Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
Wo 92~'20671 ~ ~ ~ /DB92/00356
Novel 2H-benzotb~vran derivatives subatitutod ~n the 4
~osition bY arvl or N-heteroarYl, proce~ses for their
~reParation and their use, and preDarations compr~sina
the com~ounds
Description
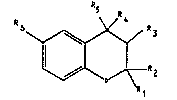
The invention relates to novel sub~tituted
benzopyran derivatives of the general formula I
R
~~ Z
in which
Rl and R" which may be identical or different, denote
hydrogen, Clc-alkyl, branched C36-alkyl, C37-cyoloalkyl or
phenyl or denote, together with the carbon atom enclosed
by them, C37-spiroalkyl,
either R3 or R~ is hydrogen, hydroxyl, Cl,-alkoxy, formyl-
oxy, Cl~-alkylcarbonyloxy, Cla-alkoxycarbonyloxy, Cl,-
monoalkylaminocarbonyloxyorCl~-dialkylaminocarbonyloxy,
where the Cl,-alkyl- or -alkoxy groups may be either
linear or branched, and the other substituent of the two
in each case is hydrogen, or R3 and R~ together form a
bond,
R5 denotes a monocyclic six-membered aryl or N-heteroaryl
group, a bicyclic aryl or N-heteroaryl group which is
composed of two fused six-membered rings, in which the
heterocycle contains one or two nitrogen atoms which
21~?~;3
-- 2
. belongs to one of the following three subgroup~ A), B) or
C), in which context in
A) the aryl or N-heteroaryl group R, carries a hydroxyl
group in the 2 position and is optionally urther sub~ti-
tuted one or two times by halogen, cyano, Cl,-alkyl or
Cl8-alkoxy, in
B) R5 is an N-heteroaryl group containing one or two N
atoms, which carries an N-oxide group in the 2 position
and is optionally substituted one or two times by halo-
gen, C~8-alkyl, Cl~-alkoxy, Cl~-mono- or Cl,-dialkylAm~no,
hydroxyl, amino, cyano, Cl~-alkoxycarbonyl, Cl~-mono- or
Cl8-dialkylaminocarbonyl, hydroxycarbonyl, aminocarbonyl
or phenyl, and in
C) Rs is an N-heteroaryl group or an N-heteroaryl N-oxide
whose basic structure derives from the group 3-pyridyl,
4-pyridyl, 5-pyrimidinyl, 3-pyridazinyl, 4-pyrimidinyl,
2-pyrazinyl, 3-quinolyl and 4-isoquinolyl and is option-
ally substituted one or two times by halogen, Cl~-alkyl,
Cl~-alkoxy, Cl~-monoalkylamino, Cl~-dialkylamino, cyano,
hydroxyl, amino or phenyl, the hydroxyl group or the
N-oxide group in this subgroup not being located in the
2 position, and
Rc is difluoromethoxy, trifluoromethoxy, trifluoroethoxy,
tetrafluoroethoxy, difluoromethylthio, difluoromethyl-
sulfinyl, difluoromethylsulfonyl, trifluoromethylthio,trifluoromethylsulfinyl, trifluoromethylsulfonyl, tri-
fluoroethylthio, trifluoroethylsulfinyl, trifluoroethyl-
sulfonyl, phosphono or Cl~-dialkoxyphosphoryl, in which
abovementioned alkyl can be straight-chain or branched,
and to their salts and acid addition salts, tautomers and
optical isomer~, to processes for their preparation, to
their use and to preparations which comprise these
compounds.
For the sake of simplicity, the compounds accord-
ing to the invention are defined in only one tautomeric
form, which is reproduced by formula 1. The invention,
however, extends to all tautomeric forms of the
compounds.
` ~ 3 ~ 21~?~9~4
Compound~ of the general formula I and their
salts and acid addition salts contain asymmetrlc carbon
and sulfur atoms. Consequently, the invent~on al~o
relates to the various optical isomers and dla~tereomers.
The racemates can ~e resolved by methods ~nown per se to
give their optical isomers.
The invention also relates to the novel compound~
of the formulae II, IV, V and XI:
RS
0 ~ 2
0~ 2
R 1R 2
- 4 -
having the abov~mentioned meanings for Rl, ~2~ R~ and R~,
-` and to the processes for their preparation. They are u~od
as precursors or intermediates in the preparation of the
end products according to the invention.
Compounds which are related in structural term~
to the compounds of the present invention are described
in ~uropean Patent Application 298 452. The compounds of
the pre~ent invention, however, are neither specifically
disclosed nor suggested.
Unless otherwise specified, the alkyl groupe and
alkyl moietie~ or al~ylene moieties of groups according
to the invention can be straight-chain or branched, and
they possess in each case preferably from 1 to 6 carbon
atoms, preferably from 1 to 4 carbon atoms and parti-
cularly l or 2 carbon atoms. The branched alkyl groups
possQss at least 3 carbon atoms. Preferred alkyl or
alkylene moieties are methyl, ethyl, n-propyl, isopropyl
and butyl and, accordingly, methylene, ethylene, n- or
isopropylene and butylene.
Cycloalkyl groups according to the invention
preferably posses~ from 3 to 7 carbon atoms, particularly
3 to 6 carbon atoms. Cyclopropyl, cyclopentyl and cyclo-
hexyl are particularly preferred.
Formyl 18 HCO-, formyloxy is HCOO-, C1,-alkyl-
carbonyloxy is Cl~-alkyl-CO-O-, Cl~-alkoxycarbonyloxy is
C1~-alkyl-O-CO-O-, C~-monoalkylaminocarbonyloxy is Cl,-
alkyl-NH-CO-O-, C1,-dialkylaminocarbonyloxy is (Cl~-
alkyl)~N-CO-O-, Cl~-alkylcarbonyl is Cl,-alkyl-CO-, Cl~-
alkoxycarbonyl is Cl,-alkoxy-CO-, C~,-monoal~ylamino-
carbonyl is Cl,-alkyl-NH-CO-, Cl,-dialkylamlnocarbonyl is
(Cl,-alkyl),N-CO-, Cl,-dialkylamino is (Cl,-alkyl),N-, Cl~-
monoalkylamino is Cl,-alkyl-NH-, halogen is fluorine,
chlorine, bromine or iodine, phosphono denotes -PO(OH) 2
and Cl,-dialkoxyphosphoryl is (Cl~-alkyl-O)~PO-.
Tho trifluoroethyl group, or trifluoroethyl as
part of other radicals according to the invention, such
as trifluoroethoxy, is preferably 2,2,2-trifluoroethyl.
The tetrafluoroethyl group, or tetrafluoroethyl
as part of other radicals according to the invention,
- 5 -
such as tetrafluoroethoxy, is proferably 1,1,2,2-totra-
fluoroethyl.
Halogen i8 preferably fluorine or chlorino.
R1 and R~ are preferably hydrogen, methyl or
ethyl, of which methyl is particularly preferrod.
R1 and R2 are very particularly preferably both
simultaneously methyl.
If R1 and R2, together with the carbon atom which
they enclose, form a spiroalkyl ring, spirocyclopentyl
and spirocyclohexyl are preferred.
R3 represents preferably hydrogen or hydroxyl if
R~ is hydrogen, and R~ represents preferably hydrogen or
hydroxyl if R3 i~ hydrogen. Preferably, R3 and R~ also
form a bond, 80 that there i8 a double bond between the
C-3 and the C-4 of the benzopyran structure.
If R3 is not hydrogen but has the meaning given
above, and does not form a bond together with R~, then
tho substituents R3 and R5 in compounds of the general
formula I are preferably trans with respect to one
a 0 another.
If R3 or R~ represent allcoxy, ethoxy and, in
particular, methoxy are preferrod.
If R3 or R, represent allcylcarbonyloxy, propionyl-
oxy and, in particular, acetoxy and formyloxy are
preferred.
R3 and R,, very particularly proferably form a bond
between the C-3 and the C-4 of the benzopyran structure.
Monocyclic or bicyclic aryl or N-heteroaryl
groups which form the basis for the subgroups A), B) and
C) are, in particular, phenyl, naphthyl, pyridyl, pyr-
imidinyl, pyridazinyl, pyrazinyl, quinolyl, isoguinolyl,
but particularly preferably are pyridyl and pyrimidinyl.
Preferred mono- or bicyclic aryl or N-heteroaryl
groups Rs of ~ubgroup A) are 2-hydroxyphenyl, 4-chloro-2-
hydroxyphenyl, 2-hydroxy-4-methylphenyl, 4-cyano-2-
hydroxyphenyl, 2-hydroxy-1-naphthyl, 1-hydroxy-2-naph-
thyl, 3-hydroxy-2-naphthyl, 2-hydroxy-3-pyridyl,
4-hydroxy-3-pyridyl, 3-hydroxy-4-pyridyl, 3-hydroxy-4-
pyridazinyl, 3-hydroxy-2-pyrazinyl and 2-hydroxy-3-
Qquinolyl .
Preferred mono- or bicycllc N-heteroaryl groupe
Rs of subgroup B) are 2-pyridyl N-oxide, 3-chloro-2-
pyridyl N-oxide, 4-chloro-2-pyridyl N-oxide, 5-chloro-2-
pyridyl N-oxide, 6-chloro-2-pyridyl N-oxide, 3-methyl-2-
pyridyl N-oxide, 4-methyl-2-pyridyl N-oxlde, 5-methyl-2-
pyridyl N-oxide, 6-methyl-2-pyridyl N-oxide, 5-phenyl-2-
pyridyl N-oxide, 4-hydroxy-2-pyridyl N-oxide, 5-hydroxy-
2-pyridyl N-oxide, 6-hydroxy-2-pyridyl N-oxide, 4-
methoxy-2-pyridyl N-oxide, 5-methoxy-2-pyridyl N-oxide,
6-methoxy-2-pyridyl N-oxide, 5-methoxycarbonyl-2-pyridyl
N-oxide, 5-cyano-2-pyridyl N-oxide, 2-pyrimidinyl 1-
oxide, 6-pyrimidinyl l-oxide, 2-pyrazinyl 1-oxide, 2-
quinolyl N-oxide and 3-isoguinolyl N-oxide.
lS Preferred mono- or bicyclic N-heteroaryl groups
Rs Of subgroup C) are 3-pyridyl, 3-pyridyl N-oxide,
4-pyridyl, 4-pyridyl N-oxide, 2-methyl-3-pyridyl, 4-meth-
yl-3-pyridyl, 5-methyl-3-pyridyl, 6-methyl-3-pyridyl,
2-methoxy-3-pyridyl, 4-methoxy-3-pyridyl, 5-methoxy-3-
pyridyl, 6-methoxy-3-pyridyl, 2-fluoro-3-pyridyl,
2-chloro-3-pyridyl, 6-chloro-3-pyridyl, 2-cyano-3-
pyridyl, 6-cyano-3-pyridyl, 6-methoxycarbonyl-3-pyridyl,
6-dimethylamino-3-pyridyl, 6-hydroxy-3-pyridyl, 3-pyrid-
azinyl, 6-methyl-3-pyridazinyl, 2-pyrazinyl, 2-pyrazinyl
4-oxido, 5-methyl-2-pyrazinyl, 6-methyl-2-pyrazinyl,
5,6-dimethyl-2-pyrazinyl, 4-pyrimidinyl, 5-pyrimidinyl,
5-pyrimldinyl 1-oxide, 3-quinolyl, 3-quinolyl N-oxide,
4-isoquinolyl and 4-ieoquinolyl N-oxide.
Compounds according to tho invention containing
substituents of subgroups B) and C) aro preferred;
particular preference is given, however, to those
containing the following eubstituents:
2-pyridyl N-oxide, 2-pyrimidinyl N-oxide, 2-pyrazinyl
l-oxide, 6-pyrimidinyl 1-oxide, 2-quinolyl N-oxide,
3-isoquinolyl N-oxide, 3-pyridyl, 3-pyridyl N-oxide,
2-fluoro-3-pyridyl, 5-pyrimidinyl, 3-pyridazinyl, 3-quin-
olyl and 4-ieoquinolyl.
Very particularly preferred subetituente R5 are
2-pyridyl N-oxide and 3-pyridyl.
~ 7 ~ 2~
If Rc is C1,-dial~oxyphosphoryl, then the alkoxy
radicals may be identical or different. Preferably they
are both simultaneously methoxy or ethoxy.
Preferred substituents R6 are difluoromethoxy,
trifluoromethoxy, trifluoromethylthio, trifluoromethyl-
sulfonyl and diethoxyphosphoryl.
Preferred compounds of the general formula I are
compounds of the general formula VI. Particularly pre-
ferred compounds of the general formula VI are those in
which Rl and R2 are both simultaneou~ly methyl and R5
represents 2-pyridyl N-oxide or 3-pyridyl.
The following compounds according to the in~en-
tion, their salt~ and acid addition salts, tautomers and
optical isomers, are preferred:
1. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-2H-
benzo~b]pyran l'-oxide
2. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-2H-
benzo[b]pyran l'-oxide
3. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyridyl)-
2H-benzo~b]pyran l'-oxide
4. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2'-pyr-
idyl)-2H-benzo[b]pyran l'-oxide
5. 6-Trifluoromethylsulfinyl-2,2-dimethyl-4-(2'-
pyridyl)-2~-benzo[b]pyran 1'-oxide
6. 6-Difluoromethylthio-2,2-dimethyl-4-(2'-pyridyl)-2H-
benzo[blpyran l'-oxide
7. 6-Difluoromethylsulfonyl-2,2-dimethyl-4-(2'-
pyridyl)-2H-benzo[b]pyran l'-oxide
8. 6-Difluoromethylsulfinyl-2,2-dimethyl-4-(2'-pyr-
idyl)-2H-benzo[b]pyran l'-oxide
9. 6-(2',2',2'-Trifluoroethoxy)-2,2-dimethyl-4-(2'-pyr-
idyl)-2H-benzo[b]pyran l'-oxide
10. 6-(2',2',2'-Trifluoroethylthio)-2,2-dimethyl-4-(2'-
pyridyl)-2H-benzo[b]pyran l'-oxide
11. 6-(2',2',2'-Trifluoroethylsulfonyl)-2,2-dimethyl-4-
(2'-pyridyl)-2H-benzo[b]pyran l'-oxide
12. 6-(2',2',2'-Trifluoroethyl~ulfinyl)-2,2-dimethyl-4-
(2'-pyridyl)-2~-benzo[b]pyran 1'-oxide
13. 6-(1',1',2',2'-Tetrafluoroethoxy)-2,2-dimethyl-4-
_ 8 - ~ ~ ~2~
(2~-pyridyl)-2H-benzolb]pyran l'-ox~de
14. 6-Diethoxyphosphoryl-2,2-dimethyl-4-(2'-pyridyl)-2R-
benzotb~pyran l~-oxide
15. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-3,4-
dihydro-2H-benzo[b]pyran l'-oxide
16. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-3,4-
dihydro-2H-benzolb]pyran l'-oxide
17. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyridyl)-
3,4-dihydro-2H-benzo[b]pyran l'-oxide
18. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2'-pyr-
idyl)-3,4-dihydro-2R-benzotb]pyran l'-oxide
19. 6-Trifluoromethylsulfinyl-2,2-dimethyl-4-(2'-pyr-
idyl)-3,4-dihydro-2H-benzotb]pyran l'-oxide
20. 6-Difluoromethylthio-2,2-dimethyl-4-(2'-pyridyl)-
3,4-dihydro-2H-benzo[b]pyran l'-oxide
21. 6-Difluoromethylsulfonyl-2,2-dimethyl-4-(2'-pyr-
idyl)-3,4-dihydro-2H-benzo[b]pyran l'-oxide
22. 6-Diethoxyphosphoryl-2,2-dimethyl-4-(2'-pyridyl)-
3,4-dihydro-2H-benzo[b]pyran l'-oxide
23. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-3,4-
dihydro-2H-benzo[b]pyran-4-ol l'-oxide
24. 6-Dlfluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-3,4-
dihydro-2H-benzo[b]pyran-4-ol l'-oxide
2S. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyridyl)-
2S 3,4-dihydro-2H-benzo[b]pyran-4-ol l'-oxide
26. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2'-pyr-
idyl)-3,4-dihydro-2H-benzo[b]pyran-4-ol l'-oxide
27. 6-Trifluoromethylsulfinyl-2,2-dimethyl-4-(2'-pyr-
idyl)-3,4-dihydro-2H-benzotb]pyran-4-ol l'-oxide
28. 6-Trifluoromethylthio-2,2-dimethyl-4-methoxy-4-(2'-
pyridyl)-3,4-dihydro-2H-benzo[b]pyran l'-oxide
29. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-methoxy-4-
(2'-pyridyl)-3,4-dihydro-2H-benzotb]pyran l'-oxide
30. 6-Trifluoromethyl~ulfinyl-2,2-dimethyl-4-methoxy-4-
(2'-pyridyl)-3,4-dihydro-2H-benzotb]pyran l'-oxide
31. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-trans-
3,4-dihydro-2H-benzo[b]pyran-3-ol l'-oxide
32. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyridyl)-
trans-3,4-dihydro-2H-benzo[b]pyran-3-ol l'-oxide
- 9 - 2 ~ s
33. 6-Trifluoromethylthlo-2,2-dimethyl-4-(2'-pyridyl)-
trans-3,4-dihydro-2~-benzolb]pyran-3-ol 1'-oxlde
34. 6-Trifluoromethylsulfonyl-2,2-d~methyl-4-(2'-
pyridyl)-trans-3,4-dihydro-2H-benzo[b~pyran-3-ol
l'-oxide
35. 6-Trifluoromethylth~o-2,2-d$methyl-4-(3'-chloro-2'-
pyridyl)-2H-benzo[b]pyran 1'-oxide
36. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(3'-chloro-
2'-pyridyl)-2H-benzotb]pyran l'-oxide
37. 6-Difluoromethoxy-2,2-dimethyl-4-(4'-chloro-2'-
pyridyl)-2H-benzo[blpyran 1'-oxide
38. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-chloro-2'-
pyridyl)-2H-benzo[b~pyran 1'-oxide
39. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(4'-chloro-
2'-pyridyl)-2H-benzolb]pyran 1'-oxide
40. 6-Difluoromethoxy-2,2-dimethyl-4-(5'-chloro-2'-
pyridyl)-2H-benzotb]pyran 1'-oxide
41. 6-Trifluoromethylthio-2,2-dimethyl-4-(5'-chloro-2'-
pyridyl)-2H-benzo[blpyran 1'-oxide
42. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-chloro-
2'-pyridyl)-2H-benzolb]pyran 1'-oxide
43. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(6'-chloro-
2'-pyridyl)-2H-benzo[b]pyran 1'-oxide
44. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-methyl-2'-
pyridyl)-2H-benzolb~pyran l'-oxide
45. 6-Difluoromethoxy-2,2-dimethyl-4-(4'-methyl-2'-
pyridyl)-2H-benzolb]pyran l'-oxide
46. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(4'-methyl-
2'-pyridyl)-2H-benzolb]pyran l'-oxide
47. 6-Difluoromethoxy-2,2-dimethyl-4-(5'-methyl-2'-
pyridyl)-2H-benzolb]pyran l'-oxide
48. 6-Trifluoromethylthio-2,2-dimethyl-4-(5'-methyl-2'-
pyridyl)-2H-benzolb~pyran l'-oxide
49. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-methyl-
2'-pyridyl)-2H-benzo[b]pyran l'-oxide
50. 6-Trifluoromethoxy-2,2-dimethyl-4-(5'-methyl-2'-
pyridyl)-2H-benzo[b]pyran l'-oxide
51. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(6'-methyl-
2'-pyridyl)-2H-benzolb]pyran l'-oxide
- lo 2 ~ ~2;~ Jil
52. 6-Trifluoromethylthio-2,2-dimethyl-4-(S'-phenyl-2'-
pyridyl)-2~-benzo[b]pyran 1'-oxide
53. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-phenyl-
2'-pyridyl)-2H-benzotb]pyran 1'-oxide
S 54. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-hydroxy-2'-
pyridyl)-2H-benzotb]pyran 1'-oxide
SS. 6-Trifluoromethylthio-2,2-dimethyl-4-(S'-hydroxy-2'-
pyridyl)-2~-benzo[b~pyran 1'-oxide
56. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-hydroxy-2'-
pyridyl)-2H-benzolb]pyran 1'-oxide
57. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-methoxy-2'-
pyridyl)-2H-benzolb]pyran 1'-oxide
58. 6-Trifluoromethylthio-2,2-dimethyl-4-(S'-methoxy-2'-
pyridyl)-2H-benzotb~pyran 1'-oxide
59. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-methoxy-2~-
pyridyl)-2H-benzotb]pyran 1'-oxide
60. 6-Difluoromethoxy-2,2-dimethyl-4-(5'-methoxy-
carbonyl-2'-pyridyl)-2H-benzotblpyran l'-oxide
61. 6-Difluoromethoxy-2,2-dimethyl-4-(5'-cyano-2'-
pyridyl)-2H-benzotb]pyran 1'-oxide
62. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyrimidinyl)-
2H-benzotb]pyran l'-oxide
63. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyrimidinyl)-
3,4-dihydro-2H-benzotb~pyran 1'-oxide
64. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyrimidinyl)-
2H-benzotb~pyran l'-oxide
65. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyrimidinyl)-
3,4-dihydro-2H-benzotb~pyran 1'-oxide
66. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(2'-pyrim-
idinyl)-2~-benzotb~pyran 1'-oxide
67. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(2'-pyrim-
idinyl)-3,4-dihydro-2H-benzolb~pyran 1'-oxide
68. 6-Difluoromethoxy-2,2-dimothyl-4-(6'-pyrimidinyl)-
3,4-dihydro-2H-benzotb~pyran 1'-oxide
69. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-pyrimidinyl)-
3,4-dihydro-2H-benzo[b~pyran 1'-oxide
70. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(6'-pyrim-
idinyl)-3,4-dihydro-2H-benzo[b]pyran l'-oxide
71. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-pyrazinyl)-3,4-
dihydro-2H-benzolblpyran l'-oxide
72. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-pyrazinyl)-
3,4-dihydro-2H-benzo[b]pyran l'-ox~de
73. 6-Trifluoromethylsulfonyl-2,2-dlmethyl-4-(2'-pyr-
azinyl)-3,4-dihydro-2H-benzolb~pyran l'-oxide
74. 6-Difluoromethoxy-2,2-dimethyl-4-(2'-gulnolyl)-2H-
benzo[blpyran l'-oxide
75. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-quinolyl)-2H-
benzo[b]pyran l'-oxide
76. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-qu~nolyl)-
2H-benzolb]pyran l'-oxide
77. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2'-quin-
olyl)-2H-benzolblpyran l'-oxide
78. 6-Trifluoromethylsulfinyl-2,2-dimethyl-4-t2'-quin-
olyl)-2H-benzolb]pyran l'-oxide
79. 6-Trifluoromethoxy-2,2-dimethyl-4-(3'-isoquinolyl)-
2H-benzolb]pyran l'-oxide
80. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-isoquin-
olyl)-2H-benzolb]pyran l'-oxide
81. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-~3'-iso-
quinolyl)-2H-benzo[blpyran l'-oxide
82. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-hydroxy-
phenyl)-2H-benzolb]pyran
83. 6-Trifluoromethylsulfonyl-2,2-dimothyl-4-(2~-
hydroxyphenyl)-2H-benzotb]pyran
84. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2~-
hydroxy-4'-chlorophenyl)-2~-benzolblpyran
85. 6-Trlfluoromethylsulfonyl-2,2-dimethyl-4-(2~-
hydroxy-4'-methylphenyl)-2H-benzolblpyran
86. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(2'-
hydroxy-4'-cyanophenyl)-2H-benzolb]pyran
87. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-hydroxy-1~-
naphthyl)-2H-benzolblpyran
88. 6-Trifluoromethylthio-2,2-dimethyl-4-(1'-hydroxy-2~-
naphthyl)-2H-benzolblpyran
89. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-hydroxy-2~-
naphthyl)-2H-benzolb]pyran
90. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-hydroxy-3'-
pyridyl)-2H-benzolblpyran
' - " ' "
:'
- 12 _ 2 ~ G.~
91. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-hydroxy-3'-
pyridyl)-2H-benzotb~pyran
92. 6-Trifluoromethylthio-2,2-dimothyl-4-(4'-hydroxy-3'-
pyridyl)-2H-benzolb~pyran
93. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-hydroxy-4'-
pyridyl)-2H-benzo[b~pyran
94. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-hydroxy-4'-
pyridazinyl)-2~-benzo[b]pyran
9S. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-hydroxy-2'-
pyrazinyl)-2H-benzo[blpyran
96. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-hydroxy-3'-
quinolyl)-2H-benzo[b]pyran
97. 6-Difluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-2H-
benzolb]pyran
98. 6-Difluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-2H-
benzolb]pyran l'-oxide
99. 6-Difluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-3,4-
dihydro-2H-benzo[b~pyran
100. 6-Trifluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-2H-
benzolb]pyran
101. 6-Trifluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-2~-
benzo[b]pyran l'-oxide
102. 6-Trifluoromethoxy-2,2-dimethyl-4-(3'-pyridyl)-3,4-
dihydro-2H-benzolb~pyran
103. 6-Trifluoromothylthio-2,2-dimothyl-4-(3'-pyridyl)-
2~-benzolb~pyran
104. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-pyridyl)-
2H-benzotb]pyran l'-oxido
105. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-pyridyl)-
3,4-dihydro-2H-benzolb]pyran
106. 6-Trifluoromothylthio-2,2-dimethyl-4-(3'-pyridyl)-
3,~4-dihydro-2H-benzotb]pyran-4-ol
107. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(3~-
pyridyl)-2H-benzolb~pyran
108. 6-Trifluoromethyleulfonyl-2,2-dimethyl-4-(3~-
pyridyl)-2H-benzolb~pyran 1'-oxide
109. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(3-
pyridyl)-3,4-dihydro-2~-benzo[b]pyran
110. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(3'-
- 13 -
pyridyl)-3,4-dihydro-2H-benzolb]pyran-4-ol
111. 6-Diethoxypho~phoryl-2,2-dimethyl-4-(3'-pyrldyl)-2H-
benzo[b]pyran
112. 6-Diethoxypho~phoryl-2,2-dimethyl-4-(3'-pyridyl)-2H-
benzo[b]pyran l'-oxide
113. 6-Diethoxyphosphoryl-2,2-dimethyl-4-(3'-pyridyl)-
3,4-dihydro-2H-benzo[b]pyran
114. 6-Difluoromethoxy-2,2-dimethyl-4-(4'-pyridyl~-2~-
benzo[b]pyran
115. 6-Trifluoromethoxy-2,2-dimethyl-4-(4'-pyridyl)-2H-
benzo[blpyran
116. 6-Trifluoromethoxy-2,2-dimethyl-4-(4'-pyridyl)-2~-
benzo[b]pyran l'-oxide
117. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-pyridyl)-
2H-benzo[b]pyran
118. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(4'-
pyridyl)-2H-benzo[b]pyran
119. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(4~-
pyridyl)-2H-benzotb]pyran l'-oxide
120. 6-Trifluoromethyl~ulfonyl-2,2-dimethyl-4-(2'-methyl-
3'-pyridyl)-2H-benzo[b]pyran
121. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(4'-methyl-
3'-pyridyl)-2H-benzolb]pyran
122. 6-Trifluoromethylsulfonyl-2,2-d~methyl-4-(5'-methyl-
3'-pyridyl)-2H-benzotb]pyran
123. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(6'-methyl-
3'-pyridyl)-2H-benzolb]pyran
124. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-methoxy-3'-
pyridyl)-2H-benzo[b]pyran
125. 6-Trifluoromothylthio-2,2-dimethyl-4-(2'-methoxy-3'-
pyridyl)-2H-benzotb]pyran-4-ol
126. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-methoxy-3~-
pyridyl)-2H-benzotb]pyran
127. 6-Trifluoromethylthio-2,2-dimethyl-4-(4'-methoxy-3'-
pyridyl)-2H-benzotb]pyran-4-ol
128. 6-Trifluoromethylthio-2,2-dimethyl-4-(5'-methoxy-3'-
pyridyl)-2H-benzo[b]pyran
129. 6-Trifluoromethylthio-2,2-dimethyl-4-(5~-methoxy-3~-
pyridyl)-2H-benzotb]pyran-4-ol
,
- 14 -
130. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-met~oxy-3'-
pyridyl)-2H-benzolb]pyran
131. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-methoxy-3'-
pyridyl)-2~-benzo[blpyran-4-ol
5132. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-fluoro-3'-
pyridyl)-2~-benzo[blpyran
133. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-fluoro-3'-
pyridyl)-2H-benzo[b]pyran-4-ol
134. 6-Trifluoromethoxy-2,2-dimethyl-4-(2'-chloro-3'-
10pyridyl)-2H-benzo[b]pyran
135. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-chloro-3'-
pyridyl)-2H-benzo[b]pyran
136. 6-Trifluoromethoxy-2,2-dimethyl-4-~2'-cyano-3'-
pyridyl)-2H-benzo[b]pyran
15137. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-cyano-3'-
pyridyl)-2H-benzo[b]pyran
138. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-methoxy-
carbonyl-3'-pyridyl)-2H-benzo[b]pyran
139. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-dimethylA~;no-
203'-pyridyl)-2H-benzo[b]pyran
140. 6-Trifluoromethoxy-2,2-dimethyl-4-(6'-hydroxy-3'-
pyridyl)-2H-benzolb]pyran
141. 6-Trifluoromethylthio-2,2-dimethyl-4-(3'-pyrid-
azinyl)-2H-benzolb]pyran
25142. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-methyl-3'-
pyridazinyl)-2H-benzo[b]pyran
143. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyrazinyl)-
2H-benzo[b]pyran
144. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyrazinyl)-
302H-benzotblpyran-4-ol
145. 6-Trifluoromethylthio-2,2-dimethyl-4-(2'-pyrazinyl)-
2H-benzo[blpyran 4'-oxide
146. 6-Trifluoromethylthio-2,2-dimethyl-4-(5'-methyl-
pyrazinyl)-2H-benzolb]pyran
35147. 6-Trifluoromethylthio-2,2-dimethyl-4-(6'-methyl-2'-
pyrazinyl)-2H-benzo[b]pyran
148. 6-Trifluoromethylthio-2,2-dimethyl-4-(5',6'-dime-
thyl-2'-pyrazinyl)-2H-benzo[b]pyran
149. 6-Trifluoromethoxy-2,2-dimethyl-4-~4'-pyrimidinyl)-
- 15 -
3,4-dihydro-2H-benzolb]pyran
150. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-pyrim-
idinyl)-2H-benzolb]pyran
151. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-pyrim-
idinyl)-2H-benzo[b]pyran-4-ol
152. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(5'-pyr~m-
idinyl)-2~-benzo[b~pyran 1'-oxlde
153. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(3'-quin-
olyl)-2H-benzo[b]pyran
154. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(3'-guin-
olyl)-2H-benzo[b]pyran 1'-oxide
155. 6-Trifluoromsthylsulfonyl-2,2-dimethyl-4-(4'-iso-
quinolyl)-2~-benzo[b]pyran
156. 6-Trifluoromethylsulfonyl-2,2-dimethyl-4-(4'-iso-
quinolyl)-2H-benzolb]pyran 1'-oxide.
The compounds 1-19, 22-27, 44-50, 62-70, 74-77,
82, 83, 90, 91, 97-113, 132, 133, 141, 150 and 151 are
particularly preferred, in particular 1-19 and 97-113,
and very particular preference is given to 1-4, 103 and
107.
The compounds of the formula I and thoir salts
and acid addition salts can be prepared by
a) preferably, for the preparation o$ a compound of the
formula I in which R5 is an N-heteroaryl group which
carriee an N-oxide group in the 2, 3 or 4 position,
oxidizing a compound of tho general formula I in which
R~, R2, R3, R~ and R6 have the abovementioned meaning and
Rs i8 a corresponding N-heteroaryl group having an N atom
in 2, 3 or 4 position, or
b) for the preparation of a compound of the formula I
corresponding to the formula VI
R5
Vl
in which R1, R2, Rs and R6 have the meaning given above and
.
.
,
,, . , , ~ ., .
~"` `` - 16 - ~ al~
R3 and R~ together form a bond, heating a compound of the
general formula II
R~ t ~ 2
having the meaning given above of Rl, R2, R5 and R6, or
eliminating water from a compound of the formula I which
S corresponds to the formula III and in which R3 denotes
hydrogen and R~ denotes hydroxyl
R~ 5 ~H
ttl
having the given meaning of Rl, R2, Rs and R6, or
c) for the preparation of a compound of the formula I
which corresponds to the general formula III and has the
meaning given abovo o$ Rl, R" Rs and R6, reacting a
compound of the general formula IV
o R 1
t~
in which Rl, R, and R6 have the meaning given above with
an organometallic compound RsM in which Rs has the above-
mentioned meaning and M represents a metal or metal
halide, preferably Li- or Mg~al, where Hal is chlorine,
bromine or iodine, or
d) for the preparation of those compounds of the
- 17 - ~ 9~
general formula I in which Rl and R~ denote hydrogen and
which come under the formula X of reaction schemes 1 and
2, in which Rl, R2, Rs and R~ have the meaning g$ven above,
cyclizing a compound of the general formula V
R5
in which R1, R2, Rs and R6 have the meaning given above, or
hydrogenating a compound of the general formula VI
R6 RS
1~ R 2
in which R~, R~, Rs and Rc have the given meaning in the
presence of a catalyst, or
e) preferably, for the preparation o$ a compound of the
general formula I in which R6 denotes difluoromethyl-
8ul finyl, difluoromethylsulfonyl, trifluoromethyl-
sulfinyl,tr$fluoromethylsulfonyl,trifluoroethylsulfinyl
or trifluoroethylsulfonyl and R1, R~, R3, R~ and R5 have
the abovementioned meaning, oxidizing a compound of the
general formula I in which R, is difluoromethylthio,
trifluoromethylthio or trifluoroethylthio, or
f) preferably, for the preparation of a compound of the
general formula I in which R5 denotes an N-heteroaromatic
radical which is substituted in the o or p position
relative to the N atom by chloro or cyano, and R3 and R~
in each case denote hydrogen or together denote a bond
and Rl, R~ and R6 have the meaning given above, reacting
a compound of the general formula I in which R5 is an
N-heteroaryl N-oxide group and Rl, R2, R3, R~ and R6 have
.
- 18 -
the appropriate meaning with a nonmetal chloride or a
8ilyl cyanide, or
g) for the preparation of a compound of tho formula XII
according to scheme 1, corresponding to the formula I in
which R3 is hydroxyl and R~ is hydrogen, and Rl, R~, R5 and
R6 have the meaning given above, epoxidizing a compound
of the general formula VI in which R~, R2~ Rs and R, have
the meaning given above to give the compound XI
(scheme 1) and then reducing the epoxide to the compound
of the formula XII (~cheme 1), or
h) for the preparation of compounds of the formula I in
which Rl, R2, ~5 and R~ have the meaning given above and R3
or R, denote ClR-alkoxy, formyloxy, Cl~-alkylcarbonyloxy,
C~-alkoxycarbonyloxy, C~-monoalkylaminocarbonyloxy or
Cl8-dialkylaminocarbonyloxy, reacting the corresponding
compounds of the formula I in which R3 or R~ denotes
hydroxyl, i.e. the compounds corresponding to formulae
III and XII (scheme 1), with corresponding alkylating
agents or acylating agents, and optionally converting
compounds obtained by the above processes into their
salts or acid addition salts or N-oxides.
For the oxidation of an N-heteroaromatic radical
Rs according to process a), reagents can be used which
are known per se. For instance, the oxidation can be
carried out using hydrogen peroxide, organic per-acids,
for example peracetic acid, perbenzoic acid, m-chloro-
perbenzoic acid, perphthalic acid, salts thereof such as
magnesium perphthalate, or using sodium metaperiodate,
sodium perborato and the like. The reaction is expedi-
ently carried out in acetic acid, in an alcohol of lowchain length, preferably in ethanol or methanol, with or
without the addition of water, in an inert organic
sol~ent, for example dichloromethane, dichloroethane and
trichloromethane, and at temperatures between 0C and the
boiling temperature of the solvent, preferably at room
temperature.
The conversion of a compound of the general
``` 21 a.~d11
- 19 -
formula II in accordance with process b) is carrled out
at the boiling temperature of a high-bolllng lnert
organic solvent, for example chlorobsnzene, 1,2-dlchloro-
benzene, N,N-diethylaniline, ethylene glycol and diphenyl
S ether, i.e. at temperatures of between approxlmately
120C and approxlmately 300C, or else ~ithout eolvent~
in this temperature range. If at least one of the substl-
tuents Rl and R2 is hydrogen, higher temperatures are
preferably necessary. Increased pre~sure may be of
advantage in this reaction.
The dehydration of the compounds of the general
formula III is carried out by methods which are known
from the literature. Examples which may be mentioned are:
sodium hydride in tetrahydrofuran or another inert
organlc solvent, preferably at the reflux temperature of
the reaction mixture; p-toluenesulfonic acid in toluene
at reflux temperature with water separation; reaction
with excess methanesulfonyl chlorlde/triethylamine in
dichloromethane or chloroform with subsequent heating at
reflux temperature; heating with anhydrous copper(II)
sulfate or potassium hydrogen sulfate at temperatures of
between 50C and 200C, preferably 100-120C.
The addition of metalated aromatic or N-hetero-
aromatic compounds RsM to chromanones of the general
formula IV in accordance with process c) is carried out
at tomperatures of between -120C and room temperature,
proferably at, initlally, approx~Ptely -78C with
subsoguent heating. Tho reaction 18 preferably carried
out in an ether such as diethyl ether, tetrahydrofuran,
1,2-dimethoxyothane or dioxano. Polar solvents such as
hexamothylphosphoric triamide or 1,3-dimethyl-2-oxohexa-
hydropyrimldino may be added.
The processes for the generation of the anions of
the aromatlc compounds R5H are known from the literature.
They can be obtained either from RsH by regiospecific
deprotonation using strong bases, for exa~ple metal alkyl
or aryl compounds, preferably compounds of lithium, for
example methyllithium, isomers of butyllithium, phenyl-
lithium or mesityllithium, or secondary metal amides, for
- 20 2 1 i3 ~ 3 -~
example lithium diisopropylamidQ (LDA) or llthium
2,2,6,6-tetramethylpiperidide (LiTMP), with the optional
addition of complexing, chelatlng or otherwise reactlon-
promoting amine compounds, for example tetramethylethy-
lenediamine (TMEDA), ethylenedlamine, hexamethylphos-
phoric triamide (HMPT), or polyamines (DMEU, DMPU, DABCO)
(for examples see, inter alia, Tetrahedron 39, (1983),
2009, Organic Reactions 26, (1979), 1, Tetrahedron
Letters 29, (1988), 773, and Synthesis 1988, 881 and the
literature cited therein), or are accessible by halogen-
metal exchange with the corresponding haloheterocycles
Rs-Hal, in which Hal preferably represents bromine or
iodine, by means of metals, metal al~yls or halo-metal
al~yls, for example lithium alkyls or Grignard reagents
by processe~ known from the literature.
The cyclization of u com~ound of the general
formula V in accordance with process d) can be effected
by reaction with an acid, preferably with an inorganic
acid, for example sulfuric acid, and in an inert organic
solvent such as a halogenated hydrocarbon, preferably
dichloromethane, dichloroethane or chloroform. The
reaction is preferably carried out at room temperature
and, advantageously, in situ.
The hydrogenation of the compounds of the general
formula VI can be effected in a known manner in the
presencQ of a noble metal catalyst, in particular a
catalyst containing metals from subgroup VIII of the
Periodic Table, especially containing palladium or
platinum, a~ the metals themselves or their oxides or
hydroxides on suitable supports, such as activated
charcoal, under hydrogen pressures from 1-250 bar and at
temperature~ of from 20C to 200C in suitable inert
organic solvents, for example methanol, ethanol, iso-
propanol, diethyl ether, tetrahydrofuran, dioxane, ethyl
acetate or hexane, with the optional addition of acids
such as hydrochloric acid or acetic acid.
The oxidation of the thioether substituents R6 in
compounds of the general formula I in accordance with
process e) can be carried out in a known manner. The
- 21 - 21~2'~
oxidizing agents used can be hydrogen peroxide, organlc
per-acids, for example peracetic acid, perbenzoic acid,
m-chloroperbenzoic acid, perphthalic acid, their salts,
for example magnesium perphthalate or inorganic ealts,
for example sodium metaperiodate, ~odium perborate,
potassium permanganate or Oxone~. The solvents u~ed are
halogenated hydrocarbons such as dichloromethane, chloro-
form and dichloroethane, or acetic acid in the case of
hydrogen peroxide, or, in the case of metal salts,
alcohols of low chain length or water or mixtures of the
two. The reaction can also be carried out in a two-phase
solvent system with phase-transfer catalysis in accor-
dance with conventional literature methods. For the
oxidation of thioether substituents R6 to sulfinyl sub-
stituents R~, it is per-acids, especially peracetic acid
and m-chloroperbenzoic acid, which are preferred: for the
oxidation to sulfone substituents R6, it is Oxone~ in
methanol/water mixtures at temperatures of between 0C
and the reflux temperature of the mixture, in particular
at room temperature. Mixtures of the sulfinyl and the
sulfonyl compound may also be obtained, which can be
separated by conventional methods such as crystallization
or chromatography. In this process, it is also posssible
to oxidize compounds of the formula I in which Rs is an
N-heteroaryl group to compounds containing R5 as the sub-
stituent which carries a corresponding N-oxide group, or
to oxidize compound~ of the formula I in which R, and R~
together form a bond to a benzopyran 3,4-epoxide. It may
thorefore, if appropriate, be of advantage to undertake
the conversion of R, into one of the precursors listed in
reaction scheme 1 or 2.
The conversion of N-oxides of N-heteroaryl
substituents R5 of the formula I in accordance with
process f) to give compounds of the general formula I
having N-heteroaryl substituents Rs~ which are ~ubsti-
tuted in the o or p position relative to the N atom by
chlorine or cyano, is carried out by methods known from
the literature. Those compounds which are used in
particular for the introduction of chlorine are chlorides
... ... ........ .
2'~''3~
- 22 -
of phosphorus, preferably phosphorus oxychlorido (Chem.
Pharm. Bull. 31, (1983), 4533; Chem. Pharm. Bull. 36,
(1988), 2244). The solvents used may be chlorinated
hydrocarbons such a~ dichloromethane, chloroform and 1,2-
dichloroethane, or ethers such a~ tetrahydrofuran,dioxane or 1,2-dimethoxyethane, preferably at the boillng
temperature of the reaction mixture. The reaction can
also be carried out without a solvent, i.e. with an
excess of chlorinating agent, at temperatures between
50C and 150C, preferably at approximately 110C, and
the mixture of o- and p-chloro compounds which is
generally formed can be resolved by chromatography. The
preferred compound for the introduction of cyano is
trimethylsilyl cyanide (J. Org. Chem. 48 (1983), 1375;
Synthesis 1983, 316; Synthesis 1984, 681: Chem. Pharm.
Bull. 33 (1985), 565; Chem. Pharm. Bull. 35 (1987),
3119), which can also be generated during the reaction in
situ from trimethylchlorosilane and sodium cyanide or
potassium cyanide, with the addition of a tertiary amine,
for example triethylamine, as base. The solvents used are
chlorinated hydrocarbons, for example dichloromethane,
dichloroethane or chloroform, or dimethylformamide or
preferably acetonitrile at reaction temperatures of
between 0C and the boiling temperature of the reaction
mixture.
The reduction of a benzopyran 3,4-epoxide in
accordance with process g) can be carried out using
various reducing agents, for example by hydrogenation,
which is preferably carried out with the same noble metal
catalysts and under similar reaction conditions as
already describod for process variant d). Another
possibility is reaction with phenyl selenide, which is
generated from diphenyl diselenide by reduction in an
alcohol of low chain length, for example ethanol, and
reacted in situ (Tetrahedron Letters 28 (1987), 4293).
Further suitable reagents are: an excess of alkali metal
iodide in a buffered solution containing acetic acid and
acetone, preferably at room temperature (Chem. Ber. 109
(1976), 3907); transition metai compounds in low
- 23 ~ 2~
oxidation states, preferably samarium(II) lodide ln
tetrahydrofuran (J. Org. Chem. Sl (1986), 2596;
Tetrahedron Letters 28 (1987), 4437) and of low
selectivity, especially when groups which are capable of
being reduced further, for example an N-oxide group, are
present in the molecule, or else with complex metal
hydrides such as lithium aluminum hydride, sodium
borohydride, sodium cyanoborohydride or sodium bi~(2-
methoxyethoxy)aluminum hydride.
The compounds of the general formula I in which
R3 or R~ is hydroxyl, and which are conse~uently obtain-
able by processes g) or c), respectively, can be reacted
according to process h), by conversion of the free
hydroxyl group, to give further compounds of the formula
I, specifically by alkylation to give ethers, or by
acylation to give carboxylates, carbonates or carbamates.
Tho methods used for this purpose are standard methods of
organic synthesis.
Alkylation can be carried out, for example, by
ao using a Cl6-alkyl iodide in an inert solvent such as
toluene or dimethylformamide in the presence of a base
such as potassium hydroxide or barium oxide.
Esterification can be aarried out by u~ing a Cl~-
acyl chloride or acyl anhydride or another activated
dorivativo of the relevant alkanoic acid, optionally with
the presence of an organic base such as pyridine or
triethylamine or of an inorganic base such as potassium
carbonato, accompanied optionally by the action of
catalycts such as 4-(N,N-dimethylamino)pyridine, or with
the Cl~-carboxylic acid with the accompanying action of
condensation reagents such as dicyclohexylcarbodiimide in
an inert solvent, if desired at elevated temperature.
Reaction to give carbonates is carried out
analogously, by reaction with Cl~-alkyl chloroformate~
under the conditions given above.
Reaction to give carbamates is carried out either
in analogy to processes described above, by reaction with
mono- or dialkylaminocarbamoyl chlorides, or by reaction
with C,~-alkyl isocyanates in an inert solvent, for
- 24 - 21~9~'~
example toluene, at temperaturee of between 0C and the
boiling temperature of the reaction mixture.
Formyloxy can be introduced by reaction with pho~gene and
subsequent hydrolysis of the corresponding chloroformate
or reaction with formic acid ln pyridine.
Compounds of the general formula I in which Rs is
a substituent of subgroup A), i.e. an aryl or N-hetero-
aryl group having a hydroxyl group in the 2 position, can
be obtained from compounds in which these groups are
protected by protective groups which are customary for
phenolic hydroxyl groups. Preferred such groups are Cl~-
alkoxy groups, particularly preferably methoxy, or
formaldehyde acatals, for example methoxymethyl.
The elimination of an ether group is carried out
in a manner known per se. The reaction can be effected,
for example, using an alkali metal al~anethiolate of low
chain length, preferably sodium methanethiolate, expe-
diently in an inert organic solvent, for example
dimethylformamide, at elevated temperature, for example
at about 100C, or el~e using other reagents, for example
lithium iodide, a silyl halide or a boron halide.
The elimination of a formaldehyde acetal can be
carried out by customary literature methods, for example
under acidic reaction conditions, using, in particular,
mineral acids such as hydrochloric acid.
The precursors which are suitable for these
described hydrolysea are obtainable by the procees
variants given above, in particular b), c) and d),
particularly preferably c). For the metalation of alkoxy
N-heteroaromatic compounds, many examples are ~nown from
the literature tcf. discussion of process variant c)).
Ortho metalations of anisole derivative~ (J. Org. Chem.
41 (1976), 3653) and of methoxymethoxybenzene derivatives
(Tetrahedron Lett. 22 ~1981), 3923; J. Org. Chem. 47
(1982), 2101 are li~ewise known.
If Rs~ specifically, is an N-heteroaromatic
radical having an N atom in the 3 position and a hydroxyl
group in the 2 position, then such a compound i~ obtain-
able from a compound of the general formula I in which R5
- 25 - 21~2~
is a heteroaromatic N-oxide with tho nitrogen in tho samo
position. ~he conversion is carried out uslng a Cl~-
carboxylic acid anhydride, preferably acetic anhydrido,
with ~ubsequent hydrolysis. It is carried out at elevated
temperature, preferably at the boiling temperature of tho
reaction mixture. The hydrolysis of the product obtained,
namely a compound of the formula I in which R5 denotes an
N-heteroaryl group having an N atom in the 3 position and
an alkanoyloxy group in the 2 position with the al~anoyl
radical of the C1~-carboxyl$c acid anhydride, can be
carried out under acidic or basic conditions by known
processes. For the acid hydrolysis, aqueous mineral acids
are used, for example hydrochloric acid, hydrobromic
acid, sulfuric acid or organic acids, for example
p-toluenesulfonic acid, expediently in an inert organic
solvent, for example dioxane or tetrahydrofuran. The
reaction preferably takes place at room temperature. The
basic hydrolysis can be carried out using an al~ali metal
hydroxide, for example sodium hydroxide, or an al~ali
metal Cl~-alkoxide, for example sodium methoxide or
sodium ethoxide, preferably in an $nert organic solvent
such as methanol or ethanol at room temperature. The
synthesis of the abovementioned 2-hydroxy-N-hetero-
aromatic compounds from N-oxides of 3-N-heteroaromatic
compounds can also be carried out via the 2-halo-3-N-
heteroaromatic compounds described by process f), the
hydrolysis of which in an acidic or in a basic reaction
medium can be carried out li~ewise as described above.
Some substituents of the aromatic and N-hetero-
aromatic radicals Rs can be converted into other func-
` tional groups by conventional, known processes. Forexample, a nitro group can be rsduced to an ~;no group.
The preferred reducing agent used is iron powder/acetic
acid. An amino group can be converted by conventional
processes, in a Sandmeyer reaction, to a cyano group, a
halogen or a hydroxyl group. Furthermore, a cyano group
can be hydrolyzed to give a carboxyl group, which can be
converted, for example, using a diazoal~ane into a
carboxylic acid ester, preferably with diazomethane to a
- 26 - 2 1 ~ 2 ~
methyl carboxylate. Other carboxylic acid derivatives can
also be obtained from the cyano group by k~own procos~es.
A halogen atom, preferably a chlorine atom, can
be converted in~o a C,~-alkoxy, Cl,-monoalkylamino or C1~-
dialkylamino group. ~he exchange can be offected in amanner known per se using an alkali metal alkoxide of the
corresponding chain length, for example using sodium
methoxide or sodium ethoxide. The reaction is preferably
carried out in an inert organic solvent, such as the
alcohol corresponding to the alkali metal al~oxide, and
preferably at the reflux temperature of the reaction
mixture.
The exchange with an alkali metal ~m~ de is
carried out in similar manner, but the inert organic
solvent used is expodiently an ether, for example tetra-
hydrofuran, dioxane or l,2-dimethoxyethane. The alkali
metal amides are preferably produced from the correspond-
ing amines using al~ali metal al~yls, for example butyl-
lithium, in the same solvent, and reacted in situ.
The resolution of diastereomers and cis/trans
mixtures can be carried out by conventional methods, for
example by chromatography or crystallization.
Compounds of the general formula I which are
obtained as racemates, or certain racemic precursors
thereof, can be resolved by known methods to give their
optical isomer~, for example by recrystallization in
optically active solvents, by microorganisms, or by
reaction with an optically active acid or base which
forms a salt or another compound with the racemic com-
pound, separation of the diastereoisomers by fractional
crystallization, and liberation of the enantiomers using
suitable agents. Examples of particularly suitable
optically active acids are the d and 1 forms of tartaric
acid, ditolyltartaric acid, malic acid, mandelic acid,
camphorsulfonic acid or pyrrolidonecarboxylic acid.
Suitable optically active bases are alpha-phenylethyl-
amine, menthylamine, ephedrine, brucine and quinine.
Preferably, a chiral compound of the general
formula I in which R5 is a basic N-heteroaromatic radical
- 27 _ 21~29~
is reacted with one of the chiral acid~ llsted above, or
a chiral compound of the general formula I which contaln~
a free hydroxyl group, especially if R3 or R~ i8 hydroxyl,
is esterified with a chiral acid.
Advantageously, the more active of tho isomers ~
isolated. According to the invention, however, it is also
pos~ible to obtain the pure enantiomers by asymmetric
synthesis.
Compounds of the general formula I in which Rs is
an N-heteroaromatic radical or contains an amino group
are basic and can therefore be converted into their acid
addition salts. Compounds of the general formula I in
which R5 is a phenolic hydroxyl group or contains a
carboxyl group aro acidic, and can therefore be converted
into their salts.
Physiologically tolerated salts or aaid addition
salts are preferred. Examples of suitable such acids are,
as regards inorganic acids, sulfuric acid or hydrohalic
acids, for example hydrochloric acid, and, as regards
ao organic acids, examples are fumaric acid, maleic acid,
citric acid and tartaric acid. In the preparation, the
alcoholic solution of a suitable acid is added to the hot
alcoholic solution of the base, and the salt is obtained
after adding ether. Preferred salts are the alkali metal,
alkaline earth metal and ammonium salts of compounds of
the formula I, which are obtained using the corresponding
bases, in particular sodium hydroxide or potassium
hydroxide.
The precursors and intermediates which are
required for the preparation of compounds of tho general
formula I and which, to the extent that they are novel,
are a further sub~ect of the present invention, are
li~ted in reaction schemes 1 and 2. In every case, the
starting compounds are phenols VII in which R6 has the
meaning given above. They are known or can be prepared by
known processes, for example by reduction of the corres-
ponding 4-substituted nitroaromatic compounds, for
example using hydrogen with Raney nickel a~ catalyst, or
using nascent hydrogen, to give the corresponding
2~ ~7~4
- 28 -
Reaction cchemo 1
R~o
~ VII
2 ~oill 2
Iv ~ ~/VIII
I x 5
5 o a ~0~1 2
tl~
2 ~ ~ ~2
Xt 1~ ~ x~ X'
- 2 9 - 2 ~ u ~
React~ on ~cheme 2
~OH 6~ O~OQ
VII X~tI
~o~ 2 ~o~ 2
XIV XV
~ 3~ $ ~
X~'l X`'t ~
- 30 -
4-substituted anilinen, which are diazotized and boiled
to give the said 4-substituted phenols~
In cases where R~ has the meaning difluoromethyl-
thio, difluoromethylsulfinyl, difluoromethylsulfonyl,
trifluoromethylthio, trifluoromethyl~ulfinyl, trlfluoro-
methylsulfonyl, 2,2,2-trifluoroothylthio, 2,2,2-tr$-
fluoroethylsulfinyl and 2,2,2-trifluoroethylsulfonyl, the
radicals mentioned can often be introduced more advan-
tageously by chemical transformations of intermediates in
which R6 has a meaning other than is mentioned above. For
example, 2H-benzo[b~pyrans of the general formula VI in
which Rc has the meaning difluoromethylsulfonyl, trifluo-
romethylsulfonyl and 2,2,2-trifluoroethylsulfonyl, are
obtained by reacting the corresponding fluoroalkyl-
sulfonyl fluorides with 2H-benzo[b]pyrans of the general
formula VI in which R6 has the meaning MgHal, where Hal
has the meaning chlorine, iodine and in particular
bromine, a~ long as Rs contains no substituents which
interfere with this reaction: i.e. R5 is unsubstituted or
is substituted, for example, by Cl~-alkyl, phenyl or Cl~-
al~oxy. It i~ also po~sible to react the Grignard
compounds of the 2H-benzo[b]pyrans described above with
disulfides of the general formula R7-S-S-R7, in which R7
has the meaning trifluoromethyl, difluoromethyl or 2,2,2-
trifluoroethyl, to give 2~-benzo[b]pyrans in which R~ has
the meaning difluoromethylthio, trifluoromethylthio and
2,2,2-trifluoroethylthio.
Starting from intermediates or end products in
which R6 contains a sulfur atom, it is also possible, in
conventional methods, by reduction and in particular
oxidation, to obtain intermediates and end products in
which the sulfur atom referred to has a different oxida-
tion state. Possible oxidizing agents have already been
listed in the description of proce~s variant e). ~or
example, 4-difluoromethylsulfonylphenol, 4-trifluoro-
methylsulfonylphenolor4-(2,2,2-trifluoroethyl~ulfonyl)-
phenol can be obtained more favorably than by known
processes by oxidizing the corresponding fluoroal~yl-
thiophenol~ with Oxone~ in methanol/water mixtures at
- 31 -
temperatures of between -10C and the reflux temperature
of the reaction mixture, preferably at temperatures
between 0C and 25C.
~pecially in cases where R, has the moaning
difluoromethylsulfinyl, trifluoromethylsulfinyl or 2,2,2-
trifluoroethylsulfinyl, it is more favorable, ~tarting
from the corre~ponding 4-fluoroal~ylthiophenols in
accordance with reaction scheme 1, first to prepare the
corresponding 6-fluoroal~ylthio-2H-benzo[b~pyrans VI in
which Rl, R, and R5 have the meaning given above, and then
to carry out the desired oxidation to give the respective
6-fluoroal~ylsulfinyl-2~-benzolb]pyrans of the general
formula VI.
In some cases it is possible to introduce Cl,-
dialkoxyphosphoryl as the substituents R~ in inter-
mediates and end products of the present invention, by
reacting corresponding compounds in which R6 is bromine
with a Cl~-trialkyl phosphite in the presence of a
nickel(II) halide, preferably nic~el(II) chloride, at
elevated temperature, preferably at approximately 180C.
Intermediates and end products in which R6 denotes phos-
phono can be obtained by conventional methods for the
elimination of phosphonic acid esters from the corres-
ponding Cl~-dialkoxypho~phoryl compounds. Li~ewise, the
converse transformation to give phosphonic acid esters
can also be carried out.
Compounds of the general formula YIII can be
obtained from the phenols VII by reaction with compounds
of the general formula Y-CRlR~-C~CH, in which Y is chlor-
ine, bromine or hydroxyl. If Y is chlorine or bromine,
` the reaction is carried out by known methods which havebeen described in many references (e.g. J. Org. Chem. 38
(1973), 3832; J. Org. Chem. 39 (1974), 881; J. Org. Chem.
37 (1972), 841; J. Ned. Chem. 26 (1983), 1582) in an
inert organic solvent in the presence of a base. Pre-
ferred reaction conditions are, for example, potassium
carbonate as base in dimethylformamide at approximately
90C or, preferably, in acetone or butanone under reflux,
or sodium hydroxide or potassium hydroxide with a phase-
3 ,~J';~
- 32 -
transfer catalyst such as trimethylbenzylamoonium
hydroxide in methanol at room temperaturo. If Y i~
hydroxyl, the reaction can be carried out in the pre~ence
of a condensation agent, for example diethyl azo-
dicarboxylate/triphenylphosphine (see Synthesis 1981, 1)in an inert organ$c solvent, for example dichloromethane.
A compound of the general formula II can be
obtained from a compound of the general formula VIII by
reacting the latter with a halogenated aromatic or
N-heteroaromatic compound R5~al, in which Hal denotes
bromine or iodine, preferably iodine. The reaction is
carried out in the prese~ce of copper(I) iodide and a
palladium compound as catalyst, preferably bis(triphenyl-
phosphine)palladium dichloride or diacetate, and a
triarylphosphine, preferably triphenylphosphine, in an
aliphatic amine of low chain length, preferably in
triethylamine or diethylamine, as solvent, at tempera-
tures between room temperature and the boiling point of
the reaction mixture, preferably at approximately 80C.
It may be advantageous to conduct the reaction in a
sealed reaction vessel, i.e. at somewhat greater than
atmospheric pressure. Such reactions are known from the
literature in large number (e.g. Heterocycles 9 (1978),
271t Synthesis I980, 627: Synthesis 1981, 364: Chem.
Pharm. Bull. 28 (1980), 3488; Synthesis 1983, 313; and J.
Org. Chem. 53 (1988), 386).
The benzo[b]pyran-4-ones of the general formula
IV can be prepared by methods known from the literature
(Angew. Chem. 94 (1982), 254), preferably by the conden-
sation of ~etones or aldehydes of the general formula
R~R~C=O with phenols of the general formula VII, which are
additionally substituted in the 2 position by an acetyl
group, or by hydrolysis of 4-bromobenzo[b~pyrans IX. This
reaction is carried out using strong inorganic acids, for
example concentrated sulfuric acid, preferably at room
temperature, or using heavy metal salts in organic acids,
preferably using mercury salts, for example mercury(II)
acetate or mercury(II) trifluoroacetate in glacial acetic
acid or trlfluoroacetic acid, or else in other inert
,," . ~ ,
. ,~ -
- , .
'
_ 33 _
organic solvents, for example acetonitrile, nitromethano
or dichloromethane, and t~mperatures between 0C and the
reflux temperature of the reaction mixture. ~he ~ddition
of a Lewis acid, for example boron trifluorido, may be
5advantageous (Tetrahedron Letters 1978, 1943; ibid. 1979,
3489).
4-Bromobenzo[b]pyrans IX can be obtained by first
cyclizing the propargyl ethers of the general formula
VIII under the reaction conditions described for proces~
b) to give benzo[b]pyrans which carry hydrogen in posi-
tions 3 and 4 and the substituents Rl, R, and Rc, having
the meaning given above, in positions 2 and 6, and then
adding bromine to the C-C double bond between the C-3 and
the C-4 of the benzopyran, preferably in an inert organic
solvent such as tetrachloromethane or chloroform at
temperatures between 0C and room temperature. ~sing
bases, the bromine in the 3 position, exclusively or
preferentially, is eliminated from the trans-3,4-dibromo-
benzo[b]pyrans which are obtained to give the compounds
of the general formula IX. The bases preferably used are
alkali metal alkoxides, for example sodium methoxide,
sodium ethoxide or potassium tert-butoxide in the corres-
ponding alcohol as solvent, tertiary amines, for example
triethylamine, diisopropylethylamine or diazabicyclo-
undecane (DBU), or alkali metal hydrides, for example
sodium hydride, in inert organic solvents such as diethyl
ether, tetrahydrouran, 1,2-dimethoxyethane, dichloro-
methane and the like. The reaction temperature is prefer-
ably room temperature or a slightly elevated temperature.
30Benzo[b~pyran-3,4-epoxides of the general formula
XI can be prepared from the benzo[b~pyrans VI by oxida-
tion in accordance with known proce~ses. If R5 is an
N-heteroaromatic radical and/or R~ is difluoromethylthio,
difluoromethylsulfinyl, trifluoromethylthio, trifluoro-
methylsulfinyl, trifluoroethylthio or trifluoroethyl-
sulfinyl, a possible secondary reaction is for oxidation
to take place additionally at the nitrogen or at the
sulfur. Reagents which can be used are per-acids, for
example peracetic acid, perbenzoic acid, perphthalic acid
- 34 -
or m-chloroperbenzoic acid in inert anhydrou~ organic
solvents such as dichloromethane, chloroform, dichloro-
ethane, diethyl ether, tetrahydrofuran or acetonitrlle,
or magnesium perphthalate in an alcohol of low chaln
length with or without the additlon of water or hydrogen
peroxide in the presence of a tungstate, preferably
sodium tungstate, in the same solvent at temperatures
between room temperature and the reflux temperature of
the reaction mixture.
The synthesis route to compounds of the general
formula V by cyclization according to process d) is
represented in reaction scheme 2. Phenols of the general
formula VII are reacted with a-halocarboxylic acid esters
HalRlR2CCOOR~, in which halogen preferably ropresents
bromine or chlorine, and R, represents an alkyl radical
of low chain length, preferably methyl or ethyl, by known
processes to givo compounds of the general formula XIII.
The base used in this reaction is an alkali metal
alkoxide of the alcohol R~OH in this latter compound as
solvent, an alkali metal hydride, for example sodium
hydride, in an ether, for example tetrahydrofuran, or in
dimethylformamide, or an alkali metal carbonate, for
example potassium carbonate in acetone or butanone as
solvent. The reaction is carried out at temperatures
between 0C and the boiling temperature of the reaction
mixture, preferably at room temperature.
The reaction of the compounds of the general
formula XIII to give the ketones of the general formula
XIV is carried out with anions of methyl heterocycles
MeR5 in which R, i~ preferably an N-heterocycle having an
N atom or an N-oxide group in the 2 position, for example
4-methylpyrimidine or 2-picoline N-oxide. The anions are
produced from the heterocycles MeR5 with strong bases,
for example with alkali metal dialkylamides of low chain
length, for example lithium diisopropylamide, or with
alkali metal al~yls ~uch as butyllithium, in inert
organic solvents such as diethyl ether or tetrahydro-
furan. The reaction is preferably carried out in the same
solvent at temperatures of between -78C and the reflux
_ 35 i~
temperature of the reaction mixture, preferably at
reduced temperature with subse~uent heating to room
temperature.
The reduction of the ketones XIV to give the
alcohols of the general formula XV can be carrlod out ln
a known manner, preferably uslng complex hydrldes, for
example sodium borohydride, in an alcohol such as
methanol or ethanol, or using lithium aluminum hydride in
diethyl ether or tetrahydrofuran as solvent, preferably
at room temperature. If it is desired to convert an
N-heterocycle Rs into its N-oxide, this is preferably
carried out at this stage of the synthesis seguence by
methods and under reaction conditions which have been
described under process a).
The conversion of the alcohols XV into compounds
of the general formula XVI in which Rg is a leaving group
is carried out by known processes. Rg is preferably
iodine, an alkanesulfonate, e.g. methanesulfonate, or an
arylsulfonate, e.g. phenylsulfonate or p-tolylsulfonate.
The sulfonates are obtained with the corresponding
sulfonyl chlorides in the presence of an acid-binding
agent, for example a tertiary am~ne such as trlethyl-
amine, diisopropylethylamine or pyridine, which can also
be used as the solvent. The reaction is otherwise carried
out in a conventional inert organic solvent, preferably
at room temperature. Compounds of the general formula XVI
in which Rg is iodine can be obtained from the above-
described sulfonatee of the general formula XVI by known
processes using an alkali metal iodide, for example
sodium iodide. The reaction is carried out in an inert
organic solvent, preferably acetone or butanone, at
elevated temperature, preferably at the reflux tempera-
ture of the reaction mixture.
The compounds of the general formula XVI are
converted into compounds of the general formula V,
passing through the stage of the olefins XVII. These can
be isolated if the elimination of HRg is carried out at
room temperature or ~lightly elevated temperature. The
reaction employs strong bases, for example sodium
r.~ ~ vi L,~L
~ 36 ~
hydride, sodium methoxide, sodium ethoxido, potassium
tert-butoxide or tertiary amine~ ~uch as dlisopropyl-
ethylamine, triethylamine or diazabicycloundecane (DB~
in inert organic solvents such as tetrahydrofuran,
diethyl ether, methanol, ethanol and tho like. The
reaction of the compounds XVII is then carried out by
heating in high-boiling aromatic hydrocarbons ~uch as
chlorobanzene or 1,2-dichlorobenzene. The direct conver-
sion of compounds XVI into compounds of the formula V is
preferably carried out at the reflux temperature of the
above reaction mixture using tertiary amines as the base.
Compounds of the formula XVII can also be obtained, as
by-products from the alcohols XV, as early as during the
preparation of the compounds of the general formula XVI.
The compounds of the formula I according to the
invention, their physiologically tolerated salts and acid
addition salts and their tautomeric and optical isomers
are therapeutic active substances; they possess a high
pharmacological activity and are valuable pharma-
ceutical~. In particular, they exhibit vasodilatory,
vasospasmolytic and, especially, broncholytic activity,
the vasospasmolytic activity being expressed tbroughout
the vascular system or else, in a more or less isolated
mannor, in defined vascular regions such as the cerebral,
coronary or peripheral vessels.
The compound~ according to the invention have, in
particular, a hypotensive activity, and can therefore be
used as antihypertensives.
Tho substances according to the invention are
notable for a considerable reduction in the arterial
blood pressure. Oral doses of from 0.01 - 10 mg/kg led to
a reduction in the blood pressure of hypertensive rats by
at least 20 %.
The substances according to the invention are
notable for their particular influence on the flow of
potassium ions in the cells. They are, in particular,
potassium-channel activators. They are suitable for the
prophylaxi~ and for the treatment of the following
diseases in warm-blooded animals, e~pecially in humans:
~ 3i~
- 37 -
1. high blood pressure, especially high artorlal blood
pressure,
2. cardiac insufficiency, coronary insufflciency and
angina pectoris,
3. occlusive arterial disease and peripheral circùl-
atory disorders,
4. cerebral insufficiency, migraine, vertigo, diseases
of the inner ear and of the auditory system,
5. increased intraocular tension, glaucoma, weak-
sightedness,
6. renal insufficiency, organic diseases of the urinarytract and of the accessory glands of the urinary
tract, impaired potency,
7. organic diseases of the gastrointestinal tract and
of the pancreas and liver,
8. deficient circulation in the scalp, hair 1088,
9. disoases of tho respiratory tract, including
bronchial asthma,
10. metabolic disease~,
11. spasmogenic diseases of the uterus, and
12. incontinenco.
Furthermore, the compounds according to the
invention promote circulation in the scalp and promote
hair growth. They also have an inhibitory action on
uterine contractions.
The compounds according to the invention exhibit
a long period of action with only a low toxicity. They
~ ~ ~ " ~ ~ ~ L~
- 38 -
are therefore particularly euitable for the treatment of
acute and chronic heart diseaee, for the therapy of high
blood pressure and cardiac in~ufficiency, and for the
treatment of asthma and of cerebral and peripheral
circulatory disorders.
The compounds of the present invention can be
administered to humans orally or parenterally in a dose
of from 0.001 to 100 mg, preferably from 0.01 to 50 mg
and particularly preferably from O.OS to 10 mg per day,
and, in particular, in subdivided doses, for example from
two to four times daily. These dosages are advantageous
for the treatment of the abovementioned diseases, in
particular heart disease, hypertension, asthma and
circulatory disorders.
In general it has proven advantageous, in the
case of intravenous human application to administer
amounts of from approximately 0.001 to 10 mg, preferably
from approximately 0.05 to 5 mg per day in order to
achieve e~fective results. In the case of oral adminis-
ao tration to humans, the dosage is from approximately O.OS
to 30 mg, preferably from 0.1 to 10 mg per day.
The dosages stated above are particularly
preferred for the treatment of hypertension.
In spite of thie, it may be necessary to depart
from the stated quantities, specifically in dependence on
body weight and/or on the nature of administration, but
also on account of the individual response to the medica-
ment and/or of the nature of its formulation and the
time, or time interval, at which administration i8 made.
For example, it may in some instancee be sufficient to
employ lese than the abovementioned minimum amount,
whereas in other cases the upper limit mentioned may be
exceeded. In the case where greater quantities are
administered, it may be advisable to divide these into a
number of individual doeee over the day.
The invention also relates to the compounde
according to the invention for treating the above-
mentioned diseases, and to methods for the treatment of
these dieeaees in which these compounde are ueed, and to
- 39 _ '~ ?J~ ~L~
their use in processes for the preparation of compo~i-
tions which comprise these compounds, for the treatment
of said diseases, and to processes for the proparation of
the compounds.
In accordance with the inventlon, pharmaceut~cal
preparations or compositions are provided which compr~se
a compound according to the invention or a pharma-
ceutically acceptable salt or acid addition salt thereof,
together if desired with a pharmaceutically acceptable
diluent or excipient.
The compounds according to the invention can be
mixed with conventional pharmaceutically acceptable
diluents or excipients and, if desired, with other
auxiliaries and can be administered, for example, orally
or parenterally. They can be administered, preferably
orally, in the form of granules, capsules, pills,
tablets, coated tablets - including film-coated tablets -
syrups, emulsions, suspensions, dispersions, aerosols and
solutions, and as liquids, or parenterally in the form of
ao solutions, emulsions or suspensions. Preparations for
oral administration may contain one or more additives
such as sweeteners, flavors, colorants and preservatives.
Tablets may contain the active substance mixed with
conventional pharmaceutically acceptable auxiliaries, for
example inert diluents such as calcium carbonate, sodium
carbonate, lactose and talc, granulating agents and
agents which promote the decomposition of the tablets on
oral administration, such as starch or alginic acid,
binders such as starch or gelatine, and lubricants such
as magnesium stearate, stearic acid and talc. Examples of
suitable excipients are lactose, gelatine, corn starch,
stearic acid, ethanol, propylene glycol, ethers of
tetrahydrofurfuryl alcohol and water.
Examples of auxiliaries which may be listed are:
water, nontoxic organic solvents such as paraffins (e.g.
petrolaum fractions), vegetable oils (e.g. groundnut/
sesame oil), alcohols (e.g. ethyl alcohol, glycerol),
glycols (e.g. propylene glycol, polyethylene glycol),
solid excipients such as natural ground minerals (e.g.
- 40 - ~ J ~
~aolins, clays, talc, chalk) and synthetic ground
minerals (e.g. highly disperse silica, silicates), sugar~
(e.g. cane sugar, lactose and dextrose), emulsifier~
(e.g. polyoxyethylene fatty acid esters, polyoxyethylene
fatty alcohol ethers, al~ylsulfonates and aryl-
sulfonates), dispersants ~e.g. lignin-sulflte wasto
liquors, methylcellulose, starch and polyvinyl-
pyrrolidone), and lubricants ~e.g. magnesium stearate,
talc, stearic acid and sodium lauryl sulfate).
The formulations are prepared, for example, by
extending the active substances with solvents and/or
exc~pients, using if desired emulsifiers and/or disper-
sants; in the case, for example, where water is used as
a diluent, organic solvents may if desired be used as
auxiliary solvents.
Administration is carried out in a conventional
manner, preferably orally or parenterally, and in
particular perlingually or intravenously. In the case of
oral administration, tablets may also of course contain,
in addition to the excipients mentioned, additives such
as sodium citrate, calcium carbonate and dicalcium
phosphate together with various adjuvants such as starch,
preferably potato starch, gelatin and the like. It is
also possible to use in addition lubricants such as
magnesium stearate, sodium lauryl sulfate and talc for
tableting. In the case of aqueous suspensions and/or
elixirs which are meant for oral administration, the
active substances can be mixed not only with the
abovementioned auxiliaries but with various taste
improvers or colorants.
In the case of parenteral administration, solu-
tions of the active substances may be employed with the
use of suitable liquid carrier materials.
Using ~nown procedures, the tablets can be coated
in order to delay their decomposition and absorption in
the gastrointestinal tract, whereby the activity of the
active substance may extend over a prolonged time period.
Likewise, the suspension may contain the active substance
mixed with auxiliaries which are conventional for the
,
- 41 ^
preparation of such compositions, for example ~usponding
agents such as methylcellulose, tragacanth or ~odlum
alginate, wetting agents such a~ lecithin, polyoxyethyl-
ene stearate and polyoxyethylenesorbltan monooleate and
preservatives such as ethyl parahydroxybenzoate. Capsulss
can contain the active substance as the ~ole component or
mixed with a solid diluent such as calcium carbonate,
calcium phosphate or kaolin. The injectable preparations
are likewise formulated in a manner ~nown per se.
The pharmaceutical preparations may contain the
active substance in an amount of from 0.1 to 90 percent
by weight, in particular from 1 to 90 percent by weight,
i.e. in quantities which are sufficient to achieve the
stated scope of dosage, the remaining percentage compris-
ing excipient or additive. With regard to the preparation
and administration, solid preparations such as tablets
and capsules are preferred. The preparations preferably
contain the active substance in an amount of from 0.05 to
10 mg.
The invention also relates to a process for the
preparation of pharmaceutical preparations, which
comprises mixing one or more compounds as claimed in
claim 1 or their physiologically tolerated salts and, if
desired, conventional excipients and/or diluents.
The examples which follow are intended to eluci-
dato the invention:
Example 1
2,2-Dimethyl-6-trifluoromethylthio-4-(2'-pyridyl)-2H-
benzolb]pyran l'-oxide
1.5 g (4.4 mmol) of 2,2-dimethyl-6-trifluoromethylthio-4-
(2'-pyridyl)-2H-benzolb~pyran and 1.4 g (2.4 mmol) of 85%
magnesium monoperoxyphthalate hexahydrate are dissolved
in a mixture of 10 ml of ethanol and 5 ml of water and
stirred for 1 h at 80C. The solution is cooled, diluted
with water and extracted twice with ether, and the
combined organic pha~es are dried over magnesium sulfate
and concentrated. Chromatography on silica gel using
' '
',~ ,
'
21~2~
- 42 -
dichloromethane/ethanol, 98:2, gave 100 mg (6.4~) of tho
pyridine N-oxide, m.p. 144-145C.
ExamDle 2
2,2-Dimethyl-6-trifluoromethoxy-4-(2'-pyridyl)-2~-benzo-
lb]pyran 1'-oxide
From 2.0 g (6.2 mmol) of 2,2-dimethyl-6-tri-
fluoromethoxy-4-(2'-pyridyl)-2H-benzo[blpyran, by a
procedure analogous to that de~cribed in Example 1,
200 mg (9.6%) of the pyridine N-oxide, m.p. 168-170C,
were obtained.
ExamDle 3
a, a -Dimethyl-6-difluoromethoxy-4-(2'-pyridyl)-2~-benzo-
tb]pyran 1'-oxido
1.9 g (6.3 mmol) of 2,2-dimethyl-6-difluorometh-
oxy-4-(2'-pyridyl)-2H-benzolb]pyran and 1.9 g (3.2 mmol)
of 85% strength magnesium monoperoxyphthalate hexahydrate
are dissolved in 10 ml of glacial acetic acid and heated
at 80C for 1 h. Tho solution is cooled, neutralized with
sodium hydrogen carbonate solution and extracted with
dichloromethane, and the organic phase i~ dried over
magnesium sulfate and concentrated. After chromatography
on silica gol using dichloromethane/ethanol, 98:2, 330 mg
(16%) of the pyridine N-oxide, m.p. 147-148C (from ethyl
acetate/petroleum ether) are obtained.
a s Example 4
2,2-Dimethyl-6-trifluoromethyl~ulfonyl-4-(2'-pyridyl)-2H-
benzolb]pyran l'-oxide
From 350 mg (0.95 mmol) of 2,2-dimethyl-6-tri-
fluoromethylsulfonyl-4-(2'-pyridyl)-2H-benzo[b]pyran
l'-oxide, by a procedure as described in Example 3,
100 mg (27~) of the pyridine N-oxide were obtained as
colorless crystals, m.p. 174-179C.
Example 5
2,2-Dimethyl-6-trifluoromethoxy-4-(3'-pyridyl)-2H-benzo-
lblpyran l'-oxide
- 43 -
7.0 g (21.8 mmol) of 2,2-dimethyl-6-trlfluoro-
methoxy-4-(3'-pyridyl)-2H-benzo[b~pyran and 7 7 g
(13.2 mmol) of 85% magnesium monoperoxyphthalate hexa-
hydrate are diseolved in a mixture of 100 ml of water and
30 ml of glacial acetic acid and stirred at room tempera-
ture for 2 h. The solution is diluted with ether and
extracted with 2 N eodium hydroxide eolution, and the
organic phase is dried over magnesium sulfate and concen-
trated. After chromatography on silica gel using
dichloromethane/ethanol, 95:5, 4.8 g (65%) of the pyri-
dine N-oxide are obtained as an oil.
Exam~le 6
2,2-Dimethyl-6-trifluoromethylsulfonyl-4-(2'-quinolyl)-
28-benzo~b]pyran l'-oxide
From 750 mg (1.8 mmol) of 2,2-dimethyl-6-tri-
fluoromethylsulfonyl-4-(2'-quinolyl)-2H-benzo[b]pyran by
a procedure analogous to that of Example 3, 110 mg (14~)
of the quinoline N-oxide (colorless crystals, m.p. 138-
140C) were obtained.
Exam~le 7
2,2-Dimethyl-6-trifluoromethoxy-4-(3'-pyridyl)-2H-benzo-
~b]pyran
3.0 g (9.3 mmol) of 1-(3'-pyridyl)-3-methyl-3-
(4'-trifluoromothoxyphenoxy)butyne are dissolved in 20 ml
of 1,2-dichlorobenzene and heated at 175C for 4 h. After
dilution with ether, the solution i8 washed with water
and the organic phase is dried over magnesium sulfate and
concentrated. After chromatography on silica gel using
chloroform as eluent, the benzopyran is isolated as an
o~l. Yield: 2.1 g (70~).
Example 8
2,2-Dimethyl-6-trifluoromethylthio-4-(3'-pyridyl)-2H-
benzo~b]pyran
From 800 mg (2.4 mmol) of 1-(3'-pyridyl)-3-
methyl-3-(4'-trifluoromethylthiophenoxy)butyne, by a
procedure ae described in the preceding examplé, 250 mg
- 44 -
(31%) of the benzo[b]pyran are obtained as an oil.
Example 9
2,2-Dimethyl-6-trifluoromethylsulfonyl-4-(3'-pyrldyl)-2~-
benzo[b~pyran
In analogy to Example 7, from 2.7 g (7.3 mmol) of
1-(3'-pyridyl)-3-methyl-3-(4'-trifluoromethylsulfonyl-
phenoxy)butyne, 1.14 g (42%) of the benzopyran were
obtained as colorless crystals, m.p. 121-122C (ethyl
acetate/petroleum ether).
Exam~le 10
2,2-Dimethyl-6-diethoxyphosphoryl-4-(3'-pyridyl)-2H-
benzo[b]pyran
In analogy to Example 7, from 600 mg (1.61 mmol)
of 3-mQthyl-1-(3'-pyridyl)-3-(4'-diethoxyphosphoryl-
phenoxy)butyne after chromatography on silica gel usingdichloromethane/ethanol, 95:5, 360 mg (60~) of the
benzopyran are obtained as an oil.
Exam~le ll
2,2-Dimethyl-6-trifluoromethylsulfonyl-4-(4'-pyridyl)-2H-
benzolb~pyran
By a procedure analogous to that described inExample 7, from 2.7 g (7.3 mmol) of 1-(4'-pyridyl)-3-
methyl-3-(4'-trifluoromethylsulfonylphenoxy)butyne,
110 mg (4~) of the substance were obtained as colorless
crystal~, m.p. 91-93C.
Example 12
2,2-Dimethyl-6-trifluoromethoxy-4-(2'-pyridyl)-3,4-
dihydro-2H-benzolb]pyran l'-oxide
a) 700 mg (2.2 mmol) of 2,2-dimethyl-6-trifluorometh-
oxy-4-(2'-pyridyl)-2H-benzolb]pyran are dissolved in a
mixture of 10 ml of ethanol and 1 ml of glacial acetic
acid, 100 mg of 10% palladium on activated charcoal are
added, and the mixture is hydrogenated overnight in an
autoclave at 50C and 50 bar. After filtering off the
catalyst, the mixture is concentrated and the re~idue is
... ..
- 45 -
taken up in dichloromethane and extracted by ~ha~lng wlth
saturated sodium hydrogen carbonate ~olution. The organlc
phase is dried over magnesium sulfate and concentrated ln
vacuo. 500 mg of crude 2,2-dimethyl-6-trifluoromothoxy-4-
(2'-pyridyl)-3,4-dihydro-2H-benzotb~pyran are obtalned,
which i8 employed further for the oxidation.
b) The crude 3,4-dihydro-2H-benzo[b~pyran 18 dissolved
in 12 ml of dichloromethane, 350 mg (1.7 mmol) of 85%
strength m-chloroperbenzoic acid are added, and the
mixture is stirred overnight. After extraction by sha~ing
with saturated sodium hydrogen carbonate solution, the
organic pha~e is dried over magnesium sulfate and concen-
trated. After chromatography on silica gel using
dichloromethane/ethanol, 95:5, 275 mg (37% based on both
reaction steps) of the pyridine N-oxide are obtained as
an oil.
ExamDle 13
2,2-Dimethyl-6-trifluoromethoxy-4-(2'-cyano-3'-pyridyl)-
2H-benzo[b~pyran
1.5 g (4.4 mmol) of 2,2-dimethyl-6-trifluorometh-
oxy-4-(3'-pyridyl)-2H-benzo[b~pyran l'-oxide are
dissolved in 10 ml of acetonitrile, 900 mg (8.9 mmol) of
triethylamine and 1.34 g (13.5 mmol) of trimethylsilyl
cyanide are added, and the mixture is boiled at reflux
overnight. ~he mixture is concentrated in vacuo, the
residue is taken up in chloroform and extracted by
sha~ing with water, and the organic phase is dried over
magnesium sul$ate. Chromatography on silica gel using
dichloromethane/ethanol, 98:2, gives 320 mg (21~) of the
substance as colorles~ crystals (m.p. 88-90C).
Example 14
2,2-Dimethyl-6-trifluoromethoxy-4-(4'-cyano-3'-pyridyl)-
2H-benzo[b]pyran
120 mg (7.8%) of the nitrile are obtained from
the batch described above, after chromatography, a~
colorless crystals (m.p. 126-127C, ethyl acetate/
- 46 - ~ & ~ ~t~3
petroleum ether).
Example 15
2,2-Dimethyl-6-trifluoromethoxy-4-(4'-chloro-3'-pyrldyl)-
2H-benzo[b]pyran
1.5 g (4.4 mmol) of 2,2-dimethyl-6-trifluoro-
methoxy-4-(3'-pyridyl)-2H-benzo[blpyran l'-oxide in
7.5 ml of phosphorus oxychloride are heated at 110C for
2 h. The mixture is cooled and concentrated in vacuo, the
re~idue is taken up in chloroform and extracted by
~haking with sodium hydrogen carbonate solution, and the
organic phase is dried over magnesium sulfate and concen-
trated. After chromatography on silica gel using
dichloromethane, 400 mg (25%) of the substance are
isolatQd as an oil.
Exam~le 16
2,2~Dimethyl-6-trifluoromethoxy-4-(2'-chloro-3'-pyridyl)-
2H-benzolb~pyran
140 mg (9~) of the substance are obtained from
the reaction mixture described above, after chromato-
ao graphy, as an oil.
Example 17
2,2-Dlmethyl-6-trifluoromethylsulfonyl-trans-3,4-dihydro-
3-hydroxy-4-(2'-pyridyl)-2H-benzolblpyran l'-oxide
a) 600 mg (1.6 mmol) of 2,2-dimethyl-6-trifluoromethyl-
sulfonyl-4-(2'-pyridyl)-2H-benzo[b]pyran and 1.3 g
(6.4 mmol) of m-chloroporbenzoic acid are dissolved in
10 ml of dichloromethane and stirred at room temperature
for 60 h. The product is extracted by sha~ing with
saturated sodium hydrogen carbonate solution, the organic
phase is dried over magnesium sulfate, concentrated, and
chromatographed on silica gel using dichloromethane/
sthanol, 98:2, and in this procedure 500 mg (77%) of
2,2-dimethyl-6-trifluoromethylsulfonyl-3,4-epoxy-4-(2~-
pyridyl)-3,4-dihydro-2H-benzolblpyran l'-oxide are
isolated as an oil.
- 47 _
b) Under an argon atmosphere, 530 mg (1.7 mmol) of
diphenyl diselenide are dissolved in 6 ml of ethanol~
130 mg (3.4 mmol) of sodium borohydride are added,
whereupon foaming occurs, and the mixture is stlrred for
a further 10 min. After neutralization of this solution
using glacial acetic acid, a solution of 500 mg
(1.25 mmol) of the above-described epoxide in 4 ml of
ethanol is added, and the mixture is boiled at reflux for
8 h. The mixture is cooled, diluted with water and
extracted twice with ethyl acetate, the organic phase i8
washed twice with saturated sodium chloride solution,
dried over magnesium sulfate and concentrated, and then
chromatographed on silica gel using dichloromethane/
ethanol, 98:2, with a rising ethanol content, affording
100 mg of the title substance.
Exam~le 18
2,2-Dimethyl-6-trifluoromethoxy-4-(4'-pyridyl)-3,4-
dihydro-2H-benzo[b]pyran 3'-oxide
a) 26.7 g (150 mmol) of 4-trifluoromethoxyphenyl and
38.0 g (195 mmol) of ethyl 2-bromo-2-methylpropionate are
dissolved in 300 ml of butanone, 30.4 g (0.22 mol) of
potassium carbonate and 1 g of potassium iodide are
added, and the mixture is boiled at reflux for 26 h.
After the addition of a further 13.1 g (67 mmol) of the
ester and 10 g (72 mmol) of potassium carbonate the
mixture is heated for a further 17 h and is cooled; the
inorganic salts are filtered off, the filtrate is concen-
trated, and the residue is chromatographed on silica gel
using dichloromethane. 31.1 g (71%) of ethyl 2-methyl-2-
(4'-trifluoromethoxyphenoxy)propionate are isolated as an
oil by this procedure.
b) 10.1 ml of diisopropylamine are dis~olved in 300 ml
of absolute tetrahydrofuran; the solution is cooled to
-78C, 45 ml (72 mmol) of a 1.6 M solution of
3S butyllithium in hexane are added dropwise under an argon
- 48 -
atmosphere, and the mixture is stirred for 15 min.
Subsequently, a solution of 5.64 g (60 mmol~ of 4-
methylpyrimidine in 120 ml of TH~ is added dropwlso, the
mixture is heated to room temperature and stirrod for 2
h at this temperature before being cooled agaln to -78C,
and 17.5 g (60 mmol) of the above-descrlbed estor in 180
ml of THF are added dropwise. The mixture i~ heated
overnight to room temperature, water is added, the
mixture is extracted with ethyl acetate, and the organic
phase is washed with saturated sodium chloride solution,
dried over magnesium sulfate and concentrated.
Chromatography of the residue on silica gel using ether
gives 9.8 g (48%) of 3-methyl-1-(4'-pyrimidinyl)-3-(4~-
trifluoromethoxyphenoxy)-2-butanone as an oil.
c) 9.8 g (28.8 mmol) of the above-doscribed ~etone are
dissolved in 250 ml of ethanol, 1.1 g (29.1 mmol) of
sodium borohydride are added, and the mixture is stirred
at room temperature overnight. The solvent is removed by
distillation, saturated sodium chloride solution is
added, the mixture is extracted with ethyl acetate, the
organ$c phaso is dried over magnesium sulfate and concen-
trated, and the residue is chromatographed on silica gel
using dichloromethane/ethanol, 98:2. Yield: 8.2 g (83%)
of 3-methyl-1-(4'-pyrimidinyl)-3-(4'-trifluorometh-
oxyphenoxy)-2-butanol.
d) 8.2 g (24.0 mmol) of the alcohol described in the
aforogoing proceduro and 8.0 g (39.4 mmol) of 85%
strength m-chloroperbenzoic acid are dissolved in 150 ml
of dichloromethane, and the solution is stirred at room
temperature for 24 h. The reaction mixture is washed
thoroughly with saturated sodium hydrogen carbonate
solution and twico with sodium chloride solution, and the
organic phase is dried over magnesium sulfate, concen-
trated, and chromatographed on silica gel using dichloro-
methane/ethanol, 96:4.
Yield: 4.3 g (50%) of 4-(3'-methyl-3'-(4n-trifluorometh-
oxyphenoxy)-2'-hydroxy)butylpyrimidine 3-oxide.
``` 21~2~
- 49 -
e) 4.2 g (11.7 mmol) of the above-de~cr~bod alcohol are
dissolved in 20 ml of dry triethylamlne, 2.6 g
(23.3 mmol) of methanesulfonyl chloride are addod, and
the mixture is heated at 60C for 30 min. The mlxture 1~
cooled, water is added, and after extractlon with
dichloromethane the organlc phase is dr$ed over magneslum
sulfate, concentrated and chromatographed on sllica ge~1
using chloroform.
Yield: 300 mg (7.5%) of 3-methyl-3-(4'-trifluoromethoxy-
phenoxy)-1-(4n-pyrimidinyl)-trans-1-butene 3~-oxide.
f) 300 mg (0.88 mmol) of the trans-butene from the
above-described reaction step in 30 ml of toluene are
heated at reflux for 28 h. The solvent is removed by
distillation in vacuo, and the residue is chromatographed
on silica gel using chloroform.
Yield: 100 mg (33%) of 1-(4'-pyrimidinyl)-1-(2n-hydroxy-
5n-trifluoromethoxyphenyl)-3-methyl-2-butene 3'-oxide.
g) 100 mg (0.29 mmol) of the phenol from the preceding
reaction step are dissolved in 10 ml of chloroform,
6 drops of concentrated sulfuric acid are added, and the
mixture is heated at 50C for 40 h. It is cooled and
extracted by shaking with sodium carbonate solution, and
tho extract is dried over magnesium sulfate and chromato-
graphed on silica gel using chloroform, affording 45 mg
(45%) of the title benzo[b~pyran as an oil.
ExamDle 19
6-Trifluoromethylsulfonyl-2,2-dimethyl-4-methoxy-4-(2'-
`pyridyl)-3,4-dlhydro-2H-benzolb]pyran 1'-oxide
a) 1.1 g (7.0 mmol) of 2-bromopyridine are dissolved in
20 ml of dry tetrahydrofuran under an argon atmosphere
and cooled to -78C, and 4.4 ml (7.0 mmol) of a 1.6 molar
solution of butyllithium in hexane are added over 20 min.
After 30 min, the mixture is heated to -60C, a solution
of 2.2 g (8.0 mmol) of 6-trifluoromethylthio-2,2-
dimethyl-3,4-dihydro-2H-benzolb~pyran-4-one in 10 ml of
T~F is in~ected rapidly, and the mixture is stirred at
so
- -
this temperature for 2 h, cooled to -78C and heated to
room temperature overnight. Saturated ammonlum chlor~do
solution is added, the mixture i8 extracted wlth ether,
and the organic phase is dried over mngnesium sulfate and
concentrated. After chromatography on silica gel uslng
dichloromethane, 330 mg ~13%) of 6-trifluoromethylthio-
2,2-dimethyl-4-(2'-pyridyl)-3,4-dihydro-2~-benzotb]pyran-
4-ol are obtained.
b) 300 mg (0.84 mmol) of the above-described alcohol
are dissolved in 10 ml of dry tetrahydrofuran under an
argon atmosphere, 30 mg (1.0 mmol) of 80% sodium hydride
are added, and the mixture i8 stirred at room temperature
for 40 min. After the addit~on of 240 mg (1.7 mmol) of
iodomethane, the mixture is stirred for a further 2 h and
wor~ed up as in a).
Yield: 300 mg (96%) of 6-trifluoromethylthio-2,2-dime-
thyl-4-methoxy-4-(2'-pyridyl)-3,4-dihydro-2H-benzotb]-
pyran.
c) 300 mg (0.81 mmol) of the above-described ether are
stirred overnight with 200 mg (1.0 mmol) of 85% strength
m-chloroperbenzoic acid in 10 ml of dichloromethane, then
the same quantity of the oxidizing agent is again added,
and the mixture is stirred for a further 24 h. Saturated
sodium hydrogen carbonate solution iB added, the mixture
is extracted by shaking with dichloromethane, the organic
phase ie dried over magnesium sulfate and concentrated,
and the residue is chromatographed on silica gel using
dichloromothane/ethanol, 98:2.
Yield: 130 mg (38%) of the title sulfone as colorless
crystals, m.p. 157-158C.
Exam~le 20
6-Trifluoromethylsulfinyl-2,2-dimethyl-4-methoxy-4-(2'-
pyridyl)-3,4-dihydro-2H-benzotb]pyran 1'-oxide
In the oxidation described in the preceding
example, 90 mg (28%) of the sulfoxide are obtained as
colorless crystals, m.p. 62-64C.
Example 21
6-Trifluoromethylthio-2,2-dimethyl-4-(2'-methoxy-3'-
pyridyl)-3,4-dihydro-2H-benzotb]pyran-4-ol
1.2 g (5.1 mmol) of 3-iodo-2-methoxypyridino are
dissolved under an argon atmocphere in 15 ml of dry
tetrahydrofuran and cooled to -90C, 3.1 ml (5.0 mmol) of
a 1.6 molar solution of butyllithium in hexane are added
dropwise, and the mixture is stirred at this temperature
for 2.5 h. The mixture is warmed to -50C, 1,5 g
(5.4 mmol) of 6-trifluoromethylthio-2,2-dimethyl-3,4-
dihydro-2H-benzo[b~pyran-4-one in 10 ml of THF are
rapidly added dropwise, the mixture ie again cooled to -
80C, and is heated to -40C over a period of 2 h.
Working up i8 carried out as in reaction a) of Example
19, and by this procedure 400 mg (20%) of the substance
are isolated as an oil.
Exam~le 22
6-Trifluoromethylthio-2,2-dimethyl-4-(2'-methoxy-3'-
pyridyl)-2H-benzolb~pyran
400 mg (1.0 mmol) of the above-described alcohol
are boiled with 490 mg (2.6 mmol) of p-toluene~ulfonic
acid monohydrate in 15 ml of toluene in a water separator
for 3 h. The mixture is cooled and worked up as in
reaction c) of Example 19, and chromatography is carried
as out using dichloromethane.
Yield: 270 mg (71%) of colorless needles, m.p. approxi-
matoly 25C.
The precursors needed for the example~ listed
above are prepared as follows:
Examplos 1'-6' were obtained by a procedure
analogous to that described in Example 7, and in some
cases were reacted further without intensive purification
operations.
Example 1'
2,2-Dimethyl-6-trifluoromethylthio-4-(2'-pyridyl)-2H-
benzolb]pyran
1.8 g (50%) of semi-crystalline substance from
- 52 ~ 2~ ~2~ ~4
3.6 g (10.7 mmol) of 1-(2'-pyridyl)-3-mothyl-3-(4'-
trifluoromethylthiophenoxy)-butyne.
ExamPle 2'
2,2-Dimethyl-6-trifluoromethoxy-4-(2'-pyridyl)-2H-benzo-
lb]pyran
2.0 g (69%) of crystals, m.p. 57-59C, from 2.9 g
(9.0 mmol) of 1-(2'-pyridyl)-3-methyl-3-(4'-trifluoro-
methoxyphenoxy)butyne.
Example 3'
2,2-Dimethyl-6-trifluoromethylsulfonyl-4-(2'-pyridyl)-2H-
benzo[b]pyran
350 mg (17%) of an oil from 2.0 g (5.4 mmol) of
1-(2'-pyridyl-3-methyl-3-(4'-trifluoromethylsulfonyl-
phenoxy)butyne.
Exam~le 4'
2,2-Dimethyl-6-difluoromethoxy-4-(2'-pyridyl)-2H-benzo-
lb]pyran
1.9 g (46%) of an oil from 4.1 g (13.5 mmol) of
1-(2'-pyridyl)-3-methyl-3-(4'-difluoromethoxyphenoxy)-
butyne.
Example 5'
2,2-Dimethyl-6-trifluoromethylsulfonyl-4-(2'-quinolyl)-
2H-benzotb]pyran
After chromatography on silica gel using
dichloromethane, 700 mg (19%) of crystals, m.p. 140-142C
(from ether/petroleum ether) were obtained, in addition
to 600 mg (16%) of 2-isopropyl-3-(2'-guinolyl)-5-tri-
fluoromethylsulfonylbenzofuran, m.p. 133-134C
(ether/petroleum ether), from 3.7 g (8.8 mmol) of 1-(2'-
quinolyl)-3-methyl-3-(4'-trifluoromethylsulfonylphenoxy)-
butyne.
ExamDle 6'
1-(2'-Pyridyl)-3-methyl-3-(4'-trifluoromethylthio-
- 53
phenoxy)butyne
3.9 g (15 mmol) of 3-methyl-3-(4'-trlfluoro-
methylthiophenoxy)butyne and 3.1 g (15 mmol) of 2-iodo-
pyridine are dis~olved in 30 ml of triethylamine, 240 mg
of bis~triphenylphosphine)pallad~um dichlorlde and 140 mg
of copper(I) iodide are added, and the mixture 1~ stirred
at 80C for 2.5 h in a screw-top test-tube. The mlxture
is cooled, diluted with ether and extracted by shaking
with water, and the organic phase is washed a number of
times with dilute hydrochloric acid and dried over
magnesium sulfate. After chromatography on silica gel
using dichloromethane/ethanol, 98:2, 3.6 g (71~) of the
propargyl ether are isolated as an oil.
Example 7'
1-(2'-Pyridyl)-3-methyl-3-(4'-trifluoromethoxyphenoxy)-
butyno
7.4 g (30 mmol) of 3-methyl-3-(4'-trifluorometh-
oxyphenoxy)butyne, 6.0 g (29.3 mmol) of 2-iodopyridine,
480 mg of bi~(triphenylphosphine)palladium dichloride and
280 mg of copper(I) iodide are dissolved in 50 ml of
triethylamine, and the solution is stirred at room
temperature for 2 h. After being worked up as described
above, 9.6 g (98%) of the propargyl ether are obtained as
an oil.
E~ample 8'
1-(2'-Pyridyl)-3-methyl-3-(4'-trifluoromethylsulfonyl-
phenoxy)butyne
From 5.0 g (17.1 mmol) of 3-methyl-3-(4~-
trifluoromethylsulfonylphenoxy)butyne and 3.8 g
(18.5 mmol) of 2-iodopyridine, by a procedure analogous
to that of Example 7' after 12 h at room temperature,
2.0 g (32%) of the substance were obtained as an oil.
Exam~le 9'
1-(2'-Pyridyl)-3-methyl-3-(4'-difluoromethoxyphenoxy)-
butyne
From 3.4 g (15 mmol) of 3-methyl-3-(4'-difluoro-
9 ~ ~L
- 54 -
methoxyphenoxy)butyne and 3.1 g (15 mmol) of 2-lodo-
pyridine, by a procedure analogous to that of Example 6',
4.1 g (90%) of the propargyl ether were obtainod a~ an
oil.
ExamDle 10'
1-(3'-Pyridyl)-3-methyl-3-(4'-trifluoromethylthiophen-
oxy)butyne
500 mg (1.9 mmol) of 3-methyl-3-(4'-trifluoro-
methylthiophenoxy)butyne and 390 mg (1.9 mmol) 3-iodo-
pyridine give, by a procedure analogous to that ofExample 6', 500 mg (77%) of the substance as an oil.
Examplo 11'
1-(3'-Pyridyl)-3-methyl-3-(4'-trifluoromethoxyphenoxy)-
butyne
12.2 g (50 mmol) of 3-methyl-3-(4'-trifluorometh-
oxyphenoxy)butyne, 9.4 g (46 mmol) of 3-iodopyridine,
700 mg of bis(triphenylphosphine)palladium dichloride and
500 ml of copper(I) iodide are dissolved in 70 ml of
triethylamine, and the solution is stirred at room
temperature for 3 h. The solution is wor~ed up as
described in Example 6' and 14.2 g (96%) of the substance
are isolated as an oil.
Example 12'
1-(3'-Pyridyl)-3-methyl-3-~4'-trifluoromethylsulfonyl-
phenoxy)butyne
From 2.7 g (9.2 mmol) of 3-methyl-3-(4'-tri-
fluorosulfonylphenoxy)butyne, 2.1 g (10.2 mmol) of
3-iodopyridine, 200 mg of bis(triphenylphosphine)-
palladium dichloride and 100 mg of copper(I) iodide in
30 ml of triethylamine, after 3 h at 50C and after being
worked up in a manner analogous to that of Example 6',
2.7 g (79%) of the substance are obtained as an oil.
Example 13'
3-Methyl-1-(3'-pyridyl)-3-(4'-diethoxyphosphorylphenoxy)-
butyne
From 1.0 g (3.37 mmol) of 3-methyl-3-(4'-dieth-
oxyphosphorylphenoxy)butyne and 640 mg (3.12 mmol) of
3-iodopyridine, in a manner analogous to that of
Example 6' after chromatography on silica gel uoing
ether, 620 mg (54%) of the substance are obtained.
ExamDle 14'
1-(4'-Pyridyl)-3-methyl-3-(4'-trifluoromethylsulfonyl-
phenoxy)butyne
From 2.9 g (9.9 mmol) of 3-methyl-3-(4'-tri-
fluoromethylsulfonylphenoxy)butyne, 2.0 g (9.9 mmol) of4-iodopyridine, 100 mg of copper(I) iodide and 150 mg of
bis(triphenylphosphine)palladium dichloride in 20 ml of
triethylamine, after 2 h at 80C and wor~ing up the
mixture as already described, 2.7 g (74%) of the pro-
pargyl ether are obtained as an oil.
Example 15'
1-(2'-Quinolyl)-3-methyl-3-(4'-trifluoromethylsulfonyl-
phenoxy)butyno
3.2 g (10.9 mmol) of 3-methyl-3-(4'-trifluoro-
methylsulfonylphenoxy)butyne, 2.6 g (10.2 mmol) of
2-iodoquinoline, 140 mg of bis(triphenylphosphine)-
palladium dichloride and 100 mg of copper(I) iodide are
dissolved in 20 ml of triethylamine, and the mixture is
heated at 50C for 10 min. Wor~ing up the mixture in a
as mannor analogous to that of ~xample 6', and chromato-
graphy on silica gel using dichloromethane, give 3.8 g
(89~) of the substance as an oil.
gxample 16'
3-Methyl-3-(4'-trifluoromethylthiophenoxy)butyne
13.8 g (0.1 mol) of dried potassium carbonate and
1.6 g (0.01 mol) of potassium iodide are suspended in a
solution of 19.4 g (0.1 mol) of 4-(trifluoromethylthio)-
phenol in 250 ml of dry butanone, and 15.4 g (0.15 mol)
of 3-chloro-3-methyl-1-butyne are added dropwise. The
mixture is then heated at reflux with stirring for
~' ~ , .
' ' ,
, , ,
- 56 _ 2 ~ 3~
20 hours. 15.4 g of 3-chloro-3-methyl-1-butyne and 13.8 g
of potas~ium carbonate are aga~n added, and the m~xture
is aga~n heated at reflux, for 40 hour~. Inorgan~c
constituents are removed by filtrat~on, tho solut~on i8
concentrated, and the residue is ta~en up in 200 ml of
methylene chloride and extracted with 1 N NaOH ~olution.
The organ~c phase is washed with water, dr~ed and concen-
trated, and the residue is filtered over silica gel.
Yield: 22 g (85~).
Exam~le 17'
3-Methyl-3-(4'-trifluoromethoxyphenoxy)butyne
69.2 g (0.5 mol) of dried potassium carbonate and
8.3 g (0.05 mol) of potassium iodide are suspended in a
solution of 90 g (0.5 mol) of 4-trifluoromethoxyphenol in
900 ml of dry acetone, and 70 g (0.68 mol) of 3-chloro-3-
methyl-1-butyne are added dropwise. The mixture is
stirred at reflux temperature for 36 hours, after which
a further 35 g (0.34 mol) of 3-chloro-3-methyl-1-butyne
are added, and the mixture is stirred at reflux tempera-
ture for a further 36 hours. The suspension is cooled,
filtered and washed with acetone. The filtrate is concen-
trated, and the residue is taken up in methylene chloride
and extracted with 1 N NaOH solution. The organic phase
is washed until neutral, dried and concentrated by
evaporation.
Yield: 67 g (55~).
Example 18'
3-Methyl-3-(4'-trifluoromethylsulfonylphenoxy)butyne
A) 1 g (4.4 mmol) of 4-trifluoromethylsulfonylphenol
together with 0.66 g of potassium carbonate, 80 mg of
potassium iodide and 2 g of 3-chloro-3-methyl-1-butyne in
13 ml of dry butanone are stirred at 80-90C under argon
for 20 hours. The mixture is allowed to cool and is
filtered, and the filtrate is concentrated by evapor-
ation. The residue is taken up in 20 ml of methylene
chloride and washed with water (2 x 20 ml), and is dried
and concentrated by evaporation. 1.2 g (93%) of the
~ ~ ~ 2 ~ ~ ~
- 57 -
butyne remain as an oil.
B) 20 g (76.8 mmol) of 3-methyl-3-(4'-trlfluoromethyl-
thiophenoxy)butyne are dissolved in a mixture of 700 21
of water and 700 ml of methanol, and 141.8 g oS Oxone~
are added. The mixture i8 stirred for 4 day~ at room
temperature, a further 500 ml of water are added, and
extraction i8 carried out three times with chloroform.
The combined organic phases are dried over magnesium
sulfate and concentrated, and the residue is chromato-
graphed on silica gel using dichloromethane.Yield: 14.0 g (62%).
Example 19'
3-Methyl-3-(4'-difluoromethoxyphenoxy)butyne
is prepared in a manner analogous to that of Example 16'.
Yield: 70%; b.p. 56-58C/0.035 mbar.
Example 20'
3-Methyl-3-(4'-diethoxyphosphorylphenoxy)butyne
is prepared, in a manner analogous to that of Example
16', from 4-diethoxyphosphorylphenol.
Yield: 78%; colorless oil after chromatography on silica
gel using ethyl acetate/hexane, 40:5.
Example 21'
4-Trifluoromethylsulfonylphenol
A) 1 g ~5.2 mmol) of 4-trifluoromethylthiophenol is
dissolved in 20 ml of methanol, and a suspension of 9.6 g
of Oxono~ in 20 ml of water is added at 0C with
stirring. After stirring for 5 days at room temperature,
the mixture is diluted with 50 ml of water and extracted
with chloroform (3 x 50 ml). After being dried and
concentrated by evaporation, 1.1 g (94%) of colorless
crystals remain, m.p. 123C (lit.: 119-120C).
B) 1 g (5.2 mmol) of 4-tr$fluoromethylthiophenol
together with 4 ml of glacial acetic acid and 4 ml of 30%
strength hydrogen peroxide are stirred at 50C for
.
- 58 -
20 hours. Subsequently, a further 2 ml of 30% ~trength
hydrogen peroxide are added, and after a further 2 hour~
at 50C the mixture i8 worked up as de~cribod above.
After chromatography on silica gel, 230 mg ~20%) of tho
phenol are obtained.
Example 22'
2,2-Dimethyl-6-trifluoromethylthio-3,4-dihydro-2H-benzo-
tb]pyran-4-one
a) 22.0 g (84.5 mmol) of 3-methyl-3-(4'-trifluoro-
methylthiophenoxybutyne are dissolved in 50 ml of 1,2-
dichlorobenzene, and the solution is heated at 180C for
2 h under an argon atmosphere and then fractionated in
vacuo, affording 13.0 g (59%) of 2,2-dimethyl-6-tri-
fluoromethylthio-2H-benzo~b~pyran (b.p. 55C/0.02 mbar).
b) 6.5 g (25.0 mmol) of 2,2-dimethyl-6-trifluoromethyl-
thio-2H-benzolb]pyran are dissolved in 25 ml of dry
chloroform and cooled to -5C. At this temperature a
solution of 4.0 g (1.27 ml; 25.0 mmol) of bromine in
20 ml of chloroform is added dropwise, and the mixture is
stirred for a further 5 min and concentrated. 2,2-Dime-
thyl-trans-3,4-dibromo-6-trifluoromethylthio-3,4-dihydro-
2H-benzotb~pyran crystallizes in colorless crystals from
cold petroleum ether.
Yield: 10.1 g (96~), m.p.: 66-67C.
c) 1.25 g (54.4 mmol) of sodium are dissolved in 20 ml
of absolute methanol and the solution is concentrated to
drynes~, the residue is ta~en up in 1,2-dimethoxyethane
(DME), and 22.5 g (53.6 mmol) of the above-dQscribed
dibromide in 60 ml of DME are added. The mixture i8 then
boiled at reflux for 40 min, water is added, the mixture
is extracted by sha~ing with ether, and the organic phase
is washed with saturated sodium chloride ~olution, dried
over magnesium sulfate and concentrated. 2,2-Dimethyl-4-
bromo-6-trifluoromethylthio-2H-benzo[b]pyran is obtained
as an oil which, on the ba~i~ of it~ lH-NMR spectrum, is
sufficiently pure for further reaction.
59 ~29~
Yield: 16.5 g (91%).
d) 4.5 g (13.3 mmol) of the above-de~cribed benzopyran
are dissolved in 10 ml of concentrated sulfuric acid, and
the solution is stirred at room temperature overn~ght.
The mixture i~ then carefully poured into sodium hydrogen
carbonate ~olution and extracted with ether, and the
organic phase is dried over magnesium sulfate and concen-
trated. After chromatography on silica gel uslng
dichloromethane, 2.0 g (55%) of the chromanone are
obtained a~ colorless crystals, m.p. 45-47C.
Exam~le 23
Production of tablets and capsules
Tablets and capsules containing the constituents
indicated below are produced by known procedures. These
tablets and capsules are suitable for the treatment of
the abovementioned diseases, in particular hypertension,
in dosage~ of in each case one tablet or capsule once
daily.
Components Weight (mg)
Tablet Capsule
6-Trifluoromethylsulfonyl-
2,2-dimethyl-4-(2'-pyridyl)-
2H-benzotb]pyran l'-oxide 0.2 0.1
Tragacanth 10
Lactose 247.5 300
Corn starch 25
Tal¢ 15
Magnesium stearate 2.5
Exam~le 24
Production of ampules
Ampules which contain the components stated below
can be produced in a ~nown manner. Glass ampules are
filled under nitrogen with a solution of the active
substance in water and 1,2-propanediol.
`` 2 1 ~
- 60 -
6-Trlfluoromethylsulfonyl-2,2-
dimethyl-4-(2'-pyrldyl)-2H-benzo-
[b]pyran 1'-oxlde 0.02 mg
1,2-Propanediol 0.8 ml
5 Dlstilled water to 2.0 ml