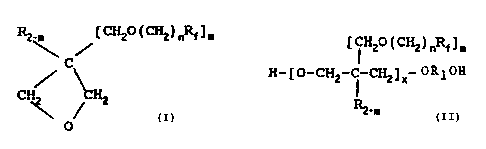Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
210 0213
PREPARATION AND POLYMERIZATION OF PERFLUOROALKOXY
ALKYLENE OXIDES TO PREPARE HYDROPHOBIC POLYETHERS
BACKGROUND OF THE INVENTION
Field Of Invention
This invention relates to oxetane monomers having
pendant perfluoroalkoxy groups, to a method for their
preparation and a method for their polymerization, and to
the hydroxy-terminated prepolymers so prepared. The
prepolymers have a polyether backbone and are useful for the
preparation of resins and coatings having hydrophobic
properties, low di-electric constants and low refractive
indices.
Brief Statement Of The Prior Art
It is known that oxetane can be substituted in the 3-
position with methyl groups containing energetic functional
groups such as nitrato, azide, nitro and difluoroamino. The
polymerization of these substituted oxetanes in the presence
of polyhydroxy aliphatic compounds produces hydroxy-
terminated polymers having a polyether backbone with pendant
energetic groups. These polymers may then be cured with
isocyanates, forming high molecular weight polymers which
are useful as binders in propellent applications as
described in U.S. Patents 4,393,199; 4,483,978; 4,707,540;
and 4,764,586.
It is desirable to incorporate fluorine in the
polyether polymers, however, the incorporation of high
percentages of fluorine in an ether backbone of a polymer
causes thermal and chemical instability, resulting in the
generation of fluoride ion or hydrogen fluoride. When the
fluorine is incorporated in polymers which lack an ether
linkage, the resulting materials have high glass transition
temperatures which limit the usefulness and applications of
the resulting polymers.
CA 02100218 2003-05-06
2
BRIEF DESCRIPTION OF THE INVENTION
Thi:; invention comprises oxetane monomers having alkoxy
side chains with a fully fluorinated omega carbon,
preferably perfluorinated alkoxy side chains, a method for
their preparation, a method for their polymerization and to
the resultant prepolymers. The monomers are prepared by the
reaction of the corresponding fluorinated alkoxides with
aryl sulf'onate or halogenated derivatives of 3-hydroxyalkyl
oxetanes. An example of the latter is the p-toluene
1.0 sulfonate: derivative of 3-hydroxymethyl-3-methyl oxetane or
the derivative of 1:~,3-bis(hydroxymethyl)-oxetane. A high
yield of the oxetanes having pendant perfluorinated alkoxy
side chains is obtai.ned.
The fluorinated alkoxy oxetane monomers of this
invention can be .r.eadily polymerized in the presence of
Lewis acid cataly::;ts to obtain high yields of hydroxy-
terminated prepolymers having molecular weights from 1,000
to about 30,000, and a polyether backbone resulting from the
opening of the oxe~tane ring. These prepolymers are useful
as hydrophobic, non-stick coatings and as precusors for
binders for propellants.
DESCRII?TION OF PREFERRED EMBODIMENT
This invention comprises the following oxetane
derivativias, a method for their preparation, a method for
their polymerization and the resulting prepolymers:
R CH2O ( CH2 ) nR~ ] m
=C
CHz CH2
O
wherein:
n is 1 to 5;
m is 1.
-35 R is hydrogen or alkyl having from 1 to 4
carbons; and
Rf is linear or branched chain perfluoro-
CA 02100218 2005-11-21
3
alkyl having from 1 to 20.carbons, a linear or
branched perfluorinated isoalkyl having from 1
to 20 carbons, or an oxa-perfluorinated
polyether, having from 4 to about 60 carbons.
The invention also comprises the polymerization of the
monomer and the resultant hydroxy-terminated prepolymers
having the structure indicated below;
~ i 2C(CH2)nRf]m
H- [ O-CHZ-C
-CHZ ] x- 0 R 10H
~
R2-w
wherein:
wherein n is 1 to 5;
m is 1;
R is hydrogen or alkyl having from.1 to 4
carbons;
R1 is alkylene or isoalkylene having 2 to about
5 carbons;
Rf is linear or branched chain perfluorinated
alkyl having from 1 to 20 carbons, or an oxa-
perfluorinated polyether having from 4 to about
60 carbons; and
x is 2 to about 250.
The oxetane derivatives are obtained by reaction of aryl
sulfonate derivatives of hydroxyalkyl oxetanes with
fluorinated alkoxides. The aryl sulfonate derivatives of the
hydroxyalkyl oxetanes have the general formula:
R2_; CH2OS02R31,
/C'
~ /cHZ
wherein:
m is 1;
R is hydrogen or alkyl having from 1 to 4
carbons; and
CA 02100218 2003-05-06
3a
wherein:
m is 1;
F is hydrcl-qen or alkyl having from 1 to 4
carbons; _~nd
CA 02100218 2005-11-21
4
R3 is monocyclic aryl having from C6 to C 10
carbons, e.g., benzyl, tolyl, xylyl, mesityl.
The preferred sulfonates are toluene sulfonates, e.g. p-
toluene sulfonate derivatives of oxetane.
The fluorinated alkoxides are obtained by the reaction
of fluorinated alcohols with sodium hydride in a suitable
solvent such as dimethylformamide. The fluorinated alcohols
which can be used have the general formula:
Rf ( CHZ ) OH
wherein:
n is 1 to 5,
Rf is linear or branched chain perfluorinated
alkyl having from 1 to 20 carbons, or an
oxa-perfluorinated polyether, having from
4 to about 60 carbons.
Examples of suitable fluorinated alcohols are: trifluoro-
ethanol, heptafluorobutanol, pentadecafluorooctanol,
tridecafluorooctanol, etc. Other useful alcohols include
fluorinated alcohols having other halogens, in the omega
position, e.g., omega-halo perfluorinated alcohols of the
following formulas:
1. HOCHZ (CF2) nCFzX and
2. HOCHZ (CFZCF) nCF2X
CF3
wherein:
X is halo, e.g., bromo, iodo, chloro or
fluoro; and
n is 2 to about 20; and
3. HOCHZ ( 0CF2CF2 ) nF, and
4. HOCHZ ( OCF2CF) n F
1
CF3
wherein n is 2 to about 20.
The fluorinated alkoxy oxetane monomers readily polymerize
in the presence of a Lewis acid catalyst and a polyhydroxy
aliphatic compound as a polymerization initiator. In the
2100213
presence of a Lewis acid, the alcohol releases a proton
which initiates the ring opening polymerization of the
oxetane which propagates through the formation of the
following intermediate:
5 i C R
' /
H-O-CH -C-CH +t C
2 L z-I/R
f f
Suitable catalysts are Lewis acids, i.e., compounds
capable of accepting a pair of electrons, examples of which
include: complexes of boron trifluoride, phosphorus penta-
fluoride, antimony pentafluoride, zinc chloride, aluminum
bromide, etc.
Suitable initiators are polyhydroxy aliphatic compounds
such as alkyl and isoalkyl polyols having from 2 to about 5
carbons and from 2 to 4 hydroxyls, e.g., ethylene glycol,
butane-l,4-diol, propylene glycol, isobutane-l,3-diol,
pentane-l,5-diol, pentaerythritol, etc.
The polymerization is conducted in the presence of a
suitable inert solvent, preferably a halogenated Cl to C5
hydrocarbon, e.g., methylene chloride, methylene bromide,
ethylene dichloride, ethylene dibromide, propylene
dichloride, Freons, fluorinated solvents, etc.
The catalyst and initiator are preferably mixed in the
solvent prior to the addition of the oxetane monomer. An
example of a preferred catalyst, initiator and solvent
combination is the boron trifluoride etherate, or boron
trifluoride tetrahydrofuranate, and butane-l,4-diol in
methylene chloride.
To this mixture the monomer is added and solution
polymerization is practiced at solution concentrations from
5 to 75 weight percent. In the polymerization, the
concentration of the catalyst and the proportions of the
initiator, e.g., butane-l,4-diol, can be varied to control
the molecular weight of the polymer, with higher proportions
of initiator resulting in lower molecular weight of the
prepolymer product. Useful proportions,of boron trifluoride
catalyst to initiator can be from about 100:1 to about 1:2.
210 C'? 1- 3
6
The polymerization terminates with the formation of the
hydroxy-terminated polymer according to the following
mechanism:
R C R R
H- [ O-CHZ-C-CHz ] n. 1- C/ + H20 H- [ O-CH2 C-CH2 ] ,-OH
Rf Rf Rf
The polymerization can be homopolymerization or
copolymerization in which a mixture of two or more of the
afore-described oxetane monomers is added to the
polymerization zone. A particularly useful copolymerization
is block polymerization in which the comonomers are
sequentially added in selected proportions to obtain block
copolymers of controlled block sizes and properties.
To prepare prepolymers with the optimum coating
properties, the oxetane monomer should have a 3-substituent
in which the Rf group has its omega carbon fully
fluorinated. One of the main applications of the
hydroxy-terminated prepolymers of this invention is in the
development of hydrophobic, non-stick coatings. The most
important criteria in the development of these coatings is
the minimization of the free surface energy of the coating,
which is a measure of the wettability of the coating and
defines critical properties, such its hydrophobicity and
adhesive characteristics. Terminal carbons which contain
hydrogen, e.g.,-CFZH or -CFH2 or -CH3, have significantly
greater surface energies (15-39 dynes/cm) than those with
fully halogenated groups, e.g., those with -CF3 groups,
which have surface energies of about 6 dynes/cm.'
Most preferably, the 3-substituent (Rf) is
perfluoroalkyl. The perfluoroalkyl group is an extremely
strong electron withdrawing group and its presence changes
the electronic and steric properties, of the oxetane
monomers. This affects the ease of their polymerization and
the functionality, molecular weight, and structure, i.e.,
cyclic or linear, of the polymer. The most useful
hydroxy-terminated prepolymers with fluorinated side chains
CA 02100218 2003-05-06
are those which are we,l.l def_ined and which have a
functionality of at least 2. Presence of rion-functional
or mono-furlctional rnater.i_als :.n the 1--,repolymers results in
coatings with poor !:nec~Iani_cal arid surface properties.
Cyclic groups, mci.i_nly cyclic tetramers and trimers,
in the polymer are r.orr-functional arid reduce the
usefulness of the prepo=i.ymers. Other non-functional
groups can be formed by counter--i.on terminations, such as
diethyl ether and fl.x.zoride ion terminations. In addition
to the role of the fluoroalkyl substituent on the oxetane
on its reactivity, other factor.s thuw, control the
formation of non- ai-id mono-furictional materials, such as
the monomer/initiator rati_c, ratio of alcohol to Lewis
acid, type of Lewis acid, react.ion temperature, solvent,
and concent.ration.
Preferably, tY:.~-a. hydroxyterminated prepolymer is one
in which rr is I and n is 2, and preferably Rf is
tridecafluorooctyl. Also prefer.ab.le is a
hydroxyter:minated p:repolymer 'tiaving a molecular weight
from 1,000 to about 30,000, and a polymer backbone
resulting from the opening of: the oxet.ane ring of oxetane
monomers substituted at ttie =, carboii position with one to
two perfluorinated a:Lkoxide groups of the formula:
C'HzO (CH2) rl"',:?5 wherein:
n is 1 or 2; and
Pf is lin(..ar or branched chain perfluorinated
alkyl having trom 1 to 20 carbcns, a linear or
branched õOerfluorinated isoalkyl having from 1
:30 to 20 carOans, or arl oxa-perfluori.nated
polyether, having from 4 i--o about 60 carbons.
CA 02100218 2003-05-06
la
In this case, n is preferably 2. Rf is preferably
tridecafluorohexyl.
The preferred perfluorinated alkoxy oxetane monomer
is one in whic~i m is -1. and n 2_s 2 arld preferably Rf is
tridecafluorohexyl.
It is also desired to use mono--3-substituted
oxetanes, i.e., the value of "m" in the empirical formula
is 1. These ar.e de;.,i.~t:-ed, since homopolymerization of di-
3-substituted oxetane monomers yields crystalline
:LO polymers. As an example, polymerization of 3, 3-bis-
(chlorometryl) oxetane yields a crystalline polymer that
melts in the neighborhood of 22J C.
The hydroxyter:'.iz.riat:.ed prepolymers prepared from the
preferred rrLono--3-substi_tut.ed oxetane monomers are
amorphous, low visco:;:i..ty oils w:~:ich are easy to process.
The prepolymers are relatively pure, whereas those derived
from di-3-substituted oxetane monomers contain large
amounts of nonfunct_orial cyclic oligomers. Also, the
desired surface proE-Derti.es of the prepolymer can be
achieved with only one fluoririated substituent in the 3
position o:f the oxe?:.ane monomer, and a second fluorinated
substituent does no'= significantly contribute to the
surface prcperties.
In the following examples, the polymerization was
practiced with bororl t:,rifluoride etY-ierate, or boron
trifluoride tetrahy-irofuranate, in butane-i,4-diol. The
initiator was prepar_ed f:rom commercial grade boron
trifluoride etherate which was distilled prior to use.
Similarly, the butarae--1,4--diol was distilled from calcium
2100213
8
hydride and stored over a 4 A molecular sieve prior to use.
The polymerizations were conducted in a 100 milliliter
glass flask which was jacketed and equipped with a
mechanical stirrer. NMR analysis of products was performed
on a Bruker MSL-300 spectrometer at 300 MHZ in
deutrochioroform solution with proton and carbon shifts in
ppm relative to tetramethylsilane and fluorine shifts
relative to fluorotrichloromethane. The infrared analysis
was done by diffuse reflectance on a Nicolet SX-5 spectro
meter on KBr. The thermal analysis was performed on a
Dupont DSC 9100 analyzer. Weight average molecular weights
were determined using a Waters gelperineation chromatograph
equipped with four columns (100 A,500 A, 103 A and 104 A), a
differential refractive index detector and a Data Module
730.
EXAMPLE 1
PREPARATION OF 3-(2,2,2-TRIFLUOROETHOXYMETHYL)-
3-METHYLOXETANE.
A dispersion of 50 weight percent (2.8 grams, 58.3
mmol) sodium hydride in mineral oil, was washed twice with
hexanes and suspended in 35 milliliters of dimethyl
formamide. Then, 5.2 grams (52 mmol) of trifluoroethanol
was added and the mixture was stirred for 45 minutes. A
solution of 10.0 grams (39 mmol) of 3-hydroxymethyl-3-
methyloxetane p-toluenesulfonate in 15 milliliters of
dimethyl formamide was added and the mixture was heated at
75-85 C for 20 hours, when 'H NMR analysis of an aliquot
sample showed that the starting sulfonate had been consumed.
The mixture was poured into 100 milliliters of ice
water and extracted with 2 volumes of inethylene chloride.
The combined organic extracts were washed twice with water,
twice with 2 weight percent aqueous hydrochloric acid,
brine, dried over magnesium sulfate, and evaporated to give
6.5 grams of 3-(2,2,2-trifluorethoxymethyl)-3-methyloxetane
as an oil containing less than 1 weight percent dimethyl
formamide. The yield of this product was 90 percent. The
oil was distilled at 30 C and 0.2 millimeters mercury
2100213
9
pressure to give 4.3 grams of analytically pure product,
corresponding to a 60 percent yield. The analyses of the
product were as follows: IR (KBr) 2960-2880, 1360-1080, 990,
840 cm'1; 'H NMR 1.33 (s,2H), 3.86 (q, J=8.8 Hz, 2 H), 4.35
(d, J=5.6Hz, 2 H), 4.51 (d, J=5.6 Hz, 2 H); 13C NMR 20.72,
39.74, 68.38 (q, J=40 Hz), 77.63, 79.41, 124 (q, J=272 Hz).
The calculated elemental analysis for C7HjjF302 is: C=45.65;
H=6.02; F=30.95. The experimental analysis found: C=45.28;
H=5.83; F=30.59.
EXAMPLE 2
PREPARATION OF 3,3-BIS-
(2,2,2-TRIFLUOROETHOXYMETHYL)OXETANE
A 50 weight percent dispersion of sodium hydride in
18.4 grams (0.383 mol) of mineral oil, was washed twice
with hexanes and was suspended in 200 milliliters of
dimethyl formamide. Then, 38.3 grams (0.383 mol)
trifluoroethanol was added dropwise over 45 minutes while
hydrogen gas evolved. The mixture was stirred for 30
minutes and a solution of 30.0 grams (0.073 mol) of 3,3-
bis(hydroxymethyl)oxetane di-p-toluenesulfonate in 50
milliliters of dimethyl formamide was added. The mixture
was heated to 75 C for 64 hours when 'H NMR analysis of an
aliquot showed that the starting sulfonate had been
consumed.
The mixture was poured into water and extracted with
two volumes of methylene chloride. The combined organic
extracts were washed with brine, 2 weight percent aqueous
hydrochloric acid, water, dried over magnesium sulfate, and
evaporated to give 17.5 grams of 3,3-bis(2,2,2-trifluoro-
ethoxymethyl)oxetane as an oil containing less than 1 weight
percent dimethyl formamide. The oil was purified by bulb-
to-bulb distillation at 42-48 C and 10.1 millimeters mercury
pressure to give 15.6 grams of analytically pure ether,
corresponding to a 79 percent yield. The analyses of the
product were as follows: IR (KBr) 2960-2800, 1360-1080, 995,
840 cm 1 ; tH NML2 3.87 (s, 4 H), 3.87 (q, J=8.8 Hz, 4 H), 4.46
(s, 4 H); 13C NMR 43.69, 68.62 (q, J 35 Hz), 73.15,
M0M
75.59, 123.87 (q, J=275 Hz); 19F-74.6 (s). The calculated
elemental analysis for C9H12Fb03 is; C=38.31; H=4.29; and
F=40.40. The experimental analyses found: C=38.30; H=4.30;
and F= 40.19.
5 EXAMPLE 3
PREPARATION OF 3-(2,2,3,3,4,4,4-HEPTAFLUORO-
BUTOXYMETHYL)-3-METHYLOXETANE
A 50 weight percent dispersion of sodium hydride 6.1
grams (127 mmol) in mineral oil, was washed twice with
10 hexanes and was suspended in 60 milliliters of dimethyl
formamide. Then 24.0 grams (120 mmol) of 2,2,3,3,4,4,4-
heptafluorobutan-l-ol was added and the mixture was stirred
for 45 minutes. A solution of 25.0 grams (97.5 mmol) of 3-
hydroxymethylp-toluenesulfonate in 15 milliliters of
dimethyl formamide was added and the mixture was heated at
75-85 C for 30 hours when 'HNMR analysis of an aliquot
showed that the starting sulfonate had been consumed.
The mixture was poured into 100 milliliters of
ice/water and extracted with two volumes of methylene
chloride. The combined organic extracts were washed twice
with water, twice with 2 weight percent aqueous hydrochloric
acid, brine, dried over magnesium sulfate, and evaporated to
give 27.5 grams of 3-(2,2,3,3,4,4,4-heptafluorobutoxyethyl)-
3-methyloxetane as an oil. The oil was distilled at 33 C
and 0.2 millimeters mercury pressure to give 12.2 grams of
analytically pure ether, corresponding to a 44 percent
yield. The experimental analyses were: IR (KBr) 2960-2880,
1280-1030, 995, 840 cm'', 'H NMR 1.31 (s, 3 H), 3,67 (s 2 H),
3.99 (t, J=13.3 Hz 2 H) 4.34 (d. J=5.7 Hz 2 H) 4.50 (5.7 Hz,
2 H); 13C NMR 20.242 (q, J=125 Hz) 39.627 (s) 67.778 (3x3.
J=145.1 Hz, J=26.1 Hz), 77.730 (t. J=142 Hz), 79.110 (t,
J=150 Hz), 108.72 (3 x 4, J=264 Hz. J=36 Hz), 114.7 (3 x 3,
J=256 Hz, J= 30 Hz), 117.58 (4 x 3, J=286.9 Hz, J=33.7 Hz);
19F -81.4, -120.6, -128.1. The calculated elemental
analysis for C9H"F7OZ is C=38.04; H=3.90; F=46.80. The
experimental analyses found: C=38.03; H=3.65; and F=46.59.
210 G21S
11
EXAMPLE 4
POI,YMERIZATION OF 3-(2,2,2-TRIFLUOROETHOXYMETHYL)-
3-METHYLOXETANE.
A solution of 34.3 milligrams (0.38 mmol) of butane-
1,4-diol and 109.7 milligrams (0.77 mmol) of boron
trifluoride etherate in 4 grams of methylene chloride was
stirred at ambient temperature for 15 minutes under nitrogen
in a dry polymerization flask. The solution was cooled to
1.5 C and a solution of 1.20 grams (6.52 mmol) of 3-(2,2,2-
trifluoroethoxymethyl)-3-methyloxetane in 1.3 grams of
methylene chloride was added. The resultant solution was
stirred for 5 hours at 1-2 C at which time 'H NMR analysis
of an aliquot indicated that the starting oxetane had been
consumed. The solution was warmed to ambient temperature
and quenched with water. The organic layer was washed with
brine, 2 weight percent aqueous hydrochloric acid, and
evaporated to give 1.053 grams of poly-3-(2,2,2-trifluoro-
ethoxymethyl)-3-methyloxetane as an oil, corresponding to a
88 percent yield. The polymer analyses were: DSC Tg -45 C,
decomposition temperature was greater than 200 C; GPC (THF)
Molecular weight Mn 7376, Mw 7951, polydispersity 1.08,
inherent viscosity 0.008; Molecular Weight by 'H NMR 6300;
'H NMR 0.95 (s, 3 H), 3.26 (m, 4 H), 3.52 (2, 2 H) 3.84 (q.
2 H); 13C NMR 17.57, 42.09, 69.30 (q, J=33 Hz), 74.42,
75.90, 125.18 (q, J=280 Hz).
EXAMPLE 5
PREPARATION OF
POLY-BIS-[3-(2,2,2-TRIFLUOROETHOXYMETHYL)OXETANE]
A solution of 33.9 milligrams (0.378 mmol) of butane-
1,4 diol and 106.3 milligrams (0.75 mmol) of boron
trifluoride etherate in 3.8 grams of inethylene chloride was
stirred at ambient temperature for 15 minutes under nitrogen
in a dry polymerization flask. The solution was cooled to
1.5 C and a solution of 1.88 grams (6.67 mmol) of bis-[3-
2,2,2-trifluoroethoxy-methyl)]oxetane in 2.3 grams of
methylene chloride was added. The resultant solution was
stirred for 16 hours at 1-2 C at which'time 1H NMR analysis
210G,'?1 (3
12
of an aliquot indicated that the starting oxetane had been
consumed.
The solution was warmed to ambient temperature and
quenched with water. The organic layer washed with brine, 2
percent aqueous hydrochloric acid, and evaporated to give
1.62 grams of polybis-[3-(2,2,2-trifluoroethoxymethyl)-
oxetane, corresponding to 85% yield. The prepolymer was a
waxy solid. The polymer analyses were: DSC Tg 54.88 C.,
mp 80.96 C (26.35 Joules/gram), decomposition temperature
was greater than 210 C; GPC (THF) Molecular weight Mn 5321,
Mw 7804, polydispersity 1.47, inherent viscosity 0.008; 1H
NMR 1.60 (m), 2.46 (s), 3.36 (s. 4 H), 3.58 (s, 4 H), 3.79
(q, 4 H); 13C NMR 45.49, 68.25 (q. J=33 Hz), 69.20. 70.97.
123.81 (q, J=280 Hz).
EXAMPLE 6
PREPARATION OF POLY-3-(2,2,3,3,4,4,4-
HEPTAFLUOROBUTOXYMETHYL)-3-METHYLOXETANE
A solution of 34.7 milligrams (0.38 mmol) of butane-
1,4-diol and 109.7 milligrams (0.77 mmol) of boron
trifluoride etherate in 3.4 grams of inethylene chloride was
stirred at ambient temperature for 15 minutes under nitrogen
in a dry polymerization flask. The solution was cooled to
1.5 C and a solution of 2.00 grams (7.08 mmol) of 3-
(2,2,3,3,4,4,4-hepta-fluorobutoxymethyl)-3-methyloxetane in
3.3 grams of methylene chloride was added. The resultant
solution was stirred for 4 hours at 1.2 C; at which time 'H
NMR analysis of an aliquot indicated that the starting
oxetane had been consumed.
The solution was warmed to ambient temperature and
quenched with water. The organic layer washed with brine, 2
percent aqueous hydrochloric acid, and evaporated to give
1.65 grams of poly-3-(2,2,3,3,4,4,4-heptafluorobutoxy-
methyl)-3-methyloxetane, corresponding to a 83% yield. The
prepolymer was an oil and had the following analyses: GPC
(THF) Molecular weight Mn 4066, Mw 5439, polydispersity
1.34, inherent viscosity 0.054. This oil was extracted
with methanol and dried to give 1.46 grams of polymer,
2100218
13
corresponding to 72% yield, and has the following analyses:
DSC Tg -45 C; GPC (THF) Molecular weight Mn 4417, Mw 5658,
polydispersity 1.28 inherent viscosity 0.056. Molecular
weight 'H MMR 8718, 'H NMR 0.93 (s, 3 H), 3.20 (m, 4 H),
3.48 (s, 2 H), 3.92 (q, J=13.6 Hz, 2 H); 13C MMR 16.14,
40.57, 67.37 (t, J=Hz), 72.89, 74.76.
EXAMPLE 7
PREPARATION OF
3-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-PENTADECAFLUORO-
OCTYLAXYMETHY)-3-METHYIAXETANE
A dispersion of 50 weight percent sodium hydride (4.0 g,
83 mmol) in mineral oil was washed with hexanes and
suspended in 200 milliliters of dimethylformamide. A
solution of 30 grams of 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-
pentadecafluorooctan-l-ol (75 mmol) in 50 milliliters of
dimethylformamide was added over a period of 3 hours, and
the resulting mixture was stirred at room temperature for
one hour. Next, a solution of 9.3 grams (77 mmol) of
3-chloromethyl-3-methyloxetane in 20 milliliters of
dimethylformamide was added and the resulting mixture was
heated at 75 C. for 16 hours. The mixture was cooled to
room temperature and slowly poured into 1 liter of ice/water
and extracted with two volumes of Freon 113. The combined
organic extracts were washed twice with water, once with 2
weight percent aqueous hydrochloric acid and once with
brine, dried over magnesium sulfate, filtered, and
evaporated to give 32 grams of crude product. The crude
product was distilled under reduced pressure to give 26.5
grams (73%) of analytically pure 3-(2,2,3,3,4,4,5,5,6,6,7,7,
8,8,8- pentadecafluorooctoxymethy)-3-methyloxetane, an oil
with a boiling point 68 to 70 C./l.6 mm-Hg. The
experimental analyses were: 1H N1KR (CDC13/Freon 113) 6 4.49
and 4.37 (AB, J=5.5 Hz, 4 H), 4.00 (triplet, J=13.2 Hz, 2
H), 3.70 (singlet, 2 H), and 1.32 (singlet, 2 H); 13C NMR 3
21.02, 40.33, 68.77 (triplet, J=146.2 Hz), 78.60, and 79.87;
19F NMR S -81.3 (3 F), -119.9 (2F), -122.6 (2 F), -123.3 (2
F), -123.5 (2 F), -123.9 (2 F) and -126.8 (2 F). The
2100218
14
elemental analysis was; Calculated for C13H11F1502: C, 32.2;
H, 2.3; F, 58.9. Found: C, 32.2; H, 2.2; F, 58.3.
EXAMPLE 8
PREPARATION OF
3-(3,3,4,4,5,5,6,6,7,7,8,8,8-TRIDECAFLUORO-
OCTYLOXYMETHYL)-3-METHYLOXETANE
In a manner similar to that described above, 12.0 grams
of 3-bromomethyl-3-methyloxetane (73 mmol) was reacted with
26.5 grams of 3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluoro-
octan-l-ol (72.7 mmol) in 300 milliliters) dimethyl-
formamide in the presence of 3.9 grams of a 50 weight
percent dispersion of sodium hydride (81 mmol) in mineral
oil at 85 C. for 24 hours to give 21.5 grams (70% yield) of
3-(3,3,4,4,5,5,6,6,7,7,8,8,8- tridecafluorooctyloxy-
methyl)-3-methyloxetane; a colorless oil with a boiling
point of 66-68 C./2-2.5 mm-Hg; 'H NMR (CDC13) d 4.50 and
4.36 (AB, J=5.5 Hz, 4 H), 3.78 (t, J=6.6 Hz, 2 H), 3.53 (s,
2 H), 2.42 (triplet of triplets, J=6.6 and 18 Hz, 2 H), and
1.31 (s, 3 H); 13c NMR (CDC13) 8 79.89, 63.31, 39.9, 31.64
(t), and 21.1 (signals due to carbons bearing fluorines are
not included due to the complex splitting patterns and
overlap of signals); 19F NMR d-81.4 (3 F), -113.8 (2 F),
-118.2 (2 F), -122.3 (2 F), -123.3 (2 F), -124.1 (2 F) and -
126.7 (2 F). The elemental analysis was: Calculated for
C13H13F1302: C, 34.8; H, 2.9; F, 55.1. Found: C, 35.1; H,
3.0; F, 54.7.
EXAMPLE 9
PURIFICATION OF COMMERCIAL FLUOROALCOHOLS
Zonyl BA-L is a narrow distribution, oligomeric mixture
of fluoroalcohols that is available from Dupont Chemicals in
pilot plant quantities. Zonyl BA-L is a yellow liquid which
by GLC is a mixture of the following oligomers:
3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctan-l-ol (C8, 60%);
3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecan-l-
ol (ClO, 26%); 3,3,4,4,5,5,6,6,7,7,8,8, 9,9,10,10,11,11,
12,12,12-heneicosafluorododecanol (C12, 6%); and various
unidentified high boiling compounds (8%). Zonyl BA-L was
2100218
washed with equal volumes of 10 weight percent aqueous
sodium thiosulfate, 10 weight percent aqueous sodium
bicarbonate (to remove HF), water, and brine, dried,
filtered, and distilled under reduced pressure (3 mm-Hg) at
5 50-100 C. to give a mixture of 69% C8, 26% C10 and 5% C12 in
83% yield.
EXAMPLE 10
PREPARATION OF A MIXTURE OF 3,3,4,4,5,5,6,6,7,7,8,8,8-
TRIDECAFLUOROOCTYLOXYMETHYL-,
10 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-
HEPTADECAFLUORODECYLOXYMETHYL-,
AND
3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-
HENEICOSAFLUORODODECYLOXYMETHYL-3-METHYLOXETANE
15 In a manner similar to that described above, a mixture
of 69% C8, 26% C10 and 5% C12 fluoroalcohols (51.6 grams,
129 mmol) was reacted with 27 grams of 3-iodomethyl-
3-methyloxetane (127 mmol) in 500 milliliters of
dimethylformamide at 85 C. for 18 hours to give 60 grams of
crude product. The crude product was fractionally distilled
through a 6" Vigerux column to yield the following
fractions: Fraction #1 (4.8 grams) was collected between
C. and 45 C. at 3.5-2.9 mm-Hg, and was a mixture of
unreacted fluoroalcohols. Fraction #2 (2.8 grams) was
25 collected at 45-71 C./0.7-3.0 mm-Hg, and was a mixture of
unreacted fluoroalcohols and fluorinated oxetane monomers.
The final fraction (49 grams, 80%), boiling at
70-85 C./0.7-0.9 mm-Hg, was a mixture of 73% 3,3,4,4,5,5,
6,6,7,7,8,8,8-tridecafluorooctyloxymethyl- 3-methyloxetane
(C8-oxetane) , 24% 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-
heptadecafluorodecyloxymethyl-3-methyloxetane (ClO-oxetane),
and 3% 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-
heneicosafluorododecyl-oxymethyl-3-methyloxetane
(C12-oxetane), a colorless oil with a boiling point of
70-85 C./0.7-0.9 mm-Hg; 'H NMR (CDC13) 6 4.50 and 4.35 (AB,
J=5.9 Hz, 4 H), 3.78 (t, J=6.6 Hz, 2 H),, 3.53 (s, 2 H), 2.42
(tt, J=6.6 and 17.6 Hz, 2 H), and 1.31 (s, 3 H); 13c NMR 6
2100218
16
21.3, 31.86 (t, J=130.1 Hz), 40.2, 63.6, 76.8, and 80.2
(signals for carbons bearing fluorine are not included due
to complex splitting patterns and overlap of signals); 19F
NMR S -81.5, -113.8, -122.3, -123.3, -124.1, -124.5, -125.8,
and -126.7.
EXAMPLE 11
PREPARATION OF
3,3-BIS(2,2,3,3,4,4,4-HEPTAFLUOROBUTOXY-
METHYL)OXETANE
In a manner similar to that described above, 155 grams
of 3,3-bis(chloromethyl)oxetane (1 mole) was reacted with
402 grams of 2,2,3,3,4,4,4-heptafluorobutan-l-ol (2.01
moles) in two liters of dimethylformamide in the presence of
100 grams of a 50 weight percent dispersion of sodium
hydride in mineral oil, (2.3 moles) at 85 C. for 16 hours to
give 340 grams (70% yield) of 3,3-bis(2,2,3,3,4,4,4-
heptafluorobutoxymethyl)oxetane, a clear colorless liquid.
Glc analysis revealed the purity of this material to be in
excess of 99%, with a boiling point of 70-72 C./1.0-1.3
mm-Hg; 1H NMR (CDC13) 6 4.44 (s, 4 H), 3.97 (t, J=13.2 Hz, 4
H), 3.86 (s, 4 H); 13C NMR 6 43.9, 68.1 (t), 73.5, and 75.6
(signals from carbons bearing fluorine are not included due
to the complex splitting patterns and overlap of signals);
19F NMR d-81.6 (3 F), -121.0 (2 F) and -128.3 (2 F). The
elemental analysis was: Calculated for C13H12F1403: C, 32.4;
H, 2.5. Found: C, 32.3; H, 2.3.
EXAMPLE 12
PREPARATION OF
3,3-BIS(2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-PENTADECA-
FLUOROOCTYLOXYMETHYL)OXETANE
in a manner similar to that described above, 15.5 grams
of 3,3-bis-(chloromethyl)oxetane (0.1 mole) was reacted with
84 grams of 2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-pentadeca-
fluorooctan-l-ol (0.21 moles) in 300 milliliters of
dimethylformamide in the presence of 10.0 grams of a 50
weight percent dispersion of sodium hydride in mineral oil
(0.23 moles) at 85 C. for 32 hours to give 58 grams (66%
210 0;~' 8
17
yield) of 3,3-bis(2,2,3,3,4,4,4-heptadecafluorooctyl-
oxymethyl)oxetane, a clear colorless liquid. An analytical
sample was prepared by chromatographing the product on a
silica gel column using methylene chloride as an eluent.
The boiling point of the liquid was 130-35 C./1.0-1.2 mm-Hg.
The analyses were: 'H NMR (CDC13) b 4.44 (s, 4 H), 3.96 (t,
J=13.4 Hz, 4 H), 3.88 (s, 4 H); 13c NMR S 44.2, 68.7 (t),
73.9, and 76.5 (signals from carbons bearing fluorine are
not included due to the complex splitting patterns and
overlap of signals); 19F NMR 6-81.5 (6 F), -119.7 (4 F),
-121.5 (4 F), -122.6 (8 F), -123.3 (4 F), and -126.8 (4 F).
The elemental analysis was: Calculated for C21H12F3003: C,
28.6; H, 1.4; F, 64.6. Found: C, 28.2; H, 1.5.
EXAMPLE 13
COPOLYMERIZATION OF 3-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-
PENTADECAFLUOROOCTYLOXYMETHYL)-3-METHYLOXETANE WITH
3-(2,2,2-TRIFLUOROETHOXYMETHYL)-3-METHYLOXETANE
A one-liter, three-necked, round-bottomed flask was
fitted with a mechanical stirrer, nitrogen inlet/outlet
tubes, a reflux condenser, a thermometer, and a constant
addition funnel. The apparatus was dried with a heat gun,
cooled under nitrogen to room temperature, and charged with
a mixture of 0.914 grams of trimethylolpropane (6.52 mmol),
3.1 grams of boron trifluoride tetrahydrofuranate (22 mmol),
160 milliliters of 1,1,2-trichlorotrifluoroethane and 30
milliliters of anhydrous methylene chloride. The mixture
was stirred at room temperature for 30 minutes, cooled to
10 C., and then treated, dropwise, with a solution of 106
grams of 3-(2,2,2-trifluoroethoxymethyl)-3-methyloxetane
(576 mmol) and 94 grams of 3-(2,2,3,3,4,4,5,5,6,6,
7,7,8,8,8-pentadecafluorooctyloxymethy)-3-methyloxetane
(195.2 mmol) in 40 milliliters of 1,1,2-trichloro-
trifluoroethane. A mild exotherm was observed on addition
of the monomer. The reaction temperature was maintained at
18 C. throughout the addition and the progress of the
reaction was monitored by NMR analysis. The mixture was
stirred at 18 C. for 2 hours and then at 25 C. for 4 hours
210 0 '18
18
at which time NMR analysis of an aliquot indicated that 98
percent of the oxetane monomers were consumed. The reaction
mixture was diluted with 50 milliliters of methylene
chloride and 50 milliliters of 1,1,2-trichlorotrifluoro-
ethane, and quenched with 50 milliliters of water. The
organic layer was separated, washed with two equal volumes
of water, and added dropwise to a 10 fold excess of methanol
at room temperature. The precipitated oil was separated and
redissolved in a 50:50 mixture of inethylene chloride and
1,1,2-trichlorotrifluoroethane and transferred to a 500-
milliliter, round-bottomed flask. The solvent was
evaporated under reduced pressure and the resulting oil was
dried at 85 C./2 mm-Hg for 16 hours to give 170 grams of a
clear, colorless, viscous oil, corresponding to 85 percent
yield. The NMR analyses of this material indicated it was a
random copolymer of the above two monomers in a 74:26 ratio.
The polymer analyses were: DSC, T9 -40 C.; GPC (THF,
polystyrene standard) Number Average Molecular weight was
6,178, Weight Average Molecular Weight was 7,286,
Polydispersity was 1.18; Number average Molecular Weight by
'H NIH2 was 7,040; Functionality was 3; Inherent viscosity
was 0.065; 'H NM'R (CDC13) S 0.94 (s, -CH3), 3.23 (m, backbone
-CH2'S) , 3.47 (s, -CH20), 3.75 (q, J=8. 6 Hz, -CHZCF3) and
3.85 (t, J=13.5 Hz, -CH2C3F7) ; IH NMR (CDC1,/Trifluoroacetic
anhydride) 8 1.00 (s, -CH3)1 3.37 (m, backbone -CH21s), 3.49
(s,-CH20), 3.78 (q, J=8.6 Hz, -CH2CF3), 3.96 (t, J=13.5 Hz,
-CHZC3F7) , and 4.30 (s, CH2OCOCF3) f 13c NMR (CDC13) d 17.1,
41.2, 41.3, 68.5 (t), 68.9 (q), 73.7, 75.3 and 75.5.
In a manner similar to that described above, random
copolymers of above monomers in 50:50, 33:67, 25:75 and
10:90 ratios were also prepared. These copolymers were
clear, colorless oils that were soluble in a solvent mixture
of methylene chloride and 1,1,2-trichlorotrifluoroethane.
210~~IS
19
EXAMPLE 14
COPOLYMERIZATION OF 3-(2,2,2-TRIFLUOROETHOXYMETHYL)-3-
METHYLOXETANE WITH
3-(2,2,3,3,4,4,4-HEPTAFLUOROBUTOXYMETHYL)-3-
METHYLOXETANE
In a manner similar to that described in Example 13 , a
solution of 35 grams of 3-(2,2,2-trifluoro ethoxymethyl)-
3-methyloxetane (190 mmol) and 183 grams of 3-(2,2,3,3,-
4,4,4-heptafluorobutoxymethyl)-3-methyloxetane (644 mmol) in
50 milliliters of 1,1,2-trichlorotri-fluoroethane was added
to a mixture of 0.390 gram of 1,4-butanediol (4.33 mmol),
1.55 grams of boron trifluoride tetrahydrofuranate (11.1
mmol), and 100 milliliters of methylene chloride at 18 C.
The mixture was stirred at 18 C. for 3 hours, quenched with
water, and precipitated into methanol to give, after drying
at 85 C./2 mm-Hg for 16 hours, 186 grams of a clear,
colorless oil, corresponding to 85 percent yield. NMR
analysis revealed that this material was a 22:78 random
copolymer of the above two monomers.
The polymer analyses were: DSC, T9= -42'C.; GPC (THF,
polystyrene standard) Number Average Molecular weight was
15,660; Weight Average Molecular Weight was 30,640;
Polydispersity was 1.96; Number Average Molecular Weight by
IH NMR was 18,400; Functionality was 2; Inherent viscosity
was 0.071; 1H NMR (CDC13/Freon 113) d 0.91 (s, CH3), 3.22 (m,
backbone -CH2'S), 3.44 (s, -CH2O), 3.79 (q, J=8.8 Hz,
-CHZCF3) and 3.86 (t, J=13.5 Hz, -CHzC3F7) ;'H NMR CDC13/Freon
113/ Trifluoroacetic anhydride) 6 0.95 (s, -CH3), 3.23 (m,
backbone -CHZ'S) , 3.46 (s,-CH2O), 3.77 (q, J=8.6 Hz, CH2CF3) ,
3.87 (t, J=13.5 Hz, -CH2C3F7) , and 4.31 (s, CHZOCOCF3) ; 13C
NMR (CDC13/Freon 113) 6 17.3, 41.6, 41.8, 68.6 (t), 69.3
(q), 74.2, 75.6, and 75.9 (signals from carbons bearing
fluorine are not included).
In a similar manner, random copolymers of above
monomers in 50:50 and 75:25 ratios were also prepared. The
copolymers were clear, colorless oils that were soluble in
tetrahydrofuran, methylene chloride and
2100213
1,1,2-trichlorotrifluoroethane (Freon 113).
EXAMPLE 15
POLYMERIZATION OF 3-(3,3,4,4,5,5,6,6,7,7,8,8,8-
TRIDECAFLUOROOCTYLOXYMETHYL)-3-METHYLOXETANE
5 In a manner similar to that described in Example VII, a
solution of 10 grams of 3-(3,3,4,4,5,5,6,6,7,7,8,8,8-
tridecafluorooctyloxymethyl)-3-methyloxetane (22.3 mmol) in
three milliliters of Freon 113 was added dropwise to a
mixture of 109 milligrams of boron trifluoride tetrahydro-
10 furanate (0.78 mmol) and 35 milligrams of 1,4-butanediol
(0.39 mmol) in methylene chloride at 10 C. The mixture was
stirred at room temperature for 24 hours, quenched with
water, and precipitated in methanol to give, after drying at
80 c/2 mm-Hg for 16 hours, 8.3 gram of poly 3-(3,3,4,4,5,5,-
15 6,6,7,7,8,8,8-tridecafluorooctyl-oxymethyl)-3-methyloxetane,
a clear colorless oil. The polymer analyses were: Inherent
viscosity (THF/30 C.) was 0.067 dL/g; GPC: Mn = 5,340, Mw =
6,620, Poly Dispersity = 1.24; DSC, Tg =-38 C.; IH NMR
(CDC13,/Freon 113) S 3.67 (t, 5.9 Hz, 2 H), 3.31 (s, 2 H),
20 3.21 (m, 4 H), 2.35 (m, 2 H), and 0.93 (s, 3 H); IH NMR
(CDC13/Freon 113) d 0.95 (s, 3 H), 2.37 (br t, J=18.3 Hz, 2
H), 3.25 (m, 4 H), 3.35 (s, 2H), 3.71 (t, 6.0 Hz, 2 H), and
4.30 (AB, -CHZOCOCF3); Number Average Molecular Weight based
on 'H NMR was 4,756; 13C NMR (CDC13/Freon 113) d 17.35, 31.75
(t) , 41.5, 63.4, 74.1 and 74.3.
EXAMPLE 16
COPOLYMERIZATION OF A MIXTURE OF
3,3,4,4,5,5,6,6,7,7,8,8,8-
TRIDECAFLUOROOCTYLOXYMETHYL-,
3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-
HEPTADECAFLUORODECYLOXYMETHYL-, AND
3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-
HENEICOSAFLUORODODECYLOXYMETHYL-3-METHYLOXETANE
In a manner similar to that described in Example 13, a
solution of 30 grams of C8- (73%), C10- (24%), and C12-(3%)
oxetane monomers (62 mmol) in 10 milliliters of Freon 113
was added dropwise to a mixture of 300'milligrams of boron
210021S
21
trifluoride tetrahydrofuranate (2.14 mmol) and 95 milligrams
of 1,4- butanediol (1.05 mmol) in 30 milliliters of
methylene chloride at 10 C. The mixture was stirred at room
temperature for 24 hours, quenched with water, and
precipitated in methanol to give, after drying at 80 C./2
mm-Hg for 16 hours, 24 grams of the title copolymer,
corresponding to 80 percent yield. The copolymer was a
colorless, viscous oil. The analysis of the copolymer was:
Inherent viscosity = 0.075 dL/g; GPC: Mn = 6,639, Mw =
9,368, Poly Dispersity = 1.41; 'H NMR (CDC13/Freon 113) S
0.95 (s, 3 H), 2.37 (br t, J=18.3 Hz, 2 H), 3.25 (m, 4 H),
3.35 (s, 2H), 3.71 (t, 6.0 Hz, 2 H), and 4.30 (AB,
-CH20COCF3); Number Average Molecular Weight based on 'H NMR
was 5,021; 13C NMR (CDC13/Freon 113) 6 17.35, 31.75 (t),
41.1, 41.5, 63.4, 74.1 and 74.3.
EXAMPLE 17
POLYMERIZATION OF
3,3-BIS(2,2,3,3,4,4,4-HEPTAFLUOROBUTOXYMETHYL)OXETANE
In a manner similar to that described in Example 13 , a
solution of 252 grams of 3,3-bis(2,2,3,3,4,4,4-heptafluoro-
butoxymethyl)oxetane (523 mmol) in 75 milliliters of Freon
113 was added to a mixture of 1.05 grams of boron
trifluoride tetrahydrofuranate (7.5 mmol) and 0.265 gram of
1,4-butanediol (2.93 mmol) in 178 milliliters of methylene
chloride at 10 C. The mixture was stirred at room
temperature for 48 hours at which time NMR analysis of an
aliquot indicated 96 percent conversion. The reaction was
quenched with water and the polymer was precipitated into
methanol to give, after drying at 80 C./2 mm-Hg for 16
hours, 211 grams of poly 3,3-bis (2,2,3,3,4,4,4-heptafluoro-
butoxymethyl)oxetane, a colorless oil in 84 percent yield.
GPC analysis of this oil revealed it was a mixture of 68%
linear and 32% cyclic materials. The cyclic product was
isolated and identified as the cyclic tetramer, a white waxy
solid with a melting point 80 C.; IH NMR 6 3.87 (t, J=13.5
Hz, 4 H) , 3.54 (s, 4H), and 3.32 (S, 4H) (No end groups
were observed on addition of trifluoroacetic anhydride); 13C
2100'-1 1~
22
NMR 6 71.2, 68.6, 68.4 (t), and 46.2 (signals due to carbons
bearing fluorine are not included).
The above oil was further purified by first dissolving
the material in methylene chloride/Freon 113 (75:25
mixture), precipitating the polymer into a 10 fold excess of
methanol, stirring the precipitated oil with tetrahydrofuran
at room temperature for 2 days, and finally separating and
drying the insoluble fraction at 85 C. at 2 mm-Hg for 16
hours. This yielded 128 grams of a clear, viscous oil,
corresponding to 51% overall yield. The oil by GPC analysis
was determined to be a mixture of 91% linear polymer and 9%
cyclic tetramer. The polymer analyses were: DSC, Tg =-43 C
gpc: Mn=5, 526, M. = 7,336, PD = 1.32; 'H NMR (CDCl3/Freon
113/TFAA) 6 3.39 (s, 4 H), 3.59 (s, 4 H), 3.87 (t, J=13.5
Hz, 4 H) and 4.40 (s, -CH2OCOCF3); Number Average Molecular
Weight based on 'H NMR = 5,200; 13C NMR (CDC13/Freon 113) 8
46.4, 68.5 (t), 70.1 and 72.1 (signals from carbons bearing
fluorines are not included).
The invention has been described with reference to the
illustrated and presently preferred embodiment. It is not
intended that the invention be unduly limited by this
disclosure of the presently preferred embodiment. Instead,
it is intended that the invention be defined, by the
compounds, products and their equivalents, and the method
steps and their equivalents, as set forth in the following
claims: