Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02130515 1998-02-19
1
(a) TITLE OF THE INVENTION
11-BENZALDOXIME-17B-METHOXY-17a-METHOXYMETHYL-
ESTRASDIENE DERIVATIVES, PROCESSES FOR THEIR PRODUCTION
AND PHARMACEUTICALS CONTAINING SUCH COMPOUNDS
(b) TECHNICAL FIELD TO WHICH THE INVENTION BELONGS
This invention relates to new 11-benzaldoxime-estradiene derivatives, to
processes
for their production, to pharmaceuticals containing these compounds, and to
uses of the
compounds.
(c) BACKGROUND ART
11B-substituted phenyl estratrienes are known. Patent specification EP 057 115
describes the production of 11B-aryl-17a-propinyl-estra-4,9-dimes, and Patent
specification DE 3 504 421 describes the reaction of 11B-(4-formylphenyl)-
estra-4,9-
dime-3-ons with hydroxylamines. Both the 11B-formyl phenylene residue and the
3-keto
group are oximated. In addition, syn-and anti-isomers are formed at C-3.
Nothing is
known as yet about any physiological effects of the described compounds.
Progesterone is secreted during menstruation, and in large amounts by the
ovary
and the placenta during pregnancy. Its regulatory significance has perhaps not
been
clarified in every respect.
What is safely known is that progesterone, together with oestrogenes, produces
the cyclic changes in the uterine mucosa during the menstrual cycle and
pregnancy.
After ovulation, an increased level of progesterone causes the uterine mucosa
to adopt
a condition that permits the embedding of an embryo (blastocyst). Conservation
of the
tissues in which the embryo grows is also dependent on progesterone.
CA 02130515 1998-02-19
2
A dramatic change in the muscular function of the uterus takes place during
pregnancy. Response of the gravid uterine muscle to hormonal and mechanical
stimuli
that induce labour in the non-gravid state is strongly reduced or is non-
existent. There
can be no doubt that progesterone has a key function here, despite the fact
that at certain
stages of pregnancy, e.g., shortly before giving birth, there is a high
reactivity even at
high blood-progesterone concentrations.
Very high progesterone levels are also reflected by other typical processes
during
pregnancy. The composition of the mammary glands and the obstruction of the
cervix
until shortly before the date of birth-giving may serve as examples of this.
Progesterone contributes subtly to controlling ovulation processes. It is
known
that high doses of progesterone have anti-ovulatory qualities. They result
from an
inhibition of the hypophyseal gonadotropin secretion which is a prerequisite
for the
maturation of the follicle and for its ovulation. On the other hand, it can be
seen that
the comparatively-small quantity of progesterone which is secreted by the
maturing
follicle plays an active part in preparing and triggering ovulation.
Hypophyseal
mechanisms (temporary, so-called positive feedback of progesterone to
gonadotropin
secretion) appear to have a great significance in this respect (See,
Loutradie, D.; Human
Reproduction 6, 1991, 1238-1240).
The doubtlessly-existing functions of progesterone in the maturing follicle
and
luteal corpus themselves have been less well analyzed. It can be assumed,
eventually,
that there are both stimulating and inhibiting effects on endocrinic functions
of the follicle
and the luteal corpus.
It may also be assumed that progesterone and progesterone receptors are of
great
importance for pathophysiological processes. Progesterone receptors have been
found
not only in endometriotic focuses, but also in tumours of the uterus, the
mamma, and the
CNS (meningiomas). The role of these receptors in conjunction with the growth
behaviour of these pathologically-relevant tissues is not necessarily
dependent on
progesterone levels in the blood. It has been proved that substances which are
characterized as progesterone antagonists, e.g., RU 486 = Mifepristone (EP-0
057 115)
and ZK 98299 = Onapristone (DE-OS-35 04 421) tend to trigger far-reaching
functional
CA 02130515 1998-02-19
3
changes even at negligible levels of progesterone in the blood. It appears to
be possible
that modifications of the transcriptional effects of the progesterone receptor
that is not
filled with progesterone are decisive in this respect (See, Chwalisz, K. et
al.,
Endocrinology, 129, 317-322, 1991).
The effects of progesterone in tissues of the genitals and in other tissue are
brought about by interaction with the progesterone receptor. In a cell,
progesterone
bonds to its receptor with high affinity. This causes changes in the receptor
protein;
conformational changes, dimerization of 2 receptor units to form one complex,
baring
of the receptor's DNA bonding place by dissociating a protein (HSP 90), and
bonding
to hormone-responsive DNA elements. Eventually, the transcription of certain
genes is
regulated. (See, Gronemeyer, H. et al., J. Steroid Biochem. Molec. Biol. 41, 3-
8,
1992) .
The effect of progesterone or progesterone antagonists does not depend only on
their concentration in the blood. The concentration of receptors in a cell is
strongly
regulated as well. Oestrogens stimulate the synthesis of progesterone
receptors in most
tissues. Progesterone inhibits the synthesis of oestrogen receptors and that
of its own
receptor. It is assumed that this interaction of oestrogens and gestagens goes
to explain
why gestagens and antigestagens can influence oestrogen-dependent processes
without
being bonded by the oestrogen receptor. These relations are naturally of great
importance for the therapeutical application of antigestagens. These
substances appear
to be appropriate for directly influencing female reproductive processes,
e.g., for
preventing nidation after ovulation, or for increasing uterine reactivity to
prostaglandins
and oxytocin in a later pregnancy, or for achieving metreurysis and cervix
softening
("maturing").
Antigestagens inhibit ovulation in various species of subhuman primates. The
mechanism of this effect has not yet been fully elucidated. Among the
hypotheses
discussed are an inhibition of gonadotropin secretion, and ovarian mechanisms
based on
disturbing para- and autocrinic functions of progesterone in the ovary.
CA 02130515 1998-02-19
4
Antigestagens are capable of modulating or weakening the effects of
oestrogens,
although the majority of them do not have any oestrogen receptor affinity at
the cyto-
plasmic level, and although they can cause an increase of the oestrogen
receptor
concentration. Similar effects in endometriotic focuses or tumorous tissue
equipped with
oestrogen and progesterone receptors justify the expectation of a favourable
influence on
pathologic conditions. Particular advantages with regard to exerting a
favourable
influence on pathologic conditions, e.g., endometriosis, might be achieved if
an inhibited
ovulation supplemented the inhibiting effects of an antigestagen acting in the
tissue.
Ovarian hormonal products and their stimulating effect on the pathologically
altered
tissue would also be reduced by inhibiting ovulation. It would be desirable to
inhibit
ovulation in severe cases of endometriosis to bring the tissue in the genital
tract which
would normally be in constant reconstruction, into a reversible state of rest.
A proposed contraception method is one according to which an antigestagen
treatment suppresses ovulation, and secretory transformation of the
endometrium is
induced by subsequent gestagen treatment. The days of treatment with
antigestagens and
gestagens and the treatment-free days result in a 28-day cycle with a regular
withdrawal
bleeding (See, Baulieu, E.E., Advances in Contraception 7, 345-51 1991).
Antigestagens can have different hormonal and anti-hormonal properties. Anti-
glucocorticoid properties are of particular therapeutical relevance. These are
unfavourable for therapeutical applications mainly aimed at inhibiting
progesterone
receptors as they have undesired side effects when applied at the dosage which
are
required for such therapy which may prevent the application of a
therapeutically sensible
dose, or require that treatment be discontinued.
(d) DESCRIPTION OF THE INVENTION
An object of a main aspect of this invention is the provision of 1113-
benzaldoxime-
estra-4,9-diene derivatives which show partial or complete reduction of
glucocorticoid
properties, which is an important prerequisite for a therapy using
antigestagens,
especially with indications that require therapy over several weeks or months.
CA 02130515 2000-07-28
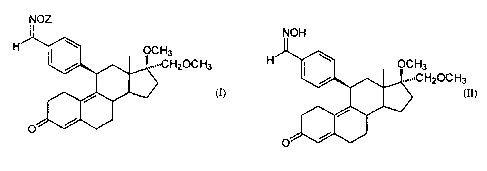
By one broad aspect, the present invention, provides a 1113-benzaldoxime-estra-
4,9-
diene derivative of the general Formula I
3
H20C H3
(I)
where Z is -CO-CH3, -CO-O-CZHS, -CO-NH-phenyl, -CO-NH-CZHS, -CO-CZHS, -CH3, or
CO-phenyl, or their pharmaceutically-acceptable salts.
By one variant thereof, the salt is derived from a physiologically-compatible
inorganic acid or organic acid. By one variation thereof, such physiologically-
compatible
acid is selected from the group consisting of hydrochloric acid, hydrobromic
acid,
phosphoric acid, sulphuric acid, oxalic acid, malefic acid, fumaric acid,
lactic acid, tartaric
acid, malic acid, citric acid, salicylic acid, adipic acid, and benzoic acid.
Preferred compounds according to aspects of this invention are the following:
1113-[4-(acetoximinomethyl)phenyl]-1713-methoxy-17a-methoxymethyl-estra-4,9-
diene-3-on;
1113 {4-[(ethoxycarbonyl)oximinomethyl]phenyl}-1713-methoxy-17a-methoxymethyl-
estra-4,9-diene-3-on;
CA 02130515 2000-07-28
6
1113-{4-[(ethylaminocarbonyl)oximinomethyl]phenyl}-1713-methoxy-17a-
methoxymethyl-estra-4,9-dime-3-on;
17(3-methoxy-17a-methoxymethyl-1113-{4-[(phenylaminocarbonyl)oximino-
methyl]phenyl}-estra-4,9-dime-3-on; and
1113-[4-(propionyloximinomethyl)phenyl]-1713-methoxy-17a-methoxymethyl-estra-
4,9-dime-3-on.
This invention, in another aspect, provides a process for producing compounds
of the
general Formula I and their pharmaceutically acceptable salts, which comprises
the
esterification, or the etherification, or the urethane formation of a compound
of the general
Formula II
(II)
3
H20C H3
CA 02130515 2000-07-28
by the reaction of a compound of the general Formula II with an esterifying,
etherifying,
or urethane-forming agent which contains the group Z, or its precursor.
By a variant thereof, the esterification reaction in carried out using an
acylating
agent. By a variation thereof, the esterification is carried out in the
presence of a base,
using an acid anhydride or an acid chloride, and preferably where the base is
pyridine. By
another variation thereof, the etherification is carried out using methyl
iodide in the
presence of a base, and preferably where the base is potassium tert.
butanolate.
By yet another variant thereof, the esterification is carried out using
diazomethane in
methanol.
By still another variant thereof, the urethane formation is achieved by
reacting with
an alkyl isocyanate, or with an aryl isocyanate, in an inert solvent. By a
variation thereof,
the inert solvent is toluene.
By a still further variant thereof, the urethane formation is achieved by
reacting a
carbamoylchloride in the presence of a base. By a variation thereof, the base
is
triethylamine.
By a variant of the above variants and variations, the process includes the
step of
forming a pharmaceutically-acceptable salt thereof. By one variation thereof,
the salt is
prepared by reacting a compound of Formula I with a pharmaceutically-
acceptable acid in
an organic solvent. By another variation thereof, the pharmaceutically-
acceptable acid is
selected from the group consisting of hydrochloric acid, hydrobromic acid,
phosphoric
acid, sulphuric acid, oxalic acid, malefic acid, fumaric acid, lactic acid,
tartaric acid, malic
acid, citric acid, salicylic acid, adipic acid, and benzoic acid. By other
variations thereof,
the solvent is a lower alcohol, e.g., methanol, ethanol, n-propanol or
isopropanol, or is a
lower ketone, e.g., acetone, methylethyl ketone or methyl isobutyl ketone, or
is an ether,
e.g., diethyl ether, tetrahydrofurane or dioxane.
By another variant thereof, the salt is prepared by converting an acid-
addition salt of
the compound of Formula I into a free base, and obtaining another acid-
addition salt by
reacting this free base with an inorganic acid or with an organic acid. By a
variation
thereof, the conversion of the acid-addition salt of the compound of Formula I
into a free
base is carried out using an alkali or an ion exchanger. By yet another
variation thereof,
the acid is selected from the group consisting of hydrochloric acid,
hydrobromic acid,
CA 02130515 2000-07-28
phosphoric acid, sulphuric acid, oxalic acid, malefic acid, fumaric acid,
lactic acid, tartaric
acid, malic acid, citric acid, salicylic acid, adipic acid, and benzoic acid.
Specific processes according to other aspects of this invention are ones
wherein group
Z is selected so as to produce 11-B-[4-(acetoximinomethyl)phenyl]-17B-methoxy-
17a-
methoxymethyl-estra-4,9-dime-3-on; or wherein group Z is selected so as to
produce 11B-
{4-[(ethoxycarbonyl)oximinomethyl]phenyl}-17B-methoxy-17a-methoxymethyl-estra-
4,9-
diene-3-on; or wherein group Z is selected so as to produce 11B-{4-
[(ethylaminocarbonyl)-
oximinomethyl}phenyl}-17B-methoxy-17a-methoxymethyl-estra-4,9-dime-3-on; or
wherein
group Z is selected so as to produce 17B-methoxy-17a-methoxymethyl-11B-{4-
[(phenyl-
aminocarbonyl)oximinomethyl]phenyl}-estra-4,9-diene-3-on; or wherein group Z
is
selected so as to produce 11B-[4(propionyloximinomethyl)phenyl]-17B-methoxy-
17a-
methoxymethyl-estra-4,9-diene-3-on.
By yet another aspect of this invention, a pharmaceutical composition is
provided
comprising a pharmaceutically-effective concentration of a compound as
described
hereinabove, or salts or mixtures thereof, in a pharmaceutically-acceptable
carrier.
By another aspect of this invention, the use is provided of a compound as
described
above, or salts or mixture thereof, as an antigestogeric agent with reduced
glucocorticoid
activity.
By another aspect of this invention, the use is provided of a compound as
described
above, or salts or mixture thereof, for the preparation of a pharmaceutical
composition for
use as an antigestogeric agent with reduced glucocorticoid activity.
The manufacturing of the compounds of the general Formula I of aspects of this
invention by esterification, etherification, or urethane formation, according
to other aspects
of this invention, may be carried out in a generally-known way using acylating
agents,
e.g., acid anhydrides or acid chlorides, in the presence of bases, preferably
pyridine; or by
etherification using methyl iodide in the presence of bases, preferably
potassium tert.
butanolate, or using diazomethane in methanol; or by urethane formation by
reacting with
alkyl or aryl isocyanates in inert solvents, preferably toluene; or by
reacting
carbamoylchlorides in the presence of bases, preferably triethylamine.
CA 02130515 1998-02-19
9
The parent compound of the general Formula II is manufactured from a 5a, l0a-
epoxide of the general Formula III
0
(III)
H3C0
H3C0
[cf., for example, Nedelec Bull. Soc. chim. France (1970), 2548].
The introduction of the phenyl residue to the 1113 position while forming a
09(10),Sa hydroxy structure of the general Formula IV (below) is achieved by a
Cu(I)
salt-catalyzed Grignard reaction (See, Tetrahedron Letters 1979, 2051) with a
p
bromobenzaldehyde ketal, preferably p-bromobenzaldehyde dimethyl ketal, at
temperatures between 0°C and 30°C.
OCHj
H3C0
H
(IV)
H3~
H3~"
OH
CA 02130515 1998-02-19
The -CHZ-0-CH3 group is introduced to the 17 position in a generally-known way
via the spiroepoxide of the general Formula V, by reacting with trimethyl
sulphonium
iodide and potassium tert. butanolate in dimethyl sulphoxide [See, Hubner et
al.; J.
prakt. Chem. 314, 667 (1972); Arzneim. Forsch. 30, 401 (1973)];
5
H CO OCH.j
3
H
(V)
H3i
H3~" '
OH
CA 02130515 1998-02-19
11
and subsequent ring opening using alcoholates [See, Ponsold et al. ; Z. Chem.
11, 106
(1971)]. The resulting 17a-CHZ-0-CH3 compounds of the general Formula VI
OCH;
H3C0
H
:H2-0-~H3
(VI)
H3~
H3C _
OH
may either be decomposed into their respective aldehydes by acid hydrolysis,
preferably
using toluene-p-sulphonic acid in acetone (See, Teutsch et al. DE 2801416); or
be
converted, following etherification of the free hydroxyl groups with alkyl
halogenides in
the presence of potassium tert. butanolate, first into Sa,1713 diethers (See,
Kasch et al.
DD 290 893), which are then transformed into their respective aldehydes by
acid
hydrolysis, preferably using toluene-p-sulphonic acid in acetone. The
aldehydes thus-
obtained are converted into compounds of the general Formula II by reacting
them with
hydroxylamine.
The resulting compound of the general Formula I according to aspects of the
invention may be converted, if required, into an acid-addition salt,
preferably a salt of
a physiologically-compatible acid. Common physiologically-compatible inorganic
acids
and organic acids are, for example, hydrochloric acid, hydrobromic acid,
phosphoric
CA 02130515 1998-02-19
12
acid, sulphuric acid, oxalic acid, malefic acid, fumaric acid, lactic acid,
tartaric acid,
malic acid, citric acid, salicylic acid, adipic acid, and benzoic acid. Other
acids that can
be used are described, for example, in Fortschritte der Arzneimittelforschung,
vol. 10,
pp. 224-225, Birkhiiuser Verlag, Basel and Stuttgart, 1966, and Journal of
Pharmaceutical Sciences, vol. 66, pp. 1-5 (1977).
Acid-addition salts are normally obtained in a generally-known way by mixing
the
free base or its solutions with the respective acid or its solutions in an
organic solvent,
for example, a lower alcohol, e.g., methanol, ethanol, n-propanol or
isopropanol, or a
lower ketone, e.g., acetone, methylethyl ketone or methyl isobutyl ketone, or
an ether,
e.g., diethyl ether, tetrahydrofurane or dioxane. Compositions of the above
mentioned
solvents may be used for improved crystallizing. In addition, physiologically-
compatible
hydrous solutions of acid-addition salts of the compound according to Formula
I
according to aspects of this invention may be produced in a hydrous acidic
solution.
The acid-addition salts of compounds of the general Formula I according to
aspects of this invention can be converted into a free base in a generally-
known way,
e.g., using alkalis or ion exchangers. Other salts can be obtained by reacting
this free
base with inorganic acids or organic acids, especially acids which are suited
for forming
pharmaceutically-acceptable salts. These and other salts of the new compounds
according
to aspects of this invention, e.g. its picrate, may be used to purify the free
base: the free
base is converted into a salt, the salt is separated, and the base is released
from the salt
again.
The present invention in yet another aspect, provides a pharmaceutical
composition, comprising a pharmaceutically-effective concentration of a
compound as
described hereinabove, or salts or mixtures thereof, in a pharmaceutically-
acceptable
carrier.
CA 02130515 1998-02-19
13
By specific variants thereof, the compound of Formula I may be one of the
following:
11B-[4-(acetoximinomethyl)phenyl]-17B-methoxy-17a-methoxymethyl-estra-4,9-dime-
3-
on; or 11B-{4-[(ethoxycarbonyl)oximinomethyl]phenyl}-17B-methoxy-17a-
methoxymethyl-estra-4,9-diene-3-on; or 11B-{4-[(ethylaminocarbonyl)oxi-
minomethyl]phenyl}-17B-methoxy-17a-methoxymethyl-estra-4,9-dime-3-on; or
17B-methoxy-17a-methoxymethyl-11B-{4-
[(phenylaminocarbonyl)oximinomethyl]phenyl}-
estra-4,9-diene-3-on; or 11B[4-(propionyloximinomethyl)phenyl)-17B-methoxy-17a-
methoxymethyl-estra-4,9-diene-3-on; or 1113-[4-(methyloximinomethyl)phenyl]-
17B-
hydroxy-17a-methoxymethyl-estra-4,9-dime-3-on; or 11B-[4-(benzoyloximino-
methyl)phenyl] -17B-hydroxy-17a-methoxymethyl-estra-4, 9-dime-3-on.
These pharmaceutical compositions of aspects of this invention are designed
for
oral, rectal, subcutaneous, intravenous or intramuscular applications, and
contain, as an
active ingredient, apart from the usual substrates and diluents, a compound
according to
the general Formula I or its acid addition salt, according to aspects of this
invention.
The pharmaceutical compositions of aspects of the invention are produced in a
known way using the usual solid or liquid substrates or diluents, and the
common
adjuvants which are used in pharmaceutical engineering, and with an
appropriate dosage
depending on the intended mode of application. Preferred formulations are
those forms
which are suitable for oral administration, for example, tablets, film
tablets, dragees,
capsules, pills, powder, solutions, suspensions, or depot forms.
Consideration may be given also to parenteral formulations, e.g., injection
solutions. Suppositories represent another form of application.
CA 02130515 1998-02-19
14
Tablets may be obtained, for example, by intermixing the active substance with
known adjuvants, for example: inert diluents, e.g., dextrose, sugar, sorbitol,
mannite,
polyvinylpyrrolidone; blasting agents, e. g. , maize starch or alginic acid;
binders, e. g. ,
starch or gelatin; lubricants, e. g. , magnesium stearate or talcum; and/or
materials by
which to produce a depot effect, e.g., carboxyl polymethylene, carboxymethyl
cellulose,
cellulose acetate phthalate or polyvinyl acetate. Tablets may consist of
several layers.
Dragees may be produced by coating cores which are manufactured in analogy
to tablet manufacture using agents which are generally applied to dragee
coating, for
example, polyvinylpyrrolidone or shellac, gum Arabic, talcum, titanium
dioxide, or
sugar. The coating of the dragee may also consist of several layers in which
the
adjuvants mentioned in the paragraph on tablets can be used.
Solutions or suspensions containing the active agent of aspects of the
invention
may additionally contain flavour-enhancing substances, e.g., saccharin,
cyclamate or
sugar, or aromatic substances, e. g. , vanillin or orange extract. They may
also contain
suspension-supporting adjuvants, e.g., sodium carboxymethyl cellulose, or
preservatives,
e.g., p-hydroxybenzoates. Capsules containing active substances may be
produced, for
example, by mixing the active substance of aspects of the invention with an
inert
substrate, e.g., lactose or sorbitol, and encapsulating such mixture in
gelatin capsules.
Appropriate suppositories may be made by mixing the active substance with
suitable substrates, e.g., neutral fats or polyethylene glycol and their
derivatives.
CA 02130515 1998-02-19
By yet another aspect of this invention, the use is provided for a compound of
Formula I, or its acid-addition salt, as an antigestagenic agent with reduced
glucocorticoid activity.
By still a further aspect of this invention the use is provided of Formula I
as
S described above, or its acid addition salt for the preparation of a
pharmaceutical
composition for use as an antigestagenic agent with reduced glucocorticoid
activity.
(e) AT LEAST ONE MODE FOR CARRYING OUT THE INVENTION
The following examples explain the invention.
Examples
180 mg of 11B-[4-(hydroximinomethyl)phenyl]-1713-methoxy-17a-methoxymethyl-
estra-
4,9-diene-3-on are acetylated in 5 ml of acetic anhydride/pyridine (l:l).
After adding
water, the batch is extracted three times with acetic ester. The organic phase
is washed
with dilute hydrochloric acid and water, dried above sodium sulphate, and
concentrated
by evaporation under reduced pressure. The yield is 172 mg of crude product
that is
purified by preparative thin-layer chromatography using silica gel PFZas+3~s
and a
toluene/acetone solvent system at a concentration of 4:1.
Yield: 115 mg of 1113-[4-(acetyloximinomethyl)phenyl]-1713-methoxy-17a-
methoxymethyl-estra-4,9-dime-3-on. The product crystallizes from acetic ester.
Melting point: 1115 - 120°C (acetic ester)
aD = -E 218° (CHC 13)
IR spectrum in KBr (cm'): 1754 (OAc);
1654 (C=C-C=C-C=0); 1602 (phenyl)
CA 02130515 1998-02-19
16
UV spectrum in MeOH: A",a,~= 271 nm E = 28 157
A",a,~= 297 nm E = 26 369
'H-NMR spectrum in CDC13 [b, ppm]: 0.511 (s, 3H, H-18); 2.227 (s, 3H,OCOCH );
3.408 (s,3H, 17a-CH20CH ); 3.386, 3.431, 3.544, 3.580 (m, 2H, CH OCH3); 4.399
(d,
1H, J=7.2 Hz, H-lla); 5.785 (s, 1H, H-4); 7.242, 7.266, 7.618, 7.647 (m, 4H,
AA'BB' system of aromatics protons); 8.315 (s, 1H, CH-NOAc)
MS m/e: 446 C~H32N04 M+ - CHZOCH3 +
2130515
17
Example 2
0.3 ml of chloroethyl formate are dripped into 210 mg of
113- [4- (hydroximinomethyl)phenyl] -17(3-methoxy-17a-
methoxymethyl-estra-4,9-dime-3-on in 5 ml of pyridine
while cooling with water. A white sediment forms. The
batch is watered after 30 minutes, which results in a
solution in which a white sediment settles down that is
filtered off by suction and washed with water. Yield
after drying: 133 mg. The aqueous phase is extracted with
chloroform, washed with dilute hydrochloric acid and
water, dried, and concentrated by evaporation under
reduced pressure. Yield: 66 mg. Both solids are united
and purified by preparative thin-layer chromatography
using silica gel PF245+366 and a toluene/acetone solvent
system at a concentration of 4:1.
Yield: 150 mg of ll~i-[4-(ethoxycarbonyloximinomethyl)-
phenyl]-17~i-methoxy-17a-methoxymethyl-estra-4,9-dime-3-
on which are recrystallized from acetone/hexane.
Melting point: 137 - 148°C
aD = + 204° (CHC13)
W spectrum in MeOH: 7~,max = 270 nm s = 27 094
Amax = 297 nm s = 25 604
1H-NMR spectrum in CDC13 [8, ppm]: 0.507 (s, 3H, H-18);
1.383 (t, 3H, J=7.0 Hz, OCH2C$3) ; 3.246 (s, 3H, 17(3-
OC$3); 3.410 (s, 3H, 17a-CH20C$3); 3.39 - 3.56 (m, 2H,
C$20CH3); 4.35 (d, 1H, J=7.0 Hz, H-lla); 5.784 (s, 1H,
H-4); 7.23, 7.26, 7.61, 7.64 (m, 4H, AA'BB' system of
aromatics protons); 8.303 (s, 1H, C~=NR)
MS m/e: 431.24701 C2gH32N03 M+ - C2H50COOH
213OSiS
18
Example 3
190 mg of 11(3- [4- (hydroximinomethyl) phenyl] -17(3-methoxy-
17a-methoxymethyl-estra-4,9-dime-3-on are suspended in
ml of toluene. 0.5 ml of phenyl isocyanate and 1 ml of
5 triethyl amine are added subsequently. The batch is
agitated at room temperature for 3 hours and refluxed for
2 hours. The white sediment is filtered off by suction,
and the solvent concentrated by evaporation under
reduced pressure. Thus 310 mg of a light brown solid are
10 obtained which is purified by preparative thin-layer
chromatography using silica gel PF245+366 and a toluene/
acetone solvent system at a concentration of 9:1.
65 mg of 17~i-methoxy-17a-methoxymethyl-11(3-{4-[(phenyl-
amino-carbonyl)oximinomethyl)]phenyl}-estra-4,9-
dime-3-one are isolated.
Melting point: 241 - 246°C (acetone)
aD = + 178° (CHC13)
W spectrum in MeOH: 7~.max = 238 nm s = 29 444
Amax = 300 nm s = 29 649
1H-NMR spectrum in CDC13 [8, ppm]: 0.474 (s, 3H, H-18);
3.245 (s, 3H, 17(3-OCH3); 3.405 (s, 3H, 17a-CH20C$3);
3.406 - 3.545 (m, 2H, ABX system, 17a-C~20CH3); 4.413 (d,
1H, J=6.8 Hz, H-lla); 5.797 (s, 1H, H-4); 7.264 (m, 5H,
aromatic), 7.272, 7.293, 7.548, 7.575 (m, 4H, AA'BB'
system of aromatics protons); 8.0 (s, 1H, C$=N-)
MS m/e: 431.24249 C2gH33N03 M+ - C6H5CN0 + H20
Example 4
708 mg of 11(3- [4- (hydroximinomethyl)phenyl] -17(3-methoxy-
17a-methoxymethyl-estra-4,9-dime-3-on are dissolved in
15 ml of toluene. 1.5 ml of ethyl isocyanate and 3 ml of
triethyl amine are added subsequently. The batch is
2130515
.~,., 19
agitated at room temperature for 6 hours and allowed to
stand overnight. Then 20 ml of aqueous ammonium solution
are added, the phases are separated, extracted with
toluene, washed in water, aqueous ammonium solution, and
water, dried above sodium sulfate and concentrated by
evaporation under reduced pressure. Thus 800 mg of a
bright yellow solid are obtained which is purified by
preparative thin-layer chromatography using silica gel 60
PF254+366 and a toluene/acetone solvent system at a
concentration of 9:1.
610 mg of 11(3-{4-[(ethylaminocarbonyl)oximinomethyl)]-
phenyl}-17(3-methoxy-17a-methoxymethyl-estra-4,9-dime-3-
on are isolated.
Melting point: 142 - 147°C photodecomposition
(ether/acetone/hexane)
1H-NMR spectrum in CDC13 [~, ppm]: 0.522 (s, 3H, H-18);
1.241 (t, 3H, J=7.5 Hz, NHCH2C~-j3) ; 3.253 (s, 3H, 17~3-
OCH3); 3.415 (s, 3H, 17a-CH20C~3); 3.366 - 3.574 (m, 4H,
ABX system, 17a-CH20CH3, NHCj~2CH3); 4.410 (d, 1H, J=7.2
Hz, H-lla); 5.790 (s, 1H, H-4); 6.238 (m, 1H, NHCO),
7.258, 7.286, 7.561, 7.589 (m, 4H, AA'BB' system of
aromatics protons); 8.294 (s, 1H, C~j=N-)
Example 5
500 mg of 11(3- [4- (hydroximinomethyl) phenyl] -17(3-methoxy-
17a-methoxymethyl-estra-4,9-dime-3-on are agitated
subject to inert gas for 2.5 hours in 4 ml of propionic
acid anhydride/ pyridine 1:1 (v: v). The mixture is poured
into iced water, and the sticky substance is extracted
with chloroform. The organic phase is washed with dilute
hydrochloric acid and water, dried above sodium sulfate,
and concentrated by evaporation under reduced pressure.
2130515
The bright yellow foam is purified using chromatography
and recrystallized from acetic ester. Yield: 306 mg of 11
(3- [4- (propionyloximinomethyl) phenyl] -17(3-methoxy-17a-
methoxymethyl-estra-4,9-dime-3-on.
5
Melting point: 110 - 114°C (acetic ester)
1H-NMR spectrum in CDC13 [8, ppm]: 0.515 (s, 3H, H-18);
1.241 (t, 3H, J=7.6 Hz, OCOCH2Cj33) ; 3.253 (s, 3H, 17(3-
OC$3); 3.415 (s, 3H, 17a-CH20C~3); 3.4 - 3.6 (m, 2H, ABX
10 system, 17a-C$20CH3); 4.128 (d, 1H, J=7.2 Hz, H-lla);
5.790 (s, 1H, H-4); 7.244, 7.271, 7.627, 7.655 (m, 4H,
AA'BB' system of aromatics protons); 8.322 (s, 1H, C~=N-)
Example 6
15 An etherial diazomethane solution is added while cooling
with ice to 170 mg of 113- [4- (hydroximinomethyl) phenyl] -
17(3-methoxy-17a-methoxymethyl-estra-4,9-diene-3-on until
the mixture takes on a slight yellow colouring. The batch
is agitated for 2 hours at 5°C, and dilute sodium
20 hydroxide solution is added. Then the mixture is
extracted with ether, washed neutrally, and dried above
sodium sulfate. The organic phase is evaporated under
reduced pressure. The yellow resin is purified by
preparative thin-layer chromatography using silica gel 60
PF254+366 and a toluene/acetone solvent system at a
concentration of 4:1.
Yield: 110 mg of 11(3- [4- (methoximinomethyl)phenyl] -17(3-
hydroxy-17a-methoxymethyl-estra-4,9-dime-3-on in the
form of colourless lamellae.
Melting point: 83 - 89°C
aD = + 197° (CHC13)
21 2130515
IR spectrum in CHC13 (cm-1): 1700 (C=NOCH3);
1649 (C=C-C=C-C=0); 1590 (aromatic)
W spectrum in MeOH: 7~,max = 275 nm s = 23 098
Amax = 300 nm s = 22 872
1H-NMR spectrum in CDC13 [8, ppm]: 0.529 (s, 3H, H-18);
3.247 (s, 3H, 17~i-OCH3); 3.408 (s, 3H, 17a-CH20C~3); 3.39
- 3.598 (m, 2H, ABX system, 17a-C$20CHg); 4.381 (d, 1H,
J=7.5 Hz, H-lia); 5.773 (s, 1H, H-4); 7.173, 7.201,
7.463, 7.491 (m, 4H, AA'BB' system of aromatics protons);
8.023 (s, 1H, C$phenyl)
Example 7
A filtered mixture of 2 ml of benzoyl chloride and 3 ml
of pyridine is added to 500 mg of 113-[4-(hydroximino-
methyl)phenyl]-17(3-methoxy-17a-methoxymethyl-estra-4,9-
diene-3-on. After 2 hours, the mixture is stirred into
150 ml of iced water. The steroid precipitates as a
sticky substance. 5 ml of hydrochloric acid are added,
and the mixture is taken up in acetic ester. The phases
are separated, the organic phase is washed in aqueous
bicarbonate solution and water, dried above sodium
sulfate, filtered off, and evaporated under reduced
pressure. The yellow oil (1.57 g) is liberated from non-
polar products using chromatography on 40 g of silica gel
60 and a toluene/acetic ester gradient. The main fraction
(0.7 g) is purified by preparative thin-layer chromato-
graphy using silica gel 60 PF254+366 and a chloroform/
acetone solvent system. 410 mg of a colourless foam is
obtained that is recrystallized from methanol.
Yield: 263 mg of 11(3- [4- (benzoyloximinomethyl)phenyl] -17(3
-methoxy-17a-methoxymethyl-estra-4,9-dime-3-on as
colourless foam.
Melting point: 115 - 122°C (methanol)
.~. 2130515
22
aD = + 216 ° ( CHC13 )
UV spectrum in MeOH: 7~.max = 279 nm s = 33 720
Amax = 299 nm s = 30 120
1H-NMR spectrum in CDC13 [b, ppm]: 0.527 (s, 3H, H-18);
3.253 (s, 3H, 17~i-OC$3); 3.415 (s, 3H, 17a-CH20C$3);
3.403 - 3.598 (m, 2H, ABX system, 17a-C$20CH3); 4.416 (d,
1H, J=7.2 Hz, H-lla); 5.792 (s, 1H, H-4); 7.299 - 7.615
(m, 5H, aromatics protons, COphenyl); 7.701, 7.729,
8.111, 8.140 (m, 4H, AA'BB' system of aromatics protons);
8.520 (s, 1H, CHphenyl)
Example 8
Measurement of bonding affinity for receptors
Receptor bonding affinity was determined by competitive
bonding of a specifically binding 3H labelled hormone
(tracer) and the compound to be tested to receptors in
the cytosol from animal target organs. It was tried to
obtain receptor saturation and a balanced reaction. The
following incubation conditions were selected:
Glucocorticoid receptor: thymus cytosol of the adrenalec-
tomized rat, thymi kept at -30°C, buffer: TED. Tracer:
3H-dexamethasone, 20 nM; reference substance:
dexamethasone.
After an incubation period of 18 hours at 0 - 4°C, bonded
and free steroid was separated by mixing in active
carbon/ dextrane (1%/0.1%), centrifuging off and
measuring the bonded 3H activity in the supernatant.
The IC5o for the compound to be tested and for the
reference substance were determined from measurements in
CA 02130515 1998-02-19
23
series of concentrations. The quotient (x 100%) is the relative motor bonding
affinity.
Example 9
Inhibition of early gravidity in the rat:
Female rats are mated in the pro-oestrus. If semen is found in the vaginal
smear on the
next day, this day is counted as day 1 (=dl) of the gravidity. Treatment with
the test
substance or vehicle is applied on d5 - d7, autopsy is carried out on d9. The
substances
are injected subcutaneously in 0.2 ml of vehicle (benzylbenzoate/castor oil 1
+ 4). The
rate of fully inhibited gravidities found in various groups can be seen from
Table 1. A
superior inhibition capability of nidation was found for J 917 and J 900 as
compared to
RU 486.
As is evident from the above examples, the 11B-substituted benzaldoxime-estra-
4,9-dienes of aspects of the invention are antigestagenic substances that
combine, if
compared with RU 486, superior in vivo acting potential (cf. Table 2) with a
significantly
reduced antigluco-corticoid activity, which has been proved by the reduced
bonding to
glucocorticoid receptors 9 (cf. Table 1).
Table 1
Receptor bonding of selected substances listed in
Examples 1 and 2
Compound Relative molar bonding affinity
acc. to (RBA) [ % ] for the glucocorticoid
Example receptor (dexamethasone =100 % )
1 (J914) 73
2 (J900) 66
compared with
RU 486
(mifepristone) 685
2K 98299
(onapristone) 39
CA 02130515 1998-02-19
24
This combination of properties of the antigestagens according to aspects of
the
invention promises superior inhibition of progesterone while at the same time
reducing
antiglucocorticoid activity. This advantage is of particular relevance for
indications that
require excellent compatibility because of the duration of treatment. During
the
menstrual cycle, uterine weight is decisively influenced by the circulating
oestrogen.
Reduced uterine weights reflect an inhibition of this oestrogenic function.
The inhibition
of uterine weight during the menstrual cycle determined in guinea pigs is
superior to RU
486 and points to (indirect) anti-oestrogenic properties of the compounds
according to
aspects of the invention. The respective effects promise the exertion of a
particularly
favourable influence on pathologically-modified tissues in which oestrogens
stimulate
growth (endometriotic focuses, myomas, mammary and genital carcinomas, benign
prostatic hypertrophy).
Table 2
Early abortive effect of RU 486 and J 914 (Example 1) and J 900 (Example 2)
in the rat after subcutaneous application from the 5th to 7th day of pregnancy
(dose 0.2
ml/animal/day in benzylbenzoate/castor oil [1 + 4 v/v])
Group, Dose complete gravid- ED 50++
substance (mg/animal/ ity inhibition+ (mg/animal
day) N* /N % day)
vehicle - 0125 0 -
RU 486 3.0 5/5 100
1.0 2/5 40 1.1
0.3 OIS 0
J 900 1.0 9/ 10 90 0. 6
0.3 0/5 0
CA 02130515 1998-02-19
J 914 3.0 5/5 100
1.0 7/10 70 0.6
0. 3 1/5 20
0.1 0/6 0
5
+ empty uteri
N number of inseminated females
N* number of females not pregnant
+ + graphic determination