Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 93/16072 2 1 ~ ;~ 1 1 2 PCI /GB93/00214
- 1 -
Benzopyran, benzoth10pyran and benzofuran der1vat~ves as 5-HT4 antagonlsts
This invention relates to the use of compounds as ~HT4 receptor antagonists
in the treatment of gastrointestinal disorders, CNS disorders including
migraine and/or cardiovascular disorders, and to certain novel compounds
having 5-HT4 receptor antagonist activity.
European ~ournal of Pharmacology 146 (1988), 187-188, and Naunyn-
&hmiedeberg's Arch. Pharmacol. (1989) 340:40~410, describe a non
classical ~hydroxytryptamine receptor, now designated th~ ~HT4 receptor, --
and that ICS 20~930, which is also a ~HT3 receptor antagonist, acts as an
antagonist at this r~ceptor.
WO 91/16045 (SmithKline and French Laboratories Limited) describes the -~
use of cardiac ~HT4 receptor antagonists in the treatment of atnal
arrhythmias and stroke. -~
: .~
EP-A-501322 (Glaxo Group Limited) describes indole derivatives having
5-tlT4 antagonist activity. -
Some 5-HT3 receptor antagonists have been disclosed as of potential use in
the treatment of oertain aspects of irritable bowel syndrome [EP-A-189002
(Sandoz Umited) and EP-A-201165 (Beecham Group p.l.c)l.
5-HT3 receptor interactions which are of potential us~ in the treatment of IBS `-
are those associated either with the visceral pain and abnormal perception of
sensation aspects of this disease, or they are related to the ability of some 5-HT3 receptor antagonists to cause constipation in volunteers.
Some ~HT3 receptor antagonists haYe been disclosed as of potential use in
the treatment of gastrointestinal disorders associated with upper gut motility
[EP-A-226266 (Glaxo Group Ltd.) and EP-A-189002 (Sandoz Limited)].
5-HT3 receptor antagonists are also well known antiemetics, such as
ondansetron, granisetron and tropisetron lDrugs of the Future 1989, 14 (9)
p.875- F.D. King snd G.J. Sang-r].
. ~
:
wo 93/l6072 ? 1 2 9 1 1 ~! - 2 - PCI`/GB93/00214
EP-A-234872 (Adria), US 48~9683 (Rorer) and EP-A-307172 (Lilly) describe
5-HT3 receptor anta~onists derived from a benzoic acid nucleus, 2,3-
disubstituted by alkyleneoxy.
5 It has now been discover~d that certain of the compounds embraced by the
general forrnulae disclosed therein, and related compounds, have ~HT4
receptor antagonist properties, and are therefore of potential use in the
treatment of IBS or atrial arrhythmias and stroke.
10 The compounds of the present invention also have a potential use in the
treatment of CNS disorders such as anxiety and/or migraine, in the treatment -~
of upper gut motility disorders and as antiemetics.
,; -
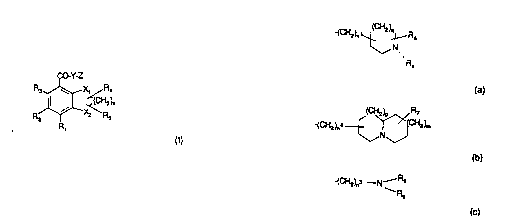
~Accordingly, the present invention provides the use of a compound of formula
15 (I) or a pharmaceutically acceptable salt thereof:
CO Y-Z
R3~ X, R4
R~X~z~R
(I) :.
20 in which X1-(CH2)X-X2 and the aromatic carbon atoms to which they are
attached form a ~7 membered ring
wherein: -
one of X1 and X2 is 0, S or CH2 and the other is CH2;
xis1,20r3;
25 R1 is hydrogen, amino, halo, C1 6 alkyl, hydroxy or C1 ~; alkoxy;
R2 is hydrogen, halo, C1 6 alkyl, C1-6 alkoxy, nitro, amino or C1 6 alkylthio;
R3 is hydrogen, ha!o, C1-6 alkyl, C1 6 alkoxy or amino;
R4 and Rs are independently hydrogen or C1 6 alkyl;
YisOorNH;
30 Z is of su~tormula (a), (b) or (c):
-(CH2)n~ R6
N
Ra
(a)
WO 93/16072 .~ ~. 2 9 ~ 1 PCI'/GB93/00214
2 n ~,NJ
(b)
~(C}'~2)r~3 --N--R8
Rg
(c) ''
whersin '''".
n1 is1,2,30r4;n~is1 or2;n3is2,3,40r5;
10 qisO,1,20r3;pisO,1 or2;misO,1 or2;
Ra is hydrogen or a lipophilic group, such as C1 -1 2 alkyl or aralkyl; sr
Ra is (CH2)rP~10 wherein r is 2 or 3 and R10 is selected from cyano, --
hydroxyl, C1-6 alkoxy, phenox~, C(O)C1-6 alkyl, COC6H5- -CR11 R12
NR11 COR1 2, SO2NP~1 1 R12 or NR1 1 S02R1 2 wherain R1 1 and R12
are hydrogen or C1-6 alkyl; and
R~;, R7 and R8 are independently hydrogen or C1 6 alkyl; and
Rg is hydrogen or C1 10 alk~
~; or a compound of formula (I) wherein the CO-Y linkage is r~placed by a ;-
heterocyclic bioisostere; -~
20 in the manufacture of a medicament for use as a ~HT4 receptor antagonist.
'~
Examplss of alkyl or alkyl containing groups include C1, C2, C3, C4, Cs, C6,
C7, Cg, Cg, C1 o, C1 1 or C1 2 branched, straight chained or cyclic alkyl, as
appropnate. C1 4 alkyl groups include methyl, ethyl ~ and iso propy!, n, i~
25 , s~ and tert-butyl. Cyclic alkyl includes cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl and cyclooctyl.
Aryl ;includes phenyl and naphthyl optionally substituted by one or more
substituents selacted from halo, C1-6 alkyl and C1-6 alkoxy.
- Halo includes fluoro, chloro, bromo and iodo,. -
A suitable bioisostsre for the amide or ester linkage containing Y in formula
(I), is of formula (d)
.
WO 93/16072 PCI`/GB93/00214
2129112 H ~J
(d)
wh~rein
the dotted circle r~presents one or two double bonds in any position in the 5-
5 member~d ring; H, ~1 and I independently represent oxygen, sulphur, nitrogenor carbon, provided that at least one of H, J and t is other than carbon; U
repr~sents nitrogen or carbon.
Suitable examples of (d) are as described for X, Y and Z in EP-A-328200
10 (Merck Sharp & Dohme Ltd.), such as an oxadiazole moiety.
Suitable examples of the Xj-(CH2)X-X2 include 0-(CH2)2-CH2, 0-(CH2)3-
CH2, ~CH2-CH2, or corresponding values wherein X1 = X2 = CH2, wherein
any o~ the methylena linkages ars optisnally mono- or di-substituted by C1 6
15 alkyl groups, such as methyl. Preferably X1-(C~2)2-X2 is ~(CH2)2-CH2-
R1 is preferably hydrogen or amino.
R2 is pr~ferably hydrogen or halo.
20R3 is preferably hydrogen or halo.
R4 and Rs are often hydrogen. When R4/Rs is C1 6 alkyl, it is o~ten methyL
In particular R4 and Rs are methyl such that the disubstituent oontaining X
25 and X2 is X1-c~cH3)2-x2-
Y is preferably O or NH.
When Z is of su~formu!a (a)l n1 is preferably 2, 3 or 4 when the azacycle is
30 attached at the nitrogen atom and nl is preferably 1 when th~ azacycl~ isattached at a carbon atom, such as the 4-position when q is 2.
:
When Z is of su~formula (b), n2 is preferably such that the number of carbon
atoms between the ester or amide linkage is ~rom 2 to 4 carbon atoms.
Suitable values for p and m include p = m = 1; p = 0, m = 1; p = 1, m = 2.
W093/16072 ~ ~ 2 3 ~ 12 i : pcr/GB93loo2l4
When ~ is of su~formula (c), n3 is preferably 2, 3 or 4.
R8 and Rg are praferabiy both alkyl, especially one of R8 and Rg is C4 or ~-
larger alkyl.
-.
Specific values of Z of particular interest are as follows: : .
. .
/ \~N" Bu (~
. . .
/~\N /~)
\ (ii) :
\/\ N~) :
(iii) ,:~.
.10 . .
~N~b BU (iv)
N (v~
/\N~ -
~ (vi) ' ~ ~
N (vii)
The invention also provides novel compounds within 10rmula (1) with side
:~ ~ 15 chains (i), (ii), (iii), (iv), (v), ~vi) or (vii). In a further aspect, the piperidine ring
- in (i),. (ii) or (iii) may be replaced by pyrrolidinyl or azetidinyl, and/or the
N^substituent in (i) or (ii) may be replaced by C3 or lar~er alkyl or optionallysubstitutedbenzyl.
wo 93/16072 ~ 1 2 9 1 1 2 PCI /GB93/00214
In an altemative aspect, the N-substituent in formula (i) or (ii) may be
replaced by (CH2)nR4, as defined in formula(l) and in relation to the specific
examples of EP-A-501322.
5 The invention also provides novel compounds within formula (I) having
X1 -(CH~)x~X2 as O-(CH2)2-CH2, in particular thoss wherein the side chain Z
is of sub-formula (a) or (c).
The pharmaceutically acceptable salts of the compounds of ~he formula (I)
10 include acid addition salts with conventional acids such as hydrochloric,
hydrobromic, boric, phosphoric, sulphuric a~ids and pharmaceutically
acceptable or~anic acids such as acetic, tartaric, maleic, citnc, succinic,
benzoic, ascorbic, methanesulphonic, a-keto glutaric, a-glycerophosphoric,
and glucose-1-phosphoric acids.
Examples of pharmaceu~ically-acceptable salts includ~ quaternary derivatives
of the compounds of formula (I) such as the compounds quatemised by
compounds RX-T wherein Rx is C1-6 alkyl, phenyl-C1 6 alkyl or C~7
cycloalkyl, and T is a radical corresponding to an anion of an acid. Suitable
20 examples Of Rx include methyl, cthyl and r~ and iso propyl; and benzyl and
phenethyl. Suitable examples of T include halide such as chloride, bromide
and iodide.
. ~
Examples of pharrnaceutically acceptable salts also include internal salts such
25 asN-oxides.
The compounds of the formula (I), their pharmaceutically acceptable salts,
(including quaternary dsrivatives and N-oxides) may also form
pharmaceutically acceptable solvates, such as hydrates, which are included
30 wherever a compound of formula (l~ or a salt thereof is herein referred to.
It will also be realised that the (CH2)n2 moiety in compounds of formula ~I) ;
wherein Z is (b), may adopt an a or ,B or configuration with respect to the
fused azabicyclic moiety.
WO 93/1 6072 .;~J 12 3 1 i 2 PCJ/GB93/00214
-7-
The compounds of formula (I) wherein CO-Y is an ester or amide linkage are
prepared by conventional coupling of the Z moiety with the appropriate acid.
Suitable methods are as described in GB 21 25398A (Sandoz Limited), GB -~
1593146A, EP-A-36269 and EP-A-289170 (Beecham Group p.l.c.). When
CO-Y is replaced by a heterocyclic bioisostere, suitable methods are
describsd in EP-A-328200 (M~rck Sharp & Dohme Limited). Refer~nce is
also made to EP-A-~01322 (Glaxo Group Limited).
Aza(bi)cyclic side chain intermediates are known compounds or may be
10 prepared according to the methods described in PCT/GB92/01519 and
/01612 (SrnithKlins Beecham p.l.c.).
- The compounds of the pr~sent invention are 5-HT4 receptor antagonists and
it is thus believed may generally be used in the treatment or prophyla~tis o~
15 gastrointestinal disorders, cardiovascular disorders and CNS disorders.
They are of potential interest in the tr0atmsnt of irritabls bowel syndrome
(IBS), in particular the diarrhoea aspects of IBS, i.e., these compounds block
the ability of 5 HT to stimulate gut motility via activation of enteric neurones.
20 In animal models of IBS, this can be conveniently measured as a reduction of
the rate of defaecation. They are also of potential use in the treatment o~
urinary incontinence which is often associated with IBS.
They may also be of potential use in other gastrointestinal disorders, such as
25 those associated with upper gut motility, and as antiemetics. In particular,
they are of potential use in the treatment of the nausea and gastric symptoms ~ -
of gastro-oesophagaal reflux disease and dyspepsia. Anti~metic activity is
determined in known animal models of cytotoxic agent induced emesis.
30 Specific cardiac ~-HT4 rcceptor antagonists which prevent atrial fibrillationand other atrial arrhythmias associated with 5-HT,- would also be expected to
reduce occurrence of stroke (see A.J. Kaumann 1990, Naumyn- 1
Schmiedeberg's Arch. Pharrnacol. 342, 619-622, for appropriate animal test
method).
Atlxiolytic activity is likely to be effected via the hippocampus (Dumais et al
1988, Mol Pharmacol., 34, 880-887). Activity can be demonstrated in
standard animal models, the social interaction test and the X-maze test.
Wo 93/1 6072 21 2 91 1~ PCl /GB93/00214
Migra,ne sufferers often undergo situations o~ anxiety and emotional stress
that precede the appearance of headache (Sachs, 1985, Migraine, Pan
Books, London). It has also been observed that during and within 48 hours of
a migraine attack, cyclic AMP levels are considerably increased in the
cerebrospinal fluid (Welch ~t al., 1976, Headache 16, 160-167). It is believed
that a migraine, including the prodomal phase and the associated incrsasad
levels of cyclic AMP are ralated to stimulation of ~-HT4 receptors, and hence
that administration of a 5-HT4 antagonist is of potential benefit in rQlieving a -
migraine attack.
The invention also provides a pharmaceutical composition comprising a
compound of tormula (I), or a pharmaceutically acceptable salt thereof, and a
pharrnaceutically acceptable carrier. -
15 Such compositions are prepared by admixture and are usually adapted for
oral or parenteral administration, and as such may be in the form of tablets,
capsules, oral liquid preparations, powders, granules, lozenges, ;;
reconstitutable powders, injectable and infusable solutions or suspensions or
suppositories. Orally administrable compositions are preferred, since they are -
more convenient for general use.
Tablets and capsules for oral administration are usually presented in a unit
dose, and contain conventional excipients such as binding agents, fillers,
diluents, tabletting agents, lubricants, disintegrants, colourants, flavourings,and wetting agents. The tablets may be coated according to well known
methods in the art, for example with an enteric coating.
.
Suitable fillers for use include cellulose, mannitol, lactose and other similar
agents. Suitable disintegrants include starch, polyvinylpolypyrrolidone and
starch derivatives such as sodium starch glycollate. Suitable lubricants ~-
include, for exàmple, magnesium stearate.
Suitable pharmaceutically acceptable wetting agents include sodium lauryl
sulphate. Oral liquid prQparations may be in the form of, for example,
aqueous or oily suspensions, solutions, emulsions, syrups, or elixirs, or may
be presented as a dry product for reconstitution with water or other suitable
vehicle betore use. Such liquid preparations may contain conventional
additives such as suspending agents, for example sorbitol, syrup, methyl
:
WC) 93/16072 PCI /GB93/00214
g
cellulose, gelatin, hydro)~yethylcellulose, carboxymethylcellulose, aluminium
stearate gel or hydrogenated edible fats, emulsifying agents, for example
l~cithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may
include edible oils), ~or example, almond oil, fractionated coconut oil, oily
5 esters such as esters of glycerine, propylene glycol, or ethyl alcohol;
pr~servatives, for example methyl or propyl p-hydroxybcnzoate or sorbic acid,
and if desired conventional flavouring or colouring agents. i-
Oral liquid preparations arc usually in the torm of aqueous or oily -~
10 suspensions, solutions, emulsions, syrups, or elixirs or are presented as a dry
product tor reconstitution with water or other suitable vehicle before use~
Such liquid preparations may contain conventional additives such as
suspending agents, emulsifying agents, non-aqueous vehicles (which may
inciude edible oils), preservatives, and flavouring or colouring agents. -.
-
The oral compositions may be prepared by conventional methods of blending,
filling or tabletting. Repeated blending operations may be used to distribute
the active agent throughout those compositions employing large quantities of - -
fillers. Such operatlons are, of oourse, conventional in the art.
For parenteral administration, fluid unit dose forms are prepared containing a - ~
compound of the present invention and a sterile vehicle. The compound, ~ -
depending on the vehicle and the concentration, can be either suspended or -
dissolved. Parenteral solutions are normally prepared by dissolving the
2~ compound in a vehicle and filter sterilising befor~ filling into a suitable vial or ~- -
ampoule and sealing. Advantageously, adjuvants such as a local ~ ;-
anaesthetic, preservatives and buffering agents are also dissolved in the
vehicle. To enhance the stability, the composition can be frozen after filling
into the vial and the water removed under vacuum.
Parenteral suspensions are prepared in substantially the same manner
exce~t that the compound is suspended in the vehicle instead of being
dissolved and sterilised by exposure of ethylene oxide before suspending in
the sterile vehicle. Advantageously, a surfactant or wetting agent is included
35 in the composition to facilitate uniform distribution of the compound of the
invention.
WO93~16072 %129 I.~L~ PCI`/GB93/W~14
- 1 0 -
The invention further provides a method of treatment or prophylaxis o~ irritablebowel syndrome, dyspepsia, atnal arrhythmias and stroke, anxiety and/or
migraine in mammals, such as humans, which comprises the administration
of an effective amount of a compound of the formula (I) or a pharmaceutically
5 acceptable salt thereof.
An amount effective to treat the disorders hereinbefore described depends on
the relative efficacies of the compounds of the invention, the nature and
severity of the disorder being treated and the weight of the mammal. ~-
10 However, a unit dose for a 70kg adult will l-.ormally contain 0.05 to 1 OOOmg for
example 0.5 to 500mg, ot the compound of the invention. Unit doses may be -
administered once or n~ore than once a day, for example, 2, 3 or 4 times a
day, more usually 1 to 3 times a day, that is in the range of approximately
0.0001 to 50mg/kg/day, more usually 0.0002 to 25 mg/kg/day.
1 5 ~:
No adverse toxicological effects are indicated within the aforementionsd
dosage ranges.
. "~ .
The invention also provides a compound of formula (I) or a pharmaceutically
acceptable salt thereof for use as an active therapeutic substance, in - ~-
particular for use in the treatment of irritable bowel syndrome, gastro- ~-
oesophageal reflux disease, dyspepsia, atrial arrhythmias and stroke, anxiety
and/or migraine.
The following Examples illustrate the preparation of compounds of formula (I);
the following descriptions relate to interrnediates.
A preferred compound corresponds to any example, but wherein there is an
amino substituent in the 4-position and a chloro substituent in the ~position ofthe benzoic acid-nucleus depicted in forrnula (I). -
.
~ ~ .
WO 93/16072 ~ 2 PCI`/GB93/00214
- 1 1 - ~.
Examples
Rl R2 R3 X1 /X2 Y ~ :
El H H HO-(CH2)2-cH2 ( )
E2 H H HO CH2-CH2 (i~
E3 H Cl H~(cH2)2-cH2
E4 H H H~CH(CH3)2-5CH2)2 (i)
E5 H Cl HO-CH(cH3)2-cH2 (i)
E6 NH2 Cl H~(cH2)2-cH2 (i)
E7 H Cl HO-(CH2)2-CH2 ( ) ;~
E8 H Cl Ho-(cH2)2-cH2 NH (i)
E9 H Cl HO-CH(cH3)2-(cH2)2 (i)
E10 H H HS-(CH2)2-CH2 (i)
E11 H Br HS-(CH2)2~H2 (i)
E12 NH2 Cl H~(cH2)2-cH2 NH (i)
.
WO 93/16072 PCI~GB93/00214
2l29l 12 12-
Example 1
(1 -Butyl~piperidyl)methyl-12tl]-3,4-dihydro-1 -benzopyran~carboxylate ` --
(E1 ) ~
~ -
To a stirr~d solution of [2H]-3,4-dihydro-1-benzopyran-8-carboxylic acid (0.39)
in dichloromethan~ (1 5ml) at 0C under nitrogen, was added oxalyl chlo~ide
- (0.16 ml) and dry dimethylformamide (3m dr~ps3. A~ter 1 hour, ths solvents
were evaporated und~r reduced prassure. The rssidual acid chloride was
dissolved in dry THF (20 ml) and added dropwise to a solution of the lithium
anion of 1-butyl-4-piperidinemethanol in dry THF, prepared by treating a
solution of l-butyl~-pipcridinemethanol (0.299) in dry THF (20ml) with ~-~
butyllithium (1.1 ml of a 1.6 molar solution in hexane), at 0C under nitrogen
for 15 minutes. Aner stirring at ambient temperature overnight, the reaction
mixture was diluted with water, the volatile solvent was evaporated under
reduced pressure, and the produet was extraeted into dichloromethane. The
organie phase was dried (Na2S04) and the solvent was evaporated under
reduced pressure. The residue was chromatographed through a short flash
silica column using an increasing proportion ot methanol in chloroform (1%, --
2%). The product was isolated as the hydrochloride salt trom IPAlEt20 to
; give the title compound (177mg). -
mp 166-8C.
1 H NMR 250MHz (CDCI3) (free base)
~: 7.62(d,1H), 7.19(d,1H), 6.85(t,1H), 4.30(t,2H), 4.15(d,2H), 3.02(d,211),
2.83(t,2H), 2.45-2.25(m,211), 2.1-1.9(m,4H), 1.9-1.65(m,3H), 1.6-1.2(m,6H), -
0.92(t,3H)
WO93J16072 ~ i 2 PCI`/GB93/00214 :~
- 13-
Example 2
(l-Butyl~piperidylpTIethyl-2,3 dihydrobenzofuran~7-carboxylate
hydrochloride (E2)
To a stirred solution of 2,3-dihydrobenzofuran-7-carboxylic acid (D1 ) (0.29) indichloromethane (15ml) at 0C under nitrogsn was added oxalyl chloride
(0.1 2ml) and dry dimethylformamide (3 drops). After 1 hour the solvents were
evaporated under reduced prsssure. Ths r~sidual acid chloride was
10 dissolved in dichloromethane (15ml) and treated with a solution of 1-buty1-4-piperidine methanol (0.219) in dichloromethane (1 Oml) and triethylamine
(0.19ml). A~ter stirring at ambient temperature ovemight, the reaction mixture ~was washed with saturated NaHC03 and the organic phase was dri~d -
(Na2S04). The solvent was evaporated under reduced pressure and the
15 residue chromatographed through a short flash silica column, eluting with
increasing proportions of m~thanol in chloroform (1%, 2%). The product was
isolated as the hydrochloride salt ~rom IPAJEt20 to give the title ~mpound
(84 mg).
20 mp 187-9C
1 H NMR 250MHz (CDCI3) (free bàse)
~: 7.72(d,1 H), 7.35(d,1 H), 6.88(t,1 H), 4.72(t,2H), 4.1 9(d,2H), 3.22(t,2H), 3.1-
25 2.9(d,2H), 2.5-2.25(m,2H), 2.1-1 .2(m, 11 H), 0.92(t,3H)
,
WO 93/16072 PCl`/GB93/00214 ::
~J 1~12
- 14-
Example 3
(l-Butyl~piperidyl)methyl-~chlor~l2H~3,Wlhydr~l-benzopyran~
carboxylat~ (E3)
Following th~ procedure outlined in Example 1 (cxcept that methyllithium was
used in a plac~ of n-butyllithium), 6~hloro-12H]-3,4~ihydro-1-benzopyran-8-
carboxylic acid (300 mg) was converted to the title compound (143 mg, 28%).
1 H NMR 250 MHz (CDCI3)
~: 7.55 (s, 1 H), 7.15 (s, 1 H), 4.29 (t, 2H), 4.15 (d, 2H), 3.05 (brd, 2H), 2.80 (t,
2H)~ 2.5-2.25 (m, 2H), 2.18-1.10 (m, 13H), 0.94 (t, 3H) --
15 Example 4
2,2-Dlmethyl-12H1~3,4-dlhydr~1-benzopyran~(1-butyl~
piperidyl)methylcarboxylate hydrochlorlde (E4)
20 To a solution of 2,2-dimethyl-[2H]-3,4-dihydro-1-benzopyran-8-carboxylic acid (EP-A-307172) (300 mg) in acetonitrile (20 ml) was added N, N'-
carbonyldiimidazole (236mg). Stirring was continued at ambient temperature
for 1 h. The solvent was concentrated in vacuo to afford crude imidazolide.
25 Methyllithium (1.5M in diethyl ether, 0.97 ml) was aWed dropwise to a cooled
(0C) solution of 1-butyl-4-piperidinemethanol (250 mg) in dry THF (10 ml)
under a nitrogen atmosphere. Stirring was continued at room temperature for
10 min. A solution of the irnidazolide in dry THF (1 Oml) was added and
stirring continued ovemight. Water was aWed and the solvent concentrat~d
30 in vacvo. The residue was partitioned between chlorofonn and watar. The
or~anic phasc was dried (Na2S04), filtered and concentrated. The residue
was chromatographed on silica using chloroform and ethanol as eluant to
affo~d pure ester (471 mg) as an oil. Treatment with ethareal HCI and
trituration o~ the resultant gum with diethyl ether gavs title compound as a :
35 solid.
`' `
WO 93116072 ~1 2 9 1 ~ 2 PCI /GB93/00214 :- -
- 15-
1 H NMR 250 MHZ (CDC13)
~: 7.60(d,1H), 7.19(d,1H), 6.82(t,1H), 3.99(d,2H), 2.93-3.03(m,2H), 2.81(t,2H),
2.28-2.37(m,2H),1.61 -2.02(m,7H),1.23-1.56(m,12H inc s,6H), 0.94(t,3H)
E~cample 5
7~1 -Butyl~piperidyl)methyl-~chloro-2,3-dlhydro-2,2-dimethyl-
benzoturancarboxylate (E5)
Following the procedure outlined in Example 4, 5-chloro-2,3~ihydro-2,2-
dimethylbenzofuran-7-carboxylic acid was converted to the title compound.
mp 225-6C (hydrochloride salt)
1 H NMR 250MHz (CDCI3) (free base)
~: 7.65(d,1 H), 7.22 (d,1 H), 4.12(d,2H), 2.92-3.11 (m,4H incl. s2H), 2.28-2.38
(m,2H),1.22-2.0 (m,17H inc s,6H), 0.92 (t,3H)
Example 6
~1-Butyl~piperidyl)methyl-5-amino~chloro-[2H]-3,Wihydro-1- -
benzopyran~carboxylate hydrochloridQ (E6)
Following the procedure outlined in Example 4, ~amino~hloro-12H]-3,4- ~;
dihydro-1-benzopyran-8-carboxylic acid (D4) (200mg) was converted to the
titl~ compound (68mg 19%).
~ ' j , ' ! '
mp 209-213~C
; ~ ~1 H NMR 250 MHz (CDCI3) (free base)
~: 7.7(s,1 H); 4.39(brs, 2H), 4.22(t,2H), 4.10(d,2H), 3.05~brd,2H), 2.6-
2.25(m,4H),2.2-1.93(m,4H),1.9-1.68(m,3H),1.65-1.2(m,6H), 0.92(t,3H)
.
WO 93/1607~ PCI /GB93/00214
Example7 212~3 il2 16 -
(1-Pip~ridy~)ethyl~chloro-12H]-3,4 dihydro-l-benzopyran~carboxylate
hydrochloride (E7)
Following the procedure outlined in Example 1, except that MeLi was used in
place of butyllithium, 6-chlor~-[2H]-3,4~ihydro-1-benzopyran-8-carboxylic
acid (390mg) was converted to the title compound (1 82mg, 28%) mp 185-
7G.
1Q
1 H NMR 250 MHz (CDCI3) (free base)
~: 7.58(s,~H), 7.15~s,1H), 4.4(t,2H), 4.29(t,2H), 2.88-2.65(m,4H), 2.6-
2.35(m,4H), 2.15-1.9(m,2H), 1.7-1.3(m,6H)
Example 8
(1-Butyl~piperidyl)methyl-6~chloro-[2H]-3,4 dihydro 1-benzopyran~
~0 carboxamlde hydrochloride (E8)
A solution of 6-chloro-12Hl-3,4-dihydro-1-benzopyran-8-carboxylic acid
(200mg) in dry dichloromethane (6ml) was treated with oxalyl chloride (0.13
ml) and dry dimethylformamide (2 drops) under nitrogen. After stirring for four ;~
25 hours at ambient temperature the solvent was evaporated under reduced
pressur~. The resulting acid chloride was dissolved in dry dichloromethane
(6ml) and added to a stirring solution of 1 -butylpiperidin-4-ylmethylamine i
(160mg) in dry dichloromethane (5ml) containing triethylamine (0.14 ml).
Af~er stirring at ambient tempera~ure ovemight under nitrogen, the reaction --~
30 mixture was washed with NaHC03 and the organic phase dried over
Na2S04. The sohient was evaporated under reduced pressure tb ~ive the
product which was purifiad using column chromatography (SiO2,
methanol/chloroform), and converted to the hydrochloride salt ~E61 ) ~1 38mg,
37%) mp 227-9C.
WO 93/16072 ~ 1 2 9 1 1 2 PCr/GB93/00214
- 17 -
H NMR 250 MHz (CDGI3) (free base)
~: 7.98(s,1H), 7.12(s,1H), 4.35(t,2H), 3.35(t,2H), 3.08(brd, 2H), 2.85(t,2H),
2.5-2. 3(m,2H), 2.2-1.92(m,4H),1.9-1.2(m,9H), 0.92(t,3H)
Example 9
6-Chloro-2,2-dimethyl-[2H]-3,4dihydro~1 -benzopyran~(l-butyl~ -~
piperidyl)methyl carboxylate hydrochloride (E9) ~ -
Following the procedure outlined in Example 4,6~hlor~2,2-dimethyl-[2H~-3,4-
dihydro-l-benzopyran-8-carboxylic acid (D5) (19) was converted to the title
compound (310mg)
1 H NMR 250 MHz (CDCI3) (free base)
, .
~: 7.55(d,1H), 7.16(d,1H), 4.13(d,2H), 2.98(bd, ~H), 2.79(t,2H), 2.32(t,2H),
1.68-2.00(m,7H),1.24-1.54(m,12H inc s,6H), 0.92(t,3H)
Example 10
(1-Butyl 1 piperidyl)methylthiochroman~carboxylate (E10)
This was pr~pared according to the general method described in Exampie 1.
Thus thiochroman-8~arboxylic acid (0.160g, 0.825 mmol) (D6) was
converted to th~ title compound (0.1459, 53%) which was subsequently
transformed into its hydrochloride salt m.pt 157-158C.
1 H I~MR (250 MHz, C:WI3) (frse base),
~; -
~ : 7.82 (d,1H), 7.17 (d,1H), 7.00 (t,1H), 4.15 (d, 2H), 3.00 (m, 4H), 2.87 (t,
2H), 2.38 (t, 2H), 2.20-1.95 (m, 5H),1.80 (m, 2H),1.50 (m, 4H),1.30 (m, 2H),
35 0.90 (t, 3H).
~:
WO 93/16072 ~ PCl`/GB93/00214
~ ~ "
- 18-
Example 1 1
~1-Butyl~piperididyl)methyl~-bromothiochroman~carboxylate (E11 )
5 This was prepared according to the general method dsscribed in Example 1.
Thus 6-bromothiochroman-8-carboxylic acid (0.3189, 1.17mmol) (D7) was
comferted to the title compound (0.21 7g, 44%) which was subsequently ~ -
transformed into its hydrochlorids salt. m.pt. 197-198. --
1 H NMR (200 MHz, CDCI3) (free base)
~: 7.90 (d, 1 H), 7.30 (d, 1 H), 4.20 (d, 2H), 3.00 (m, 4H), 2.88 (t, 2H), 2.32 (t,
2H), 2.10 (m, 2H), 2.00-1.70 (m, 4H), 1.5~1.20 (m, 7H), 0.92 (t, 3H).
1 5 ~;
Example 12
5-Amino~ butyl-4piperidyl)methyl-6-chloro-[2Hl-3,Wlhydro~
benzopyran~carboxamide (E12)
A solution of 5-amino-6-chloro-12H]-3,4-dihydro-1-benzopyran-8-carboxylic
acid (D4, 200mg ) in acétonitrile (6ml ) was treated with bis-
carbonyldiimidazole (171 mg) and the resulting solution was stirred at room
temperature for 2 hours. A solution of (1-butyl-4-piperidyl) methylamine
25 (150mg) in acetonitrile (10ml) was added and the reaction mi)nure was stirr~dat room temperature for 16 hours. The solvent was removed in vacuo and
the residue partitioned between water and dichloromethane. The
dichloromethane layer was removed and the aqueous extracted further with
dichloromethane. The organic extracts were combined and washed with
30 water, then dried (Na2SO4) and concsntrated to give a yellow gum that was
punfied by column chromatography on silica using CHCI3 with increasing
quantities of MeOH as eluant to give the title compound as an off white solid,
30mg mp65-6C.
35 1 H NMR (250 MHz CDCI3)
~: 8.0 (s,1H), 7.96-7.8 (m,1H), 4.45-4.15 (m,4H,) 3.32 (t,2H), 3.02 (d,2H), 2.53(t, 2H), 2.39 (t,2H), 2.2-1.9 (m,4H), 1.86-1.15(m,9H), 0.92 (t,3H)
WO 93~16072 ~ 1 ~ 9112 PCl/GB93/00214
- 19-
Descriptions
Description 1 (intermediate for Example 2)
[2H]-3,4Dihydro-1-benzopyran~carboxylic acid
Following the procedure outlined in EP-A-307172 Example 16, [2Hl-3,4- -
dihydro-1-benzopyran tO.85g) was converted to the title compound (D1) -;:
1 0, (0.779~
1 H NMR (200MHz)(CDCI3)
~: 8.0(d,1 H), 7.3(d~ 1 H), 7.0(t,1 H), 4.45(t,2H), 2.89(t,2H), 2.25-2.0(m,2H~
Description 2 (intermediate for Example 2)
2,3-Dihydrobenzofuran-7~carboxylic acid
Following the procedure outlined in EP-A-307172, Example 15, 2,3-
dihydrobenzofuran (0.59g) was converted to the title compound (D2) (0.41g)
1 H NMR 250MHz (CDCI3)
~: 7.82(d,1 H), 7.44(d,1 H), 6.96(t,1 H), 4.80(t,2H), 3.30(t,2H)
WO 93/16072 212 !~ I 1 2 PCI`/GB93/00214
- 20 -
Description 3 (intermediate for Example 3)
6-Chloro-[2Hl-3,4-dihydro-1-benzopyran~carboxylic acid
5 A solution of ~2H]-3,4-dihydro-1 -benzopyran-8-carboxylic acid (D1 ) (150mg) in
glacial acetic acid (1 Oml) was tr~ated with a solution of 1.3 equivalents of
chlorine ~80mg) in glacial acetic acid (2.8 mls) dropwise with ice-cooling. ~ -
A~er stirring ovemight at ambient temp~rature the solvents were evaporated
under reduced pressure, and the residue triturated with diethyl ether to give
10 th~ title compound (D33 (64mg, 36%).
1 H NMR 200 MHz (CDCI3)
~: 10.78 (brs, 1 H), 7.95 ~s, 1 H), 7.25 (s, 1 H~, 4.45 (t, 2H), 2.88 (t, 2H), 2.2~ -~
15 2.Q (m, 2H)
Description 4 (intermediate for Example 6)
20 a) Methyl(4acety1amino-2-propargyloxy)benzoate
A solution of methyl-4-acetylamino-2-hydroxybenzoate (prepared as
described in EP-A-234872) (5 9) in a mixture of dry tetrahydrofuran (100 ml)
and dry dimethylformamide (150 ml) was treated with 1 equivalent of sodium
2~ hydride (~.72g of an 80% dispersion in oil). After stirring for 1 hour under
nitrogen, 2.5 equivalents of propargylbromide (5.33 ml) were added, and the
mixture was heatsd under r~flux for three days. The solvents were
evaporated under reducsd pressure and the residue partitioned between 10%
sodium hydroxide and ethyl acetate. The organic phasa was dried over ~-
30 Na2S04 and evap~rated under r~duced pressure to give a red oil which, a~ter
trituration with 60-80 petrol-ether gavs the title compound as a light tan
powder (4.959, 84%)
1 H NMP~ 200 MHz (CDCI3)
~: 7 91 (brs, 1 H), 7.81 (d,1 H), 7.68(s,1 H), 7.05(d,1 H), 4.78(d,2H), 3.86(s,3H),
2.54(m,1 H), 2.2(s,3H)
WO 93/16072 21 2 9 1 1 2 PCI`/GB93/00214
- 21 -
b) Methyl(5~acetamido-[2H~ benzopyran)~carboxylate
A solution of methyl(4-acetamido-2-propargyloxy)benzoate (6.38g) in 1,2-
dichlorobenzene (65ml) was heated under reflux under nitrogen for 60 hours.
5 The solvent was evaporated under reduced pressure and the residue purified
on a silica column, aluting with methanol/chloroform, to give the title
compound as a tan solid (3.479, 54%)
1 H NMR 200 MHz (CDCI3)
~: 7.79(brs, 1 H), 7.64(d,1 H), 7.3(d,1 H), 6.46(d,1 H), 5.85(m,1 H), 4.75(brs,2H),
3.85(s,3H), 2.20(s,3H)
c) Methyl(5-scetamido~l2H]-3,4 dihydro-l-benzopyran)~
1 5 carboxylate
A solution of methyl (5-acetamido-12H]-1-benzopyran)-~carboxylate (D23)
(770mg) in ethanol, was hydrogenated over 10% palladium on charcoal at
atmospheric pressure for 1.25 hours. The reaction mixture was filtered and
20 the filtrate evaporated under reduced pressure to giv~ the title compound as a
white powder (670 mg, 86%)
1 H NMR 250 MHz (CDCI3)
.
25 ~: 7.68(d,1 H), 7.49(brs, 1 H), 7.1 9(brs,1 H), 4.22(t,2H), 3.88(s,3H), 2.62(t,2H),
2.20(s,3H), 2.1-1.96(m,2H)
dj Methyl(5-acetamldo-6-chloro-l2H~ dihydro-1-benzopyran~
carboxylate
Following the procedure outlined in Description 3, methyl(5-acetamido-[2H]^
3,4-dihydro-1-benzopyran)-8-carboxylate (660mg) was converted to the title
compound which was isolated as a light tan powder ~525mg, 70%)
.
35 1 H NMR Z50 MHz (CDCI3)
~: 7.7(s,1H), 7.15(s,1H), 4.29(t,2H), 3.89(s,3H), 2.70(t,2H), 2.25(s,3H), 2.05-
1 .9(m,2H)
Wo 93tl6072 PCr/GBs3/00214 ~:
2129112 -22-
e) ~Amino-6-chloro-[2H]~ dihydro-l-benzopy~n-8~carboxylic
acid :
A solution of methyl (5-acetamido-6-chloro-12H]-3,4-dihydro-1-benzowran-8- -
carboxylate (1.0159) in ethanol (20 ml) water (10 ml) and 10% sodium :;
hydroxide (30 ml) was heated under reflux for 24 hours, then cooled and
treated with concentrated hydrochloric acid (until pH 2) and the resulting ;:
pr~cipitate was filtered off to give the title compound (D4) (427mg, 52%) :~
10 -
1 H NMR 250 MHz (CDCI3)
~: 7.85(s,1 H), 4.34(t,2H), 2.54(t,2H), 2.25-2.0(m,2H) ;
Description 5 (intermediate for Example 9)
,
6-Chloro-2,2-dimethyl-[2H]-3,4dihydro-l-benzopyran~carboxylic acid
Following the procedure outlined in Description 3, 2,2~imethyl-12H]^3,4- ~-
dihydro-l-benzopyran-8-carboxylic acid (EP-A-307172) (2.419) gave the title
compound (D5) (2.689). M+ 240 (E1 ) :-
WO 93/16072 212 911 ~ PCI`IGB93/00214
- 23 -
Description 6 (intermediate for Example 10)
a) Methylthiochro Tlan~one~carboxylate
~2-carbomethoxythiophenoxy) propionic acid (6.00g, 0.025mol) (I.?IY. Still
and M.~. Thomas J.Org. Chem 1968, 2733) was added slowly to ice cooled
conc. sulphuric acid (75ml) with stirring. After 21 h, the reaction mixture was
poured into ice water and then made alkaline using solid sodium hydrogen
carbonate. The resultant suspension was then extracted with CH2CI2(3X~.
The combined organic layers were then dried (Na~SO4), and evaporated
under reduced pressure to give an orange oil which was dried in vacuo and
crystallised on standing to give (2.40g, 43/O).
b) Methylthiochroman~ol~carboxylate
Methylthiochroman-4-one-8-carboxylate (0.5009, 2.25mmol) was dissolved
with stirring in ethanol (20 ml). After 1 h, the reaction mixture was evaporatedunder reduced pressure and the residue partitioned between ethyl acetate
and water. The aqueous layer was then extracted with ethyl acetate (1 X) and
the combined organic layers were dried (Na2S04) and evaporated under
reduced pressure to give a yellow oil, which was purified by silica-gel
chromatography (1 :1 Pentane:EtOAc as eluant3 to give the title compound as
a colourless oil (0.4999, 99%)
1H NMR (200 MHz, CDCI3) S: 7.95 (dd,1H), 7.55 (dd,1H), 7.12 (t? 1H), 4.87 -
(m, 1 H), 3.92 (s, 3H) 3.23 (m,1 H), 2.88 (m,1 H), 2.37 (m,1 H), 2.08 (m,1 H),
1.92 (d,1 H).
c) Methyl-2H-thiochromene~carboxylate
-~
Mcthyl thiocliroman-4-ol-8icarboxylate (0.3379,1.50 mmol) was dissolved in
toluene (25ml) and was treated with p-toluenesulphonic acid (0.0289, 0.15
mmol). The mixture was then heated to reflux with stirring. A~ter 2h, the
reaction mixture was allowed to cool and was washed with sodium hydrogen `
carbonate solution. The aqueous layer was then extracted with EtOAc (1 X),
and the combined organic layers were dried (Na2SO4) and evaporated under -
reduced pressure to give a pale yellow oil which was purified by silica-gel
; chromatography (pentane: Et2O 2:1 as eluant) to give the title compound
WO 93/16072 2 1 2 ~ ~ 1 2 PCI/GB93/00214
- 24 -
(D2b) as a pale yellow oil (0.270g, 87%)
1H NMR (200 MHz, CDCI3, ~: 7.80 (dd, lH), 7.20 (dd, 1H), 7.10 (t, 1H), 6.52
(d, 1 H), 6.02 (m, 1 H), 3.92 (s, 3H), 3.48 (dd, 1 H).
d) Methylthiochroman~carboxylate
Methyl-2H-thiochromene-8-carboxylate (1.83g, 8.88 mmol) was dissolved in
ethanol (100ml) and treated with 10% PdC (1 .5g). Th~ mixturs was then
10 hydrog~nated at atmospheric pressurs at room temperature. Af~er 19h the
reaction mixtur~ was filtsred through celite and evaporatad under r~duced
pressure to giv~ a colourless oil which was driad in v~cuo to give the title
compound (1.259, 68%)
1 H NMR (200 Ml Iz, CDCI3), ~ 7.80 (d, 1 H), 7.62 (d, 1 H), 6.97 (t, 1 H~, 3.89 (s,
3H), 2.97 (t, 2H), 2.87 (t, 2H), 2.12 (m, 2H).
e) Thiochroman~carboxylic acid
20 Methylthiochroman-8 carboxylate (0.2209, 1.0~mmolj was dissolved in
ethanol (5ml) and treated with 10% sodi"m hydroxide solution (1 Oml). Ths
mixture was then heated to reflux with stirring. After 5h, the reaction mixture
was allowed to cool. The ethanol present was then removed by evaporation
under reduced pressure. The aqueous residue was then washed with
25 CH2CI2 ~2X) be~ore being acidified to pH1 using 5M HCI. The resultant pale
yellow prscipitate was then filter~d off and dried in vacuo to give the title
compound (0.1569, 76%) (D6).
1 H NMR (200 MHz, Ct)30D)
~: 7.80 (d, 1 H), 7.2d (d, 1 H), 7.00 (t, 1 H), 2.90 (m, 4H), 2.08 (m, 2H).
wo 93/16072 ~12 91 ~ 2 pcr/Gs93/oo2~4
- 25 -
Description 7 (intermediate for Example 11 )
a) M~thyl ~bromothiochroman~carboxylate
5 A solution of methylthiochroman-8 carboxylate (0.3009, 1.44 mmol) (D6a) in
dichloromethane (20ml) was treated with bromin~ (0.106 ml, 2.07 mmol) and
the r~action mixture left at room temperature. After 4 days the r~action
mixture was washed with sodium metabisulphile sollnion. Ths organic layer
was then dned (Na2S04) and ~vapor~ted under reduced pressure to give the
10 title compound as a colourless oil (0.3409, 82%).
1 H NMR (250 MHz, CDCI3) ~: 7.92 (d, 1 H), 7.30 (d, 1 H), 3.92 (s, 311), 3.00 (t,
2H), 2.88 (t, 2H), 2.10 (m, 2H).
b) ~Bromothiochroman~carboxylic acid -
This was prepared according to the general method described in Description
6e). Thus, methyl-6-bromo-thiochroman (0.3259, 1.1 3mmol) was converted
to the title compound (0.3069, 99%) (D7).
-:
1 H NMR (200MHz, CD3SOCD3): 13.25 (brs, 1 H), 7.82 (d, 1 H), 7.50 (d, 1 H),
2.92 (m, 4H), 2.00 (m, 2H).
:~'
.
,'
':
;; .
.
-:'
WO 93/16072 PCI/GB93/00214
212!3 1 12
- 2~ -
5~HT4 RECEPTOR ANTAGONIST ACTIVITY
1) Guinea pig colon
Male guinea-pigs, weighing 250-4009 are used. Longitudinal muscle-
myenteric plexus preparations, approximatsly 3cm long, ars obtainsd from
the distal colon region. These are suspended under a 0.59 load in isolated
tissus baths containing Krebs solution bubbled with 5% CO2 in 2 and
maintained at 37C. In all experiments, the Krebs solution also contains
methiothepin 1 o-7M and granisetron 1 o-6M to block effects at 5-HT1, 5-HT2
and 5-HT3 r~ceptors.
After construction of a simple concentration-r~sponse curve with 5-HT, using
30s contact times and a 1 5min dosing cycle, a concentration of 5-HT is ~ -
selected so as to obtain a oontraction of the muscle approximately 40-70%
maximum(10~9M approx). The tissue is then altemately dosed every 1 5min
with this cor~entration of 5-HT and then with an approximately equi-effsctive ~ i
concentration of the nicotine receptor stimulant, dimethylphenylpiperazinium
(DMPP). After obtaining consistent responses to both 5-HT and DMPP,
increasing concentrations of a putative 5-HT4 receptor antagonist are then
added to the bathing solution. The effects of this compound are then
determined as a percentage reduction of the contractions evoked by ~HT or
by DMPP. From this data, plCso values are determined, being defined as the
-log concentration of antagonist which reduces ths contraction by 50%. A ~ -
compol~nd which reduces the responss to 5-HT but not to DMPP is believed
to act as a 5-HT4 receptor antagonist.
Compounds were generally active in the range of concentrations of the order
of plCso=7 or more, E3, E8 and E12 showing particularly good activity.
- 2) Piglet Atria
Compounds were tested in the piglet spontaneous beating screen (Naunyn-
35 Schmiedeberg's Arch. Pharmacol 342, 619-622). PKB (-log10 Kg) value for
the compound of Examples 3 and 7 were 9.8 and 7.7 respectively.
O 93/16072 PCl/GB93/00214
J~l~91 ~2
- 27 -
3) Rat oesophagus
Rat oesophageal tunica muscularis mucosae is set up according to Baxter et.
aL Naunyn-Schmiedeberg's Arch. Pharmacol., 343, 439-446 (1991). The
5 inner smooth muscle tube of the muscularis mucosae is isolated and mounted
~or isometric tension recording in oxygenated (95% 2/5% CO2) Tyrodes
solution at 37C. All experiments are performcd in pargyline pre-treated
preparations (1 OO~,lM for 15 min followed by washout) and in the presence of
cocaine (30~M). Relaxant responses to 5-HT are obtainsd after pre-
contrac~ing the oesophagus tissue with carbachol (311M). ;:
4) 5-HT-induced motility in dog gastric pouch
~.
Compounds are tested in the in vivo method described in "Stimulation of ;
canine motility by BRL 24924, a new gastric prokinetic agent", Bermudez et `-~
al, J. Gastrointestinal Motility, 2(4), 281-286.
The compound E3 showed inhibition at a dose of 10~19 kg~
:-