcriptional elements in independent
5 operative association with selected ~IIUCIU1~11 DNA sequenres which are transcribed into
mRNA and trq-n~lq-ted into protein when proviral se~luenres are ~ ssed in infected
host cells. Recombinant e~l lcssion vectors used for direct transfection will include
DNA sequences enabling replication of the vector in bacterial host cells. Various
r~colubinanl e,~ s~;on vectors suitable for use in the present invention are described
10 below.
"Tr~n~ ee" means introduction of a l~,colllbinant e~ ,s~ion vector containing
a select~ble fusion gene into a cell. Tr~n~d!lchon methods may be physical in nature
(i.e., transfection), or they may rely on the use of recombinant viral vectors, such as
retroviruses, encoding DNA which can be transcribed to RNA, packaged into
15 infectious viral particles and used to infect target cells and thereby deliver the desired
genetic material (i.e., infection). Many dirr~l~"~ types of ",s.."".~ n gene transfer and
~,col..bin~-t ~A~ssion vectors have been developed (see, e.g., Miller and Calos, eds.,
"Gene Transfer Vectors for ~mm~ n Cells," Current Corn~n. Mol. Biol., (Cold
Spring Harbor Laboratory, New York, 1987)). Naked DNA can be physically
introduced into m~mm~ n cells by transfection using any one of a number of
ttochni~lues inrl~lrlin~, but not limited to, c~lrillm phr~srh~te transfection (Berman et al.,
Proc. Natl. Acad. Sci. USA 84 81: 7176, 1984), DEAE-Dextran transfection
(McCutchan et al., J. Natl. Cancer Inst. 41:351, 1986; 1 uthm~n et al., Nucl. Acids
Res. 11:1295, 1983), protoplast fusion (I)eans et al., Proc. Natl. Acad. Sci. USA 84
81: 1292, 1984), ele~;ll~olalion (Potter et al., Proc. Natl. Acad. Sci. USA 84 81:
7161, 1984), lipofection (Felgner et al., Proc. Natl. Acad. Sci. USA 84:7413, 1987),
polybrene transfection (Kawai and Nishzawa, Mol. Cell. Biol 4: 1172, 1984) and direct
gene transfer by laser mic~ui~clul~ of cell ll-e.l~bl~u~es (Tao et al., Proc. Natl. Acad.
Sci. USA 84 84:4180, 1987). Various infection techniques have been developed
which utilize ~combinalll infectious virus particles for gene delivery. This l~ senls a
plefe.l~d a~ ach to the present invention. The viral vectors which have been used in
this way include virus vectors derived from simian virus 40 (SV40; Karlsson et al.,
Proc. Natl. Acad. Sci. USA 84 82:158, 1985), adenoviruses (Karlsson et al., EMBOJ. 5:2377, 1986), adeno-associated virus (LaFace et al., Virology 162:483, 1988) and
retroviruses (Coffin, 1985, pl7-71 in Weiss et al (eds.), RNA Tumor Viruses, 2nded., Vol 2, Cold Spring Harbor La~ tol~, New York). Thus, gene transfer and
e~ ssion methods are l,u~ us but essenti~lly function to introduce and express
Wo 92/08796 ' l 2 0 9 ~ 2 2 2 pcr/uss1/o8442
genetic mqt~rial in l,.-.,,,,,~lian cells. Several of the above techniques have been used to
transduce hc.llalo~u,elic or lymphoid cells, incl~ ing calcil-m phosphate transfection
(Berman et al., supra, 1984), protoplast fusion (Deans et al., supra 1984),
elect,~pc"ation (Cann et al., Oncogene 3:123, 1988), and infection with recombinant
adenovirus (Karlsson et al., supra; Ruether et al., Mol. Cell. Biol. 6:123, 1986) adeno-
~csoci~tecl virus (LaFace et al., supra) and retrovirus vectors (Overell et al., Oncogene
4:1425, 1989). P~ aly T lymphocytes have been successfully tr~nc~uce~ by
eleeLIupulation (Cann et al., supra, 1988) and by retroviral infection (Nishihara et al.,
Cancer Res. 48:4730, 1988; Kasid et al., supra, 1990).
Construction of Selectable Fusion Genes
The selectable fusion genes of the present invention comprise a dominant
positive selectable gene fused to a negative select~ble gene. A selectable gene will
generally comprise, for example, a gene encoding a protein capable of conferring an
antibiotic resi~tance phenotype or supplying an aulo~ophic l~uire.llent (for dominant
positive selection), or activating a toxic metabolite (for negative selection). A DNA
sequence encodh~g a bif -nctional fusion protein is constructed using recolllbil-ant DNA
techniques to assemble separate DNA fr~gmen~s encoding a dominant positive
selectable gene and a negative select~ble gene into an a~pl~liate e~cpl~;ssion vector.
The 3' end of one selectable gene is ligated to the 5' end of the other selectable gene,
with the reading frames of the sequences in frame to perrnit translation of the mRNA
sequen~es into a single biologically active bifunctional fusion protein. The selectable
fusion gene is e"~l.,ssed under control of a single proll.olel.
The dominant positive selectable gene is any gene which, upon being
trans~luced into a host cell, expresses a dominant phenotype permitting positiveselection of stable tran~uct~nt~ Selection of stable tran~d~lct~nt~ can be carried out,
for eY~nlrle, using the hy~ull~cin-B phosphol.~,sr~,~.se gene (hph) which confers the
sdectable phenotype of hy~u.,.~lcil~ resict~nce (Hmr) (Santerre et al., Gene 30:147,
1984; Sugden et al., Mol. Cell. Biol. 5:410, 1985; obtainable from plasmid pHEBol,
under ATCC Accession No. 39820). Hygromycin B is an aminoglycoside antibiotic
that inhibits protein synthesis by disrupting t~nCl~tion and pn,..,o~ing mistranslation.
The hph gene confers Hmr to cells tr~n~dn~ed with the hph gene by phosphorylating
and detoxifying the antibiotic hygromycin B. Other acceptable dominant positive
select~ble genes include the following: the aminoglycoside phosphotransferase gene ;~
35 (neo or aph) from TnS which codes for resi~t~nce to the antibiotic G418 (Colbere-
Garapin et al., J. Mol. Biol. 150:1, 198; Southern and Berg, J. Mol. Appl. Genet.
1:327, 1982); the y~nthine-~uanine phosphoribosyl transferase gene (gpt) from E coli
Wo 92/08796 2 U 9 6 ~ 2 2 Pcr/US9l/08442
--
enco ling resistance to mycoph~nolic acid (Mllllig~n and Berg, Proc. Natl. Acad. Sci
USA 78:2072, 1981); the dihydrofolate reductase (DHFR) gene from murine cells orE. coli which is necessary for biosynthesis of purines and can be competitively
inhibited by the drug methotrexate (MlX) to select for cells constitutively expressing
5 increased levels of DHFR (Simonsen and Levinson, Proc. Natl. Acad. Sci. (U.S.A.
80:2495, 1983; Simonsen et al., Nucl. Acids. Res. 16:2235, 1988); the S
typhimurium hi~ti-linol dehydrogenase (hisD) gene (Hartman, et al, Proc. Natl. Acad
Sci. (USA) 85:8047, 1988); the E. coli tryptophan synthase ~ subunit (trpB) gene(Hartman et al., supra); the puromycin-N-acetyl transferase (pac) gene (Vara et al.,
Nucl. Acids Res. 14:4117, 1986); the adenosine ~le~min~e (ADA) gene (Daddona et
al., J. Biol. Chem. 259:12101, 1984); the multi-drug resist~nre (MDR) gene (Kane et
al., Gene 84:439, 1989); the mouse o~ hine deca l,o~ylase (OCD) gene (Gupba and
Coffino, J. Biol. Chem. 260:2941, 1985); the E. coli aspartate transcarbamylase
catalytic subunit (pyrB) gene (Ruiz and Wahl, Mol. Cell. Biol. 6:3050, 1986); and the
15 E. coli asnA gene, encoding asparagine sythetase (Cartier et al., Mol. Cell. Biol.
7:1623, 1987).
The negative selectable gene is any gene which, upon being tr~n~d~lce~ into a
host cell, expresses a phenotype permitting negative selection (i.e., elimin~tion) of
stable ~ hlc~ In ~leÇ~ d e~llbo~ ..L~, the negative sele~t~ble gene used in the
20 fusion genes of the present invention is the Herpes simplex virus type I thymidine
kinase (HSV-I TK) gene (Wigler et al., Cell 11:223, 1977; McKnight et al., Nucl.Acids Res. 8:5931, 1980; Preston et al., J. Virol. 38:593, 1981; Wagner et al., Proc.
Natl. Acad. Sci. USA 78:1441, 1981) which confers ganciclovir sensitivity (GCVS)(St. Clair et al., Antimicrob. Agents Chemother. 31:844, 1987). The HSV-I TK gene
25 is available from Bethesda Research Labs (Catalog No. BRL 5365 SA). Negative
selection can also be ~ Ço~ cd, for example, using the cellular hypoxanthine
phosphoribosyltransferase (HPRT) gene (Jolly et al., Proc. Natl. Acad. Sci. USA
80:477, 1983; Fenwick, "The HGPRT System", pp. 333-373, M. Gol~ s.~.~n (ed.),
Molecular Cell Genetics, (John Wiley and Sons, New York, 1985)) and the cellular30 adenine phosphoribosyltransferase (APRT) gene (Wigler et al., Proc. Natl. Acad. Sci.
USA 76: 1373, 1979; Taylor et al., "The APRT System", pp., 311-332, M. Gottesm~n(ed.), Molecular Cell Genetics, (John Wiley and Sons, New York, 1985)); and the E.
coli gpt gene (Besnard et al., Mol. Cell. Biol. 7:4139, 1987).
Due to the deg~ acy of the genetic code, there can be considerable variation in
35 nucleotide sequences encoding the same amino acid sequence; exemplary DNA
embo~l;...~ nl~ are those cc~ ding to the nucleotide sequences shown in SequenceListing No. 1. Such variants will have mo~lified DNA or amino acid sequences, having
Wo 92/08796 ~ 2 0 9 fi 2 2 2 PCr/USsl/OX442
. ,
one or more substitutions, deletion~, or ~lAition~ the net effect of which is to retain
biological activity, and may be sub~l;n~le~d for the sperific sequences disclosed herein.
The sequences of select~hle fusion genes comprising hph and TK are equivalent if they
contain all or part of the sequences of hph and HSV-I TK and are capable of
5 hybridizing to the nucleotide sequence of Sequence Listing No. 1 under moderately
stringçnt con-lition~ (50~C, 2 X SSC) and express a biologically active fusion protein.
A "biologically active" fusion protein will share sufficient amino acid sequencesimilarity with the specific e...hoA;...ç~ of the present invention disclosed herein to be
capable of conr~ g the s~l~t~hle phenotypes of the comronent selert~h. le genes.In a preferred embodiment, sequences from the bacterial hygromycin
phosphotransferase (hph) gene are fused with sequences from the HSV-I TK gene.
The resulting selectable fusion gene (referred to as the HyTK selectable fusion gene)
enco~es a bifunctional fusion protein that confers Hmr and GCVS, and provides a
means by which dominant positive and negative selectable phenotypes may be
15 expressed and reg~ teA as a single genetic entity. The HyTK select~ble fusion gene is
tll~,l~;role a useful ~dditio~ to the eYisting panel of sekct~ble genes available for use in
animal cells, becau~e it allows both ~lo~ positive and negative selection in wild-
type cells.
20 Recon~binallt Expression Vectors
The sçl~ct~ble fusion genes of the present invention are utili7ed to identify,
isolate or el;...;..~e host cells into which the select~hle fusion genes are introd~lceA The
select~ble fusion genes are introduced into the host cell by tr~n~A~Icing into the host cell
a recolllbinall~ expression vector which co~ ins the selectable fusion gene. Such host
25 cells include cell types from higher eukaryotic origin, such as l.~.n..~ n or insect
cells, or cell types from lower prokaryotic origin, such as bacterial cells, for example,
E. coli.
As inAi~te-l above, such select~ble fusion genes are preferably introduced into
a particular cell as a colllyonenl of a recQmhin~nt e~yl~;s~ion vector which is c~p~hle of
30 e~ylessing the selectable fusion gene within the cell and conferring a selectable
phenotype. Such l~co,l,bin~t e~-y~;,sion vectors generally include synthetic or natural
nucleotide sequences compri~ing the select~hle fusion gene operably linked to suitable
t-~ - ;plional or tr~n~l~tion~l control sequ~,nces, for e~mple, an origin of replication,
optional oy~ or se~luences to control transcription, a sllit~ble y~lllOt~,r and çnh~ncer
35 linked to the gene to be expressed, and other 5' or 3' flanking nontranscribed
s~uences, and 5' or 3' non~ sl~ted sequences, such as necess~ ribosome binding
sites, a polyadenylation site, splice donor and acc~ytol sites, and transcriptional
Wo 92/08796 2 0 9 6 2 2 2 Pcr/US9l/08442
termination sequences. Such regnl~tory sequences can be derived from "~"",~ n,
viral, microbial or insect genes. Nucleotide sequences are operably linked when they
are functionally related to each other. For eY~mple, a ~ Ot~ is operably linked to a
select~hle fusion gene if it controls the transcription of the select~ble fusion gene; or a
5 ribosome binding site is operably linked to a select~hle fusion gene if it is positioned so
as to permit trancl~tion of the selectable fusion gene into a single bifunctional fusion
protein. Generally, operably linked means contig~lous.
Specific ,~coll~binant eAI,Iession vectors for use with ",~,...n~ n ba~ten~l, and
yeast cellular hosts are described by Pouwels et al. (Cloning Vectors: A Laboratory
10 Manual, Elsevier, New York, 1985) and are well-known in the art. A detailed
~scli~ion of recombin~nt c..~ ssion vectors for use in animal cells can be found in
Rigby, J. Gen. Virol. 64:255, 1983; Elder et al., Ann. Rev. Genet. 15:295, 1981; and
Sublalllalli et al., Anal. Biochem. ~35:1, 1983. Appropriate recombinant t;~ ssion
vectors may also include viral vectors, in particular retroviruses (discussed in detail
below).
The select~ble fusion genes of the present invention are preferably placed underthe transcriptional control of a strong enh~n~er and promoter ~Ap~s~ion casselle.
Examples of such eA~I., ,sion c~csettes include the human cytomPg~lovirus imme~ t5-
early (HCMV-E) promoter (Boshart et al., Cell 41:521, 1985), the ~-actin promoter
(Gunning et al., Proc. Natl. Acad. Sci. (USA) 84:5831, 1987), the histone H4
promoter (Guild et al., J. Virol. 62:3795, 1988), the mouse metallothionein plulnoter
(McIvor et al., Mol. Cell. Biol. 7:838, 1987), the rat growth hormone ~.~ otel (Miller
et al., Mol. Cell. Biol. 5:431, 1985), the human adenosine de~min~ce promoter
(Hantzapoulos et al., Proc. Natl. Acad. Sci. USA 86:3519, 1989) the HSV tk plulllotel
(Tabin et al., Mol. Cell. Biol. 2:426, 1982), the a~-l an~,~ in çnh~ncer (Peng et al.,
Proc. Natl. Acad. Sci. USA 85:8146, 1988) and the immunoglobulin
enh~ncçr/~r~ oter (Bl~nkPnctein et al., Nucleic Acid Res. 16:10939, 1988), the SV40
early or late promoters, the Adenovirus 2 major late pi~ll~tel, or other viral ~r~.lllot~,ls
derived from polyoma virus, bovine papilloma virus, or other retroviruses or
adenoviruses. The pl~l,lotel and e~nh~ncer elements of i.. i-oglobulin (Ig) genes
confer marked spe~ifirity to B lymphocytes (Banerji et al., Cell 33:729, 1983; Gillies et
al., Cell 33:717, 1983; Mason et al., Cell 41 :479, 1985), while the elPmPntC controlling
tr~n~iption of the ~globin gene filnction only in el ~vid cells (van AccPn~elft et al.,
Cell 56:969, 1989). Using well-known restriction and lig~tion techniques, approl"iate
35 transcriptional control sequences can be excised from various DNA sources andintegrated in operative relationship with the intact selectable fusion genes to be
eA~Iei~sed in accordance with the present invention. Thus, many ~nc~iptional control
Wo 92~08796 ~ 2 D 9 G 2 ~ ~ Pcr/us91 /08442
sequences may be used s~lccescfully in retroviral vectors to direct the expression of
ills~ilt~ genes in ;~recl~ cells.
Retroviruses
Retroviruses can be used for highly efficient transduction of the selectable
fusion genes of the present invention into eukaryotic cells and are ~.cÇ~l.Gd for the
delivery of a select~ble fusion gene into primary cells. Moreover, retroviral integration
takes place in a controlled fashion and results in the stable integration of one or a few
copies of the new genetic il~ol...a~ion per cell.
Retroviruses are a class of viruses whose genome is in the form of RNA. The
genomic RNA of a retrovirus contains trans-acting gene sequences coding for three
viral proteins: a sLIuc~ l protein gag which ~sc~i~tes with the RNA in the core of the
virus particle; the reverse transcriptase pol which makes the DNA complement; and a
envelope glycopll~teill env which resides in the li~l, oleill envelope of the particles and
binds the virus to the surface of host cells on infection. Replication of the retrovirus is
regulated by cis-acting elcl--enls, such as the ~ olel for transcription of the proviral
DNA and other nucleotide sequences necessq~ for viral replication. The cis-acting
clc ..f ~ are present in or a~ cent to two iclentic~l untr~n~l~tYl long terminal repeats
(LTRs) of about 600 base pairs present at the 5' and 3' ends of the retroviral genome.
20 Retroviruses replicate by copying their RNA ge -o...e by reverse l,~sc-i~ion into a
double-stranded DNA interm~Ai~te7 using a virus-encoded, RNA-directed DNA
polymerase, or reverse transcriptase. The DNA interme~ te is integrated into
chlo,..osol.lal DNA of an avian or ~.~A.~ n host cell. The integr~ted retroviral DNA
is called a provirus. The provirus serves as temrl~te for the synthesis of RNA chains
25 for the formation of infectious virus particles. rOI w~d transcription of the provirus
and assembly into infectious virus particles occurs in the presence of an appl~liate
helper virus having en~log~onous trans-acting genes l~uircd for viral replirqtion
Retroviruses are used as vectors by replacing one or more of the endogenous
trans-acting genes of a proviral form of the retrovirus with a recombinant thelap~ ic
30 gene or, in the case of the present invention, a selectable fusion gene, and then
transducing the recombinant provirus into a cell. The trans-acting genes include the
gag, pol and env genes which encode, respectively, proteins of the viral core, the
el,~yllle reverse l,~s~.ip~se and con~ e .~ of the envelope protein, all of which are
l-ecess:-. y for production of intact virions. Recombinant retroviruses deficient in the
35 trans-acting gag, pol or env genes cannot synthesi7e essenti~l proteins for replication
and are accordingly replication-defective. Such replication-defective recombinant
retroviruses are prop~ ted using packaging cell lines. These packaging cell lines
Wo 92/08796 ;4 Pcr/US9l/08442
contain integrated retroviral genomes which provide all trans-acting gene sequences
necess~ry for production of intact virions. Proviral DNA sequences which are
tr~n~A-llceA into such p~c~ing cells lines are transcribed into RNA and enc~ps~ teA into
infectious virions con~ ing the selectable fusion gene (and/or thc~ ic gene), but,
5 lacking the trans-acting gene products gag, pol and env, cannot synthesize thenecess~ry gag, pol and env proteins for enc~psi-l~ting the RNA into particles for
infecting other cells. The resulting infectious retrovirus vectors can therefore infect
other cells and inte~te a select~ble fusion gene into the cellular DNA of a host cell, but
cannot replicate. Mann et al. (Cell 33:153, 1983), for example, describe the
10 develo~l,le"t of various parl~ging cell lines (e.g., ~Ir2) which can be used to produce
helper virus-free stocks of recombinant retrovirus. Encapsidation in a cell lineharboring trans-acting ele...enls encoding an ecoL.~ic viral envelope (e.g., ~r2)
provides ecoLIopic (limited host range) progeny virus. ~lt~n~tively, assembly in a cell
line cont~inil-g amphotropic p~ in~ genes (e.g., PA317, ATCC CRL 9078; Miller
and Buuill~ole, Mol. Cell. Biol. 6:2895, 1986) provides amphotropic (broad host
range) progeny virus.
Nulll~,r~us provirus constructs have been used succes~fully to express foreign
genes (see, e.g., Coffin, in Weiss et al. (eds.), RNA Tumor Viruses, 2nd Ed., Vol. 2,
(Cold Spring Harbor Laboratory, New York, 1985, pp. 17-71). Most proviral
20 elements are derived from murine retroviruses. Retroviruses adaptable for use in
accordance with the present invention can, however, be derived from any avian or..~.,....~li~n cell source. Suitable retroviruses must be capable of infecting cells which
are to be the ,~ip;ent~ of the new genetic m~teri~l to be l,nl-~ ce~ using the retroviral
vector. Examples of suitable retroviruses include avian retroviruses, such as avian
25 elyl}lloblastosis virus (AEV), avian leukosis virus (ALV), avian myeloblastosis virus
(AMV), avian s~;ol"a virus (ASV), I~ujin&l,Ji sa,cullla virus (FuSV), spleen necrosis
virus (SNV), and Rous s~.;ol"a virus (RSV); bovine leukemia virus (BLV); feline
retroviruses, such as feline l~llk~mi~ virus (FeLV) or feline s~;ol~la virus (FeSV);
murine retroviruses, such as murine lel~kemi~ virus (MuLV); mouse Ill;~llllll~ly tumor
30 virus (MMTV), and murine sa,.;ol,la virus (MSV); and ~Ihl~ate retroviruses, such as
human T-cell lymphotropic viruses 1 and 2 (HTLV-1, and -2), and simian sarccl"a
virus (SSV). Many other suitable retroviruses are known to those skilled in the art. A
taxonomy of retroviruses is provided by Teich, in Weiss et al. (eds.), RNA TumorViruses, 2d ed., Vol. 2 (Cold Spring Harbor Laboratory, New York, 1985, pp. 1-
35 160). ~er~ ,d retroviruses for use in connection with the present invention are themurine retroviruses known as Moloney murine le!lkemi~ virus (MoMLV), Moloney
murine sal.;ul,la virus (MoMSV), Harvey murine s~-,-lla virus (HaMSV) and Kirsten
Wo 92/08796 2 0 9 6 2 2 2 pcr/us91/o8442
~,_ 15
murine sa,.;uma virus (KiSV). the sequences l~uil~,d to construct a retroviral vector
from the MoMSV gel OlllC can be obtained in conjun.;~ion with a pBR322 plasmid
sequence such as pMV (ATCC 37190), while a cell line producer of KiSV in K-BALB
cells has been d~osited as ATCC CCL 163.3. A deposit of pRSVneo, derived from
S pBR322 including the RSV LTR and an intact r.eomyci.l drug resi~t~nce marker is
available from ATCC under Accession No. 37198. Plasmid pPB101 comprising the
SNV genome is available as ATCC 45012. The viral genomes of the above
retroviruses are used to construct repliç~tion-defective retrovirus vectors which are
capable of integrating their viral genomes into the c~ullloso,~al DNA of an infected
10 host cell but which, once integrated, are inc~p~ble of replication to provide infectious
virus, unless the cell in which it is introduced cont~in~ other proviral el~ encoding
r~ cl;o~l active ITans-acting viral l~lul~;ns.
The selectable fusion genes of the present invention which are tr~n~duce~ by
retroviruses are expressed by placing the selectable fusion gene under the
15 transcriptional control of the e .h .-~ and pl~lllolt,. inco ~ led in the retroviral LTR,
or by placing them under the control of a heterologous tl~ CC ;l.l;onal control sequen~es
inserted between the LTRs. Use of both heterologous transcriptional control sequences
and the LTR transcriptional control sequences enables coe~l)lession of a therapeutic
gene and a selectable fusion gene in the vector, thus allowing selection of cells
20 t;~l,r~ssing speci~lc vector sequences encoding the desired th~_l~c..lic gene product.
Ol,tairi-lg high-level e~lession may require placing the therapeutic gene and/orselect~hle fusion gene within the retrovirus under the transcriptional control of a strong
heterologous enh~ncçr and ~r~lllot,~ GA~ ssion c~csette Many dirr~,.c.-l heterologous
enh~lcers and plulllolcl~ have been used to express genes in retroviral vectors. Such
25 enhancers or pn,lllolcls can be derived from viral or cellular sources, including
...~."u~ n genomes, and are preferably consliluli~e in nature. Such heterologousswil,lional control se4uences are discussed above with r~fel~nce to recombinant
e,.~,~ssion vectors. To be G~i~ssed in the t~n~uced cell, DNA se4u~,nces introduced
by any of the above gene transfer methods are usually e~ ssed under the control of an
30 RNA polymerase II plulllolei.
Particularly pl~re,l~d recombinant expression vectors for use in retroviruses
include pLXSN, pLNCX and pLNL6, and derivatives thereof, which are described by
Miller and Rosman, Biotechniques 7:980, 1989. These vectors are capable of
G~ ,s~ing heterologous DNA under the transcriptional control of the retroviral LTR or
35 the CMV p~u~l~otei~ and the neo gene under the control of the SV40 early region
l,io"loler or the retroviral LTR. For use in the present invention, the neo gene is
replaced with the bifunctional selectable fusion genes disclosed herein, such as the
wo 92/08796 2 0 Y 6 2 2 2 Pcr/US9l/08442
16 ~
HyTK selectqhle fusion gene. Construction of useful replirqtion-defective ret~,~viruses
is a matter of routine skill. The reslllting recombinant retroviruses are capable of
;oll into the chrnmoso-mql DNA of an infected host cell, but once in~ ted, are
inclq~pqble of replication to provide infectious virus, unless the cell in which it is
introduced contains another proviral insert encoding functionally active trans-acting
viral pl~Jteil s.
Uses of Bifunctional Selectable Fusion Genes
The selectqhle fusion genes of the present invention are particularly preferred
for use in gene therapy as a means for i~n~ryil~g, i~olvting or eliminqting cells, such as
som-q-tic cells, into which the selectqble fusion genes are introduced. In gene therapy,
somatic cells are removed from a patient, tr~n~d~ceA with a recombinant e,~plession
vector containing a theldl)e.llic gene and the select~ble fusion gene of the present
invention, and then l~,;nlluduced back into the patient. Somatic cells which can be used
as vehicles for gene therapy include helllalopoietic (bone marrow-derived) cells,
keratinocytes, hepatocytes, endothelial cells and fibroblasts (Friedman, science244: 1275, 1989). Alternatively, gene therapy can be accollll~lished through the use of
injectable vectors which ~ ce somatic cells in vivo. The fe~ibility of gene transfer
in hllm~n~ has been demonstrated (Kasid et al., Proc. Natl. Acad. Sci. USA 87:473,
1990; Rosenberg et al., N. Engl. J. Med. 323:570, 1990).
The select~ble fusion genes of the present invention are particularly useful forçl;.";n~ g g~n~tir~lly m--r1ified cells in vivo. In vivo .olimin~tion of cells ~A~ ssing a
negative selectable phenotype is particularly useful in gene therapy as a means for
ablating a cell graft, thereby providing a means for reversing the gene therapy
pl~)cedulc. For ey~mple7 it has been shown that ~tlministration of the anti-herpes virus
drug ganciclovir to transgenic ~nim~l~ e"~lessing the HSV-I TK gene from an
o~lobulin plUlllOt.el results in the selective ~bl~tit n of cells e~ hlg the HSV-I
TK gene (Heyman et al., Proc. Natl. Acad. Sci (USA) 86:2698, 1989). Using the
same transgenic mice, GCV has also been shown to induce full regression of Abelson
leukemia virus-induce~l lymphomas ((Moolten et al., Hurnan Gene Therapy 1:125,
1990). In a third study, in which a murine sal.;oma (K3T3) was infected with a
retrovirus e~l.,sshlg HSV-I TK and transplanted into syngeneic mice, the tumors
dur,ed by the sal~ioma cells were co~plet~ly er~1ir~t~ following ~ n~ with GCV
(Moolten and Wells, J. Natl. Cancerlnst. 82:297, 1990).
The bifunctional selectable fusion genes of the present invention can also be
used to façilit~te gene m~li~lr~tion by homologous l~cc,lllbination. Reid et al., Proc.
Natl. Acad. Sci. USA 87:4299, 1990 has recently described a two-step procedure for
Wo 92/08796 17 PCr/USs1/08442
,_
gene l.~o lirir~l;on by homologous recomhin~tion in ES cells ("in-out" homologous
~co.~bin~tion) using the HPRT gene. Briefly, this procedure involves two steps: an
"in" step in which the HPRT gene is embedded in target gene se~u~nces, transfected
into HPRT- host cells and homologous lecolllbinants having incol~,o.~l~d the HPRT
S gene into the target locus are identified by their growth in HAT l1,r"1;ll.11 and genomic
analysis using PCR. In a second "out" step, a construct containing the desired
rep1~r~ t sequences embe~ ~ in the target gene sequences (but without the HPRT
gene) is transfected into the cells and homologous l~lllbin~lls having the repU ~emrnt
se~lu~nces (but not the HPRT gene) are icol~tçd by negative selection against HPRT+
10 cells. ~lthough this procedure allows the intro~ ction of subtle mut~tinnc into a target
gene without introducing selectable gene sequences into the target gene, it requires
positive selection of transformants in a HPRT- cell line, since the HPRT gene isrecessive for positive selection. Also, due to the inefficient expression of the HPRT
gene in ES cells, it is ..ecess..y to use a large 9-kbp HPRT mini-gene which
15 comp1ir~tes the consllu~;~ on and prop~g~tion of homologous r~combination vectors.
The selectable fusion genes of the present invention provide an improved means
whereby "in-out" homologous recombination may be pc.rolllled. Because the
select~hle fusion genes of the present invention are dolllit~allt for positive selection, any
wild-type cell may be used (i.e., one is not limited to use of cells deficient in the
20 select~ble ~henoly~e). Moreover, the size of the vector co~ init~g the sç1ect~ble fusion
gene is reduced cignifif ~nt1y relative to the large HPRT mini-gene.
By way of illustration, the HyTK select~hle fusion gene is used as follows: In
the first "in" step, the HyTK selectable fusion gene is embeclde~l in target gene
sequences, transfected into a host cell, and homologous recombinants having
25 incc,l~ ed the HyTK selfxt~bl~ fusion gene into the target locus are identifi~1 by their
growth in mrfli-1m cont~ining Hm followed by genome analysis using PCR. The
HyTK+ cells are then used in the second "out" step, in which a construct con~it-i~-g the
desired rep1.~rç...f nt se~ ences çmbel1fl~ in the target gene sequences (but without the
HyTK sele~t~hle fusion gene) is t,~n~r~t~ into the cells. Hr~mologous recombinants
30 are i~;ol7~'ed by selective ç1imin~tion of HyTK+ cells using ganciclovir followed by
~,_n~llle analysis using PCR.
Wo 92/08796 ~ - 2 0 9 6 2 2 2 Pcr/US9l/08442
18
FXAMP~ FS
F.Y~mple 1
Construction and Cha.~.cte. ;,~ n of
Plasmid Vectors Co~ g HyTK Selectable Fusion Gene
A. Construction of the Bifunctional HyTK Selectable Fusion Gene. The hph
10 and HSV-I TK genes were first placed under the regulatory control of the HCMVp~ otel in tgCMV/hygro and tgCMV/TK, respcclively. Plasmid tgCMV/hygro
(Figure 1) consists of the following ele.l~F II~: the BalI-SstII fragment containing the
HCMV IE94 promoter (Boshart et al., Cell 41:521, 1985); an oligonucleotide
coh~inillg a sequence co"rolming to a consensus translation initiation sequence for
lll~llllll~li~n cells (GCCGCCACC ATG) (Kozak, Nucl. Acids Res. 15:8125, 1987);
nucleotides 234-1256 from the hph gene (Kaster et al., Nucl. Acids Res. 11:6895,1983), encoding hy~o,llycin phospho~ sr~,."se; the BclI-BamHI fragment from the
SV40 genome (Tooze, J., ed., Molecular Biology of Tumor Viruses, 2nd Ed. DNA
Tumor Viruses. Cold Spring Harbor Laboratory, New York, 1981), cont~ining the
SV40 early region polyadenylation sequence; the NruI-AlwNI fragrnent from pML2d
(Lusky and Botchan, Nature 293:79, 1981), c~nl~ ing the ba~ teri~l replication origin;
and the AlwNI-AadI fragment from pGEM1 (Promega CoIporation), co~ ing the B-
e gene.
Plasmid tgCMV/TK (Figure 1) is similar to tgCMV/hygro, but contains
nucleotides 519-1646 from the HSV-I TK gene (Wagner et al., Proc. Natl. Acad. Sci.
USA 78:1441, 1981) in place of the hph gene.
Plasmid tgCMV/HyTK (Figure 1), con~aining the selectable fusion gene
com~Jlisiilg the hph gene and the HSV-I TK gene, was constructed by inserting the
1644-bp SpeI-ScaI fragment from tgCMV/hygro between the SpeI and MluI sites of
tgCMV/TK. Before ligation, the MluI site in the HSV-I TK gene was treated with T4
DNA polymerase to allow blunt end ligation with the ScaI site, thus preserving the
open reading f~me. Translation of this fused gene (referred to as the HyTK select~ble
fusion gene) is e~ec~ed to generate a single bifunctional fusion protein, concicting of
amino acids 1-324 from the hph protein and amino acids 10-376 from the HSV-I TK
3~ protein. The C t~ fnal 17 amino acids of the hph protein, and the N-terminal 9 amino
acids of the TK protein, are deleted in the bifunctional fusion protein.
B. Dominant Positive Selec~ion of Cells Containing the HyTk Selectable
Fusion Gene. To demonstrate that the HyTK selectable fusion gene encodes both hph
WO 92/08796 ~ 2 0 9 G 2 2 2 PCr/US91/08442
19
,_
and TK enzymatic activities, the frequencies with which tgCMV/HyTK conf~lled
hygl~ lyciil recict~nce (Hmr) (in NI~V3T3 cells and Rat-2 cells), and the ability to
grow in ,.,F.l;...,, co~ ni~g hy~ t~;nF" .~llin~.in, and thymidine (HAl~) (in Rat-
2 cells), were co~ ar~d with those of the parental pl~cmiAc, tgCMV/hygro and
S tgCMV~rK, l~ ely.
NIH/3T3 cells were grown in Dulbecco's MoAifiecl Eagle Medium (DMEM;
Gibco Labol.1tolies) supplçrnenteA with 10% bovine calf serum (Hyclone), 2 mM L-glutamine, 50 U/ml penicillin, and 50 ~Lg/ml sL~ olllycill at 37 ~C in a hnmi-lifie~l
almo~l)hF.e supplc ,u ~-led with 10% CO2. TK- Rat-2 cells (I'opp, Virology 113:408,
1981) were grown in DMEM suppl~ e~ with 10% fetal bovibe serum (Hyclone), 2
mM L-~lul~...ine, 50 U/ml penicillin, and 50 ~g/ml s~plol~lycin at 37~C in a
humidifie~ atmosphere suppk --~nte~l with 10% CO2. NI~3T3 and Rat-2 cells were
transfected with the DNA vectors described above by electroporation, as follows.Exponentially growing NIH/3T3 and Rat-2 cells were harvested by trypsinization,
15 washed free of serum, and resuspended in DMEM at a concentration of 107 cells/ml.
Supercoiled plasmid DNA (5 ~g) was added to 800 111 of cell sus~,ension (8x106 cells),
and the mixture subjected to electroporation using the Biorad Gene Pulser and
Capacitance Extender (200-300 V, 960 ~F, 0.4 cm electrode gap, at ambient
l~mp~"~ture). Following transfection, the cells were l~tul~cd to 9-cm dishes and20 grown in non-selective .n~ .... After 24 hours, the cells were trypsini7~, seeded at
6xlOs per 9-cm dish, and allowed to attach overnight. The non-selective ~ ,... was
repl3ced with selective m.otlium (containing 500 ~g/ml hy~lu~l~ycin B for NIH3T3cells, and 300 llg/ml hygromycin B or HAT for Rat-2 cells), and selection was
continue~ for applu,.;...~tely 10-12 days until colonies were evident. The plates were
25 stained with methylene blue and counlcd. The results are shown in Table 1 below.
The r,ull.~el of colonies l~polled is the average nulllb~ of colonies per 9-cm dish.
TABLE 1
Positive Selection Using HyTK Fusion Gene
NIM~T3 Cells Rat-2 Cells
No. Hmr No. Hmr No. HAl~
pl~cmi~1 Colonies Colonies Colonies
tgCMV~ygro 45 368 n.t.
tgCMV/I~ n.t. n.t. 356
tgCMV/HyTK 100 428 124
n.t.= not tested.
Wo 92/08796 2 0 ~ ~ 2 2 ~ pcr/us91/o8442
~
In both cell lines, tgCMV/HyTK gave rise to Hmr colo~ P,s at a slightly higher
rl~que.lcy than tgCMV/hygro. However, in Rat-2 cells, tgCMV/HyTK was slightly
less efficiPnt than tgCMV~TK in gellc~a~ing HATr colonies. This de- ~-o~ .dles that the
5 HyTK sel~Pct~kle fusion gene encodes both hph and TK CllL~I11aliC activities, although
with altered effi~i~Pn~iPs
C. Negative Selection of Cells Contailling the HvTK Selectable Fusion Gene.
To investigate the utility of the HyTK se1P,ct~kle fusion gene for negative selection, the
colonies reslllting from each L,~sr~;l-on (Table 1) were pooled and e~p~nded into cell
10 lines for further analysis. The Hmr NW3T3 cell pools and the Hmr and HATr Rat-2
cell pools were tested for GCVS in a short term cell proliferation assay as follows.
The transfected NW3T3 and Rat-2 cells (3 x 104 of each) were seeded into 9-
cm tissue culture dishes in complete growth m~Aillm, and allowed to attach for 4 hours.
The ..~1;l.... was then suppk--.-- -~t~A with various concwllld~ions of GCV (Syntex, Palo
Alto, CA), and the cells inr~lb~eA for an ~d-lition~l 60 hours. At this time, the "~
was removed, the attached cells were harvested by trypsini7~tion and stained with
t~ypan blue, and viable cells were counted. Cell growth was expressed as a fraction of
the cell growth observed in the absence of GCV. The results shown are the average of
tripli~te assays.
The results shown in Figure 3 demonstrate that the HyTK selectable fusion
gene confers GCVS in NW3T3 cells. The degree of inhibition of cell growth was
propc.llional to the concentration of GCV used, and approached 100% at a
conc~ lalion of 1 ~M. In contrast, NW3T3 cells ~ Ç~t~d with tgCMV/hygro were
not adversely arre ;led by GCV over the range of concentlations tested (0.03 ~M - 1.0
~lM).
The results shown in Figure 4 in-lic~te that the HyTK select~ble fusion gene is
more effective than the HSV-I TK gene for negative selectiorl in Rat-2 cells. Growth of
Rat-2 cells ~lsr~cl~ with tgCMV/HyTK was almost completely inhibited even at thelowest concentration of GCV used (0.03 IlM), whether the cells were initially selected
for Hmr or HATr. Growth of Rat-2 cells transfected with tgCMV/hygro was not
inhibitGd by GCV over the range of concentrations tested (0.03 ~lM - 1.0 IlM). The
growth of Rat-2 cells transfected with tgCMVrrK was inhibited by GCV, but the
concenl ~ ations required for growth inhibition were much higher than those required to
inhibit the growth of Rat-2 cells transfected with tgCMV/HyTK. The Rat-2 cells
llansLcl~d with tgCMV/TK were less sensitive to GCV than the Rat-2 cells transfected
with tgCMV/HyTK. This appears to conflict with the result obtained when the two
genes were used for positive selection in Rat-2 cells (Table 1), which in-lic~ted that the
21 '~ O ~
HyTK selectable fusion gene was less effective than the HSV-I TK gene in confe.-.ng
HATr. A further observation conce,ning the relative sensitivities of these cell lines to
GCV was that the NIHJ3T3 cells transfected with tgCMV/Hyl K were less sensitive to
GCV than the Rat-2 cells transfected with tgCMV/HyTK.
D. Northern Analvsis of Transfected Cell Lines. To investigate the basis for
the the dirre.e,.lial sensitivities of the Hmr and HATr NI~V3T3 and Rat-2 cell pools to
GCV (Figures 3 and 4), and the altered efficiency with which the HyTK selectablefusion gene gave rise to Hmr and HATr colonies (Table 1), Northern blots of mRNAfrom each cell pool were probed with se.lu.,nces from the hph and HSV-I TK genes, as
follows.
Polyadenylated mRNA was prepared according to standard procedures
(Ausabel et al., eds., Current protocols in Molecular Biology. Wiley, New York.,1987). RNA samples (10 llg) were electrophoresed through 1.1% agarose gels
containing formaldehyde as described (Ausabel et al., supra). Following
electrophoresis, the gels were inverted and blotted by capillary transfer in 20 x SSC
onto Duralon UV nylon membranes (Stratagene). After fixing the mRNA to the
membrane by UV-irradiation (0.12 J/cm2), the membranes were inrubat~P~ in Starlc's
buffer (50% formamide 5 x SSC, 50 mM potassium phosphate (pH 6.5), 1% SDS,
0.1% Ficoll, 0.1% PVP, 0.1% BSA, 300 ~g/ml sheared and denatured salmon sperm
DNA, 0.05% Sarkosyl) at 50 ~C for several hours. A uniforrnly labelled single
stranded ~ntisence RNA probe specific for hph was plepa,~ (Ausabel et al., supra), 1
x 106 cpm/ml were added to the hybridization mixture, and the incubation was
continued at 63 ~C for 15 h. The ~ ,n-~lane was then washed in 0.1 x SSC, 0.1% SDS
at 63 ~C, and exposed to autoradiographic film (Kodak XAR-5~. For detection of
HSV-I TK and B-~tin sequences, gel-purified restriction fr~g~nçn~s from the HSV-I
TK and B-Actin genes were ra~inl~be~ by random priming (Ausabel et al., supra).
Membranes were pre-hybridized in Starks buffer at 42~C for several hours, after which
1 x 106 cpm/ml of probc was added to the hybridization mixturc and incubation
continued at 42~C for 15 hours. The ~,.e.l,b~anes were then washed in 6 x SSC, 1%
SDS at 63~C, and exposed to anto~l;ographic film (Kodak XAR-5).*
In both Rat-2 and NI~V3T3 cells, the steady state level of mRNA ~tectcd with
the hph probe was higher in the cells transfected with tgCMV~hygro than the cells
c~e;l with tgCMV/HyTK and sPlectP~ for Hmr (Figure 5, gel A, lanes 5 and 6).
This may indicate that a higher level of expression of the hph gene is I~UU'~i tO confer
recictanre to equivalent levels of hy~. OlllyCu~ B (300 ~Lg/ml in Rat-2, and 500 llg/ml in
NI~V3T3), due to the fact that the bifunrtit)n~l fusion protein is more effective than the
hph protein at inactivating hygromycin B, or is more stable than the kph protein. This
* TradeMark
~o96222
WO 92/08796 pcr/us91 /0X442
22
co--ch~c;on is su~ ed by the results in Table 1, which show that tgCMV/HyTK gaverise to a slightly greater number of Hmr colonies in both cell lines than did
tgCMV/hygro.
The RNA Northern analysis also in-lir~ted that the Rat-2 cells transfected with
S tgCMV/TK expressed a steady state level of mRNA similar to the Rat-2 cells
transfected with tgCMV/HyTK and selecte~ for HATr (Figure 5, gel B, lanes 2 and 4).
However, tgCMV/TK gave rise to a greater number of HATr cells than did
tgCMV/HyTK (Table 1). This suggests that the HyTK sel~ct~ble fusion protein is less
effective than the HSV-I TK protein at phos~hol ylating thymidine, or is less stable than
the HSV-I TK protein.
Finally, the Rat-2 cells transfected with tgCMV/HyTK e~cpr~ssed steady state
levels of mRNA several fold higher than (when selected for Hmr: Figure 5, gel B, lane
3), or similar to (when selected for HATr; Figure 5, gel B, lane 4), the Rat-2 cells
transfected with tgCMV/TK (Figure 5, gel B, lane 2). However, both the Rat-2 cell
pools transfected with tgCMV/HyTK were over 30-fold more sensitive to GCV than
the Rat-2 cells transfected with tgCMV/TK (Figure 4). This suggests that the
bifunctional fusion protein is m~rk~-~1ly more effective than the HSV-I TK protein at
phosphorylating ganciclovir, or is m~rk5~dly more stable than the HSV-I TK protein.
The increased ability of the bifunctional fusion protein to confer GCVS, and
conco~ t decreased ability to confer HATr, suggests that the substrate affinity of the
bifunctional fusion protein is altered relative to that of the HSV-I TK protein, rather
than the stability.
Example 2
Construction and Charaet . ;~ti~-n of
Retroviral Vectors Con~ini"~ HyTK Selectable Fusion Gene
A. Construction of Retrov*al Vectors. Two retroviral expression vectors
containing the HyTK selectable fusion gene were constructed. In the first,
tgLS(+)HyTK, the HyTK selectable fusion gene was placed under the regulatory
control of the promoter present in the retroviral LTR. In the second, tgLS(-
)CMV/HyTK, the HyTK select~ble fusion gene was placed under the regulatory control
of the HCMV ~lolllOte~.
The retroviral expression vector tgLS(+)HyTK (the proviral structure of which
is shown in Figure 2) consists of the following elem~.nt~: the 5' LTR and sequences
through the PstI site at nucleotide 984 of MoMSV (Van Beveren et al., Cell 27:97,
1981); sequences from the PstI site at nucleotide 563 to nucleotide 1040 of MoMLV
9 ~
23
(ShiMick et al., Nature 293:543, 1981), inco~ ing point mutations (ATG ~ TAG)
which elimin~te the Pr65 gag translation initiation codon (Bender et al., J. Virol.
61:1639, 1987); a fragment from tgCMV/HyTK, containing the HyTK selectable
fusion gene; sequences from nucleotide 7764 and through the 3' LTR of MoMLV
S (Shinnick et al., Nature 293:543, 1981); the NruI-AlwNI fragment from pML2d
(Lusky and Botchan, Nature 293:79, 1981), containing the bacterial replication origin;
and the AlwNI-AatII fragment from pGEM1 (~UlllCg1 Corporation), containing the B-
l~t~ n~ce gene.
The retroviral expression vector tgLS(-)CMV/HyTK (the proviral structure of
which is shown in Figure 2) is similar to tgLS(+)HyTK, but carries a point mut~tion
(AGGT ~ AGGC) which elimin~t~s the MoMSV-derived splice donor sequence
(transferred from the retroviral vector, ~H [Overell et al., Mol. Cell. Biol. 8:1803,
1988]), and contains the HCMV promoter upstream of the HyTK selectable fusion
gene sequences.
The retroviral expression vector tgLS(+)HyTK/stop (the proviral structure of
which is shown in Figure 2) was derived from tgLS(+)HyTK by inserting the universal
translation terminator oligonucleotide (Pharmacia),5'-GCTTAATTAATTAAGC-3', at
the NaeI site located near the junction of the hph and HSV-I TK sequences of theHyTK selectable fusion gene.
B. Generation of Stable Cell Lines Producing Retroviral Vectors. Stable Y'~
pac~ging cell lines were gcneldt~,d which produce the above ecotropic retroviruses as
follows. Y'2 cells (Mann et al., Cell 33:153, 1983) were grown in Dulbecco's
Modified Eagle Medium (DMEM; Gibco Laboratories) suppl~ with 10% bovine
calf serum (Hyclone), 2 mM L-glutamine, 50 U/ml penicillin, and 50 ,ug/ml
~L~c~o~llycin at 37 ~C in a humidified atmosphere supplemented with 10% CO2.
PA317 cells (Miller and Buttimore, Mol. Cell. Biol. 6:2895, 1986) were grown in
DMEM supple-"~ht~,d with 10% fetal bovine serum (Hyclone),2 r,nM L-glu~min~,50
U/ml penicillin, and 50 ~lg/ml ~ o,l,ycin at 37 ~C in a h~lrnirlified atmospheresupp~ "t~cl with 10% CO2.
The retroviral expression vectors described above were first transfected into
PA317 ~llphullulJ;c p~ g,ing cells by ele~,~upol~,tion. Amphollul,ic virions produced
by the transiently tr~ncfecte~ PA317 pacl~ging cells were then used to infect the ~2
cells as follows. E~l.ol-~ .h~lly growing PA317 cells were harvested by trypsini7~tion~
washed free of serum, and resuspen-le~ in DMEM at a conce.l~ldtion of 107 cells/ml.
Supercoiled plasmid DNA (5 ,ug) was added to 800 ~11 of cell s~lcpen~ion (8x106 cells),
and the mixture subjected to electroporation using the Biorad Gene Pulser*and
Capacitance Extender (200-300 V, 960 IlF, 0.4 cm electrode gap, at ambient
*TradeMark
A
Wo 92/08796 ~ ~ Q g 6 2 22 PCr/USsl/08442
24
.~_
t~ JC~Iulc). The transfected PA317 cells were then transfared to a 9-cm tissue
culture dish CO~ g 10 ml of complete g~wth ~ l.ll supplc...~ ed with 10 mM
sodium butyrate (Sigma Chemical Co.), and allowed to attach overnight. After 15
hours, the meAi~lm was removed and replaced with fresh ,.. ~.l;.. After a further 24
5 hours, the me~ m containing transiently produced amphotropic retrovirus particles
was harvested, centrifuged at 2000 Tpm for 10 min, and used to infect the ~2 ecotropic
parl~ging cells. E~l)oncnlially dividing ~2 cells wae plated at a density of 106 cells
per 9-cm tissue culture dish, and allowed to attach overnight. The following day, the
medium was removed and replaced with serial dilutions of the virus-containing
10 supernatant (6 mVdish) in me-lillm supplem~nted with 4 ~g/ml polybrene (SigmaChPmic~l Co.). Infection of the ~2 cells by the viral particles was allowed to proceed
overnight, and then the ~upe,natant was replaced with complete growth medium.
Infected cells were selected for drug resict~nce after a further 8-24 hours of growth by
adding hyglulllycill B (Calbiochem) to a final concel,~ on of 500 ~g/ml. Colonies of
15 Hmr cells were isolated using cloning cylinders 12-14 days later, and individually
e-~p~nde~l into bulk cultures for analysis. Southern analysis (data not shown) revealed
that the proviral ~l~uclules were intact in six out of six independent clones, indicating
that the HyTK selectable fusion gene is c4~ .At;ble with the retroviral life cycle.
C. Transduction of Hmr~ HATr. and GCVS by tgLSf+)HyTK and tgLS(-)
20 CMV/HyTK Retroviral Expression Vectors. The infected ~2 clones were titered on
NIH/3T3 cells (selecting for Hmr), and on Rat-2 cells (selecting for Hrnr, or for HATr)
(Table 2), as follows. The ~2 clones ~l~luc;ng the virus were grown to conflue-nce in
9-cm tissue culture dishes, then fed with 15 ml of drug-free medium. After an
overnight incubation, aliquots of su~l,.atant were taken for assay. Exponentially
25 dividing NIH/3T3 or Rat-2 cells were harvested by trypsini7~tion and seeded at a
density of 2.5 x 104 cells per 35 mm well in ~well tissue culture trays. The following
day, the m~ m was repl~ced with serial ~iilutiolls of virus-con~;1inil-g supern~t~nt (1
mVwell) in m~ m supple .~ ~ with 4 ~lg/ml polybrene (Sigma Chemir~l Co.). All
sUp~rn~t~ntc were centrifuged at 2000 rpm for 10 min before use to remove viable30 cells. Infection was allowed to proceed overnight, and then the supernatant was
replaced with complete growth "lediulll. Infected cells were selected for drug
recict~nce after a further 8-24 hours of growth by adding hy~o~l~ycin B (Calbiochem)
to a final concentration of 500 ~lg/ml (NlH/3T3 cells) or 300 ~g/ml (Rat-2 cells), or by
adding HAT supplem~nt (Gibco) (Rat-2 cells). After a total of 12-14 days of growth,
35 cells were fixed in situ with 100% ..~ nol, and stained with methylene blue.
As shown in Table 2, below, both retroviruses con~ d Hmr (to NIEV3T3 and
Rat-2 cells) and HATr (to Rat-2 cells). All viruses were harvested from a clone of
wo 92/08,g6 2 0 9 6 2 2 2 PCr/VSsl/08442
i~lr~t~ ~2 cells.
TABLE 2
S Titers of f COll~iC retroviruses produced by ~2
pacL-~ing cells on NI~V3T3 cells and Rat-2 cells.
NIH/3T3 l~t-2
Hmr Hrnr HATr
Virus CFU/ml CFU/ml CFU/ml
tgLS(+)HyTK 1.8 x 107 1.6 x 107 4 x 106
clone 5.5
tgLS(-)CMV/HyTK 1 x 106 1 x 106 8 x 105
clone 6.2
To demonstrate that the tgLS(+)HyTK and tgLS(-)CMV/HyTK retroviruses
also conrell~,d GCVS, Nl~/3T3 cells infected with the two retroviruses, were selected
for Hmr (500 ~g/ml) for 10 days, and then pooled, expanded, and tested for GCVS in
the following long-term cell proliferation assay.
Uninfected NIH/3T3 cells and the infected NIH/3T3 cell pools were plated at
relatively low cell density (104 cells/9-cm dish) in complete growth medium and
allowed to attach for 4 hours. The ., .e l;. .", was then supplç . .~ nleA with hy~ y~ B
(500 ~g/ml), with or without 1 ~lM GCV, and the cells incubated for a period of 10
days. The mfulillm was then removed and the cells were fixed in situ with 100%
meth~nol and stained with methylene blue. The growth of both cell pools, as measured
by colony formation, was almost completely inhibited by GCV (Figure 6, plates e and
g)~ in~ic~ing that both retroviruses confe.l~,d Hmr and GCVS. Uninfected NI~U3T3were resistant to this concf.~ l;on of GCV, and grew to a cc nfl-lf nt monolayer (Figure
6, plate b), but were completely killed by 500 ~g/ml Hm (Figure 6, plate c). Colonies
of cells resistant to both Hm and GCV were obtained at a low frequency (104-10-3)
from the retrovirus-infected populations (Figure 6, plates e and g). The proviruses
present in the cells that gave rise to these colonies had likely suffered point mutations,
or very small deletions or realT~ngf ...- nl~ in the HSV-I TK moiety which elimin~ted the
ability to phosphorylate GCV. Similar results were also obtained with Rat-2 cell lines
infected with tgLS(+)HyTK or with tgLS(-)CMV/HyTK (data not shown).
WO 92/087962 D ~ 6 ~ 2 2 PCr/US91/OX442
26
F~YPnIPIe 3
Evidence for the ~uduclion of a Bifunctional Selectable Fusion Protein
In HSV-I i.,recled cells, the HSV-I TK gene normally utilizes three tr~n~l~tion
iti~tion sites, and encodes three nested polypeptides which all possess TK activity
(Haarr et al., J. Virol. 56:512, 1985). Since the HyTK selectable fusion gene retains
two of these initi~tion codons, it was conceivable that, as a result of translation
10 initi~tion at one or both of these internal AUG codons, the HyTK select~hle fusion gene
might also encode nested polypeptides pOSSGS~ g TK activity. The bif~ ;o.-~l fusion
protein, while lc~ining hph activity, might or might not possess TK activity. To rule
out this possibility, an oligonucleotide sequence, 5'-GCTTAATTAATTAAGC-3',
bearing translation termination codons in all three reading frames was introduced into
15 the HyTK select~ble fusion gene in tgLS(+)HyTK, gen~ ~i--g the construct design~ted
tgLS(+)HyTK/stop (Figure lB). The oligonucleotide was inserted at a NaeI site
dowl.s~,all. of the hph-derived sequences, but u~ ,a~ll of the two internal AUG
codons in the HSV-I TK-derived sequences of the HyTK selectable fusion gene
(Figure lB). The tgLS(+)HyTK and tgLS(+)HyTK/stop retroviral e~lession vectors
20 were transfected into ~2 cells, and the trAn~iently produced virus was used to infect
Rat-2 cells, which were then selected for Hmr or HATr (Table 3). The retroviral
c~ ession vector tgLS(+)HyTK tr~nsduced both Hmr and HATr, but retroviral
c~ ,ssion vector tgLS(+)HyTK/stop was only able to tr~n~Auce Hmr. Insertion of the
translation termin~tion codons completely abolished the ability of the retrovirus to
25 ~ ce HATr, inAi~ting that the internal translation initiation codons are not utilized
in the HyTK select~hle fusion gene, and the HyTK selectable fusion gene does indeed
encode a bifunctional fusion protein. Viruses were harvested from transiently
transfected ~2 cells.
TABLE 3~0
Titers of cCOI~u~iC retroviruses produced tr~n~i~-ntly in ~2
p~ ng cells on NI~3T3 cells and Rat-2 cells.
NIH3T3 Rat-2
Hmr Hmr HATr
Virus CFU/ml CFU/ml CFU/ml
tgLS(+)HyTK 4.5 x 104 9.5 x 103 1.1 x 104
tgLS(-)CMV/~IyTK 2.6 x 104 5.9 x 104 0
Wo 92/08796 27l~ 9 6~2 2 2 Pcr/US91/0x442
27
As d~sçribeA in the above eY~n~rles, retroviral e~ ;,sion vectors co~ ing the
HyTK select~hle fusion gene were constructed and used to demonstrate the efficacy of
the HyTK selectable fusion gene for positive and negative selectiQn in NW3T3 and- Rat-2 cells. High titer virus stocks were generated, which conferred both Hmr and
5 HATr on infected cells. Infected cells contained ul,le~,anged proviruses and were
killed (>99.9%) by GCV. The HyTK selectable fusion gene was slightly more
effective than the hph gene at conf~lfing Hmr in both NI~J3T3 and Rat-2 cells (Table
1). Genetic evidence that the HyTK selectable fusion gene encodes a bifunctionalfusion protein posse~;ng hph and HSV-I TK enzymatic activities was obtained by
10 inserting tr~nsl~tion termination codons into the HyTK selectable fusion gene (in
tgLS(+)HyTK/stop; Figure 2), downstream of the hph-derived sequences, but
U~ iLlGalll of the HSV-I TK-derived sequences. As would be expected if the HyTK
selP~ct~bl~ fusion gene enco~e~ a bifunctional fusion protein, insertion of the translation
n~lion codons left the ability of the virus to confer Hmr intact, but abolished the
15 ability of the retrovirus to tr~n~uce HATr (Table 3). When cc,lll~a~ed with the HSV-I
TK gene in Rat-2 cells, the HyTK sele~t~ble fusion gene was slightly less effective at
conferring ability to grow in HAT ms~ m (Table 1), but m~rkeAly more effective at
confe,.,ing GCVS (Figure 4). These observations cannot be explained on the basis of
the relative steady state levels of mRNA e~p,cssion (Figure 5), nor on the basis of
20 changes in the stability of the HyTK selectable fusion protein. The a~ent
contradiction might be explained by hypothesi7ing that the HSV-I TK-derived moiety
of the HyTK sele~t~ble fusion protein possesses a substrate affinity dirr~,lc;, t from that
of the wild-type HSV-I TK protein (possibly due to conformational change), with a
reduced ability to phosphorylate thymidine and an increased ability to phosphorylate
25 GCV. Altered substrate affinities have been noted previously in a number of
pathogenic drug-resistant strains of HSV-I, which encode mutant TK proteins thatexhibit a reduced ability to phosphorylate thymidine analogs, yet retain the ability to
phosphorylate thymidine (Larder et al., J. Biol. Chem. 258:2027, 1983; Palu et al.,
Virus Res. 13:303,1989; Larder and Darby, Ann~iral Res. 4:1,1984). The slightly
30 increased efficiency with which the HyTK selectable fusion gene confers Hmr, relative
to the h~h gene (Table 1), may be due to an increase in protein stability, or an increased
spe~ific activity of the phosphotransferase.
Moreover, a single approximately 76 kD protein was specifically
immllnoprecipitated by a rabbit polyclonal antiserum directed against HSV-I TK from
35 extracts of cells expressing the HyTK selectable fusion gene. Thus the phenotype
conferred by the HyTK selectable fusion gene was not due to internal translationiniti~tion in the HSV-I TK derived moiety of the gene, and the HyTK selectable fusion
PCI / US9 1 /08442
WO 92/08796 ,~
28 _
gene does indeed encode a bifiln~inn~l sek~!;3hle fusion gene.
WO 92/08796 2~0 9 6 2 2 2 P~/US91/08442
.~ 29
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: Lupton, Stephen D.
(ii) TITLE OF INVENTION: Bifunctional Selectable Fusion Genes
(iii) NUMBER OF SEQUENCES: 2
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: Immunex Corporation
(B) STREET: 51 University Street
(C) CITY: Seattle
(D) STATE: WA
(E) COUNTRY: USA
(F) ZIP: 98101
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Diskette, 3.50 inch, 800 Rb storage
(B) COMPUTER: Apple Macintosh
(C) OPERATING SYSTEM: Maintosh
(D) SOFTWARE: Microsoft Word
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE:
(C) CLASSIFICATION:
(viii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: 07/612,326
(B) FILING DATE: 13-NOV-1990
(ix) ATTORNEY/AGENT INFORMATION:
(A) NAME: Wight, Christopher L.
(B) REGISTRATION NUMBER: 31, 680
(C) REFERENCE/DOCKET NUMBER: 2702 -A
(x) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (206) 587-0430
(B) TELEFAX: (206) 587-0606
(C) TELEX: 756822
(2) INFORMATION FOR SEQ ID NO:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 2076 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: double
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
- (iii) HYPOTHETICAL: N
(iv) ANTI-SENSE: N
(ix) FEATURE:
(A) NAME/REY: CDS
WO 92/08796 2 0 9 6 2 2 2 PCI /US91/08442
.
(B) LOCATION: 1..2076
(D) OTHER INFORMATION:
(ix) FEATURE:
(A) NAME/KEY: mat_peptide
(B) LOCATION: 1..2073
(D) OTHER INFORMATION:
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:l:
ATG AAA AAG CCT GAA CTC ACC GCG ACG TCT GTC GAG AAG TTT CTG ATC 48
Met Lys Lys Pro Glu Leu Thr Ala Thr Ser Val Glu Lys Phe Leu Ile
1 5 10 15
GAA AAG TTC GAC AGC GTC TCC GAC CTG ATG CAG CTC TCG GAG GGC GAA 96
Glu Lys Phe Asp Ser Val Ser Asp Leu Met Gln Leu Ser Glu Gly Glu
GAA TCT CGT GCT TTC AGC TTC GAT GTA GGA GGG CGT GGA TAT GTC CTG 144
Glu Ser Arg Ala Phe Ser Phe Asp Val Gly Gly Arg Gly Tyr Val Leu
CGG GTA AAT AGC TGC GCC GAT GGT TTC TAC AAA GAT CGT TAT GTT TAT 192
Arg Val Asn Ser Cys Ala Asp Gly Phe Tyr Lys Asp Arg Tyr Val Tyr
CGG CAC TTT GCA TCG GCC GCG CTC CCG ATT CCG GAA GTG CTT GAC ATT 240
Arg His Phe Ala Ser Ala Ala Leu Pro Ile Pro Glu Val Leu Asp Ile
GGG GAA TTC AGC GAG AGC CTG ACC TAT TGC ATC TCC CGC CGT GCA CAG 288
Gly Glu Phe Ser Glu Ser Leu Thr Tyr Cys Ile Ser Arg Arg Ala Gln
GGT GTC ACG TTG CAA GAC CTG CCT GAA ACC GAA CTG CCC GCT GTT CTG 336
Gly Val Thr Leu Gln Asp Leu Pro Glu Thr Glu Leu Pro Ala Val Leu
100 105 110
CAG CCG GTC GCG GAG GCC ATG GAT GCG ATC GCT GCG GCC GAT CTT AGC 384
Gln Pro Val Ala Glu Ala Met Asp Ala Ile Ala Ala Ala Asp Leu Ser
115 120 125
CAG ACG AGC GGG TTC GGC CCA TTC GGA CCG CAA GGA ATC GGT CAA TAC 432
Gln Thr Ser Gly Phe Gly Pro Phe Gly Pro Gln Gly Ile Gly Gln Tyr
130 135 140
ACT ACA TGG CGT GAT TTC ATA TGC GCG ATT GCT GAT CCC CAT GTG TAT 480
Thr Thr Trp Arg Asp Phe Ile Cys Ala Ile Ala Asp Pro His Val Tyr
145 150 155 160
CAC TGG CAA ACT GTG ATG GAC GAC ACC GTC AGT GCG TCC GTC GCG CAG 528
His Trp Gln Thr Val Met Asp Asp Thr Val Ser Ala Ser Val Ala Gln
165 170 175
GCT CTC GAT GAG CTG ATG CTT TGG GCC GAG GAC TGC CCC GAA GTC CGG 576
Ala Leu Asp Glu Leu Met Leu Trp Ala Glu Asp Cys Pro Glu Val Arg
180 185 190
CAC CTC GTG CAC GCG GAT TTC GGC TCC AAC AAT GTC CTG ACG GAC AAT 624
His Leu Val His Ala Asp Phe Gly Ser Asn Asn Val Leu Thr Asp Asn
195 200 205
WO 92/08796 ' ~ 2 0'9 6 2 2 2 P~/US91/08442
.._
GGC CGC ATA ACA GCG GTC ATT GAC TGG AGC GAG GCG ATG TTC GGG GAT 672
Gly Arg Ile Thr Ala Val Ile Asp Trp Ser Glu Ala Met Phe Gly Asp
210 215 220
TCC CAA TAC GAG GTC GCC AAC ATC TTC TTC TGG AGG CCG TGG TTG GCT 720
Ser Gln Tyr Glu Val Ala Asn Ile Phe Phe Trp Arg Pro Trp Leu Ala
225 230 235 240
TGT ATG GAG CAG CAG ACG CGC TAC TTC GAG CGG AGG CAT CCG GAG CTT 768
Cys Met Glu Gln Gln Thr Arg Tyr Phe Glu Arg Arg His Pro Glu Leu
245 250 255
GCA GGA TCG CCG CGG CTC CGG GCG TAT ATG CTC CGC ATT GGT CTT GAC 816
Ala Gly Ser Pro Arg Leu Arg Ala Tyr Met Leu Arg Ile Gly Leu Asp
260 265 270
CAA CTC TAT CAG AGC TTG GTT GAC GGC AAT TTC GAT GAT GCA GCT TGG 864
Gln Leu Tyr Gln Ser Leu Val Asp Gly Asn Phe Asp Asp Ala Ala Trp
275 280 285
GCG CAG GGT CGA TGC GAC GCA ATC GTC CGA TCC GGA GCC GGG ACT GTC 912
Ala Gln Gly Arg Cys Asp Ala Ile Val Arg Ser Gly Ala Gly Thr Val
290 295 300
GGG CGT ACA CAA ATC GCC CGC AGA AGC GCG GCC GTC TGG ACC GAT GGC 9 60
Gly Arg Thr Gln Ile Ala Arg Arg Ser Ala Ala Val Trp Thr Asp Gly
305 310 315 320
TGT GTA GAA GTC GCG TCT GCG TTC GAC CAG GCT GCG CGT TCT CGC GGC 100 8
Cys Val Glu Val Ala Ser Ala Phe Asp Gln Ala Ala Arg Ser Arg Gly
325 330 335
CAT AGC AAC CGA CGT ACG GCG TTG CGC CCT CGC CGG CAG CAA GAA GCC 10 56
His Ser Asn Arg Arg Thr Ala Leu Arg Pro Arg Arg Gln Gln Glu Ala
340 345 350
ACG GAA GTC CGC CCG GAG CAG AAA ATG CCC ACG CTA CTG CGG GTT TAT 110 4
Thr Glu Val Arg Pro Glu Gln Lys Met Pro Thr Leu Leu Arg Val Tyr
355 360 365
ATA GAC GGT CCC CAC GGG ATG GGG AAA ACC ACC ACC ACG CAA CTG CTG 1152
Ile Asp Gly Pro His Gly Met Gly Lys Thr Thr Thr Thr Gln Leu Leu
370 375 380
GTG GCC CTG GGT TCG CGC GAC GAT ATC GTC TAC GTA CCC GAG CCG ATG 1200
Val Ala Leu Gly Ser Arg Asp Asp Ile Val Tyr Val Pro Glu Pro Met
385 390 395 400
ACT TAC TGG CGG GTG CTG GGG GCT TCC GAG ACA ATC GCG AAC ATC TAC 1248
Thr Tyr Trp Arg Val Leu Gly Ala Ser Glu Thr Ile Ala Asn Ile Tyr
405 410 415
ACC ACA CAA CAC CGC CTC GAC CAG GGT GAG ATA TCG GCC GGG GAC GCG 1296
Thr Thr Gln His Arg Leu Asp Gln Gly Glu Ile Ser Ala Gly Asp Ala
420 425 430
GCG GTG GTA ATG ACA AGC GCC CAG ATA ACA ATG GGC ATG CCT TAT GCC 1344
Ala Val Val Met Thr Ser Ala Gln Ile Thr Met Gly Met Pro Tyr Ala
435 440 445
W 0 92/08796 2 0 9 6~2 2 2 32 PC~r/US91/08442
GTG ACC GAC GCC GTT CTG GCT CCT CAT ATC GGG GGG GAG GCT GGG AGC 1392
Val Thr Asp Ala Val Leu Ala Pro His Ile Gly Gly Glu Ala Gly Ser
450 455 460
TCA CAT GCC CCG CCC CCG GCC CTC ACC CTC ATC TTC GAC CGC CAT CCC 1440
Ser His Ala Pro Pro Pro Ala Leu Thr Leu Ile Phe Asp Arg His Pro
465 470 475 480
ATC GCC GCC CTC CTG TGC TAC CCG GCC GCG CGG TAC CTT ATG GGC AGC 1488
Ile Ala Ala Leu Leu Cys Tyr Pro Ala Ala Arg Tyr Leu Met Gly Ser
485 490 495
ATG ACC CCC CAG GCC GTG CTG GCG TTC GTG GCC CTC ATC CCG CCG ACC 1536
Met Thr Pro Gln Ala Val Leu Ala Phe Val Ala Leu Ile Pro Pro Thr
S00 505 510
TTG CCC GGC ACC AAC ATC GTG CTT GGG GCC CTT CCG GAG GAC AGA CAC 1584
Leu Pro Gly Thr Asn Ile Val Leu Gly Ala Leu Pro Glu Asp Arg His
515 520 525
ATC GAC CGC CTG GCC AAA CGC CAG CGC CCC GGC GAG CGG CTG GAC CTG 1632
Ile Asp Arg Leu Ala Lys Arg Gln Arg Pro Gly Glu Arg Leu Asp Leu
530 535 540
GCT ATG CTG GCT GCG ATT CGC CGC GTT TAC GGG CTA CTT GCC AAT ACG 1680
Ala Met Leu Ala Ala Ile Arg Arg Val Tyr Gly Leu Leu Ala Asn Thr
545 550 555 560
GTG CGG TAT CTG CAG GGC TGC GGG TCG TGG CGG GAG GAC TGG GGA CAG 1728
Val Arg Tyr Leu Gln Gly Cys Gly Ser Trp Arg Glu Asp Trp Gly Gln
565 570 575
CTT TCG GGG ACG GCC GTG CCG CCC CAG GGT GCC GAG CCC CAG AGC AAC 1776
Leu Ser Gly Thr Ala Val Pro Pro Gln Gly Ala Glu Pro Gln Ser Asn
580 585 590
GCG GGC CCA CGA CCC CAT ATC GGG GAC ACG TTA TTT ACC CTG TTT CGG 1824
Ala Gly Pro Arg Pro His Ile Gly Asp Thr Leu Phe Thr Leu Phe Arg
595 600 605
GCC CCC GAG TTG CTG GCC CCC AAC GGC GAC CTG TAT AAC GTG TTT GCC 1872
Ala Pro Glu Leu Leu Ala Pro Asn Gly Asp Leu Tyr Asn Val Phe Ala
610 615 620
TGG GCC TTG GAC GTC TTG GCC AAA CGC CTC CGT TCC ATG CAC GTC TTT 1920
Trp Ala Leu Asp Val Leu Ala Lys Arg Leu Arg Ser Met His Val Phe
625 630 635 640
ATC CTG GAT TAC GAC CAA TCG CCC GCC GGC TGC CGG GAC GCC CTG CTG 1968
Ile Leu Asp Tyr Asp Gln Ser Pro Ala Gly Cys Arg Asp Ala Leu Leu
645 650 655
CAA CTT ACC TCC GGG ATG GTC CAG ACC CAC GTC ACC ACC CCC GGC TCC 2016
Gln Leu Thr Ser Gly Met Val Gln Thr His Val Thr Thr Pro Gly Ser
660 665 670
ATA CCG ACG ATA TGC GAC CTG GCG CGC ACG TTT GCC CGG GAG ATG GGG 2064
Ile Pro Thr Ile Cys Asp Leu Ala Arg Thr Phe Ala Arg Glu Met Gly
675 680 685
WO 92/08796 2 0 9 6 2 2 2 PCT/US91/08442
_ 33
GAG GCT AAC TGA 2076
Glu Ala Asn
690
(2) INFORMATION FOR SEQ ID NO: 2:
(i) SEQUENCE CHARACTERISTICS:
~A) LENGTH: 691 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 2:
Met Lys Lys Pro Glu Leu Thr Ala Thr Ser Val Glu Lys Phe Leu Ile
1 5 10 15
Glu Lys Phe Asp Ser Val Ser Asp Leu Met Gln Leu Ser Glu Gly Glu
Glu Ser Arg Ala Phe Ser Phe Asp Val Gly Gly Arg Gly Tyr Val Leu
Arg Val Asn Ser Cys Ala Asp Gly Phe Tyr Lys Asp Arg Tyr Val Tyr
Arg His Phe Ala Ser Ala Ala Leu Pro Ile Pro Glu Val Leu Asp Ile
Gly Glu Phe Ser Glu Ser Leu Thr Tyr Cys Ile Ser Arg Arg Ala Gln
Gly Val Thr Leu Gln Asp Leu Pro Glu Thr Glu Leu Pro Ala Val Leu
100 105 110
Gln Pro Val Ala Glu Ala Met Asp Ala Ile Ala Ala Ala Asp Leu Ser
115 120 125
Gln Thr Ser Gly Phe Gly Pro Phe Gly Pro Gln Gly Ile Gly Gln Tyr
130 135 140
Thr Thr Trp Arg Asp Phe Ile Cys Ala Ile Ala Asp Pro His Val Tyr
145 150 155 160
His Trp Gln Thr Val Met ASp Asp Thr Val Ser Ala Ser Val Ala Gln
165 170 175
Ala Leu Asp Glu Leu Met Leu Trp Ala Glu Asp Cys Pro Glu Val Arg
180 185 190
His Leu Val His Ala Asp Phe Gly Ser Asn Asn Val Leu Thr Asp Asn
195 200 205
Gly Arg Ile Thr Ala Val Ile Asp Trp Ser Glu Ala Met Phe Gly Asp
210 215 220
Ser Gln Tyr Glu Val Ala Asn Ile Phe Phe Trp Arg Pro Trp Leu Ala
225 230 235 240
WO 92/08796 ' P~/US91/08442
2û9~22~4
CYQ Met Glu Gln Gln Thr Arg Tyr Phe Glu Arg Arg HiS Pro Glu Leu
245 250 255
~la Gly Ser Pro Arg Leu Arg Ala Tyr Met Leu Arg Ile Gly Leu Asp
260 265 270
Gln Leu Tyr Gln Ser Leu Val Asp Gly Asn Phe Asp Asp Ala Ala Trp
275 280 285
Ala Gln Gly Arg Cys Asp Ala Ile Val Arg Ser Gly Ala Gly Thr Val
290 295 300
Gly Arg Thr Gln Ile Ala Arg Arg Ser Ala Ala Val Trp Thr Asp Gly
305 310 315 320
~ys Val Glu Val Ala Ser Ala Phe Asp Gln Ala Ala Arg Ser Arg Gly
325 330 335
~is Ser Asn Arg Arg Thr Ala Leu Arg Pro Arg Arg Gln Gln Glu Ala
340 345 350
Thr Glu Val Arg Pro Glu Gln Lys Met Pro Thr Leu Leu Arg Val Tyr
355 360 365
Ile Asp Gly Pro His Gly Met Gly Lys Thr Thr Thr Thr Gln Leu Leu
370 375 380
Val Ala Leu Gly Ser Arg Asp Asp Ile Val Tyr Val Pro Glu Pro Met
385 390 395 400
~hr Tyr Trp Arg Val Leu Gly Ala Ser Glu Thr Ile Ala Asn Ile Tyr
405 410 415
~hr Thr Gln His Arg Leu Asp Gln Gly Glu Ile Ser Ala Gly Asp Ala
420 425 430
Ala Val Val Met Thr Ser Ala Gln Ile Thr Met Gly Met Pro Tyr Ala
435 440 445
Val Thr Asp Ala Val Leu Ala Pro His Ile Gly Gly Glu Ala Gly Ser
450 455 460
Ser His Ala Pro Pro Pro Ala Leu Thr Leu Ile Phe Asp Arg His Pro
465 470 475 480
~le Ala Ala Leu Leu Cys Tyr Pro Ala Ala Arg Tyr Leu Met Gly Ser
485 490 495
~et Thr Pro Gln Ala Val Leu Ala Phe Val Ala Leu Ile Pro Pro Thr
500 505 510
Leu Pro Gly Thr Asn Ile Val Leu Gly Ala Leu Pro Glu Asp Arg His
515 520 525
Ile Asp Arg Leu Ala Lys Arg Gln Arg Pro Gly Glu Arg Leu Asp Leu
530 535 540
Ala Met Leu Ala Ala Ile Arg Arg Val Tyr Gly Leu Leu Ala Asn Thr
545 550 555 560
Val Arg Tyr Leu Gln Cys Gly Gly Ser Trp Arg Glu Asp Trp Gly Gln
WO 92/08796 2 0 9 6 2 2 2 PC~r/US91/08442
__ 35
565 570 575
Leu Ser Gly Thr Ala Val Pro Pro Gln Gly Ala Glu Pro Gln Ser Asn
580 585 590
Ala Gly Pro Arg Pro His Ile Gly Asp Thr Leu Phe Thr Leu Phe Arg
59s 600 605
Ala Pro Glu Leu Leu Ala Pro Asn Gly Asp Leu Tyr Asn Val Phe Ala
610 615 620
Trp Ala Leu Asp Val Leu Ala Lys Arg Leu Arg Ser Met His Val Phe
625 630 635 640
Ile Leu Asp Tyr Asp Gln Ser Pro Ala Gly Cys Arg Asp Ala Leu Leu
645 650 655
Gln Leu Thr Ser Gly Met Val Gln Thr His Val Thr Thr Pro Gly Ser
660 665 670
Ile Pro Thr Ile Cys Asp Leu Ala Arg Thr Phe Ala Arg Glu Met Gly
675 680 685
Glu Ala Asn
690